EP2616465B1 - Triazin-oxadiazole - Google Patents
Triazin-oxadiazole Download PDFInfo
- Publication number
- EP2616465B1 EP2616465B1 EP11757294.1A EP11757294A EP2616465B1 EP 2616465 B1 EP2616465 B1 EP 2616465B1 EP 11757294 A EP11757294 A EP 11757294A EP 2616465 B1 EP2616465 B1 EP 2616465B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- alkyl
- amino
- methyl
- phenyl
- triazine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(C)(*)*1C=CC(*(C2)C2*(*)N(*)c2nc(N)nc(C(N)=NO)n2)=C(*)C=C1 Chemical compound CC(C)(*)*1C=CC(*(C2)C2*(*)N(*)c2nc(N)nc(C(N)=NO)n2)=C(*)C=C1 0.000 description 2
- XORMAXFRFRFLIK-UHFFFAOYSA-N CN(c1ccccc1)c1nc(N)nc(-c2n[o]c(N3CCC(COCC(F)(F)F)CC3)n2)n1 Chemical compound CN(c1ccccc1)c1nc(N)nc(-c2n[o]c(N3CCC(COCC(F)(F)F)CC3)n2)n1 XORMAXFRFRFLIK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5386—1,4-Oxazines, e.g. morpholine spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
Definitions
- the present invention relates to new triazine-oxadiazoles; processes for the preparation of such triazine-oxadiazoles; pharmaceutical compositions comprising such triazine-oxadiazoles optionally in combination with one or more other pharmaceutically active compounds; such triazine-oxadiazoles optionally in combination with one or more other pharmaceutically active compounds as a medicament; such triazine-oxadiazoles optionally in combination with one or more other pharmaceutically active compounds for the treatment of chronic pain, such as positive symptoms of chronic pain e.g.
- the compounds of the present invention are sodium channel blockers, in particular selective inhibitors of the voltage-gated sodium channel 1.7 (Nav1.7) which is involved in pain. Since other sodium channel subtypes are involved in different essential physiological processes such as heart activity (Nav1.5), muscle contraction (Nav1.4) and CNS neurotransmission (Nav1.1, 1.2 and 1.6), selectivity for Nav1.7 is believed to be associated with the potential elimination of side effects.
- Nav1.7 voltage-gated sodium channel 1.7
- Nav1.7 blockers are described: The tarantula venom peptide Pro-TX-II is a potent inhibitor of Nav1.7 ( Schmalhofer et al, Molecular Pharmacology 2008, 74, 1476-1484 ). A series of Benzazepinone Nav1.7 blockers are described to show activity in pre-clinical pharmacological models of pain ( Williams et al, Biochemistry, 2007, 46(50), 14693-14703 ; McGowan et al., Anesth Analg, 2009, 109, 951-958 ). Amino-thiazoles and amino-pyridines are described as Nav1.7 inhibitors ( WO2007109324 ) and isoxazoles are described as Nav1.7 inhibitors ( WO2009010784 ).
- 1,2,4-Oxadiazoles as Nav1.7 inhibitors are described in WO2011/103196 and WO2010/0022055 .
- Nonsense mutations in SCN9A appear to be linked to Congenital Indifference to Pain (CIP) ( Cox et al, Nature, 2006, 444(7121), 894-898 ).
- CIP Congenital Indifference to Pain
- Patients with CIP are essentially completely indifferent to sensations that would cause pain in most individuals e.g. bone fractures, burns, dental abscesses, appendicitis and childbirth. Concurrently, they are able to distinguish between other sensations, such as thermal (hot/cold) and tactile (sharp/dull) stimuli ( Goldberg et al, Clinical Genetics, 2007, 71(4), 311-319 ).
- triazines have been reported, e.g. as kinase inhibitors by Janssen ( WO2004009562 ); as integrin inhibitors by Biochem Pharma ( WO2000075129 ).
- the invention relates to triazine-oxadiazoles of the formula (I) and/or pharmaceutically acceptable salts and/or solvates thereof, wherein R 1 is selected from
- the invention relates to triazine-oxadiazoles of the formula (I) and/or pharmaceutically acceptable salts and/or solvates thereof, wherein R 1 is selected from
- the invention relates to the use of triazine-oxadiazoles of the formula (I) and/or pharmaceutically acceptable salts and/or solvates thereof, wherein R 1 is selected from
- halogen is used herein to describe, unless otherwise stated, a group selected from fluoro (fluorine), chloro (chlorine), bromo (bromine) or iodo (iodine).
- alkyl refers to a fully saturated branched, including single or multiple branching, or unbranched hydrocarbon moiety having up to 20 carbon atoms. Unless otherwise provided, alkyl refers to hydrocarbon moieties having 1 to 16 carbon atoms, 1 to 10 carbon atoms, 1 to 7 carbon atoms, or 1 to 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2- dimethylpentyl, 2,3-dimethylpentyl, n-heptyl, n-octyl, n-nonyl, n-decyl and the like.
- alkyl groups have 1-7, more preferably 1-4 carbons.
- halo-alkyl refers to an alkyl as defined herein, that is substituted by one or more halo groups as defined herein.
- the halo-alkyl can be mono-halo-alkyl, di-halo-alkyl or poly-halo-alkyl including per-halo-alkyl.
- a mono-halo-alkyl can have one iodo, bromo, chloro or fluoro within the alkyl group.
- Di-halo-alky and poly-halo-alkyl groups can have two or more of the same halo atoms or a combination of different halo groups within the alkyl.
- the poly-halo-alkyl contains up to 12, or 10, or 8, or 6, or 4, or 3, or 2 halo groups.
- halo-alkyl include fluoro-methyl, di-fluoro-methyl, tri-fluoro-methyl, chloro-methyl, di-chloro-methyl, tri-chloro-methyl, penta-fluoro-ethyl, hepta-fluoro-propyl, di-fluoro-chloro-methyl, di-chloro-fluoro-methyl, di-fluoro-ethyl, di-fluoro-propyl, di-chloro-ethyl and dichloro-propyl.
- a per-halo-alkyl refers to an alkyl having all hydrogen atoms replaced with halo atoms.
- cycloalkyl refers to saturated or partially unsaturated monocyclic, bicyclic or tricyclic hydrocarbon groups of 3-12 carbon atoms. Unless otherwise provided, cycloalkyl refers to cyclic hydrocarbon groups having between 3 and 10 ring carbon atoms or between 3 and 7 ring carbon atoms. Exemplary monocyclic hydrocarbon groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl and cyclohexenyl.
- Exemplary bicyclic hydrocarbon groups include octahydroindyl, decahydronaphthyl, bicyclo[2.1.1]hexyl, bicyclo[2.2.1]heptyl, bicyclo[2.2.1]heptenyl, 6,6-dimethylbicyclo[3.1.1]heptyl, 2,6,6-trimethylbicyclo[3.1.1]heptyl, bicyclo[2.2.2]octyl.
- Exemplary tricyclic hydrocarbon groups include adamantyl.
- the term "cycloalkyl" preferably refers to cyclopropyl, cyclopentyl or, cyclohexyl.
- C 2 -C 7 -alkenyr refers to a linear or branched hydrocarbon group containing from 2 to 5 carbon atoms that contains at least one carbon to carbon double bond. Examples of such groups include ethenyl, propenyl, butenyl and pentenyl. Unless a particular structure is specified, the terms butenyl and pentenyl etc. include all possible E and Z isomers.
- aryl refers to 6-carbon monocyclic, 10-carbon bicyclic, 14-carbon tricyclic aromatic ring system. Examples of “aryl” are phenyl and naphthyl. As used herein, the term “aryl” preferably refers to phenyl.
- heteroaryl refers to a 4-, 5-, 6-, or 7-membered monocyclic, 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or 10-, 11-, 12-, 13-, 14- or 15-membered tricyclic unsaturated ring or ring system - carrying the highest possible number of conjugated double bonds in the ring(s), which contain at least one heteroatom selected from N, O and S, wherein the N and S can also optionally be oxidized to various oxidation states.
- 'Heteroaryl' can be attached at a heteroatom or a carbon atom.
- Heteroaryl' can include fused or bridged rings as well as spirocyclic rings.
- heteroaryl include pyridyl, quinolinyl, isoquinolinyl, pyridazinyl, pyrimidinyl, pyrazinyl, quinoxalinyl, furanyl, benzofuranyl, dibenzofuranyl, thiophenyl, benzothienyl, pyrrolyl, indolyl, pyrazolyl, indazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, imidazolyl, benzimidazolyl, oxadiazolyl-such as 1,2,5-oxadiazolyl, 1,2,4-oxadiazolyl, 1,2,3-oxadiazolyl,1,3,4-oxadiazolyl, thiadiazolyl - such as 1,2,5-thiadiazolyl, 1,
- heteroaryl preferably refers to furanyl, thiophenyl, pyridyl, thiazolyl, pyrazolyl, isoxazolyl, imidazolyl, pyrrolyl, benzofuranyl, pyrimidinyl, oxazolyl.
- heterocyclyl refers to a 4-, 5-, 6-, 7 or 8-membered monocyclic, 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or 10-, 11-, 12-, 13-, 14- or 15-membered tricyclic saturated or partially unsaturated ring or ring system, which contain at least one heteroatom selected from N, O and S, wherein the N and S can also optionally be oxidized to various oxidation states.
- 'Heterocyclyl' can be attached at a heteroatom or a carbon atom.
- 'Heterocyclyl' can include fused or bridged rings as well as spirocyclic rings.
- heterocyclyl examples include dioxolanyl, imidazolinyl, imidazolidinyl, isothiazolidinyl, isoxazolidinyl, morpholinyl, oxazolidinyl, piperidinyl, piperazinyl, pyrrolidinyl, pyrazolidinyl, thiazolidinyl, tetrahydrofuranyl, trithianyl, tetrahydropyranyl, thiamorpholinyl as well as di-, tetra-, hexa-, octa- or deca-hydro derivatives of "heteroaryl".
- heterocyclyl preferably refers to morpholinyl, piperidinyl, tetrahydrofuranyl, tetrahydropyranyl, pyrrolidinyl, dihydrobenzofuranyl.
- heterocyclyl in the context of Y preferably refers to tetrahydrofuranyl, tetrahydropyranyl, dihydrobenzofuranyl.
- heterocyclyl in the context of a heterocyclic ring formed by R 4 and R 4 ' together with the nitrogen atom to which they are attached or by R 5 and R 5 ' together with the nitrogen atom to which they are attached or by R 6 and R 6 ' together with the nitrogen atom to which they are attached, preferably refers to morpholinyl, piperidinyl, pyrrolidinyl.
- spirocyclyl refers to a "4 to 7 membered monocyclic heterocyclyl" as defined hereinbefore which is fused to a second 3 to 6 membered saturated ring via one single atom, and wherein the second ring contains 0, 1 or 2 heteroatoms independently selected from oxygen, nitrogen and sulphur.
- oxy refers to an -O- linking group.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein wherein R 1 is selected from
- 6-[5-(2-Furanyl)-1,2,4-oxadiazol-3-yl]-N2-methyl-N2-phenyl-1,3,5-triazine-2,4-diamine, (CAS-899373-19-4) is a compound from a chemical library.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 2 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 2 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 2 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 and R 2 , together are selected from -CH 2 -CH 2 - or -CH 2 -CH 2 -CH 2 -.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 3 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 3 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 3 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 3 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 3 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein wherein m is 0.
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) and/or a pharmaceutically acceptable salt and/or a solvate thereof, wherein R 1 is selected from
- the invention provides a compound of the formula (I) is selected from 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine, 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-phenyl-[1,3,5]triazine-2,4-diamine, 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-methyl-N-(3-methylphenyl)-[1,3,5]triazine-2,4-diamine, 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-methyl-N-(4-methylphenyl)-[1,3,5]triazine-2,4-diamine, 6-(5-Furan-2-yl-[1,2,4]
- an optical isomer or "a stereoisomer” refers to any of the various stereo isomeric configurations which may exist for a given compound of the present invention and includes geometric isomers. It is understood that a substituent may be attached at a chiral center of a carbon atom. Therefore, the invention includes enantiomers, diastereomers, rotamers, atropisomers or racemates of the compound. "Enantiomers” are a pair of stereoisomers that are non-superimposable mirror images of each other. A 1:1 mixture of a pair of enantiomers is a "racemic" mixture.
- Diastereoisomers are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other.
- the absolute stereochemistry is specified according to the Cahn-Ingold-Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon may be specified by either R or S.
- Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
- Certain of the compounds described herein contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers, and other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)-or (S)-.
- the present invention is meant to include all such possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures.
- Optically active (R)-and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. If the compound contains a double bond, the substituent may be E or Z configuration.
- the cycloalkyl substituent may have a cis- or trans-configuration. If the compound contains an axis of chirality, it might be present in form of atropisomers, which are conformational isomers, where rotation around a single bond is restricted. Atopisomers may be specified either as the (R a )- or (S a )-enatiomer. All rotameric forms are also intended to be included. All tautomeric forms are also intended to be included.
- the term "pharmaceutically acceptable salts” refers to salts that retain the biological effectiveness and properties of the compounds of this invention and, which typically are not biologically or otherwise undesirable.
- the compounds of the present invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide, bicarbonate/carbonate, bisulfate/sulfate, camphorsulfonate, chloride/hydrochloride, chlortheophyllonate, citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulfate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydr
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns 1 to 12 of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound, a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the too.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- any reference to the compounds or a compound of the formula (I) hereinbefore and hereinafter is to be understood as referring to the compound in free form and/or also to one or more salts thereof, as appropriate and expedient, as well as to one or more solvates, e.g. hydrates.
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms of the compounds.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, and chlorine, such as 2 H , 3 H, 11 C, 13 C, 14 C, 15 N, 18 F 31 P, 32 P, 35 S, 36 Cl, 125 I respectively.
- the invention includes various isotopically labeled compounds as defined herein, for example those into which radioactive isotopes, such as 3 H and 14 C or those into which non-radioactive isotopes, such as 13 C, are present.
- isotopically labeled compounds are useful in metabolic studies (with 14 C) , reaction kinetic studies (with, for example 2 H or 3 H ), detection or imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) including drug or substrate tissue distribution assays, or in radioactive treatment of patients.
- PET positron emission tomography
- SPECT single-photon emission computed tomography
- an 18 F or labeled compound may be particularly desirable for PET or SPECT studies.
- Isotopically labeled compounds of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the schemes or in the examples and preparations described below by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- isotopic enrichment factor means the ratio between the isotopic abundance and the natural abundance of a specified isotope.
- a substituent in a compound of this invention is denoted deuterium, such compound has an isotopic enrichment factor for each designated deuterium atom of at least 3500 (52.5% deuterium incorporation at each designated deuterium atom), at least 4000 (60% deuterium incorporation), at least 4500 (67.5% deuterium incorporation), at least 5000 (75% deuterium incorporation), at least 5500 (82.5% deuterium incorporation), at least 6000 (90% deuterium incorporation), at least 6333.3 (95% deuterium incorporation), at least 6466.7 (97% deuterium incorporation), at least 6600 (99% deuterium incorporation), or at least 6633.3 (99.5% deuterium incorporation).
- Isotopically-labeled compounds of the formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O, d 6 -acetone, d 6 -DMSO.
- Compounds of the invention i.e. compounds of the formula (I) that contain groups capable of acting as donors and/or acceptors for hydrogen bonds may be capable of forming co-crystals with suitable co-crystal formers.
- These co-crystals may be prepared from compounds of the formula (I) by known co-crystal forming procedures. Such procedures include grinding, heating, co-subliming, co-melting, or contacting in solution compounds of the or (I) I) with the co-crystal former under crystallization conditions and isolating co-crystals thereby formed.
- Suitable co-crystal formers include those described in WO 2004/078163 .
- the invention further provides co-crystals comprising a compound of the formula (I).
