ES2709110T3 - Un proceso mejorado para la preparación de sunitinib y sus sales de adición de ácido - Google Patents
Un proceso mejorado para la preparación de sunitinib y sus sales de adición de ácido Download PDFInfo
- Publication number
- ES2709110T3 ES2709110T3 ES13763825T ES13763825T ES2709110T3 ES 2709110 T3 ES2709110 T3 ES 2709110T3 ES 13763825 T ES13763825 T ES 13763825T ES 13763825 T ES13763825 T ES 13763825T ES 2709110 T3 ES2709110 T3 ES 2709110T3
- Authority
- ES
- Spain
- Prior art keywords
- sunitinib
- formula
- acid
- compound
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc1c(C=C(c2cc(F)ccc2N2)C2=O)[n]c(C)c1C(*)=O Chemical compound Cc1c(C=C(c2cc(F)ccc2N2)C2=O)[n]c(C)c1C(*)=O 0.000 description 3
- RBHBOUYXUXWCNJ-WDZFZDKYSA-N Cc1c(/C=C(/c(cc(cc2)F)c2N2)\C2=O)[nH]c(C)c1C(O)=O Chemical compound Cc1c(/C=C(/c(cc(cc2)F)c2N2)\C2=O)[nH]c(C)c1C(O)=O RBHBOUYXUXWCNJ-WDZFZDKYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C53/00—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen
- C07C53/132—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen containing rings
- C07C53/134—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen containing rings monocyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
- C07C55/06—Oxalic acid
- C07C55/07—Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/02—Dicarboxylic acids
- C07C55/10—Succinic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/235—Saturated compounds containing more than one carboxyl group
- C07C59/245—Saturated compounds containing more than one carboxyl group containing hydroxy or O-metal groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
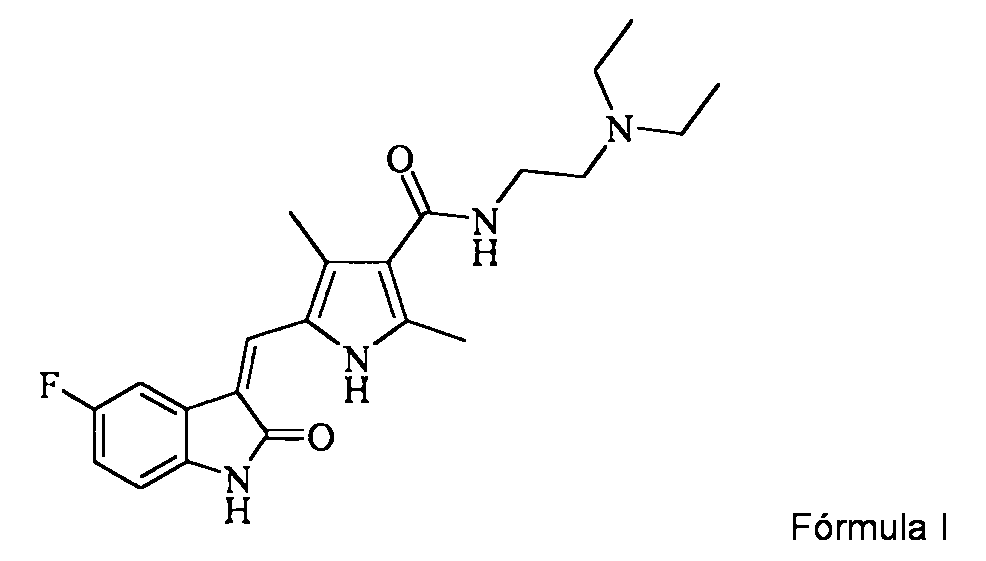
Un proceso para la preparación de sunitinib de fórmula I o una sal de adición de ácido del mismo,**Fórmula** que comprende: a) hacer reaccionar ácido 5-((Z)-(5-fluoro-2-oxoindolin-3-5 iliden)metil)-2,4-dimetil-1H-pirrol-3-carboxílico de**Fórmula** opcionalmente en un disolvente orgánico y en presencia de una base y un catalizador, con un agente de sulfonilación adecuado para obtener un compuesto activado de fórmula III**Fórmula** en donde 'X' representa un grupo alquilo o arilsulfoniloxi, y b) amidar el compuesto de fórmula III con N,N-dietiletilen diamina en un disolvente orgánico para obtener sunitinib.
Description
DESCRIPCION
Un proceso mejorado para la preparacion de sunitinib y sus sales de adicion de acido
Prioridad
Esta solicitud reivindica el beneficio bajo la solicitud provisional india n° 1083/CHE/2012, presentada el 23 de marzo de 2012, titulada "Un proceso mejorado para la preparacion de sunitinib", cuyo contenido se incorpora aqrn como referencia.
Campo de la invencion
La presente invencion se refiere en general a un proceso mejorado para la preparacion de sunitinib y sus sales de adicion de acido.
Antecedentes de la invencion
Sunitinb se describe qmmicamente como N-[2-(dietilamino)etil]-5-[(ZH5-fluoro-1,2-dihidro-2-oxo-3H-indol-3-iliden)metil]-2,4-dimetil-1H-pirrol-3-carboxamida como se representa en la formula l.

