ES2740127T3 - Tratamiento de subpoblaciones con enfermedad de Alzheimer con inmunoglobulina G combinada - Google Patents
Tratamiento de subpoblaciones con enfermedad de Alzheimer con inmunoglobulina G combinada Download PDFInfo
- Publication number
- ES2740127T3 ES2740127T3 ES14730660T ES14730660T ES2740127T3 ES 2740127 T3 ES2740127 T3 ES 2740127T3 ES 14730660 T ES14730660 T ES 14730660T ES 14730660 T ES14730660 T ES 14730660T ES 2740127 T3 ES2740127 T3 ES 2740127T3
- Authority
- ES
- Spain
- Prior art keywords
- subject
- weeks
- igg
- alzheimer
- administration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/47—Hydrolases (3) acting on glycosyl compounds (3.2), e.g. cellulases, lactases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39516—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum from serum, plasma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/06—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies from serum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/06—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies from serum
- C07K16/065—Purification, fragmentation
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6893—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids related to diseases not provided for elsewhere
- G01N33/6896—Neurological disorders, e.g. Alzheimer's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
Abstract
Composición que comprende una cantidad terapéuticamente eficaz de inmunoglobulina G (IgG) humana combinada para su uso en el tratamiento de la enfermedad de Alzheimer en un sujeto con enfermedad de Alzheimer moderadamente grave que es portador de al menos un alelo APOE4, en la que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, en la que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico, y en la que el sujeto tiene una puntuación del miniexamen de estado mental (MMSE) de desde 14 hasta 22.
Description
DESCRIPCIÓN
Tratamiento de subpoblaciones con enfermedad de Alzheimer con inmunoglobulina G combinada
Antecedentes de la invención
La enfermedad de Alzheimer (EA) es un trastorno neurodegenerativo progresivo y la principal causa de demencia en los ancianos. El aumento de la longevidad en el siglo pasado ha contribuido a un aumento exponencial de EA. Se estima que más de 5 millones de personas en los Estados Unidos (EE.UU.) actualmente padecen EA. Se pronostica que la prevalencia de la EA se triplicará en 2050 (Herbert et al., Alzheimer Dis. Asoc. Disord., 15: 169-173 (2001)). Los costes anuales del tratamiento de EA en la sociedad estadounidense se aproximan a los 100 mil millones de $ y los gastos futuros proyectados amenazan con superar el presupuesto para atención sanitaria a menos que se encuentren medios más eficaces para tratar y prevenir la EA. Actualmente, cuatro inhibidores de la acetilcolinesterasa y un antagonista del receptor del ácido N-metil-D-aspártico (NMDA) se han aprobado como tratamientos para la EA sintomática en los Estados Unidos. Estos medicamentos pueden mejorar de manera transitoria las capacidades cognitivas y ralentizar el deterioro cognitivo en pacientes con EA. Sin embargo, la mayoría de los pacientes que reciben estos agentes se deterioran por debajo de su nivel inicial antes del tratamiento en el plazo de seis a doce meses con respecto al nivel inicial de la terapia. Hay una escasez de pruebas que sugieran que estos medicamentos cambian la evolución de la neuropatología subyacente a la enfermedad de Alzheimer.
La enfermedad de Alzheimer es cada vez más prevalente en los países desarrollados, donde se ha producido un aumento de la población de personas mayores debido en parte a la mejora de la atención sanitaria. Aunque menos del 1% de la población menor de 60 años está afectada por el Alzheimer, se estima que del 25% al 33% de las personas desarrollan alguna forma de Alzheimer a la edad de 85 años. A partir de 2012, a 5,4 millones de estadounidenses se les diagnosticó Alzheimer. A medida que la esperanza de vida sigue aumentando en todo el mundo, la prevalencia de la enfermedad de Alzheimer y otras demencias relacionadas con la edad también debería seguir creciendo.
La enfermedad de Alzheimer se clasifica normalmente como de “comienzo temprano”, haciendo referencia a los casos que comienzan a manifestarse entre los 30 y 60 años de edad en los individuos afectados, y el Alzheimer más común de “comienzo tardío”, en el que los síntomas aparecen por primera vez después de la edad de 60 años. Aunque solo aproximadamente el 10% de todos los casos de Alzheimer son familiares, la enfermedad de Alzheimer de comienzo temprano se ha vinculado con mutaciones en la proteína precursora de amiloide (app), presenilina 1 (psenl) y presenilina 2 (psen2), mientras que la enfermedad de Alzheimer de comienzo tardío se ha vinculado con mutaciones en el gen de la apolipoproteína E (apoE) (Ertekin-Taner N., Neurol Clin., 25: 611-667 (2007)).
A nivel histopatológico, esta enfermedad neurodegenerativa se caracteriza por la formación de placas de amiloide, ovillos neurofibrilares, angiopatía amiloide y degeneración granulovacuolar en la corteza cerebral (Mirra et al., Arch Pathol Lab Med., 117: 132-144 (1993); Perl DP, Neurol Clin., 18: 847-864 (2000)). Las placas de amiloide características, usadas para confirmar la enfermedad de Alzheimer en la autopsia, se forman en gran parte por la deposición de un pequeño péptido beta-amiloide (Ap) derivado de la proteína precursora de amiloide (APP).
Si bien existen muchas dianas potenciales para la farmacoterapia de EA, se cree que las anomalías en la producción, el procesamiento y/o el aclaramiento cerebral del péptido beta-amiloide (Ap) se encuentran entre las etapas más importantes y más tempranas de la patogenia de EA (Selkoe DJ., Ann. Intern. Med. 2004; 140: 627 638). Se sabe que Ap forma espontáneamente agregados solubles denominados oligómeros y fibrillas insolubles que pueden formar depósitos en el cerebro. Estos agregados pueden precipitar una cascada de cambios cerebrales inflamatorios y otros reactivos que eventualmente interfieren en la transmisión sináptica y aceleran la muerte de neuronas en muchas regiones del cerebro, incluidas las redes corticales y subcorticales que ayudan a la cognición y la conducta (Selkoe DJ, citado anteriormente). El tratamiento temprano de las anomalías relacionadas con Ap podría anticiparse a elementos posteriores de la cascada patógena de EA y alterar de ese modo la evolución de la enfermedad.
Hasta la fecha, la Administración de Alimentos y Fármacos de los Estados Unidos (FDA) ha aprobado dos tipos de medicamentos para el tratamiento de la enfermedad de Alzheimer: inhibidores de la colinesterasa, incluidos Aricept® (donepezilo), Exelon (rivastigmina), Razadyne (galantamina) y Cognex (tacrina); y el inhibidor del receptor de glutamato de tipo NMDA, memantina (comercializado con varias marcas diferentes). Aunque no se ha identificado una cura para la enfermedad de Alzheimer, estas terapias sirven para aliviar síntomas cognitivos tales como pérdida de memoria, confusión y pérdida de la capacidad de pensamiento crítico en sujetos a los que se les ha diagnosticado demencia relacionada con la edad (por ejemplo, enfermedad de Alzheimer). En total, se estima que el gasto de atención sanitaria en la enfermedad de Alzheimer y las demencias relacionadas con la edad en 2012 será de 200 mil millones de $ solo en los Estados Unidos (Factsheet, Alzheimer's Association, marzo de 2012).
Además de estas terapias aprobadas, varios estudios han sugerido que la inmunoglobulina intravenosa combinada (IgIV) es eficaz en la ralentización de la progresión de los síntomas en pacientes con Alzheimer (Dodel RC et al, J
Neurol Neurosurg Psychiatry, octubre; 75(10): 1472-4 (2004); Magga J. et al., J Neuroinflammation, 7 de diciembre; 7:90 (1997); Relkin NR et al., Neurobiol Aging, 30(11): 1728-36 (2008); Puli L. et al., J. Neuroinflammation 29 de mayo; 9:105 (2012)).
El documento US 2011/251479 se refiere al uso de la monitorización por IRM de la tasa de hipertrofia ventricular como una medida objetiva con el propósito de evaluar la progresión de la enfermedad en pacientes que padecen enfermedad de Alzheimer y para determinar la eficacia terapéutica de un régimen de tratamiento para pacientes con Alzheimer.
Una revisión reciente de bapineuzumab describe un estudio de fase II con un total de 234 pacientes con diagnóstico clínico de EA probable, de 50 a 85 años con puntuaciones de MMSE de 16 a 26. Los pacientes entraron en tres cohortes de dosificación: 0,5 mg/kg, 1,0 mg/kg y 2,0 mg/kg. No surgió ningún efecto significativo de bapineuzumab en pacientes que recibieron al menos una infusión seguida de al menos una evaluación. Aunque se observaron efectos clínicos leves, estos no fueron en portadores de ApoE4. 12 de 124 pacientes tratados con bapineuzumab desarrollaron edema cerebral vasogénico, que mostró un riesgo aumentado con el aumento de las copias del gen ApoE4. (Kerchner GA, Boxer AL., Expert Opin Biol Ther. julio de 2010; 10(7):1121-30). Otra revisión que describe los efectos secundarios del bapineuzumab relacionados con el edema cerebral vasogénico, y propone que el tratamiento futuro debe incluir menores dosis, particularmente en portadores de ApoE4. (Cribbs DH., CNS Neurol Disord Drug Targets, 9(2):207-16 (2010)).
Otro estudio describe un análisis de seguridad y farmacológico de seguimiento de ponezumab después de una única infusión i.v. de 10 minutos en sujetos con EA de leve a moderada. El estudio fue un estudio de seguridad, tolerancia y farmacología de fase I, de dosis única, de aumento gradual de dosis, abierto. Los sujetos recibieron 1 mg/kg, 3 mg/kg, 5 mg/kg o 10 mg/kg (n = 3, 3, 4 y 5, respectivamente). Los sujetos tenían más de 50 años de edad y tenían puntuaciones de MMSE de 16 a 26. La función cognitiva no mostró tendencias relacionadas con el tratamiento (Burstein AH, Zhao Q, Ross J, Styren S, Landen JW, Ma WW, McCush F, Alvey C, Kupiec JW, Bednar MM., Clin Neuropharmacol., 36(1):8-13 (2013))).
El propósito del estudio descrito en Spearling. et al. fue determinar la correlación de bapineuzumab con edema vasogénico y derrames en surcos (ARIA-E) y microhemorragias y depósitos de hemosiderina (ARIA-H). Se revisaron las exploraciones mediante IRM de 262 participantes en dos estudios de fase II. El 17% de los pacientes desarrollaron ARIA-E. La aparición de ARIA-E aumentó con la dosis de bapineuzumab y la presencia de alelos ApoE4. (Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M., Lancet Neurol., 11(3):241-9 (2012)).
Farlow et al. describen un estudio que evaluó la seguridad y la tolerancia de 12 infusiones semanales de solanezumab en pacientes con EA de leve a moderada. Este fue un ensayo de fase 2, aleatorizado, doble ciego, controlado con placebo, con 52 pacientes con EA. Las cohortes fueron de 100 mg cada 4 semanas, 100 mg semanales, 400 mg cada 4 semanas o 400 mg semanales durante 12 semanas. Los pacientes tenían más de 50 años de edad con EA probable de leve a moderada con puntuaciones de MMSE de 15-26. Los pacientes revelaron hemorragias por úlceras, pancreatitis, dolor torácico, infección, punción lumbar, cefalea, hematoma subdural, síncope, agitación, quiste ovárico y úlcera en la piel. No hubo diferencias significativas entre el solanezumab y el placebo con una puntuación cognitiva de 11 puntos o de 14 puntos. (Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, DeMattos RB, Mohs R, Paul SM, Siemers ER, Alzheimer's Dement. 8(4):261-71 (2012)).
Véanse también; Landen JW, Zhao Q, Cohen S, Borrie M, Woodward M, Billing CB Jr, Bales K, Alvey C, McCush F, Yang J, Kupiec JW, Bednar MM., Clin Neuropharmacol. enero-febrero de 2013; 36(1):14-23; Uenaka K, Nakano M, Willis BA, Friedrich S, Ferguson-Sells L, Dean RA, Ieiri I, Siemers ER. Clin Neuropharmacol., 35(1):25-9 (2012); Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu Zx, Loetscher H, Santarelli L. Arch Neurol., 69(2):198-207 (2012); Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, Demattos RB. Clin Neuropharmacol., 33(2):67-7 (2012); Imbimbo BP, Ottonello S, Frisardi V, Solfrizzi V, Greco A, Seripa D, Pilotto A, Panza F., Expert Rev Clin Immunol., 8(2):135-49 (2012); Carlson C, Estergard W, Oh J, Suhy J, Jack CR Jr, Siemers E, Barakos J., Alzheimer's Dement., 7(4):396-401 (2011); Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodríguez Martínez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M, Lancet Neurol., 9(4):363-72 (2010). Blennow K, Zetterberg H, Rinne JO, Salloway S, Wei J, Black R, Grundman M, Liu E; AAB-001 201/202 Investigators, Arch Neurol., 69(8):1002-10 (2012).
Diferentes isoformas de ApoE pueden modular los niveles de Ap. Tai et al. describen un ensayo ELISA que mide el complejo apoE/Ap soluble para considerar la hipótesis de que niveles reducidos del complejo apoE/Ap soluble se correlaciona con un aumento de Ap oligomérico soluble. En preparaciones de sinaptosomas corticales humanos y de ratones, los niveles de apoE/Ap fueron menores en un modelo de ratón ApoE4 que en modelos de ratón ApoE2 y ApoE3, sugiriendo que las isoformas de apoE modulan específicamente el aclaramiento de Ap oligomérico (ApoE2
es protector). En los sinaptosomas corticales humanos, los niveles de complejo apoE/Ap eran menores en pacientes con EA, y menores en la cohorte que tiene la isoforma ApoE4 dentro del grupo de EA. Además, los niveles de Ap oligomérico aumentaron y fueron mayores en la cohorte de isoforma de ApoE4. (Tai et al., J.B.C., 288:5914-5926 (2013)).
Kim et al. sometieron a prueba la hipótesis de que los anticuerpos anti-apoI pueden tener efectos antiamiloidogénicos al unirse a apoE en placas de Ap y activar el aclaramiento de amiloide mediado por la microglía. Se generaron varios anticuerpos anti-apoE. El artículo demuestra que la inmunización pasiva frente a apoE puede atenuar la acumulación de Ap. Los anticuerpos anti-apoE aumentaron la microglía positiva para CD45 y disminuyeron las citocinas proinflamatorias. Los autores plantearon la hipótesis de que los anticuerpos anti-apoE pueden reclutar la microglía a placas que contienen apoE, lo que desencadena la fagocitosis directa y la atenuación de citocinas proinflamatorias, lo que conduce a una reducción de la acumulación de amiloide. (Kim et al. J. Exp Med 209:2149-2156 (2012)).
Por consiguiente, existe la necesidad en la técnica de métodos para tratar la enfermedad de Alzheimer en individuos que lo necesitan, que se han clasificado como positivos para ApoE4 y/o que tienen demencia moderada. También existe la necesidad en la técnica de métodos para asignar un tratamiento eficaz para la enfermedad de Alzheimer a individuos que lo necesitan, basándose en la subpoblación de la enfermedad de Alzheimer en la que se clasifica el individuo.
Breve sumario de la invención
La presente divulgación proporciona soluciones a estos y otros problemas al proporcionar métodos para el tratamiento de la enfermedad de Alzheimer en pacientes con enfermedad de Alzheimer moderada y/o que portan un alelo ApoE4 mediante la administración de inmunoglobulina G combinada. De manera ventajosa, se muestra en el presente documento que la administración de inmunoglobulina G combinada a dosis alta (por ejemplo, 400 mg/kg/2 semanas (IgIV) ralentiza la progresión de la demencia en sujetos con Alzheimer con enfermedad moderada y en sujetos con Alzheimer que portan un alelo ApoE4.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave, en el que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En otro aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer que es portador de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas Después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En otro aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave que es portador de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita para el tratamiento que comprende la administración de desde 300 mg/kg hasta 800 mg/kg de cuerpo. peso del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer en un sujeto que lo necesita que es portador de al menos un alelo APOE4 para el tratamiento que comprende la administración desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o
más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita, que es portador de al menos un alelo APOE4 para el tratamiento que comprende la administración de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 250 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 300 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 350 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 400 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 450 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 500 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 550 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 600 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 650 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 700 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 750 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada es de 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave, en el que la cantidad de IgG humana combinada es de desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave, en el que la cantidad de IgG humana combinada es de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer que es portador de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde
50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer que es portador de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave que es portadora de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar directamente al SNC una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave que es portadora de al menos un alelo APOE4, en el que la cantidad de IgG humana combinada es de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita para el tratamiento que comprende la administración directamente al SNC de desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita para el tratamiento que comprende la administración directamente al SNC de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer en un sujeto que lo necesita que es portador de al menos un alelo APOE4 para la administración directamente al SNC de desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer en un sujeto que lo necesita que es portador de al menos un alelo APOE4 para la administración directamente al SNC de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de la enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita y que es portador de al menos un alelo APOE4 para la administración del tratamiento directamente al SNC de desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En un aspecto, la divulgación proporciona un uso de una composición que comprende inmunoglobulina G (IgG) humana combinada para el tratamiento de enfermedad de Alzheimer moderadamente grave en un sujeto que lo necesita y que es portador de al menos un alelo APOE4 para la administración del tratamiento directamente al SNC de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico.
En una realización de los métodos y usos descritos anteriormente, el método comprende además la etapa de diagnosticar al sujeto con enfermedad de Alzheimer moderadamente grave antes de iniciar el régimen terapéutico. En una realización de los métodos y usos descritos anteriormente, el estado de APOE4 del sujeto se determina antes de comenzar el régimen terapéutico.
En una realización de los métodos y usos descritos anteriormente, el sujeto es homocigoto para el alelo APOE4.
En una realización de los métodos y usos descritos anteriormente, el sujeto es heterocigoto para el alelo APOE4. En una realización de los métodos y usos descritos anteriormente, el sujeto tiene un genotipo APOE4/APOE3.
En una realización de los métodos y usos descritos anteriormente, el sujeto tiene un genotipo APOE4/APOE2.
En una realización de los métodos y usos descritos anteriormente, el sujeto no tiene un alelo APOE4.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada se administra en una dosis única a lo largo del periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la cantidad de IgG humana combinada se administra en dosis múltiples a lo largo del periodo de dos semanas.
En una realización de los métodos y usos descritos anteriormente, la una o más dosis administradas durante el periodo de dos semanas son iguales.
En una realización de los métodos y usos descritos anteriormente, la una o más dosis administradas durante el periodo de dos semanas son variables.
En una realización de los métodos y usos descritos anteriormente, la dosis se administra al menos dos veces al día.