- the term "pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drugs, drug stabilizers, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, and the like and combinations thereof, as would be known to those skilled in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289- 1329 ). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
- ком ⁇ онент there is meant either a fixed combination in one dosage unit form, or a kit of parts for the combined administration where a compound of the formula (I) and a combination partner may be administered independently at the same time or separately within time intervals that especially allow that the combination partners show a cooperative, e.g. synergistic effect.
- a therapeutically effective amount of a compound of the present invention refers to an amount of the compound of the present invention that will elicit the biological or medical response of a subject, for example, reduction or inhibition of an enzyme or a protein activity, or ameliorate symptoms, alleviate conditions, slow or delay disease progression, or prevent a disease, etc.
- the term "a therapeutically effective amount” refers to the amount of the compound of the present invention that, when administered to a subject, is effective to (1) at least partially alleviating, inhibiting, preventing and/or ameliorating a condition, or a disorder or a disease (i) selected from chronic pain, such as positive symptoms of chronic pain e.g.
- a therapeutically effective amount refers to the amount of the compound of the present invention that, when administered to a cell, or a tissue, or a non-cellular biological material, or a medium, is effective to at least partially reducing or inhibiting Nav1.7.
- subject refers to an animal.
- the animal is a mammal.
- a subject also refers to for example, primates (e.g., humans), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice, fish, birds and the like.
- the subject is a primate.
- the subject is a human.
- the term “inhibit”, “inhibition” or “inhibiting” refers to the reduction or suppression of a given condition, symptom, or disorder, or disease, or a significant decrease in the baseline activity of a biological activity or process.
- the term “treat”, “treating” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat”, “treating” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat”, “treating” or “treatment” refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- “treat”, “treating” or “treatment” refers to preventing or delaying the onset or development or progression of the disease or disorder.
- a subject is "in need of” a treatment if such subject would benefit biologically, medically or in quality of life from such treatment.
- the term "a,” “an,” “the” and similar terms used in the context of the present invention are to be construed to cover both the singular and plural unless otherwise indicated herein or clearly contradicted by the context. All methods described herein can be performed in any suitable order unless otherwise indicated herein or otherwise clearly contradicted by context. The use of any and all examples, or exemplary language (e.g. "such as”) provided herein is intended merely to better illuminate the invention and does not pose a limitation on the scope of the invention otherwise claimed.
- Compounds of the present invention are either obtained in the free form, as a salt thereof, or as prodrug derivatives thereof.
- the compounds of the present invention may also form internal salts, e.g., zwitterionic molecules.
- the compounds of the present invention can also be obtained in the form of their hydrates, or include other solvents used for their crystallization.
- the invention relates in a third aspect to the manufacture of a compound of formula(I) as described above
- the compounds of formula (I) or salts thereof are prepared in accordance with processes known per se, e.g. the synthesis and structures of 4,6-disubstituted 2-(5-alkyl-1,2,4-oxadiazol-3-yl)-1,3,5-triazines have been reported ( Russian Chemical Bulletin, 2005, 54(8), 1900-1906 ), though not previously described for the manufacture of the compounds of the formula (I).
- a compound of formula (I) may be prepared according to Schemes 1 to 4.
- the invention relates to a process for manufacturing a compound of formula (I) comprising the step a) of reacting a compound of formula (IV) wherein the substituents are as defined above, with an acylating agent of formula (III), wherein the substituents are as defined above, in the presence of a base e.g. pyridine, suitably in the presence of one or more diluents, particular solvents, e.g. toluene; followed by dehydration leading to cyclization, typically induced by heating.
- a base e.g. pyridine
- diluents particular solvents
- dehydration leading to cyclization typically induced by heating.
- Typical reaction conditions for acylations and dehydrations are known in the field and may applied to the present process.
- the compound of formula (IV) is obtained comprising the step b) of reacting a compound of formula (V) wherein the substituents are as defined above, with hydroxylamine in the presence of a suitable solvent such as ethanol and water, optionally using a base e.g. sodium hydrogencarbonate if necessary with heating e.g. to reflux.
- a suitable solvent such as ethanol and water
- a base e.g. sodium hydrogencarbonate
- the compound of formula (V) is obtained comprising the step c) of reacting a compound of formula (VI) wherein the substituents are as defined above, with a reagent such as potassium cyanide or sodium cyanide in a suitable solvent such as DMF or DMSO, optionally with heating e.g. to 120 C; alternatively a reagent such as tetrabutylammonium cyanide in a solvent such as acetonitrile may be used at a temperature of 50 C, optionally in the in the presence of a co-reagent such as diazabicyclooctane. Typical reaction conditions for this reaction are known in the field and may applied to the present reaction step.
- an alkylating agent e.g. iodomethane in the presence of a base such as potassium carbonate in a suitable solvent e.g. DMF.
- the compound of formula (VI) is obtained comprising the step d) of reacting a compound of formula (VII) wherein the substituents are as defined above, first with, cyanuric chloride (VIII) at 0 C to room temperature followed by treatment with aqueous ammonia in a suitable solvent such as THF.
- aqueous ammonia in a suitable solvent such as THF.
- Typical reaction conditions are known in the field and may applied to the present reaction step.
- Amines of formula (VII) are commercially available, or may be prepared according to standard methods known to those skilled in the art.
- the invention in another embodiement (Method II), relates to a process for manufacturing a compound of formula (I) comprising the steps a) and b), wherein the compound of formula (V) is obtained comprising the step c') of reacting a compound of formula (VII) wherein the substituents are as defined above, with chlorotriazine (IX) in a suitable solvent such as DMF or DMSO in the presence of a suitable base e.g. DIPEA, optionally with heating to e.g. 90 C.
- a suitable solvent such as DMF or DMSO
- a suitable base e.g. DIPEA
- Typical reaction conditions are known in the field and may applied to the present reaction step.
- Chlorotriazine (IX) is obtained comprising the step d') of treating 6-amino-4,5-dihydro-4-oxo-1,3,5-triazine-2-carbonitrile (X) ( J. Am. Chem. Soc., 1961, 83, 1261-2 ) with a chlorinating reagent e.g. phosphoryl chloride at reflux. Typical chlorination conditions are known in the field and may applied to the present reaction step.
- the invention in another embodiment (Method 2), relates to a process for manufacturing a compound of formula (I) comprising the step a) of reacting an ester of formula (IX) with an amine of formula (X) or alcohol of formula (XI), optionally in the presence of a reagent such as trimethylaluminium diazabicyclo-octane complex (for amine (X)) or an acid or base.
- a reagent such as trimethylaluminium diazabicyclo-octane complex (for amine (X)) or an acid or base.
- Typical amide or transesterification conditions are known in the field and may applied to the present reaction step.
- Amines and alcohols of formulae (X) and (XI) are commercially available, or may be prepared according to standard methods known to those skilled in the art.
- An ester of formula (IX) is prepared in step b) by reaction of a compound of formula (IV) with ethyl oxalyl chloride.
- the invention in another embodiment (Method 3), relates to a process for manufacturing a compound of formula (I) comprising the step a) of reacting a compound of formula (XII) with an amine of formula (XIII) in the presence of a suitable base e.g. potassium carbonate in a suitable solvent such as DMF, optionally with heating.
- a suitable base e.g. potassium carbonate
- a suitable solvent such as DMF
- a compound of formula (XIII) is prepared in step b) by reaction of a compound of formula (IV) with trichloroacetic anhydride.
- the invention further includes any variant of the present processes, in which an intermediate product obtainable at any stage thereof is used as starting material and the remaining steps are carried out, or in which the starting materials are formed in situ under the reaction conditions, or in which the reaction components are used in the form of their salts or optically pure antipodes.
- Intermediates and final products can be worked up and/or purified according to standard methods, e.g. using chromatographic methods, distribution methods, (re-) crystallization, and the like.
- mixtures of isomers that are formed can be separated into the individual isomers, for example diastereoisomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereoisomers, for example analogously to the methods described herein above.
- solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2-propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2-one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydride,
- Such solvent mixtures may also be used in working up, for example by chromatography or partitioning.
- the compounds, including their salts may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
- the invention relates also to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ. All starting materials, building blocks, reagents, acids, bases, dehydrating agents, solvents and catalysts utilized to synthesize the compounds of the present invention are either commercially available or can be produced by organic synthesis methods known to one of ordinary skill in the art.
- protective groups and the manner in which they are introduced and removed are described, for example, in “ Protective Groups in Organic Chemistry”, Plenum Press, London, New York 1973 , and in “ Methoden der Organischen Chemie", Houben-Weyl, 4th edition, Vol. 15/1, Georg-Thieme-Verlag, Stuttgart 1974 and in Theodora W. Greene, "Protective Groups in Organic Synthesis", John Wiley & Sons, New York 1981 .
- a characteristic of protective groups is that they can be removed readily, i.e. without the occurrence of undesired secondary reactions, for example by solvolysis, reduction, photolysis or alternatively under physiological conditions.
- the invention relates in a fourth aspect to the the use of compounds of the present invention as pharmaceuticals.
- the compounds of formula (I) have valuable pharmacological properties, as described hereinbefore and hereinafter.
- the invention thus provides:
- the invention relates in a fifth aspect to pharmaceutical compositions comprising a compound of the present invention.
- the pharmaceutical composition can be formulated for particular routes of administration such as oral administration, parenteral administration, and rectal administration, etc.
- the pharmaceutical compositions of the present invention can be made up in a solid form (including without limitation capsules, tablets, pills, granules, powders or suppositories), or in a liquid form (including without limitation solutions, suspensions or emulsions).
- compositions can be subjected to conventional pharmaceutical operations such as sterilization and/or can contain conventional inert diluents, lubricating agents, or buffering agents, as well as adjuvants, such as preservatives, stabilizers, wetting agents, emulsifiers and buffers, etc.
- pharmaceutical compositions are tablets or gelatin capsules comprising the active ingredient together with
- compositions for oral administration include an effective amount of a compound of the invention in the form of tablets, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use are prepared according to any method known in the art for the manufacture of pharmaceutical compositions and such compositions can contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets may contain the active ingredient in admixture with nontoxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients are, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin or acacia; and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets are uncoated or coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate can be employed.
- Formulations for oral use can be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example, peanut oil, liquid paraffin or olive oil.
- compositions are aqueous isotonic solutions or suspensions, and suppositories are advantageously prepared from fatty emulsions or suspensions.
- Said compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- Said compositions are prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1-75%, or contain about 1-50%, of the active ingredient.
- compositions for transdermal application include an effective amount of a compound of the invention with a suitable carrier.
- Carriers suitable for transdermal delivery include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound of the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- compositions for topical application include aqueous solutions, suspensions, ointments, creams, gels or sprayable formulations, e.g., for delivery by aerosol or the like.
- topical delivery systems will in particular be appropriate for dermal application, e.g., for the treatment of skin cancer, e.g., for prophylactic use in sun creams, lotions, sprays and the like. They are thus particularly suited for use in topical, including cosmetic, formulations well-known in the art.
- Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- a topical application may also pertain to an inhalation or to an intranasal application. They may be conveniently delivered in the form of a dry powder (either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids) from a dry powder inhaler or an aerosol spray presentation from a pressurized container, pump, spray, atomizer or nebulizer, with or without the use of a suitable propellant.
- a dry powder either alone, as a mixture, for example a dry blend with lactose, or a mixed component particle, for example with phospholipids
- the present invention further provides anhydrous pharmaceutical compositions and dosage forms comprising the compounds of the present invention as active ingredients, since water may facilitate the degradation of certain compounds.
- Anhydrous pharmaceutical compositions and dosage forms of the invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- An anhydrous pharmaceutical composition may be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e. g., vials), blister packs, and strip packs.
- compositions and dosage forms that comprise one or more agents that reduce the rate by which the compound of the present invention as an active ingredient will decompose.
- agents which are referred to herein as “stabilizers,” include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers, etc.
- the invention thus provides
- the invention relates in a sixth aspect to combinations comprising a compound of formula (I) and one or more additional active ingredients.
- the invention thus provides
- Pain treatment as defined herein may be applied as a sole therapy or may involve, in addition to a compound according to the invention, administration of other analgesics or adjuvant therapy.
- Such therapy may for example include in combination with a compound of the present invention, one or more of the following categories of pain-relieving ingredients:
- the additional active ingredient is a hormonal medicine.
- NMR Spectroscopy was determined using either a Bruker DPX 250 MHz NMR or a Bruker DRX 500 MHz NMR. Values are reported as shifts (in ppm), with zero corresponding to tetramethylsilane as an internal standard. Chemical shifts are reported in ppm ([delta]). Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. The NMR spectra were recorded at a temperature ranging from 25 to 90 C. When more than one conformer was detected the chemical shifts for the most abundant one are reported.
- Flash silica gel chromatography was carried out on silica gel 230-400 mesh or on prepacked silica cartridges.
- 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine is commercially available ((CAS-899373-19-4) e.g. Ambinter, Aurora Fine Chemicals, Tim Tec Inc., Interchim). Alternatively it may be prepared according to the following procedure ( Method 1 ):
- Example 1 (62 mg, 50%).
- Example 42 N-Methyl-N-phenyl-6- ⁇ 5-[3-(2,2,2-trifluoro-ethoxy)-pyridin-2-yl]-[1,2,4]oxadiazol-3-yl ⁇ -[1,3,5]triazine-2,4-diamine ( Method 3 )
- Example 46 N-Methyl-N-phenyl-6-(5-piperidin-1-yl-[1,2,4]oxadiazol-3-yl)-[1,3,5]triazine-2,4-diamine ( Method 4 )
- Example 70 2- ⁇ [4-Amino-6-(5-furan-2-yl-[1,2,4]oxadiazol-3-yl)-[1,3,5]triazin-2-yl]-phenyl-amino ⁇ -ethanol ( Method 5 )
- 6-(5-Furan-2-yl-[1,2,4]oxadiazol-3-yl)-N-phenyl-[1,3,5]triazine-2,4-diamine (prepared in an analogous manner to Example 2, 100 mg, 310 ⁇ mol) and potassium carbonate (86 mg, 620 ⁇ mol) were stirred together in a sealed tube in DMF (5 ml). 2-Bromoethanol (26 ⁇ L, 310 ⁇ mol) was added and the reaction was heated at 110 C for 18 h. Potassium carbonate (43 mg, 310 ⁇ mol) and 2-bromoethanol (26 ⁇ L, 310 ⁇ mol) were added and the reaction stirred at 110 C for a further 18 h.
- Example 75 5- ⁇ 3-[4-Amino-6-(methyl-phenyl-amino)-[1,3,5]triazin-2-yl]-[1,2,4]oxadiazol-5-yl ⁇ -thiophene-2-carboxylic acid methylamide
- Example 137 ⁇ 3-[4-Amino-6-(methyl-phenyl-amino)-[1,3,5]triazin-2-yl]-[1,2,4]oxadiazol-5-yl ⁇ -[3-(4-fluoro-phenyl)-azetidin-1-yl]-methanone
- Example 143 6- ⁇ 5-[6-(2-Methoxy-ethoxymethyl)-pyridin-3-yl]-[1,2,4]oxadiazol-3-yl ⁇ -N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine ( Method 7 )
- Example 144 N-Methyl-N-phenyl-6- ⁇ 5-[6-(tetrahydro-furan-3-yloxymethyl)-pyridin-3-yl]-[1,2,4]oxadiazol-3-yl ⁇ -[1,3,5]triazine-2,4-diamine
- Example 145 Racemic N-Methyl-N-phenyl-6- ⁇ 5-[(1R,3R,5S)-8-(3,3,3-trifluoro-propyl)-8-aza-bicyclo[3.2.1]oct-3-yl]-[1,2,4]oxadiazol-3-yl ⁇ -[1,3,5]triazine-2,4-diamine (Method 8)
- Example 156 2-N-(2-Methoxyethyl)-2-N-phenyl-6- ⁇ 5-[6-(2,2,2-trifluoroethoxy)pyridin-3-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- Example 159 1-[4-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)piperidin-1-yl]-2,2,2-trifluoroethan-1-one ( Method 9 )
- Example 161 1-[4-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)piperidin-1-yl]-2-methylpropan-1-one
- Example 173 6-[5-(1-Benzylpiperidin-4-yl)-1,2,4-oxadiazol-3-yl]-2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine
- Example 178 2-N-Methyl-2-N-phenyl-6-[5-(4- ⁇ [(2,2,2-trifluoroethane)sulfonyl]methyl ⁇ piperidin-1-yl)-1,2,4-oxadiazol-3-yl]-1,3,5-triazine-2,4-diamine ( Method 10 )
- the reaction mixture was re-treated with 3-chloroperoxybenzoic acid (13.22 mg, 0.077 mmol) and stirred at room temperature for 1 h.
