EP0000009B1 - Procédé d'oxydation de quinine en quininone et quinidinone. - Google Patents
Procédé d'oxydation de quinine en quininone et quinidinone. Download PDFInfo
- Publication number
- EP0000009B1 EP0000009B1 EP78400019A EP78400019A EP0000009B1 EP 0000009 B1 EP0000009 B1 EP 0000009B1 EP 78400019 A EP78400019 A EP 78400019A EP 78400019 A EP78400019 A EP 78400019A EP 0000009 B1 EP0000009 B1 EP 0000009B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- quininone
- quinidinone
- quinine
- solution
- oxidation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 title claims description 22
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 title claims description 21
- SRFCUPVBYYAMIL-UHFFFAOYSA-N Quinidinon Natural products C1C(C(C2)C=C)CCN2C1C(=O)C1=CC=NC2=CC=C(OC)C=C21 SRFCUPVBYYAMIL-UHFFFAOYSA-N 0.000 title claims description 15
- SRFCUPVBYYAMIL-CKFHNAJUSA-N [(2r,4s,5r)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-(6-methoxyquinolin-4-yl)methanone Chemical compound C([C@H]([C@H](C1)C=C)C2)CN1[C@H]2C(=O)C1=CC=NC2=CC=C(OC)C=C21 SRFCUPVBYYAMIL-CKFHNAJUSA-N 0.000 title claims description 15
- 235000001258 Cinchona calisaya Nutrition 0.000 title claims description 11
- 229960000948 quinine Drugs 0.000 title claims description 11
- 238000007254 oxidation reaction Methods 0.000 title claims description 10
- 230000003647 oxidation Effects 0.000 title claims description 8
- 238000000034 method Methods 0.000 title claims description 5
- 238000006243 chemical reaction Methods 0.000 claims description 11
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 claims description 10
- 239000012965 benzophenone Substances 0.000 claims description 8
- 239000002904 solvent Substances 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 4
- 150000008366 benzophenones Chemical group 0.000 claims description 3
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 claims description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 claims description 2
- 239000003153 chemical reaction reagent Substances 0.000 claims description 2
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 2
- 229910052751 metal Inorganic materials 0.000 claims description 2
- 239000002184 metal Substances 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims description 2
- 238000005520 cutting process Methods 0.000 claims 1
- 150000008376 fluorenones Chemical class 0.000 claims 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- 239000000243 solution Substances 0.000 description 20
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 16
- 241000157855 Cinchona Species 0.000 description 12
- 239000002585 base Substances 0.000 description 10
- 150000002576 ketones Chemical class 0.000 description 10
- 229960001404 quinidine Drugs 0.000 description 8
- YLQWCDOCJODRMT-UHFFFAOYSA-N fluoren-9-one Chemical compound C1=CC=C2C(=O)C3=CC=CC=C3C2=C1 YLQWCDOCJODRMT-UHFFFAOYSA-N 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 229910052783 alkali metal Inorganic materials 0.000 description 4
- 150000001340 alkali metals Chemical group 0.000 description 4
- 239000006227 byproduct Substances 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- 235000021513 Cinchona Nutrition 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- 0 *C(c1ccccc1)c1ccccc1 Chemical compound *C(c1ccccc1)c1ccccc1 0.000 description 2
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 2
- 244000186140 Asperula odorata Species 0.000 description 2
- 235000008526 Galium odoratum Nutrition 0.000 description 2
- -1 aliphatic ketones Chemical class 0.000 description 2
- 229930013930 alkaloid Natural products 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000004064 recycling Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- LOUPRKONTZGTKE-FEBSWUBLSA-N (S)-[(2S,4S,5R)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-(6-methoxy-4-quinolinyl)methanol Chemical compound C1=C(OC)C=C2C([C@H](O)[C@H]3N4CC[C@]([C@H](C4)C=C)(C3)[H])=CC=NC2=C1 LOUPRKONTZGTKE-FEBSWUBLSA-N 0.000 description 1