En una realización de los métodos y usos descritos anteriormente, la dosis se administra cada día.
En una realización de los métodos y usos descritos anteriormente, la dosis se administra cada dos días.
En una realización de los métodos y usos descritos anteriormente, la dosis se administra dos veces a la semana En una realización de los métodos y usos descritos anteriormente, la dosis se administra cada semana.
En una realización de los métodos y usos descritos anteriormente, la cantidad se administra mediante administración intravenosa, administración subcutánea, administración intramuscular o administración intraperitoneal.
En una realización de los métodos y usos descritos anteriormente, la cantidad se administra mediante administración intranasal, administración intratecal, administración intracerebral, administración intracerebroventricular, administración epidural o administración espinal.
En una realización de los métodos y usos descritos anteriormente, el método comprende además administrar una cantidad terapéuticamente eficaz de una segunda composición para el tratamiento de la enfermedad de Alzheimer.
En un aspecto, la divulgación proporciona un método para seleccionar un régimen de tratamiento para un sujeto con la enfermedad de Alzheimer, comprendiendo el método las etapas de: (a) diagnosticar la gravedad de la enfermedad de Alzheimer como levemente grave, moderadamente grave o grave; (b) determinar si el sujeto porta el alelo APOE4; y (c) asignar un régimen de tratamiento que comprende la administración de inmunoglobulina humana (IgG) combinada si el sujeto tiene enfermedad de Alzheimer moderadamente grave y es portador de un alelo APOE4 o asignar un régimen de tratamiento que comprende la administración de un anticuerpo monoclonal anti-beta amiloide si el sujeto tiene la enfermedad de Alzheimer levemente grave y no es portador de un alelo APOE4.
En una realización de los métodos descritos anteriormente, el sujeto es homocigoto para el alelo APOE4.
En una realización de los métodos descritos anteriormente, el sujeto es heterocigoto para el alelo APOE4.
En una realización de los métodos descritos anteriormente, el sujeto tiene un genotipo APOE4/APOE3.
En una realización de los métodos descritos anteriormente, el sujeto tiene un genotipo APOE4/APOE2.
En una realización, la divulgación proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita, comprendiendo el método: administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con Alzheimer de moderado a moderadamente grave, en el que los pacientes con Alzheimer de moderado a moderadamente grave tienen una puntuación del miniexamen de estado mental (MMSE) de desde 14 hasta 20, de 14 a 21, de 14 a 22, de 14 a 23; de 15 a 20, de 15 a 21, de 15 a 22, de 15 a 23; de 16 a 20, de 16 a 21, de 16 a 22, o de 16 a 23, inclusive, en el que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos
semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En otras realizaciones, el sujeto es portador de al menos un alelo APOE4 y tiene una puntuación del miniexamen de estado mental (MMSE) de desde 14 hasta 20, de 14 a 21, de 14 a 22, de 14 a 23; de 15 a 20, de 15 a 21, de 15 a 22, de 15 a 23; de 16 a 20, de 16 a 21, de 16 a 22 o de 16 a 23, inclusive.
Breve descripción de los dibujos.
La figura 1 ilustra las curvas de concentración-respuesta para ELISA de anticuerpo anti-ApoE4 realizadas usando ApoE4 recombinante inmovilizado en placa para medir la unión a anticuerpo anti-ApoE4 en plasma humano combinado (1R01B00) e IgG purificada a partir de plasma sanguíneo humano combinado (LE12K246). Las lecturas de ELISA se corrigieron con blanco restando la unión de albúmina sérica humana a pocillos recubiertos de las intensidades medidas de la combinación de plasma y la unión a inmunoglobulina G.
La figura 2 es un diagrama esquemático que representa el flujo del estudio descrito en el ejemplo 2.
Las figuras 3A-3E ilustran un programa de evaluaciones realizadas durante el periodo de tiempo entre el examen de los sujetos y el mes 9 del ensayo.
La figura 4 ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 5 ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto en sujetos portadores de APOE-e4. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 6 ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto en sujetos no portadores de APOE-e4. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 7 ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto en sujetos con enfermedad de Alzheimer leve. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 8A ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto en sujetos con enfermedad de Alzheimer moderada. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 8B ilustra los cambios medios por mínimos cuadrados de la puntuación total del miniexamen de estado mental modificado con respecto al nivel inicial y los IC de 95 estimados dentro del primer modelo mixto en sujetos con enfermedad de Alzheimer moderada (MMSE < 20) que son portadores de un alelo ApoE4. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 200 mg/kg y placebo a los 18 meses.
La figura 8C ilustra los cambios medios de la puntuación total de ADAS-Cog con respecto al nivel inicial y los IC de 95 en sujetos con enfermedad de Alzheimer moderada. p1 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%), 400 mg/kg y placebo a los 18 meses. p2 = valor de p correspondiente a la comparación entre IgIV medias por mínimos cuadrados de grupos de tratamiento (10%),
200 mg/kg y placebo a los 18 meses.
La figura 9 proporciona el cambio con respecto a los datos estadísticos de nivel inicial para los análisis de ADAS-Cog, ADCS-ADL y 3MS.
La figura 10 muestra imágenes del cerebro de tomografía por emisión de positrones (PET) con [18F]-2-fluorodesoxiglucosa (18F-FDG) de sujetos tratados con placebo (10A) e IgIV a dosis alta (10B).
La figura 11 proporciona un resumen del estado de biomarcadores de obtención de imágenes para todas las cohortes y subpoblaciones de pacientes.
La figura 12 muestra imágenes de atrofia ventricular típica en los cerebros de pacientes con Alzheimer tratados con placebo (12A) e IgIV a dosis alta (12B).
La figura 13 proporciona un resumen del cambio medio con respecto al nivel inicial de los niveles de péptidos Ap42 y Ap40 en plasma para todas las cohortes y subpoblaciones de pacientes.
La figura 14 proporciona un resumen del cambio medio con respecto al nivel inicial de los niveles de anticuerpos anti-Ap42 y anti-Ap40 en plasma para todas las cohortes y subpoblaciones de pacientes.
La figura 15 proporciona un resumen de los niveles de péptido Ap42 en plasma para todas las cohortes de tratamiento.
La figura 16 proporciona un resumen de los niveles de péptido Ap40 en plasma para todas las cohortes de tratamiento.
La figura 17 proporciona un resumen del cambio medio con respecto al nivel inicial de los niveles de péptidos Ap42 y Ap40 en el LCR para todas las cohortes y subpoblaciones de pacientes.
La figura 18 proporciona un resumen de los niveles de péptido Ap42 en el LCR para todas las cohortes de tratamiento.
La figura 19 proporciona un resumen de los niveles de IgG totales en el LCR para todas las cohortes de tratamiento. La figura 20 proporciona un resumen de los niveles de anticuerpos anti-fibrillas de Ap en el LCR para todas las cohortes de tratamiento.
La figura 21 proporciona un resumen de los niveles de anticuerpos anti-oligómeros de Ap en el LCR para todas las cohortes de tratamiento
La figura 22 proporciona un resumen de los niveles de anticuerpos anti-monómeros de Ap en el LCR para todas las cohortes de tratamiento.
La figura 23 proporciona un resumen del cambio medio con respecto al nivel inicial de los niveles de IgG totales en el LCR para todas las cohortes y subpoblaciones de pacientes.
La figura 24 proporciona un resumen del cambio medio con respecto al nivel inicial de niveles de anticuerpos anti fibrillas de Ap, anti-oligómeros de Ap y anti-monómeros de Ap en el LCR para todas las cohortes y subpoblaciones de pacientes.
La figura 25 proporciona un resumen de los niveles de proteína Tau en el LCR para todas las cohortes de tratamiento.
La figura 26 proporciona un resumen del cambio medio con respecto al nivel inicial de los niveles de Tau y Tau fosforilada en el LCR para todas las cohortes y subpoblaciones de pacientes.
La figura 27 proporciona un resumen de los niveles de proteína Tau fosforilada en el LCR para todas las cohortes de tratamiento.
La figura 28 proporciona un resumen del número de sujetos con una disminución del nivel de hemoglobina después del tratamiento para todas las cohortes de tratamiento.
La figura 29 proporciona un resumen del número de efectos secundarios graves para todas las cohortes de IgIV y placebo.
La figura 30 proporciona un resumen del número de efectos secundarios no graves para todas las cohortes de IglV y placebo.
La figura 31 proporciona un resumen de los efectos secundarios adversos para todas las cohortes de tratamiento. La figura 32 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para la evaluación de ADAS-Cog.
La figura 33 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para la evaluación de 3MS.
La figura 34 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para la evaluación de CGIC.
La figura 35 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para el dibujo de reloj.
La figura 36 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para la evaluación de Trail B.
La figura 37 ilustra la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial del volumen ventricular para los sujetos que reciben IgIV a dosis alta, según lo determinado mediante IRM volumétrica.
La figura 38 proporciona un resumen del análisis de correlación de resultados realizado mediante el análisis de biomarcadores de obtención de imágenes y el análisis de criterio de valoración primario.
La figura 39 proporciona un resumen del genotipo ApoE4 y la distribución de alelos para el estudio de tratamiento con IgIV.
La figura 40A ilustra las puntuaciones medias del examen ADAS-Cog y los IC de 95 estimados dentro del primer modelo mixto en sujetos con enfermedad de Alzheimer moderada (MMSE < 22) o leve (MMSE > 23) a los 0 meses (nivel inicial), 9 meses y 18 meses de tratamiento con 400 mg/kg/2 semanas, 200 mg/kg/2 semanas y placebo. La figura 40B ilustra las puntuaciones medias del miniexamen de estado mental modificado y los IC de 95 estimados dentro del primer modelo mixto en sujetos con enfermedad de Alzheimer moderada (MMSE < 22) o leve (MMSE > 23) a los 0 meses (nivel inicial), 9 meses y 18 meses de tratamiento con 400 mg/kg/2 semanas, 200 mg/kg/2 semanas y placebo.
La figura 41 proporciona un resumen de los niveles de IgG totales en suero sanguíneo para todas las cohortes de tratamiento.
La figura 42 ilustra el cambio con respecto al nivel inicial en la puntuación de ADAS-Cog, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con puntuaciones de florbetapir iguales a o mayores de 1,2.
La figura 43 ilustra el cambio con respecto al nivel inicial en la puntuación de MMSE modificado, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con puntuaciones de florbetapir iguales o superiores a 1,2.
La figura 44 ilustra el cambio con respecto al nivel inicial en los niveles de polipéptido Ap42 del LCR, como la diferencia media con respecto al placebo, para individuos en dosis altas (0,4 g/kg) y dosis bajas (0,2 g/kg) cohortes de tratamiento de IgIV clasificadas con puntuaciones de florbetapir iguales a o mayores de 1,2.
La figura 45 ilustra el cambio con respecto al nivel inicial en la puntuación de ADAS-Cog, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 46 ilustra el cambio con respecto al nivel inicial en la puntuación de ADAS-Cog, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 47 ilustra el cambio con respecto al nivel inicial en la puntuación de ADAS-CGIC, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 48 ilustra el cambio con respecto al nivel inicial en la puntuación de ADAS-CGIC, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 49 ilustra el cambio con respecto al nivel inicial en la puntuación de MMSE modificado, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 50 ilustra el cambio con respecto al nivel inicial en la puntuación de MMSE modificado, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 51 ilustra el cambio con respecto al nivel inicial en IRM volumétrica, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 52 ilustra el cambio con respecto al nivel inicial en IRM volumétrica, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas de amiloide.
La figura 53 ilustra el cambio con respecto al nivel inicial en SUVR compuesta normalizada, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 54 ilustra el cambio con respecto al nivel inicial en SUVR compuesta normalizada, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas de amiloide.
La figura 55 ilustra el cambio con respecto al nivel inicial en los niveles de polipéptido Ap40 en plasma, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas sin placas amiloides.
La figura 56 ilustra el cambio desde la línea base en los niveles de polipéptido Ap40 en plasma, como la diferencia media con respecto al placebo, para los individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 57 ilustra el cambio con respecto al nivel inicial en los niveles de polipéptido Ap42 en plasma, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 58 ilustra el cambio con respecto al nivel inicial en los niveles de polipéptido Ap42 en LCR, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas amiloides.
La figura 59 ilustra el cambio con respecto al nivel inicial en los niveles de IgG total en LCR, como la diferencia media con respecto al placebo, para individuos en las cohortes de tratamiento con IgIV a dosis alta (0,4 g/kg) y dosis baja (0,2 g/kg) clasificadas con placas de amiloide.
Descripción detallada de la invención
Introducción
La presente divulgación describe los resultados de un estudio realizado para evaluar el nuevo uso de la inmunoglobulina G combinada, que está aprobada en los Estados Unidos para tratar diversos trastornos de inmunodeficiencia y autoinmunitarios. La IgIV es un agente biológico con propiedades antiinflamatorias e inmunomoduladoras que contiene anticuerpos de inmunoglobulina G (IgG) humana derivados del plasma sanguíneo de donantes sanos. Específicamente, la IgIV contiene anticuerpos que se unen al beta amiloide fibrilar y oligomérico, lo que respalda el uso de la IgIV como agente para la inmunoterapia pasiva para EA. La inmunización pasiva no requiere que los receptores produzcan anticuerpos por sí mismos y puede solventar de ese modo el problema de la generación inadecuada de anticuerpos por parte de personas mayores. A diferencia de la vacunación activa, la activación de células T no es necesaria para obtener todos los beneficios terapéuticos de la inmunización pasiva; por
tanto, esto puede reducir pero no eliminar por completo la posibilidad de reacciones inflamatorias reactivas. Por tanto, la inmunización pasiva podría proporcionar una alternativa segura y eficaz a la vacunación activa para el tratamiento de pacientes ancianos con EA.
Ventajosamente, se muestra en el presente documento que la administración de inmunoglobulina G combinada a determinadas subpoblaciones de pacientes con Alzheimer (por ejemplo, pacientes positivos para ApoE4 y/o con enfermedad de Alzheimer moderadamente grave) da como resultado una reducción significativa de la progresión de los síntomas de demencia.
Por ejemplo, tal como se muestra en la figura 5, la administración de 400 mg de IgG por kg de peso corporal del individuo cada dos semanas (mg/kg/2 semanas) a pacientes con Alzheimer que portan al menos un alelo ApoE4 dio como resultado una reducción estadísticamente significativa de la progresión de demencia, en comparación con pacientes que recibieron placebo (p1 = 0,012). Sin embargo, la administración sólo de 200 mg/kg/2 semanas de IgG a pacientes con Alzheimer que portaban al menos un alelo ApoE4 no dio como resultado una reducción estadísticamente significativa de la progresión de demencia, en comparación con pacientes que recibieron placebo (p1 = 0,793).
Asimismo, tal como se muestra en la figura 8, la administración de 400 mg de IgG por kg de peso corporal del individuo cada dos semanas (mg/kg/2 semanas) a pacientes con Alzheimer que tienen una enfermedad moderada (definida como tener una puntuación inicial de MMSE de entre 16 y 20) dio como resultado una clara reducción de la progresión de demencia, en comparación con pacientes que recibieron placebo, tal como se evaluó mediante evaluación cognitiva mediante 3MS (p1 = 0,067; figura 8A) y ADAS-Cog (p1 = 0,083; figura 8C). Sin embargo, la administración sólo de 200 mg/kg/2 semanas de IgG a pacientes con Alzheimer que portaban al menos un alelo ApoE4 no dio como resultado una clara reducción de la progresión de demencia, en comparación con pacientes que recibieron placebo, tal como se evaluó mediante evaluación cognitiva mediante 3MS (p2 = 0,567; figura 8A) y ADAS-Cog (p2 = 0,697; figura 8C).
Los resultados positivos observados para el tratamiento con IgIV de pacientes con enfermedad de Alzheimer moderada se vuelven más pronunciados cuando los sujetos que tienen una puntuación inicial de MMSE de 22 y/o 21 se incluyen en la cohorte de enfermedad de Alzheimer moderada. Tal como se notifica en la tabla 1, la administración de 400 mg/kg/2 semanas de IgIV redujo significativamente la progresión de demencia en pacientes a los que se les ha diagnosticado enfermedad de Alzheimer moderada, tal como se evaluó mediante evaluación cognitiva mediante ADAS-Cog (p = 0,046, MMSE < 21; p = 0,006, MMSE < 22) y 3MS (p = 0,09, MMSE < 21; p = 0,029, MMSE < 22).
Además, el análisis de los datos revela que al excluir a los sujetos a los que se les diagnosticó inicialmente con una puntuación de MMSE superior a 22 que son negativos para ApoE4, el tratamiento con IgIV a dosis alta redujo significativamente la progresión de demencia en la subpoblación. Tal como se notificó en la tabla 2 y la tabla 3, pacientes positivos para ApoE4 y/o con enfermedad de Alzheimer moderada se beneficiaron significativamente de la administración de 400 mg/kg/2 semanas de IgIV, tal como se evaluó mediante evaluación cognitiva mediante ADAS-Cog (p = 0,026 frente a placebo) y 3MS (p = 0,032 frente a placebo).
Los resultados descritos anteriormente, que tomados en conjunto sugieren que la administración de IgG a dosis alta (por ejemplo, 300 mg/kg/2 semanas de IgIV o más) a subpoblaciones de pacientes con Alzheimer que portan un alelo ApoE4 y/o que tienen enfermedad moderada, es eficaz en la ralentización de la progresión de los síntomas de demencia.
Estos datos son particularmente sorprendentes a la luz de los resultados notificados para la terapia monoclonal anti-P-amiloide y anti-ApoE4. Estos estudios, resumidos anteriormente, notifican que la terapia monoclonal es más eficaz en pacientes no portadores de ApoE4 y en aquellos con enfermedad de Alzheimer levemente grave. Además, varios estudios han notificado resultados negativos (por ejemplo, edema vasogénico y derrames en surcos) en portadores de ApoE4.
Por ejemplo, en la reunión de octubre de 2012 de la American Neurological Association (ANA), se notificó que el tratamiento con bapineuzumab (anticuerpo monoclonal anti-p-amiloide) a lo largo de 78 semanas no daba como resultado ningún cambio en la puntuación de ADAS-Cog o evaluación de discapacidad en demencia (DAD) en sujetos con ApoE4, y no proporcionó ningún efecto significativo sobre la tasa de cambio en el volumen cerebral por IRM (BBSI) en sujetos con enfermedad de Alzheimer moderada. En la misma reunión de ANA de octubre de 2012, se notificó que el tratamiento con solanezumab (anticuerpo monoclonal anti-p-amiloide) no proporcionó una reducción significativa de la puntuación ADAS-Cog para los sujetos con enfermedad de Alzheimer moderada.