- the reaction mixture was washed with a 5% aqueous solution of sodium thiosulfate and saturated aqueous solution of sodium bicarbonate.
- the organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure.
- the crude compound was purified by preparative HPLC, Method A, to afford the title compound (6.2 mg, 16%).
- Example 216 6-(5- ⁇ 4-[(Cyclopropylmethoxy)methyl]piperidin-1-yl ⁇ -1,2,4-oxadiazol-3-yl)-2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine
- Example 256 2-N-Methyl-2-N-phenyl-6- ⁇ 5-[1-(2,2,2-trifluoroethyl)-1H-pyrazol-3-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine ( Method 12 )
- Methyl 1-(2,2,2-trifluoroethyl)-1H-pyrazole-3-carboxylate (Intermediate 204, 0.312 g, 1.5 mmol) was dissolved in a mixture of THF (2 mL), MeOH (2 mL) and water (1 mL); lithium hydroxide (0.188 g, 4.5 mmol) was added and the mixture was stirred at room temperature for 16 h. The mixture was concentrated, the residue dissolved in water (1 mL) and the mixture was neutralised. This was extracted with EtOAc (2 x 10 mL), the organic extracts were dried over sodium sulfate and concentrated under vacuum.
- Example 258 3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -N-(6-methoxypyridin-3-yl)-1,2,4-oxadiazole-5-carboxamide ( Method 13 )
- Triethylamine (2 mL, 14.04 mmol) and benzyl bromide (1.2 mL, 10.0 mmol) were added to a solution of 3-oxocyclobutane-1-carboxylic acid (1.0 g, 8.77 mmol) in THF (10 mL) and the mixture was stirred at room temperature for 2 h.
- EtOAc (10 mL) was added and the mixture was washed with water followed by 1 M hydrochloric acid and then brine. The organic layer was dried over sodium sulfate and concentrated under vacuum.
- benzyl 3-oxocyclobutane-1-carboxylate 0.800 g, 3.92 mmol
- a portion of benzyl 3-oxocyclobutane-1-carboxylate (0.800 g, 3.92 mmol) was dissolved in a mixture of THF (2.5 mL) and water (2.5 mL) and cooled to 0 C.
- Sodium borohydride (0.051 g, 1.96 mmol) was added and the mixture was stirred at room temperature for 1 h. The mixture was concentrated under vacuum and EtOAc (10 mL) was added.
- Triethylamine (1.10 mL, 7.58 mmol) and 1H-benzotriazol-1-ol (0.77 g, 5.05 mmol) were added to a solution of N-[3-(dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride (0.97 g, 5.05 mmol) in DMF (10 mL) and the mixture was stirred for 20 min at room temperature.
- Picolinic acid (0.62 g, 5.05 mmol) was added and the mixture was stirred for 30 min at room temperature.
- Example 279 (Methyl 5-(3- ⁇ 4-amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)pyridine-2-carboxylate)
- Oxalyl chloride (91 ⁇ L, 1078 ⁇ mol) was added to a solution of pyridine-2,5-dicarboxylic acid 2-methyl ester (91 mg, 501 ⁇ mol) under an inert atmosphere in DCM (3 mL) and DMF (0.3 mL). The solution was stirred at room temperature for 1 h and then evaporated to dryness. The resulting solid was suspended in anhydrous pyridine (10 mL) and 4-amino-N-hydroxy-6-(methyl-phenyl-amino)-[1,3,5]triazine-2-carboxamidine (prepared in an analogous manner to Intermediate 1, 100 mg, 385 ⁇ mol) was added. The resulting solution was stirred at room temperature for 16 h under nitrogen.
- Example 280 (5-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)-N-(2,2,2-trifluoroethyl)pyridine-2-carboxamide)
- Example 282 6-[5-(3,5-Dimethoxypyridin-2-yl)-1,2,4-oxadiazol-3-yl]-2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine
- 6-[5-(3,5-Difluoropyridin-2-yl)-1,2,4-oxadiazol-3-yl]-2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine was prepared by dissolving 2-N-methyl-2-N-phenyl-6-[5-(pyrimidin-2-yl)-1,2,4-oxadiazol-3-yl]-1,3,5-triazine-2,4-diamine (Example 281, 20 mg, 52.3 ⁇ mol) and cesium carbonate (20 mg, 61.3 ⁇ mol) in MeOH (1 mL). The resulting mixture was evaporated to dryness, suspended in hot water and filtered. The white solid collected was washed further with distilled water and dried under vacuum to afford the title compound as a white solid (13 mg, 62%).
- Example 284 5-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)pyridine-2-carboxamide ( Method 15 )
- reaction mixture was then re-treated with trifluoromethanesulfonic acid 2,2,2-trifluoro-ethyl ester (56 mg, 239 ⁇ mol) and sodium hydride (60% dispersion in mineral oil, 7 mg, 159 ⁇ mol) and stirred for an additional 48 h at room temperature.
- the reaction mixture was quenched with water (20 mL) and extracted into EtOAc (3 x 25 mL). Combined organics were dried over magnesium sulfate and evaporated to dryness.
- the crude product was purified by silica chromatography (3% MeOH in DCM) and then re-purified by preparative HPLC-MS (Method B) to afford the title compound as an off white solid (5.6 mg, 15%).
- the resulting solid was dissolved in DCM (10 mL), pyrrolidine (24 ⁇ L, 333 ⁇ mol) and triethylamine (46 ⁇ L, 333 ⁇ mol) and stirred for a further 3 h at room temperature. Incomplete reaction was observed so the reaction was heated at 35 C for a further 16 h. The reaction mixture was then washed with water (3 x 25 mL) and combined organics were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by silica chromatography (5% MeOH in DCM) to afford the title compound as a beige solid (25 mg, 45%).
- reaction mixture was then cooled to 0 C and sodium borohydride (29 mg, 771 ⁇ mmol) was added.
- the reaction mixture was stirred at room temperature for 3 h and then was quenched with water (10 mL) and extracted into EtOAc (3 x 25 mL). Combined organics were dried over magnesium sulfate, evaporated and the crude product was purified by silica chromatography (2% MeOH in DCM) to afford the title compound as a beige solid (13 mg, 12%).
- Example 304 1-[5-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)pyridin-2-yl]ethan-1-one
- reaction mixture was cooled again and quenched with acetone (1 mL) and a 10% citric acid aq. solution (5 mL).
- the reaction mixture was extracted with EtOAc (3 x 50 mL) and the resultant organics washed with brine (25 mL), dried over magnesium sulfate and evaporated to dryness. 50 milligrams of the resulting crude material was purified by preparative HPLC-MS (Method C) to afford the title compound as an off white solid (21 mg).
- Example 312 2-N-(2-Methoxyethyl)-2-N-phenyl-6-(5- ⁇ 6-[(2,2,2-trifluoroethoxy)methyl]pyridin-3-yl ⁇ -1,2,4-oxadiazol-3-yl)-1,3,5-triazine-2,4-diamine ( Method 17 )
- Example 314 2-N-Methyl-2-N-phenyl-6- ⁇ 5-[1-(2,2,2-trifluoroethyl)azetidin-3-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- Example 317 2-N-Methyl-2-N-phenyl-6-[5-(6- ⁇ [(2,2,2-trifluoroethane)sulfinyl]methyl ⁇ pyridin-3-yl)-1,2,4-oxadiazol-3-yl]-1,3,5-triazine-2,4-diamine ( Method 18 )
- Example 324 2-N-Methyl-2-N-phenyl-6- ⁇ 5-[4-(3,3,3-trifluoropropylidene)piperidin-1-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- the reaction mixture was degassed by nitrogen bubbling and a 1M solution of LiHMDS in THF (2.10 mL, 2.10 mmol) was added drop wise at room temperature.
- the reaction mixture was diluted with EtOAc and filtered.
- the collected solid was taken in MeOH, filtered and wash with a large amount of MeOH and further purified by preparative HPLC, to afford the title compound (7 mg, 6%).
- Example 332 2-N-Methyl-2-N-phenyl-6-(5- ⁇ 6-[(propan-2-yl)amino]pyridin-3-yl ⁇ -1,2,4-oxadiazol-3-yl)-1,3,5-triazine-2,4-diamine ( Method 20 )
- Example 346 6-[5-(3-Fluoropyridin-2-yl)-1,2,4-oxadiazol-3-yl]-2-N-phenyl-1,3,5-triazine-2,4-diamine
- Example 358 2-N-Methyl-2-N-phenyl-6-(5- ⁇ 5-[1-(2,2,2-trifluoroethoxy)ethyl]pyridin-2-yl ⁇ -1,2,4-oxadiazol-3-yl)-1,3,5-triazine-2,4-diamine
- Example 366 2-N-Methyl-2-N-phenyl-6-(5- ⁇ 6-[2-(2,2,2-trifluoroethoxy)ethoxy]pyridin-3-yl ⁇ -1,2,4-oxadiazol-3-yl)-1,3,5-triazine-2,4-diamine
- 6-[2-(2,2,2-Trifluoroethoxy)ethoxy]pyridine-3-carboxylic acid (Intermediate 242, 0.133 g, 0.50 mmol) and 1,1'-carbonyldiimidazole (0.081 g, 0.50 mmol) were combined in pyridine (3 mL) and stirred at room temperature for 3 h.
- 4-Amino-N-hydroxy-6-[methyl(phenyl)amino]-1,3,5-triazine-2-carboximidamide was added and the mixture stirred at room temperature for 4 h before being heated to 80 C for 16 h.
- Example 384 1-[3-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)azetidin-1-yl]ethan-1-one
- Example 386 (1R,5S,6S)-6-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)-3-benzyl-3-azabicyclo[3.1.0]hexane-2,4-dione)
- Example 401 2-N-Methyl-2-N-phenyl-6- ⁇ 5-[(4- ⁇ [(2,2,2-trifluoroethyl)sulfanyl]methyl ⁇ piperidin-1-yl)carbonyl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine ( Method 21 )
- Example 414 2-N-(3-Chloro-4-fluorophenyl)-2-N-methyl-6- ⁇ 5-[6-(2,2,2-trifluoroethoxy)pyridin-3-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- the reaction mixture was stirred at room temperature for 14 h after which time further iodomethane (0.042 g, 0.304 mmol) was added and the mixture was stirred at room temperature for 56 h.
- LCMS indicated incomplete conversion so further iodomethane (0.014 g, 0.105 mmol) was added and the mixture was heated to 50 C for 12 h.
- cesium carbonate 0.032 g, 0.105 mmol
- Example 422 6-[5-(4-Aminocyclohexyl)-1,2,4-oxadiazol-3-yl]-2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine ( Method 23 )
- Example 425 N-[4-(3- ⁇ 4-Amino-6-[methyl(phenyl)amino]-1,3,5-triazin-2-yl ⁇ -1,2,4-oxadiazol-5-yl)cyclohexyl]-2-methylpropanamide ( Method 22 )
- Example 432 2-N-(6-Fluoropyridin-3-yl)-2-N-methyl-6- ⁇ 5-[6-(2,2,2-trifluoroethoxy)pyridin-3-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- 6-(2,2,2-Trifluoroethoxy)pyridine-3-carboxylic acid (prepared in an analogous manner to Intermediate 206, 0.216 g, 0.051 mmol) was added to a solution of N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (0.049 g, 0.252 mmol) in pyridine (1 mL) and the mixture was stirred at room temperature for 16 h. Further N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (0.0255 g, 0.1 mmol) was added and the mixture was stirred at 50 C for 4 h.
- Example 436 2-N-(2-Methoxyethyl)-6-[5-(3-phenoxyazetidin-1-yl)-1,2,4-oxadiazol-3-yl]-2-N-phenyl-1,3,5-triazine-2,4-diamine
- Trichloroacetic anhydride (0.181 mL, 0.989 mmol) was added to 4-amino-N'-hydroxy-6-[(2 methoxyethyl)(phenyl)amino]-1,3,5-triazine-2-carboximidamide (Intermediate 160, 0.250 g, 0.824 mmol) in toluene (10 mL) and stirred at room temperature for 5 min. Pyridine (3 mL) was added and the mixture was heated to 85 C for 1 h. The mixture was concentrated under vacuum and the residue dissolved in EtOAc. This was washed with water, saturated aq. sodium bicarbonate and then brine. The organic layer was dried over sodium sulfate and concentrated.
- Example 445 2-N-methyl-2-N-phenyl-6- ⁇ 5-[(3S)-3-(2,2,2-trifluoroethoxy)pyrrolidin-1-yl]-1,2,4-oxadiazol-3-yl ⁇ -1,3,5-triazine-2,4-diamine
- Di-tert-butyl azodicarboxylate (2.96 g, 12.83 mmol) was added to an ice cooled mixture of tert-butyl (3R)-3-hydroxypyrrolidine-1-carboxylate (2.00 g, 10.70 mmol), 2,2,2-trifluroethanol (10.70 g, 106.95 mmol) and triphenylphosphine (3.37 g, 12.83 mmol) in THF (24 mL). The mixture was stirred at 70 C for 18 h.
- Example 478 5- ⁇ 3-[4-Amino-6-(methyl-phenyl-amino)-[1,3,5]triazin-2-yl]-[1,2,4]oxadiazol-5-yl ⁇ -pyridine-2-carboxylic acid dimethylamide
- Example 481 6- ⁇ 5-[3-(Cyclopropylamino)pyridin-2-yl]-1,2,4-oxadiazol-3-yl ⁇ -2-N-methyl-2-N-phenyl-1,3,5-triazine-2,4-diamine
- 1,1'-Carbonyldiimidazole (62 mg, 0.380 mmol) was added to a solution of 3-fluoropyridine-2-carboxylic acid (50 mg, 0.354 mmol) in anhydrous pyridine (1 mL) in a sealed tube at room temperature and the mixture was stirred for 1 h.
- 4-Amino-N-hydroxy-6-(methyl-3-methylphenyl-amino)-[1,3,5]triazine-2-carboxamidine (prepared in an analogous manner to Intermediate 1, 66 mg, 0.253 mmol) was added and the mixture was stirred at room temperature for 60 min and then at 90 C for approximately 18 h.
- Example 482 6-[5-(3-Fluoropyridin-2-yl)-[1,2,4]oxadiazol-3-yl]-N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine ( Method 26 )
- N-[3-(Dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride (633 mg, 3.3 mmol) was added to a solution of 3-fluoropyridine-2-carboxylic acid (400 mg, 2.83 mmol) in anhydrous pyridine (6 mL) in a sealed tube at room temperature under nitrogen. The reaction mixture was stirred at room temperature for 1 h. 4-Amino-N-hydroxy-6-(methyl-3-methylphenyl-amino)-[1,3,5]triazine-2-carboxamidine (prepared in an analogous manner to Intermediate 1, 611 mg, 2.36 mmol) was added and the mixture was stirred at room temperature for approximately 18 h.
- N-[3-(dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride 315 mg, 1.15 mmol
- anhydrous pyridine 1 mL
- N-[3-(dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride 100 mg, 0.52 mmol
- 3-fluoropyridine-2-carboxylic acid 70 mg, 0.5 mmol
- Example 487 6- ⁇ 5-[3-(Cyclopropylmethylamino)-pyridin-2-yl]-[1,2,4]oxadiazol-3-yl ⁇ -N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine ( Method 25 )
- Example 492 6-[5-(3-Isopropoxyazetidin-1-yl)-[1,2,4]oxadiazol-3-yl]-N-methyl-N-phenyl-[1,3,5]triazine-2,4-diamine ( Method 27 )
- a base for example diisopropylethylamine or potassium carbonate
- Example 269, Example 298, Example 435, Example 485 and Example 511 do not exist.
- the reaction was treated with methanol (6 ⁇ L, 0.148 mmol), stirred for 2 h, then treated with methanol (20 ⁇ L, 0.494 mmol), stirred for 2 h, then treated with potassium tert-butoxide (50 mg, 0.446 mmol) and stirred for 18 h.
- the mixture was then concentrated in vacuo, suspended in sat. aq. sodium hydrogencarbonate solution and extracted with EtOAc (5 x 10 mL). The combined extracts were washed with brine (10 mL) and concentrated in vacuo to give the title compound was obtained (80 mg, 36%).
- 6-Chloropyridine-3-carboxylic acid (1g, 6.35 mmol) and thionyl chloride (10 mL, 138 mmol) were heated to reflux for 2 hours. The mixture was concentrated under vacuum and the residue was azeotroped with diethyl ether and evaporated. A portion of this acid chloride (0.75 g, 4.26 mmol) was then added to a solution of 4-amino-N-hydroxy-6-[methyl(phenyl)amino]-1,3,5-triazine-2-carboximidamide (prepared in an analogous manner to Intermediate 1, 0.920 g, 3.55 mmol) in pyridine (12 mL) and the mixture was stirred at room temperature overnight before being heated to 70 C for 3 h.
- 6-Chloropyridine-2-carboxylic acid (1.00 g, 6.347 mmol), 2,2,2-trifluoroethan-1-ol (0.683 mL, 9.520 mmol) and potassium hydroxide (1.424 g, 25.387 mmol) were dissolved in DMSO (25 mL) and heated at 100 C for 18 h.
- the reaction mixture was re-treated with potassium hydroxide (0.356 g, 6.347 mmol) and heated at 100 C for 18 h.
- reaction mixture was then re-treated with 2,2,2-trifluoroethan-1-ol (1.367 mL, 19.040 mmol) and potassium hydroxide (0.356 g, 6.347 mmol) and heated at 110 C for 18 h.
- the reaction mixture was then further re-treated with potassium hydroxide (0.356 g, 6.347 mmol) and 2,2,2-trifluoroethan-1-ol (0.683 mL, 9.520 mmol) and the mixture was stirred at room temperature for 72 h.
- Water (10 mL) was added and acidified to pH 1 with 1 M hydrochloric acid.
- the aqueous was extracted with EtOAc (3 x 15 mL) and the organic layer was washed with brine (15 mL).
- tert-Butyl 4-(hydroxymethyl)piperidine-1-carboxylate (0.300 g, 1.393 mmol), phenol (0.122 mL, 1.393 mmol), triphenylphosphine (0.440 g, 1.672 mmol) and N- ⁇ [(tert-butoxy)carbonyl]imino ⁇ (tert-butoxy)formamide (0.390 g, 1.672 mmol) were combined in THF (4 mL) and stirred at room temperature for 18 h. The reaction mixture was concentrated under reduced pressure and purified by FCC (EtOAc: heptane, 1:1) to afford a white solid.
- tert-Butyl 4-(hydroxymethyl)piperidine-1-carboxylate (0.300 g, 1.393 mmol), 2,2,2-trifluoroethan-1-ol (1.00 mL, 13.935 mmol), triphenylphosphine (0.440 g, 1.672 mmol) and N- ⁇ [(tert-butoxy)carbonyl]imino ⁇ (tert-butoxy)formamide (0.390 g, 1.672 mmol) were combined in THF (10 mL) and stirred at room temperature for 36 h before being heated to 75 C for 18 h.
- 2,2,2-Trifluoroethane-1-thiol (0.151 mL, 1.636 mmol) was added dropwise to a suspension of sodium hydride (60% dispersion in mineral oil, 0.065 g, 1.636 mmol) in anhydrous DMF (3 mL) at 0 C.
- a solution of tert-butyl 4-[(methanesulfonyloxy)methyl]piperidine-1-carboxylate (Intermediate 181, 0.320 g, 1.091 mmol) in DMF (4 mL) was then added dropwise and the reaction mixture was stirred at room temperature for 4 h.
- tert-Butyl 4-[(phenylsulfanyl)methyl]piperidine-1-carboxylate was prepared from tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (Intermediate 181, 0.320 g, 1.091 mmol) and thiophenol (0.117 mL, 1.145 mmol) according to the method described for Intermediate 183 to afford the title compound (0.483 g).
- tert-Butyl 4-[(propan-2-ylsulfanyl)methyl]piperidine-1-carboxylate was prepared from tert-butyl 4-(hydroxymethyl)piperidine-1-carboxylate (Intermediate 181, 0.320 g, 1.091 mmol) and sodium propanethiolate (0.161 g, 1.636 mmol) according to the method described for Intermediate 183 to afford the title compound (0.180 g, 60%).
- 1,1,1-Trifluoropropan-2-ol (0.406 mL, 4.483 mmol) was added drop wise to a suspension of sodium hydride (60% in mineral oil) (0.239 g, 5.977 mmol) in dry THF (20 mL) at 0 C. The mixture was stirred at 0 C for 15 minutes. A solution of methyl 6-(methanesulfonyloxy)methyl]pyridine-3-carboxylate (Intermediate 189, 0.733 g, 2.989 mmol) in THF (10 mL) was added drop wise. The reaction mixture was stirred at 0 C for 30 minutes and room temperature for 3 days.
- the reaction mixture was quenched with water (10 mL) and diluted with EtOAc (30 mL). The phases were separated, and the organic phase was extracted with a saturated aqueous solution of sodium bicarbonate (3 x 15 mL). The aqueous extracts were combined, washed with EtOAc (1 x 10 mL). The organic phase was discarded and the aqueous phase was acidified to pH 4 using 2N HCl and extracted with EtOAc (3 x 30 mL). The organic extracts were combined, dried over sodium sulfate, filtered and evaporated to provide a brown crude residue.
- Trifluoroacetic acid (1.15 mL) was added dropwise to a solution of tert-butyl 4-(2,2,2-trifluoroethoxy)piperidine-1-carboxylate (prepared in an analogous manner to Intermediate 203, 1.14 g, 4.0 mmol) in DCM (10 mL) at room temperature.
- the reaction mixture was stirred at room temperature for 16 h.
- the mixture was then concentrated under vacuum, the crude residue was dissolved in water and product was extracted with diethylether (2 times).
- the aqueous phase was basicified to pH 10 using solid potassium carbonate and extracted with DCM (4 times).
- the organic extracts were combined, dried over sodium sulfate and concentrated under vacuum to afford the title compound (610 mg, 83%).
- Methyl 1 H-pyrazole-3-carboxylate (1.0 g, 7.94 mmol) was dissolved in DMF (25 mL) and cesium carbonate (12.9 g, 39.7 mmol) was added. The mixture was cooled to 0 C and 2,2,2-trifluoroethyl methanesulfonate (2.4 mL, 19.8 mmol) was gradually added. The mixture was warmed to room temperature and stirred for 16 h. Water was added and the mixture was extracted with EtOAc (3 x 25 mL). The combined organic extracts were washed with brine (3 x 10 mL) and concentrated under vacuum.
- Methyl 5-hydroxypyridine-2-carboxylate (Intermediate 224, 0.610 g, 3.59 mmol) was dissolved in DMF (5 mL), cesium carbonate (1.40 g, 4.31 mmol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (1.0 g, 4.31 mmol) were added and the mixture stirred at room temperature for 18 h. The mixture was partitioned between EtOAc (50 mL) and water (15 mL) and the aqueous layer extracted with further EtOAc (2 x 20 mL). The combined organic layers were dried over sodium sulfate and concentrated under vacuum.
- 6-Hydroxypicolinic acid (0.500 g, 3.59 mmol) was dissolved in MeOH (20 mL) and cooled to 0 C under nitrogen.
- Thionyl chloride (0.651 mL, 8.98 mmol) was added dropwise, the mixture was warmed to room temperature and stirred for 2 h. TLC indicated no reaction, so further thionyl chloride (0.781 mL, 10.77 mmol) was added and the mixture heated to 75 C for 8 h. The mixture was concentrated under vacuum to afford the title compound as a white solid (0.685 g), which was used in the next step without further purification.
- NMR shows approximately 25% unreacted 5-hydroxypicolinic acid. 1H NMR (500MHZ, MeOH-d4) ⁇ ppm 8.44 (1 H, d), 8.39 (1 H, d), 8.02 (1 H, dd) and 4.09 (3H, s).
- 6-Chloronicotinic acid (0.500 g, 3.17 mmol), powdered potassium hydroxide (0.712 g, 12.69 mmol) and cyclohexanol (0.636 g, 6.35 mmol) were combined in DMSO (12 mL) and heated in a sealed tube to 100 C for 18 h and then to 120 C for 21 h.
- the mixture was acidified to pH 1 by adding 2M hydrochloric acid.
- the mixture was then left to stand at room temperature overnight and the resultant precipitate filtered off, washed with water and dried under vacuum to afford the title compound as a cream solid (0.389 g, 55%).
- 6-Chloronicotinic acid (0.500 g, 3.17 mmol), powdered potassium hydroxide (0.712 g, 12.69 mmol) and 1,1,1-trifluoropropan-2-ol (0.724 g, 6.35 mmol) were combined in DMSO (12 mL) and heated in a sealed tube to 100 C for 18 h. The mixture was acidified to pH 1 by adding 2M hydrochloric acid. The mixture was then left to stand at room temperature overnight and the resultant precipitate filtered off, washed with water and dried under vacuum to afford the title compound as a cream solid (0.527 g, 71%).
- 1 H NMR 500MHZ, MeOH-d4) ⁇ ppm 8.82 (1 H, d), 8.29 (1 H, dd), 6.94 (1 H, d), 5.96 (1 H, m) and 1.52 (3H, d).
- 3,3,3-Trifluoropropanol (0.338 g, 2.96 mmol) was dissolved in THF (5 mL) and cooled to 0 C under nitrogen. Potassium tert-butoxide (0.332 g, 2.96 mmol) was added and the mixture stirred for 5 min. Ethyl 6-chloronicotinate (0.500 g, 2.69 mmol) was added, the mixture was warmed to room temperature and stirred for 3 h. Brine (10 mL) was added and the mixture extracted with EtOAc (3 x 10 mL). The combined organic layers were dried over sodium sulfate and concentrated to yield a yellow oil.
- Oxan-4-ol (0.302 g, 2.96 mmol) was dissolved in THF (5 mL) and cooled to 0 C under nitrogen. Potassium tert-butoxide (0.332 g, 2.96 mmol) was added and the mixture stirred for 5 min. Ethyl 6-chloronicotinate (0.500 g, 2.69 mmol) was added, the mixture was warmed to room temperature and stirred for 3 h. Water (5 mL) was added and the mixture extracted with EtOAc (2 x 10 mL). The organic layers were dried over sodium sulfate and concentrated under vacuum.
- 2,2,2-Trifluoroethanol (0.133 mL, 1.49 mmol) was dissolved in DMF and cooled to 0 C under nitrogen.
- Sodium hydride (60% dispersion in mineral oil, 0.060 g, 1.49 mmol) was added and the mixture stirred for 10 min.
- tert-Butyl 4-(methanesulfonyloxy)piperidine-1-carboxylate (Intermediate 239, 0.320 g, 1.15 mmol) was added and the mixture was stirred at room temperature for 18 h. The mixture was then diluted with EtOAc (30 mL) and washed with water (10 mL), followed by saturated aq. sodium bicarbonate (10 mL) and brine (10 mL).
- Ethyl 6-[(1-methylpyrrolidin-3-yl)oxy]pyridine-3-carboxylate was prepared from 1-methylpyrrolidin-3-ol (0.161 g, 1.59 mmol) and ethyl 6-chloronicotinate (0.269 g, 1.45 mmol) according to the method described for Intermediate 236.
- the crude material was purified by FCC, eluting with a gradient of 0-20% MeOH in EtOAc to afford the title compound as a colourless oil (0.089 g, 25%).
- Methyl 6-[2-(2,2,2-trifluoroethoxy)ethoxy]pyridine-3-carboxylate (Intermediate 243, 0.453 g, 1.62 mmol) was dissolved in MeOH (5 mL), 2M aq. sodium hydroxide (1.62 mL, 3.24 mmol) was added and the mixture was stirred at room temperature for 1 h. The mixture was then concentrated under vacuum and acidified with 2M hydrochloric acid. The resultant precipitate was filtered off and dried under vacuum to afford the title compound as a white solid (0.245 g, 57%).
- tert-Butyl 4-(propan-2-ylsulfanyl)piperidine-1-carboxylate (prepared in an analogous manner to Intermediate 245, 0.160 g, 0.62 mmol) was dissolved in DCM (3 mL) and cooled to 0 C.
- m-Chloroperbenzoic acid (0.138 g, 0.62 mmol) was added, the mixture was warmed to room temperature and stirred for 17 h. Further m-chloroperbenzoic acid (0.040 g, 0.18 mmol) was added and the mixture was stirred at room temperature for 3 h.
- the mixture was then diluted with DCM (10 mL) and washed with saturated aq. sodium thiosulfate (10 mL).
- Methyl 4-[(2,2,2-trifluoroethoxy)methyl]benzoate (Intermediate 249, 1.05g, 4.23 mmol) was dissolved in MeOH (10 mL), 2M aq. sodium hydroxide (6.4 mL, 12.69 mmol) was added and the mixture was stirred at room temperature for 3 h. THF (2 mL) was then added and the mixture stirred at room temperature over the weekend. The mixture was then concentrated under vacuum and the residue acidified by adding 2M hydrochloric acid. The mixture was diluted with a little water and extracted with DCM (3 x 20 mL).
- 2,2,2-Trifluoroethanol (0.35 mL, 4.80 mmol) was dissolved in THF (2 mL) and cooled to 0 C under nitrogen.
- Sodium hydride (60% dispersion in mineral oil, 0.192 g, 4.80 mmol) was added portionwise and the mixture stirred for 10 min.
- a solution of methyl 4-(bromomethyl)benzoate in THF (3 mL) was added, the mixture was warmed to room temperature and then heated to 50 C for 3 h. The mixture was then diluted with water (15 mL) and EtOAc (20 mL). The organic layer was separated and the aqueous neutralised with 1 M hydrochloric acid. The aqueous layer was further extracted with EtOAc (2 x 20 mL).
- Powdered potassium hydroxide (712 mg, 12.7 mmol) was added to a mixture of 6-chloronicotinic acid (500 mg, 3.17 mmol) and 2,2-difluoroethanol (520 mg, 6.35 mmol) in DMSO (15 mL) and the mixture was stirred at 120 C for 14 h. An additional two equivalents of 2,2-difluoroethanol and potassium hydroxide were added and the mixture was heated for 24 h. The mixture was cooled to room temperature, diluted with water (5 mL), acidified with 1 N aq. HCl (to pH 1) and extracted with EtOAc (3 x 50 mL).
- Methanesulfonyl chloride (320 ⁇ L, 4.13 mmol) was added to a solution of [(1R,5S,6S)-3-benzyl-3-azabicyclo[3.1.0]hexan-6-yl]methanol (Intermediate 253, 600 mg, 2.95 mmol) and triethylamine (620 ⁇ L, 4.43 mmol) in DCM (20 mL) at room temperature. The mixture was stirred for 3 h and then saturated aq. ammonium chloride (20 mL) was added. The mixture was extracted with DCM (3 x 20 mL).
- Ethyl 5-benzyl-4,6-dioxo-1H,3aH,4H,5H,6H,6aH-pyrrolo[3,4-c]pyrazole-3-carboxylate (Intermediate 255, 40 g, 132.9 mmol) was heated in 500 mL flask at 190 C. After evolution of nitrogen additional ethyl 5-benzyl-4,6-dioxo-1H,3aH,4H,5H,6H,6aH-pyrrolo[3,4-c]pyrazole-3-carboxylate (3 x10 g) was added portionwise and the mixture was heated for 30 min until gas evolution stopped.
- 2,2,2-Trifluoroethane-1-thiol (471 ⁇ L, 3.41 mmol) was added dropwise to a suspension of sodium hydride (60% dispersion in mineral oil, 0.065 g, 1.636 mmol) in dry DMF (10 mL) at 0 C and the mixture was stirred for 15 min.
- Diisopropyl azodicarboxylate (1.19 g, 5.87 mmol) was added to an ice cooled mixture of tert-butyl (3R)-3-hydroxypyrrolidine-1-carboxylate (1 g, 5.34 mmol), phenol (0.5 g, 5.34 mmol) and triphenylphosphine (1.5 g, 5.8 mmol) in THF (14 mL). The mixture was stirred at room temperature for 20 h. The mixture was then concentrated under vacuum and the residue was twice triturated with diethyl ether. The resultant solid was filtered off and discarded. The filtrate was washed with 1 M aq. sodium hydroxide, dried over sodium sulfate and concentrated.
- tert-Butyl (3R)-3-phenoxypyrrolidine-1-carboxylate was prepared from tert-butyl (3S)-3-hydroxypyrrolidine-1-carboxylate (0.43 g, 2.286 mmol) according to the method described for Intermediate 270 to afford the title compound (0.312 g, 51% yield).
- Methyl 5-aminopyridine-2-carboxylate (Intermediate 168, 200 mg, 1.31 mmol) and cesium carbonate (642 mg, 1.97 mmol) were dissolved in anhydrous DMF (2 mL) and stirred under nitrogen for 10 min.
- 2,2,2-Trifluoroethyl trifluoromethanesulfonate (246 ⁇ L, 1.70 mmol) was added and the mixture was stirred at room temperature for 18 h. Further cesium carbonate (642 mg, 1.97 mmol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (246 ⁇ L, 1.70 mmol) were added and the mixture was stirred at 85 C for 6 h.
- Patch-clamp electrophysiology is considered the gold standard technique for investigation of ion channel function and pharmacology. Using this method, sodium channel currents can be measured in cells, which stably (or transiently) express the sodium channel subtype of interest. Application of compounds during such experiments provides a functional measure of the activity and potency of compounds affecting the ion channel of interest.
- Electrophysiological studies can be performed using automated or manual patch-clamp techniques.
- Automated patch-clamp instruments suitable for this work include the Ion Works HT (MDS), Ion Works Quattro (MDS), PatchLiner (Nanion technologies), Port-A-Patch (Nanion technologies), QPatch (Sophion) or any other suitable platform.
- MDS Ion Works HT
- MDS Ion Works Quattro
- PatchLiner Non-A-Patch
- QPatch Sophion
- cells expressing the voltage-gated sodium channel subtype of interest are dispensed in appropriate chips (plates with more than one recording chambers each containing one or more apertures to generate whole-cell patches), as provided by the manufacturer.
- the whole-cell perforated patch configuration is used for probing the pharmacology of compounds in automated patch-clamp electrophysiology on sodium channels.
- Extracellular and intracellular buffers for such experiments are composed according to the literature, or according to the instructions provided by the manufacturer of the instruments.
- Test solutions which contain the compounds to be tested, are applied to the cells expressing the sodium channel of interest by a pipetting system, typically integrated into the robots. Electrophysiological studies can also be performed using the whole cell configuration of the standard manual patch clamp technique as described in the literature (e.g. Pflugers Arch., 1981, 391(2), 85-100 ).
- cells that express the voltage-gated sodium channel protein of interest are exposed to the drugs by conventional microperfusion systems, or by a home-built perfusion system.
- a suitable voltage stimulus protocol is used to activate the voltage-gated sodium channels.
- a suitable voltage stimulus protocol is used to activate voltage-gated sodium channel proteins of interest.
- a typical stimulus protocol may consist of sweeps of sequential voltage pulses starting from a holding potential (i.e. -65 mV), to a more positive and depolarising test pulse potential (i.e. -10 mV) and finally to a more negative and hyperpolarising potential (i.e. -100 mV).
- a holding potential i.e. -65 mV
- a more positive and depolarising test pulse potential i.e. -10 mV
- a more negative and hyperpolarising potential i.e. -100 mV
- a stimulus protocol was designed to assess compounds due to their potential to block NaV1.7 channels primarily in inactivated state. Selectivity over other voltage-gated sodium channel members was assessed by protocols that reflect the native function of the channels (i.e. heart rate or firing of neurons).
- Electrophysiological studies can also be performed using the whole cell configuration of the standard manual patch clamp technique as described in the literature (e.g. Pflugers Arch., 1981, 391(2), 85-100 ).
- cells that express the human voltage-gated sodium channel protein of interest are exposed to the drugs by conventional microperfusion systems, or by a home-built perfusion system.
- a suitable voltage stimulus protocol is used to activate the voltage-gated sodium channels.
- a suitable voltage stimulus protocol may consist of sequential voltage pulses from a holding potential, each first hyperpolarizing the cell, then depolarizing the channels for a brief period.
- a particularly suitable holding potential may be a voltage allowing a fraction of channels to remain in an inactivated state, like -60 mV.
- a suitable hyperpolarizing voltage and duration may be -120 mV for 100 ms, and depolarization voltage and duration may be-20 mV for 25 ms, and a suitable frequency of pulses may be 0.1 Hz, but may also have other parameters. Accordingly, a stimulus protocol was designed to assess compounds due to their potential to block NaV1.7 channels primarily in inactivated state. Selectivity over other voltage-gated sodium channel members was assessed by protocols that reflect the native function of the channels (i.e. heart rate or firing of neurons).
- Compounds of formula (I) may show analgesic activity in the FCA (Freund's Complete Adjuvant) test in the rat, a model of inflammatory pain which is induced by intraplantar injection of Complete Freund's Adjuvant ( Stein et al. Pharmac. Biochem. Behav., 1988, 31, 445-451 ).
- FCA Full's Complete Adjuvant
- the analgesic effects in the model may be obtained at doses that do not produce tissue concentrations leading to conduction block in nerve fibres. Thus, the local anesthetic effect may not mask the analgesic properties of the compounds ( Scott et al., British Journal of Anaesthesia, 1988, 61, 165-8 ).
- Examples 125 and 130 were shown to be efficacious at a dose of 1 mg/kg (p.o.) and Example 67 was shown to be efficacious at a dose of 3 mg/kg (p.o.). All compounds were tested 72 h post FCA injection.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Claims (15)
- Verbindung der Formel (I) oder ein pharmazeutisch verträgliches Salz davon
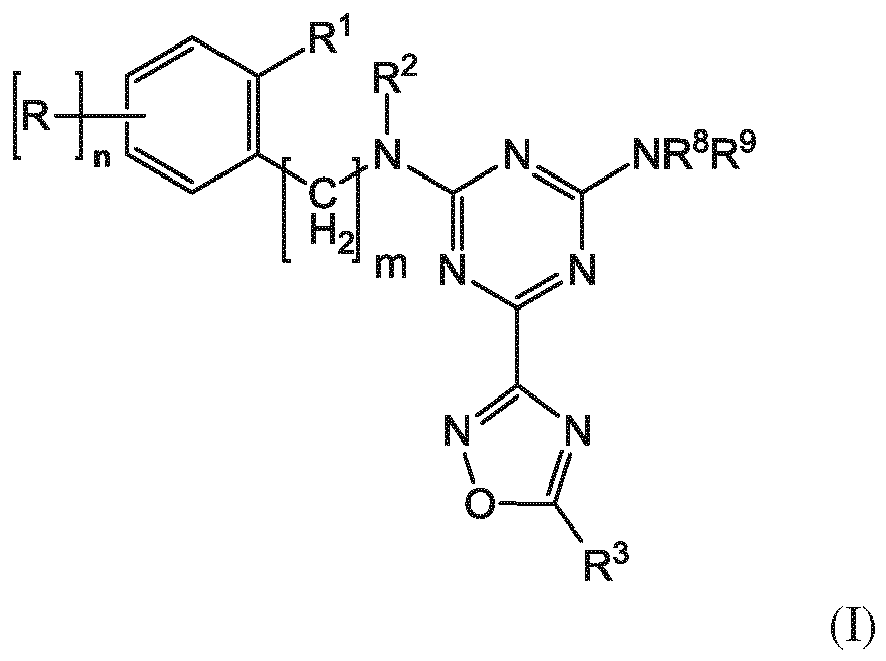
R1 ausgewählt ist aus:Wasserstoff-,Halogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-;R2 ausgewählt ist aus:Wasserstoff-,C1-C7-Alkyl-,Halogen-C2-C7-alkyl-,Amino-C2-C7-alkyl-,N-C1-C7-Alkylamino-C2-C7-alkyl-,N,N-di-C1-C7-alkylamino-C2-C7-alkyl-,Hydroxy-C2-C7-alkyl-,C1-C7-Alkoxy-C2-C7-alkyl -,C3-C10-Cycloalkyl-C1-C7-alkyl-,Cyano-C2-C7-alkyl-;oder
R1 und R2 zusammen mit den Atomen, an die sie gebunden sind, einen 4- bis 7-gliedrigen, gesättigten oder teilweise gesättigten heterocyclischen Ring bilden, der unsubstituiert ist oder mit 1 bis 3 Substituenten substituiert ist, die ausgewählt sind aus:C1-C7-Alkyl-Halogen-C1-C7-alkyl-Amino-C1-C7-alkyl-N-C1-C7-Alkylamino-C1-C7-alkyl-N,N-di-C1-C7-alkylamino-C1-C7-alkyl-Hydroxy-C1-C7-alkyl-C1-C7-Alkoxy-C1-C7-alkyl-C3-C10-Cycloalkyl-C1-C7-alkyl-Cyano-C1-C7-alkyl-;R ausgewählt ist aus:Halogen-C1-C7-Alkyl-Halogen-C1-C7-alkyl-C1-C7-Alkoxy-Cyano-Halogen-C1-C7-alkoxy-Nitro;-C(O)-O-R', wobei R' ausgewählt ist aus Wasserstoff, C1-C7-Alkyl; C3-C10-Cycloalkyl; C1-C7-Alkoxy; Halogen-C1-C7-alkylaryl; Aryl-C1-C7-alkyl-; Heteroaryl; Heteroaryl-C1-C7-alkyl-; Heterocyclyl;-S(=O)2-C1-C7-Alkyl; -S(=O)2-C3-C10-Cycloalkyl; -S(=O)2-C1-C2-Alkoxy;R3 ausgewählt ist aus:(a) -L-Y, wobei-L- ausgewählt ist aus einer direkten Bindung; -(CH2)p-, -C(O)-, -NR7-, -NR7-C(O)- oder -C(O)-NR7-,wobei p ausgewählt ist aus 1, 2 oder 3,R7 ausgewählt ist aus Wasserstoff und C1-C7-Alkyl,Y ist ausgewählt aus Cycloalkyl, Aryl, Heteroaryl, Heterocyclyl, Spirocyclyl,die unsubstituiert sind oder substituiert sind mit 1-3 Substituenten ausgewählt ausoderHalogen-;C1-C7-Alkyl-;Halogen-C1-C7-alkyl-;Halogen-C1-C7-alkyloxy-C1-C7-alkyl; Halogen-C1-C7-alkyloxy-C1-C7-alkyloxy;C1-C7-Alkoxy-; C1-C7-Alkoxy-C1-C7-alkoxy-; NC-C1-C7-Alkoxy-;C1-C7-Alkoxy- C1-C7-alkyl-;C3-C10-Cycloalkyloxy-C1-C7-alkyl-;C3-C10-Cycloalkyl-C1-C7-alkyloxy-;C3-C10-Cycloalkyloxy-;C3-C10-Cycloalkyl-NR7'- C1-C7-alkyl-, wobei R7' ausgewählt ist aus Wasserstoff und C1-C7-Alkyl ;C3-C10-Cycloalkyl- C1-C7-alkoxy- C1-C7-alkyl-;C2-C7-Alkenyl; Halogen-C2-C7-alkenyl;Hydroxy-;Hydroxy-C1-C7-alkyl-;Halogen-C1-C7-alkyloxy-;Amino-;N-C1-C7-Alkylamino-;N-Halogen-C1-C7-alkylamino-;N-Heterocyclylamino-, N-C3-C10-Cycloalkylamino-, wobei das Heterocyclyl und Cycloalkyl wahlweise substituiert sind mit Halogen-C1-C7-alkyloxy, C1-C7-Alkyl; C3-C10-Cycloalkyl und C1-C7-Alkoxy;N-C3-C10-Cycloalkyl-C1-C7-alkylamino-;N,N-di-C1-C7-alkylamino-; N,N-Dihalogen-C1-C7-alkylamino-;N,N-Diheterocyclylamino-, N,N-di-C3-C10-cycloalkylamino-, wobei das Heterocyclyl und Cycloalkyl wahlweise substituiert sind mit Halogen-C1-C7-alkyloxy, C1-C7-Alkyl; C3-C10-Cycloalkyl und C1-C7-Alkoxy;Cyano-; Oxo;C1-C7-Alkoxycarbonyl-;C1-C7-Alkoxy-C1-C7-alkoxy-C1-C7-alkyl-;Aryl; Aryl-C1-C7-alkyl-; Aryloxy;Heterocyclyl;Heterocyclyl-C1-C7-alkyl-; Heterocyclyloxy-;Heterocyclyloxy-C1-C7-alkyl-; Aryloxy-C1-C7-alkyl-; Heteroaryloxy-C1-C7-alkyl-;Hydroxycarbonyl-;-S-Halogen-C1-C7-alkyl; -S-C1-C7-Alkyl; -S-Aryl;Halogen-C1-C7-alkyl-S- C1-C7-alkyl; C1-C7-Alkyl-S-C1-C7-alkyl;-S(=O)2- C1-C7-alkyl; -S(=O)2-Halogen-C1-C7-alkyl; -S(=O)2-Aryl; -S(=O)2-Heteroaryl; -S(=O)2-NR4'R4; -S(=O)2-Heterocyclyl;Halogen-C1-C7-alkyl-S(=O)2-C1-C7-alkyl; C1-C7-Alkyl-S(=O)2-C1-C7-alkyl;-S(=O)-C1-C7-Alkyl; -S(=O)-Halogen-C1-C7-alkyl; -S(=O)-C1-C7-Alkoxy;-S(=O)-C3-C10-Cycloalkyl;-C(O)-C1-C7-Alkyl; -C(O)-Halogen-C1-C7-alkyl; -C(O)-C1-C7-Alkoxy; -C(O)-C3-C10-Cycloalkyl;-C(O)O-C1-C7-Alkyl; -C(O)O-C3-C10-Cycloalkyl; -C(O)O-Halogen-C1-C7-alkyl;-C(O)O-C1-C7-Alkoxy;-C(O)-NR4'R4 oder -NHC(O)-R4, wobeiR4 ausgewählt ist aus Wasserstoff, C1-C7-Alkyl, Halogen-C1-C7-alkyl, C3-C10-Cycloalkyl, C3-C10-Cycloalkyl-C1-C7-alkyl und C1-C7-Alkoxy;R4' ausgewählt ist aus Wasserstoff;oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen, gesättigten oder teilweise gesättigten monocyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält, und wobei der heterocyclische Ring wahlweise substituiert ist mit Aryl, Aryloxy-, C1-C7-Alkyl, Halogen-C1-C7-alkyl oder C1-C7-Alkoxy, und das Aryl wahlweise substituiert ist mit Halogen, C1-C7-Alkyl, Halogen-C1-C7-alkyl oder C1-C7-Alkoxy;(b) -C(O)-NR5'R5 oder -C(O)-O-R5, wobeim 0-1 ist;R5 und R5' wahlweise ausgewählt sind aus Wasserstoff, C1-C7-Alkyl; C3-C10-Cycloalkyl; C1-C7-Alkoxy; Halogen-C1-C7-alkylaryl; Aryl-C1-C7-alkyl-; Aryl; Heteroaryl; Heteroaryl-C1-C7-alkyl-; Heterocyclyl; Indan oder R5 und R5' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-9-gliedrigen, gesättigten oder teilweise gesättigten monocyclischen oder bicyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält;wobei das C3-C10-Cycloalkyl; Aryl, Heteroaryl, Heterocyclyl und Indan wahlweise substituiert sind mit 1 bis 3 Substituenten, die ausgewählt sind aus C1-C7-Alkyl, Halogen-C1-C7-alkyl, C1-C7-Alkoxy, Halogen-C1-C7-alkyloxy-, Halogen-C1-C7-alkyloxy- C1-C7-alkyl, C1-C7-Alkyloxy-C1-C7-alkyl und Hydroxy-C1-C7-alkyl;
n 0-2 ist;
R8 Wasserstoff ist und R9 ausgewählt ist aus Wasserstoff, C1-C7-Alkoxy-C1-C7-alkyl-, C1-C7-Alkyl, C1-C7-Alkoxy und Halogen-C1-C7-alkyl;
"Cycloalkyl" sich auf gesättigte oder teilweise ungesättigte monocyclische, bicyclische oder tricyclische Kohlenwasserstoffgruppen mit 3 bis 12 Kohlenstoffatomen bezieht;
"Aryl" sich auf 6-Kohlenstoff-monocyclische, 10-Kohlenstoff-bicyclische oder 14-Kohlenstoff-tricyclische aromatische Ringsysteme bezieht;
"Heteroaryl" sich auf einen 4-, 5-, 6- oder 7-gliedrigen monocyclischen 7-, 8-, 9-, 10-, 11- oder 12-gliedrigen bicyclischen oder 10-, 11-, 12-, 13-, 14- oder 15-gliedrigentricyclischen ungesättigten Ring oder entsprechendes Ringsystem bezieht, das die höchstmögliche Zahl von konjugierten Doppelbindungen in dem Ring/den Ringen trägt, der/die mindestens ein Heteroatom ausgewählt aus N, O und S enthält/enthalten, wobei das N und S auch wahlweise zu verschiedenen Oxidationsstufen oxidiert sein können; "Heterocyclyl" sich auf einen 4-, 5-, 6-, 7 oder 8-gliedrigen monocyclischen, 7-, 8-, 9-, 10-, 11-, oder 12-gliedrigen bicyclischen oder 10-, 11-, 12-, 13-, 14- oder 15-gliedrigen tricyclischen gesättigten oder teilweise ungesättigten Ring oder entsprechendes Ringsystem bezieht, die wahlweise mindestens ein Heteroatom ausgewählt aus N, O und S enthalten, wobei das N und S auch wahlweise zu verschiedenen Oxidationsstufen oxidiert sein können; und
"Spirocyclyl" sich auf ein "4 bis 7-gliedriges monocyclisches Heterocyclyl" wie vorstehend definiert bezieht, das über ein einzelnes Atom an einen zweiten 3- bis 6-gliedrigen gesättigten Ring kondensiert ist und wobei der zweite Ring Null, 1 oder 2 Heteroatome unabhängig ausgewählt aus Sauerstoff, Stickstoff und Schwefel enthält;
mit der Maßgabe, dass 6-[5-(2-Furanyl)-1,2,4-oxadiazol-3-yl]-N2-methyl-N2-phenyl-1,3,5-triazin-2,4-diamin und 6-[5-(2-Furanyl)-1,2,4-oxadiazol-3-yl]-N,N,N'-methyl-N'-phenyl-1,3,5-triazin-2,4-diamin ausgenommen sind. - Verbindung nach Anspruch 1 der Formel (I) oder ein pharmazeutisch verträgliches Salz davon:

R1 ausgewählt ist aus:Wasserstoff-,Halogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-;R2 ausgewählt ist aus:Wasserstoff-,C1-C7-Alkyl-,Halogen-C2-C7-alkyl-,Amino-C2-C7-alkyl-,N-C1-C7-Alkylamino-C2-C7-alkyl-,N,N-di-C1-C7-alkylamino-C2-C7-alkyl-,Hydroxy-C2-C7-alkyl-,C1-C7-Alkoxy-C2-C7-alkyl-,C3-C10-Cycloalkyl-C1-C7-alkyl-,Cyano-C2-C7-alkyl-;oder
R1 und R2 zusammen mit den Atomen, an die sie gebunden sind, einen 4- bis 7-gliedrigen, gesättigten oder teilweise gesättigten heterocyclischen Ring bilden, der unsubstituiert ist oder durch 1 bis 3 Substituenten substituiert ist, die ausgewählt sind aus:C1-C7-Alkyl-Halogen-C1-C7-alkyl-Amino-C1-C7-alkyl-N-C1-C7-Alkylamino-C1-C7-alkyl-N,N-di-C1-C7-alkylamino-C1-C7-alkyl-Hydroxy-C1-C7-alkyl-C1-C7-Alkoxy-C1-C7-alkyl-C3-C10-Cycloalkyl-C1-C7-alkyl-Cyano-C1-C7-alkyl-;R ausgewählt ist aus:Halogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-,C1-C7-Alkoxy-,Cyano-,Halogen-C1-C7-alkoxy-,Nitro;R3 ausgewählt ist aus:(a) -(CH2)p-Y, wobeip ausgewählt ist aus 0, 1, 2 oder 3, undY ist ausgewählt aus Aryl, Heteroaryl, Heterocyclyl, die unsubstituiert sind oder substituiert sind mit 1-3 Substituenten ausgewählt ausoderHalogen-;C1-C7-Alkyl-;Halogen-C1-C7-alkyl-;C1-C7-Alkoxy-;C3-C10-Cycloalkyloxy-;Hydroxy-;Halogen-C1-C7-alkyloxy-;Amino-;
N-C1-C7-Alkylamino-;
N,N-di-C1-C7-alkylamino-;
Cyano-;
C1-C7-Alkoxycarbonyl-;
Hydroxycarbonyl-;
-C(O)-NR4'R4, wobeiR4 ausgewählt ist aus Wasserstoff, C1-C7-Alkyl,R4' ausgewählt ist aus Wasserstoff;oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen, gesättigten oder teilweise gesättigten monocyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält;(b) -C(O)-NR5'R5 oder -C(O)-O-R5, wobeim 0-1 ist undR5 ausgewählt ist aus Wasserstoff, Benzyl, Indanyl, Tetrahydrofuranyl, Tetrahydropyranyl, Oxiranyl, C3-C10-Cycloallkyl, C1-C7-Alkyl,R5' ausgewählt ist aus Wasserstoff, C1-C7-Alkyl,oder R5 und R5' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen, gesättigten oder teilweise gesättigten monocyclischen oder bicyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält;
n 0-1 ist;
mit der Maßgabe, dass 6-[5-(2-Furanyl)-1,2,4-oxadiazol-3-yl]-N2-methyl-N2-phenyl-1,3,5-triazin-2,4-diamin ausgenommen ist. - Verbindung nach Anspruch 1 oder 2 oder ein pharmazeutisch verträgliches Salz davon, wobei
R1 ausgewählt ist ausWasserstoff-,Chlor-,Fluor-,Methyl-;R2 ausgewählt ist ausWasserstoff-,C1-C4-Alkyl-Halogen-C2-C4-alkyl-,N,N-di-C1-C2-alkylamino-C2-C4-alkyl-,Hydroxy-C2-C4-alkyl-,C1-C2-Alkoxy-C2-C4-alkyl-,C3-C6-Cycloalkyl-C1-C7-alkyl-. - Verbindung nach Anspruch 1 oder ein pharmazeutisch verträgliches Salz davon, wobei
R3 ausgewählt ist aus-(CH2)p-Y, wobeip ausgewählt ist aus 0, 1, 2 oder 3 undY ausgewählt ist aus Aryl, Heteroraryl, Heterocyclyl, die unsubstituiert sind oder substituiert sind mit 1-3 Substituenten ausgewählt aus:Halogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-,C1-C7-Alkoxy-,C3-C10-Cycloalkyloxy-,Hydroxy-,Halogen-C1-C7-alkyloxy-,Amino-,N-C1-C7-Alkylamino-,N,N-di-C1-C7-alkylamino-,-S(=O)2-C1-C7-Alkyl; -S(=O)2-Halogen-C1-C7-alkyl;-S(=O)-C1-C7-Alkyl; -S(=O)-Halogen-C1-C7-alkylC3-C10-Cycloalkyl- C1-C7-Alkoxy- C1-C7-Alkyl-;Cyano-C1-C7-Alkoxycarbonyl-Hydroxycarbonyl--C(O)-NR4'R4, wobeiR4 ausgewählt ist aus Wasserstoff, C1-C7-Alkyl;R4' ausgewählt ist aus Wasserstoff,oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen, gesättigten oder teilweise gesättigten, monocyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält. - Verbindung nach Anspruch 1 oder ein pharmazeutisch verträgliches Salz davon, wobei
R1 ausgewählt ist ausWasserstoff-,Fluor-;R2 ausgewählt ist ausWasserstoff-C1-C4-Alkyl-,Halogen-C2-C4-alkyl-,N,N-di-C1-C2-alkylamino-C2-C4-alkyl-,Hydroxy-C2-C4-alkyl-,C1-C2-Alkoxy-C2-C4-alkyl-,C3-C6-Cycloalkyl-Ci-C7-alkyl-;oderR1 und R2 zusammen ausgewählt sind aus-CH2-CH2- oder -CH2-CH2-CH2-;R ausgewählt ist ausHalogen-,C1-C4-Alkyl-,Halogen-C1-C4-alkyl-,C1-C4-Alkoxy-,Cyano-; undR3 ausgewählt ist aus- (CH2)p-Y, wobeip ausgewählt ist aus 0, 1, 2 oder 3, undY ausgewählt ist aus Aryl, Heteoraryl, Heterocyclyl, die unsubstituiert sind oder substituiert sind mit 1-3 Substituenten ausgewählt aus:m 0 ist undHalogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-,C1-C7-Alkoxy-,C3-C10-Cycloalkyloxy-,Hydroxy-,Halogen-C1-C7-alkyloxy-,Amino-,N-C1-C7-Alkylamino-,N,N-di-C1-C7-alkylamino-,Cyano-,C1-C7-Alkoxycarbonyl-,-C(O)-NR4'R4, wobeiR4 ausgewählt ist aus Wasserstoff, C1-C7-Alkyl;R4' ausgewählt ist aus Wasserstoff,oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen, gesättigten oder teilweise gesättigten, monocyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält,
n 0-1 ist. - Verbindung nach Anspruch 1 bis 4 oder ein pharmazeutisch verträgliches Salz davon, wobei
R3 ausgewählt ist aus:Phenyl, Furanyl, Thiophenyl, Pyridyl, Thiazolyl, Pyrazolyl, Isoxazolyl, Imidazolyl, Pyrrolyl, Benzofuranyl, Pyrimidinyl, Oxazolyl, Morpholinyl, Piperidinyl, Tetrahydrofuranyl, Tetrahydropyranyl, Pyrrolidinyl, Dihydrobenzofuranyl, die unsubstituiert sind oder durch 1-2 Substituenten substituiert sind ausgewählt ausChlor-,Brom-,Fluor-,Methyl-,Trifluormethyl-,2,2,2-Trifluorethyl-,2,2,2-Trifluorethyloxymethyl-,Cyclopropylmethoxymethyl--S(=O)2-2,2,2-Trifluorethyl-;-S(=O)2-Propyl;-S(=O)-3,3,3-Trifluorpropyl;3,3,3-Trifluorpropyloxymethyl-;Methoxy-,Cyclopentyloxy-,Trifluormethyloxy-,2,2,2-Trifluorethyloxy-,Amino-,Cyano-,Methoxycarbonyl-,-C(O)-NR4'R4, wobeiR4 ausgewählt ist aus Wasserstoff, Methyl;R4' ausgewählt ist aus Wasserstoff,oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, Morpholinyl, Piperidinyl, Pyrrolidinyl bilden. - Verbindung nach Anspruch 1, wobei die Verbindung ausgewählt ist aus
2-N-Methyl-2-N-phenyl-6-(5-{4-[(2,2,2-trifluorethoxy)methyl]piperidin-1-yl}-1,2,4-oxadiazol-3-yl)-1,3,5-triazin-2,4-diamin;
2-N-Methyl-2-N-phenyl-6-{5-[1-(propan-2-sulfonyl)piperidin-4-yl]-1,2,4-oxadiazol-3-yl}-1,3,5-triazin-2,4-diamin;
6-(5-{4-[(Cyclopropylmethoxy)methyl]piperidin-1-yl}-1,2,4-oxadiazol-3-yl)-2-N-methyl-2-N-phenyl-1,3,5-triazin-2,4-diamin;
2-N-(3-Fluorphenyl)-6-[5-(3-fluorpyridin-2-yl)-1,2,4-oxadiazol-3-yl]-1,3,5-triazin-2,4-diamin;
2-N-Methyl-2-N-phenyl-6-(5-{6-[2-(2,2,2-trifluorethoxy)ethoxy]pyudin-3-yl}-1,2,4-oxadiazol-3-yl)-1,3,5-triazin-2,4-diamin;
2-N-Methyl-2-N-phenyl-6-{5-[(2R)-1-[(2,2,2-trifluorethan)sulfonyl]pyrrolidin-2-yl]-1,2,4-oxadiazol-3-yl}-1,3,5-triazin-2,4-diamin;
2-N-Methyl-2-N-phenyl-6-(5-{6-[(3,3,3-tnfluorpropan)sulfinyl]pyridin-3-yl}-1,2,4-oxadiazol-3-yl)-1,3,5-triazine-2,4-diamin;
2-N-Methyl-2-N-phenyl-6-(5-{4-[(3,3,3-tufluorpropoxy)methyl]piperidin-1-yl}-1,2,4-oxadiazol-3-yl)-1,3,5-triazin-2,4-diamin
und
N-Methyl-N-phenyl-6-{5-[6-(2,2,2-trifluorethoxy)-pyridin-3-yl]-[1,2,4]oxadiazol-3-yl}-[1,3,5]triazin-2,4-diamin;
oder ein pharmazeutisch verträgliches Salz davon. - Verbindung nach Anspruch 1 mit der Struktur

- Verbindung nach Anspruch 1 mit der Struktur

- Verbindung nach Anspruch 1 mit der Struktur

- Verbindung der Formel (I) oder ein pharmazeutisch verträgliches Salz davon,

R1 ausgewählt ist ausWasserstoff-,Halogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-;R2 ausgewählt ist ausWasserstoff-,C1-C7-Alkyl-,Halogen-C2-C7-alkyl-,Amino-C2-C7-alkyl-,N-C1-C7-Alkylamino-C2-C7-alkyl-,N,N-di-C1-C7-alkylamino-C2-C7-alkyl-,Hydroxy-C2-C7-alkyl-,C1-C7-Alkoxy-C2-C7-alkyl-,C3-C10-Cycloalkyl-C1-C7-alkyl-,Cyano-C2-C7-alkyl-;oder
R1 und R2 zusammen mit den Atomen, an die sie gebunden sind, einen 4- bis 7-gliedrigen, gesättigten oder teilweise gesättigten, heterocyclischen Ring bilden, der unsubstituiert ist oder mit 1 bis 3 Substituenten substituiert ist, die ausgewählt sind aus:C1-C7-Alkyl-,Halogen-C1-C7-alkyl-,Amino-C1-C7-alkyl-,N-C1-C7-Alkylamino-C1-C7-alkyl-,N,N-di-C1-C7-alkylamino-C1-C7-alkyl-,Hydroxy-C1-C7-alkyl-,C1-C7-Alkoxy-C1-C7-alkyl-,C3-C10-Cycloalkyl-C1-C7-alkyl-,Cyano-C1-C7-alkyl-;R ausgewählt ist ausHalogen-,C1-C7-Alkyl-,Halogen-C1-C7-alkyl-,C1-C7-Alkoxy-,Cyano-,Halogen-C1-C7-alkoxy-,Nitro;-C(O)-O-R', wobei R' ausgewählt ist aus Wasserstoff, C1-C7-Alkyl; C3-C10-Cycloalkyl; C1-C7-Alkoxy; Halogen-C1-C7-alkylaryl; Aryl-C1-C7-alkyl-; Heteroaryl; Heteroaryl-C1-C7-alkyl-; Heterocyclyl;-S(=O)2-C1-C7-Alkyl; -S(=O)2-C3-C10-Cycloalkyl; -S(=O)2-C1-C7-Alkoxy;R3 ausgewählt ist aus(a) -L-Y, wobei-L- ausgewählt ist aus einer direkten Bindung; -(CH2)p-, -C(O)-, -NR7-, -NR7-C(O)-oder - C(O)-NR7-,wobei p ausgewählt ist aus 1, 2 oder 3,R7 ausgewählt ist aus Wasserstoff und C1-C7-Alkyl,Y ausgewählt ist aus Cycloalkyl, Aryl, Heteroaryl, Heterocyclyl, Spirocyclyl, die unsubstituiert sind oder substituiert sind durch 1-3 Substituenten ausgewählt ausoderHalogen-;C1-C7-Alkyl-;Halogen-C1-C7-alkyl-;Halogen-C1-C7-alkyloxy-C1-C7-alkyl; Halogen-C1-C7-alkyloxy-C1-C7-alkyloxy; C1-C7-Alkoxy-; C1-C7-Alkoxy-C1-C7-alkoxy-; NC-C1-C7-Alkoxy-;C1-C7-Alkoxy- C1-C7-alkyl-;C3-C10-Cycloalkyloxy-C1-C7-alkyl-;C3-C10-Cycloalkyl-C1-C7-alkyloxy-;C3-C10-Cycloalkyloxy-;C3-C10-Cycloalkyl-NR7'- C1-C7-alkyl-, wobei R7' ausgewählt ist aus Wasserstoff und C1-C7-Alkyl;C3-C10-Cycloalkyl- C1-C7-alkoxy- C1-C7-alkyl-;C2-C7-Alkenyl; Halogen-C2-C7-alkenyl;Hydroxy-;Hydroxy-C1-C7-alkyl-;Halogen-C1-C7-alkyloxy-;Amino-;N-C1-C7-Alkylamino-;N-Halogen-C1-C7-alkylamino-;N-Heterocyclylamino-, N-C3-C10-Cycloalkylamino-, wobei das Heterocyclyl und Cycloalkyl wahlweise substituiert sind mit Halogen-C1-C7-alkyloxy, C1-C7-Alkyl; C3-C10-Cycloalkyl und C1-C7-Alkoxy;N-C3-C10-Cycloalkyl-C1-C7-alkylamino-;N,N-di-C1-C7-alkylamino-; N,N-Dihalogen-C1-C7-alkylamino-;N,N-Diheterocyclylamino-, N,N-di-C3-C10-cycloalkylamino-, wobei das Heterocyclyl und Cycloalkyl wahlweise substituiert sind mit Halogen-C1-C7-alkyloxy, C1-C7-Alkyl; C3-C10-Cycloalkyl und C1-C7-Alkoxy;Cyano-; Oxo;C1-C7-Alkoxycarbonyl-;C1-C7-Alkoxy-C1-C7-alkoxy-C1-C7-alkyl-;Aryl; Aryl-C1-C7-alkyl-; Aryloxy;Heterocyclyl;Heterocyclyl-C1-C7-alkyl-; Heterocyclyloxy-;Heterocyclyloxy-C1-C7-alkyl-; Aryloxy-C1-C7-alkyl-; Heteroaryloxy-C1-C7-alkyl-;Hydroxycarbonyl-;-S-Halogen-C1-C7-alkyl; -S-C1-C7-Alkyl; -S-Aryl;Halogen-C1-C7-alkyl-S- C1-C7-alkyl; C1-C7-Alkyl-S-C1-C7-alkyl;-S(=O)2-C1-C7-Alkyl; -S(=O)2-Halogen-C1-C7-alkyl; -S(=O)2-Aryl; -S(=O)2-Heteroaryl; -S(=O)2-NR4'R4; -S(=O)2-Heterocyclyl;Halogen-C1-C7-alkyl-S(=O)2-C1-C7-alkyl; C1-C7-Alkyl-S(=O)2-C1-C7-alkyl;-S(=O)-C1-C7-Alkyl; -S(=O)-Halogen-C1-C7-alkyl; -S(=O)-C1-C7-Alkoxy;-S(=O)-C3-C10-Cycloalkyl;-C(O)-C1-C7-Alkyl; -C(O)-Halogen-C1-C7-alkyl; -C(O)-C1-C7-Alkoxy; -C(O)-C3-C10-Cycloalkyl;-C(O)O-C1-C7-Alkyl; -C(O)O-C3-C10-Cycloalkyl; -C(O)O-Halogen-C1-C7-alkyl;-C(O)O-C1-C7-Alkoxy;-C(O)-NR4'R4 oder -NHC(O)-R4, wobeiR4 ausgewählt ist aus Wasserstoff, C1-C7-Alkyl, Halogen-C1-C7-alkyl, C3-C10-Cycloalkyl, C3-C10-Cycloalkyl- C1-C7-alkyl und C1-C7-Alkoxy;R4' ausgewählt ist aus Wasserstoff;oder R4 und R4' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-7-gliedrigen gesättigten oder teilweise gesättigten monocyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält, und wobei der heterocyclische Ring wahlweise substituiert ist mit Aryl, Aryloxy-, C1-C7-Alkyl, Halogen-C1-C7-alkyl oder C1-C7-Alkoxy und das Aryl wahlweise substituiert ist mit Halogen, C1-C7-Alkyl, Halogen-C1-C7-alkyl oder C1-C7-Alkoxy;(b) -C(O)-NR5'R5 oder -C(O)-O-R5, wobeim 0-1 ist; undR5 und R5' ausgewählt sind aus Wasserstoff, C1-C7-Alkyl; C3-C10-Cycloalkyl; C1-C7-Alkoxy; Halogen-C1-C7-alkylaryl; Aryl-C1-C7-alkyl-; Aryl; Heteroaryl; Heteroaryl C1-C7-Alkyl-; Heterocyclyl; Indan; oder R5 und R5' zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen 4-9-gliedrigen, gesättigten oder teilweise gesättigten monocyclischen oder bicyclischen heterocyclischen Ring bilden, der wahlweise ein weiteres Heteroatom ausgewählt aus N, O oder S enthält;wobei das C3-C10-Cycloalkyl; Aryl, Heteroaryl, Heterocyclyl und Indan wahlweise substituiert sind mit 1 bis 3 Substituenten ausgewählt aus C1-C7-Alkyl, Halogen-C1-C7-alkyl, C1-C7-Alkoxy, Halogen-C1-C7-alkyloxy- und Hydroxy-C1-C7-alkyl;