Sunitinb (comercializado como Sutent por Pfizer) es un inhibidor oral de multicinasas y es util para el tratamiento de tumores gastrointestinales y carcinomas de celulas renales avanzados. Sunitinb esta disponible comercialmente como sal de L-malato, descrita como N-[2-(dietilamino)etil]-5-[(Z)(5-fluoro-1,2-dihidro-2-oxo-3H-indol-3-iliden)metil]-2,4-dimetil-1H-pirrol-3-carboxilamida, acido (S)-2-hidroxi butanodioico (1:1).
Sunitinb y su sal de malato se describen en la correspondiente patente de producto de EEUU n° 6.573.293 (“la patente '293"), cedida a Sugen y Pharmacia & Upjohn, en donde la patente particular '293 describe la preparacion de sunitinib por la condensacion de (2-dietilaminoetil)amida acido 5-formil-2,4-1H-pirrol-3-carboxflico con 5-fluoro-2-oxindol en etanol en presencia de pirrolidina. La secuencia de reaccion se representa esquematicamente como sigue:

La patente EEUU 2009/0247767 ("la publicacion '767") describe la preparacion de sunitinib por activacion deacido 5-((Z)-(5-fluoro-2-oxoindolm-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carboxflico al correspondiente acido clorhndrico o un derivado de carbonil dimidazol seguido de reaccion con 2-dietilaminoetilamina. La secuencia de reaccion se representa esquematicamente como sigue:

La patente '293 describe sales de sunitinib, tales como restos cargados positivamente que incluyen amonio cuaternario, sales tales como clorhidrato, sulfato, carbonato, lactato, tartrato, malato, maleato, succinato; y especies cargadas negativamente; sin embargo la patente '293 no dice nada sobre la preparacion y la naturaleza de las formas cristalinas espedficas de las sales.
La patente EEUU n°7.435.832 ("la patente '832") describe una base libre y sales de sunitinib (p.ej. acido ciclamico, acido maleico, acido bromlddrico, acido mandelico, acido tartarico, acido fumarico, acido ascorbico, acido fosforico, acido clorhfdrico, acido p-toluensulfonico, acido citrico y sales de acido malico) se habfan evaluado las propiedades relacionadas con el procesamiento de la sal y la preparacion de las composiciones farmaceuticas orales de la misma, que incluyen, por ejemplo, cristalinidad, toxicidad, higroscopicidad, estabilidad y morfologfa, perosolo la sal de malato se eligio de la evaluacion y solo dos formas cristalinas de sunitinib L malato se describieron espedficamente.
La publicacion PCT n° WO 2010/011834 describe la sal de acetato de sunitinib y sus polimorfos.
La publicacion PCT n° WO 2010/041134 describe la preparacion de malato de sunitinib a traves de la generacion de una sal de acido debil donde un acido debil es un acido mas debil que el acido malico. El acido mas debil se describe preferiblemente como acido acetico.
La publicacion PCT n° WO 2010/049449 describe sales de D-tartarato, L-tartarato y citrato de sunitinib junto con su preparacion y las correspondientes formas polimorficas.
La publicacion PCT n° WO 2011/033472 describe sal de sunitinib con un acido aquiral, en donde el acido aquiral se selecciona del grupo acido citrico, acido p-toluensulfonico, acido sulfurico, acido acetico y acido metanosulfonico.
La publicacion PCT n° W02011/100325 describe sales de sunitinib y polimorfos de estos, tales como fumarato de sunitinib y clorhidrato de sunitinib.
La publicacion PCT n°WO 2011/110199 describe la preparacion de sunitinib por una reaccion de amidacion deacido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetil-1H pirrol-3-carboxflico en presencia de trifosofonato.
La publicacion PCT n° W02012/059941 describe sales de acido protico de sunitinib, en dondeel acido protico descrito
es clorhidrato, bromhidrato, fosfato o salicilato.
Las sales a menudo mejoran las caractensticas ffsicas y biologicas de la actividad farmacologica primariasin modificar, basado en el mecanismo de accion. Por lo tanto, existe una necesidad continua de obtener nuevas sales de sunitinib que tengan propiedades ffsicas o qwmicas mejoradas. La presente invencion satisface esta necesidad al proporcionar nuevas sales de sunitinib con una solubilidad marcadamente mejorada en agua o medios acuosos como propiedad esencial de los ingredientes farmaceuticos activos. Las nuevas formas de sal o las formas polimorficas correspondientes del producto farmaceutico pueden proporcionar las propiedades deseables, tales como manejo, solubilidad incrementada, disolucion incrementada, higroscopicidad disminuida, estabilidad, almacenamiento, vida util y/o facilidad de purificacion.
Dado que sunitinib constituye un importante agente terapeutico, las formas adicionales y mejoradas para preparar sunitinib y sus sales son un gran valor para la ciencia farmaceutica. Por lo tanto, existe una necesidad en el desarrollo de un metodo consistente y novedoso y/o un proceso mejorado para preparar sunitinib y sus sales, que es comercialmente viable, mas seguro para la manipulacion, requiere menos tiempo y es mas puro.
Los metodos reportados, que involucran cloruro de tionilo asociado con ciertas desventajas, el cloruro de tionilo es una sustancia altamente reactiva, que libera gases toxicos peligrosos tales como dioxido de azufre, cloruro de azufre o cloruro de hidrogeno. Ademas, el reactivo encontrado no es ecologico. El otro proceso reportado que involucra el uso de carbonil diimidazol que se considera desventajoso es de naturaleza higroscopica, que requiere condiciones altamente anhidras. Por lo tanto, proporciona un alcance de una mejora en el proceso para la preparacion de sunitinib para escala industrial. Para superar los problemas asociados a los procedimientos informados, los inventores disenaron asf un proceso mejorado para la preparacion de sunitinib, adecuado para escala industrial con mejores resultados en el rendimiento.
Compendio de la invencion.
Un aspecto importante de la presente invencion es un proceso mejorado para la preparacion de sunitinib a traves de nuevos intermedios de acido carboxflico activado. El presente proceso implica la reaccion del acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carboxflico con un grupo de activacion adecuado, seguido de amidacion con N,N-dietil etilendiamina para obtener sunitinib.
En una realizacion, la presente invencion proporciona un proceso para la preparacion de sunitinib o sus sales de adicion de acido de formula 1,