Se ha propuesto que la inmunoglobulina G combinada (por ejemplo, IgIV) contiene anticuerpos naturales contra pamiloide. Relkin et al. 2009 (Neurobiol. Aging 30(11): 1728-36). En este estudio, la IgG humana combinada se administró por vía intravenosa (terapia con IgIV) a ocho sujetos a los que se les ha diagnosticado enfermedad de Alzheimer (EA) leve. Los pacientes recibieron terapia con IgIV durante 6 meses, se suspendió el tratamiento y luego
reanudaron el tratamiento durante 9 meses más. Se encontró que los anticuerpos contra p-amiloide en el suero de pacientes con EA aumentaron en proporción a la dosis de IgIV y los niveles en plasma de p-amiloide aumentaron transitoriamente después de cada infusión. Además, se muestra en el presente documento que una preparación comercial de inmunoglobulina G combinada también contiene anticuerpos naturales contra la proteína ApoE4. Sin pretender restringirse a ninguna teoría, la combinación de anticuerpos naturales contra diversas proteínas relacionadas con el Alzheimer que se encuentran en la inmunoglobulina G preparada a partir de plasma combinado puede contribuir a mejorar la eficacia de la IgG combinada en comparación con la terapia con anticuerpos monoclonales anti-p-amiloide y anti-ApoE en algunas cohortes.
Definiciones
Tal como se usa en el presente documento, los términos “inmunoglobulina G humana combinada” e “IgG humana combinada” se refieren a una composición que contiene inmunoglobulina G (IgG) polivalente purificada de la sangre/plasma de múltiples donantes, por ejemplo, más de cien o más de mil donantes de sangre. Normalmente, la composición será al menos el 80% de IgG (p/p, por ejemplo, peso de IgG por peso de proteína total), preferiblemente al menos el 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% o el 99% de IgG (p/p). En determinadas realizaciones, la composición de IgG humana combinada contiene inmunoglobulinas IgG intactas. En otras realizaciones, la composición combinada de IgG humana contiene fragmentos de IgG, por ejemplo las preparadas mediante el tratamiento de anticuerpos intactos con tripsina. En determinadas realizaciones, las composiciones de IgG humana combinada usadas en los tratamientos divulgados en el presente documento contienen modificaciones naturales o sintéticas, por ejemplo, modificaciones postraduccionales y/o modificaciones químicas. En una realización, la composición de inmunoglobulina G humana combinada se formula para administración intravenosa (por ejemplo, una preparación de IgIV).
Tal como se usa en el presente documento, los términos “positivo para ApoE4” y “portador de ApoE4” se usan indistintamente y se refieren a un sujeto o una población de pacientes que tiene al menos un alelo polimórfico ApoE4. Tal como se usa en el presente documento, un alelo ApoE4 se refiere a un alelo de apoE (por ejemplo, gen que codifica la secuencia de referencia de NCBI: NM_000041.2) que codifica una proteína que tiene residuos de arginina en las posiciones 112 y 158 de la proteína ApoE madura y las posiciones 130 y 176 del polipéptido precursor de ApoE (secuencia de referencia de NCBI: NP_000032.1).
Tal como se usa en el presente documento, los términos “enfermedad de Alzheimer moderada” y “enfermedad de Alzheimer moderadamente grave” se usan indistintamente y se refieren al estado de la enfermedad de Alzheimer en un sujeto o una población de pacientes con una puntuación en el miniexamen de estado mental (MMSE) de 14 a 20, inclusive. Las subpoblaciones preferidas de pacientes con Alzheimer moderado y/o moderadamente grave tienen una puntuación en el miniexamen de estado mental (MMSE) de desde 14 hasta 20, de 14 a 21, de 14 a 22, de 14 a 23; de 15 a 20, de 15 a 21, de 15 a 22, de 15 a 23; de 16 a 20, de 16 a 21, de 16 a 22 o de 16 a 23, inclusive.
En el contexto de la presente divulgación, el MMSE se emplea como una prueba a modo de ejemplo que puede usarse para identificar a un individuo que tiene enfermedad de Alzheimer moderada o moderadamente grave que probablemente responda favorablemente al tratamiento con inmunoglobulina G humana combinada. El experto en la técnica reconocerá que puede usarse una prueba distinta de MMSE para clasificar a un sujeto con enfermedad de Alzheimer moderada como candidato para el tratamiento con inmunoglobulina G humana combinada (por ejemplo, tratamiento con IgIV), en relación con los métodos descritos en el presente documento. Por ejemplo, un sujeto o una población de pacientes también se considera que tiene enfermedad de Alzheimer moderada o moderadamente grave si se ha evaluado mediante una prueba diferente (por ejemplo, a través de la miniprueba del estado mental modificada (3MS), la subescala cognitiva de la evaluación de la escala de evaluación de la enfermedad de Alzheimer (ADAS-Cog), la evaluación de ADCS-impresión clínica global de cambio (ADCS-CGIC), u otra evaluación conocida de la enfermedad de Alzheimer) que tienen una puntuación equivalente a una puntuación de MMSE correspondiente a enfermedad de Alzheimer moderada. El experto en la técnica entenderá cómo corresponder los resultados de una prueba alternativa para caracterizar a un sujeto como que tiene enfermedad de Alzheimer moderada, tal como se definió anteriormente usando la evaluación cognitiva de MMSE.
Tal como se usa en el presente documento, el término “periodo de dos semanas” se refiere a un intervalo de aproximadamente 10 a aproximadamente 18 días dentro de un ciclo de dosificación de IgG humana combinada. En una realización, un periodo de dos semanas se refiere a un intervalo de dosificación de 14 días. En otra realización, un periodo de dos semanas se refiere a un intervalo de dosificación de dos veces al mes. En otra realización, un periodo de dos semanas se refiere a un intervalo de dosificación de aproximadamente 26 veces al año. En algunas realizaciones, un periodo de dos semanas se refiere a un intervalo de dosificación de 10 días, 11 días, 12 días, 13 días, 14 días, 15 días, 16 días, 17 días, 18 días, 24 veces al año, 25 veces al año, 26 veces al año, 27 veces al año o 28 veces al año. Según lo entendido por un experto en la técnica, el periodo de dos semanas incluye límites razonables basados en el cumplimiento del paciente.
En el contexto de la presente divulgación, una dosificación de IgG humana combinada administrada por periodo de dos semanas se refiere a la cantidad total de IgG humana combinada administrada durante el periodo de dos semanas, ya se administre en una dosis única o en dosis múltiples durante el periodo de dos semanas. En una
realización, la dosificación completa se administra en una dosis única una vez durante el periodo de dos semanas. En otra realización, la dosificación se administra en dos o más dosis más pequeñas durante el periodo de dos semanas. Por ejemplo, una dosis de 400 mg/kg/2 semanas engloba una dosis única de 400 mg/kg administrada una vez durante el periodo de dos semanas, una dosificación de 200 mg/kg administrada dos veces durante el periodo de dos semanas, una dosificación de 100 mg/kg administrada cuatro veces durante el periodo de dos semanas, y otros regímenes de dosificación en los que se administran dosis múltiples que hacen un total de 400 mg/kg durante el periodo de dos semanas.
En algunas realizaciones, la cantidad de IgG humana combinada administrada por periodo de dos semanas se refiere a un promedio de IgG humana combinada administrada por periodo de dos semanas a lo largo de la duración del tratamiento. De esta manera, en algunas realizaciones, la dosis de IgG humana combinada administrada en periodos consecutivos de dos semanas varía. Por ejemplo, en una realización, se dice que un sujeto al que se administran alternativamente dosis de IgG humana combinada de 200 mg/kg/2 semanas y 600 mg/kg/2 semanas ha recibido un 400 mg/kg/periodo de 2 semanas de IgG humana combinada. En algunas realizaciones, al sujeto se le administra una dosificación repetida durante un periodo de más de 2 semanas, que promedia una dosis convencional por periodo de dos semanas. En una realización, al sujeto se le administra una dosis distribuida a lo largo de un periodo de tres semanas. En otra realización, al sujeto se le administra una dosificación distribuida a lo largo de un periodo de un mes. En otras realizaciones, al sujeto se le administra una dosificación distribuida a lo largo de 10 días, 11 días, 12 días, 13 días, 14 días, 15 días, 16 días, 17 días, 18 días, 19 días, 20 días, 21 días, 22 días. día, 23 días, 24 días, 25 días, 26 días, 27 días, 28 días, 29 días, 30 días, 31 días, 3 semanas, 4 semanas, 5 semanas, 6 semanas, 7 semanas, 8 semanas, 9 semanas, 10 semanas, 11 semanas, 12 semanas, 1 mes, 2 meses o un periodo más largo. Por ejemplo, en una realización, a un sujeto que se le administraron dosis semanales repetidas de 100 mg/kg, 200 mg/kg y 300 mg/kg de IgG humana combinada (por ejemplo, 600 mg/kg/periodo de 3 semanas) se dice que recibió 400 mg/kg/periodo de 2 semanas.
Tal como se usa en el presente documento, los términos “administración intranasal” y “administración nasal” se refieren a la administración de una composición terapéutica a la cavidad nasal de un sujeto, de tal manera que un agente terapéutico en la composición se administra directamente a uno o más epitelios ubicados en la nariz. En determinadas realizaciones, la administración intranasal se logra usando una preparación líquida (por ejemplo, una preparación acuosa), una preparación en aerosol o una preparación de polvo seco, cada una de las cuales puede administrarse a través de un dispositivo de administración nasal no invasivo propulsado externamente o autopropulsado (por ejemplo, por inhalación), o por medio de un gel, una crema, pomada, loción o pasta aplicado directamente a uno o más epitelios nasales (por ejemplo, epitelio olfativo o epitelio respiratorio nasal).
El término “tratamiento” o “terapia” significa en general obtener un efecto fisiológico deseado. El efecto puede ser profiláctico en cuanto a la prevención total o parcial de una enfermedad o un estado o síntoma de los mismos y/o puede ser terapéutico en cuanto a una cura parcial o completa para una lesión, enfermedad o un estado y/o la mejora de un efecto adverso atribuible a la lesión, enfermedad o el estado e incluye detener el desarrollo o provocar la regresión de una enfermedad o un estado. El tratamiento también puede referirse a cualquier retardo en el inicio, la mejoría de los síntomas, la mejora de la supervivencia del paciente, el aumento del tiempo o la tasa de supervivencia, la mejora de la función cognitiva, etc. El efecto del tratamiento puede compararse con un individuo o combinación de individuos que no reciben el tratamiento.
Tal como se usa en el presente documento, una “cantidad o dosis terapéuticamente eficaz” o “cantidad o dosis suficiente/eficaz” se refiere a una dosis que produce efectos para los que se administra. La dosis exacta dependerá del propósito del tratamiento, y podrá determinarla un experto en la técnica usando técnicas conocidas (véase, por ejemplo, Lieberman, Pharmaceutical Dosage Forms (vols. 1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); y Remington: The Science and Practice of Pharmacy, 20a edición, 2003, Gennaro, Ed., Lippincott, Williams & Wilkins).
En algunas realizaciones, la progresión o la gravedad de la enfermedad de Alzheimer se mide mediante una evaluación cognitiva (por ejemplo, miniexamen de estado mental (MMSE), subescala cognitiva de la escala de evaluación de la enfermedad de Alzheimer (ADAS-Cog), miniexamen de estado mental modificado (3MS), prueba de fluidez verbal o prueba neuropsicológica complementaria), una evaluación clínica, conductual y funcional (por ejemplo, estudio cooperativo de la enfermedad de Alzheimer (ADCS)-actividades de la vida diaria (ADL), ADCS-impresión clínica global de cambio (ADCS-CGIC) o evaluación del inventario neuropsiquiátrico (NPI), una evaluación de la calidad de vida (por ejemplo, calidad de vida en la enfermedad de Alzheimer (QOL-AD) de Logsdon o cuestionario de sobrecarga del cuidador), y/o una evaluación de la utilización de recursos de atención sanitaria (por ejemplo, una evaluación del inventario de uso de recursos de ADCS (ADCS-RUI)).
Tal como se usa en el presente documento, los términos “dosis” y “dosificación” se usan indistintamente y se refieren a la cantidad de principio activo administrada a un individuo en cada administración. La dosis variará dependiendo de varios factores, incluidos la frecuencia de administración; el tamaño y la tolerancia del individuo; gravedad del estado; riesgo de efectos secundarios; y la vía de administración. Un experto en la técnica reconocerá que la dosis puede modificarse dependiendo de los factores anteriores o en función del progreso terapéutico. El término “forma de dosificación” se refiere al formato particular del producto farmacéutico y depende de la vía de administración. Por
ejemplo, una forma de dosificación puede ser un líquido o polvo seco, formulado para administración intranasal. Tal como se usa en el presente documento, el término “composición de polvo seco” se refiere a una forma liofilizada o secada por pulverización de una formulación de IgG humana combinada terapéutica. En una realización, una composición de polvo seco contiene un contenido de agua residual de menos del 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, el 1% o menos.
Un “control” se usa en el presente documento, se refiere a una referencia, en general una referencia conocida, para la comparación con un grupo experimental. Un experto en la técnica entenderá qué controles son valiosos en una situación dada y será capaz de analizar datos basándose en comparaciones con valores de control. Los controles también son valiosos para determinar la significación de los datos. Por ejemplo, si los valores para un parámetro dado varían ampliamente en los controles, la variación en las muestras de prueba no se considerará significativa. Tal como se usa en el presente documento, el término “aproximadamente” indica un intervalo aproximado de más o menos el 10% de un valor especificado. Por ejemplo, la expresión “aproximadamente el 20%” engloba un intervalo del 18-22%. Tal como se usa en el presente documento, también incluye la cantidad exacta. Así, “aproximadamente el 20%” significa “aproximadamente el 20%” y también “el 20%”.
Antes de que la presente divulgación se describa con mayor detalle, debe entenderse que esta invención no está limitada a realizaciones particulares descritas ya que, por supuesto, esto puede variar. También debe entenderse que la terminología usada en el presente documento tiene el propósito de describir sólo realizaciones particulares, y no pretende ser limitativa, ya que el alcance de la presente invención estará limitado sólo por las reivindicaciones adjuntas.
Cuando se proporciona un intervalo de valores, se entiende que cada valor intermedio, hasta la décima parte de la unidad del límite inferior, a menos que el contexto indique claramente otra cosa, entre el límite superior e inferior de ese intervalo y cualquier otro valor establecido o intermedio en el intervalo establecido, se engloba dentro de la invención. Los límites superior e inferior de estos intervalos más pequeños pueden incluirse independientemente en los intervalos más pequeños y también están englobados dentro de la invención, sujetos a cualquier límite excluido específicamente en el intervalo establecido. Cuando el intervalo establecido incluye uno o ambos límites, los intervalos que excluyen cualquiera o ambos de los límites incluidos también se incluyen en la invención.
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que entiende habitualmente un experto en la técnica a la que pertenece esta invención. Aunque también pueden usarse cualquier material y método similar o equivalente a los descritos en el presente documento en la práctica o prueba de la presente invención, ahora se describen materiales y métodos representativos ilustrativos.
Se observa que, tal como se usa en el presente documento y en las reivindicaciones adjuntas, las formas en singular “un(o)”, “una” y “el/la” incluyen referentes en plural, a menos que el contexto indique claramente otra cosa. También se señala que las reclamaciones pueden redactarse para excluir cualquier elemento opcional. Como tal, esta declaración pretende servir como base de antecedentes para el uso de tal terminología exclusiva como “únicamente”, “sólo” y similares en relación con la mención de elementos de reivindicación, o el uso de una limitación “negativa”.
Fabricación de inmunoglobulina G combinada
Los productos de inmunoglobulina del plasma humano se usaron por primera vez en 1952 para tratar la inmunodeficiencia. Inicialmente, los métodos de elección fueron la administración intramuscular o subcutánea del isotipo G de inmunoglobulina (IgG) aislado del plasma. Sin embargo, los productos de IgG que podían administrarse por vía intravenosa, denominados inmunoglobulina intravenosa (IgIV), se desarrollaron posteriormente para permitir la administración de cantidades mayores de IgG necesarias para el tratamiento eficaz de diversas enfermedades. Por lo general, la IgIV contiene las inmunoglobulinas de inmunoglobulina G (IgG) combinada del plasma de múltiples donantes, por ejemplo, más de cien o más de mil donantes de sangre. Estos productos de IgG purificados se usan principalmente para tratar tres categorías principales de estados médicos: (1) inmunodeficiencias: agammaglobulinemia ligada al cromosoma X, hipogammaglobulinemia (inmunodeficiencias primarias) y estados inmunitarios comprometidos adquiridos (inmunodeficiencias secundarias), que presentan bajos niveles de anticuerpos; (2) enfermedades inflamatorias y autoinmunitarias; y (3) infecciones agudas.
La inmunoglobulina humana combinada G (IgG) se fabrica de acuerdo con diferentes procesos dependiendo del fabricante específico. Sin embargo, el origen de la mayoría de los procesos de fabricación se encuentra en la cuarta entrega de una serie de artículos fundamentales publicados sobre la preparación y las propiedades de las proteínas séricas y plasmáticas. Cohn et al. (J. Am. Chem. Soc., 1946, 68(3): 459-475). Este artículo describió por primera vez un método para el fraccionamiento alcohólico de proteínas plasmáticas (método 6), que permite el aislamiento de una fracción enriquecida en IgG a partir de plasma humano.
Los procedimientos de Cohn se desarrollaron inicialmente para obtener albúmina con una pureza relativamente alta (95%) mediante precipitación fraccionada con alcohol. Sin embargo, Oncley et al. (J. Am. Chem. Soc., 71(2): 541 550 (1949)), Deutsch et al. (J. Biol. Chem., 164, 109-118 (1946)), y Kistler y Nitschmann (Vox Sang., 7, 414-424 (1962)) se dieron cuenta de que precipitados de proteína particulares (fracción II y fracción II III) del método de Cohn podían usarse como material de partida para la purificación de composiciones de inmunoglobulinas altamente enriquecidas. Para lograr la mayor pureza y seguridad requeridas para la administración intravenosa de composiciones de IgG, se han añadido varias etapas de purificación y pulido (por ejemplo, adsorción en fase sólida, diversas técnicas cromatográficas, filtración de flujo cruzado, tratamiento con disolventes y/o detergentes, tratamiento térmico y nanofiltración) a los procesos de fabricación de IgG después de las etapas de fraccionamiento alcohólico.
