ES2760571T3 - Procedimientos de fabricación de derivados de indazol - Google Patents
Procedimientos de fabricación de derivados de indazol Download PDFInfo
- Publication number
- ES2760571T3 ES2760571T3 ES16726309T ES16726309T ES2760571T3 ES 2760571 T3 ES2760571 T3 ES 2760571T3 ES 16726309 T ES16726309 T ES 16726309T ES 16726309 T ES16726309 T ES 16726309T ES 2760571 T3 ES2760571 T3 ES 2760571T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- salt
- defined above
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000004519 manufacturing process Methods 0.000 title abstract description 10
- 125000003453 indazolyl group Chemical class N1N=C(C2=C1C=CC=C2)* 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 102
- 150000003839 salts Chemical class 0.000 claims abstract description 79
- 238000000034 method Methods 0.000 claims abstract description 45
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims abstract description 42
- 239000003054 catalyst Substances 0.000 claims abstract description 28
- 229910052763 palladium Inorganic materials 0.000 claims abstract description 21
- 125000005843 halogen group Chemical group 0.000 claims abstract description 14
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 11
- 239000003638 chemical reducing agent Substances 0.000 claims abstract description 8
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 7
- 238000010511 deprotection reaction Methods 0.000 claims abstract description 6
- 125000006239 protecting group Chemical group 0.000 claims abstract description 6
- -1 tetrahydro-2H-pyran-2-yl Chemical group 0.000 claims description 25
- 238000005271 boronizing Methods 0.000 claims description 4
- 239000000460 chlorine Substances 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 51
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 45
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 27
- 239000000243 solution Substances 0.000 description 26
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 238000002425 crystallisation Methods 0.000 description 19
- 230000008025 crystallization Effects 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000005160 1H NMR spectroscopy Methods 0.000 description 14
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 13
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 12
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 10
- 239000000543 intermediate Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 8
- 0 CCC(C1IC1C1C=CC(*)=C2)=CC1=C2*1*cc(*C)[o]1 Chemical compound CCC(C1IC1C1C=CC(*)=C2)=CC1=C2*1*cc(*C)[o]1 0.000 description 7
- 229940126062 Compound A Drugs 0.000 description 7
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- MCIDWGZGWVSZMK-UHFFFAOYSA-N 2-[6-(1h-indol-4-yl)-1h-indazol-4-yl]-5-[(4-propan-2-ylpiperazin-1-yl)methyl]-1,3-oxazole Chemical compound C1CN(C(C)C)CCN1CC1=CN=C(C=2C=3C=NNC=3C=C(C=2)C=2C=3C=CNC=3C=CC=2)O1 MCIDWGZGWVSZMK-UHFFFAOYSA-N 0.000 description 6
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 6
- 229910052757 nitrogen Chemical group 0.000 description 6
- 238000000634 powder X-ray diffraction Methods 0.000 description 6
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 6
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 5
- 125000000217 alkyl group Chemical group 0.000 description 5
- 238000004821 distillation Methods 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- WHKWMTXTYKVFLK-UHFFFAOYSA-N 1-propan-2-ylpiperazine Chemical compound CC(C)N1CCNCC1 WHKWMTXTYKVFLK-UHFFFAOYSA-N 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 239000012280 lithium aluminium hydride Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- HNVIQLPOGUDBSU-OLQVQODUSA-N (2s,6r)-2,6-dimethylmorpholine Chemical compound C[C@H]1CNC[C@@H](C)O1 HNVIQLPOGUDBSU-OLQVQODUSA-N 0.000 description 3
- QCGMEWVZBGQOFN-UHFFFAOYSA-N 1,3-oxazole-5-carboxylic acid Chemical compound OC(=O)C1=CN=CO1 QCGMEWVZBGQOFN-UHFFFAOYSA-N 0.000 description 3
- GIVPAYGOPADRAT-UHFFFAOYSA-N 2-[6-(1H-indol-4-yl)-1H-indazol-4-yl]-5-[(4-propan-2-ylpiperazin-1-yl)methyl]-1,3-oxazole methanol Chemical compound CO.CC(C)N1CCN(Cc2cnc(o2)-c2cc(cc3[nH]ncc23)-c2cccc3[nH]ccc23)CC1 GIVPAYGOPADRAT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 3
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 3
- FOVWTMIRHOADJJ-UHFFFAOYSA-N [2-[1-(oxan-2-yl)indazol-4-yl]-1,3-oxazol-5-yl]-(4-propan-2-ylpiperazin-1-yl)methanone Chemical compound C(C)(C)N1CCN(CC1)C(=O)C1=CN=C(O1)C1=C2C=NN(C2=CC=C1)C1OCCCC1 FOVWTMIRHOADJJ-UHFFFAOYSA-N 0.000 description 3
- 229960004308 acetylcysteine Drugs 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 239000001384 succinic acid Substances 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- GJVXQAIKAKUZAZ-UHFFFAOYSA-N 1,3-oxazol-5-yl-(4-propan-2-ylpiperazin-1-yl)methanone Chemical compound C(C)(C)N1CCN(CC1)C(=O)C1=CN=CO1 GJVXQAIKAKUZAZ-UHFFFAOYSA-N 0.000 description 2
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ZUVPLKVDZNDZCM-UHFFFAOYSA-N 3-chloro-2-methylaniline Chemical compound CC1=C(N)C=CC=C1Cl ZUVPLKVDZNDZCM-UHFFFAOYSA-N 0.000 description 2
- WOWFGZLCKNNPIV-UHFFFAOYSA-N 3-phenylbenzene-1,2-disulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC(C=2C=CC=CC=2)=C1S(O)(=O)=O WOWFGZLCKNNPIV-UHFFFAOYSA-N 0.000 description 2
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 2
- MFGIVPTZAMUOSU-UHFFFAOYSA-N 4-chloro-1-(oxan-2-yl)indazole Chemical compound N1=CC=2C(Cl)=CC=CC=2N1C1CCCCO1 MFGIVPTZAMUOSU-UHFFFAOYSA-N 0.000 description 2
- CQTGQYVQJOJQCM-UHFFFAOYSA-N 4-chloro-1h-indazole Chemical compound ClC1=CC=CC2=C1C=NN2 CQTGQYVQJOJQCM-UHFFFAOYSA-N 0.000 description 2
- SVLZRCRXNHITBY-UHFFFAOYSA-N 4-chloro-1h-indole Chemical compound ClC1=CC=CC2=C1C=CN2 SVLZRCRXNHITBY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- GDGWIDWBUSNQNK-UHFFFAOYSA-N CC(C)N1CCN(CC1)C(=O)C1=CN=C(O1)C1=CC(=CC2=C1C=NN2C1CCCCO1)B1OC(C)(C)C(C)(C)O1 Chemical compound CC(C)N1CCN(CC1)C(=O)C1=CN=C(O1)C1=CC(=CC2=C1C=NN2C1CCCCO1)B1OC(C)(C)C(C)(C)O1 GDGWIDWBUSNQNK-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 239000004367 Lipase Substances 0.000 description 2
- 102000004882 Lipase Human genes 0.000 description 2
- 108090001060 Lipase Proteins 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 238000005576 amination reaction Methods 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 229940114081 cinnamate Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229910052741 iridium Inorganic materials 0.000 description 2
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 2
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 229940011051 isopropyl acetate Drugs 0.000 description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 2
- 235000019421 lipase Nutrition 0.000 description 2
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 229940116351 sebacate Drugs 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 2
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical compound [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 2
- 229910000080 stannane Inorganic materials 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M trans-cinnamate Chemical compound [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 2
- IWYDHOAUDWTVEP-SSDOTTSWSA-N (R)-mandelic acid Chemical class OC(=O)[C@H](O)C1=CC=CC=C1 IWYDHOAUDWTVEP-SSDOTTSWSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- NATULDXOXIBHEB-UHFFFAOYSA-N 1H-indazole 1,3-oxazole Chemical compound N1N=CC2=CC=CC=C12.O1C=NC=C1 NATULDXOXIBHEB-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- LXFQSRIDYRFTJW-UHFFFAOYSA-M 2,4,6-trimethylbenzenesulfonate Chemical compound CC1=CC(C)=C(S([O-])(=O)=O)C(C)=C1 LXFQSRIDYRFTJW-UHFFFAOYSA-M 0.000 description 1
- HHQMHPHKZAACBP-UHFFFAOYSA-N 2-ethyl-1,3-oxazole-5-carboxylic acid Chemical compound CCC1=NC=C(C(O)=O)O1 HHQMHPHKZAACBP-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- NPAXPTHCUCUHPT-UHFFFAOYSA-N 3,4,7,8-tetramethyl-1,10-phenanthroline Chemical compound CC1=CN=C2C3=NC=C(C)C(C)=C3C=CC2=C1C NPAXPTHCUCUHPT-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-M 4-aminosalicylate(1-) Chemical compound NC1=CC=C(C([O-])=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-M 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- LSJOYRIJHIYLLP-UHFFFAOYSA-N CO[Ir] Chemical class CO[Ir] LSJOYRIJHIYLLP-UHFFFAOYSA-N 0.000 description 1
- MHXHNLNQRNPZOP-UHFFFAOYSA-N CO[Ir].C1CC=CCCC=C1 Chemical compound CO[Ir].C1CC=CCCC=C1 MHXHNLNQRNPZOP-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229910002483 Cu Ka Inorganic materials 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 101100456896 Drosophila melanogaster metl gene Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- BKLOVTNYVMYOLE-UHFFFAOYSA-N benzoic acid 2-[6-(1H-indol-4-yl)-1H-indazol-4-yl]-5-[(4-propan-2-ylpiperazin-1-yl)methyl]-1,3-oxazole Chemical compound OC(=O)c1ccccc1.CC(C)N1CCN(Cc2cnc(o2)-c2cc(cc3[nH]ncc23)-c2cccc3[nH]ccc23)CC1 BKLOVTNYVMYOLE-UHFFFAOYSA-N 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000004851 cyclopentylmethyl group Chemical group C1(CCCC1)C* 0.000 description 1
- SZSMWWNXCCLLEK-UHFFFAOYSA-N dhp 3,4-dihydro-2h-pyran Chemical compound C1COC=CC1.C1COC=CC1 SZSMWWNXCCLLEK-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- KRMORCCAHXFIHF-UHFFFAOYSA-N ethyl 1,3-oxazole-5-carboxylate Chemical compound CCOC(=O)C1=CN=CO1 KRMORCCAHXFIHF-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 150000002473 indoazoles Chemical class 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- WPEQESCHNHMPFW-UHFFFAOYSA-N methanol;1,3-oxazole Chemical compound OC.C1=COC=N1 WPEQESCHNHMPFW-UHFFFAOYSA-N 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- NLUPPCTVKHDVIQ-GASCZTMLSA-N n-[5-[4-[5-[[(2r,6s)-2,6-dimethylmorpholin-4-yl]methyl]-1,3-oxazol-2-yl]-1h-indazol-6-yl]-2-methoxypyridin-3-yl]methanesulfonamide Chemical compound C1=C(NS(C)(=O)=O)C(OC)=NC=C1C1=CC(C=2OC(CN3C[C@@H](C)O[C@@H](C)C3)=CN=2)=C(C=NN2)C2=C1 NLUPPCTVKHDVIQ-GASCZTMLSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 125000004304 oxazol-5-yl group Chemical group O1C=NC=C1* 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- CKMXAIVXVKGGFM-UHFFFAOYSA-M p-cumate Chemical compound CC(C)C1=CC=C(C([O-])=O)C=C1 CKMXAIVXVKGGFM-UHFFFAOYSA-M 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003906 phosphoinositides Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-M sulfamate Chemical compound NS([O-])(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-M 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Virology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Un procedimiento de preparación de un compuesto de fórmula (IV) o una sal del mismo**Fórmula** en la que R1 es**Fórmula** y R2 es**Fórmula** o R1 es**Fórmula** y R2 es**Fórmula** procedimiento que comprende: (a) hacer reaccionar un compuesto de fórmula (VII) o una sal del mismo**Fórmula** en la que R1a es**Fórmula** con un compuesto de fórmula (X) o una sal del mismo**Fórmula** en la que X es halógeno y P es un grupo de protección, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XI) o una sal del mismo**Fórmula** en la que R1a y P son como se definieron anteriormente, (b) hacer reaccionar un compuesto de fórmula (XI) o una sal del mismo con un agente de borilación en presencia de un sistema de catalizador para dar un compuesto de fórmula (XII) o una sal del mismo**Fórmula** en la que R1a y P son como se definieron anteriormente, (c) hacer reaccionar un compuesto de fórmula (XII) o una sal del mismo con un compuesto de fórmula (XIV) o una sal del mismo R2-X1 (XIV) en la que R2 es como se definió anteriormente y X1 es halógeno, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XIII) o una sal del mismo**Fórmula** en la que R1a, R2 y P son como se definieron anteriormente, y (d) hacer reaccionar un compuesto de fórmula (XIII) o una sal del mismo con un agente de reducción seguido por desprotección.
Description
DESCRIPCIÓN
Procedimientos de fabricación de derivados de indazol
Campo de la invención
La presente invención se dirige a procedimientos novedosos para preparar compuestos y sales de los mismos, compuestos que son inhibidores de la actividad o función de la fosfoinositida 3'OH quinasa isoforma delta (PI3K8) e intermedios novedosos.
Antecedentes de la invención
La solicitud de patente internacional PCT/EP2010/055666 (número de publicación WO2010/125082) describe compuestos que tienen la fórmula general (I):