n 0-2 ist;
R8 Wasserstoff ist und R9 ausgewählt ist aus Wasserstoff, C1-C7-Alkoxy-C1-C7-alkyl-, C1-C7-Alkyl, C1-C7-Alkoxy und Halogen-C1-C7-alkyl;
zur Verwendung als Medikament. - Pharmazeutische Zusammensetzung, umfassend eine therapeutisch wirksame Menge einer Verbindung der Formel (I) nach einem der Ansprüche 1 bis 10 und einen oder mehrere pharmazeutische verträgliche Träger/Hilfsstoffe.
- Kombination, insbesondere pharmazeutische Kombination, umfassend eine therapeutisch wirksame Menge einer Verbindung der Formel (I) nach einem der Ansprüche 1 bis 10 und ein oder mehrere therapeutisch aktive Mittel und speziell schmerzstillende Mittel.
- Verbindung der Formel (I) nach einem der Ansprüche 1 bis 10 zur Verwendung bei der Behandlung von chronischem Schmerz.
- Verbindung der Formel (I) nach einem der Ansprüche 1 bis 10 zur Verwendung bei der Behandlung einer krankhaften Störung oder Erkrankung, ausgewählt aus chronischem Schmerz, wie beispielsweise Positivsymptomen von chronischem Schmerz; z.B. Parästhesie, Dynästhesie, Hyperalgesie, Allodynie und spontaner Schmerz, sowie Negativsymptomen, wie beispielsweise Sensibilitätsverlust.
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL11757294T PL2616465T3 (pl) | 2010-09-13 | 2011-09-13 | Triazyno-oksadiazole |
| SI201130707T SI2616465T1 (sl) | 2010-09-13 | 2011-09-13 | Triazin-oksadiazoli |
| HRP20160094TT HRP20160094T1 (hr) | 2010-09-13 | 2011-09-13 | Triazin-oksadiazoli |
| CY20161100095T CY1117182T1 (el) | 2010-09-13 | 2016-02-03 | Τριαζιν-οξαδιαζολες |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US38215810P | 2010-09-13 | 2010-09-13 | |
| PCT/EP2011/065868 WO2012035023A1 (en) | 2010-09-13 | 2011-09-13 | Triazine-oxadiazoles |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2616465A1 EP2616465A1 (de) | 2013-07-24 |
| EP2616465B1 true EP2616465B1 (de) | 2015-11-04 |
Family
ID=44651777
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP11757294.1A Active EP2616465B1 (de) | 2010-09-13 | 2011-09-13 | Triazin-oxadiazole |
Country Status (27)
| Country | Link |
|---|---|
| US (2) | US8895733B2 (de) |
| EP (1) | EP2616465B1 (de) |
| JP (1) | JP2013537180A (de) |
| KR (1) | KR101843600B1 (de) |
| CN (1) | CN103221408A (de) |
| AU (1) | AU2011303978B2 (de) |
| BR (1) | BR112013005889A2 (de) |
| CA (1) | CA2812081A1 (de) |
| CL (1) | CL2013000685A1 (de) |
| CO (1) | CO6690768A2 (de) |
| CR (1) | CR20130111A (de) |
| CU (1) | CU20130036A7 (de) |
| DK (1) | DK2616465T3 (de) |
| EA (1) | EA026132B1 (de) |
| ES (1) | ES2559449T3 (de) |
| HR (1) | HRP20160094T1 (de) |
| HU (1) | HUE027256T2 (de) |
| MX (1) | MX2013002864A (de) |
| NZ (1) | NZ608116A (de) |
| PE (1) | PE20131377A1 (de) |
| PH (1) | PH12013500473A1 (de) |
| PL (1) | PL2616465T3 (de) |
| PT (1) | PT2616465E (de) |
| RS (1) | RS54544B1 (de) |
| SG (1) | SG188438A1 (de) |
| SI (1) | SI2616465T1 (de) |
| WO (1) | WO2012035023A1 (de) |
Families Citing this family (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112013005889A2 (pt) * | 2010-09-13 | 2020-08-25 | Novartis Ag | triazinaoxadiazóis, composição farmacêutica que os compreende e uso dos mesmos |
| CN104093716B (zh) | 2011-10-31 | 2017-06-23 | 克赛农制药股份有限公司 | 联芳醚磺酰胺化合物及其作为治疗剂的用途 |
| BR112014010271A2 (pt) | 2011-10-31 | 2017-04-18 | Xenon Pharmaceuticals Inc | compostos de benzenossulfonamida e seu uso como agentes terapêuticos |
| SG11201408284VA (en) | 2012-05-22 | 2015-02-27 | Xenon Pharmaceuticals Inc | N-substituted benzamides and their use in the treatment of pain |
| KR101663436B1 (ko) | 2012-07-06 | 2016-10-06 | 제넨테크, 인크. | N-치환된 벤즈아미드 및 이의 사용 방법 |
| CA2892045C (en) | 2012-11-21 | 2022-05-31 | Ptc Therapeutics, Inc. | Substituted reverse pyrimidine bmi-1 inhibitors |
| BR112015022488A2 (pt) | 2013-03-14 | 2017-07-18 | Genentech Inc | triazolopiridinas substituídas e métodos de uso das mesmas |
| US9493429B2 (en) | 2013-03-15 | 2016-11-15 | Genentech, Inc. | Substituted benzoxazoles and methods of use thereof |
| AU2013399092A1 (en) | 2013-08-30 | 2016-03-17 | Ptc Therapeutics, Inc. | Substituted pyrimidine Bmi-1 inhibitors |
| EP3071553A4 (de) | 2013-11-21 | 2017-08-02 | PTC Therapeutics, Inc. | Substituierte pyridin- und pyrazin-bmi-1-hemmer |
| WO2015076801A1 (en) * | 2013-11-21 | 2015-05-28 | Ptc Therapeutics, Inc. | Substituted triazine bmi-1 inhibitors |
| MX2016006936A (es) | 2013-11-27 | 2016-10-05 | Genentech Inc | Benzamidas sustituidas y metodos para usarlas. |
| CN106715418A (zh) | 2014-07-07 | 2017-05-24 | 基因泰克公司 | 治疗化合物及其使用方法 |
| CA2986045A1 (en) | 2015-05-22 | 2016-12-01 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| MA42683A (fr) | 2015-08-27 | 2018-07-04 | Genentech Inc | Composés thérapeutiques et leurs méthodes utilisation |
| PE20181003A1 (es) | 2015-09-28 | 2018-06-26 | Genentech Inc | Compuestos terapeuticos y sus metodos de uso |
| MA43304A (fr) | 2015-11-25 | 2018-10-03 | Genentech Inc | Benzamides substitués utiles en tant que bloqueurs de canaux sodiques |
| EP3854782A1 (de) | 2016-03-30 | 2021-07-28 | Genentech, Inc. | Substituierte benzamide und verfahren zur verwendung davon |
| JOP20190072A1 (ar) * | 2016-10-13 | 2019-04-07 | Glaxosmithkline Ip Dev Ltd | مشتقات 1، 3 سيكلوبوتان ثنائي الاستبدال أو آزيتيدين كمثبطات للإنزيم المخلق للبروستاجلاندين d المكون للدم |
| CR20190236A (es) | 2016-10-17 | 2019-09-09 | Genentech Inc | Compuestos terapéuticos y métodos para utilizarlos |
| CN110099898B (zh) * | 2016-10-24 | 2023-07-25 | 优曼尼蒂治疗公司 | 化合物及其用途 |
| CA3049010A1 (en) | 2017-01-06 | 2018-07-12 | Yumanity Therapeutics, Inc. | Methods for the treatment of neurological disorders |
| EP3601273B1 (de) | 2017-03-24 | 2021-12-01 | Genentech, Inc. | 4-piperidin-n-(pyrimidin-4-yl)chroman-7-sulfonamid-derivate als natriumkanalinhibitoren |
| CA3083000A1 (en) | 2017-10-24 | 2019-05-02 | Yumanity Therapeutics, Inc. | Compounds and uses thereof |
| MX2020006587A (es) | 2017-12-22 | 2020-12-10 | Ravenna Pharmaceuticals Inc | Derivados de aminopiridina como inhibidores de fosfatidilinositol fosfato cinasa. |
| WO2019165290A1 (en) | 2018-02-26 | 2019-08-29 | Genentech, Inc. | Pyridine-sulfonamide compounds and their use against pain and related conditions |
| BR112020019191A2 (pt) | 2018-03-23 | 2021-01-05 | Yumanity Therapeutics, Inc. | Compostos e seus usos |
| US10947251B2 (en) | 2018-03-30 | 2021-03-16 | Genentech, Inc. | Therapeutic compounds and methods of use thereof |
| TW202003490A (zh) | 2018-05-22 | 2020-01-16 | 瑞士商赫孚孟拉羅股份公司 | 治療性化合物及其使用方法 |
| WO2020055544A2 (en) | 2018-08-17 | 2020-03-19 | Ptc Therapeutics, Inc. | Method for treating pancreatic cancer |
| KR20220007845A (ko) | 2019-01-24 | 2022-01-19 | 유마니티 테라퓨틱스, 인크. | 화합물 및 이의 용도 |
| TW202112767A (zh) | 2019-06-17 | 2021-04-01 | 美商佩特拉製藥公司 | 作為磷脂酸肌醇磷酸激酶抑制劑之胺基吡啶衍生物 |
| EA202192047A1 (ru) | 2019-11-13 | 2021-12-08 | Юманити Терапьютикс, Инк. | Соединения и их применение |
| WO2021195360A1 (en) * | 2020-03-27 | 2021-09-30 | Landos Biopharma, Inc. | Plxdc2 ligands |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT72878B (en) | 1980-04-24 | 1983-03-29 | Merck & Co Inc | Process for preparing mannich-base hydroxamic acid pro-drugs for the improved delivery of non-steroidal anti-inflammatory agents |
| EP1187825A1 (de) | 1999-06-07 | 2002-03-20 | Shire Biochem Inc. | Thiophen integrin inhibitoren |
| EP1546121B1 (de) | 2002-07-18 | 2012-08-29 | Janssen Pharmaceutica NV | Substituierte triazin-kinaseinhibitoren |
| CA2514733A1 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| AR045010A1 (es) * | 2003-07-02 | 2005-10-12 | Vertex Pharma | Pirimidinas utiles como moduladores de canales ionicos cuya activacion es controlada por tension |
| WO2007109324A2 (en) | 2006-03-21 | 2007-09-27 | Xenon Pharmaceuticals, Inc. | Potent and selective nav 1.7 sodium channel blockers |
| US20080227799A1 (en) | 2006-07-11 | 2008-09-18 | Liotta Dennis C | CXCR4 Antagonists Including Heteroatoms for the Treatment of Medical Disorders |
| US8193194B2 (en) * | 2007-05-25 | 2012-06-05 | Vertex Pharmaceuticals Incorporated | Ion channel modulators and methods of use |
| US20090005381A1 (en) | 2007-06-26 | 2009-01-01 | Philip Manton Brown | Methods of treating serotonin-mediated diseases and disorders |
| WO2009010784A1 (en) | 2007-07-13 | 2009-01-22 | Astrazeneca Ab | New compounds 955 |
| WO2009091388A2 (en) | 2007-12-21 | 2009-07-23 | Progenics Pharmaceuticals, Inc. | Triazines and related compounds having antiviral activity, compositions and methods thereof |
| GB0805477D0 (en) | 2008-03-26 | 2008-04-30 | Univ Nottingham | Pyrimidines triazines and their use as pharmaceutical agents |
| US8236880B2 (en) | 2008-04-23 | 2012-08-07 | Taylor Made Golf Company, Inc. | Compositions comprising an amino triazine and ionomer or ionomer precursor |
| US8633233B2 (en) | 2008-08-06 | 2014-01-21 | Hydra Biosciences, Inc. | Methods and compositions for treating anxiety |
| WO2010022055A2 (en) * | 2008-08-20 | 2010-02-25 | Amgen Inc. | Inhibitors of voltage-gated sodium channels |
| EP2536689A1 (de) * | 2010-02-17 | 2012-12-26 | Amgen Inc. | Arylcarboxamidderviate als natriumkanalhemmer zur schmerzbehandlung |
| BR112013005889A2 (pt) * | 2010-09-13 | 2020-08-25 | Novartis Ag | triazinaoxadiazóis, composição farmacêutica que os compreende e uso dos mesmos |
-
2011
- 2011-09-13 BR BR112013005889-7A patent/BR112013005889A2/pt not_active IP Right Cessation
- 2011-09-13 PH PH1/2013/500473A patent/PH12013500473A1/en unknown
- 2011-09-13 RS RS20160045A patent/RS54544B1/sr unknown
- 2011-09-13 WO PCT/EP2011/065868 patent/WO2012035023A1/en not_active Ceased
- 2011-09-13 KR KR1020137009331A patent/KR101843600B1/ko not_active Expired - Fee Related
- 2011-09-13 CA CA2812081A patent/CA2812081A1/en not_active Abandoned
- 2011-09-13 SI SI201130707T patent/SI2616465T1/sl unknown
- 2011-09-13 ES ES11757294.1T patent/ES2559449T3/es active Active
- 2011-09-13 CN CN2011800544823A patent/CN103221408A/zh active Pending
- 2011-09-13 JP JP2013527645A patent/JP2013537180A/ja active Pending
- 2011-09-13 HU HUE11757294A patent/HUE027256T2/en unknown
- 2011-09-13 SG SG2013016829A patent/SG188438A1/en unknown
- 2011-09-13 AU AU2011303978A patent/AU2011303978B2/en not_active Ceased
- 2011-09-13 MX MX2013002864A patent/MX2013002864A/es not_active Application Discontinuation
- 2011-09-13 EP EP11757294.1A patent/EP2616465B1/de active Active
- 2011-09-13 DK DK11757294.1T patent/DK2616465T3/da active
