EP0843684B1 - Supports solides universels et leurs procedes d'utilisation - Google Patents
Supports solides universels et leurs procedes d'utilisation Download PDFInfo
- Publication number
- EP0843684B1 EP0843684B1 EP97922372A EP97922372A EP0843684B1 EP 0843684 B1 EP0843684 B1 EP 0843684B1 EP 97922372 A EP97922372 A EP 97922372A EP 97922372 A EP97922372 A EP 97922372A EP 0843684 B1 EP0843684 B1 EP 0843684B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- oligonucleotide
- reagent
- solid support
- cleaving
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 C[C@]1O[C@](C)(CO*OCC*)C(C*=C)C1O Chemical compound C[C@]1O[C@](C)(CO*OCC*)C(C*=C)C1O 0.000 description 5
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates generally to the fields of chemistry and biology. More particularly, the present invention is directed to compositions and methods for use in the synthesis of oligonucleotides (e.g., DNA and RNA sequences).
- oligonucleotides e.g., DNA and RNA sequences.
- oligonucleotides are synthesized utilizing a building block approach which involves the sequential addition of nucleotides onto a growing oligonucleotide chain immobilized on to a solid support. Because every DNA oligonucleotide may have any of 4 different initial nucleotides, it is necessary to maintain a supply of 4 different nucleoside (A, C, G and T) loaded solid supports to be able to synthesize any given DNA sequence. In the case of DNA synthesis, the first nucleoside from the 3' end of the DNA sequence is typically preloaded on the solid support through an ester linkage.
- the sequence that is to be synthesized contains a T nucleoside at the 3' end
- a T support is employed and the balance of the nucleotides in the DNA sequence added thereto (for example, using an automated DNA synthesizer).
- the oligonucleotide is cleaved from the solid support through the hydrolysis of the ester linkage.
- an additional 4-different nucleoside loaded solid supports must be available to the user. Similar considerations apply if any specialty modified nucleoside is desired at the 3' end.
- the new oligonucleotides can be released from the glass, deprotected and cleaved from the uridylyl terminus in one reaction. Since the uridyl functionality is cleaved from the solid support in this cleaving reaction, the support is not available for subsequent oligonucleotide syntheses.
- Crea and Horn suggested a similar approach which involved preparing the dimer 5'-O-p-chlorophenylphospho-2'(3')-O-acetyluridilyl-[2'(3')-3']-5'-O-dimethoxytritylthymidine p-chlorophenylester and attaching the dimer to cellulose via a phosphate linkage.
- the 5' position of the thymidine is available for oligonucleotide attachment and synthesis. [R. Crea & T. Horn, Nucleic Acids Research 8 , 2331 (1980)].
- Schwartz et al. attached an adapter, 2'(3')-O-dimethoxytrityl-3'(2')-O-benzoyluridine-5'-O-(2-cyanoethyl N,N-diisopropylphosphoramidite, to a thymidine derivatized polystyrene and synthesized an oligonucleotide from the O-dimethoxytrityl position of the uridine.
- a support for oligonucleotide synthesis is described in WO 93/20091, which carries a homocyclic or heterocyclic ring containing a first and second carbon wherein: (I) the first carbon atom is substituted by a nucleophilic group or a group convertible to same on treatment with a base; (ii) the second carbon atom is substituted by a hydroxy group, a hydroxy group protected by an acid labile protecting group, or a phosphate group substituted by an oligonucleotide; and (iii) the first and second carbon atoms are directly connected by a covalent bond; this support being suitable for use in oligonucleotide synthesis.
- Reagents and methods for cleaving and deprotecting insolubilised and protected synthetic oligonucleotides are described in US 5 348 868.
- a reagent comprising methylamine and t-butylamine is indicated as useful in suppressing transamination events.
- oligonucleotide polymeric support system is described in WO 85/01051 as featuring a universal primer which allows chain elongation-in either the 3' or 5' direction, and is capable of withstanding mildly basic and acidic conditions, and which possesses selectively oxidisable substituents, whereby the desired oligonucleotide is releasable from the polymeric support.
- a process for solid support nucleic acid synthesis, and compounds useful as solid supports therein are described in WO 95/01987.
- a mineral or an organic polymer, bound by a bivalent hydrocarbon radical to an epoxy or glycol-type group is used as a solid support, said group comprising two adjacent saturated carbon atoms on which an -OH and a nucleophilic group are substituted.
- the present invention provides methods and compositions which overcome prior art problems associated with the solid support synthesis of oligonucleotides.
- the methods and compositions of the present invention provide a single reusable universal solid support suitable for step-wise oligonucleotide synthesis and the subsequent single step cleavage and deprotection of the synthesized oligonucleotide.
- the present invention provides cleaving reagents suitable for removing oligonucleotides from the solid support to which they are synthesized.
- the cleaving reagents of the present invention are volatile and thus do not require subsequent time consuming reagent removal processes.
- the cleaving reagents of the present invention remove synthesized oligonucleotides in a substantially reduced length of time.
- the present invention provides an oligonucleotide synthesis reagent having the following general formula: SS - R 6 - O -R 3 wherein SS is a solid support; R 6 is where R 5 is hydrogen or alkyl and R 4 is a phosphate protecting group; and R 3 is a ring moiety having vicinal groups -XR 1 and -YR 2 wherein each of X and Y is independently selected from the group consisting of O, S and NH and one of R 1 and R 2 is a blocking moiety and the other is hydrogen or a hydroxy protecting group.
- R 3 is a sugar moiety with -XR 1 and -YR 2 occupying the second and third carbon positions of the sugar ring.
- R 3 has the structure wherein B is a purine or pyrimidine base; each of X and Y and R 1 and R 2 are as described above.
- Preferred R 1 or R 2 blocking moieties are alkylcarbonyl or arylcarbonyl which can be prepared by forming an ester blocking group at one of two vicinal hydroxy functionalities.
- Preferred embodiments of R 6 are the phosphoramidite linkages and the oxidized form, phosphoramidate linkages, characterized when R 6 is respectively and phosphate protecting group R 4 is a cyanoethyl moiety.
- these reagents are useful as universal solid supports in the synthesis of oligonucleotides where the synthesis takes place at the vicinal R 1 or R 2 position having the hydroxy protecting group; the other R 1 or R 2 position being blocked with the arylcarbonyl or alkylcarbonyl blocking moiety.
- the post synthesis release of oligonucleotides synthesized using the oligonucleotide synthesis reagents of the present invention is substantially faste than the release of oligonucleotides utilizing prior art solid support reagents.
- the oligonucleotides are released at the vicinal group position of the ring moiety.
- the link between the ring moiety and the solid support is a phosphoramidate or phosphoramidite the release does not effect the solid support bond, and oligonucleotide reagent is suitable for repeat synthesis procedures.
- cleaving methods and cleaving reagents for releasing oligonucleotides from a solid support are provided.
- the cleaving reagents and releasing methods described herein are applicable to oligonucleotides attached to a variety of solid supports including ester linked oligonucleotides, phosphate linked oligonucleotides and phosphoramidate or phosphoramidite linked oligonucleotides.
- Cleaving methods of the present invention involve contacting a solid support bearing an oligonucleotide with a cleaving reagent of the present invention which comprises a mixture of a first compound which includes methylamine and/or ammonium hydroxide and a second compound which can be a secondary amine and/or a tertiary amine.
- the cleaving reagent is in the range of about 1:9 (v/v) of 40 wt% aqueous methylamine:23 - 25 wt% aqueous trimethyl amine to about 9:1 (v/v) 40 wt% aqueous methylamine:23 - 25 wt% aqueous trimethyl amine.
- the present invention provides improvements in compositions and methods for solid support based oligonucleotide synthesis. More particularly, the present invention provides oligonucleotide synthesis reagents in which a ring moiety is linked to a solid support via a phosphoramidate linkage.
- the ring moiety has vicinally positioned functionalities, one of which is the site for the step wise synthesis of oligonucleotides and the other of which is blocked during the synthesis but is unblocked to become active during the cleaving process.