en la que
R1' es heteroarilo bicíclico de 9 o 10 miembros en el que el heteroarilo bicíclico de 9 o 10 miembros contiene de uno a tres heteroátomos independientemente seleccionados de oxígeno y nitrógeno y se sustituye opcionalmente con alquilo Ci-6, cicloalquilo C3-6, halo, -CN o -NHSO2R5', o
piridinilo opcionalmente sustituido con uno o dos sustituyentes independientemente seleccionados de alquilo C1-6 , -OR6', halo y -NHSO2R7';
R2' y R3', junto con el átomo de nitrógeno al que se adhieren, se unen para formar un heterociclilo de 6 o 7 miembros en el que el heterociclilo de 6 o 7 miembros contiene opcionalmente un átomo de oxígeno o un átomo de nitrógeno adicional y se sustituye opcionalmente por uno o dos sustituyentes independientemente seleccionados de alquilo C1-6 ;
R4' es hidrógeno o metilo;
R6' es hidrógeno o alquilo C1-4 ; y
R5' y R7' son cada uno independientemente alquilo C1-6, o fenilo opcionalmente sustituido con uno o dos sustituyentes independientemente seleccionados de halo;
y sales de los mismos.
Los ejemplos del documento WO2010/125082 describen la preparación de 6-(1H-indol-4-il)-4-(5-{[4-(1-metiletil)-1 -piperazinil]metil}-1,3-oxazol-2-il)-1H-indazol que se puede representar por la fórmula (II):

en lo sucesivo denominado “Compuesto A” y sus sales clorhidrato, y la preparación de N-[5-[4-(5-{[(2R,6S)- 2,6-dimetil-4-morfolinil]metil}-1,3-oxazol-2-il)-1H-indazol-6-il]-2-(metiloxi)-3-piridinil]metanosulfonamida que se puede representar por la fórmula (III):

en lo sucesivo denominado “Compuesto B” y la sal de (R) -mandelato del mismo.
La solicitud de patente internacional PCT/EP2011/068604 (número de publicación WO2012/055846) describe un polimorfo novedoso del Compuesto A y sales del Compuesto A y polimorfos del mismo, procedimientos para su preparación, composiciones farmacéuticas que los comprenden y su uso en el tratamiento de diversos trastornos.
La solicitud de patente internacional PCT/EP2011/065419 (número de publicación WO2012/032067) describe polimorfos novedosos y sales del Compuesto B, procedimientos para su preparación, composiciones farmacéuticas que los comprenden y su uso en el tratamiento de diversos trastornos.
El documento WO2012/032065 describe una ruta alternativa (Ruta B) a los Compuestos A y B.
Los procedimientos para preparar los Compuestos A y B descritos en los documentos WO2010/125082, WO2012/055846 y WO2012/032067 no son adecuados para fabricar los compuestos a escala comercial. Por ejemplo, uno de los procedimientos descritos utiliza un reactivo de estannano en la etapa de acoplamiento de oxazol-indazol clave. Los reactivos de Estannano son normalmente tóxicos y perjudiciales para el medio ambiente. En el documento WO2012/055846 se describe un procedimiento alternativo que evita el uso de un reactivo de estannano. Sin embargo, este procedimiento requiere un número significativo de etapas, comienza con materiales de partida costosos y tiene un bajo rendimiento general.
Por lo tanto, sigue subsistiendo la necesidad de desarrollar un procedimiento mejorado para preparar los Compuestos A y B que pueda ser adecuado para uso a escala comercial.
Sumario de la invención
La presente invención proporciona procedimientos novedosos para preparar compuestos de fórmula (IV) y sales de los mismos

en la que R1 y R2 son como se definen a continuación, e intermedios novedosos.
Descripción detallada de la invención
La presente invención proporciona una serie de aspectos que se refieren a un procedimiento que se resume en el Esquema 1 a continuación:
Por lo tanto, en un aspecto, la presente invención proporciona un procedimiento para preparar compuestos de fórmula (IV) y sales de los mismos

en la que
R1 es

y
R2 es

o
R1 es

y
R2 es

procedimiento que comprende:
(a) hacer reaccionar un compuesto de fórmula (VII) o una sal del mismo
en la que R1a es

con un compuesto de fórmula (X) o una sal del mismo

en la que X es halógeno y P es un grupo de protección, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XI) o una sal del mismo

en la que R1a y P son como se definieron anteriormente,
(b) hacer reaccionar un compuesto de fórmula (XI) o una sal del mismo con un agente de borilación en presencia de un sistema de catalizador para dar un compuesto de fórmula (XII) o una sal del mismo

en la que R1a y P son como se definieron anteriormente,
(c) hacer reaccionar un compuesto de fórmula (XII) o una sal del mismo con un compuesto de fórmula (XIV) o una sal del mismo
R2-X1 (XIV)
en la que R2 es como se definió anteriormente y X1 es halógeno, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XIII) o una sal del mismo

en la que R18, R2 y P son como se definieron anteriormente, y
(d) hacer reaccionar un compuesto de fórmula (XIII) o una sal del mismo con un agente de reducción seguido por desprotección.
La presente invención proporciona procedimientos para fabricar compuestos de fórmula IV que comprenden todas las cuatro etapas a, b, c y d.
En una realización, R1 es

y R2 es

tal que el compuesto de fórmula (IV) o sal del mismo producido por el procedimiento de la invención es el Compuesto A o una sal del mismo.
En una realización adicional, R1 es

