ES2757560T3 - Composiciones terapéuticas para el tratamiento del virus de la inmunodeficiencia humana - Google Patents
Composiciones terapéuticas para el tratamiento del virus de la inmunodeficiencia humana Download PDFInfo
- Publication number
- ES2757560T3 ES2757560T3 ES16798063T ES16798063T ES2757560T3 ES 2757560 T3 ES2757560 T3 ES 2757560T3 ES 16798063 T ES16798063 T ES 16798063T ES 16798063 T ES16798063 T ES 16798063T ES 2757560 T3 ES2757560 T3 ES 2757560T3
- Authority
- ES
- Spain
- Prior art keywords
- tablet
- formula
- compound
- pharmaceutically acceptable
- layer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- SOLUWJRYJLAZCX-LYOVBCGYSA-N OC1=C(C(N2[C@@H](C3)O[C@H]4C[C@@H]2CC4)=O)N3C=C(C(NCc(c(F)cc(F)c2)c2F)=O)C1=O Chemical compound OC1=C(C(N2[C@@H](C3)O[C@H]4C[C@@H]2CC4)=O)N3C=C(C(NCc(c(F)cc(F)c2)c2F)=O)C1=O SOLUWJRYJLAZCX-LYOVBCGYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/537—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2095—Tabletting processes; Dosage units made by direct compression of powders or specially processed granules, by eliminating solvents, by melt-extrusion, by injection molding, by 3D printing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Oncology (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Biophysics (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una tableta que comprende 50 mg del compuesto de Fórmula I:**Fórmula** o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma.
Description
DESCRIPCIÓN
Composiciones terapéuticas para el tratamiento del virus de la inmunodeficiencia humana
CAMPO TÉCNICO
[0001] Se proporcionan formulaciones farmacéuticas adecuadas para el tratamiento de infecciones virales tales como VIH, en particular formas de dosificación oral sólidas que incluyen el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida.
ANTECEDENTES
[0002] El virus de la inmunodeficiencia humana tipo 1 (VIH-1), la infección es una enfermedad potencialmente mortal y seria de gran importancia pública para la salud, con aproximadamente 35 millones de personas infectadas en todo el mundo (Programa Conjunto de Naciones Unidas sobre VIH/SIDA (ONUSIDA). Global report: UNAIDS report on the global AIDS epidemic, 2013). Estándar de atención para el tratamiento de la infección por VIH-1 utiliza la terapia antirretroviral combinada (ART) para suprimir la replicación viral por debajo de los límites detectables, aumentar el recuento de células CD4 y detener la progresión de la enfermedad.
[0003] También existe la necesidad de medicamentos para atender a poblaciones con opciones de tratamiento limitadas (p. ej., niños, mujeres y ancianos). En ciertas situaciones, estas poblaciones pueden tener dificultades para mantener el tratamiento debido a la carga de la píldora (cantidad de píldoras que se deben tomar cada día, así como a diferentes combinaciones de píldoras) o al tamaño de las píldoras, una vez que se coformulan en una composición multidrogas. Por ejemplo, actualmente no hay una combinación de dosis fija registrada para una dosis diaria (es decir, QD) para niños muy pequeños (p. ej., menores de 12 años).
[0004] Un objetivo de la terapia antirretroviral es lograr la supresión viral en el paciente infectado por el VIH. Las pautas de tratamiento publicadas por el Departamento de Salud y Servicios Humanos de los Estados Unidos establecen que el logro de la supresión viral requiere el uso de terapias combinadas, es decir, varios medicamentos de al menos dos o más clases de medicamentos. Además, las decisiones sobre el tratamiento de pacientes infectados por el VIH son complicadas cuando el paciente requiere tratamiento para otras afecciones médicas (p. ej., metformina, rifampicina, antivirales contra el VHC, anticonceptivos hormonales, etc.). Debido a que el estándar de atención requiere el uso de múltiples medicamentos diferentes para suprimir el VIH, así como para tratar otras afecciones que el paciente puede estar experimentando, el potencial de interacción farmacológica es un criterio para la selección de un régimen farmacológico. Como tal, existe la necesidad de terapias antirretrovirales que tengan un potencial disminuido para las interacciones farmacológicas (p. ej., las que afectan a los transportadores (p. ej., OCT-2) o activan los receptores (p. ej., PXR).
[0005] El documento WO 2015/196116 se refiere a sodio (2R, 5S, 13aR)-7,9-dioxo-10-((2,4,6-trifluorobencilo) carbamoil)-2,3,4,5,7,9,13,13a-octahidro-2, 5-metanopirido[1 ’,2’ :4,5]pirazino[2,1-b][1,3]oxazepin-8-olato.
[0006] El documento WO 2015/196137 se refiere a formas cristalinas y cocristales de (2R, 5S, 13aR)-8-hidroxi-7,9-dioxo-N-(2,4,6-trifluorobencilo)-2,3,4,5,7,9,13,13a-octahidro-2,5-metanpirido[1 ’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida, y formulaciones farmacéuticas, y usos terapéuticos de las mismas. El documento citado también se refiere a formas cristalinas de sodio (2R, 5S, 13aR)-7,9-dioxo-10-((2,4,6-triiluorobencilo)carbamoílo)-2,3,4,5,7,9,13,13a-octahidro-2,5-metanopirido[1 ’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-8-olato Forma I.
[0007] WO 2014/100323 se refiere a compuestos para su uso en tratamiento de infección por el virus de inmunodeficiencia humana (VIH).
[0008] WO 2013/116720 se refiere al uso de la forma de hemifumarato de {9-[(R)-2-[[(S)-[[(S)-1-(isopropoxicarbonilo)etilo]amino]fenoxifosfinilo]metoxi]propilo]adenina}(hemifumarato de tenofovir alafenamida) en combinación con cobicistat.
[0009] WO 2015/022351 se refiere a una combinación farmacéutica que comprende (a) dolutegravir, (b) emtricitabina y (c) tenofovir o un profármaco de la misma, en donde los compuestos están presentes en una relación en peso específica y/o en cantidades específicas, para usar como medicamento, en el tratamiento de una infección/enfermedad viral como una infección por VIH.
[0010] Thomson Reuters Drug News (2011) "Gilead and Tibotec finalize agreement on single-tablet HIV regimen" informaron un acuerdo de licencia entre Gilead Sciences y Tibotec Pharmaceuticals para el desarrollo y comercialización de un régimen de una sola tableta que combina Prezista (darunavir) con Emtriva (emtricitabina), GS-7340 y cobicistat.
RESUMEN
[0011] La presente invención se define por las reivindicaciones adjuntas.
[0012] Una realización de la invención proporciona un comprimido que comprende 50 mg del compuesto de Fórmula I:

0 una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma.
[0013] En otra realización de la invención, se proporciona un comprimido de la invención para uso en el tratamiento terapéutico de una infección por VIH.
[0014] Todas las composiciones y formas de dosificación oral la presente invención incluyen un compuesto de Fórmula I, (2R, 5S, 13aR)-8-hidroxi- 7,9-dioxo-N-(2,4,6-trifluorobencilo)-2, 3,4,5,7,9,13,13a-octahidro-2,5-metanopirido[1’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida, que tiene la siguiente estructura: o una sal farmacéuticamente aceptable del mismo.
[0015] En ciertas realizaciones, la sal farmacéuticamente aceptable del compuesto de fórmula I es un compuesto de Fórmula II, sodio (2R, 5S, 13aR)-7,9-dioxo-10-((2,4,6-trifluorobencilo)carbamoílo)-2,3,4,5,7,9,13,13a-octahidro-2,5-metanopirido[1’,2’:4,5]pirazino[2,1-b] [1,3] oxazepina-8-olato, que tiene la siguiente estructura: [0016] los inventores han formulado con éxito una forma de dosificación oral que contiene el compuesto de Fórmula I, tenofovir alafenamida y emtricitabina. Esta forma de dosificación oral es adecuada para su uso en medicina, y en particular en el tratamiento de infecciones virales como el VIH.
[0017] En un aspecto, se proporciona una forma de dosificación oral sólida que comprende el compuesto de Fórmula 1 o una sal farmacéuticamente aceptable del mismo, tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y se proporciona emtricitabina o una sal farmacéuticamente aceptable del mismo. En ciertas realizaciones, la forma de dosificación comprende 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina. Por ejemplo, en ciertas realizaciones, la forma de dosificación comprende 50 mg del compuesto de Fórmula I como una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida como una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina. En ciertas realizaciones, la forma de dosificación comprende 52 mg del compuesto de Fórmula II, 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina.
[0018] En otro aspecto, una forma de dosificación oral sólida que comprende 75 mg del compuesto de Fórmula I como una farmacéuticamente sal aceptable del mismo, se proporciona 25 mg tenofovir alafenamida como una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina. En ciertas realizaciones, se proporciona una forma de dosificación oral sólida que comprende 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina. En ciertas realizaciones, se proporciona una forma de dosificación oral sólida que comprende 78 mg del compuesto de Fórmula II, 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina.
[0019] Los inventores han encontrado que es posible formular formas de dosificación orales sólidas que son farmacéuticamente aceptables (es decir, farmacológicamente eficaces y físicamente aceptables) mientras que la reducción del total cantidad de excipientes necesarios para lograr un perfil farmacocinético aceptable. Por consiguiente, en un aspecto se proporciona una forma de dosificación oral sólida, que comprende 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo., en donde la forma de dosificación tiene un peso total de menos de 850 mg (p. ej., menos de 800 mg o menos de 730 mg o menos de 700 mg).
[0020] En otro aspecto se proporciona una forma sólida de dosificación oral, que comprende 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal sal farmacéuticamente aceptable del mismo, en donde la forma de dosificación tiene un peso total de menos de 850 mg (p. ej., menos de 800 mg o menos de 700 mg).
[0021] En otro aspecto, un comprimido recubierto que comprende 50 mg del compuesto de Fórmula I o una sal farmacéuticamente sal aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable se proporciona de la misma.
[0022] En otro aspecto, un comprimido recubierto que comprende 75 mg del compuesto de Fórmula I o una sal farmacéuticamente sal aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable se proporciona de la misma.
[0023] En otro aspecto, un comprimido que comprende 52 mg del compuesto de Fórmula II, 28 mg tenofovir alafenamida se proporciona hemifumarato, y 200 mg de emtricitabina.
[0024] En otro aspecto, un comprimido que comprende 78 mg del compuesto de Fórmula II, 28 mg tenofovir alafenamida se proporciona hemifumarato, y 200 mg de emtricitabina.
[0025] En otro aspecto, que comprende una tableta (a) 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) 25 mg tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o se proporciona una sal farmacéuticamente aceptable de la misma, en la que (a) y (b) están segregados, y en el que la tableta tiene un peso total de menos de aproximadamente 1,5 g (p. ej., menos de aproximadamente 1 g). Típicamente, (a) y (b) están presentes dentro de capas separadas en una tableta multicapa.
[0026] En otro aspecto, que comprende una tableta (a) 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) 25 mg tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o se proporciona una sal farmacéuticamente aceptable de la misma, en la que (a) y (b) están segregados, y en el que la tableta tiene un peso total de menos de aproximadamente 1,5 g (p. ej., menos de aproximadamente 1 g). Típicamente, (a) y (b) están presentes dentro de capas separadas en una tableta multicapa.
[0027] En otro aspecto, un comprimido que comprende 6,5 a 11,0% p/p del compuesto de Fórmula I o una sal farmacéuticamente sal aceptable del mismo, 3,0-4,5% p/p tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 25- Se proporciona 30 % p/p de emtricitabina o una sal farmacéuticamente aceptable de la misma, donde los porcentajes en peso denotan una proporción de la tableta completa. En algunas realizaciones, (a) el compuesto de Fórmula I está presente a partir del compuesto de Fórmula II y/o (b) la tenofovir alafenamida está presente como hemifumarato de tenofovir alafenamida.
[0028] En otro aspecto, un comprimido que comprende 9,5 a 11,5% p/p del compuesto de Fórmula I o una sal farmacéuticamente sal aceptable del mismo, 2,5 a 4,5% p/p tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y se proporciona 26-33 % p/p de emtricitabina o una sal farmacéuticamente aceptable de la misma, donde los porcentajes en peso denotan una proporción de la tableta completa. En algunas realizaciones, (a) el compuesto de Fórmula I está presente a partir del compuesto de Fórmula II y/o (b) la tenofovir alafenamida está presente como hemifumarato de tenofovir alafenamida.
[0029] Los inventores han encontrado que el uso de una combinación de dosis fija puede ayudar en la consecución de los parámetros farmacocinéticos apropiados y/o la estabilidad de la tableta adecuada. Además, el uso de una tableta multicapa como un tipo particular de combinación de dosis fija también puede proporcionar beneficios farmacocinéticos y/o de estabilidad. Por consiguiente, en otro aspecto, una tableta de combinación de dosis fija que comprende (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) emtricitabina o una sal farmacéuticamente aceptable del mismo es previsto. Además, se proporciona un comprimido multicapa que comprende (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) emtricitabina o una sal farmacéuticamente aceptable del mismo.
[0030] En otro aspecto, se proporciona un kit que comprende (a) un comprimido que comprende el compuesto de Fórmula I o una sal farmacéuticamente sal aceptable del mismo, tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y emtricitabina o una sal farmacéuticamente aceptable del mismo, y (b) un desecante (p. ej., gel de sílice).
[0031] También se proporcionan métodos de producción de formas de dosificación orales sólidas tales como comprimidos, como se discute con más detalle a continuación.
[0032] Además, se proporcionan métodos para tratar a los pacientes, que también se discute en más detalle a continuación.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
[0033]
La Figura 1 muestra los resultados de los estudios realizados en las Formulaciones F1, F2 y F3 para evaluar la disolución de 78 mg de Compuesto de Fórmula II como agente único en comparación con una bicapa que usa fluido intestinal simulado en ayunas como medio de disolución.
La Figura 2 muestra los resultados de los estudios realizados en las Formulaciones F1 y F2 para evaluar la disolución de 78 mg del Compuesto de Fórmula II como agente único en comparación con una doble capa que usa fluido intestinal simulado alimentado como medio de disolución.
La Figura 3 muestra los resultados de los estudios realizados en las Formulaciones F1, F2, F4, F5 y F6 para evaluar la disolución de 78 mg del Compuesto de Fórmula II en tabletas que contienen varios excipientes.
La Figura 4 muestra los resultados de los estudios realizados en las Formulaciones F7 y F8 para evaluar la disolución de 52 mg del Compuesto de Fórmula II como agente único en comparación con una doble capa que usa fluido intestinal simulado en ayunas como medio de disolución.
La Figura 5 es un diagrama de flujo que ilustra la preparación de una formulación de tableta que contiene el compuesto de Fórmula II.
La Figura 6 es un diagrama de flujo que ilustra la preparación de una formulación de tableta que contiene el compuesto de Fórmula II, emtricitabina y hemifumarato de tenofovir alafenamida.
DESCRIPCIÓN DETALLADA
[0034] Típicamente, las formas de dosificación oral descritas aquí comprenden tres ingredientes farmacéuticos activos: el compuesto de fórmula I (o una sal farmacéuticamente aceptable del mismo), tenofovir alafenamida (o una sal farmacéuticamente aceptable del mismo), y emtricitabina (o una sal farmacéuticamente sal aceptable del mismo).
(2R,5S, 13aR)-8-Hidroxi-7,9-dioxo-N-(2,4,6-trifluorohencilo)-2,3,4,5,7,9,13,13a-octahidro-2,5-metanopirido[1’,2 ’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida
[0035] (2R, 5S, 13aR)-8-hidroxi-7,9-dioxo-N-(2,4,6-trifluorobencilo)-2,3,4,5,7,9,13,13a-octahidro-2,5-metanopirido[1’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida (Fórmula I), es un potente inhibidor de la integrasa del VIH con actividad in vitro contra el VIH-1 de tipo salvaje. Tiene la siguiente fórmula (ver WO2014/100323):

[0036] Su nombre IUPAC es (2R, 5S, 13aR)-8-hidroxi-7,9-dioxo-N-(2,4,6-trifluorobencilo)-2,3,4,5,7,9,13,13aoctahidro-2,5-metanopirido[1’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida. Su nombre CAS es 2,5-metanopirido[1 ’,2’:4,5]pirazino[2,1-b][1,3]oxazepina-10-carboxamida, 2,3,4,5,7,9,13,13a-octahidro-8-hidroxi-7,9-dioxo-N-[(2,4,6-trifluorofenilo)metilo]-, (2R, 5S, 13aR). El compuesto de Fórmula I es un ácido débil con pKa de 8,6. La solubilidad acuosa del compuesto del ácido libre de Fórmula I depende del pH, y la solubilidad aumenta con el aumento del pH, con un máximo a pH 10,5. La estabilidad química del compuesto de Fórmula I también depende del pH, con una estabilidad máxima a pH4El compuesto de Fórmula I se considera un compuesto BCS Clase 2, con baja solubilidad y alta permeabilidad.
[0037] Las formas de dosificación orales sólidas dan a conocer en el presente documento incluyen el compuesto de Fórmula I, por lo general en la forma de una sal farmacéuticamente aceptable. El compuesto de Fórmula I puede estar presente dentro de una forma de dosificación oral en forma solvatada o no solvatada, y las referencias a la "Fórmula I" incluyen ambas formas. Típicamente, el compuesto de Fórmula I es en la forma de compuesto de Fórmula II, que tiene la fórmula a continuación:
[0038] En ciertas realizaciones específicas, sólida oral se proporcionan formas de dosificación que contienen 50 mg del compuesto de Fórmula I, p. ej., como aproximadamente 52 mg del compuesto de Fórmula II.
[0039] En ciertas realizaciones específicas, las formas de dosificación oral sólidas que contienen 75 mg del compuesto de Fórmula I, p. ej., como de aproximadamente 78 mg del compuesto de Fórmula II, se proporcionan.
[0040] Como se usa en el presente documento, y en ausencia de una referencia específica a una sal aceptable particular, farmacéuticamente y/o solvato del compuesto de Fórmula I (p. ej., Fórmula II), cualquier dosis, ya sea expresado en p. ej., miligramos o como un % en peso, debe tomarse como referencia a la cantidad del compuesto de ácido libre de Fórmula I, es decir, la cantidad de:

[0041] Por ejemplo, por lo tanto, una referencia a "50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable y/o solvato del mismo” significa una cantidad del compuesto de Fórmula I o una sal y/o solvato farmacéuticamente aceptable del mismo que proporciona la misma cantidad del compuesto de Fórmula I que 50 mg del compuesto de ácido libre de Fórmula I.
[0042] Por ejemplo, por lo tanto, una referencia a "75 mg del compuesto de Fórmula I o una sal y/o solvato farmacéuticamente aceptable del mismo" significa una cantidad del compuesto de Fórmula I o una sal y/o solvato farmacéuticamente aceptable del mismo que proporciona el misma cantidad del compuesto d de Fórmula I como 75 mg del compuesto de ácido libre de Fórmula I.
Tenofovir alafenamida
[0043] Tenofovir alafenamida (TAF) es un inhibidor de la transcriptasa inversa de nucleótidos que tiene la siguiente fórmula (véase el documento WO02/08241 A2):

[0044] Su nombre IUPAC es (S)-¡soprop¡lo-2-(((SH(((fíJ-1-(6-am¡no-9H-pur¡na-9-ilo)propano-2-ilo)oxi)metilo)(fenoxi)fosforilo)amino)propanoato. También se conoce como {9-[(R)-2-[[(S)-[[(S)-1-(isopropoxicarbonilo)etilo]amino]fenoxifosfinilo]-metoxi]propilo]adenina}. Tenofovir alafenamida es una base débil, con un pKa de 3,9. Su solubilidad aumenta con la disminución del pH, con una solubilidad máxima de aproximadamente pH 3. Tenofovir alafenamida se considera un compuesto BCS Clase 3, con una solubilidad de alto equilibrio y una permeabilidad aparente más baja.
[0045] Las formas de dosificación orales sólidas aquí descritas incluyen tenofovir alafenamida, por lo general en forma de una sal farmacéuticamente sal aceptable. Tenofovir alafenamida puede estar presente dentro de una forma de dosificación oral en forma solvatada o no solvatada, y las referencias a "tenofovir alafenamida" incluyen ambas formas. En particular, el tenofovir alafenamida puede asociarse con el fumarato, como monofumarato o hemifumarato. Típicamente, tenofovir alafenamida es en forma de tenofovir hemifumarato alafenamida que tiene la fórmula a continuación (véase el documento WO 2013/025788 A1):

[0046] Como se usa en el presente documento, y en ausencia de una referencia específica a una sal particular farmacéuticamente aceptable y/o solvato de tenofovir alafenamida, cualquier dosificación, ya sea expresada en, p. ej., miligramos o como % en peso, debe tomarse como referencia a la cantidad de tenofovir alafenamida, es decir, la cantidad de:

[0047] Por ejemplo, una referencia a "25 mg tenofovir alafenamida o una sal farmacéuticamente aceptable y/o solvato del mismo' significa una cantidad de tenofovir alafenamida o una sal farmacéuticamente aceptable y/o solvato del mismo que proporciona la misma cantidad de tenofovir alafenamida como 25 mg de tenofovir alafenamida base libre.
[0048] La cantidad de tenofovir alafenamida en una forma de dosificación oral sólida proporcionada en este documento está generalmente entre 10 mg y 30 mg, por ejemplo dentro del intervalo de 20 mg a 30 mg, y más típicamente entre 24 mg y 2 8 mg. En ciertas realizaciones específicas, se proporcionan formas farmacéuticas orales sólidas que contienen 25 mg de tenofovir alafenamida, p. ej., como aproximadamente 28 mg de hemifumarato de tenofovir alafenamida.
Emtricitabina
[0049] La emtricitabina (FTC) es un inhibidor de la transcriptasa inversa de nucleósidos que tiene la fórmula siguiente:

[0050] Su nombre IUPAC es 4-amino-5-fluoro-1-[(2 R, 5 Sj-2-(hidroximetilo)-1,3-oxatiolano-5-ilo]-1,2-dihidropirimidina-2-ona. También se conoce como 5-fluoro-1-[(2R, 5S)-2-(hidroximetilo)-1,3-oxatiolano-5-ilo]citosina. Actualmente está autorizado como parte de EMTRIVA® (emtricitabina 200 mg), TRUVADA® (emtricitabina 200 mg, tenofovir disoproxilo fumarato 300 mg), ATRIPLA® (emtricitabina 200 mg, efavirenz 600 mg, tenofovir disoproxilo fumarato 300 mg) y STRIBILD® (emtricitabina 200 mg, cobicistat 150 mg, tenofovir disoproxilo fumarato 300 mg, elvitegravir 150 mg) y COMPLERA®/EVIPLERA® (rilpivirina 25 mg, emtricitabina 200 mg, tenofovir disoproxilo fumarato 300 mg). La emtricitabina es una base libre, que exhibe un pKa de 2,65. La solubilidad se mejora en condiciones ácidas. Se considera un compuesto BCS Clase 1, con alta solubilidad y alta permeabilidad.
[0052] Las formas de dosificación orales sólidas descritas en este documento incluyen emtricitabina, opcionalmente como una sal farmacéuticamente aceptable. La emtricitabina puede estar presente dentro de una forma de dosificación oral en forma solvatada o no solvatada, y las referencias a "emtricitabina" incluyen ambas formas. Típicamente, la emtricitabina está presente como una base libre.
[0053] Como se usa en el presente documento, y en ausencia de una referencia específica a una sal particular farmacéuticamente aceptable y/o solvato de emtricitabina, cualquier dosis, ya sea expresado en p. ej., miligramos o como un % en peso, se debe tomar como una referencia a la cantidad de emtricitabina, es decir, la cantidad de:

[0054] Por ejemplo, una referencia a "200 mg de emtricitabina o una sal y/o solvato farmacéuticamente aceptable de la misma" significa una cantidad de emtricitabina o una sal y/o solvato farmacéuticamente aceptable de la misma que proporciona la misma cantidad de emtricitabina como 200 mg de emtricitabina base libre.
[0055] La cantidad de emtricitabina en una forma de dosificación oral sólida proporcionada en este documento es generalmente entre 180 mg y 220 mg, por ejemplo entre 190 mg y 210 mg, y más típicamente entre 195 mg y 205 mg. En ciertas específicas formas de realización, formas de dosificación oral sólidas que contienen 200 mg de emtricitabina se proporcionan.
Formas sólidas orales de dosificación
[0056] Los inventores han formulado con éxito el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida en una sola forma de dosificación estable que es farmacológicamente eficaz y físicamente aceptable. Las formas de dosificación oral sólidas descritas en el presente documento están destinadas al uso farmacéutico en sujetos humanos. Por consiguiente, deben tener un tamaño y peso apropiados para la administración humana oral (p. ej., deben tener un peso total de menos de aproximadamente 1,5 g, p. ej., menos de aproximadamente 1,0 g), además de ser terapéuticamente eficaces.
[0057] En ciertas realizaciones, las formulaciones de los tres ingredientes activos en una forma de dosificación oral sólida que tiene un están dentro peso total de menos de aproximadamente 1,0 g, por ejemplo menos de aproximadamente 800 mg, o incluso menos de aproximadamente 750 mg, o incluso menos de 700 mg. Esto es ventajoso dado que TRIUMEQ® (sulfato de abacavir equivalente a 600 mg de abacavir, dolutegravir sódico equivalente a 50 mg de dolutegravir y 300 mg de lamivudina) tiene un peso total de más de aproximadamente 1000 mg, basado en el peso del activo ingredientes en cada tableta (debido a la cantidad de excipientes que se requieren para producir una tableta farmacéuticamente aceptable). La provisión de una forma de dosificación relativamente pequeña (en particular una tableta) representa una ventaja clínica porque puede esperarse que aumente la conveniencia del paciente y, por lo tanto, el cumplimiento en comparación con las formas de dosificación más grandes que son más gravosas para que los pacientes traguen. En realizaciones específicas, la forma de dosificación oral sólida descrita en el presente documento tiene un peso total de entre 700 y 750 mg. En ciertas realizaciones, la forma de dosificación oral sólida descrita en el presente documento tiene un peso total de entre 700 y 725 mg, o aproximadamente 700 mg. En realizaciones específicas, la forma de dosificación oral sólida descrita en el presente documento tiene un peso total de entre aproximadamente 50 y aproximadamente 750 mg, entre aproximadamente 100 y aproximadamente 750 mg, entre aproximadamente 200 y aproximadamente 750 mg, o entre aproximadamente 250 y aproximadamente 750 mg. Las formas de dosificación descritas actualmente pueden comprender menos de 600 mg de excipientes, tales como menos de 500 mg de excipientes o menos de 450 mg de excipientes. Por ejemplo, las formas de dosificación oral sólidas descritas en el presente documento pueden comprender entre 300 y 600 mg de excipientes, o entre 350 mg y 500 mg de excipientes, o entre 400 mg y 500 mg de excipientes. Más típicamente, las formas de dosificación oral sólida descritas en el presente documento comprenden entre 425 mg y 450 mg de excipientes. En tales realizaciones, las formas de dosificación comprenden como ingredientes activos (a) 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo. En ciertas realizaciones, las formas de dosificación comprenden como ingredientes activos (a) 52 mg del compuesto de Fórmula II, (b) 28 mg de hemifumarato de tenofovir alafenamida y (c) 200 mg de emtricitabina. En algunas realizaciones, las formas de dosificación comprenden como ingredientes activos (a) 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo. En ciertas realizaciones, las formas de dosificación comprenden como ingredientes activos (a) 78 mg del compuesto de Fórmula II, (b) 28 mg de hemifumarato de tenofovir alafenamida y (c) 200 mg de emtricitabina.
[0058] Las formas de dosificación oral sólidas descritas en el presente documento típicamente estarán en forma de una tableta de combinación de dosis fija. Esto se debe a que los inventores han descubierto que el uso de tabletas combinadas de dosis fija puede ayudar a optimizar las propiedades farmacocinéticas de los ingredientes activos, particularmente la exposición total del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, medido por el área bajo la curva (AUC) y Cmax. En realizaciones particulares, las formas de dosificación oral sólidas descritas en el presente documento están en forma de una tableta multicapa. En ciertas realizaciones, el uso de combinaciones de dosis fija, p. ej., tabletas multicapa, puede afectar el perfil de disolución de uno o más de los ingredientes activos dentro de la forma de dosificación y, por lo tanto, es probable que tenga un impacto en la farmacocinética in vivo de forma de dosificación. En particular, se ha observado que la disolución del compuesto de Fórmula I (p. ej., como Fórmula II) varía dependiendo de si la tableta está en una formulación de combinación de dosis
fija con tenofovir alafenamida y emtricitabina y/o si la tableta es una monocapa o tableta multicapa. También se ha observado que la presencia de ciertos excipientes en la formulación de tableta multicapa (o la ausencia de otros) afecta el perfil de disolución de uno o más de los ingredientes activos dentro de la forma de dosificación. La provisión de una tableta con parámetros farmacocinéticos particulares, p. ej., parámetros farmacocinéticos es una ventaja particular proporcionada por la presente descripción.
[0059] En una realización, que comprende un comprimido multicapa (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) emtricitabina o una sal farmacéuticamente aceptable del mismo se previsto. Típicamente, cada capa contiene al menos uno de (a), (b) y (c). Por ejemplo, en ciertas realizaciones, la tableta comprende una primera capa que comprende (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, y una segunda capa que comprende (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y además comprende (c) emtricitabina o una sal farmacéuticamente aceptable de la misma. En tales realizaciones, típicamente la primera capa está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y/o la segunda capa está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, la primera capa está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo), y la segunda capa está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo).
[0060] Una realización particular proporciona un comprimido, en el que la primera capa comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., Fórmula II) y está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo), y la segunda capa comprende tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable de la misma y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo). En una realización particular, se proporciona una tableta, en la que la primera capa comprende 52 mg del compuesto de Fórmula II y está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo), y la segunda capa comprende 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo), en donde la primera capa tiene un peso total de menos de aproximadamente 400 mg, tal como aproximadamente 325 mg, y la segunda capa tiene un peso total de menos de aproximadamente 450 mg, tal como aproximadamente 380 mg. En una realización, la capa que contiene tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo no contiene lactosa y/o almidón. En una realización, la capa que contiene el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo no contiene lactosa, crospovidona y/o estearilfumarato de sodio.
[0061] Una realización particular proporciona un comprimido, en el que la primera capa comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., Fórmula II) y está sustancialmente libre de emtricitabina o una sal farmacéuticamente sal aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de emtricitabina o una sal farmacéuticamente aceptable de la misma), y (b) la segunda capa comprende tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable de la misma y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo). En una realización particular, se proporciona una tableta, en la que (a) la primera capa comprende 52 mg del compuesto de Fórmula II y está sustancialmente libre de emtricitabina o una sal farmacéuticamente aceptable de la misma (p. ej., la primera capa contiene menos del 1% en peso de emtricitabina o una sal farmacéuticamente aceptable del mismo), y (b) la segunda capa comprende 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo), en el que la primera capa tiene un peso total de menos de aproximadamente 400 mg, tal como aproximadamente 325 mg, y la segunda capa tiene un peso total de menos de aproximadamente 450 mg, tal como aproximadamente 380 mg. En una realización, la capa que contiene emtricitabina o una sal farmacéuticamente aceptable de la misma no contiene lactosa y/o almidón. En una realización, la capa que contiene el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo no contiene lactosa, crospovidona y/o estearilfumarato de sodio.
[0062] Una realización particular proporciona un comprimido, en el que la primera capa comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., Fórmula II) y está sustancialmente libre de tenofovir alafenamida y emtricitabina o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de tenofovir alafenamida y emtricitabina o una sal farmacéuticamente aceptable de los mismos), y (b) la segunda capa comprende tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos y
emtricitabina o una sal farmacéuticamente aceptable de los mismos y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo). En una realización particular, se proporciona una tableta, en la que (a) la primera capa comprende 52 mg del compuesto de Fórmula II y está sustancialmente libre de tenofovir alafenamida y emtricitabina o una sal farmacéuticamente aceptable de la misma (p. ej., la primera capa contiene menos de 1% en peso de tenofovir alafenamida y emtricitabina o una sal farmacéuticamente aceptable de los mismos), y (b) la segunda capa comprende 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo), en donde la primera capa tiene un peso total de menos de aproximadamente 400 mg, tal como aproximadamente 325 mg, y la segunda capa tiene un peso total de menos de aproximadamente 450 mg, tal como aproximadamente 380 mg. En una realización, la capa que contiene tenofovir alafenamida y emtricitabina o una sal farmacéuticamente aceptable de la misma no contiene lactosa y/o almidón. En una realización, conteniendo la capa el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo no contiene lactosa, crospovidona y/o estearilfumarato de sodio.
[0063] En una realización particular, se proporciona un comprimido, en el que (a) la primera capa comprende 78 mg del compuesto de la Fórmula II y está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos de 1% en peso de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo), y (b) la segunda capa comprende 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej. la segunda capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo), en donde la primera capa tiene un peso total de menos de aproximadamente 400 mg, tal como aproximadamente 355 mg, y la segunda capa tiene un peso total de menos de aproximadamente 450 mg, tal como aproximadamente 380 mg. En una realización, la capa que contiene tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo no contiene lactosa y/o almidón.
[0064] A menos que se especifique lo contrario, los términos "primera capa", "segunda capa", "tercera capa" y así sucesivamente no especifican un orden particular u orientación de formulaciones de tableta multicapa descritas en el presente documento. Por el contrario, estos términos se utilizan para distinguir las secciones de la composición entre sí y para especificar las características o componentes de cada sección o compartimento. A modo de ejemplo, en una realización, se proporciona una tableta en la que una primera capa comprende 52 mg del compuesto de Fórmula II y está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo (p. ej., la primera capa contiene menos del 1% en peso de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo), y (b) la segunda capa comprende 28 mg de hemifumarato de tenofovir alafenamida y 200 mg de emtricitabina y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., la segunda capa la capa contiene menos del 1% en peso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo), en donde la primera capa tiene un peso total de menos de aproximadamente 400 mg, tal como aproximadamente 325 mg, y la segunda capa tiene un total peso de menos de aproximadamente 450 mg, tal como aproximadamente 380 mg. La primera capa se puede sintetizar primero o se puede sintetizar en segundo lugar. La primera capa puede estar en la parte inferior o en la parte superior o en un lado. El término "primera capa" no limita el orden y la orientación.
[0065] Los comprimidos descritos en este documento son típicamente comprimidos de liberación inmediata. En una realización, se proporciona una tableta que libera al menos el 50% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo en aproximadamente 20 minutos, medido usando el aparato USP II, en 333 ml de fluido intestinal simulado en ayunas, pH 6,5, a 37°C y velocidad de paleta de 100 rpm. En ciertas realizaciones, las tabletas descritas en este documento liberan al menos el 60% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo en 20 minutos, medido usando el aparato USP II, en 333 ml de fluido intestinal simulado en ayunas 50 mM, a 37°C y velocidad de paleta de 100 rpm. En algunas realizaciones, se proporciona una tableta que libera al menos el 70% del compuesto de Fórmula I en 60 minutos, medido usando el Aparato USP II, en 333 ml de fluido intestinal simulado en ayunas a 37°C y una velocidad de paleta de 100 rpm.
[0066] Las tabletas descritas en el presente documento generalmente tendrán una dureza dentro del intervalo de 14 20 kP y, en ciertas realizaciones específicas, tendrán una dureza de 17 kP. La dureza se puede evaluar convenientemente al conducir un yunque para comprimir una tableta a una velocidad de carga constante hasta que se fracture, operando de acuerdo con USP <1217> (utilizando, p. ej., un medidor de dureza TBH 220, ERWEKA GmbH, Heusenstamm Alemania).
[0067] Las tabletas descritas en el presente documento generalmente tendrán una friabilidad de <1% en peso. La friabilidad se puede evaluar de acuerdo con USP <1216>.
[0068] El núcleo de un comprimido proporcionado en el presente documento pueden tener una dureza de entre 14-20 kP y una friabilidad de <1% en peso.
[0069] Las tabletas típicamente incluirán uno o más excipientes. Los excipientes deben ser compatibles con los otros ingredientes de la formulación y fisiológicamente inocuos para el receptor de los mismos. Los expertos en la técnica de formulación de tabletas conocen bien ejemplos de excipientes adecuados y pueden encontrarse, p. ej., en Handbook of Pharmaceutical Excipients (eds. Rowe, Sheskey & Quinn), 6a edición 2009. Como se usa en el presente documento, el término "excipientes" pretende referirse, entre otros, a agentes basificantes, solubilizantes, deslizantes, rellenos, aglutinantes, lubricantes, diluyentes, conservantes, agentes tensioactivos y/o agentes dispersantes. El término también incluye agentes tales como agentes edulcorantes, agentes aromatizantes, agentes colorantes, agentes conservantes y agentes de recubrimiento. Dichos componentes generalmente estarán presentes mezclados dentro de la tableta.
[0070] Los ejemplos de solubilizantes incluyen, pero no se limitan a, tensioactivos iónicos (incluyendo tanto iónicos y tensioactivos no iónicos) tales como laurilsulfato de sodio, de cetiltrimetilamonio bromuro, polisorbatos (como el polisorbato 20 u 80), poloxámeros (como el poloxámero 188 o 207) y macrogols. En una realización particular, una tableta que comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, incluye un polisorbato, en particular el polisorbato 20. En ciertas realizaciones específicas, la cantidad de polisorbato 20 en una tableta descrita en el presente documento es inferior a aproximadamente 5 mg, tal como menos de aproximadamente 1 mg o aproximadamente 0,5 mg.
[0071] Los ejemplos de lubricantes, deslizantes y ayudas de flujo incluyen, pero no se limitan a, estearato de magnesio, estearato de calcio, ácido esteárico, aceite vegetal hidrogenado, palmitostearato de glicerilo, behenato de glicerilo, estearilo fumarato de sodio, dióxido de silicio coloidal y talco. La cantidad de lubricante en una tableta está generalmente entre aproximadamente 0,5-5% en peso. En ciertas realizaciones, la cantidad de lubricante en una tableta es aproximadamente 1,5% en peso. En ciertas realizaciones específicas, las tabletas descritas en el presente documento incluyen estearato de magnesio. En ciertas otras realizaciones, las tabletas descritas en el presente documento no incluyen fumarato de estearilo de sodio. En ciertas realizaciones, la tableta incluye menos de aproximadamente 10 mg de estearato de magnesio, o menos de aproximadamente 7,5 mg de estearato de magnesio. En ciertas realizaciones, la tableta incluye menos de aproximadamente 9 mg de estearato de magnesio, o menos de aproximadamente 8,75 mg de estearato de magnesio. En ciertas realizaciones, la tableta incluye aproximadamente 5 mg a aproximadamente 10 mg de estearato de magnesio, o aproximadamente 6 mg a aproximadamente 9 mg de estearato de magnesio, o aproximadamente 7 mg a aproximadamente 9 mg de estearato de magnesio, o aproximadamente 8 mg a aproximadamente 9 mg de estearato de magnesio, o aproximadamente 8,1 mg, aproximadamente 8,2 mg, aproximadamente 8,3 mg, aproximadamente 8,4 mg, aproximadamente 8,5 mg, aproximadamente 8,6 mg, aproximadamente 8,7 mg, aproximadamente 8,8 mg o aproximadamente 8,9 mg de estearato de magnesio.
[0072] Los ejemplos de desintegrantes incluyen, pero no se limitan a, almidones, celulosas, PVP reticulado (crospovidona), almidón glicolato de sodio, croscarmelosa de sodio, etc. En ciertas realizaciones, las tabletas descritas en el presente documento incluyen croscarmelosa de sodio. En ciertas otras realizaciones, las tabletas descritas en el presente documento no incluyen crospovidona. En ciertas realizaciones, la tableta incluye menos de aproximadamente 50 mg de croscarmelosa de sodio, o menos de aproximadamente 25 mg de croscarmelosa de sodio. En ciertas realizaciones, la tableta incluye aproximadamente 30 mg a aproximadamente 60 mg de croscarmelosa de sodio, o aproximadamente 40 mg a aproximadamente 60 mg de croscarmelosa de sodio, o aproximadamente 45 mg a aproximadamente 55 mg de croscarmelosa de sodio, o aproximadamente 45 mg, aproximadamente 46 mg, aproximadamente 47 mg, aproximadamente 48 mg, aproximadamente 49 mg, aproximadamente 50 mg, aproximadamente 51 mg, aproximadamente 52 mg o aproximadamente 53 mg, o aproximadamente 54 mg, o aproximadamente 55 mg de croscarmelosa sódica.
[0073] Los ejemplos de cargas (también conocidas como agentes de carga o diluyentes) incluyen, pero no se limitan a, almidones, maltodextrinas, polioles (como la lactosa) y celulosas. En ciertas realizaciones, las tabletas proporcionadas en el presente documento pueden ser microcristalina celulosa. En ciertas otras realizaciones, las tabletas proporcionadas en el presente documento no contienen lactosa. En ciertas realizaciones, las tabletas proporcionadas en el presente documento incluyen menos de aproximadamente 300 mg de microcristalina celulosa, en particular menos de aproximadamente 250 mg de microcristalina celulosa, y/o menos de aproximadamente 225 mg de microcristalina celulosa. En ciertas realizaciones, las tabletas proporcionadas en el presente documento incluyen menos de aproximadamente 500 mg de microcristalina celulosa, o menos de aproximadamente 450 mg de microcristalina celulosa, o menos de aproximadamente 400 mg de microcristalina celulosa, o menos de aproximadamente 375 mg de microcristalina celulosa. En ciertas realizaciones, las tabletas proporcionadas en el presente documento incluyen aproximadamente 250 mg a aproximadamente 500 mg de microcristalina celulosa, o aproximadamente 300 mg a aproximadamente 450 mg de microcristalina celulosa, o aproximadamente 300 mg a aproximadamente 400 mg de microcristalina celulosa, o aproximadamente 325 mg a aproximadamente 375 mg de microcristalina celulosa, o aproximadamente 350 mg a aproximadamente 370 mg de microcristalina celulosa. En ciertas realizaciones, las tabletas proporcionadas en el presente documento incluyen aproximadamente 300 mg, o aproximadamente 310 mg, o aproximadamente 320 mg, o aproximadamente 330 mg, o aproximadamente 340 mg, o aproximadamente 350 mg, o aproximadamente 360 mg, o aproximadamente 370 mg, o aproximadamente 380 mg, o aproximadamente 390 mg, o aproximadamente 400 mg de microcristalina celulosa.
[0074] Los ejemplos de aglutinantes incluyen, pero no se limitan a, PVP reticulado, HPMC, sacarosa, almidones, etc.
[0075] En ciertas realizaciones, los comprimidos proporcionados en este documento están no recubiertos. En ciertas otras realizaciones, las tabletas proporcionadas en el presente documento están recubiertas (en cuyo caso incluyen un recubrimiento). Aunque se pueden usar tabletas no recubiertas, es más habitual en el entorno clínico proporcionar una tableta recubierta, en cuyo caso se puede usar un recubrimiento no entérico convencional. Los recubrimientos de película son conocidos en la técnica y pueden estar compuestos de materiales poliméricos hidrófilos, pero no se limitan a materiales de polisacáridos, tales como hidroxipropilmetilcelulosa (HPMC), metilcelulosa, hidroxietilcelulosa (HEC), hidroxipropilcelulosa (HPC), alcohol poli(vinílico)-co-etilenglicol) y otros polímeros solubles en agua. Aunque en ciertas realizaciones el material soluble en agua incluido en el recubrimiento de película de las realizaciones descritas en el presente documento incluye un único material polimérico, en ciertas otras realizaciones se forma usando una mezcla de más de un polímero. En ciertas realizaciones, el revestimiento es amarillo o marrón. Los recubrimientos adecuados incluyen, pero no se limitan a, recubrimientos de película polimérica tales como los que comprenden alcohol polivinílico, p. ej., 'Opadry® II' (que incluye PVA parcialmente hidrolizado, dióxido de titanio, macrogol 3350 (PEG) y talco, con colorantes opcionales como hierro óxido (p. ej., óxido de hierro rojo u óxido de hierro negro) o carmín índigo u óxido de hierro amarillo o amarillo FD&C # 6). La cantidad de recubrimiento está generalmente entre aproximadamente el 2-4% del peso del núcleo, y en ciertas realizaciones específicas, aproximadamente el 3%. A menos que se indique específicamente lo contrario, cuando la forma de dosificación está recubierta, debe entenderse que una referencia al % en peso de la tableta significa la de la tableta total, es decir, que incluye el recubrimiento.
Farmacocinética
[0076] En ciertas realizaciones, las composiciones farmacéuticas descritas en el presente documento dan como resultado un aumento de la exposición sistémica (AUCinf, Cmax) para el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En realizaciones particulares, las formulaciones de tabletas multicapa descritas en el presente documento dan como resultado una exposición sistémica aumentada para el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo en comparación con una tableta de agente único formulación del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En ciertas realizaciones, la formulación de tableta multicapa da como resultado un aumento de al menos aproximadamente 20% en la exposición sistémica del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo en comparación con una formulación de tableta de un solo agente del compuesto de Fórmula I o una sustancia farmacéuticamente sal aceptable de los mismos. En algunas realizaciones, el aumento de la exposición sistémica es al menos aproximadamente el 25% o al menos aproximadamente el 30%. En algunas realizaciones, el aumento de la exposición sistémica es de aproximadamente el 30%.
Cmax
[0077] Cmax es la concentración plasmática/sérica máxima observada del fármaco.
[0078] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionar una Cmax en plasma del compuesto de Fórmula I en pacientes en ayunas de aproximadamente 5300 a aproximadamente 8900 ng/ml, p. ej., aproximadamente 7100 ng/ml.
[0079] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente sal aceptable los mismos proporcionan una Cmax en plasma de emtricitabina en pacientes en ayunas de aproximadamente 1700 a aproximadamente 2800 ng/ml, p. ej., aproximadamente 2300 ng/ml.
[0080] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan una Cmax en plasma de tenofovir alafenamida en pacientes en ayunas de aproximadamente 190 a aproximadamente 320 ng/ml, p. ej., aproximadamente 250 ng/ml.
[0081] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan una Cmax en plasma del Compuesto de Fórmula I de aproximadamente 4200 ng/ml a aproximadamente 8000 ng/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0082] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal
farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionar una Cmax en plasma del compuesto de Fórmula I en pacientes en ayunas de aproximadamente 4200 ng/ml a aproximadamente 6500 ng/ml, o de aproximadamente 4700 ng/ml a aproximadamente 5300 ng/ml, o de aproximadamente 4700 ng/ml a aproximadamente 5800 ng/ml, o desde aproximadamente 5000 ng/ml hasta aproximadamente 5500 ng/ml.
[0083] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionar una Cmax en plasma del compuesto de Fórmula I en pacientes alimentados de aproximadamente 4500 ng/ml a aproximadamente 8000 ng/ml, o de aproximadamente 4800 ng/ml a aproximadamente 7900 ng/ml, o de aproximadamente 5300 ng/ml a aproximadamente 6900 ng/ml, o desde aproximadamente 5600 ng/ml hasta aproximadamente 6600 ng/ml.
[0084] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan una Cmax en plasma de emtricitabina de aproximadamente 1770 ng/ml a aproximadamente 2800 ng/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0085] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan un plasma Cmax de emtricitabina en pacientes en ayunas de aproximadamente 1770 ng/ml a aproximadamente 2800 ng/ml, o de aproximadamente 2000 ng/ml a aproximadamente 2600 ng/ml, o de aproximadamente 2000 ng/ml a aproximadamente 2500 ng/ml, o de aproximadamente 2100 ng/ml a aproximadamente 2400 ng/ml.
[0086] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionar una Cmax en plasma máxima de emtricitabina en pacientes alimentados de aproximadamente 1000 ng/ml a aproximadamente 3000 ng/ml, o de aproximadamente 1500 ng/ml a aproximadamente 2000 ng/m de aproximadamente 1700 ng/ml a aproximadamente 2200 ng/ml L, o de aproximadamente 1800 ng/ml a aproximadamente 2100 ng/ml.
[0087] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionan una Cmax en plasma de tenofovir alafenamida de aproximadamente 185 ng/ml a aproximadamente 315 ng/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0088] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un plasma Cmax de tenofovir alafenamida en pacientes en ayunas de aproximadamente 185 ng/ml a aproximadamente 315 ng/ml, o de aproximadamente 200 ng/m, hasta aproximadamente 300 ng/ml, o desde aproximadamente 210 ng/ml hasta aproximadamente 290 ng/ml, o desde aproximadamente 220 ng/ml hasta aproximadamente 275 ng/ml, o desde aproximadamente 230 ng/ml hasta aproximadamente 265 ng/ml, o de aproximadamente 240 ng/ml a aproximadamente 260 ng/ml.
[0089] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan una Cmax en plasma de tenofovir alafenamida en pacientes alimentados de aproximadamente 150 ng/ml a aproximadamente 350 ng/ml, o de aproximadamente 185 ng/ml a aproximadamente 300 ng/m de aproximadamente 210 ng/ml a aproximadamente 280 ng/ml L, o de aproximadamente 250 ng/ml a aproximadamente 265 ng/ml.
AUCinf
[0090] AUCinf es el área bajo la concentración de plasma/suero versus curva de tiempo extrapolada a tiempo infinito, calculada como AUC0-último (Cúltimo/Az).
[0091] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que
contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionan un AUCinf de plasma del compuesto de Fórmula I en pacientes alimentados de aproximadamente 117000 a aproximadamente 196000 h • ng/ml, p. ej., aproximadamente 157000 hmg/ml.
[0092] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan un plasma AUCinf de emtricitabina en pacientes alimentados de aproximadamente 8700 a aproximadamente 14500hmg/ml, p. ej., aproximadamente 2300hmg/mL.
[0093] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionan un AUCinf en plasma de tenofovir alafenamida en pacientes alimentados de aproximadamente 150 y 260 hmg/ml, por ejemplo, aproximadamente 210 hmg/ml.
[0094] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCinf de plasma del Compuesto de Fórmula I de aproximadamente 84450 hmg/ml a aproximadamente 141000 hmg/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0095] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUC en plasma inf del compuesto de Fórmula I en pacientes en ayunas de aproximadamente 84450 hmg/ml a aproximadamente 141000 hmg/ml, o de aproximadamente 90000 hmg/ml a aproximadamente 135000 hmg/ml, o de aproximadamente 95000 hmg/ml a aproximadamente 130000 hmg/ml, o de aproximadamente 100000 hmg/ml a aproximadamente 125000 hmg/ml, o de aproximadamente 110000 hmg/ml a aproximadamente 120000 hmg/ml.
[0096] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionar un AUC inf de plasma del compuesto de Fórmula I en pacientes alimentados de aproximadamente 100000 hmg/ml a aproximadamente 200000 ng/ml, o de aproximadamente 112000 hmg/ml a aproximadamente 175000 ng/ml, o de aproximadamente 126000 hmg/ml a aproximadamente 155000 ng/ml, o de aproximadamente 133000 hmg/ml a aproximadamente 147000 ng/ml.
[0097] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUC inf de plasma de emtricitabina de aproximadamente 8100 hmg/ml a aproximadamente 13600 hmg/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0098] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente sal aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan un AUC inf de plasma de emtricitabina en pacientes en ayunas de aproximadamente 8100 hmg/ml a aproximadamente 13600 hmg/ml, o de aproximadamente 8700 hmg/ml a aproximadamente 13000 hmg/ml, o de aproximadamente 92000 hmg/ml a aproximadamente 12500 hmg/ml, o de aproximadamente 9700 hmg/ml a aproximadamente 12000 hmg/ml, o de aproximadamente 10000 hmg/ml a aproximadamente 11400 hmg/ml.
[0099] En realizaciones particulares, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un AUCinf en plasma de emtricitabina en pacientes alimentados de aproximadamente 7500 hmg/ml a aproximadamente 15000 ng/ml, o de aproximadamente 8300 hmg/ml hasta aproximadamente 14000 ng/ml, o desde aproximadamente 9500 hmg/ml hasta aproximadamente 12000 ng/ml, o desde aproximadamente 9900 hmg/ml hasta aproximadamente 11600 ng/ml.
[0100] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene
50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUC inf en plasma de tenofovir alafenamida de aproximadamente 200 hmg/ml a aproximadamente 500 hmg/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0101] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable de los mismos proporcionan un AUC inf en plasma de tenofovir alafenamida en pacientes en ayunas de aproximadamente 200 hmg/ml a aproximadamente 265 hmg/ml, o de aproximadamente 200 hmg/ml a aproximadamente 300 hmg/ml, o de aproximadamente 210 hmg/ml a aproximadamente 290 hmg/ml, o de aproximadamente 220 hmg/ml a aproximadamente 270 hmg/ml, o de aproximadamente 230 hmg/ml a aproximadamente 265 hmg/mL.
[0102] En realizaciones particulares, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionar un AUCinf de plasma de tenofovir alafenamida en pacientes alimentados de aproximadamente 200 hmg/ml a aproximadamente 500 ng/ml, o de aproximadamente 230 hmg/ml a aproximadamente 400 ng/ml, o de aproximadamente 260 hmg/ml a aproximadamente 350 ng/ml, o de aproximadamente 275 hmg/ml a aproximadamente 370 ng/ml.
AUCúltimo
[0103] AUCúltimo es el área bajo la concentración de plasma/suero versus curva de tiempo desde el tiempo cero hasta la última concentración cuantificable.
[0104] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCúltimo en plasma del compuesto de Fórmula I en pacientes alimentados de aproximadamente 114000 a aproximadamente 190000 hmg/ml, p. ej., aproximadamente 152000 hmg/ml.
[0105] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCúltimo en plasma de emtricitabina en pacientes alimentados de aproximadamente 8600 a aproximadamente 14000 hmg/ml, p. ej., aproximadamente 11000 hmg/ml.
[0106] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 75 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCúltimo en plasma de tenofovir alafenamida en pacientes alimentados de aproximadamente 150 y 260 hmg/ml, p. ej., aproximadamente 210 hmg/ml.
[0107] En ciertas realizaciones específicas, una composición farmacéutica que comprende un comprimido que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCúltimo en plasma del compuesto de Fórmula I de aproximadamente 81700 hmg/ml a aproximadamente 140000 hmg/ml, independientemente de si el sujeto fue alimentado o en ayunas.
[0108] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un AUCúltimo en plasma del compuesto de Fórmula I en pacientes en ayunas de aproximadamente 81700 hmg/ml a aproximadamente 140000 hmg/ml, de aproximadamente 87000 hmg/ml a aproximadamente 131000 hmg/ml, de aproximadamente 92000 hmg/ml a aproximadamente 130000 hmg/ml, de aproximadamente 98100 hmg/ml a aproximadamente 120000 hmg/ml, de aproximadamente 104000 hmg/ml a aproximadamente 115000 hmg/ml.
[0109] En realizaciones particulares, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionar un AUCúltimo en plasma del compuesto de Fórmula I en pacientes alimentados de aproximadamente 100000 hmg/ml a aproximadamente 200000 ng/ml, o de aproximadamente 108000 hmg/ml a
aproximadamente 170000 ng/ml, o de aproximadamente 122000 hmg/ml a aproximadamente 150000 ng/ml, o de aproximadamente 128000 hmg/ml a aproximadamente 142000 ng/ml.
[0110] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un AUCúltimo de plasma de emtricitabina de aproximadamente 7500 hmg/ml a aproximadamente 15000 hmg/ml, independientemente de si el sujeto fuera alimentado o en ayunas.
[0111] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable los mismos proporcionan un AUCúltimo en plasma de la emtricitabina en pacientes en ayunas de aproximadamente 8000 hmg/ml a aproximadamente 13400 hmg/ml, de aproximadamente 8500 hmg/ml a aproximadamente 12800 hmg/ml, de aproximadamente 9000 hmg/ml a aproximadamente 12300 hmg/ml, de aproximadamente 9500 hmg/ml a aproximadamente 11000 hmg/ml, de aproximadamente 10000 hmg/ml a aproximadamente 11200 hmg/ml.
[0112] En realizaciones particulares, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan un AUCúltimo en plasma de emtricitabina en pacientes alimentados de aproximadamente 7500 hmg/ml a aproximadamente 15000 ng/ml, o de aproximadamente 8000 hmg/ml a aproximadamente 14000 ng/ml, o de aproximadamente 9000 hmg/ml a aproximadamente 12000 ng/ml, o de aproximadamente 9700 hmg/ml a aproximadamente 11300 ng/ml.
[0113] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, proporcionan un AUCúltimo de plasma de tenofovir alafenamida de aproximadamente 165 hmg/ml a aproximadamente 400 hmg/ml, independientemente de si el sujeto se alimentó o ayunó.
[0114] En ciertas realizaciones específicas, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, 200 mg de emtricitabina o una sal farmacéuticamente aceptable del mismo, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un AUCúltimo en plasma del tenofovir alafenamida en pacientes en ayunas de aproximadamente 165 hmg/ml a aproximadamente 390 hmg/ml, de aproximadamente 186 hmg/ml a aproximadamente 227 hmg/ml, de aproximadamente 196 hmg/ml a aproximadamente 217 hmg/ml.
[0115] En realizaciones particulares, una composición farmacéutica que comprende una tableta que contiene 50 mg del compuesto de Fórmula I o una sal farmacéuticamente aceptable de la misma, 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma, y 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo proporcionan un AUCúltimo en plasma de tenofovir alafenamida en pacientes alimentados de aproximadamente 200 hmg/ml a aproximadamente 400 ng/ml, o de aproximadamente 230 hmg/ml a aproximadamente 390 ng/ml, o de aproximadamente 260 hmg/ml a aproximadamente 345 ng/ml, o de aproximadamente 275 hmg/ml a aproximadamente 330 ng/ml.
Cúltimo
[0116] Cúltimo es la última concentración de plasma/suero cuantificable observada del fármaco.
[0117] Cmax, Cúltimo, AUCinf y AUCúltimo son parámetros farmacocinéticos estándar que se pueden estimar manualmente o usando un software de modelado bien conocido en la técnica, como el paquete Pharsight WinNonlin que usa un modelo no compartimental. La base general para el cálculo de estas cantidades es bien conocida (p. ej., ver Rowland y Tozer (2010) Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications ISBN 978-0781750097, o Jambhekar & Breen (2012) Basic Pharmacokinetics ISBN 978-0853699804). Típicamente, los parámetros se evaluarán como el promedio (p. ej., media geométrica o aritmética) dentro de un grupo de al menos 12 (y normalmente entre 24 y 36) adultos humanos sanos. Los parámetros deben medirse de acuerdo con las normas y prácticas que serían aceptables para una agencia reguladora farmacéutica como la FDA, EMA, MHLW o WHO. Los valores pueden estar basados en mediciones tomadas en intervalos apropiados siguientes el momento de la ingestión de la tableta, por ejemplo cada hora, o en intervalos de muestreo cada vez más escaso, tal como 1,3, 5, 7, 9, 11, 13, 15, 20, y 24 horas después de la ingestión. Se pueden evaluar después de una dosis única de medicamento o en estado estacionario, pero generalmente se evaluarán después de una dosis única.
[0118] Es bien conocido en las técnicas de biodisponibilidad y bioequivalencia cómo determinar si una tableta particular cumple con los requisitos reglamentarios para una biodisponibilidad equivalente y bioequivalencia farmacocinética, p. ej., ver: Niazi (2014) Handbook of Bioequivalence Testing, 2a Edición, ISBN 978-1482226379;
Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations FDA March 2003; and Guideline On The Investigation Of Bioequivalence, EMEA 2010 CPMP/EWP/QWP/1401/98 Rev. 1/Corr **. Para garantizar el poder estadístico, se realizará un estudio para medir los valores de Cmax, AUCúltimo y AUCinf en múltiples sujetos, p. ej., en un grupo de al menos 12 (y normalmente entre 24 y 36) adultos humanos sanos.
[0119] Debido a que determinar los valores de Cmax, AUCúltimo y AUCinf es necesariamente destructivo, estos parámetros no se determinarán directamente para la forma de dosificación (en particular la tableta) en cuestión, sino para una forma de dosificación realizada por el mismo proceso de fabricación con los mismos componentes. Por lo tanto, se puede preparar un lote de una forma de dosificación (p. ej., tabletas) mediante un proceso particular, y el intervalo de confianza del 90% de Cmax, AUCúltimo y AUCinf se evaluará en una muestra de esas tabletas. Si estos valores cumplen el requisito del 80-125% mencionado anteriormente, las tabletas hechas por el proceso de fabricación en cuestión son tabletas de la presente invención.
[0120] Un comprimido de combinación de dosis fija está provisto que comprende (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej. Fórmula II), (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) emtricitabina o una sal farmacéuticamente aceptable del mismo.
[0121] En una realización, se proporciona una tableta multicapa, que comprende (a) el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo (p. ej., Fórmula II), (b) tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) emtricitabina o una sal farmacéuticamente aceptable de la misma.
[0122] En una realización, la tableta multicapa descrita en el presente documento comprende (a) una primera capa que comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable de la misma, (b) una segunda capa que contiene tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) comprende además emtricitabina o una sal farmacéuticamente aceptable de la misma.
[0123] En una realización de la tableta multicapa descrita en el presente documento, (a) la primera capa está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y /o (b) la segunda capa está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0124] En una realización de la tableta multicapa descrita en el presente documento, (a) la primera capa está sustancialmente libre de emtricitabina o una sal farmacéuticamente aceptable de la misma, y/o (b) la segunda capa está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0125] En una realización de la tableta multicapa descrita en el presente documento, (a) la primera capa está sustancialmente libre de emtricitabina o una sal farmacéuticamente aceptable de la misma y tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y/o (b) la segunda capa es sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0126] En una realización, la tableta multicapa descrita en el presente documento comprende (a) una primera capa que comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, (b) una segunda capa que contiene emtricitabina o una sal farmacéuticamente aceptable del mismo, y (c) comprende además tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo.
[0127] En una realización de la tableta multicapa descrita en este documento, (a) la primera capa está sustancialmente libre de emtricitabina o una sal farmacéuticamente aceptable de la misma, y /o (b) la segunda capa está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0128] En una realización de la tableta multicapa descrita en el presente documento, (a) la primera capa comprende el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo y está sustancialmente libre de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (b) el la segunda capa comprende tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable de la misma y está sustancialmente libre del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0129] En una realización de la tableta multicapa descrita en el presente documento, la primera capa está sustancialmente libre de emtricitabina.
[0130] En una realización, la tableta multicapa descrita en el presente documento comprende 50 6 mg del compuesto de Fórmula I. En una realización, la tableta multicapa descrita en el presente documento comprende 200 20 mg de emtricitabina. En una realización, la tableta multicapa descrita en el presente documento comprende 25 3 mg de tenofovir alafenamida.
[0131] En una realización, el comprimido multicapa descrito en este documento comprende 75 6 mg del compuesto de Fórmula I. En una realización, el comprimido de múltiples capas descritas en este documento comprende 200 20
mg de emtricitabina. En una realización, la tableta multicapa descrita en el presente documento comprende 25 3 mg de tenofovir alafenamida.
[0132] En una realización, la tableta multicapa descrita en el presente documento comprende 52 6 mg del compuesto de Fórmula II. En una realización, la tableta multicapa descrita en el presente documento comprende 200 20 mg de emtricitabina. En una realización, la tableta multicapa descrita en el presente documento comprende 28 3 mg de hemifumarato de tenofovir alafenamida.
[0133] En una realización, la tableta multicapa descrita en el presente documento comprende 78 6 mg del compuesto de Fórmula II. En una realización, la tableta multicapa descrita en el presente documento comprende 200 20 mg de emtricitabina. En una realización, la tableta multicapa descrita en el presente documento comprende 28 3 mg de hemifumarato de tenofovir alafenamida.
[0134] En una realización, una primera capa de la tableta multicapa descrita en el presente documento comprende uno o más excipientes, por ejemplo uno o más diluyentes, desintegrantes, aglutinantes o lubricantes.
[0135] En una realización, una primera capa de la tableta multicapa comprende croscarmelosa de sodio. En una realización, una primera capa de la tableta multicapa comprende croscarmelosa de sodio, microcristalina celulosa y estearato de magnesio.
[0136] En una realización se proporciona un comprimido en donde menos de aproximadamente 25 por ciento en peso de una primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que menos del 20 por ciento en peso de una primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que menos del 16 por ciento en peso de una primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que aproximadamente 5 a aproximadamente 20 por ciento en peso, o aproximadamente 10 a aproximadamente 18 por ciento en peso, o aproximadamente 14 a aproximadamente 18 por ciento en peso de una primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que aproximadamente el 16 por ciento en peso de una primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0137] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en el que el peso total de la primera capa es al menos aproximadamente 290 mg.
[0138] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 300 mg.
[0139] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 310 mg.
[0140] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 320 mg.
[0141] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 330 mg.
[0142] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 340 mg.
[0143] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 350 mg.
[0144] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 360 mg.
[0145] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en la que el peso total de la primera capa es al menos aproximadamente 290 mg y es inferior a aproximadamente 360 mg.
[0146] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto deFórmula II y en donde el peso total de la primera capa es al menos aproximadamente 300 mg y es inferior a aproximadamente 350 mg.
[0147] En una realización, se proporciona una tableta en la que la primera capa comprende 52 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 310 mg y es inferior a aproximadamente 330 mg.
[0148] En una realización, la primera capa de la tableta multicapa tiene un peso total de 323 75 mg, o 323 25 mg, o 323 10 mg, o 323 mg.
[0149] En una realización, la primera capa de la tableta multicapa comprende