- LOUPRKONTZGTKE-AFHBHXEDSA-N (r)-[(2r,4s,5r)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-(6-methoxyquinolin-4-yl)methanol Chemical compound C([C@H]([C@H](C1)C=C)C2)CN1[C@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-AFHBHXEDSA-N 0.000 description 1
- 229910000497 Amalgam Inorganic materials 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- AEFOLTVWSRMXMW-LWINWCPBSA-N Cinchonidinone Chemical compound C1=CC=C2C(C(=O)[C@@H]3CC4CCN3C[C@@H]4C=C)=CC=NC2=C1 AEFOLTVWSRMXMW-LWINWCPBSA-N 0.000 description 1
- AEFOLTVWSRMXMW-UHFFFAOYSA-N Cinchonidinone Natural products C1=CC=C2C(C(=O)C3CC4CCN3CC4C=C)=CC=NC2=C1 AEFOLTVWSRMXMW-UHFFFAOYSA-N 0.000 description 1
- AEFOLTVWSRMXMW-IXRXBBNISA-N Cinchoninone Chemical compound C1=CC=C2C(C(=O)[C@H]3CC4CCN3C[C@@H]4C=C)=CC=NC2=C1 AEFOLTVWSRMXMW-IXRXBBNISA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000002983 circular dichroism Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- DKRSEIPLAZTSFD-UHFFFAOYSA-N d-quinotoxine Natural products C12=CC(OC)=CC=C2N=CC=C1C(=O)CCC1CCNCC1C=C DKRSEIPLAZTSFD-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- MJGFBOZCAJSGQW-UHFFFAOYSA-N mercury sodium Chemical compound [Na].[Hg] MJGFBOZCAJSGQW-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- 150000008584 quinuclidines Chemical class 0.000 description 1
- 150000005838 radical anions Chemical class 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910001023 sodium amalgam Inorganic materials 0.000 description 1
- 238000009331 sowing Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- DKRSEIPLAZTSFD-LSDHHAIUSA-N viquidil Chemical compound C12=CC(OC)=CC=C2N=CC=C1C(=O)CC[C@@H]1CCNC[C@@H]1C=C DKRSEIPLAZTSFD-LSDHHAIUSA-N 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
- C07D453/04—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems having a quinolyl-4, a substituted quinolyl-4 or a alkylenedioxy-quinolyl-4 radical linked through only one carbon atom, attached in position 2, e.g. quinine
Definitions
- the present invention relates to an oxidation process of the Oppenauer type with a new reagent and its application to the manufacture of quininone and quinidinone, intermediates for the synthesis of quinidine.
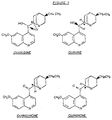
- the quininone and quinidinone differentiated by the asymmetric carbon isomerism at C s of the quinuclidine nucleus are generally two forms in equilibrium, which explains the phenomena of mutarotation in solution hitherto observed.
- the quininone formed is found in the presence of variable amounts of impurities, mainly: quinine, quinidine, epiquinine, epiquinidine, quinicine (quinotoxin ), which has the effect of reducing the reaction yield, the by-products formed having to be separated and purified before being recycled (it is noted in particular that the presence - even in small proportions - of epibases considerably reduces the crystallization yields of quinine and quinidine from their saturated solutions.
- impurities mainly: quinine, quinidine, epiquinine, epiquinidine, quinicine (quinotoxin )
- the reaction being carried out in enolic form it is necessary to introduce a base of PK sufficient to enolate all of the ketone forms contributing to the reaction.
- a base of PK sufficient to enolate all of the ketone forms contributing to the reaction.
- An amount of 2 to 7 equivalent moles has proved necessary in the various experiments described in the literature.
- the bases used, because of their PKI are the alcoholates, and mainly the sodium, potassium and aluminum alcoholates, methyl, ethyl, isopropyl and tert-butyl alcohols.
- the object of the invention is mainly to use a base having the ideal characteristics PK and compatibility which is an "in situ" derivative of the ketone used as proton acceptor.
- Ketyls are prepared by simple addition of the alkali metal to a solution of ketone in an appropriate aromatic solvent.
- a “Ketyl” obtained by interaction of an alkali metal with a diphenylketone, more particularly with benzophenone or fluorenone, will therefore be used as the quinine oxidant.