- the oligonucleotide synthesis reagent of the present invention is a universal solid support in that a single oligonucleotide reagent is useful for the synthesis of any oligonucleotide having any initial nucleoside, thus precluding the need to maintain a variety of different suitably derivatized solid supports. Moreover, because during typical cleaving processes the phosphoramidate bond of the present oligonucleotide reagent remains stable, synthesis reagents of the present invention can be used for subsequent oligonucleotide syntheses.
- two hydroxyls are linked to adjacent carbon atoms, preferably in a cis-orientation.
- one of the two hydroxyls or hydroxyl equivalents is blocked; preferably, hydroxyl is blocked in the form of an ester derivative of the hydroxyl or hydroxyl equivalent.
- Compositions in which either one of the hydroxyls or equivalent or a mixture of both hydroxyls or equivalents may suitably be employed.
- the remaining unblocked vicinal site is used to grow the oligonucleotide chain. Typically, this site is protected with a suitable protecting group, for example DMT, (dimethoxytrityl), and then deprotected just prior to adding the initial nucleotide in the synthesis.
- the blocked vicinal site is unblocked or hydrolysed to liberate the hydroxyl or equivalent.
- the hydroxyl or equivalent then makes an intramolecular attack on the adjacent phosphate group of the first nucleotide of the oligonucleotide chain, forming a cyclic phosphate. This results in the release of the oligonucleotide molecule from the solid support.
- Whatever nucleoside is first added to the solid support during the oligonucleotide synthesis becomes the 3'-end terminal nucleoside of the synthesized molecule (when the oligonucleotide is synthesized in the conventional 3' to 5' direction).
- oligonucleotide synthesis reagents having the general formula I SS - R 6 - O -R 3 wherein SS is a solid support; R 6 is where R 5 is hydrogen or alkyl and R 4 is a phosphate protecting group; and R 3 is wherein B is a purine or pyrimidine base; each of X and Y is independently selected from the group consisting of O, S and NH; one of R 1 and R 2 is an alkylcarbonyl or arylcarbonyl group and the other of R 1 and R 2 is hydrogen or a protecting group suitable for protecting -O, -S, or -NH.
- B is preferably uridine. independently selected from the group consisting of O, S and NH and one of R 1 and R 2 is a blocking moiety and the other is hydrogen or a hydroxy protecting group suitable for protecting -OH, -SH, or NH 2 .
- R 6 is a phosphoramidite or its oxidized form, phosphoramidate
- R 5 is preferably hydrogen. This is because these oligonucleotide synthesis reagents are generally prepared using a primary amine. However, those skilled in the art will also appreciate that R 5 can be alkyl because the phosphoramidate can be prepared using secondary amines.
- Phosphate protecting group R 4 is suitably any group capable of protecting the phosphorous of the phosphoramidate or phosphoramidite from cleaving or reacting during oligonucleotide synthesis.
- cyanoethyl moieties are preferred phosphate protecting groups for their stability under oligonucleotide synthesis conditions and their ease of removal with ammonia or methylamine.
- the phosphoramidate or phosphoramidite linkage of the utilized in the present invention need not be deprotected.
- alkyl moieties generally or aryl containing moieties are also suitable phosphate protecting groups R 4 .
- R 3 is preferably a ring moiety and -XR 1 and YR 2 are oriented in space in a fixed cis position.
- 23 is preferably a sugar with-XR 1 and -YR 2 occupying the second and third carbon positions of the sugar ring.
- nucleosides, or sugars having an attached purine or pyrimidine base are particularly preferred ring moieties.
- X and Y are preferably O (oxygen).
- O oxygen
- R 1 or R 2 One of the vicinal positions must be bloked from participating in the oligonucleotide synthesis by a suitable blocking group, R 1 or R 2 . Because, as described below, the unblocked X or Y is active in the final oligonucleotide cleaving step, the blocking group must be easily removed under cleaving reaction conditions but stable under those conditions typically found in oligonucleotide synthesis. For this reason one of R 1 or R 2 is preferably an alkylcarbonyl or arylcarbonyl.
- the hydroxyl-protecting group associated with the R 1 or R 2 which is not a blocking group is suitably any protecting group which is easily removed so that the protected group is available as the site for the introduction of a first nucleoside during the initiation of oligonucleotide synthesis.
- the 4,4'-dimethoxytrityl (DMT) group is particularly preferred.
- suitable groups include, but are not limited to, the following: 4,4',4"-tris-(benzyloxy)trityl (TBTr); 4,4',4"-tris-(4,5-dichlorophthalimido)trityl (CPTr); 4,4',4"-tris(levulinyloxy)trityl (TLTr); 3-(imidazolylmethyl)-4,4'-dimethoxytrityl (IDTr); pixyl (9-phenylxanthen-9-yl); 9-(p-methoxyphenyl)xanthen-9-yl (Mox); 4-decyloxytrityl (C 10 Tr); 4-hexadecyloxytrityl (C 16 Tr); 9-(4-octadecyloxyphenyl)xanthene-9-yl (C 18 Px); 1,1-bis-(4-methoxyphenyl)-1'-pyrenyl methyl (BMPM);
- B represents a pyrimidine or purine base.
- Preferred for use in accordance with the present invention are those bases characteristic of guanine, adenine, thymine and cytosine; however, other purine or pyrimidine bases as may be employed in the synthesis of nucleotide analogs may alternatively be used as group B.
- oligonucleotide synthesis reagents of the present invention have the following structures:
- a solid support may be selected from controlled pore glass, copolymers of ethylene and acrylate, copolymers of ethylene and methacrylate, polystyrene, and nylon.
- the solid support comprises a functional group for attachment of a suitable phosphoramidite to form a phosphoramidate linkage, substituted phosphoramidite linkage or suitable ester linkage thereto by routine methods; common functional groups include hydroxyl, sulfhydryl and amino.
- CPG Non, R. T.
- the solid support can be derivatized with a spacer linkage having a suitable functional group for forming the linkage to the vicinal reactive groups.
- spacer linkages are favored by some because the can act as a leash in distancing the solid support.
- the oligonucleotide synthesis reagent of the present invention can be used in any oligonucleotide synthesis method capable of utilizing an unprotected or protect -OH, -SH, -NH 2 include those methods using phosphoramidite reagents, the most widely used coupling chemistry for synthesis of oligonucleotides.
- other coupling chemistries are equally suitable for use, such as H-phosphonate chemistry [U.S. Patent No. 4,959,463; Froehler, B. C. et al., Nucleic Acids Research 14 , 5399 (1986)] and triester chemistry [Stec, W. J.
- oligonucleoside methylphosphonates Agarwal, S. & Goodchild, J., Tetrahedron Letters 28 , 3539 (1987)
- oligonucleoside phosphorothioates Beaucage, S.L. et al., J. Am.
- Synthesis on a solid support in the manner described herein also provides the option of generating a solid support tethered oligonucleotide.
- the support tethered oligonucleotide can be used in a variety of applications, such as DNA affinity extractions and reverse blot hybridization.
- the solid support is treated at room temperature with methylamine for about 1 hour to remove protecting groups from the heterocyclic amino groups of purine and pyrimidine bases contained in the nucleosides. This treatment releases only a minor amount of the oligonucleotide from the solid support, leaving the major portion of the oligonucleotide intact on the solid support. This provides biologically active oligonucleotide still attached to the solid support.
- the adjacent hydroxyl is protected by a group such as silyl.
- the oligonucleotides stay bound to the support after the treatment with, for example, CH 3 NH 2 .
- the silyl group can be removed , for example by tetrabutylammonium fluoride, thus liberating the hydroxyl; under basic conditions, the liberated hydroxyl attacks the adjacent phosphate, thus releasing the DNA from the solid support.
- cleaving an oligonucleotide from a solid support and reagents useful for cleaving oligonucleotides from a solid support to which they have been synthesized.
- the cleaving reagents and cleaving methods of the present invention have application in oligonucleotide synthesis systems wherein the oligonucleotide is synthesized in the step-wise addition of nucleoside from one hydroxyl or equivalent thereof of a vicinal diol pair or functional equivalent thereof.
- the oligonucleotide reagent has utility in cleaving oligonucleotides synthesized utilizing the oligonucleotide synthesis reagent of the present invention.