[0150] En una realización, la primera capa de la tableta multicapa comprende:

[0151] En una realización, la primera capa de la tableta multicapa comprende:

[0152] En una realización, la primera capa de la tableta multicapa consiste en:

[0153] En una realización, la primera capa de la tableta multicapa consiste en:

[0154] En una realización, la primera capa de la tableta multicapa consiste en:

[0155] En una realización, la primera capa de la tableta multicapa incluye:

[0156] En una realización, la primera capa de la tableta multicapa incluye:

[0157] En una realización, la primera capa de la tableta multicapa consiste en:

[0158] En una realización, la primera capa de la tableta multicapa incluye:

[0159] En una realización, la primera capa de la tableta multicapa incluye:

[0160] En una realización se proporciona un comprimido que menos de aproximadamente 30 por ciento en peso de la primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable de los mismos. En una realización, se proporciona una tableta en la que menos del 25 por ciento en peso de la primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que menos del 22 por ciento en peso de la primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que aproximadamente del 15 al 27 por ciento en peso, o del 17 al 25 por ciento en peso, o del 19 al 23 por ciento en peso de la primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona una tableta en la que aproximadamente el 21 por ciento en peso de la primera capa es el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.
[0161] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 320 mg.
[0162] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 330 mg.
[0163] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 340 mg.
[0164] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 350 mg.
[0165] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 360 mg.
[0166] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 370 mg.
[0167] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 380 mg.
[0168] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 390 mg.
[0169] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en donde el peso total de la primera capa es al menos aproximadamente 320 mg y es inferior a aproximadamente 390 mg.
[0170] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en la que el peso total de la primera capa es al menos aproximadamente 330 mg y es inferior a aproximadamente 380 mg.
[0171] En una realización, se proporciona una tableta en la que la primera capa comprende 78 2,8 mg del compuesto de Fórmula II y en la que el peso total de la primera capa es al menos aproximadamente 350 mg y es inferior a aproximadamente 360 mg.
[0172] En una realización, la primera capa de la tableta multicapa tiene un peso total de 353 75 mg, o 353 25 mg, o 353 10 mg, o 353 mg.

[0173] En una realización, la primera capa de la tableta multicapa comprende:
[0174] En una realización, la primera capa de la tableta multicapa comprende:

[0175] En una realización, la primera capa de la tableta multicapa comprende:

[0176] En una realización, la primera capa de la tableta multicapa consiste en:

[0177] En una realización, la primera capa de la tableta multicapa consiste en:

[0178] En una realización, la primera capa de la tableta multicapa consiste en:

[0179] En una realización, la primera capa de la tableta multicapa consiste en:

[0180] En una realización, la segunda capa de la tableta multicapa no comprende lactosa. En una realización, la segunda capa de la tableta multicapa no comprende almidón. En una realización, la segunda capa de la tableta multicapa no comprende lactosa ni almidón.
[0181] En una realización, la segunda capa de la tableta multicapa comprende uno o más excipientes, p. ej., uno o más diluyentes, desintegrantes, aglutinantes o lubricantes.
[0182] En una realización, la segunda capa de la tableta multicapa comprende microcristalina celulosa y croscarmelosa sódica.
[0183] En una realización, la segunda capa de la tableta multicapa comprende microcristalina celulosa, croscarmelosa estearato de sodio y magnesio.
[0184] En una realización, la segunda capa de la tableta multicapa comprende 20-40 mg de croscarmelosa de sodio. En una realización, la segunda capa de la tableta multicapa comprende 105-125 mg de microcristalina celulosa. En una realización, la segunda capa de la tableta multicapa comprende 1-8 mg de estearato de magnesio.
[0185] En una realización, la segunda capa de la tableta multicapa no comprende lactosa. En una realización, la segunda capa de la tableta multicapa no comprende almidón. En una realización, la segunda capa de la tableta multicapa no comprende lactosa ni almidón.
[0186] En una realización, la segunda capa de la tableta multicapa tiene un peso total de menos de 600 mg, o menos de 500 mg, o menos de 400 mg, o menos de 380 mg. En una realización, la segunda capa de la tableta multicapa tiene un peso total de 377 mg 50 mg o 377 mg 25 mg, o 377 mg 5 mg, o 377 mg.
[0187] En una realización, más del 50% en peso de la segunda capa del comprimido multicapa es emtricitabina o una sal del mismo y tenofovir alafenamida o una sal del mismo. En una realización, más del 55% en peso de la segunda capa de la tableta multicapa es emtricitabina o una sal del mismo y tenofovir alafenamida o una sal del mismo. En una realización, más del 60% en peso de la segunda capa de la tableta multicapa es emtricitabina o una sal del mismo y tenofovir alafenamida o una sal del mismo. En una realización, aproximadamente 55% a aproximadamente 65% en peso de la segunda capa de la tableta multicapa es emtricitabina o una sal de la misma y tenofovir alafenamida o una sal de la misma. En una realización, más del 60% en peso de la segunda capa de la tableta multicapa es emtricitabina y hemifumarato de tenofovir alafenamida. En una realización, aproximadamente 55% a aproximadamente 65% en peso de la segunda capa de la tableta multicapa es emtricitabina y hemifumarato de tenofovir alafenamida.
[0188] En una realización, la segunda capa de la tableta multicapa contiene menos de 250 mg de excipientes, p. ej., menos de 200 mg o menos de 150 mg.
[0189] En una realización, al menos el 50% en peso de la segunda capa de la tableta multicapa es emtricitabina. En una realización, al menos el 53% en peso de la segunda capa de la tableta multicapa es emtricitabina. En una realización, aproximadamente 45% a aproximadamente 65% en peso de la segunda capa de la tableta multicapa es emtricitabina. En una realización, aproximadamente 50% a aproximadamente 60% en peso de la segunda capa de la tableta multicapa es emtricitabina. En una realización, aproximadamente 51% a aproximadamente 53% en peso de la segunda capa de la tableta multicapa es emtricitabina.
[0190] En una realización, al menos 4% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización, al menos el 6% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización, al menos el 7% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización, de aproximadamente 5% a aproximadamente 10% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización, aproximadamente del 6% a aproximadamente el 9% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización, aproximadamente del 7% a aproximadamente el 8% en
peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida. En una realización,
aproximadamente el 7% en peso de la segunda capa de la tableta multicapa es hemifumarato de tenofovir alafenamida.
[0191] En una realización, menos del 20% en peso de la segunda capa de la tableta multicapa es croscarmelosa
sódica. En una realización, menos del 10% en peso de la segunda capa de la tableta multicapa es croscarmelosa de
sodio. En una realización, aproximadamente 5% a aproximadamente 10% en peso de la segunda capa de la tableta
multicapa es croscarmelosa de sodio. En ciertas realizaciones, el uso de aproximadamente 5 a 7% (p. ej.,
aproximadamente 6%) de croscarmelosa sódica en peso de la segunda capa puede proporcionar una disolución
mejorada del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, en relación con otros
desintegrantes, p. ej., polivinilpirrolidona (crospovidona) En ciertas realizaciones, el uso de aproximadamente 5 a 10%
de croscarmelosa sódica en peso de la segunda capa puede proporcionar una disolución mejorada del compuesto de
Fórmula I o una sal farmacéuticamente aceptable del mismo, en relación con otros desintegrantes, p. ej.,
polivinilpirrolidona (crospovidona).
[0192] En una realización, menos de 50% en peso de la segunda capa del comprimido multicapa es microcristalina
celulosa. En una realización, menos del 40% en peso de la segunda capa de la tableta multicapa es microcristalina
celulosa. En una realización, menos del 31% en peso de la segunda capa de la tableta multicapa es microcristalina
celulosa.
[0193] En una realización, menos del 5% en peso de la segunda capa de la tableta multicapa es estearato de
magnesio. En una realización, menos del 3% en peso de la segunda capa de la tableta multicapa es estearato de magnesio. En una  realización, menos del 2% en peso de la segunda capa de la tableta multicapa es estearato de magnesio. En una realización, aproximadamente 0,5% a aproximadamente 1,5% en peso de la segunda capa de la tableta multicapa es estearato de magnesio. En ciertas realizaciones, el uso de aproximadamente 1% a
realización, menos del 2% en peso de la segunda capa de la tableta multicapa es estearato de magnesio. En una realización, aproximadamente 0,5% a aproximadamente 1,5% en peso de la segunda capa de la tableta multicapa es estearato de magnesio. En ciertas realizaciones, el uso de aproximadamente 1% a
aproximadamente 2% (p. ej., aproximadamente 1,5%) de estearato de magnesio en peso de la segunda capa puede
proporcionar una disolución mejorada del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo,
en relación con otros lubricantes p. ej., estearilo fumarato de sodio.
[0194] En una realización, el peso total de la segunda capa es inferior al 150% del peso total de la primera capa. En
una realización, el peso total de la segunda capa es inferior al 125% del peso total de la primera capa. En una
realización, el peso total de la segunda capa es inferior al 116% del peso total de la primera capa. En una realización,
el peso total de la segunda capa es inferior al 106% del peso total de la primera capa.
[0195] En una realización, la segunda capa de la tableta multicapa comprende:

[0196] En una realización, la segunda capa de la tableta multicapa comprende:

[0197] En una realización, la segunda capa de la tableta multicapa consiste en:

[0198] En una realización, la segunda capa de la tableta multicapa consiste en:

[0199] En una realización, la segunda capa de la tableta multicapa consiste en:

[0200] En una realización, la segunda capa de la tableta multicapa consiste en:

[0201] En una realización, la segunda capa de la tableta multicapa incluye:

[0202] En una realización, la segunda capa de la tableta multicapa incluye:

[0203] En una realización, la segunda capa de la tableta multicapa incluye:

[0204] En una realización, la segunda capa de la tableta multicapa incluye:

[0205] En una realización, la segunda capa de la tableta multicapa incluye:

[0206] En una realización, la segunda capa de la tableta multicapa incluye:

[0207] En una realización, la segunda capa de la tableta multicapa incluye:

[0208] En una realización, la segunda capa de la tableta multicapa incluye:

[0209] En una realización de las formulaciones de tabletas multicapa descritas en el presente documento, la primera capa está en contacto con la segunda capa.
[0210] En una realización, la primera capa se produce primero, seguida de la segunda capa. Es decir, en una realización, la primera capa se prepara y se presiona en una primera capa, seguida de la segunda capa que se prepara y se presiona con la primera capa en una tableta multicapa. En una realización, la segunda capa se produce primero,
seguida de la primera capa. Es decir, en una realización, la segunda capa se prepara y se presiona en una segunda capa, seguida de la primera capa que se prepara y se presiona con la segunda capa en una tableta multicapa.
[0211] Como se usa en el presente documento, cuando se describen las tabletas multicapa descritas en el presente documento, los términos "primera capa" y "segunda capa" no pretenden indicar el método por el cual se producen las tabletas, en particular el orden en que se obtienen las capas.
[0212] En ciertas realizaciones, la tableta multicapa comprende además capas adicionales. En ciertas realizaciones, la capa o capas adicionales están situadas entre la primera y la segunda capa. En ciertas realizaciones, la capa o capas adicionales están ubicadas a ambos lados de la primera y/o segunda capa, de modo que son una capa exterior de la mesa y/o están dispuestas entre la primera y/o segunda capa y una capa de revestimiento. En algunas realizaciones, la capa o capas adicionales encapsulan las capas primera y segunda.
[0213] En una realización, la tableta multicapa comprende además una tercera capa que está entre y que separa la primera capa y la segunda capa. En una realización, la tercera capa de la tableta multicapa comprende monohidrato de lactosa, o microcristalina celulosa, o una mezcla de los mismos.
[0214] En una realización, la tableta multicapa comprende además un recubrimiento de película. En una realización, la tableta multicapa comprende además de aproximadamente 14 mg a aproximadamente 28 mg de un recubrimiento de película. En una realización, la tableta multicapa comprende además de aproximadamente 19 mg a aproximadamente 23 mg de un recubrimiento de película. En una realización, la tableta multicapa comprende además aproximadamente 21 mg de un recubrimiento de película. En una realización, el recubrimiento de película comprende alcohol polivinílico, polietilenglicol, talco, dióxido de titanio y óxido de hierro negro. En una realización, el recubrimiento de película consiste en 21 7 mg de Opadry II Brown 85F165072. En una realización, el recubrimiento de película consiste en 21 7 mg de Opadry II Yellow 85F92259.
[0215] En una realización, la tableta multicapa incluye además un recubrimiento de película. En una realización, la tableta multicapa comprende además de aproximadamente 1,9% a aproximadamente 3,9% p/p de un recubrimiento de película. En una realización, la tableta multicapa comprende además de aproximadamente 2,5% a aproximadamente 3,5% p/p de un recubrimiento de película. En una realización, la tableta multicapa comprende además aproximadamente el 3% de un recubrimiento de película. En una realización, el recubrimiento de película incluye alcohol polivinílico, polietilenglicol, talco, dióxido de titanio, óxido de hierro rojo y óxido de hierro negro. En una realización, el recubrimiento de película incluye 36% - 40% de alcohol polivinílico, 18% - 22% de polietilenglicol, 13% - 16% de talco, 20% - 24% de dióxido de titanio, 2% - 3% de óxido de hierro rojo y 0,5% - 0,7% de óxido de hierro negro.
[0216] En una realización, se proporciona una tableta que comprende aproximadamente 6,4-8,5% p/p del compuesto de Fórmula II, aproximadamente 25-32% p/p mg de emtricitabina, aproximadamente 3,5-4,5% p/p mg de hemifumarato de tenofovir alafenamida, aproximadamente 46-57% p/p mg de microcristalina celulosa, aproximadamente 5,9-8,5% p/p mg de croscarmelosa sódica y aproximadamente 1,0-2,0% p/p mg de estearato de magnesio.
[0217] En una realización, se proporciona una tableta que comprende 52 6 mg del compuesto de Fórmula II, 200 20 mg de emtricitabina, 28 3 mg de hemifumarato de tenofovir alafenamida, 360 30 mg de microcristalina celulosa, 50 8 mg de croscarmelosa de sodio, y 10,5 3 mg de estearato de magnesio y en el que el peso total de la tableta es al menos aproximadamente 685 mg.
[0218] En una realización, se proporciona una tableta que comprende 52 6 mg del compuesto de Fórmula II, 200 10 mg de emtricitabina, 28 1,5 mg de hemifumarato de tenofovir alafenamida, 360 15 mg de microcristalina celulosa, 50 4 mg de Croscarmelosa de sodio, y 10,5 1,5 mg de estearato de magnesio y en el que el peso total de la tableta es al menos aproximadamente 685 mg y es inferior a aproximadamente 715 mg.
[0219] En una realización, se proporciona una tableta que comprende 52 6 mg del compuesto de Fórmula II, 200 20 mg de emtricitabina, 28 3 mg de hemifumarato de tenofovir alafenamida, 360 30 mg de microcristalina celulosa, 50 8 mg de croscarmelosa sódica, y 8,6 3 mg de estearato de magnesio y en el que el peso total de la tableta es al menos aproximadamente 685 mg.
[0220] En una realización, se proporciona una tableta que comprende 52 6 mg del compuesto de Fórmula II, 200 10 mg de emtricitabina, 28 1,5 mg de hemifumarato de tenofovir alafenamida, 360 15 mg de microcristalina celulosa, 50 4 mg de croscarmelosa sódica, y 8,6 1,5 mg de estearato de magnesio y en el que el peso total de la tableta es al menos aproximadamente 685 mg y es inferior a aproximadamente 715 mg.
[0221] En una realización, la tableta tiene un peso total de 700 75 mg, o 700 25 mg, o 700 10 mg, o 700 mg. En una realización, la tableta no está revestida y tiene un peso total de aproximadamente 700 75 mg, o aproximadamente 700 25 mg, o aproximadamente 700 10 mg, o aproximadamente 700 mg. En una realización, la tableta tiene un peso total de aproximadamente 720 75 mg, o aproximadamente 720 25 mg, o aproximadamente 720 10 mg, o aproximadamente 720 mg, o aproximadamente 721 mg, o aproximadamente 722 mg, o
aproximadamente 723 mg, o aproximadamente 724 mg, o aproximadamente 725 mg, o aproximadamente 726 mg, o aproximadamente 727 mg, o aproximadamente 728 mg, o aproximadamente 729 mg, o aproximadamente 730 mg.
[0222] En una realización, se proporciona una tableta que comprende 80 6 mg del compuesto de Fórmula II, 200 20 mg de emtricitabina, 28 3 mg de hemifumarato de tenofovir alafenamida, 360 30 mg de microcristalina celulosa, 50 8 mg de croscarmelosa sódica, y 11 3 mg de estearato de magnesio y en el que el peso total de la tableta es de al menos aproximadamente 715 mg.
[0223] En una realización, se proporciona una tableta que comprende 78 2,3 mg del compuesto de Fórmula II, 200 10 mg de emtricitabina, 28 1,5 mg de hemifumarato de tenofovir alafenamida, 361 15 mg de microcristalina celulosa, 51 4 mg de croscarmelosa sódica, y 11 1,5 mg de estearato de magnesio y en el que el peso total de la tableta es de al menos aproximadamente 715 mg y es inferior a aproximadamente 745 mg.
[0224] En una realización, la tableta tiene un peso total de 730 75 mg, o 730 25 mg, o 730 10 mg, o 730 mg. En una realización, la tableta tiene un peso total de aproximadamente 750 75 mg, o aproximadamente 750 25 mg, o aproximadamente 750 10 mg, o aproximadamente 750 mg, o aproximadamente 751 mg, o aproximadamente 752 mg, o aproximadamente 753 mg, o aproximadamente 754 mg, o aproximadamente 755 mg.
[0225] En una realización, se proporciona una tableta que comprende:

[0226] En una realización, se proporciona una tableta que comprende:

[0227] En una realización, se proporciona una tableta que incluye:

[0228] En una realización, se proporciona una tableta que incluye:
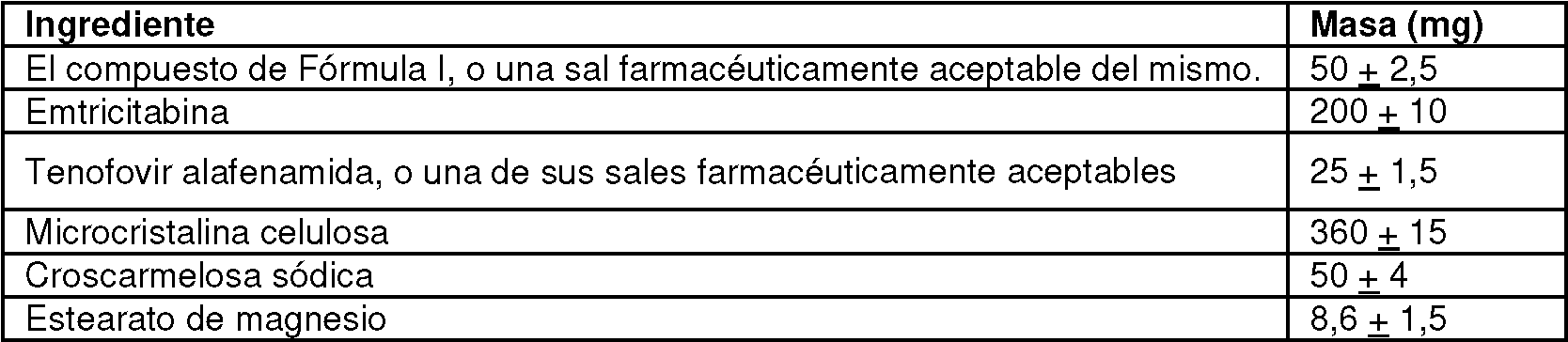
[0229] En una realización, se proporciona una tableta que incluye:

[0230] En una realización, se proporciona una tableta que incluye:

[0231] En una realización, se proporciona una tableta que incluye:

[0232] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

[0233] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

[0234] En una realización, una tableta es provisto que comprende una primera capa que incluye:

y una segunda capa que incluye:

[0235] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

[0236] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

[0237] En una realización, un se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

[0238] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

[0239] En una realización, se proporciona una tableta que comprende:

[0240] En una realización, se proporciona una tableta que comprende:

[0241] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

[0242] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

[0243] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0244] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0245] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y
opcionalmente un recubrimiento de película.
[0246] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0247] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0248] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0249] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0250] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0251] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0252] En una realización, se proporciona una tableta que comprende una primera capa que incluye:
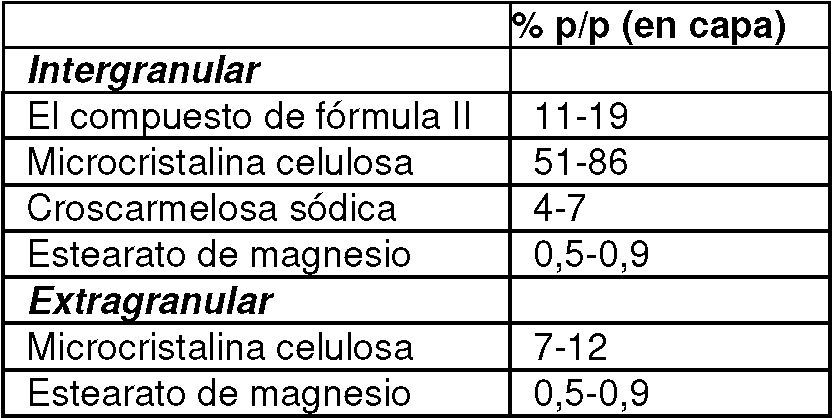
y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0253] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0254] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0255] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0256] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0257] En una realización, se proporciona una tableta que comprende una primera capa que incluye:
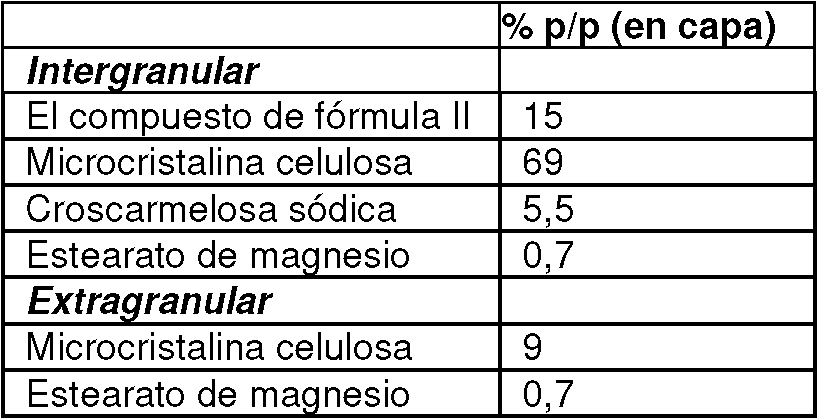
y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0258] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0259] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película.
[0260] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

y opcionalmente un recubrimiento de película.
[0261] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

y opcionalmente un recubrimiento de película.
[0262] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en:

y un recubrimiento de película que consiste en 21 mg de Opadry II Brown 85F165072 (una combinación de alcohol polivinílico, polietilenglicol (PEG), talco, dióxido de titanio, óxido de hierro rojo y óxido de hierro negro).
[0263] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y un recubrimiento de película que consiste en 21 mg de Opadry II Brown 85F165072 (una combinación de alcohol polivinílico, polietilenglicol (PEG), talco, dióxido de titanio, óxido de hierro rojo y óxido de hierro negro).
[0264] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película (p. ej., Opadry II Brown 85F165072 (una combinación de alcohol polivinílico, polietilenglicol (PEG), talco, dióxido de titanio, óxido de hierro rojo y óxido de hierro negro).
[0265] En una realización, se proporciona una tableta que comprende una primera capa que incluye:

y una segunda capa que incluye:

y opcionalmente un recubrimiento de película (p. ej., Opadry II Brown 85F165072 (a combinación de alcohol polivinílico, polietilenglicol (PEG), talco, dióxido de titanio, óxido de hierro rojo y óxido de hierro negro).
[0266] En una realización, se proporciona una tableta que comprende una primera capa que consiste en:

y una segunda capa que consiste en: y un recubrimiento de película que consiste en 21,9 mg de Opadry II Yellow 85F92259 (una combinación de alcohol polivinílico, polietilenglicol (PEG), talco, dióxido de titanio y amarillo de hierro).
[0267] En una realización, se proporciona una composición que incluye aproximadamente 75 mg de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), aproximadamente 25 mg de tenofovir alafenamida, o una sal farmacéuticamente aceptable de los mismos, y alrededor de 200 mg de emtricitabina, o una sal farmacéuticamente aceptable del mismo, en el que la AUCúltima media de cada componente después de una sola dosis administrada a un ser humano es de aproximadamente 81700 h-ng/ml a aproximadamente 140000 h-ng/mL del compuesto de Fórmula I, de aproximadamente 7500 h-ng/ml a aproximadamente 15000 h-ng/ml de emtricitabina, y de aproximadamente 165 h-ng/ml a aproximadamente 400 h-ng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa.
[0268] En una realización, se proporciona una composición que incluye un compuesto de 50 mg de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), 25 mg de tenofovir alafenamida, o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina, o una sal farmacéuticamente aceptable de la misma, en el que la AUCúltima media de cada componente después de una dosis única es de aproximadamente 87000 h-ng/ml a aproximadamente 131000 h-ng/ml del compuesto de Fórmula I, de aproximadamente 8500 h -ng/ml a aproximadamente 12800 h-ng/ml de emtricitabina, y de aproximadamente 186 h-ng/ml a aproximadamente 227 hng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0269] En una realización, se proporciona una composición que incluye un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), tenofovir alafenamida, o una sal farmacéuticamente aceptable del mismo, y emtricitabina, o un fármaco farmacéuticamente sal aceptable del mismo, en la que el AUCinf medio de cada componente después de una dosis única administrada a un humano es de aproximadamente 84450 h-ng/ml a aproximadamente 141000 h-ng/ml del compuesto de Fórmula I, de aproximadamente 8100 h-ng/ml a aproximadamente 136000 h-ng/ml de emtricitabina, y de aproximadamente 200 hng/ml a aproximadamente 500 h-ng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0270] En una realización, se proporciona una composición que incluye un compuesto de Fórmula I (p. ej., un compuesto de Fórmula II), tenofovir alafenamida y emtricitabina, en donde la Cmax media de cada componente
después de una dosis única es de aproximadamente 90.000 h-ng/mL a aproximadamente 135.000 h-ng/mL del compuesto de Fórmula I, de aproximadamente 8700 h-ng/mL a aproximadamente 13.000 h-ng/mL de emtricitabina, y de aproximadamente 200 h-ng/mL a aproximadamente 300 h-ng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0271] En una realización, se proporciona una composición que incluye un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), tenofovir alafenamida, o una sal farmacéuticamente aceptable del mismo, y emtricitabina, o un fármaco farmacéuticamente sal aceptable del mismo, en donde la Cmax media de cada componente después de una dosis única administrada a un humano es de aproximadamente 4200 ng/ml a aproximadamente 8000 mg/ml del compuesto de Fórmula I, de aproximadamente 1770 ng/ml a aproximadamente 2800 ng/ml de emtricitabina y de aproximadamente 185 ng/ml a aproximadamente 315 ng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0272] En una realización, se proporciona una composición que incluye un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), tenofovir alafenamida, o una sal farmacéuticamente aceptable del mismo, y emtricitabina, o un fármaco farmacéuticamente sal aceptable del mismo, en donde la Cmax media de cada componente después de una dosis única es de aproximadamente 4700 ng/ml a aproximadamente 5300 ng/ml del compuesto de Fórmula I, de aproximadamente 2000 ng/ml a aproximadamente 2600 ng/ml de emtricitabina, y de aproximadamente 200 ng/ml a aproximadamente 300 ng/ml de tenofovir alafenamida. En ciertas realizaciones, la composición se administra al ser humano en estado de ayuno. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0273] En una realización, se proporciona una composición que incluye un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo (p. ej., un compuesto de Fórmula II), tenofovir alafenamida, o una sal farmacéuticamente aceptable del mismo, y emtricitabina, o un fármaco farmacéuticamente sal aceptable del mismo, en la que el AUCúltimo medio, AUCinf y/o Cmax de cada componente después de una dosis única administrada a un humano en el estado alimentado es 80-125%, 80-120%, 85-115%, 90 -110%, o 95-105% del AUCúltimo medio, AUCinf y/o Cmax de cada componente después de una dosis única administrada a un humano en ayunas. En ciertas realizaciones, la composición es una tableta. En ciertas realizaciones, la tableta es una tableta bicapa. En ciertas realizaciones, la tableta es una tableta de doble capa, con el Compuesto de Fórmula I en una capa y emtricitabina y tenofovir alafenamida en la otra capa. En ciertas realizaciones, el sujeto está en ayunas. En otras realizaciones, el sujeto es alimentado.
[0274] En ciertas realizaciones, el perfil farmacocinético de las composiciones descritas en el presente documento está dentro de intervalos aceptables, independientemente de si el sujeto ha comido antes de tomar el medicamento. Por consiguiente, en ciertas realizaciones, las composiciones descritas en el presente documento pueden tomarse independientemente del consumo de alimentos por parte del sujeto. En ciertas realizaciones, la ingesta de alimentos es una comida baja en grasas, moderada o alta en grasas.
[0275] En una realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que el AUCúltima medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente El 160% del AUCúltimo medio es de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 145% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 135% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCúltima medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 125% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas.
[0276] En ciertas realizaciones, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I,
25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro del 60% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro del 45% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro del 35% del AUCúltimo medio de una dosis única después de la administración a pacientes en ayunas. En otras realizaciones, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que el AUCúltimo medio de una dosis única después de la administración a pacientes alimentados está dentro del 25% del AUCúltimo medio de un dosis única después de la administración a pacientes en ayunas.
[0277] En una realización, se proporciona una composición que comprende 50 mg de compuesto de fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCinf medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente el 100% a aproximadamente el 160% de la AUCinf. media de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCinf medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 150% de la media de AUCinf de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que el AUCinf medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 135% del AUCinf medio de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCinf medio de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 125% de la media de AUCinf de una dosis única después de la administración a pacientes en ayunas.
[0278] En ciertas realizaciones, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCinf medio de una dosis única después de la administración a pacientes alimentados está dentro del 60% del AUCinf media de una dosis única después de la administración a pacientes en ayunas. En otra realización, una composición que comprende 50 mg de compuesto de Fórmula I, 25 mg tenofovir alafenamida, y 200 mg de emtricitabina está proporcionado, en el que el AUCinf medio de una dosis única después de la administración a los pacientes alimentados está dentro del 50% del AUCinf medio de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde el AUCinf. medio de una dosis única después de la administración a pacientes alimentados está dentro del 35% del AUCinf medio de una dosis única después de la administración a pacientes en ayunas. En otras realizaciones una composición que comprende 50 mg de compuesto de Fórmula I, 25 mg de tenofovir alafenamida, y 200 mg de emtricitabina se proporciona, en el que el AUCinf medio de una dosis única después de su administración a pacientes alimentados está dentro de 25% del AUCinf medio de una dosis única después de la administración a pacientes en ayunas.
[0279] En una realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 160% de la Cmax media de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 130% de Cmax media de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 120% de la Cmax media de una dosis única después de la administración a pacientes en ayunas. En otra realización, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro de aproximadamente 100% a aproximadamente 115% de Cmax media de una dosis única después de la administración a pacientes en ayunas.
[0280] En ciertas realizaciones, se proporciona una composición que comprende 50 mg de Compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro del 60% de la media Cmax de una dosis única después de la administración a pacientes en ayunas. En otra realización, una composición que comprende 50 mg de compuesto de Fórmula I, 25 mg tenofovir alafenamida, y 200 mg de emtricitabina está proporcionado, en el que la media Cmax de una
dosis única después de la administración a los pacientes alimentados está dentro de 30% de la media Cmax de una dosis única después de la administración a pacientes en ayunas. En una realización adicional, se proporciona una composición que comprende 50 mg de compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en donde la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro del 20% de la Cmax media de una dosis única después de la administración a pacientes en ayunas. En otras realizaciones, se proporciona una composición que comprende 50 mg de compuesto de Fórmula I, 25 mg de tenofovir alafenamida y 200 mg de emtricitabina, en la que la Cmax media de una dosis única después de la administración a pacientes alimentados está dentro del 15% de la Cmax media de un dosis única después de la administración a pacientes en ayunas.
Métodos de fabricación
[0281] También se proporcionan métodos para producir las composiciones y formas de dosificación (en particular tabletas) descritas en el presente documento. En algunas realizaciones, el método comprende (a) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como una primera capa, y (b) comprimir el tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable del mismo como una segunda capa. Por ejemplo, en algunas realizaciones, el método comprende (a) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como una primera capa, seguido de (b) comprimir el tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o un fármaco farmacéuticamente sal aceptable del mismo como una segunda capa. En otras realizaciones, el método comprende (a) comprimir el tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable del mismo seguido de (b) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como otra capa. En otras realizaciones, el método comprende (a) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como una capa seguida de (b) comprimir el tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable del mismo como otra capa. La primera capa y la segunda capa pueden comprimirse por separado y combinarse posteriormente. Sin embargo, más típicamente, una primera capa se forma por compresión y, posteriormente, una segunda capa se comprime sobre la primera capa. En ciertas realizaciones, la elección del orden de las capas en la formación de tabletas multicapa puede tener un impacto en las propiedades de las tabletas (p. ej., la adhesión de las capas dentro de la tableta).
[0282] En algunas realizaciones, se proporciona una tableta en la que la primera capa se puede obtener mediante un método de (a) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como una primera capa, y (b) comprimir la tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable de la misma como una segunda capa. En otras realizaciones, se proporciona una tableta en la que la segunda capa se puede obtener mediante un método de (a) comprimir el compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo como una primera capa, y (b) comprimir la tenofovir alafenamida o una sustancia sal farmacéuticamente aceptable del mismo y emtricitabina o una sal farmacéuticamente aceptable de la misma como una primera capa.
[0283] En ciertas realizaciones, los métodos incluirán una etapa de recubrimiento de los núcleos de la tableta después de la compresión, p. ej., con un recubrimiento de película como se describe anteriormente.
[0284] En general, los métodos de formación de tabletas son bien conocidos en la técnica de la farmacia. Las técnicas y formulaciones generalmente se encuentran en Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA).
[0285] Se puede hacer una tableta por compresión o moldeo, opcionalmente con uno o más excipientes. Las tabletas comprimidas se pueden preparar comprimiendo en una máquina adecuada el ingrediente activo en una forma que fluye libremente, tal como un polvo o gránulos, opcionalmente mezclados con excipientes.
Métodos terapéuticos
[0286] Las formas de dosificación oral sólida (en particular tabletas) descritas en el presente documento se usan para el tratamiento de infección por VIH (p. ej., infección por VIH-1). En ciertas realizaciones, las formas de dosificación oral sólida (en particular tabletas) descritas en el presente documento se usan para la profilaxis previa a la exposición (PrEP) para reducir el riesgo de VIH-1 adquirido sexualmente.
[0287] En consecuencia, se proporcionan métodos para tratar a un sujeto infectado con VIH, que comprende administrar una forma de dosificación oral sólida descrita en el presente documento (en particular una tableta) al sujeto. De manera similar, se proporciona una forma de dosificación oral sólida (en particular una tableta) para usar en tales métodos de tratamiento. También se proporciona el uso del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y emtricitabina o una sal farmacéuticamente aceptable del mismo, en la fabricación de una forma de dosificación oral descrita en este documento (en particular un tableta) para el tratamiento de la infección por VIH.
[0288] En ciertas realizaciones, las formas de dosificación oral sólida (en particular tabletas) descritas en el presente documento se usan para la profilaxis previa a la exposición (PrEP) para reducir el riesgo de VIH-1 adquirido sexualmente. Por consiguiente, se proporcionan métodos para prevenir la infección en un sujeto en riesgo de infección con VIH, que comprende administrar una forma de dosificación oral sólida descrita en el presente documento (en particular una tableta) al sujeto. De manera similar, se proporciona una forma de dosificación oral sólida descrita en el presente documento (en particular una tableta) para usar en tales métodos de tratamiento.
[0289] En ciertas realizaciones, se proporciona un método para tratar a un sujeto con VIH con las composiciones descritas en el presente documento, en el que el método no aumenta el riesgo de una interacción fármaco-fármaco en el paciente. En ciertas realizaciones, se proporciona un método para tratar a un sujeto con VIH con las composiciones descritas en el presente documento, en el que el método disminuye el riesgo de una interacción fármaco-fármaco en el paciente. Ejemplos de interacciones farmacológicas incluyen interacciones entre el compuesto de Fórmula I, tenofovir alafenamida o emtricitabina con rifampicina, metformina, uno o más anticonceptivos hormonales o un agente antiviral del VHC (p. ej., uno o más de sofosbuvir, ledipasvir, velpatasvir, voxilaprevir, grazoprevir, boceprevir, elbasvir, dasabuvir, ombitasvir, paritaprevir, ABT-530 y ABT-493). En ciertas realizaciones, se proporciona un método para tratar a un sujeto con VIH, en el que al sujeto se le administra una o las composiciones descritas en el presente documento, en el que el sujeto también se está tratando con rifampicina, metformina o un agente antiviral del VHC (p. ej., uno o más de sofosbuvir, ledipasvir, velpatasvir, voxilaprevir, grazoprevir, boceprevir, elbasvir, dasabuvir, ombitasvir, paritaprevir, ABT-530 y ABT-493).
[0290] Los métodos implican administrar una forma de dosificación oral descrita en el presente documento (en particular una tableta) al sujeto, típicamente un humano, y generalmente implicarán administraciones repetidas, típicamente una vez al día. El tratamiento puede ser profiláctico o terapéutico.
General
[0291] El término "alimentado" en relación con la administración de una forma de dosificación oral sólida a un sujeto humano significa la administración de la forma de dosificación por vía oral en condiciones de alimentación (comida moderada en grasa), p. ej., la administración dentro de aproximadamente 30 minutos del consumo humano de una comida estandarizada de aproximadamente 300 a 600 calorías y aproximadamente 10 a aproximadamente 15 gramos de grasa. En algunas realizaciones del documento, "alimentado" se refiere a la administración dentro de aproximadamente 30 minutos del consumo humano de una comida rica en grasas.
[0292] El término "sustancialmente libre" en relación con la presencia de un componente dado dentro de, p. ej., una composición significa que menos del 5% en peso de la composición (p. ej., menos del 1% en peso de la composición) es ese componente dado. La palabra "sustancialmente" no excluye "completamente", p. ej., una composición que está "sustancialmente libre" de Y puede estar completamente libre de Y. Cuando sea necesario, la palabra "sustancialmente" puede omitirse de la definición de la invención.
[0293] El término "segregado" tal como se usa en relación con ciertos componentes (p. ej., A y B) dentro de una tableta significa que esos componentes son físicamente discretos de manera que la presencia de un componente (p. ej., A) no afecta sustancialmente la estabilidad en el almacenamiento del (los) otro(s) componente(s) (p. ej. B) del cual está segregado. Típicamente, cuando los componentes se segregan en una tableta, entonces estarán presentes en capas separadas en una tableta multicapa. A modo de ejemplo, los componentes A y B pueden estar presentes en capas separadas en una tableta multicapa, en donde (a) la capa que contiene el componente A está sustancialmente libre de componente B y (b) la capa que contiene el componente B está sustancialmente libre de componente A Las capas separadas pueden estar en contacto entre sí o pueden estar separadas, p. ej., por una o más capas adicionales.
[0294] El término "comprende" y sus variaciones, tales como "comprende" y "que comprende", deben interpretarse en un sentido abierto e inclusivo, es decir, como "incluido, pero no limitado a".
[0295] El término "entre" con referencia a dos valores incluye esos dos valores, p. ej., el intervalo "entre" 10 mg y 20 mg abarca, p. ej., 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 y 20 mg.
[0296] El término "aproximadamente" usado en relación con una cantidad incluye el valor establecido y tiene el significado dictado por el contexto (p. ej., incluye el grado de error asociado con la medición de la cantidad particular). Por ejemplo, en cierto ejemplo no limitativo, el término "aproximadamente" en relación con un valor numérico x se refiere a x + 10%, x + 5%, o x + 1%.
[0297] "% p/p" significa el peso de un componente como un porcentaje del peso total de, p. ej., una capa o forma de dosificación en la que está presente el componente. Por ejemplo, una composición que comprende "5% p/p X" se refiere a una composición en la que el peso del componente X es el 5% del peso total de la composición.
[0298] La referencia a lo largo de esta especificación a "una realización" o "una realización" significa que una característica, estructura o característica particular descrita en relación con la realización está incluida en al menos una realización proporcionada en este documento. Por lo tanto, las apariencias de las frases "en una realización" o
"en una realización" en varios lugares a lo largo de esta especificación no se refieren necesariamente a la misma realización. Además, las características, estructuras o características particulares se pueden combinar de cualquier manera adecuada en una o más realizaciones.
[0299] El término "farmacéuticamente aceptable" con respecto a una sustancia se refiere a esa sustancia que generalmente se considera segura y adecuada para su uso sin toxicidad indebida, irritación y/o respuesta alérgica, proporcional a una relación beneficio/riesgo razonable. "Farmacéuticamente aceptable" con respecto a los excipientes incluye, sin limitación, cualquier adyuvante, vehículo, excipiente, deslizante, agente edulcorante, diluyente, conservante, tinte/colorante, potenciador del sabor, tensioactivo, agente humectante, agente dispersante, agente de suspensión, estabilizador, agente isotónico, solvente o emulsionante que ha sido aprobado por la Administración de Drogas y Alimentos de los Estados Unidos como aceptable para uso en humanos o animales domésticos.
[0300] "Sal farmacéuticamente aceptable" se refiere a una sal de un compuesto que es farmacéuticamente aceptable y que posee (o puede convertirse en una forma que posee) la actividad farmacológica deseada del compuesto original. Dichas sales incluyen sales de adición de ácido formadas con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico o ácido fosfórico; o formado con ácidos orgánicos como ácido acético, ácido bencenosulfónico, ácido benzoico, ácido canforsulfónico, ácido cítrico, ácido etanosulfónico, ácido fumárico, ácido glucoheptónico, ácido glucónico, ácido láctico, ácido maleico, ácido malónico, ácido mandélico, ácido metanosulfónico, ácido 2-naftalenosulfónico, ácido oleico, ácido palmítico, ácido propiónico, ácido esteárico, ácido succínico, ácido tartárico, ácido p-toluenosulfónico o ácido trimetilacético, y sales formadas cuando un protón ácido presente en el compuesto original es reemplazado por un ion metálico, p. ej., un ion de metal alcalino, un ion alcalinotérreo o un ion de aluminio; o coordina con una base orgánica tal como dietanolamina, trietanolamina o N-metilglucamina. También se incluyen en esta definición las sales de amonio y de amonio sustituido o cuaternizado. Se pueden encontrar listas no limitantes representativas de sales farmacéuticamente aceptables en SM Berge et al., J. Pharma Sci., 66 (1), 1-19 (1977), y Remington: The Science and Practice of Pharmacy, R. Hendrickson., ed., 21a edición, Lippincott, Williams & Wilkins, Filadelfia, PA, (2005), en p. 732, Tabla 38-5.
[0301] Como se usa en el presente documento, el término "sales" incluye cocristales. El término "cocristal" se refiere a un compuesto cristalino que comprende dos o más componentes moleculares, p. ej., en donde la transferencia de protones entre los componentes moleculares es parcial o incompleta.
[0302] El término "solvato" significa un complejo molecular que comprende un compuesto y una o más moléculas solventes farmacéuticamente aceptables. Los ejemplos de moléculas solventes incluyen agua y alcoholes C1-6, p. ej., etanol. Cuando el solvato es agua, se puede usar el término "hidrato".
[0303] "Tratar" y "tratamiento" de una enfermedad incluyen lo siguiente:
(1) prevenir o reducir el riesgo de desarrollar la enfermedad, es decir, hacer que los síntomas clínicos de la enfermedad no se desarrollen en un sujeto que pueda estar expuesto o predispuesto a la enfermedad pero aún no experimenta o muestra síntomas de la enfermedad,
(2) inhibir la enfermedad, es decir, detener o reducir el desarrollo de la enfermedad o sus síntomas clínicos, y
(3) aliviar la enfermedad, es decir, causando regresión de la enfermedad o sus síntomas clínicos.
[0304] El término "cantidad efectiva" se refiere a una cantidad que puede ser efectiva para obtener la respuesta biológica o médica deseada, incluyendo la cantidad de un compuesto que, cuando se administra a un sujeto para tratar una enfermedad, es suficiente para efectuar dicho tratamiento para la enfermedad. La cantidad efectiva variará dependiendo del compuesto, la enfermedad y su gravedad y la edad, peso, etc. del sujeto a tratar. La cantidad efectiva puede incluir un intervalo de cantidades.
EJEMPLOS
[0305] Los siguientes ejemplos se proporcionan con fines ilustrativos, no limitativos. Los ejemplos 1,2, 2a, 3, 4, 5 y la formulación de tableta F8 del ejemplo 7 se proporcionan como ejemplos de referencia.
Ejemplo 1 - Compuesto de tabletas de agente único de Fórmula II
[0306] Se preparó una formulación (tableta F1) del compuesto de Fórmula II por granulación en seco. La figura 5 es un diagrama de flujo que ilustra la preparación de esta formulación. La composición de la formulación de agente único se muestra en la tabla a continuación:

[0307] En los estudios farmacocinéticos del Ejemplo 3, los comprimidos de la Formulación F1 se recubrieron con película con 12 mg de Opadry II Amarillo 85F92259. El peso total de las tabletas recubiertas con película fue de 312 mg.
[0308] El compuesto de fórmula II se mezcló con excipientes intergranulares (lactosa, microcristalina celulosa, y crospovidona), mezcló adicionalmente con la parte intergranular del estearilo fumarato de sodio), compactado con rodillo, molido, y mezclado final con el estearilo fumarato de sodio para producir una mezcla final de polvo para la compresión. La mezcla final de polvo tenía un tamaño medio de partícula de 347 pM con un índice de compresibilidad del 21%. La mezcla de polvo final se comprimió en núcleos de tabletas, que se recubrieron con película como se describe anteriormente hasta un aumento de peso diana del 4%.
E je m p lo 2 - C o m p u e s to de F ó rm u la I I / ta b le ta s b ic a p a e m tr ic ita b in a / te n o fo v ir a la fe n a m id a
[0309] Una formulación de bicapa (comprimido F2) del compuesto de Fórmula II, emtricitabina, y tenofovir hemifumarato alafenamida se preparó usando el método descrito en el Ejemplo 8. La Figura 6 es un diagrama de flujo que ilustra la preparación de tabletas bicapa. La composición de la formulación se resume en la siguiente tabla:

[0310] En los estudios farmacocinéticos del Ejemplo 3, los comprimidos de la Formulación F2 se recubrieron con película con 21,9 mg de Opadry II Amarillo 85F92259 (representaron un aumento de peso del 3%). El peso total de los comprimidos recubiertos con película fue de 752 mg.
[0311] Las tabletas se fabricaron usando una granulación en seco mediante un proceso de compactación por rodillo. Se fabricó una mezcla en polvo final de emtricitabina/tenofovir alafenamida mediante granulación en seco de emtricitabina y tenofovir alafenamida con excipientes (microcristalina celulosa, croscarmelosa de sodio, estearato de magnesio intergranular), seguido de la mezcla con lubricante extragranular (estearato de magnesio extragranular). La mezcla final en polvo del Compuesto de Fórmula I se fabricó por granulación en seco del componente activo con excipientes (microcristalina celulosa, croscarmelosa sódica, estearato de magnesio intergranular) seguido de una mezcla con la carga extragranular y lubricante (microcristalina celulosa y estearato de magnesio extragranular). En este estudio, la mezcla de polvo final de emtricitabina/tenofovir alafenamida se comprimió como capa 1 y la mezcla de polvo final del Compuesto de Fórmula I se comprimió como capa 2 para proporcionar un núcleo de tableta de bicapa final. El núcleo estaba recubierto con película como se describió anteriormente.
E je m p lo 2A C o m p rim id o s de e m tr ic ita b in a /te n o fo v ir a la fe n a m id a
[0312] La composición de los comprimidos de combinación de dosis fija de emtricitabina/tenofovir alafenamida (F/TAF) utilizados en estudios posteriores se muestra en la siguiente tabla:
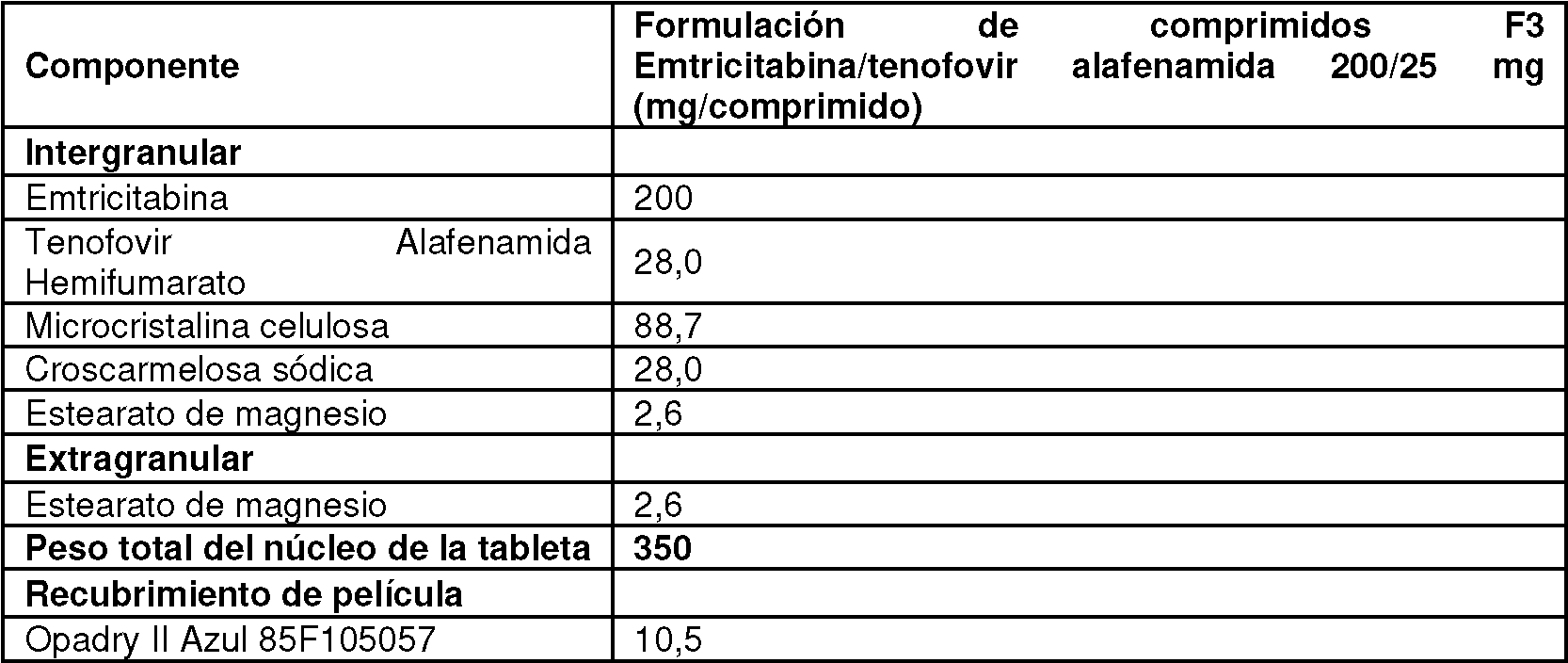
[0313] Emtricitabina y hemifumarato de tenofovir alafenamida se mezclaron con microcristalina celulosa y croscarmelosa de sodio, seguido de lubricación con estearato de magnesio. La premezcla de compactación por rodillo se compactó por rodillo y se molió usando un molino oscilante. Los gránulos resultantes se lubricaron con estearato de magnesio y se comprimieron en núcleos de tabletas de 350 mg que posteriormente se recubrieron con película.
Ejemplo 3 - Estudios farmacocinéticos
[0314] Se llevaron a cabo estudios para evaluar los perfiles farmacocinéticos de las formulaciones F1, F2 y F3 de los Ejemplos 1, 2 y 2A. Se realizó un estudio cruzado aleatorio, abierto, de período múltiple, de secuencia fija para evaluar la biodisponibilidad relativa de la formulación de tabletas bicapa F2 en comparación con la de la formulación de tabletas de agente único F1 coadministrada simultáneamente con una tableta de combinación de dosis fija que contiene emtricitabina y tenofovir alafenamida (formulación de tableta F3). La biodisponibilidad se evaluó en sujetos sanos.
Diseño del estudio y duración del tratamiento
[0315] Se administraron tres dosis únicas de las siguientes formulaciones de tabletas por vía oral durante un máximo de 21 días de duración total del estudio:
(a) una tableta combinada de dosificación fija que contiene emtricitabina y tenofovir alafenamida (200/25 mg - Tableta F3) y una tableta de agente único que contiene el compuesto de Fórmula I (75 mg - Tableta F1) administrado simultáneamente, en ayunas (dosificado el día 1; días 2-8 lavado);
(b) una tableta de combinación de dosis fija que contiene el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida (75/200/25 mg - Tableta F2) administrado en ayunas (dosificado el día 9; días 10-16 lavado); y
(c) una tableta de combinación de dosis fija que contiene el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida (75/200/25 mg - Tableta F2) administrado en condiciones de alimentación con una comida rica en grasas (dosificado el día 17; día 21 descarga).
Criterios de evaluación
[0316] Se calcularon los siguientes parámetros farmacocinéticos plasmáticos: Cmax, AUCúltimo y AUCinf.
Métodos estadísticos
[0317] Farmacocinética: las concentraciones plasmáticas y los parámetros PK se enumeraron y resumieron por analito y grupo de tratamiento usando estadística descriptiva. Además, se ajustó un análisis paramétrico de varianza utilizando un modelo de efectos mixtos apropiado para un diseño cruzado a la transformación logarítmica natural de los parámetros PK (AUCinf, AUCúltimo y Cmax). Se construyeron intervalos de confianza (IC) bilaterales del 90% para la relación de medias geométricas de mínimos cuadrados (GLSM) de cada parámetro PK para el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida.
Resultados
Disposición del sujeto y demografía
[0318] Un total de 28 sujetos fueron asignados al azar y recibieron al menos 1 dosis del fármaco del estudio.
[0319] Los resultados para la primera cohorte del estudio clínico se resumen en la tabla a continuación. Esta tabla presenta los valores medios (% CV), las relaciones de la media de mínimos cuadrados geométricos (GLSM) y los valores IC del 90% de los parámetros PK de plasma AUCúltimo, AUCinf y C máx. para cada ingrediente activo, después de la administración de la formulación de tabletas bicapa F2 o la formulación de tabletas de agente único F1, coadministrado con una combinación de dosis fija de emtricitabina y tenofovir alafenamida en ayunas.

[0320] La exposición total del compuesto de Fórmula I, en condiciones de ayuno, fue aproximadamente un 30% mayor cuando se dosificó en la formulación de tableta bicapa F2, que contenía dos agentes terapéuticos adicionales (TAF y FTC), en comparación con la formulación de tabletas de agente único F1, co-dosificados con una tableta de combinación de dosis fija que contiene TAF y FTC (F3). En condiciones de ayuno, el AUCinf y Cmax del compuesto de Fórmula I fueron 27% y 31% más altos, respectivamente, después de la administración de la formulación de tabletas bicapa F2 que después de la administración de la formulación de tabletas de agente único F1 con la combinación de dosis fija de F/TAF (F3, 200/25 mg). La exposición a emtricitabina y tenofovir alafenamida fue similar después de la administración de la formulación de tabletas bicapa F2 o la formulación de tabletas de agente único F1 con F/TAF FDC (F3, 200/25 mg).
[0321] En base a los datos de este estudio, se desarrolló una nueva formulación de dosis fija del compuesto de Fórmula I, emtricitabina y tenofovir alafenamida.
[0322] El efecto de los alimentos sobre la farmacocinética del compuesto de Fórmula I también se examinó en un periodo 2 no aleatorizado, de etiqueta abierta, cruzado, de secuencia fija, y la cohorte de dosis única que consiste en 8 sujetos únicos. En este estudio, se administró una dosis única del compuesto de Fórmula I en dos períodos con secuencia fija en condiciones de ayuno y alimentación, respectivamente. Se requirió un lavado de al menos cinco vidas medias entre las secuencias en ayunas y alimentadas.
[0323] El compuesto de la Fórmula I se preparó de acuerdo a por ciento en peso de la formulación del comprimido del Ejemplo 1, una dosis equivalente a 100 mg del compuesto de Fórmula I y las cantidades de excipiente ajustadas en consecuencia para llegar a los porcentajes en peso designados (100 mg/tableta). Se administró una única tableta que contenía el compuesto de Fórmula I (100 mg/tableta) con aproximadamente 240 ml de agua después de un ayuno nocturno (sin comida ni bebida, excepto agua, durante al menos 10 horas) el día 1. Los sujetos continuaron ayunando hasta 4 horas después de la dosis o hasta la recolección de la muestra de sangre de 4 horas después de la dosis. El día 9, se administró una única tableta que contenía el compuesto de Fórmula I (100 mg/tableta) con aproximadamente 240 ml de agua después de un ayuno nocturno (sin alimentos ni bebidas, excepto agua, durante al menos 10 horas) y dentro de los 5 minutos de los sujetos terminaron un desayuno estándar alto en grasas (aproximadamente 800 calorías con 50% de calorías provenientes de grasas).
[0324] La evaluación del efecto de los alimentos sobre compuesto de Fórmula I se obtuvo mediante la comparación de AUCinf, AUC0-último, y Cmax en los Días 1 y 9 en condiciones de alimentación y en ayunas. Los parámetros PK de transformación logarítmica natural (es decir, AUCinf, AUC0-last, y Cmax) se evaluaron usando un modelo de efectos mixtos con alimentos como un efecto fijo y el sujeto como un efecto aleatorio. Se construyó el IC del 90% para las razones geométricas de mínimos cuadrados (GLSM) para rápido versus alimentado. La comparación estadística de los parámetros farmacocinéticos del compuesto de Fórmula 1 después de la administración de una dosis única de esta formulación de tableta en estados alimentados y en ayunas se presenta en la tabla a continuación.

[0325] La tabla anterior presenta las relaciones de GLSM y los intervalos de confianza (IC) del 90% asociados para los tratamientos de prueba (alimentados) frente a los de referencia (en ayunas) para los parámetros primarios de PK en plasma del compuesto de Fórmula I. Administración de una dosis única del compuesto de Fórmula I 100 mg con alimentos (desayuno rico en calorías/grasas) aumentó los valores de GLSM de Cmax y AUCinf 101% (IC 90% de relación GLSM 165,93% a 242,74%) y 84% (90% IC del índice GLSM 152,05% a 222,59%), respectivamente. No hubo cambios aparentes en el aclaramiento y t1/2 después de la administración con alimentos, lo que indica que los alimentos mejoraron la biodisponibilidad del compuesto de Fórmula I al mejorar su solubilidad y/o absorción.
[0326] El efecto de los alimentos sobre la PK del compuesto de fórmula I, emtricitabina y tenofovir alafenamida cuando se administra como la tableta de formulación F2 se evaluó en el estudio clínico descrito anteriormente (el estudio aleatorio, abierto, de período múltiple, cruzado de secuencia fija en el que participaron 28 sujetos que compararon la formulación de tableta bicapa F2 con la de la formulación de tableta de agente único F1 coadministrada simultáneamente con una tableta de combinación de dosis fija que contiene emtricitabina y tenofovir alafenamida (formulación de tableta F3).
[0327] Los valores medios (% CV), proporciones GLSM, y el 90% de los valores de IC del plasma para parámetros PK AUCúltimo, AUCinf, y Cmax para el compuesto de Fórmula I, emtricitabina, y tenofovir alafenamida después de la administración de la formulación F1 de tableta en condiciones de ayuno o con una comida rica en grasas se presentan en la siguiente tabla.

[0328] En comparación con la administración en ayunas, la administración de la formulación del comprimido F2 con una comida rica en grasas dio como resultado un AUCinf de 46% más alto y un Cmax de 27% más alto para el compuesto de Fórmula I. El impacto de los alimentos sobre la exposición a emtricitabina y tenofovir alafenamida fue similar al observado previamente.
[0329] Los datos presentados en las dos tablas anteriores también demuestran que el efecto de los alimentos sobre la exposición total del compuesto de Fórmula I se reduce en comprimido de dos capas de formulación F2, en relación con el comprimido de agente único de formulación F1. La relación media geométrica de AUCinf para el compuesto de Fórmula I en tabletas de agente único de formulación de F1 fue de 1,84, mientras que la relación geométrica media de AUCinf para el compuesto de Fórmula I en la formulación de tabletas de doble capa F2 fue 1,46. En comparación, la proporción media geométrica de AUCinf para emtricitabina en 0,91 y 1,00 para emtricitabina y 1,75 y 1,58 para tenofovir alafenamida en cada formulación respectivamente (comparando las exposiciones de emtricitabina y tenofovir alafenamida de la tableta de formulación de F2 con la tableta de formulación de F1 combinada con la tableta de formulación de F3). La relación media geométrica de Cmax para tabletas de agente único de formulación de F1 fue 2,01, mientras que la relación media geométrica de Cmax para tabletas de doble capa de formulación de F2 fue 1,27. En comparación, las proporciones medias geométricas de Cmax para emtricitabina y tenofovir alafenamida en las tabletas de formulaciones de F1 y F2 fueron 0,85 y 0,83, respectivamente. Estos datos indican que se puede tomar una formulación de tableta combinada de dosificación fija del compuesto de Fórmula I, emtricitabina y tenofovir alafenamida sin tener en cuenta los alimentos.
Ejemplo 4 - Estudios de disolución
[0330] Se llevaron a cabo estudios para evaluar los perfiles de disolución de las tabletas F1, F2 y F3-A. La disolución del compuesto de Fórmula II se midió usando el Aparato USP II, en 250 ml de fluido intestinal simulado en estado de ayuno (FaSSIF) a pH 6,5, a 37°C y una velocidad de paleta de 100 rpm. Se preparó FaSSIF añadiendo polvo de fluido intestinal simulado (SIF) (Biorelevant.com) a tampón fosfato de pH 6,5 a aproximadamente 4,48 g/L. Una vez que se disolvió el polvo, el volumen de la solución resultante se duplicó. En el caso de tabletas de agente único de formulación F1, la disolución del compuesto de Fórmula II se midió en presencia de tableta de combinación de dosis fija de formulación F3 del Ejemplo 2A que contiene emtricitabina y hemifumarato de tenofovir alafenamida. Los resultados se muestran en la Figura 1. Estos datos muestran que la formulación de bicapa (tableta F2) exhibió una disolución mejorada del compuesto de Fórmula II, en comparación con la formulación de agente único (tableta F1 combinada con F3). La tableta de formulación de F3-A se preparó como una formulación de agente único de acuerdo con el proceso descrito en el Ejemplo 1 de acuerdo con la tabla a continuación.

(Continuación)

[0331] Como se muestra arriba, la tableta F3-A usó la composición (selección de excipientes y cantidades) del compuesto de la capa de fórmula II del ejemplo 2 (F2). La Figura 1 demuestra además que la formulación de tableta F2 exhibió una disolución mejorada del compuesto de fórmula II en comparación con las formulaciones de tableta F1 y F3-A. El porcentaje total del compuesto de Fórmula II disuelto de la formulación F1 y F3-A fue comparable a aproximadamente el 60%. La cantidad total del compuesto de Fórmula II disuelto de la formulación F2 fue aproximadamente un 10% mayor.
[0332] En un segundo estudio, la disolución del compuesto de Fórmula II se midió usando fluido intestinal simulado en estado alimentado (FeSSIF) como medio de disolución. FeSSIF se preparó agregando polvo de fluido intestinal simulado (SIF) (Biorelevant.com) a tampón fosfato de pH 5,0 a aproximadamente 22,405 g/L. Una vez que se disolvió el poder, el volumen de la solución resultante se duplicó. En este estudio, la disolución del compuesto de Fórmula II se midió utilizando el Aparato USP II, en 250 ml de fluido intestinal simulado alimentado a pH 5,5, a 37°C y una velocidad de paleta de 100 rpm. Al igual que en el estudio anterior que usaba fluido intestinal simulado en ayunas, la formulación de tabletas de agente único F1 se probó en presencia de una segunda formulación de tabletas que contenía emtricitabina y hemifumarato de tenofovir alafenamida (tableta F3). Los resultados se muestran en la Figura 2. Estos datos también muestran que la formulación de bicapa (tableta F2) exhibió una disolución mejorada del compuesto de Fórmula II, en comparación con la formulación de agente único (tableta F1).
E je m p lo 5 - E s tu d io s de e x c ip ie n te s
[0333] Para evaluar adicionalmente el efecto de excipientes específicos sobre la disolución del compuesto de Fórmula I, se prepararon tres tabletas bicapa adicionales de formulaciones F4, F5 y F6. La tableta de formulación de F4 es similar a la tableta de formulación F2, con croscarmelosa de sodio reemplazada por crospovidona. Asimismo, las tabletas de formulación F5 son similares a las tabletas de formulación F2, con el estearato de magnesio reemplazado por estearilo fumarato de sodio. Finalmente, las tabletas de formulación F6 son similares a las tabletas de formulación F2, con croscarmelosa de sodio y estearato de magnesio reemplazado por crospovidona y estearilfumarato de sodio, respectivamente. Las tabletas F4, F5 y F6 se prepararon usando el método descrito en el Ejemplo 8.

[0334] La Figura 3 muestra los resultados de los estudios de disolución realizados en las formulaciones de tabletas F4, F5 y F6, así como las formulaciones de tabletas F1 y F2. Estos datos demuestran que la sustitución de cada excipiente mejora la disolución del compuesto de Fórmula I.
Ejemplo 6 - Compuesto de Fórmula I/tabletas de bicapa de emtricitabina/tenofovir alafenamida - dosis de 50 mg
[0335] Como resultado de la mayor exposición
in vivo
del compuesto de Fórmula I que se observó en los estudios farmacocinéticos, se preparó una formulación bicapa (tableta F7) del compuesto de Fórmula II, emtricitabina y hemifumarato de tenofovir alafenamida incorporando una dosis más baja del compuesto de Fórmula I. La tableta de formulación F7 fue preparada como se describió anteriormente, es decir, usando el método descrito en el Ejemplo 8. La composición de la formulación se resume en la tabla a continuación:

Ejemplo 7 - Compuesto de tabletas de agente único de Fórmula I (50 mg)
[0336] Se realizaron estudios de disolución comparando las Dosis de 50 mg en tableta bicapa F7 con un comprimido de agente único de 50 mg.
[0337] Se preparó una formulación de 50 mg (tableta F8) del compuesto de Fórmula II de manera similar a la tableta de formulación F1 del Ejemplo 1. La composición de la formulación de agente único se muestra en la tabla a continuación:

[0338] La Figura 4 muestra la disolución de 50 mg de tableta bicapa (combinación de dosis fija) de formulación F7 y 50 mg de tableta de agente único de formulación F8. La disolución del compuesto de Fórmula II se midió usando el Aparato USP II, en 333 ml de FaSSIF de pH 6,5, a 37°C y una velocidad de paleta de 100 rpm. Se probaron dos tabletas de cada tableta de formulación F7 y F8. Como en el Ejemplo 3, en el caso de la tableta de agente único de formulación F8, se midió la disolución del compuesto de Fórmula II en presencia de la tableta de formulación de combinación de dosis fija del Ejemplo 2A que contiene emtricitabina y hemifumarato de tenofovir alafenamida. Estos datos muestran que la formulación de bicapa de 50 mg (tableta F7) exhibió una disolución mejorada del compuesto de Fórmula II, en comparación con la formulación de agente único (tableta F8).
Ejemplo 8 - proceso de fabricación
[0339] El procedimiento de fabricación/envasado para el compuesto de Fórmula II/emtricitabina/tabletas de hemifumarato de tenofovir alafenamida se divide en cinco procesos unitarios:
1. mezcla del compuesto de la sustancia farmacológica de Fórmula II con excipientes intergranulares, compactación por rodillo o slugging, molienda y mezcla con excipientes extragranulares para producir la mezcla en polvo final para el compuesto de Fórmula II;
2. mezcla de emtricitabina y sustancias de drogas de hemifumarato de tenofovir alafenamida con excipientes intergranulares, granulación en seco, molienda y mezcla con excipientes extragranulares para producir una mezcla final de polvo de emtricitabina/tenofovir alafenamida hemifumarato;
3. compresión de la tableta para producir núcleos de tabletas bicapa;
4. recubrimiento de película de tableta para producir tabletas recubiertas de película; y
5. embalaje.
[0340] Los pasos del proceso de fabricación para producir el medicamento final se detallan a continuación.
Mezcla en polvo final para el compuesto de Fórmula II (dispensado, mezcla, granulación en seco, molienda, mezclado final)
[0341]
1. Pesar el compuesto de Fórmula II y los excipientes (microcristalina celulosa y croscarmelosa sódica). Corregir el peso del compuesto de Fórmula II basado en el factor de contenido de fármaco (DCF), con una reducción concomitante en el peso de la microcristalina celulosa.
2. Mezclar la porción intergranular de estearato de magnesio con la batidora y mezclar.
3. Granular en seco la mezcla resultante usando un compactador de rodillos o golpear la mezcla resultante y moler.
4. Agregar microcristalina celulosa extragranular y estearato de magnesio y mezclar.
Mezcla final en polvo de emtricitabina/tenofovir alafenamida hemifumarato (dispensación, mezcla, granulación en seco, molienda, mezcla final)
[0342]
5. Pesar sustancias y excipientes de emtricitabina y tenofovir alafenamida hemifumarato (microcristalina celulosa y croscarmelosa sódica). Ajustar el peso de las sustancias farmacológicas de emtricitabina y hemifumarato de tenofovir alafenamida en función de su DCF correspondiente, con un ajuste concomitante al peso de la microcristalina celulosa.
6. Mezclar la sustancia farmacológica de emtricitabina y hemifumarato de tenofovir alafenamida, microcristalina celulosa y croscarmelosa de sodio en una batidora y mezclar.
7. Mezclar la porción intergranular de estearato de magnesio con la batidora y mezclar.
8. Granular en seco la mezcla resultante usando un compactador de rodillos o golpear la mezcla resultante y moler.
9. Mezclar en la porción extragranular de estearato de magnesio.
Comprimido
[0343] 10. Comprimir la mezcla final de polvo del compuesto de Fórmula II como la primera capa y la mezcla final de polvo de emtricitabina/tenofovir alafenamida como segunda capa, con una fuerza de compresión principal apropiada para lograr una dureza diana de 17 kP (rango: 14 a 20 kP). Para formulaciones de tabletas que contienen 50 mg del compuesto de Fórmula II, la mezcla en polvo final se comprime a un peso de capa diana de 323 mg usando un peso total de tableta diana de 700 mg. Para formulaciones de tabletas que contienen 75 mg del compuesto de Fórmula II, la mezcla de polvo final se comprime a un peso de capa diana de 353 mg usando un peso total de tableta diana de 730 mg.
Revestimiento de película
[0344] 11. Preparar una suspensión de Opadry® II Brown 85F165072 (para comprimidos que contienen 50 mg de compuesto de la Fórmula I) o Opadry® II Yellow 85F92259 (para comprimidos que contienen 75 mg de compuesto de la Fórmula I). Cubra con película los núcleos de la tableta para lograr el aumento de peso diana de la tableta del 3% (rango 2-4%). Secar las tabletas recubiertas con película antes de enfriar y descargar.
Ejemplo 9 - Estudios farmacocinéticos - cohorte 2
[0345] El estudio farmacocinético del Ejemplo 3 se continuó con un segundo grupo de tratamiento (cohorte) para evaluar el perfil farmacocinético de la formulación F7 del Ejemplo 7 en comparación con el de la formulación F2. Diseño del estudio y duración del tratamiento
[0346] Se administraron cuatro dosis únicas de las siguientes formulaciones de tabletas una vez al día por vía oral durante 36 días de duración total del estudio:
(a) una tableta combinada de dosificación fija que contiene emtricitabina y tenofovir alafenamida (200/25 mg - Tableta F3) y una tableta de agente único que contiene el compuesto de Fórmula I (75 mg - Tableta F1) administrado simultáneamente, en ayunas (Tratamiento A) (dosificado en el día 1; días 2-8 lavado);
(b) una tableta combinada de dosis fija que contiene el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida (50/200/25 mg - Tableta F2) administrado en ayunas (Tratamiento D) (dosificado el día 9; días 10-16 lavado); y
(b) una tableta combinada de dosis fija que contiene el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida (50/200/25 mg - Tableta F2) administrado en condiciones de alimentación con una comida rica en grasas (Tratamiento E) (dosificado en el día 17; días 18-24 lavado);
(c) una tableta combinada de dosis fija que contiene el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida (50/200/25 mg - Tableta F2) administrado en condiciones de alimentación con una comida moderada en grasas (Tratamiento F) (dosificado el día 25; día 29 descarga).
Criterios de evaluación
[0347] Se calcularon los siguientes parámetros farmacocinéticos plasmáticos: AUCúltimo, AUCinf, %AUCexp, Cmax, Cúltimo, Tmax, Túltimo, CL/F, Vz/F, y t1/2.
Métodos estadísticos
[0348] Farmacocinética: los datos de concentración de sujeto individual y los parámetros PK de sujeto individual para cada analito (compuesto de Fórmula 1, FTC y TAF) se enumeraron y resumieron usando estadísticas descriptivas por grupo de tratamiento (cohorte) y tratamiento. Las estadísticas de resumen se determinaron para los datos de concentración de sujetos individuales por punto de tiempo, cohorte y tratamiento y los parámetros de PK de los sujetos individuales por cohorte y tratamiento. Además, la media geométrica, el intervalo de confianza (IC) del 95%, y la media y la desviación estándar (DE) de los valores naturales transformados logarítmicamente se presentaron para los datos de parámetros PK individuales del sujeto. El tamaño de la muestra para cada punto de tiempo se basó en el número de sujetos con datos de concentración no perdidos en ese punto de tiempo. El número de sujetos con una concentración por debajo del nivel de cuantificación (BLQ) se presentó para cada punto de tiempo.
[0349] Se realizaron comparaciones estadísticas de los parámetros de PK transformados logarítmicamente naturales para cada analito y comparación de tratamiento de interés. El modelado estadístico se basó en el conjunto de análisis PK para el analito bajo evaluación. Para cada analito, todos los sujetos con datos disponibles para el parámetro PK bajo evaluación se incluyeron en el modelado. Las comparaciones estadísticas que utilizaron el Tratamiento A como referencia solo utilizaron datos de esta cohorte, no de la cohorte anterior del Ejemplo 3 (es decir, el Tratamiento A no se combinó). Para cada analito y cada parámetro PK, se ajustó un modelo ANOVA de efectos mixtos paramétricos (teoría normal) a los valores de transformación logarítmica natural del parámetro PK bajo evaluación. El modelo estadístico incluyó el tratamiento como un efecto fijo y el sujeto como un efecto aleatorio.
[0350] Un total de 27 sujetos completaron el segundo grupo de tratamiento del estudio.
Resultados
[0351] Los parámetros farmacocinéticos de plasma para cada analito se presentan a continuación. Esta tabla se presenta los valores medios (% CV), proporciones geométricas medias de mínimos cuadrados (GLSM), y valores de IC al90% de los parámetros de plasma PK AUCúltimo, AUCinf, y Cmax para cada ingrediente activo, después de la administración de la formulación de comprimido bicapa F7 y formulación de tabletas monocapa F1 coadministradas con la formulación F/TAF F3, en condiciones de ayuno.

[0352] En particular, estos datos confirmaron que la exposición del compuesto de Fórmula I en la Formulación F7 fue proporcionalmente menor que la observada en la Formulación F2 y fue consistente con las exposiciones predichas para la dosis revisada. Estos datos fueron consistentes con el modelo de datos clínicos anteriores que confirmaron que estas exposiciones proporcionaron eficacia antiviral contra el VIH. La exposición a FTC y TAF fue similar entre la Formulación F7 y la Formulación F2 coadministradas con F3; La Cmax de TAF fue ligeramente inferior (aproximadamente un 16%) para la formulación F7 que para la administración conjunta de F2 y F3.
[0353] El efecto de los alimentos sobre la farmacocinética del compuesto de Fórmula I, emtricitabina y tenofovir alafenamida cuando se administra en la formulación F7 también se examinó en un grupo de tratamiento adicional del estudio clínico descrito anteriormente.
[0354] Los valores de la media (% CV), proporciones GLSM, y valores de IC al 90% del plasma para parámetros PK de AUCúltimo, AUCinf, y Cmax para el compuesto de Fórmula I, emtricitabina y tenofovir alafenamida después de la administración de la tableta de formulación F7 en condiciones de ayunas o con una comida rica en grasas y con una comida moderada en grasas se presentan en la siguiente tabla.

[0355] En comparación con la administración en ayunas, la administración de la tableta de formulación F7 con una comida rica en grasas dio como resultado un AUCinf de 24% más alto y un 13% más alto Cmax para el compuesto de Fórmula I. La administración de una comida moderada en grasas dio como resultado un AUCinf un 24% más alto y una Cmax un 20% más alta para el compuesto de Fórmula I. El impacto de los alimentos en la exposición a emtricitabina y tenofovir alafenamida fue similar a lo observado previamente.
[0356] Estos datos confirman que se puede tomar una tableta de formulación de combinación fija dosificada del compuesto de Fórmula I, emtricitabina y tenofovir alafenamida sin tener en cuenta los alimentos.
Ejemplo 10 - Estudios de compresión
[0357] Para investigar más a fondo las propiedades de las composiciones, los siguientes estudios investigaron la compresibilidad de la mezcla y la adhesión de la capa.
[0358] Primero, se prepararon varias composiciones de mezcla final que incluyen el Compuesto de Fórmula I (como una sal de sodio, es decir, el Compuesto de Fórmula II) con niveles variables de lactosa intergranular o extragranular que se muestra en la tabla a continuación.
Compuesto de Fórmula II: mezclas finales de polvo que contienen lactosa
Componentes

Intergranular % p/p
Compuesto de Fórmula II 16,24 16,24 16,24 16,24 14,86 14,86 Microcristalina celulosa 46,25 56,25 56,25 56,25 69,12 60,63 Lactosa 20,00 N/A N/A N/A 0,00 8,50 Croscarmelosa sódica 6,00 6,00 6,00 6,00 5,49 5,49
Estearato de magnesio 0,75 0,75
0,75 0,75  0,75
0,75  0,75
0,75  0,69
0,69  0,69
0,69
Extragranular % p/p
Microcristalina celulosa 10,00 10,00 5,00 15,00 9,15 9,15
Lactosa N/A 10,00 15,00 5,00 N/A N/A
Estearato de magnesio 0,75 0,75 0,75 0,75 0,69 0,69
Peso de la capa (mg)  323
323  323 323 323
323 323 323  353
353  353
353
[0359] A continuación, las composiciones de mezcla final del compuesto de Fórmula II con cantidades variables de estearato de magnesio que van desde 0,75% a 0,5% se prepararon como se muestra en la tabla siguiente:
Compuesto de Fórmula I: Mezclas finales de polvo con diferentes niveles de estearato de magnesio
Componentes

Intergranular % p/p
Compuesto de Fórmula II 16,24 16,24 16,24 16,24 16,24 16,24 Microcristalina celulosa 66,25 66,30 66,40 66,40 66,50 66,75 Croscarmelosa sódica 6,00 6,00 6,00 6,00 6,00 6,00 Estearato de magnesio  0,75
0,75  0,70
0,70  0,60
0,60  0,60
0,60  0,50
0,50  0,50 Extragranular % p/p
0,50 Extragranular % p/p
Microcristalina celulosa 10,00 10,00 10,00 10,00 10,00 10,00 Estearato de magnesio 0,75 0,75 0,75 0,75 0,75 0,50,5 Peso de la capa (mg)  323
323  323
323  323
323  323
323  323
323  323
323
[0360] Finalmente, las composiciones de mezcla final que contienen emtricitabina y tenofovir alafenamida se prepararon con cantidades variables de estearato de magnesio que varían de 0,75% a 0,5% como se muestra en la tabla a continuación:
Composiciones cuantitativas de mezclas finales de polvos F/TAF
Componentes