y R2 es

tal que el compuesto de fórmula (IV) o sal del mismo producido por el procedimiento de la invención es el Compuesto B o una sal del mismo.
Las sales de los Compuestos A y B que se pueden preparar de acuerdo con la invención incluyen clorhidrato, bromhidrato, nitrato, metilnitrato, sulfato, bisulfato, sulfamato, fosfato, acetato, hidroxiacetato, fenilacetato, propionato, butirato, isobutirato, valerato, maleato, hidroximaleato, acrilato, fumarato (por ejemplo el hemi fumarato), malato, tartrato, citrato, salicilato, p-aminosaliciclato, glicolato, lactato, heptanoato, ftalato, oxalato, succinato (por ejemplo el hemi succinato), benzoato, o-acetoxibenzoato, clorobenzoato, metilbenzoato, dinitrobenzoato, hidroxibenzoato, metoxibenzoato, naftoato, hidroxinaftoato, mandelato, tannato, formato, estearato, ascorbato, palmitato, oleato, piruvato, pamoato (por ejemplo el hemi pamoato), malonato, laurato, glutarato, glutamato, estolato, metanosulfonato (mesilato), etanosulfonato (esilato), 2-hidroxietanosulfonato, bencenosulfonato (besilato), paminobencenosulfonato, p-toluenosulfonato (tosilato), naftaleno-2-sulfonato, naftalenodisulfonato (por ejemplo el hemi naftalenodisulfonato), mesitilenosulfonato, bifenildisulfonato (por ejemplo el hemi bifenildisulfonato), cinnamato (por ejemplo el hemi cinnamato), sebacato (por ejemplo el hemi sebacato), piromelitato (por ejemplo el hemi piromelitato) y bencenodiacrilato (por ejemplo sal de hemi bencenodiacrilato).
En una realización, se puede utilizar el procedimiento de la presente invención para preparar la sal de hemi succcinato del Compuesto A. En otra realización, el procedimiento de la presente invención se puede utilizar para preparar la sal de benzoato del Compuesto A. La sal de benzoato del Compuesto A tiene ciertas propiedades que pueden hacerla particularmente adecuada para el desarrollo como un fármaco inhalado, en particular, tiene una solubilidad adecuada en fluido pulmonar simulado, buena estabilidad química y física, e higroscopicidad adecuada.
También se incluyen dentro del ámbito de la divulgación cualquier solvato, por ejemplo, hidratos, complejos y formas polimórficas de benzoato de 6-(1H-indol-4-il)-4-(5-{[4-(1 -metiletil)-1-piperazinil]metil}-1,3-oxazol-2-il)-1H-indazol. El benzoato de 6-(1H-Indol-4-il)-4-(5-{[4-(1-metiletil)-1-piperazinil]metil}-1,3-oxazol-2-il)-1H-indazol puede existir en forma cristalina o no cristalina, o como una mezcla de las mismas. Para las sales que están en forma cristalina, el experto en la técnica apreciará que se pueden formar solvatos farmacéuticamente aceptables en los que las moléculas de disolvente se incorporan a la red cristalina durante la cristalización. Los solvatos pueden involucrar disolventes no acuosos tales como etanol, isopropanol, DMSO, ácido acético, etanolamina y EtOAc, o pueden involucrar agua como el disolvente que se incorpora a la red cristalina. Los solvatos en los que el agua es el disolvente que se incorpora en la red cristalina se denominan normalmente “hidratos”. Los hidratos incluyen hidratos estequiométricos, así como composiciones que contienen cantidades variables de agua. Como apreciará el experto, la cantidad de agua puede depender de las condiciones, por ejemplo, la humedad. Por ejemplo, a medida que disminuye la humedad, la cantidad de agua puede disminuir y a medida que aumenta la humedad, puede aumentar la cantidad de agua. Dichas variaciones en la cantidad de agua se incluyen dentro del ámbito de la divulgación.
En otro aspecto, la presente invención proporciona un procedimiento para preparar compuestos de fórmula (XI) y sales de los mismos

en la que R1a es

y P es un grupo de protección, procedimiento que comprende hacer reaccionar un compuesto de fórmula (VII) o una sal del mismo

en la que R1a es como se definió anteriormente, con un compuesto de fórmula (X) o una sal del mismo

en la que X es halógeno y P es como se definió anteriormente, en presencia de un catalizador de paladio.
En una realización, R1a es

En una realización adicional, R1a es

El grupo de protección P puede ser cualquier grupo de protección adecuado descrito en, por ejemplo, Protective Groups in Organic Synthesis por Wuts y Greene (John Wiley & Sons). En una realización, el grupo de protección es tetrahidro- 2H-piran-2-ilo.
En una realización, X es cloro. En una realización adicional, X es bromo.
El catalizador de paladio utilizado en la formación del compuesto de fórmula (XI) descrito anteriormente puede ser cualquier complejo de catalizador de paladio adecuado, por ejemplo, un complejo de paladio con un ligando adecuado. El ligando puede ser, por ejemplo, un ligando Buchwald tal como XPhos (2-diciclohexilfosfino-2',4',6'-triisopropilbifenil) o RuPhos (2-diciclohexilfosfino-2’,6'-dMsopropoxibifenil). En una realización, el catalizador de paladio es un complejo de paladio con XPhos.
El compuesto de fórmula (VII) o sal del mismo se puede preparar a partir de ácido oxazol-5-carboxílico de etilo, el compuesto de fórmula (V) o una sal del mismo

mediante hidrólisis para dar ácido oxazol-5-carboxílico, el compuesto de fórmula (VI) o una sal del mismo
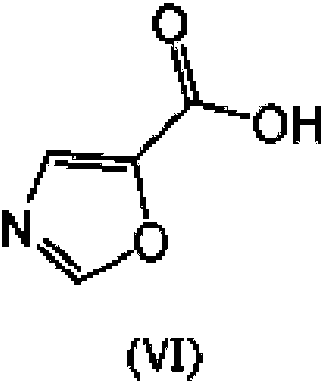
seguido por aminación con 1-(isopropil)piperazina o (2R,6S)-2,6-dimetilmorfolina.
Alternativamente, el compuesto de fórmula (VII) o una sal del mismo se puede preparar directamente mediante tratamiento de ácido oxazol-5-carboxílico de etilo con 1-(isopropil)piperazina o (2R,6S)-2,6-dimetilmorfolina en presencia de una enzima. En una realización, la enzima es lipasa liofilizada TL.
El compuesto de fórmula (X) o sal del mismo se puede preparar al hacer reaccionar un compuesto de fórmula (VIII) o una sal del mismo

en la que X es como se definió anteriormente, con nitrito de isoamilo, seguido por protección.
Alternativamente, en otro aspecto, la presente divulgación proporciona un procedimiento para preparar compuestos de fórmula (XI) y sales de los mismos

en la que R1a es y P son como se definieron anteriormente, procedimiento que comprende hacer reaccionar un compuesto de fórmula (Va) o una sal del mismo

en la que R3 es alquilo C1-6 , tal como etilo (para dar un compuesto de fórmula (V) como se definió anteriormente), con un compuesto de fórmula (X) o una sal del mismo

en la que X es halógeno y P es como se definió anteriormente, en presencia de un catalizador de paladio, seguido por hidrólisis y aminación con 1-(isopropil)piperazina o (2R, 6S)-2,6-dimetilmorfolina.
En otro aspecto, la presente invención proporciona un procedimiento para preparar compuestos de fórmula (XII) y sales de los mismos

en la que R1a y P son como se definieron anteriormente,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XI) o una sal del mismo

en la que R1a y P son como se definieron anteriormente, con un agente de borilación en presencia de un sistema de catalizador.
El agente de borilación utilizado de acuerdo con la presente invención puede ser cualquier agente de borilación adecuado tal como pinacolborano.
El sistema de catalizador utilizado de acuerdo con la presente invención puede ser cualquier sistema de catalizador adecuado tal como un sistema de catalizador de iridio, níquel, hierro, rodio o cobalto. Sistemas de catalizadores adecuados se describen en, por ejemplo, Chem. Commun., 2015, 51, 6508-6511; J. Am. Chem. Soc., 2013, 135, 17258-17261; y J. Am. Chem. Soc., 2014, 136, 4133-4136. En una realización, el sistema de catalizador es un sistema de catalizador de iridio, por ejemplo, dímero de (1,5-ciclooctodieno)(metoxi) iridio (I).
En otro aspecto, la presente invención proporciona un procedimiento para preparar compuestos de fórmula (XIII) y sales de los mismos

en la que R1a, R2 y P son como se definieron anteriormente,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XII) o una sal del mismo

en la que R1a y P son como se definieron anteriormente, con un compuesto de fórmula (XIV) o una sal del mismo
R2-X1 (XIV)
en la que R2 es como se definió anteriormente y X1 es halógeno, en presencia de un catalizador de paladio.
En una realización, X1 es cloro. En una realización adicional, X1 es bromo.
El catalizador de paladio utilizado en la formación del compuesto de fórmula (XIII) descrito anteriormente puede ser cualquier complejo de catalizador de paladio adecuado, por ejemplo, un complejo de paladio con un ligando adecuado. El ligando puede ser, por ejemplo, un ligando Buchwald tal como XPhos (2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo) o triciclohexilfosfina. En una realización, el catalizador de paladio es un complejo de paladio con XPhos.
En un aspecto adicional, la presente invención proporciona un procedimiento para preparar compuestos de fórmula (IV) y sales de los mismos

en la que R1 y R2 son como se definieron anteriormente,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XIII) o una sal del mismo

en la que R1a, R2 y P son como se definieron anteriormente, con un agente de reducción seguido por desprotección. El agente de reducción puede ser cualquier agente de reducción adecuado tal como un hidruro, por ejemplo, hidruro de litio y aluminio, borohidruro de sodio, hidruro de diisobutilaluminio (DIBAL) o bis(2-metoxietoxi)hidruro de aluminio y sodio (Red-AI). En una realización, el agente reductor es hidruro de litio y aluminio.
Como lo apreciará un experto en la técnica, las condiciones requeridas para la desprotección dependerán de la naturaleza del grupo de protección. Si el grupo de protección utilizado es tetrahidro-2H-piran-2-ilo, se puede eliminar en condiciones ácidas. Los compuestos intermedios de fórmulas (VII), (XI), (XII) y (XIII) son novedosos y, por lo tanto, los compuestos y sus sales forman otro aspecto de la invención.
En una realización, la presente invención proporciona un compuesto de fórmula (VII) o una sal del mismo en la que R1a es

En otra realización, la presente invención proporciona un compuesto de fórmula (XI) o una sal del mismo en la que R1a es

y P es tetrahidro-2H-piran-2-ilo.
En otra realización, la presente invención proporciona un compuesto de fórmula (XII) o una sal del mismo en la que R1a es

y P es tetrahidro-2H-piran-2-ilo.
En otra realización, la presente invención proporciona un compuesto de fórmula (XIII) o una sal del mismo en la que R1a es

R2 es

y P es tetrahidro-2H-piran-2-ilo.
Una realización particular de la presente invención se muestra en el Esquema 2 a continuación y se describe en detalle en los siguientes Ejemplos, que están destinados solo a la ilustración y no están destinados a limitar el alcance de la invención de ninguna manera.

Como se utiliza en el presente documento, los símbolos y convenciones utilizados en estos procedimientos, esquemas y ejemplos son consistentes con los utilizados en la literatura científica contemporánea, por ejemplo, el Journal of the American Chemical Society o el Journal of Biological Chemistry. A menos que se indique lo contrario, todos los materiales de partida se obtuvieron de proveedores comerciales y se utilizaron sin purificación adicional. Específicamente, se pueden utilizar las siguientes abreviaturas en los ejemplos y en toda la especificación:
Ac Acetilo
COD 1,5-Ciclooctadieno
CPME Éter de Ciclopentil metilo
DHP 3,4-Dihidro-2H-pirano
DMSO Sulfóxido de dimetilo
Et Etilo
EtOAc Acetato de etilo
G Gramos
H Hora(s)
IPA Isopropanol
iPr Isopropilo
HPLC Cromatografía líquida de alto rendimiento
Kg Kilogramos
L Litro
Me Metilo
Mg Miligramos
MIBK Metil isobutil cetona
Min Minuto(s)
ml Mililitros
mol Moles
mmol Milimoles
Pin Pinacol
RuPhos 2-diciclohexilfosfino-2',6'-diisoproxiproxifenilproxiproxi
Rt Tiempo de retención
TFA Ácido trifluoroacético
THF Tetrahidrofurano
TMS Trimetilsilano
XPhos 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo
XRPD Difracción de rayos X en polvo
Ejemplos
Intermedio 1
Ácido oxazol-5-carboxílico

Una solución acuosa de monohidrato de hidróxido de litio (124,5 kg de una solución preparada a partir de 49,44 kg de monohidrato de hidróxido de litio disuelto en 319 kg de agua, 398 mol) se agregó a una solución de 5-oxazolcarboxilato de etilo (54 kg, 382,7 mol) en agua (54 kg) manteniendo la temperatura por debajo de 25 °C. La reacción se agitó durante 6,5 h y luego se agregó HCl acuso concentrado (64,8 kg) manteniendo la temperatura por debajo de 25 °C, la cristalización se enfrió a 5 °C y se mantuvo durante 1 h. El producto se filtró, se lavó con agua
fría (88 kg), luego con isopropanol (171 kg) y se secó bajo vacío a 50 °C para dar el compuesto del título (37,88 kg, 87,5 %).
1H RMN (400 MHz, DMSO-da) 8 ppm 13,68 (br. s., 1 H), 8,59 (s, 1 H), y 7,88 (s, 1 H).
Intermedio 2
(4-Isoprop¡lp¡perazin-1-¡l)(oxazo)-5-¡l)metanona

Procedimiento A
Se agregó cloruro de oxalilo (47,7 kg, 375,8 mol) a una solución de ácido oxazol-5-carboxílico (32,88 kg, 290,8 mol) en acetato de isopropilo (144 kg) manteniendo la temperatura a 52-58 °C. La temperatura se aumentó a 58,5 °C, se agitó durante 5 h y luego se enfrió a 20 °C. La mezcla de reacción se agregó a una solución de 1-(isopropiOpiperazina (41 kg, 319,8 mol) y carbonato de potasio (118,4 kg) en acetato de isopropilo (348 kg) y agua (103 kg) manteniendo la temperatura por debajo de 25 °C. La reacción se agitó durante 15 mins, la temperatura se aumentó a 33 °C y la fase orgánica se lavó con agua (191 kg), se concentró bajo presión reducida a 95 l y se enfrió a 20 °C. Se agregó n-Heptano (157 kg) y la cristalización se agitó durante 2 h, y el producto se filtró, se lavó con nheptano (157 kg) y se secó bajo vacío 40 °C para dar el compuesto del título (57,14 kg, 88,0 %).
1H RMN (400 MHz, CDCla-d) 8 ppm 7,94 (s, 1 H), 7,56 (s, 1 H), 3,91-3,73 (m, 4 H), 2,75 (spt., J=6,5 Hz, 1 H), 2,63 2,52 (m, 4 H) y 1,06 (d, J=6,6 Hz, 6 H).
Procedimiento B
Se agregaron 1-Isopropilpiperazina (1,06 g, 8,24 mmol) y etil-oxazol-5-carboxilato (1,16 g, 8,24 mmol) a una suspensión de tamices moleculares de 5 A (8,0 g) en éter de ciclopentilo metilo (40 ml) y se agitó durante 1 h a 55 °C. Se agregó lipasa liofilizada TL (2,0 g), la mezcla de reacción se agitó durante 28,75 h, luego se filtró a través de papel de fibra de vidrio, se lavó a través de con éster de ciclopentil metilo ( 3 x 6 ml). El filtrado combinado y los lavados se concentraron bajo presión reducida y el residuo crudo se volvió a suspender en metil ciclohexano (6 ml), se filtró, se lavó con metil ciclohexano ( 2 x 5 ml), y se secó bajo vacío para dar el compuesto del título (1,40 g, 76 %).
Intermedio 3
4-Cloro-1H-indazol
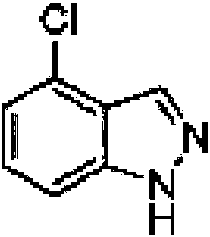
Se agregó anhídrido acético (69 kg, 675,9 mol) a una suspensión agitada de 3-cloro-2-met¡lan¡l¡na (30 kg, 211,9 mol), acetato de potasio (25 kg, 254,7 mol) y metiltetrahidrofurano (302 l) a 25 °C y luego se agitó durante 2 h. Se agregó nitrito de isopentilo (44,5 kg, 379,9 mol) a la suspensión y los contenidos se calentaron a 73 °C durante 18 h. La suspensión se enfrió a 20 °C, luego se agregó agua (90 L), y la reacción se enfrió a 5 °C. Se agregó una solución acuosa de NaOH (105 l de un 32 % p/p), la solución se calentó a 40 °C y se agitó durante 2 h. La fase acuosa inferior se eliminó y la capa orgánica se lavó con agua (150 L) y luego solución salina (18 kg en 90 l de agua). La capa orgánica se concentró a 90 l mediante destilación atmosférica y un intercambio disolvente a n-heptano realizado mediante destilación atmosférica para un volumen final de 240 l. La suspensión se enfrió a 7 °C, se agitó durante 2 h y el sólido se aisló mediante filtración. La torta se lavó con heptano (2 x 60 l) y se secó bajo vacío para dar el compuesto del título (23,6 kg, 73 %).1
1H RMN (400MHz, MeOD-d4) 8 = 8,08 (s, 1H), 7,48 (d, J=8,6 Hz, 1H), 7,33 (dd, J=7,5, 8,4 Hz, 1H), 7,14 (d, J=7,3 Hz, 1H) HPLC r . t - 1,94 min.
Intermedio 4
4-Cloro-1-(tetra h¡dro-2H-p¡ran-2-¡l)-1H-¡ndazol

Se agregó ácido trifluoroacético (3,5 kg, 30,7 mol) a una solución de 4-cloro-1H-indazol (23 kg, 15,1 mol) y 3,4-dihidro-2H-piran (43,1 kg, 512,7 mol) en acetato de etilo (235 l). La mezcla de reacción se calentó a 80 °C durante 4 h, luego se enfrió a 23 °C, se agregó trietilamina (3,2 kg, 31,6 mol) y se agitó durante 30 min. El disolvente se intercambió a isopropanol utilizando una destilación atmosférica para un volumen final de 138 L. La solución se enfrió a 55 °C y se agregó agua (138 l) manteniendo la temperatura. La solución se enfrió a 42 °C y se sembró (8 g), luego se enfrió a 30 °C, se mantuvo durante 10 h, y se agregó agua (46 l), se enfrió a 18 °C, y la suspensión se agitó durante 2 h luego se filtró, se lavó con 6:1 v/v de agua/2-propanol (2 x 46 l) y se secó bajo vacío a 50 °C para dar el compuesto del título (28,8 kg, 80,7 %).
1H RMN (500MHz, DIVISOR) 8 = 8,18 (s, 1H), 7,74 (d, J=8,5 Hz, 1H), 7,42 (dd, J=7,5, 8,4 Hz, 1H), 7,27 (d, J=7,3 Hz, 1H), 5,88 (dd, J=2,5, 9,5 Hz, 1H), 3,90 - 3,85 (m, 1H), 3,79 - 3,70 (m, 1H), 2,44 - 2,35 (m, 1H), 2,07 - 1,95 (m, 2H), 1,81 -1,69 (m, 1H), 1,64-1,53 (m, 2H).
Intermedio 5
(4-Isoprop¡)p¡peraz¡n-1-¡l)(2-(1-(tetrah¡dro-2H-p¡ran-2-il)-1H-¡ndazol-4-¡l)oxazol-5-¡l)metanona

Procedimiento A
Cloruro de paladio (0,74 kg, 4,2 mol) y XPhos (4,36 kg, 9,1 mol) se suspendió en éter de ciclopentilo metilo (372 l). Se agregaron 4-Cloro-1-(tetrahidro-2H-piran-2-il)-1H-indazol (36 kg, 152,1 mol), (4-Isopropilpiperazin-1-il)(oxazol-5-il)metanona (34,78 kg, 155,8 mol) y carbonato de potasio (325 de malla, 35,64 kg, 257,9 mol) y se enjuagó con éter de ciclopentilo metilo (2,58 kg). Se agregó una solución de ácido piválico (9,294 kg, 91,0 mol) disuelta en éter de ciclopentilo metilo (10 l) seguida por un enjuague de éter de ciclopentilo metilo (10 l). La reacción se desgasificó en vacío y se rellenó con nitrógeno tres veces, luego se calentó a reflujo durante 5 h y los contenidos se enfriaron a 40 °C. La mezcla de reacción se lavó con agua (144 l) luego con 5 % p/v de solución acuosa de cloruro de sodio (151,2 kg) y la fase orgánica se concentró a presión atmosférica a 288 l. La mezcla de reacción se filtró en otro recipiente y el filtro se lavó con éter de ciclopentilo metilo (36 l), luego se destiló a 108 l bajo presión atmosférica. Se agregó metil ciclohexano (162 l) al recipiente manteniendo la temperatura a 75 °C, los contenidos luego se enfriaron a 62-65 °C y la cristalización se sembró con (4-isopropilpiperazin-1-il)(2-(1-(tetrahidro-2H-piran-2-il)-1H-indazol-4-il)oxazol-5-il)metanona (90 g) se suspendió en metil ciclohexano congelado (0,52 l). La cristalización se mantuvo a 62 °C durante 30 mins, se enfrió a 7 °C y luego se mantuvo a 7 °C durante la noche. El producto se filtró, se lavó con metil ciclohexano (2 x 72 l) y se secó en un horno de vacío a 50 °C para dar el compuesto del título (57,2 kg, 88,9 %).1
1H RMN (400MHz, DMSO-d6) 8 ppm 8,59 (s, 1H), 8,00 (d, J=8,6 Hz, 1 H), 7,95 (s, 1H), 7,91 (d, J =7,3 Hz, 1 H),7,62 (dd, J=7,3, 8,3 Hz, 1H), 5,97 (dd, J=2,0, 9,5 Hz, 1H), 4,03 - 3,85 (m, 1H), 3,84 - 3,70 (m, 1H), 3,66 (br. s., 4H), 2,72 (spt., J=6,5 Hz, 1 H), 2 ,57-2,40 (m, 5H), 2,11 - 1,96 (m, 2H), 1,85- 1,68 (m, 1H), 1,68 - 1,47 (m, 2H), 0,99 (d, J=6,4 Hz, 6 H)
Procedimiento B
4-Cloro-1-(tetrahidro-2H-piran-2-il)-1H-indazol (10 g, 42,2 mmol), (4-Isopropilpiperazin-1-il)(oxazol-5-il)metanona (9,67 g, 43,3 mmol), carbonato de potasio (325 de malla, 9,93 g, 71,8 mmol) y ácido piválico (2,59 g, 25,3 mmol) se
suspendieron en CPME (90 ml). La reacción se agitó durante 10 mins a temperatura ambiente y luego se desgasificó en vacío y se rellenó con nitrógeno tres veces. Se agregaron cloruro de paladio (206 mg, 1,16 mmol) y XPhos (1,21 g, 2,53 mmol) y se enjuagaron con CPME (10 ml). La mezcla de reacción se desgasificó en vacío y se rellenó con nitrógeno tres veces, luego se calentó a reflujo durante 5 h y los contenidos se enfriaron de nuevo a 50 °C. La mezcla de reacción se lavó con 3 % p/p de solución acuosa de cloruro de sodio (30 ml), luego con 20 % p/p de solución acuosa de cloruro de sodio (30 ml) y la fase orgánica se concentró a presión atmosférica hasta 80 ml. La mezcla de reacción se enfrió a temperatura ambiente y se mantuvo durante la noche. La mezcla de reacción luego se filtró en otro recipiente y el filtro se lavó con CPME (20 ml), luego se destiló a 30 ml bajo presión atmosférica. Los contenidos se enfriaron a 80 °C y se agregó metil ciclohexano (45 ml) al recipiente manteniendo la temperatura a 75 °C, los contenidos luego se enfriaron a 62-65 °C y la cristalización se sembró con (4-isopropilpiperazin-1-il)(2-(1 -(tetrahidro-2H-piran-2-il)-1H-indazol-4-il)oxazol-5-il)metanona. La cristalización se mantuvo a 63 °C durante 30 mins, se enfrió a 5 °C durante 6 h y luego se mantuvo a 5 °C durante la noche. El producto se filtró, se lavó con metil ciclohexano congelado (2 x 20 ml) y se secó en un horno de vacío a 50 °C para dar el compuesto del título (13,52 g, 76 %).
1H RMN (400MHz, DMSO-d6) 8 ppm 8,59 (s, 1H), 8,00 (d, J=8,6 Hz, 1 H), 7,95 (s, 1H), 7,91 (d, J=7,3 Hz, 1 H),7,62 (dd, J=7,3, 8,3 Hz, 1H), 5,97 (dd, J=2,0, 9,5 Hz, 1H), 4,03 - 3,85 (m, 1H), 3,84 - 3,70 (m, 1H), 3,66 (br. s., 4H), 2,72 (spt., J=6,5 Hz, 1 H), 2 ,57-2,40 (m, 5H), 2,11 - 1,96 (m, 2H), 1,85- 1,68 (m, 1H), 1,68 - 1,47 (m, 2H), 0,99 (d, J=6,4 Hz, 6 H)
Intermedio 6
(4-Isoprop¡lp¡peraz¡n-1-¡l)(2-(1-(tetrah¡dro-2H-p¡ran-2-¡l)-6-(4.4.5.5-tetramet¡l-1.3,2-d¡oxaborolan-2-¡l)-1H-¡ndazol-4-il)oxazol-5-il)metanona

Se agregó pinacolborano (40,80 kg, 318,8 mol) a una solución agitada de (4-isopropilpiperazin-1-il)(2-(1-(tetrahidro-2H-piran-2-il)-1H-indazol-4-il)oxazol-5-il)metanona (54,00 kg, 127,5 mol), dímero (1,5-ciclooctadieno)(metoxi)iridio (I) (0,864 kg, 1,30 mol) y 3,4,7,8-tetrametil-1,10-fenantrolina (1,51 kg, 6,39 mol) en THF (243,2 kg) a 20 °C. La reacción se calentó a reflujo durante 8 h, luego se enfrió a 20 °C y se transfirió en un recipiente que contiene isopropanol (253,8 kg), se lavó a través de con THF (23,8 kg). El disolvente se destiló a 270 l a 200 mbar e se agregó isopropanol (253,8 kg) y luego se volvió a destilar a 324 l a 100 mbar. La cristalización se calentó a 40 °C durante 4 h, se enfrió a 20 °C, se agitó durante la noche, se filtró, se lavó con isopropanol (84,8 kg), y se secó en un horno de vacío a 50 °C para dar el compuesto del título (57,35 kg, 82,8 %).1
1H RMN (400MHz, CDCla-d) 8 ppm 8,70 (s, 1H), 8,40 (s, 1H), 8,17 (s, 1H), 7,74 (s, 1H), 5,85 (dd, J=2,4, 9,5 Hz, 1H), 4,11 - 4,03 (m, 1H), 3,97 - 3,77 (m, 5H), 2,78 (spt., J=6,4 Hz, 1 H), 2,70 - 2,59 (m, 5H), 2,24 - 2,14 (m, 1H), 2,14 -2,03 (m, 1H), 1,86- 1,72 (m, 2H), 1,72- 1,62 (m, 1H), 1,40 (s, 12H), 1,08 (d, J=6,4 Hz, 6H).
Intermedio 7
(2-(6-(1H-¡ndol-4-¡l)-1-(tetrah¡dro-2H-p¡ran-2-¡l)-1H-¡ndazol-4-¡l)oxazol-5-il)(4-¡soprop¡lp¡peraz¡n-1-¡l)metanona

