-
FACHGEBIET
DER ERFINDUNG
-
Die
Erfindung betrifft ein chemisch-enzymatisches Verfahren zur Modifizierung
von polymeren Kohlenhydratmaterialien, insbesondere zur Verwendung
einer aktivierten Polymergrenzfläche,
um spezifische chemische Gruppen auf die Oberfläche eines polymeren Kohlenhydratmaterials
anzuhängen,
um die physikalisch-chemischen Eigenschaften des Materials zu verändern, sowie
Materialien, die durch dieses Verfahren hergestellt werden, und
Produkte, die diese Materialien umfassen.
-
TECHNISCHER
HINTERGRUND UND STAND DER TECHNIK
-
Fast
alle Zellulosematerialien, die in den Papier-, Karton- und Textilindustrien
verwendet werden, werden entweder vor (z.B. Holzstoff, Baumwollfasern,
etc.) oder nach der Erzeugung des Produkts in seiner endgültigen dreidimensionalen
Form (z.B. Papierblätter,
gewölbter
Karton, Gewebe, etc.) chemisch behandelt, um die Oberflächeneigenschaften
dieser Materialien zu verändern.
Die Behandlung der Zellulosematerialien mit chemischen Zusatzstoffen
an verschiedenen Zeitpunkten des Herstellungsprozesses führt zu dramatischen Veränderungen
in den Eigenschaften der Faseroberfläche. Zum Beispiel wird Carboxymethylzellulose,
ein anionisches Zellulosederivat, zu Holzstoff zugegeben, um die
Retention von allgemein verwendeten kationischen Füllstoffen
und Leimungsmitteln zu erhöhen.
-
Auf
die gleiche Weise werden organische Leimungsmittel wie z.B. Alkylketendimer
und Alkylbernsteinsäureanhydrid
während
der Erzeugung von Papierblättern
zugegeben, um die Hydrophobizität
und die Wirkung der Blattbedruckbarkeit zu erhöhen. Bei der Verwendung von
Zellulosematerialien als Verpackungsmittel für Flüssigkeiten und Lebensmittel
werden Papier und Karton häufig
mit einem Thermoplastik wie z.B. Polyethylen laminiert, um eine
undurchdringbare Grenzschicht für
die wässrigen
Lösungen
zur Verfügung
zu stellen. Sowohl Textilwaren als auch Papierblätter werden routinemäßig gefärbt oder
bedruckt, was ein weiteres Beispiel einer Modifikation der Oberfläche ist.
Viele der derzeitigen Technologien bei Wertpapieren und Verpackungen
(Banknoten, verfolgbare Dokumente und Verpackungen) sind auf die
Oberflächenbehandlungen
mit spezifischen Chemikalien angewiesen, die später analysiert werden können, um
die Authentizität
zu bestimmen. Des weiteren besitzen zellulosehaltige Materialien
ein großes
Potential in der Polymerindustrie für einen weiten Anwendungsbereich
wie z.B. als Füllstoffe,
Laminate und Panelprodukte, holzpolymere Verbundstoffe, polymere
Verbundstoffe, Legierungen und Mischungen und Zellulosederivate
(zellulosehaltige Stoffe).
-
Aufgrund
einiger Engpässe
bei ihrer, Herstellung oder Leistung ist die Verwendung von solchen
Verbundstoffen jedoch immer noch begrenzt. In den vergangenen Jahren
stieg der Bedarf für
oberflächenmodifizierte
Füllstoffe,
welche die Eigenschaften des ursprünglichen Polymers verbessern
und die Kosten oder das Gewicht des fertigen Produkts vermindern.
Die Optimierung der Bindung zwischen den beiden Flächen der
Faser und der Polymergrundsubstanz ist ein wichtiger Aspekt in Bezug
auf die optimale mechanische Leistung und Beständigkeit der faserverstärkten Verbundstoffe.
Die Qualität
der Faser-Grundsubstanz-Grenzfläche
ist für
die Anwendbarkeit von natürlichen
Fasern als Verstärkung
für Kunststoffe
von Bedeutung. Weil die Fasern und die Grundsubstanzen chemisch
unterschiedlich sind, wird eine starke Haftung an ihren Grenzflächen für einen
wirksamen Transfer von Belastungs- und Bindungsverteilung überall an
der Grenzfläche
benötigt.
Neue Verfahren zur Derivatisierung von Zellulose werden daher benötigt, um
die Adhäsion
der Faser-Grundsubstanz für
die Polymerverarbeitung, für
Klebestoffe und für
neue Verbundmaterialien zu verbessern.
-
Der
derzeitig erhältlichen
Technologie zur Modifikation der Oberfläche einer Zellulosefaser durch
physikalische oder chemische Behandlungen fehlt ein hohes Maß an Kontrolle
in der Art, durch welche die Wirkstoffe auf die Faseroberfläche eingearbeitet
werden. Eine besonders ernste Schwäche der direkten chemischen
Modifizierung von Zellulose ist, dass die meisten chemischen Substanzen
in die Faserstruktur eindringen und dass die chemischen Modifizierungen
innerhalb der Faser stattfinden, was zu dem Verlust der Struktur und
der Eigenschaften der Faser führt.
Als Katalysatoren sind Enzyme hochspezifisch in ihrer Wirkungsweise und
bieten daher eine attraktive Alternative zu den herkömmlichen
Verfahren. Zusätzlich
bedeutet die proteinöse
Natur dieser Katalysatoren, dass sie leicht biologisch abbaubar
und umweltfreundlich sind. Des weiteren sind Enzyme naturgemäß oberflächenaktiv
und überwinden
daher das Problem des Eindringens in die Faserstruktur.
-
Abbauenden
Enzymen, die den Zusammenbruch ihrer Substrate durch die Spaltung
von chemischen Bindungen katalysieren, wurde über die letzten 15 Jahre für die Behandlung
von Zellulosematerialien eine erhöhte Aufmerksamkeit geschenkt,
insbesondere in der Faserstoff- und Papierindustrie. Ligninasen
werden zum Beispiel verwendet, um die Bleichfähigkeit und den Weißgehalt
von Faserstoffen durch das Entfernen von Lignin zu verbessern, während eine
Behandlung mit Xylanase das Entfernen von erneut gefälltem Lignin
während
des Siedeverfahrens erleichtert. Gleichermaßen werden Cellulasen in der
Textilindustrie im großen
Maße verwendet,
um die Fertigstellung von Bekleidung zu bewirken. Die Behandlung
von Jeansstoff mit Cellulose ersetzte zum Beispiel weitgehend das
mechanische Rotieren mit Bimssteinen, um so genannte „Stone-washed"-Effekte zu verursachen.
Cellulasen sind ebenfalls ein wichtiger Bestandteil bei einer Anzahl von
Waschdetergenzien, in denen sie als Mittel zur Verhinderung der
Fusselbildung durch das enzymatische Abschneiden von ausgefransten
Baumwollfasern wirken.
-
Trotz
der weltverbreiteten Verwendung von abbauenden Enzymen bei der Modifikation
von Zellulosefasern, ist die Verwendung von Enzymen, die in die
entgegengesetzte Richtung wirken, das heißt in Richtung der Synthese,
wenig entwickelt. Dies liegt hauptsächlich daran, dass nur wenig über die
Enzyme bekannt ist, die für
die Synthese von Polysacchariden wie z.B. Zellulose und Hemizellulose
verantwortlich sind. Die Mehrzahl dieser Enzyme, die als Nucleotid-Zucker-abhängige Transferasen
bekannt sind, sind zellmembrangebunden, wodurch ihre Isolation und
Charakterisierung schwierig ist. Des weiteren ist die Herstellung
der aktivierten Zuckermoleküle
teuer. Die Verwendung von Nucleotid-Zucker-abhängigen Transferasen bei der
Modifikation von Zellulosefasern ist des weiteren durch die Tatsachen
begrenzt, dass die chemische Modifikation des Zuckerrings der natürlichen
Substrate, die letztendlich in die wachsende Polysaccharidkette
eingebaut werden, nicht toleriert wird. Neben Transferasen können Glycosylhydrolasen
wie z.B. bestimmte β-Glycosidasen
oder Cellulasen ebenfalls zur Synthese von Kohlenhydrat verwendet
werden, wobei ein beibehaltender Reaktionsmechanismus verwendet
wird, wenn Wasser von dem Reaktionsgemisch ausgeschlossen wird.
Wenn die Reaktion in organischen Lösungsmitteln durchgeführt wird,
katalysieren diese Enzyme Transglycosylierungsreaktionen, die zu
einer Erzeugung anstelle eines Abbaus von glykosidischen Bindungen
führt.
-
Die
Notwendigkeit, organische Lösungsmittel
zu verwenden, ist jedoch ein deutlicher Nachteil dieses Verfahrens.
Des weiteren können
beibehaltende Glycosydasen gentechnisch hergestellt werden, um ihre
katalytischen Nucleophile zu entfernen. Ein solches Enzym kann kein
kovalentes Enzym-Substrat-Zwischenprodukt
erzeugen, das für
die Hydrolyse benötigt
wird, aber es kann stattdessen die Kondensierung von geeigneten
Empfänger-
und Spenderzuckern katalysieren, wenn der Spenderzucker fluoriert
ist, um den Übergangsstatus
der Reaktion nachzuahmen. Wie mit den Nucleotid abhängigen Glycosyltransferasen
ist der Nachteil die Notwendigkeit von aktivierten Substraten, welche
die Verwendung dieser Technologie in Anwendungen mit einem großen Maßstab begrenzen.
-
Daher
besteht ein Bedarf, Verfahren für
das Einbringen einer großen
Auswahl von chemischen Gruppen mit verschiedenen Funktionalitäten auf
polymere Kohlenhydratmaterialen zu entwickeln und insbesondere auf
Zellulosefasern, ohne die Unversehrtheit der Faserstruktur zu gefährden. Idealerweise
sollte das Verfahren ein oder mehrere Enzyme umfassen, die ohne
eine hydrolytische oder eine andere abbauende Aktivität sind, da
diese gegen die Versuche wirken würde, chemische Gruppen auf
den Fasern anzuhängen.
-
EP 562 832 offenbart ein
Gen, das eine Endoxyloglucan-Transferase codiert, und schlägt dieses
Gen für
die Verwendung bei der Regulierung der Morphologie einer Pflanze
vor. Die Offenbarung erwähnt
ebenfalls ein Verfahren zum Umlagern von Xyloglucan-Molekülen, das
die Spaltung einer D-Glycosylbindung in einem Xyloglucan-Molekül durch
die Verwendung einer Endoglucan-Transferase und die Bindung des
so erhaltenen reduzierenden Endes des Xyloglucan-Molekül-Segments
an die D-Glucose des nicht reduzierenden Endes eines anderen Xyloglucan-Moleküls umfasst.
Durch die mehrfache Wiederholung können Xyloglucan-Moleküle mit einer
willkürlichen
Struktur aufgebaut werden, von denen behauptet wird, dass sie für die Synthese
von chimären
Polysacchariden anwendbar sind.
-
U.S.
5,968,813 (eine Weiterführung
von PCT/DK96/00538, veröffentlicht
als WO 97/23683) offenbart ein Verfahren zur Verbesserung der Stärkeeigenschaften
von Zellulosematerialien, gemäß der ein
Zellulosematerial mit einer Xyloglucan-Endotransglycosylase (XET) in einem
wässrigen
Medium in Kontakt gebracht wird. Die Behandlung mit XET soll die
Vernetzung zwischen den Zellulosefasern erhöhen, wodurch die Stärke und/oder
Beibehaltung der Form des Zellulosematerials verbessert wird.
-
ZUSAMMENFASSUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft ein Verfahren zur Modifizierung eines
polymeren Kohlenhydratmaterials (PCM), wobei das Verfahren einen
Bindungsschritt umfasst, in dem eine chemische Gruppe mit einer
gewünschten
Funktionalität
an das Kohlenhydratmaterial mit Hilfe eines Kohlenhydrat-Bindemoleküls gebunden wird,
welches die chemische Gruppe trägt
und ein lösliches
Kohlenhydratpolymer (SCP) umfasst, das eine Hemizellulose umfasst,
wobei das Bindemolekül
an das PCM binden kann.
-
In
einer Ausführungsform
umfasst das Verfahren die Schritte, ein Kohlenhydratpolymerfragment
(CPF) zur Verfügung
zu stellen, das eine chemische Gruppe mit einer gewünschten
Funktionalität
umfasst, die typischerweise hydrophob, geladen, reaktiv sein kann.
Das CPF wird mit einem löslichen
Kohlenhydratpolymer (SCP) unter Bedingungen in Kontakt gebracht,
die zu der Erzeugung eines Komplexes zwischen dem CPF und mindestens
einem Teil des SCP führt.
Das PCM wird schließlich
modifiziert, indem man das SCP an das PCM binden lässt.
-
Die
vorliegende Erfindung betrifft des weiteren Materialien und Zusammensetzungen,
die durch das Verfahren hergestellt werden, und Materialien und
Zusammensetzungen, die den Komplex aus dem PCM und einem SCP umfassen,
das eine chemische Gruppe mit einer gewünschten Funktionalität umfasst.
-
AUSFÜHRLICHE BESCHREIBUNG DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft ein Verfahren zur Modifizierung eines
polymeren Kohlenhydratmaterials (PCM), wobei das Verfahren einen
Bindungsschritt umfasst, in dem eine chemische Gruppe mit einer
gewünschten
Funktionalität
an das Kohlenhydratmaterial mit Hilfe eines Kohlenhydrat-Bindemoleküls gebunden wird,
welches die chemische Gruppe trägt
und ein lösliches
Kohlenhydratpolymer (SCP) umfasst, das eine Hemizellulose umfasst,
wobei das Kohlenhydrat-Bindemolekül an das PCM binden kann.
-
Eine
Ausführungsform
dieses Verfahrens wird in 1 dargestellt,
wobei das nicht modifizierte PCM (1) und das Kohlenhydrat-Bindemolekül (CLM)
(2) gezeigt werden, wobei das CLM (2) mindestens
einen Teil eines SCP (3) und eine chemische Gruppe (5)
umfasst und gegebenenfalls mit einem Kohlenhydratpolymerfragment
(CPF) (4), das die chemische Gruppe umfasst, als Komplex
vorliegt. Weil das Kohlenhydrat-Bindemolekül an das PCM binden kann, wird
die Bindung stattfinden, wenn es mit dem PCM in Kontakt gebracht wird.
-
In
einer anderen Ausführungsform
der Erfindung umfasst das Verfahren die Schritte:
- (i)
Bereitstellen eines Kohlenhydratpolymerfragments (CPF), das eine
chemische Gruppe mit einer gewünschten
Funktionalität
umfasst,
- (ii) Inkontaktbringen des CPF, das die chemische Gruppe umfasst,
mit einem löslichen
polymeren Kohlenhydrat (SCP) unter Bedingungen, die zu der Erzeugung
eines Komplexes führen,
der aus dem CPF, das die chemische Gruppe umfasst, und dem SCP besteht,
wobei CPF und SCP zusammen das Kohlenhydrat-Bindemolekül (CLM)
erzeugen, und
- (iii) Inkontaktbringen des CLM mit dem PCM, um unter Bedingungen
modifiziert zu werden, in denen das CLM an das PCM bindet, um das
modifizierte polymere Kohlenhydratmaterial zu erhalten.
-
Der
Ausdruck "polymere
Kohlenhydratmaterialien",
der mit "PCM" abgekürzt wird,
betrifft ein Material, das ein wasserunlösliches, polymeres Kohlenhydratmaterial
und/oder ein wasserlösliches
polymeres Kohlenhydratmaterial umfasst. Das PCM kann jedes Material
sein, das vollständig
oder teilweise aus sich wiederholenden Einheiten von einem oder
von mehreren Monosacchariden aufgebaut ist. Solche PCMs sind häufig Verbundstoffe
mit zwei oder mit mehreren unterschiedlichen Arten von polymeren
Kohlenhydraten oder ein Kohlenhydratpolymer und ein anderes Polymer
wie z.B. ein Protein. Das PCM kann ein Chitin umfassen, das ein Polymer
von N-Acetylglycosamin ist, welches häufig Komplexe mit Proteinen
oder anderen Polysacchariden wie z.B. Mannan erzeugt.
-
Das
PCM kann ebenfalls Zellulose umfassen. Zellulose kann ein Homopolymer
aus β-1,4-gebundenen Glucoseeinheiten
sein. Die langen Homopolymere der Glucose (z.B. 8–15000 Glucoseeinheiten)
sind aufeinander durch Wasserstoffbindungen aufgestapelt, wodurch
sie ein unlösliches
Material bilden. Solche Zellulosematerialien können vollständig kristallin sein oder sie
können
in einer ungeordneten, amorphen Form vorliegen oder sie können ein
Gemisch aus den beiden Formen sein. Sie können ebenfalls durch ein erstes
Löslichmachen
des unlöslichen
Zellulosematerials und der anschließenden Regeneration hergestellt
werden, um ein unlösliches
Zellulosematerial mit einer unterschiedlichen Kettenorganisation
(Zellulose II) zu erzeugen.