Intergranular % p/p
Tenofovir Alafenamida Hemifumarato 7,5 7,5 7,5 7,5
Emtricitabina 53,1 53,1 53,1 53,1
Microcristalina celulosa 29,9 30,4 30 30,2
Croscarmelosa sódica 8,0 8,0 8,0 8,0
Estearato de magnesio  0,75
0,75  0,50
0,50  0,70
0,70  0,60
0,60
Extragranular % p/p
Estearato de magnesio 0,75 0,50 0,70 0,60
Peso de capa (mg)  377
377  377
377  377
377  377
377
[0361] Se estudió la alteración del contenido de estearato de magnesio para evaluar el impacto sobre el compuesto de Fórmula I y la compresibilidad de la mezcla de emtricitabina/tenofovir alafenamida y la adhesión de la capa. Las formulaciones también fueron monitoreadas para filmar y pegar.
[0362] Se prepararon catorce combinaciones de las mezclas de polvo final descritas en las tablas anteriores tal como se resume en la siguiente tabla. Se determinó la fuerza crítica de apisonamiento para las catorce combinaciones.
Estudio de compresión Capa crítica 1 Resultados de precompresión

[0363] La combinación A representa el orden de capas y la composición utilizada como línea base para la comparación de otras composiciones. La tableta de formulación de cada capa corresponde a la capa correspondiente de tableta de formulación F7. Un objetivo era investigar si era posible mejorar la fuerza crítica de apisonamiento de 1,0 kN observada para la Combinación A.
[0364] Las combinaciones B - G representan estudios en los que la lactosa se incluyó en la capa que incluye el Compuesto de Fórmula I para investigar evaluar la adhesión de la capa. La combinación G incluía un 20% de lactosa en la porción intergranular, sin nada en la porción extragranular. La relación de lactosa a microcristalina celulosa varió de 1:1, 3:1, 1:3 en las combinaciones C, D y E, respectivamente. La combinación G incluyó 20 mg de lactosa en la porción intergranular. Se observó delaminación a 1,5 kN para las combinaciones B - G. Como resultado, no se investigaron más las fuerzas de apisonamiento inferiores. Cuando se invirtió el orden de las capas (Combinación H), la delaminación no ocurrió a 1,0 kN pero se observó a 1,5 kN usando las composiciones de capa de la Combinación A.
[0365] En las Combinaciones I - N, se investigó la variación de la cantidad de estearato de magnesio en cada capa para determinar el efecto sobre la compresibilidad y la adhesión de la capa. El estearato de magnesio intergranular fue 0,75%, 0,70% y 0,60% en la capa de Compuesto de Fórmula I de las Combinaciones I, J y K, respectivamente. El contenido total de estearato de magnesio en la capa F/TAF de cada una de las combinaciones I, J y K fue de 1,0%, 1,4% y 1,2 %%, respectivamente. El estearato de magnesio intergranular fue 0,6% y 0,5% en la capa de Compuesto de Fórmula I de las Combinaciones L y M, respectivamente (con 0,75% de estearato de magnesio extragranular en cada una). La combinación N tiene el nivel general más bajo de estearato de magnesio en ambas capas (1,0% en cada capa).
[0366] Se encontró que la reducción de estearato de magnesio de 0,75% a 0,5% en cada una de las porciones intergranulares y extragranulares de la capa de emtricitabina/tenofovir alafenamida aumentó la fuerza de estampación crítica a 2,0 kN (combinación I). La fuerza crítica de apisonamiento podría mejorarse aún más a 2,5 kN cuando el estearato de magnesio se redujo a 0,5% extragranular y/o intergranular en la capa que contiene el Compuesto de Fórmula I (combinaciones M y N). Sin embargo, a pesar de aumentar la fuerza crítica de apisonamiento, la disminución del estearato de magnesio al 0,5% en cada componente de la mezcla en polvo que contiene el Compuesto de Fórmula 1 tuvo un efecto negativo en la adherencia. Los cambios intermedios en el estearato de magnesio (Combinaciones J - L) no tuvieron mejoría con respecto a la combinación 1. Como resultado, el nivel de estearato de magnesio disminuyó solo en la capa de emtricitabina/tenofovir alafenamida (combinación de formulación I). Finalmente se determinó una fuerza de apisonamiento de capa 1 crítica de 1377 N. La siguiente tabla resume la composición de la Formulación I.
[0367] Por lo tanto, la adición de lactosa intergranular o extragranular a la capa que contiene el Compuesto de Fórmula I tuvo poco efecto sobre la adhesión de la capa. La disminución del estearato de magnesio de cada una de las mezclas finales de polvo mejoró la compresibilidad.
Composición cuantitativa de la formulación combinada I

Claims (1)
- REIVINDICACIONES1. Una tableta que comprende 50 mg del compuesto de Fórmula I:
 o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma.2. La tableta de la reivindicación 1, en donde la tableta comprende 50 mg del compuesto de Fórmula I como una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida como una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina.3. La tableta de la reivindicación 1 o 2, en donde la tableta comprende 52 mg de la sal de sodio del compuesto de Fórmula I y 28 mg de tenofovir alafenamida hemifumarato.4. La tableta de una cualquiera de las reivindicaciones anteriores, en la que la tableta tiene un peso total de:(i) menos de 1,0 g;(ii) menos de 800 mg; o(iii) menos de 730 mg.5. La tableta de una cualquiera de las reivindicaciones anteriores, en donde la tableta es una tableta recubierta. 6. La tableta de una cualquiera de las reivindicaciones anteriores que comprende (a) 50 mg del compuesto de Fórmula I:
o una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma.2. La tableta de la reivindicación 1, en donde la tableta comprende 50 mg del compuesto de Fórmula I como una sal farmacéuticamente aceptable del mismo, 25 mg de tenofovir alafenamida como una sal farmacéuticamente aceptable del mismo y 200 mg de emtricitabina.3. La tableta de la reivindicación 1 o 2, en donde la tableta comprende 52 mg de la sal de sodio del compuesto de Fórmula I y 28 mg de tenofovir alafenamida hemifumarato.4. La tableta de una cualquiera de las reivindicaciones anteriores, en la que la tableta tiene un peso total de:(i) menos de 1,0 g;(ii) menos de 800 mg; o(iii) menos de 730 mg.5. La tableta de una cualquiera de las reivindicaciones anteriores, en donde la tableta es una tableta recubierta. 6. La tableta de una cualquiera de las reivindicaciones anteriores que comprende (a) 50 mg del compuesto de Fórmula I: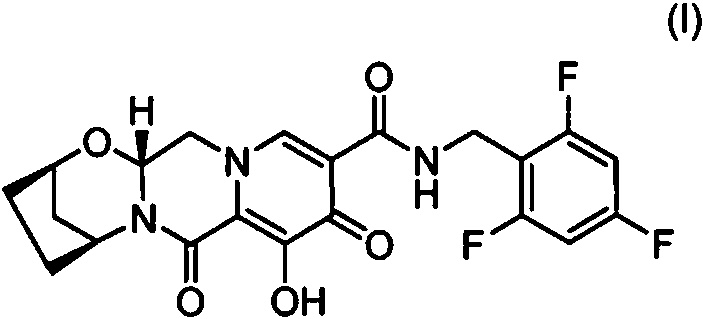 o una sal farmacéuticamente aceptable del mismo, (b) 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma, en donde la tableta es una tableta multicapa.7. La tableta multicapa de la reivindicación 6, en la que cada capa contiene al menos uno de (a), (b) y (c).8. El comprimido de una cualquiera de las reivindicaciones anteriores, en el que el comprimido contiene menos del 15% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.9. El comprimido de una cualquiera de las reivindicaciones anteriores, en el que el comprimido contiene menos del 11% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.10. Una tableta de acuerdo con una cualquiera de las reivindicaciones anteriores para su uso en el tratamiento terapéutico de una infección por VIH.
o una sal farmacéuticamente aceptable del mismo, (b) 25 mg de tenofovir alafenamida o una sal farmacéuticamente aceptable del mismo, y (c) 200 mg de emtricitabina o una sal farmacéuticamente aceptable de la misma, en donde la tableta es una tableta multicapa.7. La tableta multicapa de la reivindicación 6, en la que cada capa contiene al menos uno de (a), (b) y (c).8. El comprimido de una cualquiera de las reivindicaciones anteriores, en el que el comprimido contiene menos del 15% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.9. El comprimido de una cualquiera de las reivindicaciones anteriores, en el que el comprimido contiene menos del 11% del compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo.10. Una tableta de acuerdo con una cualquiera de las reivindicaciones anteriores para su uso en el tratamiento terapéutico de una infección por VIH.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562253042P | 2015-11-09 | 2015-11-09 | |
| US201662399999P | 2016-09-26 | 2016-09-26 | |
| PCT/US2016/060989 WO2017083304A1 (en) | 2015-11-09 | 2016-11-08 | Therapeutic compositions for treatment of human immunodeficiency virus |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2757560T3 true ES2757560T3 (es) | 2020-04-29 |
Family
ID=57346085
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16798063T Active ES2757560T3 (es) | 2015-11-09 | 2016-11-08 | Composiciones terapéuticas para el tratamiento del virus de la inmunodeficiencia humana |
Country Status (28)
| Country | Link |
|---|---|
| US (3) | US10548846B2 (es) |
| EP (2) | EP3346995B1 (es) |
| JP (1) | JP6621933B2 (es) |
| KR (4) | KR20240095320A (es) |
| CN (2) | CN113546052A (es) |
| AU (2) | AU2016354007C9 (es) |
| BR (1) | BR102016026127A2 (es) |
| CA (1) | CA2948021C (es) |
| CL (1) | CL2018001199A1 (es) |
| CO (1) | CO2018004776A2 (es) |
| CR (1) | CR20180253A (es) |
| EA (1) | EA201890654A1 (es) |
| EC (1) | ECSP18033723A (es) |
| ES (1) | ES2757560T3 (es) |
| HK (2) | HK1256093A1 (es) |
| IL (1) | IL258459A (es) |
| MX (1) | MX2018005729A (es) |
| NZ (1) | NZ741957A (es) |
| PE (1) | PE20181207A1 (es) |
| PH (1) | PH12018501001A1 (es) |
| PL (1) | PL3346995T3 (es) |
| PT (1) | PT3346995T (es) |
| SG (1) | SG11201802983TA (es) |
| SI (1) | SI3346995T1 (es) |
| SV (1) | SV2018005682A (es) |
| TW (2) | TW202220660A (es) |
| UY (1) | UY36981A (es) |
| WO (1) | WO2017083304A1 (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102406288B1 (ko) | 2012-12-21 | 2022-06-13 | 길리애드 사이언시즈, 인코포레이티드 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| NO2717902T3 (es) | 2014-06-20 | 2018-06-23 | ||
| TW201613936A (en) | 2014-06-20 | 2016-04-16 | Gilead Sciences Inc | Crystalline forms of(2R,5S,13aR)-8-hydroxy-7,9-dioxo-n-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| CN107427529A (zh) | 2014-12-26 | 2017-12-01 | 埃莫里大学 | N4‑羟基胞苷和衍生物及与其相关的抗病毒用途 |
| KR20240095320A (ko) | 2015-11-09 | 2024-06-25 | 길리애드 사이언시즈, 인코포레이티드 | 인간 면역결핍 바이러스의 치료를 위한 치료 조성물 |
| JOP20190024A1 (ar) | 2016-08-26 | 2019-02-19 | Gilead Sciences Inc | مركبات بيروليزين بها استبدال واستخداماتها |
| EP3664896A4 (en) * | 2017-08-09 | 2021-06-09 | VIIV Healthcare Company | COMBINATIONS, USES AND CORRESPONDING TREATMENTS |
| JP2020530024A (ja) * | 2017-08-09 | 2020-10-15 | ヴィーブ ヘルスケア カンパニー | 組み合わせ並びにその使用及び治療 |
| KR102626210B1 (ko) | 2017-12-07 | 2024-01-18 | 에모리 유니버시티 | N4-하이드록시사이티딘 및 유도체 및 이와 관련된 항-바이러스 용도 |
| ES2962605T3 (es) | 2018-02-26 | 2024-03-20 | Gilead Sciences Inc | Compuestos de pirrolizina sustituidos como inhibidores de la replicación del VHB |
| EP3852761A1 (en) * | 2018-09-19 | 2021-07-28 | Gilead Sciences, Inc. | Integrase inhibitors for the prevention of hiv |
| CN111096954B (zh) * | 2018-10-29 | 2022-09-16 | 江苏豪森药业集团有限公司 | 一种用于抗病毒感染的药物组合物及制备方法 |
| EP3653629A1 (en) * | 2018-11-16 | 2020-05-20 | Sandoz AG | Acid addition salts of an integrase strand transfer inhibitor |
| CN111686082A (zh) * | 2019-03-11 | 2020-09-22 | 苏州特瑞药业有限公司 | 一种富马酸磷丙替诺福韦制剂及其制备方法 |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1331208A (zh) | 2000-06-30 | 2002-01-16 | 上海博德基因开发有限公司 | 一种新的多肽——人肿瘤坏死因子受体21.45和编码这种多肽的多核苷酸 |
| KR100749160B1 (ko) | 2000-07-21 | 2007-08-14 | 길리애드 사이언시즈, 인코포레이티드 | 포스포네이트 뉴클레오티드 유사체의 전구 약물의 제조방법 |
| MY169670A (en) | 2003-09-03 | 2019-05-08 | Tibotec Pharm Ltd | Combinations of a pyrimidine containing nnrti with rt inhibitors |
| AU2003267098B2 (en) | 2002-09-11 | 2008-11-20 | Merck & Co., Inc. | Dihydroxypyridopyrazine-1,6-dione compounds useful as HIV integrase inhibitors |
| SI1583542T1 (sl) | 2003-01-14 | 2008-12-31 | Gilead Sciences Inc | Sestavki in postopki za kombinacijsko antivirusnoterapijo |
| WO2006030807A1 (ja) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有するカルバモイルピリドン誘導体 |
| DK1874117T3 (da) | 2005-04-28 | 2013-09-23 | Viiv Healthcare Co | Polycyklisk carbamoylpyridonderivat med hiv-integrasehæmmende aktivitet |
| TWI471145B (zh) | 2005-06-13 | 2015-02-01 | Bristol Myers Squibb & Gilead Sciences Llc | 單一式藥學劑量型 |
| CA2635468C (en) | 2005-12-30 | 2016-08-09 | Gilead Sciences, Inc. | Methods for improving the pharmacokinetics of hiv integrase inhibitors |
| WO2008043829A2 (en) | 2006-10-14 | 2008-04-17 | Boehringer Ingelheim International Gmbh | Method for treating hiv-1 infection by the combined administration of nevirapine, tenofovir and emtricitabine |
| BRPI1008664A2 (pt) | 2009-02-06 | 2016-03-08 | Gilead Sciences Inc | comprimidos para a terapia de combinação |
| SG190333A1 (en) | 2010-11-19 | 2013-06-28 | Gilead Sciences Inc | Therapeutic compositions comprising rilpivirine hcl and tenofovir disoproxil fumarate |
| EP2729130B1 (en) | 2011-07-07 | 2017-09-06 | Janssen Sciences Ireland UC | Darunavir combination formulations |
| HUE031253T2 (en) | 2011-08-16 | 2017-07-28 | Gilead Sciences Inc | Tenofovir alafenamid hemifumarate |
| JP6059255B2 (ja) * | 2012-02-03 | 2017-01-11 | ギリアード サイエンシス インコーポレーテッド | ウイルス感染の処置における使用のためのテノホビルアラフェンアミドヘミフマル酸塩とコビシスタットを含む併用療法 |
| AU2012327170A1 (en) | 2012-02-03 | 2013-08-22 | Gilead Sciences, Inc. | Therapeutic compounds |
| US20140221355A1 (en) | 2012-12-21 | 2014-08-07 | Gilead Sciences, Inc. | Polycyclic-carbamoylpyridone compounds and their pharmaceutical use |
| KR102406288B1 (ko) * | 2012-12-21 | 2022-06-13 | 길리애드 사이언시즈, 인코포레이티드 | 폴리시클릭-카르바모일피리돈 화합물 및 그의 제약 용도 |
| NO2865735T3 (es) * | 2013-07-12 | 2018-07-21 | ||
| CA2920811A1 (en) * | 2013-08-14 | 2015-02-19 | Ratiopharm Gmbh | Medicament comprising a pharmaceutical combination of dolutegravir, emtricitabine and tenofovir |
| WO2015039348A1 (en) | 2013-09-23 | 2015-03-26 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds useful as hiv integrase inhibitors |
| TWI677489B (zh) | 2014-06-20 | 2019-11-21 | 美商基利科學股份有限公司 | 多環型胺甲醯基吡啶酮化合物之合成 |
| NO2717902T3 (es) * | 2014-06-20 | 2018-06-23 | ||
| TW201613936A (en) | 2014-06-20 | 2016-04-16 | Gilead Sciences Inc | Crystalline forms of(2R,5S,13aR)-8-hydroxy-7,9-dioxo-n-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| CA2921336A1 (en) | 2015-06-30 | 2016-12-30 | Gilead Sciences, Inc. | Pharmaceutical formulations |
| CA2990210C (en) | 2015-06-30 | 2021-01-12 | Gilead Sciences, Inc. | Pharmaceutical formulations comprising tenofovir and emtricitabine |
| KR20240095320A (ko) * | 2015-11-09 | 2024-06-25 | 길리애드 사이언시즈, 인코포레이티드 | 인간 면역결핍 바이러스의 치료를 위한 치료 조성물 |
| KR102100436B1 (ko) | 2018-10-30 | 2020-04-13 | 울산과학기술원 | 강자성 원소 치환형 상온 다강성 물질 및 그 제조 방법 |
-
2016
- 2016-11-08 KR KR1020247017929A patent/KR20240095320A/ko unknown
- 2016-11-08 TW TW110129951A patent/TW202220660A/zh unknown
- 2016-11-08 BR BR102016026127A patent/BR102016026127A2/pt not_active Application Discontinuation
- 2016-11-08 AU AU2016354007A patent/AU2016354007C9/en active Active
- 2016-11-08 KR KR1020237001933A patent/KR20230015512A/ko not_active IP Right Cessation
- 2016-11-08 PL PL16798063T patent/PL3346995T3/pl unknown
- 2016-11-08 CN CN202110669391.6A patent/CN113546052A/zh active Pending
- 2016-11-08 WO PCT/US2016/060989 patent/WO2017083304A1/en active Application Filing
- 2016-11-08 EP EP16798063.0A patent/EP3346995B1/en active Active
- 2016-11-08 TW TW105136333A patent/TWI737647B/zh active
- 2016-11-08 MX MX2018005729A patent/MX2018005729A/es unknown
- 2016-11-08 NZ NZ741957A patent/NZ741957A/en unknown
- 2016-11-08 KR KR1020207025458A patent/KR102606625B1/ko active IP Right Grant
- 2016-11-08 CA CA2948021A patent/CA2948021C/en active Active
- 2016-11-08 JP JP2018543287A patent/JP6621933B2/ja active Active
- 2016-11-08 US US15/346,335 patent/US10548846B2/en active Active
- 2016-11-08 ES ES16798063T patent/ES2757560T3/es active Active
- 2016-11-08 PE PE2018000720A patent/PE20181207A1/es unknown
- 2016-11-08 EA EA201890654A patent/EA201890654A1/ru unknown
- 2016-11-08 EP EP19186760.5A patent/EP3632415A1/en active Pending
- 2016-11-08 SI SI201630440T patent/SI3346995T1/sl unknown
- 2016-11-08 CN CN201680065167.3A patent/CN108348473B/zh active Active
- 2016-11-08 CR CR20180253A patent/CR20180253A/es unknown
- 2016-11-08 PT PT167980630T patent/PT3346995T/pt unknown
- 2016-11-08 KR KR1020187015929A patent/KR102153996B1/ko active IP Right Grant
- 2016-11-08 SG SG11201802983TA patent/SG11201802983TA/en unknown
- 2016-11-09 UY UY0001036981A patent/UY36981A/es not_active Application Discontinuation
-
2018
- 2018-03-29 IL IL258459A patent/IL258459A/en unknown
- 2018-05-02 EC ECIEPI201833723A patent/ECSP18033723A/es unknown
- 2018-05-03 CO CONC2018/0004776A patent/CO2018004776A2/es unknown
- 2018-05-03 SV SV2018005682A patent/SV2018005682A/es unknown
- 2018-05-03 CL CL2018001199A patent/CL2018001199A1/es unknown
- 2018-05-08 PH PH12018501001A patent/PH12018501001A1/en unknown
- 2018-11-27 HK HK18115158.6A patent/HK1256093A1/zh unknown
- 2018-12-13 HK HK18116014.8A patent/HK1256903A1/zh unknown
-
2020
- 2020-02-12 AU AU2020200995A patent/AU2020200995B9/en active Active
- 2020-08-06 US US16/987,081 patent/US20210052502A1/en not_active Abandoned
-
2022
- 2022-05-06 US US17/738,803 patent/US11744802B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2757560T3 (es) | Composiciones terapéuticas para el tratamiento del virus de la inmunodeficiencia humana | |
| ES2945345T3 (es) | Formulaciones farmacéuticas | |
| US20170027967A1 (en) | Pharmaceutical formulations | |
| US20180085387A1 (en) | Therapeutic compositions for treatment of human immunodeficiency virus | |
| US20230285307A1 (en) | Dispersible Tablet Formulations Comprising Dolutegravir | |
| US20230233470A1 (en) | Formulations | |
| OA18732A (en) | Therapeutic compositions for treatment of human immunodeficiency virus. |