- 2011-09-13 PE PE2013000506A patent/PE20131377A1/es not_active Application Discontinuation
- 2011-09-13 PT PT117572941T patent/PT2616465E/pt unknown
- 2011-09-13 EA EA201390381A patent/EA026132B1/ru not_active IP Right Cessation
- 2011-09-13 NZ NZ608116A patent/NZ608116A/en not_active IP Right Cessation
- 2011-09-13 HR HRP20160094TT patent/HRP20160094T1/hr unknown
- 2011-09-13 PL PL11757294T patent/PL2616465T3/pl unknown
-
2013
- 2013-03-13 CU CU2013000036A patent/CU20130036A7/es unknown
- 2013-03-13 CR CR20130111A patent/CR20130111A/es unknown
- 2013-03-13 CL CL2013000685A patent/CL2013000685A1/es unknown
- 2013-03-13 CO CO13050505A patent/CO6690768A2/es unknown
- 2013-10-15 US US14/054,221 patent/US8895733B2/en not_active Expired - Fee Related
-
2014
- 2014-10-08 US US14/509,705 patent/US20150025057A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| PT2616465E (pt) | 2016-03-09 |
| US20150025057A1 (en) | 2015-01-22 |
| MX2013002864A (es) | 2013-08-29 |
| PH12013500473A1 (en) | 2013-05-06 |
| CA2812081A1 (en) | 2012-03-22 |
| RS54544B1 (sr) | 2016-06-30 |
| CU20130036A7 (es) | 2013-05-31 |
| KR101843600B1 (ko) | 2018-03-29 |
| HRP20160094T1 (hr) | 2016-02-26 |
| CO6690768A2 (es) | 2013-06-17 |
| AU2011303978A1 (en) | 2013-03-21 |
| EP2616465A1 (de) | 2013-07-24 |
| CL2013000685A1 (es) | 2013-10-04 |
| SI2616465T1 (sl) | 2016-02-29 |
| PE20131377A1 (es) | 2013-11-30 |
| SG188438A1 (en) | 2013-05-31 |
| DK2616465T3 (da) | 2016-02-01 |
| WO2012035023A1 (en) | 2012-03-22 |
| ES2559449T3 (es) | 2016-02-12 |
| EA026132B1 (ru) | 2017-03-31 |
| BR112013005889A2 (pt) | 2020-08-25 |
| NZ608116A (en) | 2014-05-30 |
| JP2013537180A (ja) | 2013-09-30 |
| US8895733B2 (en) | 2014-11-25 |
| PL2616465T3 (pl) | 2016-04-29 |
| AU2011303978B2 (en) | 2014-07-03 |
| HUE027256T2 (en) | 2016-08-29 |
| KR20130105657A (ko) | 2013-09-25 |
| EA201390381A1 (ru) | 2013-08-30 |
| CR20130111A (es) | 2013-05-29 |
| US20140051676A1 (en) | 2014-02-20 |
| CN103221408A (zh) | 2013-07-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2616465B1 (de) | Triazin-oxadiazole | |
| US12187731B2 (en) | Compounds useful as inhibitors of ATR kinase | |
| JP7028861B2 (ja) | ピリジル置換のインドール化合物 | |
| EP2953940B1 (de) | Piperidin-1-yl- und azepin-1-yl-carboxylate als muskarin-m4-rezeptoragonisten | |
| JP2021519337A (ja) | Ikarosの分解のためのセレブロン結合剤 | |
| CA2962578A1 (en) | Aminotriazine derivatives useful as tank-binding kinase inhibitor compounds | |
| JPWO2014109414A1 (ja) | 含窒素複素環化合物またはその塩 | |
| JP7730407B2 (ja) | 4-(イミダゾ[1,2-a]ピリジン-3-イル)-ピリミジン誘導体 | |
| TW201605824A (zh) | 哌啶衍生物 | |
| WO2013012827A1 (en) | Diaminocyclohexane compounds and uses thereof | |
| TW202227404A (zh) | 可用於治療與5-ht2a血清素受體相關疾病之作為5-ht2a血清素受體調節劑之異噁唑衍生物 | |
| TW201336840A (zh) | 三□-□二唑 | |
| WO2026011019A1 (en) | Pyrrolidinyl compounds as inhibitors of cgas |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20130415 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA |
|
| RAX | Requested extension states of the european patent have changed |
Extension state: BA Payment date: 20130415 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1183306 Country of ref document: HK |
|
| 17Q | First examination report despatched |
Effective date: 20140424 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| INTG | Intention to grant announced |
Effective date: 20150105 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| INTG | Intention to grant announced |
Effective date: 20150601 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR |
|
| AX | Request for extension of the european patent |
Extension state: BA |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 759108 Country of ref document: AT Kind code of ref document: T Effective date: 20151115 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602011021210 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: TUEP Ref document number: P20160094 Country of ref document: HR |
|
| REG | Reference to a national code |
Ref country code: RO Ref legal event code: EPE |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: T3 Effective date: 20160129 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2559449 Country of ref document: ES Kind code of ref document: T3 Effective date: 20160212 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: TRGR |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: T1PR Ref document number: P20160094 Country of ref document: HR |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: FP |
|
| REG | Reference to a national code |
Ref country code: PT Ref legal event code: SC4A Free format text: AVAILABILITY OF NATIONAL TRANSLATION Effective date: 20160202 |
|
| REG | Reference to a national code |
Ref country code: NO Ref legal event code: T2 Effective date: 20151104 |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: FG4A Ref document number: E011630 Country of ref document: EE Effective date: 20160201 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20151104 |
|
| REG | Reference to a national code |
Ref country code: GR Ref legal event code: EP Ref document number: 20160400205 Country of ref document: GR Effective date: 20160414 |
|
| REG | Reference to a national code |
Ref country code: SK Ref legal event code: T3 Ref document number: E 20515 Country of ref document: SK |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602011021210 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 6 |
|
| REG | Reference to a national code |
Ref country code: HU Ref legal event code: AG4A Ref document number: E027256 Country of ref document: HU |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SM Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20151104 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20160805 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 7 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: UEP Ref document number: 759108 Country of ref document: AT Kind code of ref document: T Effective date: 20151104 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: ODRP Ref document number: P20160094 Country of ref document: HR Payment date: 20180824 Year of fee payment: 8 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: PLFP Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 20180824 Year of fee payment: 8 Ref country code: DE Payment date: 20180828 Year of fee payment: 8 Ref country code: MC Payment date: 20180829 Year of fee payment: 8 Ref country code: LT Payment date: 20180904 Year of fee payment: 8 Ref country code: FR Payment date: 20180828 Year of fee payment: 8 Ref country code: IE Payment date: 20180911 Year of fee payment: 8 Ref country code: IT Payment date: 20180919 Year of fee payment: 8 Ref country code: NO Payment date: 20180913 Year of fee payment: 8 Ref country code: RO Payment date: 20180828 Year of fee payment: 8 Ref country code: EE Payment date: 20180831 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: PL Payment date: 20180822 Year of fee payment: 8 Ref country code: LV Payment date: 20180904 Year of fee payment: 8 Ref country code: CZ Payment date: 20180828 Year of fee payment: 8 Ref country code: HR Payment date: 20180824 Year of fee payment: 8 Ref country code: GR Payment date: 20180829 Year of fee payment: 8 Ref country code: IS Payment date: 20180823 Year of fee payment: 8 Ref country code: DK Payment date: 20180911 Year of fee payment: 8 Ref country code: CH Payment date: 20180913 Year of fee payment: 8 Ref country code: GB Payment date: 20180912 Year of fee payment: 8 Ref country code: NL Payment date: 20180912 Year of fee payment: 8 Ref country code: HU Payment date: 20180906 Year of fee payment: 8 Ref country code: SI Payment date: 20180831 Year of fee payment: 8 Ref country code: RS Payment date: 20180906 Year of fee payment: 8 Ref country code: FI Payment date: 20180910 Year of fee payment: 8 Ref country code: SE Payment date: 20180910 Year of fee payment: 8 Ref country code: BG Payment date: 20180831 Year of fee payment: 8 Ref country code: AT Payment date: 20180828 Year of fee payment: 8 Ref country code: TR Payment date: 20180906 Year of fee payment: 8 Ref country code: BE Payment date: 20180823 Year of fee payment: 8 Ref country code: SK Payment date: 20180903 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: PT Payment date: 20180910 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CY Payment date: 20180829 Year of fee payment: 8 Ref country code: MT Payment date: 20180918 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20181001 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: MK Payment date: 20180829 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: AL Payment date: 20180921 Year of fee payment: 8 |
|
| REG | Reference to a national code |
Ref country code: HR Ref legal event code: PBON Ref document number: P20160094 Country of ref document: HR Effective date: 20190913 Ref country code: DE Ref legal event code: R119 Ref document number: 602011021210 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: EE Ref legal event code: MM4A Ref document number: E011630 Country of ref document: EE Effective date: 20190930 |
|
| REG | Reference to a national code |
Ref country code: FI Ref legal event code: MAE |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1183306 Country of ref document: HK |
|
| REG | Reference to a national code |
Ref country code: DK Ref legal event code: EBP Effective date: 20190930 Ref country code: LT Ref legal event code: MM4D Effective date: 20190913 Ref country code: NO Ref legal event code: MMEP |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LV Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190914 Ref country code: FI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: RO Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| REG | Reference to a national code |
Ref country code: SE Ref legal event code: EUG |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: MM Effective date: 20191001 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: RS Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200316 Ref country code: CY Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: CZ Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: SK Ref legal event code: MM4A Ref document number: E 20515 Country of ref document: SK Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20191001 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200401 Ref country code: GR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200416 Ref country code: PT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200422 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: LT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: EE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: NO Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 |
|
| REG | Reference to a national code |
Ref country code: BE Ref legal event code: MM Effective date: 20190930 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MM01 Ref document number: 759108 Country of ref document: AT Kind code of ref document: T Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: HR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: BG Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20200531 Ref country code: SI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190914 Ref country code: HU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190914 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 Ref country code: SK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| REG | Reference to a national code |
Ref country code: SI Ref legal event code: KO00 Effective date: 20200722 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: DK Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190930 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20210128 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190914 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IS Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20191001 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20190913 Ref country code: AL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20190913 |