Procedimiento A
Se agregó 4-Cloroindol (13,8 kg, 91,04 mol) a una solución agitada de (4-isopropilpiperazin-1-il)(2-(1 -(tetrahidro-2H-piran-2-il)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-indazol-4-il)oxazol-5-il)metanona (50 kg, 91,00 mol), acetato de paladio (0,2 kg, 0,89 mol), XPhos (0,65 kg, 1,36 mol), y fosfato de potasio (46,4 kg, 218,6 mol) en MIBK (176 kg) y agua (250 kg). La mezcla de reacción se desgasificó bajo vacío 3 x, luego se calentó a 82 °C durante 78 mins, y se agregó MIBK (300l). Las fases se mezclaron durante 30 mins, luego se separaron y la fase orgánica se lavó con una solución acuosa elaborada a partir de carbonato de potasio (2,75 kg), N-acetil cisteína (5,2 kg) y agua (250 kg), luego agua (250 kg) y se destiló a 300 L a presión atmosférica. La cristalización se enfrió a 20 °C, se agitó durante 5 h, se filtró, se lavó con MIBK (80 kg) y se secó en el horno de vacío a 50 °C para dar el compuesto del título (36,15 kg, 73,7 %).
1H RMN (400 MHz, CDCls-d) 8 ppm 8,72 (s, 1 H), 8,38 (br. s, 1 H), 8,35 (d, J=1,0 Hz, 1 H), 8,06 (s, 1 H), 7,81 (s, 1 H), 7,52-7,46 (m, 1 H), 7,36-7,30 (m, 3 H), 6,79-6,75 (m, 1 H), 5,84 (dd, J=9,17, 2,32 Hz, 1 H), 4,10-4,04 (m, 1 H), 3.88 (br. s., 4 H), 3,84-3,72 (m, 1 H), 2,76 (spt., J=6,5 Hz, 1 H), 2,68-2,59 (m, 5 H), 2,30-2,12 (m, 2 H), 1,86-1,73 (m, 2 H), 1,73-1,64 (m, 1 H), 1,07 (d, J=6,60 Hz, 6 H).
Procedimiento B
4-Cloroindol (8,71 kg, 57,46 mol) se agregó a una solución agitada de (4-isopropilpiperazin-1-il)(2-(1-(tetrahidro- 2H-piran-2-il)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-indazol-4-il)oxazol-5-il)metanona (34,55 kg, 62,88 mol), y fosfato de potasio (32,1 kg, 151,2 mol) en MIBK (139,2 kg) y agua (172,8 kg). La mezcla de reacción se desgasificó bajo vacío 3 x, y luego se agregaron acetato de paladio (0,14 kg, 0,62 mol) y XPhos (0,45 kg, 0,94 mol) enjuagados con MIBK (1 kg). La reacción se calentó a reflujo durante 1 h. La reacción se enfrió a menos de 40 °C y se agregó N-acetil cisteína (3,59 kg), seguida por MIBK (138,2 kg). Las fases se calentaron a 82 °C, se mezclaron, luego se separaron y la fase orgánica se lavó con una solución acuosa elaborada a partir de carbonato de potasio (1,9 kg), N-acetil cisteína (3,59 kg), MIBK (27,6 kg) y agua (173,8 kg), luego agua (172,8 kg) y se destiló a 155 L a presión atmosférica. La cristalización se enfrió a 20 °C, se agitó durante 22 h, se filtró, se lavó con MIBK (2 x 55,3 kg) y se secó en un horno de vacío a 50 °C para dar el compuesto del título 24,72 kg, 73 %).1
1H RMN (400 MHz, CDCls-d) 8 ppm 8,72 (s, 1 H), 8,38 (br. s, 1 H), 8,35 (d, J=1,0 Hz, 1 H), 8,06 (s, 1 H), 7,81 (s, 1 H), 7,52-7,46 (m, 1 H), 7,36-7,30 (m, 3 H), 6,79-6,75 (m, 1 H), 5,84 (dd, J=9,17, 2,32 Hz, 1 H), 4,10-4,04 (m, 1 H), 3.88 (br. s., 4 H), 3,84-3,72 (m, 1 H), 2,76 (spt., J=6,5 Hz, 1 H), 2,68-2,59 (m, 5 H), 2,30-2,12 (m, 2 H), 1,86-1,73 (m, 2 H), 1,73-1,64 (m, 1 H), 1,07 (d, J=6,60 Hz, 6 H).
Ejemplo 1
Solvato de 2-(6-(1H-Indol-4-¡l)-1H-¡ndazol-4-¡l)-5-((4-¡soprop¡lp¡perazin-1-¡l)met¡l)oxazol metanol
Procedimiento A
Se agregó cloruro de aluminio (2,94 kg, 22 mol) a THF (216 kg) y se agitó hasta que se disolvió. Se agregó (2-(6-(1H-Indol-4-il)-1-(tetrahidro-2H-piran-2-il)-1H-indazol-4-il)oxazol-5-il)(4-isopropilpiperazin-1-il)metanona (34,75 kg, 64,5 mol) al recipiente lavado con THF (0,5 kg) y los contenidos se enfriaron a 0 °C. Se agregó una solución de hidruro de litio y aluminio en THF (10 % p/p, 22,6 kg, 59,6 mol) manteniendo la temperatura a menos de 20 °C, seguido de un lavado de línea con THF (0,9 kg). La reacción se agitó durante 30 mins y luego se agregó EtOAc (15,6 kg) seguido por agitación de 1 h. Se agregó una solución de trietanolamina (2,36 kg) en THF (2,1 l) seguida por trietanolamina (36,8 kg) y luego NaOH acuoso (15 % p/p, 121 kg). Las fases se separaron y la fase orgánica se lavó con NaOH acuoso (15 % p/p, 121 kg), se agregó THF (17 kg), y el disolvente se destiló a 104 l a presión atmosférica. Una destilación de volumen constante (104 l) se realizó al agregar MeOH (174 l), y luego se agregó MeOH (193,3 kg). Se agregó clorotrimetilsilano (35,8 kg, 329,5 mol) y la reacción se calentó a 50 °C durante 3 h, y luego se agregó trietilamina (35,8 kg, 353,8 mol) y la cristalización se enfrió a 20 °C, se filtró, se lavó con MeOH (2 x 55 kg) y se secó en el horno de vacío a 50 °C para dar el compuesto del título (24,80 kg, 81 %).
1H RMN (400MHz, DMSO-da) 5 ppm 13,43 (br. s., 1H), 11,36 (br. s., 1H), 8,61 (s, 1H), 8,09 (d, J=1,2 Hz, 1H), 7,92 (m, 1H), 7,51 - 7,46 (m, 2H), 7,32 (s, 1H), 7,28 - 7,22 (m, 2H), 6,61 (m, 1H), 4,09 (q, J=5,3 Hz, 1H), 3,73 (s, 2H), 3,18 (d, J=4,9 Hz, 3H), 2,60 (br. s., 1H), 2,56 - 2,40 (m, 4H), 0,94 (d, J=6,4 Hz, 6H).
Procedimiento B
Se agregó cloruro de aluminio (4,25 g, 31,8 mmol) a THF (250 ml) y se agitó hasta que se disolvió. Se agregó (2-(6-(1H-Indol- 4-il)-1-(tetrahidro-2H-piran-2-il)-1H-indazol-4-il)oxazol-5-il)(4-isopropilpiperazin-1-il)metanona (50 g, 93 mmol) al recipiente lavado con THF (100 ml) y los contenidos se enfriaron a 0-5 °C. Se agregó una solución de hidruro de litio y aluminio en THF (10 % p/p, 35,6 ml, 85 mmol) manteniendo la temperatura a menos de 20 °C. La reacción se agitó durante 105 mins y luego se agregó EtOAc (25 ml) seguido por agitación de 1 h. Se agregó trietanolamina (50,5 ml) seguida de NaOH acuoso (15 % p/p, 150 ml). Las fases se separaron y la fase orgánica se lavó con NaOH acuoso (15 % p/p, 150 ml), y el disolvente se destiló a 150 ml a presión atmosférica. Una destilación de volumen constante (150 ml) se realizó al agregar MeOH (250 ml). En un recipiente separado, se agregó clorotrimetilsilano (59,3 ml, 464 mol) con cautela a MeOH (400 ml). La reacción se calentó a 50-55 °C y la solución de metanol del sustrato se agregó durante aproximadamente 2 h. La reacción se agitó a 55 °C durante 3,25 h y luego se agregó trietilamina (71,2 ml, 511 mmol) manteniendo la temperatura a menos de 60 °C y la cristalización se mantuvo durante 2 h a 50 °C, se enfrió a 20 °C durante 2 h, se mantuvo a 20 °C durante 2 días, se filtró, y se lavó con MeOH (2 x 100 ml) y se secó en el horno de vacío a 50 °C para dar el compuesto del título (35,4 g, 81 %).
1H RMN (400MHz, DMSO-d6) 5 ppm 13,43 (br. s., 1H), 11,36 (br. s., 1H), 8,61 (s, 1H), 8,09 (d, J=1,2 Hz, 1H), 7,92 (m, 1H), 7,51 - 7,46 (m, 2H), 7,32 (s, 1H), 7,28 - 7,22 (m, 2H), 6,61 (m, 1H), 4,09 (q, J=5,3 Hz, 1H), 3,73 (s, 2H), 3,18 (d, J=4,9 Hz, 3H), 2,60 (br. s., 1H), 2,56 - 2,40 (m, 4H), 0,94 (d, J=6,4 Hz, 6H).
Ejemplo 2
Hemi-succinato de 2-(6-(1H-Indol-4-¡l)-1H-¡ndazol-4-¡l)-5-((4-¡soprop¡lp¡perazin-1-¡l)met¡l)oxazol