que comprende:
a) hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetil-1H-pirrol-3-carboxflico de formula II

con un agente de activacion adecuado para obtener un compuesto activado de formula III,

donde 'X' representa un grupo de activacion, siempre que 'X' no sea cloro ni imidazol,
b) amidar el compuesto de formula III con N,N-dietil etilen diamina en un disolvente organico para obtener sunitinib.
En una segunda realizacion, la presente invencion proporciona un procedimiento para la preparacion de sunitinib o sus sales de adicion de acido de formula I, que comprende:
a) hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carbox^lico de formula II con un agente de sulfonilacion adecuado para obtener un compuesto activado de formula III, en donde 'X' representa un grupo sulfoniloxi,
b) amidar el compuesto de formula III con N,N-dietil etilen diamina en un disolvente organico para obtener sunitinib.
En una tercera realizacion, la presente invencion proporciona un procedimiento para la preparacion de compuesto de formula III,

en donde 'X' representa un grupo sulfoniloxi, que comprende: hacer reaccionar el acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carboxflico de formula II con un agente de sulfonilacion adecuado.
En una cuarta realizacion, la presente invencion proporciona un compuesto de formula III

en donde 'X' representa un grupo sulfoniloxi.
En una quinta realizacion, la presente invencion proporciona un compuesto deformula III, en donde 'X' representa un grupo sulfoniloxi seleccionado de compuestos de sulfoniloxi tales como alquilo o aril sulfoniloxi, en donde alquilo representa alquilo de cadena lineal o ramificada C-4 y arilo representa alquilo sustituido o fenilo no sustituido.
En una sexta realizacion, la presente invencion proporciona un compuesto de formula III, en donde 'X' representa un grupo p-tolueno sulfoniloxi.
En una septima realizacion, la presente invencion proporciona un proceso para la preparacionde sunitinib o sus sales
de adicion de acido de la formula I, que comprende:
a) hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolm-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carboxflico de formula II con cloruro de p-tolueno sulfonilo para obtener un compuesto de tolueno sulfoniloxi activado de formula III,
b) amidar el compuesto de formula III con N,N-dietil etilen diamina en un disolvente organico para obtener sunitinib,
c) convertir el sunitinib en sus sales de adicion de acido.
Tambien se describen sales de adicion de acido de sunitinib, en donde los acidos se seleccionan del grupo que consiste en acido malonico, acido oxalico, acido ferulico, acido succmico acido p-cumarico, acido sinapico, acido cafeico, acido malnco, acido fumarico y acido fosforico.
Tambien se describe una composicion farmaceutica que comprende una cantidad terapeuticamente eficaz de sunitinib y sus sales de adicion de acido preparadas por el proceso de la invencion.
Breve descripcion de los dibujos
El dibujo adjunto, que se incorpora y forma parte de esta memoria descriptiva, ilustra la realizacion de la invencion y junto con la descripcion, sirve para explicar los principios de la invencion.
La figura 1 muestra el patron de PXRD del malato de sunitinib.
Descripcion detallada de la invencion
La presente invencion abarca un proceso mejorado para la preparacion de sunitinib a traves de nuevos intermedios de acido carboxflico activado. El proceso presente implica la reaccion del acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2, 4-dimetil-1H pirrol-3-carboxflico con un grupo de activacion adecuado, seguido de amidacion con N,N-dietil etilen diamina para obtener sunitinib.
En una realizacion, la presente invencion proporciona un compuesto de formula III

en donde 'X' representa un grupo sulfoniloxi.
En una realizacion preferida, la presente invencion proporciona un compuesto de formula III, en donde 'X' representa un grupo de activacion tal como compuestos de sulfoniloxi seleccionados de alquilo o aril sulfoniloxi, en donde alquilo representa cadena lineal C1-4 o ramificada de alquilo y arilo representa alquilo sustituido o fenilo no sustituido.
En otra realizacion preferida, la presente invencion proporciona un compuesto de formula III, donde 'X' representa un grupo p-tolueno sulfoniloxi.
El compuesto de formula I se puede usar para preparar sunitinib y sus sales de adicion de acido que tienen la siguiente formula:

En otra realizacion, la presente invencion proporciona un procedimiento para la preparacion de sunitinib o sus sales de adicion de acido de formula I,

que comprende:
a) hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetil-1H-pirrol 3-carboxflico de formula II

con un agente de sulfonilacion para obtener un compuesto activado de fórmula III,