- the methods and cleaving reagents of the present invention are equally useful for cleaving oligonucleotides attached to solid supports through ester linkages and phosphate linkages.
- the cleaving methods of the present invention involve contacting the solid support bearing an oligonucleotide with a cleaving reagent of the present invention which includes a mixture of an amine selected from the group consisting of tertiary amines and secondary amines and a base selected from the group consisting of ammonium hydroxide and a primary amine.
- a cleaving reagent of the present invention which includes a mixture of an amine selected from the group consisting of tertiary amines and secondary amines and a base selected from the group consisting of ammonium hydroxide and a primary amine.
- a most preferred cleaving reagent is a solution of trimethylamine and methylamine.
- Additional secondary and tertiary amines suitable in the practice of the present invention include a variety of amines (bases with higher pKa's) such as triethylamine, n-propylamine, diisopropylamine, diisopropylethylamine, dimethylamine, diethylamine, piperidine, N-methylpiperidine and N-methylpyrrolidine.
- Preferred tertiary amines include trimethylamine, triethylamine, N-methylpyrrolidine, and diisopropylethylamine.
- the amount of ammonium hydroxide or primary amine and the amount of secondary amine or tertiary amine in the cleaving reagent can vary considerably.
- a ratio of methylamine or ammonium hydroxide to a secondary amine or tertiary amine is within the range of about 1:100 to about 100:1.
- Preferred concentration ratios and constituents are at least 9 parts of an aqueous methylamine solution to one part organic base having a basicity greater than ammonia.
- the most preferred cleaving reagent is 1 part of 40 wt% aqueous methylamine to 1 part of 3 - 25 wt% trimethylamine.
- aqueous methylamine is 40 wt% methylamine and typical concentrations of trimethylamine are in the range of 23 - 25 wt%.
- reference to a 1:1 v/v solution of methylamine and trimethylamine refers to a 1:1 v/v of a 40 wt% methylamine and about 24 wt% solution of trimethylamine.
- inorganic salts in cleaving reaction mixtures in order to enhance the cleaving kinetics.
- Such salts are typically salts of Li or Na, however, other cations are also suitable, including Mn and Mg. It has been discovered that when 0.5 M LiCl is utilized in a cleaving reagent of 1:1 methylamine:trimethylamine the amount of oligonucleotide cleaved after 15 minutes at 65°C increases from about 45% (in the absence of LiCl) to about 65 minutes.
- the rate of cleavage of the oligonucleotide from the solid support is affected by the nature of a bond between the solid support and the oligonucleotide synthesis site. More particularly, cleaving reactions involving oligonucleotides synthesized at vicinal sites bound to a solid support through ester linkages first involve hydrolysis of the ester linkage and the subsequent cleaving of the oligonucleotide from the vicinal site. Relative to the use of an aliphatic ester linkage to attach the binding group comprising a vicinal diol group to the solid support, use of phosphoramidate linkages facilitate faster cleavage kinetics.
- ester linkage (as described in deBear et al.), the ester is first hydrolyzed in about 5 minutes. This releases the oligonucleotide and linking group from the solid support. Detachment of the linking group from the oligonucleotide then requires additional incubation for about 4 hours at 65°C.
- the oligonucleotide is released from the vicinal reactive site and the phosphoramidate bond remains intact still attached to the support.
- This cleaving reaction can be accomplished utilizing a 1:1 v/v methylamine/trimethylamine cleaving reagent, (as described above) in approximately 90 minutes at 65°C.
- Significant amounts of the oligonucleotide are released from the linkage in a substantially less amount of time. That is, about 60% of the oligonucleotide will release after a reaction time of 15 minutes. This suggests that the phosphoramidate group of the linking group is accelerating the cleavage of the vicinal diol system from the nucleic acid.
- methylamine is about 10 times more reactive than ammonia in causing the hydrolysis of esters by virtue of its higher nucleophilicity. Since methylamine showed only about a twofold acceleration of kinetics, it was suspected that methylamine may be acting merely as a stronger base than ammonia, and not as a stronger nucleophile. This suggests that a stronger base might be needed to activate one hydroxyl of the vicinal diol system to make an attack on the adjacent hydroxyl.
- organic bases such as trimethylamine, triethylamine, n-propylamine, diisopropylamine, diisopropylethylamine, dimethylamine, diethylamine, piperidine, N-methylpiperidine and N-methylpyrrolidine were employed as a 1:1 mixture with methylamine.
- Methylamine was used to remove protecting groups from the heterocyclic amino groups of the oligonucleotide and to remove phosphate protecting group of the nucleosides in the oligonucleotide being synthesized.
- the cleavage time has been reduced to anywhere between 3 hours and 7 hours.
- the method of the present invention is suitable for cleaving oligonucleotides from virtually any type of vicinal diol or equivalent system used for oligonucleotide synthesis.
- Such systems may be described by the general formula in which S is a solid support, A is a linking group, X and Y are independently selected from the group consisting of -OH, -SH and -NH 2 , and R 9 , R 10 and R 11 are substituents which do not interfere with the oligonucleotide synthesis reaction.
- amino or thiol groups under certain conditions may provide advantages in terms of offering more stability and/or providing faster kinetics of release of nucleic acids from the solid support.
- two of R 9 , R 10 and R 11 together comprise a single moiety which (in combination with the carbons bearing the X and Y substituents) form a ring system.
- the kinetics of cleavage is faster with a uridine system as compared to a glycerol system, probably due to the free rotation around the glycerol C-C bond, which may be less conducive to the formation of the cyclic phosphate.
- the hydroxyls are locked in a cis-configuration which is conducive to the formation of the cyclic phosphate.
- a wide variety of other vicinal diol systems such as, for example, dihydroxycyclopentane, dihydroxycyclohexane and anhydroerythritol would also be suitable.
- the blocking moiety at a vicinal position of the oligonucleotide synthesis reagents of the present invention or other relevant solid support systems does not effect the overall cleavage time utilizing the reagents of the present invention. That is, the cleaving time does not have any bearing on whether the group was protected by, e.g., an acetyl group or a benzoyl group.
- the ester hydrolysis i.e., removal of the protecting group from the hydroxyl
- the attack of the liberated hydroxyl on the adjacent phosphorous of the synthesized oligonucleotide is a rate limiting slower step.
- the residue was purified using silica gel (pre-heated at 100-120° C) column-chromatography.
- the column was packed with the pre-heated silica gel in ethyl acetate/diisopropylamine, 95/5 v/v and eluted with ethyl acetate.
- the fractions which contained the product were collected, evaporated under reduced pressure and dried using high vacuum to provide the title compound as a white solid (0.6 g, 69% yield).
- the 1 H-NMR in CDCl 3 was as follows: ⁇ 1.19 (m,12H, 2CH(C H 3 ) 2 ), 1.97 (d, 3H, COC H 3 ), 2.46 (m, 4H, C H 2 C H 2 CN), 3.63 (m, 4H, C 5' C H 2 and 2 x CH(C H 3 ) 2 ), 3.77 (d, 6H, 2 x OC H 3 ), 4.13 (m, 1H, C 4' H ), 4.29 (m, 1H, C 3' H ), 5.0 (m, 1H, C 2' H ), 5.69 (m, 1H, C 1' H ), 6.50 (m, 1H, C 5 H ) and 6.75-7.72 (m, 14H, C 6 H and aromatic protons of DMT).
- the 31 P-NMR data in CDCl 3 was as follows: ⁇ 147.98 and 148.56 ppm.
- the resin was suspended in 10 ml of dry pyridine; DMAP (250 mg, 0.2 M) was added, followed by addition of Ac 2 O (2.5 ml, 2.5 M) to cap unreacted amino groups, and the reaction mixture was shaken at room temperature for 5 hrs.
- the resin was filtered, washed with dry pyridine (10 ml), CH 3 CN (10 ml) and diethyl ether (10 ml) and dried in vacuo for 2 hr.
- the hydroxy group loading of the solid support was found to be in the range of 200-250 ⁇ moles/g as determined by measurement (at A 500 nm) to calculate the amount of dimethoxytrityl cation released.
- Example 7 was suspended in dry acetonitrile (3 ml).
