EP2921497B1 - Composé de silicone et utilisation de celui-ci - Google Patents
Composé de silicone et utilisation de celui-ci Download PDFInfo
- Publication number
- EP2921497B1 EP2921497B1 EP15159516.2A EP15159516A EP2921497B1 EP 2921497 B1 EP2921497 B1 EP 2921497B1 EP 15159516 A EP15159516 A EP 15159516A EP 2921497 B1 EP2921497 B1 EP 2921497B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- compound
- group
- formula
- carbon atoms
- following formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims description 163
- 229920001296 polysiloxane Polymers 0.000 title claims description 76
- 229920000642 polymer Polymers 0.000 claims description 44
- 238000000034 method Methods 0.000 claims description 38
- 125000004432 carbon atom Chemical group C* 0.000 claims description 26
- 239000002253 acid Substances 0.000 claims description 16
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical group CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 15
- 238000006243 chemical reaction Methods 0.000 claims description 14
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 13
- 150000004820 halides Chemical class 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- 125000002947 alkylene group Chemical group 0.000 claims description 7
- 239000002516 radical scavenger Substances 0.000 claims description 6
- 125000001246 bromo group Chemical group Br* 0.000 claims description 2
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 125000002346 iodo group Chemical group I* 0.000 claims description 2
- 239000000178 monomer Substances 0.000 description 52
- 239000000203 mixture Substances 0.000 description 31
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- -1 bis(trimethylsiloxy)methylsilyl Chemical group 0.000 description 27
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 239000000047 product Substances 0.000 description 16
- 239000011541 reaction mixture Substances 0.000 description 15
- 239000002904 solvent Substances 0.000 description 15
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 14
- 230000000052 comparative effect Effects 0.000 description 14
- 239000001301 oxygen Substances 0.000 description 14
- 229910052760 oxygen Inorganic materials 0.000 description 14
- 230000035699 permeability Effects 0.000 description 14
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 12
- 239000000243 solution Substances 0.000 description 11
- 238000007259 addition reaction Methods 0.000 description 9
- 150000002430 hydrocarbons Chemical class 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 8
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 125000001153 fluoro group Chemical group F* 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 6
- 238000006459 hydrosilylation reaction Methods 0.000 description 6
- 238000005191 phase separation Methods 0.000 description 6
- 229910052697 platinum Inorganic materials 0.000 description 6
- 239000003505 polymerization initiator Substances 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 229940125904 compound 1 Drugs 0.000 description 5
- 230000005484 gravity Effects 0.000 description 5
- 150000002500 ions Chemical group 0.000 description 5
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 239000012295 chemical reaction liquid Substances 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethylcyclohexane Chemical compound CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 4
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- URZHQOCYXDNFGN-UHFFFAOYSA-N 2,4,6-trimethyl-2,4,6-tris(3,3,3-trifluoropropyl)-1,3,5,2,4,6-trioxatrisilinane Chemical compound FC(F)(F)CC[Si]1(C)O[Si](C)(CCC(F)(F)F)O[Si](C)(CCC(F)(F)F)O1 URZHQOCYXDNFGN-UHFFFAOYSA-N 0.000 description 3
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 239000007795 chemical reaction product Substances 0.000 description 3
- 210000004087 cornea Anatomy 0.000 description 3
- 230000008034 disappearance Effects 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000003999 initiator Substances 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 description 3
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 238000006116 polymerization reaction Methods 0.000 description 3
- MJYFYGVCLHNRKB-UHFFFAOYSA-N 1,1,2-trifluoroethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(F)(F)CF MJYFYGVCLHNRKB-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- HWSSEYVMGDIFMH-UHFFFAOYSA-N 2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOC(=O)C(C)=C HWSSEYVMGDIFMH-UHFFFAOYSA-N 0.000 description 2
- RIWRBSMFKVOJMN-UHFFFAOYSA-N 2-methyl-1-phenylpropan-2-ol Chemical compound CC(C)(O)CC1=CC=CC=C1 RIWRBSMFKVOJMN-UHFFFAOYSA-N 0.000 description 2
- GCYHRYNSUGLLMA-UHFFFAOYSA-N 2-prop-2-enoxyethanol Chemical compound OCCOCC=C GCYHRYNSUGLLMA-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 239000005046 Chlorosilane Substances 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical group C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 2
- 239000004721 Polyphenylene oxide Chemical group 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 229910020171 SiOLi Inorganic materials 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- YCUVUDODLRLVIC-UHFFFAOYSA-N Sudan black B Chemical compound C1=CC(=C23)NC(C)(C)NC2=CC=CC3=C1N=NC(C1=CC=CC=C11)=CC=C1N=NC1=CC=CC=C1 YCUVUDODLRLVIC-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 239000003849 aromatic solvent Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 235000007215 black sesame Nutrition 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 239000012263 liquid product Substances 0.000 description 2
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 2
- 229920001515 polyalkylene glycol Polymers 0.000 description 2
- 229920000570 polyether Chemical group 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- XLPJNCYCZORXHG-UHFFFAOYSA-N 1-morpholin-4-ylprop-2-en-1-one Chemical compound C=CC(=O)N1CCOCC1 XLPJNCYCZORXHG-UHFFFAOYSA-N 0.000 description 1
- QTKPMCIBUROOGY-UHFFFAOYSA-N 2,2,2-trifluoroethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC(F)(F)F QTKPMCIBUROOGY-UHFFFAOYSA-N 0.000 description 1
- TZJQCUDHKUWEFU-UHFFFAOYSA-N 2,2-dimethylpentanenitrile Chemical compound CCCC(C)(C)C#N TZJQCUDHKUWEFU-UHFFFAOYSA-N 0.000 description 1
- BOZPHKBMANPZNL-UHFFFAOYSA-N 2-[3-[chloro(dimethyl)silyl]propoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCC[Si](C)(C)Cl BOZPHKBMANPZNL-UHFFFAOYSA-N 0.000 description 1
- XUDBVJCTLZTSDC-UHFFFAOYSA-N 2-ethenylbenzoic acid Chemical compound OC(=O)C1=CC=CC=C1C=C XUDBVJCTLZTSDC-UHFFFAOYSA-N 0.000 description 1
- XMLYCEVDHLAQEL-UHFFFAOYSA-N 2-hydroxy-2-methyl-1-phenylpropan-1-one Chemical compound CC(C)(O)C(=O)C1=CC=CC=C1 XMLYCEVDHLAQEL-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- FRIBMENBGGCKPD-UHFFFAOYSA-N 3-(2,3-dimethoxyphenyl)prop-2-enal Chemical compound COC1=CC=CC(C=CC=O)=C1OC FRIBMENBGGCKPD-UHFFFAOYSA-N 0.000 description 1
- OKQXCDUCLYWRHA-UHFFFAOYSA-N 3-[chloro(dimethyl)silyl]propyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCC[Si](C)(C)Cl OKQXCDUCLYWRHA-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- BESKSSIEODQWBP-UHFFFAOYSA-N 3-tris(trimethylsilyloxy)silylpropyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCC[Si](O[Si](C)(C)C)(O[Si](C)(C)C)O[Si](C)(C)C BESKSSIEODQWBP-UHFFFAOYSA-N 0.000 description 1