Procedimiento A
Se disolvieron solvato de 2-(6-(1H-Indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol metanol (729,1 g, 1,54 mol) y ácido succínico (109,6 g, 0,93 mol) en 10 % p/p de agua en DMSO (4,35 l) a 85 °C. La solución se filtró en otro recipiente, la línea se lavó con 10 % p/p de agua caliente en DMSO (725 ml) y la solución se mantuvo a 85 °C. Se agregó agua (725 ml) manteniendo la temperatura a 85 °C, luego los contenidos se enfriaron a 40 °C durante 2 h, y la cristalización se sembró con hemi-succinato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol (3,63 g) suspendido en 25 % p/p de agua en DMSO (54 ml), seguido por un lavado de línea de 25 % p/p de agua en DMSO (18,5 ml). La suspensión se envejeció durante 30 mins a 40 °C y luego se agregó agua (1,81 l) durante 1 h, los contenidos se agitaron a 40 °C durante 1 h, luego la cristalización se enfrió a 20 °C durante 1 h, luego se envejeció durante 17 h, se filtró, se lavó con 40 % p/p de agua en DMSO (1,45 l), agua (1,45 l), isopropanol (2 x 1,45 l) y se secó bajo vacío a 50 °C para dar el compuesto del título (626 g, 81 %).
H RMN (400MHz, DMSO-d6) 5 ppm 13,43 (1H, br s), 11,36 (1H, s), 10,00 (br. s, 1H) 8,60 (1H, d, J = 0,9 Hz), 8,08 (1H, d, J = 1,3 Hz), 7,90-7,92 (1H, m), 7,47-7,50 (1H, m), 7,47 (1H, t, J = 2,8 Hz), 7,41 (1H, m), 7,24 (1H, t, J = 7,3 Hz), 7,23 (1H, dd, J = 7,3, 1,7 Hz), 6,59-6,62 (1H, m), 2,66 (1H, m), 2,40-2,61 (8H, m), 2,38 (2H, s), 1,22 (6H, d, J = 6,5 Hz).
Procedimiento B
Se disolvieron solvato de 2-(6-(1H-Indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol metanol (10,0 kg, 21,2 mol) y ácido succínico (1,50 kg, 12,7 mol) en DMSO (50 l) a 85 °C. La solución se filtró en otro recipiente, la
línea se lavó con DMSO caliente (10 l) y la solución se mantuvo a 85 °C. Se agregó agua (17 l) manteniendo la temperatura a 82-88 °C, luego los contenidos se enfriaron a 40 °C a 0,2 °C/min, y la cristalización se sembró con hemi-succinato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1 -il)metil)oxazol (50 g) se suspendió en 25 % p/p de agua en DMSO (500 ml), seguido por un lavado de línea de 25 % p/p de agua en DMSO (500 ml). La suspensión se envejeció durante 30 mins luego se agregó agua (30 l) durante 8 h, los contenidos se agitaron durante 1 h, luego la cristalización se enfrió a 20 °C durante 1 h, luego se envejeció durante 14 h, se filtró, se lavó con 40 % p/p de agua en DMSO (40 l), agua (40 l), isopropanol (3 x 60 l) como lavados de suspensión y se secó bajo vacío a 50 °C para dar el compuesto del título (7,25 kg, 68 %).
1H RMN (400MHz, DMSO-da) 8 ppm 13,43 (1H, br s), 11,36 (1H, s), 10,00 (br. s, 1H) 8,60 (1H, d, J = 0,9 Hz), 8,08 (1H, d, J = 1,3 Hz), 7,90-7,92 (1H, m), 7,47-7,50 (1H, m), 7,47 (1H, t, J = 2,8 Hz), 7,41 (1H, m), 7,24 (1H, t, J = 7,3 Hz), 7,23 (1H, dd, J = 7,3, 1,7 Hz), 6,59-6,62 (1H, m), 2,66 (1H, m), 2,40-2,61 (8H, m), 2,38 (2H, s), 1,22 (6H, d, J = 6,5 Hz).
Procedimiento C
Se disolvieron solvato de 2-(6-(1H-Indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol metanol (629,3 g, 1,33 mol) y ácido succínico (94,4 g, 0,80 mol) en DMSO (2,83 l) a 85 °C. La solución se filtró en otro recipiente, se lavó la línea con DMSO caliente (629 ml) y la solución se mantuvo a 85 °C. Se agregó agua (1,132 l) manteniendo la temperatura a 85 °C, luego los contenidos se enfriaron a 70 °C durante 1 h, y la cristalización se sembró con hemisuccinato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol (3,147 g) se suspendió en 25 % v/v de agua en DMSO (31,5 ml), seguido por un lavado de línea de 25 % p/p de agua en DMSO (31,5 ml). La suspensión se envejeció durante 120 mins luego se agregó agua (1,573 l) durante 2,5 h, los contenidos se agitaron durante 0,5 h, luego la cristalización se enfrió a 20 °C durante 3 h, luego se envejeció durante 17 h, se filtró, se lavó con 40 % v/v de agua en DMSO (2,52 l), agua (2,52 l), isopropanol (2 x 2,52 l) y se secó bajo vacío a 50 °C para dar el compuesto del título (501,4 g, 75 %).
1H RMN (700MHz, DMSO-d6) 8 ppm 13,43 (1H, br s), 11,36 (1H, s), 10,00 (br. s, 1H) 8,60 (1H, d, J = 0,9 Hz), 8,08 (1H, d, J = 1,3 Hz), 7,90-7,92 (1H, m), 7,47-7,50 (1H, m), 7,47 (1H, t, J = 2,8 Hz), 7,41 (1H, m), 7,24 (1H, t, J = 7,3 Hz), 7,23 (1H, dd, J = 7,3, 1,7 Hz), 6,59-6,62 (1H, m), 3,75 (2H, s), 2,66 (1H, m), 2,40-2,61 (8H, m), 2,38 (2H, s), 1,22 (6H, d, J = 6,5 Hz).
Ejemplo 3
Benzoato de 2-(6-(1H-Indol-4-¡l)-1H-¡ndazol-4-¡l)-5-((4-¡soprop¡lp¡perazin-1-¡l)met¡l)oxazol

Se disolvieron 2-(6-(1H-Indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol (28,55 g, 64,8 mmol) y ácido benzoico (8,326 g, 68,2 mmol) en DMSO (57 ml) y se calentaron a 40 °C antes de agregar isopropanol (57 ml). La cristalización se sembró con benzoato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol* (183,6 mg) y se agitó durante 20 mins, se agregó isopropanol (114 ml), se enfrió a 20 °C y se envejeció durante la noche, luego se filtró, se lavó con isopropanol (171 ml) y se secó bajo vacío a 40 °C para dar el compuesto del título (25,95 g, 71 %).
1H RMN (400MHz, DMSO-d6) 8 = 13,41 (br. s., 1H), 11,36 (br. s., 1H), 8,60 (d, J=0,7 Hz, 1H), 8,08 (d, J=1,0 Hz, 1H), 8,00 - 7,86 (m, 3H), 7,64 - 7,57 (m, 1H), 7,52 - 7,45 (m, 5H), 7,32 (s, 1H), 7,28 - 7,21 (m, 2H), 6,61 (br. s., 1H), 3,74 (s, 2H), 2,63 (sept, J=6,6 Hz, 1H) 2,55 - 2,41 (m, 8H), 0,95 (d, J=6,6 Hz, 6H).
Los datos de difracción de rayos X en polvo (XRPD) se adquirieron en un difractómetro de polvo PANalytical X'Pert Pro, modelo PW3040/60 utilizando un detector X'Celerator. Las condiciones de adquisición fueron: radiación: Cu Ka, tensión del generador: 40 kV, corriente del generador: 45 mA, ángulo de inicio: 2,0° 20, ángulo final: 40,0° 20, tamaño de etapa: 0,0167° 20, tiempo por etapa: 31,75 segundos. La muestra se preparó al montar unos pocos miligramos de muestra sobre una oblea de silicio (placa de fondo cero), dando como resultado una fina capa de polvo.
Los datos de XRPD para un polimorfo de benzoato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol se muestran en la Figura 1.
Los ángulos XRPD característicos y los espacios d para la forma de estado sólido se resumen en la Tabla 1. Las posiciones de los picos se midieron utilizando el software X'Pert Highscore.
Tabla 1

Específicamente, la semilla de benzoato de 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol se obtuvo al agregar acetonitrilo como disolvente a 2-(6-(1H-indol-4-il)-1H-indazol-4-il)-5-((4-isopropilpiperazin-1-il)metil)oxazol (19 mg) seguido por un equivalente de ácido benzoico en THF (3M).
Claims (11)
1. Un procedimiento de preparación de un compuesto de fórmula (IV) o una sal del mismo

en la que
R1 es

y
R2 es

o
R1 es

y
R2 es

procedimiento que comprende:
(a) hacer reaccionar un compuesto de fórmula (VII) o una sal del mismo

en la que R1a es

con un compuesto de fórmula (X) o una sal del mismo

en la que X es halógeno y P es un grupo de protección, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XI) o una sal del mismo

en la que R1a y P son como se definieron anteriormente,
(b) hacer reaccionar un compuesto de fórmula (XI) o una sal del mismo con un agente de borilación en presencia de un sistema de catalizador para dar un compuesto de fórmula (XII) o una sal del mismo

en la que R1a y P son como se definieron anteriormente,
(c) hacer reaccionar un compuesto de fórmula (XII) o una sal del mismo con un compuesto de fórmula (XIV) o una sal del mismo
R2-X1 (XIV)
en la que R2 es como se definió anteriormente y X1 es halógeno, en presencia de un catalizador de paladio para dar un compuesto de fórmula (XIII) o una sal del mismo

en la que R1a, R2 y P son como se definieron anteriormente, y
(d) hacer reaccionar un compuesto de fórmula (XIII) o una sal del mismo con un agente de reducción seguido por desprotección.
2. Un procedimiento de acuerdo con la reivindicación 1 en el que R1 es

y R2 es

3. Un procedimiento de acuerdo con la reivindicación 1 en el que X y X1 son cada uno independientemente cloro.
4. Un procedimiento de preparación de un compuesto de fórmula (XI) o una sal del mismo

en la que R1a es

y P es un grupo de protección,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (VII) o una sal del mismo

en la que R1a es como se definió anteriormente, con un compuesto de fórmula (X) o una sal del mismo

en la que X es halógeno y P es como se definió anteriormente, en presencia de un catalizador de paladio.
5. Un procedimiento de preparación de un compuesto de fórmula (XII) o una sal del mismo

en la que R1a es

y P es un grupo de protección,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XI) o una sal del mismo