- the solid support was filtered, washed with dry CH 3 CN (2 x 3 ml) and then suspended in 3 ml of I 2 solution (0.3% I 2 in THF/pyridine/water - 93:5:2) and the reaction mixture was allowed to shake at room temperature for 30 min.
- the solid support was filtered, washed with THF (2 x 5 ml) and CH 3 CN ( 2 x 5 ml) and dried in vacuo for 1-2 hr.
- To the solid support was added 3 ml of 17% N-methylimidazole in THF, followed by addition of 3 ml of 10% Ac 2 O, 10% lutidine, 80% THF v/v; the reaction mixture was shaken at room temperature for 30 min.
- the solid support was filtered, washed with CH 3 CN (2 x 5 ml) and diethyl ether (2 x 5ml) and dried in vacuo. Loading of the uridine moiety was found to be 20.2 ⁇ moles/g as determined by measurement (at A 500 nm) of the amount of dimethoxytrityl cation released.
- The- solid support was filtered, washed with dry CH 3 CN (2 x 6 ml) and then suspended in 6 ml of I 2 solution (0.3 % I 2 in THF/pyridine/water - 93:5:2) and the reaction mixture was allowed to shake at room temperature for 30 min.
- the solid support was filtered, washed with THF (2 x 6ml), CH 3 CN (2 x 5 ml) and dried in vacuo for 2 hr.
- To the solid support was added 5 ml of (17% N-methylimidazole in THF), followed by addition of 5 ml of (10% Ac 2 O, 10% lutidine, 80% THP, v/v) and the reaction mixture shaken at room temperature for 30 min.
- the solid support was filtered, washed with CH 3 CN (2 x 10 ml) and diethyl ether (2 x 5 ml) and dried in vacuo. Loading of the uridine moiety was found to be in the range of 45-55 ⁇ moles/g as determined by measurement (at A 500 nm) of the amount of dimethoxytrityl cation released.
- Toyopearl-AF-amino-65-resin (1 g, 300 mmoles/g amino group, TosoHaas, Philadelphia, PA) was suspended in dry CH 3 CN (6 ml).
- 2'(3')-O-acetyl-2'(3')-O-(4,4'-dimethoxytrityl)-uridine-5'-O-( ⁇ -cyanoethyl-N,N-diisopropyl)-phosphoramidite 39.4 mg, 0.05 mmole
- 2 ml of 0.5 M solution of tetrazole in CH 3 CN was shaken at room temperature for 30 min.
- the solid support was filtered, washed with dry CH 3 CN (2 x 6 ml) and suspended in 6 ml of I 2 solution (0.3% I 2 in THF/pyridine/water - 93:5:2) and the reaction mixture was shaken at room temperature for 30 min.
- the solid support was filtered, washed with THF (2 x 6 ml) and CH 3 CN (2 x 5 ml) and dried in vacuo for 2 hr.
- To the solid support was added 5 ml of 17% N-methylimidazole in THF, followed by addition of 5 ml of a solution of 10% Ac 2 O, 10% lutidine and 80% THF, v/v; the reaction mixture was shaken at room temperature for 30 min.
- the solid support was filtered, washed with CH 3 CN (2 x 10 ml) and diethyl ether (2 x 5 ml) and dried in vacuo.
- the loading of uridine moiety on the resin was determined by measuring trityl release of a small aliquot in 2.5% dichloroacetic acid in dichloromethane at 500 nm in a spectrophotometer. The loading in this example was 45 ⁇ moles/g.
- CPG-LCAA resin (Sigma, 500 mg, ⁇ 60 ⁇ moles/g amino group) was suspended in dry CH 3 CN (3ml). 2'(3')-O-acetyl-2'(3')-O-(4,4'-dimethoxytrityl)-uridine-5'-O-( ⁇ -cyanoethyl-N,N-diisopropyl)-phosphoramidite (39.4 mg, 0.05 mmole) was added, followed by addition of 2 ml of a 0.5 M solution of tetrazole in CH 3 CN, and the reaction mixture was shaken at room temperature for 30 min.
- the solid support was filtered; washed with dry CH 3 CN (2 x 3 ml) and then suspended in 5 ml of I 2 (0.3% I 2 in THF/pyridine/water - 93:5:2) and the reaction mixture was shaken at room temperature for 30 min.
- the solid support was filtered, washed with THF (2 x 5 ml) and CH 3 CN (2 x 5 ml) and dried under vacuum for 2 hr.
- To the solid support was added 3 ml of 17% N-methylimidazole in THF, followed by addition of 3 ml of a solution of 10% Ac 2 O, 10% lutidine and 80% THF, v/v; the reaction mixture was shaken a room temperature for 30 min.
- the solid support was filtered, washed with CH 3 CN (2 x 10 ml) and diethyl ether (2 x 5 ml) and dried in vacuo.
- the loading of uridine on the support was 8.1 ⁇ moles/g as determined by A 500 nm of dimethoxytrityl cation released.
- the solid support was filtered, washed with pyridine (10 ml), MeOH (2 x 10 ml) and diethyl ether (10 ml) and dried in vacuo.
- the 2',3'-orthoester intermediate was hydrolysed to yield a mixture of 2',3'-acetates by treatment with 80% aqueous acetic acid (10 ml) at room temperature for 4 hr.
- the solid support was washed with MeOH (2 x 10 ml) and diethyl ether (10 ml) and dried in vacuo for 3 hr.
- the solid support (1 g) was suspended in dry CH 2 Cl 2 (10 ml), collidine (0.66 ml, 0.5 M) was added followed by addition of tetrabutylammonium perchlorate (0.17 g, 0.05 M) and dimethoxytrityl chloride (0.169 g, 0.05 M). The reaction mixture was shaken at room temperature for 1 hr. The solid support was filtered, washed with CH 2 Cl 2 (2 x 10 ml), MeOH (2 x 10 ml), diethyl ether (2 x 10 ml) and dried under vacuum for 3 hr. Loading of the solid support with uridine moiety was found to be 22.35 ⁇ moles/g as determined by A 500 nm of dimethoxy trityl cation released.
- the solid support was filtered, washed extensively with DMF (2 x 10 ml) and diethyl ether (2 x 5 ml) and dried under vacuum for 1 hr.
- the solid support was resuspended in dry pyridine (10 ml), DMAP (250 mg, 0.2 M) was added followed by the addition of acetic anhydride (2.5 ml, 2.5 M) and the reaction mixture was shaken at room temperature for 16 hrs.
- the resin was filtered, washed with CH 3 CN (2 x 10 ml) and diethyl ether (2 x 10 ml) and dried in vacuo for 2 hr.
- the loading of the resin with uridine moiety was found to be 48.8 ⁇ moles/g as determined by A 500 nm of dimethoxytrityl cation released.
- Fractogel 65F-resin (5 g dry weight, TosoHaas, Philadelphia, PA) was suspended in dry CH 3 CN (50 ml); 1',1'-carbonyldiimidazole (8.1 g, to make 1M solution) was added and the reaction mixture was shaken at room temperature for 4 hr.
- the solid support was filtered on a sintered glass funnel and washed with dry CH 3 CN (2 x 50 ml) and then resuspended in dry CH 2 Cl 2 (50 ml). 1,12-diaminododecane (10 g, to make 1M solution) was added and the resultant mixture was shaken at room temperature overnight (16-20 hr).
- n-Propylamine (4.1 ml, to make 1M solution) was added to the above mixture and the solution was shaken for an additional one hour.
- the resin was filtered and washed successively with dry CH 2 Cl 2 (5 x 50 ml), acetone (5 x 50 ml) and CH 3 CN (5 x 20 ml), the last filtrate wash showing a negative ninhydrin test; the resin was then washed with diethyl ether (2 x 20 ml) and dried under vacuum for about 3 hr.
- the amino group content on the solid support was found to be in the range of 300-400 ⁇ moles/g as determined by picric acid assay. [J. M. Stewart & J. D.
- Fractogel 65F-resin (5 g, dry weight) with 1,12-diaminododecane was carried out following the procedure of Example 16.
- the amino group content on the solid support was found to be 330 ⁇ moles/g as determined by picric acid assay.
