ES2765013T3 - Procedimiento para preparar derivados de quinolina - Google Patents
Procedimiento para preparar derivados de quinolina Download PDFInfo
- Publication number
- ES2765013T3 ES2765013T3 ES12783763T ES12783763T ES2765013T3 ES 2765013 T3 ES2765013 T3 ES 2765013T3 ES 12783763 T ES12783763 T ES 12783763T ES 12783763 T ES12783763 T ES 12783763T ES 2765013 T3 ES2765013 T3 ES 2765013T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mixture
- formula
- acid
- polar aprotic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- KDXROTVXLGAFGW-UHFFFAOYSA-N OC(C1(CC1)C(Nc1ccccc1)=O)=O Chemical compound OC(C1(CC1)C(Nc1ccccc1)=O)=O KDXROTVXLGAFGW-UHFFFAOYSA-N 0.000 description 3
- ADVVEQBFECDPKP-UHFFFAOYSA-N CC(C1)C(F)=CC=C1NC(C1(CC1)C(O)=O)=O Chemical compound CC(C1)C(F)=CC=C1NC(C1(CC1)C(O)=O)=O ADVVEQBFECDPKP-UHFFFAOYSA-N 0.000 description 1
- SIDZXXQYQLBOHZ-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N)c1)c1F SIDZXXQYQLBOHZ-UHFFFAOYSA-N 0.000 description 1
- CXQHYVUVSFXTMY-UHFFFAOYSA-N COc(c(OCCCN1CCOCC1)c1)cc2c1nccc2Oc(ccc(NC(C1(CC1)C(Nc(cc1)ccc1F)=O)=O)c1)c1F Chemical compound COc(c(OCCCN1CCOCC1)c1)cc2c1nccc2Oc(ccc(NC(C1(CC1)C(Nc(cc1)ccc1F)=O)=O)c1)c1F CXQHYVUVSFXTMY-UHFFFAOYSA-N 0.000 description 1
- DDYIDQLZMNTOCH-UHFFFAOYSA-N O=C(C1(CC1)C(Nc(cc1)ccc1F)=O)Nc(cc1)ccc1F Chemical compound O=C(C1(CC1)C(Nc(cc1)ccc1F)=O)Nc(cc1)ccc1F DDYIDQLZMNTOCH-UHFFFAOYSA-N 0.000 description 1
- PFMAFXYUHZDKPY-UHFFFAOYSA-N OC(C1(CC1)C(Nc(cc1)ccc1F)=O)=O Chemical compound OC(C1(CC1)C(Nc(cc1)ccc1F)=O)=O PFMAFXYUHZDKPY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
- C07D215/233—Oxygen atoms attached in position 2 or 4 only one oxygen atom which is attached in position 4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/58—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/59—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by halogen atoms or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
- C07D239/90—Oxygen atoms with acyclic radicals attached in position 2 or 3
Abstract
Un proceso para preparar un compuesto de Fórmula A: **(Ver fórmula)** en la que R2 es H, F, Cl, o Br; que comprende (a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar, en la que el disolvente aprótico polar es acetato de isopropilo, a una temperatura de 23 a 27 °C; y (b) añadir **(Ver fórmula)** y una base de amina terciaria a la mezcla de la etapa (a) para proporcionar el compuesto de Fórmula A.
Description
DESCRIPCIÓN
Procedimiento para preparar derivados de quinolina
CAMPO DE LA INVENCIÓN
[0001] La presente descripción se refiere a un procedimiento para preparar compuestos útiles para modular la actividad enzimática de las cinasas de proteínas. Más específicamente, la presente descripción se refiere a un procedimiento para reparar compuestos útiles para modular actividades celulares tales como la proliferación, la diferenciación, la muerte celular programada, la migración y la quimioinvasión.
ANTECEDENTES DE LA INVENCIÓN
[0002] La modulación (particularmente la inhibición) de la proliferación celular y de la angiogénesis, dos procesos celulares clave necesarios para el crecimiento y la supervivencia tumorales (Matter A. Drug Disc Technol 2001 6, 1005-1024) es un objetivo atractivo para el desarrollo de fármacos de molécula pequeña. Una terapia antiangiogénica representa una estrategia potencialmente importante para el tratamiento de tumores sólidos y de otras enfermedades asociadas a una vascularización desregulada, incluyendo enfermedad coronaria isquémica, retinopatía diabética, psoriasis y artritis reumatoide. Asimismo, son deseables agentes antiproliferativos para ralentizar o detener el crecimiento de los tumores.
[0003] Un objetivo de este tipo para la modulación con moléculas pequeñas de la actividad antiangiogénica y antiproliferativa es la c-Met. La cinasa c-Met es el miembro prototipo de una subfamilia de cinasas de tirosina del receptor heterodimérico (RTK) que incluyen Met, Ron y Sea. La expresión de la c-Met se produce en una gran diversidad de tipos celulares que incluyen células epiteliales, endoteliales y mesenquimatosas, donde la activación del receptor induce la migración, la invasión, la proliferación celular y otras actividades biológicas asociadas con "el crecimiento celular invasivo". Como tal, la transducción de señales a través de la activación del receptor c-Met es responsable de muchas de las características de las células tumorales.
[0004] N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida y N-[3-fluoro-4-({6-(metiloxi)-7-[(3-morfolin-4-ilpropil)oxi]quinolin-4-il}oxi)fenil]-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida son dos inhibidores de molécula pequeña de c-Met que actualmente se encuentran en investigación clínica como tratamientos para una diversidad de cánceres. Por consiguiente, existe la necesidad continua de procesos nuevos y eficientes para realizar estas dos prometedoras terapias contra el cáncer.
[0005] O. Saavedra et al., Bioorganic & Medicinal Chemistry Letters, vol. 19, n.° 24, 15 de diciembre de 2009, páginas 6836-6839, describen la preparación de una serie de inhibidores de molécula pequeña a base de tieno[3,2-ó]piridina de c-Met y VEGFR2 que tienen la funcionalidad M3-arilmalonamida. El documento también muestra la preparación de la monoanilida de ácido 1,1-ciclopropanodicarboxílico haciendo reaccionar el ácido libre con cloruro de tionilo y posteriormente con anilina y trietilamina en THF a 0 °C.
RESUMEN DE LA INVENCIÓN
[0006] Estas y otras necesidades se satisfacen mediante la presente invención que se refiere, en un primer aspecto, a un proceso para preparar un compuesto de Fórmula A:

A
en la que R2 es H, F, Cl, o Br;
que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar, en la que el disolvente aprótico polar es acetato de isopropilo, a una temperatura de 23 a 27 °C; y
(b) añadir

y una base de amina terciaria a la mezcla de la etapa (a) para proporcionar el compuesto de Fórmula A.
[0007] En un segundo aspecto, la invención proporciona un proceso para preparar un compuesto de Fórmula I:

en la que:
R1 es halo;
R2 es halo;
R3 es alquilo (C1 -C6) o alquilo (C1 -C6) opcionalmente sustituido con heterocicloalquilo, en la que "heterocicloalquilo" significa un grupo monocíclico monovalente saturado o parcialmente insaturado de 3 a 8 átomos en el anillo, o un grupo bicíclico condensado monovalente saturado o parcialmente insaturado de 5 a 12 átomos en el anillo en el que uno o más heteroátomos en el anillo se seleccionan independientemente de -O-, -S(O)n-, -N=, y -N(Ry)-, siendo los átomos del anillo restantes carbono, y en los que uno o dos átomos de carbono en el anillo pueden reemplazarse por un grupo -C(O)-, -C(S)-, o C(=NH)-;
n es 0, 1, o 2;
Ry es hidrógeno, alquilo, hidroxi, alcoxi, acilo o alquilsulfonilo;
R4 es alquilo (C1 -C6) y
Q es CH o N;
que comprende:
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar, en la que el disolvente aprótico polar es acetato de isopropilo, a una temperatura de 23 a 27 °C;
(b) añadir

y una base de amina terciaria a la mezcla de la etapa (a) para formar un compuesto de Fórmula A:

A
(c) acoplar un compuesto de Fórmula A con una amina de Fórmula B para formar un compuesto de Fórmula I:

[0008] Más generalmente, la presente descripción proporciona un proceso para preparar un compuesto de Fórmula A:

en la que R2 es H, F, Cl, o Br;
que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; y (b) añadir

y una base de amina terciaria a la mezcla de la etapa (a).
[0009] El compuesto de Fórmula A se usa para formar un compuesto de Fórmula I:

en la que:
R1 es halo;
R2 es halo;
R3 es alquilo (C1-C6) o alquilo (C1-C6) opcionalmente sustituido con heterocicloalquilo;
R4 es alquilo (C-i-Ca); y Q es CH o N.
[0010] En una realización de la descripción, el compuesto de Fórmula I es el compuesto 1:

Compuesto 1
o una sal farmacéuticamente aceptable del mismo. El Compuesto 1 se conoce como N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi} fenil)-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida. El documento WO 2005/030140 describe la síntesis de N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida (Ejemplo 12, 37, 38, y 48) y también describe la actividad terapéutica de esta molécula para inhibir, regular y/o modular la transducción de señales de las cinasas (Ensayos, Tabla 4, entrada 289). El Ejemplo 48 está en el párrafo en el documento WO 2005/030140.
[0011] En otra realización de la descripción, el compuesto de Fórmula I es el compuesto 2:

Compuesto 2
o una sal farmacéuticamente aceptable del mismo. El Compuesto 2 se conoce como N-[3-fluoro-4-({6-(metiloxi)-7-[(3-morfolin-4-ilpropil)oxi] quinolin-4-il}oxi)fenil]-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida. El documento WO 2005 030140 describe la síntesis del Compuesto (I) (Ejemplos 25, 30, 36, 42, 43 y 44) y también describe la actividad terapéutica de esta molécula para inhibir, regular, y/o modular la transducción de señales de las cinasas (Ensayos, Tabla 4, entrada 312). Se ha medido que el Compuesto 2 tiene un valor de CI50 de c-Met de aproximadamente 0,6 nanomolar (nM). El documento PCT/US09/064341, que reivindica la prioridad de la solicitud provisional de EE.UU.
61/199.088, depositada el 13 de noviembre de 2008, describe una síntesis ampliada del compuesto 2.
[0012] Por lo tanto, en otro aspecto, la descripción se refiere a un proceso para preparar un compuesto de Fórmula I como se ha definido anteriormente:

(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; (b) añadir

y una base de amina terciaria a la mezcla de la etapa (a) para formar un compuesto de Fórmula A; y

(c) acoplar un compuesto de Fórmula A con una amina de Fórmula B para formar un compuesto de Fórmula I

[0013] El Compuesto de Fórmula B puede prepararse como se describe en el documento WO 2005/030140, como se ha mencionado previamente. Los enfoques alternativos a la síntesis del compuesto de Fórmula I, el compuesto A, el compuesto B, y los Compuestos 1 y 2 se describen en las solicitudes adicionales PCT/2009/643411 y PCT/US2010/021194.
[0014] El proceso de monoamidación descrito y reivindicado en el presente documento presenta varias ventajas de procesamiento significativas. Las estrategias anteriores para la elaboración del Compuesto A requerían la mezcla de ácido 1,1-ciclopropanodicarboxílico con trietil amina, y a continuación la adición de cloruro de tionilo seguido de la anilina. La reacción era típica e indeseablemente exotérmica. Los inventores encontraron que la exotermia se
eliminó mediante la reordenación de la secuencia de adición de los reactivos. Los tiempos de reacción se reducen significativamente, y el producto resultante no requiere ninguna purificación adicional. Además, el proceso como se describe en el presente documento es altamente selectivo para la formación del producto de monoamidación del Compuesto A.

sobre la bis-amida

La bis-amida, si está presente, se elimina fácilmente mediante el uso de las condiciones de aislamiento desarrolladas por los inventores.
[0015] El proceso como se describe en el presente documento es generalizable para la monoamidación selectiva de ácidos de dicarboxílicos simétricos mediante el uso de un conjunto de aminas primarias o secundarias. Por lo tanto, en otro aspecto, la descripción proporciona un proceso para la elaboración de una mono-amida a partir del ácido dicarboxílico correspondiente, que comprende:
(a) poner en contacto un ácido dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; y
(b) añadir una base de amina primaria y una amina terciaria a la mezcla resultante.
[0016] Hay muchos aspectos y realizaciones diferentes de la descripción que se describen en el presente documento a continuación, y cada aspecto y cada realización no es limitante con respecto al alcance de la descripción. Los términos "aspectos" y "realizaciones" no pretenden ser limitantes independientemente de donde aparezcan los términos "aspecto" o "realización" en esta memoria descriptiva. El término de transición "que comprende", como se usa en el presente documento, que es sinónimo de "que incluye", "que contiene", o "caracterizado por", es inclusivo o abierto y no excluye ningún elemento no citado adicional.
Descripción detallada de la invención
Abreviaturas y definiciones
[0017] Las siguientes abreviaturas y términos tienen los significados indicados en toda su extensión:

(continuación)

(continuación)

[0018] El símbolo significa un enlace simple; "=" significa un doble enlace.
[0019] Cuando se representan o describen estructuras químicas, salvo que explícitamente se indique de otro modo, se asume que todos los carbonos tienen sustitución de hidrógeno para conformar una valencia de cuatro. Por ejemplo, en la estructura de la izquierda del siguiente esquema de reacción hay nueve hidrógenos implícitos. Los nueve hidrógenos se representan en la estructura de la derecha. A veces, un átomo en particular de una estructura se describe en una fórmula textual como portador de hidrógeno o hidrógenos como una sustitución (hidrógeno definido expresamente), por ejemplo, -CH2CH2-. El experto en la técnica comprenderá que las técnicas descriptivas mencionadas anteriormente son comunes en la técnica química para proporcionar brevedad y simplicidad a la descripción de estructuras de otro modo complejas.