-
In
den Zellwänden
von Pflanzen erzeugt die Zellulose Komplexe mit anderen löslichen
Polysacchariden der Zellwand wie z.B. mit der Hemizellulose und
dem Pektin. Beispiele von PCMs, die Zellulose- und/oder Zellulose/Hemizellulose-Verbundstoffe umfassen,
sind Zellulosefasern, Zellulosemikrofibrillen (Pflanzenhaare), Papier-
und Zellstoffprodukte und zellulosehaltige Textilwaren.
-
Wie
aus der Beschreibung und den Beispielen ersichtlich sein wird, betrifft
der Ausdruck PCM alle Strukturen in kleinen Polymeren (z.B. in Dimensionen,
die kleiner als ein nm sind), großen Polymeren (z.B. in Dimensionen
von 0,1-1000 nm), Aggregaten von Polymeren (z.B. in Dimensionen
von 1-10.000 nm), Fasern (z.B. in Dimensionen von 0,1-100.000 μm), Aggregaten
von Fasern (z.B. in Dimensionen von 0,00001-1000 m).
-
Der
Ausdruck "Zellulosefaser" betrifft eine Pflanzenzelle,
die aus einer äußeren primären Zellwand
besteht, die in einer dickeren und komplexeren sekundären Zellwand
eingekapselt ist. Die wesentliche Faserkomponente ist Zellulose,
welche die tragende Komponente der Zellwände der Pflanze ist. In Abhängigkeit
von verschiedenen Aufschlusssequenzen können die Zellstofffasern das
Material der primären
Zellwand enthalten oder nicht. Der Ausdruck "Zellulosemikrofibrillen" betrifft die elementaren
Einheiten des Zellulosekristalls, die durch Pflanzen oder durch
andere Organismen hergestellt werden. Zellulosemikrofibrillen können aus
zellulosehaltigen Pflanzenfasern hergestellt werden oder einfacher
aus Kulturen von Bakterien, die Zellulose synthetisieren, wie. z.B.
Acetobacter xylinum spp.
-
Im
Zusammenhang mit der vorliegenden Erfindung können die Zellulosefasern aus
einer einjährigen Pflanze
wie zum Beispiel Flachs, Hanf oder Getreide oder aus einer mehrjährigen Pflanze
wie z.B. Baumwolle, Pappel, Birke, Weide, Eukalyptus, Lärche, Kiefer
oder Fichte extrahiert werden. Zellulosemikrofibrillen können von Bakterienkulturen
wie z.B. Acetobacter xylinum spp. erhalten werden. Ein Papier- oder Zellstoffprodukt kann
aus jedem zellulosehaltigen Material, das auf dem Fachgebiet bekannt
ist, bestehen. Dieses schließt
Materialien wie z.B. Holz- oder Faserstofffasern, verschiedene chemische
Faserstoffe, mechanische oder thermomechanische Faserstoffe, Flusenfaserstoffe,
Filterpapiere, Feinpapiere, Zeitungspapier, regenerierte Zellulosematerialien,
Deckenkartons, Taschentücher
und andere Hygieneprodukte, Sack- und Packpapiere, andere Verpackungsmaterialien,
Presspan und Hartpappe sowie die Oberflächen von festen Holzprodukten
oder Holz- und Faserverbundstoffe ein, ist aber nicht darauf beschränkt.
-
Weitere
Beispiele von PCM umfassen polymere Kohlenhydratmaterialien, die
in medizinischen Anwendungen verwendet werden, wie z.B. als Membrane,
Gele, Kügelchen,
die in der Diagnostik oder in der Separationstechnologie verwendet
werden, und Membrane, die in elektronischen Anwendungen verwendet
werden. Das Faserprodukt kann im Zusammenhang mit der vorliegenden
Erfindung auch eine neue Art von Verbundstoff mit anderen natürlichen
oder synthetischen Polymeren oder Materialien sowie elektronischen
Verbindungen sein.
-
Im
Zusammenhang mit der vorliegenden Erfindung ist ein Zellulosestoff
jeder zellulosehaltige Stoff, der auf dem Fachgebiet bekannt ist,
wie z.B. Baumwolle, Viskose, Kupfer-, Acetat- und Triacetatfasern,
Modal, Kunstseide, Leinen, Tencel®, etc.,
oder Gemische davon oder Gemische von jedem dieser Fasern oder Gemische
von jedem dieser Fasern zusammen mit synthetischen Fasern oder Wolle
wie z.B. als Gemische von Baumwolle und Elasthan (Strechtjeansstoff),
Tencel® und
Wolle, Viskose und Polyester, Baumwolle und Polyester und Baumwolle
und Wolle.
-
Der
Ausdruck "lösliche Kohlenhydratpolymere", der mit (SCP) abgekürzt wird,
betrifft Polymere, die ein oder mehrere verschiedene Monosaccharide
oder deren Derivate betreffen, die in wässrigen oder organischen Lösungsmittel
gelöst
werden können.
Beispiele schließen
Polysaccharide ein, die als Hemizellulose klassifiziert werden (solche
Kohlenhydratpolymere, die nicht nur aus β(1-4)-gebundenen Glucoseeinheiten
zusammengesetzt sind, das heißt
Zellulose), Pektine (Polyuronsäuren
und -ester) und Stärken
(α(1-4)-gebundene Polyglucose
mit oder ohne α(1-6)-Seitenkettenverzweigung).
Xyloglucan, das ein Polysaccharid ist, das aus einem β(1-4)-gebundenen
Polyglucose-Grundgerüst
zusammengesetzt ist, das mit α(1-6)-Xyloseresten
dekoriert ist, die selber weiter mit anderen Sacchariden wie z.B.
Fucose und Arabinose substituiert sein können, ist ein Beispiel für ein solches
SCP, insbesondere einer Hemizellulose. In einer bevorzugten Ausführungsform
ist das SCP in der Lage, an das PCM zu binden, z.B. über eine
oder über
mehrere Wasserstoffbindungen, eine ionische Interaktion, eine oder
mehrere kovalente Bindungen, van-der-Waals-Kräfte oder über jede Kombination von diesen.
In einer anderen Ausführungsform
der vorliegenden Erfindung kann das SCP ein CPF gemäß der nachfolgenden
Beschreibung sein.
-
Der
Ausdruck "Kohlenhydratpolymerfragmente", der mit "CPF" abgekürzt wird,
betrifft Moleküle,
die enzymatisch oder chemisch hergestellte Fragmente der SCPs sein
können.
Beispiele von solchen Fragmenten umfassen jede Anzahl der sich wiederholenden
Einheiten der SCPs. Geeignete Fragmente können daher von 2 bis zu etwa
5000 Monosaccharideinheiten in dem Polymer-Grundgerüst wie z.B.
etwa 2- 10, 4-10,
3-100, 11-15, 20-25, 26-40, 41-60, 61-100, 101-200, 201-300, 301-400,
401-500, 501-1000, 1001-2000, 2001-3000, 3001-4000 oder 4001-5000 Monosaccharideinheiten
enthalten. Das CPF kann des weiteren Seitenketten von unterschiedlicher
Länge und
Zusammensetzung umfassen. Spezifische Beispiele schließen Xylogluco-Oligosaccharide
(XGO) ein, wie z.B. solche, die eine Struktur besitzen, wie sie
in 4 beschrieben wird, oder ein Fragment davon, oder
solche, die mit einem oder mit mehreren Fucosylresten oder mit anderen
Monosacchariden weiter modifiziert sind, sind aber nicht darauf
beschränkt.
-
XGOs
sind im Allgemeinen gemäß des Nomenklatursystems,
das in Fry et al. (1993), Physiologia Plantarum 89, 1-3 zusammengefasst
wird, benannt, wobei unter anderem G einen nicht substituierten
beta-Glycopyranolsylrest darstellt, X eine Xylopyranosyl-alpha(1-6)-glucopyranosyleinheit
darstellt, L eine Galactopyranosylbeta(1-2)xylopyranosyl-alpha(1-6)glycosyleinheit
darstellt, F eine Fucopyranosylalpha(1-2)-galactopyranosyl-beta(1-2)-xylopyranosyl-alpha(1-6)-glycosyleinheit
darstellt. Diese verschiedenen Einheiten können über eine beta(1-4)-Bindung zwischen
den Glycopyranosyleinheiten verbunden sein, um ein beta(1-4)-Glucan-Polysaccharid-Grundgerüst zu erzeugen.
Unter Verwendung dieser Nomenklatur sind die XGOs, die im Allgemeinen
nach der Spaltung von Xyloglucan der Tamarinde mit Endoglucanase
isoliert werden, XXXG, XLXG, XXLG und XLLG (vergl. 4).
Wenn die Glucose (G) des reduzierenden Endes dieser Oligosaccharide
in der reduzierten Alditolform vorliegt, wird diese Einheit durch "Gol" dargestellt. Die
reduzierten (Alditol)-Derivate der vorstehend beschriebenen Oligosaccharide
von dem Xyloglucan der Tamarinde werden daher zum Beispiel als XXXGol,
XLXGol, XXLGol und XLLGol bezeichnet.
-
Im
Zusammenhang mit der vorliegenden Erfindung betrifft der Ausdruck "chemische Gruppe" jede chemische radikale
Gruppe (R-) mit einem möglichen
Interesse für
die Aktivierung oder Modifikation der unlöslichen polymeren Kohlenhydratoberflächen. Die
Aktivierung der unlöslichen
polymeren Kohlenhydratoberflächen
wird als eine Modifikation definiert, die es ermöglicht, weitere chemische oder
enzymatische Reaktionen durchzuführen,
während
die Modifikation der Oberflächen
als eine Behandlung definiert wird, die als solche ausreichend ist,
um die funktionellen Eigenschaften zu verändern.
-
Beispiele
von chemischen Gruppen, die für
eine solche Aktivierung oder Modifikation geeignet sind, können ionische
Gruppen (kationische, wie z.B. quaternäre Aminogruppen, Ammoniumgruppen,
Carbokationen, Sulfoniumgruppen oder Metallkationen, etc.; anionische,
wie z.B. Alcoxide, Thiolate, Phosphonate, Carbanionen, Carboxylate,
Boronate, Sulfonate, Bunte-Salze, etc., oder zwitterionische, wie
z.B. Aminosäuren, Ylide
oder andere Kombinationen von anionischen und kationischen Gruppen
auf dem gleichen Molekül)
oder deren nicht ionisierten Konjugatsäuren oder -basen (wie es geeignet
ist), hydrophobe Gruppen (Alkyl-Kohlenwasserstoffe, wie z.B. fetthaltige
Acyl- oder Alkylgruppen und ungesättigte Derivate oder Perfluor-Alkane
oder Aryl-Kohlenwasserstoffe, wie z.B. aromatische oder polyzyklische
aromatische Kohlenwasserstoffe oder Heterozyklen), ungeladene hydrophile
Gruppen (z.B. Polyether, wie z.B. Polyethylenglykol), potentielle
reaktive Gruppen wie z.B. solche, die elektrophile Atome enthalten
(wie z.B. Carbonylverbindungen, Carbokationen, Alkylhalide, Acetate,
etc.), Nucelophile (z.B. Stickstoff, Schwefel, Sauerstoff, Carbanione,
etc.) oder Monomere für
Polymerisationsreaktionen (freie Radikale, wie z.B. Acrylamid, Brombutyrat,
Vinyl, Styrol, etc. oder ansonsten z.B. nucleophile oder elektrophile
Reagenzien), chromophore oder fluorophore Gruppen (Pigmente, Farbstoffe
oder optische Aufheller, wie z.B. C.1-Farbstoffe, Fluorescein, Sulfo-Rhodamin,
Pyren), Biotin, radioaktive Isotope, Vorläufer freier Radikale und stabile
freier Radikal-Gruppen (z.B. TEMPO), Nucleinsäuresequenzen, Aminosäuresequenzen,
Proteine oder proteinbindende Mittel (z.B. Affinitätsliganden,
Biotin, Avidin, Streptavidin, Kohlenhydrate, Antikörper oder
Enzymsubstrate oder deren Analoge), Rezeptoren, Hormone, Vitamine
und Arzneistoffe einschließen.
-
Der
Ausdruck "Kohlenhydrat-Bindemoleküle", der mit "CLM" abgekürzt wird,
betrifft ein Molekül
oder Komplex, das/der mindestens einen Teil eines SCP gemäß der vorstehenden
Beschreibung und eine chemische Gruppe enthält. In einer bevorzugten Ausführungsform
ist das CLM in der Lage, an das PCM zu binden, z.B. über eine
oder mehrere Wasserstoffbindungen, eine ionische Interaktion, eine
oder mehrere kovalente Bindungen, van-der-Waals-Kräfte oder über jede
Kombination von diesen.
-
Beispiele
9, 13, 15, 16, 18a und 30 zeigen die Anwendung einer chemischen
Gruppe, Sulfo-Rhodamin, die ein Chromophor, Fluorophor oder ein
Zwitterion ist, zur Modifikation von Zellulosematerialien. Chromophore
Gruppen sind im Allgemeinen als Pigmente bekannt, Fluorophore werden
als optische Aufheller in Textilien und bei anderen Applikationen
verwendet und ionische Verbindungen wirken als Retentionshilfen
bei der Herstellung von Papier. Beispiele 10, 14, 17, 18b und 21
zeigen Modifikationen von zellulosehaltigen Fasern mit Fluorescein,
das ebenfalls chromophor, fluorophor und anionisch über einen
weiten pH-Bereich ist und daher Anwendungen haben wird, bei denen
diese Gruppen erwünscht
sind. Beispiele 8, 19 und 20 stellen Verfahren zum Einbringen einer
Aminogruppe auf der Faseroberfläche
dar, die über
einen weiten pH-Bereich kationisch ist und daher als ein Ionenaustauschmittel
geeignet ist und die ebenfalls die Retention bei der Herstellung
von Papier erhöhen
kann.
-
Des
weiteren ist die Aminogruppe an sich reaktiver als die chemischen
Gruppen, die schon in den zellulosehaltigen Fasern vorhanden sind,
und kann daher für
die Bindung von einer großen
Auswahl von anderen chemischen Stoffen auf die Faseroberfläche verwendet
werden. Der Einbau von Radioaktivität wird in dem Beispiel 11 und
in dem XET-Enzymtest dargestellt, der im Teil "a" unter
dieser Überschrift
beschrieben wird. Die Radioaktivität kann für Suchanwendungen und Studien
der Fasermorphologie verwendet werden. Reaktionen zum Einbau von
Alkylketten werden in den Beispielen 22, 24 und 25 beschrieben.
Insbesondere zeigen die Beispiele 24 und 25, wie Alkenylbernsteinsäureanhydrid,
ein typisches Mittel zur Hydrophobisierung von Papier, spezifisch
an die Faseroberfläche
gebunden werden kann, wobei die Retention dieser Gruppe möglicherweise
erhöht
wird.
-
Die
Beispiele 23 und 26 zeigen, dass die nicht fluorophoren, aromatischen
Gruppen an die Faser gebunden werden können; wobei das zweite Beispiel
eine Cinnamoyl-Gruppe einbringt, die Polymerisierungsreaktionen
eingehen kann, wie z.B. solche, die Polystyrol und Lignin herstellen.
Die Bromisobutyryl-Gruppe, die gebunden wird, wie im Beispiel 27
beschrieben wird, ist ebenfalls ein anderer Initiator für Polymerisierungsreaktionen
durch freie Radikale. Wie in Beispiel 31 beschrieben wird, können solche
Gruppen verwendet werden, um auf Zellulose basierende Pfropf-Kopolymere
herzustellen, die hochqualitative Faser-Grundsubstanz-Grenzflächen mit
einer sehr starken, beidseitigen Adhäsion, aber nur eine geringe
oder keine schädigende
Wirkung auf die Faser/Zellulosestruktur besitzen. Das Einbringen
von Biotin in Beispiel 28 ermöglicht die
direkte Bindung von Avidin-Protein-Konjugaten
auf die Faseroberfläche,
was weitverbreitet ist und verwendet werden kann, um eine Enzym-
und Protein-Bindungsaktivität
auf der Faser einzuführen.
-
Die
Zugabe einer Thiol- (oder Sulfhydryl-) Gruppe ermöglicht eine
hohe Spezifität
und, was besonders wichtig ist, die reversible Bindung von anderen
Thiolen über
die Erzeugung von Disulfidbindungen, wie in den Beispielen 29 und
30 beschrieben wird. Eine große
Anzahl von chemischen Gruppen kann spezifisch durch dieses Verfahren
eingeführt
und anschließend
entfernt werden, wenn diese nicht länger erwünscht sind. Schließlich zeigen
alle Beispiele, die vorstehend erwähnt werden, dass eine große Vielfalt
von Amin-reaktiven chemischen Gruppen verwendet werden können, um
andere Funktionalitäten
einzuführen,
was Sulfonylchloride, Isothiocyanate, Isocyanate, Säureanhydride,
aktivierte Carboxyl-Verbindungen (sogar solche, die in situ hergestellt
werden) und Thioester einschließt,
aber nicht darauf begrenzt ist. Viele dieser chemischen Stoffe können verwendet
werden, um unsere Reaktionen durchzuführen, die zum Beispiel die
Faser-Faser-Bindung oder die Reaktivität der Zellulose mit anderen
Materialien verstärken.
-
Das
CLM kann durch eine organische oder chemische Synthese und/oder
durch die Verwendung der katalytischen Aktivität bestimmter Enzyme hergestellt
werden. Eine Ausführungsform
der Herstellung eines CLM unter Verwendung eines Enzyms und eines
CPF wird in 2 dargestellt. Das SCP (8)
wird mit einem Enzym (7) und einem CPF (4), das
eine chemische Gruppe (5) umfasst, in Kontakt gebracht.
In dieser Ausführungsform
spaltet das Enzym (7) das SCP und führt stattdessen das CPF mit
der chemischen Gruppe ein, was zu dem Produkt CLM (2) führt. Das
CLM kann eine oder mehrere chemische Gruppen umfassen.
-
In
einer Ausführungsform
der vorliegenden Erfindung kann das CLM unter Verwendung eines Enzyms hergestellt
werden, das in der Lage ist, native oder chemisch modifizierte Mono-
oder Oligosaccharide an die Enden von Oligo- oder Polysacchariden
zu transferieren. Solche Enzyme schließen Enzyme, die eine hohe Transglykosylierungsaktivität, aber
eine niedrige hydrolytische Aktivität besitzen, Glycosylhydrolasen
mit einer hohen innewohnenden Transglykosylierungsaktivität, Enzyme,
die biotechnologisch hergestellt wurden, um deren Transglykosylierungsaktivität zu erhöhen, und
Glycosyltransferasen, die Nucleotidzucker als Substrate verwenden,
ein, sind aber nicht darauf beschränkt.
-
In
einer Ausführungsform
der vorliegenden Erfindung kann das Enzym als jedes Enzym definiert
werden, das, wenn es mit einem geeigneten Glycosyl-Spendersubstrat (z.B.
Xyloglucan) in der Gegenwart oder beim Fehlen von einem Mono-, Oligo-
oder Polysaccharid-Akzeptorsubstrat (z.B. XGO) unter geeigneten
Bedingungen zur Erhaltung der Enzymaktivität untersucht wird, eine Rate
des Einbringens des Akzeptorsubstrats in das Spendersubstrat zeigt,
die mindestens 10% der Hydrolyserate beträgt, wie z.B. mindestens 15%,
20%, 25%, 30%, 40%, 50%, 75%, 100%, 200%, 300%, 400%, 500%, 600%,
700%, 800%, 900%, wie z.B. mindestens 1000% der Hydrolyserate. Die
Analyse, die zur Bestimmung der Raten des Einbringens des Akzeptorsubstrats
in das Spendersubstrat verwendet wird, kann die radiometrische Analyse
und/oder die kolorimetrische Analyse zur Bestimmung der Enzymaktivität sein,
die hierin beschrieben werden.
-
Repräsentative
Beispiele von Enzymen mit einer natürlich hohen Transglycosylierungsaktivität schließen Amylosucrasen
(Skov et al., 2001, J Biol Chem 276:25273-8) und die Cyclodextrin-Glycosyltransferase (Ven
der Veen et al., 2000, Biochim Biophys Acta 1543:336-360) ein, sind
aber nicht darauf beschränkt.
Viele Glycosylhydrolasen, die über
einen Retentionsmechanismus wirken, besitzen eine Transglycosylierungsaktivität, die durch
die Verwendung von organischen Lösungsmitteln
verstärkt
werden kann. Beispiele von solchen Hydrolasen schließen einige
Cellulasen und Mannasen (Kwon et al., 2002, Biosci Biotechnol Biochem 66:110-6;
Harjunpaa et al., 1999, FEBS Lett 443:149-53), Xylanasen (Christakopoulos
et al., 1996, Carbohydr Res 289:91-104) und Chitinasen (Sasaki et
al., 2002, J Biochem (Tokyo) 131:557-64) ein. Beispiele von Enzymen,
die gentechnisch zur Erhöhung
der Transglycosylierungsaktivität
hergestellt werden, sind so genannte auf zurückhaltende Glycosylhydrolasen
basierende Glycosynthasen (Meyer et al., 2001, Chem Biol 8:437-43; Fairweather
et al., 2002, Chembiochem 3:866-73). Beispiele für diese Arten von Enzymen werden
zur Synthese von Designer-Oligosacchariden für akademische, industrielle
und potentiell therapeutische Zwecke verwendet.
-
Vorzugsweise
wird ein Enzym ausgewählt,
das eine hohe Transglycosylierungsaktivität besitzt, und am stärksten bevorzugt,
auch für
alle praktischen Zwecke, wird ein Enzym, das eine niedrige oder
nicht detektierbare hydrolytische oder eine andere abbauende Aktivität besitzt.
Vorzugsweise werden keine Nucleotidzucker oder organische Lösungsmittel
benötigt,
um die Transglycosylierungsaktivität zu fördern. Ein Beispiel für solche transglycosylierenden
Enzyme ist die Xyloglucanendotransglycosylase, ein Enzym, das von
Pflanzen bekannt ist.
-
Stephen
C. Frey et al. weisen in Biochem J 15 (1992) 282, S. 821-828 zum
Beispiel darauf hin, dass XET für
das Spalten und die Wiedervereinigung von intermikrofibrillären Xyloglucanketten
verantwortlich ist und dass XET daher das Lockern der Wände hervorruft,
das für
die Ausdehnung der Pflanzenzelle benötigt wird. Man nimmt an, dass
XET in allen Pflanzen vorhanden ist, insbesondere in allen Landpflanzen.
XET wurde aus dikotylen Pflanzen, monokotylen Pflanzen extrahiert,
insbesondere aus monokotylen grasartigen Pflanzen und aus monokotylen
Liliengewächsen
und ebenfalls aus einem Moos und einem Lebermoos. XET kann ebenfalls
von einer Pflanze erhalten werden, wie in Beispiel 1 (Blumenkohl)
und in Beispiel 5 (Suspensionskultur von hybriden Espenzellen) beschrieben
wird, oder es kann erhalten werden, wie in Fry et al. (vorstehend)
beschrieben wird.
-
In
einer anderen Ausführungsform
wird das transglycosylierende Enzym durch die aerobe Kultivierung von
einem Wirtsorganismus hergestellt, der mit der genetischen Information
transformiert wurde, die das transglycosylierende Enzym codiert.
Der Wirtsorganismus kann eine Pflanze sein, insbesondere Tabak,
Mais oder eine hybride Espe, Pilze, insbesondere Hefen wie z.B.
Pichia pastoris oder Saccharomyces cerevisiae, filamentöse Pilze
wie z.B. Trichoderma reesei oder Aspergilli, welche die geeigneten
genetischen Informationen besitzen, die für die Expression des heterologen
Proteins in dem in Frage kommenden Wirt benötigt wird. Solche Transformanten
können
durch Verfahren, die auf dem Fachgebiet bekannt sind, hergestellt
und kultiviert werden.
-
Gene:
Das Gen, welches das transglycosylierende Enzym codiert, kann aus
der Natur von einem Organismus, der ein geeignetes transglycosylierendes
Enzym exprimiert, z.B. eine Pflanze oder ein Mikroorganismus, erhalten
werden. Das Gen kann ebenfalls mittels Gentechnik basierend auf
dem zur Verfügung
stehenden Wissen über
natürlich
vorkommende Enzyme hergestellt werden und es kann durch Deletion,
Substitution oder Addition von Sequenzinformationen wie z.B. codierenden
Regionen und Promotoren modifiziert werden. Das XET-Gen kann zum
Beispiel von Blumenkohl (Beispiel 3), von hybrider Espe (Beispiel
4) oder wie in
EP 562 836 offenbart
wurde, erhalten werden.
-
Wirtszellen:
Die Wirtszellen, die das so erhaltene DNA-Konstrukt umfassen, können unter
Verwendung von Verfahren erhalten werden, die dem Fachmann bekannt
sind.
-
Die
Wirtszelle ist vorzugsweise eine eukaryontische Zelle, insbesondere
eine Pflanzenzelle wie z.B. eine Suspension von Pappel- oder Tabakzellen
oder eine Gewebekultur oder die Blätter oder die Samen dieser Pflanzen
und ähnlicher
Pflanzen. Die Pflanzenzellen können
durch Agrobacterium vermitteltem Gentransfer oder durch die Verwendung
einer Particle-Gun in einer Weise, die an sich bekannt ist, transformiert
werden. Die Wirtszelle kann ebenfalls Hefe oder eine filamentöse Pilzzelle
oder eine Bakterienzelle sein. Insbesondere kann die Zelle einer
Art von Trichoderma angehören,
vorzugsweise Trichoderma harzianum oder Trichoderma reesei, oder
einer Art von Aspergillus, am stärksten
bevorzugt Aspergillus oryzae oder Aspergillus niger. Pilzzellen
können
durch ein Verfahren transformiert werden, das die Erzeugung eines
Protoplasten und die Transformation des Protoplasten gefolgt durch
die Regeneration der Zellwand in einer Weise, die an sich bekannt ist,
beinhaltet.
-
Die
Verwendung von Aspergillus als ein Wirtsmikroorganismus wurde unter
anderem in
EP 238 023 (Novo
Nordisk A/S) beschrieben und die Verwendung von Trichoderma wurde
unter anderem in EP0244234 A2 04-11-1987 [1987/45], EP0244234 A3
12-10-1988 [1988/41], EP0244234 B1 21-07-1993 [1993/29], EP0244234
B2 07-11-2001 [2001/45] beschrieben; wobei deren Inhalt durch Bezugnahme
hiermit aufgenommen wird. Die Wirtszelle kann ebenfalls eine Hefezelle
sein, z.B. ein Stamm von Saccharomvces, insbesondere von Saccharomvces
cerevisiae, oder ein Stamm von Pichia sp. wie z.B. Pichia pastoris
oder Kluyveromyces sp. wie z.B. Kluyvermyces lactis. Die Wirtszelle
kann ebenfalls ein Bakterium sein wie zum Beispiel das Gram-positive
Bakterium Bacillus subtilis oder Gram-negative Bakterien wie z.B.
E. coli. Die Transformation der Bakterien kann zum Beispiel durch
eine Protoplastentransformation oder durch die Verwendung von kompetenten
Zellen in einer Weise, die an sich bekannt ist, durchgeführt werden.
-
Gemäß der Erfindung
kann ein transglycosylierendes Enzym von einer dikotylen Pflanze
oder einer monokotylen Pflanze erhalten werden, insbesondere einer
dikotylen Pflanze, die aus der Gruppe bestehend aus den nachfolgenden
Pflanzenfamilien Blumenkohl, Sojabohne, Tomate, Kartoffel, Raps,
Sonnenblume, Baumwolle, Tabak und Pappel ausgewählt ist, oder einer monokotylen
Pflanze, die aus der Gruppe bestehend aus Weizen, Reis, Korn, Schilfrohr
und Zuckerrohr ausgewählt
ist. Ein Beispiel von solchen Enzymen ist jedes Enzym, das durch
eine der Sequenzen SEQ ID Nr.: 1, 2, 3 oder durch ein funktionelles
Homolog davon codiert wird. Mit einem funktionellen Homolog ist
hierin eine Sequenz gemeint, die eine Homologie mit einem Enzym zeigt,
das durch eine der Sequenzen SEQ ID Nr.: 1, 2, 3 codiert wird, wobei
die Homologie mindestens 50% wie z.B. mindestens 60%, 70%, 80%,
90%, 95%, 99% oder 100% beträgt.
-
Das
funktionelle Homolog kann in einer anderen Ausführungsform ein Enzym sein,
das durch eine Nucleinsäuresequenz
codiert wird, wobei die Nucleinsäuresequenz
eine Homologie mit mindestens einer der Sequenzen in SEQ ID Nr.:
1, 2, 3 von mindestens 50% wie z.B. mindestens 60%, 70%, 80%, 90%,
95%, 99% oder 100% besitzt.
-
In
dem vorliegenden Kontext bedeutet der Ausdruck "Homologie" ein quantitatives Maß des Grades der
Homologie zwischen zwei Aminosäuresequenzen
von gleicher Länge
oder zwischen zwei Nucleotidsequenzen von gleicher Länge. Wenn
die zwei Sequenzen, die verglichen werden sollen, nicht von gleicher
Länge sind,
müssen
sie in der best möglichen
Passform angeordnet werden. Die Sequenzidentität kann als
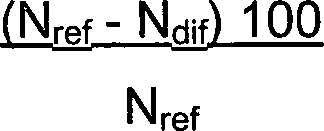
berechnet werden, wobei Ndif
die Gesamtzahl der nicht identischen Reste in den zwei Sequenzen
bedeutet, wenn diese angeordnet sind, und wobei Nref die Anzahl
der Reste in einer der Sequenzen bedeutet. Die DNA-Sequenz AGTCAGTC
wird daher eine Sequenzidentität
von 75% mit der Sequenz AATCAATC besitzen (Ndif = 2 und Nref = 8).
Eine Lücke
wird als Nicht-Identität
des spezifischen Rests/der spezifischen Reste gewertet, das heißt die DNA-Sequenz
AGTGTC wird eine Sequenzidentität
von 75% mit der DNA-Sequenz AGTCAGTC besitzen (Ndif = 2 und Nref
= 8). Die Sequenzidentität
kann in einer anderen Ausführungsform
durch das BLAST-Programm berechnet werden, z.B. das BLASTP-Programm
(Pearson & Lipman
(1988) (www.ncbi.nlm.nih.gov/cgi-bin/BLAST). In einem Aspekt der
Erfindung wird das Anordnen mit dem allgemeinen Anordnungsalgorithmus
mit Standardparameter durchgeführt,
wie von Huang & Miller
(1991) beschrieben wurde, erhältlich
bei http://www.ch.embnet.org/software/LALIGN_form.html.
-
In
einer anderen Ausführungsform
kann das Enzym ein Enzym sein, das eine niedrigere Sequenzhomologie
mit den Sequenzen besitzt, das aber so aufgebaut wurde, dass es
eine transglycosylierende Aktivität besitzt.
-
Die
hier genannten Erfinder haben die Erfindung unter anderem auf die
Tatsachen begründet,
dass Xyloglucan, das in den primären
Zellwänden
von Pflanzenfasern auf natürliche
Weise vorkommt, in der Lage ist, starke Wasserstoffbindungen mit
Zellulose zu erzeugen, und dass die endogene XET-Aktivität von Pflanzen zum Einbringen
von radioaktiven und fluorszierenden XGOs an die Xyloglucan-Komponente
von Suspensionskulturen von Pflanzenzellen führt (vrgl. Biochem J. 279,
1991, S. 529-535 und Plant Cell Physiol 40, 1999, S. 1172-1176).
-
Die
Erfinder haben anschließend
nachgewiesen, dass isolierte Xyloglucanpolymere chemisch und/oder
enzymatisch modifiziert werden können,
um eine große
Anzahl von verschiedenen chemischen Gruppen zu enthalten, und dass
solche chemisch modifizierten Xyloglucanpolymere als eine Grenzfläche für das Einbringen
von neuen chemischen Gruppen auf die zellulosehaltigen Faseroberflächen verwendet
werden können.
Ein deutlicher Vorteil des Verfahrens ist, dass die Verwendung von
solchen Grenzflächen-Polymeren den
nachfolgenden Verlust der Faserstruktur vermeidet, ein Ergebnis,
auf das ansonsten im Allgemeinen mit der direkten chemischen Modifikation
von Zellulose gestoßen
wird. Die Erfinder haben nachgewiesen, dass ein transglycosylierendes
Enzym wie z.B. XET als Empfängerzucker
Oligosaccharide, die verschiedene Arten von chemischen Gruppen enthalten,
verwenden kann.
-
Da
Wasser für
solche Enzyme nicht in Konkurrenz als ein Empfänger der Transglycosylierung
steht, können
sie in wässrigen
Lösungen
verwendet werden, um effizient die chemischen Gruppen auf die Grenzflächenpolymere
wie z.B. Xyloglucan einzuführen,
die entweder in Lösung
oder gebunden an ein anderes polymeres Material wie z.B. Zellulose
vorliegen. Des weiteren haben die Erfinder nachgewiesen, dass Xyloglucan, sogar
wenn es chemisch modifiziert ist, fest an die Oberfläche von
Zellulose bindet und dass die eingeführten chemischen Gruppen, auch
wenn sie an den porösen
Oberflächen
von zellulosehaltigem Material über
XG gebunden sind, trotzdem weiter für chemische Reaktionen zugänglich sind.
Die Erfinder haben anschließend nachgewiesen,
dass es überraschenderweise
möglich
war, durch die Zugabe eines transglycosylierenden Enzyms und von
chemisch modifizierten CPFs zu den SCPs viele verschiedene neue
chemische Gruppen mit einer erwünschten
Funktionalität
auf die Oberflächen
des PCM mit einer hohen Ausbeute zu binden.
-
Das
PCM, das modifiziert werden soll, kann von einer Pflanze abgeleitet
werden, die aus der Gruppe ausgewählt ist bestehend aus einer
monokotylen Pflanze wie z.B. eine Pflanze der Familie Gramineae
und einer dikotylen Pflanze wie z.B. einer Pflanze, die aus der
Gruppe bestehend aus angiospermen Pflanzen (Hartholz), zapfentragenden
Pflanzen (Weichholz) und Pflanzen, die der Familie Gossypium angehören, ausgewählt ist.
-
Das
PCM kann in der Form von zellulosehaltigen Pflanzenfasern oder in
der Form von zellulosehaltigen Mikrofibrillen, die von zellulosehaltigen
Pflanzenfasern oder von einem Bakterium stammen, vorliegen.
-
Das
SCP kann einen Teil des PCM, das modifiziert werden soll, erzeugen,
weswegen der Schritt des Einbringens des CPF, das eine chemische
Gruppe mit einer gewünschten
Funktionalität
umfasst, in das SCP, z.B. unter Verwendung eines Enzyms, direkt
auf dem PCM-SCP-Komplex erfolgen kann. Das Prinzip wird in 3 dargestellt.
Im oberen Teil von 3 ist der PCM-SCP-Komplex (9)
zu sehen, der das PCM (1) und das SCP (2), das
Enzym (7) und ein CPF (4) umfasst, das eine chemische
Gruppe (5) mit einer gewünschten Funktionalität umfasst.
Im mittleren Teil von 3 bindet das Enzym (7)
an das SCP (2) des PCM-SCP-Komplexes (9) und kann
einen Zwischenkomplex (10) erzeugen. In dem Prozess, der
zu dem unteren Teil von 3 führt, spaltet das Enzym (7)
das SCP (2) und bringt (12) das CPF (4),
das eine chemische Gruppe mit einer gewünschten Funktionalität (5)
umfasst, ein. (12) ist das SCP-Fragment, das durch das
SCP abgespalten wurde.
-
In
einer anderen Ausführungsform
muss das SCP nicht an dem PCM gebunden sein, um modifiziert zu werden.
In dem Fall kann das SCP modifiziert sein, um die chemische Gruppe
zu umfassen, und das Produkt der SCP-Modifikation, CLM, wird anschließend mit
dem PCM in Kontakt gebracht. In einer anderen Ausführungsform
wird das SCP zuerst mit dem SCP ohne PCM in Kontakt gebracht. Wenn
der SCP-PCM-Komplex
erzeugt wurde, kann der Schritt des Einbringens des CPF, das eine
chemische Gruppe mit einer gewünschten
Funktionalität
umfasst, in das SCP, z.B. unter Verwendung eines Enzyms, direkt
auf dem PCM-SCP-Komplex erfolgen.
-
Es
ist möglich,
das PCM mit einem Gemisch von chemischen Gruppen zu modifizieren,
z.B. durch das Durchführen
der hierin beschriebenen Verfahren in der Sequenz und/oder durch
die Verwendung eines Gemisches von CLMs, die verschiedene chemische
Gruppen umfassen, und durch die Bindung des Gemisches von CLMs,
die verschiedene chemische Gruppen umfassen, an das PCM in einem
Verfahrensschritt.
-
Sowohl
das SCP als auch das CPF können
chemische Gruppen enthalten.
-
Das
CPF kann von Xyloglucan abgeleitet sein und es kann von 3 bis etwa
100, einschließlich
von 4 bis 10 Monosaccharideinheiten des Polymergrundgerüsts enthalten.
-
In
einer Ausführungsform
wird das CPF, das die chemische Gruppe umfasst, mit dem löslichen
polymeren Kohlenhydrat (SCP) in der Gegenwart eines Enzyms, das in
der Lage ist, die Erzeugung des Komplexes, der aus dem CPF besteht,
das die chemische Gruppe umfasst, in Kontakt gebracht und ist mindestens ein
Teil des SCP. Das Enzym kann in der Lage sein, native oder chemisch
modifizierte Mono- oder
Oligosaccharide auf Oligo- und/oder Polysaccharide zu transferieren.
In einer Ausführungsform
kann das Enzym eine transglycosylierende Aktivität besitzen.
-
In
einer anderen Ausführungsform
zeigt das Enzym eine Rate des Einbringens des Empfängersubstrats
in das Spendersubstrat, die mindestens 10% der Hydrolyserate beträgt, wie
z.B. mindestens 15%, 20%, 25%, 30%, 40%, 50% oder 75%, wie z.B.
mindestens 100% beträgt,
wenn die Untersuchung mit einem geeigneten Glycosyl-Spendersubstrat
in Gegenwart und beim Fehlen eines Mono-, Oligo- oder Polysaccharid-Empfängersubstrats
unter geeigneten Bedingungen zur Erhaltung der Enzymaktivität durchgeführt wird.
-
Die
Analyse zur Evaluierung der Rate des Einbringens des Empfängersubstrats
in das Spendersubstrat kann eine Analyse sein, die aus den nachfolgenden
Schritten besteht:
- i) Inkubieren von 0,1 mg
Xyloglucan, 0,1 mg Xyloglucan-Oligosaccharide (Gemisch von XXXG,
XLXG, XXLG und XLLG; 15:7:32:46 Gewichtsverhältnis) in 200 μl 40 mM Citratpuffer,
pH-Wert 5,5 für
30 Minuten bei 30°C.
- ii) Beenden der Reaktion mit 100 μl 1 M HCl.
- iii) Einstellen der Ionenstärke
durch Zugabe von 800 μl
20% Na2SO4 und 200 μl einer I2 (0,5% I2, 1% KI, w/w)-Lösung.
- iv) Messen der optischen Dichte bei 620 nm.
- v) Durchführen
der Schritte i)-iv) ohne Zugabe der Xyloglucan-Oligosaccharide (XGO)
aus Schritt i)
- vi) Berechnen der Zunahme der optischen Dichte in Prozent zwischen
der Inkubation mit XGO und der Inkubation ohne XGO.
-
Das
Enzym kann von der Gruppe bestehend aus einer Transglycosylase,
einer Glycosylhydrolase, einer Glycosyltransferase ausgewählt sein.
Das Enzym kann ein Wildtyp-Enzym oder ein funktional und/oder strukturell
modifiziertes Enzym sein, das von einem solchen Wildtyp-Enzym abgeleitet
ist. In einer Ausführungsform
ist das Enzym eine Xyloglucan-Endotransglycosylase (XET, EC 2.4.1.207).
-
Das
Enzym, das eine Transglycosylierungsaktivität besitzt, kann von einer Pflanze
abgeleitet sein, einschließlich
einer Pflanze, die zu der Familie Brassica gehört, oder einer Pflanze einer
Populus-Art, oder es kann mittels einer rekombinanten Technik hergestellt
werden.
-
In
einer Ausführungsform
kann die chemische Gruppe, die eine gewünschte Funktionalität besitzt, ausgewählt sein
aus einer Gruppe bestehend aus einer ionischen Gruppe, einer hydrophoben
Gruppe, einer ungeladenen hydrophilen Gruppe, einer reaktiven Gruppe,
einem Nucleophil, einem polymerisierbaren Monomer, einer chromophoren
Gruppe, einer fluorophoren Gruppe, Biotin, einem radioaktiven Isotop,
einem Vorläufer
freier Radikale, einer stabilen freien Radikal-Gruppe, einem Protein und einem proteinbindenden
Mittel.
-
Eine
Ausführungsform
der vorliegenden Erfindung betrifft ein Verfahren, in dem das erhaltene
modifizierte PCM relativ zu dem nicht modifizierten PCM veränderte Oberflächeneigenschaften
wie z.B. veränderte Stärkeeigenschaften,
eine veränderte
Oberflächenspannung,
veränderte
wasserabstoßende
Eigenschaften, eine veränderte
Reaktivität,
veränderte
optische Eigenschaften oder Kombinationen von diesen besitzt.
-
Ein
anderer Aspekt der Erfindung ist ein modifiziertes polymeres Kohlenhydratmaterial
(mPCM), das durch das Verfahren von jedem hierin beschriebenen Verfahren
erhältlich
ist, wobei chemische Gruppen mit einer gewünschten Funktionalität an das
Material gebunden sind, indem die Bindung durch ein Kohlenhydrat-Bindemolekül vermittelt
wird, das eine chemische Gruppe und ein SCP, das eine Hemizellulose
ist, umfasst, wobei das Kohlenhydrat-Bindemolekül an das PCM binden kann. Das
mPCM kann in der Form von zellulosehaltigen Pflanzenfasern oder
von zellulosehaltigen Mikrofibrillen, die von einer zellulosehaltigen
Pflanzenfaser oder von einem Bakterium abstammen, vorliegen.
-
Die
chemischen Gruppen des mPCM können
reaktive Gruppen sein, die andere funktionale Gruppen binden können, und
das mPCM kann daran zwei oder mehrere verschiedene Arten von chemischen
Gruppen gebunden haben.
-
Ein
Aspekt der Erfindung ist ein Verbundmaterial, das die Materialien
umfasst, die hierin beschrieben werden.
-
Ein
anderer Aspekt der Erfindung betrifft die Verwendung des mPCM oder
der Verbundmaterialien bei der Herstellung von Papierblättern, Wellpappe,
Webware, Hilfsmitteln in einem diagnostischen oder chemischen Assay
oder Verfahren, Verpackungsmittel für Flüssigkeiten und Lebensmittel,
Papier und Karton, die häufig
mit einem Thermoplastik wie z.B. Polyethylen laminiert sind, um
eine undurchdringbare Grenzschicht für die wässrigen Lösungen zur Verfügung zu
stellen, Textilien, Wertpapieren, Banknoten, verfolgbaren Dokumenten,
Füllstoffen,
Laminaten und Panelprodukten, holzpolymeren Verbundstoffen, polymeren
Verbundstoffen, Legierungen und Mischungen oder Zellulosederivaten
(zellulosehaltigen Stoffen).
-
Gemäß der vorliegenden
Erfindung können
neue chemische Gruppen dem PCM, das ein innewohnendes geeignetes
SCP enthält,
durch die Verwendung des transglycosylierenden Enzyms zugefügt werden,
um die chemisch modifizierten CPFs an das SCP, das in den zellulosehaltigen
Materialien enthalten ist, zu binden. In dem vorliegenden Zusammenhang
bedeutet der Ausdruck "innewohnend", dass das PCM ein
SCP vor der Modifikation umfasst. Gemäß der vorliegenden Erfindung
können
neue chemische Gruppen ebenfalls dem PCM, das kein innewohnendes
SCP enthält,
zugegeben werden, indem zuerst ein transglycosylierendes Enzym verwendet
wird, um die chemisch modifizierten CPFs an das SCP in Lösung zu
binden, gefolgt durch die Sorption des modifizierten SCP an das
PCM.
-
Gemäß einer
spezifischen Ausführungsform
der vorliegenden Erfindung können
neue chemische Gruppen an Zellulosematerialien, die kein innewohnendes
Xyloglucan enthalten, gebunden werden, indem zuerst das XET-Enzym
verwendet wird, um die chemisch modifizierten XGOs an Xyloglycan
(XG) in Lösung
zu binden, gefolgt durch die Sorption des modifizierten XG an die
Zellulosematerialien.
-
Gemäß der vorliegenden
Erfindung erhält
ein PCM eine veränderte
Oberflächenchemie
und/oder eine verbesserte chemische Reaktivität nach der Behandlung mit chemisch
modifizierten CPFs, die an ein SCP unter Verwendung des transglycosylierenden
Enzyms gebunden werden. Die SCPs, welche die chemisch reaktiven
Gruppen tragen, werden fest an die Oberflächen des PCM binden, wodurch
die chemische Reaktivität der
Oberflächen
erhalten wird. Die chemische Reaktivität an sich oder wenn sie durch
weitere chemische und/oder polymerisierende Reaktionen modifiziert
wurde, beeinflusst die Oberflächeneigenschaften
des PCM. Des weiteren wird die Dichte der chemisch reaktiven Gruppen
durch die Veränderung
der Konzentrationen des transglycosylierenden Enzyms und/oder der
CPFs und/oder der Reaktionszeit kontrolliert, wie in den Beispielen
14a und 14b gezeigt wird. Die Oberflächeneigenschaften können durch
jedes Verfahren, das auf dem Fachgebiet bekannt ist, gemessen werden,
wie in den beigefügten
Beispielen, z.B. Beispiel 16b, gezeigt wird.
-
In
der Natur arbeiten transglycosylierende Enzyme wie z.B. das XET-Enzym
in vivo in der lebenden Pflanze, so dass das Enzym offensichtlich
in einer wässrigen
Umgebung arbeiten kann. Das Verfahren gemäß der Erfindung kann daher
in einer wässrigen
Lösung
durchgeführt
werden oder es kann in Wasser in der Gegenwart von bestimmten Komponenten
wie z.B. einem Puffer und/oder einem Netzmittel und/oder einem Stabilisator
und/oder einem Polymer und/oder einer organischen Komponente, welche
die Aktivität
des Wassers vermindert, wie z.B. DMSO, durchgeführt werden.
-
Der
Puffer kann geeigneterweise ein Phosphat, Borat, Citrat, Acetat,
Adipat, Triethanolamin, Monoethanolamin, Diethanolamin, Carbonat
(insbesondere Alkalimetall, Erdalkalimetall, insbesondere Natrium-
oder Kaliumcarbonat oder Ammonium- und HCL-Salze), Diamin, insbesondere
Diaminoethan, Imidazol, Tris oder Aminosäurepuffer sein.
-
Das
Netzmittel dient der Verbesserung der Benetzbarkeit des PCM. Das
Netzmittel ist vorzugsweise von einer nichtionischen Art der Grenzflächenaktivierung.
Der Stabilisator kann ein Mittel zur Stabilisierung des Enzyms sein.
-
Es
wird im Allgemeinen geeignet sein, das Reaktionsmedium, das z.B.
das PCM, das CLM und gegebenenfalls eine oder mehrere Komponenten
umfasst, die von der Gruppe bestehend aus einem SCP, das eine chemische
Gruppe umfassen kann oder nicht, einem CPF, das chemische Gruppen
und ein Enzym umfasst, ausgewählt
sind, für
einen Zeitraum von mindestens einigen Minuten in Abhängigkeit
von den Reaktionsbedingungen zu inkubieren. Eine Inkubationszeit
von etwa einer Minute bis zu 20 Stunden wie z.B. etwa 2-5 Minuten,
5-7 Minuten, 7-10 Minuten, 10-15 Minuten, 15-20 Minuten, 20-30 Minuten,
30-40 Minuten, 40-60 Minuten, 1-2 Stunden, 2-4 Stunden, 4-6 Stunden,
6-8 Stunden, 8-10 Stunden, 10-12 Stunden, 12-14 Stunden, 14-16 Stunden,
16-18 Stunden oder 18-20 Stunden wird im Allgemeinen geeignet sein.
Insbesondere eine Inkubationszeit von 30 Minuten bis zu 10 Stunden
wird häufig
bevorzugt werden. Die Inkubationszeit wird vorzugsweise mit einem
Zeitabstand begrenzter als +/– 5
Stunden kontrolliert, wie z.B. begrenzter als +/- 2 Stunden, +/– 1 Stunde,
+/– 45
Minuten, +/– 30
Minuten, +/– 15
Minuten, +/– 10
Minuten, +/– 5
Minuten, +/– 2
Minuten, +/– 1
Minute, +/– 30
Sekunden, +/– 10
Sekunden, +/– 1
Sekunden, +/– 0,1
Sekunden, +/– 0,01
Sekunden.
-
Die
Temperatur des Reaktionsmediums in dem Verfahren der Erfindung kann
geeigneterweise in dem Bereich von –5-100°C liegen, wie z.B. 0-5, 5-10,
10-15, 15-20, 20-25, 25-30, 30-33, 33-36, 36-38, 38-40, 40-50, 50-60,
60-70, 70-80, 80-90 oder 90-100°C.
In Ausführungsformen,
in denen das Reaktionsgemisch ein Enzym umfasst, sollte die Temperatur
des Reaktionsgemisches während
der Inkubation vorzugsweise in der Nähe der Temperatur liegen, die
einen optimalen Umsatz während
der Inkubation erzeugt. Die bevorzugte Temperatur sollte weniger
als 10°C
von der Temperatur entfernt sein, die einen optimalen Umsatz während der
Inkubation erzeugt, wie z.B. weniger als 10°C, 8°C, 6°C, 4°C, 2°C, 1°C, 0,5°C oder 0,1°C.
-
Für die Bindung
des modifizierten oder nicht modifizierten SCP an das PCM wird es
im Allgemeinen geeignet sein, das Gemisch für einen Zeitraum von mindestens
einigen Minuten in Abhängigkeit
von den Reaktionsbedingungen zu inkubieren. Eine Inkubationszeit
von etwa einer Minute bis zu 48 Stunden wird im Allgemeinen geeignet
sein, insbesondere eine Inkubationszeit von 30 Minuten bis zu 10
Stunden wird häufig
bevorzugt. Die Inkubationslösung
kann geeigneter Weise in einem pH-Bereich von 2-11 gepuffert sein, vorzugsweise
bei einem pH-Wert von 5-8, mit einer Pufferkonzentration zwischen
0 und 5 M, vorzugsweise 0,0-0,1 M. Die Temperatur des Reaktionsmediums
in diesem Verfahren kann geeigneterweise in dem Bereich von 10-100°C in Abhängigkeit
der Stabilität
der individuellen Komponenten in dem Gemisch sein.
-
Die
Erfindung wird nun ausführlicher
mit Bezug auf die begleitenden Zeichnungen beschrieben, wobei:
-
1 das
Prinzip der Modifizierung eines polymeren Kohlenhydratmaterials
darstellt,
-
2 das
Prinzip der Einfügung
eines löslichen
Kohlenhydratpolymers, das eine chemische Gruppe in einem polymeren
Kohlenhydratmaterial umfasst, darstellt,
-
3 das
Prinzip der Modifizierung von PCM, das ein SCP vor der Modifikation
umfasst, darstellt,
-
4 Beispiele
von Xyloglucan-Oligosaccharidstrukturen (XGO-7 (XXXG), XGO-8 (XLXG,
XXLG) und XGO-9 (XLLG)) zeigt,
-
5 die
Zeitabhängigkeit
der Herstellung von XG-FITC zeigt,
-
6 die
Abhängigkeit
der Herstellung von XG-FITC von der Menge des Enzyms in der Reaktion
darstellt,
-
7 ein
Bild von XG-FITC-behandeltem Papier mittels konfokaler Fluoreszenzmikroskopie
zeigt,
-
8 ein
Photo der Aufnahme von Fluorescein in das mit XG-NH2 behandelte
Papier zeigt,
-
9 die
relativen Mengen der Aminogruppen darstellt, die auf der Oberfläche von
XG-NH2 behandeltem, zellulosehaltigem Papier
nach der Behandlung mit verschiedenen Amino-reaktiven Reagenzien
vorhanden sind,
-
10 die
Reaktivität
von Papier mit und ohne Modifikationen durch XG-NH2 auf
FITC darstellt und
-
11 die
Reaktion von Thiol behandeltem Papier mit Sulfo-Rhodamin-Methanthiosulfonat
darstellt.
-
BEISPIELE
-
MATERIAL UND VERFAHREN
-
Bestimmung
der Enzymaktivität
und insbesondere der XET-Aktivität
-
a. Radiometrische Analyse
-
Die
hier genannten Erfinder haben ein modifiziertes Analyseverfahren
entwickelt, das dem von Steele, N et al. (Phytochemistry, 2000,
54, 667-680) ähnlich
ist und das verwendet wurde, wie nachfolgend beschrieben wird. [1-3H]-XLLGol (300 μl, 0,36 μmol in H2O)
wurde zu dem nicht radioaktiven xgo-9-Alditol (700 μl, 8,6 μmol) in 50
mM Citratphosphatpuffer, pH-Wert 5,5, zugegeben. Wenn diese Stammlösung in
der Analyse verwendet wurde, wurde sie in Puffer auf eine Konzentration
von 2,24 μmol/ml
(3,1 mg/ml) verdünnt.
Die radioaktive XLLGol-Stammlösung
(10 μl,
2,24 μmol/ml)
wurde zu dem Xyloglucan (10 μl,
3,0 mg/ml in Puffer) zugegeben. Die verdünnte Enzymlösung (10 μl) wurde zugegeben und das Reaktionsgemisch
wurde bei 25°C
für 30 min
inkubiert. Die Reaktion wurde anschließend mit einer 50% Lösung Ameisensäure in Wasser
(20 μl)
beendet. Das Reaktionsgemisch (40 μl) wurde auf rundem Whatman-3MM-Chromatographiepapier
(20 mm Durchmesser) getrocknet. Die runden Papiere wurden für 4 Stunden
unter fließendem
Wasser gewaschen, in einem 65°C
warmen Ofen getrocknet und der radioaktive Einschluss wurde in Szintillationsgefäßen, die
den Ready-safe-Szintillationscocktail (6 ml, Beckman Coulter AB,
Bromma, Schweden) enthielten, mit einem Packard Tricarb 1500 Szintillationsmessgerät untersucht.
Es erfolgte keine Elution der Radioaktivität aus dem Papier in die Szintillationsflüssigkeit.
Leerwerte wurden durch die Zugabe von Säure zu der Reaktion vor der
Zugabe des Enzyms gemessen, die Kontrolle der insgesamt zugegebenen
Radioaktivität
wurde dadurch gemessen, dass die runden Papiere der Kontrolle nicht
gewaschen wurden. Ein Maß dafür, wie das
Filterpapier die Analyse beeinflusst, wurde durch den Vergleich
der Kontrollen mit der Szintillationsmessung von dem Kontrollgemisch
ohne dem Filterpapier erhalten.
-
b. Kolorimetrische Analyse
-
Die
Enzymaktivität
wurde gemäß einem
modifiziertem Protokoll, das auf das Protokoll von Slulová et al.
(1995), Anal Biochem 229, 80-85 basierte, gemessen. XET wurde mit
0,1 mg Xyloglucan, 0,1 mg Xyloglucan-Oligosaccharide (Gemisch von
XXXG, XLXG, XXLGund XLLG; 15:7:32:46) in 200 μl 40 mM Citratpuffer, pH-Wert 5,5 für 30 Minuten
bei 30°C
inkubiert. Die Analyse wurde mit der Zugabe von 100 μl 1 M HCl
beendet, die Ionenstärke
wurde durch Zugabe von 800 μl
20% Na2SO4 und 200 μl einer I2 (0,5% I2, 1% KI,
w/w)-Lösung eingestellt.
Die optische Dichte wurde bei 620 nm gemessen. Für die Zwecke dieses Dokuments
wird eine Einheit der Enzymaktivität als 0,1 Einheiten der Veränderung
der optischen Dichte (nach der Korrektur der Hintergrundshydrolyse) über einen
Zeitraum von 30 Minuten definiert.
-
BEISPIEL 1
-
EXTRAKTION VON XET AUS
BLUMENKOHL
-
Die
Extraktion aus Blumenkohl wurde durch die Homogenisierung der Blumenkohlröschen im
eiskalten Citratpuffer (0,35 M, pH-Wert 5,5, 10 mM CaCl2 enthalten)
und durch das Filtern des Gemisches durch Miracloth durchgeführt. Das
Filtrat wurde mit ultrareinem Wasser (18 MΩ.cm) verdünnt, bis die Leitfähigkeit
der Lösung
die gleiche war, wie die von 0,1 M Ammoniumacetatpuffer, pH-Wert
5,5. Die Lösung
wurde anschließend
mit dem SP-Fast-Flow-Kationenaustauscher (Amersham Biosciences,
Schweden) vorsichtig für
1 Stunde bei 4°C
gerührt.
Das SP-FF-Gel wurde auf einem Glasfrittenfilter gesammelt und mit
0,1 M Ammoniumacetat, pH-Wert 5,5, gewaschen, bis das Filtrat klar
war. Das Gel wurde in eine Säule
gegeben und die gebundenen Proteine wurden mit einem linearen Gradienten
von 0 bis 1,0 M NaCl in 0,1 M Ammoniumacetat, pH-Wert 5,5 über 10 Säulenvolumen
eluiert. Fraktionen, welche die XET-Aktivität enthielten, wurden vereinigt
und mit Ammoniumsulfat (1 M) gemischt. Die Probe wurde auf eine
Resource-ISO-Säule
(1 ml, Amersham Biosciences, Schweden) gegeben und anschließend mit
einem linearen Gradienten von 1,0 M bis 0 Ammoniumsulfat in Ammoniumacetat,
pH-Wert 5,5, über
20 Säulenvolumen
eluiert. Die Fraktionen, welche die XET-Aktivität enthielten, wurden vereinigt
und mittels SDS-PAGE und Silberfärbung
auf ihre Reinheit untersucht. Das Gel zeigte nur eine einzelne Bande,
von der durch Immunoblotting nachgewiesen wurde, dass es sich um
XET handelt.
-
BEISPIEL 2
-
EXTRAKTION VON XET AUS
EINER ZELLSUSPENSIONSKULTUR EINER HYBRIDEN ESPE, Populus tremula
x tremuloides Mich.
-
Espen-XET
wurde durch die Homogenisierung des Materials einer granulösen Zellkultur
in eiskaltem Citratpuffer (0,35 M, pH-Wert 5,5, 10 mM CaCl2 enthalten), durch das Rühren des Gemisches für 2 Stunden bei
4°C und
durch das Filtern durch Miracloth extrahiert. Das Filtrat wurde
mit ultrareinem Wasser (18 MΩ.cm) verdünnt, bis
die Leitfähigkeit
der Lösung
die gleiche war, wie die von 0,1 M Ammoniumacetatpuffer, pH-Wert 5,5.
Die Lösung
wurde anschließend
mit dem SP-Fast-Flow-Kationenaustauscher
(Amersham Biosciences, Schweden) vorsichtig für 1 Stunde bei 4°C gerührt. Das
SP-Trisacryl-Gel wurde gesammelt und mit 0,1 M Ammoniumacetat, pH-Wert
5,5, durch einem Glasfrittenfilter gewaschen, bis das Filtrat klar
war. Das Gel wurde in eine Säule
gegeben und die gebundenen Proteine wurden mit einem linearen Gradienten
von 0,0 bis 1,0 M NaCl in 0,1 M Ammoniumacetat, pH-Wert 5,5 über 10 Säulenvolumen
eluiert. Fraktionen, welche die XET-Aktivität enthielten, wurden vereinigt,
der Puffer wurde durch 0,1 M Ammoniumacetat pH-Wert 5,5 in einer
Sephadex G-25 Größenausschlusssäule ausgetauscht
und auf eine Resource-S-Austauschsäule (1 ml, Pharmacia) geladen.
Die gebundenen Proteine wurden mit einem linearen Gradienten von
0,0 bis 1,0 M NaCl in 0,1 M Ammoniumacetat, pH-Wert 5,5, über 10 Säulenvolumen
eluiert. Die Fraktionen, welche die XET-Aktivität enthielten, wurden vereint,
auf eine Sephacryl S200-Säule
(120 ml, Amersham Biosciences, Schweden) gegeben und mit 2 Säulenvolumen
0,1 M Ammoniumacetat, pH-Wert 5,5, eluiert. Fraktionen, die der
letzten Spitze entsprachen, welche die größte Menge der XET-Aktivität enthielten,
wurden vereint und auf eine Resource-S-Säule (1 ml, Amersham Biosciences,
Schweden) gegeben. Die Fraktionen wurden anschließend mit
einem linearen Gradienten von 0,0 bis 0,5 M NaCl in 0,1 M Ammoniumacetat,
pH-Wert 5,5, in einem Volumen, das mehr als 10 Säulenvolumen entsprach, eluiert.
Fraktionen, welche die XET-Aktivität enthielten, wurden vereinigt
und es wurde mittels SDS-PAGE gezeigt, dass sie homogen waren.
-
BEISPIEL 3
-
REINIGUNG DES REKOMBINANTEN
XET AUS DER KULTIVIERUNG VON Pichia pastoris
-
Zellen
von Pichia pastoris-Kulturen, die mit dem genetischen Material transformiert
wurden, das XET (vergl. Beispiele 4 und 5) codierte, zeigten im
Allgemeinen die höchste
XET-Aktivität
im Kulturmedium 3 Tage nach einer Induktion mit Methanol. Diese
Hefezellen wurden durch Zentrifugation geerntet und die Kulturmedien
wurde des weiteren durch einen 0,45 μm-Filter filtriert und anschließend konzentriert
und durch Ultrafiltration entsalzt. Das XET wurde durch eine zweiphasige
Kationenaustausch-Chromatographie gereinigt. Das konzentrierte Kulturfiltrat
(in einem Puffer von 0,1 M Ammoniumacetat, pH-Wert 5,5) wurde zuerst
auf eine SP-Trisacryl-Säule
aufgetragen und anschließend
durch einen linearen Gradienten von 0 bis 1 M NaCl in 0,1 M Ammoniumacetat,
pH-Wert 5,5, eluiert. Die Fraktionen, welche die XET-Aktivität enthielten,
wurden vereint, in 0,1 M Ammoniumacetat, pH-Wert 5,5, entsalzt und
auf eine Resource-S-Säule
aufgetragen, anschließend durch
denselben linearen Salzgradienten, der in der ersten Phase der Kationenaustausch-Chromatographie verwendet
wurde, eluiert. Die Homogenität
des Proteins wurde durch SDS-PAGE und Silberfärbung untersucht. Es wurde
nur eine einzelne Bande mit einem Molekulargewicht von etwa 32 kDa
detektiert und von dieser wurde durch Immunoblotting bestätigt, das
es sich um XET handelt. Es wurde gezeigt, dass das Protokoll für die Expression
von allen Sequenzen SEQ ID Nr.: 1, 2, 3, welche die verschiedenen
Isoenzyme von XET codieren, erfolgreich war.
-
BEISPIEL 4
-
ISOLIERUNG DES GENS, WELCHES
DAS XET VON BLUMENKOHL CODIERT
-
Die
cDNA, die dem XET-Gen entsprach, wurde durch die Extraktion der
RNA durch das Zerreiben von frischem Blumenkohlgewebe unter flüssigem Stickstoff
(N
2) und der Lyse der Zellen unter denaturierenden
Bedingungen isoliert. Die lysierte Zellprobe wurde anschließend durch
eine QIA-Shredder-Säule
zentrifugiert, um das unlösliche
Material zu entfernen. Die RNA wurde anschließend selektiv an eine RNAeasy-Membran
gebunden, mit Puffer gewaschen und schließlich in Wasser eluiert. Die
XET cDNA wurde unter Verwendung einer zweiphasigen Polymerasen-Kettenreaktion (PCR)
gemäß dem Protokoll,
das auf dem Fachgebiet bekannt ist, hergestellt. Der erste Strang
der cDNA wurde bei 55°C
für eine
Stunde synthetisiert, wobei 1 μg
RNA und ein Oligo-dT
(18)-Primer mit der
reversen Transkriptase verwendet wurden. Die Matrizen für die degenerierten
Primer für
die spezifische PCR-Reaktion wurden durch die N-terminale Sequenzierung
des Blumenkohl-XET-Proteins
erhalten, welche eine nachfolgende Sequenz anzeigte:
-
Die
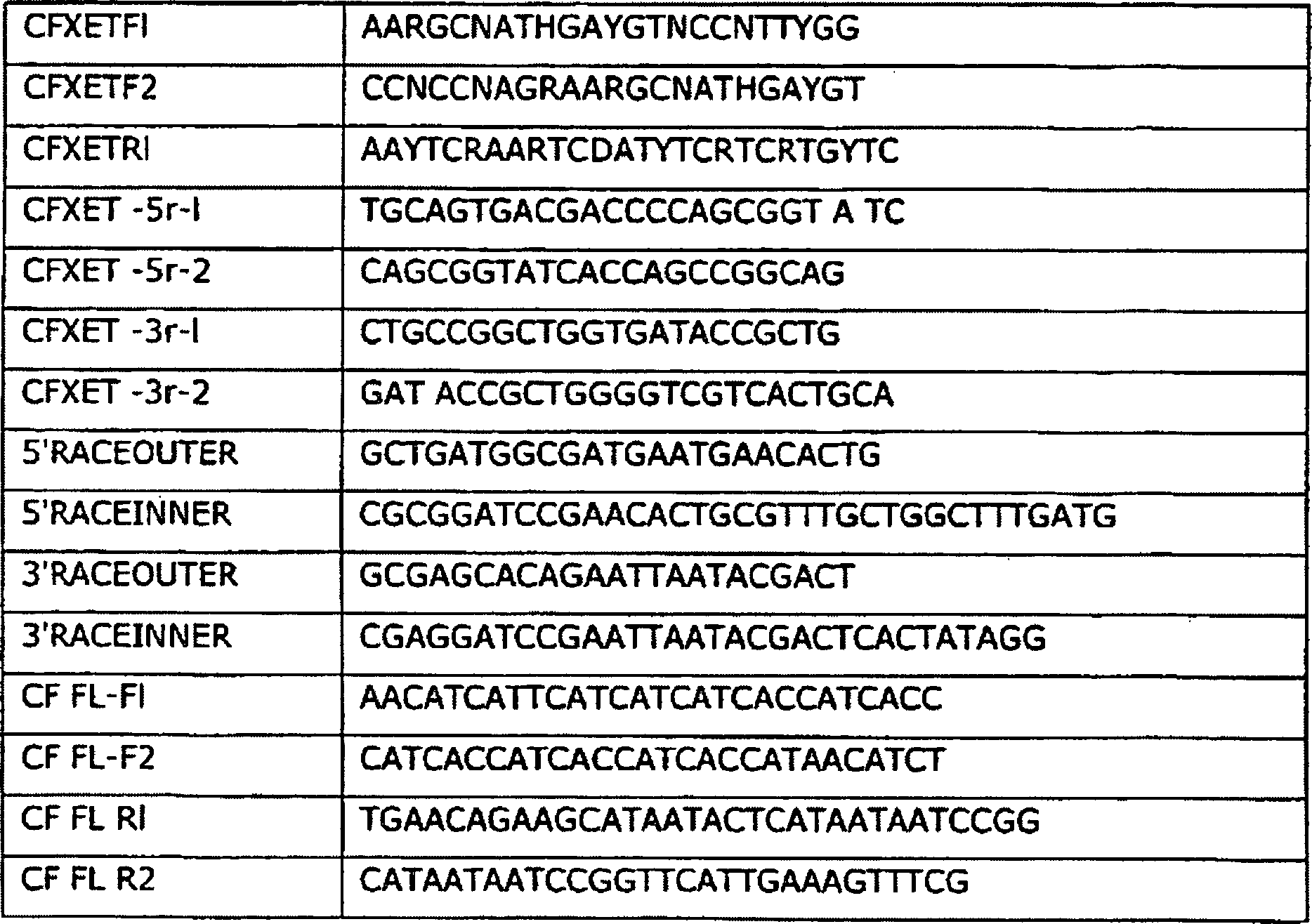
Primersequenzen aller Primer, die verwendet wurden, werden in Tabelle
1 gezeigt. Der reverse Primer CFXETR1 und die Nested-Primer CFXET
F1 wurden für
eine zweiphasige Nested-PCR verwendet, was zu einem PCR-Produkt
mit dem korrekten Molekulargewicht und der Sequenz, die dem XET
in der Glycosylhydrolase-/Transglycosylase Familie 16 entsprach,
führte.
Die cDNA mit vollständiger
Länge wurde
anschließend
amplifiziert, wobei eine Serie von degenerierten Nested-Primern
(Tabelle 1) verwendet wurden, gefolgt von der Sequenzbestimmung
der cDNA mit vollständiger
Länge (SEQ
ID Nr.: 1). Tabelle
1. In den Beispielen verwendeten Primer
- IBU-Code für gemischte Basen: M = A +
C, R = A + G, W = A + T, S = G + C, Y = C + T, V = A + G + C, N
= A + C + T, B = G + T + C, N = A + G + C + T, K = G + T.
-
Die
Primer CFXETFI, CFXETF2 und CFXETRI sind genspezifische, degenerierte
Primer für
die RT-PCR. CFXET-5r-I, CFXET-5r-2, CFXET-3r-I und CFXET-3r-2 sind genspezifische,
interne Primer, während 5'RACE GUTER, 5'RACE INNER, 3'RACE GUTER und 3'RACE INNER komplementäre Primer
zu den Adaptoren der RLM-RACE
sind. CF-FL-FI, CF-FL-F2, CF-FL-RI sind CF-FL-R2 genspezifische
Primer für
die Amplifikation der cDNA mit vollständiger Länge.
-
Beispiel 5
-
ISOLIERUNG DES GENS, WELCHES
DAS XET VON HYBRIDER ESPE CODIERT
-
Die
cDNA, die das Pappel-XET codiert, wurde zum Beispiel aus einer kambialen
EST-Bibliothek einer hybriden Espe isoliert, die aufgebaut war,
wie in Hertzberg et al., 1998, beschrieben wurde. Die Anmerkungen der
Bibliothek offenbarten drei Sequenzen, die den XET-ähnlichen
Enzymen entsprachen. Die Sequenzierung der vollständigen Länge von
einem der Klone zeigte, dass er eine cDNA-Kopie des XET-Enzyms in vollständiger Länge enthielt,
das als XET16A (SEQ ID Nr.: 3) bezeichnet wurde, während ein
anderer Klon einem zweiten XET-Enzym in vollständiger Länge entsprach, das als XET16C
(SEQ ID Nr.: 2) bezeichnet wurde.
-
BEISPIEL 6
-
EXTRAKTION VON XYLOGLUCAN
AUS DEM SAMENKERNPULVER DER TAMARINDE
-
Das
Verfahren von Edwards et al. (Planta 1985, 163, 133-140) wurde modifiziert,
wie nachfolgend beschrieben wird. NaBH4 (0,75
g) wurde in 2,0 M NaOH (1,5 L) gelöst. Das entölte Samenkernpulver der Tamarinde
(30 g) wurde langsam zu der Lösung
unter kräftigem
Rühren
(Schaufelrührer)
zugegeben, um ein Verklumpen zu verhindern. Das Gemisch wurde anschließend auf
90°C erhitzt
und bei dieser Temperatur für
1 Stunde unter kontinuierlichem Rühren gehalten. Nach einer unvollständigen Abkühlung wurden
die Feststoffe über
Glasfasern abgefiltert und verworfen. Nach einer weiteren Abkühlung wurde
das Filtrat durch die langsame Zugabe von Eisessig (300 ml) angesäuert, gefolgt
von der langsamen Zugabe von EtOH (3,0 I), um das Xyloglucan als
eine farblose, gelatineartige Masse zu präzipitieren. Die Feststoffe
wurden durch die Filterung über
ein Baumwolltuch gesammelt und das Filtrat wurde anschließend verworfen.
Das Xyloglucan wurde anschließend
in reinem Wasser (1,5 L, 18 MΩ.cm)
unter leichtem Erhitzen gelöst
und durch die langsame Zugabe von EtOH (3,0 L) erneut präzipitiert.
Die feste Masse wurde erneut durch Filtration durch ein Baumwolltuch
gesammelt, das per Hand ausgewrungen wurde, um das überschüssige Filtrat
freizusetzen. Die Feststoffe wurden anschließend unter vermindertem Druck
(Ölpumpe)
getrocknet und in einer Haushaltskaffeemühle (Braun) gemahlen, um ein
feines Pulver (17 g) zu erhalten.
-
BEISPIEL 7
-
ENDOGLUCANASE VERMITTELTE
HERSTELLUNG VON XYLOGLUCAN-OLIGOSACCHARIDEN
-
Xyloglucan
(3 g) wurde in 200 ml aufbereitetem Wasser (18 MΩ.cm) bei 50°C unter starkem Rühren gelöst. Nach
dem Abkühlen
auf 30°C
wurde Cellulase (30 mg, 4 E/mg, von T. reesei, Fluka) zugegeben
und die Lösung
wurde bei dieser Temperatur übernacht
gehalten. Aktivierter Kohlenstoff (3 g) wurde anschließend zugegeben
und das Gemisch wurde für
15 Minuten gerührt.
Nach der Zugabe von Acetonitril (200 ml) wurde das Gemisch durch
einen Celite-Filter auf einem Glasfaserfilterpapier (Whatman GF/A)
gefiltert. Das Filtrat wurde anschließend unter Vakuum (Wasserentlüfter) konzentriert
und das restliche Lösungsmittel
wurde mit einer starken Vakuum(Öl)-pumpe
entfernt. Das Gemisch von Xyloglucan-Oligosacchariden (XXXG, XLXG, XXLG und
XLLG im molaren Verhältnis
von 15:7:32:46, wie durch eine Hochleistungs-Anionenaustausch-Chromatographie
mit Wechselstromnachweis, HPAEC-PAD bestimmt wurde) wurde durch
eine semipräparative
HPLC auf einer Amid-80-Säule
(TosoHaas, 21,5 mm × 300
mm, Eluent 55:45 Acetonitril-Wasser) fraktioniert, wenn dies benötigt wurde.
XLXG und XXLG waren unter diesen Bedingungen nicht lösbar. Massenspektrometrie
durch Elektrospray-Ionisation (Micromass Q-TOF2) wurde verwendet,
um die Identität
der Oligosaccharide zu bestätigen.
-
Xyloglucan
(3 g) wurde in 200 ml reinem Wasser (18 MΩ.cm) bei 50°C unter starkem Rühren gelöst. Nach
dem Abkühlen
auf 30°C
wurde Cellulase (30 mg, 4 E/mg, von Trichderma reesei, Fluka) zugegeben
und die Lösung
wurde bei dieser Temperatur übernacht
gehalten. Beta-Galactosidase (150 mg, 9 E/mg gegen Lactose, von
Aspergillus oryzae, Sigma G-5160) wurde anschließend zugegeben und die Lösung wurde
für 1 Stunde
bei Raumtemperatur gerührt.
Die Lösung
wurde für
3 min gekocht, gefolgt durch die schnelle Abkühlung vor der Zugabe von aktiviertem
Kohlenstoff (3 g). Das Gemisch wurde anschließend für 15 min bei Raumtemperatur
gerührt.
Nach der Zugabe von Acetonitril (200 ml) wurde das Gemisch durch
einen Celite-Filter auf einem Glasfaserfilterpapier (Whatman GF/A)
gefiltert. Das Filtrat wurde anschließend unter Vakuum (Wasserentlüfter) konzentriert
und das restliche Lösungsmittel
wurde mit einer starken Vakuum(Öl)-pumpe
entfernt. Das Gemisch von Xyloglucan-Oligosacchariden wurde anschließend durch
eine semipräparative
HPLC auf einer Amid-80-Säule
(TosoHaas, 21,5 mm × 300
mm, Eluent 55:45 Acetonitril-Wasser) fraktioniert. Massenspektrometrie
durch Elektrospray-Ionisation (Micromass Q-TOF2) wurde verwendet,
um die Identität
der Oligosaccharide zu bestätigen.
-
BEISPIEL 8
-
HERSTELLUNG VON AMINOALDITOL-DERIVATEN
VON XYLOGLUCO-OLIGOSACCHARIDEN
(XGO-NH2)
-
Xyloglucan-Oligosaccharide
(2,4 g, 1,9 mmol, Gemisch von XXXG, XLXG, XXLG und XLLG) wurden in
einer gesättigten
Ammoniumhydrogencarbonatlösung
(50 ml) gelöst.
Natriumcyanoborohydrid (2,4 g, 39 mmol) wurde anschließend zugegeben
und die Reaktion wurde bei Raumtemperatur in der Dunkelheit gerührt. Nach
sieben Tagen wurde die Reaktion gefiltert und Essigsäure wurde
zugegeben, bis die Lösung
einen pH-Wert von 2 erreichte. Nach der Konzentration unter Vakuum
wurde das unbearbeitete Produkt erneut in 75 ml Wasser gelöst und in
10 Portionen auf eine P2-Säule
(BioRad, Bio-Gel P2, 5 cm × 22
cm) aufgetragen. Die Fraktionen von jedem Säulendurchlauf, die das XGO-NH2 enthielten und die eine niedrige Leitfähigkeit
aufwiesen, wurde vereint und bis zur Trockenheit konzentriert (Ausbeute 1,31
g, 51%). Die Massenspektrometrie durch Elektrospray-Ionisation (Micromass
Q-TOF2) wurde verwendet,
um die Identität
der modifizierten Oligosaccharide zu bestätigen.
-
BEISPIEL 9
-
HERSTELLUNG VON SULFO-RHODAMIN-DERIVATEN
VON XYLOGLUCO-OLIGOSACCHARIDEN (XGO-SR)
-
XGO-Aminoalditole
(XGO-NH2, 0,5 g, 0,4 mM, Gemisch von XXXG-NH2, XLXG-NH2, XXLG-NH2 und XLLG-NH2 wurden in 3% wässriges Natriumtetraborat (30
ml) gelöst.
Sulfo-Rhodamin B-Säurechlorid
(192 mg, 0,3 mM, Fluka 86186) wurde in Dimethylformamid (DMF, 1
ml) gelöst
und tropfenweise in die gerührte
Flüssigkeit
zugegeben. Die Reaktion wurde durch TLC (5:4:1 Chloroform: Methanol:
Wasser) überwacht
und nach sieben Tagen bis zur Trockenheit konzentriert. Das unbearbeitete
Produkt wurde durch eine Flash-Chromatographie auf Kieselsäuregel gereinigt
(schrittweise Elution mit 55:45:5 und 5:4:1 Chloroform: Methanol:
Wasser). Um kleinste Mengen von Kieselsäure aus dem Produkt zu entfernen,
wurde das Material auf eine Säule
(Supelclean ENVI-18 SPE-Gefäß, 6 ml,
Supelco, Bellefonte, PA, USA) für
eine Umkehrphasen-Chromatographie geladen und durch einen abgestuften
Gradienten aus voll entsalztem Wasser, 10% wässrigem Acetonitril und 20%
wässrigem
Acetonitril eluiert (Ausbeute: 20 mg, 2,7%).
-
BEISPIEL 10
-
HERSTELLUNG VON FLUORESCEIN-DERIVATEN
VON XYLOGLUCO-OLIGOSACCHARIDEN (XGO-FITC)
-
Fluorescein-Isothiocyanat-Isomer
I (FITC, 12 mg, 0,03 mmol, Fluka 46952) wurde zu einer Lösung von XGO-Oligoalditolen
(XGO-NH2, 45 mg, 0,036 mmol, Gemisch von
XXXG-NH2, XLXG-NH2,
XXLG-NH2 und XLLG-NH2)
in Natriumbicarbonatpuffer (100 mM, pH-Wert 9,0, 20 ml) gegeben.
Die Reaktion wurde durch TLC (70:30:1 Acetonitril: Wasser: Essigsäure) überwacht
und wurde nach dem Rühren
für 24
Stunden unter Vakuum bis zur Trockenheit konzentriert. Das unbearbeitete
Produkt wurde in 1,5 ml ultrareinem Wasser erneut gelöst, auf
eine P2-Säule
(BioRad, Bio-Gel P2, 1,6 cm × 50
cm) gegeben und mit 10 mM wässrigem
Ammoniumbicarbonat mit einer Flussrate von 0,2 ml/min eluiert. Alle
Fraktionen wurden durch TLC (70:30:1 Acetonitril: Wasser: Essigsäure) überwacht,
welches zeigte, dass das nicht umgesetzte FITC und XGO-NH2 erfolgreich von dem erwünschten Produkt getrennt wurde.
Fraktionen, die XGO-FITC enthielten (nachgewiesen durch einen angeschlossenen
UV-Detektor und TLC) und die eine niedrige Leitfähigkeit zeigten, wurden vereint
und unter Vakuum konzentriert, um einen orangefarbenen Feststoff
zu erhalten (Ausbeute: 34 mg, 60%). Die Massenspektrometrie durch
Elektrospray-Ionisation (Micromass Q-TOF2) wurde verwendet, um die
Identität
der modifizierten Oligosaccharide zu bestätigen.
-
BEISPIEL 11
-
SYNTHESE VON RADIOAKTIVEN
UND NICHT RADIOAKTIVEN ALDITOL-DERIVATEN
VON XLLG-XYLOGLUCO-OLIGOSACCHARID ([1-3H]-XLLGol
UND [1-1H]-XLLGol)
-
Das
Xylogluco-Oligosaccharid XLLG (8,6 μmol) wurde in aufbereitetem
Wasser (250 μl,
18 MΩ.cm) gelöst, das
auf einen pH-Wert von 11,5 mit NAOH eingestellt wurde. NaB3H4 (8,2 μmol, 3,76
GBq) wurde anschließend
zugegeben und man ließ die
Reaktion übernacht
bei Raumtemperatur stehen. Die Reaktion wurde durch die vorsichtige
Zugabe von Eisessig gestoppt, bis der pH-Wert der Lösung etwa
4 betrug. Man ließ die Lösung anschließend für einen
Zeitraum von 30 min stehen, um zu ermöglichen, dass das Tritiumgas
durch den Abzug entlüftet
wurde. Salze wurden aus dem Produkt durch eine Gelfiltrations-Chromatographie
auf Bio-Gel P-2-Harz (BioRad, Lagervolumen 20 ml) mit gereinigtem
Wasser (18 MΩ.cm)
als Eluent entfernt. Fraktionen mit etwa 1 ml Volumen wurden gesammelt.
Die Fraktionen wurden durch eine Flüssigkeitsszintillationsmessung
und Dünnschichtchromatographie
(Kieselsäuregel,
7:3 Acetonitril-Wasser als Eluent, Ammoniummolybdat/Schwefelsäure-Färbung) analysiert.
Die Fraktionen, die sowohl radioaktiv waren als auch ein Produkt mit
einem Rf enthielten, der identisch zu XLLG war, wurden vereinigt.
Ein qualitativer Tollens-Test für
die Reduzierung von Zucker auf diesem Produkt war negativ, was darauf
hinweist, dass die Reduktionsreaktion vollständig abgelaufen war. Das Produkt
besaß eine
Radioaktivität
von 115270 Bq/μl.
-
Die
Synthese von nicht radioaktivem XLLGol wurde in einer Weise durchgeführt, die
identisch zu der war, die vorstehend beschrieben wurde, mit der
Ausnahme, dass NaB3H4 durch
NaBH4 als Reduktionsmittel ausgetauscht
wurde. Das Verdampfen der Chromatographielösung ergab das Produkt als
ein weißes
Pulver, das ein Proton im NMR-Spektrum ergab, das identisch zu dem
war, von dem zuvor berichteten wurde (York W. et al., Carbohydrate
Research, 1990, 200, 9-31). Die durchschnittliche Ausbeute der drei
Reaktionen (50 (+/–) 3%)
wurde verwendet, um die Ausbeute der radioaktiven Synthese zu schätzen, die
eine Konzentration von 1,21 × 10–9 mol/L
und eine spezifische Aktivität
von 95 MBq/μl
anzeigte.
-
BEISPIEL 12
-
HERSTELLUNG VON REGENERIERTEN
ZELLULOSEMEMBRANEN
-
a. Herstellung von regenerierten
Zellulosemembranen aus Kupferammonium-Lösung
-
Gemäß des Verfahrens
von Okajima [Okajima K (1995), Polymer Journal 27(11), 1113-1122]
wurden 10 g Zellulose (Whatman Nr. 1 Filterpapier, UK) in einem
Gemisch von 65 g NH4OH (20%) gelöst, 12 g
frisch hergestelltes Cu(OH)2, 8 g 10% (w/v)
NaOH und 30 g Wasser gelöst,
um eine klare blaue, visköse
Lösung
bei 4°C
zu ergeben. Die Lösung
wurde auf eine Glasplatte gegossen, um eine Stärke von 0,3 mm zu erhalten,
und anschließend
in Eindickungsbäder
gegeben, die bei 4°C
in 10% wässrigem
NaOH gehalten wurden, gefolgt von 4% wässriges H2SO4 für
jeweils 5 Minuten. Die so erhaltenen Regenerat-Zellulosefilme wurden
unter fließendem
Wasser gewaschen und auf einer Glasplatte bei Raumtemperatur getrocknet.
-
b. Herstellung von regenerierter
Zellulosemembran aus wässriger
NaOH/Harnstoff-Lösung
-
Bemliese® Vliesstoff,
der aus Baumwoll-Linterstoffen in Kupferammonium-Lösung
(DP = 650, Asahi Chemical Industry Co. Lid, Japan) hergestellt wurde,
wurde als Quelle der Zellulose verwendet. 10 g Bemliese® Vliesstoff
wurde in 200 ml 6 Gew.-% NaOH/4 Gew.-% wässrige Harnstofflösung gelöst, um eine
klare Zelluloselösung
bei 4°C
zu erhalten. Die Lösung
wurde auf eine Glasplatte gegossen, um eine Stärke von 0,5 mm zu erhalten,
anschließend
sofort in eine 5 Gew.-% wässrige
H2SO4-Lösung getaucht,
um das Festwerden für
5 min bei 4°C
zu ermöglichen.
Die so erhaltenen Membrane wurden mit fließendem Wasser gewaschen und
an der Luft auf einer Glasplatte bei Raumtemperatur getrocknet.
-
BEISPIEL 13
-
XET VERMITTELTER EINBAU
VON SULFO-RHODAMIN MODIFIZIERTEN XYLOGLUCAN-OLIGOSACCHARIDEN (XGO-SR)
IN XYLOGLUCAN IN LÖSUNG
-
Das
Fluorophor Sulfo-Rhodamin wurde chemisch in das reduzierende Ende
von Xyloglucan-Oligosacchariden eingebaut, um XGO-Rhodamin herzustellen,
wie in dem Beispiel 8. A beschrieben wurde. Ein Gemisch (4 ml) von
Xyloglucan (XG, 0,5 mg/ml), XGO-Rhodamin (0,5 mg/ml) und XET (0,025
mg/ml) in Ammoniumacetatpuffer (50 mM, pH-Wert 5,5) wurde bei Raumtemperatur
(22°C) für 10 min
inkubiert. Die Reaktion wurde durch das Eluieren des Reaktionsgemisches
durch eine HiTrap SP FF-Säule
(Amersham Bioscience, Schweden) zur Entfernung des XET-Enzyms beendet.
Etwa 25% des zugegebenen Enzyms wurde mit XGO-Rhodamin modifiziert, wie durch die
kolorimetrische Analyse von Kooiman [Kooiman P (1960), Recl Trav Chim
Pay-Bas, 79, 675-678] bestimmt wurde.
-
BEISPIEL 14
-
XET VERMITTELTER EINBAU
VON FLUORESCEIN MODIFIZIERTEN XYLOGLUCAN-OLIGOSACCHARIDEN (XGO-FITC)
IN XYLOGLUCAN IN LÖSUNG
-
Fluorescein-Isothiocyanat
Isomer I wurde chemisch in das reduzierende Ende von Xyloglucan-Oligosacchariden
eingebaut, um XGO-FITC herzustellen, wie in dem Beispiel 10 beschrieben
wurde. Die Wirkungen der Konzentration des XET-Enzyms und der Reaktionszeit
auf den Einbau von XGO-FITC in Xyloglucan in Lösung wurde analysiert, wie
nachfolgend beschrieben wird.
-
a. Zeitabhängigkeit
-
Proben,
die ein Gemisch (200 μl
Gesamtvolumen) von Xyloglucan (XG, 1 mg/ml), XGO-FITC (0,5 mg/ml)
und XET (8 Einheiten) in Citratpuffer (20 mM, pH-Wert 5,5) enthielten, wurden bei 30°C für 5, 10,
20, 40, 60, 120, 180, 300 und 360 min inkubiert. Zum gegebenen Zeitpunkt
wurde jede Reaktion durch das Erhitzen auf 75°C für 5 min beendet. Nach dem Abkühlen auf
Raumtemperatur wurden 400 μl
Ethanol zugegeben und das Gemisch wurde bei 12000 g für 5 min
bei 4°C
zentrifugiert, um das modifizierte und nicht modifizierte XG zu
präzipitieren,
während
das XGO-FITC in Lösung
blieb. Sowohl das Präzipitat
als auch der Überstand wurden
unter Vakuum getrocknet und erneut getrennt in je 200 μl Wasser
gelöst.
0,019; 0,025; 0,031; 0,035; 0,038; 0,041; 0,042; 0,043; 0,044 mg
XGO-FITC wurden jeweils in das reduzierende Ende von Xyloglucan
in den 5, 10, 20, 40, 60, 120, 180, 300 und 360 min-Proben eingebaut,
wie durch die UV-Absorption bei 495 nm der erneut gelösten Präzipitatlösung bestimmt
wurde, wobei eine Standardkurve verwendet wurde, die von XGO-FITC-Lösungen mit
einer steigenden Konzentration abgeleitet wurde. Die Ergebnisse
werden in 5 aufgezeichnet.
-
b. Enzymabhängigkeit
-
Ein
Gemisch (200 μl
Gesamtvolumen) von Xyloglucan (XG, 1 mg/ml), XGO-FITC (0,5 mg/ml)
in Citratpuffer (20 mM, pH-Wert 5,5) wurde mit abnehmenden Mengen
von XET (32,0; 16,0; 14,4; 12,8; 9,6; 6,4; 4,8; 3,2; 1,6 und 0,8
Einheiten) bei 30°C
für 40
min inkubiert. Zu diesem Zeitpunkt wurden die Reaktionsgemische genauso
behandelt, wie in Beispiel 13b beischrieben wurde. Unter Verwendung
dieses Verfahrens wurde gezeigt, dass 0,042; 0,038; 0,038; 0,037;
0,034; 0,031; 0,027; 0,022; 0,015 und 0,009 mg XGO-FITC in das reduzierende
Ende von Xyloglucan in den Proben, die jeweils 32,0; 16,0; 14,4;
12,8; 9,6; 6,4; 4,8; 3,2; 1,6 und 0,8 Einheiten des XET-Enzyms enthielten,
eingebaut wurden. Die Ergebnisse werden in 6 aufgezeichnet.
-
BEISPIEL 15
-
ADSORPTION VON SULFO-RHODAMIN
MODIFIZIERTEN XYLOGLUCANEN AUF ZELLULOSEMATERIALIEN
-
Zellulosehaltige
Materialien (0,1 g Munktell-Filterpapierstreifen) wurden in die
Lösung
getaucht, die das Rhodamin modifizierte XG enthielt (4 ml, hergestellt
gemäß dem Verfahren
in Beispiel 10), und wurden übernacht
(ca. 15 Stunden) in einem End-Over-End-Mixer bei Raumtemperatur
geschüttelt.
Das Binden von XG an die zellulosehaltigen Fasern (11,4 mg XG/g
Zellulose) wurde durch den Rückgang
von XG aus der Lösung
analysiert, wie durch das kolorimetrische Verfahren von Kooiman
bestimmt wurde. Das zellulosehaltige Material wurde anschließend aus
der ursprünglichen
Lösung
entfernt und wiederholt mit ultrareinem Wasser in einem End-Over-End-Mixer gewaschen,
um überschüssiges XGO-Rhodamin
zu entfernen. Nach dem intensiven Waschen wurde die Adsorption von
Rhodamin-XG auf der Zellulose ebenfalls als eine intensive rosa
Färbung
unter Umgebungslicht und als eine starke Fluoreszenz unter UV-Licht
beobachtet. Kontrollproben, die unter identischen Bedingungen behandelt
wurden, die aber nur unmodifiziertes Xyloglucan und XGO-SR enthielten, waren
nach dem Waschen farblos.
-
BEISPIEL 16
-
XET VERMITTELTER EINBAU
VON CHEMISCH MODIFIZIERTEN XYLOGLUCAN-OLIGOSACCHARIDEN IN XYLOGLUCAN.
DAS ZUVOR AN ZELLULOSEFASERN ANGELAGERT WURDE
-
XG
(0,5 mg/ml) wurde mit zellulosehaltigen Fasern übernacht (15 h, leichtes Mischen
im End-Over-End-Mixer) inkubiert, um zunächst das XG an die Zellulose anzulagern.
Die XG-Zellulose wurde anschließend
mit einem Gemisch von XGO-Rhodamin
(0,1 mg/ml) und XET (0,025 mg/ml) in einem 50 mM Ammoniumacetatpuffer,
pH-Wert 5,5 behandelt. Nach dem Mischen in einem End-Over-End-Mixer bei
Raumtemperatur für
4 Stunden wurde die Probe intensiv mit ultrareinem Wasser gewaschen.
Der kovalente Einbau der fluoreszierenden Oligosaccharide wurde
durch eine starke rosa Färbung
auf den Zellulosefasern nachgewiesen, die ebenfalls eine starke
Fluoreszenz unter UV-Licht zeigten.
-
BEISPIEL 17
-
ADSORPTION VON FLUORESCEIN
MODIFIZIERTEM XYLOGLUCAN (XG-FITC)
AUF ZELLULOSEHALTIGEM PAPIER
-
- a. Ein Gemisch (4 ml Gesamtvolumen) von Xyloglucan
(XG, 0,5 mg/ml), XGO-FITC (0,5 mg/ml) und XET (0,025 mg/ml) in Ammoniumacetatpuffer
(25 mM, pH-Wert 5,5) wurde bei Raumtemperatur (22°C) für 10 min
inkubiert. Die Reaktion wurde durch das Eluieren des Reaktionsgemisches
durch eine HiTrap SP FF-Säule
(Amersham Bioscience, Schweden) zur Entfernung des XET-Enzyms beendet.
Zellulosehaltiges Material (0,1 g Whatman Nr. 1 Filterpapierstreifen)
wurden anschließend
in die Lösung
eingetaucht und in einem End-Over-End-Mixer für 15 Stunden bei Raumtemperatur
geschüttelt.
Die Bindung von XG an die zellulosehaltigen Fasern (12,6 mg XG/g
Zellulose) wurde durch den Rückgang
von XG aus der Lösung
analysiert, wie durch das kolorimetrische Verfahren von Kooiman
bestimmt wurde. Das zellulosehaltige Material wurde anschließend aus
der ursprünglichen
Lösung
entfernt und wiederholt mit ultrareinem Wasser in einem End-Over-End-Mixer gewaschen,
um überschüssiges XGO-FITC
zu entfernen. Die Adsorption von XG-FITC auf der Zellulose wurde
als eine intensive rosa Färbung
unter Umgebungslicht und als eine starke Fluoreszenz unter UV-Licht
beobachtet. Kontrollproben, zu denen kein XET-Enzym zu der XG/XGO-FITC-Lösung zugegeben
wurden, waren nach dem Waschen farblos und zeigten keine Fluoreszenz.
- b. Ein Gemisch (200 μl)
von Xyloglucan (XG, 1 mg/ml), XGO-FITC (0,5 mg/ml) und XET (8 Einheiten)
in Citratpuffer (20 mM, pH-Wert 5,5) wurde bei 30°C für 60 min
inkubiert. Die Reaktion wurde durch die Erhitzung auf 75°C für 5 min
beendet. 100 μl
dieser Lösung
wurden in ultrareinem Wasser auf 500 μl verdünnt und zellulosehaltiges Material
(Whatman Nr. 1 Filterpapierscheiben, 1,5 cm Durchmesser, 15,4 mg)
wurde in die Lösung
eingetaucht, gefolgt durch das Schütteln in einem End-Over-End-Mixer
für 15
Stunden bei Raumtemperatur. Das Filterpapier wurde anschließend entfernt
und mit ultrareinem Wasser (2 × 1
ml) gewaschen. Die Menge des XGO-FITC, die in XG eingebaut wurde
und anschließend
an das Filterpapier gebunden wurde (0,0232 mg), wurde durch den
Rückgang
von XGO-FITC aus der Lösung
(einschließlich
der Waschlösung)
analysiert, wie durch die Adsorption bei 495 nm in 0,1 M Natriumbicarbonat
gegen eine Standardkurve von XGO-FITC bestimmt wurde. Um die Menge
des FITC modifizierten Xyloglucans (XG-FITC) auf der Papieroberfläche direkt
zu quantifizieren, wurde das Papier sowohl mit einer CCD-Kamera
während einer
Fluoreszenzanregung (Fujifilm Imager) und mit einem Desktop-Scanner
abgebildet. Man konnte zeigen, dass die Intensität des blauen Kanals eine lineare
Korrelation mit der Menge des auf dem Papier adsorbierten XG-FITC
zeigte, wenn es in einem Fullcolor RGB Modus gescannt wurde. Des
weiteren konnte gebundenes XG-FITC von dem Papier mit 2 M wässriger
NaOH extrahiert und anschließend
durch eine UV-Adsorption
bei 495 nm in 0,1 M Natriumbicarbonat quantifiziert werden. Man
konnte zeigen, dass die Behandlung von XG-FITC mit 2 M NaOH keine
Wirkung auf die UV-Adsorption
oder der Fluoreszenzemission und dem Spektrum der Anregung der Verbindung
hatte. Die Bilder der konfokalen Mikroskopie zeigten, dass das Fluorophor
spezifisch auf der Faseroberfläche
lokalisiert war, und wiesen nach, dass das Signal trotz der Porosität des Materials
deutlich detektierbar war.
-
Ein
Bild einer konfokalen Fluoreszenzmikroskopie eines XG-FITC behandelten
Papiers wird in 7 gezeigt. Die hellen Bereiche
deuten auf eine hohe relative Intensität der Fluoreszenz hin und die
dunklen Bereiche deuten auf eine niedrigere relative Intensität der Fluoreszenz
hin.
-
BEISPIEL 18
-
ADSORPTION VON SULFO-RHODAMIN
MODIFIZIERTEM (XG-SR) UND FLUORESCEIN MODIFIZIERTEM (XG-FITC) XYLOGLUCAN
AUF ZELLULOSEMEMBRANEN
-
- a. Regenerierte Zellulosemembranen (0,05 g)
wurden in eine Lösung
getaucht, die das Sulfo-Rhodamin modifizierte XG (XG-SR, 4 ml, hergestellt
gemäß dem Verfahren
in Beispiel 13) enthielt, und wurde für 15 Stunden in einem End-Over-End-Mixer bei Raumtemperatur
geschüttelt.
Das Binden von XG-SR an die regenerierte Zellulosemembran wurde
durch den Rückgang
von XG-SR aus der Lösung
auf 0,3 mg/g bestimmt, wobei das kolorimetrische Verfahren von Kooiman
verwendet wurde. Das zellulosehaltige Material wurde anschließend aus
der ursprünglichen
Lösung
entfernt und wiederholt mit ultrareinem Wasser in einem End-Over-End-Mixer gewaschen,
um das überschüssige XGO-Rhodamin
zu entfernen. Die Adsorption von XG-Rhodamin auf der Zellulose wurde
als eine intensive rosa Färbung
unter Umgebungslicht und als eine starke Fluoreszenz unter UV-Licht
beobachtet. Die konfokale Fluoreszenzmikroskopie zeigte, dass XG-SR
auf den Membranoberflächen
lokalisiert war.
- b. Ein Gemisch (200 μl)
von Xyloglucan (XG, 1 mg/ml), XGO-FITC (0,5 mg/ml) und XET (2 μg) in Citratpuffer (20
mM, pH-Wert 5,5) wurde bei 30°C
für 40
min inkubiert. Die Reaktion wurde durch das Erhitzen auf 75°C für 5 min
beendet. Die regenerierten Zellulosemembrane (0,05 g) wurden in
die Lösung
getaucht. Die Menge des XGO-FITC, die in XG eingebaut wurde und
anschließend
an die Membran gebunden wurde, wurde durch den Rückgang von XGO-FITC aus den
Lösungen
(einschließlich
der Waschlösungen)
analysiert, wie durch die UV-Adsorption
bei 495 nm in 0,1 M Natriumbicarbonat gegen eine Standardkurve von
XGO-FITC bestimmt wurde. Konfokale Fluoreszenzmikroskopie zeigte,
dass XG-FITC ausschließlich auf
den Membranoberflächen
gebunden war.
-
BEISPIEL 19
-
HERSTELLUNG VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2)
-
Eine
typische Reaktion bestehend aus 10 mg Xyloglucan der Tamarindus
indica, 3,75 mg Amino-modifizierte Xyloglucan-Oligosaccharide (Gemisch
von XXXG-NH2, XLXG-NH2,
XXLG-NH2, XLLG-NH2,
hergestellt gemäß Beispiel
8) und 182 Einheiten XET (49 μg
Protein, Bradford-Assay) wurden in 20 mM Citratpuffer, pH-Wert 5,5,
für 30
Minuten bei 30°C
inkubiert. Das Enzym wurde durch das Erhitzen auf 75°C für zehn Minuten
deaktiviert. Die kolorimetrische Analyse von Sulová et al.
(1995), Anal Biochem 229, 80-85, zeigte typischerweise eine Veränderung
von 0,4 Einheiten der optischen Dichte bei 620 nm nach der Inkubation,
was vergleichbar mit dem war, wenn XGO-FITC als ein Substrat unter ähnlichen
Bedingungen verwendet wurde.
-
BEISPIEL 20
-
ADSORPTION DES AMINO-MODIFIZIERTEN
XYLOGLUCANS (XG-NH2)AUF ZELLULOSEHALTIGEM
PAPIER
-
Amino-modifiziertes
Xyloglucan (XG-NH2, hergestellt, wie in
Beispiel 19 beschrieben wurde) wurde 1:1 mit Wasser verdünnt und
mit einem Blatt Filterpapier (Whatman Nr. 1, 1,5 cm Durchmesser,
15 mg) in einem Glassgefäß übernacht
bei Raumtemperatur unter kreisförmigen
Schütteln
inkubiert. Das Papier wurde intensiv mit ultrareinem Wasser gewaschen.
Typischerweise wurden 70 bis 80% des Aminomodifizierten Xyloglucans an
das Papier adsorbiert, wie durch das kolorimetrische Verfahren von
Kooimann bestimmt wurde. Der Gehalt der Aminogruppen auf dem Papier
wurde mit Ninhydrin quantifiziert, wie von Sarin et al. (1981),
Anal Biochem 17, 147-157, beschrieben wurde, was typischerweise
70-80 nmol der nachgewiesenen Aminogruppen pro Blatt des Papiers
ergab.
-
BEISPIEL 21
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN
-
DER OBERFLÄCHE VON
ZELLULOSEHALTIGEM PAPIER ADSORBIERT WAR, MIT FLUORESCEIN-ISOTHIOCYANAT
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit Fluorescein-Isothiocyanat, Isomer
I (0,6 mg) in 500 μl
0,1 M NaHCO3 übernacht bei Raumtemperatur
in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde intensiv mit 0,1 M NaHCO3 und
ultrareinem Wasser gewaschen. Papier, das in dieser Weise behandelt
wurde, erschien intensiv gelb unter Umgebungslicht und zeigte eine
starke Fluoreszenz. Der Grad der Modifikation wurde quantifiziert,
wie in Beispiel 17 b dargestellt wurde. Kontrollproben des Papiers,
die in der gleichen Weise behandelt wurden, denen aber kein XG-NH2 zugegeben wurde, waren farblos und zeigten
keine Fluoreszenz.
-
Ein
Foto von dem Ergebnis des Einbaus von Fluorescein in das Papier,
das mit XG-NH2 behandelt und mit FITC umgesetzt
wurde, wird in 8 gezeigt. Die linke Filterscheibe,
die dunklere, ließ man
mit FITC reagieren, wohingegen die rechte Filterscheibe identisch
zu der linken behandelt wurde, außer dass kein XG-NH2 vor der Reaktion mit FITC gebunden wurde.
Von 8 wird deutlich, dass das FITC an der Filterscheibe
gebunden ist.
-
BEISPIEL 22
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT ESSIGSÄURE
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit 3,75 mM Essigsäureanhydrid
und 11 mM Triethylamin in 2 ml wasserfreies Methanol übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln inkubiert. Das
Papier wurde anschließend
mit Methanol gewaschen, gefolgt von einem Überschuss an Wasser. Die Menge
der Aminogruppen, die durch den quantitativen Ninhydrin-Assay [Sarin
et al. (1981), Anal Biochem 117, 147-157] nachgewiesen wurde, nahm
um 84% im Vergleich zu einer Kontrollprobe ab. Acetyliertes Papier,
das auf dieser Weise hergestellt wurde, wurde ebenfalls mit Fluorescein-Isothiocyanat
in eine Weise umgesetzt, die in Beispiel 20 dargestellt wurde, und
wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 100% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 23
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT PHENYLISOCYANAT
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit einer 1 M
Phenylisocyanat-Lösung
in Methanol (2 ml) übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde anschließend mit Methanol (3 × 5 ml,
in Gefäßen) gewaschen,
gefolgt von einem Überschuss
(1 L, auf einer Glasfritte) von Wasser. Die Menge der Aminogruppen,
die durch den quantitativen Ninhydrin-Assay [Sarin et al. (1981),
Anal Biochem 117, 147-157]
nachgewiesen wurde, nahm um 70% im Vergleich zu einer Kontrollprobe,
ab. Papier, das auf dieser Weise hergestellt wurde, wurde ebenfalls
mit Fluorescein-Isothiocyanat
in eine Weise umgesetzt, die in Beispiel 20 dargestellt wurde, und
wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 64% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 24
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT ALKENYLBERNSTEINSÄUREANHYDRID (ASA) IN DIMETHYLSULFOXID
DMSO
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit einer 4 mM
ASA-Lösung in
DMSO (2 ml) übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde zweimal mit 10 ml 2-Propanol, zweimal
mit 10 ml Methanol und zweimal mit 10 ml Wasser in dem Gefäß und anschließend mit
1 L gereinigtem Wasser auf einer Glasfritte gewaschen. Die Menge
der nachgewiesenen Aminogruppen nahm um 63% im Vergleich zu einer
unbehandelten Kontrollprobe ab. Papier, das auf dieser Weise hergestellt
wurde, wurde ebenfalls mit Fluorescein-Isothiocyanat in eine Weise
umgesetzt, die in Beispiel 21 dargestellt wurde, und wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 63% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 25
-
REAKTION VON AMINO-MODIFIZIERTEM
XYlOGLUCAN (XG-NH2)DAS AN DER OBERFLÄCHE VON
ZELLULOSEHALTIGEM PAPIER ADSORBIERT WAR, MIT BERNSTEINSÄUREANHYDRID
IN METHANOL (MeOH)
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit einer 1,5
M Bernsteinsäureanhydrid-Lösung in
wasserfreiem Methanol (2 ml) übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde zweimal mit 10 ml Methanol, zweimal
mit 10 ml gereinigtem Wasser in dem Gefäß und anschließend mit
1 L gereinigtem Wasser auf einer Glasfritte gewaschen. Die Menge
der nachgewiesenen Aminogruppen nahm um 48% im Vergleich zu einer
unbehandelten Kontrollprobe ab. Papier, das auf dieser Weise hergestellt wurde, wurde
ebenfalls mit Fluorescein-Isothiocyanat in eine Weise umgesetzt,
die in Beispiel 21 dargestellt wurde, und wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 38% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 26
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT N-CINNAMOYL-IMIDAZOL
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit 1 M N-Cinnamoyl-Imidazol
in Dimethylsulfoxid (2 ml) übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde anschließend mit 2-Propanol (2 × 5 ml),
Methanol (2 × 5
ml) und einem Überschuss
von Wasser (1 L) gewaschen. Die Menge der Aminogruppen, die durch
den quantitativen Ninhydrin-Assay [Sarin et al. (1981), Anal Biochem
117, 147-157] nachgewiesen wurde, nahm um 65% im Vergleich zu einer
Kontrollprobe ab. Papier, das auf dieser Weise hergestellt wurde,
wurde ebenfalls mit Fluorescein-Isothiocyanat
in eine Weise umgesetzt, die in Beispiel 21 dargestellt wurde, und
wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 84% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 27
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT BROM-ISOBUTTERSÄURE
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit einer Lösung, die 1
M Brom-Isobuttersäure
und 1 M 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid enthielt, in
MeOH (2 ml) übernacht
bei Raumtemperatur in Glasgefäßen unter kreisförmigen Schütteln inkubiert.
Das Papier wurde mit Methanol (2 × 10 ml) und ultrareinem Wasser
(2 × 10 ml)
in dem Gefäß und anschließend mit
1 L ultrareinem Wasser auf einer Glasfritte gewaschen. Die Menge
der nachgewiesenen Aminogruppen nahm um 50% im Vergleich zu einer
unbehandelten Kontrollprobe ab. Papier, das auf dieser Weise hergestellt
wurde, wurde ebenfalls mit Fluorescein-Isothiocyanat in eine Weise umgesetzt,
die in Beispiel 21 dargestellt wurde, und wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 79% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
BEISPIEL 28
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR MIT BIOTIN-3-SULFO-N-HYDROXYCUCCINIMIDESTER
(SUCCINIMIDYL-BIOTIN)
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit einer Lösung, die
180 μM Succinimidyl-Biotin-Lösung in
10 mM NaHCO3 (2 ml) enthielt, übernacht
bei Raumtemperatur in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde viermal mit 10 ml gereinigtem Wasser
in dem Gefäß und anschließend mit
1 L gereinigtem Wasser auf einer Glasfritte gewaschen. Die Menge
der nachgewiesenen Aminogruppen nahm um 71% im Vergleich zu einer
unbehandelten Kontrollprobe ab. Papier, das auf dieser Weise hergestellt
wurde, wurde ebenfalls mit Fluorescein-Isothiocyanat in eine Weise umgesetzt,
die in Beispiel 21 dargestellt wurde, und wurde gemäß Beispiel
17 b quantifiziert, wodurch gezeigt wurde, dass 57% der Aminogruppen
im Vergleich zu einer Kontrolle aus nicht modifiziertem Amino-Papier
umgesetzt wurden.
-
Das
biotinylierte Papier und eine Kontrollprobe wurde übernacht
mit 500 μl
einer 0,1% BSA-Lösung mit
einem End-Over-End-Mixer inkubiert, um eine unspezifische Bindung
von Proteinen an das Papier zu blockieren. Das Papier wurde zweimal
mit 1 ml Wasser gewaschen und mit 10 μg Streptavidin-alkalische Phosphatase-Konjugat
in 100 mM Tris, pH-Wert 9,5 für
15 min bei Raumtemperatur inkubiert. Das Papier wurde viermal mit
1 ml des gleichen Tris-Puffers gewaschen. Das Papier wurde anschließend mit
18 μg 5 Brom-4-Chlor-3-Indolylphosphat/Nitro
blue (BCIP/NBT) in 320 μl
Tris-Puffer, pH-Wert 9,5 für
5 min inkubiert. Das Papier wurde anschließend zweimal mit 20 μl gereinigtem
Wasser gewaschen und getrocknet. Das Papier wurde für eine Bildanalyse
nach dem Scannen auf einem Desktop-Bild-Scanner verwendet. Das sichtbare blaue
Papier war 40% stärker
gefärbt
als die Kontrolle, das nicht biotinylierte Amino-Papier.
-
BEISPIEL 29
-
REAKTION VON AMINO-MODIFIZIERTEM
XYLOGLUCAN (XG-NH2), DAS AN DER OBERFLÄCHE VON ZELLULOSEHALTIGEM
PAPIER ADSORBIERT WAR, MIT γ-THIOBUTYROLACTON
-
Zellulosehaltiges
Papier (Whatman Nr. 1, 1,5 cm Durchmesser, 15 mg), das hergestellt
wurde, wie in Beispiel 20 beschrieben wurde, wurde mit γ-Thiobutyrolacton
(87 μl,
1 mol) in einem Gemisch von Ethanol (98%, 500 μl) und einer wässrigen
Lösung
von Natriumbicarbonat (0,1 M, 500 μl) übernacht bei Raumtemperatur
in Glasgefäßen unter
kreisförmigen
Schütteln
inkubiert. Das Papier wurde zweimal mit 5 ml einer wässrigen
Lösung
von 0,1 M Natriumbicarbonat und schließlich mit etwa 1 L reinem Wasser
gewaschen. Die Menge der nachgewiesenen Aminogruppen nahm um 52%
im Vergleich zu einer unbehandelten Kontrollprobe ab.
-
9 zeigt
die relativen Mengen von Aminogruppen auf der Oberfläche von
XG-NH2-modifiziertem zellulosehaltigen Papier
nach der Behandlung mit verschiedenen aminoreaktiven Reagenzien.
Hellgraue Säulen
betreffen die Bestimmung mittels der quantitativen Ninhydrin-Analyse,
wohingegen die dunkelgrauen Säulen
die Quantifizierung durch die Reaktion mit FITC, gefolgt durch eine
quantitative Bildanalyse, betreffen. 10 zeigt
die Reaktivität
von Papier mit und ohne Modifikation durch XG-NH2 gegenüber FITC.
Der "Leerwert" ist ein im Handel
erhältliches
Filterpapier, das mit FITC behandelt wurde, "XGN" ist
ein im Handel erhältliches
Filterpapier, das mit XG-NH2, gefolgt von
FITC behandelt wurde; "1" ist ein im Handel
erhältliches
Filterpapier, das mit XG-NH2, gefolgt von
Essigsäureanhydrid
und anschließend
mit FITC behandelt wurde.
-
BEISPIEL 30
-
REAKTION VON SULFO-RHODAMIN-METHANTHIOSULFONAT
MIT DEN THIOL-GRUPPEN, DIE AUF DER OBERFLÄCHE VON ZELLULOSEHALTIGEM PAPIER
EINGEBRACHT WURDEN
-
Die
Hälfte
des trockenen zellulosehaltigem Papier (7,7 mg), das hergestellt
wurde, wie in Beispiel 29 beschrieben wurde, wurde mit 2 ml 10 mM
Dithiothreitol (DTT) in einer wässrigen
Lösung
von 0,1 M NaHCO3 unter Argon in einem Glasgefäß für 2 Stunden
unter gelegentlichem Schütteln
behandelt. Nachdem das Papier dreimal mit 2 ml entgastem, ultrareinem
Wasser unter Argon gewaschen wurde, wurde 1 ml 1 mM Sulfo-Rhodamin-Methanthiosulfonat
in einer Lösung
von DMSO/H2O (1:9) zugegeben und man ließ die Reaktion
für 2 Stunden
unter gelegentlichen Rühren
ablaufen. Das Papier wurde mit DMSO gewaschen, um das nicht umgesetzte
Sulfo-Rhodamin-Methanthiosulfonat zu entfernen, mit ultrareinem
Wasser gewaschen und getrocknet.
-
Das
Papier zeigte eine intensive rosa Färbung unter Umgebungslicht
und eine starke Fluoreszenz unter UV-Licht, während das leere Papier, das
in der gleichen Weise behandelt wurde, farblos war. Die Hälfte des rosafarbenen
Papiers (ca. 3 mg) wurde mit 500 μl
10 mM Dithiothreitol (DTT) in einer wässrigen Lösung von 0,1 M NaHCO3 erneut behandelt, um das Disulfid gebundene
Sulfo-Rhodamin-Methanthiosulfonat
von der Papieroberfläche
zu entfernen. Die Lösung
des Überstands
wurde mittels UV-Adsorption bei 565 nm quantifiziert. Das Papier
wurde 3 mal mit 1 ml entgastem Wasser unter Argon gewaschen und
man ließ es
erneut mit 1 ml 1 mM Sulfo-Rhodamin-Methanthiosulfonat in einer
Lösung
eines DMSO/H2O (1:9)-Gemisches für 2 Stunden unter
gelegentlichen Rühren
reagieren. Nach dem Waschen mit DMSO und Wasser zeigte das Papier
erneut eine intensive rosa Färbung
unter Umgebungslicht und eine starke Fluoreszenz unter UV-Licht,
während
das leere Papier, das in der gleichen Weise behandelt wurde, farblos
war.
-
11 zeigt
das Ergebnis einer Reaktion von thioliertem Papier mit Sulfo-Rhodamin-Methanthiosulfonat.
Die Proben auf der linken Seite zeigen in jeder Reihe XG-NH2-behandeltes Papier, während solche auf der rechten
Seite XG-NH2-behandelts Papier zeigen, das anschließend mit
Thiobutyrolacton behandelt wurde, um eine Thiol (-SH)-Gruppe einzubringen.
Obere Reihe: Proben nach der Behandlung mit Sulfo-Rhodamin-Methanthiosulfonat.
Mittlere Reihe: Anteile von Proben aus der oberen Reihe nach dem
Waschen mit der DTT-Lösung.
Untere Reihe: DTT gewaschene Proben, die man erneut mit Sulfo-Rhodamin-Methanthiosulfonat reagieren
ließ.
-
BEISPIEL 31
-
RADIKALE POLYMERISIERUNG
DURCH ATOMTRANSFER UNTER VERWENDUNG EINES GEBUNDENEN INITIATORS
MIT XG AUF DER OBERFLÄCHE
VON ZELLULOSEHALTIGEM MATERIAL
-
Die
radikale Polymerisierung durch Atomtransfer von Zellulosepapieroberflächen bei
Umgebungstemperatur wurde beschrieben [Carlmark und Malström, 1002,
J Am Chem Soc 124: 900-901], wenn jedoch große Ladungsmengen des Initiators
verwendet werden, ist die Integrität des Papiers stark gefährdet. Wir
haben die gleiche Polymerisierungsreaktion mit einem Initiator durchgeführt, der
auf der Zelluloseoberfläche über Xyloglucan
immobilisiert war, wie in Beispiel 27 beschrieben wurde, ein Verfahren,
das große
Mengen des Initiators auf der Papieroberfläche ohne die Degradation der
Papierstruktur ergab. Das aufgepfropfte Kopolymer, das so hergestellt
wurde, zeigte eine dramatisch verstärkte Faser-Polymer-Bindung im Vergleich zu den
Kontrollproben, bei dem der Initiator nicht auf der Faseroberfläche gebunden
war.
-
Obwohl
die Erfindung mit Bezug auf ihre bevorzugten Ausführungsformen
beschrieben wurde, welche die besten Verfahren darstellen, die den
Erfindern zur Zeit bekannt sind, sollte es selbstverständlich sein,
dass verschiedene Veränderungen
und Modifikationen gemacht werden können, die dem Fachmann offensichtlich sein
werden, ohne den Umfang der Erfindung, wie er in den hierin beigefügten Ansprüchen dargelegt
wurde, zu verlassen. SEQUENZPROTOKOLL