- the solid support (1 g) was suspended in dry CH 2 Cl 2 (20 ml); p-nitrophenyl chloroformate (4.02 g, to make 1 M solution) was added followed by the addition of collidine (3.95 ml, to make 1.5 M solution) and the reaction mixture was shaken at room temperature overnight (16-20 hr). The solid support was filtered, washed with dry CH 2 Cl 2 (5 x 20 ml) and dried in vacuo for 2 hr.
- the solid support (1 g) was then suspended in dry CH 2 Cl 2 (10 ml). 2',3'-O-methoxyethylidene uridine (3 g, to make 1 M solution) was added followed by the addition of TEA (1.52 ml, to make 1 M solution) and the reaction mixture was shaken at room temperature for 24 hr. The reaction mixture was quenched by addition of n-propylamine (0.82 ml, to make 1 M solution) and the shaking was continued for another 1 hr. The solid support was filtered, washed with CH 2 Cl 2 (2 x 10 ml), CH 3 OH (2 x 10 ml) and diethyl ether (2 x 10 ml) and dried under vacuum for 2 hr.
- the 2',3'-orthoester intermediate was hydrolysed to yield a mixture of 2', 3'-acetates by treating the solid support with 80% aqueous acetic acid (10 ml) at room temperature for 4 hr.
- the solid support was filtered, washed with CH 3 OH (2 x 10 ml) and diethyl ether (10 ml) and dried in vacuo for 3 hr.
- the solid support (1 g) was then suspended in dry CH 2 Cl 2 (10 ml) and collidine (0.66 ml, 0.05 M) was added, followed by the addition of tetrabutylammonium perchlorate (0.17 g, 0.05 M) and dimethoxytrityl chloride (0.169 g, 0.5 M). The reaction mixture was shaken at room temperature for 1 hr. The solid support was filtered and washed with CH 2 Cl 2 (2 x 10 ml), CH 3 OH (2 x 10 ml) and diethyl ether (2 x 10 ml) and dried under vacuum for 3 hr. Loading of the. solid support with uridine moiety was found in the range of 20-30 mmoles/g as determined by A 500 nm of dimethoxytrityl cation released.
- the following describes the synthesis of an oligonucleotide and the subsequent cleaving of the oligonucleotide using a cleaving reagent of present invention.
- oligonucleotide of sequence: 5' TCC.ATG.GCA.ACT.GTC.AAG.GCA.CTG.GCT.CGT.AGC.CTA.CTG.GCT.TG A.CCG.TAA 3' (SEQ ID NO: 1) was assembled on the solid support of Example 15, using an Oligo-1000 DNA Synthesizer (Beckman Instruments, Brea, CA) and a standard synthetic protocol. After removing the last trityl group, the oligomer having a 3'-terminal uridine moiety was cleaved from the solid support by contacting the solid support with a solution of CH 3 NH 2 /Me 3 N at room temperature for 5 minutes.
- the solution was a 1:1, v/v, of 40 % aqueous methyamine and 24 wt% aqueous trimethylamine.
- the methylamine/trimethylamine solution containing the cleaved oligonucleotide was heated at 65°C for 3 hr.
- the reagent was evaporated to dryness using a speed vac concentrator.
- the residue was dissolved in double distilled water (200 ml) and the product was analyzed by capillary gel electrophoresis.
- the P/ACE electropherogram revealed a high quality product which was identified by comparing its elution time with same sequence independently synthesized on a conventional CPG-A support.
- the following describes the synthesis an oligonucleotide utilizing an oligonucleotide synthesis reagent of the present invention and cleaving reagents of the present invention.
- the solid support was dried and placed in a screw cap vial with 300 ml of MeNH 2 /Me 3 N (1:1, v/v as described in Example 18)) and heated at 65°C (water bath) for 90 min.
- the reagent (MeNH 2 /Me 3 N) was carefully decanted to another vial and evaporated to dryness using a speed vac concentrator.
- the residue was dissolved in double distilled water (500 ml) and the total A 260 nm was calculated.
- the product was analyzed by capillary gel electrophoresis.
- the P/ACE electropherogram revealed a good quality product. This product was identified by comparing its elution time with the same sequence independently synthesized on a regular CPG - T support.
- a 35 mer of the sequence 5' GAT.GCC.AGT.TCG.GTC.ATA. CAC.GTA.GTA.CTA.CGA.CC 3' was synthesized on the solid support of Example 11 and analyzed by capillary electrophoresis and by reverse phase HPLC as 5'-DMT oligonucleotides.
- RNA synthesis was 12 minutes. The last DMT group was. left on the oligonucleotide.
- RNA was deprotected and cleaved from the support using a 1:1 v/v solution of CH 3 NH 2 /(CH 3 ) 3 N for 4 hours at 65° C.
- the 1:1 solution was prepared using a 40% w/v aqueous methylamine solution and a 23%-25% by wt. aqueous trimethylamine solution.
- the 2'-protecting group was removed using 1.0 M tetrabutylammonium fluoride in THF for 15 hrs at room temperature.
- the oligoribonucleotide was desalted using a Nap-10 column (Pharmacia LKB Biotechnology, Piscataway, NJ).
- the resultant product after HPLC purification with the DMT group still in place was chemically and physically indistinguishable from a 10 mer synthesized using a commercially available CPG support.
- HPLC retention time 21.40 minutes. Conditions: C18 Microsorb MV (Rainin) 5 m particles, 4.6 mm x 25 cm. Bottle A: 0.1 M ammonium acetate (pH 6.9); Bottle B: Acetonitrile. Flow rate: 1 ml/min., 0-25 min gradient to 50% B, 25-27 min at 50% B. 27030 min gradient to 0% B, 30-32 min at 0% B.
- oligonucleotide RNA using an oligonucleotide synthesis reagent of the present invention and cleaving reagents of the present invention.
- the oligoribonucleotide was desalted using Sep-Pak (C18 cartridges from Millipore). Samples were lyophilized and analyzed by capillary electrophoresis on a Beckman P/ACE 2000.
- the capillary gel column was a Beckman Instruments U100P Urea Gel column, having a 37 cm overall length. A Beckman Tris-Borate, 7M Urea Buffer Gel Buffer Kit was used according to manufacturer's instructions.
- the absorbences of the oligoribonucleotides were in the range of 1.0 to 2 OD260 nm/ml, depending upon the quality and length of oligoribonucleotide. Injection was at 10 kV for 5 sec, while separation was at 11 kV for 30-60 min, depending upon the length.
- a 25 mer phosphorothioate of sequence 5' AGT.CAG.TCA.GTC.AGT.CAG.TCA.GTC.T 3' was synthesized on the solid support prepared in Example 15. The synthesis was performed using 0.2 ⁇ mole scale (loading 20 ⁇ moles/g) on an Oligo-1000. After the synthesis, the oligo was cleaved and deprotected using the MeNH 2 /Me 3 N (1:1) solution prepared in Example 20 for 4 hrs at 65°C. The sample was analyzed by HPLC and CE. HPLC retention time for the phosphorothioate having final DMT in place was 17.23 minutes under the conditions described in Example 20.
- CE retention time was 27.40 minutes for the phosphorothioate with the final DMT removed; the conditions were the same as in Example 21.
- the resultant product was chemically and physically indistinguishable from a 25 mer synthesized on commercially available CPG support.
- Example 22 Following the procedure described in Example 22, an oligophosphorothioate of sequence 5' AGT.CAG.TCA.GTC.AGT.CAG.TCA.GTC.T 3' (SEQ ID NO: 6) was synthesized on the solid support prepared in Example 12. Cleavage and deprotection was carried out with the MeNH 2 /Me 3 N (1:1 v/v) reagent of Example 20 for 2 hrs at room temperature. HPLC retention time of cleaved phosphorothioate having the final DMT in place was 17.22 minutes. The CE retention time for the phosphorothioate having the final DMT removed was 26.64. The conditions were the same as in Example 20 for HPLC and Example 21 for CE.
- a 15 mer of sequence 5' GAC.CAG.TAC.TCA.CGA 3' was assembled on CPG solid support from Example 15 and on a regular CPG-A support having a succinyl linker.
- the oligomer synthesized on the solid support of Example 15 was cleaved and deprotected with the CH 3 NH 2 /Me 3 N (1:1 v/v) solution prepared in Example 20 at 65°C for 4 hr.
- the oligomer synthesized on regular CPG-A support was cleaved and deprotected with ammonia at 65°C for 3 hr.
- the crude oligomers were subjected to enzyme digestion using phosphodiesterase I (Sigma) reconstituted with 5 ml of 40 mM Tris, 10 mM MgCl 2 , pH 7.5 (using 0.01 U per assay) and alkaline phosphatase (Sigma) from E. coli 200u/1.8 ml (using 0.2 u per assay).
- phosphodiesterase I Sigma
- alkaline phosphatase Sigma
- HPLC column C18 Ultrasphere (Beckman) 5 m particles, 4.6 mm x 25 cm.
- a 35 mer of sequence 5' GAT.GCC.AGT.TCG.GTC.ATA.CAC.GTA.GTA.CTA.CGA.CC 3' was synthesized on 0.2 ⁇ mole (loading 20 ⁇ moles/g) of CPG-T and on the prepared as described in Example 15.
- An Oligo-1000 DNA Synthesizer was utilized. After synthesis, cleavage and deprotection was done using the MeNH 2 /Me 3 N (1:1, v/v) as described in Example 20 for 4 hrs at 65°C. 1 OD 260 nm of sample was taken and mixed with 25 ml of snake venom phosphodiesterase and 2 ml of alkaline phosphatase.
- Example 15 Support T-CPG support A 9 9.14 8.65 C 9 8.63 9.26 G 8 8.20 8.90 T 9 9.02 8.91
- the oligonucleotide was cleaved from the solid support using MeNH 2 /Me 3 N (1:1, v/v) prepared as described in Example 20 at 65°C for 2 hr.
- the solid support was washed with distilled water (1 ml) and treated with (Co)triethylenetetramine(OH)H 2 O] +2 as described in Y. Matsumoto et al., Chemistry Letters 427 (1990).
- To a 0.05 M solution of [Co(triethylenetetramine)Cl 2 ]Cl (15.57 mg/l ml water) prepared according to the literature procedure [J. Chin and X.
- the solid support was then suspended in 1 ml of dry CH 2 Cl 2 . Collidine (1.3 ml, 0.001 M) was added, followed by the addition of tetrabutylammonium perchlorate (0.34 mg, 0.001 M) and dimethoxytrityl chloride (0.338 mg, 0.001 M). The reaction mixture was shaken at room temperature for 15 min. The solid support was filtered, washed with CH 2 Cl 2 (1ml), CH 3 OH (1 ml), diethyl ether (1 ml) and dried under vacuum for 1 hr.
- the solid support was then suspended in dry pyridine (1 ml). DMAP (1.25 mg, 0.001 M) was added, followed by the addition of acetic anhydride (12.5 ml, 0.0125 M) and the reaction mixture was shaken at room temperature for 15 min. The solid support was filtered, washed with dry CH 3 CN (1 ml) and diethyl ether (1 ml) and dried under vacuum for 30 min.
- the above synthesized phosphoramidite compounds were used to prepare oligonucleotide reagents of the present invention by attaching the phosphoramidites to a variety of solid supports to form a phosphoramidate linkage between an oligonucleotide synthesis site and the solid support.
- CPG and Fractogel having surface amino groups were reacted with the phosphoramidites.
- After treating this reaction product with 80% aqueous acetic acid the 2',3'-O-cyclic orthoester functionality opened to provide the oligonucleotide synthesis reagent of Example 11.
- glycerol type compound for use in preparing an oligonucleotide reagent suitable for synthesizing oligonucleotides which are subsequently cleaved from the solid support with cleaving reagents of the present invention.
- the above prepared compound was used to prepare a oligonucleotide synthesis reagent by reacting it with a standard succinylated CPG and Fractogel solid supports. Subsequently contacting the reacted solid support with 80% aqueous acetic acid at room temperature provide a reagent having a glycerol moiety attached to the support through an ester linkage.
- the 35-mer prepared using the solid support describe in Example 25 was utilized to study the effectiveness of various cleaving reagents.
- the solid support containing the synthesized 35-mer was contacted with each cleaving reagent for 30 minutes at 65°C and the % of oligonucleotide actually detached from the solid support was measured.
- ammonia, methylamine and trimethylamine are gases at standard temperature and pressure. They are readily available in aqueous solutions which were the forms utilized in preparing the solutions mentioned above. The solutions used were 40 wt% aqueous methylamine and 23 - 25 wt% trimethylamine. Thus, the 1:1 methylamine:trimethylamine are about 20 wt% methylamine and in about 12 wt% trimethylamine. Similarly, the ammonium hydroxide NH 4 OH was prepared using concentrated ammonium hydroxide or about 28 wt% to 30 wt%.
- the above described cleaving reagent containing methylamine and trimethylamine vary in concentrations of from about 0.4 wt% to 39.6 wt% methylamine and from about 0.24 wt% to about 22.5 wt% trimethylamine.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
Claims (27)
- Réactif de synthèse d'oligonucléotide comprenant une composition possédant la formule :dans laquelle R5 est de l'hydrogène ou un alkyle et R4 est un groupe protecteur phosphoreux ; et R3 est un groupe caractéristique en anneau possédant la structure :
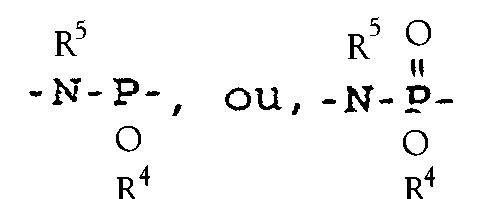 dans laquelle B est une base purique ou une base pyrimidique, chacun de X et Y est indépendamment choisi parmi O, S et NH et l'un de R1 et R2 est un groupe caractéristique de blocage pouvant être retiré par un réactif choisi parmi l'hydroxyde d'ammonium et le méthylamine, et l'autre est de l'hydrogène ou un groupe de protection hydroxy convenant pour protéger -OH, -SH ou NH2.
dans laquelle B est une base purique ou une base pyrimidique, chacun de X et Y est indépendamment choisi parmi O, S et NH et l'un de R1 et R2 est un groupe caractéristique de blocage pouvant être retiré par un réactif choisi parmi l'hydroxyde d'ammonium et le méthylamine, et l'autre est de l'hydrogène ou un groupe de protection hydroxy convenant pour protéger -OH, -SH ou NH2.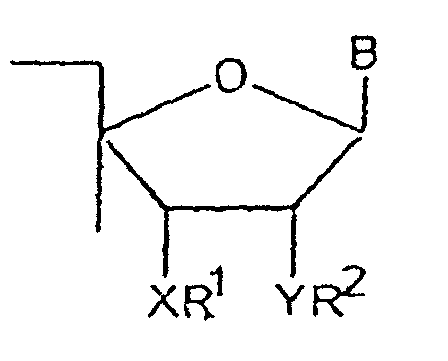
- Réactif d'oligonucléotide selon la revendication 1, dans lequel B est un uridyle.
- Réactif de synthèse d'oligonucléotide selon la revendication 1, dans lequel l'un de R1 et R2 est choisi parmi l'acétyle, le benzoyle et le FMOC.
- Réactif de synthèse d'oligonucléotide selon la revendication 1, dans lequel l'un de R1 et R2 est du diméthoxytrityle.
- Réactif de synthèse d'oligonucléotide selon la revendication 1, dans lequel le support solide est choisi parmi le verre à porosité contrôlée, les copolymères d'éthylène et d'acrylate, les copolymères d'éthylène et de méthacrylate, le polystyrène et ne nylon.
- Réactif de synthèse d'oligonucléotide comprenant un composé possédant la formule :dans laquelle SS est un support solide, chacun de X et Y est indépendamment choisi parmi O, S et NH, et l'un de R1 et R2 est un groupe alkylcarbonyle ou arylcarbonyle, et l'autre est de l'hydrogène ou un groupe de protection hydroxy.

- Réactif de synthèse d'oligonucléotide selon la revendication 6, dans lequel chacun de X et Y est O, et l'un de R1 et R2 est un acétyle, et l'autre est du diméthoxytrityle.