[0020] Si un grupo "R" está representado "flotando" en un sistema anular, como por ejemplo en la fórmula:

entonces, a menos que se defina de otro modo, un sustituyente "R" puede residir en cualquier átomo del sistema anular, asumiendo el reemplazo de un hidrógeno representado, implícito o definido expresamente de uno de los átomos del anillo, siempre que se forme una estructura estable.
[0021] Si un grupo "R" está representado flotando en un sistema anular condensado, como por ejemplo en las fórmulas:

entonces, a menos que se defina de otro modo, un sustituyente "R" puede residir en cualquier átomo del sistema anular condensado, asumiendo el reemplazo de un hidrógeno representado (por ejemplo, el -NH- en la fórmula
anterior), un hidrógeno implícito (por ejemplo, como la fórmula anterior, donde los hidrógenos no se muestran pero se entiende que están presentes), o un hidrógeno definido expresamente (por ejemplo, cuando en la fórmula anterior, "Z" equivale a =CH-) de uno de los átomos del anillo, siempre que se forme una estructura estable. En el ejemplo representado, el grupo "R" puede residir en cualquiera de los anillos de 5 miembros o 6 miembros del sistema anular condensado. Cuando se representa que un grupo "R" existe en un sistema anular que contiene carbonos saturados, como por ejemplo, en la fórmula:

donde, en este ejemplo, "y" puede ser más de uno, asumiendo que cada uno reemplaza un hidrógeno actualmente representado, implícito o definido expresamente del anillo; entonces, a menos que se defina de otro modo, cuando la estructura resultante sea estable, dos "R" pueden residir en el mismo carbono. Un ejemplo sencillo es cuando R es un grupo metilo; puede existir un dimetilo geminal en un carbono del anillo representado (un carbono "anular"). En otro ejemplo, dos R en el mismo carbono, incluyendo ese carbono, pueden formar un anillo, creando así una estructura de un anillo espirocíclico (un grupo "espirociclilo") con el anillo representado como, por ejemplo, en la fórmula.

[0022] "Alquilo (C1-C6)" o "alquilo" significa un grupo hidrocarburo lineal o ramificado que tiene de uno a seis átomos de carbono. Algunos ejemplos de grupos alquilo inferior incluyen metilo, etilo, propilo, isopropilo, butilo, sbutilo, f-butilo, isobutilo, pentilo, hexilo, y similares. "Alquilo C6" se refiere, por ejemplo, a n-hexilo, iso-hexilo, y similares.
[0023] "Heterocicloalquilo" significa un grupo monocíclico monovalente saturado o parcialmente insaturado de 3 a 8 átomos en el anillo o un grupo bicíclico condensado monovalente saturado o parcialmente insaturado de 5 a 12 átomos en el anillo, en el que uno o más, por ejemplo, uno, dos, tres o cuatro heteroátomos en el anillo se seleccionan independientemente de -O-, -S(O)n-(n es 0, 1, o 2), -N=, -N(Ry)-(donde Ry es hidrógeno, alquilo, hidroxi, alcoxi, acilo, o alquilsulfonilo), siendo los átomos del anillo restantes carbono. Uno o dos átomos de carbono del anillo pueden estar reemplazados por un grupo -C(O)-, -C(S)-, o -C(=NH)-. El radical bicíclico condensado incluye sistemas anulares de puente. A menos que se indique de otro modo, la valencia del grupo puede estar situada en cualquier átomo de cualquier anillo del radical, cuando las normas de valencia lo permitan. En particular, cuando el punto de valencia está situado en un átomo de nitrógeno, Ry está ausente. En otra realización el término heterocicloalquilo incluye, pero sin limitación, azetidinilo, pirrolidinilo, 2-oxopirrolidinilo, 2,5-dihidro-1 H-pirrolilo, piperidinilo, 4-piperidonilo, morfolinilo, piperazinilo, 2-oxopiperazinilo, tetrahidropiranilo, 2-oxopiperidinilo, tiomorfolinilo, tiamorfolinilo, perhidroazepinilo, pirazolidinilo, imidazolinilo, imidazolidinilo, dihidropiridinilo, tetrahidropiridinilo, oxazolinilo, oxazolidinilo, isoxazolidinilo, tiazolinilo, tiazolidinilo, quinuclidinilo, isotiazolidinilo, octahidroindolilo, octahidroisoindolilo, decahidroisoquinolilo, tetrahidrofurilo, y tetrahidropiranilo, y los derivados de los mismos, y N-óxido o un derivado protegido de los mismos.
[0024] "Halógeno" o "halo" se refiere a flúor, cloro, bromo o yodo.
[0025] "Rendimiento", para cada una de las reacciones descritas en el presente documento, se expresa como un porcentaje del rendimiento teórico.
[0026] "Paciente", para los propósitos de la presente descripción, incluye seres humanos y otros animales, particularmente mamíferos, y otros organismos. Por lo tanto, los procesos son aplicables tanto en terapia humana como en aplicaciones veterinarias. En otra realización, el paciente es un mamífero, y en otra realización el paciente es un ser humano.
[0027] Una "sal farmacéuticamente aceptable" de un compuesto significa una sal que es farmacéuticamente aceptable y que posee la actividad farmacológica deseada del compuesto precursor. Se entiende que las sales farmacéuticamente aceptables no son tóxicas. Puede encontrarse información adicional sobre sales farmacéuticamente aceptables adecuadas en Remington's Pharmaceutical Sciences, 17a ed., Mack Publishing Company, Easton, PA, 1985, o S. M. Berge, et al., "Pharmaceutical Salts", J. Pharm. Sci., 1977;66:1-19.
[0028] Ejemplos de sales de adición de ácidos farmacéuticamente aceptables incluyen aquellas formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, y similares; así como ácidos orgánicos tales como ácido acético, ácido trifluoroacético, ácido propiónico, ácido hexanoico, ácido ciclopentanopropiónico, ácido glicólico, ácido pirúvico, ácido láctico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido málico, ácido cítrico, ácido benzoico, ácido cinámico, ácido 3-(4-hidroxibenzoil)benzoico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2etanodisulfónico, ácido 2-hidroxietanosulfónico, ácido bencenosulfónico, ácido 4-clorobencenosulfónico, ácido 2-naftalenosulfónico, ácido 4-toluenosulfónico, ácido canforsulfónico, ácido glucoheptónico, ácido 4,4'-metilenbis-(3-hidroxi-2-eno-1-carboxílico), ácido 3-fenilpropiónico, ácido trimetilacético, ácido butilacético terciario, ácido lauril sulfúrico, ácido glucónico, ácido glutámico, ácido hidroxinaftoico, ácido salicílico, ácido esteárico, ácido mucónico, ácido p-toluenosulfónico y ácido salicílico, y similares.
[0029] "Profármaco" se refiere a compuestos que son transformados (típicamente rápidamente) in vivo para producir el compuesto parental de las fórmulas anteriores, por ejemplo, mediante hidrólisis en la sangre. Algunos ejemplos habituales incluyen, pero sin limitación, las formas de éster y amida de un compuesto con una forma activa portadora de un resto de ácido carboxílico. Ejemplos de ésteres farmacéuticamente aceptables de los compuestos de esta descripción incluyen, pero sin limitación, ésteres de alquilo (por ejemplo, con entre aproximadamente uno y aproximadamente seis carbonos), el grupo alquilo es una cadena lineal o ramificada. Los ésteres aceptables también incluyen ésteres de cicloalquilo y ésteres de arilalquilo tales como, pero sin limitación, bencilo. Ejemplos de amidas farmacéuticamente aceptables de los compuestos de esta descripción incluyen, pero sin limitación, amidas primarias y alquil amidas secundarias y terciarias (por ejemplo, con entre aproximadamente uno y aproximadamente seis carbonos). Las amidas y los ésteres de los compuestos de la presente descripción pueden prepararse según procesos convencionales. Se proporciona un análisis exhaustivo de profármacos en T. Higuchi y V. Stella, "Pro-drugs as Novel Delivery Systems", Vol 14 of the A.C.S. Symposium Series, y en Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
[0030] "Cantidad terapéuticamente eficaz" es una cantidad de un compuesto de la descripción, que cuando se administra a un paciente, mejora un síntoma de la enfermedad. Una cantidad terapéuticamente eficaz pretende incluir una cantidad de un compuesto en solitario o junto con otros principios activos eficaces para modular la c-Met, y/o el VEGFR2, o eficaces para tratar o prevenir el cáncer. La cantidad de un compuesto de la descripción que constituye una "cantidad terapéuticamente eficaz" variará dependiendo del compuesto, de la patología y su gravedad, la edad del paciente a tratar, y similares. La cantidad terapéuticamente eficaz puede determinarse por el experto en la técnica considerando su conocimiento y esta descripción.
[0031] "Tratar" o "tratamiento" de una enfermedad, trastorno o síndrome, como se usa en el presente documento, incluye (i) prevenir la aparición de la enfermedad, trastorno o síndrome en un ser humano, es decir, hacer que los síntomas clínicos de la enfermedad, trastorno o síndrome no se desarrollen en un animal que pueda estar expuesto o esté predispuesto a la enfermedad, trastorno o síndrome, pero que aún no haya experimentado ni muestre los síntomas de la enfermedad, trastorno o síndrome; (ii) inhibir la enfermedad, trastorno o síndrome, es decir, detener su desarrollo; y (iii) aliviar la enfermedad, trastorno o síndrome, es decir, provocar la regresión de la enfermedad, trastorno o síndrome. Como se conoce en la técnica, pueden ser necesarios ajustes para una administración sistémica frente a localizada, la edad, el peso corporal, la salud general, el sexo, la dieta, el tiempo de administración, las interacciones farmacológicas y la gravedad de la afección, y pueden averiguarse con la experiencia rutinaria.
Proceso
[0032] En un aspecto, esta descripción se refiere a un proceso para preparar un compuesto de Fórmula A:

en la que R2 es H, F, Cl, o Br; que comprende
(a) poner en contacto ácido 1,1-ciclopropanodicarboxílico con cloruro de tionilo en un disolvente aprótico polar; y (b) añadir