- the reaction of quinine and "Ketyl” is carried out in a solvent medium; said solvent is selected from aromatic hydrocarbons, C 6 -C 9 saturated cyclic hydrocarbons, C 6 -C 9 or aliphatic hydrocarbons corresponding to naphtha or gas oil cuts.
- the xylene solution is treated with water (20 ml), then extracted with 100 ml of sulfuric acid diluted to 20%.
- the cold sulfuric solution is neutralized by adding ammonia.
- the crystals that form are composed of quinidinone; as quininone and quinidinone are in equilibrium in the oil obtained, the precipitation of quinidinone has the consequence of shifting said equilibrium towards the production of quinidinone.
- the cold sulfuric solution is neutralized by adding ammonia. Decanting oil. It is separated and crystallized by seeding and appropriate treatment.
- the mixed solutions are kept at reflux for 5 hours.
- a “Ketyl” fluorenone solution is produced from 9 g of fluorenone in anhydrous toluene.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Quinoline Compounds (AREA)
Description
- La présente invention concerne un procédé d'oxydation de type Oppenauer par un réactif nouveau et son application à la fabrication de quininone et de quinidinone, intermédiaires de synthèse de la quinidine.
- Rabe et col. en 1909 [Ann. 364, 346] avaient déjà proposé une préparation de quininone et de quinidinone à partir de quinine ou de quinidine. L'oxydation s'effectuait avec de très faibles rendements.
- Woodward [JACS 67, 1425 (1945)] et Rabe [Ber 51, 446 (1918)] ont réalisé par la suite des synthèses permettant d'isoler un produit avec un bon rendement.
- Sueles les études plus récentes par dichroisme circulaire de la structuree des produits isolés a permis de définir que le produit isolé - le plus souvent à caractère huileux - et présentant en solution une mutarotation était en fait un mélange de deux isomères : la quininone et la quinidinona (par analogie avec la structure des deux alcaloïdes naturels du quinquina : la quinine et la quinidine).
- On a pu mettre en évidence, en particulier grâce aux travaux de synthèse de USKOKOVIC (USP 3 753 992 - 1973), que la seule forme cristalline, p.f. -98 à 101°C avait la structure de la quinidinone. La quininone ne pouvant être isolée que sous forme d'une huile.
- Les structures sont indiquées sur la figure 1..
- La quininone et la quinidinone que se différencient par l'isomérie au carbone asymétrique en Cs du noyau quinuclidine sont en général deux formes en équilibre, ce qui explique les phénomènes de mutarotation en solution jusqu'ici constatés.
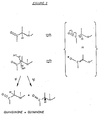
- En présence d'une base de PK suffisant à énoliser la cétone, l'équilibre entre la quininone et la quinidinone s'opère entre les formes indiquées à la figure 2.
- Ce mécanisme explique les différences de rendements obtenus et le manque de définition des caractères physicochimiques du produit communément appelé "quininone" qui n'est en fait que le mélange des deux stéréo-isomères présentant le phénomène de mutarotation en solution.
- La plupart du temps, le rendement d'oxydation s'évalue par dosage spectrophotométrique UV à 360 nm.
- L'oxydation de Oppenauer s'effectue dans le cas des alcanoïdes du quinquina seulement en présence de cétone de la famille des benzophénones.
- Cette cétone, jouant le rôle d'accepteur de protons, doit remplir un certain nombre de conditions strictes en ce qui concerne:
- - le potentiel d'oxydation,
- - la réactivité à basse température,
- - les contraintes d'encombrement stériques,
- - l'absence de réactions secondaires.
- De nombreux dérivés de benzophénone ont été testés et ont donné des résultats sensiblement équivalents. Les autres cétones aromatiques ou aliphatiques (acétone, cyclohexanone, acétophénone, etc.,) sont sans activité dans le cas des produits qui nous intéressent.
- Pour des raisons économiques, seules la benzophénone et la fluorénone sont pratiquement utilisées.