en donde 'X' representa un grupo sulfoniloxi,
b) amidar el compuesto de formula III con N,N-dietil etilen diamina en un disolvente organico para obtener sunitinib.
La reaccion del acido 5-[(Z)-(5-fluoro-2-oxo-1,2-dihidro-3H-indol-3-iliden)metil]-2,4-dimetiMH-pirrol-3-carboxflico de
formula II con un agente de sulfonilacion da como resultado la formacion de un nuevo derivado de acido carbox^lico activado (III), que sirve como proposito de un mejor grupo saliente. El proceso con estos reactivos demostro ser un proceso mas rapido, facil, rentable, con procedimientos de trabajo mas simples con una gran mejora en los rendimientos.
El material de partida acido 5-[(Z)-(5-fluoro-2-oxo-1,2-dihidro-3H-indol-3-iliden)metil]-2,4-dimetiMH-pirrol-3-carboxflico de formula II es conocido en la tecnica y se puede preparar por cualquier metodo conocido, por ejemplo, el compuesto de partida de formula II se puede sintetizar como se describe en la patente de EEUU n° 6.573.293.
La etapa a) de la reaccion de formula II con un agente de sulfonilacion implica al menos una base, un disolvente organico y opcionalmente un catalizador.
El grupo sulfonilo adecuado para su uso en la presente memoria puede ser, por ejemplo, un grupo de activacion son compuestos de sulfonilo. En una realizacion preferida, los compuestos de sulfonilo tales como alquilo o aril sulfonilo, en donde alquilo representa alquilo y arilo de cadena lineal o ramificada C1-4 representa fenilo sustituido con alquilo o sin sustituir; mas preferiblemente p-tolueno sulfonilo.
El agente de sulfonilacion incluye, pero no se limita a, haluro de p-tolueno sulfonilo tal como cloruro de p-tolueno sulfonilo y similares.
Una base adecuada para su uso en la presente memoria puede ser, por ejemplo, una base organica tal como una amina primaria, secundaria o terciaria. Ejemplos representativos de tales aminas incluyen, pero no se limitan a, trietilamina, tributilamina, diisopropiletilamina, dietilamina, N-metilmorfolina, piridina, N,N-dimetilanilina, N,N-dietilanilina y similares y mezclas de estos. Alternativamente, se puede usar una base inorganica e incluye un alcali carbonato de metal tal como carbonato de litio, carbonato de sodio, carbonato de potasio y similares; bicarbonato de metal alcalino tal como bicarbonato de litio, bicarbonato de sodio, bicarbonato de potasio y similares; hidruro de metal alcalino tal como hidruro de litio, hidruro de sodio, hidruro de potasio y similares; hidroxido de metal alcalino tal como hidroxido de litio, hidroxido de sodio, hidroxido de potasio y similares; alcoxido de metal alcalino tales como metoxido de litio, metoxido de sodio, etoxido de sodio, t-butoxido de potasio y similares; y mezclas de estos. Se prefieren las aminas organicas (particularmente piridina).
Un catalizador adecuado para su uso en la presente memoria puede ser, por ejemplo, N,N-dimetil amino piridina.
La reaccion se lleva a cabo ventajosamente en un disolvente organico adecuado. Los disolventes organicos adecuados incluyen, pero no se limitan a, amidas tales como formamida, dimetilformamida, dimetilacetamida, hexametilfosforico triamida y similares y mezclas de estos. Preferiblemente, el disolvente organico adecuado es dimetil formamida o dimetil acetamida es mas preferida. En una realizacion preferida de la presente invencion, el disolvente organico adecuado es dimetil formamida. En general, la cantidad de disolvente organico empleado en la reaccion puede variar de aproximadamente 5 volumenes a aproximadamente 30 volumenes y preferiblemente de aproximadamente 5 volumenes a aproximadamente 20 volumenes.
Tfpicamente, la reaccion se mantiene, preferiblemente con agitacion para permitir la formacion del compuesto activado de formula III. Preferiblemente, la reaccion se mantiene durante un penodo de aproximadamente 1 hora a aproximadamente 12 horas, mas preferiblemente, durante aproximadamente 1 hora a aproximadamente 3 horas. Preferiblemente, la reaccion se mantiene a una temperatura inferior a aproximadamente la temperatura ambiente, preferiblemente a aproximadamente 10°C a 20°C.
El compuesto activado resultante de formula III de la invencion se puede procesar adicionalmente directamente en el mismo recipiente de reaccion para formar sunitinib de formula l. Alternativamente, el compuesto activado resultante de formula III se puede aislar del medio de reaccion eliminando el disolvente al vacfo para obtener el residuo por cualquier metodo conocido en la tecnica, al final de la reaccion y seguido de cristalizacion opcional en compuesto solido. La etapa de concentracion puede ser, por ejemplo, destilacion, evaporacion, secado rotativo (como con Buchi Rotavapor), secado por congelacion, secado en lecho fluidizado, secado instantaneo, secado instantaneo por centrifugacion y similares, preferiblemente destilacion al vacfo.
El compuesto activado de formula III preparado usando el proceso de la presente invencion se puede convertir en sunitinib y sus sales de adicion de acido, de la siguiente manera:
La etapa b) del proceso anterior se puede llevar a cabo agregando una cantidad suficiente de N,N-dietil etilen diamina al producto resultante para llevar la reaccion de amidacion en un disolvente organico.
El disolvente organico utilizado en la presente memoria puede incluir, entre otros, amidas tales como formamida, dimetilformamida, dimetilacetamida, triamida hexametilfosforica y similares y mezclas de estos. Preferiblemente, el disolvente organico adecuado es dimetilformamida o dimetil acetamida es mas preferido. En una realizacion preferida de la presente invencion, el disolvente organico adecuado es dimetilformamida.
Tfpicamente, la reaccion se mantiene, preferiblemente bajo agitacion para permitir la formacion de sunitinib. Preferiblemente, la reaccion se mantiene durante un penodo de aproximadamente 1 hora a aproximadamente 16
horas, mas preferiblemente, durante aproximadamente 1 hora a aproximadamente 5 horas. Preferiblemente, la reaccion se mantiene a una temperatura inferior a 50°C; mas preferiblemente menos de aproximadamente 20°C.
El sunitinib obtenido despues se puede recuperar. El proceso de recuperacion de sunitinib puedecomprenden agregar agua a la mezcla de reaccion para precipitar sunitinib, filtrar el sunitinib precipitado y secar u opcionalmente extraer el sunitinib de la capa acuosa con un disolvente organico, por ejemplo un disolvente inmiscible con agua seleccionado de acetato de etilo, cloruro de metileno y similares; despues se concentra el disolvente para obtener sunitinib.
El sunitinib asf obtenido se puede purificar para minimizar el contenido de desetil sunitinib de formula Id, disolviendo el sunitinib en un disolvente organico tal como formamida, dimetilformamida, dimetilacetamida, hexametilfosforico triamida y similares y mezclas de estos, siendo preferible la dimetilformamida El disolvente se puede calentar para obtener una disolucion a una temperatura de aproximadamente la temperatura ambiente a aproximadamente la temperatura de reflujo, preferiblemente a aproximadamente 80°C. La reaccion se puede tratar con una base adecuada y una fuente de yoduro de etilo.