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical group CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 229910018557 Si O Inorganic materials 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 229940117913 acrylamide Drugs 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 150000007824 aliphatic compounds Chemical class 0.000 description 1
- 150000001346 alkyl aryl ethers Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000012159 carrier gas Substances 0.000 description 1
- QABCGOSYZHCPGN-UHFFFAOYSA-N chloro(dimethyl)silicon Chemical compound C[Si](C)Cl QABCGOSYZHCPGN-UHFFFAOYSA-N 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- JJRDHFIVAPVZJN-UHFFFAOYSA-N cyclotrisiloxane Chemical compound O1[SiH2]O[SiH2]O[SiH2]1 JJRDHFIVAPVZJN-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000007033 dehydrochlorination reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000010550 living polymerization reaction Methods 0.000 description 1
- 238000003754 machining Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- VHRYZQNGTZXDNX-UHFFFAOYSA-N methacryloyl chloride Chemical compound CC(=C)C(Cl)=O VHRYZQNGTZXDNX-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- OVHHHVAVHBHXAK-UHFFFAOYSA-N n,n-diethylprop-2-enamide Chemical compound CCN(CC)C(=O)C=C OVHHHVAVHBHXAK-UHFFFAOYSA-N 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- GKWCCSUCDFFLBP-UHFFFAOYSA-N oxirane Chemical group C1CO1.C1CO1 GKWCCSUCDFFLBP-UHFFFAOYSA-N 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Inorganic materials [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 1
- 125000005401 siloxanyl group Chemical group 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 238000012719 thermal polymerization Methods 0.000 description 1
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
- C07F7/0838—Compounds with one or more Si-O-Si sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F299/00—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers
- C08F299/02—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates
- C08F299/08—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates from polysiloxanes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/04—Polysiloxanes
- C08L83/08—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen and oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/10—Block- or graft-copolymers containing polysiloxane sequences
- C08L83/12—Block- or graft-copolymers containing polysiloxane sequences containing polyether sequences
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B1/00—Optical elements characterised by the material of which they are made; Optical coatings for optical elements
- G02B1/04—Optical elements characterised by the material of which they are made; Optical coatings for optical elements made of organic materials, e.g. plastics
- G02B1/041—Lenses
- G02B1/043—Contact lenses
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/12—Polysiloxanes containing silicon bound to hydrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/20—Polysiloxanes containing silicon bound to unsaturated aliphatic groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/22—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen and oxygen
- C08G77/24—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen and oxygen halogen-containing groups
Definitions
- the present invention relates to a compound which is useful as starting materials for preparing ophthalmic devices such as contact lenses, intraocular lenses and artificial corneas, hereinafter also referred to as ophthalmic monomer, and a method for the preparation thereof.
- the present invention relates to a compound which has a specific number of silicone atoms and a specific number of fluorine atoms, is copolymerizable with the other polymerizable monomer such as a (meth)acryl monomer to provide a polymer having high transparency, oxygen permeability and excellent stain resistance and being suitable for ophthalmic uses, and a method for preparing the silicone compound.
- silicone compounds are known as an ophthalmic monomer.
- TRIS 3-[tris(trimethylsiloxy)silyl] propyl methacrylate
- hydrophilic monomers such as 2-hydroxyethyl methacrylate (HEMA). Therefore, when TRIS is copolymerized with a hydrophilic monomer, there is such a disadvantage that a transparent polymer is not obtained.
- SiGMA described above has good compatibility with hydrophilic monomers such as HEMA.
- the copolymers obtained from SiGMA have relatively high oxygen permeability and high hydrophilicity. Recently, higher oxygen permeability is required for an ophthalmic polymer so as to be used continuously on eyes for a longer term. Polymers obtained from SiGMA do not have sufficient oxygen permeability.
- Patent Literature 1 describes a compound represented by the following formula (a).
- the weight ratio of the Si-containing moiety i.e. bis(trimethylsiloxy)methylsilyl
- the weight ratio of the Si-containing moiety, i.e. tris(trimethylsiloxy)silyl, to the whole molecule is 60 %.
- the compound represented by the formula (a) thus has the higher weight ratio of the Si-containing moiety and, therefore, gives higher oxygen permeability to ophthalmic devices.
- Patent Literature 2 describes that the compound represented by the aforesaid formula (a) is prepared by a reaction of a corresponding epoxy precursor and methacrylic acid. There is such a problem such that many side reactions occur and the physical properties of the resulting copolymers vary.
- Japanese Patent No. 4882136 Paten Literature 3, describes a compound represented by the following formula (e) and an ophthalmic lens prepared from a polymer having repeating units derived from the compound.
- the polymer obtained by polymerization using the aforesaind compound as a monomer component has poor mechanical strength, and a reactivity of the polymerization of the compound is poor. Further, a stain resistance of the polymer obtained is insufficient.
- a silicone having a tetrameric or more structure is thought to be preferable in term of oxygen permeability and, in particular, a silicone having a tetrameric or pentameric structure is thought to be more preferable in order to balance between oxygen permeability and strength of the copolymer. Therefore, development of a method for preparing a silicone monomer having a tetrameric or more structure with a high purity is desired.
- Patent Literature 4 describes a method for the preparation of a silicone compound represented by a following formula (b), comprising steps of anion-polymerizing a cyclic siloxane in the presence of a lithium trialkylsilanolate as an initiator and, then, reacting the reaction product with a chlorosilane having a (meth)acryl group, such as 3-(2-methacryloyloxy ethoxy) propyl dimethyl chlorosilane.
- a chlorosilane having a (meth)acryl group such as 3-(2-methacryloyloxy ethoxy) propyl dimethyl chlorosilane.
- silicone compound obtained in the afore-mentioned method is mixed with a hydrophilic monomer, such as 2-hydroxyethyl methacrylate, turbidity occurs sometimes. Further, a ratio of terminals of the silicone chain blocked with the chlorosilane is not high.