- Dans ces conditions, l'oxydation de la quinine et de la quinidine en quininone déjà décrite [WOODWARD, VENDLER, JACS 67, 1245 (1945)] [WARNHOFF, REYNOLDS, J. Org. Chem. 28, 1431 (1963)] ne s'effectue qu'en présence de 5 moles équivalents de cétone accepteur. Lorsque des quantités inférieures sont mises en présence, les conditions de l'équilibre OPPENAUER-MEERWEIN-POONDORFF sont réalisées et la quininone formée se trouve en présence de quantités variables d'impuretés, principalement: quinine, quinidine, épiquinine, épiquinidine, quinicine (quinotoxine), ce qui a pour effet de réduire le rendement de la réaction, les sous-produits formés devant être séparés et purifiés avant d'être recyclés (on note en particulier que la présence - même en faibles proportions - d'épibases diminue considérablement les rendements de cristallisation de la quinine et de la quinidine à partir de leurs solutions saturées.
- La réaction s'effectuant sous forme énolique, il est nécessaire d'introduire une base de PK suffisant à énoliser l'ensemble des formes cétoniques concourant à la réaction. Une quantité de 2 à 7 moles équivalents s'est avérée nécessaire dans les diverses expériences décrites dans la littérature. Les bases employées, du fait de leur PKI sont les alcoolates, et principalement les alcoolates de sodium, de potassium et d'aluminium, des alcools méthylique, éthylique, isopropylique et tert-butylique.
- Ces produits coûteux, dangereux d'emploi, présentent de plus l'inconvénient de libérer un alcool primaire en quantité importante.
- La présence de cet alcool a deux inconvénients principaux:
- ―entrant en réaction, il provoque des réductions parasites de la quininone formée en sous-produits secondaires gênants (déplacement de l'equilibre vers les bases et épibases);
- -il nécessite des traitements de purification coûteux pour le recyclage des solvants (distillation fractionée).
- L'objet de l'invention est principalement d'utiliser une base présentant les caractéristiques idéales de PK et de compatibilité qui soit un dérivé "in situ" de la cétone utilisée comme accepteur de protons.
- En effet, on connâit des anions radicalaires formés par addition d'un atome de métal alcalin (Me) sur une cétone de la série des benzophénones [BENT, HARRISSON, JACS 66, 969 (1944)].
- Il s'agit d'un anion radiculaire présentant une tautomérie entre deux formes principples:

- Pour la fluorénone, l'équilibre peut se représenter de faton analogue:

- Les "Ketyl" sont préparés par simple addition du métal alcalin à une solution de cétone dans un solvant aromatiques approprié.
- On peut également faire réagir un amalgame (par exemple un amalgame de sodium).
- On utilisera donc comme oxydant de la quinine un "Kétyl" obtenu par interaction d'un métal alcalin sur une diphénylcétone, plus particulièrement sur Ia benzophénone ou la fluorénone.
- On constate que l'alcaldide base, mis en présence de la solution de "Kétyl",, réagit très rapidement pour donner la forme cétonique désirée avec un rendement quantitatif. On note l'absence de sous-produits et d'impuretés dans le milieu réactionnel.
- Le réaction de la quinine et du "Kétyl" est réalisée dans un milieu solvant; ce solvant est choisi parmi les hydrocarbures aromatiques en C6―C9, les hydrocarbures cycliques saturés en C6―C9 ou les hydrocarbures aliphatiques correspondant à des coupes naphta ou gas-oil.
- Les avantages de cette invention sont multiples:
- - obtention avec un excellent rendement de la cétone désirée,
- - simplification de la réaction qui s'opère dans des conditions optimales de durée et de température,
- - remplacement d'un alcoolate coûteux et dangereux par l'emploi d'un métal alcalin (principalement de sodium) d'usage répandu dans l'industrie chimique et bon marché,
- - élimination des problèmes de recyclage de solvants, car il ne se forme plus une mole d'alcool aliphatique par mole d'alcoolate engagé.
- L'ensemble de ces propriétés a permis, comme on le verra dans les exemples décrivant ci-après, à titre non limitatif, les applications de l'invention, d'obtenir dans de bonnes conditions économiques la quininone et la quinidinone à partir de mélanges d'épibases considérées jusqu'ici comme des sous-produits fatals de l'hémisynthèse des alcaloïdes du quinquina (en particulier de la quinidine).