La base adecuada se puede usar en la presente memoria, incluye cualquier base como se describe anteriormente, prefiriendose la diisopropiletilamina.
El sunitinib obtenido despues se puede recuperar. El proceso de recuperacion de sunitinib puede comprender agregar agua a la mezcla de reaccion y extraer el sunitinib de la capa de agua con un disolvente organico, por ejemplo un disolvente inmiscible con agua seleccionado de acetato de etilo, cloruro de metileno y similares; seguido de la concentracion del disolvente para obtener un residuo, que se trata adicionalmente con un disolvente adecuado tal como metanol, isopropanol, acetona y similares y mezclas de estos. Esto puede permitir un alto nivel de pureza del sunitinib resultante a partir del sunitinib bruto, por ejemplo, una pureza de al menos aproximadamente 95%, preferiblemente al menos aproximadamente 98% y mas preferiblemente al menos aproximadamente 99,5% y menos de aproximadamente 0,1% de desetil sunitinib.
El sunitinib recuperado despues se puede convertir en sal de sunitinib, preferiblemente, en malato de sunitinib. La conversion se puede hacer haciendo reaccionar sunitinib base con un acido, preferiblemente acido malico. Cuando el acido es acido malico, la conversion se puede hacer, por ejemplo, de acuerdo con el proceso descrito en la publicacion de EEUU n° 2003/0069298 incorporado en la presente memoria por referencia.
Alternativamente, el sunitinib recuperado despues se puede convertir en otras sales de adicion de acido farmaceuticamente aceptables del mismo. Las sales de adicion de acido farmaceuticamente aceptables incluyen, pero no se limitan a acido malonico, acido oxalico, acido ferulico, acido succmico, acido p-cumarico, acido sinapico, acido cafeico, acido malnco, acido fumarico y acido fosforico.
La presente invencion proporciona un procedimiento para la preparacion de sales de adicion de acido de sunitinib del mismo, que comprende:
a) proporcionar una base de sunitinib obtenida mediante el proceso descrito anteriormente, disuelta enuno o mas solventes organicos, tales como alcoholes Cm , seleccionados entre metanol, etanol, n-propanol, isopropanol, n-butanol, isobutanol, t-butanol y similares, y mezclas de estos; disolventes halogenados tales como cloruro de metileno, cloroformo y similares y mezclas de estos; nitrilos tales como acetonitrilo, propionitrilo, benzonitrilo y similares y mezclas de estos; cetonas tales como acetona, metil etil cetona, metil isobutil cetona y similares y mezclas de estos; preferiblemente, los disolventes organicos se seleccionan de metanol, isopropanol, acetonitrilo, cloruro de metileno.
b) calentar la disolucion para disolver la base libre de sunitinib. Tfpicamente, la disolucion se calienta a una temperatura de al menos aproximadamente 30°C a aproximadamente reflujo. Preferiblemente, la disolucion se calienta de aproximadamente 25°C a aproximadamente 85°C, y mas preferiblemente de aproximadamente 25°C a aproximadamente 65°C.
c) tratar la disolucion resultante con un acido seleccionado del grupo que consiste en acido malonico, acido oxalico, acido ferulico, acido succmico, acido p-cumarico, acido sinapico, acido cafeico, acido malnco, acido fumarico y acido fosforico.
d) aislar las sales formadas de sunitinib mediante cualquier tecnica convencional, por ejemplo, concentrada sometiendo la disolucion a calentamiento, enfriando la disolucion a precipitacion, cristalizacion, precipitacion con disolvente, secado por pulverizacion, secado por congelacion, evaporador de pelfculafina agitado (ATFE) y similar.
El producto resultante se puede secar adicionalmente opcionalmente. El secado se puede llevar a cabo adecuadamente en un secador de bandejas, horno de vado, horno de aire, secador de lecho fluidizado, secador de destello, secador de flash y similares. El secado se puede llevar a cabo a una temperatura en el intervalo de aproximadamente 30°C a aproximadamente 50°C. El secado se puede llevar a cabo durante cualquier tiempo deseado hasta que se alcance la pureza requerida del producto, por ejemplo, un penodo de tiempo que oscila entre aproximadamente 1 hora y aproximadamente 10 horas.
La presente invencion proporciona sales de adicion de acido de sunitinib que incluye sal de malato preparada usando el proceso de la invencion que tiene una pureza qmmica mayor o igual a aproximadamente el 97%, segun se mide por HPLC, preferiblemente aproximadamente 98% segun se mide por HPLC, y mas preferiblemente aproximadamente 99,5%, segun se mide por HPLC y sustancialmente libre de desetil sunitinib de formula Id; en donde la palabra "sustancialmente libre" se refiere a sales de sunitinib, preferiblemente sal de malato de sunitinib que tiene menos de aproximadamente 0,1%, de formula Id, segun se mide por HPLC, mas preferiblemente menos de aproximadamente 0,05% de formula Id, segun se por HPLC.
En una realizacion preferida de la invencion, sunitinib se prepara segun el esquema 1:

En otra realizacion, las sales de adicion de acido de sunitinib de la presente invencion se caracterizan por un patron de difraccion de rayos X en polvo (XRD). La difraccion de rayos X en polvo se puede medir con un difractometro de rayos X en polvo equipado con un anodo de Cu ([A] = 1,54 Angstrom), fuente de rayos X operada a 30kV, 15 mA y se utiliza un filtro de Ni para eliminar la radiacion de K-beta. La calibracion de dos theta se realiza utilizando un estandar NIST SRM 640c Si. La muestra se analizo utilizando los siguientes parametros del instrumento: intervalo de medicion = 3-45°20; ancho de banda = 0,020°; y velocidad de escaneo = 5°/minuto.
Ejemplos.
Habiendo descrito la invencion con referencia a ciertas realizaciones preferidas, otras realizaciones seran evidentes para un experto en la tecnica a partir de la consideracion de la memoria descriptiva. La invencion se define adicionalmente por referencia a los siguientes ejemplos que describen en detalle la preparacion de la composicion y metodos de uso de la invencion. Sera evidente para los expertos en la materia que se pueden realizar muchas modificaciones, tanto de los materiales como de los metodos, sin apartarse del alcance de la invencion.
Ejemplo 1: Preparacion de base de sunitinib.
Una mezcla de acido 5-((Z)-(5-fluoro-2- oxoindolin-3-iliden)metil)-2,4-dimetil-1H-pirrol-3-carboxflico (70 g) en DMF (1.050 ml) se enfrio a 15-19°C. A esta mezcla se anadio cloruro de tolueno sulfonilo (133,4 g) en DMF (350 ml) en 15 min. Se anadio 4- dimetilamino piridina (81,3 g) en DMF (350 ml) en 15 minutos y se anadio piridina (36,9 g) en 15 minutos a 15-19°C. La mezcla de reaccion se agito a la misma temperatura durante un penodo de 60-90 minutos. La
masa de reaccion se enfrio a 13-16°C. A la mezcla de reaccion anterior se anadio N,N-dietil etilen diamina (81,3 g) en DMF (350 ml) en 10 min a 13-16°C. La masa de reaccion se mantuvo bajo agitacion durante un penodo de 30 minutes a la misma temperatura. El progreso de la reaccion se controlo mediante HPLC con un Ifmite de acido 5-((Z)-(5-fluoro-2-oxoindoIin-3-iIiden)metiI)-2, 4-dimetiI-1H-pirroI-3-carbox^Iico no mas de 5,0%. Despues de la finalizacion de la reaccion se cargaron 28 ml de agua de reaccion y se destilo DMF a vacte. Se cargo 1.050 ml de disolucion de bicarbonato de sodio al 5% con agitacion. El pH de la disolucion anterior se ajusto a 12-13 con disolucion de NaOH al 50%. Se cargo acetato de etilo y se agito durante 5 min. La capa organica se separo y se trato con NaOH al 1%, y la disolucion de salmuera seguido de la separacion de la fraccion de acetato de etilo y la destilacion se retiro bajo vacte por debajo de 50°C. El residuo se trato con metanol al 30% en agua para aislar el material. Se trato con metanol para obtener base de sunitinib. El solido obtenido se seco al vacte a 55-65°C durante un penodo de 5 horas. Rendimiento: 65,6 g de base de sunitinib con ~0,5% de etil sunitinib.
Ejemplo 2: Purificacion de base de sunitinib.
El sunitinib obtenido en la reaccion anterior (con ~0,5% de desetil sunitinib) se disolvio en DMF a temperatura ambiente y se calento a 80°C para obtener una disolucion transparente. La temperatura del sistema se enfrio a 15-20°C en un penodo de 30 minutos seguido por adicion de diisopropil etil amina (5,32 g) durante 10 min. La masa de reaccion se mantuvo durante un penodo de 15 minutos con agitacion y se anadio yoduro de etilo (6,43 g) durante un penodo de 15 minutos. La masa de reaccion se agito durante un penodo de 8 horas a temperatura ambiente y el progreso de la reaccion se controlo mediante HPLC (contenido de impureza de desetil NMT 0,1%). Despues de completada la reaccion (1.640 ml) se anadio disolucion de bicarbonato de sodio al 5% (262 ml) NaOH 50% y acetato de etilo. La fraccion organica se separo y se trato con agua y disolucion de salmuera. La fraccion de acetato de etilo se separo y se destilo para obtener el residuo. El residuo se trato con metanol para obtener una base de sunitinib cruda. El crudo anterior se trato con acetona. El solido obtenido se filtro y se seco a vacte durante un penodo de 5 horas a 55-60°C para obtener sunitinib (48 g, menos de 0,1% desetil sunitinib).
Ejemplo 3: Preparacion de sunitinib L-malato:
Sunitinib (10 g) se suspendio en metanol (1.540 ml) bajo agitacion y se anadio acido L-malico (3,36 g) a una temperatura de 25-30°C. La masa de reaccion se agito para obtener una disolucion clara. Se filtro y el metanol se elimino por destilacion a vacte por debajo de 40°C. Se cargo acetonitrilo (50 ml) y el disolvente se elimino por destilacion. Se enfrio a 25-30°C. Se agrego 380 ml de acetonitrilo a la masa y se mantuvo a 45-50°C bajo agitacion durante un penodo de 15 minutos. La masa de reaccion se enfrio a 30-35°C. El solido obtenido se filtro y se lavo con acetonitrilo (10 ml). El compuesto resultante se seco por succion y se seco al vacte durante un penodo de 4-5 horas a 50°C para obtener sunitinib L-malato. (12,0 g; rendimiento 90%).
Pureza mediante HPLC: 98,5%; contenido de acido L-malico: 24-26% (p/p);
DSC: inicio 195°C y finalizacion 198,77°C; TGA: sin perdida observada hasta 200°C;
Contenido de humedad: 0,79%.
IR (cm-1): 3328, 3233, 3073, 2984, 2885, 1673, 1635, 1574, 1529, 1496, 1477, 1439, 1322, 1278, 1256, 1196, 807, 791.
El XRPD se describe en la figura 01.
Claims (6)
1. Un proceso para la preparación de sunitinib de fórmula I o una sal de adición de ácido del mismo,
que comprende:
a) hacer reaccionar acido 5-((ZH5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetil-1H-pirrol-3-carbox^lico de formula II