y una base de amina terciaria a la mezcla de la etapa (a).
[0033] En el proceso, el ácido 1,1-ciclopropanodicarboxílico se combina con un disolvente aprótico polar para
formar una mezcla. En una realización de la descripción, el disolvente aprótico polar se selecciona del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, acetato de isopropilo, acetona, dimetilformamida, acetonitrilo, y dimetilsulfóxido, o combinaciones de los mismos. En otra realización de la descripción, el disolvente aprótico polar se selecciona del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, acetato de isopropilo, acetona, dimetilformamida, y acetonitrilo, o combinaciones de los mismos. En otra realización de la descripción, el disolvente aprótico polar se selecciona del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, y acetato de isopropilo, o combinaciones de los mismos. En una realización de la descripción, el disolvente aprótico polar es acetato de isopropilo.
[0034] El volumen de disolvente aprótico polar usado variará dependiendo de la escala de la reacción. Típicamente, se usan aproximadamente 5-10 volúmenes de ácido aprótico polar con respecto al volumen de ácido 1,1-ciclopropanodicarboxílico que se usa. Más típicamente, se usan 6-9 volúmenes de ácido aprótico polar. Más típicamente, se usan 7,5-8,5 volúmenes de ácido aprótico polar. Preferentemente, se usan aproximadamente 8 volúmenes del ácido aprótico polar.
[0035] A continuación, se añade cloruro de tionilo a la mezcla que comprende ácido 1,1-ciclopropanodicarboxílico y el ácido aprótico polar. Se usa un exceso molar de cloruro de tionilo con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico que se usa. Típicamente, se usan aproximadamente de 1,01 a 1,5 equivalentes molares de cloruro de tionilo con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico que se usan. Más típicamente, se usan aproximadamente de 1,01 a 1,2 equivalentes molares de cloruro de tionilo. Más típicamente, se usan aproximadamente de 1,01 a 1,1 equivalentes molares de cloruro de tionilo. Más típicamente, se usan aproximadamente 1,05 equivalentes molares de cloruro de tionilo.
[0036] La mezcla que comprende ácido 1,1-ciclopropanodicarboxílico, cloruro de tionilo, y el disolvente aprótico polar se sacude o se agita de otro modo durante 2 a 24 horas. La "temperatura ambiente" significa generalmente que no se emplea ningún dispositivo de calentamiento externo, tal como una funda calefactora, un revestimiento calefactor, o similares, para aumentar la temperatura de la mezcla. Típicamente, la temperatura es aproximadamente de 23 a 27 °C. Más típicamente, la temperatura es aproximadamente de 24 a 26 °C. Típicamente, la temperatura es aproximadamente 25 °C. La agitación a temperatura ambiente continúa típicamente durante aproximadamente 6 a 16 horas. Más típicamente, la agitación continúa durante aproximadamente 13-15 horas a aproximadamente 25 °C.
[0037] A continuación, a la mezcla se le añade una mezcla de una anilina opcionalmente sustituida

y una base de amina terciaria en un disolvente aprótico polar. Típicamente, la anilina opcionalmente sustituida es 4-fluoroanilina.
[0038] Se usa un exceso molar de anilina con respecto al número de moles de ácido 1,1 ciclopropanodicarboxílico. Típicamente, se usan aproximadamente de 1,01 a 1,5 equivalentes molares de anilina con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico que se usan. Más típicamente, se usan aproximadamente de 1,01 a 1,2 equivalentes molares de anilina. Más típicamente, se usan aproximadamente de 1,05 a 1,15 equivalentes molares de anilina. Más típicamente, se usan aproximadamente 1,1 equivalentes molares de anilina.
[0039] La base de amina terciaria es típicamente una trialquil amina, en la que los grupos alquilo son iguales o diferentes y pueden ser lineales o ramificados. El uso de bases de trialquil amina se conoce bien por el experto, y muchas están disponibles comercialmente, tales como trietilamina, diisopropiletil amina, o similares. Típicamente, la base de amina terciaria es trietil amina. Se usa un exceso molar de la base de amina terciaria con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico. Típicamente, se usan aproximadamente de 1,01 a 1,5 equivalentes molares de una base de amina terciaria con respecto al número de moles de 1,1-ciclopropanodicarboxílico que se usan. Más típicamente, se usan aproximadamente de 1,01 a 1,2 equivalentes molares de una base de amina terciaria. Más típicamente, se usan aproximadamente de 1,05 a 1,15 equivalentes molares de anilina. Más típicamente, se usan aproximadamente 1,1 equivalentes molares de una base de amina terciaria.
[0040] La anilina opcionalmente sustituida y la base de amina terciaria se combinan típicamente en un disolvente aprótico polar antes de añadirse a la mezcla de ácido 1,1-ciclopropanodicarboxílico/cloruro de tionilo/acetato de isopropilo. El disolvente aprótico polar que se usa es típicamente el mismo que el disolvente que se usa para formar la mezcla de ácido 1,1-ciclopropanodicarboxílico, y se selecciona del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, acetato de isopropilo, acetona, dimetilformamida, acetonitrilo, y dimetilsulfóxido, o combinaciones de los mismos. En otra realización de la descripción, el disolvente aprótico polar se selecciona del
grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, acetato de isopropilo, acetona, dimetilformamida, y acetonitrilo, o combinaciones de los mismos. En otra realización de la descripción, el disolvente aprótico polar se selecciona del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, y acetato de isopropilo, o combinaciones de los mismos. En una realización de la descripción, el disolvente aprótico polar es acetato de isopropilo.
[0041] El volumen de disolvente aprótico polar usado para formar la mezcla de anilina/base de amina terciaria variará dependiendo de la escala de la reacción. Típicamente, se usan aproximadamente 1-5 volúmenes de ácido aprótico polar con respecto al volumen de la anilina opcionalmente sustituida que se usa. Más típicamente, se usan 1,5-3 volúmenes de ácido aprótico polar. Más típicamente, se usan aproximadamente 2 volúmenes de ácido aprótico polar.
[0042] La mezcla combinada resultante se deja mezclar a temperatura ambiente durante 0,5 a 5 horas, y más preferiblemente de 1 a 3 horas. Más típicamente, la mezcla se deja mezclar durante 2 horas.
[0043] La mezcla, que en este punto es típicamente una suspensión que comprende el Compuesto A, se inactiva a continuación mediante tratamiento con una base acuosa concentrada tal como NaOH acuoso 5 N, KOH, o K3PO4, o similares. En una realización de la descripción, la base es NaOH. La cantidad de base acuosa empleada para inactivar la reacción variará dependiendo de la escala de la reacción. Para la escala descrita anteriormente, se usan típicamente aproximadamente 4-6 volúmenes de NaOH 5 N. La fase orgánica de la mezcla bifásica resultante se extrae entonces con múltiples lavados de NaOH 0,5 N, y las fases acuosas se combinan. Los extractos básicos combinados se extraen de nuevo con un disolvente aprótico tal como heptano. A continuación, las bases acuosas combinadas se acidifican posteriormente con un ácido mineral acuoso tal como HCl, H2SO4, o similares. Típicamente, el ácido usado es un 30 por ciento de HCl en agua. El ácido se añade a las fases acuosas combinadas para formar una suspensión. A continuación, se aísla el Compuesto A mediante filtración.
[0044] En una realización adicional de la descripción, se proporciona un proceso para preparar un compuesto de Fórmula A:

en la que R2 es H, F, Cl, o Br; que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a temperatura ambiente; y
(b) añadir una mezcla que comprende

y trietil amina en acetato de isopropilo a la mezcla resultante.
[0045] En una realización adicional de la descripción, se proporciona un proceso para preparar un compuesto de Fórmula A:

en la que R2 es H, F, Cl, o Br; que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a
temperatura ambiente;
(b) añadir

y una trietil amina a la mezcla;
(c) inactivar la mezcla de la etapa (b) con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A en una base acuosa diluida
(e) acidificar la mezcla con HCl; y
(f) aislar el Compuesto A por filtración.
[0046] En una realización adicional de la descripción, se proporciona un proceso para preparar un compuesto de Fórmula A-1:

A-1
que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a temperatura ambiente; y
(b) añadir una mezcla que comprende 4-fluoroanilina y una trietil amina en acetato de isopropilo a la mezcla resultante.
[0047] En una realización adicional de la descripción, se proporciona un proceso para preparar un compuesto de Fórmula A-1:

A-1
que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a temperatura ambiente;
(b) añadir una mezcla que comprende 4-fluoroanilina y una trietil amina en acetato de isopropilo a la mezcla resultante;
(c) inactivar la mezcla con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A-1 en una base acuosa diluida;
(e) acidificar la mezcla con HCl; y
(f) aislar el Compuesto A por filtración.
[0048] En otra realización, la descripción se refiere a un proceso para preparar el Compuesto 1:

Compuesto 1
que comprende las etapas de:
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; (b) añadir 4-fluoroanilina y trietil amina a la mezcla de la etapa (a) para formar un compuesto de Fórmula A; y

A-1
(c) acoplar un compuesto de Fórmula A-1 con una amina de Fórmula B-1 para formar el Compuesto 1.

[0049] En otra realización, la descripción se refiere a un proceso para preparar el Compuesto 1:

Compuesto 1
que comprende las etapas de:
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; (b) añadir 4-fluoroanilina y trietil amina a la mezcla de la etapa (a) para formar un compuesto de Fórmula A;

A-1
(c) inactivar la mezcla con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A-1 en una base acuosa diluida;
(e) acidificar la mezcla con HCl;
(f) aislar el compuesto de Fórmula A-1 por filtración; y
(g) acoplar un compuesto de Fórmula A-1 con una amina de Fórmula B-1 para formar el Compuesto 1.

[0050] En otra realización, la descripción se refiere a un proceso para preparar el Compuesto 2:

(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; (b) añadir 4-fluoroanilina y trietil amina a la mezcla de la etapa (a) para formar un compuesto de Fórmula A; y

A-1
(c) acoplar un compuesto de Fórmula A-1 con una amina de Fórmula B-2 para formar el Compuesto 1.

[0051] En otra realización, la descripción se refiere a un proceso para preparar el Compuesto 1:

(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar; (b) añadir 4-fluoroanilina y trietil amina a la mezcla de la etapa (a) para formar un compuesto de Fórmula A;
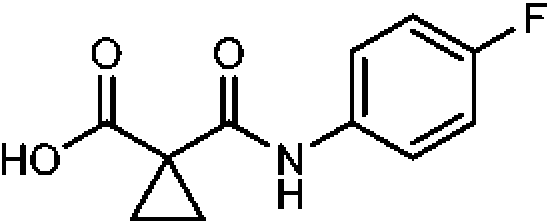
A-1
(c) inactivar la mezcla con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A-1 en una base acuosa diluida;
(e) acidificar la mezcla con HCl;
(f) aislar el compuesto de Fórmula A-1 por filtración; y
(g) acoplar un compuesto de Fórmula A-1 con una amina de Fórmula B-1 para formar el Compuesto 1.

[0052] Como se describe en el presente documento, la reacción de cloruro de tionilo con ácido 1,1-ciclopropanodicarboxílico en un disolvente aprótico polar como se describe en el presente documento, ofrece una ventaja significativa sobre los procesos anteriores ya que la reacción no es exotérmica. Una variante de reacción anterior en la que SOCh se añadió a una mezcla de ácido 1,1-ciclopropanodicarboxílico y Et3N en tetrahidrofurano era muy exotérmica. El proceso como se describe en el presente documento es digno de atención ya que los ácidos carboxílicos no se convierten normalmente en los correspondientes cloruros de acilo cuando se tratan con SOCh a temperatura ambiente.
[0053] El proceso como se describe en el presente documento es altamente selectivo para la formación del producto de monoamidación del Compuesto A sobre la bis-amida.