- On met en suspension 2,4 g de sodium dans 40 ml de xylène anhydre au reflux, afin d'obtenir une bonne dispersion.
- Après refroidissement à 90°C, on introduit lentement 36 g de benzophénone anhydre.
- La solution prend alors la couleur bleu-vert, caractéristique du "Kétyl" (présence d'électrons).
- Dans un second réacteur, on dissout l'ébullition 13 g de quinine base anhydre dans 50 ml de xylène.
- On coule cette solution dans la solution précédente. Après 60 minutes au reflux, la réaction est complète.
- Lâ solution xylénique est traitée à l'eau (20 ml), puis extraite par 100 ml d'acide sulfurique dilué à 20%.
- On neutralise par addition d'ammoniaque la solution sulfurique froide.
- Il se sépare une huile qui cristallise lentement après ensemencement.
- Les cristaux qui se forment sont composés de quinidinone; comme la quininone et la quinidinone sont en équilibre dans l'huile obtenue, la précipitation de la quinidinone a pour conséquence de déplacer ledit équilibre vers la production de quinidinone.
- Poids obtenu: 12,2 g (94% de la théorie) de quinidinone
- Chromatographie en couches minces
- Eluant acétone 80/DMF 20, 1 tache, Rf: 0,8
- On met en suspension 24 g de sodium dans 40 ml de toluène anhydre, en chauffant, pour obtenir par fusion une fine division du métal.
- En parallèle, on dissout 360 g de benzophénone anhydre dans 450 ml de toluène sec. Ces deux solutions sont mélangées et maintenues au reflux pendant 15 minutes.
- 130 g de quinine base anhydre sont dissous à l'ébullition dans 500 ml de toluène anhydre. Cette solution est versée à chaud dans la solution de benzophénone "Kétyl". Après 30 minutes au reflux, la réaction est complète.
- La solution toluénique refroidie à 50°C est lavée à l'eau, puis extraite par 700 ml d'acide sulfurique dilué.
- La solution sulfurique froide est neutralisée par addition d'ammoniaque. Une huile décante. Elle est séparée et cristallisée par ensemencement et traitement approprié.
- Poids d'huile obtenu: 124 g de quininone, p.f. 102°C
- CCM 1 tache, Rf 0,8
- A une solution de fluorénone "Kétyl," préparée à partir de 180 g de fluorénone et de 130 g de sodium dans le xylène anhydre, on ajoute une solution de 65 g de mélange, de composition suivante:

- Les solutions mélangées sont maintenues au reflux pendant 5 heures.
- La réaction est alors complète.
- On obtient après traitement approprié 55 g d'un produit contenant:
- quininone + quinidinone: 39 g
- cinchoninone + cinchonidinone: 16 g
- On réalise une solution de fluorénone "Kétyl" à partir de 9 g de fluorénone dans le toluène anhydre.
- A cette solution, on ajoute à chaud une solution de 37 g de quinidine base anhydre dans 150 ml de toluène anhydre.
- Après 15 minutes de reflux, la réaction est complète.
- Après traitement approprié, on obtient 36 g de quinidinone pure (CCM) (97% de la théorie).