opcionalmente en un disolvente organico y en presencia de una base y un catalizador, con un agente de sulfonilacion adecuado para obtener un compuesto activado de formula III

en donde 'X' representa un grupo alquilo o arilsulfoniloxi, y
b) amidar el compuesto de formula III con N,N-dietiletilen diamina en un disolvente organico para obtener sunitinib.
2. El proceso de la reivindicacion 1, en el que en el grupo alquilo o arilsulfoniloxi, alquilo representa alquilo de cadena lineal C1-4 o ramificada y arilo representa un grupo fenilo alquilo sustituido o no sustituido con alquilo.
3. El proceso segun la reivindicacion 1, en el que en la reaccion de la etapa a) la base se selecciona del grupo que consiste en trietilamina, tributilamina, diisopropiletilamina, dietilamina, N-metilmorfolina, piridina, N,N-dimetilanilina, N, N dietilanilina y mezclas de estos, en donde los disolventes organicos en la reaccion de la etapa a) y en la reaccion de la etapa b) se seleccionan, independientemente, del grupo que consiste en formamida, dimetilformamida, dimetilacetamida, hexametilfosforo triamida y sus mezclas, y en donde el catalizador en la reaccion de la etapa a) es N,N-dimetilaminopiridina.
4. Un compuesto de formula III

en donde 'X' representa un grupo alquilo o aril sulfoniloxi.
5. Un proceso para la preparacion del compuesto de formula III segun la reivindicacion 4 o una sal farmaceuticamente aceptable del mismo que comprende la etapa de hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carbox^lico de formula II con un agente de sulfonilacion adecuado en un disolvente organico en presencia de una base y un catalizador.
6. Un proceso para la preparación de sunitinib malato de fórmula I según la reivindicación 1,

que comprende
a) hacer reaccionar acido 5-((Z)-(5-fluoro-2-oxoindolin-3-iliden)metil)-2,4-dimetiMH-pirrol-3-carboxflico de formula II

Formula II
con cloruro de p-tolueno sulfonilo en presencia de piridina y N,N-dimetilamino piridina en dimetilformamida para obtener el compuesto de formula III,