Típicamente, se forma menos del 5 por ciento, o más típicamente menos del 1 por ciento, de la bis-amida a través del proceso como se reivindica en el presente documento, según se evidencia mediante un análisis por HPLC de las muestras de control del proceso. Además, la bis-amida, si está presente, normalmente se elimina completamente usando las condiciones de aislamiento.
[0054] Ventajosamente, el proceso descrito también acorta considerablemente el tiempo necesario para la producción de un lote del Compuesto A. Actualmente, el proceso para la producción a gran escala del Compuesto A requiere varios días y una posterior purificación mediante recristalización. Usando el proceso mejorado, se espera que el tiempo de producción típico sea de uno-dos días, y no requiera una recristalización adicional.
Procedimientos experimentales
[0055] La presente descripción, que incluye la invención, se ilustra adicionalmente mediante los siguientes ejemplos en el Esquema de reacción 1 y la descripción de los mismos, y no deben interpretarse como limitantes de la descripción, incluyendo la invención, en el alcance de los procedimientos descritos en ellos. Los expertos en la técnica reconocerán que los materiales de partida pueden modificarse y que pueden emplearse etapas adicionales para producir los compuestos descritos en el presente documento, según se demuestra mediante los siguientes ejemplos. Los expertos en la técnica reconocerán también que puede ser necesario utilizar diferentes disolventes o reactivos para conseguir algunas de las transformaciones anteriores.
[0056] Salvo que se especifique de otro modo, todos los reactivos y disolventes son de calidad comercial estándar y se usan sin purificación adicional. La atmósfera apropiada para realizar la reacción, por ejemplo, en aire, nitrógeno, argón, y similares, será evidente para los expertos en la técnica.
Preparación de ácido 1-(4-fluorofenilcarbamoil)ciclopropanocarboxílico (Compuesto A-1)
[0057]

[0058] El ácido 1,1-ciclopropanodicarboxílico de partida se trató con cloruro de tionilo (1,05 equivalentes) en aproximadamente 8 volúmenes de acetato de isopropilo a 25 °C durante 5 horas. A continuación, la mezcla resultante se trató con una solución de 4-fluoroanilina (1,1 equivalentes) y trietilamina (1,1 equivalentes) en acetato de isopropilo (2 volúmenes) durante 1 hora. La suspensión de producto se inactivó con una solución 5 N de NaOH (5 volúmenes) y la fase acuosa se descartó. La fase orgánica se extrajo con una solución 0,5 N de NaOH (10 volúmenes) y el extracto básico se lavó con heptano (5 volúmenes) y posteriormente se acidificó con una solución al 30 % de HCl para dar una suspensión. El Compuesto A-1 se aisló por filtración.
[0059] El Compuesto A-1 se preparó en una báscula de 1,00 kg usando ácido 1,1-ciclopropanodicarboxílico como el reactivo limitante para proporcionar 1,32 kg del Compuesto A-1 (77 % de rendimiento aislado; 84 % de equilibrio de masa) con un 99,92 % de pureza (HPLC) y un 100,3 % de ensayo.
Preparación de N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida (Compuesto 1) y la sal (L)-malato de la misma.
[0060] Una ruta sintética que puede usarse para la preparación de N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida y la sal (L)-malato de la misma se representa en el Esquema 1.
Esquema 1

[0061] Otra ruta sintética que puede usarse para la preparación de N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)
N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida y la sal (L)-malato de la misma se representa en el Esquema 2.
Esquema 2

Preparación de 4-Cloro-6,7-dimetoxi-quinolina
[0062] Un reactor se cargó secuencialmente con 6,7-dimetoxi-quinol-4-ol (47,0 kg) y acetonitrilo (318,8 kg). La mezcla resultante se calentó a aproximadamente 60 °C y se añadió oxicloruro de fósforo (POCl3, 130,6 kg). Después de la adición de POCl3, la temperatura de la mezcla de reacción se elevó a aproximadamente 77 °C. La reacción se consideró completa (aproximadamente 13 horas) cuando quedo menos del 3 % del material de partida (análisis por cromatografía líquida de alto rendimiento [HPLC] en proceso). La mezcla de reacción se enfrió de aproximadamente 2 a 7 °C y a continuación se inactivó en una solución enfriada de diclorometano (DCM, 482,8 kg), NH4OH al 26 % (251,3 kg), y agua (900 l). La mezcla resultante se calentó a aproximadamente 20 a 25 °C, y las fases se separaron. La fase orgánica se filtró a través de un lecho de AW hyflo supercel NF (Celite; 5,4 kg), y el lecho de filtro se lavó con DCM (118,9 kg). La fase orgánica combinada se lavó con salmuera (282,9 kg) y se mezcló con agua (120 l). Las fases se separaron y la fase orgánica se concentró mediante destilación al vacío con la eliminación de disolvente (aproximadamente 95 l de volumen residual). Se cargó DCM (686,5 kg) en el reactor que contenía la fase orgánica y se concentró por destilación al vacío con la eliminación de disolvente (aproximadamente 90 l de volumen residual). Después, se cargó metil t-butil éter (MTBE, 226,0 kg) y la temperatura de la mezcla se ajustó de -20 a -25 °C y se mantuvo durante 2,5 horas dando como resultado un precipitado sólido, que después se filtró y se lavó con n-heptano (92,0 kg), y se secó en un filtro a aproximadamente 25 °C en una atmósfera de nitrógeno para proporcionar el compuesto del título (35,6 kg).
Preparación de 4-(6,7-Dimetoxi-quinolin-4-iloxi)-fenilamina
[0063] Se cargó 4-aminofenol (24,4 kg) disuelto en N,N-dimetilacetamida (DMA, 184,3 kg) en un reactor que contenía 4-cloro-6,7-dimetoxiquinolina (35,3 kg), t-butóxido de sodio (21,4 kg), y DMA (167,2 kg) a 20 - 25 °C. A continuación, esta mezcla se calentó a 100 - 105 °C durante aproximadamente 13 horas. Después de que se considerase completa la reacción según se determinó usando un análisis por HPLC en proceso (quedaba menos del 2 % de material de partida), el contenido del reactor se enfrió de 15 a 20 °C y se cargó agua (enfriada previamente, de 2 a 7 °C, 587 l) a una velocidad para mantener una temperatura de 15 a 30 °C. El precipitado sólido resultante se filtró, se lavó con una mezcla de agua (47 l) y DMA (89,1 kg) y finalmente con agua (214 l). A continuación, la torta de filtró se secó a aproximadamente 25 °C en un filtro para producir 4-(6, 7-dimetoxi-quinolin-4-iloxi)-fenilamina en bruto (59,4 kg en húmedo, 41,6 kg en seco calculados basándose en LOD). Se calentó a reflujo 4-(6, 7 - dimetoxi-quinolin-4-iloxi)-fenilamina en bruto (aproximadamente 75 °C) en una mezcla de tetrahidrofurano (THF, 211,4 kg) y DMA (108,8
kg) durante aproximadamente 1 hora, a continuación se enfrió de 0 a 5 °C y se maduró durante aproximadamente 1 hora después de lo cual el sólido se filtró, se lavó con THF (147,6 kg) y se secó en un filtró al vacío a aproximadamente 25 °C para producir 4-(6,7-dimetoxi-quinolin-4-iloxi)-fenilamina (34,0 kg).
Preparación alternativa de 4-(6,7-Dimetoxi-quinolin-4-iloxi)-fenilamina
[0064] Se cargaron 4-cloro-6,7-dimetoxiquinolina (34,8 kg) y 4-aminofenol (30,8 kg) y terc-pentóxido de sodio (1,8 equivalentes) 88,7 kg, 35 por ciento en peso en THF) en un reactor, seguido de N,N-dimetilacetamida (DMA, 293,3 kg). A continuación, esta mezcla se calentó de 105 a 115 °C durante aproximadamente 9 horas. Después de considerar la reacción completa según se determinó usando un análisis por HPLC en proceso (quedó menos del 2 % de material de partida), el contenido del reactor se enfrió de 15 a 25 °C y se añadió agua (315 kg) durante un periodo de dos horas, mientras se mantuvo al temperatura entre 20 y 30 °C. Después, la mezcla de reacción se agitó durante una hora más de 20 a 25 °C. El producto en bruto se recogió por filtración y se lavó con una mezcla de 88 kg de agua y 82,1 kg de DMA, seguido de 175 kg de agua. El producto se secó en un filtro deshidratador durante 53 horas. El LOD mostró menos del 1 % p/p.
[0065] En un procedimiento alternativo, se usaron 1,6 equivalente de terc-pentóxido de sodio y la temperatura de reacción se aumentó de 110 a 120 °C. Además, la temperatura enfriada se aumentó de 35 a 40 °C y la temperatura de partida de la adición de agua se ajustó de 35 a 40 °C, con una exotermia permitida a 45 °C.
Preparación de cloruro de 1-(4-Fluoro-fenilcarbamoil)-ciclopropanocarbonilo
[0066] Se añadió cloruro de oxalilo (12,6 kg) a una solución de ácido 1-(4-fluoro-fenilcarbamoil)-ciclopropanocarboxílico (22,8 kg) en una mezcla de THF (96,1 kg) y N, N-dimetilformamida (DMF; 0,23 kg) a tal velocidad que la temperatura del lote no excediera de 25 °C. Esta solución se usó en la siguiente etapa sin procesamiento adicional.
Preparación alternativa de cloruro de 1-(4-Fluoro-fenilcarbamoil)-ciclopropanocarbonilo
[0067] Un reactor se cargó con ácido 1-(4-fluoro-fenilcarbamoil)-ciclopropanocarboxílico (35 kg), 344 g de DMF, y 175 kg de THF. La mezcla de reacción se ajustó de 12 a 17 °C y a continuación en la mezcla de reacción se cargaron 19.9 kg de cloruro de oxalilo durante un periodo de 1 hora. La mezcla de reacción se dejó en agitación de 12 a 17 °C durante 3 a 8 horas. Esta solución se usó en la siguiente etapa sin procesamiento adicional.
Preparación de [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil]-amida (4- fluoro-fenil)-amida del ácido ciclopropan-1,1-dicarboxílico
[0068] La solución de la etapa anterior que contenía cloruro de 1-(4-fluoro-fenilcarbamoil)-ciclopropanocarbonilo se añadió a una mezcla del compuesto 4-(6,7-dimetoxi-quinolin-4-iloxi)-fenilamina (23,5 kg) y carbonato potásico (31,9 kg) en THF (245,7 kg) y agua (116 l) a tal velocidad que la temperatura del lote no excediera de 30 °C. Cuando la reacción se completó (en aproximadamente 20 minutos), se añadió agua (653 l). La mezcla se agitó de 20 a 25 °C durante aproximadamente 10 horas, lo que dio como resultado la precipitación del producto. El producto se recuperó por filtración, se lavó con una solución prefabricada de THF (68,6 kg) y agua (256 l), y se secó en primer lugar en un filtro en una atmósfera de nitrógeno a aproximadamente 25 °C y a continuación a aproximadamente 45 °C al vacío para proporcionar el compuesto del título (41,0 kg, 38,1 kg, calculado basándose en LOD).
Preparación alternativa de [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil] -amida (4-fluoro-fenil)-amida del ácido ciclopropan-1,1-dicarboxílico
[0069] Un reactor se cargó con 4-(6,7-dimetoxi-quinolin-4-iloxi)-fenilamina (35,7 kg, 1 equivalente), seguido de 412.9 kg de THF. En la mezcla de reacción se cargó una solución de 48,3 K2CO3 en 169 kg de agua. La solución de cloruro de ácido descrita en la Preparación alternativa de cloruro de 1-(4-Fluoro-fenilcarbamoil)-ciclopropanocarbonilo anterior se transfirió al reactor que contenía 4-(6,7-dimetoxi-quinolin-4-iloxi)-fenilamina mientras se mantuvo la temperatura entre 20 a 30 °C durante un mínimo de dos horas. La mezcla de reacción se agitó de 20 a 25 °C durante un mínimo de tres horas. A continuación, la temperatura de reacción se ajustó de 30 a 25 °C, y la mezcla se agitó. La agitación se detuvo y las fases de la mezcla se dejaron separar. La fase acuosa inferior se eliminó y se descartó. A la fase orgánica superior restante se le añadieron 804 kg de agua. La reacción se dejó en agitación de 15 a 25 °C durante un mínimo de 16 horas.
[0070] El producto precipitó. El producto se filtró y se lavó con una mezcla de 179 kg de agua y 157,9 de THF en dos porciones. El producto en bruto se secó al vacío durante al menos dos horas. A continuación, el producto seco se recogió en 285,1 kg de THF. La suspensión resultante se transfirió al recipiente de reacción y se agitó hasta que la suspensión se convirtió en una solución transparente (disuelta), que requirió calentamiento de 30 a 35 °C durante aproximadamente 30 minutos. A continuación, se añadieron 456 kg de agua a la solución, así como 20 kg de SDAG1 etanol (etanol deuterado con metanol durante dos horas). La mezcla se agitó de 15 a 25 °C durante al menos 16 horas. El producto se filtró y se lavó con una mezcla de 143 kg de agua y 126,7 de THF en dos porciones. El producto se secó a un punto de referencia de temperatura máximo de 40 °C.
[0071] En un procedimiento alternativo, la temperatura de reacción durante la formación de cloruro de ácido se ajustó de 10 a 15 °C. La temperatura de recristalización se cambió de 15 a 25 °C a de 45 a 50 °C durante 1 hora y a continuación se enfrió de 15 a 25 °C durante 2 horas.
Preparación de [4-(6,7-dimetoxi- quinolin-4-iloxi)-fenil]-amida (4- fluoro-fenil)-amida del ácido ciclopropan-1,1-dicarboxílico, XL184 sal (L) malato
[0072] Se cargaron [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil]-amida (4-fluoro-fenil)-amida del ácido ciclopropan-1.1- dicarboxílico (1-5; 13,3 kg), ácido L-málico (4,96 kg), metil etil cetona (MEK; 188,6 kg) y agua (37,3 kg) en un reactor y la mezcla se calentó a reflujo (aproximadamente 74 °C) durante aproximadamente 2 h. La temperatura del reactor se redujo de 50 a 55 °C, y el contenido del reactor se filtró. Estas etapas secuenciales descritas anteriormente se repitieron dos veces más partiendo de cantidades similares de 1-5 (13,3 kg), ácido L-málico (4,96 kg), MEK (198,6 kg), y agua (37,2 kg). El filtrado combinado se secó azeotrópicamente a presión atmosférica usando MEK (1133,2 kg) (volumen residual aproximado 711 L; KF < 0,5 % p/p) a aproximadamente 74 °C. La temperatura del contenido del reactor se redujo de 20 a 25 °C y se mantuvo durante aproximadamente 4 horas, dando como resultado un precipitado sólido que se filtró, se lavó con MEK (448 kg) y se secó al vacío a 50 °C para proporcionar el compuesto del título (45,5 kg).
Preparación alternativa de [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil] -amida (4-fluoro-fenil)-amida del ácido ciclopropan-1,1-dicarboxílico, sal (L) malato
[0073] Se cargaron [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil]-amida (4-fluoro-fenil)-amida del ácido ciclopropan-1.1- dicarboxílico (47,9 kg), ácido L-málico (17,2), 658,2 kg de metil etil cetona, y 129,1 kg de agua (37,3 kg) en un reactor y la mezcla se calentó de 50 a 55 °C durante aproximadamente 1 a 3 horas, y a continuación de 55 a 60 °C durante 4 a 5 horas más. La mezcla se aclaró por filtración a través de un cartucho de 1 |jm. La temperatura del reactor se ajustó de 20 a 25 °C y se destiló al vacío con un vacío de 20 a 27 kPa (150 a 200 mg Hg) con una temperatura de camisa máxima de 55 °C al intervalo en volumen de 558 a 731 l.
[0074] La destilación al vacío se realizó dos veces más con la carga de 380 kg y 380,2 kg de metil etil cetona, respectivamente. Después de la tercera destilación, el volumen del lote se ajustó a 18 v/p de [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil]-amida (4-fluoro-fenil)-amida del ácido ciclopropan-1,1- dicarboxílico cargando 159,9 kg de metil etil cetona para dar un volumen total de 880 l. Se realizó una destilación al vacío adicional ajustando 245,7 de metil etil cetona. La mezcla de reacción se dejó con agitación moderada de 20 a 25 °C durante al menos 24 horas. El producto se filtró y se lavó con 415,1 kg de metil etil cetona en tres porciones. El producto se secó al vacío con el punto de referencia de temperatura de camisa a 45 °C.
[0075] En un procedimiento alternativo, el orden de la adición se cambió de manera que una solución de 17,7 kg de ácido L-málico disuelto en 129,9 kg de agua se añadiera a [4-(6,7-dimetoxi-quinolin-4-iloxi)-fenil]-amida (4-fluorofenil)-amida del ácido ciclopropan-1,1-dicarboxílico (48,7 kg) en metil etil cetona (673,3 kg).
Preparación del Compuesto 2
[0076] El Compuesto 2 se preparó según se proporcionó en el Esquema 3 y los ejemplos experimentales adjuntos.
Esquema 3