Los actuales fabricantes de IgG se basan en general en un precipitado de fracción II III del método 6 de Cohn o en un precipitado A de Kistler-Nitschmann como material de partida para el procesamiento posterior. Ambas fracciones se forman mediante un proceso en dos etapas en el que proteínas como el fibrinógeno y el factor XIII se retiran mediante una etapa de precipitación inicial (precipitación de fracción I) realizada a un alto pH (7,2) con una baja concentración de etanol (8-10% v/v), seguido de una segunda etapa de precipitación en la que se precipita IgG del sobrenadante de la fracción I a pH 6,8 con etanol al 20-25% (v/v) (fracción II III) o a pH 5,85 con etanol al 19% (v/v) (precipitado A), mientras que la albúmina y una porción significativa de A1PI permanecen en el sobrenadante. Estos métodos, al tiempo que sentaban las bases para una industria completa de proteínas de la sangre derivadas del plasma, no fueron capaces de proporcionar preparaciones de IgG con una pureza suficientemente alta para el tratamiento prolongado de varias enfermedades relacionadas con el sistema inmunitario, incluido el síndrome de Kawasaki, la púrpura trombocitopénica inmunitaria e inmunodeficiencias primarias, sin una aparición indebida de efectos secundarios graves. Como tal, se desarrollaron metodologías adicionales que emplean diversas técnicas, tales como cromatografía de intercambio iónico, para proporcionar formulaciones de IgG de mayor pureza. Hoppe et al. (Munch Med Wochenschr, (34):1749-1752 (1967)), Falksveden (patente sueca n.° 348942), y Falksveden y Lundblad (Methods of Plasma Protein Fractionation, 1980) fueron de los primeros en emplear la cromatografía de intercambio iónico con este propósito.
Administración
En un aspecto, la presente invención proporciona un método para tratar la enfermedad de Alzheimer en un sujeto que lo necesita mediante la administración de una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave y/o que porta un alelo apoE4.
En una realización, el método incluye administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada a un sujeto con enfermedad de Alzheimer moderadamente grave y/o que porta un alelo ApoE4, en el que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. En algunas realizaciones, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
En algunas realizaciones, la inmunoglobulina G (IgG) humana combinada se administra al sujeto por una vía sistémica. Los ejemplos no limitativos de administración sistémica incluyen administración intravenosa, administración subcutánea, administración intramuscular o administración intraperitoneal.
En algunas realizaciones, cuando se administra de forma sistémica, la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG). En una realización, al sujeto se le administran de forma sistémica desde 400 mg/kg hasta 800 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 300 mg/kg hasta 700 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 400 mg/kg hasta 700 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 300 mg/kg hasta 600 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 400 mg/kg hasta 600 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 300 mg/kg hasta 500 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica desde 400 mg/kg hasta 500 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran de forma sistémica aproximadamente 300, 350, 400, 450, 500, 550, 600, 650, 700, 750 u 800 mg/kg/2 semanas de IgG.
En algunas realizaciones, cuando se administra de forma sistémica, la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 300 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG). En una realización, al sujeto se le administran de forma sistémica aproximadamente 200 mg/kg/2 semanas. En una realización, al sujeto se le administran de forma sistémica aproximadamente 250 mg/kg/2 semanas.
En una realización, el método incluye administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada directamente al SNC de un sujeto con enfermedad de Alzheimer moderadamente grave y/o que porta un alelo ApoE4, en el que la cantidad de IgG humana combinada es de desde 400 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. Los ejemplos no limitativos de administración directamente al SNC incluyen administración intranasal, administración intratecal, administración intracerebral, administración intracerebroventricular, administración epidural o administración espinal.
En una realización, el método incluye administrar una cantidad terapéuticamente eficaz de una composición que comprende inmunoglobulina G (IgG) humana combinada directamente al SNC de un sujeto con enfermedad de Alzheimer moderadamente grave y/o que porta un alelo ApoE4, en el que la cantidad de IgG humana combinada es de desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas, y en el que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico. Los ejemplos no limitativos de administración directamente al SNC incluyen administración intranasal, administración intratecal, administración intracerebral, administración intracerebroventricular, administración epidural o administración espinal.
En general, cuando se administra por vía subcutánea, la dosificación de inmunoglobulina G (IgG) humana combinada se aumenta para tener en cuenta una menor biodisponibilidad. En una realización, cuando se administra por vía subcutánea, la dosificación de IgG humana combinada aumenta en desde el 25% hasta el 50%, en comparación con una dosificación convencional usada para la administración intravenosa. En una realización, cuando se administra por vía subcutánea, la dosificación de IgG humana combinada aumenta en desde el 30% hasta el 45%, en comparación con una dosificación convencional usada para la administración intravenosa. En una realización específica, cuando se administra por vía subcutánea, la dosificación de IgG humana combinada aumenta en aproximadamente el 37%, en comparación con una dosificación convencional usada para la administración intravenosa.
En una realización, cuando se administra por vía subcutánea, la cantidad de IgG humana combinada es de desde
375 mg/kg hasta 1.000 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG).
En una realización, al sujeto se le administran por vía subcutánea desde 500 mg/kg hasta 1.000 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 375 mg/kg hasta 875 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 500 mg/kg hasta
875 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 375 mg/kg hasta 750 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde
500 mg/kg hasta 750 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 375 mg/kg hasta 625 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 500 mg/kg hasta 625 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea aproximadamente 375, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950 ó 1.000 mg/kg/2 semanas de IgG.
En una realización, cuando se administra por vía subcutánea, la cantidad de IgG humana combinada es de desde
400 mg/kg hasta 1.100 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG).
En una realización, al sujeto se le administran por vía subcutánea desde 550 mg/kg hasta 1.100 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 400 mg/kg hasta 950 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 550 mg/kg hasta
950 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 400 mg/kg hasta 825 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde
550 mg/kg hasta 825 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 400 mg/kg hasta 675 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 550 mg/kg hasta 675 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea aproximadamente 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 950, 1.000, 1.050 ó 1.100 mg/kg/2 semanas de IgG.
En una realización, cuando se administra por vía subcutánea, la cantidad de IgG humana combinada es desde
450 mg/kg hasta 1.200 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG).
En una realización, al sujeto se le administran por vía subcutánea desde 600 mg/kg hasta 1.200 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 450 mg/kg hasta 1.050 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 600 mg/kg hasta 1.050 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 450 mg/kg hasta 900 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde
600 mg/kg hasta 900 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 450 mg/kg hasta 750 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea desde 600 mg/kg hasta 750 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran por vía subcutánea aproximadamente 450, 500, 550, 600, 650, 700, 750, 800, 850, 950, 1.000, 1.050, 1.100, 1.150 ó 1.200 mg/kg/2 semanas de IgG.
En una realización, la biodisponibilidad de la IgG humana combinada administrada por vía subcutánea puede
aumentarse mediante la administración conjunta de un agente de permeación, por ejemplo, una hialuronidasa tal
como PH20 (véase, las publicaciones de solicitud PCT números WO 2011/034604 y Wo 2009/117085. Un experto
en la técnica será capaz de determinar fácilmente una dosificación apropiada de agente de permeación (por
ejemplo, una hialuronidasa) para la administración conjunta con la IgG humana combinada.
Por tanto, en una realización, la IgG humana combinada se formula conjuntamente con el agente de permeación
(por ejemplo, una hialuronidasa). En otra realización, la IgG humana combinada y el agente de permeación (por
ejemplo, una hialuronidasa) se formulan por separado y se mezclan antes de la administración subcutánea. En otra
realización, la IgG humana combinada y el agente de permeación (por ejemplo, una hialuronidasa) se formulan y
administran por separado (por ejemplo, el agente de permeación se administra directamente antes o después de la
administración de la IgG humana combinada).
En una realización, cuando se administra conjuntamente por vía subcutánea con un agente de permeación (por
ejemplo, una hialuronidasa), la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de
peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG). En una realización, al sujeto se le
administran conjuntamente por vía subcutánea un agente de permeación (por ejemplo, una hialuronidasa) y desde
400 mg/kg hasta 800 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran conjuntamente por vía
subcutánea un agente de permeación (por ejemplo, una hialuronidasa) y de desde 300 mg/kg hasta 700 mg/kg/2
semanas de IgG. En una realización, al sujeto se le administran conjuntamente por vía subcutánea un agente de
permeación (por ejemplo, una hialuronidasa) y desde 400 mg/kg hasta 700 mg/kg/2 semanas de IgG. En una
realización, al sujeto se le administran conjuntamente por vía subcutánea un agente de permeación (por ejemplo,
una hialuronidasa) y desde 300 mg/kg hasta 600 mg/kg/2 semanas de IgG. En una realización, al sujeto se le
administran conjuntamente por vía subcutánea un agente de permeación (por ejemplo, una hialuronidasa) y desde
400 mg/kg hasta 600 mg/kg/2 semanas de IgG. En una realización, al sujeto se le administran conjuntamente por vía
subcutánea un agente de permeación (por ejemplo, una hialuronidasa) y desde 300 mg/kg hasta 500 mg/kg/2
semanas de IgG. En una realización, al sujeto se le administran conjuntamente por vía subcutánea un agente de
permeación (por ejemplo, una hialuronidasa) y desde 400 mg/kg hasta 500 mg/kg/2 semanas de IgG. En una
realización, al sujeto se le administran conjuntamente por vía subcutánea un agente de permeación (por ejemplo,
una hialuronidasa) y aproximadamente 300, 350, 450, 500, 550, 600, 650, 700, 750 u 800 mg/kg/2 semanas de IgG.
En una realización, cuando se administra conjuntamente por vía subcutánea con un agente de permeación (por
ejemplo, una hialuronidasa), la cantidad de IgG humana combinada es de desde 200 mg/kg hasta 300 mg/kg de
peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de IgG). En una realización, al sujeto se le
administran conjuntamente por vía subcutánea un agente de permeación (por ejemplo, una hialuronidasa) y
aproximadamente 200 ó 250 mg/kg/2 semanas de IgG.
En general, cuando se administra directamente al sistema nervioso central, la dosificación de inmunoglobulina G
(IgG) humana combinada se puede reducir en un factor de desde aproximadamente 2 hasta 20, preferiblemente en
un factor de desde aproximadamente 4 hasta aproximadamente 10 (por ejemplo, aproximadamente 6 veces). En
algunas realizaciones, cuando se administra directamente al SNC, la cantidad de IgG humana combinada es de
desde 50 mg/kg hasta 400 mg/kg de peso corporal del sujeto por periodo de dos semanas (mg/kg/2 semanas de
IgG). En una realización, al sujeto se le administran desde 50 mg/kg hasta 350 mg/kg/2 semanas de IgG
directamente al SNC. En una realización, al sujeto se le administran desde 50 mg/kg hasta 300 mg/kg/2 semanas de
IgG directamente al SNC. En una realización, al sujeto se le administran desde 50 mg/kg hasta 250 mg/kg/2
semanas de IgG directamente al SNC. En una realización, al sujeto se le administran desde 50 mg/kg hasta
200 mg/kg/2 semanas de IgG directamente al SNC. En una realización, al sujeto se le administran desde 50 mg/kg
hasta 150 mg/kg/2 semanas de IgG directamente al SNC. En una realización, al sujeto se le administran desde
50 mg/kg hasta 100 mg/kg/2 semanas de IgG directamente al SNC. En algunas realizaciones, al sujeto se le
administran aproximadamente 50, 75, 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 350, 375 ó 400 mg/kg/2
semanas de IgG directamente al SNC.
En algunas realizaciones, cuando se administra directamente al SNC, la cantidad de IgG humana combinada es de
desde 1 mg hasta 400 mg de dosis total por periodo de dos semanas. En una realización, al sujeto se le administran
desde 1 mg hasta 350 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 300 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 250 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 200 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 150 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 100 mg de dosis total de IgG directamente a una realización, al sujeto se le ad desde 1 mg hasta 50 mg de dosis total de IgG directamente al SNC. En algunas realizaciones, al sujeto se le
una realización, al sujeto se le ad desde 1 mg hasta 50 mg de dosis total de IgG directamente al SNC. En algunas realizaciones, al sujeto se le
administran aproximadamente 50 mg, 75 mg, 100 mg, 125 mg, 150 mg, 175 mg, 200 mg, 225 mg, 250 mg, 275 mg,
300 mg, 325 mg, 350 mg, 375 mg o 400 mg de dosis total de IgG por periodo de 2 semanas directamente al SNC.
En algunas realizaciones, cuando se administran dosificaciones múltiples al sujeto durante el periodo de dos
semanas, cada dosis individual será igual. En estas realizaciones, las dosificaciones individuales serán
inversamente proporcionales al número de administraciones. Por ejemplo, para administrar una cantidad total de 400 mg/kg de IgG en dos administraciones a lo largo del periodo de dos semanas, se usan dos dosificaciones de 200 mg/kg. Mientras que para administrar los mismos 400 mg/kg de IgG en cuatro administraciones a lo largo del periodo de dos semanas, se usan cuatro dosificaciones de 100 mg/kg.
En algunas realizaciones, cuando se administran dosis múltiples al sujeto a lo largo del periodo de dos semanas, cada dosis individual variará. En una realización, se administra una primera dosis alta al comienzo del periodo de dos semanas y posteriormente se administran una o más dosificaciones más pequeñas. Por ejemplo, para administrar una cantidad total de 400 mg/kg a lo largo del periodo de dos semanas, se administra una dosis inicial de 200 mg/kg al comienzo del periodo y posteriormente se administran diez dosis de 20 mg/kg.
Al administrar dosificaciones múltiples a lo largo del periodo de dos semanas, determinados parámetros farmacocinéticos pueden controlarse a lo largo del periodo de dos semanas. Por ejemplo, en una realización, un médico estabiliza el AUC (área bajo la curva) de IgG humana combinada en el paciente mediante la administración, o la prescripción de la administración, de una o más dosificaciones de mantenimiento a lo largo del periodo de dos semanas. Asimismo, en algunas realizaciones, la biodisponibilidad, Cmáx (concentración pico), Tmáx (tiempo para lograr Cmáx), Cmín (la concentración menor o valle) y/o la fluctuación pico-valle de la IgG se controla mediante la administración de dosis múltiples y/o la variación de la dosis a lo largo del periodo de dos semanas.
En una realización particular, la IgG humana combinada puede administrarse en combinación con otro tratamiento para una demencia relacionada con la edad, por ejemplo, enfermedad de Alzheimer. En determinadas realizaciones, el tratamiento para una demencia relacionada con la edad administrada conjuntamente con IgG humana combinada es la administración de un inhibidor de la colinesterasa (por ejemplo, ARICEPT (donepezilo), EXELON (rivastigmina), RAZADYNE (galantamina) o COGNEX (tacrina), o un inhibidor del receptor de glutamato de tipo NMDA (por ejemplo, memantina).
En otras realizaciones, la segunda terapia es levodopa (L-DOPA). La segunda terapia también puede ser un agonista de la dopamina. Los ejemplos no limitativos de agonistas de la dopamina incluyen bromocriptina, pergolida, pramipexol, ropinirol, piribedil, cabergolina, apomorfina y lisurida. La segunda terapia puede ser un inhibidor de la MAO-B. Ejemplos no limitativos de inhibidores de la MAO-B son la selegilina y la rasgilina. Las segundas terapias de adición pueden incluir amantaína, composiciones anticolinérgicas, clozapina, modafinilo y fármacos antiinflamatorios no esteroideos.
En otras realizaciones, la segunda terapia es CAMPATH (alemtuzumab), ZENAPX (daclizumab), rituximab, dirucotida, BHT-3009, cladribina, fumarato de dimetilo, estriol, laquinimod, interferón p-1a pegilado, minociclina, estatinas, temsirolimús, teriflunomida y naltexona a dosis baja.
En una realización, el segundo agente terapéutico se formula conjuntamente con la IgG humana combinada (por ejemplo, en la misma composición). En otra realización, el segundo agente terapéutico se administra en una formulación diferente de la IgG humana combinada (por ejemplo, en una segunda composición). En una realización, la segunda composición que contiene el segundo agente terapéutico se administra al mismo tiempo que la composición combinada de IgG humana (por ejemplo, procediendo de inmediado, inmediatamente después o en una mezcla). En otra realización, la segunda composición que contiene el segundo agente terapéutico se administra en un momento diferente, y/o mediante un régimen terapéutico diferente, que la composición de IgG humana combinada.
En determinadas realizaciones, la segunda terapia es psicoterapia. Ejemplos no limitativos de psicoterapia son la intervención psicosocial, la intervención conductual, la terapia de reminiscencia, la terapia de validación, la psicoterapia de apoyo, la integración sensorial, la terapia de presencia simulada, el reentrenamiento cognitivo y las terapias orientadas a la estimulación tales como el arte, la música, las mascotas, el ejercicio y las actividades recreativas.
Además, dos o más terapias secundarias pueden combinarse con IgG terapéutica. Por ejemplo, la IgG combinada terapéutica se puede combinar con memantina y donepezilo.
Administracion intranasal
La administración intranasal de productos terapéuticos se ha convertido en un método cada vez más explorado para administrar agentes terapéuticos al cerebro porque elude la barrera hematoencefálica (BHE) y es un método de administración localizado y no invasivo. Además, la administración intranasal ofrece las ventajas con respecto a métodos de administración más tradicionales (por ejemplo, administración intravenosa, subcutánea, transmucosa oral, oral o rectal), de administración simple, que proporciona un inicio rápido de la acción y evita el metabolismo de primer paso. Desafortunadamente, la administración intranasal sólo ha demostrado ser eficaz para el transporte de pequeñas moléculas y, en cierta medida, proteínas de fusión de Fc más pequeñas, al cerebro. La administración de moléculas más grandes, tales como anticuerpos intactos, aún no se ha demostrado. Se cree que la dificultad para transportar proteínas más grandes se debe a la permeabilidad limitada de las uniones estrechas presentes en los
epitelios olfativos, lo que probablemente excluye las moléculas globulares que tienen un radio hidrodinámico de más de 3,6 A (Stevenson BR, et al., Mol Cell Biochem., 83(2):129-45 (1988)).
La patente estadounidense n.° 5,624,898 concedida a Frey describe composiciones y métodos para transportar agentes neurológicos, que fomentan el crecimiento y la supervivencia de las células nerviosas o aumentan la actividad de células funcionales, al cerebro por medio de la ruta neural olfatoria. Los agentes neurológicos de la patente '898 se transportan al cerebro por medio del sistema nervioso, en lugar del sistema circulatorio, de modo que los agentes potencialmente terapéuticos que son incapaces de atravesar la barrera hematoencefálica pueden administrarse a neuronas dañadas en el cerebro. Las composiciones descritas en la patente '898 incluyen un agente neurológico en combinación con un vehículo y/o aditivo farmacéutico que fomenta la transferencia del agente dentro del sistema olfativo. La patente '898 no enseña la administración intranasal de inmunoglobulinas humanas combinadas.
Las publicaciones PCT WO 2006/091332 y WO 2009/058957, ambas de Bentz et al, describen métodos para la administración de polipéptidos terapéuticos al cerebro fusionando el polipéptido a un anticuerpo o fragmento de anticuerpo y administrando la proteína de fusión resultante por vía intranasal. Aunque los ejemplos de las publicaciones PCT '332 y '957 sugieren que “cuerpos miméticos” (“mimetibodies”) de fusión de Fc pueden administrarse por vía intranasal, ninguna publicación demuestra la administración de anticuerpos intactos más grandes. De hecho, la publicación PCT '957, publicada tres años después de la publicación PCT '332, afirma que “[e]n estudios de administración publicados hasta la fecha, la eficacia de la administración intranasal al SNC ha sido muy baja y la administración de grandes macromoléculas globulares, tales como anticuerpos y sus fragmentos, no ha sido demostrado “. La publicación PCT '957 pretende resolver este problema a través del uso de un potenciador de la permeabilidad (por ejemplo, fluidificantes de membrana, moduladores de unión estrecha y ácidos grasos de longitud de cadena media y sales y ésteres de los mismos, tal como se describe a continuación), lo que potencia la administración intranasal al sistema nervioso central. Ninguna publicación PCT enseña la administración intranasal de inmunoglobulinas humanas combinadas.
La publicación PCT WO 2003/028668 de Barstow et al., describe el tratamiento de las enfermedades mediadas por el sistema inmunitario mediante administración alimentaria (es decir, administración al tracto digestivo) de inmunoglobulinas combinadas. Aunque la publicación PCT '668 define la administración alimentaria de inmunoglobulinas combinadas como que incluye la administración nasal, es en el contexto de administrar la composición al tracto digestivo. La publicación PCT '668 no enseña la administración de inmunoglobulinas humanas combinadas al cerebro mediante administración intranasal.
La publicación PCT WO 2001/60420 de Adjei et al., describe formulaciones en aerosol de polipéptidos terapéuticos, incluidas inmunoglobulinas, para la administración pulmonar. Estas composiciones aerosolizables se formulan de tal manera que después de la inhalación oral o nasal, el agente terapéutico se administra de manera eficaz a los pulmones del paciente. La publicación PCT '420 no enseña la administración de agentes terapéuticos al cerebro mediante administración intranasal.
La administración intranasal (i.n.) es un modo ventajoso de administrar un medicamento al cerebro porque no es invasivo y existe una conexión directa entre el sistema olfativo y el cerebro. La administración intranasal de IgG (IgIN) para tratar enfermedades neurológicas es particularmente ventajosa debido a que la conexión directa entre el sistema olfativo y el cerebro evita los problemas de administración asociados con la barrera hematoencefálica (BHE) y minimiza la exposición sistémica al fármaco, lo que minimiza los efectos secundarios del fármaco. Además, la administración i.n. permite composiciones tales como polvos, gránulos, disoluciones, pomadas y cremas, obviando así la necesidad de administración intravenosa e intramuscular. Por ejemplo, cuando un fármaco se administra por vía intranasal, se transporta a través de la mucosa nasal y a lo largo de la ruta neural olfatoria. El fármaco puede administrarse solo o puede combinarse con una o más moléculas transportadoras para fomentar el transporte a través de la mucosa nasal y a lo largo de la ruta neural olfatoria. El fármaco también puede administrarse en combinación con un potenciador de la absorción. Los potenciadores de la absorción fomentan la absorción del fármaco a través de la mucosa nasal y a lo largo de la ruta neural olfatoria. Además, pueden añadirse moléculas adicionales para facilitar el transporte del fármaco a través de la ruta neural olfatoria.
La administración i.n. también puede usarse para administrar medicamentos terapéuticos al cerebro a través de la ruta del trigémino. Específicamente, la administración i.n. puede usarse para administrar IgG a través de la ruta del trigémino. Los nervios olfativos y del trigémino reciben altas concentraciones de un fármaco con administración i.n. porque el nervio trigémino inerva el pseudoepitelio olfativo y respiratorio absorbente. Estos nervios pueden transportar el fármaco al cerebro y otras estructuras conectadas. Por ejemplo, los ramos del nervio trigémino alcanzan directa o indirectamente el seno maxilar, el tronco encefálico, el rombencéfalo, la lámina cribrosa, el prosencéfalo (por ejemplo, la corteza y el diencéfalo), las estructuras bucofaciales (por ejemplo, los dientes, el músculo masetero y la articulación temporomandibular), el mesencéfalo y el cerebelo, la médula espinal cervical, médula espinal torácica y médula espinal lumbar. Por consiguiente, puede transportarse IgIN a través de la ruta del trigémino para alcanzar y tratar enfermedades neurológicas.
Pueden usarse muchos tipos de dispositivos de administración intranasal para practicar los métodos proporcionados
en el presente documento. En algunas realizaciones, el dispositivo de administración es un dispositivo intranasal para la administración de líquidos. Los ejemplos no limitativos de dispositivos útiles para la administración de composiciones líquidas (por ejemplo, composiciones líquidas de IgG combinada) incluyen dispositivos de vapor (por ejemplo, inhaladores de vapor), dispositivos de goteo (por ejemplo, catéteres, cuentagotas de dosis única, cuentagotas de dosis múltiples y pipetas de dosis unitarias), dispositivos mecánicos de bomba de pulverización (por ejemplo, frascos de presión, bombas de pulverización de dosis medida de dosis múltiples y bombas de pulverización de dosis única/doble), bombas de pulverización bidireccionales (por ejemplo, dispositivos de administración nasal accionados por la respiración), atomizadores/sistemas de pulverización accionados por gas (por ejemplo, inhaladores de dosis medida accionados por propelente de nitrógeno o HFA de dosis única o dosis múltiples, incluidos los inhaladores de velocidad circunferencial y tradicionales), y nebulizadores/atomizadores eléctricos (por ejemplo, nebulizadores de membrana pulsante, nebulizadores mecánicos vibrantes y nebulizadores mecánicos de mano). En algunas realizaciones, el dispositivo de administración es un dispositivo intranasal para la administración de polvos o geles. Los ejemplos no limitativos de dispositivos útiles para la administración de composiciones en polvo (por ejemplo, composiciones de IgG combinada liofilizadas o secadas de otro modo) incluyen pulverizadores mecánicos de polvo (por ejemplo, dispositivos de pulverización de polvo basados en cápsulas accionados manualmente y dispositivos de pulverización de polvo accionados manualmente, dispositivos de administración de gel accionados manualmente), inhaladores accionados por la respiración (por ejemplo, inhaladores nasales de dosis únicas o múltiples e inhaladores nasales de dosis únicas o múltiples basados en cápsulas) e insufladores (por ejemplo, dispositivos de administración nasal accionados por la respiración). En algunas realizaciones, las inmunoglobulinas humanas combinadas se administran preferentemente en el área olfativa, ubicada en el tercio superior de la cavidad nasal y, en particular, en el epitelio olfativo. Las fibras del nervio olfativo son axones amielínicos de las células receptoras olfativas, que se encuentran en el tercio superior de la cavidad nasal. Las células receptoras olfativas son neuronas bipolares con prominencias cubiertas por cilios similares a pelos que se sobresalen en la cavidad nasal. En el otro extremo, los axones de estas células se acumulan en agregados y entran en la cavidad craneal en el techo de la nariz. Rodeados por un delgado tubo de piamadre, los nervios olfativos atraviesan el espacio subaracnoideo que contiene LCR y entran en las caras inferiores de los bulbos olfativos. Una vez que la inmunoglobulina humana combinada se dispensa en la cavidad nasal, la inmunoglobulina puede transportarse a través de la mucosa nasal y al bulbo olfativo y áreas interconectadas del cerebro tales como la formación del hipocampo, los núcleos amigdalinos, el núcleo basal de Meynert, el locus cerúleo, el tronco encefálico, y similares (por ejemplo, Johnson et al., Molecular Pharmaceutics (2010) 7(3):884-893).
En determinadas realizaciones, la inmunoglobulina humana combinada se administra a tejido inervado por el nervio trigémino. El nervio trigémino inerva los tejidos de la cabeza de un mamífero (por ejemplo, humano) incluida la piel de la cara y el cuero cabelludo, los tejidos orales y los tejidos que rodean el ojo. El nervio trigémino tiene tres ramos principales, el nervio oftálmico, el nervio maxilar y el nervio mandibular. En algunas realizaciones, los métodos proporcionados en el presente documento incluyen la administración dirigida de inmunoglobulina humana combinada a uno o más de estos ramos del trigémino, es decir, la ruta del trigémino. En algunas realizaciones, los métodos proporcionados en el presente documento incluyen la administración dirigida de inmunoglobulina humana combinada al seno maxilar, alcanzándose de ese modo el tronco encefálico, el rombencéfalo, la lámina cribosa, el prosencéfalo (por ejemplo, la corteza y el diencéfalo, mesencéfalo, cerebelo, médula espinal cervical, médula espinal torácica y médula espinal lumbar a través de la ruta del trigémino. En determinadas realizaciones, los métodos proporcionados en el presente documento incluyen la administración dirigida de inmunoglobulina humana combinada para el tratamiento de un trastorno del SNC (por ejemplo, enfermedad de Alzheimer).
En algunas realizaciones, la inmunoglobulina humana combinada se administra a tejidos nasales inervados por el nervio trigémino, por ejemplo, a tejidos nasales incluyendo los senos nasales, los dos tercios inferiores de la cavidad nasal y el tabique nasal. En determinadas realizaciones, la inmunoglobulina humana combinada se dirige a los dos tercios inferiores de la cavidad nasal y/o el tabique nasal.
En algunas realizaciones, la inmunoglobulina humana combinada se administra a uno o ambos senos maxilares del individuo. Los métodos y dispositivos para la administración al seno maxilar se conocen en la técnica, por ejemplo, véase la publicación de solicitud de patente estadounidense número 2011/0151393.
El seno maxilar está en comunicación de fluido con la cavidad nasal del paciente y comprende los senos maxilares derecho e izquierdo. Cada seno maxilar se comunica con la fosa nasal correspondiente a través del orificio del seno maxilar. El volumen máximo del seno maxilar en adultos es de aproximadamente 4 a 15 ml, aunque los senos individuales pueden comprender volúmenes fuera de este intervalo.
La ruta desde las fosas nasales al orificio correspondiente del seno maxilar y, en última instancia, al seno maxilar correspondiente, permite insertar un dispositivo en la fosa nasal hasta el orificio del seno maxilar, con lo cual puede administrarse al menos una cantidad o dosis eficaz de las inmunoglobulinas humanas combinadas y suministrarse al seno maxilar. La ruta hasta el seno maxilar es tortuosa y requiere: atravesar la narina, moverse a través de la región entre los cornetes inferior y medio, navegar por y hacia el hiato semilunar, desplazarse de manera superior hacia la abertura del seno maxilar, resistir la acción ciliada del orificio/tubo que pasa al seno maxilar y que se mueve, en última instancia, hacia el propio seno.
Dado que el nervio trigémino atraviesa el seno maxilar, las inmunoglobulinas humanas combinadas en el seno maxilar después de la administración en el mismo se moverán a lo largo del nervio trigémino hacia estructuras inervadas por el nervio trigémino. De este modo, la IgG humana combinada administrada a uno o ambos senos maxilares se administra al cerebro a través del nervio trigémino.
En una realización, el dispositivo de administración intranasal no invasivo administra una gota de líquido de una composición de IgG humana combinada en la cavidad nasal de un sujeto. En una realización particular, el dispositivo de administración intranasal no invasivo administra una gota de líquido de IgG humana combinada directamente a un epitelio nasal del sujeto. En una realización más específica, el dispositivo de administración intranasal no invasivo administra una gota de líquido de IgG humana combinada directamente al epitelio olfativo del sujeto. En una realización, se administra la gota de líquido inclinando la cabeza del sujeto hacia atrás y administrando la gota en una narina del sujeto. En otra realización, se administra la gota de líquido insertando la punta de un dispositivo de administración intranasal no invasivo en una narina del sujeto y rociando o pulverizando la gota en la cavidad nasal del sujeto.
En otra realización, el dispositivo de administración intranasal no invasivo administra un líquido o un aerosol de polvo de una composición de IgG humana combinada a la cavidad nasal de un sujeto. En una realización particular, el dispositivo de administración intranasal no invasivo administra un líquido o un aerosol de polvo de IgG humana combinada directamente a un epitelio nasal del sujeto. En una realización más específica, el dispositivo de administración intranasal no invasivo administra un líquido o un aerosol de polvo de IgG humana combinada directamente al epitelio olfativo del sujeto.
En otra realización, el dispositivo de administración intranasal no invasivo administra una composición de polvo seco de composición de IgG humana combinada a la cavidad nasal de un sujeto. En una realización particular, el dispositivo de administración intranasal no invasivo administra una composición de polvo seco de IgG humana combinada directamente a un epitelio nasal del sujeto. En una realización más específica, el dispositivo de administración intranasal no invasivo administra una composición de polvo seco de IgG humana combinada directamente al epitelio olfativo del sujeto.
En una realización, el dispositivo intranasal es un dispositivo desechable de un solo uso. En otra realización, el dispositivo intranasal es un dispositivo de uso múltiple o repetido. En determinadas realizaciones, el dispositivo de un solo uso o de uso múltiple se mide previamente. En una realización específica, el dispositivo de un solo uso o de uso múltiple está precargado. En determinadas realizaciones, el dispositivo de uso múltiple o repetitivo es recargable. En determinadas realizaciones, el dispositivo se recarga insertando una composición de IgG humana combinada en una cámara del dispositivo. En otras realizaciones, una cámara del dispositivo de uso múltiple o repetido diseñado para contener la composición combinada de IgG humana se reemplaza por una nueva cámara precargada.
Los ejemplos no limitativos de dispositivos de administración intranasal comerciales incluyen la bomba de pulverización nasal Equadel® (Aptar Pharma), el dispositivo de polvo seco Solovent (BD Technologies), el dispositivo de administración nasal de fármacos Unidose (Consort Medical PLC), el nebulizador nasal NasoNeb® (Medlnvent, LLC), el dispositivo de administración nasal VeriDoser® (Mystic Pharmaceuticals), el dispositivo de administración nasal VRx2™ (Mystic Pharmaceuticals), el dispositivo nasal DirectHaler™ (Direct-Haler A/S), el inhalador de polvo seco de un solo uso TriViar™ (Trimel Pharmaceuticals), el sistema de administración de aerosol SinuStar™ (Pari USA), la bomba Aero (Aero Pump GmbH), el dispositivo de administración nasal Fit-Lizer™ (Shin Nippon Biomedical Laboratories), el dispositivo LMA MAD Nasal™ (LMA North America, Inc.), el sistema de administración de gel bioadhesivo intranasal Compleo (Trimel Pharmaceuticals), el dispositivo de administración olfatoria presurizado (POD) Impel (Impel Neuropharma), el atomizador electrónico ViaNase™ (Kurve Technology, Inc.), el dispositivo de administración de polvo OptiNose (OptiNose US Inc.) y el dispositivo de administración de líquido Optinose (OptiNose US Inc.)
Ejemplos
Ejemplo 1 - Presencia de anticuerpos anti-ApoE4 en IgG humana combinada
El gen de la apolipoproteína E (apoE) ha sido ligado genéticamente a la aparición de la enfermedad de Alzheimer (Ertekin-Taner N., Neurol Clin., 25:611-667 (2007)). Además, el polimorfo ApoE4 (una importante isoforma del gen apoE, caracterizada por los residuos R112 y R158) se ha indicado en la etiología de la enfermedad de Alzheimer, donde puede desempeñar un papel en la modulación diferencial de los niveles de p-amiloide (Ap) a través de la formación de un complejo ApoE4-Ap. Varios investigadores, observando estas correlaciones, han explorado el uso de anticuerpos monoclonales anti-ApoE4 para el tratamiento de la enfermedad de Alzheimer (Tai et al., J. Biol Chem. 22 de febrero de 2013; 288(8):5914-26; Kim et al., J. Exp. Med. 19 de noviembre de 2012; 209(12):2149-56). Se realizaron ELISA con anticuerpo anti-ApoE para determinar si los anticuerpos anti-ApoE4 están presentes en las preparaciones de inmunoglobulina G derivadas de plasma disponibles comercialmente. En resumen, se determinó el contenido de anticuerpos anti-ApoE4 en plasma humano combinado (1R01B00) y un producto comercial líquido de
IgIV al 10% comercial (LE12K246) preparado a partir de plasma humano combinado. Tal como se muestra en la figura 1, se detectaron anticuerpos anti-ApoE4 tanto en plasma humano combinado (círculos) como en el producto de IgIV (triángulos), con un enriquecimiento de varias veces en la preparación de IgIV final.
Ejemplo 2 - Administración de inmunoglobulina G humana combinada para el tratamiento de la enfermedad de Alzheimer
Se realizó un estudio aleatorizado, doble ciego, controlado con placebo, con dos ramas, paralelo, sobre la seguridad y la eficacia de la administración de inmunoglobulina G intravenosa (IgIV) para el tratamiento de la enfermedad de Alzheimer de leve a moderada. El objetivo principal del estudio fue determinar si la IgIV, el tratamiento al 10% o bien a una dosis de 400 mg/kg de peso corporal (PC)/2 semanas o bien 200 mg/kg de PC/2 semanas durante 18 meses ralentiza la velocidad o previene la progresión de los síntomas de demencia en sujetos con enfermedad de Alzheimer (EA) de leve a moderada en comparación con placebo, tal como se mide mediante la subescala cognitiva de la escala de evaluación de la enfermedad de Alzheimer (ADAS-Cog) y el estudio cooperativo de la enfermedad de Alzheimer (ADCS) -Actividades de la vida diaria (ADL).
Otros objetivos del estudio incluyeron: si la IgIV, el tratamiento al 10% o bien a una dosis de 400 mg/kg de peso corporal/2 semanas o bien de 200 mg/kg de peso corporal/2 semanas durante 9 meses da como resultado una velocidad de progresión significativamente más lenta de los síntomas de demencia en sujetos con EA de leve a moderada en comparación con placebo, basándose en ADAS-Cog y ADCS-ADL; comparar el efecto de 400 mg/kg de PC/2 semanas con 200 mg/kg de PC/2 semanas sobre la velocidad de progresión de los síntomas de demencia tal como se determina mediante ADAS-Cog y ADCS-ADL a los 9 y 18 meses; evaluar el efecto de la IgIV, tratamiento del 10% durante 9 y 18 meses sobre medidas adicionales incluyendo ADCS-Impresión clínica global de cambio (ADCS-CGIC), el miniexamen de estado mental modificado (3MS) y las pruebas neuropsicológicas complementarias (cognición), el inventario neuropsiquiátrico (NPI) (conducta) y calidad de vida en la enfermedad de Alzheimer de Logsdon (QOL-AD) (calidad de vida); evaluar el impacto farmacoeconómico a corto plazo de la IgIV, administración al 10% durante 9 y 18 meses como farmacoterapia adicional en la EA de leve a moderada usando ADCS-inventario de uso de recursos (ADCS-RUI); para evaluar el impacto de la IgIV, tratamiento al 10% en sujetos con EA de leve a moderada durante 9 y 18 meses sobre la calidad de vida de sus cuidadores mediante el cuestionario de sobrecarga del cuidador; evaluar la seguridad y la tolerancia de dos dosis de IgIV, administrada al 10% cada semana durante 9 y 18 meses en sujetos con EA de leve a moderada; y evaluar un panel de plasma, líquido cefalorraquídeo (LCR) y biomarcadores de obtención de imágenes como medio para determinar si la IgIV, al 10% tiene efectos anti-amiloide y si los cambios en los biomarcadores del nivel inicial a los 9 meses predicen la subsiguiente estabilización o mejora en las medidas de resultados cognitivos, conductuales y funcionales a los 18 meses.
En resumen, 390 sujetos con EA probable con demencia de gravedad de leve a moderada fueron reclutados y se aleatorizaron a 44 centros dentro del consorcio ADCS en los Estados Unidos y Canadá. En el examen, cada sujeto se sometió a un miniexamen de estado mental (MMSE), así como a evaluaciones físicas, neurológicas y de analítica. Una vez determinada la elegibilidad, se realizaron evaluaciones clínicas y cognitivas de nivel inicial, así como evaluaciones de biomarcadores/obtención de imágenes, antes de la aleatorización.
Los criterios de inclusión clave para el estudio incluyeron que los sujetos (hombres y mujeres) tenían entre 50 y 89 años de edad en el momento del examen, se les había diagnosticado probablemente la enfermedad de Alzheimer, tenían demencia de leve (definida como MMES de 21-26) a moderada (definida como MMSE de 16-20) en el momento del examen, habían recibido dosis estables de medicación para la EA (inhibidor de la acetilcolinesterasa y/o memantina) durante al menos 12 semanas antes del examen, y tenían un cuidador capaz (compañero de estudio) que podía ayudar a facilitar la participación del sujeto.
Los criterios de exclusión clave para el estudio incluyeron que los sujetos tenían demencia no debida a Alzheimer, que residían actualmente en un centro especializado de enfermería, tenían problemas cardiovasculares clínicamente significativos (por ejemplo insuficiencia cardiaca congestiva, trastorno de la coagulación, hipertensión no controlada, angina inestable reciente o infarto de miocardio), tenían trombosis central o periférica reciente o enfermedad tromboembólica, tenían hallazgos específicos en una IRM cerebral (por ejemplo, 2 o más microhemorragias, accidente cerebrovascular grave o múltiples lagunas), tenían un traumatismo craneal reciente con pérdida de consciencia, contusión (cerebral) o lesión abierta en la cabeza, tenían un trastorno de convulsiones incontroladas (por ejemplo, >2 crisis episódicas al año a pesar del tratamiento adecuado con fármacos antiepilépticos), tenían una puntuación de Hachinski modificado > 4 en el momento del examen, tenían un tumor maligno, con la excepción de lo siguiente: carcinoma de células basales o células escamosas de la piel tratado adecuadamente, carcinoma in situ de cuello uterino y cáncer de próstata estable que no requiere tratamiento, tiene un trastorno autoinmunitario o neuroinmunológico activo, tuvo una depresión mayor no tratada, psicosis u otro(s) trastorno(s) psiquiátrico(s) importante(s), tuvo diabetes mal controlada (HbA1c > 7,0%), tuvo una enfermedad renal activa, tuvo otras alteraciones de la analítica clínicamente significativas (incluidos niveles de viscosidad plasmática anómalamente altos; serología positiva para VHB, VHC o VIH) tienen una deficiencia de IgA grave (<7 mg/dl), recibieron tratamiento con IgIV en los 5 años antes del examen, habían recibido tratamiento con cualquier producto(s) biológico(s) en investigación (por ejemplo, inmunización activa o inmunoterapias pasivas con anticuerpos monoclonales o
policlonales) para la EA en cualquier momento, o cualquier fármaco en investigación para la EA en el plazo de los 3 meses anteriores al examen, o estaban participando actual o recientemente en cualquier otro estudio con fármacos o dispositivos en investigación.
Los sujetos que cumplieron con los criterios de elegibilidad y completaron con éxito las evaluaciones de nivel inicial se aleatorizaron en una razón de 1:1:1 para recibir infusiones intravenosas (i.v.) de cualquiera de dos dosis de IgIV, al 10% o placebo (0,25% de albúmina humana) cada dos semanas durante 70 semanas (un total de 36 infusiones) como adición a la farmacoterapia para la EA aprobada por la Administración de Alimentos y Fármacos (FDA) convencional. Los grupos de tratamiento se asignaron de la siguiente manera: Grupo 1: IgIV, al 10% 400 mg/kg de PC/2 semanas; Grupo 2: IgIV, al 10% 200 mg/kg de PC/2 semanas; y grupo 3: placebo (0,25% de albúmina humana) en una dosis de: 4 ml/kg de PC/2 semanas, o 2 ml/kg de PC/2 semanas.
Las infusiones intravenosas de IgIV, al 10% o placebo se administraron cada dos semanas durante 70 semanas (un total de 36 infusiones), seguidas de un periodo de seguimiento de 6 semanas sin IgIV, tratamiento al 10%/ con placebo. Se estudiaron dos niveles de dosis de IgIV, 10% (400 mg/kg de PC/2 semanas y 200 mg/kg de PC/2 semanas). Para mantenerse ciegos, la mitad de los sujetos con placebo recibieron un alto volumen de infusión (4 ml/kg de PC/2 semanas) y la otra mitad un bajo volumen de infusión (2 ml/kg de PC/2 semanas) para igualar los 400 mg/kg y 200 mg/kg IgIV, dosis al 10%, respectivamente. En la figura 2 se proporciona un diagrama esquemático que representa el flujo del estudio.
El estudio tenía la potencia para comparar los cambios medios con respecto al nivel inicial en ADAS-Cog y ADCS-ADL a los 18 meses entre los 0,4 g/kg de PC de IgIV, el grupo al 10% y el grupo de placebo usando un modelo de análisis de covarianza (ANCOVA) que tenía en cuenta el efecto del tratamiento y valor de nivel inicial como covariable. Se hicieron las siguientes suposiciones para el cálculo del tamaño de muestra: la desviación estándar (DE) de la puntuación de cambio a los 18 meses es de 8 para ADAS-Cog y de 11 para ADCSADL, la correlación de la puntuación de cambio con el nivel inicial es de 0,75 para ADAS-Cog y de 0,79 para ADCS-ADL.
86 sujetos evaluables por rama proporcionan un 80% de potencia para detectar una diferencia media de 3,24 puntos en ADAS-Cog y una diferencia media de 4,52 puntos en ADCS-ADL entre los 0,4 g/kg de PC de IgIV, el grupo al 10% y los grupos de placebo, a un nivel de significación del 5%. Considerando una tasa de exclusión del 33%, aproximadamente 385 sujetos se aleatorizarán a uno de los tres grupos (dos grupos de tratamiento y un grupo de placebo) con una razón de aleatorización de 1:1:1.
Se monitorizaron los sujetos durante el transcurso del ensayo mediante la evaluación periódica de diversas evaluaciones cognitivas, evaluaciones clínicas, conductuales y funcionales, evaluaciones de calidad de vida y una evaluación de la utilización de recursos de atención sanitaria. En las figuras 3A-3E se proporciona un programa de las evaluaciones.
De forma global, el tratamiento de pacientes con Alzheimer mediante la administración de 400 mg/kg/2 semanas de IgIV dio como resultado una progresión reducida de demencia, tal como se mide mediante el análisis del miniexamen de estado mental modificado (3MS), en comparación con la administración de placebo (p1 = 0,206) y la administración de 200 mg/kg/2 semanas de IgIV (figura 4).
También se clasificaron los pacientes en subpoblaciones de individuos con enfermedad de Alzheimer leve (definida como MMSE de 21-26), individuos con enfermedad de Alzheimer moderada (definida como MMSE de 16-20), individuos que eran portadores de Apo4E (por ejemplo, que tenían al menos un alelo Apo4E) e individuos que eran negativos para Apo4E (por ejemplo, que no tienen alelos ApoE), para un análisis de subpoblaciones adicional. El genotipo ApoE4 y la distribución de alelos para este estudio se proporcionan en la figura 39.
A pesar de que el estudio no tenía potencia para comparar los cambios medios con respecto al nivel inicial en ADAS-Cog, ADCS-ADL y 3MS a los 18 meses entre las subpoblaciones de 0,4 g/kg de PC de IgIV, el grupo al 10% y las subpoblaciones del grupo placebo usando un modelo de análisis de covarianza (ANCOVA), que tiene en cuenta el efecto del tratamiento y el valor de nivel inicial como covariable, se observaron varios resultados significativos al analizar los datos de subpoblaciones de estado de enfermedad y de estado de ApoE4.
Sorprendentemente, la administración de 400 mg/kg/2 semanas de IgIV a portadores de ApoE4 (por ejemplo, sujetos que tienen al menos un alelo Apo4E) dio como resultado una reducción mucho mayor de la progresión de demencia, en comparación con los sujetos que recibieron placebo (p = 0,012) o 200 mg/kg/2 semanas de IgIV, que toda la cohorte de Alzheimer (por ejemplo, sin separación por subpoblación). Esto se ilustra en la figura 5, que muestra las puntuaciones promedio del miniexamen de estado mental modificado (3MS) en los meses 9 y 18 del ensayo. El tratamiento con 200 mg/kg/2 semanas de IgIV no redujo la progresión de demencia en los portadores de ApoE4.
A la inversa, la administración de 200 mg/kg/2 semanas de IgIV y 400 mg/kg/2 semanas de IgIV a sujetos negativos para ApoE4 (por ejemplo, sujetos que no tienen un alelo Apo4E) no ralentizó la progresión de demencia, medida mediante análisis de miniexamen de estado mental modificado (3MS), en comparación con la administración de
placebo (figura 6).
Además, la administración de 200 mg/kg/2 semanas de IglV y 400 mg/kg/2 semanas de IglV a sujetos a los que se les ha diagnosticado enfermedad leve (por ejemplo, sujetos con una puntuación de MME de 21-26) no ralentizó la progresión de demencia, medida mediante análisis del miniexamen de estado mental modificado (3MS), en comparación con la administración de placebo (figura 7).
Sin embargo, la administración de 400 mg/kg/2 semanas de IgIV a sujetos a los que se les ha diagnosticado enfermedad moderada (por ejemplo, sujetos con una puntuación de MME de 16-20) dio como resultado una mayor reducción de la progresión de demencia, en comparación con los sujetos que recibieron placebo (p = 0,067) o 200 mg/kg/2 semanas de IgIV, que toda la cohorte de Alzheimer (por ejemplo, sin separación por subpoblación). Esto se ilustra en la figura 8, que muestra las puntuaciones promedio del miniexamen de estado mental modificado (3MS) en los meses 9 y 18 del ensayo (figura 8a ) y las puntuaciones ADAS-Cog cada tres meses durante el ensayo (figura 8C). El tratamiento con 200 mg/kg/2 semanas de IgIV no redujo la progresión de demencia en sujetos con enfermedad moderada, tal como se evaluó mediante 3MS o ADAS-Cog. La figura 8B muestra que el efecto positivo del tratamiento con IgIV a dosis alta también es estadísticamente significativo entre los portadores de Apo4E con enfermedad de Alzheimer moderada (p = 0,011).
Tal como se muestra en la figura 9, el tratamiento con IgIV a dosis alta (por ejemplo, 400 mg/kg/2 semanas de IgIV) también disminuyó la progresión de demencia en pacientes con enfermedad moderada (por ejemplo, sujetos con una puntuación de MME de 16-20), tal como se evaluó mediante la subescala cognitiva de la escala de evaluación de la enfermedad de Alzheimer (ADAS-Cog).
Las figuras 32 a 36 proporcionan representaciones gráficas de la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al valor inicial para los sujetos que recibieron IgIV a dosis alta y placebo a los 18 meses para exámenes mediante ADAS-Cog, 3MS, CGIC, dibujo de reloj y Trail B, respectivamente. El miniexamen de estado mental (MMSE) es un instrumento de examen cognitivo que se valida y se usa ampliamente en la práctica clínica y, a menudo, se emplea como medida de la gravedad de los síntomas en los estudios con fármacos para la EA. El MMSE proporciona una calificación compuesta de 30 puntos para orientación espaciotemporal, memoria verbal, atención simple, memoria de trabajo, nombramiento, repetición, comprensión, escritura y habilidades de construcción. Las puntuaciones oscilan desde 0 hasta 30, indicando los valores más bajos un mayor deterioro. Los sujetos con puntuaciones de MMSE de 16-26 inclusive fueron elegibles para este estudio. El MMSE se realizó en el examen para confirmar la elegibilidad. Las puntuaciones de MMSE posteriores al examen derivaron del examen de 3MS realizado en el nivel inicial, durante las visitas de 9 M y 18 M y en la visita de fin del estudio. El MMSE proporciona una métrica con la que están familiarizados muchos médicos en ejercicio y se incluyó como medida de seguridad. Para su revisión, véase, Folstein MF, Folstein SE, McHugh PR. “Mini-mental state” A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res., 12:189-198 (1975). El miniexamen de estado mental modificado (3MS) es una herramienta de examen cognitivo validada exhaustiva que conserva la brevedad, la facilidad de administración y la puntuación objetiva del MMSE, pero proporciona un intervalo más amplio y una puntuación más refinada. Las puntuaciones oscilan desde 0 hasta 100, indicando los valores más bajos un mayor deterioro. El 3MS se realizó en el nivel inicial, durante las visitas de 9 M y 18 M, y en la visita de fin del estudio. El 3MS proporciona una métrica con la que están familiarizados muchos médicos en ejercicio y se incluirá en los análisis secundarios. Para su revisión, véase, Teng EL, Chui HC. The Modified Mini Mental State (3MS) examination, J. Clin. Psychiatry, 48: 314-318 (1987).
La subescala cognitiva de la escala de evaluación de la enfermedad de Alzheimer (ADAS-Cog) está validada y se usa ampliamente como medida de resultado cognitivo primario en los estudios de farmacoterapia para la EA. Este es un instrumento psicométrico que evalúa la memoria (recuerdo de palabras, reconocimiento de palabras), atención, razonamiento (siguiendo órdenes), lenguaje (nombramiento, comprensión), orientación, praxis ideacional (introducir una carta en un sobre) y praxis de construcción (copiar diseños geométricos). La puntuación está en el intervalo de 0 a 70, indicando una puntuación más alta un mayor deterioro. Esta prueba la aplicaron evaluadores experimentados en cada sitio en el nivel inicial, cada 3 meses durante las visitas 3 M, 6 M, 9 M, 12 M, 15 M y 18 M, y en la visita de fin del estudio, o en la visita de terminación temprana. El ADASCog fue la principal medida de resultado cognitivo para este estudio. Para su revisión, véase, Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am. J. Psychiatry 141:1356-1364 (1984).
ADCS-Actividades de la vida diaria (ADCS-ADL) es una herramienta validada para evaluar las actividades básicas e instrumentales de la vida diaria basándose en una entrevista estructurada de 23 puntos del cuidador o compañero de estudio calificado. La escala tiene un intervalo de 0 a 78, indicando las puntuaciones más bajas un mayor deterioro. El ADCS-ADL fue la medida primaria del estado funcional de los sujetos en este estudio y se evaluó en el nivel inicial, durante las visitas de 9 M y 18 M, y en la visita de fin del estudio. Para su revisión, véase, Galasko D, Bennett D, Sano M et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease, The Alzheimer's Disease Cooperative Study, Alzheimer Dis. Assoc. Disord.; 11 supl. 2:S33-S39 (1997).
Ejemplo 3 - Análisis de la administración de IglV en sujetos con enfermedad de Alzheimer moderada
Los resultados del estudio de tratamiento con IglV presentado en el ejemplo 2 se reevaluaron usando criterios modificados para definir la enfermedad de Alzheimer leve y moderada. Se encontró que al aumentar la potencia del estudio (por ejemplo, el número de individuos en la cohorte de enfermedad moderada) al incluir pacientes adicionales con enfermedad de Alzheimer avanzada que se clasificaron originariamente como que tenían enfermedad moderada, que el tratamiento con IgIV a dosis alta de sujetos con enfermedad moderada tiene un efecto estadísticamente significativo.
El estudio presentado en el ejemplo 2 definió que los sujetos con enfermedad de Alzheimer moderada tenían una puntuación de MMSE de 20 o menos (por ejemplo, eficazmente MMSE = 16-20, inclusive porque no se admitieron en el estudio individuos con una puntuación de MMSE inferior a 16). Las evaluaciones cognitivas iniciales de sujetos que tenían enfermedad moderada, usando los exámenes cognitivos ADAS-Cog y 3MS, sugirieron una tendencia positiva en la ralentización de la progresión de la enfermedad con la administración de IgIV a dosis alta (p = 0,083 y p = 0,067 para los exámenes de ADAS-Cog y 3MS, respectivamente). Sin embargo, tal como se muestra en la tabla 1, el análisis de los datos usando cohortes de enfermedad moderada redefinidas, incluidos los sujetos con puntuaciones de MMSE de 21 y 22, muestra que los sujetos con enfermedad de Alzheimer moderada (por ejemplo, MMSE de 14 a 22, inclusive) se benefician significativamente del tratamiento con IgIV a alta dosis. Estos datos indican que todos los individuos con enfermedad de Alzheimer moderada, independientemente del estado de ApoE4, pueden beneficiarse de un tratamiento con IgIV a alta dosis.
Tabla 1. Diferencia en el cambio en la puntuación del examen ADAS-Cog y 3MS con respecto al nivel inicial en pacientes con enfermedad de Alzheimer moderada tratados con IgIV a dosis alta (0,4 g/kg/2 semanas) en comparación con el placebo.

Este resultado positivo se ilustra en la figura 40, que notifica las puntuaciones promedio de ADAS-Cog (figura 40A) y 3MS (figura 40B) tomadas cada tres meses durante el ensayo para pacientes a los que se les ha diagnosticado enfermedad de Alzheimer moderada (MMSE < 22) y leve (MMSE > 23). Tal como se muestra en la figura 40, el tratamiento de pacientes con enfermedad moderada con 400 mg/kg/2 semanas de IgIV redujo la progresión de demencia en sujetos con enfermedad de Alzheimer moderada, pero no leve. El tratamiento con 200 mg/kg/2 semanas de IgIV no redujo la progresión de demencia en sujetos con enfermedad leve o moderada, tal como se evaluó mediante ADAS-Cog o 3MS.