en donde 'X' representa el grupo p-tolueno sulfoniloxi,
b) amidar el compuesto de formula III con N,N-dietiletilen diamina en dimetilformamida para obtener
base de sunitinib base, y
c) tratar la base de sunitinib con acido malico para obtener sal de malato de sunitinib.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1083CH2012 | 2012-03-23 | ||
| PCT/IB2013/000459 WO2013140232A1 (en) | 2012-03-23 | 2013-03-22 | An improved process for the preparation of sunitinib and its acid addition salts thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2709110T3 true ES2709110T3 (es) | 2019-04-15 |
Family
ID=49221916
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13763825T Active ES2709110T3 (es) | 2012-03-23 | 2013-03-22 | Un proceso mejorado para la preparación de sunitinib y sus sales de adición de ácido |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9206163B2 (es) |
| EP (1) | EP2828251B1 (es) |
| CN (1) | CN104114550A (es) |
| ES (1) | ES2709110T3 (es) |
| WO (1) | WO2013140232A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2017026119A1 (ja) * | 2015-08-10 | 2018-05-24 | 大日本住友製薬株式会社 | 5−(チアゾール−4−イル)インドリン−2−オン誘導体の精製方法 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE122008000002I1 (de) | 2000-02-15 | 2008-04-17 | Sugen Inc | Pyrrol substituierte indolin-2-on protein kinase inhibitoren |
| WO2002081466A1 (en) * | 2001-04-09 | 2002-10-17 | Sugen, Inc. | Prodrugs of 3-(pyrrol-2-ylmethylidene)-2-indolinone derivatives |
| EP2332934B1 (en) | 2001-08-15 | 2017-03-01 | Pharmacia & Upjohn Company LLC | Processes for the preparation of crystals including a malic acid salt of n-[2-(diethylamino)ethyl]-5-[(5-fluoro-1,2-dihydro-2-oxo-3h-indole-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide. |
| WO2005023765A1 (en) | 2003-09-11 | 2005-03-17 | Pharmacia & Upjohn Company Llc | Method for catalyzing amidation reactions by the presence of co2 |
| US20060009510A1 (en) | 2004-07-09 | 2006-01-12 | Pharmacia & Upjohn Company Llc | Method of synthesizing indolinone compounds |
| CN101497607B (zh) * | 2008-01-29 | 2012-11-28 | 上海百灵医药科技有限公司 | 舒尼替尼的合成方法 |
| EP2274303B1 (en) | 2008-03-31 | 2012-08-29 | Teva Pharmaceutical Industries Ltd. | Processes for preparing sunitinib and salts thereof |
| AU2009269768A1 (en) * | 2008-07-10 | 2010-01-14 | Generics [Uk] Limited | Processes for the preparation of crystalline forms of sunitinib malate |
| US8618309B2 (en) | 2008-07-24 | 2013-12-31 | Teva Pharmaceutical Industries Ltd. | Sunitinib and salts thereof and their polymorphs |
| CN101333215B (zh) * | 2008-07-29 | 2011-01-12 | 南京工业大学 | 一种舒尼替尼碱的合成方法 |
| WO2010041134A1 (en) | 2008-10-10 | 2010-04-15 | Medichem, S.A. | Process for preparing a 3-pyrrole subsituted 2-indolinone malate salt |
| EP2181991A1 (en) | 2008-10-28 | 2010-05-05 | LEK Pharmaceuticals D.D. | Novel salts of sunitinib |
| EP2186809A1 (en) * | 2008-11-13 | 2010-05-19 | LEK Pharmaceuticals D.D. | New crystal form of sunitinib malate |
| EP2896618A1 (en) * | 2009-01-02 | 2015-07-22 | Hetero Research Foundation | Polymorphs of sunitinib malate |
| CA2774634A1 (en) | 2009-09-16 | 2011-03-24 | Ranbaxy Laboratories Limited | Salts of sunitinib |
| US20120271056A1 (en) * | 2009-11-12 | 2012-10-25 | Ranbaxy Laboratories Limited | Process for the preparation of crystalline form i of l-malic acid salt of sunitinib |
| WO2011061613A1 (en) * | 2009-11-19 | 2011-05-26 | Ranbaxy Laboratories Limited | Process for the preparation of crystalline form ii of l-malic acid salt of sunitinib |
| WO2011100325A2 (en) | 2010-02-09 | 2011-08-18 | Sicor Inc. | Polymorphs of sunitinib salts |
| WO2011110199A1 (en) * | 2010-03-10 | 2011-09-15 | Synthon B.V. | A process for amidation of pyrrole carboxylate compounds |
| WO2012059941A1 (en) | 2010-11-04 | 2012-05-10 | Ind-Swift Laboratories Limited | Process for preparation of sunitinib malate and salts thereof |
| CN102002021A (zh) * | 2010-12-07 | 2011-04-06 | 合肥华方医药科技有限公司 | 一种瑞格列奈的新合成方法 |
-
2013
- 2013-03-22 ES ES13763825T patent/ES2709110T3/es active Active
- 2013-03-22 US US14/381,546 patent/US9206163B2/en not_active Expired - Fee Related
- 2013-03-22 EP EP13763825.0A patent/EP2828251B1/en active Active
- 2013-03-22 CN CN201380009303.3A patent/CN104114550A/zh active Pending
- 2013-03-22 WO PCT/IB2013/000459 patent/WO2013140232A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| US20150025252A1 (en) | 2015-01-22 |
| EP2828251B1 (en) | 2018-10-31 |
| US9206163B2 (en) | 2015-12-08 |
| CN104114550A (zh) | 2014-10-22 |
| EP2828251A1 (en) | 2015-01-28 |
| WO2013140232A1 (en) | 2013-09-26 |
| EP2828251A4 (en) | 2015-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10738038B2 (en) | Co-crystals of SGLT2 inhibitors, process for their preparation and pharmaceutical compositions thereof | |
| US20200331859A1 (en) | Salt of omecamtiv mecarbil and process for preparing salt | |
| WO2011158248A2 (en) | Process for preparation of posaconazole and crystalline polymorphic form v of posaconazole | |
| US20210371397A1 (en) | Synthesis of omecamtiv mecarbil | |
| JP6837480B2 (ja) | モノバクタム抗生物質の塩および固体形態 | |
| TWI585073B (zh) | 擔子菌截短側耳素(pleuromutilin)化合物的結晶形態、含彼之藥學組成物、及彼之製法 | |
| ES2879294T3 (es) | Formas polimórficas de Belinostat y procesos para la preparación de las mismas | |
| WO2017009784A1 (en) | Solid state forms of trisodium salt of valsartan/sacubitril complex and sacubitril | |
| ES2791187T3 (es) | Síntesis de dabigatrán | |
| TW201718516A (zh) | 組蛋白去乙醯酶抑制劑之晶形 | |
| ES2709110T3 (es) | Un proceso mejorado para la preparación de sunitinib y sus sales de adición de ácido | |
| US10301353B2 (en) | Co-crystal of carfilzomib with maleic acid and process for the preparation of pure carfilzomib | |
| WO2014034626A1 (ja) | N-[2-({2-[(2S)-2-シアノピロリジン-1-イル]-2-オキソエチル}アミノ)-2-メチルプロピル]-2-メチルピラゾロ[1,5-a]ピリミジン-6-カルボキサミドの結晶 | |
| JP5460209B2 (ja) | 4−アミノ−5−クロロ−2−エトキシ−n−〔[4−(4−フルオロベンジル)−2−モルホリニル]メチル〕ベンズアミドの精製方法 | |
| CN109970620B (zh) | 一种制备沙格列汀中间体的方法 | |
| US20110281928A1 (en) | Process for the preparation of zofenopril and its pharmaceutically acceptable salts thereof | |
| JP2005526800A (ja) | (2S)−N−5−[アミノ(イミノ)メチル]−2−チエニルメチル−1−(2R)−2−[(カルボキシルメチル)アミノ]−3,3−ジフェニルプロパノイル−2−ピロリジンカルボキサミド・nH2Oの新しい結晶形 | |
| CN110143963B (zh) | 具有杀菌消毒活性的哒嗪类化合物及其制备方法和应用 | |
| CA2902436C (en) | Salt of omecamtiv mecarbil and process for preparing salt | |
| WO2026032320A1 (zh) | 异噁唑啉类药物中间体的制备方法 | |
| ES2291101B1 (es) | Procedimiento para la preparacion de valaciclovir. | |
| US20130317214A1 (en) | Method for producing maleate using wet crystal | |
| CN102791700A (zh) | 用于制备雷尼酸锶多晶型的方法 | |
| WO2005090285A1 (ja) | トリテルペン誘導体の新規結晶 | |
| HK1219484B (zh) | 心肌肌球蛋白激动剂的盐和制备盐的方法 |