- Patent Literature 5 describes a method for preparing a silicone compound represented by the following formula (c) by esterifying (meth)acrylic acid or transesterifying (meth)acrylate with an organopolysiloxane having a hydroxyl group at the one terminal, wherein r is an integer of 3 or larger.
- r is an integer of 3 or larger.
- the esterification ratio is insufficient, the blocked terminal ratio is low, and the compound has broad distribution of a polymerization degree of the silicone moiety.
- Patent Literature 6 describes a method for preparing a silicone monomer represented by the following formula (d) by esterifying an organopolysiloxane having a hydroxyl group at the one terminal and a (meth)acrylic acid halide: wherein m is one value out of the integers of from 3 to 10, n is one value out of 1 and 2, R 1 is only one out of alkyl groups having 1 to 4 carbon atoms, and R 2 is only one out of a hydrogen atom and a methyl group, and more than 95 weight % of the compound is one kind of compound having the specific one structure, i.e., each one value of m, n, R 1 and R 2 .
- a monomeric compound having a fluorinated hydrocarbon group was developed in order to increase oxygen permeability of its polymer or add stain resistance to its polymer.
- Japanese National Phase Publication No. 2003-516562 describes a method for copolymerizing a hydrophilic monomer, a monomer having tris(siloxysilyl) group and a monomer having a fluorinated hydrocarbon group.
- Japanese Patent Application Laid-Open No. 2008-274278 and Japanese National Phase Publication No. 2013-507652 describe a fluorine-containing silicone monomer having a siloxane chain to which a fluorinated hydrocarbon group bonds as a side chain and a polymerizable group, represented by the following formula.
- R 1 is, independently of each other, an alkyl group having 1 to 6 carbon atoms or -R 4 -CF 3
- R 4 is, independently of each other, an alkenyl group having 1 to 6 carbon atoms
- R 2 is, independently of each other, an alkenyl group having 1 to 6 carbon atoms or a fluorine-containing alkenyl group having 1 to 6 carbon atoms
- R 3 is a group selected from the group consisting of a monovalent linear or branched alkyl group, a siloxane chain having 1 to 30 Si-O units, a phenyl group, a benzyl group, a linear or branched hetero atom-containing group, or a combination of these
- m is 1 to 6
- n is 0 to 14
- p is 1 to 14
- a total of n and p is 15 or less
- Y is a divalent connecting group
- a is 0 or 1
- q is 1 to 3
- the monomers described in Patent Literature 7 are less compatible with each other and the polymer obtained becomes cloudy and cause microphase separation. Further, the monomer described in Patent Literatures 8 and 9 does not have a hydrophilic group at a part bonding the (meth)acryl group and the siloxanyl group, so that the monomer is less compatible with hydrophilic monomers.
- Patent Literatures 8 and 9 describe that the aforesaid compound is prepared by subjecting a fluorinated hydrocarbon-containing cyclotrisiloxane to a living polymerization with alkyl lithium or lithium alkyl dimethyl silanolate as an initiator, and capping the terminal with methacryloxypropyl dimethyl chlorosilane after all of the cyclosiloxane monomer reacts.
- control of the number of siloxane repeating units having a fluorinated hydrocarbon group is difficult and, therefore, a product obtained is a mixture of compounds having various amounts of fluorine atoms. Further, the amount of fluorine atoms is too large, the compatibility between the compound and the other monomers is worse, a polymer obtained becomes cloudy and microphase separation occurs.
- One of the purposes of the present invention is to provide a compound which is a polymerizable monomer having a specific number of silicon atoms and a specific number of fluorine atoms, has a higher purity, is suitable as an ophthalmic monomer, is well compatible with another (meth)acryl monomer, and provides a polymer having excellent stain resistance and to provide a method for preparing the compound.
- the present inventors have made research to solve the afore-mentioned problems and found that a compound represented by the following formula (1) is well compatible with other (meth)acryl monomers and provides a colorless, transparent and excellently stain resistant polymer.
- the present invention provides a compound represented by the following formula (1): wherein m is an integer of from 2 to 10, n is an integer of from 1 to 3, R 1 is, independently of each other, an alkyl group having 1 to 6 carbon atoms, and R 2 is, independently of each other, an alkylene group having 1 to 6 carbon atoms or a fluoroalkylene group having 1 to 6 carbon atoms, R 3 is an alkyl group having 1 to 4 carbon atoms, and R 4 , R 5 and R 6 are, independently of each other, a hydrogen atom or a methyl group, wherein an amount of one kind of compound having each one value of m and n in the formula (1) is more than 95 mass % of a total mass of the compound.
- formula (1) wherein m is an integer of from 2 to 10, n is an integer of from 1 to 3, R 1 is, independently of each other, an alkyl group having 1 to 6 carbon atoms, and R 2 is, independently of each other, an alkylene group having 1 to 6
- the present invention provides a method for preparing the compound, a polymer having repeating units derived from the aforesaid compound and an ophthalmic device composed of the polymer.
- the present silicone compound has higher oxygen permeability, has one kind of specific structure at a high ratio, and is well compatible with other (meth)acryl monomers to thereby provide a colorless and transparent polymer. Further, the present compound has the specific amount of fluorine atoms to thereby provide a polymer having increased stain resistance.
- the present method comprises a reaction of a silicone compound having a hydroxyl group and an acid chloride or an addition reaction of a silicone compound having a hydrosilyl group and a (meth)acryl compound having a terminal-unsaturated hydrocarbon group and a polyether structure.
- the present method provides a compound having one kind of specific structure at a high ratio. Accordingly, the present compound and the present method are useful for preparing ophthalmic devices.
- Figure 1 is a chart of 1 H-NMR spectra of the silicone compound prepared in Example 1.
- the present silicone compound is represented by the aforesaid formula (1), which has a silicon chain structure having a fluorinated hydrocarbon side group and a hydrophilic polyether structure in the spacer part which connects a (meth)acryl structure and the siloxane structure.
- the present compound is well compatible with other polymerizable monomers and provides a colorless and transparent polymer having a higher oxygen permeability and an increased stain resistance.
- m is an integer of from 2 to 10, preferably 3 to 7, more preferably 3. If m is smaller than the lower limit, the oxygen permeability of the polymer is worse. If m is larger than the upper limit, the hydrophilicity of the polymer is worse.
- R 5 is, independently of each other, a hydrogen atom or a methyl group.
- n is an integer of from 1 to 3.