Claims (3)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR7718398 | 1977-06-15 | ||
| FR7718398A FR2394545A1 (fr) | 1977-06-15 | 1977-06-15 | Procede de fabrication de quininone et de quinidinone, intermediaires de synthese de la quinidine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0000009A1 EP0000009A1 (fr) | 1978-12-20 |
| EP0000009B1 true EP0000009B1 (fr) | 1980-07-23 |
Family
ID=9192140
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78400019A Expired EP0000009B1 (fr) | 1977-06-15 | 1978-06-14 | Procédé d'oxydation de quinine en quininone et quinidinone. |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP0000009B1 (fr) |
| DE (1) | DE2860032D1 (fr) |
| FR (1) | FR2394545A1 (fr) |
| IE (1) | IE46935B1 (fr) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1165604B (de) * | 1957-12-18 | 1964-03-19 | Chininfabrik Braunschweig Buch | Verfahren zur sterischen Umlagerung von Chinaalkaloiden |
| FR2332278A1 (fr) * | 1975-11-19 | 1977-06-17 | Nativelle Sa Ets | Procede de transformation stereochimique des alcaloides du quinquina |
| FR2332279A1 (fr) * | 1975-11-19 | 1977-06-17 | Nativelle Sa Ets | Procede d'oxydation des alcaloides du quinquina |
-
1977
- 1977-06-15 FR FR7718398A patent/FR2394545A1/fr not_active Withdrawn
-
1978
- 1978-06-14 EP EP78400019A patent/EP0000009B1/fr not_active Expired
- 1978-06-14 DE DE7878400019T patent/DE2860032D1/de not_active Expired
- 1978-06-15 IE IE120478A patent/IE46935B1/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP0000009A1 (fr) | 1978-12-20 |
| DE2860032D1 (en) | 1980-11-13 |
| IE781204L (en) | 1978-12-15 |
| IE46935B1 (en) | 1983-11-02 |
| FR2394545A1 (fr) | 1979-01-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CH616161A5 (fr) | ||
| EP0000009B1 (fr) | Procédé d'oxydation de quinine en quininone et quinidinone. | |
| EP0001029B1 (fr) | Procédé de préparation d'oximes en 3 de stéroides | |
| CA1231939A (fr) | PROCEDE DE PREPARATION DE DERIVES DE LA SERIE DE LA 17.alpha.-HYDROXY 19-NOR-PROGESTERONE | |
| EP0013995B1 (fr) | Procédé pour la préparation de cétones macrocycliques acétyleniques | |
| CH642373A5 (fr) | Compose pyrannique, procede pour sa preparation et son utilisation a titre d'intermediaire de synthese. | |
| EP0294258A1 (fr) | Dérivés d'hydrazine, procédé d'obtention et compositions pharmaceutiques les contenant | |
| FR2510991A1 (fr) | Procede pour la preparation de methacrylamides et d'acrylamides n-substitues | |
| EP0184572B1 (fr) | Procédé de préparation d'acides alpha-hydroxy-alcanoiques | |
| FR2759079A1 (fr) | Procede de preparation d'anthraquinones substituees et application a la preparation de rheines | |
| EP0000302B1 (fr) | Nouveau procédé de préparation de la quinidine. | |
| EP0362309B1 (fr) | (ethylenedioxo-3,3 cyclohexyl)-4 acetophenone et derives de ce compose, procedes pour leur preparation et utilisation de ces composes | |
| CH648483A5 (fr) | Composition pharmaceutique contre l'arthrite rhumatoide. | |
| CH623818A5 (en) | Process for producing derivatives of thiochroman | |
| EP0082782B1 (fr) | Procédé de préparation d'halogénoacétals éthyléniques | |
| BE884977A (fr) | Nouveau n-phenyl-benzamides utiles dans le traitement de l'inflammation et de la douleur des mammiferes | |
| EP0184573A1 (fr) | Procédé de préparation d'acides alpha-hydroxy-alcanoiques | |
| FR2494696A1 (fr) | Nouveau procede de preparation de steroides 3-amines et leurs sels | |
| CH662571A5 (fr) | Procede de preparation de derives de la prednisone et produits intermediaires de ce procede. | |
| CH330478A (fr) | Procédé de préparation de nouveaux thiodérivés de colchicéines | |
| BE561749A (fr) | ||
| CH421098A (fr) | Procédé de préparation de stéroïdes fluorés | |
| BE525128A (fr) | ||
| BE837643A (fr) | Composes pour la reduction ou la prevention des dommages causes au foie par l'alcool | |
| BE460230A (fr) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): BE CH DE FR GB LU NL SE |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): BE CH DE FR GB LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 2860032 Country of ref document: DE Date of ref document: 19801113 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19810614 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19810615 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19810630 Ref country code: CH Effective date: 19810630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19820101 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee |
Effective date: 19820101 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19820501 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19830331 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881117 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GR Payment date: 19910619 Year of fee payment: 5 |
|
| EUG | Se: european patent has lapsed |
Ref document number: 78400019.2 Effective date: 19820119 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |