ES2816204T3 - Antagonistas del receptor p2x7 derivados de N-[2-(4-fenoxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida y N-[2-(4-benciloxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida sustituidas - Google Patents
Antagonistas del receptor p2x7 derivados de N-[2-(4-fenoxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida y N-[2-(4-benciloxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida sustituidas Download PDFInfo
- Publication number
- ES2816204T3 ES2816204T3 ES17755110T ES17755110T ES2816204T3 ES 2816204 T3 ES2816204 T3 ES 2816204T3 ES 17755110 T ES17755110 T ES 17755110T ES 17755110 T ES17755110 T ES 17755110T ES 2816204 T3 ES2816204 T3 ES 2816204T3
- Authority
- ES
- Spain
- Prior art keywords
- thiazol
- ethyl
- piperidin
- methyl
- quinoline
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Ophthalmology & Optometry (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Epidemiology (AREA)
- Psychiatry (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de la siguiente fórmula (I) o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** incluyendo cualquier forma estereoquímicamente isomérica del mismo, en la que: n es 0 o 1; R1 es alquilo C1-C4 (opcionalmente sustituido con hidroxilo o halógeno), preferiblemente metilo, fluorometilo, difluorometilo, trifluorometilo; cada uno de R2, R3 y R4 es independientemente hidrógeno, halógeno o los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros que contiene un átomo de nitrógeno, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno; cada uno de R5 y R6 es hidrógeno o halógeno, a condición de que al menos uno de R5 y R6 sea halógeno.
Description
DESCRIPCIÓN
Antagonistas del receptor p2x7 derivados de N-[2-(4-fenoxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida y N-[2-(4-benciloxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida sustituidas
La presente invención está relacionada con nuevos compuestos de fenoxi y benciloxipiperidina sustituidos de fórmula (I) que tienen propiedades antagonistas del receptor P2X7 (P2X7), composiciones farmacéuticas que comprenden estos compuestos, procesos químicos para preparar estos compuestos y su uso en el tratamiento o profilaxis de enfermedades asociadas con la actividad del receptor P2X7 en animales, en particular humanos.
El P2X7 pertenece a la familia de los receptores ionotrópicos P2X. El P2X7 es activado por nucleótidos extracelulares, especialmente adenosina trifosfato (ATP). El P2X7 se distingue de otros miembros de la familia P2X por la localización específica (SNC y células inmunocompetentes en particular), por las altas concentraciones de ATP (en el rango mM) requeridas para activarlo y por su capacidad de formar un poro grande durante estimulación prolongada o repetida. El P2X7 es un canal iónico dependiente de ligando y está presente en una variedad de tipos de células, en gran parte de las que se sabe que están involucradas en el proceso inflamatorio y/o inmune, específicamente, macrófagos, mastocitos y linfocitos (T y B). La activación del receptor P2X7 por nucleótidos extracelulares, por ejemplo, ATP, conduce a la liberación de interleucina-1p (1L-1p) y la formación de células gigantes (macrófagos/células microgliales), desgranulación (mastocitos) y desprendimiento de L-selectina (linfocitos). Los receptores P2X7 también se encuentran en células presentadoras de antígenos (APC), queratinocitos, células acinares salivales (células parótidas), hepatocitos, eritrocitos, células eritroleucémicas, monocitos, fibroblastos, células de médula ósea, neuronas y células mesangiales renales. También se sabe que el receptor P2X7 es un sensor de dolor en el sistema nervioso. Experimentos con ratones deficientes en P2X7 demuestran el papel de P2X7 en el desarrollo del dolor ya que estos ratones estaban protegidos del desarrollo del dolor inflamatorio inducido por adyuvante y el dolor neuropático inducido por la ligadura nerviosa parcial. También hay cada vez más pruebas de que P2X7 o sus efectores posteriores, como IL-1p, están involucrados en la fisiopatología de varios trastornos neurológicos, como la enfermedad de Alzheimer (J.I. Diaz-Hernandez et al., Neurobiol. Aging 2012, 1816-1828: La inhibición in vivo de P2X7 reduce las placas Ap en AD a través de GSK3p). Se cree que P2X7 tiene una función importante en la neurotransmisión dentro del SNC a través de su activación en neuronas y glándulas postsinápticas y/o presinápticas. Han surgido datos utilizando la hibridación in situ de que el ARNm del receptor P2X7 está ampliamente distribuido en todo el cerebro de la rata. Específicamente, se encontraron áreas de alta expresión de ARNm de P2X7 en el núcleo olfativo anterior, la corteza cerebral, la corteza piriforme (Pir), el núcleo septal lateral (LS), las capas de células piramidales del hipocampo de CA1, CA3, CA4, núcleos pontinos, núcleo cuneado externo, y núcleo vestibular medio. Las señales de hibridación P2X7 también se observaron en las neuronas motoras del núcleo motor trigémino, el núcleo facial, el núcleo hipogloso y el asta anterior de la médula espinal.
Por lo tanto, existe una justificación terapéutica para el uso de antagonistas de P2X7 en el tratamiento de una variedad de estados de enfermedad. Estos estados incluyen, entre otros, enfermedades asociadas con el SNC, como la enfermedad de Alzheimer, enfermedad de Parkinson, enfermedad de Huntington, esclerosis lateral amiotrófica, lesión de la médula espinal, isquemia cerebral, traumatismo craneoencefálico, meningitis, trastornos del sueño, trastornos del estado de ánimo y ansiedad, neuroinflamación inducida por VIH y dolor neuropático e inflamatorio crónico. Además, trastornos inflamatorios periféricos y las enfermedades autoinmunes, que incluyen, entre otros, artritis reumatoide, osteoartritis, psoriasis, dermatitis alérgica, asma, enfermedad pulmonar obstructiva crónica, hiperreactividad de las vías respiratorias, choque séptico, bronquitis, glomerulonefritis, síndrome del intestino irritable, enfermedad del hígado graso, fibrosis hepática, lesiones cutáneas, enfisema pulmonar, distrofia muscular, fibrosis, aterosclerosis, lesiones por quemaduras, enfermedad de Crohn, colitis ulcerosa, degeneración macular relacionada con la edad, crecimiento y metástasis de células malignas, síndrome de Sjogren, leucemia mioblástica, diabetes, osteoporosis, enfermedad de corazón isquémico, son todas ejemplos en los que se ha implicado el involucramiento de los receptores P2X7. En vista de la importancia clínica del P2X7, la identificación de compuestos que modulan la función del receptor P2X7 representa una vía atractiva hacia el desarrollo de nuevos agentes terapéuticos.
Los inhibidores de P2X7 se describen en diversas solicitudes de patentes, tales como:
WO2004/099146 que divulga inhibidores de benzamida del receptor P2X7 y su uso en el tratamiento de enfermedades inflamatorias.
El documento WO2009/108551 que divulga análogos de heteroarilamida y su uso en condiciones mediadas por el receptor P2X7.
El documento WO2009/132000 que divulga antagonistas del receptor P2X7 sustituido con quinolina e isoquinolina y su uso en condiciones mediadas por el receptor P2X7.
El documento WO2015/119018 que divulga derivados de tiazol y oxazol como antagonistas del receptor P2X7 y su uso en condiciones mediadas por el receptor P2X7.
Sin embargo, todavía existe una necesidad insatisfecha de compuestos que puedan antagonizar eficazmente P2X7 y que puedan administrarse en los diferentes órganos diana que sean sitios de una patología mediada por P2X7, incluido el cerebro. Dichos compuestos se proporcionan en este documento.
A continuación, se presentan diversas realizaciones de la invención;
La presente invención se refiere a compuestos de tiazol de la siguiente fórmula (I) o una sal farmacéuticamente aceptable de los mismos:
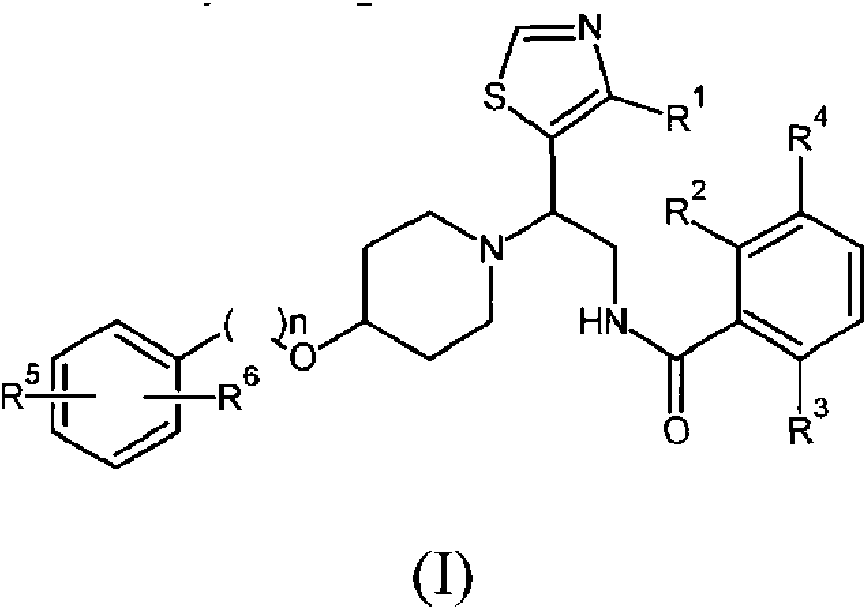
incluyendo cualquier forma estereoquímicamente isomérica de los mismos, en la que
n es 0 o 1;
R1 es alquilo C1-C4 (opcionalmente sustituido con hidroxilo o halógeno), preferiblemente metilo, fluorometilo, difluorometilo, trifluorometilo;
cada uno de R2, R3 y R4 es independientemente hidrógeno, halógeno o los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros que contiene un átomo de nitrógeno, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno o halógeno a condición de que al menos uno de R5 y R6 sea halógeno;
Como se usan en las definiciones anteriores:
Los términos "halo", "halógeno" y "haluro", que pueden usarse indistintamente, se refieren a un sustituyente fluoro, cloro, bromo o yodo.
El término "formas estereoquímicamente isoméricas" como se usa aquí anteriormente define todas las formas isoméricas posibles que pueden poseer los compuestos de fórmula (I). A menos que se mencione o se indique lo contrario, la designación química de los compuestos denota la mezcla de todas las formas estereoquímicamente isoméricas posibles, conteniendo dichas mezclas todos los diastereómeros y enantiómeros de la estructura molecular básica. Más en particular, los centros estereogénicos pueden tener la configuración R o S; los sustituyentes en radicales cíclicos (parcialmente) saturados bivalentes pueden tener la configuración cis o trans. Las formas estereoquímicamente isoméricas de los compuestos de fórmula (I) obviamente están comprendidas dentro del alcance de esta invención.
La configuración estereoquímica absoluta de los compuestos de fórmula (I) y de los intermedios utilizados en su preparación puede ser determinada fácilmente por los expertos en la técnica utilizando procedimientos bien conocidos tales como, por ejemplo, difracción de rayos X.
Además, algunos compuestos de fórmula (I) y algunos de los intermedios utilizados en su preparación pueden exhibir polimorfismo. Debe entenderse que la presente invención abarca cualquier forma polimórfica que posea propiedades útiles en el tratamiento de las afecciones indicadas anteriormente.
Las sales farmacéuticamente aceptables como se mencionó anteriormente en el presente documento pretenden comprender las formas de sal de adición ácida no tóxicas terapéuticamente activas que los compuestos de fórmula (I) pueden formar. Estas sales de adición ácida farmacéuticamente aceptables pueden obtenerse convenientemente tratando la forma de base con dicho ácido apropiado. Los ácidos apropiados comprenden, por ejemplo, ácidos inorgánicos tales como ácidos hidrohálicos, por ejemplo, ácido clorhídrico o bromhídrico, ácidos sulfúrico, nítrico, fosfórico y similares; o ácidos orgánicos como, por ejemplo, acético, propanoico, hidroxiacético, láctico, pirúvico, oxálico (es decir, etanodioico), malónico, succínico (es decir, ácido butanodioico), maleico, fumárico, málico,
tartárico, cítrico, metanosulfónico, trifluorometanosulfónico, etanosulfónico, bencenosulfónico, p-toluenosulfónico, adámico, salicílico, p-aminosalicílico, pamoico y ácidos similares.
Por el contrario, dichas formas de sal se pueden convertir mediante tratamiento con una base apropiada en la forma de base libre.
Los compuestos de fórmula (I) pueden existir tanto en formas no solvatadas como solvatadas. El término 'solvato' se usa en el presente documento para describir una asociación molecular que comprende un compuesto de la invención y una o más moléculas de solvente farmacéuticamente aceptables, por ejemplo, agua o etanol. El término "hidrato" se usa cuando dicho disolvente es agua.
Una realización preferida de la invención se refiere a compuestos de Fórmula (I) como se definió anteriormente en los que:
n es 0 o 1;
R1 es metilo o difluorometilo;
cada uno de R2, R3 y R4 independientemente es hidrógeno, flúor, cloro o los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros que contiene un átomo de nitrógeno, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno, fluoro o cloro a condición de que al menos uno de R5 y R6 sea halógeno; Otra realización de la invención se refiere a compuestos de Fórmula (I) como se definió anteriormente en los que: n es 0 o 1;
R1 es metilo o difluorometilo;
cada uno de R2, R3 y R4 es independientemente hidrógeno, fluoro o cloro, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno, fluoro o cloro a condición de que al menos uno de R5 y R6 sea halógeno; Otra realización de la invención se refiere a compuestos de Fórmula (I) como se definió anteriormente en los que: n es 0 o 1;
R1 es metilo o difluorometilo;
R3 es hidrógeno y los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros, en el que el anillo heterocíclico de seis miembros junto al grupo fenilo forman un anillo de quinolina.
Más preferiblemente, un compuesto de fórmula (I) de acuerdo con esta invención se selecciona del grupo que consiste en:

continuación

Los compuestos de fórmula (I) generalmente se pueden preparar haciendo reaccionar un compuesto de fórmula (II):

en la que los significados de n, R1, R5 y R6 son como se definieron anteriormente, con un compuesto de fórmula (III)
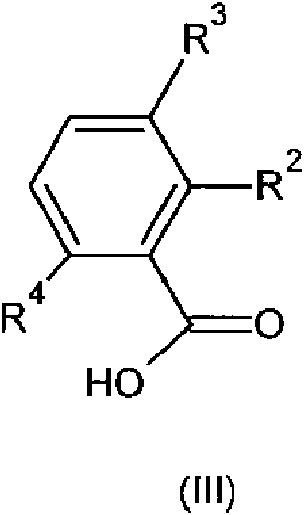
en la que los significados de R2, R3 y R4 son como se definieron anteriormente; o
con un compuesto de fórmula (IIIa):

en la que los significados de R2, R3 y R4 son como se definieron anteriormente; y W es un grupo saliente adecuado; y opcionalmente convertir el compuesto obtenido de fórmula (I) en una sal de adición del mismo, y/o preparar formas estereoquímicamente isoméricas del mismo.
La reacción de un compuesto de fórmula (II) con un compuesto de fórmula (III), se puede llevar a cabo en al menos un disolvente inerte a la reacción y opcionalmente en presencia de al menos un reactivo de acoplamiento adecuado y/o una base adecuada del mismo. Puede ser conveniente activar el ácido carboxílico de fórmula (III) agregando una cantidad efectiva de un promotor de reacción. Ejemplos no limitantes de tales promotores de reacción incluyen carbonildiimidazol, N,N'-diciclohexil-carbodiimida o 1-(3-dimetilaminopropil)-3-etilcarbodiimida, hidroxibenzotriazol, benzotriazolil-oxitris(dimetilamino)-fosfoniohexafluorofosfato, tetrapirrolidinofosfoniohexa-fluorofosfato, bromotripirrolidinofosfonio hexafluorofosfato, o un derivado funcional del mismo, tal como se describe por D. Hudson, (J. Org. Chem. (1988), 53, 617).
W en el compuesto de Fórmula (IIIa) es un grupo saliente apropiado tal como, por ejemplo, halo, por ejemplo, fluoro, cloro, bromo, yodo o, en algunos casos, W también puede ser un grupo sulfoniloxi, por ejemplo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi y grupos salientes reactivos similares. La reacción de un compuesto de fórmula (II) con un compuesto de fórmula (III), se puede realizar en un disolvente inerte a la reacción tal como, por ejemplo, acetonitrilo, dimetil acetamida, N-metil-pirrolidona o DMF, y opcionalmente en la presencia de una base
adecuada como, por ejemplo, carbonato de sodio, carbonato de potasio o trietilamina. La agitación puede mejorar la tasa de la reacción. La reacción puede llevarse a cabo convenientemente a una temperatura que oscila entre la temperatura ambiente y la temperatura de reflujo de la mezcla de reacción.
Los compuestos de fórmula (III) y (IIIa) son conocidos en la técnica o pueden prepararse siguiendo los procesos informados en los ejemplos.
Los compuestos de fórmula (II) se pueden preparar de acuerdo con el siguiente esquema:

Las aminas primarias (II) se pueden obtener por reducción de los derivados de nitrilo respectivos (IV) en una reacción de formación de enlaces nitrógeno-hidrógeno. Los ejemplos no limitantes de dicha reacción incluyen la reducción con:
- hidrógeno o una fuente de hidrógeno en presencia de un metal como níquel, platino, paladio y cobalto o un derivado del mismo como Ni-Raney, óxido de platino, óxido de paladio o cobalto Raney como catalizador;
- un hidruro tal como hidruro de litio y aluminio, hidruro de diisobutilaluminio (DIBAL), hidruro de boro o un derivado funcional del mismo.
La reacción puede realizarse en un disolvente adecuado, tal como metanol, tetrahidrofurano, ácido acético, éter dietílico, tolueno o solución de amoníaco metanólico, preferiblemente a temperaturas entre -78 °C y temperatura ambiente.
Los compuestos de fórmula (IV), en donde R1, R5 y R6 son como se definen en la fórmula (I), pueden prepararse a partir de aldehidos (VI) mediante una reacción de condensación de Strecker con el respectivo intermedio heterociclilo (VII) en presencia de una fuente de cianuro (V), por ejemplo, TMSCN o un derivado funcional del mismo, en un disolvente tal como AcOH o MeCN, preferiblemente a temperaturas entre 0 °C y TA.

Como alternativa, los compuestos de fórmula (II) también se pueden preparar mediante un procedimiento de dos pasos como se informó anteriormente. Reacción de compuestos de fórmula (IV) con un reactivo reductor, preferiblemente borohidruro de sodio en presencia de hexahidrato de cloruro de níquel (II) o hexahidrato de cloruro de cobalto (II) y Boc2O en un solvente como MeOH, preferiblemente a temperaturas entre 0°C y RT, produce la amina primaria protegida con Boc con la fórmula (VIII). La desprotección con un ácido adecuado, preferiblemente TFA, da los compuestos (II).
Ejemplos de compuestos de fórmula (VI) se representan en el siguiente esquema:

La agitación puede mejorar la tasa de la reacción de condensación de Strecker. Los materiales de partida y algunos de los intermedios son compuestos conocidos y están disponibles comercialmente o pueden prepararse de acuerdo con procedimientos de reacción convencionales generalmente conocidos en la técnica.
Dicho proceso además comprende opcionalmente una reacción asimétrica usando síntesis basada en auxiliares quirales (usando carbohidratos, amina quiral o cetimina cíclica) y/o síntesis asimétrica catalítica de Strecker (usando guanidina, base de Schiff quiral o catalizador basado en BINOL).
Los compuestos de fórmula (I) como se prepararon en los procesos descritos anteriormente pueden sintetizarse en forma de mezclas racémicas de enantiómeros que pueden separarse entre sí siguiendo procedimientos de resolución conocidos en la técnica. Los compuestos de fórmula (I) que se obtienen en forma racémica se pueden convertir en las formas de sal diastereoméricas correspondientes mediante reacción con un ácido quiral adecuado. Dichas formas de sal diastereoméricas se separan posteriormente, por ejemplo, por cristalización selectiva o fraccionada y los enantiómeros se liberan allí mediante álcali. Una forma alternativa de separar las formas enantioméricas de los compuestos de fórmula (1) implica la cromatografía líquida usando una fase estacionaria quiral. Dichas formas estereoquímicamente isoméricas puras también pueden derivarse de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida apropiados, a condición de que la reacción se produzca estereoespecíficamente. Preferiblemente, si se desea un estereoisómero específico, dicho compuesto se sintetizará por procedimientos de preparación estereoespecíficos. Estos procedimientos emplearán ventajosamente materiales de partida enantioméricamente puros.
Los compuestos de fórmula (I), las sales farmacéuticamente aceptables y sus formas estereoisoméricas poseen propiedades antagonistas del receptor P2X7 como se demuestra en los ejemplos farmacológicos. Otros ejemplos de reacciones de transformación de grupo conocidas en la técnica para convertir compuestos de fórmula (I) en otros compuestos de fórmula (I) son la hidrólisis de ésteres carboxílicos en el correspondiente ácido o alcohol carboxílico; hidrólisis de amidas a los correspondientes ácidos carboxílicos o aminas; los alcoholes pueden convertirse en ésteres y éteres; las aminas primarias pueden convertirse en aminas secundarias o terciarias; los enlaces dobles se
pueden hidrogenar al enlace sencillo correspondiente. Los materiales de partida y algunos de los intermedios son compuestos conocidos y están disponibles comercialmente o pueden prepararse de acuerdo con procedimientos de reacción convencionales generalmente conocidos en la técnica. Los compuestos de fórmula (I) preparados en los procesos descritos anteriormente pueden sintetizarse en forma de mezclas racémicas de enantiómeros que pueden separarse entre sí siguiendo procedimientos de resolución conocidos en la técnica. Los compuestos de fórmula (I) que se obtienen en forma racémica se pueden convertir en las formas de sal diastereoméricas correspondientes mediante reacción con un ácido quiral adecuado. Dichas formas de sal diastereoméricas se separan posteriormente, por ejemplo, por cristalización selectiva o fraccionada y los enantiómeros se liberan de allí mediante álcali. Una forma alternativa de separar las formas enantioméricas de los compuestos de fórmula (I) implica la cromatografía líquida usando una fase estacionaria quiral. Dichas formas estereoquímicamente isoméricas puras también pueden derivarse de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida apropiados, a condición de que la reacción se produzca estereoespecíficamente. Preferiblemente, si se desea un estereoisómero específico, dicho compuesto se sintetizará por procedimientos de preparación estereoespecíficos. Estos procedimientos emplearán ventajosamente materiales de partida enantioméricamente puros. En la preparación de los compuestos de fórmula I y los materiales de partida y/o intermedios descritos aquí, puede ser útil proteger ciertos grupos que son sensibles a las condiciones de reacción. La evaluación de la utilidad de la protección opcional, así como la selección del agente protector adecuado, de acuerdo con la reacción llevada a cabo en la preparación de los compuestos de la invención y el grupo funcional a proteger, están dentro del conocimiento común de la persona experta. La eliminación de los grupos protectores opcionales se lleva a cabo de acuerdo con técnicas convencionales. Para una referencia general sobre el uso de grupos protectores en la química orgánica, véase Theodora W. Greene and Peter G.M. Wuts " Protective groups in organic synthesis ", John Wiley & Sons, Inc., II Ed., 1991.
La preparación de las sales de los compuestos de fórmula I se lleva a cabo de acuerdo con procedimientos conocidos. Por lo tanto, los presentes compuestos de fórmula (I) son útiles como medicina especialmente en el tratamiento de una afección o enfermedad mediada por el receptor P2X7, en particular la actividad antagonista del receptor P2X7. Posteriormente, los presentes compuestos pueden usarse para la fabricación de un medicamento para el tratamiento de una afección o enfermedad mediada por la actividad del receptor P2X7, en particular la actividad antagonista del receptor P2X7.
La presente invención también proporciona el uso de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo para la fabricación de un medicamento para el tratamiento de afecciones o enfermedades seleccionadas de afecciones o enfermedades mediadas por el receptor P2X7. En una realización, la presente invención proporciona un compuesto de fórmula (I) para su uso como medicamento o para el tratamiento de afecciones o enfermedades seleccionadas de afecciones o enfermedades mediadas por el receptor P2X7. Además, la presente invención también proporciona un procedimiento de tratamiento de una afección mediada por la actividad del receptor P2X7, en un sujeto mamífero, cuyo procedimiento comprende administrar a un mamífero que necesita dicho tratamiento una cantidad terapéuticamente efectiva de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo. En vista de los mecanismos de acción descritos anteriormente, los compuestos de la invención son útiles para el tratamiento de trastornos neurodegenerativos de diversos orígenes tales como la enfermedad de Alzheimer y otras afecciones de demencia tales como el cuerpo de Lewys, demencia frontotemporal y taupatías; esclerosis lateral amiotrófica, esclerosis múltiple, enfermedad de Parkinson y otros síndromes parkinsonianos; neuroinflamación inducida por VIH; temblores esenciales; otras degeneraciones espinocerebelares y neuropatía de Charcot-Marie-Toot. Los compuestos de la invención también son útiles para el tratamiento de afecciones neurológicas tales como epilepsia, incluyendo convulsiones parciales simples, convulsiones parciales complejas, convulsiones generalizadas secundarias, que incluyen además convulsiones por ausencia, convulsiones mioclónicas, convulsiones clónicas, convulsiones tónicas, convulsiones tónicas clónicas y convulsiones atónicas.
Los compuestos de la invención también son útiles para el tratamiento de trastornos cognitivos y de trastornos psiquiátricos. Los trastornos psiquiátricos incluyen, entre otros, depresión mayor, distimia, manía, trastorno bipolar (como trastorno bipolar tipo I, trastorno bipolar tipo II), trastorno ciclotímico, ciclismo rápido, ciclismo ultradiano, manía, hipomanía, esquizofrenia, trastornos esquizofreniformes, trastornos esquizoafectivos, trastornos de personalidad, trastornos de atención con o sin comportamiento hiperactivo, trastornos delirantes, trastornos psicóticos breves, trastornos psicóticos compartidos, trastorno psicótico debido a una afección médica general, trastornos psicóticos inducidos por sustancias o un trastorno psicótico no especificado, trastornos de ansiedad tales como trastorno de ansiedad generalizada, trastornos de pánico, trastorno de estrés postraumático, trastornos de control de impulsos, trastornos fóbicos, estados disociativos y, además, fumar, drogadicción y alcoholismo. En particular trastornos bipolares, psicosis, ansiedad y adicción.
Los compuestos de la presente invención son útiles en la prevención o el tratamiento del dolor neuropático. Los síndromes de dolor neuropático incluyen, entre otros: neuropatía diabética; ciática; dolor de espalda baja no específico; dolor de esclerosis múltiple; fibromialgia; neuropatía relacionada con el VIH; neuralgia, tal como neuralgia posherpética y neuralgia del trigémino, neuralgia de Morton, causalgia; y dolor resultante de trauma físico, amputación, miembro fantasma, cáncer, toxinas o afecciones inflamatorias crónicas; dolor central como el observado en los síndromes talámicos, las formas mixtas centrales y periféricas de dolor, como los síndromes complejos de dolor regional (SDRC), también llamadas distrofias simpáticas reflejas.
Los compuestos de la invención también son útiles para el tratamiento del dolor crónico. El dolor crónico incluye, entre otros, dolor crónico causado por inflamación o una afección relacionada con la inflamación, osteoartritis, artritis reumatoide, lesión o trauma agudo, dolor de la parte superior de la espalda o dolor de la parte baja de la espalda (como resultado de una enfermedad de la columna vertebral primaria, sistemática o regional como radiculopatía), dolor óseo (debido a osteoartritis, osteoporosis, metástasis óseas o razones desconocidas), dolor pélvico, dolor asociado a la lesión de la médula espinal, dolor cardíaco en el pecho, dolor torácico no cardíaco, dolor central posterior a accidente cerebrovascular, dolor miofascial, dolor de células falciformes, dolor por cáncer, enfermedad de Fabry, dolor por SIDA, dolor geriátrico o dolor causado por dolor de cabeza, síndrome de la articulación temporomandibular, gota, fibrosis o síndromes de salida torácica, en particular artritis reumatoide y osteoartritis. Los compuestos de la invención también son útiles en el tratamiento del dolor agudo causado por una lesión aguda, enfermedad, lesiones de medicina deportiva, síndrome del túnel carpiano, quemaduras, esguinces y distensiones musculoesqueléticas, tensión muscular, síndromes de dolor cervicobraquial, dispepsia, úlcera gástrica, úlcera duodenal, dismenorrea, endometriosis o cirugía (como cirugía de corazón abierto o bypass), dolor postoperatorio, dolor por cálculos renales, dolor de vesícula biliar, dolor por cálculos biliares, dolor obstétrico o dolor dental.
Los compuestos de la invención también son útiles en el tratamiento de dolores de cabeza tales como migraña, cefalea tensional, migraña transformada o cefalea evolutiva, cefalea de racimo, así como trastornos de cefalea secundaria, tales como los derivados de infecciones, trastornos metabólicos. u otras enfermedades sistémicas y otras cefaleas agudas, hemicránea paroxística y similares, que resultan de un empeoramiento de las cefaleas primarias y secundarias mencionadas anteriormente.
Los compuestos de la invención también son útiles en el tratamiento de enfermedades tales como vértigo, tinnitus, espasmo muscular y otros trastornos que incluyen, entre otros, enfermedades cardiovasculares (tales como arritmia cardíaca, infarto cardíaco o angina de pecho, hipertensión, isquemia cardíaca, isquemia cerebral) trastornos endocrinos (como acromegalia o diabetes insipidus) enfermedades en las que la fisiopatología del trastorno implica una secreción celular excesiva o hipersecretora o inapropiada de una sustancia endógena (como la catecolamina, una hormona o un factor de crecimiento).
Los compuestos de la invención también son útiles en el tratamiento selectivo de enfermedades hepáticas, tales como enfermedades hepáticas inflamatorias, por ejemplo, hepatitis viral crónica B, hepatitis viral crónica C, lesión hepática alcohólica, cirrosis biliar primaria, hepatitis autoinmune, fibrosis hepática, esteatohepatitis no alcohólica y rechazo de trasplante de hígado.
Los compuestos de la invención inhiben procesos inflamatorios que afectan a todos los sistemas del cuerpo. Por lo tanto, son útiles en el tratamiento de procesos inflamatorios del sistema músculoesquelético de los cuales la siguiente es una lista de ejemplos, pero no incluye todos los trastornos diana: afecciones artríticas como espondilitis anquilosante, artritis cervical, fibromialgia, gota, artritis reumatoide juvenil, artritis lumbosacra, osteoartritis, osteoporosis, artritis psoriásica, enfermedad reumática; trastornos que afectan la piel y tejidos relacionados: eccema, psoriasis, dermatitis y afecciones inflamatorias como quemaduras solares; trastornos del sistema respiratorio: asma, rinitis alérgica y síndrome de dificultad respiratoria, trastornos pulmonares en los que está involucrada la inflamación, como asma y bronquitis; enfermedad pulmonar obstructiva crónica; trastornos de los sistemas inmunitario y endocrinológico: periartritis nodosa, tiroiditis, anemia aplásica, esclerodermia, miastenia gravis, esclerosis múltiple y otros trastornos desmielinizantes, encefalomielitis, sarcoidosis, síndrome nefrítico, síndrome de Bechet, polimiositis, gingivitis.
Los compuestos de la invención también son útiles en el tratamiento de trastornos del tracto gastrointestinal (GI) tales como trastornos inflamatorios del intestino que incluyen pero no se limitan a colitis ulcerosa, enfermedad de Crohn, ileitis, proctitis, enfermedad celíaca, enteropatías, colitis microscópica o colágena, gastroenteritis eosinofílica, o pouchitis que resulta después de la proctocolectomía y la anastomosis posileonatal, y el síndrome del intestino irritable, incluidos los trastornos asociados con dolor abdominal y/o molestias abdominales, como piloroespasmo, indigestión nerviosa, colon espástico, colitis espástica, intestino espástico, neurosis intestinal, colitis funcional, colitis mucosa, colitis laxante y dispepsia funcional; pero también para el tratamiento de gastritis atrófica, gastritis varialoforme, colitis ulcerosa, ulceración péptica, pirosis y otros daños en el tracto gastrointestinal, por ejemplo, por Helicobacter pylori, enfermedad por reflujo gastroesofágico, gastroparesia, como gastroparesia diabética; y otros trastornos intestinales funcionales, como la dispepsia no ulcerosa (NUD); emesis, diarrea e inflamación visceral. Los compuestos de la invención también son útiles en el tratamiento de trastornos del tracto genitourinario tales como vejiga hiperactiva, prostatitis (prostatitis crónica bacteriana y no bacteriana crónica), prostadinia, cistitis intersticial, incontinencia urinaria e hiperplasia prostática benigna, anexidades, inflamación pélvica, bartolinitis y vaginitis. En particular, vejiga hiperactiva e incontinencia urinaria.
Los compuestos de la invención también son útiles en el tratamiento de enfermedades oftálmicas tales como retinitis, retinopatías, uveítis y lesiones agudas en el tejido ocular, degeneración macular relacionada con la edad o glaucoma, conjuntivitis.
Los compuestos de la invención también son útiles en el tratamiento de trastornos alimentarios tales como la anorexia nerviosa, incluidos los tipos de restricción de subtipos y el tipo de atracones/purga; bulimia nerviosa, incluidos los subtipos tipo de purga y no purga; obesidad; trastornos alimenticios compulsivos; trastorno por atracón; y trastorno alimentario no especificado de otra manera.
Los compuestos de la invención también son útiles en el tratamiento de dermatitis alérgica, hiperreactividad de las vías respiratorias, enfermedad pulmonar obstructiva crónica (EPOC), bronquitis, choque séptico, síndrome de Sjogren, glomerulonefritis, aterosclerosis, crecimiento y metástasis de células malignas, leucemia mioblástica, diabetes, meningitis, osteoporosis, quemaduras, cardiopatía isquémica, accidente cerebrovascular, enfermedad vascular periférica, venas varicosas, glaucoma.
El término "tratar" y "tratamiento", como se usa en el presente documento, se refiere al tratamiento curativo, paliativo y profiláctico, que incluye revertir, aliviar, inhibir el progreso o prevenir la enfermedad, trastorno o afección a la que se aplica dicho término, o uno o más síntomas de dicha enfermedad, trastorno o afección.
Además, la presente invención proporciona composiciones farmacéuticas que comprenden al menos un vehículo farmacéuticamente aceptable y una cantidad terapéuticamente efectiva de un compuesto de fórmula (I).
Para preparar las composiciones farmacéuticas de esta invención, una cantidad efectiva del compuesto particular, en forma de sal de adición de básica o ácido, como ingrediente activo se combina en una mezcla íntima con al menos un vehículo farmacéuticamente aceptable, vehículo que puede tomar una amplia variedad de formas dependiendo de la forma de preparación deseada para la administración. Estas composiciones farmacéuticas están deseablemente en forma de dosificación unitaria adecuada, preferiblemente, para administración oral, administración rectal, administración percutánea o inyección parenteral.
Por ejemplo, en la preparación de las composiciones en forma de dosificación oral, puede emplearse cualquiera de los vehículos farmacéuticos líquidos habituales, como por ejemplo agua, glicoles, aceites, alcoholes y similares en el caso de preparaciones de líquidos o líquidos tales como suspensiones, jarabes, elixires y soluciones; o vehículos farmacéuticos sólidos tales como almidones, azúcares, caolín, lubricantes, aglutinantes, agentes desintegrantes y similares en el caso de polvos, píldoras, cápsulas y tabletas. Debido a su fácil administración, las tabletas y las cápsulas representan la forma unitaria de dosificación oral más ventajosa, en cuyo caso obviamente se emplean vehículos farmacéuticos sólidos. Para las composiciones de inyección parenteral, el vehículo farmacéutico comprenderá principalmente agua estéril, aunque pueden incluirse otros ingredientes para mejorar la solubilidad del ingrediente activo.
Las soluciones inyectables pueden prepararse, por ejemplo, usando un vehículo farmacéutico que comprende una solución salina, una solución de glucosa o una mezcla de ambos. Las suspensiones inyectables también pueden prepararse usando vehículos líquidos apropiados, agentes de suspensión y similares. En composiciones adecuadas para administración percutánea, el vehículo farmacéutico puede comprender opcionalmente un agente potenciador de la penetración y/o un agente humectante adecuado, opcionalmente combinado con proporciones menores de aditivos adecuados que no causan un efecto perjudicial significativo para la piel. Dichos aditivos pueden seleccionarse para facilitar la administración del ingrediente activo a la piel y/o ser útiles para preparar las composiciones deseadas. Estas composiciones tópicas se pueden administrar de varias maneras, por ejemplo, como un parche transdérmico, mediante pipeta dermatológica o una pomada. Las sales de adición de los compuestos de fórmula (1), debido a su mayor solubilidad en agua sobre la forma de base correspondiente, son obviamente más adecuadas en la preparación de composiciones acuosas.
Es especialmente ventajoso formular las composiciones farmacéuticas de la invención en forma de unidad de dosificación para facilitar la administración y la uniformidad de la dosificación.
La "forma de unidad de dosificación" como se usa en el presente documento se refiere a unidades físicamente discretas adecuadas como dosificaciones unitarias, cada unidad que contiene una cantidad predeterminada de ingrediente activo calculada para producir el efecto terapéutico deseado en asociación con el vehículo farmacéutico requerido. Ejemplos de tales formas unitarias de dosificación son tabletas (incluyendo tabletas ranuradas o recubiertas), cápsulas, píldoras, paquetes de polvo, obleas, soluciones o suspensiones inyectables, cucharaditas, cucharadas y similares, y múltiplos segregados de las mismas.
Para la administración oral, las composiciones farmacéuticas de la presente invención pueden tomar la forma de formas de dosis sólidas, por ejemplo, tabletas (formas tanto tragables como masticables), cápsulas o cápsulas de gel, preparadas por medios convencionales con excipientes y vehículos farmacéuticamente aceptables tales como agentes aglutinantes (por ejemplo, almidón de maíz pregelatinizado, polivinilpirrolidona, hidroxipropilmetilcelulosa y similares), agentes de relleno (por ejemplo, lactosa, celulosa microcristalina, fosfato de calcio y similares), lubricantes (por ejemplo, estearato de magnesio, cuento, sílice y similares), agentes desintegrantes (por ejemplo, almidón de patata, almidón glicolato de sodio y similares), agentes humectantes (por ejemplo, laurilsulfato de sodio) y similares. Dichas tabletas también pueden recubrirse por procedimientos bien conocidos en la técnica.
Las preparaciones líquidas para administración oral pueden tomar la forma de, por ejemplo, soluciones, jarabes o suspensiones, o pueden formularse como un producto seco para mezclar con agua y/u otro vehículo líquido
adecuado antes de su uso. Dichas preparaciones líquidas pueden prepararse por medios convencionales, opcionalmente con otros aditivos farmacéuticamente aceptables tales como agentes de suspensión (por ejemplo, jarabe de sorbitol, metilcelulosa, hidroxipropilmetilcelulosa o grasas comestibles hidrogenadas), agentes emulsionantes (por ejemplo, lecitina o acacia), vehículos no acuosos (por ejemplo, aceite de almendra, ésteres oleosos o alcohol etílico), edulcorantes, sabores, agentes de enmascaramiento y conservantes (por ejemplo, phidroxibenzoatos de metilo o propilo o ácido sórbico).
Los edulcorantes farmacéuticamente aceptables útiles en las composiciones farmacéuticas de la invención comprenden preferiblemente al menos un edulcorante intenso tal como aspartamo, acesulfamo de potasio, ciclamato de sodio, alitame, un edulcorante de dihidrocalcona, monelina, esteviósido sucralosa (4,1',6'- tricloro-4, 1',6'-trideoxigalactosacarosa) o, preferiblemente, sacarina, sacarina de sodio o calcio, y opcionalmente al menos un edulcorante a granel como sorbitol, manitol, fructosa, sacarosa, maltosa, isomalt, glucosa, jarabe de glucosa hidrogenada, xilitol, caramelo o miel. Los edulcorantes intensos se usan convenientemente en bajas concentraciones. Por ejemplo, en el caso de la sacarina de sodio, dicha concentración puede variar de aproximadamente 0,04 % a 0,1 % (peso/volumen) de la formulación final. El edulcorante a granel puede usarse eficazmente en concentraciones mayores que varían de aproximadamente 10 % a aproximadamente 35 %, preferiblemente de aproximadamente 10 % a 15 % (peso/volumen). Los sabores farmacéuticamente aceptables que pueden enmascarar los ingredientes de sabor amargo en las formulaciones de baja dosis comprenden preferiblemente sabores de frutas tales como sabor a cereza, frambuesa, grosella negra o fresa. Una combinación de dos sabores puede dar muy buenos resultados. En las formulaciones de dosis altas, se pueden requerir sabores farmacéuticamente aceptables más fuertes, como Caramel Chocolate, Mint Cool, Fantasy y similares.
Cada sabor puede estar presente en la composición final en una concentración que varía de aproximadamente 0,05 % a 1 % (peso/volumen). Se usan ventajosamente combinaciones de dichos sabores fuertes. Preferiblemente, se usa un sabor que no sufre ningún cambio o pérdida de sabor y/o color en las circunstancias de la formulación.
Los compuestos de fórmula (I) pueden formularse para administración parenteral mediante inyección, convenientemente inyección intravenosa, intramuscular o subcutánea, por ejemplo, mediante inyección en bolo o infusión intravenosa continua. Las formulaciones para inyección pueden presentarse en forma de dosificación unitaria, por ejemplo, en ampollas o envases multidosis, incluido un conservante añadido. Pueden tomar formas tales como suspensiones, soluciones o emulsiones en vehículos aceitosos o acuosos, y pueden contener agentes de formulación tales como agentes isotonizantes, de suspensión, estabilizantes y/o dispersantes. Alternativamente, el ingrediente activo puede estar presente en forma de polvo para mezclar con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso.
Los compuestos de fórmula (I) también pueden formularse en composiciones rectales tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorios convencionales tales como manteca de cacao y/u otros glicéridos.
Los expertos en el tratamiento de enfermedades vinculadas a la mediación de los canales iónicos activados por ligando determinarán fácilmente la cantidad terapéuticamente efectiva de un compuesto de fórmula (I) a partir de los resultados de la prueba presentados a continuación. En general, se contempla que una dosis terapéuticamente efectiva será de aproximadamente 0,001 mg/kg a aproximadamente 50 mg/kg de peso corporal, más preferiblemente de aproximadamente 0,01 mg/kg a aproximadamente 10 mg/kg de peso corporal del paciente a ser tratado Puede ser apropiado administrar la dosis terapéuticamente efectiva en forma de dos o más subdosis a intervalos apropiados a lo largo del día. Dichas subdosis pueden formularse como formas de dosificación unitarias, por ejemplo, cada una conteniendo de aproximadamente 0,1 mg a aproximadamente 1000 mg, más particularmente de aproximadamente 1 a aproximadamente 500 mg, del ingrediente activo por forma de dosificación unitaria.
Como se usa en el presente documento, una "cantidad terapéuticamente efectiva" de un compuesto, es la cantidad de un compuesto que, cuando se administra a un individuo o animal, da como resultado un nivel suficientemente alto de ese compuesto en el individuo o animal para causar una respuesta antagonista discernible del receptor P2X7. La dosis exacta y la frecuencia de administración dependen del compuesto particular de fórmula (I) utilizado, la afección particular que se está tratando, la gravedad de la afección que se está tratando, la edad, el peso y la condición física general del paciente particular también como de otro medicamento que el paciente pueda estar tomando, como es bien sabido por los expertos en la materia. Además, dicha "cantidad terapéuticamente efectiva" puede reducirse o aumentarse dependiendo de la respuesta del paciente tratado y/o dependiendo de la evaluación del médico que prescribe los compuestos de la presente invención. Los rangos de cantidad diaria efectiva mencionados anteriormente son, por lo tanto, solo pautas.
Nomenclatura y estructuras
En general, la nomenclatura utilizada en esta aplicación se basa en ChemSketch™ (ACDLabs) y se genera de acuerdo con la nomenclatura sistemática IUPAC. Las estructuras químicas mostradas aquí se prepararon usando ISIS® versión 2,2. Cualquier valencia abierta que aparezca en un átomo de carbono, oxígeno, azufre o nitrógeno en las estructuras de la presente memoria indica la presencia de un átomo de hidrógeno a menos que se indique lo
contrario. Cuando se muestra un anillo de heteroarilo que contiene nitrógeno con una valencia abierta en un átomo de nitrógeno y se muestran variables como R1, R2, R3, etc. en el anillo de heteroarilo, dichas variables pueden unirse o unirse al nitrógeno de valencia abierta. Cuando existe un centro quiral en una estructura, pero no se muestra una estereoquímica específica para el centro quiral, la estructura abarca ambos enantiómeros asociados con el centro quiral. Cuando una estructura mostrada en el presente documento puede existir en múltiples formas tautoméricas, la estructura abarca todos los tautómeros. Los átomos representados en la estructura en el presente documento pretenden abarcar todos los isótopos naturales de tales átomos. Así, por ejemplo, está previsto que los átomos de hidrógeno representados en el presente documento incluyan deuterio y tritio, y se entiende los átomos de carbono incluyen isótopos 13C y 14C.
Abreviaturas
Las abreviaturas que pueden usarse en la descripción de los Esquemas y los Ejemplos que siguen son:
AcOH: ácido acético
Anh: anhidro
AcONa: acetato de sodio
Boc: carbonato de tert-butilo
Boc2O: dicarbonato de di-tert-butilo
CC: cromatografía en columna
DAST: trifluoruro de dietilaminosulfuro
DCM: diclorometano
DEA: dietilamina
DIAD: diisopropilazodicarboxilato
DIBAL: hidruro de diisobutilaluminio
DIPEA: diisopropiletilenamina
DMAP: dimetilaminopiridina
DMF: dimetilformamida
DMSO: dimetilsulfóxido
Et2O: éter dietílico
EtOAc: acetato de etilo
EtOH: etanol
ESI: ionización por electroaspersión
HBTU: hexafluorofosfato de N,N,N',N'-tetrametil-O-(1H-benzotriazol-1-il)uronio;
H: hora;
Hrs: horas
M: molar
MeCN: acetonitrilo
MeOH: metanol
Min: minuto(s)
Ni-Raney: Níquel-Raney
RMN: resonancia magnética nuclear
rt: temperatura ambiente
TFA: ácido trifluoroacético
THF: tetrahidrofurano;
TLC: cromatografía de capa fina
TMSCN: trimetilsililcianuro;
UPLC-MS: cromatografía líquida de ultrarrendimiento-espectrometría de masas
XPhos: 4,5-bis(difenilfosfino)-9,9-dimetilxanteno.
Parte experimental
Los siguientes ejemplos ilustran la presente invención. A menos que se indique explícitamente lo contrario, todos los detalles (especialmente porcentajes y cantidades) están referidos al peso.
Ejemplos de síntesis
A. Síntesis de los intermedios Ib-ld

Se añadió cloruro de sulfurilo (1,23 ml, 15,2 mmol, 1,01 eq) gota a gota a 0 °C a 4,4-difluoroacetoacetato de etilo (2,5 g, 15,0 mmol, 1 eq) en atmósfera de nitrógeno, y se agitó durante la noche a temperatura ambiente. La reacción se diluyó con EtOAc (20 ml) y se vertió en una mezcla de hielo/agua (20 ml). La capa orgánica se secó sobre Na2SO4 anhidro, filtrado y evaporado dando 3,2 g de crudo en 2-cloro-4,4-difluoroacetoacetato como un aceite amarillo. El crudo se disolvió en etanol (10 ml), se trató con tiourea (3,2 g, 30 mmol, 2 eq) y se calentó en un reactor de microondas durante 1 h a 100°C. Luego, el disolvente se eliminó al vacío y el residuo se repartió en NaHCO3 saturado (10 ml) y EtOAc (10 ml). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SO4, anhidro filtrado y evaporado. El crudo se trató con éter dietílico, se filtró y se secó al vacío, dando 1,37 g (rendimiento 41 %) del intermedio 1b como un sólido amarillo.

El producto intermedio 1b (1,37 g, 6,16 mmol, 1 eq) se disolvió en dioxano (35 ml), se añadió nitrito de isoamilo (2,24 ml, 16,64 mmol, 2,7 eq) y la mezcla de reacción se calentó durante 1 hora a 80 °C. El disolvente se eliminó por evaporación a presión reducida, y el residuo se purificó por cromatografía ultrarrápida sobre gel de sílice (EtOAc/éter de petróleo 10/90) dando el intermedio 1c (1,02 g, rendimiento 80 %) como un sólido amarillo.

El intermedio 1c (0,758 g, 3,66 mmol, 1 eq) se disolvió en DCM seco (18,5 mL) bajo atmósfera de argón y se enfrió a -75°C. Se añadió gota a gota hidruro de diisobutil aluminio 1M en DCM (4,1 ml, 4,1 mmol, 1,12 eq) y la mezcla de reacción se agitó a -70 °C. Después de 1,5 h, se añadió gota a gota hidruro de diisobutil aluminio 1M en DCM (2,5 ml, 2,5 mmol, 0,68 eq) y la mezcla de reacción se agitó adicionalmente durante 1 h a -70°C. La reacción se calentó a 0 °C y se trató con agua (0,264 ml), NaOH al 15 % (0,264 ml) y agua (0,66 ml) en este orden. Luego se agitó durante 5 minutos a 0 °C, luego durante 30 minutos a temperatura ambiente. Se añadieron secuencialmente agua (0,24 ml) seguido de NaOH al 15 % (0,130 ml), y la reacción se agitó a temperatura ambiente hasta que se formó un precipitado. La mezcla se filtró y luego el disolvente se concentró. El residuo se purificó por cromatografía ultrarrápida sobre gel de sílice (DCM/éter de petróleo 80/20 ^ 100 % DCM) produciendo un aceite amarillo (0,34 mg, rendimiento 40 %) que contenía el producto intermedio 1d (pureza “ 70 %), que se usó como tal.
B. Síntesis de intermedios: aaminonitrilos
Materiales de partida
Todos los derivados sustituidos de 4-feniloxi o 4-benciloxi-piperidina y 4-metil-1,3-tiazol-5-carbaldehído, utilizados como materiales de partida, se adquirieron de proveedores de productos químicos:

(continuación)

Procedimiento general
Se suspendió un hidrocloruro de derivado de piperidina 4-sustituido (1 eq) en 2-3 ml de DCM y se añadió TEA (1,1-2 eq). La mezcla se agitó durante varios minutos, el disolvente se evaporó en un evaporador rotatorio y el residuo se secó al vacío durante 15 minutos a 40°C. La amina libre de hidrocloruro (1 eq), tiazolii-aidehído (180-300 mg, 1,2-1,5 eq) y AcONa (3,5 eq) se disolvieron en AcOH glacial (5-8 mL). La mezcla se agitó a temperatura ambiente bajo
argón durante 3 horas y luego se enfrió a 0 °C. Se añadió TMSCN (3-12 eq) gota a gota y la mezcla se dejó calentar a temperatura ambiente y se agitó durante 1-3 días. Mientras tanto, si fuera necesario de acuerdo con el análisis LC-MS, se añade TMSCN (3-6 eq) (hasta 12 eq de TMSCN) y la reacción se agitó durante 24 h. Luego, el disolvente se evaporó en un evaporador rotatorio a 40-45°C. Se añadió una solución saturada de NaHCO3 (20-50 ml) al residuo. Si fuera necesario, se añade NaHCO3 sólido y agua para aumentar el pH a 8. La mezcla se extrajo con DCM (5 ml x 3- 5). Las fases orgánicas combinadas se secaron (Na2SO4 anhidro) y se evaporaron. El crudo se purificó por cromatografía instantánea (SO2) con una mezcla de hexano/acetona (0-> 30 %) dando el a-aminonitrilo puro (rendimiento 28-68 %).
Mediante este procedimiento, los intermedios A0018_42_01 (rendimiento 54 %), A0018_42_02 (rendimiento 55 %), A0018_42_03 (rendimiento 55 %), A0018_42_04 (rendimiento 42 %), A0018_42_05 (rendimiento 63 %), A0018_42_06 (rendimiento 50 %), A0018_41_01 (rendimiento 45 %), A0018_41_02 (rendimiento 60 %), A0018_41_03 (rendimiento 46 %), A0018_41_04 (rendimiento 68 %), A0018_41_05 (rendimiento 65 %), A0018_41_06 (rendimiento 43 %), A00FF_42_04 (rendimiento 30 %), A00FF_42_05 (rendimiento 56 %), A00FF_42_06 (rendimiento 36 %), A00FF_41_01 (rendimiento 31 %), A00FF_41_02 (rendimiento 57 %), A00FF_41_03 (rendimiento 53 %), A00FF_41_04 (rendimiento 53 %), A00Ff_41_05 (rendimiento 28 %) a partir de 4- metil-1,3-tiazol-5-carbaldehído o, respectivamente, 4-difluorometil-1,3-tiazol-5-carbaldehído y 4-(4-fluorobenciloxipiperidina), 4-(4-clorobenciloxipiperidina), 4-(3-fluorobenciloxipiperidina), 4-(2-fluorobenciloxipiperidina), 4-(3,5-difluorobenciloxipiperidina), 4-(3,4-difluorobenciloxipiperidina), 4-(4-fluorofeniloxipiperidina), 4-(4-clorofeniloxipiperidina), 4-(3-fluorofeniloxipiperidina), 4-(2-fluorofeniloxipiperidina), 4 (3,5-difluorofeniloxipiperidina) o 4-(3,4-difluorofeniloxipiperidina).

(continuación)

C. Preparación de diaminas (procedimiento general)
Se disolvió un derivado de cianuro (100-270 mg, 1 eq) en DCM seco bajo atmósfera de argón y se enfrió en un baño de hielo-sal. Se añadió lentamente una solución 1M de DIBAL (3 eq) en DCM (en porciones, 1 eq cada 30 minutos) y la mezcla se agitó durante una hora adicional. A esta solución (a 0°C), se añadió agua (1 ml) gota a gota y la mezcla se agitó hasta que se completó la formación de precipitado. Se eliminó el DCM al vacío y el residuo se suspendió en AcOEt. El sólido obtenido se filtró y se lavó 4-6 veces con AcOEt. Las capas orgánicas combinadas se secaron sobre Na2SO4, se evaporaron para producir un residuo aceitoso y se secaron durante no menos de 1 hora al vacío a 38-40°C. El producto crudo (68-97 % de rendimiento) se usó sin purificación adicional en la siguiente etapa sintética. Usando este procedimiento:
el intermedio A0017_59_01 (rendimiento 94 %) se preparó a partir de A0018_42_01;
el intermedio A0017_59_02 (rendimiento 83 %) se preparó a partir de A0018_42_02;
el intermedio A0017_59_03 (rendimiento 87 %) se preparó a partir de A0018_42_03;
el intermedio A0017_59_04 (rendimiento 97 %) se preparó a partir de A0018_42_04;
el intermedio A0017_59_05 (rendimiento 65 %) se preparó a partir de A0018_42_05;
el intermedio A0017_59_06 (rendimiento 88 %) se preparó a partir de A0018_42_06;
el intermedio A0017_58_01 (rendimiento 62 %) se preparó a partir de A0018_41_01;
el intermedio A0017_58_02 (rendimiento del 95 %) se preparó a partir de A0018_41_02;
el intermedio A0017_58_03 (rendimiento 67 %) se preparó a partir de A0018_41 03;
el intermedio A0017_58_04 (rendimiento 89 %) se preparó a partir de A0018_41_04;
el intermedio A0017_58_05 (rendimiento 80 %) se preparó a partir de A0018_41_05;
el intermedio A0017_58_06 (rendimiento 95 %) se preparó a partir de A0018_41_06;
el intermedio A00FF_59_04 (rendimiento 87 %) se preparó a partir de A00FF_42_04;
el intermedio A00FF_59_05 (rendimiento 81 %) se preparó a partir de A00FF_42_05;
el intermedio A00FF_59_06 (rendimiento 80 %) se preparó a partir de A00FF_42_06;
el intermedio A00FF_58_01 (rendimiento 89 %) se preparó a partir de A00FF_41_01;
el intermedio A00FF_58_02 (rendimiento 94 %) se preparó a partir de AOOFF_41_02;
el intermedio A00FF_58_03 (rendimiento 88 %) se preparó a partir de A00FF_41_03;
el intermedio A00FF_58_04 (rendimiento 92 %) se preparó a partir de A00FF_41_04 el intermedio A00FF_58_05 (rendimiento 93 %) se preparó a partir de A00FF_41_05.


D. Procedimiento general para la síntesis de compuestos finales.
Preparación de los compuestos 1-38
Una mezcla de ácido carboxílico (25-115 mg, 1 eq), HATU (1,1eq) o EDCl (1eq)/HOBt (1 eq) y DIPEA (2-3 eq) en DMF anhidro o DCM (1-2 ml) se agitó durante 10-30 minutos bajo atmósfera de argón. Luego, se añadió una solución de amina cruda (1 equiv.) en DCM o DMF anhidro (1-3 ml) y la mezcla de reacción se agitó durante la noche. Se añadieron agua y NaHCO3 saturado y el producto se extrajo 4-6 veces con DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se evaporaron. El producto se purificó mediante FCC (SiO2, DCM-> AcOEt-> 0-10 MeOH/AcOEt) o SFC (5-10 % MeOH/ScCO2) y las fracciones con el producto deseado se evaporaron y se secaron a alto vacío durante 16-72 h. Rendimientos 8-71 %.
Usando este procedimiento los compuestos:
El compuesto 1 (rendimiento 19 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_59_01 en DMF+DCM, purificación por SFC;
El compuesto 2 (rendimiento 19 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_59_02 en DMF, purificación por SFC;
El compuesto 3 (rendimiento del 23 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, EDCl/HOBt y A0017_59_03 en Dc M, purificación por SFC;
El compuesto 4 (rendimiento 30 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_59_04 en DMF, purificación por SFC;
El compuesto 5 (rendimiento 16 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_59_05 en DMF, purificación por FCC seguido de SFC;
El compuesto 6 (rendimiento 12-18 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, EDCl/HOBt o HATU y A0017_59_06 en DCM o DMF+DCM, purificación por SFC;
El compuesto 7 (rendimiento 16 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_01 en DMF+DCM, purificación por SFC;
El compuesto 8 (rendimiento 13-16 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_02 en d Mf+DCM o DMF, purificación por FCC precedida por SFC;
El compuesto 9 (rendimiento 48 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_03 en DMF, purificación por FCC;
El compuesto 10 (rendimiento 12 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_04 en Dm F, purificación por fCc ;
El compuesto 11 (rendimiento 8 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_05 en DCM, purificación por SFC precedida por FCC;
El compuesto 12 (rendimiento 29 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A0017_58_06 en Dm F+DCM, purificación porSFC;
El compuesto 13 (rendimiento 28 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A001759_01 en DCM, purificación por FCC;
El compuesto 14 (rendimiento 25 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_59_02 en DMF, purificación por FCC;
El compuesto 15 (rendimiento 32 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_59_03 en DMF+DCM, purificación por FCC;
El compuesto 16 (rendimiento 31 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_59_04 en DMF+DCM, purificación por FCC;
El compuesto 17 (rendimiento 29 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_59_05 en DMF, purificación por FCC;
El compuesto 18 (rendimiento del 20 %) se preparó a partir de ácido quinolin-5-carboxílico, EDCl/HOBt y A0017_59_06 en dCm , purificación por FcC;
El compuesto 19 (rendimiento 28 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_58_01 en DMF, purificación por FCC;
El compuesto 20 (rendimiento 27 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_58_02 en DMF, purificación por FCC;
El compuesto 21 (rendimiento 71 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A0017_58_03 en DMF, purificación por FCC;
El compuesto 22 (rendimiento 25 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A001758_04 en DMF, purificación por FCC;
El compuesto 23 (rendimiento 15 %) se preparó a partir de ácido quinolin-5-carboxílico, EDCl/HOBt y A0017_58_05 en DCM, purificación por FCC;
El compuesto 24 (rendimiento 17-27 %) se preparó a partir de ácido quinolin-5-carboxílico, EDCl/HOBt o HATU y A0017_58_06 en DCM o DMF+DCM, purificación por FCC;
El compuesto 25 (rendimiento 30 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_59_04 en DMF+DCM, purificación porSFC;
El compuesto 26 (rendimiento 19 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_59_05 en DMF+DCM, purificación por dos FCC individuales;
El compuesto 27 (rendimiento 26 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_59_06 en DMF+DCM, purificación porSFC;
El compuesto 28 (rendimiento 19 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_58_02 en DMF+DCM, purificación porSFC;
El compuesto 29 (rendimiento 22 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_58_03 en DMF+DCM, purificación porSFC;
El compuesto 30 (rendimiento 30 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_58_04 en DMF+DCM, purificación porSFC;
El compuesto 31 (rendimiento 29 %) se preparó a partir de ácido 2-cloro-6-fluorobenzoico, HATU y A00FF_58_05 en DMF+DCM, purificación porSFC;
El compuesto 32 (rendimiento del 30 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_59_04 en DMF+DCM, purificación porSFC;
El compuesto 33 (rendimiento 31 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_59_05 en DMF+DCM, purificación por SFC;
El compuesto 34 (rendimiento 34 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_58_01 en DMF+DCM, purificación por SFC precedida por FCC;
El compuesto 35 (rendimiento 19 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_58_02 en DMF+DCM, purificación por FCC precedida por SFC;
El compuesto 36 (rendimiento 22 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_58_03 en DMF+DCM, purificación porSFC;
El compuesto 37 (rendimiento 22 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF58_04 en DMF+DCM, purificación por SFC
El compuesto 38 (rendimiento 35 %) se preparó a partir de ácido quinolin-5-carboxílico, HATU y A00FF_58_05 en DMF+DCM, purificación porSFC.
La Tabla 1 enumera los compuestos finales que se prepararon y probaron de acuerdo con el procedimiento experimental descrito para el Ejemplo 1.
Tabla 1

continuación

continuación

continuación

continuación

continuación

continuación
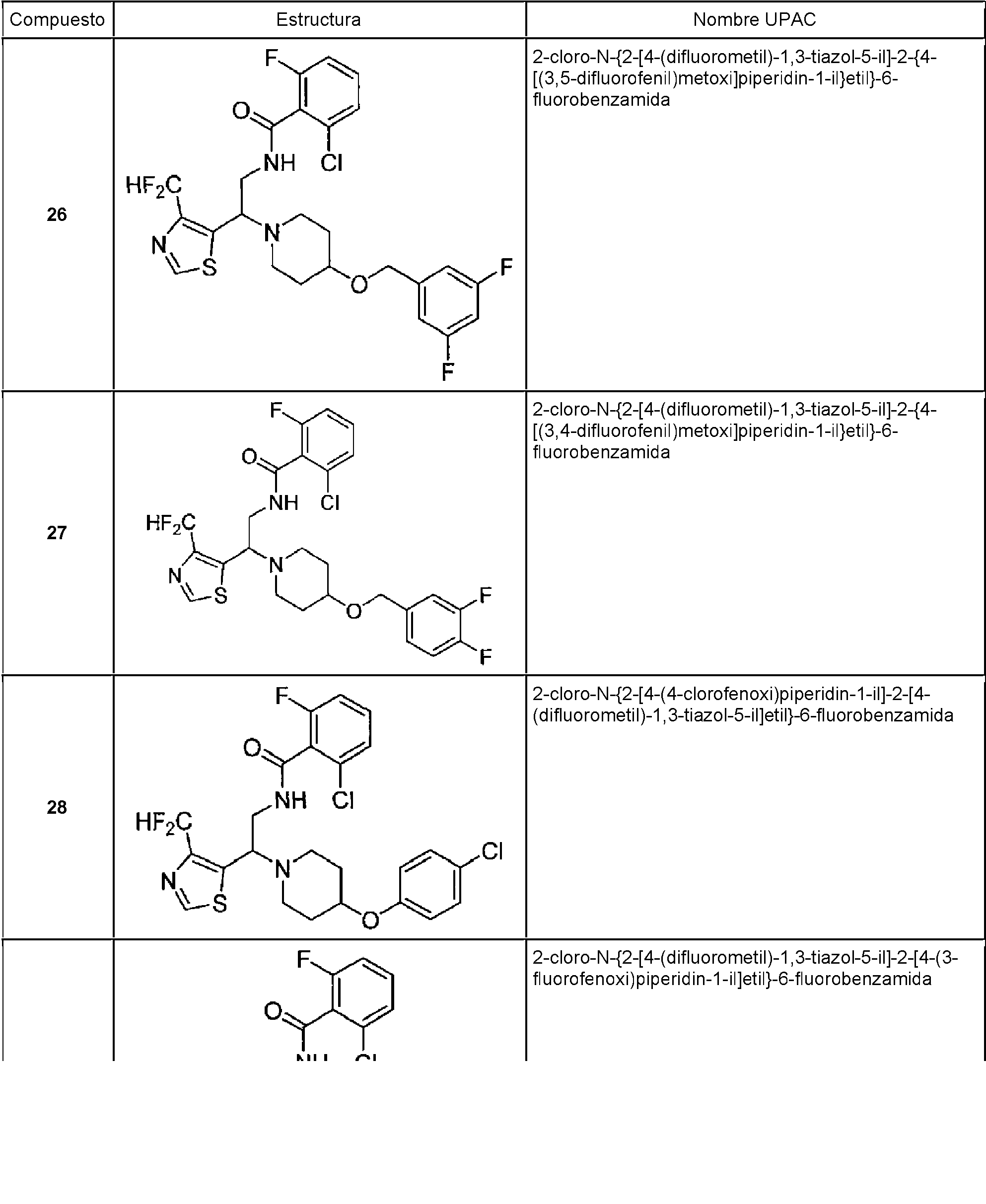
continuación


Sistema de purificación
Cromatografía instantánea (FCC)
Se realizaron separaciones de FCC en Interchim puriFlash®430, Interchim puriFlash®450 o Interchim puriFlash® 4250-250 equipados con detector UV. Tipo de columnas de sílice: Interchim puriFlash® SiHP (sílice de alto rendimiento) 50 |jm, 4-25 g.
Cromatografía de fluidos supercríticos (SFC):
Se realizaron separaciones de FCC en el sistema Waters Prep 100q SFC equipado con detectores de fotodiodo y MS QDa. Tipo de columna de sílice: Viridis Prep Silica 2-EP (2-Etilpiridina) o Bd , 19x100 mm, 5 jm. Procedimiento utilizado: disolvente (A) CO2, disolvente (B) metanol; condiciones de gradiente de 5 %-10 % de B en 8 minutos; ABPR 120 bar; T = 40°C.
Parte analítica
Procedimiento general de LCMS
La medición de HPLC se realizó usando un módulo Dionex Ultimate 3000 que comprende una bomba cuaternaria con desgasificador, un inyector automático, un horno de columna (ajustado a 25°C), un detector de matriz de diodos DAD (usualmente longitud de onda utilizada 200 nm) y una columna Kinetex XB C184,6x50 mm 2,6 jm. Un caudal de eluato fue de 0,5 ml/min. Se utilizaron dos fases móviles, la fase móvil A: solución de ácido fórmico en agua al 0,1 % (MiliQ); fase móvil B: solución de ácido fórmico al 0,1 % en acetonitrilo (HPLC J.T.Baker), y se emplearon para ejecutar condiciones de gradiente de 20 % de B a 80 % en 6,7 minutos, mantener 80 % de B durante 1,3 minutos, condiciones de gradiente de 80 % B al 95 % en 0,3 minutos, mantenga el 95 % B y las condiciones de gradiente al 20 % B en 0,5 minutos y mantenga estas condiciones durante 2 minutos para reequilibrar la columna. Se usó un volumen de inyección de 1,0 jl. El flujo de la columna se dividió en un espectrómetro MS. El detector de MS (HCT Bruker) se configuró con una fuente de ionización por electroaspersión. Los espectros de masas se adquirieron escaneando de 100 a 1000 Da. El voltaje de la aguja capilar fue de 4 kV en modo de ionización positiva y la temperatura de la fuente se mantuvo a 365°C. Se utilizó nitrógeno como gas nebulizador, el flujo fue de 9,0 l/min. La adquisición de datos se realizó con el Programa de Análisis de Datos Bruker.
Tabla 2: Tiempo de retención (Rt) en minutos, [M+H]+ pico para el procedimiento LCMS.
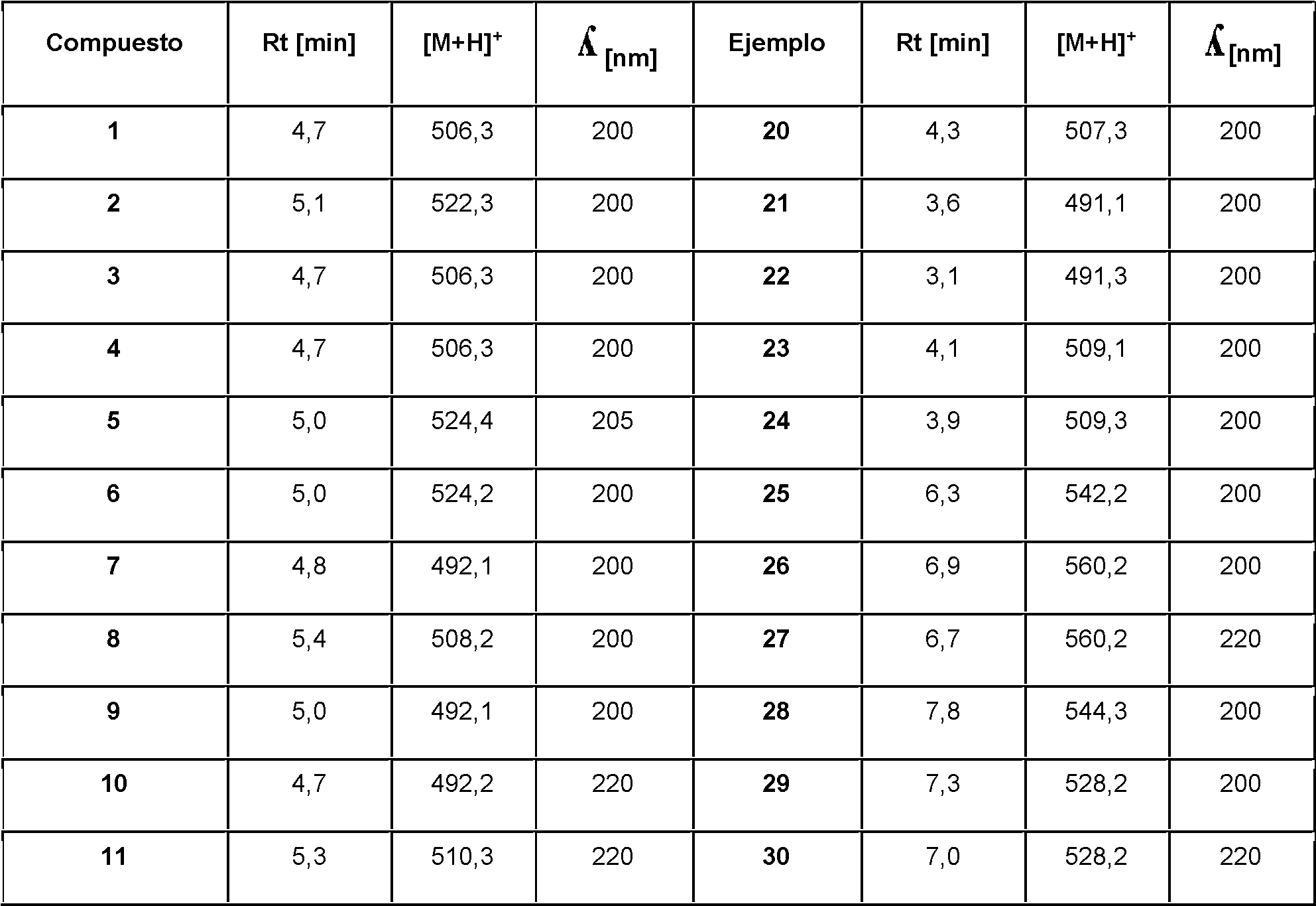
(continuación)

Caracterización por RMN
Los espectros de 1H RMN y 13C RMN se registraron en un espectrómetro Bruker Avance II1HD 400 MHz usando CDCl3 o CD3OD como disolvente. Los cambios químicos (8) se informan en partes por millón (ppm) en relación con la señal residual de los solventes no completamente deuterados, escogidos para 1H RMN asignado como 7,26 ppm para CHCh y 3,31 ppm para CHD2OD o en relación con la señal de los solventes deuterados para 13C RMN asignado como 77,16 ppm para CHChy 49,00 ppm para CD3OD.

(continuación)

(continuación)

(continuación)


(continuación)

(continuación)

(continuación)

Ejemplos farmacológicos
Se encontró que los compuestos de la invención son activos en un ensayo de canal P2X7 humano mediante sujeción-parche automatizada.
Para controlar directamente el bloque del canal P2X7, se desarrolló e implemento un ensayo electrofisiológico en el instrumento de electrofisiología automatizado QPatchl6X.
Las células HEK-293 que expresan los canales P2X7 se cultivaron en EMEM modificado.
72 horas antes del experimento, se sembraron 5 millones de células en matraces T225. Justo antes del experimento, las células se lavaron dos veces, se separaron del matraz con tripsina-EDTA, se volvieron a suspender en la solución de suspensión y se colocaron en el QPatch 16x.
Los compuestos (20 mM en un DMSO al 100 %) almacenados a -20°C se prepararon el día del experimento (una primera dilución 1:20 en DMSO al 100 % para preparar una solución madre 1 mM, luego una solución 1 microM en solución externa una dilución en serie 1:10).
Los experimentos estándar de sujeción de voltaje en célula completa se realizaron a temperatura ambiente. Para estos experimentos se utilizó la tecnología de múltiples agujeros y se muestrearon datos a 2 KHz.
La solución intracelular contenía (mM) CsF 135, NaCl 10, EGTA 1, HEPES 10, (pH 7,2 con CsOH) mientras que la extracelular contenía (mM) NaCl 145, KCl 4, MgCh 0,5, CaCfe 1, HEPES, Glc 10 (pH 7,4 con NaOH).
Después del establecimiento del sello y el paso en toda la configuración de la celda, las celdas se mantuvieron a -80 mV. Se evocó la corriente P2XR7 aplicando 100 microM de BzATP solo (4 veces) y luego en presencia de concentraciones crecientes del compuesto bajo investigación (1, 10, 100 y 1000 nM).
Los períodos de preincubación de 5 a 8 contienen concentraciones crecientes del compuesto de interés (1, 10, 100 y 1000 nM), como se ilustra en la Figura.
Se midió y normalizó la corriente interna máxima evocada por BzATP en ausencia o en presencia de concentraciones crecientes de los compuestos bajo investigación. El posible efecto modulador se midió como % de control y como IC50 determinó ajustar los datos de las curvas de dosis-respuesta con la siguiente ecuación:
Y=100/(1+10A((LogIC50-X)*Pendiente de Hill)) donde:
X: log de concentración
Y: respuesta normalizada, 100 % hasta 0 %, disminuyendo a medida que X aumenta.
Log IC50: mismas unidades de registro que X
Pendiente de Hill: factor de pendiente o HS, sin unidades
Tabla 3

continuación

Se encontró que los compuestos de la invención eran inhibidores de P2X7 de rata usando un kit de ensayo de calcio sin lavado Screen Quest ™ Fluo-8.
El influjo de Ca2+ se midió en células HEK-293 transfectadas de forma estable con el receptor usando el kit de ensayo de calcio sin lavado Screen Quest™ Fluo-8 (AAt Bioquest®). En resumen, una vez dentro de las células, los grupos bloqueadores lipófilos de Fluo-8 son escindidos por esterasas celulares no específicas, lo que da como resultado un colorante fluorescente cargado negativamente que permanece dentro de las células. Su fluorescencia aumenta al unirse al calcio. Cuando las células HEK-293/P2X7 se estimularon con BzATP, el Ca2+ entró en las células y aumentó la fluorescencia de Fluo-8 NW. El espectro de absorción de colorante era compatible con excitación a 488 nm por fuentes de láser de argón y su longitud de onda de emisión estaba en el rango de 515-575 nm.
Para analizar de forma rutinaria los compuestos, las células HEK-293 transfectadas de manera estable con P2X7R de rata se sembraron durante la noche en medio de crecimiento a 10000, 15000 o 20000 células/pocillo en una placa de 384 pocillos, de acuerdo con el nivel de respuesta después de la descongelación. 24 horas después, el medio se retiró y las células se precargaron a temperatura ambiente durante 1 hora con 20 pl/p de Fluo-8 NW preparado en Tyrode 0,3 mM sin Ca2+/Mg2+.
Los compuestos de la invención se ensayaron a 8 concentraciones (4 repeticiones para cada concentración): 10 3,16-1-0,316-0,1-0,0316-0,01 y 0,00316 pM, en la misma placa.
Los compuestos se ensayaron en FLIPRTETRA de acuerdo con el siguiente procedimiento:
• primera inyección en FLIPRTETRA de 10 pL de compuesto de prueba 3x (en el tampón Tyrode 0,3 mM Ca2+/Mg2+ -libre+ DMSO 0,5 % concentración final)
• 5' incubación
• segunda inyección en FLIPRTETRA de 15 pL de 3x BzATP a ~ECs0 (en el tampón Tyrode 0,3 mM Ca2+/Mg2+ -libre BSA 0,0003 % concentración final)
• Registro de fluorescencia durante 3'
Entre una placa y las siguientes, las puntas se lavaron exhaustivamente con agua, luego con DMSO al 100 % y finalmente con agua para evitar el arrastre dentro de las puntas.
El efecto de los compuestos de prueba se midió como porcentaje de inhibición frente a un antagonista de referencia y los valores de IC50 se calcularon en concordancia.
Tabla 4

continuación

continuación

Se descubrió que los compuestos de la presente invención eran inesperadamente más potentes que un ejemplo muy cercano de WO2O15/118019 como se informa en la Tabla 5.
Tabla 5

Evaluación in vitro de compuestos de prueba para la estabilidad metabólica utilizando hígado humano Microsomas.
Sistema de prueba de Microsomas humanos (ratón) de hígado
Concentración del compuesto de prueba: 1 pM
Puntos de tiempo: 0, 5, 10, 30 y 60 minutos.
Concentración final de proteína: 1 mg/ml
Número de réplicas: dos
Tampón de fosfato de potasio pH 7,4100 mM
Punto final: % restante del compuesto de prueba, semivida, Clint
Bioanálisis por LC-MS/MS
Preparación y dilución del compuesto de prueba:
Se preparó una solución madre 10 mM del compuesto de prueba en DMSO y se diluyó con agua: acetonitrilo (1:1) a una concentración de 1 mM. La concentración de trabajo de 100 pM se preparó por dilución adicional con agua:acetonitrilo (1:1).
Preparación del tampón fosfato de potasio pH 7,4:
Se agregarán 100 ml de agua Milli Q a K2HPO4 (1,398 g) y KH2PO4 (0,27 g) para obtener un pH final de 7,4.
Procedimiento de ensayo
Mezcla de preincubación: 2,5 pL de compuesto de prueba 75 pl de microsomas de hígado a 3,33 mg/ml 85 pl de tampón de fosfato de potasio 100 mM (preincubar durante 10 minutos a 37°C)
60 min sin cofactor: 32,5 pL de mezcla de preincubación 17,5 pL de tampón de fosfato de potasio 100 mM (Incubar durante 60 min a 37°C)
Muestra de 0 min: 16,25 pL de mezcla de preincubación 200 pL de acetonitrilo que contiene el patrón interno 8,75 pL de mezcla de incubación de cofactor 62 pL de cofactor (2,5 mM) mezcla de incubación restante (Incubado durante 60 min a 37°C).
Preparación de la muestra: 25 pL de mezcla de incubación 200 pL de acetonitrilo que contiene el patrón interno Vortex 5 min a 1200 rpm Centrifugadora 10 min a 4000 rpm. Diluir el sobrenadante 2 veces con agua e inyectarlo en LC-MS/MS.
Bioanálisis: procedimiento LC-MS/MS Se utilizarán condiciones de gradiente genéricas LC y los detalles se proporcionarán en el informe.
Cálculo: % restante de la sustancia de prueba = [Área de pico en el tiempo en min/Área de pico en 0 min]* 100 Kel: Pendiente obtenida de la gráfica de log % restante vs.tiempo (min) Vida media: t-i/2 (min) = 0,693/Kel
Eliminación intrínseca (gl/min/mg) = ABS (Kel/Concentración de proteína)*1000
Se encontró inesperadamente que los ejemplos de la presente invención son 2-4 veces más estables en las pruebas de estabilidad microsómica del hígado humano que un compuesto muy similar ejemplificado en el documento WO2015/118019 como se informa en la Tabla 6.
Tabla 6

Claims (10)
1. Un compuesto de la siguiente fórmula (I) o una sal farmacéuticamente aceptable del mismo:

incluyendo cualquier forma estereoquímicamente isomérica del mismo, en la que:
n es 0 o 1 ;
R1 es alquilo C1-C4 (opcionalmente sustituido con hidroxilo o halógeno), preferiblemente metilo, fluorometilo, difluorometilo, trifluorometilo;
cada uno de R2, R3 y R4 es independientemente hidrógeno, halógeno o los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros que contiene un átomo de nitrógeno, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno o halógeno, a condición de que al menos uno de R5 y R6 sea halógeno.
2. El compuesto de Fórmula (I) de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, en la que:
n es 0 o 1 ;
R1 es metilo o difluorometilo;
cada uno de R2, R3 y R4 es independientemente hidrógeno, flúor, cloro o los grupos R2 y
R4, tomados juntos, forman un anillo heterocíclico de seis miembros que contiene un átomo de nitrógeno a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno, fluoro o cloro a condición de que al menos uno de R5 y R6 sea halógeno.
3. El compuesto de fórmula (I) de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, en la que:
n es 0 o 1 ;
R1 es metilo o difluorometilo;
cada uno de R2, R3 y R4 es independientemente hidrógeno, fluoro o cloro, a condición de que al menos uno de R2, R3 y R4 no sea hidrógeno;
cada uno de R5 y R6 es hidrógeno, fluoro o cloro a condición de que al menos uno de R5 y R6 sea halógeno;
4. El compuesto de fórmula (I) de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, en la que:
n es 0 o 1 ;
R1 es metilo o difluorometilo;
R3 es hidrógeno y los grupos R2 y R4, tomados juntos, forman un anillo heterocíclico de seis miembros, en el que el anillo heterocíclico de seis miembros junto al grupo fenilo forman un anillo de quinolina.
5. Un compuesto de fórmula (I) según la reivindicación 1, o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, seleccionado del grupo:
2-doro-6-fluoro-N-(2-{4-[(4-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)benzamida
2-doro-N-(2-{4-[(4-dorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)-6-fluorobenzamida
2-doro-6-fluoro-N-(2-{4-[(3-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)benzamida
2-doro-6-fluoro-N-(2-{4-[(2-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)benzamida
2-doro-N-(2-{4-[(3,5-difluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)-6-fluorobenzamida 2-doro-N-(2-{4-[(3,4-difluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)-6-fluorobenzamida 2-doro-6-fluoro-N-{2-[4-(4-fluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}benzamida
2-doro-N-{2-[4-(4-dorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}-6-fluorobenzamida
2-doro-6-fluoro-N-{2-[4-(3-fluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}benzamida
2-doro-6-fluoro-N-{2-[4-(2-fluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}benzamida
2-doro-N-{2-[4-(3,5-difluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}-6-fluorobenzamida
2-doro-N-{2-[4-(3,4-difluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}-6-fluorobenzamida
N-(2-{4-[(4-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-(2-{4-[(4-dorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-(2-{4-[(3-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-(2-{4-[(2-fluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-(2-{4-[(3,5-difluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-(2-{4-[(3,4-difluorofenil)metoxi]piperidin-1-il}-2-(4-metil-1,3-tiazol-5-il)etil)quinolin-5-carboxamida
N-{2-[4-(4-fluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}quinolina-5-carboxamida
N-{2-[4-(4-clorofenoxi) piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etM}quinolina-5-carboxamida
N-{2-[4-(3-fluorofenoxi) piperidin-1 -il]-2-(4-metil-1,3-tiazol-5-il)etil}quinolina-5-carboxamida
N-{2-[4-(2-fluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}quinolina-5-carboxamida
N-{2-[4-(3,5-difluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}quinolina-5-carboxamida
N-{2-[4-(3,4-difluorofenoxi)piperidin-1-il]-2-(4-metil-1,3-tiazol-5-il)etil}quinolina-5-carboxamida
2-cloro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-{4-[(2-fluorofenil)metoxi]piperidin-1-il}etil}-6 -fluorobenzamida 2-doro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-[(3,5-difluorofenil)metoxi]piperidin-1-il}etil}-6-fluorobenzamida 2-doro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-{4-[(3,4-difluorofenil)metoxi-1-il} etil}-6-fluorobenzamida 2-doro-N-{2-[4-(4-dorofenoxi)piperidin-1-il]-2-[4-(difluorometil)-1,3-tiazol-5-il]etil}-6-fluorobenzamida 2-cloro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(3-fluorofenoxi)piperidin-1-il]etil}-6-fluorobenzamida 2-cloro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(2-fluorofenoxi)piperidin-1-il]etil}-6-fluorobenz
2-cloro-N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(3,5-difluorofenoxi)piperidin-1-il]etil}-6-fluorobenzamida N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-{4-[(2-fluorofenil)metoxi]piperidin-1-il}etil}quinolina-5-carboxamida N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-{4-[(3,4-difluorofenil)metoxi]piperidin-1-il}etil}quinolina-5-carboxamida N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(4-fluorofenoxi)piperidin-1-il]etil}quinolin-5-carboxamida
N-{2-[4-(4-clorofenoxi)piperidin-1-il]-2-[4-(difluorometil)-1,3-tiazol-5-il]etil}quinolin-5-carboxamida
N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(3-fluorofenoxi)piperidin-1-il]etil}quinolin-5-carboxamida
N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(2-fluorofenoxi)piperidin-1-il]etil}quinolin-5-carboxamida
N-{2-[4-(difluorometil)-1,3-tiazol-5-il]-2-[4-(3,5-difluorofenoxi)piperidin-1-il]etil}quinolin-5-carboxamida
6. Unprocedimiento de preparación de un compuesto de fórmula (I) como se define en la reivindicación 1 que comprende los pasos de:
i) hacer reaccionar un compuesto de fórmula (II):

en la que los significados de n, R1, R5 y R6 son como se definieron anteriormente, con un compuesto de fórmula (III)

en la que los significados de R2, R3 y R4 son como se definieron anteriormente; o
con un compuesto de fórmula (IIIa):

en la que los significados de R2, R3 y R4 son como se definieron anteriormente; y W es un grupo saliente adecuado;
y opcionalmente convertir el compuesto obtenido de fórmula (I) en una sal del mismo, y/o preparar formas estereoquímicamente isoméricas del mismo.
7. Una formulación farmacéutica que comprende un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, y un diluyente y/o vehículo farmacéuticamente aceptable.
8. Un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 5, o una sal farmacéuticamente aceptable del mismo que incluye cualquier forma estereoquímicamente isomérica del mismo, para su uso como medicamento.
9. Un compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 5 para su uso en la prevención y/o tratamiento de afecciones o enfermedades seleccionadas de afecciones o enfermedades mediadas por el receptor P2X7.
10. Un compuesto de fórmula (I) de acuerdo con la reivindicación 9 para su uso en la prevención y/o tratamiento de trastornos neurodegenerativos, cognitivos, psiquiátricos, dolor neuropático, dolor crónico, procesos inflamatorios del sistema musculoesquelético, trastornos del tracto gastrointestinal, trastornos del tracto genitourinario, enfermedades oftálmicas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16186655.3A EP3290416A1 (en) | 2016-08-31 | 2016-08-31 | Substituted n-[2-(4-phenoxypiperidin-1-yl)-2-(1,3-thiazol-5-yl)ethyl]benzamide and n-[2-(4-benzyloxypiperidin-1-yl)-2-(1,3-thiazol-5-yl)ethyl]benzamide derivatives and their use as p2x7 receptor antagonist |
| PCT/EP2017/070163 WO2018041563A1 (en) | 2016-08-31 | 2017-08-09 | Substituted n-[2-(4-phenoxypiperidin-1-yl)-2-(1,3-thiazol-5-yl)ethyl]benzamide and n-[2-(4-benzyloxypiperidin-1-yl)-2-(1,3-thiazol-5-yl)ethyl]benzamide derivatives p2x7 receptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2816204T3 true ES2816204T3 (es) | 2021-03-31 |
Family
ID=56852153
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17755110T Active ES2816204T3 (es) | 2016-08-31 | 2017-08-09 | Antagonistas del receptor p2x7 derivados de N-[2-(4-fenoxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida y N-[2-(4-benciloxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida sustituidas |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US10669267B2 (es) |
| EP (2) | EP3290416A1 (es) |
| JP (1) | JP6957595B2 (es) |
| CN (1) | CN109689650B (es) |
| CA (1) | CA3031354A1 (es) |
| DK (1) | DK3507287T3 (es) |
| ES (1) | ES2816204T3 (es) |
| HU (1) | HUE050040T2 (es) |
| IL (1) | IL264708B (es) |
| PL (1) | PL3507287T3 (es) |
| PT (1) | PT3507287T (es) |
| RU (1) | RU2755705C2 (es) |
| SI (1) | SI3507287T1 (es) |
| WO (1) | WO2018041563A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4015039A1 (en) | 2020-12-18 | 2022-06-22 | AXXAM S.p.A. | Heterocyclic derivatives as p2x7 receptor antagonists |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10237722A1 (de) * | 2002-08-17 | 2004-08-19 | Aventis Pharma Deutschland Gmbh | Indol- oder Benzimidazolderivate zur Modulation der IKappaB-Kinase |
| ES2396565T3 (es) | 2003-05-12 | 2013-02-22 | Pah Usa 15 Llc | Inhibidores benzamida del receptor P2X7 |
| WO2009108551A2 (en) | 2008-02-25 | 2009-09-03 | H. Lundbeck A/S | Heteroaryl amide analogues |
| ATE533762T1 (de) | 2008-04-22 | 2011-12-15 | Janssen Pharmaceutica Nv | Chinolin- oder isochinolinsubstituierte p2x7- antagonisten |
| TWI612034B (zh) * | 2012-12-12 | 2018-01-21 | 愛杜西亞製藥有限公司 | 作爲p2x7受體拮抗劑之吲哚羧醯胺衍生物 |
| JP6285736B2 (ja) | 2014-02-04 | 2018-02-28 | 株式会社ソニー・インタラクティブエンタテインメント | 情報処理装置および入力デバイスの割当方法 |
| EP2905282A1 (en) | 2014-02-05 | 2015-08-12 | AXXAM S.p.A. | Substituted thiazole or oxazole as P2X7 receptor antagonists |
-
2016
- 2016-08-31 EP EP16186655.3A patent/EP3290416A1/en not_active Withdrawn
-
2017
- 2017-08-09 CA CA3031354A patent/CA3031354A1/en active Pending
- 2017-08-09 DK DK17755110.8T patent/DK3507287T3/da active
- 2017-08-09 CN CN201780049645.6A patent/CN109689650B/zh active Active
- 2017-08-09 SI SI201730370T patent/SI3507287T1/sl unknown
- 2017-08-09 PT PT177551108T patent/PT3507287T/pt unknown
- 2017-08-09 PL PL17755110T patent/PL3507287T3/pl unknown
- 2017-08-09 HU HUE17755110A patent/HUE050040T2/hu unknown
- 2017-08-09 US US16/327,854 patent/US10669267B2/en active Active
- 2017-08-09 RU RU2019106209A patent/RU2755705C2/ru active
- 2017-08-09 JP JP2019503211A patent/JP6957595B2/ja active Active
- 2017-08-09 WO PCT/EP2017/070163 patent/WO2018041563A1/en active Search and Examination
- 2017-08-09 ES ES17755110T patent/ES2816204T3/es active Active
- 2017-08-09 EP EP17755110.8A patent/EP3507287B1/en active Active
-
2019
- 2019-02-07 IL IL264708A patent/IL264708B/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| RU2019106209A (ru) | 2020-10-01 |
| CA3031354A1 (en) | 2018-03-08 |
| IL264708B (en) | 2021-04-29 |
| CN109689650A (zh) | 2019-04-26 |
| SI3507287T1 (sl) | 2020-09-30 |
| DK3507287T3 (da) | 2020-08-24 |
| PT3507287T (pt) | 2020-09-04 |
| US10669267B2 (en) | 2020-06-02 |
| US20190194180A1 (en) | 2019-06-27 |
| CN109689650B (zh) | 2022-11-25 |
| EP3507287B1 (en) | 2020-07-15 |
| EP3507287A1 (en) | 2019-07-10 |
| HUE050040T2 (hu) | 2020-11-30 |
| JP2019524750A (ja) | 2019-09-05 |
| RU2755705C2 (ru) | 2021-09-20 |
| WO2018041563A1 (en) | 2018-03-08 |
| EP3290416A1 (en) | 2018-03-07 |
| RU2019106209A3 (es) | 2020-10-01 |
| JP6957595B2 (ja) | 2021-11-02 |
| PL3507287T3 (pl) | 2020-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK3102573T3 (en) | Substituted thiazole or oxazole P2X7 receptor antagonists | |
| EP3821947A1 (en) | Heterocyclic trpml1 agonists | |
| ES2816204T3 (es) | Antagonistas del receptor p2x7 derivados de N-[2-(4-fenoxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida y N-[2-(4-benciloxipiperidin-1-il)-2-(1,3-tiazol-5-il)etil]benzamida sustituidas | |
| ES2911049T3 (es) | Antagonistas heterocíclicos de P2X7 | |
| CN114929681B (zh) | 二环cx3cr1受体激动剂 | |
| EP3290417A1 (en) | 2-chloro-n-[2-(1,3-thiazol-5-yl)ethyl]-5-(5-fluoropyrimidin-2-yl)-benzamides and their use as p2x7 receptor antagonists | |
| WO2023031319A1 (en) | 2,4-dihydro-3h-1,2,4-triazol-3-one p2x7 antagonists | |
| WO2022129365A1 (en) | Heterocyclic derivatives as p2x7 receptor antagonists | |
| TW202115023A (zh) | 新型細胞凋亡訊號調節激酶1抑制劑 |