[0077] En el Esquema 3, Xb es Br o Cl. Para los nombres de los intermedios descritos en la descripción del Esquema 3 a continuación, Xb se denomina halo, en el que este grupo halo para estos intermedios significa Br o Cl.
Preparación de 1-[5 metoxi-4 (3-halo propoxi)- 2 nitro-fenil]- etanona
[0078] Se cargó agua (70 l) a la solución de 1-[4-(3-halo propoxi)- 3-metoxi fenil] etanona (tanto los compuestos de bromo como de cloro están disponibles comercialmente). La solución se enfrió a aproximadamente 4 °C. Se añadió ácido sulfúrico concentrado (129,5 kg) a tal velocidad que la temperatura del lote no excediera de aproximadamente
18 °C. La solución resultante se enfrió a aproximadamente 5 °C y se añadió ácido nítrico al 70 por ciento (75,8 kg) a tal velocidad que la temperatura del lote no excediera de aproximadamente 10 °C. Se cargaron cloruro de metileno, agua y hielo en un reactor separado. A continuación, la mezcla de reacción ácida se añadió en esta mezcla. La capa de cloruro de metileno se separó, y la capa acuosa se extrajo de nuevo con cloruro de metileno. Las capas de cloruro de metileno combinadas se lavaron con una solución acuosa de bicarbonato de potasio y se concentraron por destilación al vacío. Se añadió 1-butanol y la mezcla se concentró de nuevo por destilación al vacío. La solución resultante se agitó a aproximadamente 20 °C, durante lo cual el producto cristalizó. Los sólidos se recogieron por filtración, se lavaron con 1-butanol para proporcionar el compuesto del compuesto del título, que se aisló como una torta húmeda con disolvente, y se usó directamente en la siguiente etapa. 1H RMN (400 MHz, DMSO-d6): 57,69 (s, 1H), 7,24 (s, 1H); 4,23 (m, 2H), 3,94 (s, 3H), 3,78 (t)-3,65 (t) (2H), 2,51 (s, 3H), 2,30-2,08 (m, 2H) LC/MS Calc. para [M(Cl)+H]+ 288,1, observado 288,0; Calc. para [M(Br)+H]+ 332,0, 334,0, observado 331,9, 334,0.
Preparación de 1-[5-metoxi-4-(3-morfolin-4-il-propoxi)-2-nitro-fenil]-etanona
[0079] La torta húmeda de disolvente aislada en la etapa anterior se disolvió en tolueno. Se añadió una solución de yoduro de sodio (67,9 kg) y carbonato de potasio (83,4 kg) a esta solución, seguido de bromuro de tetrabutilamonio (9,92 kg) y morfolina (83,4 kg). La mezcla de 2 fases resultante se calentó a aproximadamente 85 °C durante aproximadamente 9 horas. A continuación, la mezcla se enfrió a temperatura ambiente. La capa orgánica se eliminó. La capa acuosa se extrajo de nuevo con tolueno. Las capas combinadas de tolueno se lavaron secuencialmente con dos porciones de tiosulfato de sodio acuoso saturado seguido de dos porciones de agua. La solución resultante del compuesto del título se usó en la siguiente etapa sin procesamiento adicional. 1H RMN (400 MHz, DMSO-d6): 57,64 (s, 1H), 7,22 (s, 1H), 4,15 (t, 2H), 3,93 (s, 3H), 3,57 (t, 4H), 2,52 (s, 3H), 2,44-2,30 (m, 6H), 1,90 (quin, 2H); LC/MS Calc. para [M+H]+ 339,2, observado 339,2.
Preparación de 1-[2-amino-5-metoxi-4-(3-morfolin-4-il-propoxi)-fenil]-etanona
[0080] La solución de la etapa anterior se concentró a presión reducida a aproximadamente la mitad del volumen original. Se añadieron etanol y Pd al 10 por ciento/C (50 por ciento mojado por agua, 5,02 kg); la suspensión resultante se calentó a aproximadamente 48 °C, y se añadió una solución acuosa de ácido fórmico (22,0 kg) y formiato de potasio (37,0 kg). Cuando se completó la adición y la reacción se consideró completa por cromatografía de capa fina (TLC), se añadió agua para disolver las sales del subproducto. La mezcla se filtró para eliminar el catalizador insoluble. El filtrado se concentró a presión reducida y se añadió tolueno. La mezcla se hizo básica (pH de aproximadamente 10) mediante la adición de carbonato de potasio acuoso. La capa de tolueno se separó y la capa acuosa se extrajo de nuevo con tolueno. Las fases de tolueno combinadas se secaron sobre sulfato de sodio anhidro. El agente de secado se eliminó por filtración y la solución resultante se usó en la siguiente etapa sin procesamiento adicional. 1H RMN (400 MHz, DMSO-d6): 57,11 (s, 1H), 7,01 (s a, 2H), 6,31 (s, 1H), 3,97 (t, 2H), 3,69 (s, 3H), 3,57 (t, 4H), 2,42 (s, 3H), 2,44-2,30 (m, 6H), 1,91 (quin, 2H LC/MS Calc. para [M+H]+ 309,2, observado 309,1.
Preparación de 6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin- 4-ol, sal de sodio
[0081] Se añadió una solución de etóxido de sodio (85,0 kg) en etanol y formiato de etilo (70,0 kg) a la solución de la etapa anterior. La mezcla se calentó a aproximadamente 44 °C durante aproximadamente 3 horas. La mezcla de reacción se enfrió a aproximadamente 25 °C. Se añadió metil t-butil éter (MTBE) lo que hizo que el producto precipitara. El producto se recogió por filtración y la torta se lavó con MTBE y se secó a presión reducida a temperatura ambiente. El producto seco se molió a través de un tamiz de malla para proporcionar 60,2 kg del compuesto del título.
1H RMN (400 MHz, DMSO-d6): 511,22 (s a, 1H), 8,61 (d, 1H), 7,55 (s, 1H), 7,54 (s, 1H), 7,17 (d, 1H), 4,29 (t, 2 H), 3,99 (m, 2H), 3,96 (s, 3H), 3,84 (t, 2H), 3,50 (d, 2H), 3,30 (m, 2H), 3,11 (m, 2H), 2,35 (m, 2H), LC/MS Calc. para [M+H]+ 319,2, observado 319,1.
Preparación de 4-cloro-6-metoxi-7-(3 morfolin-4-il)-quinolina
[0082] Se añadió oxicloruro de fósforo (26,32 kg) a una solución de 6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin-4-ol (5,00 kg) en acetonitrilo que se calentó de 50 a 55 °C. Cuando la adición se completó, la mezcla se calentó a reflujo (aproximadamente 82 °C) y se mantuvo a esa temperatura, con agitación durante aproximadamente 18 horas, momento en el que se muestreó para el análisis por HPLC en proceso. La reacción se consideró completa cuando no quedó más del 5 por ciento de material de partida. A continuación, la mezcla de reacción se enfrió de 20 a 25 °C y se filtró para eliminar los sólidos. A continuación, el filtrado se concentró para dar un residuo. Se añadió acetonitrilo y la solución resultante se concentró para dar un residuo. Se añadió cloruro de metileno al residuo y la solución resultante se inactivó con una mezcla de cloruro de metileno e hidróxido de amonio acuoso. La mezcla de dos fases resultante se separó, y la capa acuosa se extrajo de nuevo con cloruro de metileno. Las soluciones de cloruro de metileno combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron para dar un sólido. Los sólidos se secaron de 30 a 40 °C a presión reducida para proporcionar el compuesto del título (1,480 kg). 1H RMN (400 MHz, DMSO-d6): 58,61 (d, 1H), 7,56 (d, 1H), 7,45 (s, 1H), 7,38 (s, 1H), 4,21 (t, 2 H), 3,97 (s, 3H), 3,58 (m, 2H), 2,50-2,30 (m, 6H), 1,97 (quin, 2H) LC/MS Calc. para [M+H]+ 458,2, observado 458,0.
Preparación de 4-(2-fluoro-4-nitro-fenoxi)-6-metoxi-7-(3-morfolin-4-il propoxi)quinolina
[0083] Una solución de 4-cloro-6-metoxi-7-(3 morfolin-4-il)-quinolina (2,005 kg, 5,95 mol) y 2 fluoro-4-nitrofenol (1,169 kg, 7,44 mol) en 2,6-lutidina se calentó de 140 a 145 °C, con agitación, durante aproximadamente 2 horas, momento en el que se muestreó para el análisis por HPLC en proceso. La reacción se consideró completa cuando quedó menos del 5 por ciento de material de partida. La mezcla de reacción se enfrió a continuación a aproximadamente 75 °C y se añadió agua. Se añadió carbonato de potasio a la mezcla, que a continuación se agitó a temperatura ambiente durante una noche. Los sólidos que precipitaron se recogieron por filtración, se lavaron con carbonato de potasio acuoso y se secaron de 55 a 60 °C a presión reducida para proporcionar el compuesto del título (1,7 kg). 1H RMN (400 MHz, DMSO-d6): 58,54 (d, 1H), 8,44 (dd, 1H), 8,18 (m, 1H), 7,60 (m, 1H), 7,43 (s, 1H), 7,42 (s, 1H), 6,75 (d, 1H), 4,19 (t, 2H), 3,90 (s, 3H), 3,56 (t, 4H), 2,44 (t, 2H), 2,36 (m, 4H), 1,96 (m, 2H). LC/MS Calc. para [M+H]+ 337,1, 339,1, observado 337,0, 339,0.
Preparación de 3-fluoro-4-[6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin-4-iloxi]-fenilamina
[0084] Un reactor que contenía 4-(2-fluoro-4-nitro-fenoxi)-6-metoxi-7-(3-morfolin-4-il propoxi)quinolina (2,5 kg) y paladio al 10 por ciento sobre carbono (50 por ciento mojado por agua, 250 g) en una mezcla de etanol y agua que contenía ácido clorhídrico concentrado (1,5 l) se presurizó con gas hidrógeno (aproximadamente 280 kPa (40 psi)). La mezcla se agitó a temperatura ambiente. Cuando la reacción se completó (típicamente 2 horas), según se evidenció por un análisis por HPLC en proceso, el hidrógeno se ventiló y el reactor se sometió a inertización con argón. La mezcla de reacción se filtró a través de un lecho de Celite® para eliminar el catalizador. Se añadió carbonato de potasio al filtrado hasta que el pH de la solución fue de aproximadamente 10. La suspensión resultante se agitó de 20 a 25 °C durante aproximadamente 1 hora. Los sólidos se recogieron por filtración, se lavaron con agua y se secaron de 50 a 60 °C a presión reducida para proporcionar el compuesto del título (1,164 kg). 1H RMN (400 MHz, DMSO-d6): 5 8,45 (d, 1H), 7,51 (s, 1H), 7,38 (s, 1H), 7,08 (t, 1H), 6,55 (dd, 1H), 6,46 (dd, 1H), 6,39 (dd, 1H), 5,51 (br. s, 2H), 4,19 (t, 2H), 3,94 (s, 3H), 3,59 (t, 4H), 2,47 (t, 2H), 2,39 (m, 4H), 1,98 (m, 2H). LC/MS Calc. para [M+H] 428,2, observado 428,1.
Preparación de cloruro de 1-(4-fluoro-fenilcarbamoil)-ciclopropanocarbonilo
[0085] Se añadió lentamente cloruro de oxalilo (291 ml) a una solución enfriada (aproximadamente 5 °C) de ácido 1-(4-fluoro-fenilcarbamoil)-ciclopropanocarboxílico en THF a tal velocidad que la temperatura del lote no excediera de 10 °C. Cuando la adición se completó, el lote se dejó calentar a temperatura ambiente y se mantuvo con agitación durante aproximadamente 2 horas, momento en el que el análisis por HPLC en proceso indicó que la reacción se había completado. La solución se usó en la siguiente etapa sin procesamiento adicional.
Preparación de {3-fluoro-4-[6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin-4-ilamino]fenil}-amida-(4 fluorofenil)-amida del ácido ciclopropan-1,1-dicarboxílico
[0086] La solución de la etapa anterior se añadió a una mezcla de 3-fluoro-4-[6-metoxi-7-(3-morfolin-4-ilpropoxi)-quinolin-4-iloxi]-fenilamina (1160 kg) y carbonato de potasio (412,25 g) en THF y agua a tal velocidad que la temperatura del lote se mantuvo de aproximadamente 15 a 21 °C. Cuando la adición se completó, el lote se calentó a temperatura ambiente y se mantuvo con agitación durante aproximadamente 1 hora, momento en el que el análisis por HPLC en proceso indicó que la reacción se había completado. Se añadieron una solución acuosa de carbonato de potasio y acetato de isopropilo al lote. La mezcla de dos fases resultante se agitó, y a continuación las fases se dejaron separar. La fase acuosa se volvió a extraer con acetato de isopropilo. Las capas combinadas de acetato de isopropilo se lavaron con agua seguido de cloruro de sodio acuoso y a continuación se suspendieron con una mezcla de sulfato de magnesio y carbón activado. La suspensión se filtró sobre Celite®, y el filtrado se concentró para dar un aceite a aproximadamente 30 °C al vacío para proporcionar el compuesto del título, que se llevó a la siguiente etapa sin procesamiento adicional. 1H RMN (400 MHz, DMSO-d6): 510,41 (s, 1H), 10,03 (s, 1H), 8,47 (d, 1H), 7,91 (dd, 1H), 7,65 (m, 2H), 7,53 (m, 2H), 7,42 (m, 2H), 7,16 (t, 2H), 6,41 (d, 1H), 4,20 (t, 2H), 3,95 (s, 3H), 3,59 (t, 4H), 2,47 (t, 2H), 2,39 (m, 4H), 1,98 (m, 2H), 1,47 (m, 4H). LC/MS Calc. para [M+H] 633,2, observado 633,1.
Preparación de la sal bisfosfato de {3-fluoro-4-[6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin-4-ilamino]fenil}-amida (4-fluorofenil)-amida del ácido ciclopropan-1,1-dicarboxílico
[0087] Se disolvió {3-fluoro-4-[6-metoxi-7-(3-morfolin-4-il-propoxi)-quinolin-4-ilamino] fenil}-amida-(4 fluoro fenil)-amida del ácido ciclopropan-1,1-dicarboxílico de la etapa anterior en acetona y agua. Se añadió ácido fosfórico (85 %, 372,48 g) a tal velocidad que la temperatura del lote no excediera de 30 °C. El lote se mantuvo a aproximadamente de 15 a 30 °C con agitación durante 1 hora, tiempo durante el cual el producto precipitó. Los sólidos se recogieron por filtración, se lavaron con acetona, y se secaron a aproximadamente 60 °C al vacío para proporcionar el compuesto del título (1,533 kg). El compuesto del título tiene un valor de CI50 de c-Met de menos de 50 nM. La sal bisfosfato no se muestra en el esquema 3. 1H RMN (400 MHz, DMSO-d6): (difosfato) 510,41 (s, 1H), 10,02 (s, 1H), 8,48 (d, 1H), 7,93 (dd, 1H), 7,65 (m, 2H), 7,53 (d, 2H), 7,42 (m, 2H), 7,17 (m, 2H), 6,48 (d, 1H), 5,6 (s a, 6H), 4,24 (t, 2H), 3,95 (s, 3H), 3,69 (s a, 4H), 2,73 (s a, 6H), 2,09 (t, 2H), 1,48 (d, 4H).
Procedimiento para acoplamiento directo
[0088]

[0089] Se añadió ferc-butóxido de sodio sólido (1,20 g; 12,5 mmol) a una suspensión de la cloroquinolina (3,37 g; 10 mmol) en dimetilacetamida (35 ml), seguido de 2-fluoro-4-hidroxianilina sólida. La mezcla de reacción de color verde oscuro se calentó de 95 a 100 °C durante 18 horas. El análisis por HPLC mostró que daba aproximadamente el 18 por ciento de material de partida y aproximadamente el 79 por ciento de producto. La mezcla de reacción se enfrió por debajo de 50 °C, se añadieron ferc-butóxido de sodio adicional (300 mg; 3,125 mmol) y anilina (300 mg; 2,36 mmol), y el calentamiento de 95 a 100 °C se reanudó. El análisis por HPLC después de 18 horas reveló que quedaba menos del 3 por ciento de material de partida. La reacción se enfrió por debajo de 30 °C, y se añadió hielo-agua (50 ml) mientras se mantuvo la temperatura por debajo de 30 °C. Después de la agitación durante 1 hora a temperatura ambiente, el producto se recogió por filtración, se lavó con agua (2 x 10 ml) y se secó al vacío en el embudo de filtro, para producir 4,11 g del producto acoplado en forma de un sólido de color castaño (96 % de rendimiento; 89 %, corregido para el contenido de agua). 1H RMN y MS: consistente con el producto; 97,8 % de LCAP; aproximadamente el 7 por ciento en peso de agua por KF.
Preparación de la forma hidrato del Compuesto 2
[0090] El hidrato del Compuesto 2 se preparó añadiendo 4,9614 g del Compuesto 1 y 50 ml de n-propanol a un vaso de precipitados de 250 ml. La suspensión se calentó a 90 °C con agitación mediante una barra de agitación magnética a 200 rpm. Después de 2 horas, los sólidos se disolvieron completamente en una solución de color ámbar. En los puntos temporales de 1 hora y 2 horas se añadieron 10 ml de n-propanol para compensar los efectos de la evaporación, y el volumen de la solución se devolvió a 50 ml. A continuación, la solución se filtró en caliente a través de un filtro de fibra de vidrio de 1,6 pm. La solución se dejó secar entonces durante una noche en el vaso de precipitados para dar un polvo, que después se disolvió de nuevo en 150 ml de una mezcla 1:1 de acetona y agua y se suspendió durante una noche (16 horas) con una tapa de aluminio para evitar la evaporación. A continuación, los sólidos suspendidos se recogieron mediante filtración al vacío. El peso final recuperado fue de 3,7324 g (75 % de rendimiento). Este lote se almacenó en condiciones ambientales durante varios días antes del análisis.
[0091] Las determinaciones del contenido en agua mediante Karl Fisher se realizaron mediante el uso de un procedimiento estándar. El contenido en agua se midió con un coulómetro Brinkmann KF1V4 Metrohm 756 equipado con un agitador 703 Ti y mediante el uso del reactivo Hydranal Coulomat AG. Las muestras se introdujeron en el recipiente en forma de sólidos. Se usaron aprox. 30-35 mg de muestra por valoración. Una muestra cristalina del Compuesto (I) preparado en el Ejemplo 1.1.2 se midió por duplicado, y se encontró que tenía un contenido promedio de agua del 2,5 % p/p, concordando cada duplicado en un 0,1 %.
[0092] Se llevó a cabo un estudio de absorción gravimétrica de vapor (GVS) mediante el uso de un procedimiento estándar. Las muestras se analizaron en un analizador de absorción dinámica de vapor (Surface Measurement Systems) con un programa informático DVSCFR. El tamaño de las muestras era típicamente de 10 mg. Se realizó una isoterma de absorción-desorción de humedad, como se expone a continuación. El experimento de isoterma estándar, realizado a 25 °C, es un análisis de dos ciclos, que comienza con un 40 % de HR (humedad relativa), aumentando la humedad a un 90 % de HR, disminuyendo la humedad a un 0 % de HR, aumentando de nuevo la humedad a un 90 % de HR, y finalmente disminuyendo la humedad a un 0 % de HR a intervalos del 10 % de HR. El Compuesto 1 cristalino preparado en el Ejemplo 1.1.1 mostró una ganancia de peso del 2,5 % a 25 °C y una humedad del 90 %. Las curvas de absorción y desorción de GVS mostraron signos de que el hidrato se comporta como un desolvato isomorfo (Stephenson, G. A.; Groleau, E. G.; Kleeman, R. L.; Xu, W.; Rigsbee, D. R. J. Pharm. Sci.
1998, 87, 536-42).
[0093] El patrón de difracción de polvo de rayos X del hidrato cristalino del Compuesto 1 preparado anteriormente se adquirió usando un difractrómetro PANalytical X'Pert Pro. La muestra se aplanó suavemente en un portamuestras con inserto de silicio con fondo cero. Se usó un rango de barrido continuo a 20 de 2° a 50° con una fuente de radiación de CuKa y una potencia del generador de 40 kV y 45 mA. Se usó un tamaño de salto de 20 de 0,017 grados/salto con un tiempo de salto de 40,7 segundos. Las muestras se rotaron a 30 rpm. Los experimentos se realizaron a temperatura ambiente y a la humedad ambiental. El documento WO 2011/112896 muestra el patrón XRPD para el hidrato cristalino N-[3-fluoro-4-({6-(metiloxi)-7-[(3-morfolin-4-ilpropil)oxi]quinolin-4-il}oxi)fenil]-N'-(4-fluorofenil)ciclopropan-1,1-dicarboxamida. Los siguientes picos a un °20 0,1 °20 experimental se identificaron en el
patrón XRPD: 6,6, 9,0, 10,2, 12,0, 12,2, 13,1, 13,3, 14,6, 15,6, 16,2, 17,0, 17,1, 17,4, 18,2, 18,4, 18,7, 20,0, 20,3, 20,8, 21,7, 22,1, 23,1,23,4, 23,8, 24,2, 24,5, 25,0. Únicamente se proporcionan los picos por debajo de 25° 20, ya que se prefieren estos generalmente para la identificación de las formas farmacéuticas cristalinas. La lista completa de picos, o un subconjunto de los mismos, puede ser suficiente para caracterizar el hidrato del Compuesto 1.
[0094] Se adquirieron termogramas de DSC mediante el uso de un calorímetro diferencial de barrido TA Instruments Q2000. Se pesó una masa de muestra de 2,1500 mg del hidrato cristalino del Compuesto 1 directamente en un crisol de aluminio para DSC. El crisol se cerró herméticamente aplicando presión con la mano y empujando cada parte del crisol contra la otra (también conocida como configuración de tapa suelta). La temperatura se aumentó de 25 °C a 225 °C a 10 °C/minuto. Se midió un pico de temperatura de fusión de 137,4 °C y un flujo de calor de 44,2 J/g para la endotermia de fusión. Después del evento de fusión, se produce una recristalización en una forma anhidra, que a continuación se funde a 194,1 °C.
[0095] Se adquirieron termogramas de TGA mediante el uso de un analizador termogravimétrico TA Instruments Q500. El crisol de muestra se taró, y se pusieron sobre el crisol 9,9760 miligramos del hidrato cristalino del Compuesto (I). La temperatura se aumentó de 25 °C a 300 °C a 10 °C/minuto. Se observó una pérdida de peso del 2,97 % hasta 160 °C, con una pérdida de peso adicional más allá de los 200 °C debida a la descomposición. Preparación del hidrato cristalino del Compuesto 2 con diferentes estados de hidratación.
[0096] Se tomaron cinco alícuotas de 150 mg del lote de hidrato cristalino preparado anteriormente, y se colocaron en viales con tapón de rosca de 10 ml. Con los tapones de los viales quitados, estas alícuotas se almacenaron cada una en cámaras con desecante (Dri-Rite®, silicato tricálcico, HR al 2-3 %), bromuro de litio saturado (6 % de HR), cloruro de litio saturado (11 % de HR), cloruro de magnesio saturado (33 % de HR) y cloruro sódico saturado (75 % de HR). Las muestras se eliminaron después de 2 semanas y se sellaron inmediatamente con un tapón para su análisis, y se caracterizaron.
[0097] La descripción anterior se ha descrito con cierto detalle a modo de ilustración y ejemplo, con propósitos de claridad y comprensión. La descripción, incluyendo la invención, se ha descrito con referencia a diversas realizaciones y técnicas específicas y preferidas. Será obvio para un experto en la técnica que los cambios y modificaciones pueden ponerse en práctica dentro del alcance de las reivindicaciones adjuntas. Por lo tanto, debe entenderse que la descripción anterior pretende ser ilustrativa y no restrictiva. Por lo tanto, el alcance de la invención no debe determinarse con referencia a la descripción anterior, sino que debe determinarse con referencia a las siguientes reivindicaciones adjuntas.
Claims (13)
1. Un proceso para preparar un compuesto de Fórmula A:

en la que R2 es H, F, Cl, o Br;
que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar, en la que el disolvente aprótico polar es acetato de isopropilo, a una temperatura de 23 a 27 °C; y
(b) añadir

y una base de amina terciaria a la mezcla de la etapa (a) para proporcionar el compuesto de Fórmula A.
2. El proceso de la reivindicación 1, en el que se usaron de 5 a 10 volúmenes de disolvente aprótico polar con respecto al volumen de ácido 1,1-ciclopropanodicarboxílico que se usó.
3. El proceso de la reivindicación 1, en el que se usaron de 1,01 a 1,2 equivalentes molares de cloruro de tionilo.
4. El proceso de la reivindicación 1, en el que la mezcla de la etapa (a) se agita a 24-26 °C durante 6 a 16 horas.
5. El proceso de la reivindicación 1, en el que

y la base de amina terciaria se añaden como una mezcla en un disolvente aprótico polar, seleccionada del grupo que consiste en diclorometano, tetrahidrofurano, acetato de etilo, acetato de isopropilo, acetona, dimetilformamida, acetonitrilo, y dimetilsulfóxido, o combinaciones de los mismos, a la mezcla de la etapa (a).
6. El proceso de la reivindicación 5, en el que se usan de 1,01 a 1,5 equivalentes molares de anilina con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico que se usan, y se usan de 1,01 a 1,5 equivalentes molares de base de amina terciaria con respecto al número de moles de ácido 1,1-ciclopropanodicarboxílico que se usan.
7. El proceso de la reivindicación 5, en el que el disolvente aprótico polar en la etapa (b) es acetato de isopropilo, y se usan 1,5-3 volúmenes de acetato de isopropilo.
8. El proceso de la reivindicación 1, en el que la mezcla resultante de la etapa (b) se deja en agitación durante 0,75 a 4 horas a temperatura ambiente.
9. El proceso de la reivindicación 1, que comprende además inactivar la mezcla de la etapa (b) con una base acuosa concentrada, en el que la base acuosa se selecciona del grupo que consiste en NaOH, KOH, o K3PO4.
10. El proceso de cualquiera de las reivindicaciones 1-9 que comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a una
temperatura de 23 a 27 °C;
(b) añadir

y trietil amina a la mezcla de la etapa (a);
(c) inactivar la mezcla con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A en una base acuosa diluida;
(e) acidificar la mezcla con HCl; y
(f) aislar el Compuesto A por filtración.
11. El proceso de la reivindicación 10, en el que el compuesto de Fórmula A es el compuesto de Fórmula A-1:

y el proceso comprende
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en acetato de isopropilo a una temperatura de 23 a 27 °C;
(b) añadir una mezcla que comprende 4-fluoroanilina y una trietil amina en acetato de isopropilo a la mezcla de la etapa (a);
(c) inactivar la mezcla de la etapa (b) con hidróxido sódico acuoso concentrado;
(d) extraer el compuesto A-1 en una base acuosa diluida;
(e) acidificar la mezcla de la etapa (d) con HCl; y
(f) aislar el Compuesto A por filtración; y
en el que el Compuesto del producto de Fórmula A-1 está contaminado con el 5 por ciento o menos de la bisamida:

12. Un proceso para preparar un compuesto de Fórmula I:

en la que:
R1 es halo;
R2 es halo;
R3 es alquilo (C-i-Ca) o alquilo (C-i-Ca) opcionalmente sustituido con heterocicloalquilo, en la que "heterocicloalquilo" significa un grupo monocíclico monovalente saturado o parcialmente insaturado de 3 a 8 átomos en el anillo, o un grupo bicíclico condensado monovalente saturado o parcialmente insaturado de 5 a 12 átomos en el anillo en el que uno o más heteroátomos en el anillo se seleccionan independientemente de -O-, -S(O)n-, -N=, y -N(Ry)-, siendo los átomos del anillo restantes carbono, y en los que uno o dos átomos de carbono en el anillo pueden reemplazarse por un grupo -C(O)-, -C(S)-, o C(=NH)-;
n es 0, 1, o 2;
Ry es hidrógeno, alquilo, hidroxi, alcoxi, acilo o alquilsulfonilo;
R4 es alquilo (C1 -C6); y
Q es CH o N;
que comprende:
(a) poner en contacto ácido 1,1-ciclopropano dicarboxílico con cloruro de tionilo en un disolvente aprótico polar, en la que el disolvente aprótico polar es acetato de isopropilo, a una temperatura de 23 a 27 °C;
(b) añadir

y una base de amina terciaria a la mezcla de la etapa (a) para formar un compuesto de Fórmula A:

(c) acoplar un compuesto de Fórmula A con una amina de Fórmula B para formar un compuesto de Fórmula I:

13. El proceso de la reivindicación 12, en el que el compuesto de Fórmula I es el compuesto 1:

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161549312P | 2011-10-20 | 2011-10-20 | |
| PCT/US2012/061320 WO2013059788A1 (en) | 2011-10-20 | 2012-10-22 | Process for preparing quinoline derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2765013T3 true ES2765013T3 (es) | 2020-06-05 |
Family
ID=47146705
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12783763T Active ES2765013T3 (es) | 2011-10-20 | 2012-10-22 | Procedimiento para preparar derivados de quinolina |
Country Status (24)
| Country | Link |
|---|---|
| US (3) | US9365516B2 (es) |
| EP (1) | EP2768796B1 (es) |
| JP (2) | JP6208140B2 (es) |
| KR (1) | KR102075371B1 (es) |
| CN (2) | CN104395284A (es) |
| AR (1) | AR088483A1 (es) |
| AU (2) | AU2012325768B2 (es) |
| BR (1) | BR112014009302B1 (es) |
| CA (1) | CA2852771C (es) |
| DK (1) | DK2768796T3 (es) |
| EA (1) | EA031485B1 (es) |
| ES (1) | ES2765013T3 (es) |
| GE (1) | GEP20207110B (es) |
| HK (1) | HK1201517A1 (es) |
| HU (1) | HUE048023T2 (es) |
| IL (1) | IL232139A (es) |
| IN (1) | IN2014CN02971A (es) |
| MX (1) | MX343288B (es) |
| PL (1) | PL2768796T3 (es) |
| PT (1) | PT2768796T (es) |
| TW (2) | TWI642650B (es) |
| UA (1) | UA116876C2 (es) |
| WO (1) | WO2013059788A1 (es) |
| ZA (1) | ZA201402579B (es) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI619495B (zh) | 2010-07-16 | 2018-04-01 | 艾克塞里克斯公司 | C-met調節劑醫藥組合物 |
| EP2621481B2 (en) | 2010-09-27 | 2022-10-19 | Exelixis, Inc. | Dual inhibitors of met and vegf for the treatment of castration-resistant prostate cancer and osteoblastic bone metastases |
| AR085155A1 (es) | 2011-02-10 | 2013-09-11 | Exelixis Inc | Procesos para preparar compuestos de quinolina y composiciones farmaceuticas que contienen dichos compuestos |
| US20120252840A1 (en) | 2011-04-04 | 2012-10-04 | Exelixis, Inc. | Method of Treating Cancer |
| WO2012151326A1 (en) | 2011-05-02 | 2012-11-08 | Exelixis, Inc. | Method of treating cancer and bone cancer pain |
| AU2012312364B2 (en) | 2011-09-22 | 2017-11-09 | Exelixis, Inc. | Method for treating osteoporosis |
| TWI642650B (zh) * | 2011-10-20 | 2018-12-01 | 艾克塞里克斯公司 | 用於製備喹啉衍生物之方法 |
| EP2844254A1 (en) | 2012-05-02 | 2015-03-11 | Exelixis, Inc. | A dual met - vegf modulator for treating osteolytic bone metastases |
| MX366003B (es) | 2013-03-15 | 2019-06-24 | Exelixis Inc | Metabolitos de n-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-n' -(4-fluorofenil)ciclopropan-1,1-dicarboxamida. |
| EP2981263B1 (en) | 2013-04-04 | 2022-06-29 | Exelixis, Inc. | Cabozantinib dosage form and use in the treatment of cancer |
| CN104649969B (zh) * | 2013-11-22 | 2019-02-12 | 广东东阳光药业有限公司 | 一种替尼类药物的盐及其制备方法 |
| KR102354963B1 (ko) | 2014-02-14 | 2022-01-21 | 엑셀리시스, 인코포레이티드 | N-{4-[(6,7-다이메톡시퀴놀린-4-일)옥시]페닐}-n'-(4-플루오로페닐) 사이클로프로판-1,1-다이카복스아마이드의 결정질 고체 형태, 제조 방법 및 사용 방법 |
| CN106255499A (zh) | 2014-03-17 | 2016-12-21 | 埃克塞里艾克西斯公司 | 卡博替尼制剂的给药 |
| CN104788372B (zh) * | 2014-07-25 | 2018-01-30 | 上海圣考医药科技有限公司 | 一种氘代卡博替尼衍生物、其制备方法、应用及其中间体 |
| MA40386A (fr) | 2014-07-31 | 2016-02-04 | Exelixis Inc | Procédé de préparation de cabozantinib marqué au fluor-18 et d'analogues de celui-ci |
| MA40457A (fr) | 2014-08-05 | 2017-06-14 | Exelixis Inc | Combinaison de médicaments pour traiter le myélome multiple |
| US10745346B2 (en) * | 2015-09-02 | 2020-08-18 | Kuen-Feng Chen | Agonists of protein tyrosine phosphatase SHP-1 |
| RU2748549C2 (ru) | 2016-04-15 | 2021-05-26 | Экселиксис, Инк. | Способ лечения почечно-клеточного рака с использованием n-(4-(6,7-диметоксихинолин-4-илокси)фенил)-n'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, (2s)-гидроксибутандиоата |
| CN109836382B (zh) * | 2017-11-29 | 2021-11-05 | 江苏豪森药业集团有限公司 | 苹果酸卡博替尼及其中间体的制备方法 |
| CN111757735B (zh) | 2018-01-26 | 2023-09-22 | 埃克塞里艾克西斯公司 | 用于治疗激酶依赖性病症的化合物 |
| CN112142619B (zh) * | 2020-10-20 | 2023-01-10 | 浙江工业大学 | 一种1,1-环丙烷二羧酸酰胺类化合物及其制备方法和应用 |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005030140A2 (en) | 2003-09-26 | 2005-04-07 | Exelixis, Inc. | C-met modulators and methods of use |
| CA2572331A1 (en) | 2004-07-02 | 2006-02-09 | Exelixis, Inc. | C-met modulators and method of use |
| EP1874759A4 (en) | 2005-04-06 | 2009-07-15 | Exelixis Inc | C-MET MODULATORS MODULATORS AND METHODS OF USE |
| CN111643496A (zh) | 2006-12-14 | 2020-09-11 | 埃克塞利希斯股份有限公司 | 使用mek抑制剂的方法 |
| UY31800A (es) | 2008-05-05 | 2009-11-10 | Smithkline Beckman Corp | Metodo de tratamiento de cancer usando un inhibidor de cmet y axl y un inhibidor de erbb |
| US8445509B2 (en) * | 2008-05-08 | 2013-05-21 | Takeda Pharmaceutical Company Limited | Fused heterocyclic derivatives and use thereof |
| UY32142A (es) | 2008-09-26 | 2010-05-31 | Smithkline Beckman Corp | Preparación de una quinoliniloxidifenilciclopropanodicarboxamida |
| CN102282134B (zh) | 2008-11-13 | 2015-04-01 | 埃克塞里艾克西斯公司 | 喹啉衍生物制备方法 |
| JP2012511017A (ja) | 2008-12-04 | 2012-05-17 | エグゼリクシス, インコーポレイテッド | キノリン誘導体の調製方法 |
| EP2387563B2 (en) | 2009-01-16 | 2022-04-27 | Exelixis, Inc. | Malate salt of n- (4- { [ 6, 7-bis (methyloxy) quinolin-4-yl]oxy}phenyl-n' - (4 -fluorophenyl) cyclopropane-1,1-dicarboxamide, and crystalline forms thereof for the treatment of cancer |
| CA2768370A1 (en) | 2009-07-17 | 2011-01-20 | Exelixis, Inc. | Crystalline forms of n-[3-fluoro-4-({6-(methyloxy)-7-[(3-morpholin-4-ylpropyl)oxy]-quinolin-4-yl}oxy)phenyl]-n'-(4-fluorophenyl)cyclopropane-1, 1-dicarboxamide |
| UA108618C2 (uk) | 2009-08-07 | 2015-05-25 | Застосування c-met-модуляторів в комбінації з темозоломідом та/або променевою терапією для лікування раку | |
| EA201290906A1 (ru) | 2010-03-12 | 2013-03-29 | Экселиксис, Инк. | Кристаллогидратные формы n-[3-фтор-4-({6-(метилокси)-7-[(3-морфолин-4-илпропил)окси]хинолин-4-ил}окси)фенил]-n'-(4-фторфенил)циклопропан-1,1-дикарбоксамида |
| US20120070368A1 (en) | 2010-04-16 | 2012-03-22 | Exelixis, Inc. | Methods of Using C-Met Modulators |
| WO2012009723A1 (en) | 2010-07-16 | 2012-01-19 | Exelixis, Inc. | C-met modulator pharmaceutical compositions |
| TWI619495B (zh) | 2010-07-16 | 2018-04-01 | 艾克塞里克斯公司 | C-met調節劑醫藥組合物 |
| EP2621481B2 (en) | 2010-09-27 | 2022-10-19 | Exelixis, Inc. | Dual inhibitors of met and vegf for the treatment of castration-resistant prostate cancer and osteoblastic bone metastases |
| JP2013540759A (ja) | 2010-09-27 | 2013-11-07 | エクセリクシス, インク. | 去勢抵抗性前立腺癌および造骨性転移の治療のためのmetおよびvegfの二元阻害薬 |
| WO2012044577A1 (en) | 2010-09-27 | 2012-04-05 | Exelixis, Inc. | Dual inhibitors of met and vegf for the treatment of castration resistant prostate cancer and osteoblastic bone metastases |
| WO2012071321A1 (en) | 2010-11-22 | 2012-05-31 | Glaxosmithkline Llc | Method of treating cancer |
| AR085155A1 (es) | 2011-02-10 | 2013-09-11 | Exelixis Inc | Procesos para preparar compuestos de quinolina y composiciones farmaceuticas que contienen dichos compuestos |
| US20120252840A1 (en) | 2011-04-04 | 2012-10-04 | Exelixis, Inc. | Method of Treating Cancer |
| WO2012151326A1 (en) | 2011-05-02 | 2012-11-08 | Exelixis, Inc. | Method of treating cancer and bone cancer pain |
| TW201306842A (zh) | 2011-06-15 | 2013-02-16 | Exelixis Inc | 使用pi3k/mtor吡啶並嘧啶酮抑制劑及苯達莫司汀及/或利妥昔單抗治療惡性血液疾病之組合療法 |
| AU2012312364B2 (en) | 2011-09-22 | 2017-11-09 | Exelixis, Inc. | Method for treating osteoporosis |
| TWI642650B (zh) * | 2011-10-20 | 2018-12-01 | 艾克塞里克斯公司 | 用於製備喹啉衍生物之方法 |
| EA201490944A1 (ru) | 2011-11-08 | 2014-10-30 | Экселиксис, Инк. | Двойной ингибитор met и vegf для лечения рака |
| EP2844254A1 (en) | 2012-05-02 | 2015-03-11 | Exelixis, Inc. | A dual met - vegf modulator for treating osteolytic bone metastases |
| US20140121239A1 (en) | 2012-09-07 | 2014-05-01 | Exelixis, Inc. | Method of treating lung adenocarcinoma |
-
2012
- 2012-10-22 TW TW106121791A patent/TWI642650B/zh active
- 2012-10-22 MX MX2014004583A patent/MX343288B/es active IP Right Grant
- 2012-10-22 CA CA2852771A patent/CA2852771C/en active Active
- 2012-10-22 PL PL12783763T patent/PL2768796T3/pl unknown
- 2012-10-22 EA EA201490831A patent/EA031485B1/ru unknown
- 2012-10-22 UA UAA201405355A patent/UA116876C2/uk unknown
- 2012-10-22 EP EP12783763.1A patent/EP2768796B1/en active Active
- 2012-10-22 PT PT127837631T patent/PT2768796T/pt unknown
- 2012-10-22 BR BR112014009302A patent/BR112014009302B1/pt active IP Right Grant
- 2012-10-22 AR ARP120103944 patent/AR088483A1/es not_active Application Discontinuation
- 2012-10-22 GE GEAP201213471A patent/GEP20207110B/en unknown
- 2012-10-22 CN CN201280054241.3A patent/CN104395284A/zh active Pending
- 2012-10-22 DK DK12783763.1T patent/DK2768796T3/da active
- 2012-10-22 WO PCT/US2012/061320 patent/WO2013059788A1/en active Application Filing
- 2012-10-22 TW TW101139005A patent/TWI619694B/zh active
- 2012-10-22 ES ES12783763T patent/ES2765013T3/es active Active
- 2012-10-22 KR KR1020147012435A patent/KR102075371B1/ko active IP Right Grant
- 2012-10-22 US US14/353,251 patent/US9365516B2/en active Active
- 2012-10-22 CN CN201910783833.2A patent/CN110511158A/zh active Pending
- 2012-10-22 IN IN2971CHN2014 patent/IN2014CN02971A/en unknown
- 2012-10-22 HU HUE12783763A patent/HUE048023T2/hu unknown
- 2012-10-22 JP JP2014537359A patent/JP6208140B2/ja active Active
- 2012-10-22 AU AU2012325768A patent/AU2012325768B2/en active Active
-
2014
- 2014-04-09 ZA ZA2014/02579A patent/ZA201402579B/en unknown
- 2014-04-22 IL IL232139A patent/IL232139A/en active IP Right Grant
-
2015
- 2015-02-26 HK HK15101949.3A patent/HK1201517A1/xx unknown
-
2016
- 2016-04-20 US US15/133,708 patent/US9969692B2/en active Active
-
2017
- 2017-06-07 JP JP2017112355A patent/JP2017222648A/ja active Pending
- 2017-11-06 AU AU2017254982A patent/AU2017254982A1/en not_active Abandoned
-
2018
- 2018-04-17 US US15/955,246 patent/US20180230100A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2765013T3 (es) | Procedimiento para preparar derivados de quinolina | |
| JP6513598B2 (ja) | キノリン化合物およびそのような化合物を含有する医薬組成物の調製方法 | |
| ES2402524T3 (es) | Sal de malato de la N-(4-{[6,7-bis(metiloxi)quinolin-4-il]oxi}fenil)-N'-(4-fluorofenil)ciclopropano-1,1-dicarboxamida y formas cristalinas de la misma para el tratamiento del cáncer | |
| US20180072748A1 (en) | Crystal of 6,7-unsaturated-7-carbamoyl morphinan derivative and method for producing the same | |
| AU2011307306A1 (en) | Dual inhibitors of MET and VEGF for the treatment of castration resistant prostate cancer and osteoblastic bone metastases | |
| KR20120051702A (ko) | N-〔3-플루오로-4-({6-(메틸옥시)-7-〔(3-모르폴린-4-일프로필)옥시〕퀴놀린-4-일}옥시)페닐〕-n''-(4-플루오로페닐)시클로프로판-1,1-디카르복사미드의 결정형 | |
| TW200922589A (en) | Novel preparation for external use | |
| US20150133460A1 (en) | Lactate salt of 4-[6-methoxy-7-(3-piperidin-1-yl-propoxy)quinazolin-4-yl]piperazine-1-carboxylic acid(4-isopropoxyphenyl)-amide and pharmaceutical compositions thereof for the treatment of cancer and other diseases or disorders | |
| NZ624328B2 (en) | Process for preparing quinoline derivatives | |
| NZ723880B2 (en) | Process for preparing quinoline derivatives | |
| CN111278808B (zh) | 2-(5-(4-(2-吗啉代乙氧基)苯基)吡啶-2-基)-n-苄基乙酰胺的固体形式 |