-
Gebiet der
Erfindung
-
Die
vorliegende Erfindung betrifft neue Verbindungen mit negativen Retinoidhormon-
und/oder Retinoidantagonist-artigen biologischen Aktivitäten. Insbesondere
betrifft die Erfindung 4-Aryl-substituierte Benzopyran-Derivate.
Diese neuen Verbindungen besitzen Retinoidantagonist-artige Aktivität und sind
nützlich
zur Behandlung oder Prävention
von durch Retinoid und Vitamin A und Vitamin A-Vorläufer induzierte
Toxizität
in Säugetieren
und als Zusatz zur Behandlung von Säugetieren mit Retinoiden zur
Prävention
oder Linderung ungewollter oder ungewünschter Nebenwirkungen. Die
Erfindung betrifft ferner die Verwendung von negativen Retinoidhormonen
zur Erhöhung
der biologischen Aktivitäten
anderer Retinoide und Steroidhormone und Inhibierung der Grundaktivität von freien
Retinolsäurerezeptoren.
-
Hintergrund
der Erfindung
-
Verbindungen,
die Retinoid-artige Aktivität
besitzen, sind allgemein fachbekannt und werden in zahlreichen US-
und anderen Patenten und in wissenschaftlichen Veröffentlichungen
beschrieben. Es ist allgemein bekannt und fachlich akzeptiert, daß Retinoid-artige
Aktivität
nützlich
zur Behandlung von Säugetieren
ist, einschließlich
Menschen, um die Symptome zu heilen oder zu lindern, die mit zahlreichen
Krankheiten und Zuständen
verbunden sind.
-
Es
ist bekannt, daß Retinoide
(Vitamin A und seine Derivate) breite Aktivitäten, einschließlich Wirkungen
auf die Zellproliferation und -differenzierung, in einer Vielzahl
biologischer Systeme besitzen. Diese Aktivität hat Retinoide nützlich in
der Behandlung einer Vielzahl von Krankheiten gemacht, einschließlich dermatologischer
Störungen
und Krebs. Der Stand der Technik hat eine große Anzahl chemischer Verbindungen
entwickelt, die Retinoid-artige biologische Aktivität besitzen,
und es existiert umfangreiche Patent- und chemische Literatur, die
solche Verbindungen beschreibt. Die relevante Patentliteratur schließt die US-PSen
4,980,369, 5,006,550, 5,015,658, 5,045,551, 5,089,509, 5,134,159,
5,162,546, 5,234,926, 5,248,777, 5,264,578, 5,272,156, 5,278,318,
5,324,744, 5,346,895, 5,346,915, 5,348,972, 5,348,975, 5,380,877,
5,399,561, 5,407,937 (überschrieben
auf den gleichen Rechtsnachfolger wie die vorliegende Anmeldung)
und darin zitierte Patente und Veröffentlichungen ein, die insbesondere
Chroman-, Thiochroman- und 1,2,3,4-Tetrahydrochinolin-Derivate beschreiben
oder betreffen, die Retinoid-artige biologische Aktivität besitzen.
Zusätzlich
sind mehrere Anmeldungen anhängig,
die auf den Rechtsnachfolger der folgenden Anmeldung übertragen
wurden, und die auf weitere Verbindungen mit Retinoid-artiger Aktivität gerichtet
sind.
-
Die
US-PSen 4,740,519 (Shroot et al.), 4,826,969 (Maignan et al.), 4,326,055
(Loeliger et al.), 5,130,335 (Chandraratna et al.), 5,037,825 (Klaus
et al.), 5,231,113 (Chandraratna et al.), 5,324,840 (Chandraratna),
5,344,959 (Chandraratna), 5,130,335 (Chandraratna et al.), EP-A-0
176 034 (Wuest et al.), EP-A-0 350 846 (Klaus et al.), EP-A-0 176
032 (Frickel et al.), EP-A-0 176 033 (Frickel et al.), EP-A-0 253
302 (Klaus et al.), EP-A-0 303 915 (Bryce et al.), GB-A-2190378 (Klaus
et al.), DE-A-3715955 (Klaus et al.), DE-A-3602473 (Wuest et al.)
und die Artikel J. Amer. Acad. Derm. 15: 756–764 (1986) (Sporn et al.),
Chem. Pharm. Bull. 33: 404–407
(1985) (Shudo et al.), J. Med. Chem. 31: 2182–2192 (1988) (Kagechika et
al.), Chemistry and Biology of Synthetic Retinoids, CRC Press Inc.
1990, S. 334–335,
354 (Dawson et al.) beschreiben oder betreffen Verbindungen, die
eine Tetrahydronaphthyl-Einheit einschließen und Retinoid-artige oder
verwandte biologische Aktivität
besitzen. US-PS 4,391,731 (Boller et al.) beschreibt Tetrahydronaphthalin-Derivate, die nützlich in
Flüssigkristallzusammensetzungen
sind.
-
Ein
Artikel von Kagechika et al., J. Med. Chem. 32: 834 (1989), beschreibt
bestimmte 6-(3-Oxo-1-propenyl)-1,2,3,4-tetramethyl-1,2,3,4-tetrahydronaphthalin-Derivate
und verwandte Flavon-Verbindungen mit Retinoid-artiger Aktivität. Die Artikel
von Shudo et al., Chem. Pharm. Bull. 33: 404 (1985), und von Jetten
et al., Cancer Research 47: 3523 (1987), beschreiben oder betreffen
weitere 3-Oxo-1-propenyl-Derivate
(Chalcon-Verbindungen) und ihre Retinoid-artige oder verwandte biologische
Aktivität.
-
Unglücklicherweise
verursachen Verbindungen mit Retinoidartiger Aktivität (Retinoide)
ebenfalls eine Anzahl ungewünschter
Nebenwirkungen bei therapeutischen Dosisniveaus, einschließlich Kopfschmerz,
Teratogenese, mukokutane Toxizität,
muskoskeletale Toxizität,
Dyslipidämien,
Hautreizung, Kopfschmerz und Lebertoxizität. Diese Nebenwirkungen beschränken die
Akzeptanz und den Nutzen von Retinoiden zur Behandlung von Krankheit.
-
Es
ist jetzt allgemeines Fachwissen, daß zwei Haupttypen von Retinoidrezeptoren
in Säugetieren
(und anderen Organismen) existieren. Die zwei Haupttypen oder -familien
von Rezeptoren werden als die RARs bzw. RXRs bezeichnet. Innerhalb
jedes Typs gibt es Untertypen: in der RAR-Familie werden die Untertypen als
RAR-α, RAR-β und RAR-γ bezeichnet,
in RXR sind die Untertypen: RXR-α,
RXR-β und
RXR-γ. Beide
Familien von Rezeptoren sind Transkriptionsfaktoren, die voneinander
auf Basis ihrer Ligandenbindungsspezifitäten unterschieden werden können. All-trans-RA
(ATRA) bindet und aktiviert eine Klasse von Retinolsäurerezeptoren
(RARs), die RAR-α,
RAR-β und
RAR-γ einschließt. Ein
unterschiedlicher Ligand, 9-cis-RA (9C-RA), bindet und aktiviert
sowohl die RARs als auch die Mitglieder der Retinoid-X-Rezeptor-(RXR)-Familie.
-
Es
ist ebenfalls fachlich etabliert, daß die Verteilung der zwei Retinoidrezeptor-Haupttypen
und der verschiedenen Untertypen nicht gleichförmig in den verschiedenen Geweben
und Organen von Säugetierorganismen
ist. Außerdem
ist es allgemein fachlich akzeptiert, daß viele ungewollte Nebenwirkungen
von Retinoiden durch einen oder mehrere der RAR-Rezeptoruntertypen
vermittelt werden. Entsprechend wird unter Verbindungen mit Agonist-artiger
Aktivität
an Retinoidrezeptoren die Spezifität oder Selektivität für eine(n)
der Haupttypen oder -familien und sogar Spezifität oder Selektivität für einen
oder mehrere Untertypen innerhalb einer Familie von Rezeptoren als
eine wünschenswerte
pharmakologische Eigenschaft betrachtet.
-
Erst
vor kurzem wurden auf diesem Gebiet Verbindungen entwickelt, die
an RAR-Rezeptoren ohne Auslösen
der Reaktion oder Reaktionen binden, die durch Agonisten der gleichen
Rezeptoren ausgelöst
werden. Die Verbindungen oder Mittel, die an RAR-Rezeptoren ohne
Auslösen
einer "Retinoid"-Reaktion binden, können deshalb
die Aktivität
von RAR-Agonisten in biologischen Tests und Systemen blockieren
(in geringerem oder höherem
Ausmaß).
Insbesondere beschreibt in bezug auf die wissenschaftliche und Patentliteratur auf
diesem Gebiet WO 94/14777 bestimmte heterocyclische Carbonsäure-Derivate,
die an RAR-Retinoidrezeptoren binden und gemäß der Anmeldung nützlich zur
Behandlung bestimmter Krankheiten oder Zustände sein sollen, wie z.B. Akne,
Psoriasis, rheumatoide Arthritis und virale Infektionen. Eine ähnliche
Offenbarung wird im Artikel von Yoshimura et al. gemacht, J. Med.
Chem. 38: 3163–3173
(1995). Kaneko et al., Med. Chem. Res. 1: 220–225 (1991); Apfel et al.,
Proc. Natl. Acad. Sci. USA 89: 7129–7133 Augusty 1992 Cell Biology; Eckhardt
et al., Toxicology Letters 70: 299–308 (1994); Keidel et al.,
Molecular and Cellular Biology 14: 287–298 (1994); und Eyrolles et
al., J. Med. Chem. 37: 1508–1517
(1994), beschreiben Verbindungen, die Antagonist-artige Aktivität an einem
oder mehreren der RAR-Retinoid-Untertypen besitzen.
-
Zusätzlich zu
unerwünschten
Nebenwirkungen der Therapie mit Retinoidverbindungen tritt gelegentlich
ein ernsthafter medizinischer Zustand auf, der durch eine Vitamin
A- oder Vitamin A-Vorläufer-Überdosis verursacht
wird, was entweder aus der übermäßigen Einnahme
von Vitaminergänzungsmitteln
oder dem Verzehr der Leber bestimmter Fische und Tiere resultiert,
die hohe Mengen des Vitamins enthalten. Die mit dem Hypervitaminose
A-Syndrom beobachteten chronischen oder akuten Toxizitäten schließen Kopfschmerz, Hautablösung, Knochentoxizität, Dyslipidämien etc.
ein. In den vergangenen Jahren ist es offensichtlich geworden, daß die mit
Vitamin A-Analoga,
d.h. Retinoiden, beobachteten Toxizitäten im wesentlichen diejenigen des
Hypervitaminose-A-Syndroms wiederholen, was eine gemeinsame biologische
Ursache nahelegt, d.h. RAR-Aktivierung. Diese Toxizitäten werden
derzeit hauptsächlich
durch stützende
Maßnahmen
und durch Verzichten von weiterem Kontakt mit dem verursachenden
Mittel behandelt, seien es Leber, Vitaminergänzungsmittel oder Retinoide.
Während
sich einige der Toxizitäten
im Verlauf der Zeit auflösen,
sind andere (z.B. vorzeitiger Epiphysenschluß) permanent.
-
Allgemein
gesprochen sind spezifische Gegengifte die beste Behandlung für Vergiftung
durch pharmakologische Mittel, aber nur etwa zwei Dutzend Chemikalien
oder Klassen von Chemikalien aus den Tausenden, die es gibt, besitzen
spezifische bekannte Gegengifte. Ein spezifisches Gegengift würde offensichtlich
von Wert in der Behandlung von Hypervitaminose A und Retinoidtoxizität sein.
Da zunehmend wirksame Retinoide klinisch verwendet werden, könnte ein
spezifisches Gegengift für
Retinoidvergiftung tatsächlich
lebensrettend sein.
-
Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung umfaßt
Verbindungen der Formel (1):
worin R
2 H,
Niederalkyl mit 1 bis 6 Kohlenstoffen, F, Cl, Br, I, CF
3,
fluorsubstituiertes Alkyl mit 1 bis 6 Kohlenstoffen, OH, SH, Alkoxy
mit 1 bis 6 Kohlenstoffen oder Alkylthio mit 1 bis 6 Kohlenstoffen
ist;
m eine ganze Zahl mit einem Wert von 0 bis 3 ist;
R
3 H, Niederalkyl mit 1 bis 6 Kohlenstoffen
oder F ist;
o eine ganze Zahl mit einem Wert von 0 bis 3 ist;
s
eine ganze Zahl mit einem Wert von 1 bis 3 ist;
R
8 H,
eine Alkyl-Gruppe mit 1 bis 10 Kohlenstoffen oder Trimethylsilylalkyl,
worin die Alkyl-Gruppe 1 bis 10 Kohlenstoffe aufweist, oder eine
Cycloalkyl-Gruppe mit 5 bis 10 Kohlenstoffen ist oder R
8 Phenyl
oder Niederalkylphenyl ist;
R
15 unabhängig H,
F, Cl, Br, I, NO
2, N(R
8)
2, COR
8, NR
8CON(R
8)
2,
OCOR
8, OR
8, CN,
eine Alkyl-Gruppe mit 1 bis 10 Kohlenstoffen, eine fluorsubstituierte
Alkyl-Gruppe mit 1 bis 10 Kohlenstoffen, eine Alkenyl-Gruppe mit 1
bis 10 Kohlenstoffen und 1 bis 3 Doppelbindungen, eine Alkinyl-Gruppe mit 1 bis
10 Kohlenstoffen und 1 bis 3 Dreifachbindungen oder eine Trialkylsilyl-
oder Trialkylsilyloxy-Gruppe ist, worin die Alkyl-Gruppen unabhängig 1 bis
6 Kohlenstoffe aufweisen;
r eine ganze Zahl mit einem wert
von 0 bis 5 ist und
die CONH-Gruppe in der 6- oder 7-Position
des Benzopyrans ist,
oder ein pharmazeutisch akzeptables Salz
der Verbindung.
-
Die
Verbindungen der vorliegenden Erfindung sind nützlich zur Prävention
bestimmter ungewünschter Nebenwirkungen
von Retinoiden, die zur Behandlung oder Prävention von bestimmten Krankheiten
oder Zuständen
verabreicht werden. Für
diesen Zweck können
die Verbindungen der Erfindung mit Retinoiden gemeinsam verabreicht
werden. Die Verbindungen der vorliegenden Erfindung sind ebenfalls
nützlich
in der Behandlung von akuter oder chronischer Toxizität, die aus
einer Überdosis
oder Vergiftung durch Retinoidwirkstoffe oder Vitamin A resultiert.
-
Die
vorliegende Erfindung betrifft zusätzlich die Verwendung von RAR-Antagonisten
zur Blockierung aller oder einiger RAR-Rezeptororte in biologischen Systemen,
einschließlich
Säugetieren,
zur Prävention oder
Verminderung der Wirkung von RAR-Agonisten auf die Rezeptororte.
Insbesondere betrifft die vorliegende Erfindung die Verwendung von
RAR-Antagonisten zur (a) Prävention
und (b) Behandlung von chronischer oder akuter Retinoidtoxizität (einschließlich durch
Vitamin A oder Vitamin A-Vorläufer)
und von Nebenwirkungen der Retinoidtherapie.
-
In
einem besonderen Aspekt der vorliegenden Erfindung wird ein Verfahren
zur Behandlung eines pathologischen Zustands in einem Säugetier
bereitgestellt. Die behandelten Zustände sind mit einer Retinolsäurerezeptor-Aktivität verbunden.
Dieses Verfahren beinhaltet das Verabreichen eines Retinoidantagonisten oder
negativen Hormons, der/das an einen der folgenden Retinolsäurerezeptor-Untertypen
binden kann, an das Säugetier:
RARα,
RARβ und
RARγ.
Der Antagonist oder das negative Hormon wird in einer pharmazeutisch wirksamen
Menge zur Bereitstellung eines therapeutischen Nutzen gegen den
pathologischen Zustand im Säugetier
verabreicht.
-
Als
Gegengift für
akute oder chronische Retinoid- oder Vitamin A-Vergiftung kann der
RAR-Antagonist an ein Säugetier
enteral, d.h. durch intragastrale Intubation oder als Lebensmittel/Wasser-Gemisch,
oder parenteral, z.B. intraperitoneal, intramuskulär, subkutan,
topisch etc., verabreicht werden. Die einzige Anforderung für den Verabreichungsweg
ist, daß er
die Übertragung
des Antagonisten auf das Zielgewebe erlauben muß. Der RAR-Antagonist kann als solcher oder in
Kombination mit Exzipienten formuliert werden. Der RAR-Antagonist
braucht nicht in Lösung
in der Formulierung sein, zum Beispiel im Falle von enteraler Verwendung.
-
Als
Zusatz zur Therapie mit Retinoiden und zur Prävention einer oder mehrerer
der Nebenwirkungen des Retinoidwirkstoffs, der verabreicht wird,
kann der RAR-Antagonist
in ähnlicher
Weise enteral oder parenteral verabreicht werden. Der RAR-Antagonist
und RAR-Agonist brauchen nicht auf dem gleichen Verabreichungsweg
verabreicht werden. Der Schlüssel
ist, daß ausreichende
Mengen des RAR-Antagonisten
kontinuierlich im interessierenden Gewebe während des Kontakts mit dem
RAR-Agonisten vorhanden sind. Zur Prävention von Retinoidtoxizität ist es
am besten, daß der
RAR-Antagonist gleichzeitig oder vor der Behandlung mit dem RAR-Agonisten
verabreicht wird. In vielen Situationen wird der RAR-Antagonist
auf einem anderen Weg als der Agonist verabreicht werden. Zum Beispiel
können
ungewünschte
Hautwirkungen eines enteral verabreichten Retinoids durch einen
RAR-Antagonisten verhindert oder gelindert werden, der topisch verabreicht
wird.
-
Ein
Verfahren zur Potenzierung einer pharmakologischen Aktivität eines
Steroidsuperfamilie-Rezeptoragonisten beinhaltet die gleichzeitige
Verabreichung an das Säugetier,
zusammen mit dem Steroidsuperfamilie-Rezeptoragonisten, einer Zusammensetzung,
die eine pharmazeutische wirksame Dosis eines negativen Retinoidhormons
umfaßt,
um die pharmakologische Aktivität
des Steroidsuperfamilie-Rezeptoragonisten
zu potenzieren. Die pharmakologische Aktivität ist in einem Reportergen-trans-Aktivierungstest
in vitro meßbar, wie
zum Beispiel durch Messung der Anti-AP-1-Aktivität. Die zu potenzierende pharmakologische
Aktivität kann
eine antiproliferative Aktivität
sein, wie zum Beispiel Aktivität
des Typs, der im Netzhautpigmentepithel meßbar ist. Der Steroidsuperfamilie-Rezeptoragonist
kann jeder der folgenden sein: ein Retinoidrezeptoragonist, ein
Vitamin D-Rezeptoragonist,
ein Glukokortikoidrezeptoragonist, ein Schilddrüsenhormonrezeptoragonist, ein
Peroxisomproliferatoraktivierter Rezeptoragonist oder ein Östrogenrezeptoragonist.
-
Der
Retinoidrezeptor-Agonist kann ein RAR-Agonist sein, wie zum Beispiel
all-trans-Retinolsäure
oder 13-cis-Retinolsäure.
Der Retinoidrezeptor-Agonist kann ebenfalls ein RXR-Agonist sein.
Ein bevorzugter Vitamin D-Rezeptoragonist ist 1,25-Dihydroxyvitamin
D3. Ein bevorzugter Glukokortikoidrezeptor-Agonist ist Dexamethason.
Ein bevorzugter Schilddrüsenhormonrezeptor-Agonist
ist 3,3',5-Triiodthyronin.
Das negative Retinoidhormon ist ein RAR-spezifisches negatives Retinoidhormon,
das bevorzugt eine Dissoziationskonstante von weniger als oder etwa
gleich 30 nM hat. Verweise auf das RAR-spezifische negative Retinoidhormon schließen AGN
193109, AGN 193385, AGN 193389 und AGN 193871 ein. Die Zusammensetzung,
die eine pharmazeutisch wirksame Dosis eines negativen Retinoidhormons
umfaßt,
kann zum gleichen Zeitpunkt wie der Steroidsuperfamilie-Agonist
koverabreicht werden und vor der Koverabreichung kombiniert werden.
Diese können
ebenfalls als getrennte Zusammensetzungen koverabreicht werden.
-
Kurze Beschreibung
der Zeichnungen
-
1 zeigt
die chemische Struktur von AGN 193109.
-
2A–2F sind
eine Reihe von Liniendiagrammen, die zeigen, daß AGN 193109 die ATRA-abhängige Transaktivierung
an den RARs inhibiert. 2A und 2B stellen
die Aktivität
am RAR-α-Rezeptor dar; 2C und 2D stellen
die Aktivität
am RAR-β-Rezeptor
dar; 2E und 2F stellen
die Aktivität am
RAR-γ-Rezeptor
dar. In 2A, 2C und 2E stellen
offene Kästchen
die Retinolsäurebehandlung dar,
und gefüllte
Kreise stellen die AGN 193109-Behandlung dar. In 2B, 2D und 2F stellen
die einzelnen Linien die Luciferase-Aktivität dar, gemessen nach Behandlung
mit 10–8 M
ATRA und variablen Konzentrationen von AGN 193109.
-
3A und 3B sind
Liniendiagramme, die die Luciferase-Aktivität darstellen, detektiert in CV-1-Zellen,
die mit Reporterplasmid ERE-tk-Luc und Expressionsplasmid ER-RAR-α transfiziert
und mit ATRA (3A) oder AGN 193109 (3B) bei verschiedenen Konzentrationen stimuliert
wurden. Die Datenpunkte stellen den Mittelwert ± Standardabweichung für drei unabhängige Luciferasebestimmungen
dar. Die Ergebnisse der unter Verwendung unterschiedlicher Mengen
von kotransfiziertem ER-RAR-α (0,05,
0,1 und 0,2 μg/Vertiefung)
durchgeführten
Transfektionen sind in jeder Figur angegeben.
-
4A und 4B sind
Liniendiagramme, die die Luciferase-Aktivität in CV-1-Zellen darstellen, transfiziert
mit Reporterplasmid ERE-tk-Luc und Expressionsplasmid ER-RAR-β und stimuliert
mit ATRA (4A) oder AGN 193109 (4B) bei verschiedenen Konzentrationen. Die Datenpunkte
stellen den Mittelwert ± Standardabweichung
von drei unabhängigen
Luciferasebestimmungen dar. Die Ergebnisse der unter Verwendung
unterschiedlicher Mengen von kotransfiziertem ER-RAR-β (0,05,
0,1 und 0,2 μg/Vertiefung) durchgeführten Transfektionen
sind in jeder Figur angegeben.
-
5A und 5B sind
Liniendiagramme, die die Luciferase-Aktivität darstellen, detektiert in CV-1-Zellen,
die mit Reporterplasmid ERE-tk-Luc und Expressionsplasmid ER-RAR-γ transfiziert
und mit ATRA (3A) oder AGN 193109 (3B) bei verschiedenen Konzentrationen stimuliert
wurden. Die Datenpunkte stellen den Mittelwert ± Standardabweichung für drei unabhängige Luciferasebestimmungen
dar. Die Ergebnisse der unter Verwendung unterschiedlicher Mengen
von kotransfiziertem ER-RAR-γ (0,05,
0,1 und 0,2 μg/Vertiefung)
durchgeführten
Transfektionen sind in jeder Figur angegeben.
-
6 zeigt
ATRA- und AGN 193109-Dosisreaktionen von CV-1-Zellen, die mit dem ERE-tk-Luc-Reporterplasmid
und entweder dem chimären
ER-RXR-α-Rezeptor-Expressionsplasmid
allein oder in Kombination mit dem RAR-γ-VP-16-Expressionsplasmid kotransfiziert
wurden. Mit ER-RXR-α kotransfizierte
Zellen wurden mit ATRA (Kästchen)
und AGN 193109 (Rauten) behandelt. Mit der Kombination aus ER-RXR-α und RAR-γ-VP-16 kotransfizierte
Zellen wurden mit ATRA (Kreise) oder AGN 193109 (Dreiecke) behandelt.
-
7 zeigt
ein Liniendiagramm, das die Messungen der Luciferase-Aktivität darstellt,
aufgezeichnet in Lysaten von CV-1-Zellen, die mit dem ERE-tk-Luc-Reporter-
und ER-RAR-γ-Expressionskonstrukt
transfiziert und dann mit ATRA mit 10–8 M
und den Testverbindungen mit den auf der horizontalen Achse angegebenen
Konzentrationen behandelt wurden. Die Testverbindungen waren AGN
193109 (Kästchen),
AGN 193357 (offene Rauten), AGN 193385 (Kreise), AGN 193389 (Dreiecke),
AGN 193840 (schraffierte Kästchen)
und AGN 192870 (gefüllte
Rauten).
-
8 zeigt
ein Liniendiagramm, das die Messungen der Luciferase-Aktivität darstellt,
aufgezeichnet in Lysaten von CV-1-Zellen, die mit den ERE-tk-Luc-Reporter-
und RAR-γ-VP-16- und ER-RXR-α-Expressionskonstrukten
transfiziert und dann mit den Testverbindungen in den auf der horizontalen
Achse angegebenen Konzentrationen behandelt wurden. Die Testverbindungen
waren ATRA (offene Kästchen),
AGN 193109 (offene Kreise), AGN 193174 (offene Dreiecke), AGN 193199
(schraffierte Kästchen),
AGN 193385 (schraffierte Kreise), AGN 193389 (umgedrehte Dreiecke),
AGN 193840 (diagonal gefüllte
Kästchen)
und AGN 193871 (halbgefüllte
Rauten).
-
9A, 9B und 9C skizzieren
schematisch einen Mechanismus, durch den AGN 193109 die Wechselwirkung
zwischen dem RAR (schattierter Kasten) und negativen Koaktivatorproteinen
(–) modulieren können, veranschaulicht
im Zusammenhang eines Transaktivierungstests. 9A zeigt, daß negative
Koaktivatorproteine und positive Koaktivatorproteine (+) in einem
Bindungsgleichgewicht mit dem RAR stehen. In Abwesenheit eines Liganden
resultiert eine Transkription des Reportergens auf Basisniveau.
Wie in 9B veranschaulicht wird, fördert die
Zugabe eines RAR-Agonisten
die Assoziation von positiven Koaktivatorproteinen mit dem RAR und
führt zu
einer aufrequlierten Reportergen-Transkription.
Wie in 9C veranschaulicht wird, fördert die
Zugabe von AGN 193109 die Assoziation von negativen Koaktivatorproteinen
mit dem RAR und verhindert die Reportergen-Transkription.
-
10 ist ein Balkendiagramm, das die Inhibierung
von TPA-induzierter
Str-AP1-CAT-Expression als Funktion der AGN 191183-Konzentration
(10–10 bis
10–12 M)
zeigt, wobei die AGN 193109-Konzentration konstant auf 10–8 M
gehalten wird. Die Ergebnisse aus Versuchen, die mit AGN 191183
allein durchgeführt
wurden, sind als schraffierte Balken gezeigt, während gestreifte Balken die
Ergebnisse aus der Behandlung mit der Kombination aus AGN 193109
und AGN 191183 darstellen.
-
11 skizziert schematisch einen Mechanismus, durch
den AGN 193109 die Aktivitäten
von RARs und anderen Mitgliedern der Kernrezeptorfamilie potenzieren
kann. Wie im Diagramm veranschaulicht wird, besitzen eingeführte RARs
(offene Rechtecke mit AB-C-DEF-Domänen) eine erhöhte Empfindlichkeit
gegenüber
RAR-Liganden im Anti-AP1-Test, weil das negative Koaktivatorprotein
(ncp), das in beschränkender
Zufuhr vorhanden ist, auf RARs abgesondert wird und dadurch zu zwei
Populationen führt:
RAR + ncp und RAR – ncp.
RAR – ncp
hat eine erhöhte
Empfindlichkeit gegenüber
Liganden. Nicht-RAR-Kernfaktoren
(schattierte Rechtecke mit AB-C-DEF-Domänen) besitzen eine erhöhte Empfindlichkeit
gegenüber
verwandten Liganden, weil ncp auf den RAR durch die Aktivität von AGN
193109 abgesondert wurde. Die modularen Domänen der Kernrezeptoren werden
unter Verwendung von Standardnomenklatur als "AB" (ligandenunabhängige Transaktivierungsdomäne), "C" (DNA-Bindungsdomäne) und "DEF" (ligandenregulierte
Transaktivierungsdomäne und
Dimerisierungsdomäne)
bezeichnet.
-
12 ist ein Liniendiagramm, das die Wirkung von
AGN 193109 auf die 1,25-Dihydroxyvitamin D3-Dosisreaktion
in CV-1-Zellen zeigt, die mit dem MTV-DR3-Luc-Reporterplasmid transfiziert
wurden. Transfektanten wurden mit 1,25-Dihydroxyvitamin D3 (gefüllte
Kästchen),
1,25-Dihydroxyvitamin D3 und 10–8 M
AGN 193109 (gefüllte
Dreiecke) und 1,25-Dihydroxyvitamin D3 und
10–7 M
AGN 193109 (gefüllte
Kreise) behandelt.
-
13 ist ein Balkendiagramm, das die Wirkung der
AGN 193109-Koverabreichung (10 nM) auf die 1,25-Dihydroxyvitamin
D3-vermittelte Inhibierung von TPA-induzierter Str-AP1-CAT-Aktivität zeigt.
Gefüllte
Balken stellen die Inhibierung von CAT-Aktivität in mit 1,25-Dihydroxyvitamin
D3 allein behandelten transfizierten Zellen
dar. Leere Balken stellen die Inhibierung von CAT-Aktivität in mit
der Kombination aus 1,25-Dihydroxyvitamin D3 und
AGN 193109 behandelten transfizierten Zellen dar.
-
14 ist ein Liniendiagramm, das die Wirkung von
AGN 193109 allein und in Kombination mit AGN 191183 auf HeLa-Zellen zeigt, die
mit RAR-γ und
dem auf RAR reagierenden MTV-TREp-Luc-Reporterkonstrukt transfiziert
wurden. Die im Diagramm veranschaulichten Wirkstoffbehandlungen
sind: AGN 193109 allein (Kästchen),
AGN 193109 in Kombination mit AGN 191183 mit 10–10 M
(Rauten) und AGN 193109 in Kombination mit AGN 191183 mit 10–9 M.
-
15 ist ein Liniendiagramm, das zeigt, daß ECE16-1-Zellen als Reaktion
auf EGF (gefüllte
Kästchen),
aber nicht als Reaktion auf definiertes Medium allein (leere Kreise)
wuchsen. Mit AGN 193109 allein behandelte Zellen werden durch die
ausgefüllten
Dreiecke dargestellt. Die ausgefüllten
Kreise stellen Ergebnisse dar, die für Zellen erhalten wurden, die
mit 10 nM AGN 191183 und 0–1000
nM AGN 193109 behandelt wurden.
-
16 ist ein Balkendiagramm, das die Wirkung von
AGN 193109 allein auf die Proliferation von CaSki-Zellen in Gegenwart
oder Abwesenheit der AGN 191183-Retinoidagonisten zeigen. Alle Probengruppen erhielten
20 ng/ml von epidermalem Wachstumsfaktor (EGF) mit Ausnahme der
im definierten Medium (DM) allein vermehrten Probe (leere Balken).
Gestreifte Balken stellen Proben dar, die in Abwesenheit von AGN 193109
vermehrt wurden. Ausgefüllte
Balken stellen Proben dar, die in Gegenwart von 1000 nM AGN 193109 vermehrt
wurden. Die im Verfahren verwendeten Konzentrationen von AGN 191183
sind auf der horizontalen Achse gezeigt.
-
17 ist eine Dosis-Reaktions-Kurve, die zeigt,
daß AGN
193109 die antiproliferative Aktivität von ATRA auf Netzhautpigmentepithel-(RPE)-Zellen
potenzierte. Mit ATRA allein behandelte Proben sind durch ausgefüllte Kästchen dargestellt.
Mit der Kombination aus ATRA und AGN 193109 (10–7 M)
behandelte Proben sind durch ausgefüllte Kreise dargestellt. Die
zur Behandlung der verschiedenen Proben verwendete ATRA-Konzentration
ist auf der horizontalen Achse angegeben.
-
18 ist eine Dosis-Reaktions-Kurve, die zeigt,
daß sowohl
13-cis-RA als auch ATRA das RPE-Zellwachstum hemmten, und daß AGN 193109
die antiproliferative Aktivität
von 13-cis-RA potenzierte. Die in der Dosisreaktion gezeigten verschiedenen
Probenbehandlungen schlossen 13-cis-RA allein (gefüllte Kästchen), 13-cis-RA
in Kombination mit AGN 193109 (10–6 M)
(gefüllte
Kreise), 13-cis-RA in Kombination mit AGN 193109 (10–8 M)
(gefüllte
Dreiecke) und ATRA (gefüllte
Rauten) ein. Die in den Probenbehandlungen verwendeten Konzentrationen
von 13-cis-RA und ATRA sind auf der horizontalen Achse gezeigt.
-
19 ist eine Dosis-Reaktions-Kurve, die zeigt,
daß AGN
193109 die antiproliferative Aktivität von Dexamethason in primären RPE-Zellkulturen
potenzierte. Die in der Dosisreaktion gezeigten verschiedenen Probenbehandlungen
schlossen ATRA (gefüllte
Kästchen),
Dexamethason allein (gefüllte
Kreise), Dexamethason in Kombination mit AGN 193109 (10–8 M)
(gefüllte
Dreiecke) und Dexamethason in Kombination mit AGN 193109 (10–6 M)
(gefüllte
Rauten) ein. Die in den Probenbehandlungen verwendeten Konzentrationen von
Dexamethason und ATRA sind auf der horizontalen Achse gezeigt.
-
20 ist eine Dosis-Reaktions-Kurve, die zeigt,
daß AGN
193109 die antiproliferative Aktivität von Schilddrüsenhormon
(T3) in primären
RPE-Zellkulturen potenzierte. Die in der Dosisreaktion gezeigten
verschiedenen Probenbehandlungen schlossen ATRA (gefüllte Kästchen),
T3 allein (gefüllte
Kreise), T3 in Kombination mit AGN 193109 (10–8 M)
(gefüllte
Dreiecke) und T3 in Kombination mit AGN 193109 (10–6 M)
(gefüllte Rauten)
ein. Die in den Probenbehandlungen verwendeten Konzentrationen von
T3 und ATRA sind auf der horizontalen Achse gezeigt.
-
Ausführliche
Beschreibung der Erfindung
-
Definitionen
-
Für die Zwecke
der vorliegenden Erfindung wird ein RAR-Antagonist als eine Chemikalie definiert,
die an einen oder mehrere der RAR-Untertypen mit einem Kd-wert von weniger als 1 mikromolar (Kd < 1 μM) bindet, die
aber keine signifikante Transkriptionsaktivierung der durch den
RAR-Untertypen regulierten Gene in einem Rezeptor-Kotransfektionstest verursacht.
Herkömmlich
sind Antagonisten chemische Mittel, die die Aktivitäten von
Agonisten hemmen. Daher wird die Aktivität eines Rezeptorantagonisten
herkömmlich
aufgrund seiner Fähigkeit
zur Inhibierung der Aktivität
eines Agonisten gemessen.
-
Ein
RAR-Agonist wird als eine Chemikalie definiert, die an einen oder
mehrere RAR-Rezeptoruntertypen mit einem Kd-Wert
von weniger als 1 mikromolar (Kd < 1 μM) bindet
und Transkriptionsaktivierung der durch den RAR-Untertyp regulierten
Gene in einem Rezeptor-Kotransfektionstest verursacht. Der Begriff "RAR-Agonist" schließt Chemikalien
ein, die andere Rezeptoren zusätzlich
zu RARs binden oder aktivieren können,
z.B. RXR-Rezeptoren.
-
Wie
hier verwendet, ist ein negatives Hormon oder ein inverser Agonist
ein Ligand für
einen Rezeptor, der den Rezeptor veranlaßt, einen inaktiven Zustand
relativ zu einem Basiszustand anzunehmen, der in Abwesenheit jedes
Liganden auftritt. Während
daher ein Antagonist die Aktivität
eines Agonisten hemmen kann, ist ein negatives Hormon ein Ligand,
der die Konformation des Rezeptors in Abwesenheit eines Agonisten
verändern
kann. Das Konzept eines negativen Hormons oder inversen Agonisten
wurde von Bond et al. untersucht, Nature 374: 272 (1995). Insbesondere
schlugen Bond et al. vor, daß unkomplexierter β2-Adrenozeptor in
einem Gleichgewicht zwischen einer inaktiven Konformation und einer
spontan aktiven Konformation existiert. Es wird vorgeschlagen, daß Agonisten
den Rezeptor in einer aktiven Konformation stabilisieren. Umgekehrt
wird angenommen, daß inverse
Agonisten eine inaktive Rezeptorkonformation stabilisieren. Während daher
ein Antagonist seine Aktivität
aufgrund der Hemmung eines Agonisten zeigt, kann ein negatives Hormon zusätzlich seine
Aktivität
in Abwesenheit eines Agonisten durch Hemmung der spontanen Umwandlung
eines unkomplexierten Rezeptors zu einer aktiven Konformation zeigen.
Nur eine Untergruppe von Antagonisten wird als negative Hormone
wirken. Wie hier offenbart wird, ist AGN 193109 sowohl ein Antagonist
als auch ein negatives Hormon. Bis heute wurde nicht gezeigt, daß andere
Retinoide negative Hormonaktivität
besitzen.
-
Wie
hier verwendet, bezeichnet die Koverabreichung (gleichzeitige Verabreichung)
von zwei pharmakologisch wirksamen Verbindungen die Übertragung
von zwei getrennten chemischen Einheiten, ob in vitro oder in vivo.
Die Koverabreichung bezeichnet die gleichzeitige Übertragung
von getrennten Mitteln; die gleichzeitige Übertragung einer Mischung von
Mitteln; sowie die Übertragung
eines Mittels gefolgt von Übertragung des
zweiten Mittels. In allen Fällen
sollen Mittel, die koverabreicht werden, in Verbindung miteinander
wirken.
-
Der
Begriff Alkyl bezeichnet und umfaßt jede und alle Gruppen, die
als normales Alkyl, verzweigtkettiges Alkyl und Cycloalkyl bekannt
sind. Der Begriff Alkenyl bezeichnet und umfaßt normale Alkenyl-, verzweigtkettige
Alkenyl- und Cycloalkenyl-Gruppen mit einem oder mehreren Orten
der Ungesättigtheit.
In ähnlicher
Weise bezeichnet der Begriff Alkinyl und umfaßt normale Alkinyl- und verzweigtkettige
Alkinyl-Gruppen mit einer oder mehreren Dreifachbindungen.
-
Niederalkyl
bezeichnet die oben definierte breite Definition von Alkyl-Gruppen
mit 1 bis 6 Kohlenstoffen im Fall von normalem Niederalkyl und nach
Bedarf 3 bis 6 Kohlenstoffen für
niedere verzweigtkettige und Cycloalkyl-Gruppen. Niederalkenyl wird
in ähnlicher
Weise mit 2 bis 6 Kohlenstoffen für normale Niederalkenyl-Gruppen
und 3 bis 6 Kohlenstoffen für
verzweigtkettige und Cycloniederalkenyl-Gruppen definiert. Niederalkinyl wird
ebenfalls in ähnlicher
Weise mit 2 bis 6 Kohlenstoffen für normale Niederalkinyl- Gruppen und 4 bis
6 Kohlenstoffen für
verzweigtkettige Niederalkinyl-Gruppen definiert.
-
Der
Begriff "Ester", wie hier verwendet,
bezeichnet und umfaßt
jede Verbindung, die in die Definition dieses Begriffes fällt, wie
er klassisch in der organischen Chemie verwendet wird. Er schließt organische
und anorganische Ester ein.
-
Wenn
in dieser Anmeldung nichts anderes angegeben ist, stammen bevorzugte
Ester aus den gesättigten
aliphatischen Alkoholen oder Säuren
mit 10 oder weniger Kohlenstoffatomen oder den cyclischen oder gesättigten
aliphatischen cyclischen Alkoholen und Säuren mit 5 bis 10 Kohlenstoffatomen.
Besonders bevorzugte aliphatische Ester sind diejenigen, die aus
Niederalkylsäuren
und -alkoholen stammen. Ebenfalls bevorzugt sind die Phenyl- oder
Niederalkylphenylester.
-
Amid
hat die Bedeutung, die diesem Begriff in der organischen Chemie
klassisch zugewiesen wird. In diesem Fall schließt er die unsubstituierten
Amide und alle aliphatischen und aromatischen mono- und disubstituierten
Amide ein. Wenn in dieser Anmeldung nichts anderes angegeben ist,
sind bevorzugte Amide die mono- und disubstituierten Amide, die
aus den gesättigten
aliphatischen Resten mit 10 oder weniger Kohlenstoffatomen oder
den cyclischen oder gesättigten
aliphatisch-cyclischen Resten mit 5 bis 10 Kohlenstoffatomen stammen.
Besonders bevorzugte Amide sind diejenigen, die aus substituierten
und unsubstituierten Niederalkylaminen stammen. Ebenfalls bevorzugt
sind mono- und disubstituierte Amide, die aus den substituierten
und unsubstituierten Phenyl- oder Niederalkylphenylaminen stammen.
Unsubstituierte Amide sind ebenfalls bevorzugt.
-
Acetale
und Ketale schließen
die Reste der Formel -CK ein, worin K (-OR)2 ist.
Hier ist R Niederalkyl. Ebenfalls kann K -OR7O-
sein, worin R7 Niederalkyl mit 2–5 Kohlenstoffatomen
ist, geradkettig oder verzweigt.
-
Ein
pharmazeutisch akzeptables Salz kann für beliebige Verbindungen in
dieser Verbindung mit einer Funktionalität hergestellt werden, die ein
Salz bilden kann, zum Beispiel mit einer Säurefunktionalität. Ein pharmazeutisch
akzeptables Salz ist jedes Salz, das die Aktivität der Stammverbindung beibehält und keine
nachteilige oder ungünstige
Wirkung auf den Patienten, an den es verabreicht wird, und im Zusammenhang
ausübt, in
dem es verabreicht wird.
-
Pharmazeutisch
akzeptable Salze können
aus organischen oder anorganischen Basen stammen. Das Salz kann
ein ein- oder mehrwertiges Ion sein. von besonderem Interesse sind
die anorganischen Ionen Natrium, Kalium, Calcium und Magnesium.
Organische Salze können
mit Aminen hergestellt werden, insbesondere Ammoniumsalze, wie z.B.
Mono-, Di- und Trialkylamine oder Ethanolamine. Salze können ebenfalls
mit Coffein, Tromethamin und ähnlichen
Molekülen
gebildet werden. wenn es einen Stickstoff gibt, der ausreichend
basisch ist, um Säureadditionssalze
bilden zu können,
dann können
solche mit beliebigen anorganischen oder organischen Säuren oder
einem Alkylierungsmittel wie Methyliodid gebildet werden. Bevorzugte Salze
sind diejenigen, die mit anorganischen Säuren wie Salzsäure, Schwefelsäure oder
Phosphorsäure
gebildet werden. Jede einer Anzahl von einfachen organischen Säuren, wie
Mono-, Di- oder Trisäure,
kann ebenfalls verwendet werden.
-
Einige
der Verbindungen der vorliegenden Erfindung können trans- und cis-Isomere
(E und Z) aufweisen. Zusätzlich
können
die Verbindungen der vorliegenden Erfindung ein oder mehrere chirale
Zentren enthalten und können
deshalb in enantiomeren und diastereomeren Formen existieren. Der
Umfang der vorliegenden Erfindung soll alle solche Isomere als solche sowie
Mischungen aus cis- und trans-Isomeren, Mischungen aus Diastereomeren
sowie racemische Mischungen aus Enantiomeren (optische Isomere)
umfassen. In der vorliegenden Anmeldung ist eine Mischung solcher
Isomere oder eines der Isomere beabsichtigt, wenn keine spezifische
Erwähnung
der Konfiguration (cis, trans oder R oder S) einer Verbindung (oder
eines asymmetrischen Kohlenstoffs) erfolgt.
-
Aryl-substituierte
Benzopyran-Derivate mit Retinoidantagonist-artiger biologischer
Aktivität
-
Die
an den aromatischen Teil der Benzopyran-Einheit der Verbindungen
der Formel (1) gebundene R2-Gruppe ist bevorzugt
H, F oder CF3. R3 ist
bevorzugt Wasserstoff oder Methyl, noch mehr bevorzugt Wasserstoff.
-
Die
vorliegend am meisten bevorzugten Verbindungen der Erfindung sind
in Tabelle 1A unter Bezugnahme auf Formel 5c gezeigt.
-
-
-
Biologische Aktivität, Verabreichungsmodi
-
Wie
oben festgestellt wurde, sind die Verbindungen der vorliegenden
Erfindung Antagonisten für
einen oder mehrere RAR-Rezeptoruntertypen. Dies bedeutet, daß die Verbindungen
der Erfindung an einen oder mehrere RAR-Rezeptoruntertypen binden,
aber nicht die Reaktion auslösen,
die durch Agonisten der gleichen Rezeptoren ausgelöst wird.
Einige der Verbindungen der vorliegenden Erfindung sind Antagonisten
für alle
drei RAR-Rezeptoruntertypen (RAR-α,
RAR-β und
RAR-γ),
und diese werden als "RAR-Panantagonisten" bezeichnet. Einige
andere sind Antagonisten für
nur einen oder zwei der RAR-Rezeptoruntertypen.
Einige Verbindungen im Umfang der vorliegenden Erfindung sind partielle
Agonisten für
einen oder zwei RAR-Rezeptoruntertypen und Antagonisten für die verbleibenden
Untertypen. Die Verbindungen der Erfindung binden nicht an RXR-Rezeptoren
und sind deshalb weder Agonisten noch Antagonisten für RXR.
-
Abhängig vom
Ort und der Natur der ungewünschten
Nebenwirkungen, die unterdrückt
oder gelindert werden sollen, können
in Übereinstimmung
mit der Erfindung verwendete Verbindungen Antagonisten für nur einen
oder zwei der RAR-Rezeptoruntertypen
sein. Einige in Übereinstimmung
mit der Erfindung verwendete Verbindungen können partielle Agonisten für einen
oder zwei RAR-Rezeptoruntertypen und Antagonisten für die verbleibenden
Untertypen sein. Solche Verbindungen sind allgemein gesprochen in Übereinstimmung
mit der Erfindung verwendbar, falls die antagonistische Wirkung
auf denjenigen RAR-Rezeptoruntertyp (oder die Untertypen) besteht,
der (die) hauptsächlich
verantwortlich für
die Überdosisvergiftung
oder für
die ungewünschte
Nebenwirkung oder Nebenwirkungen ist (sind). In diesem Zusammenhang
wird festgestellt, daß allgemein
gesprochen eine Verbindung als Antagonist für einen gegebenen Rezeptoruntertyp
betrachtet wird, falls die Verbindung in den nachfolgend beschriebenen Kotransfektionstests
keine signifikante Transkriptionsaktivierung des Rezeptor-regulierten
Reportergens verursacht, aber dennoch an den Rezeptor mit einem Kd-Wert von weniger als ca. 1 μM bindet.
-
Ob
eine Verbindung ein RAR-Antagonist ist und deshalb in Übereinstimmung
mit der vorliegenden Erfindung verwendet werden kann, kann in den
folgenden Tests untersucht werden.
-
Ein
chimärer
Transaktivierungstest, der auf Agonist-artige Aktivität in den
RAR-α-,
RAR-β-,
RAR-γ- und
RXR-α-Rezeptoruntertypen
untersucht, und der auf der von P. L. Feigner und M. Holm veröffentlichten
Arbeit beruht, Focus Band 11, Nr. 2 (1989), wird im Detail in WO
94/17796 beschrieben, veröffentlicht
am 18. August 1994. Letztere Veröffentlichung
ist das PCT-Gegenstück
der US-Anmeldung Nr. 08/016,404, eingereicht am 11. Februar 1993,
die als US-PS 5,455,265 erteilt wurde. Eine Verbindung sollte keine
signifikante Aktivierung eines Reportergens durch einen gegebenen
Rezeptoruntertyp (RAR-α,
RAR-β oder
RAR-γ) in
diesem Test verursachen, um sich als RAR-Antagonist mit Nutzen in
der vorliegenden Erfindung zu qualifizieren.
-
Ein
Holorezeptor-Transaktivierungstest und ein Ligandenbindungstest,
die die Antagonist/Agonist-artige Aktivität der Verbindungen der Erfindung
bzw. ihre Fähigkeit
zur Bindung an die verschiedenen Retinoidrezeptoruntertypen messen,
werden in WO 93/11755 (insbesondere auf den Seiten 30–33 und
37–41)
beschrieben, veröffentlicht
am 24. Juni 1993. Eine Beschreibung des Holorezeptor-Transaktivierungstests
wird ebenfalls nachfolgend bereitgestellt.
-
Holorezeptor-Transaktivierungstest
-
CV1-Zellen
(5000 Zellen/Vertiefung) wurden mit einem RAR-Reporterplasmid MTV-TREp-LUC (50 ng) neben
einem der RAR-Expressionsvektoren
(10 ng) in einem automatisierten Format mit 96 Vertiefungen durch
das Calciumphosphat-Verfahren von Heyman et al. transfiziert, Cell
68: 397–406.
Für RXR-α- und RXR-γ-Transaktivierungstests
wurde ein RXR-reaktives Reporterplasmid CRBPII-tk-LUC (50 ng) neben
den entsprechenden RXR-Expressionsvektoren (10 ng) im wesentlichen
wie von Heyman et al. (siehe oben s.o.) und Allegretto et al. beschrieben
verwendet, J. Biol. Chem. 268: 26625–26633. Für RXR-β-Transaktivierungstests wurde
ein RXR-reaktives
Reporterplasmid CPRE-tk-LUC (50 mg) neben RXR-β-Expressionsvektor (10 mg) wie oben beschrieben
verwendet. Diese Reporter enthalten DRI-Elemente aus humanem CRBPII
bzw. bestimmte DRI-Elemente aus Promotor (siehe Mangelsdorf et al.,
The Retinoids: Biology, Chemistry and Medicine, S. 319–349, Raven
Press Ltd., New York, und Heyman et al., s.o.). Ein β-Galactosidase-Expressionsvektor (50
ng) wurde als interne Kontrolle in den Transfektionen verwendet,
um auf Variationen in der Transfektionseffizienz zu normalisieren.
Die Zellen wurden in 3-facher Ausführung für 6 Stunden transfiziert, gefolgt
von Inkubation mit Retinoiden für
36 Stunden, und die Extrakte wurden auf Luciferase- und β-Galactosidase-Aktivitäten getestet.
Das ausführliche
experimentelle Verfahren für
Holorezeptor-Transaktivierungen wurde in Heyman et al. (s.o.) und
Allegretto et al. (s.o.) beschrieben. Die in diesem Test erhaltenen
Ergebnisse werden als EC50-Werte ausgedrückt, ebenso
wie im chimären
Rezeptor-Transaktivierungstest.
Die Ergebnisse des Ligandenbindungstests werden als Kd-Werte
ausgedrückt
(siehe Cheng et al., Biochemical Pharmacology 22: 3099–3108).
-
Eine
Verbindung sollte keine signifikante Aktivierung eines Reportergens
durch einen gegebenen Rezeptoruntertyp (RAR-α, RAR-β oder RAR-γ) im Holorezeptor-Transaktivierungstest
verursachen, um sich als RAR-Antagonist mit Nutzen in der vorliegenden
Erfindung zu qualifizieren. Letztlich sollte eine Verbindung an wenigstens
einen der RAR-Rezeptoruntertypen
im Ligandenbindungstest mit einem Kd-Wert
von weniger als ca. 1 mikromolar (Kd < 1 μM) binden,
um als Antagonist für
den gebundenen Rezeptoruntertyp funktionieren zu können, vorausgesetzt
der gleiche Rezeptoruntertyp wird nicht signifikant durch die Verbindung
aktiviert.
-
Tabelle
5A zeigt die Ergebnisse des Ligandenbindungstests für bestimmte
exemplarische Verbindungen der Erfindung.
-
Tabelle
5A Ligandenbindungstest
-
Wie
aus den in Tabelle 5A zusammengefaßten Testergebnissen ersichtlich
ist, sind die darin angegebenen exemplarischen Verbindungen der
Erfindung Antagonisten für
die RAR-Rezeptoruntertypen,
aber haben keine Affinität
für RXR-Rezeptoruntertypen
(andere Verbindungen der Erfindung können Antagonisten für einige,
aber nicht alle RAR-Rezeptoruntertypen
und Agonisten für
die verbleibenden RAR-Untertypen
sein). Aufgrund dieser Eigenschaft können die Verbindungen der Erfindung
zur Blockierung der Aktivität
von RAR-Agonisten in biologischen Tests verwendet werden. In Säugetieren,
einschließlich
Menschen, können
die Verbindungen der Erfindung mit RAR-Agonisten koverabreicht werden
und mittels der pharmakologischen Selektivität oder ortsspezifischer Übertragung
vorzugsweise die ungewünschten
Wirkungen von RAR-Agonisten verhindern. Die Verbindungen der Erfindung
können
ebenfalls zur Behandlung von akuter oder chronischer Vitamin A-Überdosis
verwendet werden, die entweder aus der der übermäßigen Einnahme von Vitamin
A-Ergänzungsmitteln
oder aus dem Verzehr der Leber bestimmter Fische und Tiere resultiert,
die hohe Mengen von Vitamin A enthalten. Weiterhin können die
Verbindungen der Erfindung ebenfalls zur Behandlung von akuter oder
chronischer Toxizität
verwendet werden, die durch Retinoidwirkstoffe verursacht wird.
Es ist fachbekannt, daß die
bei Hypervitaminose A-Syndrom beobachteten Toxizitäten (Kopfschmerz,
Hautablösung,
Knochentoxizität,
Dyslipidämien) ähnlich oder
identisch zu Toxizitäten
sind, die bei anderen Retinoiden beobachtet werden, was eine gemeinsame
biologische Ursache nahelegt, nämlich
RAR-Aktivierung. Weil die Verbindungen der vorliegenden Erfindung
die RAR-Aktivierung blockieren, sind sie geeignet zur Behandlung
der vorhergehenden Toxizitäten.
-
Die
Verbindungen der Erfindung können
durch RAR-agonistische Retinoide indizierte Hautreizung im wesentlichen
verhindern, wenn die Verbindung der Erfindung topisch auf die Haut
koverabreicht wird. In ähnlicher
Weise können
die Verbindungen der Erfindung topisch auf die Haut verabreicht
werden, um Hautreizung bei Patienten oder Tieren zu blockieren,
denen RAR-agonistische Verbindungen systemisch verabreicht werden.
Die Verbindungen der Erfindung können
die Erholung von vorbestehender Retinoidtoxizität beschleunigen, durch koverabreichte
Retinoide verursachte Hypertriglyceridämie blockieren und durch einen
RAR-Agonisten (Retinoid) induzierte Knochentoxizität blockieren.
-
Allgemein
gesprochen können
die antagonistischen Verbindungen für therapeutische Anwendungen in
Säugetieren
in Übereinstimmung
mit der vorliegenden Erfindung enteral oder topisch als Gegengift
für Vitamin
A, Vitamin A-Vorläufer
oder Gegengift für
Retinoidtoxizität,
die aus einer Überdosis
oder einem anhaltenden Kontakt resultiert, nachdem die Aufnahme
des verursachenden Faktors (Vitamin A-Vorläufer oder anderes Retinoid)
abgesetzt wurde, verabreicht werden. Alternativ werden die antagonistischen
Verbindungen mit Retinoidwirkstoffen in Übereinstimmung mit der Verbindung
in Situationen koverabreicht, in denen das Retinoid einen therapeutischen
Nutzen liefert, und wenn der koverabreichte Antagonist eine oder
mehrere ungewünschte
Nebenwirkungen des Retinoids lindert oder eliminiert. Für diesen
Typ der Anwendung kann der Antagonist in einer ortsspezifischen
weise verabreicht werden, zum Beispiel als topisch aufgetragene
Creme oder Lotion, während
das koverabreichte Retinoid enteral gegeben werden kann.
-
Für therapeutische
Anwendungen in Übereinstimmung
mit der vorliegenden Erfindung werden die antagonistischen Verbindungen
in pharmazeutische Zusammensetzungen eingearbeitet, wie zum Beispiel
Tabletten, Pillen, Kapseln, Lösungen,
Suspensionen, Cremes, Salbengrundlagen, Gele, Salben, Lotionen und dgl.,
wobei solche pharmazeutisch akzeptablen Exzipienten und Träger verwendet
werden, die als solche fachbekannt sind. Zum Beispiel wird die Herstellung
topischer Formulierungen allgemein beschrieben in "Remington's Pharmaceutical
Science", Auflage
17, Mack Publishing Company, Easton, Pennsylvania. Zur topischen Anwendung
können
die antagonistischen Verbindungen ebenfalls als Puder oder Spray
verabreicht werden, insbesondere in Aerosolform. Falls der Wirkstoff
systemisch verabreicht werden soll, kann er als Pulver, Pille, Tablette
oder dgl. oder als Sirup oder Elixier, das zur oralen Verabreichung
geeignet ist, konfektioniert werden. Zur intravenösen oder
intraperitonealen Verabreichung wird die antagonistische Verbindung
als Lösung
oder Suspension zubereitet werden, die durch Injektion verabreicht
werden kann. In bestimmten Fällen kann
es nützlich
sein, die antagonistischen Verbindungen durch Injektion zu formulieren.
In bestimmten Fällen
kann es nützlich
sein, die antagonistischen Verbindungen in Zäpfchenform oder als Formulierung
mit verlängerter
Freisetzung zur Deponierung unter der Haut oder zur intramuskulären Injektion
zu formulieren.
-
Die
antagonistischen Verbindungen werden in einer therapeutisch wirksamen
Dosis in Übereinstimmung
mit der Erfindung verabreicht werden. Eine therapeutische Konzentration
wird diejenige Konzentration sein, die eine Reduktion des besonderen
Zustands (wie Toxizität
aufgrund von Retinoid- oder Vitamin A-Kontakt oder Nebenwirkung
von Retinoidwirkstoff) bewirkt oder seine Ausdehnung verlangsamt.
Es versteht sich, daß die
antagonistischen Verbindungen bei Koverabreichung zur Blockierung
der Retinoid-induzierten Toxizität
oder Nebenwirkungen in Übereinstimmung
mit der Erfindung in einer prophylaktischen Weise verwendet werden,
um das Einsetzen eines besonderen Zustands zu verhindern, wie zum
Beispiel eine Hautreizung.
-
Eine
nützliche
therapeutische oder prophylaktische Konzentration wird von Zustand
zu Zustand variieren und kann in bestimmten Fällen mit der Schwere des behandelten
Zustands und dem Ansprechen des Patienten auf die Behandlung variieren.
Entsprechend wird keine einzelne Konzentration einheitlich nützlich sein,
sondern wird eine Modifikation in Abhängigkeit von den Einzelheiten
der chronischen oder akuten Retinoidtoxizität oder des verwandten behandelten
Zustands erfordern. Zu solchen Konzentrationen kann man durch routinemäßiges Experimentieren
gelangen. Jedoch wird vorhergesehen, daß eine Formulierung, die zwischen
0,01 und 1,0 mg von antagonistischer Verbindung pro Milliliter Formulierung
enthält,
eine therapeutisch wirksame Konzentration für die topische Anwendung darstellen
wird. Bei systemischer Verabreichung würde man erwarten, daß eine Menge zwischen
0,01 und 5 mg pro kg Körpergewicht
pro Tag ein therapeutisches Ergebnis bewirken würde.
-
Die
Basis für
den Nutzen von RAR-Antagonist zur Prävention oder Behandlung von
RAR-Agonist-induzierter Toxizität
ist die kompetitive Hemmung der Aktivität von RAR-Rezeptoren durch
RAR-Agonisten. Die Hauptunterscheidung zwischen diesen zwei Anwendungen
von RAR-Antagonisten ist die Gegenwart oder Abwesenheit von vorbestehender
Retinoidtoxizität.
Die meisten der unmittelbar nachfolgend beschriebenen Beispiele
betreffen die Verwendung von Retinoiden zur Prävention von Retinoidtoxizität, aber
die hier beschriebenen allgemeinen Verfahren sind ebenso auf die
Behandlung von vorbestehender Retinoidtoxizität anwendbar.
-
Beschreibung von Experimenten,
die die Verwendung von RAR-Antagonisten
zur Prävention
oder Behandlung von Retinoidtoxizität und/oder Nebenwirkungen von
Retinoidwirkstoffen zeigen
-
Beispiel 1: Durch topisch
eingesetzten Agonisten induzierte Hautreizung wird mit topisch eingesetztem
Antagonisten behandelt
-
Die
Verbindung 4-[(E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethylnaphthalin-2-yl)propen-1-yl]benzoesäure, bezeichnet
als AGN 191183, ist im Stand der Technik als wirksamer RAR-Agonist
bekannt (siehe zum Beispiel die Beschreibung und 2b aus US-PS 5,324,840). (Die "AGN"-Nummer ist eine zufällig zugeordnete
Referenznummer, die durch den betrieblichen Rechtsnachfolger der
vorliegenden Erfindung zur Identifizierung von Verbindungen verwendet
wird).
-
4-[(5,6-Dihydro-5,5-dimethyl-8-(phenyl)-2-naphthalinyl)ethinyl]benzoesäure (AGN
192869, ebenfalls als Verbindung 60a bezeichnet) ist eine Verbindung,
deren Herstellung nachfolgend beschrieben wird. Diese Verbindung
ist ein RAR-Antagonist.
-
Eine
durch einen RAR-Agonisten, AGN 191183, topisch verabreicht, induzierte
Hautreizung kann durch einen RAR-Antagonisten,
AGN 192869, ebenfalls topisch an haarlose Mäuse verabreicht, blockiert
werden.
-
Insbesondere
wurde die Hautreizung auf einer halbquantitativen Skala durch die
tägliche
subjektive Auswertung von Hautabblättern und Abrasionen gemessen.
Eine einzelne Zahl, die topische Reizungsbewertung, faßt die in
einem Tier induzierte Hautreizung im Verlauf eines Experiments zusammen.
Die topische Reizungsbewertung wird wie folgt berechnet. Die topische
Reizungsbewertung ist die algebraische Summe einer verbunden Abblätterbewertung
und einer verbundenen Abrasionsbewertung. Die verbundenen Bewertungen reichen
von 0–9
und 0–8
für Abblättern bzw.
Abrasionen und berücksichtigen
die maximale Schwere, die Zeit des Einsetzens und die beobachtete
durchschnittliche Schwere des Abblätterns und der Abrasionen.
-
Die
Schwere des Abblätterns
wird auf einer Skala mit 5 Punkten bewertet, und die Schwere der
Abrasionen wird auf einer Skala mit 4 Punkten bewertet, wobei höhere Bewertungen
eine größere Schwere
widerspiegeln. Die Komponente der maximalen Schwere der verbundenen
Bewertungen wäre
die höchste
tägliche Schwerebewertung,
die einem gegebenen Tier während
des Beobachtungsverlaufs zugewiesen wird.
-
Für die Komponente
der verbundenen Bewertung für
die Zeit des Einsetzens wird eine Bewertung im Bereich von 0 bis
4 wie folgt zugeordnet:
-
Tabelle
6 Zeit
bis zum Erscheinen von Abblättern
oder Abrasionen der Schwere 2 oder höher
-
Die
Komponente der verbundenen Bewertung der durchschnittlichen Schwere
ist die Summe der Bewertungen von täglichem Abblättern oder
Abrasion geteilt durch die Anzahl von Beobachtungstagen. Der erste Behandlungstag
wird nicht gezählt,
da die Wirkstoffverbindung noch keine Möglichkeit hatte, zur Zeit der
ersten Behandlung zu wirken.
-
Zur
Berechnung der verbundenen Abblätter-
und Abrasionsbewertungen werden die Bewertungen der durchschnittlichen
Schwere und der Zeit des Einsetzens summiert und durch 2 geteilt.
Das Ergebnis wird zur Bewertung der maximalen Schwere addiert. Die
verbundenen Abblätter-
und Abrasionsbewertungen werden dann summiert, um die gesamte topische
Reizungsbewertung zu ergeben. Jedes Tier erhält eine topische Reizungsbewertung,
und die Werte werden als Mittelwert ± Standardabweichung der individuellen
Bewertungen einer Gruppe von Tieren ausgedrückt. Werte werden zur nächsten ganzen
Zahl gerundet.
-
Weibliche
haarlose Mäuse
[Crl:SKH1-hrBR] (8–12
Wochen alt, n = 6) wurden topisch für 5 aufeinanderfolgende Tage
mit Aceton, AGN 191183, AGN 192869 oder einer Kombination aus AGN
192869 und 191183 behandelt. Die Dosen der jeweiligen Verbindungen
sind in Tabelle 7 angegeben. Die Behandlungen werden auf die Rückenhaut
in einem Gesamtvolumen von 4 ml/kg (~0,1
ml) aufgetragen. Die Mäuse
wurden täglich
beobachtet und auf Abblättern
und Abrasionen bis zu und einschließlich 3 Tage nach der letzten
Behandlung, d.h. Tag 8, bewertet.
-
Tabelle
7 Experimenteller
Aufbau und Ergebnisse, Beispiel 1
-
Die
topischen Reizungsbewertungen für
Beispiel 1 sind in Tabelle 7 angegeben. Weder Aceton (Träger) noch
AGN 192869 (Antagonist) bei einer Dosis von 1,5 mg/kg/d (Gruppe
F) verursachte eine beobachtbare topische Reizung. AGN 191183, der
RAR-Agonist, verursachte eine mäßige topische
Reizung bei einer Dosis von 0,025 mg/kg/d. Jedoch wurde die durch
AGN 191183 induzierte topische Reizung in einer dosisabhängigen Weise
durch AGN 192869 gehemmt, mit einer beinahe vollständigen Aufhebung
der Reizung in Gegenwart eines 50-fachen molaren Überschusses
von AGN 192869. Dies zeigt, daß ein
topischer RAR-Antagonist die durch einen topischen RAR-Agonisten
verursachte Hautreizung blockiert. Eine vollständige Blockade von durch RAR-Agonist
induzierter Hautreizung kann mit geringeren Molverhältnissen
von Antagonist zu Agonist erreicht werden, wenn der RAR-Agonist
wirksamer ist, wie zum Beispiel die Verbindung 4-[(5,6- Dihydro-5,5-dimethyl-8-(4-methylphenyl)-2-naphthalinyl)ethinyl]benzoesäure (AGN
193109, ebenfalls in dieser Anmeldung als Verbindung 60 bezeichnet).
-
Beispiel 2: Durch oral
eingesetzten Agonisten induzierte Hautreizung wird mit topisch eingesetztem
Antagonisten blockiert
-
Der
wirksame RAR-Agonist AGN 191183 (4-[(E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethylnaphthalin-2-yl)propen-1-yl]benzoesäure) und
der wirksame RAR-Antagonist 4-[(5,6-Dihydro-5,5-dimethyl-8-(4-methylphenyl)-2-naphthalinyl)ethinyl]benzoesäure (AGN
193109, Verbindung 60) wurden in diesem Beispiel verwendet, und
das Körpergewicht
der Versuchstiere (Mäuse)
wurde als Marker des systemischen RAR-Agonistkontakts verwendet.
-
Gruppen
von weiblichen haarlosen Mäusen
(8–12
Wochen alt, n = 6) wurden durch intragastrale Intubation mit Maisöl oder AGN
191183 (0,26 mg/kg), suspendiert in Maisöl (5 ml/kg), behandelt. Die
Mäuse wurden
gleichzeitig auf der Rückenhaut
mit Träger
(97,6% Aceton/2,4% Dimethylsulfoxid) oder Lösungen von AGN 193109 in Träger (6 ml/kg)
topisch behandelt. Spezifische Dosen für die unterschiedlichen Behandlungsgruppen
sind in Tabelle 8 angegeben. Die Behandlungen wurden täglich für 4 aufeinanderfolgende
Tage verabreicht. Die Mäuse
wurden gewogen und auf topische Reizung täglich wie in Beispiel 1 beschrieben
bis zu und einschließlich
1 Tag nach der letzten Behandlung bewertet. Die prozentuale Körpergewichtsveränderung wird
durch Subtrahieren des Körperendgewichts
(Tag 5) vom Körperanfangsgewicht
(Tag 1), Dividieren durch das Körperanfangsgewicht
und Multiplizieren mit 100% berechnet. Topische Reizungsbewertungen
werden wie in Beispiel 1 beschrieben berechnet.
-
Topische
Reizungsbewertungen und Gewichtsverlust für die unterschiedlichen Gruppen
sind in Tabelle 8 angegeben. Kombinierte Behandlung mit den topischen
und oralen Trägern,
d.h. Aceton bzw. Maisöl,
verursachte keine topische Reizung oder Gewichtsverlust. In ähnlicher
Weise führte
die kombinierte Behandlung mit dem oralen Träger und dem topischen Antagonisten
AGN 193109 zu keiner topischen Reizung oder Gewichtsverlust. Orales
AGN 191183 als solches induzierte einen substantiellen Gewichtsverlust
und substantielle Hautreizung. Durch AGN 191183 induzierte Hautreizung
wurde wesentlich reduziert bei Kombination mit der niedrigeren Dosis
von AGN 193109 und vollständig
blockiert bei der höheren
Dosis von AGN 193109. Durch AGN 191183 induzierter Gewichtsverlust
wurde ebenfalls in einer dosisabhängigen Weise durch topisches
AGN 193109 blockiert, aber die Blockade war nicht vollständig. Daher
blockiert topisches AGN 193109 vorzugsweise die dermale Toxizität von AGN
191183. Vermutlich wurden geringe Mengen von AGN 193109 systemisch
absorbiert und blockierten somit partiell den durch AGN 191183 induzierten
Gewichtsverlust. Jedoch wäre
eine solche Absorption wahrscheinlich noch geringer in einer Art
mit weniger durchlässiger
Haut, wie zum Beispiel Menschen. Alternativ könnte die Inhibierung der Gewichtsverlusts
durch AGN 193109 auf der Linderung der durch AGN 191183 induzierten
Hautreizung beruhen.
-
Tabelle
8 Experimenteller
Aufbau und Ergebnisse, Beispiel 2
-
Somit
zeigt Beispiel 2, daß topisch
verabreichte RAR-Antagonisten
zur vorzugsweisen Blockierung der Hautreizung verwendet werden können, die
durch einen oral verabreichten RAR-Agonisten induziert wird.
-
Beispiel 3: Topisch eingesetzter
Antagonist beschleunigt die Erholung von vorbestehender Retinoidtoxizität
-
In
diesem Beispiel wird Gewichtsverlust durch topische Behandlung mit
dem RAR-Agonisten AGN 191183 induziert, und dann werden die Versuchstiere
topisch mit entweder Träger
oder dem RAR-Antagonisten AGN 193109 behandelt.
-
Weibliche
haarlose Mäuse
(8–12
Wochen alt, n = 5) wurden topisch mit AGN 191183 (0,13 mg/kg/d) in
Träger
(97,6% Aceton/2,4% DMSO, 4 ml/kg) täglich für 2 Tage behandelt. Gruppen
der gleichen Mäuse
(n = 5) wurden dann topisch entweder mit Träger oder AGN 193109 in Träger (4 ml/kg)
täglich
für 3 aufeinanderfolgende
Tage, beginnend am Tag 3, behandelt. Die Mäuse wurden an den Tagen 1–5 und am
Tag 8 gewogen. Die Körpergewichte
werden als Mittelwert ± Standardabweichung
ausgedrückt.
Mittelwerte wurden statistisch unter Verwendung eines ungepaarten
zweiseitigen t-Tests verglichen. Unterschiede wurden bei P < 0,05 als signifikant
betrachtet.
-
Tabelle
9 Ergebnisse,
Beispiel 3
-
Der
Zeitverlauf der Körpergewichte
in Beispiel 3 ist in Tabelle 9 angegeben. Körpergewichte beider Gruppen
von Mäusen
waren parallel an den Tagen 2 und 3 als Ergebnis der Behandlung
mit AGN 191183 an den Tagen 1 und 2 verringert. Körpergewichte
in den zwei Gruppen waren nicht signifikant unterschiedlich an den
Tagen 1, 2 oder 3. Jedoch erhöhte
die Behandlung mit AGN 193109 signifikant das Körpergewicht relativ zur Behandlung
mit Träger
an den Tagen 4 und 5. Diese Daten zeigten, daß die Erholung von durch AGN 191183
induziertem Körpergewichtsverlust
durch anschließende
Behandlung mit AGN 193109 beschleunigt wurde. Körpergewichte waren nicht signifikant
unterschiedlich zwischen den zwei Gruppen von Mäusen am Tag 8, was zeigt, daß eine vollständige Erholung
in beiden Gruppen erreichbar war, vorausgesetzt ausreichend Zeit
wurde zugestanden. Somit sind RAR-Antagonisten wirksam in der Linderung
von durch RAR-Agonisten induzierter Toxizität, selbst falls die durch RAR-Agonist
induzierte Toxizität
der Behandlung mit RAR-Antagonist vorangeht, d.h. im Vergiftungsszenario
mit RAR-Agonist.
-
Beispiel 4: Oral eingesetzter
Antagonist blockiert durch oral koverabreichten Retinoidagonisten
induzierte Hypertriglyceridämie
-
5-[(E)-2-(5,6,7,8-Tetrahydro-3,5,5,8,8-pentamethylnaphthalin-2-yl)propen-1-yl]-2-thiophencarbonsäure ist
ein bekannter RAR/RXR-Panagonist (siehe US-PS 5,324,840, Spalte
32) und wird als AGN 191659 bezeichnet. Diese Verbindung wurde oral
verwendet, um akute Hypertriglyceridämie in Ratten zu induzieren,
und AGN 193109 (Verbindung 60) wurde oral koverabreicht, um die
durch AGN 191659 induzierte Hypertriglyceridämie zu blockieren.
-
Männliche
Fischer-Ratten (6–7
Wochen alt, n = 5) wurden durch intragastrale Intubation mit Maisöl (Träger), AGN
191659, AGN 193109 oder einer Kombination aus AGN 191659 und AGN
193109 behandelt. AGN 191659 und AGN 193109 wurden als feine Suspensionen
in Maisöl
gegeben. Der experimentelle Aufbau, einschließlich der Dosen, wird in Tabelle
10 angegeben.
-
Blut
wurde aus der unteren Hohlvene unter Narkose mit Kohlendioxid entnommen.
Serum wurde vom Blut durch Zentrifugieren mit geringer Geschwindigkeit
abgetrennt. Gesamtserumtriglyceride (Triglyceride plus Glycerin)
wurden mit einem standardmäßigen spektrophotometrischen
Endpunkttest gemessen, der als Kit kommerziell erhältlich ist
und an ein Plattenformat mit 96 Vertiefungen angepaßt wurde.
Serumtriglyceridspiegel werden als Mittelwert ± Standardabweichung ausgedrückt. Mittelwerte
wurden statistisch durch einfache Variansanalyse gefolgt vom Dunnett-Test
verglichen, falls signifikante Unterschiede festgestellt wurden.
Unterschiede wurden bei P < 0,05
als signifikant betrachtet.
-
Wie
in Tabelle 10 gezeigt wird, verursacht AGN 191659 selbst eine signifikante
Anhebung von Serumtriglyceriden relativ zur Behandlung mit Träger. AGN
193109 selbst erhöhte
Serumtriglyceride nicht signifikant. Bedeutend ist, daß die Kombination
aus AGN 193109 und AGN 191659 bei Molverhältnissen von 1:1 und 5:1 Serumtriglyceride
auf Spiegel reduzierte, die nicht signifikant unterschiedlich von
der Kontrolle waren.
-
Tabelle
10 Experimenteller
Aufbau und Ergebnisse, Beispiel 4
-
Beispiel
4 zeigt, daß ein
RAR-Antagonist zur Blockierung von Hypertriglyceridämie verwendet
werden kann, die durch ein koverabreichtes Retinoid induziert wird.
-
Beispiel 5: Parenteral
eingesetzter Antagonist blockiert durch parenteral koverabreichten
Retinoidagonisten induzierte Knochentoxizität
-
Beispiel
5 zeigt, daß RAR-Antagonisten
die durch einen RAR-Agonisten
induzierte Knochentoxizität blockieren
können.
In diesem Beispiel wird AGN 193109 verwendet, um einen durch einen
koverabreichten RAR-Agonisten, AGN 191183, in Meerschweinchen verursachten
vorzeitigen Epiphysenschluß zu
blockieren.
-
Gruppen
von männlichen
Hartley-Meerschweinchen (~3 Wochen alt,
n = 4) wurden intraperitoneal osmotische Pumpen implantiert, die
Träger
(20% Dimethylsulfoxid, 80% Polyethylenglykol-300), AGN 191183 (0,06
mg/ml) oder AGN 191183 (0,06 mg/ml) in Kombination mit AGN 193109
(0,34 mg/ml) enthielten. Die osmotischen Pumpen werden vom Hersteller
konstruiert, um ca. 5 μl
Lösung
pro Stunde kontinuierlich für
14 Tage zu übertragen.
-
Die
Tiere wurden durch Einatmen von Kohlendioxid 14 Tage nach der Implantation
getötet.
Das linke Schienbein wurde entfernt und in 10%iges gepuffertes Formalin
gegeben. Die Schienbeine wurden durch Kontakt mit einer Ameisensäure/Formalin-Lösung für 3–4 Tage
entkalkt, und Paraffinschnitte wurden präpariert. Die Schnitte wurden
mit Hämatoxylin
und Eosin durch Standardverfahren angefärbt. Die proximale Epiphysenplatte
des Schienbeins wurde untersucht und als geschlossen oder nicht
geschlossen bewertet. Epiphysenschluß wird für diesen Zweck als jede Unterbrechung
der Kontinuität
des Knorpels der Epiphysenwachstumsplatte definiert, d.h. Austausch
durch Knochen und/oder Fibroblastengewebe.
-
Keines
der vier mit Träger
behandelten Meerschweinchen zeigte einen Epiphysenschluß am Ende
des Experiments. Dies war erwartet, da die proximale Epiphysenplatte
des Meerschweinchen-Schienbeins sich normalerweise nicht schließt, bis
die Tiere wenigstens 10 Monate alt sind. Alle vier der mit AGN 191183
behandelten Meerschweinchen zeigten einen partiellen oder vollständigen Epiphysenschluß. Jedoch
zeigte keines der mit der Kombination aus AGN 191183 und AGN 193109
behandelten Meerschweinchen einen Epiphysenschluß. Deshalb blockiert AGN 193109
bei einem 5-fachen molaren Überschuß vollständig die
durch AGN 191183 induzierte Knochentoxizität, wenn diese Verbindungen
parenteral koverabreicht wurden.
-
RAR-antagonistische
Verbindungen
-
Die
Verbindungen 4-[(5,6-Dihydro-5,5-dimethyl-8-(4-methylphenyl)-2-naphthalinyl)ethinyl]benzoesäure (AGN
193109, Verbindung 60) und 4-[(5,6-Dihydro-5,5-dimethyl-8-(phenyl)-2-naphthalinyl)ethinyl]benzoesäure (AGN
192869, Verbindung 60a) sind Referenzbeispiele für RAR-Antagonisten, die in
den oben beschriebenen Tierversuchen zur Blockierung von RAR-Rezeptoren in Übereinstimmung
mit der vorliegenden Erfindung verwendet wurden.
-
Syntheseverfahren – Aryl-substituierte
Verbindungen
-
Die
exemplarischen RAR-antagonistischen Verbindungen der Formel (1)
können
durch die hier veranschaulichten chemischen Synthesereaktionen hergestellt
werden. Der Synthesechemiker wird leicht einsehen, daß die hier
dargestellten Bedingungen spezifische Ausführungsformen sind, die für jede und
alle durch Formel (1) dargestellten Verbindungen verallgemeinert
werden können.
-
-
-
(a)
MeMgCl/THF/dann H2SO4/H2O/100°C
(100%); (b) CH3COCl/AlCl3/CH2Cl2 (54%); (c) NaOCl/NaOH/Dioxan/65°C dann EtOH/H2SO4/Δ (100%);
(d) Br2/HOAc (100%); (e) CrO3/HOAc/Ac2O (76%); (f) p-Tolyl-MgBr/THF dann p-TsOH·H2O/Toluol/Δ (25%);
(g) NaOH/EtOH/H2O (98%); (h) EDC/DMAP/DMF/2-F-4-H2NC5H3CO2Et (21,63%) oder 2,6-Difluor-4-H2NC6H2CO2Et
(22,33%); (i) NaOH/EtOH/H2O (4,86%; 5,70%).
-
Reaktionsschema 6B
-
Reaktionsschema
6B offenbart einen Syntheseweg zu spezifischen bevorzugten Benzopyran-(Chromen)-Verbindungen
der Erfindung, worin unter Bezugnahme auf Formel (1) die Z-Gruppe auch CONH
ist (Amid-Derivate). In Übereinstimmung
mit diesem Schema wird Dihydrocumarin (Verbindung 213) einer Grignard-Reaktion
mit Methylmagnesiumchlorid unterworfen, gefolgt von Erwärmen einer
tertiären
Alkoholzwischenstufe mit Säure,
um 2,2-Dimethylchroman (Verbindung 214) zu liefern. Verbindung 214
wird zu 6-Acetyl-2,2-dimethylchroman (Verbindung 215) in einer Friedel-Crafts-Reaktion
umgewandelt, und letzteres wird mit NaOCl in Gegenwart von Natriumhydroxid
oxidiert und danach verestert, um Ethyl-2,2-dimethylchroman-6-carboxylat (Verbindung
216) zu ergeben. Verbindung 216 wird mit Brom in Eisessig behandelt,
um Ethyl-8-brom-2,2-dimethylchroman-6-carboxylat
(Verbindung 217) zu ergeben. Verbindung 217 wird mit Chromtrioxid
zur Bildung einer Ketonfunktion in der 4-Position des Chromanrings
oxidiert und liefert Ethyl-8-brom-2,2-dimethyl-4-chromanon-6-carboxylat (Verbindung
218). Reaktion von Verbindung 218 mit dem aus 4-Bromtoluol hergestellten
Grignard-Reagens und anschließendes
Erwärmen
mit p-TSOH zur Vervollständigung
der Dehydratisierung des intermediären tertiären Alkohols liefert Ethyl-8-Brom-2,2-dimethyl-4-tolyl-4(2H)-chroman-6-carboxylat
(Verbindung 219). Verseifung von Verbindung 219 mit Base ergibt
die entsprechende freie Carbonsäure,
8-Brom-2,2-dimethyl-4-tolyl-4(2H)-chroman-6-carbonsäure (Verbindung 220).
Verbindung 220 wird mit Ethyl-2-fluor-4-aminobenzoat und Ethyl-2,6-difluor-4-aminobenzoat
in Gegenwart von 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
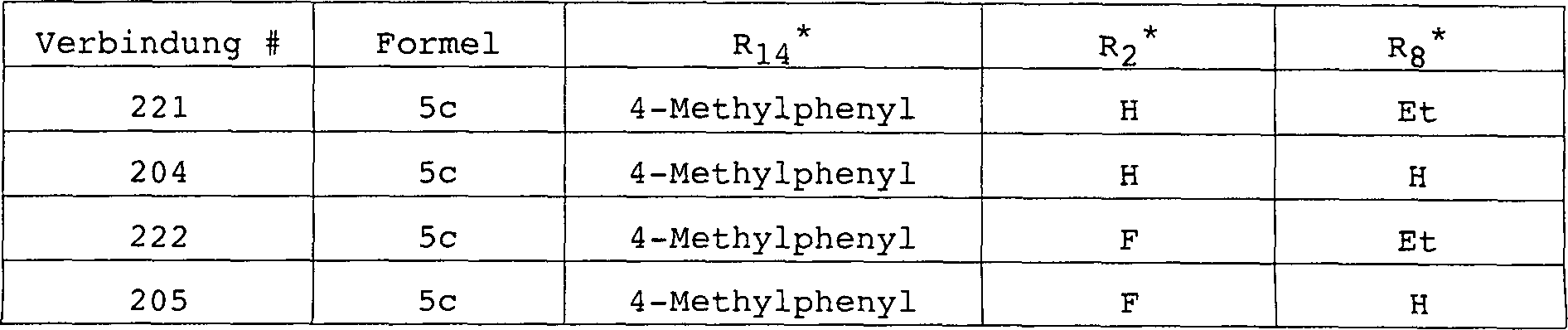
(EDC) und 4-Dimethylaminopyridin (DMAP) kondensiert, um Ethyl-2-fluor-4-[[(8-brom-2,2-dimethyl-4-tolyl-6-chromanyl)carbonyl]amino]benzoat (Verbindung
221) bzw. Ethyl-2,6-difluor-4-[[(8-brom-2,2-dimethyl-4-tolyl-6-chromanyl)carbonyl)amino]benzoat (Verbindung
222) zu liefern. Verseifung von Verbindungen 221 und 222 liefert
die entsprechenden freien Carbonsäuren, 2-Fluor-4-[[(8-brom-2,2-dimethyl-4-tolyl-6-chromanyl)carbonyl]amino]benzoesäure (Verbindung 204)
bzw. 2,6-Difluor-4-[[(8-brom-2,2-dimethyl-4-tolyl-6-chromanyl)carbonyl]amino]benzoesäure (Verbindung 205).
-
Spezifische
Beispiele
-
2,2-Dimethylchroman (Verbindung
214)
-
Zu
einer Lösung
aus Dihydrocumarin (Verbindung 213, 5,0 g, 33,78 mmol) in trockenem
THF (50 ml) bei 0°C
wurde Methylmagnesiumchlorid (33,7 ml, 3 M Lösung in THF, 0,1 mol) getropft.
Die Reaktionsmischung wurde langsam auf Raumtemperatur erwärmt und über Nacht
gerührt.
Extraktion (Et2O), Trocknen (Na2SO4) und Aufkonzentrieren unter reduziertem
Druck lieferte ein Diol (6,25 g) als farblosen Feststoff. Das Diol
wurde mit 15%igem wäßrigem H2SO4 (50 ml) in Benzol
(15 ml) kombiniert und die resultierende Mischung für 2 Stunden
auf 105°C
erwärmt.
Nach Abkühlen
auf Raumtemperatur wurde die Mischung mit Et2O
extrahiert, getrocknet (Na2SO4)
und unter reduziertem Druck auf konzentriert, um 6,0 g (100%) der
Titelverbindung als hellgelbe Flüssigkeit
zu ergeben.
1H-NMR δ: 7,08 (2H, m), 6,80 (2H, m),
2,79 (2H, t, J = 6,8 Hz), 1,81 (2H, t, J = 6,8 Hz), 1,35 (6H, s).
-
6-Acetyl-2,2-dimethylchroman
(Verbindung 215)
-
Zu
einer Lösung
aus 2,2-Dimethylchroman (Verbindung 214, 15 g, 19,4 mmol) in trockenem
CH2Cl2 (100 ml)
wurde Acetylchlorid (1,52 ml, 21,3 mmol) gefolgt von AlCl3 (2,84 g, 21,3 mmol) gegeben. Die Reaktionsmischung
wurde bei Raumtemperatur für
30 Minuten gerührt
und dann in Eiswasser gegossen. Extraktion (CH2Cl2), Trocknen (Na2SO4) und Auf konzentrieren unter reduziertem
Druck lieferte ein gelbes Öl.
Säulenchromatographie
(EtOAc:Hexan 1:9) lieferte 2,15 g (54%) der Titelverbindung als
gelben Feststoff.
1H-NMR δ: 7,73 (2H,
m), 6,80 (1H, d, J = 8,3 Hz), 2,83 (2H, t, J = 6,9 Hz), 2,54 (3H,
s), 1,84 (2H, t, J = 6,9 Hz), 1,36 (6H, s).
Analyse (C13H16)O2 C,
H
-
Ethyl-2,2-dimethylchroman-6-carboxylat
(Verbindung 216)
-
Eine
Lösung
aus 6-Acetyl-2,2-dimethylchroman (Verbindung 215, 2,15 g, 10,5 mmol)
und NaOH (2,0 g, 50 mmol) in Dioxan (50 ml) und Bleiche (50 ml,
5,25% NaOCl) wurde für
4 Tage auf 65°C
erwärmt.
Nach Abkühlen
auf Raumtemperatur wurde Na2SO3 hinzugegeben,
bis eine Teilmenge dieser Lösung
farblos blieb, wenn sie mit einem Tropfen einer Lösung aus
I2 in CCl4 behandelt
wurde. Die Lösung
wurde mit H2SO4 angesäuert (pH
4) und mit Ether extrahiert. Die vereinigten organischen Schichten
wurden getrocknet (Na2SO4)
und unter reduziertem Druck aufkonzentriert. Säulenchromatographie (EtOAc:Hexan 1:3)
lieferte 1,98 g (92%) 2,2-Dimethylchroman-5-carbonsäure als
hellbraunen Feststoff.
1H-NMR δ: 7,78 (2H,
m), 6,82 (1H, d, J = 8,3 Hz), 2,83 (2H, t, J = 6,7 Hz), 1,85 (2H,
t, J = 6,7 Hz), 1,37 (6H s).
-
Zu
einer Lösung
aus diesem Feststoff (1,64 g, 7,94 mmol) in EtOH (50 ml) wurde H2SO4 (0,5 ml) gegeben.
Die Mischung wurde über
Nacht auf 95°C
erwärmt,
auf Raumtemperatur abgekühlt
und unter reduziertem Druck auf konzentriert. Säulenchromatographie (EtOAc:Hexan,
1:5) lieferte 1,91 g (100%) der Titelverbindung als farblosen Feststoff.
1H-NMR δ:
7,79 (2H, m), 6,79 (2H, d, J = 8,3 Hz), 4,34 (2H, q, J = 7,2 Hz),
2,82 (2H, t, J = 6,7 Hz), 1,84 (2H, t, J = 6,7 Hz), 1,38 (3H, t,
J = 7,2 Hz), 1,36 (6H, s).
Analyse (C14H18O3·0,25H2O) C, H.
-
Ethyl-8-brom-2,2-dimethylchroman-6-carboxylat
(Verbindung 217)
-
Zu
einer Lösung
aus Ethyl-2,2-dimethylchroman-6-carboxylat (Verbindung 216, 500
mg, 2,14 mmol) in HOAc (4 ml) wurde Br2 (0,11
ml, 2,14 mmol) gegeben. Die Reaktionsmischung wurde bei Raumtemperatur über Nacht
gerührt,
und dann wurde ein Luftstrom durch die Reaktionsmischung zur Entfernung
des überschüssigen Br2 geleitet. Entfernung des Lösungsmittels
unter reduziertem Druck und Säulenchromatographie unter
Verwendung des Rückstands
(EtOAc:Hexan 1:9) lieferte 741 mg (100%) der Titelverbindung als Öl.
1H-NMR δ:
8,05 (1H, d, J = 2,2 Hz), 7,73 (1H, d, J = 2,2 Hz), 4,34 (2H, q,
J = 7,1 Hz), 2,84 (2H, t, J = 6,7 Hz), 1,85 (2H, t, J = 6,7 Hz),
1,40 (6H, s), 1,38 (3H, t, J = 7,2 Hz).
Analyse (C14H17BrO3·0,2H2O) C, H.
-
Ethyl-8-Brom-2,2-dimethyl-4-chromanon-6-carboxylat
(Verbindung 218)
-
Zu
einer Lösung
aus HOAc (15 ml) und Ac2O (7,5 ml) bei 0°C wurde CrO3 (1,18 g, 12 mmol) in kleinen Portionen
gegeben. Diese Lösung
wurde für
15 Minuten gerührt
und mit Benzol (10 ml) verdünnt,
und eine Lösung
aus Ethyl-B-Brom-2,2-dimethylchroman-6-carboxylat
(Verbindung 217, 741 mg, 2,38 mmol in Benzol (5 ml) wurde langsam über mehrere
Minuten hinzugegeben. Nach Rühren
bei 0°C
für 4 Stunden
wurde die Reaktionsmischung auf Eis gegossen, mit EtOAc extrahiert,
die vereinigten organischen Schichten wurden getrocknet (Na2SO4) und unter reduziertem
Druck aufkonzentriert, um ein Öl
zu ergeben. Säulenchromatographie (EtOAc:Hexan,
1:9) liefert 589 mg (76%) der Titelverbindung als farblosen Feststoff.
1H-NMR δ:
8,5 (1H, d, J = 2,1 Hz), 8,40 (1H, d, J = 2,1 Hz), 4,37 (2H, q,
J = 7,1 Hz), 2,80 (2H, s), 1,54 (6H, s), 1,40 (3H, t, J = 7,1 Hz);
Analyse
(C14H15BrO4) C, H, N.
-
Ethyl-8-brom-2,2-dimethyl-4-(4-methylphenyl)-4(2H)-chroman-6-carboxylat (Verbindung
219)
-
Zu
einer Lösung
aus Ethyl-8-brom-2,2-dimethyl-4-chromanon-6-carboxylat (Verbindung 218, 589 mg, 1,81
mmol) in THF (20 ml) unter Argongas bei –78°C wurde p-Tolylmagnesiumbromid
(2,16 ml, 2,72 mmol, 1 M Lösung
in Ether) gegeben. Die Reaktionsmischung wurde langsam auf Raumtemperatur
erwärmt
und über Nacht
gerührt.
Gesättigte
NH4Cl-Lösung
wurde zur Reaktion bei 0°C
gegeben und die resultierende Mischung mit Ether extrahiert. Die
vereinigten organischen Schichten wurden mit gesättigtem NaCl gewaschen, über Na2SO4 getrocknet und
unter reduziertem Druck aufkonzentriert. Säulenchromatographie (EtOAc:Hexan,
1:3) lieferte 297 mg einer Mischung aus der Titelverbindung und
dem intermediären
tertiären
Alkohol als Öl.
Diese Mischung wurde mit p-TsOH (20 mg) in trockenem Toluol (15
ml) vereinigt und für
30 Minuten auf 110°C
erwärmt.
Nach Abkühlen
auf Raumtemperatur wurde das Lösungsmittel
unter reduziertem Druck entfernt, und das Produkt (182 mg, 25%)
wurde durch Säulenchromatographie
(EtOAc:Hexan 1:3) isoliert.
1H-NMR δ: 8,1 (1H,
d, J = 1,7 Hz), 7,68 (1H, d, J = 1,7 Hz), 7,22 (4H, s), 5,67 (1H,
s), 4,29 (2H, q, J = 7,1 Hz), 2,41 (3H, s), 1,56 (6H, s), 1,33 (t,
J = 7,1 Hz);
MS (EI) m/e 402, 400, 387, 385, 359, 357.
Analyse
(C21H21BrO3·0,45H2O) C, H.
-
8-brom-2,2-dimethyl-4-(4-methylphenyl)-4(2H)-chroman-6-carbonsäure (Verbindung
220)
-
Zu
einer Lösung
aus Ethyl-8-brom-2,2-dimethyl-4-(4-methylphenyl)-4(2H)-chroman-6-carboxylat
(Verbindung 219, 189 mg, 0,51 mmol) in EtOH (15 ml) und THF (10
ml) wurde 20%iges wäßriges NaOH
(2 ml) gegeben. Die Reaktionsmischung wurde für 2 h auf 45°C erwärmt, auf
Raumtemperatur abgekühlt
und mit 10%igem wäßrigem HCl
auf pH 4 angesäuert.
Extraktion (EtOAc), Trocknen (MgSO4) und
Auf konzentrieren unter reduziertem Druck lieferte die Titelverbindung
(171 mg) (98%) als farblosen Feststoff.
1H-NMR δ: 8,15 (d,
J = 2,0 Hz, 1H), 7,71 (d, J = 2,0 Hz, 1H), 7,21 (m, 4H), 5,69 (s,
1H), 2,42 (s, 3H), 1,58 (s, 6H).
-
Ethyl-2-fluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chromanyl)carbonyl]amino]benzoat
(Verbindung 221)
-
Zu
einer Lösung
aus 8-Brom-2,2-dimethyl-4-(4-methylphenyl)-4(2H)-chroman-6-carbonsäure (Verbindung
220, 100 mg, 0,29 mmol) ind CH2Cl2 (8 ml) wurden DMAP (87 mg, 0,69 mmol),
Ethyl-4-amino-2-fluorbenzoat (53 mg, 0,29 mmol) und EDC (72 mg,
0,37 mmol) gegeben. Die Reaktionsmischung wurde bei Raumtemperatur über Nacht
gerührt
und dann zur Trockene auf konzentriert. Säulenchromatographie (EtOAc:Hexan, 1:3)
lieferte 98 mg (63%) der Titelverbindung als farblosen Feststoff.
1H-NMR δ:
7,99 (1H, bs), 7,89 (1H, t, J = 7,5 Hz), 7,88 (1H, d, J = 2,1 Hz),
7,65 (1H, dd, J = 12,8, 2,1 Hz), 7,25 (1H, dd, J = 7,5 Hz, 2,1 Hz),
7,19 (4H, s), 5,70 (1H, s), 4,36 (2H, q, J = 7,2 Hz), 2,38 (3H,
s), 1,56 (6H, s), 1,39 (3H, t, J = 7,2 Hz)
MS (EI) m/e 537,
537, 524, 526, 357, 355, 312.
Analyse (C28H25BrFNO4) C, H, N.
-
2-Fluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chromanyl)carbonyl]amino]benzoesäure (Verbindung 204)
-
Zu
einer Lösung
aus Ethyl-2-fluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chromanyl)carbonyl]amino]benzoat
(Verbindung 221, 135 mg, 0,25 mmol) in EtOH (5 ml) wurde 20%iges
wäßriges NaOH
(2 ml) gegeben. Die Reaktionsmischung wurde bei Raumtemperatur über Nacht
gerührt
und mit 10%igem wäßrigem HCl
auf pH 4 angesäuert.
Das EtOH wurde unter reduziertem Druck entfernt, und Ethylacetat
und Wasser wurden zum Rückstand
hinzugegeben. Die organische Schicht wurde abgetrennt und mit gesättigtem NaHCO3 und gesättigtem
wäßrigem NaCl
gewaschen und über
MgSO4 getrocknet.
-
Aufkonzentrieren
unter reduziertem Druck lieferte 110 mg (86%) der Titelverbindung
als farblosen Feststoff.
1H-NMR δ (Aceton-d6): 8,09 (1H, d, J = 2,1 Hz), 7,91 (1H, t,
J = 7,5 Hz), 7,68 (1H, d, J = 2,1 Hz), 7,83 (1H, dd, J = 12,8, 2,1
Hz), 7,50 (1H, dd, J = 7,5 Hz, 2,1 Hz), 7,27 (4H, s), 5,87 (1H,
s), 2,37 (3H, s), 1,56 (6H, s);
MS (EI) m/e 511, 509, 496,
494, 452, 450, 357, 355, 312.
Analyse (C26H21BrFNO4·0,6H2O) C, H, N.
-
Ethyl-2,6-difluor-4-[[(8-Brom-2,2-dimethyl-4-(4-methylphenyl)-6-chroman)carbonyl]amino]benzoat
(Verbindung 222)
-
Unter
Verwendung des gleichen Verfahrens wie für die Synthese von Ethyl-2-fluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chromanyl)carbonyl]amino]benzoat
(Verbindung 221) lieferte die Reaktion von 8-Brom-2,2-dimethyl-4-(4-methylphenyl)-4(2H)-chroman-6-carbonsäure (Verbindung
220, 100 mg, 0,29 mmol) mit Ethyl-2,6-difluor-4-aminobenzoat in
Gegenwart von DMAP und EDC in CH2Cl2 53 mg (33%) der Titelverbindung als farbloses Öl.
1H-NMR δ:
7,87 (1H, d, J = 2,1 Hz), 7,47 (1H, d, J = 2,1 Hz), 7,28 (2H, d,
J = 8,7 Hz), 7,19 (4H, m), 5,70 (1H, s), 4,38 (2H, q, J = 7,2 Hz),
2,41 (3H, s), 1,57 (6H, s), 1,38 (3H, t, J = 7,2 Hz).
MS (EI)
m/e 557, 555, 542, 540, 448, 446.
Analyse (C28H24BrF2NO4)
C, H, N.
-
2,6-Difluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chroman)carbonyl]amino]benzoesäure (Verbindung
205)
-
Unter
Verwendung des gleichen Verfahrens wie für die Synthese von 2-Fluor-4-[[(8-Brom-2,2-dimethyl-4-(4-methylphenyl)-6-chromanyl)carbonyl]amino]benzoesäure (Verbindung
204) lieferte die Behandlung von Ethyl-2,6-difluor-4-[[(8-brom-2,2-dimethyl-4-(4-methylphenyl)-6-chroman)carbonyl]amino]benzoat
(Verbindung 222, 53 mg, 0,1 mmol) mit wäßrigem NaH in EtOH 35 mg (70%)
der Titelverbindung als farblosen Feststoff.
1H-NMR δ (Aceton-d6):
9,93 (1H, bs), 8,07 (1H, d, J = 2,1 Hz), 7,66 (1H, d, J = 2,1 Hz),
7,54 (2H, d, J = 9,9 Hz), 7,27 (4H, s), 5,88 (1H, s), 2,38 (3H,
s), 1,56 (6H, s).
MS (EI) m/e 514, 512, 485, 483, 470, 468.
Analyse
(C26H20BrF2NO4) C, H, N.
-
Verfahren
zur Potenzierung von Kernrezeptoragonisten
-
Übersicht
und Einleitung
-
Wir
haben festgestellt, daß eine
Untergruppe von Retinoidantagonisten, die negative Hormonaktivität aufweisen,
zur Potenzierung der biologischen Aktivitäten anderer Retinoide und Hormone
der Steroidrezeptorsuperfamilie verwendet werden können. Diese
anderen Retinoide und Hormone der Steroidrezeptorsuperfamilie können entweder
endogene Hormone oder Pharmazeutika sein. So können zum Beispiel bestimmte Aktivitäten von
pharmazeutischen Retinoidagonisten, wenn sie in Kombination mit
einem negativen Retinoidhormon verwendet werden, aktiver bei der
Ausübung
spezifischer biologischer Wirkungen gemacht werden. Vorteilhaft
kann dieser Kombinationsansatz zur Wirkstoffverabreichung ungewünschte Nebenwirkungen
pharmazeutischer Retinoide minimieren, weil niedrigere Dosierungen
der pharmazeutischen Retinoide mit verbesserter Wirksamkeit verwendet
werden können.
-
Insbesondere
haben wir festgestellt, daß AGN
193109, ein synthetisches Retinoid mit der in 1 gezeigten
Struktur, einzigartige und unerwartete pharmakologische Aktivitäten aufweist.
AGN 193109 weist eine hohe Affinität für die RAR-Unterklasse von Kernrezeptoren auf,
ohne diese Rezeptoren zu aktivieren oder die Transkription von Retinoid-reaktiven
Genen zu stimulieren. Stattdessen hemmt AGN 193109 die Aktivierung von
RARs durch Retinoidagonisten und verhält sich deshalb als Retinoidantagonist.
-
Zusätzlich haben
wir festgestellt, daß negative
Retinoidhormone ohne Koverabreichung eines Retinoidagonisten oder
Steroidhormons zur Bekämpfung
bestimmter Krankheitssymptome verwendet werden können. Insbesondere können die
hier offenbarten negativen Retinoidhormone die hochgradige Basistranskription von
Genen abregulieren, die auf unkomplexierte RARs ansprechen. Falls
zum Beispiel eine unkontrollierte zelluläre Proliferation aus der Aktivität von Genen
resultiert, die auf unkomplexierte RARs ansprechen, dann kann diese
Genaktivität
durch die Verabreichung eines negativen Retinoidhormons reduziert
werden, das RARs inaktiviert. Entsprechend kann die zelluläre Proliferation,
die von der Aktivität
unkomplexierter RARs abhängt, durch
das negative Hormon gehemmt werden. Die Inhibierung von unkomplexierten
RARs kann nicht durch Verwendung herkömmlicher Antagonisten erreicht
werden.
-
Signifikanterweise
haben wir festgestellt, daß AGN
193109 sowohl die RAR-Basisaktivität unterdrücken als auch manchmal die
Aktivitäten
anderer Retinoid- und Hormonagonisten der Steroidrezeptor-Superfamilie
potenzieren kann. Im Zusammenhang der Erfindung wird ein Hormonagonist
als durch ein negatives Hormon wie AGN 193109 potenziert betrachtet, falls
in Gegenwart des negativen Hormons eine reduzierte Konzentration
des Agonisten im wesentlichen die gleiche quantitative Reaktion
auslöst
wie diejenige, die mit dem Agonisten allein erhältlich ist. Die quantitative
Reaktion kann zum Beispiel in einem Reportergentest in vitro gemessen
werden. So wird ein therapeutisches Retinoid, das eine gewünschte Reaktion
bei Verwendung in einer besonderen Dosierung oder Konzentration
ausübt,
durch AGN 193109 potenziert, falls in Kombination mit AGN 193109
eine geringere Dosierung oder Konzentration des therapeutischen
Retinoids verwendet werden kann, um den im wesentlichen gleichen
Effekt wie eine höhere
Dosierung oder Konzentration des therapeutischen Retinoids zu erzeugen,
wenn das therapeutische Retinoid allein verwendet wird. Die Liste
von Agonisten, die durch Koverabreichung mit AGN 193109 potenziert
werden können,
schließt
RAR-Agonisten, Vitamin D-Rezeptoragonisten,
Glukokortikoidrezeptoragonisten und Schilddrüsenhormonrezeptoragonisten
ein. Insbesondere schließen
spezifische Agonisten, die durch Koverabreichung potenziert werden
können,
die folgenden ein: ATRA, 13-cis-Retinolsäure, den
synthetischen RAR-Agonisten AGN 191183, 1,25-Dihydroxyvitamin D3, Dexamethason und Schilddrüsenhormon
(3,3'5-Triiodthyronin).
Ebenfalls offenbart wird hier ein Verfahren, das zur Identifizierung
anderer Hormone verwendet werden kann, die durch Koverabreichung
mit AGN 193109 potenziert werden können.
-
Somit
verhält
sich AGN 193109 in einer Weise, die nicht für einen einfachen Retinoidantagonisten
vorhergesehen wird, sondern als ein negatives Hormon, das die Aktivitäten verschiedener
Mitglieder der Familie von Kernrezeptoren potenzieren kann. Wir
offenbaren ebenfalls einen möglichen
Mechanismus, der sowohl die negative Hormonaktivität als auch
die Fähigkeit
von AGN 193109 zur Potenzierung der Aktivitäten anderer Kernrezeptorliganden
berücksichtigen
kann. Dieser Mechanismus beinhaltet Elemente, von denen bekannt
ist, daß sie
an Retinoid-abhängigen
Signalleitungswegen teilnehmen, und beinhaltet zusätzlich eine
neue negative regulatorische Komponente.
-
Die
Durchschnittsfachleute werden einsehen, daß RARs, die hochaffine Ziele
für die
AGN 193109-Bindung sind, Transkriptionsfaktoren sind, die die Expression
einer Vielzahl von Retinoid-reaktiven Genen regulieren. cis-regulatorische DNA-Bindungsorte
für die
RARs wurden in der Nähe
von Genen identifiziert, die transkriptionell in einer Retinoid-abhängigen Weise
reguliert werden. Die RAR-Bindung an solche DNA-Orte, bekannt als
Retinolsäure-Response-Elemente (RAREs),
ist gut definiert. Bedeutsam ist, daß die RAREs Heterodimere binden,
die aus einem RAR und einem RXR bestehen. Die RXR-Komponente des
Heterodimers funktioniert zur Förderung
einer hochaffinen Wechselwirkung zwischen dem RAR/RXR-Heterodimer
und dem RARE (Mangelsdorf et al., The Retionoid Receptors, "The Retinoids: Biology,
Chemistry and Medicine",
2. Auflage, Hrsg. Sporn et al., Raven Press, Ltd., New York 1994).
-
Wie
nachfolgend ausgeführt
wird, stimmen unsere Befunde, die die negative Hormonaktivität von AGN 193109
betreffen, mit einem Mechanismus überein, der die Wechselwirkung
eines mutmaßlichen
negativen Koaktivatorproteins (NCP) mit dem RAR beinhaltet. Gemäß dem vorgeschlagenen
Mechanismus wird diese Wechselwirkung durch AGN 193109 stabilisiert.
-
Unsere
Ergebnisse zeigten ferner, daß AGN
193109 die intrazelluläre
Verfügbarkeit
von NCP zur Wechselwirkung mit anderen Kernrezeptoren als RARs modulieren
kann, die durch AGN 193109 besetzt werden. Es folgt daraus, daß AGN 193109
transkriptionelle regulatorische Reaktionswege potenzieren kann,
die Kernrezeptoren involvieren, die mit den RARs die Fähigkeit
zur Bindung des NCP teilen. In dieser Hinsicht weist AGN 193109
die Fähigkeit
auf, eine Vielzahl von Kernrezeptor-Reaktionswegen zu modulieren,
eine Aktivität,
die nicht für
einen herkömmlichen
Retinoidantagonisten vorhergesagt würde. Entsprechend ist AGN 193109
nützlich
als ein Mittel zur Potenzierung der Aktivität von Kernrezeptorliganden,
einschließlich
sowohl endogener Hormone als auch verschriebener Therapeutika. Diese
spezifische Ausführungsform
veranschaulicht das allgemeinere Prinzip, daß jedes negative Hormon für einen
Kernrezeptor die Aktivität
anderer Kernrezeptoren potenzieren wird, die kompetitiv das NCP
binden.
-
Obwohl
andere Materialien und Methoden, die ähnlich oder äquivalent
zu den hier beschriebenen sind, in der Praxis oder Untersuchung
der vorliegenden Erfindung verwendet werden können, werden jetzt die bevorzugten
Verfahren und Materialien beschrieben. Allgemeine Verweise auf Verfahren,
die zur Durchführung der
verschiedenen Nukleinsäuremanipulationen
und hier beschriebenen Verfahren verwendet werden können, können gefunden
werden in "Molecular
Cloning: A Laboratory Manual" (Sambrook
et al., Hrsg., Cold Spring Harbor Lab Publ. 1989) und in "Current Protocols
in Molecular Biology" (Ausubel
et al., Hrsg., Greene Publishing Associates und Wiley-Interscience
1987). Eine Beschreibung der Experimente und Ergebnisse, die zur Schaffung
der vorliegenden Erfindung führten,
folgt.
-
Beispiel
6 beschreibt die Verfahren, die verwendet wurden um zu zeigen, daß AGN 193109
jeden von drei RARs mit hoher Affinität band, aber in der Aktivierung
der Retinoid-abhängigen Genexpression
versagte.
-
Beispiel 6
-
AGN 193109 bindet RARs
mit hoher Affinität,
aber transaktiviert nicht die Retinoid-abhängige Genexpression
-
Humane
RAR-α-,
RAR-β- und
RAR-γ-Rezeptoren
wurden separat als rekombinante Proteine unter Verwendung eines
Baculovirus-Expressionssystems
exprimiert, im wesentlichen gemäß dem von
Allegretto et al. beschriebenen Verfahren, J. Biol. Chem. 268: 26625
(1993). Die rekombinanten Rezeptorproteine wurden separat zur Bestimmung
von AGN 193109-Bindungsaffinitäten
eingesetzt, wobei der [3H]-ATRA-Verdrängungstest
verwendet wurde, beschrieben von Heyman et al., Cell 68: 397 (1992).
Dissoziationskonstanten (Kd) wurden gemäß dem von Cheng et al. beschriebenen
Verfahren bestimmt, Biochemical Pharmacology 22: 3099 (1973).
-
AGN
193109 wurde ebenfalls auf seine Fähigkeit zur Transaktivierung
von RARs in CV-1-Zellen getestet, die transient mit RAR-Expressionsvektoren
und einem Retinoid-reaktiven
Reportergenkonstrukt kotransfiziert worden waren. Rezeptorexpressionsvektoren
pRShRAR-α (Giguere
et al., Nature 330: 624 (1987)), pRShRAR-β (Benbrook et al., Nature 333:
669 (1988)) und pRShRAR-γ (Ishikawa
et al., Mol. Endocrinol. 4: 837 (1990)) wurden separat mit dem ΔMTV-TREp-Luc-Reporterplasmid kotransfiziert.
Die Verwendung dieses Luciferase-Reporterplasmids wurde von Heyman
et al. offenbart, Cell 68: 397 (1992). Das ΔMTV-TREp-Luc-Plasmid ist im
wesentlichen identisch mit dem ΔMTV-TREp-CAT-Reporterkonstrukt,
beschrieben von Umesono et al., Nature, 336: 262 (1988), ausgenommen
daß das
Chloramphenicolacetyltransferase-(CAT)-Reportergen durch eine Polynukleotidsequenz
substituiert war, die Leuchtkäferluciferase
codiert. Die Transfektion von CV-1-Zellen aus Meerkatze wurde unter Verwendung
des Calciumphosphat-Kopräzipitationsverfahrens
durchgeführt,
beschrieben in "Molecular
Cloning: A Laboratory Manual" (Sambrook
et al., Hrsg., Cold Spring Harbor Lab Publ. 1989). CV-1-Zellen wurden
mit einer Dichte von 4 × 104/Vertiefung in Mehrfachvertiefungsplatten
mit 12 Vertiefungen ausplattiert und mit einem Calciumphosphatpräzipitat,
das 0,7 μg Reporterplasmid
und 0,1 μg
Rezeptorplasmid enthielt, gemäß Standardlaborverfahren
transient transfiziert. Die Zellen wurden nach 18 Stunden gewaschen,
um das Präzipitat
zu entfernen, und erneut mit Dulbeccos modifiziertem Eagle-Medium (DMEM) (Gibco)
gefüttert,
das 10% mit Aktivkohle extrahiertem fötalem Rinderserum (Gemini Bio-Products)
enthielt. Die Zellen wurden mit Träger allein (Ethanol) oder AGN
193109 (10–9 bis
10–6 M)
für 18
Stunden behandelt. Zelllysate wurden in 0,1 M KPO4 (pH
7,8), 1,0% TRITON X-100, 1,0 mM DTT, 2 mM EDTA hergestellt. Luciferase-Aktivität wurde
wie von de Wet et al. beschrieben, Mol. Cell. Biol. 7: 725 (1987),
unter Verwendung von Leuchtkäferluciferin
(Analytical Luminescence Laboratory) und eines Plattenluminometers
mit 96 Vertiefungen von EG & G
Berthold gemessen. Die angegebenen Luciferasewerte stellten den
Mittelwert ± Standardabweichung
von Bestimmungen in dreifacher Ausführung dar.
-
Die
in Tabelle 11 angegebenen Ergebnisse zeigten, daß AGN 193109 jeden aus RAR-α, RAR-β und RAR-γ mit hoher
Affinität
band, aber nicht die Retinoid-abhängige Genexpression aktivierte.
Insbesondere band AGN 193109 jeden der drei Rezeptoren mit Kd-Werten
im Bereich von 2 bis 3 nM. Trotz dieser engen Bindung versagte AGN
193109 in der Aktivierung der Genexpression bei Vergleich mit durch
ATRA stimulierten Induktionen. Entsprechend war die halbmaximale
effektive Konzentration von AGN 193109 (EC50)
unmeßbar.
Obwohl nicht in der Tabelle dargestellt, stellten wir ebenfalls
fest, daß AGN
193109 keine meßbare
Affinität für die RXRs
hatte.
-
Tabelle
11 AGN
193109-Bindung und -Transaktivierung der RARs
-
Beispiel
7 beschreibt die Verfahren, die verwendet wurden um zu zeigen, daß AGN 193109
ein Antagonist der ATRA-abhängigen
Genexpression ist.
-
Beispiel 7
-
AGN 193109-abhängige Inhibierung
der RAR-Transaktivierung durch ATRA
-
Die
Fähigkeit
von AGN 193109, der durch ATRA-vermittelten RAR-Aktivierung entgegenzuwirken, wurde
in CV-1-Zellen untersucht, die durch das Calciumphosphat-Kopräzipitationsverfahren
von Sambrook et al. kotransfiziert worden waren (Molecular Cloning:
A Laboratory Manual, Cold Spring Harbor Lab Publ., 1989). Eurkaryontische
Expressionsvektoren pRShRAR-α (Giguere
et al., Nature 330: 624 (1987)), pRShRAR-β (Benbroock et al., Nature 333:
669 (1988)) und pRShRAR-γ (Ishikawa
et al., Mol. Endocrinol. 4: 837 (1990)) wurden mit dem von Hollenberg
et al. beschriebenen Δ-MTV-Luc-Reporterplasmid
(Cell 55: 899 (1988)) kotransfiziert. Bemerkenswert enthielt das
Reporterplasmid zwei Kopien des TRE-Palindrom-Response-Elements.
Calciumphosphat-Transfektionen wurden genau wie in Beispiel 6 beschrieben
durchgeführt.
Die Zellen wurden mit Träger
allein (Ethanol), ATRA (10–9 bis 10–6 M),
AGN 193109 (10–9 bis 10–6 M)
oder 10–8 M
ATRA in Kombination mit AGN 193109 (10–9 bis
10–6 M)
für 18
Stunden behandelt. Zelllysate und Messungen der Luciferase-Aktivität wurden
ebenfalls wie in Beispiel 6 durchgeführt.
-
Die
Ergebnisse dieser Verfahren sind in den 2A bis 2F dargestellt,
worin Luciferasewerte den Mittelwert ± Standardabweichung von Bestimmungen
in dreifacher Ausführung
darstellen. Insbesondere zeigten die in den 2A, 2C und 2C dargestellten
Ergebnisse, daß die
Stimulation von transfizierten Zellen mit ATRA zu Dosis-Reaktions-Zunahmen
der Luciferase-Aktivität
führten.
Dies bestätigte,
daß ATRA
jeden der drei RARs im experimentellen System aktiviert und eine
Vergleichsbasis zum Nachweis der Aktivität eines Antagonisten lieferte.
Die in den 2B, 2D und 2F dargestellten
graphischen Ergebnisse zeigten, daß die gleichzeitige Behandlung
von transfizierten Zellen mit 10 nM ATRA und zunehmenden Konzentrationen
von AGN 193109 zu einer Inhibierung der Luciferase-Aktivität führte. Insbesondere
ergaben gleiche Dosen von AGN 193109 und ATRA eine mehr als 50%ige
Inhibierung relativ zu ATRA allein für alle drei RAR-Untertypen. Der Vergleich
der ATRA-Dosis-Reaktion in Gegenwart unterschiedlicher Konzentrationen von
AGN 193109 zeigte, daß ATRA
durch AGN 193109 kompetitiv gehemmt wurde. Bemerkenswert stellt
die horizontale Achse auf allen der in 2 gezeigten
Diagramme den Logarithmus der Retinoidkonzentration dar. Diese Ergebnisse
bewiesen, daß AGN
193109 ein wirksamer RAR-Antagonist war.
-
Als
nächstes
führten
wir Experimente durch, um den der antagonistischen Aktivität von AGN
193109 zugrundeliegenden Mechanismus aufzuklären. Die Durchschnittsfachleute
werden einsehen, daß angenommen
wird, daß die
Kernrezeptoraktivierung eine Konformationsveränderung des Rezeptors beinhaltet,
die durch Ligandenbindung induziert wird. Tatsächlich haben die Ergebnisse
von Proteaseschutztests bestätigt, daß nukleäre Hormonagonisten
und -antagonisten dazu führen,
daß Rezeptorproteine unterschiedliche
Konformationen annehmen (Keidel et al., Mol. Cell. Biol. 14: 287
(1994); Allan et al., J. Biol. Chem. 267: 19513 (1992)). Wir verwendeten
einen solchen Test um zu bestimmen, ob AGN 193109 und ATRA dazu
führten,
daß RAR-α unterschiedliche
Konformationen annimmt. AGN 193583, ein RAR-α-selektiver
Antagonist, wurde als Positivkontrolle eingeschlossen, von der bekannt
ist, daß sie
ein Antagonist-spezifisches
Muster von Proteaseempfindlichkeit verleiht.
-
Beispiel
8 beschreibt ein Verfahren, das zu Detektion von Konformationsveränderungen
in RAR-α verwendet
wurde, die aus einer AGN 193109-Bindung resultierten. Wie nachfolgend
dargestellt wird, zeigten die Ergebnisse dieses Verfahrens unerwartet,
daß AGN
193109 zu einem Muster der Trypsin-Empfindlichkeit führt, die im wesentlichen identisch
mit der durch ATRA, einem RAR-Agonisten, induzierten und ungleich
derjenigen war, die durch einen modellmäßigen RAR-Antagonisten induziert wird. Dieser
Befund legte nahe, daß AGN
193109 Eigenschaften besitzt, die sich von anderen Retinoidantagonisten
unterscheiden.
-
Beispiel 8
-
Proteaseschutzanalyse
-
Ein
Plasmid, das mit Vektor pGEM3Z (Pharmacia) konstruiert wurde und
die RAR-α-cDNA
enthielt (Giguere et al., Nature 330: 624 (1987)), wurde im Zusammenhang
mit dem TNT-gekuppelten
Retikulozytenlysat in vitro Transkription-Translations-System (Promega) verwendet,
um [35S]-Methionin-markiertes RAR-α herzustellen. Eine beschränkte proteolytische
Verdauung des markierten Proteins RAR-α wurde gemäß dem von Keidel et al. beschriebenen
Verfahren durchgeführt,
Mol. Cell. Biol. 14: 287 (1994). Teilmengen des Retikulozytenlysats,
das [35S]-Methionin-markiertes RAR-α enthielt,
wurden mit entweder ATRA, AGN 193583 oder AGN 193109 auf Eis für 45 Minuten
in einem Gesamtvolumen von 9 μl
inkubiert. Die Retinoidendkonzentration für alle Versuche betrug 100
nM für
ATRA und AGN 193109 und 1000 nM für AGN 193583. Der Unterschied zwischen
den Endkonzentrationen der Retinoide beruhte auf dem etwa 10-fachen
Unterschied der relativen Affinitäten von ATRA und AGN 193109
(mit einem Kd-Wert
an RAR-α von
2 bzw. 10 nM) und AGN 193583 (mit einem Kd-Wert an RAR-α von ≥ 100 nM). Nach der Ligandenbindung
wurde 1 μl
von entsprechend konzentriertem Trypsin zur Mischung gegeben, um
Endkonzentrationen von 25, 50 oder 100 μg/ml zu ergeben. Die Proben
wurden bei Raumtemperatur für
10 Minuten inkubiert und die Trypsinverdauung durch Zugabe von SDS-Probenpuffer beendet.
Die Proben wurden der Polyacrylamid-Gelelektrophorese unterworfen und gemäß Standardverfahren
autoradiographiert.
-
Sowohl
der Agonist als auch Antagonist führte zu besonderen Mustern
der Trypsinempfindlichkeit, die sich von dem Ergebnis unterschieden,
das durch Verdauung des unkomplexierten Rezeptors erhalten wurde. Die
autoradiographischen Ergebnisse zeigten, daß Trypsinkonzentrationen von
25, 50 und 100 μg/ml
das radiomarkierte RAR-α vollständig in
10 Minuten bei Raumtemperatur in Abwesenheit von zugegebenem Retinoid verdauten.
Die Vorbindung von ATRA führte
zum Auftreten von zwei proteaseresistenten Hauptarten. Vorbindung
des RAR-α-selektiven Antagonisten
AGN 193583 führte
zu einer proteaseresistenten Spezies, die ein geringeres Molekulargewicht
als diejenige hatte, die aus der ATRA-Vorbindung resultierte. Dieses Ergebnis zeigte,
daß ein
Retinoidagonist und -antagonist zu Konformationsveränderungen
führte,
die aufgrund von veränderten
Trypsinempfindlichkeiten detektierbar waren. Überraschend führte die
Vorbindung von AGN 193109 zu einem Proteaseschutzmuster, das ununterscheidbar
von demjenigen war, das durch Vorbindung von ATRA erzeugt wurde.
-
Die
oben dargestellten Ergebnisse bestätigten, daß AGN 193109 den RAR-α band und
seine Konformation veränderte.
Interessanterweise ähnelte
die Natur dieser Konformationsveränderung stärker derjenigen, die aus der
Bindung eines Agonisten (ATRA) resultierte, als der Veränderung,
die durch eine Antagonistenbindung (AGN 193583) erzeugt wurde. Ersichtlich
war der Mechanismus des AGN 193109-abhängigen Antagonismus besonders.
-
Wir
erwogen mögliche
Mechanismen, die die antagonistische Aktivität von AGN 193109 erklären könnten. Insbesondere
verwendeten wir einen standardmäßigen Gel-Shift-Test
zur Untersuchung, ob AGN 193109 die RAR/RXR-Heterodimerbildung störte oder
die Wechselwirkung zwischen RAR und seinem erkennenden DNA-Bindungsort
inhibierte.
-
Beispiel
9 beschreibt einen gelelektrophoretischen Mobilitäts-Shift-Test,
der verwendet wurde um zu zeigen, daß AGN 193109 weder die RAR/RXR-Dimerisierung
hemmte noch die Bindung von Dimeren an eine Ziel-DNA hemmte.
-
Beispiel 9
-
Gel-Shift-Analyse
-
In
vitro translatiertes RAR-α wurde
im wesentlichen wie in Beispiel 8 beschrieben erzeugt, außer daß das 35S-markierte Methionin ausgelassen wurde.
In vitro translatiertes RXR-α wurde
in ähnlicher
Weise unter Verwendung eines Vektors auf pBluescript(II)(SK)-Basis,
der die RXR-α-cDNA
enthielt, die von Mangelsdorf et al. beschrieben wurde, Nature 345:
224–229
(1990), als Matrize zur Erzeugung von in-vitro-Transkripten hergestellt.
Den markierten RAR-α und
RXR-α allein
oder in Kombination oder vorgebunden mit AGN 193109 (10–6 M)
entweder allein oder in Kombination ließ man mit einer endmarkierten
DR-5 RARE doppelsträngigen Sonde
mit der Sequenz 5'-TCAGGTCACCAGGAGGTCAGA-3' (SEQ ID NO: 1) wechselwirken.
Die Bindungsmischung wurde an einem nicht-denaturierenden Polyacrylamidgel
elektrophoretisch behandelt und gemäß Standardlaborverfahren autoradiographiert.
Eine einzelne verzögerte
Spezies, die auf dem Autoradiogramm erschien und allen Bahnen auf
dem Gel gemeinsam war, stellte einen undefinierten Sonden-bindenden
Faktor dar, der im Retikulozytenlysat vorhanden war. Nur die RAR/RXR-Kombination
führte
zu einer Retinoidrezeptor-spezifischen verzögerten Spezies. Weder RAR allein
noch RXR allein band die Sonde, um diese verschobene Spezies zu
erzeugen. Die Gegenwart von AGN 193109 verminderte diese Wechselwirkung
nicht.
-
Diese
Ergebnisse zeigten, daß AGN
193109 weder die Homo- noch die Heterodimerisierungseigenschaften
von RAR-α wesentlich
veränderte.
Ferner inhibierte AGN 193109 nicht die Wechselwirkung von Rezeptordimeren
mit einem DNA-Segment, das die erkennende Bindungsstelle enthielt.
-
Angesichts
der besonderen Eigenschaften, die AGN 193109 kennzeichneten, schritten
wir mit der Untersuchung fort, ob dieser Antagonist zusätzlich die
Aktivität
von unkomplexierten RARs inhibieren könnte. Das zur Durchführung dieser
Bestimmung verwendete Rezeptor/Reporter-System wies vorteilhaft
eine hochgradige konstitutive Aktivität in Abwesenheit von zugegebenem
Retinoidagonist auf. Insbesondere setzten diese Verfahren das ER-RAR
chimäre
Rezeptor- und ERE-tk-Luc-Reportersystem
ein. Das ERE-tk-Luc-Plasmid schließt die Region –397 bis –87 der Östrogen-reaktiven
5'-flankierenden Region
des Xenopus Vitellogenin-A2-Gens ein, beschrieben von Klein-Hitpass
et al., Cell 46: 1053–1061
(1986), ligiert stromaufwärts
des HSV-Thymidinkinase-Promotors
und Luciferase-Reportergens von Plasmid tk-Luc. Die ER-RAR chimären Rezeptoren
bestanden aus der Östrogenrezeptor-DNA-Bindungsdomäne, fusioniert
an die "D-E-F"-Domäne der RARs.
Die Fachleute erkennen an, daß diese "D-E-F"-Domäne unter
Bindung von Retionoid funktioniert, um eine Retinoid-induzierbare
Transaktivierungsfunktion bereitzustellen und einen Kontaktort für die Heterodimerisierung
mit RXR bereitzustellen. Somit war die Luciferaseexpression in diesem
Reportersystem abhängig von
der Aktivierung des transfizierten chimären Rezeptorkonstrukts.
-
Beispiel
10 beschreibt das zur Demonstration verwendete Verfahren, daß AGN 193109
die unkomplexierten RARs zuordnungsfähige Grundgenaktivität inhibierte.
Diese Verfahren wurden in Abwesenheit von zugegebenem Retinoidagonist
durchgeführt.
Die nachfolgend dargestellten Ergebnisse lieferten den ersten Hinweis,
daß AGN
193109 eine negative Hormonaktivität aufwies.
-
Beispiel 10
-
Repression
von Grundgenaktivität
eines Retinoid-regulierten Reporters in transient kotransfizierten
Zellinien
-
CV-1-Zellen
wurden mit dem ERE-tk-Luc-Reporterplasmid und entweder ER-RAR-α, ER-RAR-β- oder ER-RAR-γ-Expressionsplasmiden
kotransfiziert. Das ERE-tk-Luc-Plasmid enthielt das auf Östrogen
reagierende Promotorelement des Xenopus laevis-Vitellogenin-A2-Gens
und war im wesentlichen identisch mit dem Reporterplasmid, das von
Klein-Hitpass et al. beschrieben wird, Cell 46: 1053 (1986), außer daß das CAT-Reportergen durch
eine Polynukleotidsequenz substituiert war, die Luciferase codiert.
Die in der Kotransfektion eingesetzen, den chimären ER-RAR-α-, ER-RAR-β- und ER-RAR-γ-Rezeptor codierenden
Polynukleotide wurden von Graupner et al. beschrieben, Biochem.
Biophys. Res. Comm. 179: 1554 (1991). Diese Polynukleotide wurden
in den von Ellis et al. beschriebenen pECE-Expressionsvektor ligiert,
Cell 45: 721 (1986), und unter der Transkriptionskontrolle des SV-40- Promotors exprimiert.
Calciumphosphat-Transfektionen wurden genau wie in Beispiel 6 beschrieben
unter Verwendung von 0,5 μg/Vertiefung
von Reporterplasmid und entweder 0,05 μg, 0,10 μg oder 0,2 μg/Vertiefung von Rezeptorplasmid
durchgeführt.
Zellen wurden mit Träger
allein (Ethanol), ATRA (10–9 bis 10–6 M)
oder AGN 193109 (10–9 bis 10–6 M)
für 18
Stunden behandelt. Zelllysate und Messungen der Luciferase-Aktivität wurden
wie in Beispiel 6 beschrieben durchgeführt.
-
Die
in den 3A, 4A und 5A dargestellten
Ergebnisse bestätigten,
daß ATRA
die Luciferaseexpression in allen Transfektanten stark induzierte.
Die Basisniveau-Expression von Luciferase für die drei transfizierten chimären RAR-Isoformen reichte
von ca. 7000 bis 40000 relativen Lichteinheiten (rlu) und war etwas
von der in der Transfektion verwendeten Menge an Reporterplasmid
abhängig.
Somit waren die drei chimären
Rezeptoren wie erwartet durch ATRA aktivierbar. Insbesondere banden
alle drei Rezeptoren ATRA und aktivierten die Transkription des
auf dem ERE-tk-Luc-Plasmid
beherbergten Luciferase-Reportergens.
-
Die 3B, 4B und 5B stellen
die in Abwesenheit von exogenem Retinoidagonisten erhaltenen Dosis-Reaktions-Kurven
für AGN
193109 dar. Interessanterweise war ER-RAR-α (3B)
im wesentlichen unbeeinflußt
durch AGN 193109, während
die chimären
ER-RAR-β-
und ER-RAR-γ-Rezeptoren
(4B bzw. 5B)
eine auf die AGN 193109-Dosis reagierende Abnahme der Luciferase-Reporteraktivität aufwiesen.
-
Wir
untersuchten ferner die negative Hormonaktivität von AGN 193109 durch Untersuchung
seiner Fähigkeit,
die Genexpression zu unterdrücken,
die durch einen chimären
RAR-γ-Rezeptor
vermittelt wird, der manipuliert worden war, um eine konstitutive
Transkriptionsaktivatordomäne
zu besitzen. Insbesondere verwendeten wir einen konstitutiv aktiven chimären RAR-γ-Rezeptor,
der an die saure Aktivatordomäne
von HSV VP-16 fusioniert war, als RAR-γ-VP-16 bezeichnet, in zwei Arten
von Luciferase-Reportersystemen. Das erste bestand aus dem ERE-tk-Luc-Reporter,
kotransfiziert mit ER-RARs und ER-RXR-α. Das zweite verwendete den ΔMT-TREp-Luc-Reporter
anstelle des ERE-tk-Luc-Reporters.
-
Beispiel
11 beschreibt das Verfahren, das verwendet wurde um zu zeigen, daß AGN 193109
die Aktivität
einer Transkriptionsaktivatordomäne
eines RAR unterdrücken
konnte. Die nachfolgend dargestellten Ergebnisse bewiesen, daß AGN 193109
die RAR-abhängige
Genexpression in Abwesenheit eines Agonisten unterdrücken konnte,
und bestätigten,
daß AGN
193109 negative Hormonaktivität
aufwies.
-
Beispiel 11
-
Unterdrückung von
RAR-VP-16-Aktivität
in transient transfizierten Zellen
-
CV-1-Zellen
wurden transient gemäß der in
Beispiel 6 beschriebenen Calciumphosphat-Konräzipitationstechnik kotransfiziert,
wobei 0,5 μg/Vertiefung
des ERE-tk-Luc-Luciferase-Reporterplasmids,
0,1 μg/Vertiefung
des chimären
ER-RXR-α-Reporterexpressionsplasmids
und entweder 0 μg
oder 0,1 μg/Vertiefung
des RAR-γ-VP-16-Expressionsplasmids
verwendet wurden. Der chimäre
Rezeptor ER-RXR-α bestand
aus der Hormonbindungsdomäne
(Aminosäuren
181 bis 458) von RXR-α (Mangelsdorf
et al., Nature 345: 224–229 (1990)),
fusioniert an die Östrogenrezeptor-DNA-Bindungsdomäne (Graupner
et al., Biochem. Biophys. Res. Comm. 179: 1554 (1991)), und wurde
aus dem Expressionsvektor pECE auf SV-40-Basis, beschrieben von Ellis
et al., Cell 45: 721 (1986), exprimiert. RAR-γ-VP-16 ist identisch mit dem
VP16RAR-γ1-Expressionsplasmid,
das von Nagpal et al. beschrieben wurde, EMBO J. 12: 2349 (1993),
dessen Offenbarung hier durch Verweis eingeführt wird, und codiert ein chimäres Protein,
in dem die Aktivierungsdomäne
des VP-16-Proteins von HSV an den Amino-Terminus von RAR-γ voller Länge fusioniert
ist. 18 Stunden nach der Transfektion wurden die Zellen mit phosphatgepufferter
Kochsalzlösung
(PBS) gespült
und mit DMEM (Gibco-BRL) gefüttert,
das 10% FBS (Gemini Bio-Products) enthielt, das mit Aktivkohle extrahiert
worden war, um Retinoide zu entfernen. Die Zellen wurden mit einer
angemessenen Verdünnung
von AGN 193109 oder ATRA in Ethanol als Träger oder mit Ethanol allein
für 18
Stunden behandelt, dann mit PBS gespült und unter Verwendung von
0,1 M KPO4 (pH 7,8), 1,0% TRITON X-100,
1,0 mM DTT und 2 mM EDTA lysiert. Luciferase-Aktivität wurde
gemäß dem von
de Wet et al. beschriebenen Verfahren gemessen, Mol. Cell. Biol.
7: 725 (1987), wobei Leuchtkäferluciferin (Analytical
Luminescence Laboratory) und ein Luminometer EG&G Berthold mit 96 Vertiefungen verwendet wurden.
Die Luciferase-Werte stellten den Mittelwert ± Standardabweichung von Bestimmungen
in dreifacher Ausführung
dar.
-
Wie
in 6 gezeigt wird, wiesen CV-1-Zellen, die mit dem
ERE-tk-Luc-Reporterkonstrukt und dem chimären ER-RAR-α-Expressionsplasmid transfiziert wurden,
eine schwache Aktivierung von Luciferase-Aktivität durch ATRA auf, wahrscheinlich
aufgrund von Isomerisierung von ATRA zu 9C-RA, dem natürlichen
Liganden für
die RXRs (Heyman et al., Cell 68: 397 (1992)). Mit der gleichen
Mischung aus Reporter und chimären
Reporterplasmiden transfizierte Zellen, die aber mit AGN 193109
behandelt wurden, wiesen keine Wirkung auf die Luciferase-Aktivität auf. Da
AGN 193109 nicht an die RXRs bindet, wurde dieses letztere Ergebnis
erwartet. CV-1-Zellen, die in ähnlicher
Weise mit dem ERE-tk-Luc-Reporter transfiziert waren, aber mit Substitution
eines chimären
ER-RAR-Rezeptorexpressionsplasmids für ER-RXR-α, wiesen eine robuste Induktion
von Luciferase-Aktivität
nach ATRA-Behandlung
auf.
-
Im
Gegensatz führte
der Einschluß des
RAR-γ-VP-16-Expressionsplasmids
mit den ER-RXR-α-
und ERE-tk-Luc-Plasmiden
in der Transfektionsmischung zu einer signifikanten Zunahme der
Luciferase-Basisaktivität
gemäß Messung
in Abwesenheit von zugegebenem Retinoid. Diese Zunahme der Luciferase-Basisaktivität, die für die ER-RXR-α/RAR-γ-VP-16-Kotransfektanten
beobachtet wurde, zeigte bei Vergleich mit den unter Verwendung
von Zellen erhaltenen Ergebnissen, die allein mit ER-RXR-α transfiziert
waren, daß rekombinante
ER-RXR-α-
und RAR-γ-VP-16-Proteine
heterodimerisieren konnten. Die Wechselwirkung des Heterodimers
mit dem cis-regulatorischen Östrogen-reaktiven
Element führte
zu einer Ausrichtung der VP-16-Aktivierungsdomäne auf die Promotorregion des
ERE-tk-Luc-Reporters. Die Behandlung von derart dreifach transfizierten
Zellen mit ATRA führte
zu einer mäßigen Zunahme
der Luciferase-Aktivität
gegenüber
dem hohen Basisniveau. Jedoch führte
die Behandlung der dreifachen Transfektanten mit AGN 193109 zu einer
dosisabhängigen
Abnahme der Luciferase-Aktivität.
In bedeutender Weise zeigt 6, daß die AGN
193109-Behandlung von
Zellen, die mit ER-RXR-α und
RAR-γ-VP-16
kotransfiziert waren, zu einer Unterdrückung der Luciferase-Aktivität führte, wobei
eine maximale Inhibierung bei ca. 10–8 M
AGN 193109 auftrat.
-
Unsere
Beobachtung, daß AGN
193109 die konstitutive Transkriptionsaktivierungsfunktion von RAR-γ-VP-16 in
Gegenwart eines RXR unterdrückte,
wurde durch ein Modell erklärt,
in dem die Bindung von AGN 193109 an den RAR eine Konformationsveränderung
des RAR induzierte, die eine negative Konformation stabilisiert,
die die Bindung eines trans-wirkenden negativen Koaktivatorproteins
erleichtert. Wenn der AGN 193109/RAR-Komplex durch das NCP gebunden
wird, kann der RAR die Transkription von Genen nicht aufregulieren,
die gewöhnlich
auf aktivierte RARs reagieren. Unser Modell schlägt ferner vor, daß das intrazelluläre Reservoir
von NCP in einer einschränkenden
Konzentration in bestimmten Zusammenhängen vorliegt und aufgrund
von AGN 193109-stimulierter
Komplexierung mit RARs erschöpft
werden kann.
-
Die
in 6 dargestellten Ergebnisse zeigten zusätzlich,
daß selbst
bei 10–6 M
AGN 193109 die ER-RXR-α-
und RAR-γ-VP-16-Proteine wechselwirken
konnten, um Heterodimere zu bilden, die kompetent zur Aktivierung
der Transkription des Reportergens sind. Insbesondere ergaben Zellen,
die mit ER-RXR-α und RAR-γ-VP-16 transfiziert
und mit AGN 193109 mit einer Konzentration (10–8–10–6 M)
behandelt wurden, die ausreichend zur Bereitstellung maximaler Inhibierung
war, Ablesungen der Luciferase-Aktivität von ca. 16 000 rlu. Umgekehrt
wiesen Zellen, die nur mit ER-RXR-α transfiziert und dann mit AGN
193109 bei einer Konzentration von so hoch wie 10–6 M
behandelt wurden, Luciferase-Expressionsgrade von nur ca. 8000 rlu
auf. Die Tatsache, daß ein
höheres
Maß an
Luciferase-Aktivität
in Zellen erhalten wurde, die sowohl ER-RXR-α als auch RAR-γ-VP-16 exprimierten,
selbst in Gegenwart von 10–6 M AGN 193109, zeigte
die Dauerhaftigkeit einer Wechselwirkung zwischen den zwei rekombinanten
Rezeptoren. Die Unterdrückung
der RAR-γ-VP-16-Aktivität durch
AGN 193109 legte nahe, daß die
Modulierung der NCP-Wechselwirkung
mit der VP-16-Aktivierung kodominant sein kann. Entsprechend erkannten
wir, daß es
möglich
sein kann, die Expression von Genen zu modulieren, die gewöhnlich nicht
durch Retinoide in einer AGN 193109-abhängigen Weise reguliert werden.
-
Kandidaten
für AGN
193109-regulierbare Gene schließen
diejenigen ein, die durch Transkriptionsfaktorkomplexe aktiviert
werden, die aus Nicht-RAR-Faktoren bestehen, die mit RARs assoziieren
oder heterodimerisieren, worin der Nicht-RAR-Faktor keinen RAR-Agonisten
zur Aktivierung erfordert. Während
die Stimulation mit einem RAR-Agonisten im wesentlichen keine Wirkung
auf die Expression solcher Gene haben kann, kann die Verabreichung
mit AGN 193109 die Bildung von inaktiven Transkriptionskomplexen
fördern,
die AGN 193109/RAR/NCP umfassen. Entsprechend kann die Zugabe des
negativen AGN 193109-Retionoidhormons die Transkription eines ansonsten
Retinoid-unempfindlichen Gens abregulieren.
-
Der
gleiche Mechanismus kann die Beobachtung erklären, daß AGN 193109 die Aktivität des Gewebetransglutaminase-(TGase)-Gens in HL-60-Zellen
unterdrücken
kann. Ein Retionoid-Reponse-Element,
das aus drei kanonischen Retinoid-Halborten im Abstand von 5 und
7 Basenpaaren besteht, wurde in der Transkriptionskontrollregion
dieses Gens identifiziert. Während
TGase durch RXR-selektive Agonisten induziert werden kann, reagiert
sie nicht auf RAR-selektive Agonisten. Das TGase-Retinoid-Response-Element
wird durch ein RAR/RXR-Heterodimer
gebunden (Davies et al., im Druck). Interessanterweise kann AGN
193109 die durch RXR-Agonisten induzierte TGase-Aktivität unterdrücken. Diese
AGN 193109-vermittelte
Unterdrückung kann
durch die Fähigkeit
dieses negativen Hormons zur Komplexierung von NCPs mit der RAR-Komponente des Heterodimers
erklärt
werden, wodurch die Aktivität
des assoziierten RXR unterdrückt
wird.
-
Wir
haben ebenfalls Ergebnisse erhalten, die Schlußfolgerungen stützen, die
identisch mit den in Beispiel 11 dargestellten sind, indem RAR-γ-VP-16 und
Expressionskonstrukte und das ΔMTV-TREp-Luc-Reporterplasmid
anstelle der RAR-γ-VP-16-
und ER-RXR-α-Expressionskonstrukte
in Kombination mit dem ERE-tk-Luc-Reporterplasmid eingesetzt wurden. Übereinstimmend
mit den oben dargestellten Ergebnissen stellten wir fest, daß die RAR-γ-VP-16-Aktivität am ΔMTV-TREp-Luc-Reporter
durch AGN 193109 inhibiert wurde. Deshalb unterdrückte AGN
193109 die RAR-γ-VP-16-Aktivität, wenn
dieser chimäre
Reporter direkt an ein Retinolsäurerezeptor-Response-Element
gebunden war anstelle einer indirekten Bindung an ein Östrogen-Response-Element
in der Promotorregion des Reporterplasmids. Diese Befunde zeigten,
daß ein
Test zur Identifizierung von Mitteln mit negativer Hormonaktivität nicht
durch die Verwendung eines besonderen Reporterplasmids beschränkt werden
braucht. Stattdessen beinhaltet das kritische Merkmal, das durch
ein experimentelles System verkörpert
wird, das zur Identifizierung von negativen Retionoidhormonen nützlich ist,
die Detektion der Fähigkeit
einer Verbindung, die Aktivität
eines RAR zu unterdrücken,
das manipuliert wurde, um eine konstitutive Transkriptionsaktivierungsdomäne zu enthalten.
-
Allgemein
können
negative Retinoidhormone als die Untergruppe von Retinoidverbindungen
identifiziert werden, die innerhalb einer transfekzierten Zelle
die Basisniveauexpression eines Reportergens unterdrücken, das
transkriptionsmäßig auf
die direkte oder indirekte Bindung durch einen Retinoidrezeptor
oder einen chimären
Rezeptor reagiert, der wenigstens die Domänen des Retinoidrepeztors einschließt, die
sich C-terminal
zur DNA-Bindungsdomäne
dieses Rezeptors befinden. Dieser Ansatz wurde für ein Durchmusterungsverfahren
angepaßt,
das nützlich
zur Identifizierung von negativen Retinoidhormonen ist. In den verschiedenen
Ausführungsformen
des erfundenen Durchmusterungsverfahrens ist die Struktur des Rezeptors, für den ein
negatives Hormon gesucht wird, variabel. Insbesondere kann der Retinoidrezeptor
entweder vom RAR- oder RXR-Untertyp sein. Der Rezeptor kann gegebenenfalls
manipuliert werden, um eine konstitutive Transkriptionsaktivatordomäne einzuschließen. Der
zur Durchmusterung auf negative Hormone verwendete Retinoidrezeptor
enthält
gegebenenfalls eine heterologe DNA-Bindungsdomäne als Ersatz für die DNA-Bindungsdomäne, die
endogen für
den nativen Rezeptor ist. Wenn jedoch ein zweiter Rezeptor im Durchmusterungsverfahren
verwendet wird, und wenn der zweite Rezeptor mit dem Retinoidrezeptor
dimerisieren kann, für
den ein negatives Hormon gesucht wird, dann braucht dieser Retinoidrezeptor
keine DNA-Bindungsdomäne
erfordern, weil er mit der Transkriptionskontrollregion des Reportergens
indirekt durch Dimerisierung mit dem zweiten Rezeptor verbunden
werden kann, der selbst an die Transkriptionskontrollregion gebunden
ist.
-
In
der Durchführung
des Durchmusterungsverfahrens wird die Fähigkeit einer Verbindung zur
Unterdrückung
der Basisexpression eines Reporters typischerweise in einem in-vitro-Test gemessen.
Die Basisexpression stellt das Grundlinienniveau der Reporterexpression
in transfizierten Zellen unter Bedingungen dar, unter denen kein
exogen zugegebener Retinoidagonist vorhanden ist. Gegebenenfalls
können
Schritte unternommen werden, um endogene Retinoidliganden aus der
Umgebung der transfizierten Zellen über Verfahren wie Aktivkohleextraktion
des Serums zu entfernen, das zur Kultivierung der Zellen in vitro
verwendet wird.
-
Beispiele
für Reportergene,
die nützlich
im Zusammenhang mit dem Durchmusterungsverfahren sind, schließen diejenigen
ein, die Luciferase, beta-Galactosidase, Chloramphenicolacetyltransferase
oder Zelloberflächenantigene
codieren, die durch immunochemische Mittel detektiert werden können. In
der Praxis wird die Natur des Reportergens als nicht kritisch für die Durchführbarkeit
des Verfahrens erwartet. Jedoch muß die regulatorische Transkriptionsregion
des Reporterkonstrukts ein oder mehrere cis-regulatorische Elemente
einschließen,
die Ziele für
Transfektionsfaktoren sind, für
die negative Hormone gesucht werden. Falls man zum Beispiel RAR-negative
Hormone zu identifizieren wünscht,
dann könnte
die regulatorische Transkriptionsregion des Reporterkonstrukts ein
cis-regulatorische Element enthalten, das durch ein RAR-haltiges
Protein gebunden werden kann. In diesem Beispiel sollte es eine Übereinstimmung
zwischen der DNA-Bindungsdomäne des
RAR und dem cis-regulatorischen Element der regulatorischen Transkriptionsregion
des Reporterkonstrukts geben. Falls somit ein chimäres RAR
mit einer konstitutiven Transkriptionsaktivatordomäne und einer DNA-Bindungsdomäne, die
cis-regulatorische Östrogen-reaktive
Elemente binden kann, im Durchmusterungsverfahren eingesetzt wird,
dann sollte die regulatorische Transkriptionsregion des Reporterkonstrukts
ein Östrogen-reaktives
Element enthalten.
-
Beispiele
für cis-regulatorische
Elemente, die direkt Retinoidrezeptoren (RAREs) binden, die nützlich im
Zusammenhang mit dem Reporter-Assay sind, werden von Mangelsdorf
et al. offenbart, The Retinoid Receptors, The Retionoids: Biology,
Chemistry and Medicine, 2. Auflage, Hrsg. Sporn et al., Raven Press
Ltd., New York (1994), dessen Offenbarung oben durch Verweis eingeführt wurde.
Beispiele für
cis-regulatorische Elemente, die indirekt chimäre Rezeptoren binden, schließen DNA-Bindungsorte
für jedes
DNA-Bindungsprotein
ein, für
das die DNA-Bindungsdomäne
des Proteins in einen chimären
Rezeptor eingeführt
werden kann, der aus dieser DNA-Bindungsdomäne besteht, die an einen Retinoidrezeptor
gebunden ist. Spezifische Beispiele für heterologe DNA-Bindungsdomänen, die
in chimäre
Rezeptoren eingebaut werden können
und die heterologe cis-regulatorische Elemente erkennen werden,
schließen
diejenigen ein, die Östrogen-reaktive
Elemente erkennen. Deshalb braucht der Retinoidrezeptor-Anteil eines
chimären
Rezeptors, der nützlich
im Zusammenhang mit dem Durchmusterungsverfahren ist, nicht die
DNA-Bindung des Retinoidrezeptors enthalten, sondern muß zumindest
die Ligandenbindungsdomäne
des Retinoidrezeptors enthalten.
-
Ein
weiteres Beispiel für
indirekte Retinoidrezeptorbindung an das cis-regulatorische Element
schließt die
Verwendung eines Proteins ein, das das cis-regulatorische Element
binden und mit einem Retinoidrezeptor dimerisieren kann. In diesem
Fall assoziiert der Retinoidrezeptor mit dem cis-regulatorischen Element nur durch Assoziation
mit dem für
die DNA-Bindung verantwortlichen Protein. Ein Beispiel für ein solches
System würde
die Verwendung eines Fusionsproteins einschließen, das aus einer heterologen
DNA-Bindungsdomäne besteht,
die an einen RXR fusioniert ist, der zumindest die Domäne des RXR
enthält,
die für
die Dimerisierung mit RARs verantwortlich ist. Gleichzeitig eingeführte RARs
können
mit einem solchen Fusionsprotein dimerisieren, das an das cis-regulatorische Element
gebunden ist. Wir sehen voraus, daß jedes ein cis-regulatorisches
Element bindende Protein, das mit RARs dimerisiert, um zu einer
indirekten Assoziierung des RAR mit dem cis-regulatorischen Element
zu führen,
ebenfalls zur Verwendung mit dem negativen Hormon-Durchmusterungsverfahren
geeignet sein wird.
-
In
einer bevorzugten Ausführungsform
des Durchmusterungsverfahrens werden negative Retinoidhormone als
diejenigen Retinoide identifiziert, die die Basisexpression eines
manipulierten RAR-Transkriptionsfaktors mit erhöhter Basisaktivität unterdrücken. Obwohl
es nicht wesentlich zur Durchführung
des Durchmusterungsverfahrens ist, schloß der im folgenden Beispiel
eingesetzte manipulierte RAR eine konstitutive Transkriptionsaktivierungsdomäne ein.
Die Verwendung dieses chimären
Rezeptors lieferte vorteilhaft ein Mittel, durch das die Basisexpression
eines Reportergens in Abwesenheit von Retinoid erhöht werden
konnte. Obwohl wir eine transiente Transfektion in den oben ausführlich dargestellten
Verfahren eingesetzt haben, wären auch
stabil transfizierte Zellinien, die konstitutiv den chimären Rezeptor
exprimieren, nützlich
im Zusammenhang mit dem Durchmusterungsverfahren.
-
Wie
im folgenden Beispiel offenbart wird, war ein chimärer Retinoidrezeptor
mit einer konstitutiven Transkriptionsaktivatordomäne heterodimerisierbar
mit einem zweiten Rezeptor, der manipuliert wurde, um eine DNA-Bindungsdomäne zu enthalten,
die spezifisch für
ein auf Östrogen
reagierendes cis-regulatorische Element ist. In diesem Fall assoziiert
der chimäre
Retinoidrezeptor mit einer konstitutiven Transkriptionsaktivatordomäne mit der
cis-regulatorischen
Region, die die Reportergenexpression kontrolliert, indirekt über Bindung
an einen zweiten Rezeptor, der eine DNA-Zielsequenz bindet. Insbesondere
wurde der zweite Rezeptor manipuliert, um eine DNA-Bindungsdomäne zu enthalten,
die ein auf Östrogen
reagierendes Element erkennt. Vorteilhaft reagierte des Reportergen
mit einem auf Östrogen
reagierenden Element in der Stromaufwärts-Promotorregion nicht auf Retinoidagonisten
in Abwesenheit des transfizierten chimären Rezeptors mit der konstitutiven
Transkriptionsaktivatordomäne.
Entsprechend wurde die gesamte Reportergenaktivität den transfizierten
Rezeptoren zugewiesen. Die Kombinationsverwendung aus der DNA-Bindungsdomäne des auf Östrogen reagierenden
Elements und dem cis-regulatorischen Element des auf Östrogen
reagierenden Elements sollen allein veranschaulichend sein. Die
Durchschnittsfachleute werden erkennen, daß andere Kombinationen aus manipulierten
Rezeptoren mit Spezifität
für nicht-RARE
cis-regulatorische Elemente ebenfalls nützlich in der Durchführung des
erfundenen Durchmusterungsverfahrens sein werden.
-
Zellen,
die im Zusammenhang mit dem Durchmusterungsverfahren nützlich sind,
werden eukaryontische Zellen sein, die transfiziert werden können. Die
Zellen können
tierische Zellen sein, wie humane, Primaten- oder Nagetierzellen.
Wir haben sehr gute Ergebnisse unter Verwendung von CV-1-Zellen
erreicht, aber erwarten vernünftigerweise,
daß andere
kultivierte Zellinien ebenfalls erfolgreich verwendet werden könnten. Jedes
einer Anzahl herkömmlicher
fachbekannter Transfektionsverfahren kann zur Einführung eines
Expressionskonstrukts verwendet werden, das den chimären Retinoidrezeptor
mit einer konstitutiven Transkriptionsaktivatordomäne codiert.
-
Die
konstitutive Transkriptionsaktivatordomäne wird aus einer Anzahl von
Aminosäuren
bestehen, die wahrscheinlich einen insgesamt sauren Charakter haben
werden, wie er durch eine negative Ladung unter neutralen pH-Bedingungen
dargestellt wird. Zum Beispiel kann die konstitutive Transkriptionsaktivatordomäne eine
Aminosäuresequenz
aufweisen, die ebenfalls in viralen Transkriptionsfaktoren gefunden
wird. Ein Beispiel für
einen viralen Transkriptionsfaktor mit einer konstitutiven Transkriptionsaktivatordomäne ist das
Herpes simplex-Virus 16. Jedoch wären auch andere virale oder
synthetische Transkriptionsaktivatordomänen nützlich in der Konstruktion
von Expressionskonstrukten, die den chimären Retinoidrezeptor mit einer
konstitutiven Transkriptionsaktivatordomäne codieren.
-
Wie
nachfolgend beschrieben wird, haben wir ein verallgemeinertes Durchmusterungsverfahren
entwickelt, das nützlich
zur Identifizierung von negativen Retinoidhormonen ist. Dieses Durchmusterungsverfahren
stellt ein Mittel zur Unterscheidung von einfachen Antagonisten
gegenüber
negativen Hormonen bereit. Tabelle 12 führt mehrere Retinoidverbindungen
auf, die hochwirksame Affinität
für RAR-γ aufweisen, jedoch mit Ausnahme
von ATRA diesen Rezeptor in einem transienten Kotransfektions-Transaktivierungs-Test
nicht aktivierten. Wir haben deshalb diese Verbindungen zur Bestimmung
untersucht, welche davon RAR-γ-Antagonisten
waren und welche von diesen Antagonisten, falls überhaupt, eine negative Hormonaktivität aufwiesen.
-
Beispiel
12 beschreibt das zur Identifizierung von Retinoidverbindungen,
die Antagonisten waren, verwendete Verfahren und die Untergruppe
von Antagonisten, die eine negative Hormonaktivität aufwiesen.
-
Beispiel 12
-
Test auf negative
Retinoidhormone
-
4 × 10
4 CV-1-Zellen wurden durch das Calciumphosphat-Kopräzipitationsverfahren,
beschrieben in Molecular Cloning: A Laboratory Manual (Sambrook
et al., Hrsg., Cold Spring Harbor Lab Publ. 1989) unter Verwendung
von 0,5 μg
ERE-tk-Luc-Reporterplasmid
und 0,1 μg
ER-RAR-γ (Graupner
et al., Biochem. Biophys. Res. Comm. 179: 1554 (1991)) chimärem Expressionsplasmid
transfiziert. Nach 18 Stunden wurden die Zellen mit PBS gespült und mit
DMEM (Gibco-BRL) gefüttert,
das 10% mit Aktivkohle extrahiertes FBS (Gemini Bio-Products) enthielt.
Die Zellen wurden mit 10
–8 M ATRA in Ethanol
oder mit Ethanol allein behandelt. Zusätzlich wurden ATRA-behandelte
Zellen mit 10
–9,
10
–8,
10
–7 oder
10
–6 M
der in Tabelle 12 aufgeführten
Verbindungen behandelt. Nach 18 Stunden wurden die Zellen in PBS
gespült
und in 0,1 M KPO
4 (pH 7,8), 1,0% TRITON
X-100, 1,0 mM DTT und 2 mM EDTA lysiert. Luciferase-Aktivitäten wurden
wie von de Wet et al. beschrieben gemessen, Mol. Cell. Biol. 7:
725 (1987). Tabelle
12
- a Relative Affinität (Kd) bestimmt durch Konkurrenz
der 3H-ATRA-Bindung an Baculovirus-exprimiertes
RAR-γ und
Anwendung der Cheng-Prussof-Gleichung
- b EC50 gemessen in CV-1-Zellen, die
mit ΔMTV-TREp-Luc
und RS-RAR-γ kotransfiziert
waren. "kA" bedeutet keine Aktivität.
-
Wie
durch die teilweise in 7 und in Tabelle 12 dargestellten
Ergebnisse gezeigt wird, waren alle in Tabelle 12 aufgeführten Verbindungen,
mit Ausnahme von ATRA, Retinoidantagonisten an RAR-γ.
-
Die
in Tabelle 12 identifizierten RAR-γ-Antagonisten wurden als nächstes zur
Bestimmung durchmustert, ob sie auch negative Retinoidhormone waren,
falls überhaupt.
4 × 104 CV-1-Zellen
wurden gemäß dem Calciumphosphat-Verfahren,
beschrieben in Molecular Cloning: A Laboratory Manual (Sambrook
et al., Hrsg., Cold Spring Harbor Lab Publ. 1989), unter Verwendung
von 0,5 μg
ERE-tk-Luc-Reporterplasmid und 0,1 μg ER-RXR-α (Graupner et al., Biochem.
Biophys. Res. Comm. 179: 1554 (1991)) und 0,2 μg RAR-γy-VP-16 (Nagpal et al., EMBO
J. 12: 2349 (1993)) chimären
Expressionsplasmiden transfiziert. Nach 18 Stunden wurden die Zellen
mit PBS gespült
und mit DMEM (Gibco-BRL) gefüttert,
das 10% mit Aktivkohle extrahiertes FBS (Gemini Bio-Products) enthielt.
Die Zellen wurden mit 10–9, 10–8,
10–7 oder
10–6 M
von jeder der in Tabelle 12 aufgeführten Verbindungen behandelt.
Die Behandlung mit Ethanol als Träger allein diente als Negativkontrolle.
Nach 18 Stunden wurden die Zellen in PBS gespült und in 0,1 M KPO4 (pH 7,8), 1,0% TRITON X-100, 1,0 mM DTT
und 2 mM EDTA lysiert. Luciferase-Aktivitäten wurden wie zuvor von de
Wet et al. gemessen, Mol. Cell. Biol. 7: 725 (1987).
-
Wie
in 8 gezeigt ist, konnten die Retinoidantagonisten
aus Tabelle 12 in zwei Klassen aufgrund ihrer Wirkung auf die konstitutive
Transkriptionsaktivierungsfunktion des RAR-γ-VP-16 chimären Retinoidrezeptor unterteilt
werden. Eine Gruppe, die AGN 193174, AGN 193199 und AGN 193840 einschloß, unterdrückte nicht
die RAR-γ-VP-16-Aktivität, obwohl
sie ATRA-Antagonisten waren. Im Gegensatz wiesen AGN 193109, AGN
193385, AGN 193389 und AGN 193871 eine dosisabhängige Unterdrückung der
konstitutiven RAR-γ-VP-16-Aktivität auf. Während die
Verbindungen beider Gruppen RAR-γ-Antagonisten
waren, wiesen daher nur diejenigen der zweiten Gruppe eine negative
Hormonaktivität
auf. Dieser Test unterscheidet vorteilhaft negative Retinoidhormone
von einfachen Retinoidantagonisten.
-
Die
vorhergehenden experimentellen Ergebnisse bewiesen, daß AGN 193109
die Kriterien erfüllt,
die ein negatives Hormon definieren. Insbesondere zeigten die in
Beispiel 11 dargestellten Ergebnisse, daß AGN 193109 die Fähigkeit
hatte, eine inhibitorische Aktivität an den RARs selbst in Abwesenheit
von exogen hinzugegebenen Retinoidliganden auszuüben. Als solche besaß diese
Verbindung biologische Aktivitäten,
die nicht von der Blockade der Wechselwirkung zwischen den RARs
und Agonisten wie ATRA und AGN 191183 abhing. Diese Befunde führten uns
zu der Schlußfolgerung,
daß AGN
193109 die Wechselwirkungen zwischen RARs und NCPs stabilisierte.
Wie in 9 schematisch dargestellt wird,
existieren NCP/RAR/PCP-Wechselwirkungen in einem Gleichgewichtszustand.
Ein Agonist dient zur Erhöhung
der PCP-Wechselwirkungen und Verringerung der NCP-Wechselwirkungen,
während
ein inverser Agonist oder negatives Hormon NCP-Wechselwirkungen
stabilisiert und PCP-Wechselwirkungen
verringert. Wie zuvor angezeigt wurde, legten unsere experimentellen
Ergebnisse nahe, daß die
intrazelluläre
Verfügbarkeit
von NCP für
andere Rezeptoren durch AGN 193109-Verabreichung moduliert werden
kann. Insbesondere fanden wir, daß AGN 193109 die Komplexierung
von NCP mit RARs fördern
kann, wodurch das intrazelluläre
Reservoir von NCP reduziert wird, das zur Wechselwirkung mit anderen
Transkriptionsfaktoren als den RARs verfügbar ist.
-
Als
nächstes
untersuchten wir die Wirkung von AGN 193109 auf die Agonisten-vermittelte
Inhibierung der AP-1-abhängigen
Genexpression. In Endocr. Rev. 14: 651 (1993) offenbarte Pfhal,
daß Retinoidagonisten die
Genexpression durch einen Mechanismus abregulieren können, der
die Inhibierung von AP-1-Aktivität
beinhaltet. Wir postulierten, daß AGN 193109 einen von zwei
Effekten aufweisen könnte,
wenn es in Kombination mit einem Retinoidagonisten in einem Modellsystem
verwendet wird, das zur Messung von AP-1-Aktivität entwickelt wurde. Zunächst könnte AGN
193109 merklich der Wirkung des Agonisten entgegengewirkt haben, wodurch
die Agonisten-abhängige Inhibierung
der AP-1-Aktivität
aufgehoben wird. Alternativ könnte
AGN 193109 die Aktivität
des Agonisten gesteigert haben, wodurch die Agonisten-abhängige Inhibierung
der AP-1-Aktivität
erhöht
wird.
-
Beispiel
13 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die Anti-AP-1-Aktivität
eines Retinoidagonisten potenzierte. Wie nachfolgend offenbart wird,
inhibierte der AGN 191183-Retinoidagonist schwach die AP-1-abhängige Genexpression.
Die Kombination aus AGN 193109 und dem Retinoidagonisten inhibierte
stark die AP-1-abhängige
Genexpression. Als solches hatte AGN 193109 im wesentlichen keine
Anti-AP-1-Aktivität.
-
Beispiel 13
-
AGN 193109 potenziert
die Anti-AP-1-Aktivität
eines Retinoidagonisten
-
HeLa-Zellen
wurden mit 1 μg
des Str-AP1-CAT-Reportergenkonstrukts
und 0,2 μg
von Plasmid pRS-hRARα,
beschrieben von Giguere et al., Nature 33: 624 (1987), unter Verwendung
von LIPOFECTAMINE (Life Technologies, Inc.) transfiziert. Str-AP1-CAT
wurde durch Klonieren eines DNA-Fragments,
das den Positionen –84
bis +1 des Stromelysin-1-Promotors
aus Ratte entspricht (Matrisian et al., Mol. Cell. Biol. 6: 1679 (1986)),
zwischen den HindII-BamHI-Orten von pBLCAT3 (Luckow et al., Nucl.
Acids Res. 15: 5490 (1987)) hergestellt. Diese Sequenz des Stromelysin-1-Promotors
enthält
ein AP1-Motiv als einziges Verstärkerelement (Nicholson
et al., EMBO J. 9: 4443 (1990)). Die Promotorsequenz wurde durch
Verschmelzen von zwei synthetischen Oligonukleotiden mit den folgenden
Sequenzen hergestellt: 5'-AGAAGCTTATGGAAGCAATTATGAGTCAGTTTGCGGGTGACT
CTGCAAATACTGCCACTCTATAAAAGTTGGGCTCAGAAAGGTGGACCTCGAGGATCC AG-3' (SEQ ID NO: 2) und
5'-CTGGATCCTCGAGGTCCACCTTTCTGAGCCCAACTTTTATAGAGTGGCAGTATTTGCA
GAGTCACCCGCAAACTGACTCATAATTGCTTCCATAAGCTTCT-3' (SEQ ID NO: 3). Verfahren, die die
Transfektion, Behandlung mit geeigneten Verbindungen und Messung
der CAT-Aktivität beinhalten,
wurden wie von Nagpal et al. beschrieben durchgeführt, J.
Biol. Chem. 270: 923 (1995), dessen Offenbarung hier durch Verweis
eingeführt
wird.
-
Die
Ergebnisse dieser Verfahren zeigten an, daß AGN 193109 die Anti-AP-1-Aktivität des Retinoidagonisten
AGN 191183 potenzierte. Insbesondere inhibierte AGN 191183 im Konzentrationsbereich
von 10–12 bis
10–10 M
die TPA-induzierte
Str-AP1-CRT-Expression nicht. Behandlung mit AGN 193109 im Konzentrationsbereich
von 10–10 bis
10–8 M
inhibierte die AP-1-vermittelte Reporteraktivität nicht wesentlich. Jedoch
zeigten die in 10 dargestellten Ergebnisse
an, daß die
Stimulierung der Transfektanten mit der Kombination aus AGN 193109
(10–8 M)
und AGN 191183 im Konzentrationsbereich von 10–12 bis
10–10 M
wesentlich die TPA-induzierte Str-AP1-CAT-Expression um einen Betrag
von 12 bis 21% wesentlich inhibierte. Deshalb potenzierte AGN 193109
die Anti-AP-1-Aktivität
von AGN 191183 unter Bedingungen, bei denen dieser Retinoidagonist
gewöhnlich
die AP-1-Aktivität
nicht hemmte.
-
Wir
argumentierten, daß AGN
193109 die Agonisten-vermittelte Unterdrückung von AP-1-Aktivität durch
einen Mechanismus potenzierte, der wahrscheinlich eine AGN 193109-abhängige Komplexierung
von NCPs an RARs beinhaltete. RARs gehören zu einer Superfamilie von
Kernrezeptoren, die ebenfalls Rezeptoren für 1,25-Dihydroxyvitamin D3, Glukokortikoid, Schilddrüsenhormon, Östrogen
und Progesteron einschließt. Es
war eine vernünftige
Annahme, daß die
Fähigkeit
zur Bindung von NCPs von unterschiedlichen Vertretern der Kernrezeptor-Superfamilie geteilt
werden kann. Dies führte
uns zur Spekulation, daß AGN
193109 die Anti-AP-1-Aktivität
eines oder mehrerer der Liganden potenzieren könnte, die mit dieser Superfamilie
von Kernrezeptoren wechselwirkt.
-
Die
im vorhergehenden Beispiel dargestellten Ergebnisse zeigten deutlich,
daß AGN
193109 die Anti-AP-1-Aktivität
eines Retinoidagonisten potenzierte. Insbesondere verringerte AGN
193109 die Grenzwertdosis, bei der die Anti-AP-1-Aktivität von AGN 191183 detektiert
werden konnte. Da AGN 193109 im wesentlichen keine Anti-AP-1-Aktivität als solches
hat, war seine Wirkung auf Kernrezeptor-Agonisten synergistisch.
Wir stellten ebenfalls fest, daß das
negative AGN 193109-Hormon die Anti-AP-1-Aktivität von 1,25-Dihydroxyvitamin
D3, dem natürlichen Liganden für den Vitamin
D3-Rezeptor, potenzierte.
-
Der
beobachtete Synergismus zwischen AGN 193109 und AGN 191183 im vorhergehenden
Beispiel implizierte notwendigerweise, daß die Anti-AP-1-Aktivität des Retinoidagonisten
und die AGN 193109-vermittelte Potenzierung dieser Aktivität aus unterschiedlichen
Mechanismen resultieren müssen.
Falls die Mechanismen der Wirkung der zwei Mittel identisch wäre, dann
folgt, daß die
Wirksamkeit der Kombination aus AGN 193109 und dem Agonisten additiv
sein würde.
Stattdessen wurde gezeigt, daß die
Kombination wirksamer als jedes Mittel allein war, eine Wirkung,
die man nicht vor diesem Befund vorhergesagt hätte.
-
Signifikanterweise
wurde die AGN 193109-vermittelte Potenzierung des RAR-Agonisten
unter Verwendung eines ca. 100-fachen molaren Überschusses von AGN 193109
gegenüber
derjenigen des Retinoidagonisten durchgeführt. Entsprechend sollte die
Mehrheit der RARs durch AGN 193109 gebunden sein, was zu sehr wenigen
RARs führt,
die zur Agonistenbindung verfügbar
sind. Trotz dieser Tatsache war die Population von RARs, die nicht
durch AGN 193109 gebunden waren, in der Lage, Retinoidagonisten
zu binden und kräftig
eine Agonisten-abhängige Reaktion
kräftig
zu stimulieren, die als Inhibierung der Reportergenexpression meßbar war.
Somit legen unsere Daten eine mögliche
Heterogenität
der RARs nahe, die durch AGN 193109 induziert werden.
-
Die
negative Hormonaktivität
von AGN 193109, die dessen Fähigkeit
zur Förderung
der Wechselwirkung von RARs und NCPs zugewiesen wird, lieferte eine
Basis zum Verständnis
des Synergismus zwischen AGN 193109 und Retinoidagonisten. Unsere
Ergebnisse waren vollständig übereinstimmend
mit einem Modell, in dem eine AGN 193109-Behandlung von Zellen die
Bindung von RARs und NCPs fördert,
wodurch die Anzahl von freien NCPs und freien RARs innerhalb der
Zelle reduziert wird. Dies führt
zur Erzeugung von zwei Populationen von RARs, die funktionell unterschiedlich
sind. Die erste Population wird durch RARs dargestellt, die mit
NCPs assoziiert sind. Solche AGN 193109/RAR/NCP-Komplexe können nicht
durch Retinoidagonisten aktiviert werden. Die zweite Population
besteht aus RARs, die nicht durch NCP gebunden sind und zur Wechselwirkung
mit Agonisten verfügbar
bleiben. Diese letztere Population wird als "RAR*" bezeichnet,
um freie RARs in einer Umgebung anzuzeigen, die im wesentlichen
von NCP entvölkert
ist.
-
Die
RAR*s besitzen verringerte Wahrscheinlichkeiten der Assoziierung
mit NCP durch Gleichgewichtsbindung und besitzen eine erhöhte Empfindlichkeit
gegenüber
Retinoidagonisten, zum Beispiel meßbar als Anti-AP-1-Aktivität. Dies
ist so, weil das Reservoir an PCP nicht erschöpft ist, während das intrazelluläre Reservoir
an NCP aufgrund der AGN 193109-Verabreichung
erschöpft
ist. Entsprechend können
freie RAR*s einen Retinoidagonisten binden und mit PCP-Faktoren
in einer Umgebung wechselwirken, die im wesentlich von NCP entvölkert ist.
Die Fähigkeit
von AGN 193109 zur Erhöhung
der Empfindlichkeit anderer Kernrezeptoren gegenüber ihren jeweiligen Agonisten
kann der Fähigkeit
dieser unterschiedlichen Kernrezeptoren zugewiesen werden, mit den
gleichen NCPs wechselzuwirken, die mit AGN 193109/RAR- Komplexen wechselwirken.
Dieses Modell der AGN 193109-vermittelten
Modulierung von NCP-Verfügbarkeit
für Mitglieder
der Kernrezeptorfamilie ist schematisch in 11 dargestellt.
-
Dieses
mechanistische Modell führte
uns zu der Vorhersage, daß AGN
193109 die Aktivitäten
von anderen Kernrezeptorliganden als Retinoidagonisten modulieren
könnte.
Wie im folgenden Beispiel veranschaulicht wird, bestätigten wir,
daß AGN
193109 die Aktivität
von 1,25-Dihydroxyvitamin D3 in einem in-vitro-Transaktivierungstest
potenzierte.
-
Beispiel
14 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die Aktivität
von 1,25-Dihydroxyvitamin
D3 in einem Transaktivierungstest steigerte.
-
Beispiel 14
-
AGN 193109 potenziert
die 1,25-Dihydroxyvitamin D3-Aktivität
-
HeLa-Zellen
wurden unter Verwendung des kationischen Liposom-vermittelten Transfektionsverfahrens
transfiziert, das beschrieben wird von Felgner et al., Proc. Natl.
Acad. Sci. USA 84: 7413 (1987). 5 × 104 Zellen
wurden in Mehrfachvertiefungsplatten mit 12 Vertiefungen ausplattiert
und in mit 10% FBS ergänztem DMEM
kultiviert. Die Zellen wurden in serumfreien Medium unter Verwendung
von 2 μg/Vertiefung
von LIPOFECTAMINE-Reagens (Life Technologies, Inc.) mit 0,7 μg des Reporterplasmids
MTV-VDRE-Luc, das zwei Kopien des 1,25-Dihydroxyvitamin D3-Response-Elements 5'-GTACAAGGTTCACGAGGTTCACGTCTTA-3' (SEQ ID NO: 4) aus
dem Osteopontingen der Maus enthält
(Ferrara et al., J. Biol. Chem. 269: 2971 (1994)), ligiert in das
Reporterplasmid ΔMTV-Luc (Heyman et al.,
Cell 68: 397 (1992)), und 0,3 μg
des Plasmids pGEM3Z (Pharmacia, Inc.) als Träger-DNA kotransfiziert, um
die Endkonzentration von DNA auf 1,0 μg pro Vertiefung zu bringen.
Nach sechs Stunden Transfektion wurden die Zellen mit Wachstumsmedium
gefüttert, das
mit Aktivkohle extrahiertes FBS in einer Endkonzentration von 10%
enthielt. 18 Stunden nach der Transfektion wurden die Zellen mit
Träger
allein (Ethanol) oder AGN 193109 in Ethanol mit einer Endkonzentration von
entweder 10–8 oder
10–7 M
behandelt. Sechs Stunden später
wurde 1,25-Dihydroxyvitamin D3 in Ethanol auf
eine Endkonzentration von 10–10 bis 10–7 M
hinzugegeben. 18 Stunden nach der 1,25-Dihydroxyvitamin D3-Behandlung
wurden die Zellen lysiert und geerntet. Die Luciferase-Aktivität wurde
wie oben beschrieben gemessen. Dieses experimentelle System erlaubte
ein zweckmäßiges Verfahren
zur Überwachung
und Quantifizierung der 1,25-Dihydroxyvitamin
D3-abhängigen
Genexpression.
-
Die
in 12 dargestellten Ergebnisse zeigten, daß bei Vergleich
mit dem Ergebnis, das unter Verwendung von 1,25-Dihydroxyvitamin
D3 allein erhalten wurde, das mit 1,25-Dihydroxyvitamin
D3 zusammen verabreichte AGN 193109 die
Dosis-Reaktions-Kurve zur Linken verschob. Dies bestätigte, daß AGN 193109 die
Wirksamkeit von 1,25-Dihydroxyvitamin D3 im
In-vitro-Transaktivierungstest potenzierte. Insbesondere veranschaulicht 12 graphisch, daß eine AGN 193109-Konzentration von
so niedrig wie 10–100
nM das 1,25-Dihydroxyvitamin D3 ca. 10-fach
aktiver machte. Während
eine Konzentration von 1,25-Dihydroxyvitamin D3 von
10–8 M
erforderlich war, um eine Luciferase-Expression von ca. 2000 rlu
zu erzeugen, war nur 1/10 so viel 1,25-Dihydroxyvitamin D3 erforderlich, um die gleiche Luciferaseleistung
zu erzeugen, wenn das Vitamin zusammen mit AGN 193109 bei einer
Konzentration von 10–8 bis 10–7 M
gleichzeitig verabreicht wurde. Obwohl dies nicht im Diagramm in 12 gezeigt ist, wurden im wesentlichen identische
Ergebnisse unter Verwendung von AGN 193109-Konzentrationen von 10–9 M
und 10–8 M
erhalten. Somit reduziert die Koverabreichung mit AGN 193109 wesentlich
die Menge von 1,25-Dihydroxyvitamin D3,
die zur Erzeugung einer ähnlichen
Wirkung in Abwesenheit des negativen Hormons erforderlich war.
-
Interessanterweise
gab es eine gleichzeitige Abnahme der Fähigkeit von AGN 193109 zur
Potenzierung der Aktivität
von 1,25-Dihydroxyvitamin D3, wenn das obige
Verfahren mit Kotransfektion eines Vitamin D-Rezeptor-(VDR)-Expressionsplasmids
wiederholt wurde. Wir interpretierten dieses Ergebnis als Hinweis, daß eine Überexpression
von VDRs die Fähigkeit
von AGN 193109 zur Potenzierung der Aktivität von 1,25-Dihydroxyvitamin
D3 beeinflussen könnte. Deshalb kann die intrazelluläre Konzentration
eines Ligandenrezeptors, die sich in einer gewebespezifischen Weise
unterscheiden kann, die Fähigkeit
von AGN 193109 zur Potenzierung der Aktivität eines Liganden beeinflussen,
der den Rezeptor bindet. Dies stimmte erneut überein mit einem Modell, in
dem titrierbare NCPs zur Regulierung der Vitamin D3-Reaktion
beitrug, und stützte
das oben dargestellte Modell.
-
Wie
im folgenden Beispiel veranschaulicht wird, bestätigten wir auch, daß AGN 193109
die Anti-AP-1-Aktivität
von 1,25-Dihydroxyvitamin D3 potenzierte.
Unser Modell zur Aktivität
der AGN 193109-Wirkung erläutert
diese Beobachtung durch Hervorrufung, daß NCPs kräftig mit RARs in Gegenwart
dieses Wirkstoffs assoziieren. Endogene Vitamin D-Rezeptoren, die
in HeLa-Zellen vorhanden sind, wurden wahrscheinlich empfindlicher
gegenüber
dem 1,25-Dihydroxyvitamin D3-Liganden gemacht,
mit dem Ergebnis der Steigerung der Fähigkeit dieses Liganden zur
Inhibierung der Expression aus dem Str-AP1-CAT-Reporter.
-
Beispiel
15 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die Anti-AP-1-Aktivität
von 1,25-Dihydroxyvitamin D3 potenzierte.
-
Beispiel 15
-
AGN 193109 potenziert
die Anti-AP-1-Aktivität
von 1,25-Dihydroxyvitamin D3
-
HeLa-Zellen
wurden mit 1 μg
Str-AP1-CAT unter Verwendung von LIPOFECTAMINE gemäß dem von Nagpal
et al. beschriebenen Verfahren transfiziert, J. Biol. Chem. 270:
923 (1995). Die transfizierten Zellen wurden mit AGN 193109 allein
(10–9 bis
10–7 M),
1,25-Dihydroxyvitamin D3 allein (10–12 bis
10–7 M)
oder 1,25-Dihydroxyvitamin D3 (10–12 bis
10–7 M)
in Gegenwart von 10–8 M AGN 193109 behandelt.
-
Die
Ergebnisse dieser Verfahren zeigten, daß AGN 193109 die Fähigkeit
von 1,25-Dihydroxyvitamin D3 zur Hemmung
von TPA-induzierter
AP-1-Aktivität
potenzierte. Bei alleiniger Verwendung im Konzentrationsbereich
von 10–9 bis
10–7 M
besaß AGN
193109 keine nachweisbare Anti-AP-1-Aktivität. Die in 13 dargestellten Ergebnisse zeigten, daß 1,25-Dihydroxyvitamin
D3 die TPA-stimulierte Aktivität nur im
Konzentrationsbereich von 10–8 und 10–7 M
unterdrückte.
Die Analyse der 1,25-Dihydroxyvitamin D3-vermittelten
Repression der TPA-stimulierten CAT-Aktivität in Gegenwart von 10–8 M
AGN 193109 zeigte, daß Anti-AP-1-Aktivität bei 10–10 und
10–9 M
1,25-Dihydroxyvitamin D3 und eine Zunahme
der Aktivität
bei 10–8 und
10–7 M
Dosen im Vergleich zu der 1,25-Dihydroxyvitamin D3-Behandlung
allein detektierbar war. Diese AGN 193109-abhängige Modulation der 1,25-Dihydroxyvitamin
D3-vermittelten Anti-AP-1-Aktivität war übereinstimmend
mit unserem Modell, in dem die NCP-Komplexierung an RARs das NCP unverfügbar zur
Wechselwirkung mit anderen Vertretern der Kernrezeptorfamilie machte.
Entsprechend wurden die Rezeptoren empfindlicher für die 1,25-Dihydroxyvitamin
D3-Behandlung gemacht.
-
Die
der RAR-vermittelten Transaktivierung und Anti-AP-1-Aktivität zugrundeliegenden
Mechanismen sind wahrscheinlich verschieden. Diese Schlußfolgerung
beruhte auf unserer Beobachtung, daß hohe Dosen von AGN 193109
die Transaktivierung ohne substantielle Inhibierung von Anti-AP-1-Aktivität hemmten.
Wir wollten deshalb einen weiteren Hinweis zur Stützung unseres
Modells zur RAR*-Bildung gewinnen, die durch AGN 193109-Behandlung
vermittelt wird. Um dies zu erreichen, untersuchten wir, ob AGN
193109 die Aktivität des
RAR-spezifischen Agonisten AGN 191183 in einem In-vitro-Transaktivierungstest
potenzieren konnte.
-
Beispiel
16 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die Aktivität
des RAR-spezifischen Agonisten AGN 191183 potenzierte. Die Ergebnisse
dieses Verfahrens zeigten, daß unter
besonderen Umständen
AGN 193109 die Wirksamkeit des RAR-spezifischen Retinoids steigerte, und
lieferten einen starken Hinweis, daß AGN 193109 die RAR*-Bildung fördert.
-
Beispiel 16
-
Potenzierung der Retionoidwirksamkeit
durch AGN 193109-Koverabreichung
-
HeLa-Zellen
wurden unter Verwendung des von Felgner et al. beschriebenen kationischen
Liposom-vermittelten Transfektionsverfahrens transfiziert, Proc.
Natl. Acad. Sci. USA 84: 7413 (1987). 5 × 104 Zellen
wurden in Mehrfachvertiefungsplatten mit 12 Vertiefungen ausplattiert
und in mit 10% FBS ergänztem DMEM
kultiviert. Die Zellen wurden in serumfreies Medium unter Verwendung
von LIPOFECTAMINE-Reagens (2 μg/Vertiefung,
Life Technologies, Inc.) mit 0,7 μg
des Reporterplasmids MTV-TREp-Luc, das zwei Kopien des TREpal-Response-Elements
5'-TCAGGTCATGACCTGA-3' (SEQ ID NO: 5) enthält, inseriert
in das Reporterplasmid ΔMTV- Luc (Heyman et al.,
Cell 68: 397 (1992)), und 0,1 μg
des RAR-γ-Expressionsplasmids
pRShRAR-γ (Ishikawa
et al., Mol. Endocrinol. 4: 837 (1990)) kontransfiziert. Nach 6
Stunden Transfektion wurden die Zellen mit Wachstumsmedium gefüttert, das
mit Aktivkohle extrahiertes FBS in einer Endkonzentration von 10%
enthielt. 18 Stunden nach der Transfektion wurden die Zellen mit
Träger
allein (Ethanol) oder AGN 193109 in Ethanol mit einer Endkonzentration
von 10–11 bis
10–8 M
behandelt. 6 Stunden später
wurde AGN 191183 in Ethanol auf eine Endkonzentration von entweder
0, 10–10 oder
10–9 M
hinzugegeben. Die Zellen wurden nach 18 Stunden der AGN 191183-Behandlung
geerntet, und die Luciferase-Aktivität wurde wie oben beschrieben
gemessen.
-
Vorläufige Experimente
zeigten, daß 10–9 M
AGN 193109 relativ ineffektiv in der Inhibierung der Reaktion auf
10–8 M
AGN 191183 in HeLa-Zellen war. Dies war im Gegensatz zur Fähigkeit
von 10–9 M
AGN 193109 zur Inhibierung von 10–8 M
ATRA in CV-1-Zellen (2).
-
Die
in 14 dargestellten Ergebnisse stützten die Vorhersage, daß AGN 193109
die Bildung von RAR* stimuliert. Übereinstimmend mit unserer
Charakterisierung der Antagonist- und
negativen Hormon-Aktivitäten
von AGN 193109 führte
die Behandlung mit AGN 193109 zu einer zweiphasigen Dosis-Reaktions-Kurve.
Die niedrigsten Dosen von AGN 193109 (10–11 und
10–10 M)
führten
zu einer Stimulierung der Luciferase-Aktivität gegenüber derjenigen von AGN 191183
allein. Diese Wirkung legt nahe, daß RAR*s durch AGN 193109 erzeugt
werden. Überraschenderweise
wurde dies ebenfalls für
die AGN 193109-Behandlung
allein beobachtet, was nahelegt, daß RAR*s auf einen endogenen
Liganden reagieren können.
AGN 191183 ist ein synthetischer Retinoidagonist und aktiviert wie
ATRA die Transkription durch die RARs. Die Substitution von AGN 191183
für ATRA
in Beispiel 7 würde
qualitativ ähnliche
Ergebnisse ergeben (d.h. AGN 193109 würde der Wirkung von 10 nM AGN 191183
entgegenwirken). Beispiel 16 veranschaulicht, daß, obwohl AGN 193109 als ein Antagonist
von RAR-Agonisten funktionieren kann, die Dosierungsbedingungen
leicht identifiziert werden könnten,
mit denen eine AGN 193109-Koverabreichung
die durch einen RAR-Agonisten vermittelte Aktivierung potenziert.
Es ist wichtig festzustellen, daß die in Beispiel 16 verwendeten
Dosen der Verbindungen wesentlich geringer als die Dosen sind, die
im in Beispiel 7 beschriebenen Verfahren eingesetzt wurden. Wir schlugen
vor, daß eine
AGN 193109-Behandlung zu RAR-Heterogenität, RARs gegenüber RAR*s,
führen könnte. Diese
offensichtliche Heterogenität
(d.h. Fähigkeit
zur Potenzierung) scheint unterschiedliche Fenster in der Transaktivierung
gegenüber
der AP-1-Unterdrückung
zu besitzen. Der Grund, daß die
Kurven zweiphasig sind, ist der, daß bei zunehmenden Mengen von
AGN 193109 proportional weniger RAR zur Bindung des Agonisten verfügbar ist.
Dies scheint nicht der Fall für
die AP-1-Unterdrückung zu
sein, und wir konnten nur spekulieren, daß dieser Unterschied zwei unterschiedliche
Mechanismen für
die Transaktivierung und AP-1-Unterdrückung durch die gleichen Rezeptorspezies
reflektieren muß.
-
Klinische
Ergebnisse haben bestätigt,
daß einige
Retinoide nützlich
zur Inhibierung des Wachstums von prämalignen und malignen zervikalen
Läsionen
sind. Exemplarische Untersuchungen, die diese Schlußfolgerung
stützen,
wurden von Graham et al., West. J. Med. 145: 192 (1986), von Lippman
et al., J. Natl. Cancer Inst. 84: 241 (1992), und von Weiner et
al., Invest. New Drugs 4: 241 (1986)) veröffentlicht.
-
Ähnliche
Schlußfolgerungen
werden durch die Ergebnisse von In-vitro-Untersuchungen gestützt, die kultivierte
Zellen zur Quantifizierung der antiproliferativen Wirkungen von
verschiedenen Retinoiden verwendeten. Insbesondere setzten Agarwal
et al., Cancer Res. 51: 3982 (1991), die ECE16-1-Zellinie ein, um die frühen Stadien
von zervikaler Dysplasie zu modulieren, und zeigten, daß Retinolsäure die
vom epidermalen Wachstumsfaktor (EGF) abhängige zelluläre Proliferation
hemmen konnte.
-
Beispiel
17 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 der Aktivität
des AGN 191183-Retinoidagonisten
entgegenwirken kann, der die Proliferation der ECE16-1-Zellinie hemmte.
-
Beispiel 17
-
AGN 193109 wirkt der antiproliferativen
Wirkung von Retionoiden in ECE16-1-Zellen entgegen
-
ECE16-1-Zellen
wurden mit einer Dichte von 1 × 104 Zellen pro cm2 in
komplettes Medium geimpft, das DMEM:F12 (3:1), nichtessentielle
Aminosäuren,
5% FBS, 5 μg/ml
Transferrin, 2 nM 3,3',5-Triiodthyronin (Schilddrüsenhormon
oder "T3"), 0,1 nM Choleratoxin,
2 mM L-Glutamin, 1,8 × 10–4 M
Adenin und 10 ng/ml EGF enthielt. Man ließ die Zellen über Nacht
an den Platten anhaften und wechselte dann zu definiertem Medium,
das DMEM:F12 (3:1), 2 mM L-Glutamin, nichtessentielle Aminosäuren, 0,1%
Rinderserumalbumin, 1,8 × 10–4 M
Adenin, 5 μg/ml
Transferrin, 2 nM T3, 50 μg/ml Ascorbinsäure, 100 μg/ml Streptomycin,
100 Einheiten/ml Penicillin und 50 μg/ml Gentamicin enthielt. Definiertes
Medium (DM) wurde mit 10 ng/ml EGF ergänzt. EGF-behandelte Zellen
erhielten 10 nM des AGN 191183-Retinoidagonisten in Kombination
mit entweder 0, 0,1, 1,0, 10, 100 oder 1000 nM AGN 193109 oder 1000
nM AGN 193109 allein. Nach 3 Tagen Behandlung wurden die Zellen
wie von Hembree et al. beschrieben geerntet, Cancer Res. 54: 3160
(1994), und die Zellzahlen wurden unter Verwendung eines COULTER-Zählers bestimmt.
-
Die
in 15 dargestellten Ergebnisse zeigten, daß sich ECE16-1-Zellen
als Reaktion auf EGF vermehrten, aber nicht im definierten Medium
allein. Dies bestätigte
die Befunde, die von Adreatta-van Leyen et al., J. Cell. Physio.
160: 265 (1994), und von Hembree et al., Cancer Res. 54: 3160 (1994),
veröffentlicht
wurden. Die Zugabe von 10 nM AGN 191183 und 0 nM AGN 193109 inhibierte
vollständig
die EGF-vermittelte Proliferation. Daher war AGN 191183 ein hochwirksames
antiproliferatives Retinoid. Die Erhöhung der AGN 193109-Konzentration von
0 auf 10 nM wirkt der AGN 191183-vermittelten
Wachstumsinhibierung um etwa 50% entgegen. Ein 10-facher molarer Überschuß von AGN
193109 kehrte die antiproliferative Wirkung von AGN 191183 vollständig um.
Die Behandlung von Zellen mit 1000 nM AGN 193109 allein hatte keine
Wirkung auf die EGF-vermittelte Proliferationszunahme. Diese Ergebnisse
bewiesen, daß AGN
193109 der antiproliferativen Wirkung eines Retinoids entgegenwirkte,
aber im wesentlichen keine antiproliferative Aktivität als solches
hatte, wenn es zur Behandlung von Zellen verwendet wurde, die zervikales
Epithel darstellen, das empfindlich gegenüber Wachstumsinhibierung durch
Retinoide wie AGN 191183 ist. Bemerkenswert gab es keinen Beweis,
daß AGN
193109 die antiproliferative Aktivität des AGN 191183-Agonisten
unter Verwendung des ECE16-1-Modellsystems potenzierte.
-
Im
Gegensatz zum durch die ECE16-1-Zellinie dargestellten Modellsystem
gibt es andere Beispiele, in denen die mit zervikaler Dysplasie
assoziierte zelluläre
Proliferation nicht durch Retinoidagonisten gehemmt werden kann.
Zum Beispiel beschrieben Agarwal et al., Cancer Res. 54: 2108 (1994),
die Verwendung von CaSki-Zellen als ein Modell für Zervixtumore, die nicht auf
Retinoidtherapie ansprechen. Wie nachfolgend offenbart wird, hatte
die Retinoidbehandlung anstelle der Inhibierung der Zellproliferation
im wesentlichen keine Wirkung auf die Wachstumsrate von CaSki-Zellen. Das folgende
Beispiel ist auf die Wirkung des negativen AGN 193109-Hormons auf
die Proliferationsraten dieser Zellinie gerichtet. Die Ergebnisse
bewiesen unerwartet, daß AGN
193109 die Proliferation von Zervixtumorzellen inhibieren kann,
die nicht auf die antiproliferativen Wirkungen von Retinoidagonisten
ansprechen.
-
Beispiel
18 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 das Wachstum einer Zervixtumorzellinie inhibierte, die nicht
auf die antiproliferativen Wirkungen anderer Retinoide wie AGN 191183
ansprach. Signifikanterweise zeigte AGN 193109 antiproliferative
Aktivität
in Abwesenheit von zugegebenem Retinoid.
-
Beispiel 18
-
AGN 193109 inhibiert die
Proliferationsrate der aus Zervixkarzinom stammenden CaSki-Zellinie
-
Wir
untersuchten die Wirkung von EGF auf die CaSki-Zellproliferation, entweder allein oder
in Kombination mit dem AGN 191183-Retinoidagonisten und/oder dem
negativen AGN 193109-Hormon bei einer Konzentration von 10–6 M.
Zellproliferationstests wurden wie oben für Untersuchungen beschrieben
durchgeführt, die
ECE16-1-Zellen beinhalteten. EGF wurde zu den Retinoid-behandelten
Kulturen zum Erhalt einer Endkonzentration von 20 ng/ml gegeben.
Die Zellen wurden mit AGN 191183 (10–10 bis
10–6 M)
in Gegenwart oder Abwesenheit von 10–6 M
AGN 193109 für
insgesamt 3 Tage behandelt. Das Medium wurde jeden Tag gegen frisches
Medium und nach Bedarf gegen jede der zwei Retinoidverbindungen
ausgetauscht. Zellzahlen wurden unter Verwendung eines COULTER-Zählers wie
oben beschrieben bestimmt.
-
Die
in 16 dargestellten Ergebnisse zeigten, daß CaSki-Zellen substantiell
refraktär
für die
Wirkungen eines Retinoidagonisten waren, und das AGN 193109 eine antiproliferative
Aktivität
in Abwesenheit von zugegebenem Retinoid aufwies. Die Gegenwart von
EGF im Kulturmedium stimulierte das CaSki-Zellwachstum. Diese Schlußfolgerung
beruhte auf dem Vergleich des gestreiften Balkens, der kein AGN
191183 darstellt, und dem leeren Balken, der definiertes Wachstumsmedium
("DM") allein darstellt.
Die AGN 191183-Behandlung
hatte keine antiproliferative Aktivität auf die CaSki-Tumorzellinie.
Wir ließen
jede schwache Zunahme der zellulären
Proliferationsrate unberücksichtigt,
die mit dem Retinoidagonisten assoziiert war, weil eine 10000-fache
Zunahme der Retinoidagonist-Konzentration mit einer nur ungefähr 20%igen
Zunahme der Proliferationsrate verbunden war. Somit hatte der AGN
191183-Agonist im wesentlichen keine Wirkung auf die Proliferationsrate
von CaSki-Zellen.
-
Die
in 16 dargestellten Ergebnisse zeigten auch, daß AGN 193109
die Proliferation der zervikalen CaSki-Epithelzellinie hemmte. Diese Schlußfolgerung
beruhte auf dem Vergleich der Messungen, die als schwarzer Balken "0" AGN 191183 und als gestreifter Balken "0" AGN 191183 erscheinen. Somit konnte
AGN 193109 eine biologische Reaktion in Abwesenheit von zugegebenem
Retinoidagonisten stimulieren, wenn es zur Behandlung von Zervixtumorzellen
verwendet wurde, die nicht durch Retinoidagonisten wie AGN 191183 wachstumsgehemmt
wurden.
-
Unser
Befund, daß das
negative AGN 193109-Hormon die zelluläre Proliferation hemmen konnte, stimmte
mit einem Modell überein,
in dem unkomplexierter RAR die Expression von Genen vermittelt,
die für die
Proliferation erforderlich sind. Während ein RAR-Agonist wie AGN
191183 im wesentlichen keine Wirkung hatte oder vielleicht die zelluläre Proliferation
schwach förderte,
hatte AGN 193109 eine antiproliferative Wirkung. Das negative AGN
193109-Hormon band wahrscheinlich RARs, wodurch die NCP-Assoziation
gefördert und
das Annehmen einer inaktiven Konformation durch die RARs verursacht
wurde. Gemäß unserem
Modell unterdrückte
dies die Genaktivität,
die positiv durch unkomplexierte RARs reguliert wurde. Diese Fähigkeit
von AGN 193109 zur Abregulierung der Aktivität von unkomplexierten RARs
resultierte wahrscheinlich aus seiner Fähigkeit zur Förderung
der Assoziation von RARs und NCPs.
-
Die
Durchschnittsfachleute werden einsehen, daß einige Retinoidagonisten
zur Kontrolle der unerwünschten
Konsequenzen von Zellwachstum nützlich
sind, das der Netzhautablösung
folgt. Nach Netzhautablösung
entdifferenziert, wuchert und wandert das Netzhautpigmentepithel
(RPE) in den Unternetzhautraum. Dieser Prozeß kann negativ den Erfolg chirurgischer
Operationen beeinträchtigen,
die auf die erneute Netzhautanbringung gerichtet sind. Campochiaro
et al., Invest. Opthal. & Vis.
Sci. 32: 65 (1991), haben gezeigt, daß RAR-Agonisten wie ATRA eine
antiproliferative Wirkung auf das Wachstum von primären humanen RPE-Kulturen
aufweisen. Es wurde ebenfalls gezeigt, daß Retinoidagonisten das Auftreten
von Netzhautablösung
nach chirurgischer Netzhautwiederanheftung verringern (Fekrat et
al., Opthalmology 102: 412 (1994)). Wie im folgenden Beispiel offenbart
wird, analysierten wir die Fähigkeit
des negativen AGN 193109-Hormons zur Unterdrückung des Wachstums im primären humanen
RPE-Kulturen.
-
Beispiel
19 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die antiproliferative Wirkung eines Retinoidantagonisten
in einer primären
Kultur von humanem Netzhautpigmentepithel potenziert.
-
Beispiel 19
-
AGN 193109 potenziert
die antiproliferative Aktivität
von ATRA
-
Primäre Kulturen
von humanem Netzhautpigmentepithel (RPE) wurden gemäß dem von
Campochiaro et al. beschriebenen Verfahren eingerichtet, Invest.
Opthal. & Vis.
Sci. 32: 65 (1991). 5 × 104 Zellen wurden in 16 mm-Vertiefungen von
Mehrfachvertiefungsplatten mit 24 Vertiefungen in DMEM (Gibco) ausplattiert,
das 5% FBS enthielt. Die Zellen wurden zum Schein mit Ethanolträger allein,
ATRA (10–10 bis
10–6 M)
in Ethanol, AGN 193109 (10–10 bis 10–6 M)
in Ethanol oder ATRA (10–10 bis 10–6 M)
und 10–6 M
AGN 193109 behandelt. Die Zellen wurden alle zwei Tage für insgesamt
5 Behandlungstage mit frischem Medium gefüttert, das die entsprechenden
Konzentrationen dieser Verbindungen enthielt. Die Zellen wurden
aus den Platten durch vorsichtige Verdauung mit Trypsin entfernt,
und die Zellzahl wurde mit einem elektronischen Zellzähler gezählt.
-
Die
in 17 dargestellten Ergebnisse zeigten, daß AGN 193109
dramatisch die antiproliverative Aktivität von ATRA auf RPE-Zellen potenzierte.
Die Behandlung von primären
RPE-Zellen mit ATRA
führte
zu einer dosisabhängigen
Abnahme der RPE-Zellproliferation mit einer ca. 40%igen Abnahme
bei 10–6 M
ATRA relativ zu Kontrollkulturen. AGN 193109-Behandlung veränderte nicht substantiell die
Wachstumsrate der RPE-Zellen bei jeder im Verfahren untersuchten
Konzentration. Unerwartet hatte die Kombination aus ATRA (10–11 bis
10–6 M)
und 10–6 M
AGN 193109 eine stärkere
antiproliferative Aktivität
als ATRA allein. Somit potenzierte die gleichzeitige Behandlung
mit AGN 193109 die antiproliferative Wirkung von ATRA. Insbesondere zeigten
die in der Figur gezeigten Ergebnisse, daß die antiproliferative Wirkung
von 10–8 M
ATRA unter Verwendung von nur 10–10 M
ATRA in Kombination mit 10–7 AGN 193109 erhältlich war.
Somit steigerte das negative AGN 193109-Hormon vorteilhaft die antiproliferative
Aktivität
von ATRA um das ca. 100-fache.
-
In
einem unabhängigen
Experiment zeigte ein Vergleich der antiproliferativen Wirkung von
ATRA (10–11 bis
10–6 M)
mit derjenigen von ATRA und 10–6 M AGN 193109 erneut
die scheinbare Zunahme der Empfindlichkeit von primären RPE-Zellen auf ATRA in
Gegenwart von AGN 193109. In diesem System funktionierte AGN 193109
weder als Retinoidantagonist noch wies es eine antiproliferative
Wirkung bei alleiniger Verwendung auf. Jedoch potenzierte die gleichzeitige
Verabreichung von AGN 193109 die antiproliverative Aktivität des Retinoidagonisten.
-
AGN
193109 wurde auf seine Fähigkeit
zur Potenzierung der antiproliferativen Wirkung von 13-cis-Rentinolsäure (13-cis-RA) in primären RPE-Kulturen
unter Verwendung der oben beschriebenen Bedingungen und Techniken
zur Messung der RPE-Zellproliferation
untersucht. Bemerkenswert ist 13-cis-RA klinisch signifikant. Insbesondere
ist 13-cis-RA nützlich
in der Behandlung verschiedener Krankheitszustände, die Akne (Peck et al.,
N. Engl. J. Med. 300: 329 (1977); Jones et al., Br. J. Dermatol.
108: 333 (1980)) und Schuppenzellkarzinom der Haut und Zervix einschließen, in
Kombinationsbehandlung mit Interferon 2α (Lippman et al., J. Natl. Cancer
Inst. 84: 241 (1992); Moore et al., Seminars in Hematology 31: 31
(1994)).
-
Die
in 18 dargestellten Ergebnisse zeigten, daß sowohl
13-cis-RA (10–12 bis
10–6 M)
als auch ATRA (10–12 bis 10–6 M)
wirksam das RPE-Zellwachstum hemmten. Bemerkenswert war das 13-cis-Isomer
ca. zwei Größenordnungen
weniger wirksam in diesem Test im Vergleich zu ATRA. Ähnlich zu
den Ergebnissen, die unter Verwendung der Koverabreichung von AGN
193109 und ATRA erhalten wurden (oben), erhöhte die Koverabreichung von
AGN 193109 (entweder 10–8 oder 10–6 M)
mit 13-cis-RA (10–12 bis 10–6 M)
dramatisch die Wirksamkeit von 13-cis-RA in der Vermittlung der
Unterdrückung
der RPE-Zellproliferation. Im Gegensatz zur Behandlung mit 13-cis-RA
allein steigerte die Koverabreichung von AGN 193109 die Wirksamkeit
von 13-cis-RA. Somit potenzierte AGN 193109 die antiproliferative
Aktivität
von 13-cis-RA.
-
Als
nächste
untersuchten wir die Fähigkeit
von AGN 193109 zur Potenzierung der Aktivitäten von anderen Kernrezeptorhormonen
in primären
RPE-Zellkulturen. Dexamethason, ein synthetischer Glukokortikoidrezeptoragonist,
ist ein Vertreter einer Klasse von Verbindungen, die klinisch wegen
ihrer hochwirksamen entzündungshemmenden
und immunsuppressiven Eigenschaften verwendet werden. Schilddrüsenhormon
(T3; 3,3',5'-Triiodthyronin) ist ein natürlicher
Schilddrüsenhormon-Rezeptoragonist,
der primär
zur Hormonersatztherapie in der Behandlung von Hypothyroidismus
verwendet wird. Die Verfahren, die in diesen Experimenten verwendet
wurden, waren identisch zu denjenigen, die oben für Verfahren
unter Einsatz von ATRA und 13-cis-RA beschrieben wurden.
-
Die
Ergebnisse dieser Verfahren zeigten, daß die Koverabreichung von AGN
193109 und den Kernrezeptoragonisten die antiproliferativen Aktivitäten der
Kernrezeptoragonisten potenzierte. Insbesondere zeigten die in 19 dargestellten Ergebnisse, daß die Einzelmittelbehandlung
von RPE-Zellen mit entweder Dexamethason (10–11 bis
10–6 M)
oder ATRA (10–12 bis
10–6 M)
die RPE-Zellproliferation im wesentlichen nicht hemmen konnte. Jedoch
unterdrückte
die Behandlung von RPE-Zellen mit Dexamethason (10–11 bis
10–6 M) und
entweder 10–8 oder
10–6 M
AGN 193109 die RPE-Zellproliferation
in einem Ausmaß,
das etwa der durch Behandlung mit ATRA verursachten Inhibierung
entsprach. In ähnlicher
Weise zeigten die in 20 dargestellten Ergebnisse,
daß AGN
193109 die antiproliferative Aktivität von Schilddrüsenhormon
potenzierte. Ähnlich zu
den unter Verwendung von Dexamethason erhaltenen Ergebnissen war
die Proliferation der RPE-Zellen refraktär für Einzelmittelbehandlung mit
Schilddrüsenhormon
(10–11 bis
10–6 M).
Jedoch hemmte die gleichzeitige Behandlung von RPE-Zellen mit Schilddrüsenhormon
(10–11 bis
10–6 M)
und AGN 193109 (entweder 10–8 oder 10–6 M)
die RPE-Zellproliferation in einer Schilddrüsenhormon-abhängigen Weise.
Wir schlußfolgerten,
daß AGN
193109 primäre
RPE-Kulturen empfindlich für
die antiproliferativen Wirkungen dieser Kernrezeptoragonisten machte.
Der Mechanismus, durch den AGN 193109 diese Wirkungen vermittelte,
beinhaltete wahrscheinlich eine Modulierung von NCP/RAR-Wechselwirkungen.
-
Wir
untersuchten zusätzlich
die Wirkung von AGN 193109 auf die Expression von Markergenen in
anderen experimentellen Systemen, die empfindlich auf Retinoidagonisten
waren. Sowohl die MRP8- als auch Stromelysin-Gene sind dafür bekannt,
daß sie
durch Retinoidagonisten in einer Vielzahl biologischer Systeme inhibiert
werden. Zum Beispiel haben Wilkinson et al., J. Cell Sci. 91: 221
(1988), und Madsen et al., J. Invest. Dermatol. 99: 299 (1992),
offenbart, daß die
MRP8-Genexpression
bei Psoriasis erhöht
ist. Umgekehrt wurde die MRP8-Genexpression durch den Retinoidagonisten
AGN 190168 bei humaner psoriatischer Haut (Nagpal et al., eingereicht
1995), bei humanen Keratinozyten-Raft-Kulturen (Chandraratna et
al., J. Invest. Dermatol. 102: 625 (1994)) und bei kultivierten
Keratinozyten der Vorhaut des menschlichen Neugeborenen (Thacher
et al., J. Invest. Dermatol. 104: 594 (1995)) unterdrückt. Nagpal
et al., J. Biol. Chem. 270: 293 (1995), haben offenbart, daß Stromelysin-mRNA-Spiegel
durch Retinoidagonisten wie AGN 190168 in kultivierten Keratinozyten
der Vorhaut des menschlichen Neugeborenen unterdrückt wurden.
Wir analysierten die regulierte Expression dieser Gene im Anschluß an Behandlung
von kultivierten Keratinozyten der Vorhaut des menschlichen Neugeborenen
mit entweder dem AGN 191183-Retinoidagonisten oder AGN 193109.
-
Beispiel
20 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die MRP-8-Expression in kultivierten Keratinozyten inhibierte.
-
Beispiel 20
-
AGN 193109 inhibiert die
MRP-8-Expression in Keratinozyten
-
Primäre Vorhautkeratinozyten
wurden gemäß dem von
Nagpal et al. beschriebenen Verfahren isoliert, J. Biol. Chem. 270:
923, (1995) und in Keratinozyten-Wachstumsmedium (KGM) kultiviert,
das von Clonetics erworben wurde. Nach 3 Tagen Behandlung mit AGN
191183 (10–7 M)
oder AGN 193109 (10–6 M) wurde die zelluläre Gesamt-RNA
aus behandelten und Kontroll-Keratinozyten
gemäß Standardverfahren
isoliert. Die mRNA wurde zu cDNA revers transkribiert, die dann
als Matrize in einem PCR-Amplifikationsprotokoll unter Verwendung
von Primern diente, die spezifisch für das Glyceraldehydphosphatdehydrogenase-(GAPDH)-Haushaltsgen
oder MRP-8 waren. Die GAPDH-Primer hatten die Sequenzen 5'-CCACCCATGGCAAATTCCATGGCA-3' (SEQ ID NO: 6) und
5'-TCTAGACGGCAGGTCAGGTCCACC-3' (SEQ ID NO: 7).
Die MRP-8-Primer
hatten die Sequenzen 5'-ACGCGTCCGGAAGACCTGGT-3' (SEQ ID NO: 8) und
5'-ATTCTGCAGGTACATGTCCA-3' (SEQ ID NO: 9).
Eine Teilmenge aus der MRP-8-Amplifikationsreaktion (10 μl) wurde nach
jedem Durchlauf der PCR-Amplifikation beginnend bei 12 Durchläufen und
endend bei 21 Durchläufen entfernt.
In ähnlicher
Weise wurde eine Teilmenge der GAPDH-Amplifikationsreaktion nach jedem PCR-Durchlauf
beginnend bei 15 Durchläufen
und endend bei 24 Durchläufen
entfernt. Die Proben wurden auf 2% Agarosegelen elektrophoretisch
behandelt und die getrennten Amplifikationsprodukte durch Ethidiumbromidanfärbung detektiert.
Die Anfärbungsintensität der Amplifikationsprodukte
diente als quantitatives Maß für die Menge
von Ausgangs-mRNA, die spezifisch für den gegebenen Primersatz
ist.
-
Die
Ergebnisse dieses Verfahrens zeigten, daß sowohl AGN 191183 als auch
AGN 193109 unabhängig
die MRP-8-Expression in Keratinozyten hemmten. Die Intensität des angefärbten GAPDH-Amplifikationsprodukts
war im wesentlichen äquivalent
in den Bahnen des Gels, das Ausgangsmaterial darstellte, das aus Kontroll-,
mit AGN 191183 und mit AGN 193109 behandelten Keratinozyten isoliert
wurde. Schwache Banden, die das GAPDH-Amplifikationsprodukt darstellen, waren
zuerst in den Bahnen detektierbar, die den Proben entsprechen, die
nach 18 Durchläufen
PCR-Amplifikation entfernt wurden. Die äquivalenten Anfärbungsintensitäten unter
den verschiedenen Bahnen des Gels zeigten, daß äquivalente Massen von Ausgangsmaterial
für alle
Proben verwendet wurden. Entsprechend waren Unterschiede in den
Intensitäten
der angefärbten
Banden, die MRP-8-Amplifikationsprodukte darstellen, indikativ für Unterschiede
der MRP-8-mRNA-Expression unter
den verschiedenen Ausgangsproben. Wie erwartet wurde das MRP-8-amplifizierte
Signal in mit AGN 191183 (10–7 M) behandelten Kulturen
relativ zu einer unbehandelten Kontrolle inhibiert. Die Behandlung
von kultivierten Keratinozyten mit AGN 193109 (10–6 M)
unterdrückte
ebenfalls die MRP8-Expression gemäß Bewertung durch die geringere
Intensität
des angefärbten
Amplifikationsprodukts.
-
Wie
im folgenden Beispiel veranschaulicht wird, inhibierte AGN 193109
auch die Expression eines zweiten Markergens in Keratinozyten. Nagpal
et al., J. Biol. Chem. 270: 923 (1995), offenbarten, daß die Stromelysin-mRNA-Expression
durch RAR-spezifische
Agonisten in kultivierten Keratinozyten der Vorhaut des neugeborenen
Menschen abreguliert wurde. Nicholson et al. (EMBO J. 9: 4443 (1990))
offenbarten, daß ein AP-1-Promotorelement
eine Rolle in der retinoidabhängigen
negativen Regulation des Stromelysin-1-Gens spielte. Daher war es
von Interesse zu bestimmen, ob AGN 193109 die Expression dieses
Gens verändern konnte.
-
Beispiel
21 beschreibt die Verfahren, die verwendet wurden um zu zeigen,
daß AGN
193109 die Stromelysin-1-Genexpression in Abwesenheit eines exogen
hinzugegeben Retinoidagonisten hemmte.
-
Beispiel 21
-
AGN 193109 hemmt die Stromelysin-1-Expression
in kultivierten Keratinozyten
-
Primäre Vorhaut-Keratinozyten
wurden entweder zum Schein oder für 24 Stunden mit dem RAR-Agonisten
AGN 191183 (10–7 M) oder AGN 193109
(10–6 M)
behandelt. Gesamt-RNA, hergestellt aus zum Schein behandelten und
mit Retinoid behandelten Keratinozyten, wurde revers transkribiert,
und die resultierende cDNA wurde unter Verwendung von β-Actin- oder
Stromelysin-1-Oligonukleotidprimern genau wie von Nagpal et al.
beschrieben PCR-amplifiziert, J. Biol. Chem. 270: 923 (1995)), dessen
Offenbarung hier durch Verweis eingeführt wird. Eine Probe (10 μl) aus der
PCR-Amplifikationsreaktion wurde nach jedem dritten Durchlauf entfernt,
beginnend ab 18 Durchläufen
der PCR-Amplifikation. Die Probe wurde elektrophoretisch an einem 2%
Agarosegel behandelt und nach Ethidiumbromid-Anfärbung detektiert.
-
Die
Ergebnisse dieser Verfahren zeigten, daß AGN 193109 die Stromelysin-1-Genexpression
in Abwesenheit eines exogen hinzugegebenen Retinoidagonisten hemmte.
Insbesondere waren Ethidium-angefärbte Banden, die β-Actin-Amplifikationsprodukte
darstellen, leichter in den Agarosegelen nach 18 Durchläufen von
PCR detektierbar.
-
Während alle
Bandenintensitäten
mit zusätzlichen
Durchläufen
der Amplifikationsreaktion zunahmen, waren angefärbte Banden etwas weniger intensiv
in den Banden, die die AGN 191183-behandelten Zellen darstellen. Dies
zeigte, daß eine
geringfügig
geringere Menge von RNA in den Ausgangsproben vorhanden sein mußte, die
den mit AGN 191183 behandelten Zellen entsprachen. Die Ergebnisse
zeigten auch, daß Stromelysin-1-mRNA
in zum Schein behandelten Keratinozyten detektierbar war, beginnend
bei 33 Durchläufen
der PCR-Amplifikation.
Wie erwartet wurde die Stromelysin-1-mRNA-Expression nach Behandlung mit AGN 191183
(10–7 M)
inhibiert, gemäß Bewertung
durch die schwächere
Bandenintensität
bei Vergleich mit Proben, die aus zum Schein behandelten Proben
stammten. Bei Normalisierung auf die Intensitäten der β-Actin-Amplifikationsprodukte und übereinstimmend
mit den in Messungen der MRP-8-Expression erhaltenen Ergebnissen
führte
die Behandlung von Keratinozyten mit AGN 193109 (10–6 M)
zu einer Abregulation der Stromelysin-1-mRNA-Spiegel. Tatsächlich war
die durch AGN 193109-Behandlung stimulierte Abregulation ununterscheidbar
von der durch Behandlung von Keratinozyten mit dem RAR-Agonisten
AGN 191183 verursachten Abregulation.
-
Wie
hier offenbart wird, kann AGN 193109 jeden von drei möglichen
Effekten in bezug auf die Modulation der Aktivität eines gleichzeitig verabreichten
Agonisten aus der Steroidsuperfamilie aufweisen. Erstens kann AGN
193109 keine Wirkung aufweisen. Zweitens kann AGN 193109 der Wirkung
des Agonisten entgegenwirken, was dadurch zu einer Abnahme der Aktivität des Agonisten
führt.
Schließlich
kann AGN 193109 die Aktivität
des Agonisten potenzieren, was dadurch zu einer Stimulation des
durch den Agonisten erzeugten gemessenen Effekt führt.
-
Verbindungen
mit Aktivitäten,
die durch AGN 193109 moduliert werden können, schließen Retinoidrezeptoragonisten
und Agonisten ein, die an andere Vertreter der Steroidrezeptor-Superfamilie binden.
Diese letztere Kategorie von Agonisten schließt Vitamin D-Rezeptoragonisten,
Glukokortikoid-Rezeptoragonisten und
Schilddrüsenhormon-Rezeptoragonisten
ein. Peroxisomproliferator-aktivierte Rezeptoren, Östrogen-Rezeptor und Orphan-Rezeptoren
mit derzeit unbekannten Liganden können ebenfalls durch AGN 193109
potenziert werden. Für
den Fall, daß der
Agonist aus der Steroidsuperfamilie ein RAR-Agonist ist, kann AGN 193109
entweder der Aktivität
des Agonisten entgegenwirken oder sie potenzieren. Für den Fall,
daß der
Agonist, der in Kombination mit AGN 193109 verwendet wird, eine
Verbindung ist, die an einen anderen Kernrezeptor als einen RAR
binden kann, wird die Koverabreichung von AGN 193109 entweder keine
Wirkung haben oder wird das System für den Agonisten sensibilisieren,
so daß die
Aktivität
des Agonisten potenziert wird.
-
Ein
verallgemeinertes exemplarisches Verfahren zur Bestimmung, welche
der drei möglichen
Aktivitäten
AGN 193109 in einem besonderen System haben wird, folgt. Diese Beschreibung
veranschaulicht jedes der möglichen
Ergebnisse für
die AGN 193109-Koverabreichung mit einem Agonisten aus der Steroidrezeptor-Superfamilie.
Biologische Systeme, die nützlich
zur Bewertung der Fähigkeit
von AGN 193109 zur Modulation der Aktivität eines Kernrezeptoragonisten
sind, schließen
ein (aber sind nicht beschränkt
auf): etablierte Gewebekulturzellinien, viral transformierte Zellinien,
ex-vivo primäre Kulturzellen
und in-vivo Untersuchungen, die lebende Organismen verwenden. Die
Messung der biologischen Wirkung von AGN 193109 in solchen Systemen
könnte
die Bestimmung eines jeden einer Vielzahl biologischer Endpunkte
einschließen.
Diese Endpunkte schließen
ein: Analyse der zellulären
Proliferation, Analyse des programmierten Zelltods (Apoptose), Analyse
des Differenzierungszustand von Zellen über Genexpressionstests, Analyse
der Fähigkeit
von Zellen zur Bildung von Tumoren in nackten Mäusen und Analyse der Genexpression
nach transienter oder stabiler Einführung von Reportergenkonstrukten.
-
Für illustrative
Zwecke wird eine als mRNA "X" bezeichnete mRNA-Spezies
aus Gen "X" in primären kultivierten "Y"-Zellen exprimiert, die aus dem Organ "Z" isoliert wurden. Unter Standardkulturbedingungen, worin
verschiedene genetische Marker der "Y"-Zellen
aufrechterhalten werden, einschließlich der Expression von Gen "X", führt
die Zugabe eines Retinoidagonisten zu einer Abnahme der Häufigkeit
von "X"-mRNA. Die Analyse der Gen-X-Expression
kann über
Isolierung von zellulärer
mRNA und Messung der Häufigkeit
von X-mRNA-Spiegeln über Polymerasekettenreaktion,
Ribonukleaseschutz oder RNA-Blotting-Verfahren, wie zum Beispiel
Northern-Analysen,
bewertet werden. Nach Isolation aus Organ Z werden primäre Y-Zellen
in einem geeigneten Wachstumsmedium kultiviert. Die primären Kulturen
werden dann in Gewebekulturplatten zur Expansion der Zellpopulation
plattiert. Dieser Schritt erleichtert die Trennung der Zellen in
vier Probengruppen, so daß verschiedene
Dosen des Retinoidagonisten und von AGN 193109 übertragen werden können. Die
erste Gruppe wird eine Kontrolle sein, die nur Träger erhält. Die
zweite Gruppe wird den RAR-Agonisten, Retinolsäure, übertragen in Ethanol, in Mengen
erhalten, die ausreichend sind, um Endkonzentrationen im Bereich von
10–11 bis
10–6 M
bereitzustellen. Es kann sein, daß die niedrigste Dosis empirisch
bestimmt werden muß, abhängig von
der Empfindlichkeit des Systems. Solche Bestimmungen fallen in den
Umfang von Routineexperimenten für
einen Durchschnittsfachmann. Die dritte Gruppe wird sowohl den Kernrezeptoragonisten
in den gleichen Dosen, die zur Behandlung der Zellen von Gruppe
2 verwendet wurden, als auch eine konstante Dosis von AGN 193109
erhalten. Die Dosis von AGN 193109, die zur Behandlung der Zellen
von Gruppe 3 verwendet wird, muß auch
empirisch bestimmt werden, aber sollte nahe der Affinitätskonstante
(Kd) von AGN 193109 für die
RAR-Untertypen sein (d.h. wenigstens 10–8 M).
Die vierte Gruppe wird AGN 193109 in Dosen einschließen, die
minimal diejenige einschließen,
die zur Agonist-Koverabreichung in Gruppe 3 verwendet wurde. Eine
Alternative zu diesem Dosierungsschema würde AGN 193109 für den im
vorhergehenden Beispiel beschriebenen Retinoidagonisten austauschen,
wie in Gruppe 2 angegeben, und eine konstante Dosis von Retinoidagonist
anstelle von AGN 193109, wie in Gruppen 3 und 4 angegeben. Nach
einem geeigneten Inkubationszeitraum sollten die Zellen in einer
Weise geerntet werden, die geeignet zur Bestimmung des biologischen
Endpunkts ist, der als ein Indikator für Agonistenaktivität gemessen
wird.
-
Zum
Beispiel würde
die Analyse der Wirkung von AGN 193109 auf die Retinolsäure-abhängige Regulation
der Genexpression den Vergleich der Häufigkeit der mRNA-Spezies X
in der mRNA-Ansammlung
beinhalten, die aus Zellen geerntet wurde, die gemäß jedem
der vier oben beschriebenen Protokolle behandelt wurden. Aus Kontrollzellen
stammende RNA wird zur Bestimmung der Basislinienexpression von
X-mRNA dienen und wird einen Zustand darstellen, der fehlender Unterdrückung entspricht.
Ein Vergleich dieser Niveaus mit demjenigen, das in der mRNA-Ansammlung gemessen
wird, die aus mit Retinolsäure
behandelten Zellen stammt, wird die Bestimmung der Wirkung dieses
Agonisten auf die Genexpression erlauben. Quantifizierte Niveaus
der Unterdrückung
von spezifischen mRNAs, die aus Retinolsäurebehandlung resultiert, können dann mit
mRNA-Häufigkeiten
aus Zellen verglichen werden, die parallel mit entweder AGN 193109
allein oder AGN 193109 in Kombination mit Retinolsäure behandelt
wurden. Während
dieses verallgemeinerte Beispiel eine Analyse der Wirkung von koverabreichtem
AGN 193109 auf die Expression eines Gens veranschaulicht, das durch
einen Retinoidagonisten unterdrückt
wird, könnte
das Beispiel alternativ die Analyse der Wirkung von koverabreichtem
AGN 193109 auf ein Gen beschrieben haben, das durch einen Retinoidagonisten induziert wurde.
Das kritische Merkmal zur Bestimmung, ob sich AGN 193109 als Agonist,
negatives Hormon oder ohne Wirkung in einem besonderen System verhalten
wird, wird den quantitativen Vergleich der Größe der Wirkung in Gegenwart
und Abwesenheit von AGN 193109 beinhalten.
-
Ein
Beispiel, in dem AGN 193109 die Aktivität eines koverabreichten Agonisten
potenzierte, wäre
ein Fall, in dem eine gleichzeitige Behandlung mit AGN 193109 und
Retinolsäure
zu einem Grad der X-mRNA-Expression führte, die stärker unterdrückt relativ
zu dem Niveau ist, das in mit Retinolsäure allein behandelten Zellen
gemessen wird. Insbesondere würde
ein Vergleich der Dosis-Reaktions-Kurve der biologischen Wirkung
(d.h. Unterdrückung
von X-mRNA-Häufigkeit),
aufgetragen auf der Y-Achse, gegen die Dosis des Agonisten (logarithmischer
Maßstab)
auf der X-Achse einen Vergleich der Agonist-vermittelten Unterdrückung von X-mRNA-Häufigkeit in Gegenwart und Abwesenheit
von gleichzeitiger AGN 193109-Behandlung erlauben. Die Fähigkeit
von AGN 193109 zur Sensibilisierung der biologischen Reaktion auf
den Agonisten, wodurch die Aktivität des Agonisten potenziert
wird, wird durch eine Verschiebung in der Dosis-Reaktions-Kurve nach links angezeigt.
Insbesondere würde
in Gegenwart von AGN 193109 weniger Agonist erforderlich sein, um
die gleiche biologische Wirkung zu erhalten, die unter Verwendung
des Agonisten allein erhältlich
ist.
-
Ein
Beispiel für
einen AGN 193109-vermittelten Antagonismus eines koverabreichten
Agonisten würde
ein Fall sein, in dem die gleichzeitige Behandlung mit AGN 193109
und Retinolsäure
zu einem Grad der X-mRNA-Expression führte, der weniger unterdrückt im Vergleich
zu demjenigen ist, der in mit Retinolsäure allein behandelten Zellen
gemessen wird. Ein Vergleich der Dosis-Reaktions-Kurven der X-mRNA-Unterdrückung gegen
die logarithmische Dosis des Agonisten in Gegenwart und Abwesenheit
von AGN 193109 wird eine Verschiebung nach rechts in der Dosis-Reaktions-Kurve
zeigen. Insbesondere wird in Gegenwart von AGN 193109 mehr Agonist
nötig sein,
um die gleiche biologische Wirkung zu erhalten, die mit einer Einzelmittelbehandlung
mit dem Agonisten allein erhältlich
ist.
-
Die
obigen Beispiele, worin AGN 193109 entweder einen Antagonismus oder
eine Potenzierung vermittelt, beschreiben experimentelle Ergebnisse
für die
Koverabreichung von AGN 193109 mit einem Retinoidagonisten. Falls
jedoch der mit AGN 193109 gleichzeitig verabreichte Agonist ein
Agonist ist, der einen anderen Vertreter der Steroidrezeptor-Superfamilie
als einen RAR binden und aktivieren kann, dann wird es möglich, daß AGN 193109
anstelle dem Agonisten entgegenzuwirken keine Wirkung auf die Aktivität des Agonisten
haben würde.
Falls die gleichzeitige Behandlung mit AGN 193109 und mit einem
solchen Agonisten zu einem Grad der mRNA-Expression führt, der
gleich demjenigen ist, der in mit Agonist allein behandelten Zellen gemessen
wird, dann wird die Fähigkeit
von AGN 193109 zur Beeinflussung der Verfügbarkeit von NCPs über Förderung
von RAR:NCP-Assoziationen in diesem System stumm sein. Dies wäre ein Beispiel,
in dem AGN 193109 keine Wirkung auf einen koverabreichten Agonisten
hat.
-
Beispiel für Antagonismus
-
Das
im obigen verallgemeinerten Beispiel zur Bestimmung der Wirkung
von AGN 193109, das mit einem Retinoidagonisten koverabreicht wird,
offenbarte Verfahren wird durch das in Beispiel 7 beschriebene Verfahren
exemplarisch dargestellt. Mit einem der drei Retinolsäurerezeptoren
und dem Retinoidagonist-induzierbaren MTV-TREp-Luc-Reporterkonstrukt
kotransfizierte CV-1-Zellen wurden entweder mit Ethanol (Kontrolle, Gruppe
1), AGN 193109 in Endkonzentrationen von 10–9 bis
10–6 M
(Gruppe 2), AGN 193109 in Endkonzentrationen von 10–9 bis
10–6 M,
koverabreicht mit Retinolsäure
mit 10–8 M
(Gruppe 3), oder Retinolsäure
(10–8 M, Gruppe
4) behandelt. Der Vergleich der Luciferaseaktivität von Gruppe
1 mit derjenigen von Gruppe 4 erlaubte die Bestimmung des Retinoidagonist-induzierten
Expressionsgrads des Luciferase-Reportergens
in Abwesenheit von zugegebenem AGN 193109. Der Vergleich der Luciferase-Reportergenexpression
in Zellen der Gruppe 3 mit derjenigen, die in Zellen der Gruppe
4 gemessen wurde, zeigte, daß sich
AGN 193109 als ein Antagonist des Retinoidagonisten in diesem System
verhielt.
-
Beispiel für Antagonismus
-
Das
im verallgemeinerten Beispiel zur Bestimmung der Wirkung von AGN
193109, das mit einem Retinoidagonisten gleichzeitig verabreicht
wurde, offenbarte Verfahren wurde in ähnlicher Weise verwendet, um in
Beispiel 17 zu bestimmen, daß AGN
193109 als ein Antagonist einer Retinoidagonist-vermittelten Unterdrückung von
EGF-stimulierter zellulärer
Proliferation in ECE-16-1-transformierten zervikalen Epithelzellen
funktionierte. In diesem Verfahren schlossen Behandlungen von ECE-16-1-Zellen
eine Kontrollprobe, behandelt mit EGF allein (Gruppe 1), eine Probe,
die mit der Kombination aus EGF und AGN 193109 in einer Endkonzentration
von 10–6 M
behandelt wurde (Gruppe 2), eine Probe, die mit der Kombination
aus EGF und AGN 193109 mit Endkonzentrationen von 10–10 bis
10–6 M
behandelt wurde, koverabreicht mit einer einzelnen Dosis des Retinoidagonisten
AGN 191183 mit 10–8 M (Gruppe 3), und
eine Probe ein, die mit der Kombination aus EGF und AGN 191183 mit
10–8 M
behandelt wurde (Gruppe 4). Nach drei Tagen Behandlung wurden die
zellulären
Proliferationsraten bestimmt. Die Bestimmung, daß die Zellen stimuliert worden
waren, um sich durch EGF zu vermehren, war möglich, weil eine zusätzliche
Kontrollbehandlung eingeschlossen wurde, worin Zellen definierten
Medium ausgesetzt waren, das kein EGF enthielt. Der Vergleich der
Zellzahl in Gruppe 1 mit der Zellzahl in Gruppe 4 erlaubte die Bestimmung,
daß der
RAR-Agonist AGN 191183 die EGF-stimulierte Proliferation von ECE-16-1-Zellen
unterdrückte.
Der Vergleich von Gruppe 3 mit Gruppe 4 zeigte, daß AGN 193109 der
Aktivität
des RAR-Agonisten in diesem System entgegenwirkte.
-
Beispiel für Potenzierung
-
Das
im verallgemeinerten Beispiel zur Bestimmung der Wirkung von AGN
193109, das mit einem Retinoidagonisten gleichzeitig verabreicht
wurde, offenbarte Verfahren wurde auch in Beispiel 14 verwendet
um zu bestimmen, daß AGN
193109 die Aktivität
eines Kernrezeptoragonisten in HeLa-Zellen potenzierte, die mit dem
1,25-Dihydroxyvitamin D3-induzierbaren MTV-VDRE-Luc-Reportergen
transfiziert waren. Behandlungen von transfizierten Zellen schlossen
Träger
allein (Kontrolle, Gruppe 1), 1,25-Dihydroxyvitamin D3 in
Endkonzentrationen von 10–10 bis 10–7 M
(Gruppe 2), 1,25-Dihydroxyvitamin
D3 in Endkonzentrationen von 10–10 bis 10–7 M,
gleichzeitig verabreicht mit AGN 193109 in einer Endkonzentration
von entweder 10–8 oder 10–7 M (Gruppe
3), und AGN 193109 als Einzelmittelbehandlung bei einer Endkonzentration
von entweder 10–8 oder 10–7 M
(Gruppe 4) ein. Ein Vergleich der in Gruppe 1 (Kontrolle) gemessen
Luciferaseaktivität
der Zellen mit derjenigen von Zellen der Gruppe 2 erlaubte die Bestimmung,
daß die
1,25-Dihydroxyvitamin
D3-stimulierte Luciferaseaktivität dosisabhängig war.
Ein Vergleich der in Zellen der Gruppe 4 (AGN 193109-Einzelmittelbehandlung)
gemessen Luciferaseaktivität
mit derjenigen, die in Zellen der Gruppe 3 gemessen wurde (AGN 193109-Koverabreichung),
erlaubte in ähnlicher
Weise die Bestimmung der dosisabhängigen 1,25-Dihydroxyvitamin D3-stimulierten
Luciferaseaktivität
in Gegenwart einer gegebenen Konzentration von AGN 193109. In diesem
Fall stellte der Nullwert die Luciferaseaktivität in mit AGN 193109 allein
behandelten Zellen (Gruppe 4) dar. Ein solches Dosierungsschema
erlaubte den Vergleich der drei 1,25-Dihydroxyvitamin D3-Dosis-Reaktions-Kurven.
Ein Vergleich der Dosis-Reaktions-Kurve von 1,25-Dihydroxyvitamin
D3 in Abwesenheit von AGN 193109 mit der
Kurve, die die Koverabreichung von AGN 193109 (entweder 10–8 oder
10–7 M)
darstellte, zeigte die Potenzierung der Agonistenaktivität, wie sie
durch eine Verschiebung der halbmaximalen Reaktion nach links widergespiegelt
wird.
-
Beispiel für Potenzierung
-
Das
im verallgemeinerten Beispiel zur Bestimmung der Wirkung von AGN
193109, das mit einem Retinoidagonisten koverabreicht wurde, offenbarte
Verfahren wurde ferner zur Bestimmung in Beispiel 19 verwendet,
das AGN 193109 die antiproliferative Aktivität eines RAR-Agonisten in primären Kulturen
von humanen Netzhautpigmentepithelzellen potenzierte. Die Behandlungen
von Zellen schlossen ein: Ethanolträger allein (Gruppe 1), Retinolsäure in Endkonzentrationen
von 10–10 bis
10–6 M
(Gruppe 2), Retinolsäure
in Endkonzentrationen von 10–10 bis 10–6 M,
koverabreicht mit 10–6 M AGN 193109 (Gruppe
3), und AGN 193109 allein in Endkonzentrationen von 10–10 bis
10–6 M
(Gruppe 4). Ein Vergleich der Testergebnisse, die unter Verwendung
der Zellen der Gruppen 1 und 2 erhalten wurden, erlaubte die Bestimmung
der dosisabhängigen
Inhibierung der Proliferation dieser Zellen durch Retinolsäure. In ähnlicher
Weise erlaubte ein Vergleich der Ergebnisse, die unter Verwendung
der Zellen der Gruppe 3 erhalten wurden, mit denjenigen der Gruppe
1 die Bestimmung der dosisabhängigen
Inhibierung der Proliferation dieser Zellen durch Retinolsäure in Gegenwart von
gleichzeitig verabreichtem AGN 193109. Gruppe 4 zeigte die Unfähigkeit
von AGN 193109 zur wesentlichen Veränderung der Proliferationsrate
dieser Zellen bei Verwendung als Einzelbehandlungsmittel. Ein Vergleich
der Dosis-Reaktions-Kurven
der Retinolsäure-vermittelten
Unterdrückung
der in den Gruppen 2 und 3 erzeugten zellulären Proliferation lieferte die
Basis für
die Schlußfolgerung,
daß AGN
193109 primäre RPE-Zellen
für die
antiproliferativen Wirkungen des RAR-Agonisten sensibilisierte, wodurch die
Aktivität
des RAR-Agonisten
potenziert wurde.
-
Wie
oben angegeben wurde, zeigten Agarwal et al., Cancer Res. 54: 2108
(1994), daß das
CaSki-Zellwachstum anders als das Wachstum von HPV unsterblich gemachten
ECE-16-1-Zellen nicht durch Behandlung mit Retinoidagonisten inhibiert
wurde. Wie hier offenbart wird, haben wir unerwartet festgestellt,
daß das CaSki-Zellwachstum
durch AGN 193109 in Abwesenheit eines Retinoidagonisten inhibiert
wurde. Das folgende Beispiel veranschaulicht, wie AGN 193109 zur
Inhibierung des Wachstums von CaSki-Zelltumoren in vivo verwendet
werden kann.
-
Beispiel 22
-
Inhibierung von Tumorwachstum
von CaSki-Zellen in nackten Mäusen
nach Verabreichung von AGN 193109
-
1 × 106 CaSki-Zellen werden in jede eine Gruppe
von nackten Mäusen
injiziert. Die Tumorbildung wird unter Verwendung von Techniken
bewertet, die einem Durchschnittsfachmann vertraut sein werden.
Nach der Injektion werden die Mäuse
statistisch in Kontroll- und Testgruppen unterteilt. Die Kontrollgruppe
erhält
ein Plazebo. Der Testgruppe wird AGN 193109 verabreicht. Die Tiere,
denen das Plazebo verabreicht wurde, erhalten eine Magensondierung
mit Maisöl.
Die Testtiere erhalten 20 μmol/kg
AGN 193109 in Maisöl
täglich
für den
Behandlungszeitraum. Das Tumorvolumen wird in Kubikmillimeter unter
Verwendung von skalierten Schublehren gemessen. Das Tumorvolumen
wird als Funktion der Zeit aufgetragen. Mäuse, die AGN 193109 erhalten,
weisen Tumore auf, die signifikant in ihrer Wachstumsrate im Vergleich
zu Tumoren bei den Kontrollmäusen
reduziert sind, gemäß Bewertung
nach Tumorgröße und -anzahl über den
Untersuchungszeitraum. Dieses Ergebnis liefert einen In-vivo-Beweis,
daß AGN
193109 das Wachstum eines fortgeschrittenen Zervixkarzinoms inhibiert,
das resistent gegen Therapie ist, die die Verabreichung eines Retinoidagonisten
umfaßt.
-
Wie
oben gezeigt wurde, sind CaSki-Zellen ein Modell für Zervixtumoren,
die nicht auf eine Retinoidagonist-Therapie ansprechen. Jedoch haben
wir hier offenbart, daß das
CaSki-Zellwachstum
durch AGN 193109 in Abwesenheit von Behandlung mit einem Retinoidagonisten
inhibiert wurde. Die Fähigkeit
von AGN 193109 zur Inhibierung der Proliferation von CaSki-Zellen legte nahe,
daß AGN
193109 verwendet werden könnte,
um Zervixkarzinome therapeutisch zu behandeln, die unempfindlich
gegenüber
Retinoidagonist-Therapie sind. Das folgende Beispiel veranschaulicht
ein Verfahren, das verwendet werden kann, um das therapeutische
Potential von AGN 193109 in der Behandlung eines Zervixkarzinoms
zu bewerten.
-
Beispiel 23
-
Bewertung des therapeutischen
Potentials von AGN 193109 bei Patienten mit Zervixkarzinom
-
Ein
Patient mit einem fortgeschrittenen Zervixkarzinom wird zuerst identifiziert.
Eine zervikale Biopsie wird gemäß Verfahren
erhalten, die einem Durchschnittsfachmann vertraut sein werden.
Zellen aus dem explantierten Tumor werden in Gewebekultur gemäß Standardtechniken
vermehrt, um ausreichende Zellzahlen bereitzustellen, um eine Unterteilung
in drei Probengruppen zu erlauben. Die von Agarwal et al. beschriebenen Kulturbedingungen,
Cancer Res. 54: 2108 (1994), werden für diesen Zweck eingesetzt.
Die erste Gruppe wird als Kontrolle reserviert und erhält nur Träger (Ethanol).
Die zweite Gruppe wird mit dem RAR-Agonisten Retinolsäure in einer
Konzentration von 10–10 bis 10–6 M
behandelt. Die dritte Gruppe wird mit AGN 193109 in Dosen im Bereich
von 10–10 bis
10–6 M
behandelt. Die Zellen werden mit frischem Wachstumsmedium täglich gefüttert und
mit den oben beschriebenen Retinoiden nach Bedarf für jede Probengruppe
versehen. Zellen werden nach drei Tagen unter Verwendung eines elektrischen
Zellzählers
gezählt.
Ein Vergleich der Zellzahl in Kontrollkulturen mit der Zellzahl
in mit Retinolsäure
behandelten Kulturen zeigt, daß ein
RAR-Agonist die Wachstumsrate der kultivierten Zervixkarzinomzellen
nicht substantiell inhibiert. Im Gegensatz weisen mit AGN 193109
behandelte Zellen eine dosisabhängige
Abnahme der Zellzahl bei Vergleich mit Zellzählungen in der Kontrollgruppe
auf. Dieses Ergebnis, wonach eine AGN 193109-Behandlung die kultivierte
Zervixkarzinomzellproliferation inhibiert, zeigt, daß AGN 193109
ein nützliches
Therapeutikum zur Behandlung von Zervixkarzinompatienten mit metastatischer
Krankheit sein wird.
-
Zervixkarzinompatienten,
die eine Operation zur Entfernung von primären Tumoren erfahren haben und
eine metastatische Erkrankung aufweisen, werden in einer randomisierten
klinischen Untersuchung aufgenommen um zu versuchen, den therapeutischen
Nutzen von AGN 193109 in dieser Indikation zu zeigen. Die Patienten
werden in zwei Gruppen unterteilt. Die erste Gruppe ist eine Kontrollgruppe,
während
Mitglieder der zweiten Gruppe mit AGN 193109 behandelt werden. AGN
193109 wird mit einem pharmazeutisch akzeptablen Exzipienten kombiniert,
um eine zur systemischen Verabreichung geeignete Zusammensetzung
herzustellen, alles gemäß Techniken,
die einem Durchschnittsfachmann vertraut sein werden. Der Kontrollgruppe wird
eine Placeboformulierung verabreicht, und der experimentellen Gruppe
wird die Formulierung verabreicht, die das negative AGN 193109-Hormon
enthält.
Die Dosierung der Patienten erfolgt bei der maximalen tolerierten
Dosis und wird jeden zweiten Tag für einen Zeitraum von drei Monaten
bis zu einem Jahr durchgeführt.
Das Ergebnis der Untersuchung wird über Messung des krankheitsfreien Überlebens über die Zeit
quantifiziert. Individuen, die AGN 193109 erhalten, zeigen eine
signifikante Zunahme des krankheitsfreien Überlebens, einschließlich einer überproportionalen
Anzahl von Patienten, die eine vollständige Remission ihrer metastatischen
Krankheit zeigen. Dieses Ergebnis zeigt, daß AGN 193109 therapeutischen
Nutzen zur In-vivo-Behandlung von Zervixkarzinomen besitzt, die
nicht auf die antiproliferativen Wirkungen von Retinoidagonisten
wie Retinolsäure
ansprechen.
-
Wie
oben offenbart wurde, potenzierte AGN 193109 die antiproliferative
Aktivität
von RAR-Agonisten in primären
Kulturen von humanen Netzhautpigmentepithelzellen. Entsprechend
wird vernünftigerweise
von der Koverabreichung von AGN 193109 mit einem RAR-Agonisten in
vivo erwartet, daß sie
den therapeutischen Index des Agonisten erhöht, weil eine geringere Menge
des RAR-Agonisten erforderlich sein wird, um den gleichen therapeutischen
Endpunkt zu erhalten. Zusätzlich
wurde gezeigt, daß AGN
193109 primäre
Kulturen von humanen Netzhautpigmentepithelzellen für die antiproliferativen
Wirkungen von Glukokortikoid- und Schilddrüsenhormon-Rezeptoragonisten sensibilisiert. Das
folgende Kaninchenmodell von PVR wird in zwei getrennten Untersuchungen
verwendet, um den erhöhten
therapeutischen Index zu zeigen, der durch Koverabreichung von AGN
193109 mit einem RAR-Agonisten (13-cis-Retinolsäure) bzw. einem Schilddrüsenhormon-Rezeptoragonisten
erhalten wird. Bemerkenswert wurde das Kaninchenmodell der erneuten
Netzhautablösung,
das von Sen et al. veröffentlicht
wurde, Arch. Ophthalmol. 160: 1291 (1988), verwendet um zu zeigen, das
Retinoidagonisten, die die Proliferation von primären RPE-zellen
in vitro inhibieren, auch die Häufigkeit
von Netzhautablösung
in vivo inhibieren (Araiz et al., Invest. Ophthalmol. 34: 522 (1993)).
Deshalb wurde in bezug auf ihre Verwendung als Therapeutika in der
Prävention
von Netzhautablösung
bereits eine Korrelation zwischen den Aktivitäten von Retinoidagonisten in
vitro und in vivo entwickelt. Die folgenden Beispiele veranschaulichen,
wie AGN 193109 in therapeutischen Anwendungen eingesetzt werden
kann, die auf die Prävention
von Netzhausablösung
gerichtet sind.
-
Beispiel 24
-
Verwendung von AGN 193109
zur Erhöhung
des therapeutischen Potentials von Rezeptoragonisten der Steroidsuperfamilie
in der Behandlung von proliferativer Vitreoretinopathie (PVR)
-
In
einer ersten Studie werden humane RPE-Zellen in den Glaskörper des
Kaninchenauges gemäß dem von
Sen et al. beschriebenen Verfahren injiziert, Arch. Ophthalmol.
106: 1291 (1988). Nach der intravitrealen Injektion werden die Kaninchen
in fünf
Gruppen unterteilt. Die erste Gruppe (Kontrolle) wird allein Träger durch
intravitreale Injektion erhalten. Die zweite Gruppe erhält Retinolsäure als
Einzelmittelbehandlung (100 μg)
durch intravitreale Injektion. Die dritte Gruppe erhält AGN 193109
als Einzelmittelbehandlung (100 μg) durch
intravitreale Injektion. Die vierte Gruppe erhält durch intravitreale Injektion
den RAR-Agonisten (Retinolsäure)
in einer Dosis von einem Zehntel der an Gruppe 2 verabreichten Menge
(10 μg).
Die fünfte
Gruppe erhält
die Kombination aus AGN 193109 (100 μg) und Retinolsäure (10 μg) durch
intravitreale Injektion. Die Tiere erhalten eine einzelne intravitreale
Injektion der entsprechenden Behandlung einen Tag nach der intravitrealen Injektion
von humanen RPE-Zellen. Die Kaninchen werden durch indirekt Ophthalmoskopie
an den Tagen 7, 14 und 28 untersucht und auf Häufigkeit und Schwere von traktionaler
Netzhautablösung
bewertet. Kaninchen aus der Gruppe, der 100 μg Retinolsäure injiziert wurde, weisen
eine signifikant reduzierte Häufigkeit
und Schwere von Netzhautablösung
im Vergleich zu Kontrollkaninchen oder Kaninchen auf, die entweder
AGN 193109 oder Retinolsäure (10 μg) allein
erhielten. Kaninchen in der Gruppe, der die Kombination aus AGN 193109
und Retinolsäure
(10 μg)
verabreicht wurde, weisen eine signifikant reduzierte Häufigkeit
und Schwere von Netzhautablösung
im Vergleich zu denjenigen in den Gruppen aus entweder Kontrolle,
AGN 193109 oder Retinolsäure
(10 μg)
auf. Dieses Ergebnis zeigt, daß AGN
193109 den therapeutischen Index des RAR-Agonisten Retinolsäure in einem
in vivo-Modell von PVR verbessert.
-
In
einer zweiten Studie werden die Kaninchen zuerst mit einer Injektion
von humanen RPE-Zellen in den Glaskörper des Auges versehen und
dann in vier Gruppen unterteilt. Die erste Gruppe (Kontrolle) erhält allein
Träger
durch intravitreale Injektion. Die zweite Gruppe erhält Schilddrüsenhormon
als Einzelmittelbehandlung (100 μg)
durch intravitreale Injektion. Der dritten Gruppe wird AGN 193109
als Einzelmittelbehandlung (100 μg)
durch intravitreale Injektion verabreicht. Der vierten Gruppe wird
die Kombination aus AGN 193109 (100 μg) und Schilddrüsenhormon
(100 μg)
verabreicht. Die Kaninchen werden durch indirekte Ophthalmoskopie
an den Tagen 7, 14 und 28 untersucht und auf Häufigkeit und Schwere der traktionalen
Netzhautablösung
bewertet. Ein Vergleich der Häufigkeit
und Schwere der Netzhautablösung
in den vier Gruppen zeigt, daß eine
Einzelmittelbehandlung mit entweder AGN 193109 oder Schilddrüsenhormon
die Netzhautablösung
im Vergleich mit den Kontrollkaninchen nicht hemmt. Im Gegensatz
weist die Gruppe von Kaninchen, der die Kombination aus AGN 193109
und Schilddrüsenhormon
verabreicht wurde, ein/eine signifikant reduzierte s) Auftreten
und Schwere von Netzhautablösung
auf. Dieses Ergebnis zeigt, daß AGN
193109 den therapeutischen Index von Schilddrüsenhormon in einem In-vivo-Modell von PVR
verbessert.
-
Das
folgende Beispiel veranschaulicht, wie AGN 193109 zur Steigerung
des therapeutischen Index eines RAR-Agonisten verwendet werden kann,
der zur Behandlung von menschlichen Patienten nach Netzhautanheftungsoperation
verwendet wird.
-
Beispiel 25
-
Erhöhung des therapeutischen Index
des RAR-Agonisten 13-cic-Retinolsäure
-
Eine
Gruppe von erwachsenen Freiwilligen mit aus PVR resultierender Netzhautablösung wird
zuerst identifiziert. Die Individuen erfahren eine chirurgische
Reparatur der Ablösungen
unter Verwendung von Techniken, die fachlicher Standard sind. Die
Patienten werden dann in fünf
Gruppen unterteilt. Die Kontrollgruppe besteht aus Patienten, die
eine chirurgische Reparatur der Netzhautablösung erfahren und keinerlei
Retinoid-Verbindung erhalten. Die zweite Gruppe erhält oral
40 mg 13-cis-Retinolsäure
zweimal täglich
für vier
Wochen postoperativ. Die dritte Gruppe erhält oral 40 mg AGN 103109 zweimal
täglich
für 4 Wochen
postoperativ. Die vierte Gruppe erhält oral 4 mg 13-cis-Retinolsäure zweimal
täglich
für vier
Wochen postoperativ. Die fünfte Gruppe
erhält
oral 40 mg AGN 193109 in Kombination mit oral 4 mg 13-cis-Retinolsäure zweimal
täglich
für vier
Wochen postoperativ. Das Behandlungsprotokoll und die Bewertung
der Wirkstoffeffizienz werden im wesentlichen wie von Fekrat et
al. beschrieben durchgeführt,
Ophthalmology 102: 412 (1995).
-
Die
Häufigkeit
und Schwere der erneuten Netzhautablösung bei postoperativen Patienten
in allen fünf Gruppen
wird über
einen Zeitraum von neun Monaten unter Verwendung von ophthalmologischen
Untersuchungstechniken überwacht,
die den Durchschnittsleuten vertraut sein werden. Patienten, die
oral 40 mg 13-cis-Retinolsäure
erhalten, weisen ein signifikant reduziertes Auftreten von erneuter
Netzhautablösung
im Vergleich mit Kontrollpatienten, Patienten, die oral 4 mg 13-cis-Retinolsäure zweimal
täglich
erhalten, oder Patienten auf, die oral 40 mg AGN 193109 zweimal
täglich
erhalten. Die Untersuchung der Patientengruppe, die die Kombination
aus oral 40 mg AGN 193109 und oral 4 mg 13-cis-Retinolsäure zweimal
täglich
für vier
Wochen postoperativ erhält,
zeigt, daß das
therapeutische Ergebnis dieser Patientengruppe gleich oder besser als
diejenigen Patienten ist, die oral 40 mg 13-cis-Retinolsäure zweimal täglich für 4 Wochen
postoperativ erhalten. Dieses Ergebnis zeigt, daß das negative AGN 193109-Hormon den therapeutischen
Index eines RAR-Agonisten aufgrund der Verringerung der Häufigkeit
und Schwere von erneuter Netzhautablösung bei PVR-Patienten verbessert.
-
Verallgemeinerter
Test zur Identifizierung von negativen Kernrezeptorhormonen
-
Wir
haben oben gezeigt, daß AGN
193109 als negatives Hormon funktionieren kann, das die Basistranskriptionsaktivität von RAR-Kernrezeptoren
unterdrücken
kann. Ferner haben wir einen Test beschrieben, der CV-1-Zellen verwendet,
die mit dem ERE-tk-Luc-Luciferase-Reporterplasmid
und den ER-RXR-α-
und RAR-γ-VP-16-Rezeptorexpressionsplasmiden
kotransfiziert wurden, zur Unterscheidung von RAR-Liganden, die
einfache Antagonisten sind, von denjenigen mit negativer Hormonaktivität.
-
Wir
haben schlußgefolgert,
daß negative
RAR-Hormone die Unterdrückung
von RAR-vermittelter Transkriptionsaktivität vermitteln, indem eine erhöhte Wechselwirkung
zwischen dem RAR und den NCPs gefördert wird. Ferner haben wir
gezeigt, daß AGN
193109 die Wirkungen von Agonisten von anderen Kernrezeptoren in
einer Weise potenzieren kann, die mit dem gegenseitigen Teilen von
NCPs zwischen Vertretern der Steroidsuperfamilie von Kernrezeptoren übereinstimmt.
Als solche können
Liganden geschaffen und durchmustert werden, um Verbindungen mit
negativer Hormonaktivität
an diesen Nicht-RAR-Kernrezeptoren zu
identifizieren.
-
Unser
Verfahren zur negativen RAR-Hormondurchmusterung auf Basis der Verwendung
von CV-1-Zellen, die mit dem ERE-tk-Luc-Luciferase-Reporterplasmid und den ER-RXR-α- und RAR-γ-VP-16-Rezeptorexpressionsplasmiden
kotransfiziert wurden, kann allgemein so angepaßt werden, daß die RAR-γ-Einheit
des RAR-γ-VP-16-Plasmids
zu derjenigen von Peroxisomproliferator-aktivierten Rezeptoren (PPAR), Vitamin
D-Rezeptor (VDR), Schilddrüsenhormonrezeptor
(T3R) oder jedem anderen Kernrezeptor der Steroidsuperfamilie, der
mit RXR heterodimerisieren kann, umgewandelt wird. CR-1-Zellen,
die mit solchen Plasmiden kotransfiziert werden, würden hohe
Basisspiegel von Luciferaseaktivität exprimieren. Liganden, die
die Ligandenbindungsdomäne
des für
die RAR-γ-Einheit
substituierten Rezeptors binden können, können leicht auf negative Hormonaktivität durch
Messung ihrer Fähigkeit
zur Unterdrückung
von Luciferaseaktivität
durchmustert werden.
-
Für Kernrezeptoren
der Steroidsuperfamilie, die nicht mit RXR heterodimerisieren (z.B.
Glukokortikoid- und Östrogenrezeptoren),
kann das gleiche Endergebnis unter Verwendung von GR-VP-16- oder ER-VP16-Rezeptoren
und von einem Luciferase-Reporterplasmid, das aus dem entsprechenden
Glukokortikoid- oder Östrogen-Response-Element
besteht, fusioniert an ein heterologes Promotorelement und ein Luciferase-
oder anderes Reportergen, erreicht werden. Ein wesentliches Merkmal
eines verallgemeinerten negativen Hormondurchmusterungstests ist
der Einschluß wenigstens
der Ligandenbindungsdomäne
des besonderen Kernrezeptors, für
den inverse Agonisten zu durchmustern sind, und ein Verfahren zur
Lokalisierung der Kernrezeptor-Ligandenbindungsdomäne für den Promotor
eines Reportergens. Dies könnte
unter Verwendung des natürlichen
DNA-Bindungsortes der Rezeptoren oder alternativ durch Konstruktion
eines chimären Rezeptors
mit einer heterologen DNA-Bindungsdomäne und entsprechender Verwendung
eines Reportergens, das unter der Kontrolle eines DNA- regulatorischen Elements
steht, das durch die heterologe DNA-Bindungsdomäne erkannt wird, erreicht werden.
In einer bevorzugten Ausführungsform
würde das
Plasmid, das den Kernrezeptor exprimiert, für den inverse Agonisten zu
durchmustern sind, diesen Kernrezeptor als ein Fusionsprotein exprimieren,
das eine konstitutive Aktivierungsdomäne enthält, wie zum die HSV VP-16-Aktivierungsdomäne, um eine
hohe Basisaktivität
bereitstellen zu können.
Diese hohe Basisaktivität
würde effektiv die
Testempfindlichkeit erhöhen,
was dadurch eine Analyse der Kernrezeptorliganden erlauben würde, die
die Basistranskriptionsaktivität
in Abwesenheit von zugegebenem Kernrezeptoragonisten unterdrücken.
-
Das
folgende Beispiel veranschaulicht ein Verfahren, das zur Durchmusterung
auf Verbindungen mit negativer Hormonaktivität am Schilddrüsenhormonrezeptor
verwendet werden kann.
-
Beispiel 26
-
Verfahren
zur Identifizierung von negativen Hormonen am Schilddrüsenhormonrezeptor
-
CV-1-Zellen
werden mit dem Luciferase-Reporterplasmid ERE-tk-Luc und den Plasmiden ER-RXR-α und T3R-VP-16
kotransfiziert. T3R-VP-16 ist identisch mit dem Plasmid RAR-γ-VP-16, außer daß die RAR-γ-Einheit
von RAR-γ-VP-16
durch die Schilddrüsenhormonrezeptor-cDNA
ersetzt wurde. Als solches exprimiert T3R-VP-16 ein Fusionsprotein,
das die Aktivierungsdomäne
von HSV VP-16 im Raster mit dem N-Terminus des Schilddrüsenhormonrezeptors
enthält.
Standardmäßige Transfektions-
und Zellkulturverfahren werden für
diesen Zweck eingesetzt. Nach der Transfektion werden die Zellen
gespült
und mit Wachstumsmedium gefüttert,
das 10% fötales
Kälberserum
enthält,
das mit Aktivkohle extrahiert wurde. Die Zellen werden mit Träger allein
(Ethanol), Schilddrüsenhormon (10–9 bis
10–10 M)
oder Verbindung TR-1 (10–9 bis 10–6 M)
behandelt. TR-1 ist ein synthetischer Schilddrüsenhormonrezeptorligand, der
eine starke Affinität
für den
Schilddrüsenhormonrezeptor
in kompetitiven Bindungsstudien aufweist, der aber nicht transfizierten
Schilddrüsenhormonrezeptor
in transienten Kotransfektionstransaktivierungstests, die ein auf
Schilddrüsenhormon
reagierendes Reportergen und ein Schilddrüsenhormonrezeptor-Expressionsplasmid
verwenden, aktiviert. Ferner kann TR-1 der Schilddrüsenhormon-vermittelten Transaktivierung
entgegenwirken und ist als solches ein Schilddrüsenrezeptorantagonist.
-
Die
Analyse der Luciferaseaktivität
aus CV-1-Zellen, die mit ERE-tk-Luc, ER-RXR-α und T3R-VP-16 transfiziert
wurden, zeigt ein hohes Basisniveau von Luciferasereporteraktivität in mit
Träger
behandelten Zellen. Die mit Schilddrüsenhormon behandelten Zellen
zeigen eine schwache Zunahme von Luciferaseaktivität in einer
dosisabhängigen
Weise. Die mit TR-1 behandelten Zellen weisen eine dosisabhängige Abnahme
der Luciferaseaktivität
auf. Dies zeigt, daß TR-1
eine inverse Schilddrüsenrezeptor-Agonistenaktivität aufweist, vermutlich
aufgrund der zunehmenden Wechselwirkung eines NCP mit dem Schilddrüsenhormonrezeptor.
-
Die
Proliferationsrate von humanen primären Netzhautpigmentepithelzellen
wird durch Behandlung mit RAR-Agonisten
unterdrückt.
Der therapeutische Wert dieser Beobachtung wurde in der postoperativen Verwendung
der Retinoidtherapie nach Netzhautwiederanheftungsoperation gezeigt.
Wir haben oben gezeigt, daß AGN
193109 RAR-negatives Hormon primäre
RPE-Zellen für
die antiproliferative Wirkung von ATRA und 13-cis-Retinolsäure bei
Koverabreichungsverfahren sensibilisieren kann. Ferner wurde ebenfalls
gezeigt, daß AGN
193109 RPE-Zellen für
die antiproliferativen Wirkungen anderer Kernrezeptoragonisten sensibilisiert. Insbesondere
sensibilisiert AGN 193109 RPE-Zellen
für die
antiproliferativen Wirkungen des Glukokortikoidagonisten Dexamethason
und des Schilddrüsenhormonagonisten
3,3',5-Triiodthyronin,
T3. Diese Daten stimmten mit unserem Arbeitsmodell überein,
worin AGN 193109 die Verfügbarkeit
von NCPs modulierte, die zwischen den Vertretern der Kernrezeptorfamilie
geteilt wurden. Die Behandlung von RPE-Zellen mit dem inversen Schilddrüsenhormonrezeptor-Agonisten
TR-1 wird in ähnlicher
Weise die Verfügbarkeit
von geteilten NCPs verändern,
so daß die
Koverabreichung mit einem Nicht-Schilddrüsenrezeptor-Agonisten, wie zum Beispiel mit dem
RAR-Agonisten 13-cis-Retinolsäure, zu
einer erhöhten
antiproliferativen Wirkung auf die RPE-Kulturen im Vergleich zu
13-cis-Retinolsäure
als Einzelmittelbehandlung führen
wird.
-
Das
folgende Beispiel veranschaulicht ein Verfahren, das verwendet werden
kann, um primäre RPE-Zellen
empfindlicher gegenüber
der antiproliferativen Aktivität
eines RAR-Agonisten
zu machen. Bemerkenswert veranschaulicht dieses Beispiel weiterhin,
wie die Aktivität
von RAR-Agonisten durch Koverabreichung mit einem negativen Hormon
potenziert werden kann.
-
Beispiel 27
-
Sensibilisierung von primären Netzhautpigmentepithelzellen
für die
antiproliferativen Wirkungen von RAR-Agonisten durch Koverabreichung
des TR-1 inversen Schilddrüsenhormonagonisten
-
Humane
primäre
RPE-Zellen werden gemäß Standardverfahren
erhalten und kultiviert. Die kultivierten Zellen werden in vier
Gruppen unterteilt und wie folgt behandelt. Gruppe 1 erhält allein
Träger
(Ethanol). Gruppe 2 wird mit 13-cis-Retinolsäure in Konzentrationen im Bereich
von 10–11 bis
10–6 M
behandelt. Gruppe 3 wird mit dem inversen Schilddrüsenhormonagonisten
TR-1 mit Konzentrationen im Bereich von 10–11 bis
10–1 M
behandelt. Gruppe 4 wird mit 13-cis-Retinolsäure mit
Konzentrationen im Bereich von 10–11 bis
10–6 M
TR-1 gleichzeitig behandelt. Die Zellen werden erneut mit frischem
Wachstumsmedium gefüttert
und erneut mit der entsprechenden Verbindung alle zwei Tage für insgesamt
fünf Behandlungstage
behandelt. Die Proliferationsrate über die Dauer des Experiments
wird durch Messung der Zellzahl in den Kulturen unter Verwendung
eines elektrischen Zellzählers
quantifiziert.
-
Mit
TR-1-behandelte Zellen (Gruppe 3) weisen Raten der zellulären Proliferation
auf, die im wesentlichen die gleiche wie von Kontrollzellen (Gruppe
1) sind, und es gibt keine Wirkung dieses inversen Agonisten auf
die gemessene Wachstumsrate der Kulturen. Mit 13-cis-Retinolsäure behandelte
Zellen (Gruppe 2) weisen eine dosisabhängige Abnahme der Zellzahl
auf. Ein Vergleich der dosisabhängigen
Abnahme der zellulären Proliferation
der Zellen der Gruppe 4 (13-cis-RA- und TR-1-Koverabreichung) mit
der in Gruppe 3 erhaltenen zeigt die Fähigkeit der Koverabreichung
des inversen Schilddrüsenhormon-Rezeptoragonisten
TR-1, RPE-Kulturen
für die
antiproliferative Wirkung von 13-cis-Retinolsäure zu sensibilisieren, gemäß Messung durch
die Verschiebung der Dosis-Reaktions-Kuve dieses RAR-Agonisten zur
linken Seite in Gruppe 4 im Vergleich zu Zellen der Gruppe 2.
-
-
-
-