- Réactif de synthèse d'oligonucléotide selon la revendication 6, dans lequel le support solide est choisi parmi le verre à porosité contrôlée, les copolymères d'éthylène et d'acrylate, les copolymères d'éthylène et de méthacrylate, le polystyrène, le polypropylène et le nylon.
- Réactif de synthèse d'oligonucléotide comprenant ce qui suit :dans laquelle SS est un support solide, chacun de X et Y est O, et l'un de R1 et R2 est un groupe alkylcarbonyle ou arylcarbonyle et l'autre est du diméthoxytrityle.
- Utilisation d'un réactif de clivage d'oligonucléotide comprenant un premier composé choisi parmi le méthylamine et l'hydroxyde d'ammonium, et un deuxième composé choisi parmi des amines secondaires et des amines tertiaires, pour débloquer ou hydrolyser un site vicinal bloqué d'un réactif de synthèse d'oligonucléotide de la formule I (telle que définie dans la revendication 1).
- Utilisation d'un réactif de clivage d'oligonucléotide selon la revendication 10, dans lequel ledit premier composé est un méthylamine.
- Utilisation d'un réactif de clivage d'oligonucléotide selon la revendication 10, dans lequel ledit amine secondaire est choisi parmi le diméthylamine, le diisopropylamine, et le diéthylamine.
- Utilisation d'un réactif de clivage selon la revendication 10, dans lequel ledit amine tertiaire est choisi parmi une triméthylamine aqueuse, le N-méthylpyrrolidine, un triéthylamine, et un diisopropyléthylamine.
- Utilisation d'un réactif de clivage d'oligonucléotide aqueux selon la revendication 10, ledit réactif comprenant un méthylamine et un amine tertiaire choisi parmi une triméthylamine, un triéthylamine, et le N-méthylpyrrolidine.
- Utilisation d'un réactif de clivage d'oligonucléotide aqueux selon la revendication 14, comprenant en outre un alcool alkylique inférieur.
- Utilisation d'un réactif de clivage d'oligonucléotide aqueux selon la revendication 14, dans lequel ledit méthylamine est présent à une concentration située entre 4% par poids et 39,6% par poids et dans lequel ledit amine tertiaire est présent à une concentration située entre 0,24% par poids et 22% par poids.
- Utilisation d'un réactif de clivage d'oligonucléotide selon la revendication 16, dans lequel ledit amine tertiaire est une triméthylamine.
- Utilisation d'un réactif de clivage d'oligonucléotide selon la revendication 17, dans lequel ledit méthylamine est présent de 0% par poids à 19,6% par poids et dans lequel ladite triméthylamine est présente de 0,24% par poids à 19,2% par poids.
- Procédé pour cliver un oligonucléotide à partir d'un support solide, ledit procédé comprenant les étapes suivantes :prévoir un oligonucléotide relié à un support solide via un groupe caractéristique en anneau, ledit groupe caractéristique en anneau et le support solide étant représentés par la formule I (définie dans la revendication 1), ledit oligonucléotide étant relié audit groupe caractéristique en anneau de manière vicinale par rapport à un groupe réactif bloqué ; etmettre en contact ledit oligonucléotide avec un réactif comprenant un méthylamine et un deuxième composé choisi parmi des amines secondaires et des amines tertiaires.
- Procédé selon la revendication 19, dans lequel ledit oligonucléotide est relié audit groupe caractéristique en anneau de manière vicinale par rapport à un groupe réactif bloqué choisi parmi l'alkylcarbonyle et l'arylcarbonyl et dans lequel ladite étape de mise en contact comprend le déblocage dudit groupe réactif.
- Procédé selon la revendication 20, dans lequel ledit groupe caractéristique en anneau est relié audit support solide via une liaison choisie parmi le groupe caractéristique phosphate, le groupe caractéristique phosphoramidate, et le groupe caractéristique carbamate.
- Procédé selon la revendication 19, dans lequel ledit réactif comprend 4% par poids à 36% par poids de méthylamine et dans lequel ledit composé est une triméthylamine présente à une concentration située entre 2,4% par poids et 22,5% par poids.
- Procédé selon la revendication 21, dans lequel ladite liaison est choisie parmi le groupe caractéristique phosphoramidite et phosphoramidate.
- Procédé pour synthétiser un oligonucléotide, ledit procédé comprenant les étapes suivantes :prévoir un réactif de synthèse d'oligonucléotide selon la revendication 1, etcoupler séquentiellement les unités monomères nucléosides protégées afin de fournir un oligonucléotide.
- Procédé pour synthétiser un oligonucléotide selon la revendication 24, dans lequel ledit réactif de synthèse estdans laquelle SS est un support solide, chacun de X et Y est O, et l'un de R1 et R2 est un groupe alkylcarbonyle ou arylcarbonyle et l'autre est un groupe. de protection hydroxy.

- Procédé selon la revendication 24, dans lequel ledit réactif de synthèse est :dans laquelle SS est un support solide, chacun de X et Y est chacun O, et l'un de R1 et R2 est un groupe alkylcarbonyle ou arylcarbonyle et l'autre est du diméthoxytrityle.

- Utilisation d'un réactif de clivage d'oligonucléotide selon la revendication 26, dans lequel le réactif comprend en outre LiCl.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/636,113 US5869696A (en) | 1996-04-22 | 1996-04-22 | Universal solid supports and methods for their use |
| US636113 | 1996-04-22 | ||
| PCT/US1997/006648 WO1997040458A2 (fr) | 1996-04-22 | 1997-04-21 | Supports solides universels et leurs procedes d'utilisation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0843684A2 EP0843684A2 (fr) | 1998-05-27 |
| EP0843684B1 true EP0843684B1 (fr) | 2003-08-20 |
Family
ID=24550496
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP97922372A Expired - Lifetime EP0843684B1 (fr) | 1996-04-22 | 1997-04-21 | Supports solides universels et leurs procedes d'utilisation |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US5869696A (fr) |
| EP (1) | EP0843684B1 (fr) |
| JP (1) | JP2000500158A (fr) |
| DE (1) | DE69724218T2 (fr) |
| WO (1) | WO1997040458A2 (fr) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4503828B2 (ja) * | 1998-02-11 | 2010-07-14 | ユニバーシティー オブ ヒューストン | 光生成試薬を用いる化学反応および生化学反応のための方法および装置 |
| DE69814097T2 (de) * | 1998-12-02 | 2004-04-08 | Council Of Scientific And Industrial Research | Universale Polymere Träger zur Synthese von Oligonukleotiden |
| US6465628B1 (en) * | 1999-02-04 | 2002-10-15 | Isis Pharmaceuticals, Inc. | Process for the synthesis of oligomeric compounds |
| WO2001016166A2 (fr) | 1999-08-27 | 2001-03-08 | The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Polypeptides comprenant des domaines de recepteur d'interleukine-6 se liant a un ligand, acides nucleiques correspondants, anticorps, compositions, et leurs procedes d'utilisation |
| AU2001272714A1 (en) * | 2000-06-13 | 2001-12-24 | Proligo Llc | Universal solid supports for solid phase oligosynthesis and methods for their preparation and use |
| US6768005B2 (en) | 2000-12-20 | 2004-07-27 | Avecia Limited | Process |
| KR20040016826A (ko) * | 2000-12-05 | 2004-02-25 | 아베시아 리미티드 | 포스포로티오에이트 올리고뉴클레오티드의 제조방법 |
| US7211654B2 (en) | 2001-03-14 | 2007-05-01 | Regents Of The University Of Michigan | Linkers and co-coupling agents for optimization of oligonucleotide synthesis and purification on solid supports |
| CA2445838C (fr) * | 2001-04-30 | 2011-11-22 | Avecia Biotechnology Inc | Immobilisation d'oligonucleotides sur des supports solides |
| US20040006176A1 (en) * | 2001-11-29 | 2004-01-08 | Irm Llc, A Delaware Limited Liability Company | Nucleoside analog libraries |
| US7034147B2 (en) * | 2001-11-29 | 2006-04-25 | Irm Llc | Nucleoside analog libraries |
| GB0209539D0 (en) * | 2002-04-26 | 2002-06-05 | Avecia Ltd | Monomer Polymer and process |
| US20040242897A1 (en) * | 2002-07-31 | 2004-12-02 | Guzaev Andrei P. | Universal support media for synthesis of oligomeric compounds |
| US6653468B1 (en) | 2002-07-31 | 2003-11-25 | Isis Pharmaceuticals, Inc. | Universal support media for synthesis of oligomeric compounds |
| AU2003256857A1 (en) * | 2002-08-08 | 2004-02-25 | Dharmacon, Inc. | Short interfering rnas having a hairpin structure containing a non-nucleotide loop |
| US7615629B2 (en) * | 2002-12-31 | 2009-11-10 | Sigma-Aldrich Co. | Methods and compositions for the tandem synthesis of two or more oligonucleotides on the same solid support |
| US7202264B2 (en) | 2003-01-31 | 2007-04-10 | Isis Pharmaceuticals, Inc. | Supports for oligomer synthesis |
| US20050130736A1 (en) * | 2003-12-12 | 2005-06-16 | Lottofone, Inc. | Prepaid wagering card |
| US20090036660A1 (en) * | 2007-07-31 | 2009-02-05 | Joel Myerson | Methods and compositions for generating mixtures of nucleic acid molecules |
| US20090075840A1 (en) * | 2007-09-18 | 2009-03-19 | Joel Myerson | Methods And Compositions For Generating Mixtures Of Nucleic Acid Molecules |
| KR100886139B1 (ko) | 2007-11-13 | 2009-02-27 | 주식회사 삼천리제약 | 올리고뉴클레오타이드의 제조방법 |
| JP5438922B2 (ja) * | 2008-06-25 | 2014-03-12 | 日東電工株式会社 | 核酸の製造方法 |
| KR20160150051A (ko) * | 2015-06-18 | 2016-12-28 | 닛토덴코 가부시키가이샤 | Rna 올리고뉴클레오티드 절단 방법 |
| JP7286320B2 (ja) * | 2016-03-13 | 2023-06-05 | ウェイブ ライフ サイエンシズ リミテッド | ホスホラミダイト及びオリゴヌクレオチド合成のための組成物並びに方法 |
| US11873316B2 (en) | 2016-11-23 | 2024-01-16 | Wave Life Sciences Ltd. | Compositions and methods for phosphoramidite and oligonucleotide synthesis |
| CN111704644B (zh) * | 2020-08-18 | 2020-12-04 | 苏州金唯智生物科技有限公司 | 一种氨解液及氨解方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2068132A (en) * | 1933-04-19 | 1937-01-19 | Ig Farbenindustrie Ag | Production of monomethylamine and dimethylamine |
| US2547064A (en) * | 1946-11-29 | 1951-04-03 | Ici Ltd | Separation of methyl amines |
| US2657237A (en) * | 1948-08-06 | 1953-10-27 | Southern Production Company In | Process for the separation and recovery of methyl amines |
| US3271455A (en) * | 1961-09-22 | 1966-09-06 | Dow Chemical Co | Process for separating methylamines |
| DE3481060D1 (de) * | 1983-09-02 | 1990-02-22 | Molecular Biosystems Inc | Oligonukleotid-polymer-tragsystem. |
| US4552957A (en) * | 1984-02-17 | 1985-11-12 | Texaco Inc. | Method of separating primary amines from tertiary amines using non-polar hydrocarbon solvent with or without polyhydroxylic compound |
| US5189221A (en) * | 1990-11-19 | 1993-02-23 | Texaco Chemical Company | Amine separation process |
| GB9207380D0 (en) * | 1992-04-03 | 1992-05-13 | Ici Plc | Compounds |
| US5348868A (en) * | 1992-04-24 | 1994-09-20 | Beckman Instruments, Inc. | Methods and reagents for cleaving and deprotecting oligonucleotides |
| FR2707296B1 (fr) * | 1993-07-09 | 1995-09-29 | Genset Sa | Procédé de synthèse d'acides nucléiques sur support solide et composés utiles notamment comme support solide dans ledit procédé. |
-
1996
- 1996-04-22 US US08/636,113 patent/US5869696A/en not_active Expired - Lifetime
-
1997
- 1997-04-21 WO PCT/US1997/006648 patent/WO1997040458A2/fr active IP Right Grant
- 1997-04-21 DE DE69724218T patent/DE69724218T2/de not_active Expired - Lifetime
- 1997-04-21 JP JP9538250A patent/JP2000500158A/ja active Pending
- 1997-04-21 EP EP97922372A patent/EP0843684B1/fr not_active Expired - Lifetime
Non-Patent Citations (1)
| Title |
|---|
| SCHWARTZ ET AL: "A Universal Adapter for Chemical Synthesis of DNA or RNA on any Single Type of Solid Support", TETRAHEDRON LETTERS, vol. 36, no. 1, 2 January 1995 (1995-01-02), pages 27 - 30, XP004028936 * |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69724218T2 (de) | 2004-06-03 |
| WO1997040458A3 (fr) | 1997-12-04 |
| US5869696A (en) | 1999-02-09 |
| EP0843684A2 (fr) | 1998-05-27 |
| WO1997040458A2 (fr) | 1997-10-30 |
| JP2000500158A (ja) | 2000-01-11 |
| DE69724218D1 (de) | 2003-09-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0843684B1 (fr) | Supports solides universels et leurs procedes d'utilisation | |
| US6590093B1 (en) | Orthoester protecting groups | |
| RU2079508C1 (ru) | Способ соединения нуклеозидов 3'-5'-межнуклеотидным силильным звеном | |
| US5932718A (en) | Oligonucleotides having modified internucleoside linkages or terminal amino group | |
| US5614622A (en) | 5-pentenoyl moiety as a nucleoside-amino protecting group, 4-pentenoyl-protected nucleotide synthons, and related oligonucleotide syntheses | |
| AU777049B2 (en) | Xylo-LNA analogues | |
| EP2253639A1 (fr) | Kits et utilisations impliquant des oligonuclétides modifiées par LNA | |
| MXPA96004355A (en) | Oligonucleotides and used modified intermediaries in nucleic acids therapeuti | |
| KR20080059323A (ko) | 폴리뉴클레오티드 표지 시약 | |
| WO1995026972A1 (fr) | Oligonucleotides modifies et intermediaires utiles dans les therapies fondees sur l'utilisation d'acide nucleique | |
| AU767509B2 (en) | Method for deprotecting oligonucleotides | |
| US5623068A (en) | Synthesis of DNA using substituted phenylacetyl-protected nucleotides | |
| US6605708B1 (en) | Building blocks with carbamate internucleoside linkages and oligonucleotides derived therefrom | |
| US5674856A (en) | Modified oligodeoxyribonucleoditides | |
| US6531589B1 (en) | Base protecting groups and synthons for oligonucleotide synthesis | |
| US4419509A (en) | Process for de-cyanoethylating blocked nucleotides | |
| WO1996039414A1 (fr) | Groupes nouveaux de protection des bases pendant la synthese d'oligonucleotides | |
| US6509459B1 (en) | Base protecting groups and rapid process for oligonucleotide synthesis | |
| AU2002325599B2 (en) | Oligonucleotide analogues | |
| EP0839829A2 (fr) | Support solide universelle pour röactifs oligonucléotides | |
| WO1998033806A1 (fr) | Groupes de protection de bases et procede de synthese d'oligonucleotides | |
| GB2104523A (en) | Process for de-cyanoethylating blocked nucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): DE FR GB IT |
|
| 17P | Request for examination filed |
Effective date: 19980418 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: BECKMAN COULTER, INC. |
|
| 17Q | First examination report despatched |
Effective date: 19991004 |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): DE FR GB IT |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED. Effective date: 20030820 Ref country code: FR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20030820 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REF | Corresponds to: |
Ref document number: 69724218 Country of ref document: DE Date of ref document: 20030925 Kind code of ref document: P |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20040524 |
|
| EN | Fr: translation not filed | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 20160427 Year of fee payment: 20 Ref country code: GB Payment date: 20160427 Year of fee payment: 20 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R071 Ref document number: 69724218 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: PE20 Expiry date: 20170420 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF EXPIRATION OF PROTECTION Effective date: 20170420 |