Tal como se notificó en la tabla 2 y la tabla 3, el análisis adicional de los datos recopilados para el estudio notificado en el ejemplo 1 muestra que el tratamiento de sujetos a los que se les ha diagnosticado inicialmente enfermedad de Alzheimer moderada y/o que portan un alelo ApoE4 con dosis alta (400 mg/kg/2 semanas), pero no con dosis baja (200 mg/kg/2 semanas), la IgIV redujo la progresión de demencia, tal como se evaluó mediante ADAS-Cog (p = 0,026 frente a placebo) o 3MS (p = 0,032 frente a placebo), respectivamente. De manera similar, el tratamiento de la misma cohorte de pacientes con dosis alta (400 mg/kg/2 semanas), pero no con dosis baja (200 mg/kg/2 semanas), la IgIV durante 18 meses ralentizó la reducción de la puntuación de fluidez verbal FAS con respecto al nivel inicial (p = 0,031 frente a placebo; tabla 4) y la puntuación en parte B de prueba de unir los puntos con respecto al nivel inicial (p = 0,079; tabla 5).
Tabla 2. Diferencia en el cambio en la puntuación del examen de ADAS-Cog con respecto al nivel inicial, excluyendo a los sujetos con MMSE > 22 que son negativos para ApoE4, tratados con IgIV a dosis alta (0,4 g/kg/2 semanas), IgIV a dosis baja (0,2 g/kg/2 semanas) y placebo.
Visita 0,4 g/kg 0,2 g/kg Placebo
N Media D.E. N Media D.E. N Media D.E. Mes 18 94 7,3 8,08 87 9,2 8,44 83 9,5 9,30 p = 0,026
frente a


Tabla 3. Diferencia en el cambio en la puntuación del examen de 3MS con respecto al nivel inicial, excluyendo a los sujetos con MMSE > 22 que son negativos para ApoE4, tratados con IgIV a dosis alta (0,4 g/kg/2 semanas), IgIV a dosis baja (0,2 g/kg/2 semanas) y placebo.

Tabla 4 Diferencia en el cambio en la puntuación de fluidez verbal FAS con respecto al nivel inicial, excluyendo a los sujetos con MMSE > 22 que son negativos para ApoE4, tratados con IgIV a dosis alta (0,4 g/kg/2 semanas), IgIV a dosis baja (0,2 g/kg/2 semanas) y placebo.
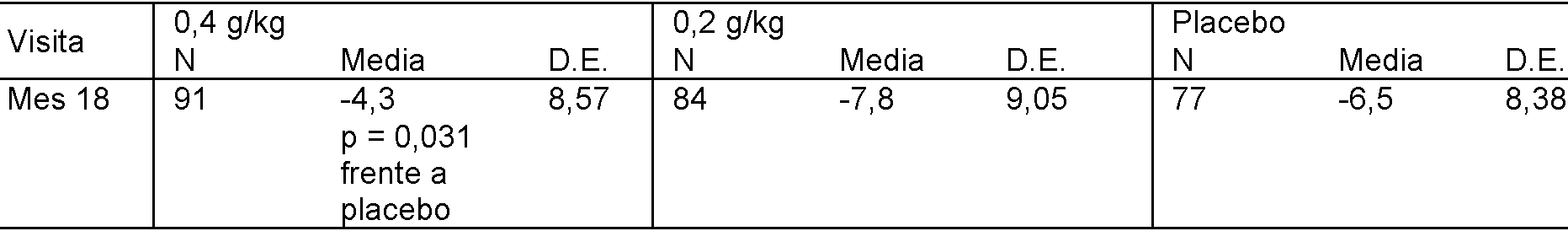
Tabla 5 Diferencia en el cambio en la puntuación de la parte B de la prueba de unión de puntos (Trail B), excluyendo a los sujetos con MMSE> 22 que son negativos para ApoE4, tratados con IgIV a dosis alta (0,4 g/kg/2 semanas), IgIV a dosis baja (0,2 g/kg/2 semana) y placebo.

De forma global, este estudio muestra que los sujetos con enfermedad de Alzheimer moderadamente grave y los sujetos que son portadores de ApoE4 pueden beneficiarse de la terapia con IgIV. Los portadores de ApoE4 a los que se les ha diagnosticado enfermedad de Alzheimer moderadamente grave parecen ser los que más se benefician de la terapia con IgIV, tal como se mide mediante los exámenes de 3MS y ADAS-Cog. Esto es sorprendente dado que estudios anteriores han sugerido que la presencia de un alelo ApoE4 limita la eficacia y la seguridad terapéutica. Por ejemplo, el alelo ApoE4 se ha asociado fuertemente con la incidencia de edema vasogénico, que no se observó en este estudio. Estos resultados sugieren que la terapia con IgIV se basa en un mecanismo de acción diferente al de la terapia con anticuerpos monoclonales.
Ejemplo 4 - Análisis de biomarcadores en sujetos con Alzheimer a los que se les administró IgIV o placebo
Para evaluar adicionalmente la eficacia de la administración de inmunoglobulina G intravenosa (IgIV) para el tratamiento y/o el control de la enfermedad de Alzheimer, se investigaron los niveles de biomarcadores de los sujetos que participaron en el estudio descrito en el ejemplo 2. Los resultados de estos análisis refuerzan aún más la conclusión de que la administración de altas dosis de IgIV (por ejemplo, 0,3-0,8 g/kg/2 semanas) es beneficiosa para los sujetos con enfermedad moderada, y especialmente para los portadores del gen ApoE4.
Los biomarcadores que se investigaron en el estudio incluyeron: los niveles de Ap40 y AI342 en plasma y líquido cefalorraquídeo (LCR) a los 9 y 18 meses; títulos de anticuerpos anti-Ap40 y anti-Ap42 en plasma y líquido cefalorraquídeo (LCR) a los 9 y 18 meses (incluidos los anticuerpos anti-monómero, anti-oligómero y anti-fibrilla); niveles de proteína tau total y fosforilada en el LCR a los 9 y 18 meses; IRM volumétrica, incluyendo hiperplasia ventricular, volumen ventricular total, así como atrofia total del cerebro y del hipocampo a los 9 y 18 meses; metabolismo cerebral de glucosa usando obtención de imágenes mediante tomografía por emisión de positrones (PET) de [18F]-2-fluorodesoxiglucosa (18F-FDG) a los 9 meses; y deposición de amiloide cerebral usando obtención de imágenes mediante PET de [18F]-lorpiramina (18F-AV-45) a los 18 meses. Los resultados de LCR, FDG-PET y AV-45 PET se midieron solo en el subgrupo de sujetos (objetivo de 40 sujetos en cada grupo de tratamiento para los subestudios de LCR y FDG-PET, y objetivo de 33 sujetos en cada grupo de tratamiento para el subestudio AV-45 PET).
Tomografía por emisión de positrones de 18F-FDG para el metabolismo cerebral de glucosa
A los 6 meses, se encontró que los sujetos con Alzheimer tratados con IglV a dosis alta (0,4 kg/2 semanas) mostraron una mejoría en el metabolismo cerebral de glucosa en ambos hemisferios del cerebro. Normalmente, el metabolismo cerebral de glucosa disminuye en del 10% al 20% por año en pacientes con Alzheimer no tratados. En la figura 10A se muestra un patrón temporoparietal y prefrontal típico de hipometabolismo de glucosa, obtenido como imagen mediante obtención de imágenes de tomografía por emisión de positrones de (18F)-2-fluorodesoxiglucosa (18F-FDG) convencional, para sujetos tratados con placebo. En cambio, los sujetos a los que se administró IgIV a dosis alta (0,4 g/kg/2 semanas) mostraron una mejoría en ambos hemisferios. En la figura 10B se muestra un patrón temporoparietal y prefrontal típico de hipometabolismo de glucosa, con obtención de imágenes como antes, para sujetos a los que se administró IgIV a dosis alta. En la figura 11 se muestra un resumen del metabolismo de glucosa en todas las cohortes y subpoblaciones de pacientes.
IRM volumétrica
La pérdida neuronal en el envejecimiento normal provoca atrofia cerebral. Esto se recrudece en pacientes con Alzheimer, donde la degeneración neuronal en la enfermedad de Alzheimer (EA) provoca atrofia cerebral acelerada. Debido a que el cráneo es un espacio cerrado, la atrofia cerebral provoca una hiperplasia progresiva de los ventrículos cerebrales llenos de líquido. Por tanto, la tasa de hiperplasia ventricular a lo largo del tiempo proporciona una medida objetiva de la velocidad de progresión de la enfermedad de Alzheimer.
Se determinó el volumen ventricular mediante IRM volumétrica para un subconjunto de sujetos incluidos en el estudio descrito en el ejemplo 2. A los 18 meses, se encontró una tendencia positiva para los sujetos portadores de ApoE4 con enfermedad de Alzheimer moderada que recibieron IgIV a dosis alta (0,4 g/kg/2 semanas) (p = 0,140), tal como se muestra en la tabla 2, a continuación. En las figuras 12A y 12B, se muestran imágenes que muestran la atrofia ventricular típica en los cerebros de los sujetos con Alzheimer de los grupos de tratamiento con placebo e IgIV a dosis alta, respectivamente. En la figura 11 se muestra un resumen del cambio medio con respecto al nivel inicial de la IRM volumétrica (normalizada con el volumen intracraneal de nivel inicial) en todas las cohortes y subpoblaciones de pacientes. En la figura 37 se proporciona una representación gráfica de la media y los intervalos de confianza del 95% para las diferencias entre los cambios con respecto al nivel inicial para sujetos que reciben IgIV a dosis alta y placebo a los 18 meses, tal como se mide mediante IRM volumétrica.
Tabla 6 Cambios en el volumen ventricular con respecto al nivel inicial a los 18 meses en sujetos portadores de ApoE4 con enfermedad de Alzheimer moderada.

Deposición de amiloide cerebral usando obtención de imágenes mediante PET de r18F1-florpiramina (18F-AV-45) El florbetapir es un compuesto radiofarmacéutico para exploración mediante PET que contiene el radionúclido flúor-18, aprobado recientemente por la FDA como herramienta de diagnóstico para la enfermedad de Alzheimer, que se une a los agregados de amiloide en el cerebro. La unión a florbetapir se analizó en un subgrupo de sujetos incluidos en el estudio descrito en el ejemplo 2. Tal como se muestra en la tabla 3 a continuación, las tomografías PET mostraron disminuciones de la unión a florbetapir en todos los sujetos que recibieron tratamiento con IgIV a dosis alta (0,4 g/kg/2 semanas) que fueron aún más pronunciadas en los portadores de ApoE4 que recibieron IgIV a dosis alta. Estas disminuciones son indicativas de disminuciones de la carga de amiloide en los cerebros de estos sujetos. En la figura 11 se muestra un resumen del cambio medio con respecto al nivel inicial de la unión a florbetapir en todas las cohortes y subpoblaciones de pacientes que recibieron 0,4 g/kg/2 semanas de IgIV.
Tabla 7 Cambios en la obtención de imágenes con florpiramina con respecto al nivel inicial a los 18 meses en sujetos portadores de ApoE4 con enfermedad de Alzheimer moderada.

Niveles de proteína AB40 y anticuerpos anti-AB40 en plasma
Se determinaron los niveles en plasma del péptido Ap40 y del anticuerpo anti-Ap40 para los sujetos incluidos en el estudio descrito en el ejemplo 2. De manera global, no hubo cambios significativos en los niveles en plasma del péptido Ap40 o del anticuerpo anti-Ap40 en ninguna de las cohortes de tratamiento. En las figuras 13 y 14, respectivamente, se encuentran resúmenes de cambio medio con respecto al nivel inicial de los niveles en plasma de péptido Ap40 y de anticuerpos anti-Ap40 para todas las cohortes y subpoblaciones de pacientes.
Niveles de proteína AP42 y anticuerpos anti-AP42 en plasma
Se determinaron los niveles en plasma de péptido Ap42 y anticuerpos anti-Ap42 para los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, no hubo cambios significativos en los niveles en plasma de anticuerpos anti-Ap42 en ninguna de las cohortes de tratamiento. Sin embargo, tal como se muestra en la figura 15, los niveles en plasma del péptido Ap42 disminuyeron en cohortes de pacientes que recibieron 0,2 g/kg/2 semanas de IgIV (disminución media del 9%) y 0,4 g/kg/2 semanas de IgIV (disminución media del 21%). En las figuras 13 y 14, respectivamente, se encuentran resúmenes del cambio medio con respecto al nivel inicial de los niveles en plasma de péptido Ap40 y de anticuerpos anti-Ap40 para todas las cohortes y subpoblaciones de pacientes.
Tomados en conjunto, los datos anteriores demuestran que el tratamiento con IgIV reduce significativamente los niveles en plasma de péptido Ap42 de una manera dependiente de la dosis. El efecto opuesto se observa en los niveles en plasma de péptidos Ap40, que están aumentados en las cohortes de pacientes tratados con IgIV.
Niveles de péptido Ap40 en líquido cefalorraquídeo (LCR)
Se determinaron los niveles de péptido Ap40 en el LCR de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, se observó una pequeña disminución de los niveles de péptido Ap40 en el LCR para todas las cohortes, tal como se observa en los datos presentados en la figura 16. En la figura 17, se encuentra un resumen del cambio medio con respecto al nivel inicial de los niveles en LCR del péptido Ap40 para todas las cohortes y subpoblaciones de pacientes.
Niveles de péptido AB42 en líquido cefalorraquídeo (LCR)
Se determinaron los niveles de péptido Ap42 en el LCR de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, se observó una disminución moderada de los niveles de péptido Ap42 en el LCR para todas las cohortes, tal como se observa en los datos presentados en la figura 18. En las figuras 17, se encuentra un resumen del cambio medio con respecto al nivel inicial de los niveles en LCR del péptido Ap42 para todas las cohortes y subpoblaciones de pacientes.
Niveles de IgG en líquido cefalorraquídeo (LCR)
Se determinaron los niveles de IgG en el LCR de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, se observó un aumento moderado de los niveles de IgG en el LCR en sujetos que recibieron IgIV a dosis baja y un aumento mayor de los niveles de IgG en el LCR en sujetos que recibieron IgIV a dosis alta, tal como se observa en la figura 19. Se observó poco o ningún aumento de IgG en el LCR en los sujetos que recibieron placebo. Estos datos concuerdan con el paso de IgG a través de la barrera hematoencefálica. Curiosamente, antes del inicio del tratamiento con IgIV, los niveles iniciales de IgG en el LCR fueron significativamente menores (p = 0,038 (prueba de la t); tabla 8) en portadores de ApoE4 (2,2 mg/ml) que en sujetos negativos para ApoE4 (2,7 mg/ml).
Tabla 8 Niveles iniciales de IgG en el líquido cefalorraquídeo (LCR) de pacientes con Alzheimer positivos para ApoE4 y negativos para ApoE4.
Asimismo, se observaron aumentos moderados y grandes de los anticuerpos anti-fibrilla de Ap y anti-oligómero de Ap en el LCR de sujetos que recibieron IgIV a dosis baja y alta, tal como se muestra en las figuras 20 y 21, respectivamente. Se observaron aumentos similares de los niveles en LCR de anticuerpo anti-monómero de Ap en los grupos de tratamiento con IgIV, sin embargo, también se observó un aumento de los niveles en LCR de anticuerpo anti-monómero de Ap en sujetos que recibieron placebo, tal como se muestra en la figura 22. Se encuentran resúmenes del cambio medio con respecto al nivel inicial de IgG total y los subtipos de anticuerpos anti-Ap para todas las cohortes y subpoblaciones de pacientes en las figuras 23 y 24, respectivamente.
Tomados en conjunto, estos datos muestran que la IgG atraviesa la barrera hematoencefálica después de la administración intravenosa. Se observaron aumentos dependientes de la dosis de IgG total, anticuerpos antioligómero de Ap y anticuerpos anti-fibrillas de Ap en el LCR de pacientes en las cohortes de tratamiento con IgIV. Niveles de proteína tau en líquido cefalorraquídeo (LCR)
Tau es una proteína del axón que fomenta el ensamblaje y la estabilidad de microtúbulos y el transporte de vesículas. En el LCR, la tau se considera un biomarcador posterior usado para monitorizar los efectos sobre procesos patógenos posteriores tales como degeneración neuronal o la formación de ovillos intraneuronales de manera posterior al efecto primario anticipado de la intervención anti-Ap. Se determinaron los niveles de Tau en el LCR de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, no se observó ningún cambio significativo en el nivel de tau en LCR en ninguna de las cohortes de tratamiento, tal como se muestra en la figura 25. En la figura 26, se encuentra un resumen del cambio medio con respecto al nivel inicial de tau para todos los grupos y subpoblaciones de pacientes.
La tau fosforilada es insoluble, carece de afinidad por los microtúbulos y se autoasocia para dar estructuras de filamentos helicoidales apareados. El aumento de los niveles de tau fosforilada en el LCR se correlaciona con el deterioro cognitivo de la EA. Se determinaron los niveles de tau fosforilada en el LCR de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, no hubo ningún cambio significativo en el nivel de tau en LCR en ninguna de las cohortes de tratamiento, tal como se muestra en la figura 27. En la figura 27, se encuentra un resumen del cambio medio con respecto al nivel inicial para los niveles de LCR de tau fosforilada para todas las cohortes y subpoblaciones de pacientes.
Niveles de IgG en suero
Se determinaron los niveles de IgG en el suero sanguíneo de los sujetos incluidos en el estudio descrito en el ejemplo 2. De forma global, se observó un aumento moderado de los niveles en suero de IgG en sujetos que recibieron IgIV a dosis baja y un aumento mayor de los niveles en suero de IgG en sujetos que recibieron IgIV a dosis alta, tal como se observa en la figura 41. No se observó un aumento de la IgG en LCR en los sujetos que recibieron placebo.
Correlaciones de biomarcadores de obtención de imágenes con criterios de valoración primarios
Se encontraron correlaciones significativas entre los resultados del tratamiento con IgIV medidos usando biomarcadores de obtención de imágenes y evaluaciones cognitivas primarias. Tal como se muestra en la figura 38, las correlaciones más fuertes se observaron entre la evaluación del volumen ventricular mediante obtención de imágenes de IRM y ADCS-Cog o ADCS-ADL. También se observaron fuertes correlaciones entre la obtención de imágenes mediante PET AV-45 y la evaluación ADCS-ADL.
Ejemplo 5 - Perfil de seguridad del tratamiento con IgIV para la enfermedad de Alzheimer
De forma global, el estudio de tratamiento con IgIV para la enfermedad de Alzheimer descrito en el ejemplo 2 tuvo un excelente perfil de seguridad.
Las disminuciones de los niveles de hemoglobina y los signos clínicos de hemólisis se etiquetan como acontecimientos adversos para todos los medicamentos de IgIV vendidos comercialmente. Sin embargo, tal como se muestra en la figura 28, hubo un pequeño aumento de la aparición de niveles disminuidos de hemoglobina en el sujeto a quien se administró IgIV a dosis baja y alta. No hubo evidencia de hemólisis en ninguno de los sujetos. Además, los niveles de LDH de cada sujeto estaban dentro de un intervalo normal. Además, no hubo un aumento global de los efectos secundarios graves (figura 29) y solo un pequeño aumento de los acontecimientos adversos no graves (figura 30). Hubo un pequeño aumento de la aparición de una erupción que requería terapia, tal como se muestra en la figura 31.
Ejemplo 6 - Análisis de la administración de IgIV en sujetos clasificados con puntuaciones de florbetapir. > 1,2" Para identificar adicionalmente subpoblaciones de pacientes con Alzheimer que se beneficiarán del tratamiento con inmunoglobulina G humana combinada, los resultados del estudio de tratamiento con IgIV presentado en el ejemplo 2 se reevaluaron con respecto a la puntuación de florbetapir de los pacientes. Ventajosamente, se encontró que el tratamiento con IgG combinada a dosis alta (0,4 g/kg/2 semanas) proporcionó un beneficio terapéutico a los pacientes con una puntuación de florbetapir > 1,2. Estos resultados se contrastan con el tratamiento con IgG a dosis baja (0,2 g/kg/2 semanas) y placebo, que mostraron poco o ningún beneficio.
Las figuras 42 y 43 evidencian que la demencia progresó más lentamente en pacientes con puntuaciones de florbetapir > 1,2 que recibieron terapia con IgIV a dosis alta que en pacientes con puntuaciones de florbetapir <1,2
que recibieron terapia con IglV a dosis baja. Específicamente, se realizó un seguimiento de la progresión de demencia en pacientes que recibieron IgIV a dosis alta y baja durante un periodo de 18 meses usando los exámenes de ADAS-Cog (figura 42) y MMSE modificado (figura 43). La progresión de demencia ralentizada se demuestra por las puntuaciones más bajas de ADAS-Cog (figura 42) y las puntuaciones más altas de MMSE modificado (figura 43) para la cohorte de IgIV a dosis alta, normalizada para la cohorte de placebo, en comparación con la cohorte de IgIV a dosis alta. Estos resultados muestran que el tratamiento con IgG a dosis baja proporcionó poco o ningún beneficio, en comparación con el placebo, mientras que IgG a dosis alta redujo la progresión de demencia en pacientes con puntuaciones de florbetapir > 1,2. La figura 44 muestra que la administración de IgIV a dosis alta, pero no de dosis bajas, redujo los niveles de péptido Ap42 en LCR en pacientes con puntuaciones de florbetapir > 1,2.
Tomados en conjunto, estos resultados muestran que los pacientes con Alzheimer con una puntuación de florbetapir > 1,2 pueden beneficiarse de la terapia con IgIV a dosis alta, por ejemplo, de 300 mg de IgG/kg a 800 mg de IgG/kg de peso corporal del sujeto por periodo de dos semanas.
Ejemplo 7 - Análisis de la administración de IgIV en sujetos con y sin placas amiloides
Para identificar adicionalmente subpoblaciones de pacientes con Alzheimer que se beneficiarán del tratamiento con inmunoglobulina G humana combinada, los resultados del estudio de tratamiento con IgIV presentado en el ejemplo 2 se reevaluaron con respecto a si los pacientes presentaban o no placas amiloides. Ventajosamente, se encontró que el tratamiento con IgG combinada a dosis alta (0,4 g/kg/2 semanas) proporcionaba un beneficio terapéutico a los pacientes sin placas amiloides.
Por ejemplo, las figuras 45, 47 y 49 evidencian que la demencia progresó más lentamente en sujetos sin placas amiloides que recibieron terapia con IgIV a dosis alta que en sujetos sin placas amiloides que recibieron terapia con IgIV a dosis baja. Específicamente, se realizó un seguimiento de la progresión de demencia en pacientes que recibieron IgIV a dosis alta y baja durante un periodo de 18 meses mediante los exámenes de ADAS-Cog (figura 45), ADAS-CGIC (figura 47) y MMSE modificado (figura 49). La progresión de demencia ralentizada se demuestra con las puntuaciones más bajas de ADAS-Cog y ADAS-CGIC (figuras 45 y 47, respectivamente) y las puntuaciones de MMSE modificado más altas (figura 49) para la cohorte de IgIV a dosis alta, normalizada para la cohorte de placebo, en comparación con la cohorte de IgIV a dosis baja. Estos resultados muestran que el tratamiento con IgG a dosis baja proporcionó poco o ningún beneficio, en comparación con el placebo, mientras que IgG a dosis alta redujo la progresión de demencia en sujetos sin placas amiloides.
Se muestran evidencias adicionales de los resultados beneficiosos por el volumen ventricular reducido (figura 51), determinado mediante IRM volumétrica tal como se describió anteriormente, y un aumento de la razón de valor de captación compuesta normalizada (SUVR; figura 53) en pacientes con Alzheimer sin placas amiloides que recibieron terapia con IgG a dosis alta (0,4 g/kg/2 semanas). De manera acorde a estos hallazgos, la administración de IgG a dosis alta redujo el cambio con respecto al nivel inicial para los niveles de péptido Ap42 en plasma en pacientes con Alzheimer sin placas amiloides (figura 55).
Tomados en conjunto, estos resultados muestran que los pacientes con Alzheimer sin placas amiloides pueden beneficiarse de la terapia con IgIV a dosis alta, por ejemplo, de 300 mg de IgG/kg a 800 mg de IgG/kg de peso corporal del sujeto por periodo de dos semanas.
Claims (15)
- REIVINDICACIONESi. Composición que comprende una cantidad terapéuticamente eficaz de inmunoglobulina G (IgG) humana combinada para su uso en el tratamiento de la enfermedad de Alzheimer en un sujeto con enfermedad de Alzheimer moderadamente grave que es portador de al menos un alelo APOE4,en la que la cantidad de IgG humana combinada es de desde 300 mg/kg hasta 800 mg/kg de peso corporal del sujeto por periodo de dos semanas,en la que la cantidad se administra en una o más dosis durante el periodo de dos semanas después del inicio de un régimen terapéutico, yen la que el sujeto tiene una puntuación del miniexamen de estado mental (MMSE) de desde 14 hasta 22.
- 2. Composición para su uso según la reivindicación 1, en la que el sujeto es homocigoto para el alelo APOE4.
- 3. Composición para su uso según la reivindicación 1, en la que el sujeto es heterocigoto para el alelo APOE4.
- 4. Composición para su uso según la reivindicación 3, en la que el sujeto tiene un genotipo APOE4/APOE3.
- 5. Composición para su uso según la reivindicación 3, en la que el sujeto tiene un genotipo APOE4/APOE2.
- 6. Composición para su uso según cualquiera de las reivindicaciones 1 a 5, en la que la cantidad de IgG humana combinada se administra en una dosis única durante el periodo de dos semanas.
- 7. Composición para su uso según cualquiera de las reivindicaciones 1 a 5, en la que la cantidad de IgG humana combinada se administra en dosis múltiples durante el periodo de dos semanas.
- 8. Composición para su uso según la reivindicación 7, en la que la dosis se administra cada semana.
- 9. Composición para su uso según cualquiera de las reivindicaciones 1 a 8, en la que la cantidad de IgG humana combinada es de 400 mg/kg de peso corporal del sujeto por periodo de dos semanas.
- 10. Composición para su uso según cualquiera de las reivindicaciones 1 a 8, en la que la cantidad de IgG humana combinada es de 600 mg/kg de peso corporal del sujeto por periodo de dos semanas.
- 11. Composición para su uso según cualquiera de las reivindicaciones 1 a 8, en la que la cantidad de IgG humana combinada es de 800 mg/kg de peso corporal del sujeto por periodo de dos semanas.
- 12. Composición para su uso según cualquiera de las reivindicaciones 1 a 11, en la que la cantidad de IgG humana combinada se administra mediante administración intravenosa, administración subcutánea, administración intramuscular o administración intraperitoneal.
- 13. Composición para su uso según cualquiera de las reivindicaciones 1 a 11, en la que la cantidad de IgG humana combinada se administra mediante administración intranasal, administración intratecal, administración intracerebral, administración intracerebroventricular, administración epidural o administración espinal.
- 14. Composición para su uso según cualquiera de las reivindicaciones 1 a 13, en la que al sujeto también se le administra una cantidad terapéuticamente eficaz de una segunda composición para el tratamiento de la enfermedad de Alzheimer.
- 15. Composición para su uso según cualquiera de las reivindicaciones 1-14, en la que el sujeto tiene una puntuación del miniexamen de estado mental (MMSE) desde 14 hasta 20.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361855062P | 2013-05-06 | 2013-05-06 | |
| US201361833447P | 2013-06-10 | 2013-06-10 | |
| US201361844732P | 2013-07-10 | 2013-07-10 | |
| US201361886464P | 2013-10-03 | 2013-10-03 | |
| PCT/US2014/036848 WO2014182631A1 (en) | 2013-05-06 | 2014-05-05 | Treatment of alzheimer's disease subpopulations with pooled immunoglobulin g |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2740127T3 true ES2740127T3 (es) | 2020-02-05 |
Family
ID=50943566
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14730660T Active ES2740127T3 (es) | 2013-05-06 | 2014-05-05 | Tratamiento de subpoblaciones con enfermedad de Alzheimer con inmunoglobulina G combinada |
Country Status (12)
| Country | Link |
|---|---|
| US (3) | US20140328856A1 (es) |
| EP (2) | EP2994160B1 (es) |
| JP (1) | JP6424210B2 (es) |
| AU (4) | AU2014262890B2 (es) |
| DK (1) | DK2994160T3 (es) |
| ES (1) | ES2740127T3 (es) |
| HR (1) | HRP20191383T1 (es) |
| HU (1) | HUE044737T2 (es) |
| LT (1) | LT2994160T (es) |
| PL (1) | PL2994160T3 (es) |
| PT (1) | PT2994160T (es) |
| WO (1) | WO2014182631A1 (es) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE063369T2 (hu) | 2014-03-21 | 2024-01-28 | Alzheon Inc | Vegyületek Alzheimer-kór kezelésében történõ alkalmazásra APOE4+/+ betegekben |
| US10568568B2 (en) * | 2014-08-27 | 2020-02-25 | Capnia, Inc. | Methods for immune globulin administration |
| MA41115A (fr) * | 2014-12-02 | 2017-10-10 | Biogen Int Neuroscience Gmbh | Procédé de traitement de la maladie d'alzheimer |
| KR102412997B1 (ko) * | 2015-09-10 | 2022-06-23 | 알제온, 인크. | 특정 환자 집단에서 신경퇴행성 장애를 치료하는 방법 |
| EP3405489A1 (en) * | 2016-01-20 | 2018-11-28 | Genentech, Inc. | High dose treatments for alzheimer's disease |
| EP3484502B1 (en) * | 2016-08-18 | 2021-08-25 | Alkahest, Inc. | Blood plasma fractions as a treatment for aging-associated cognitive disorders |
| BR112019022402A2 (pt) * | 2017-04-26 | 2020-05-19 | Alkahest Inc | regime de dosagem para tratamento de comprometimentos cognitivo e motor com plasma sanguíneo e produtos de plasma sanguíneo |
| JOP20200041A1 (ar) | 2017-08-22 | 2020-02-20 | Biogen Ma Inc | تركيبات صيدلية تحتوي على أجسام مضادة لبيتا نشوي |
| FI128916B (en) * | 2018-02-05 | 2021-03-15 | Vetcare Oy | Health-promoting composition and method for its preparation |
| NL2024431B1 (en) | 2019-12-11 | 2021-09-07 | Sulfateq Bv | Compounds for treatment of alzheimer’s disease |
| EP4236988A1 (en) * | 2020-10-30 | 2023-09-06 | Rush University Medical Center | Intranasal immunotherapy for the treatment of alzheimer's disease |
| WO2023150483A1 (en) * | 2022-02-03 | 2023-08-10 | Eli Lilly And Company | Regional tau imaging for diagnosing and treating alzheimer's disease |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE348942B (es) | 1970-06-02 | 1972-09-18 | Statens Bakteriologiska Labor | |
| US5624898A (en) | 1989-12-05 | 1997-04-29 | Ramsey Foundation | Method for administering neurologic agents to the brain |
| US6585957B1 (en) | 2000-01-25 | 2003-07-01 | Aeropharm Technology Incorporated | Medicinal aerosol formulation |
| US20030099635A1 (en) | 2001-10-04 | 2003-05-29 | Protein Therapeutics, Inc. | Use of oral gammaglobulin for the treatment of immune-mediated diseases |
| US20090136505A1 (en) | 2005-02-23 | 2009-05-28 | Johanna Bentz | Intranasal Administration of Active Agents to the Central Nervous System |
| CA2598666A1 (en) | 2005-02-23 | 2006-08-31 | Alza Corporation | Intranasal administration of active agents to the central nervous system |
| JP5697847B2 (ja) * | 2006-01-30 | 2015-04-08 | グリフオルス・セラピユーテイクス・インコーポレーテツドGrifols Therapeutics,Inc. | IgMを使用する治療方法および予防方法 |
| JO3076B1 (ar) * | 2007-10-17 | 2017-03-15 | Janssen Alzheimer Immunotherap | نظم العلاج المناعي المعتمد على حالة apoe |
| TWI532498B (zh) | 2008-03-17 | 2016-05-11 | 巴克斯特保健公司 | 供免疫球蛋白及玻尿酸酶之皮下投藥之用的組合及方法 |
| KR101441768B1 (ko) | 2009-09-17 | 2014-09-17 | 박스터 헬쓰케어 에스에이 | 히알루로니다아제 및 면역글로불린의 안정한 복합제제, 및 그 사용방법 |
| US8622993B2 (en) | 2009-12-18 | 2014-01-07 | Healthpartners Research Foundation | Device and method for delivering therapeutic substances to the maxillary sinus of a patient |
| US8066993B2 (en) * | 2010-04-13 | 2011-11-29 | Norman R Relkin | Use of ventricular enlargement rate in intravenous immunoglobulin treatment of Alzheimer's disease |
| CA2799162A1 (en) * | 2010-05-13 | 2011-11-17 | The University Of Kentucky Research Foundation | Treatment of mci and alzheimer's disease |
| ES2959479T3 (es) * | 2010-09-17 | 2024-02-26 | Takeda Pharmaceuticals Co | Estabilización de inmunoglobulinas a través de formulación acuosa con histidina a pH ácido débil a neutro |
| JP2014507649A (ja) * | 2011-01-18 | 2014-03-27 | バクスター・インターナショナル・インコーポレイテッド | ヒト血液中の抗βアミロイド抗体の測定 |
| US20120251524A1 (en) * | 2011-04-01 | 2012-10-04 | Baxter Healthcare S.A. | Use of cytokine levels in intravenous immunoglobulin treatment of alzheimer's disease |
| AU2012311483B2 (en) * | 2011-09-24 | 2016-03-10 | Csl Behring Gmbh | Combination therapy using immunoglobulin and C1-inhibitor |
-
2014
- 2014-05-05 US US14/270,192 patent/US20140328856A1/en not_active Abandoned
- 2014-05-05 LT LTEP14730660.9T patent/LT2994160T/lt unknown
- 2014-05-05 ES ES14730660T patent/ES2740127T3/es active Active
- 2014-05-05 DK DK14730660.9T patent/DK2994160T3/da active
- 2014-05-05 HU HUE14730660 patent/HUE044737T2/hu unknown
- 2014-05-05 PT PT14730660T patent/PT2994160T/pt unknown
- 2014-05-05 EP EP14730660.9A patent/EP2994160B1/en active Active
- 2014-05-05 AU AU2014262890A patent/AU2014262890B2/en active Active
- 2014-05-05 JP JP2016512995A patent/JP6424210B2/ja active Active
- 2014-05-05 EP EP19175106.4A patent/EP3552624A1/en active Pending
- 2014-05-05 PL PL14730660T patent/PL2994160T3/pl unknown
- 2014-05-05 WO PCT/US2014/036848 patent/WO2014182631A1/en active Application Filing
-
2019
- 2019-01-14 US US16/247,141 patent/US20190144530A1/en not_active Abandoned
- 2019-04-09 AU AU2019202459A patent/AU2019202459B2/en active Active
- 2019-07-31 HR HRP20191383TT patent/HRP20191383T1/hr unknown
-
2020
- 2020-10-19 AU AU2020257034A patent/AU2020257034A1/en not_active Abandoned
-
2021
- 2021-04-19 US US17/234,658 patent/US20210238267A1/en active Pending
-
2024
- 2024-02-19 AU AU2024201051A patent/AU2024201051A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20191383T1 (hr) | 2020-02-07 |
| AU2014262890B2 (en) | 2019-01-31 |
| AU2019202459B2 (en) | 2020-07-23 |
| PL2994160T3 (pl) | 2019-10-31 |
| US20210238267A1 (en) | 2021-08-05 |
| EP3552624A1 (en) | 2019-10-16 |
| HUE044737T2 (hu) | 2019-11-28 |
| AU2019202459A1 (en) | 2019-05-02 |
| AU2024201051A1 (en) | 2024-03-14 |
| AU2014262890A1 (en) | 2015-11-26 |
| JP2016518411A (ja) | 2016-06-23 |
| JP6424210B2 (ja) | 2018-11-14 |
| WO2014182631A1 (en) | 2014-11-13 |
| EP2994160A1 (en) | 2016-03-16 |
| AU2020257034A1 (en) | 2020-11-12 |
| US20140328856A1 (en) | 2014-11-06 |
| US20190144530A1 (en) | 2019-05-16 |
| PT2994160T (pt) | 2019-08-07 |
| LT2994160T (lt) | 2019-08-26 |
| EP2994160B1 (en) | 2019-07-03 |
| DK2994160T3 (da) | 2019-08-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2740127T3 (es) | Tratamiento de subpoblaciones con enfermedad de Alzheimer con inmunoglobulina G combinada | |
| US11851481B2 (en) | Treatment of central nervous system disorders by intranasal administration of immunoglobulin g | |
| AU2021200093B2 (en) | Treatment and prevention of alzheimer's disease (ad) | |
| US20240148782A1 (en) | Treatment and prevention of alzheimer's disease (ad) |