- the present compound has a (poly)alkyleneoxide structure to thereby has the good hydrophilicity. If n is zero, the hydrophilicity is worse. If n is larger than 3, the compound does not have one kind of a specific structure at a high ratio and a polymer thereof has poor durability and mechanical strength.
- n is 1 or 2 and the silicone compound preferably has an ethylene oxide structure, an ethylene oxide-ethylene oxide structure, an ethylene oxide-propylene oxide structure or a propylene oxide-ethylene oxide structure.
- the compound has good balance of hydrophilicity.
- preferred is the compound having an ethylene oxide structure whose n is 1 and R 5 is a hydrogen atom. If the compound has too many propylene oxide structures, the hydrophobicity of the polymer obtained is too high and the hydrophilicity is poorer.
- R 1 is, independently of each other, an alkyl group having 1 to 6 carbon atoms, such as a methyl group, an ethyl group, a propyl group, a butyl group, a pentyl group and a hexyl group. Among these, a methyl group is preferable.
- R 2 is, independently of each other, an alkylene group having 1 to 6 carbon atoms or a fluoroalkylene group having 1 to 6 carbon atoms.
- alkylene group include a methylene group, an ethylene group, a propylene group, a butylene group, a pentylene group and a hexylene group.
- fluoroalkylene group include 2,2-difluoroethylene, 3,3-difluoropropylene, 3,3,4,4-tetrafluorobutylene, 3,3,4,4,5,5-hexafluorohexylene and 3,3,4,4,5,5,6,6-octafluorohexylene.
- an ethylene group is preferable.
- R 3 is an alkyl group having 1 to 4 carbon atoms, preferably a butyl group, and R 4 and R 6 are, independently of each other, a hydrogen atom or a methyl group.
- the present method of the invention provides one kind of compound which is represented by the formula (1) and has one specific structure having each specific one value of m and n at a high ratio, as will described below.
- a high ratio means that an amount of the aforesaid one kind of compound having an each specific one value of m and n, based on a total amounts of the compound represented by the formula (1), is more than 95 mass %, preferably 97 mass % or more, further preferably 99 mass % or more.
- One kind of compound having one specific structure is particularly a compound having an each specific one value of R 1 , R 2 , R 3 , R 4 , R 5 and R 6 and a specific one kind of (poly)alkyleneoxide structure.
- the ratio is determined in gas chromatography, hereinafter referred to as "GC".
- GC gas chromatography
- any turbidity does not occur and a transparent polymer is obtained, because the starting compound has a high ratio of one specific structure. If the ratio is less than 95 mass %, for instance, other compounds having different values of m are contained in an amount of more than 5 mass %, a mixture of the present silicone compound and a non-silicone monomer is turbid and does not provide a colorless and transparent polymer.
- the molecular weight is 910 and a content of siloxanes is approximately 50 mass %, based on the total mass of the compound other than the fluoromethyl group and a content of fluorine atoms is approximately 25 mass %, based on the total mass of the compound. That is, the compound comprises a large amount of Si atoms, whereby a polymer obtained therefrom has high oxygen permeability. Further, the compound has a desired amount of a fluorine atom and, therefore, stain resistance of a polymer is improved.
- the present invention further provides methods for preparing the afore-mentioned compound represented by the formula (1).
- One of the present methods comprises a step of reacting a silicone compound represented by the following formula (2) : wherein m, n, R 1 , R 2 , R 3 , R 4 and R 5 are as defined above; with (meth)acryl acid halide represented by the following formula (3): wherein X is a Cl, Br or I atom and R 6 is as defined above.
- the reaction is preferably carried out in such a manner that the acid halide represented by the formula (3), preferably an acid chloride, is slowly added to a solution of the polyorganosiloxane represented by the formula (2) in toluene or hexane to be allowed to react at a temperature of from 0 to 50 degrees C under cooling, for instance, in a water bath.
- the acid halide represented by the formula (3) preferably an acid chloride
- the amount of the acid halide (3) is 1 to 3 moles, preferably 1.05 to 2 moles, per mole of the polyorganosiloxane represented by the formula (2). If the amount is smaller than the lower limit, the polyorganosiloxane (2) would remain unreacted in the reaction product and a high ratio of one specific structure of the formula (1) is not attained. If the amount is larger than the upper limit, this is economically disadvantageous.
- the aforesaid reaction is preferably carried out in the presence of an acid scavenger.
- an acid scavenger On account of the acid scavenger, a higher yield is obtained.
- the acid scavenger include amines such as triethylamine and pyridine, preferably triethylamine.
- An amount of the acid scavenger may be 1 mole to 2 moles, per mole of the (meth)acryl acid halide represented by the formula (3).
- a purity of the (meth)acryl acid halide have an influence on a ratio of one specific compound in the silicone compound represented by the formula (1).
- the (meth)acryl acid halide preferably has a higher purity.
- commercial (meth)acryl acid halide having a purity of 99 % or more is preferable. Almost no side reaction occurs in the reaction with an acid chloride.
- a preferred embodiment in the present method is such that a peak of the unreacted silicone compound represented by the formula (2) is monitored in GC during the reaction; and after disappearance of the peak is confirmed in GC, water is added to the reaction mixture and stirred. Then, the reaction mixture is left standing to cause phase separation into an aqueous phase and an organic phase. The organic phase is washed with water several times and, then, a solvent in the organic phase is stripped off. According to this manner, a compound having one specific structure at more than 95 mass % in GC is obtained. The ratio is decided by a percentage area of the peak in GC. When a flame ionization detector (FID) is used, an area of the peak is proportional to the number of its carbon atoms. Therefore, a percentage area is almost equal to its mass percentage.
- FID flame ionization detector
- the silicone compound represented by the aforesaid formula (2) is prepared by an addition reaction of a polyorganohydrogen siloxane represented by the following formula (4): wherein m, R 1 , R 2 and R 3 are as defined above; with a compound represented by the following formula (5), hereinafter referred to as "allyl ether compound”: wherein n, R 4 and R 5 are as defined above.
- This addition reaction may be carried out in any conventional manners.
- the reaction is carried out in the presence of a hydrosilylation catalyst such as platinum group metal compounds.
- a solvent may be used.
- the solvent include aliphatic or aromatic solvents such as hexane, methylcyclohexane, ethylcyclohexane and toluene; and alcoholic solvents such as ethanol and IPA.
- a ratio of the aforesaid compounds to be used may be according to conventional manners.