en la que R1a y P son como se definieron anteriormente, con un agente de borilación en presencia de un sistema de catalizador.
6. Un procedimiento de preparación de un compuesto de fórmula (XIII) o una sal del mismo

en la que R1a es
y R2 es

o R1a es

y R2 es
y P es un grupo de protección,
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XII) o una sal del mismo

en la que R1a y P son como se definieron anteriormente, con un compuesto de fórmula (XIV) o una sal del mismo
R2-X1 (XIV)
en la que R2 es como se definió anteriormente y X1 es halógeno, en presencia de un catalizador de paladio.
7. Un procedimiento de preparación de un compuesto de fórmula (IV) y o una sal del mismo

en la que
R1 es

y
R2 es

o
R1 es

y
R2 es
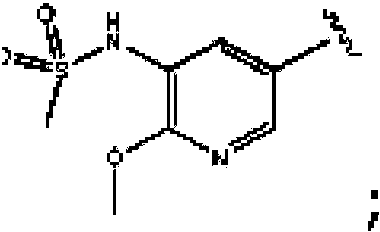
procedimiento que comprende hacer reaccionar un compuesto de fórmula (XIII) o una sal del mismo

en la que
R1a es

y R2 es

o
R1a es

y R2 es

y P es un grupo de protección, con un agente de reducción seguido por desprotección.
8. Un compuesto de fórmula (VII) o una sal del mismo

en la que R1a es

9. Un compuesto de fórmula (XI) o una sal del mismo

en la que R1a es

y P es tetrahidro-2H-piran-2-ilo.
10. Un compuesto de fórmula (XII) o una sal del mismo

en la que R1a es

y P es tetrahidro-2H-piran-2-ilo.
11. Un compuesto de fórmula (XIII) o una sal del mismo

en la que R1a es

R2 es

y P es tetrahidro-2H-piran-2-ilo.
Ċ
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1509492.3A GB201509492D0 (en) | 2015-06-02 | 2015-06-02 | Novel processes |
| PCT/EP2016/062250 WO2016193255A1 (en) | 2015-06-02 | 2016-05-31 | Processes to make indazole derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2760571T3 true ES2760571T3 (es) | 2020-05-14 |
Family
ID=53677608
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16726309T Active ES2760571T3 (es) | 2015-06-02 | 2016-05-31 | Procedimientos de fabricación de derivados de indazol |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US10266525B2 (es) |
| EP (1) | EP3303329B1 (es) |
| JP (1) | JP6389967B2 (es) |
| KR (1) | KR20180011115A (es) |
| CN (1) | CN107660204B (es) |
| AU (1) | AU2016273752B2 (es) |
| BR (1) | BR112017023486B1 (es) |
| CA (1) | CA2983966A1 (es) |
| CY (1) | CY1122343T1 (es) |
| DK (1) | DK3303329T3 (es) |
| ES (1) | ES2760571T3 (es) |
| GB (1) | GB201509492D0 (es) |
| HR (1) | HRP20192117T1 (es) |
| HU (1) | HUE047523T2 (es) |
| ME (1) | ME03653B (es) |
| PL (1) | PL3303329T3 (es) |
| PT (1) | PT3303329T (es) |
| RS (1) | RS59639B1 (es) |
| RU (1) | RU2017134467A (es) |
| SI (1) | SI3303329T1 (es) |
| WO (1) | WO2016193255A1 (es) |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8575162B2 (en) * | 2009-04-30 | 2013-11-05 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| WO2012032065A1 (en) * | 2010-09-08 | 2012-03-15 | Glaxo Group Limited | Indazole derivatives for use in the treatment of influenza virus infection |
| HUE026059T2 (en) | 2010-09-08 | 2016-05-30 | Glaxosmithkline Ip Dev Ltd | N- [5- [4- (5 - {[(2R, 6S) -2,6-dimethyl-4-morpholinyl] methyl} -1,3-oxazol-2-yl) -1H-indazol-6-yl ] Polymorphs and salts of 2 - (methyloxy) -3-pyridinyl] methanesulfonamide |
| GB201018124D0 (en) * | 2010-10-27 | 2010-12-08 | Glaxo Group Ltd | Polymorphs and salts |
| CA2925064A1 (en) | 2013-10-17 | 2015-04-23 | Glaxosmithkline Intellectual Property Development Limited | Pi3k inhibitor for treatment of respiratory disease |
-
2015
- 2015-06-02 GB GBGB1509492.3A patent/GB201509492D0/en not_active Ceased
-
2016
- 2016-05-31 DK DK16726309T patent/DK3303329T3/da active
- 2016-05-31 RS RS20191554A patent/RS59639B1/sr unknown
- 2016-05-31 US US15/578,258 patent/US10266525B2/en active Active
- 2016-05-31 ME MEP-2019-326A patent/ME03653B/me unknown
- 2016-05-31 PT PT167263094T patent/PT3303329T/pt unknown
- 2016-05-31 JP JP2017562313A patent/JP6389967B2/ja active Active
- 2016-05-31 EP EP16726309.4A patent/EP3303329B1/en active Active
- 2016-05-31 ES ES16726309T patent/ES2760571T3/es active Active
- 2016-05-31 AU AU2016273752A patent/AU2016273752B2/en not_active Ceased
- 2016-05-31 WO PCT/EP2016/062250 patent/WO2016193255A1/en active Application Filing
- 2016-05-31 PL PL16726309T patent/PL3303329T3/pl unknown
- 2016-05-31 RU RU2017134467A patent/RU2017134467A/ru not_active Application Discontinuation
- 2016-05-31 CA CA2983966A patent/CA2983966A1/en not_active Abandoned
- 2016-05-31 CN CN201680029002.0A patent/CN107660204B/zh active Active
- 2016-05-31 HU HUE16726309A patent/HUE047523T2/hu unknown
- 2016-05-31 BR BR112017023486-6A patent/BR112017023486B1/pt active IP Right Grant
- 2016-05-31 SI SI201630555T patent/SI3303329T1/sl unknown
- 2016-05-31 KR KR1020177033696A patent/KR20180011115A/ko unknown
-
2019
- 2019-11-26 HR HRP20192117TT patent/HRP20192117T1/hr unknown
- 2019-12-04 CY CY20191101273T patent/CY1122343T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| RU2017134467A (ru) | 2019-07-15 |
| JP2018518477A (ja) | 2018-07-12 |
| BR112017023486B1 (pt) | 2023-02-14 |
| US10266525B2 (en) | 2019-04-23 |
| CA2983966A1 (en) | 2016-12-08 |
| US20180155334A1 (en) | 2018-06-07 |
| EP3303329A1 (en) | 2018-04-11 |
| ME03653B (me) | 2020-07-20 |
| SI3303329T1 (sl) | 2020-02-28 |
| AU2016273752B2 (en) | 2018-07-05 |
| WO2016193255A1 (en) | 2016-12-08 |
| GB201509492D0 (en) | 2015-07-15 |
| HUE047523T2 (hu) | 2020-04-28 |
| JP6389967B2 (ja) | 2018-09-12 |
| CY1122343T1 (el) | 2021-01-27 |
| CN107660204B (zh) | 2021-04-13 |
| HRP20192117T1 (hr) | 2020-02-21 |
| BR112017023486A2 (pt) | 2018-07-24 |
| PL3303329T3 (pl) | 2020-03-31 |
| RS59639B1 (sr) | 2020-01-31 |
| KR20180011115A (ko) | 2018-01-31 |
| AU2016273752A1 (en) | 2017-10-19 |
| RU2017134467A3 (es) | 2019-10-11 |
| DK3303329T3 (da) | 2019-12-02 |
| EP3303329B1 (en) | 2019-09-18 |
| PT3303329T (pt) | 2019-12-16 |
| CN107660204A (zh) | 2018-02-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN104672225B (zh) | 二氢嘧啶衍生物的制备方法及其中间体 | |
| RU2728827C2 (ru) | Кристаллическая форма ингибитора тирозинкиназы брутона и способ её получения | |
| JP2022516469A (ja) | ユビキチン特異的プロテアーゼ1を阻害するための組成物 | |
| CN106046022A (zh) | 具有hiv整合酶抑制活性的化合物的制造方法 | |
| TR201807861T4 (tr) | Kanser tedavisinde kullanım için bileşikler yapmak için yeni işlem. | |
| BR112020026783A2 (pt) | Inibição da proteína de ligação creb (cbp) | |
| JP2015518011A (ja) | ピロロ[2,1−f][1,2,4]トリアジン誘導体およびその抗腫瘍用途 | |
| CN110225904B (zh) | 制备细胞毒性苯二氮䓬衍生物的方法 | |
| DK2991984T3 (en) | PALLADIUM-CATALYST COUPLING OF PYRAZOLAMIDES | |
| ES2760571T3 (es) | Procedimientos de fabricación de derivados de indazol | |
| JP2023502123A (ja) | S1p1モジュレーター化合物及び化合物を調製する方法 | |
| CN104804001B (zh) | 4‑取代吡咯并[2,3‑d]嘧啶化合物及其用途 | |
| US9969735B2 (en) | Process for making tricyclic lactam compounds | |
| CN112794851B (zh) | 3-(吡啶-3基)-7-氮杂吲哚衍生物PI3Kδ抑制剂及其制备方法与应用 | |
| CN117247374A (zh) | 一种bcl-xl抑制剂及其制药用途 | |
| CN115362150A (zh) | Enpp1抑制剂及其组合物和用途 | |
| CN115340502A (zh) | Bcl-xl抑制剂及其制备方法和用途 | |
| JP2014185091A (ja) | 3−アミン−4−ピペリドン誘導体の製造方法 | |
| NZ621837B2 (en) | Piperazine- substituted benzothiophene derivatives as antipsychotic agents |