- the amount of the allyl ether compound may be 1.2 moles or more, preferably 1.5 moles or more, per mole of the polyorganohydrogen siloxane. The upper limit of the amount may be usually 5 moles or less, particularly 3 moles or less, but is not limited to them.
- the allyl ether compound represented by the aforesaid formula (5) is preferably represented by the following formulas.
- the allyl ether compound is optionally diluted with a solvent to which, then, a hydrosilylation catalyst of platinum family is added. Any conventional hydrosilylation catalysts of platinum family may be used and not limited to any particular one.
- the polyorganohydrogen siloxane is added dropwise to the mixture to react at room temperature or a higher temperature. After the completion of the addition, the reaction mixture is held under heating, until disappearance of the peak of the raw material, polyorganohydrogen siloxane, is confirmed, for instance, in GC. After the end point of the reaction is confirmed in GC, the unreacted polyorganohydrogen siloxane does not remain in a product, so that a silicone compound obtained has one specific structure at a higher ratio.
- the aforesaid addition reaction may be conducted in one step.
- an excessive allyl ether compound is removed from the reaction liquid.
- the reaction liquid is subjected to stripping under a reduced pressure, or washed with ion exchanged water or an aqueous sodium sulfate solution to extract the allyl ether compound into an aqueous phase.
- a proper amount of solvent such as toluene and hexane, may preferably be used to attain clear phase separation.
- the solvent is stripped off from the organic phase under a reduced pressure, whereby the silicone compound represented by the aforesaid formula (2) and having a high ratio of one specific structure such as more than 95 mass %, even approximately 97 mass % or more, further approximately 99 mass % or more, is obtained.
- the silicone compound may be distilled twice or more to further increase the ratio.
- a high ratio means that an amount of the aforesaid one kind of compound having each specific one value of m and n, based on a total amounts of the compound represented by the formula (2), is more than 95 mass %, preferably 97 mass % or more, further preferably 99 mass % or more.
- the one specific structure means one kind of compound having each one value of m and n, particularly, one kind of compound having each one value of m, n, R 1 , R 2 , R 3 and R 4 and one kind of (poly)alkylene oxide structure.
- the polyorganohydrogen siloxane represented by the aforesaid formula (4) may be prepared in known manners.
- the compound (4) wherein m is 3, R 1 is a methyl group, R 2 is an ethylene group and R 3 is a butyl group may be prepared in the following manner.
- BuMe(CF 3 CH 2 CH 2 )SiOLi is synthesized using BuLi.
- 1,3,5-Tris(3,3,3-trifluoropropyl)-1,3,5-trimethylcyclo trisiloxane is subjected to a ring-opening reaction using the BuMe(CF 3 CH 2 CH 2 )SiOLi as an initiator and, then, the reaction is terminated with dimethylchlorosilane.
- a mixture of compounds having m of 2 to 5 is obtained.
- the mixture is distillated to collect a fraction at 146 degrees C and 84 Pa to obtain a compound whose m is 3, at a ratio of 98 mass % or higher in the fraction obtained.
- the distillation may be carried out after the mixture is addition reacted with the allyl ether compound represented by the formula (5).
- the product of the addition reaction has a higher boiling point. Therefore, the former manner is preferred.
- a silicone compound (2) having one specific structure is obtained at a higher ratio.
- the silicone compound (2) may be prepared also by the steps of subjecting the allyl ether to a silylation to provide a silyl ester with a silylating agent such as hexamethyldisilazane, addition reacting the compound obtained in the aforesaid manners and, then, hydrolyzing the silyl ester.
- a silylating agent such as hexamethyldisilazane
- Another method of the present methods for preparing the aforesaid formula (1) comprises a step of reacting a polyorganohydrogen siloxane represented by the following formula (4): wherein m, R 1 , R 2 and R 3 are as defined above; with a compound represented by the following formula (6): wherein n, R 4 and R 5 are as defined above.
- This addition reaction may be carried out in any conventional manners.
- the reaction is carried out in the presence of a hydrosilylation catalyst such as platinum group metal compounds.
- a solvent may be used.
- the solvent include aliphatic or aromatic solvents such as hexane, methylcyclohexane, ethylcyclohexane and toluene; and alcoholic solvents such as ethanol and IPA.
- the amount of the compound represented by the formula (6) may be 1.2 moles or more, preferably 1.5 moles or more, per mole of the polyorganohydrogen siloxane.
- the upper limit of the amount may be usually 5 moles or less, particularly 3 moles or less, but is not limited to them.
- the compound represented by the aforesaid formula (6) is preferably these represented by the following formulas.
- the compound represented by the formula (6) is optionally diluted with a solvent to which, then, a hydrosilylation catalyst of platinum family is added. Any conventional hydrosilylation catalysts of platinum family may be used and not limited to any particular one.
- the polyorganohydrogen siloxane is added dropwise to the mixture to react at room temperature or a higher temperature. After the completion of the addition, the reaction mixture is held under heating, until disappearance of the peak of the raw material, polyorganohydrogen siloxane, is confirmed, for instance, in GC. After the end point of the reaction is confirmed in GC, the unreacted polyorganohydrogen siloxane does not remain in a product, so that a silicone compound obtained has one specific structure at a higher ratio.
- the aforesaid addition reaction may be conducted in one step.
- an excessive amount of the compound represented by the formula (6) is removed from the reaction liquid.
- the reaction liquid is subjected to stripping under a reduced pressure, or washed with ion exchanged water or an aqueous sodium sulfate solution to extract the compound represented by the formula (6) into an aqueous phase.
- a proper amount of solvent such as toluene and hexane, may preferably be used to attain clear phase separation.
- the compound represented by the formula (6) is stripped off from the reaction product under a reduced pressure, whereby the silicone compound represented by the aforesaid formula (1) and having a high ratio of one specific structure such as more than 95 mass %, even approximately 97 mass % or more, further approximately 99 mass % or more, is obtained.
- the silicone compound of the present invention is well compatible with other compounds having a group polymerizable with the silicone compounds, such as compounds having a (meth)acryl group, hereinafter referred to as "polymerizable monomer". Therefore, the silicone compound copolymerizes with the polymerizable monomer to provide a colorless and transparent polymer.
- the silicone compound has a fluorinated hydrocarbon atom, so that it provides a polymer having excellent stain resistance. Further, the silicone compound is well compatible with a fluorinated substituent group-containing (meth)acryl monomer, so that stain resistance of the polymer obtained is increased.
- polymerizable monomer examples include acryl monomers such as (meth)acrylic acid, methyl (meth)acrylate, ethyl (meth)acrylate, (poly)ethylene glycol dimethacrylate, polyalkylene glycol mono(meth)acrylate, polyalkylene glycol monoalkyl ether (meth)acrylate, trifluoroethyl (meth)acrylate, 2-hydroxyethyl (meth)acrylate, and 2,3-dihydroxypropyl (meth)acrylate; acrylic acid derivatives such as N, N-dimethyl acrylamide, N, N-diethyl acrylamide, N-acryloyl morpholine, and N-methyl (meth)acrylamide; other ethylenically unsaturated aliphatic or aromatic compound such as crotonic acid, cinnamic acid, and vinyl benzoic acid; and silicone compounds having polymerizable groups such as a (meth)acryl group. These may be used singly or
- the copolymerization of the present compound and the other polymerizable monomer mentioned just above may be carried out in conventional known manners.
- known polymerization initiator such as thermal polymerization initiators or photo polymerization initiators may be used.
- the polymerization initiator include 2-hydroxy-2-methyl-1-phenyl-propane-1-one, azobis isobutyronitrile, azobis dimethylvaleronitrile, benzoyl peroxide, tert-butyl hydroperoxide, and cumene hydroperoxide.
- the polymerization initiator may be used singly or two or more of them may be used in combination.
- the amount of the polymerization initiator is 0.001 to 2 parts by mass, preferably 0.01 to 1 part by mass, relative to 100 parts by mass of a total amount of the polymerizable components.
- a polymer having a unit derived from the compound in the present invention has high oxygen permeability and excellent durability of mechanical strength in a buffered phosphate solution and stain resistance. Therefore, the present compounds are suitable as materials for preparing ophthalmic devices such as contact lenses, intraocular lenses and artificial corneas.
- a method for preparation of the ophthalmic device with the present polymer may be any conventional ones. For instance, a machining method and a molding method may be used for forming lenses such as contact lenses and intraocular lenses.
- a viscosity was determined by a Cannon-Fenske viscosimeter and a specific gravity was as determined by a hydrometer.
- a refraction index was as determined by a digital refractometer RX-5000, ex Atago Co., Ltd.
- 1 H-NMR analysis was conducted by JNM-ECP500, ex JEOL Ltd. with deuterochloroform as a measuring solvent.
- a ratio of a compound was determined by gas chromatography, i.e. GC. Conditions in GC were as follows.
- the reaction mixture was held at 15 degrees C for one hour, to which a mixture of 337.4g (0.72 mol) of 1,3,5-tris(3,3,3-trifluoropropyl)-1,3,5-trimethylcyclo trisiloxane and 270 g of tetrahydrofuran was then added dropwise over two hours at the internal temperature of 0 to 5 degrees C.
- the reaction mixture was aged at the internal temperature of 0 to 5 degrees C for two hours and, then further at the internal temperature of 20 to 25 degrees C for one hour.
- the mixture was distillated to collect a fraction at 146 degrees C and 84 Pa to obtain 255 g of a product with a yield of 47.8 %(0.34 mol).
- 1 H-NMR analysis showed that the product was a compound represented by the following formula (7).
- the ratio of the compound represented by the following formula (7) in the obtained product was 98.4 mass %, as determined in GC, the viscosity was 11 mm 2 /s at 25 degrees C, the specific gravity was 1.144 at 25 degrees C and the refraction index was 1.3810.
- the reaction mixture was held at 100 degrees C for one hour and, then, analyzed in GC.
- the peak of the compound represented by the aforesaid formula (7) disappeared, which means that the reaction completed.
- 100 Grams of ion exchanged water were added to the reaction mixture with stirring to wash it and, then, left standing to cause phase separation.
- the aqueous phase containing the excessive ethylene glycol monoallyl ether was removed.
- the organic phase was similarly washed twice with each 100 g of ion exchanged water and, then, the toluene in the organic phase was stripped off under a reduced pressure to obtain 193.7 g (0.23 mol) of a colorless and transparent liquid, silicone compound represented by the following formula (9).
- the yield was 92 %.
- the ratio of the silicone compound represented by the following formula (9) in the obtained product was 98.1 mass %, as determined in GC.
- the water bath was removed and the reaction mixture was held at room temperature, while monitoring the peak of the silicone compound represented by the formula (9) in GC. Ten hours later, the intensity of the peak of the silicone compound fell down below the detection limit by GC and, then, 250 g of ion exchanged water was added to the reaction mixture to wash it. The reaction mixture was left standing to cause phase separation. The aqueous phase was discarded. The organic phase was washed twice with water. The solvent, hexane, was stripped off from the organic phase under a reduced pressure to obtain 163.8 g (0.18 mol) of a colorless and transparent liquid product with a yield of 90 %.
- silicone compound 1 a silicone compound represented by the following formula (10), hereinafter referred to as silicone compound 1.
- the ratio of the silicone compound represented by the following formula (10) in the product was 97.8 mass %, as determined in GC, the viscosity was 21.6 mm 2 /s at 25 degrees C, the specific gravity was 1.139 at 25 degrees C and the refraction index was 1.4037.
- the ratio of the silicone compound represented by the aforesaid formula (10) in the obtained product was 98.0 mass %, as determined in GC, the viscosity was 21.6 mm 2 /s at 25 degrees C, the specific gravity was 1.139 at 25 degrees C and the refraction index was 1.4037.
- Example 9 The procedures of Example 9 described in Japanese Patent Application Laid-Open No. 2008-274278 , Patent Literature 8, were repeated to synthesize a polysiloxane represented by the following formula (12).
- the obtained product was a mixture of a compound whose m was 0, a compound whose m was 3, a compound whose m was 6, and a compound whose m was 9, hereinafter referred to as silicone compound 2.
- the obtained compound was a colorless and transparent liquid.
- the ratio of the silicone compound represented by the aforesaid formula (13) in the obtained product was 98.3 mass %, as determined in GC.
- Example 2 The procedures of Example 2 were repeated, except that 40.3 g (0.32 mol) of allyl methacrylate was used instead of 54.4 g (0.32 mol) of the compound represented by the aforesaid formula (11) to obtain 165.3 g (0.194 mol) of a colorless and transparent liquid product with a yield of 97 %.
- 1 H-NMR analysis showed that the obtained compound in the product was a silicone compound represented by the following formula (14), hereinafter referred to as silicone compound 4.
- the ratio of the silicone compound represented by the formula (14) in the obtained product was 98.7 mass %, as determined in GC, the viscosity was 18.4 mm 2 /s at 25 degrees C, the specific gravity was 1.143 at 25 degrees C and the refraction index was 1.4046.
- silicone compound 1 prepared in Example 1 35 parts by mass of N, N-dimethyl acryl amide, 1 part by mass of triethylene glycol dimethacrylate, 5 parts by mass of trifluoroethyl methacrylate and 0.5 part by mass of Darocur 1173, ex Ciba Specialty Chemicals Inc., were mixed with stirring to obtain monomer mixture 1.
- silicone compound 1 prepared in Example 1 60 Parts by mass of silicone compound 1 prepared in Example 1, 40 parts by mass of N, N-dimethyl acryl amide, 1 part by mass of triethylene glycol dimethacrylate and 0.5 part by mass of Darocur 1173, ex Ciba Specialty Chemicals Inc., were mixed with stirring to obtain monomer mixture 2.
- the each mixture was deaerated in an argon atmosphere.
- the mixture obtained was poured into a mold having two pieces of quartz glass plates which faced each other.
- the mixture was irradiated with light from an extra high pressure mercury lamp for one hour to obtain a film having a thickness of approximately 0.3 mm.
- the appearance of the film was observed visually.
- the results are as shown in Table 1.
- Two films for each one mixture were prepared in the same manner as in 2) above. Any water on the surface of the films was wiped off. Then, one of the twos was soaked in a buffered phosphate solution, PBS(-), at 37 degrees C for 24 hours. The film after soaked and another film without being soaked were cut into test samples having a dumbbell shape of a width of 2.0 mm. The top and the bottom of the test sample was held by a jig and pulled at a constant speed. Tensile strength and elongation at break were determined with a tensile tester AGS-50NJ, ex Shimadzu Corporation.
- Example 3 Example 4 Comparative Example 1 Comparative Example 2 Comparative Example 3 Monomer mixture 1 2 3 4 5
- Compatibility Colorless and transparent Colorless and transparent Turbid Slightly turbid Slightly turbid
- Appearance of the film Colorless and transparent Colorless and transparent Cloudy Slightly cloudy Slightly cloudy
- Water contact angle, ° 49 47 59 64 53 (4) Stain resistance Good Good Bad Bad Bad (5) Durability of a mechanical strength Good Good Bad Bad Bad Bad
- the compounds used in Comparative Examples 1 and 3 were less compatible with the other (meth)acryl monomers and did not provide a colorless and transparent polymer.
- the compound used in Comparative Example 2 did not have any fluorinated hydrocarbon group and, therefore, was less compatible with trifluoroethyl(meth)acrylate and did not provide a colorless and transparent polymer.
- the polymers obtained from the monomer mixtures in Comparative Examples 1 to 3 had poor water wettability (hydrophilicity), stain resistance and durability of mechanical strength.
- the silicone compound of the present invention is well compatible with the other (meth)acryl monomer and provides a colorless and transparent polymer. Further, the silicone compound is well compatible with a fluorinated (meth)acryl monomer, too.
- the silicone compound provides a polymer having excellent water wettability, stain resistance and durability of mechanical strength. Further, as shown in Example 4 of Table 1, the present silicone compound provides, on account of its fluorinated hydrocarbon group, a polymer having an excellent stain resistance without other fluorinated monomer.
- the present silicone compound provides a polymer having excellent hydrophilicity, stain resistance and durability of mechanical strength. Further, the present method provides a compound having one specific structure at a high ratio. Accordingly, the present compound and the present method are useful for preparing ophthalmic devices such as contact lenses, intraocular lenses and artificial corneas.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Eyeglasses (AREA)
- Materials For Medical Uses (AREA)
- Silicon Polymers (AREA)
Claims (12)
- Composé représenté par la formule (1) suivante

- Composé selon la revendication 1, dans lequel m dans la formule (1) vaut 3.
- Polymère ayant des motifs répétitifs dérivés du composé selon la revendication 1 ou 2 et des motifs répétitifs dérivés d'au moins un autre composé ayant un groupe qui est polymérisable avec ledit composé.
- Dispositif ophtalmique composé du polymère selon la revendication 3.
- Procédé pour préparer un composé représenté par la formule (1) suivante :


avec un halogénure d'acide (méth)acrylique représenté par la formule (3) suivante :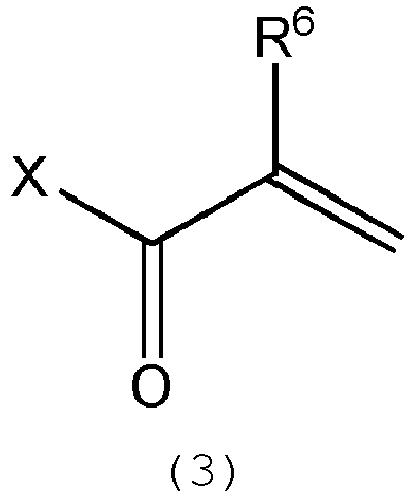
- Procédé selon la revendication 5, dans lequel ladite réaction est effectuée en présence d'un piégeur d'acide.
- Procédé selon la revendication 6, dans lequel le piégeur d'acide est la triéthylamine.
- Procédé selon l'une quelconque des revendications 5 à 7, lequel procédé comprend une étape de réaction d'un polyorganohydrogénosiloxane représenté par la formule (4) suivante :


avec un composé représenté par la formule (5) suivante :
- Procédé selon la revendication 8, dans lequel la quantité d'un type de composé ayant chacun une certaine valeur de m et n dans la formule (2) est supérieure à 95 % en masse par rapport à la masse totale du composé.
- Procédé pour préparer un composé représenté par la formule (1) suivante :


avec un composé représenté par la formule (6) suivante :
- Procédé selon l'une quelconque des revendications 5 à 10, dans lequel la quantité d'un type de composé ayant chacun une certaine valeur de m et n dans la formule (2) est supérieure à 95 % en masse par rapport à la masse totale du composé.
- Procédé selon l'une quelconque des revendications 5 à 11, dans lequel m dans la formule (1) vaut 3.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014057845A JP6147211B2 (ja) | 2014-03-20 | 2014-03-20 | 眼科デバイス製造用モノマー |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2921497A1 EP2921497A1 (fr) | 2015-09-23 |
| EP2921497B1 true EP2921497B1 (fr) | 2017-07-05 |
Family
ID=52684123
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP15159516.2A Active EP2921497B1 (fr) | 2014-03-20 | 2015-03-17 | Composé de silicone et utilisation de celui-ci |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9353135B2 (fr) |
| EP (1) | EP2921497B1 (fr) |
| JP (1) | JP6147211B2 (fr) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6491311B2 (ja) * | 2014-03-17 | 2019-03-27 | ダウ シリコーンズ コーポレーション | フッ素化化合物、該化合物を含む硬化性組成物、及び硬化物 |
| JP2017105753A (ja) * | 2015-12-04 | 2017-06-15 | 信越化学工業株式会社 | 両末端に異なる官能基を有する直鎖オルガノポリシロキサン、及びその製造方法 |
| JP7108558B2 (ja) * | 2019-02-08 | 2022-07-28 | 信越化学工業株式会社 | (メタ)アクリルシリコーン化合物の製造方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5978236A (ja) | 1982-10-28 | 1984-05-07 | Toagosei Chem Ind Co Ltd | ビニル重合反応性直鎖シリコ−ン高分子量モノマ−の製造法 |
| US4665147A (en) * | 1983-06-30 | 1987-05-12 | Loctite Corporation | Novel methacrylated siloxanes |
| EP0130731A3 (fr) * | 1983-06-30 | 1986-03-26 | Loctite Corporation | Siloxanes méthacrylés |
| JP3186574B2 (ja) * | 1995-04-05 | 2001-07-11 | 信越化学工業株式会社 | 被覆組成物 |
| JP2001055446A (ja) | 1999-08-18 | 2001-02-27 | Chisso Corp | ポリオルガノシロキサンおよびその製造方法 |
| US6649722B2 (en) | 1999-12-10 | 2003-11-18 | Novartis Ag | Contact lens |
| JP4882136B2 (ja) | 2000-05-12 | 2012-02-22 | 東レ株式会社 | モノマー、ポリマーおよびそれを用いた眼用レンズ |
| JP4802569B2 (ja) | 2005-06-23 | 2011-10-26 | 東レ株式会社 | シリコーンモノマーの製造方法 |
| JP2007186709A (ja) | 2007-03-05 | 2007-07-26 | Toray Ind Inc | 眼用レンズ用モノマー、眼用レンズ用ポリマーおよびコンタクトレンズ |
| JP2008255328A (ja) * | 2007-03-14 | 2008-10-23 | Kanto Denka Kogyo Co Ltd | 含フッ素ランダム共重合体及びその製造方法 |
| US7799888B2 (en) * | 2007-04-27 | 2010-09-21 | Gelest, Inc. | Low molecular weight siloxanes with one functional group |
| JP4646152B2 (ja) * | 2008-05-27 | 2011-03-09 | 信越化学工業株式会社 | 眼科デバイス製造用モノマー |
| GB0917806D0 (en) * | 2009-10-12 | 2009-11-25 | Sauflon Cl Ltd | Fluorinated silicone hydrogels |
-
2014
- 2014-03-20 JP JP2014057845A patent/JP6147211B2/ja active Active
-
2015
- 2015-03-16 US US14/658,799 patent/US9353135B2/en active Active
- 2015-03-17 EP EP15159516.2A patent/EP2921497B1/fr active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2921497A1 (fr) | 2015-09-23 |
| US20150266905A1 (en) | 2015-09-24 |
| US9353135B2 (en) | 2016-05-31 |
| JP6147211B2 (ja) | 2017-06-14 |
| JP2015183017A (ja) | 2015-10-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3085727B1 (fr) | Silicone et son utilisation | |
| JP6037454B2 (ja) | 眼科デバイス製造用モノマー | |
| JP6267101B2 (ja) | 眼科デバイス製造用モノマー | |
| EP2921497B1 (fr) | Composé de silicone et utilisation de celui-ci | |
| EP2955208B1 (fr) | Composé de silicone et utilisation de celui-ci | |
| JP6045515B2 (ja) | 眼科デバイス製造用モノマー | |
| EP2955207B1 (fr) | Composé de silicone et utilisation de celui-ci | |
| EP2952515B1 (fr) | Composé de silicone et utilisation de celui-ci | |
| JP6305323B2 (ja) | 共重合体および眼科デバイス | |
| JP6356628B2 (ja) | 眼科デバイス製造用モノマー、その重合体、及び眼科デバイス | |
| JP6305324B2 (ja) | 共重合体および眼科デバイス | |
| JP6073207B2 (ja) | 眼科デバイス製造用モノマー | |
| JP6037453B2 (ja) | 眼科デバイス製造用モノマー | |
| WO2020246313A1 (fr) | Siloxane et procédé de production associé | |
| JP6282218B2 (ja) | 眼科デバイス製造用モノマー | |
| JP6335836B2 (ja) | 眼科デバイス製造用モノマー、その重合体、及び眼科デバイス | |
| JP6195418B2 (ja) | 眼科デバイス製造用モノマー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA ME |
|
| 17P | Request for examination filed |
Effective date: 20160317 |
|
| RBV | Designated contracting states (corrected) |
Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| 17Q | First examination report despatched |
Effective date: 20160905 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: EXAMINATION IS IN PROGRESS |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: GRANT OF PATENT IS INTENDED |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: G02B 1/04 20060101ALI20170111BHEP Ipc: C08F 299/08 20060101ALI20170111BHEP Ipc: C08L 83/08 20060101ALI20170111BHEP Ipc: C07F 7/18 20060101AFI20170111BHEP Ipc: C08G 77/12 20060101ALI20170111BHEP Ipc: C08L 83/12 20060101ALI20170111BHEP Ipc: C08G 77/08 20060101ALI20170111BHEP Ipc: C08G 77/20 20060101ALI20170111BHEP Ipc: C08G 77/24 20060101ALI20170111BHEP |
|
| INTG | Intention to grant announced |
Effective date: 20170201 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: SHIN-ETSU CHEMICAL CO., LTD |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE PATENT HAS BEEN GRANTED |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 906537 Country of ref document: AT Kind code of ref document: T Effective date: 20170715 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602015003376 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MP Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 906537 Country of ref document: AT Kind code of ref document: T Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: NO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20171005 Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: NL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 4 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20171006 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20171105 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20171005 Ref country code: ES Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: RS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602015003376 Country of ref document: DE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| 26N | No opposition filed |
Effective date: 20180406 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| REG | Reference to a national code |
Ref country code: BE Ref legal event code: MM Effective date: 20180331 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180317 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180317 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180331 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180331 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180331 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20180317 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT; INVALID AB INITIO Effective date: 20150317 Ref country code: MK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20170705 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20170705 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20240130 Year of fee payment: 10 Ref country code: GB Payment date: 20240201 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20240213 Year of fee payment: 10 |