CN1665799A - 运动内酯化合物 - Google Patents
运动内酯化合物 Download PDFInfo
- Publication number
- CN1665799A CN1665799A CN038150530A CN03815053A CN1665799A CN 1665799 A CN1665799 A CN 1665799A CN 038150530 A CN038150530 A CN 038150530A CN 03815053 A CN03815053 A CN 03815053A CN 1665799 A CN1665799 A CN 1665799A
- Authority
- CN
- China
- Prior art keywords
- substituted
- unsubstituted
- compound
- ethyl
- erythromycin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 91
- 238000000034 method Methods 0.000 claims abstract description 50
- 238000002360 preparation method Methods 0.000 claims abstract description 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 9
- 230000030135 gastric motility Effects 0.000 claims abstract 2
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 claims description 67
- 229960003276 erythromycin Drugs 0.000 claims description 56
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 31
- 239000000203 mixture Substances 0.000 claims description 20
- 108090000623 proteins and genes Proteins 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 16
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 16
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 125000000623 heterocyclic group Chemical group 0.000 claims description 12
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 12
- 125000006730 (C2-C5) alkynyl group Chemical group 0.000 claims description 11
- 101001110310 Lentilactobacillus kefiri NADP-dependent (R)-specific alcohol dehydrogenase Proteins 0.000 claims description 8
- 150000002148 esters Chemical group 0.000 claims description 8
- 150000003839 salts Chemical group 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 7
- 125000000217 alkyl group Chemical group 0.000 claims description 6
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 6
- 238000012239 gene modification Methods 0.000 claims description 5
- 238000010353 genetic engineering Methods 0.000 claims description 5
- 230000005017 genetic modification Effects 0.000 claims description 5
- 235000013617 genetically modified food Nutrition 0.000 claims description 5
- 239000000651 prodrug Chemical group 0.000 claims description 5
- 229940002612 prodrug Drugs 0.000 claims description 5
- 125000005865 C2-C10alkynyl group Chemical group 0.000 claims description 4
- 101001014220 Monascus pilosus Dehydrogenase mokE Proteins 0.000 claims description 4
- 101000573542 Penicillium citrinum Compactin nonaketide synthase, enoyl reductase component Proteins 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 208000018556 stomach disease Diseases 0.000 claims description 2
- 125000006729 (C2-C5) alkenyl group Chemical group 0.000 claims 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims 4
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims 2
- 241000171345 Escallonia rubra Species 0.000 claims 2
- 230000018044 dehydration Effects 0.000 claims 2
- 238000006297 dehydration reaction Methods 0.000 claims 2
- OSFBJERFMQCEQY-UHFFFAOYSA-N propylidene Chemical compound [CH]CC OSFBJERFMQCEQY-UHFFFAOYSA-N 0.000 claims 2
- 238000012258 culturing Methods 0.000 claims 1
- 201000010099 disease Diseases 0.000 abstract description 6
- 230000001771 impaired effect Effects 0.000 abstract description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 66
- -1 lactone compound Chemical class 0.000 description 37
- 239000000047 product Substances 0.000 description 34
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- URLVCROWVOSNPT-XOTOMLERSA-N (2s)-4-[(13r)-13-hydroxy-13-[(2r,5r)-5-[(2r,5r)-5-[(1r)-1-hydroxyundecyl]oxolan-2-yl]oxolan-2-yl]tridecyl]-2-methyl-2h-furan-5-one Chemical compound O1[C@@H]([C@H](O)CCCCCCCCCC)CC[C@@H]1[C@@H]1O[C@@H]([C@H](O)CCCCCCCCCCCCC=2C(O[C@@H](C)C=2)=O)CC1 URLVCROWVOSNPT-XOTOMLERSA-N 0.000 description 22
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- QUHYUSAHBDACNG-UHFFFAOYSA-N acerogenin 3 Natural products C1=CC(O)=CC=C1CCCCC(=O)CCC1=CC=C(O)C=C1 QUHYUSAHBDACNG-UHFFFAOYSA-N 0.000 description 19
- URLVCROWVOSNPT-QTTMQESMSA-N desacetyluvaricin Natural products O=C1C(CCCCCCCCCCCC[C@@H](O)[C@H]2O[C@@H]([C@@H]3O[C@@H]([C@@H](O)CCCCCCCCCC)CC3)CC2)=C[C@H](C)O1 URLVCROWVOSNPT-QTTMQESMSA-N 0.000 description 19
- 239000000243 solution Substances 0.000 description 19
- 230000033001 locomotion Effects 0.000 description 17
- 210000002784 stomach Anatomy 0.000 description 17
- 230000008569 process Effects 0.000 description 16
- 239000011347 resin Substances 0.000 description 16
- 229920005989 resin Polymers 0.000 description 16
- IDRYSCOQVVUBIJ-PPGFLMPOSA-N erythromycin B Chemical group O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@H]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)C)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 IDRYSCOQVVUBIJ-PPGFLMPOSA-N 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- 241000187560 Saccharopolyspora Species 0.000 description 13
- 238000010520 demethylation reaction Methods 0.000 description 13
- 239000002585 base Substances 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 229930006677 Erythromycin A Natural products 0.000 description 11
- 238000010828 elution Methods 0.000 description 10
- 101150086043 eryA gene Proteins 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- IDRYSCOQVVUBIJ-UHFFFAOYSA-N Erythromycin-B Natural products CC1C(OC2C(C(CC(C)O2)N(C)C)O)C(C)(O)CC(C)C(=O)C(C)C(O)C(C)C(CC)OC(=O)C(C)C1OC1CC(C)(OC)C(O)C(C)O1 IDRYSCOQVVUBIJ-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 150000002596 lactones Chemical class 0.000 description 9
- 206010021518 Impaired gastric emptying Diseases 0.000 description 8
- 240000008042 Zea mays Species 0.000 description 8
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 8
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 8
- 230000001580 bacterial effect Effects 0.000 description 8
- 238000005119 centrifugation Methods 0.000 description 8
- 235000005822 corn Nutrition 0.000 description 8
- 238000006471 dimerization reaction Methods 0.000 description 8
- 239000012634 fragment Substances 0.000 description 8
- 208000001288 gastroparesis Diseases 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 235000019439 ethyl acetate Nutrition 0.000 description 7
- 229960001398 flurithromycin Drugs 0.000 description 7
- 239000006260 foam Substances 0.000 description 7
- 208000021302 gastroesophageal reflux disease Diseases 0.000 description 7
- 235000013372 meat Nutrition 0.000 description 7
- 235000014347 soups Nutrition 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 229920001353 Dextrin Polymers 0.000 description 6
- 239000004375 Dextrin Substances 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 6
- 238000000586 desensitisation Methods 0.000 description 6
- 235000019425 dextrin Nutrition 0.000 description 6
- ZFBRGCCVTUPRFQ-HWRKYNCUSA-N erythronolide B Chemical compound CC[C@H]1OC(=O)[C@H](C)[C@@H](O)[C@H](C)[C@@H](O)[C@](C)(O)C[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@H]1C ZFBRGCCVTUPRFQ-HWRKYNCUSA-N 0.000 description 6
- ZFBRGCCVTUPRFQ-UHFFFAOYSA-N erythronolide-B Natural products CCC1OC(=O)C(C)C(O)C(C)C(O)C(C)(O)CC(C)C(=O)C(C)C(O)C1C ZFBRGCCVTUPRFQ-UHFFFAOYSA-N 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- 239000011630 iodine Substances 0.000 description 6
- 238000002156 mixing Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 239000013612 plasmid Substances 0.000 description 6
- 238000002390 rotary evaporation Methods 0.000 description 6
- 101150028073 rplD gene Proteins 0.000 description 6
- 230000001954 sterilising effect Effects 0.000 description 6
- 238000004659 sterilization and disinfection Methods 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 5
- 101100313763 Arabidopsis thaliana TIM22-2 gene Proteins 0.000 description 5
- 206010003591 Ataxia Diseases 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 229950002013 berythromycin Drugs 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 239000003120 macrolide antibiotic agent Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 235000012424 soybean oil Nutrition 0.000 description 5
- 239000003549 soybean oil Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 102000057413 Motilin receptors Human genes 0.000 description 4
- 108700040483 Motilin receptors Proteins 0.000 description 4
- 241000187747 Streptomyces Species 0.000 description 4
- 206010047700 Vomiting Diseases 0.000 description 4
- 230000000845 anti-microbial effect Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 230000000875 corresponding effect Effects 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 230000017858 demethylation Effects 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 230000008673 vomiting Effects 0.000 description 4
- HQZOLNNEQAKEHT-UHFFFAOYSA-N (3R,4S,5R,6S,7S,9R,11R,12S,13R,14R)-14-ethyl-4,6,12-trihydroxy-3,5,7,9,11,13-hexamethyloxacyclotetradecane-2,10-dione Natural products CCC1OC(=O)C(C)C(O)C(C)C(O)C(C)CC(C)C(=O)C(C)C(O)C1C HQZOLNNEQAKEHT-UHFFFAOYSA-N 0.000 description 3
- YGXCJEZVERXINS-JCTYMORFSA-N (3R,4S,5S,6R,7R,9R,10S,11S,12R,13S,14R)-6-[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-14-ethyl-7,10,12,13-tetrahydroxy-4-[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-3,5,7,9,11,13-hexamethyl-oxacyclotetradecan-2-one Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)[C@@H](O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 YGXCJEZVERXINS-JCTYMORFSA-N 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- JFVYXJKGJMUGRG-KJPZRSJGSA-N Erythromycin a enol ether Chemical group O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C2=C(C)C[C@](O2)(C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 JFVYXJKGJMUGRG-KJPZRSJGSA-N 0.000 description 3
- 201000005081 Intestinal Pseudo-Obstruction Diseases 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 230000002354 daily effect Effects 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 201000006549 dyspepsia Diseases 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 150000002084 enol ethers Chemical class 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 238000000855 fermentation Methods 0.000 description 3
- 230000004151 fermentation Effects 0.000 description 3
- 210000000232 gallbladder Anatomy 0.000 description 3
- 238000005227 gel permeation chromatography Methods 0.000 description 3
- KDCIHNCMPUBDKT-UHFFFAOYSA-N hexane;propan-2-one Chemical compound CC(C)=O.CCCCCC KDCIHNCMPUBDKT-UHFFFAOYSA-N 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 230000033444 hydroxylation Effects 0.000 description 3
- 238000005805 hydroxylation reaction Methods 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 210000005070 sphincter Anatomy 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000002769 thiazolinyl group Chemical group 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- KQRXEQGYUDDPNW-QXRSSOOUSA-N (e)-but-2-enedioic acid;(2s,4r,5r,8r,9s,10s,11r,12r)-5-ethyl-9-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-11-[(2s,3r,4s,6r)-3-hydroxy-6-methyl-4-[methyl(propan-2-yl)amino]oxan-2-yl]oxy-4-methoxy-2,4,8,10,12,14-hexamethyl-6,15-dioxabicycl Chemical compound OC(=O)\C=C\C(O)=O.O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@](C(=O)[C@@H](C)C2=C(C)C[C@](O2)(C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C(C)C)O)[C@H]1C)(C)OC)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1.O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@](C(=O)[C@@H](C)C2=C(C)C[C@](O2)(C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C(C)C)O)[C@H]1C)(C)OC)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 KQRXEQGYUDDPNW-QXRSSOOUSA-N 0.000 description 2
- BDKLKNJTMLIAFE-UHFFFAOYSA-N 2-(3-fluorophenyl)-1,3-oxazole-4-carbaldehyde Chemical compound FC1=CC=CC(C=2OC=C(C=O)N=2)=C1 BDKLKNJTMLIAFE-UHFFFAOYSA-N 0.000 description 2
- DIOQKPOBSJVSJS-UHFFFAOYSA-N 3,6-Dideoxy-3-dimethylamino-beta-D-glucose Natural products CC1OC(O)C(O)C(N(C)C)C1O DIOQKPOBSJVSJS-UHFFFAOYSA-N 0.000 description 2
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- VRJHQPZVIGNGMX-UHFFFAOYSA-N 4-piperidinone Chemical compound O=C1CCNCC1 VRJHQPZVIGNGMX-UHFFFAOYSA-N 0.000 description 2
- 241000186361 Actinobacteria <class> Species 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- WGLYHYWDYPSNPF-RQFIXDHTSA-N Apramycin sulfate Chemical compound OS(O)(=O)=O.O([C@H]1O[C@@H]2[C@H](O)[C@@H]([C@H](O[C@H]2C[C@H]1N)O[C@@H]1[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O1)O)NC)[C@@H]1[C@@H](N)C[C@@H](N)[C@H](O)[C@H]1O WGLYHYWDYPSNPF-RQFIXDHTSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 206010010774 Constipation Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000007882 Gastritis Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 102000004867 Hydro-Lyases Human genes 0.000 description 2
- 108090001042 Hydro-Lyases Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 206010021333 Ileus paralytic Diseases 0.000 description 2
- IJUPCLYLISRDRA-UHFFFAOYSA-N Mycaminose Natural products CC(O)C(O)C(N(C)C)C(O)C=O IJUPCLYLISRDRA-UHFFFAOYSA-N 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010039710 Scleroderma Diseases 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000187432 Streptomyces coelicolor Species 0.000 description 2
- 241000701955 Streptomyces virus phiC31 Species 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- DPDMMXDBJGCCQC-UHFFFAOYSA-N [Na].[Cl] Chemical compound [Na].[Cl] DPDMMXDBJGCCQC-UHFFFAOYSA-N 0.000 description 2
- 206010000059 abdominal discomfort Diseases 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 2
- 235000011130 ammonium sulphate Nutrition 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229950006334 apramycin Drugs 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 230000001977 ataxic effect Effects 0.000 description 2
- XYOVOXDWRFGKEX-UHFFFAOYSA-N azepine Chemical compound N1C=CC=CC=C1 XYOVOXDWRFGKEX-UHFFFAOYSA-N 0.000 description 2
- 230000003570 biosynthesizing effect Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- DCSUBABJRXZOMT-IRLDBZIGSA-N cisapride Chemical compound C([C@@H]([C@@H](CC1)NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)OC)N1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-IRLDBZIGSA-N 0.000 description 2
- 229960005132 cisapride Drugs 0.000 description 2
- DCSUBABJRXZOMT-UHFFFAOYSA-N cisapride Natural products C1CC(NC(=O)C=2C(=CC(N)=C(Cl)C=2)OC)C(OC)CN1CCCOC1=CC=C(F)C=C1 DCSUBABJRXZOMT-UHFFFAOYSA-N 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000009849 deactivation Effects 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 229930193775 erythronolide Natural products 0.000 description 2
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 208000024798 heartburn Diseases 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 238000011081 inoculation Methods 0.000 description 2
- 239000002054 inoculum Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 235000012204 lemonade/lime carbonate Nutrition 0.000 description 2
- 229940041033 macrolides Drugs 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 230000004118 muscle contraction Effects 0.000 description 2
- IJUPCLYLISRDRA-ULAWRXDQSA-N mycaminose Chemical compound C[C@@H](O)[C@@H](O)[C@H](N(C)C)[C@@H](O)C=O IJUPCLYLISRDRA-ULAWRXDQSA-N 0.000 description 2
- MHWLWQUZZRMNGJ-UHFFFAOYSA-N nalidixic acid Chemical class C1=C(C)N=C2N(CC)C=C(C(O)=O)C(=O)C2=C1 MHWLWQUZZRMNGJ-UHFFFAOYSA-N 0.000 description 2
- 230000003533 narcotic effect Effects 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- 201000007620 paralytic ileus Diseases 0.000 description 2
- 230000002572 peristaltic effect Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 208000024981 pyrosis Diseases 0.000 description 2
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 229940087562 sodium acetate trihydrate Drugs 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 229930188070 thiostrepton Natural products 0.000 description 2
- 238000009423 ventilation Methods 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- UTTLXGDLSXWDJI-OTELOYJSSA-N (2r,3r,4s,5r,8r,9s,10s,11r,12s)-5-ethyl-11-[(2s,3r,4s,6r)-4-[ethyl(methyl)amino]-3-hydroxy-6-methyloxan-2-yl]oxy-3,4-dihydroxy-9-[(2r,4r,5s,6s)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy-2,4,8,10,12,14-hexamethyl-6,15-dioxabicyclo[10.2.1]pentadec-1(14) Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C2=C(C)C[C@@](O2)(C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)CC)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 UTTLXGDLSXWDJI-OTELOYJSSA-N 0.000 description 1
- IWTSXJNGTTXMFK-KTQUSEMZSA-N (2r,3s,4r,5r,8r,9s,10s,11r,12r)-5-ethyl-11-[(2s,3r,4s,6r)-4-[ethyl(methyl)amino]-3-hydroxy-6-methyloxan-2-yl]oxy-3-hydroxy-9-[(2s,4s,6s)-4-methoxy-4,6-dimethyloxan-2-yl]oxy-2,4,8,10,12,14-hexamethyl-6,15-dioxabicyclo[10.2.1]pentadec-1(14)-en-7-one Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@H]([C@H](O)[C@@H](C)C2=C(C)C[C@](O2)(C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)CC)O)[C@H]1C)C)CC)[C@H]1C[C@@](C)(OC)C[C@H](C)O1 IWTSXJNGTTXMFK-KTQUSEMZSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- KFXDYZXVIHNBFD-UHFFFAOYSA-N 2-(thian-2-ylsulfonyl)thiane Chemical compound C1CCCSC1S(=O)(=O)C1CCCCS1 KFXDYZXVIHNBFD-UHFFFAOYSA-N 0.000 description 1
- IQRUSQUYPCHEKN-UHFFFAOYSA-N 2-iodobutane Chemical compound CCC(C)I IQRUSQUYPCHEKN-UHFFFAOYSA-N 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- SUBDBMMJDZJVOS-UHFFFAOYSA-N 5-methoxy-2-{[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl}-1H-benzimidazole Chemical compound N=1C2=CC(OC)=CC=C2NC=1S(=O)CC1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-UHFFFAOYSA-N 0.000 description 1
- LVRVABPNVHYXRT-BQWXUCBYSA-N 52906-92-0 Chemical compound C([C@H](N)C(=O)N[C@H](C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(O)=O)C(C)C)C1=CC=CC=C1 LVRVABPNVHYXRT-BQWXUCBYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- FVQADOUNIPIICR-UHFFFAOYSA-N C1=CN=C(C(=N1)S(=O)(=O)C2=NC=CN=C2S(=O)(=O)O)S(=O)(=O)O Chemical compound C1=CN=C(C(=N1)S(=O)(=O)C2=NC=CN=C2S(=O)(=O)O)S(=O)(=O)O FVQADOUNIPIICR-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- ZOYWWAGVGBSJDL-UHFFFAOYSA-N D-desosamine Natural products CC1CC(N(C)C)C(O)C(O)O1 ZOYWWAGVGBSJDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- DKMROQRQHGEIOW-UHFFFAOYSA-N Diethyl succinate Chemical compound CCOC(=O)CCC(=O)OCC DKMROQRQHGEIOW-UHFFFAOYSA-N 0.000 description 1
- 206010063655 Erosive oesophagitis Diseases 0.000 description 1
- MWFRKHPRXPSWNT-UHFFFAOYSA-N Erythromycin-C Natural products CC1C(OC2C(C(CC(C)O2)N(C)C)O)C(C)(O)CC(C)C(=O)C(C)C(O)C(O)(C)C(CC)OC(=O)C(C)C1OC1CC(C)(O)C(O)C(C)O1 MWFRKHPRXPSWNT-UHFFFAOYSA-N 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 206010017918 Gastroenteritis viral Diseases 0.000 description 1
- 206010052105 Gastrointestinal hypomotility Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000218924 Micromonospora melanospora Species 0.000 description 1
- 102000002419 Motilin Human genes 0.000 description 1
- 101800002372 Motilin Proteins 0.000 description 1
- 208000016285 Movement disease Diseases 0.000 description 1
- YQLFLCVNXSPEKQ-UHFFFAOYSA-N Mycarose Natural products CC1OC(O)CC(C)(O)C1O YQLFLCVNXSPEKQ-UHFFFAOYSA-N 0.000 description 1
- 125000004633 N-oxo-pyridyl group Chemical group 0.000 description 1
- 206010061876 Obstruction Diseases 0.000 description 1
- 241000315040 Omura Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 239000001888 Peptone Substances 0.000 description 1
- 108010080698 Peptones Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000235070 Saccharomyces Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 241000187559 Saccharopolyspora erythraea Species 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 241000186988 Streptomyces antibioticus Species 0.000 description 1
- 241001655322 Streptomycetales Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NSFFHOGKXHRQEW-UHFFFAOYSA-N Thiostrepton B Natural products N1C(=O)C(C)NC(=O)C(=C)NC(=O)C(C)NC(=O)C(C(C)CC)NC(C(C2=N3)O)C=CC2=C(C(C)O)C=C3C(=O)OC(C)C(C=2SC=C(N=2)C2N=3)NC(=O)C(N=4)=CSC=4C(C(C)(O)C(C)O)NC(=O)C(N=4)CSC=4C(=CC)NC(=O)C(C(C)O)NC(=O)C(N=4)=CSC=4C21CCC=3C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 238000007171 acid catalysis Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 125000005236 alkanoylamino group Chemical group 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960003403 betaine hydrochloride Drugs 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- HOPSCVCBEOCPJZ-UHFFFAOYSA-N carboxymethyl(trimethyl)azanium;chloride Chemical compound [Cl-].C[N+](C)(C)CC(O)=O HOPSCVCBEOCPJZ-UHFFFAOYSA-N 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 150000005676 cyclic carbonates Chemical class 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- VTJCSBJRQLZNHE-CSMHCCOUSA-N desosamine Chemical compound C[C@@H](O)C[C@H](N(C)C)[C@@H](O)C=O VTJCSBJRQLZNHE-CSMHCCOUSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- AIUDWMLXCFRVDR-UHFFFAOYSA-N dimethyl 2-(3-ethyl-3-methylpentyl)propanedioate Chemical class CCC(C)(CC)CCC(C(=O)OC)C(=O)OC AIUDWMLXCFRVDR-UHFFFAOYSA-N 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- DGXRZJSPDXZJFG-UHFFFAOYSA-N docosanedicarboxylic acid Natural products OC(=O)CCCCCCCCCCCCCCCCCCCCC(O)=O DGXRZJSPDXZJFG-UHFFFAOYSA-N 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- CLQUUOKNEOQBSW-KEGKUKQHSA-N erythromycin D Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@H]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)C)CC)[C@H]1C[C@@](C)(O)[C@@H](O)[C@H](C)O1 CLQUUOKNEOQBSW-KEGKUKQHSA-N 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 238000003810 ethyl acetate extraction Methods 0.000 description 1
- OBNCKNCVKJNDBV-UHFFFAOYSA-N ethyl butyrate Chemical compound CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 1
- FKRCODPIKNYEAC-UHFFFAOYSA-N ethyl propionate Chemical compound CCOC(=O)CC FKRCODPIKNYEAC-UHFFFAOYSA-N 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 230000003137 locomotive effect Effects 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- JYAQWANEOPJVEY-LYFYHCNISA-N mycarose Chemical compound C[C@H](O)[C@H](O)[C@](C)(O)CC=O JYAQWANEOPJVEY-LYFYHCNISA-N 0.000 description 1
- 229960000210 nalidixic acid Drugs 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000002981 neuropathic effect Effects 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000006916 nutrient agar Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229940049547 paraxin Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 235000019319 peptone Nutrition 0.000 description 1
- 229940066779 peptones Drugs 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000008855 peristalsis Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229930001119 polyketide Natural products 0.000 description 1
- 150000003881 polyketide derivatives Chemical class 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229940089505 prilosec Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000002325 prokinetic agent Substances 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 238000011218 seed culture Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- RPLOPBHEZLFENN-HTMVYDOJSA-M sodium;4-[(2r,3r)-2-[(2,2-dichloroacetyl)amino]-3-hydroxy-3-(4-nitrophenyl)propoxy]-4-oxobutanoate Chemical compound [Na+].[O-]C(=O)CCC(=O)OC[C@@H](NC(=O)C(Cl)Cl)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 RPLOPBHEZLFENN-HTMVYDOJSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 208000027765 speech disease Diseases 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000005622 tetraalkylammonium hydroxides Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 125000001730 thiiranyl group Chemical group 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- WCNFFKHKJLERFM-UHFFFAOYSA-N thiomorpholinyl sulfone group Chemical group N1(CCSCC1)S(=O)(=O)N1CCSCC1 WCNFFKHKJLERFM-UHFFFAOYSA-N 0.000 description 1
- ZCAGUOCUDGWENZ-UHFFFAOYSA-N thiomorpholinyl sulfoxide group Chemical group N1(CCSCC1)S(=O)N1CCSCC1 ZCAGUOCUDGWENZ-UHFFFAOYSA-N 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- NSFFHOGKXHRQEW-AIHSUZKVSA-N thiostrepton Chemical compound C([C@]12C=3SC=C(N=3)C(=O)N[C@H](C(=O)NC(/C=3SC[C@@H](N=3)C(=O)N[C@H](C=3SC=C(N=3)C(=O)N[C@H](C=3SC=C(N=3)[C@H]1N=1)[C@@H](C)OC(=O)C3=CC(=C4C=C[C@H]([C@@H](C4=N3)O)N[C@H](C(N[C@@H](C)C(=O)NC(=C)C(=O)N[C@@H](C)C(=O)N2)=O)[C@@H](C)CC)[C@H](C)O)[C@](C)(O)[C@@H](C)O)=C\C)[C@@H](C)O)CC=1C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-AIHSUZKVSA-N 0.000 description 1
- 229940063214 thiostrepton Drugs 0.000 description 1
- NSFFHOGKXHRQEW-OFMUQYBVSA-N thiostrepton A Natural products CC[C@H](C)[C@@H]1N[C@@H]2C=Cc3c(cc(nc3[C@H]2O)C(=O)O[C@H](C)[C@@H]4NC(=O)c5csc(n5)[C@@H](NC(=O)[C@H]6CSC(=N6)C(=CC)NC(=O)[C@@H](NC(=O)c7csc(n7)[C@]8(CCC(=N[C@@H]8c9csc4n9)c%10nc(cs%10)C(=O)NC(=C)C(=O)NC(=C)C(=O)N)NC(=O)[C@H](C)NC(=O)C(=C)NC(=O)[C@H](C)NC1=O)[C@@H](C)O)[C@](C)(O)[C@@H](C)O)[C@H](C)O NSFFHOGKXHRQEW-OFMUQYBVSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 239000006216 vaginal suppository Substances 0.000 description 1
- 229940120293 vaginal suppository Drugs 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 239000002912 waste gas Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/06—Anti-spasmodics, e.g. drugs for colics, esophagic dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/44—Preparation of O-glycosides, e.g. glucosides
- C12P19/60—Preparation of O-glycosides, e.g. glucosides having an oxygen of the saccharide radical directly bound to a non-saccharide heterocyclic ring or a condensed ring system containing a non-saccharide heterocyclic ring, e.g. coumermycin, novobiocin
- C12P19/62—Preparation of O-glycosides, e.g. glucosides having an oxygen of the saccharide radical directly bound to a non-saccharide heterocyclic ring or a condensed ring system containing a non-saccharide heterocyclic ring, e.g. coumermycin, novobiocin the hetero ring having eight or more ring members and only oxygen as ring hetero atoms, e.g. erythromycin, spiramycin, nystatin
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Public Health (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Hospice & Palliative Care (AREA)
- Otolaryngology (AREA)
- Nutrition Science (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Cephalosporin Compounds (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description


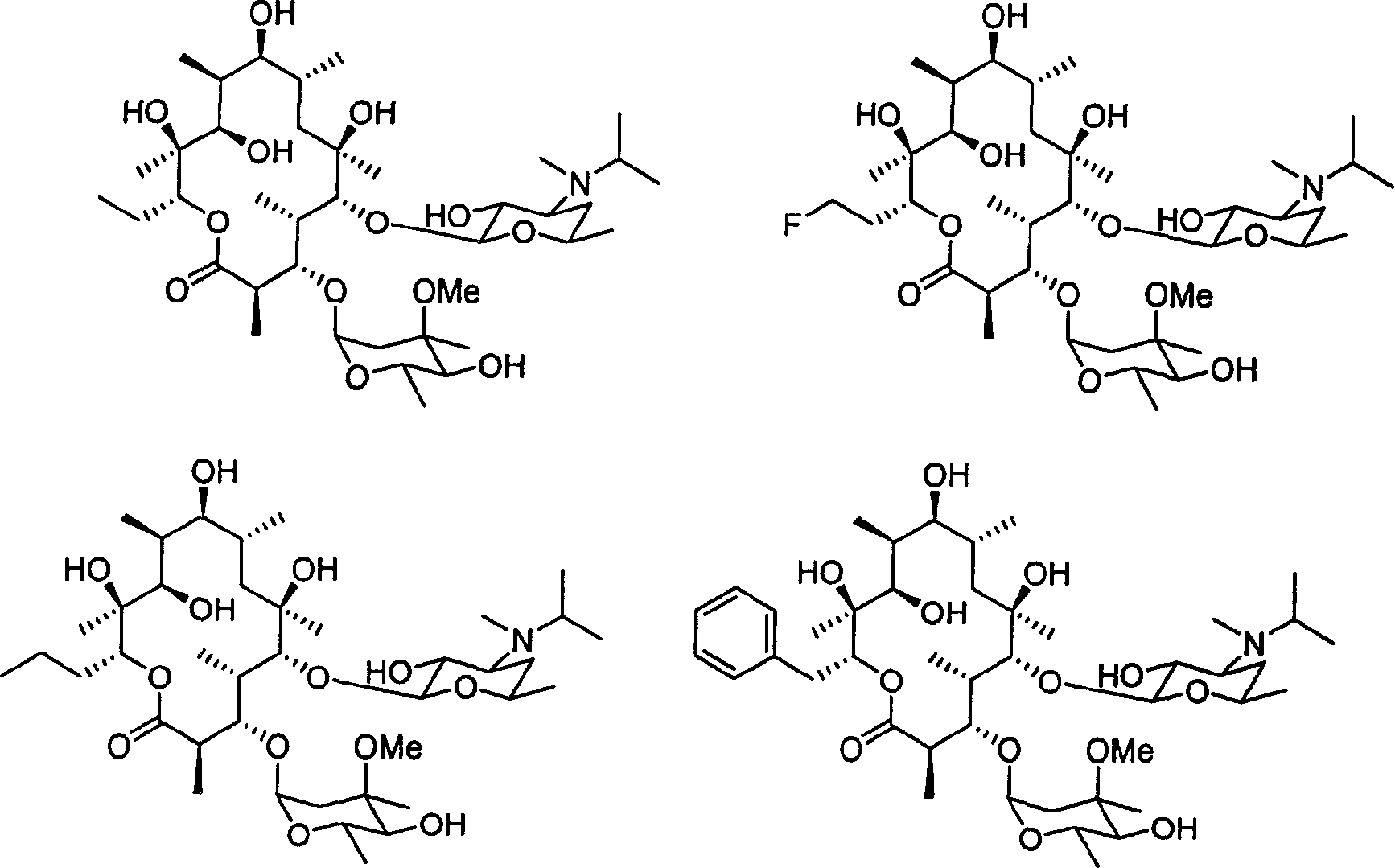


| 表1促胃动素受体活性 | |||||
| 化合物编号 | R1 | R2 | R3 | R4 | EC50(μM) |
| 红霉素A1A1BCDEFGHJ | CH3CH2CH3CH2CH3CH2FCH2CH2FCH2CH2FCH2CH2CH3CH2CH2CH3CH2CH2CH3CH2CH2CH3CH2 | CH3CH3CH(CH3)2CH3CH2CH3CH(CH3)2CH3CH(CH3)2C(CH3)CH2CH3CH(CH3)2 | OHOHOHOHOHOHOHOHOHH | OHOHOHOHOHOHOHOHOHH | 1.12.50.49520.528.61.43.30.16 |
| 表2对于ATCC 6301的体内最小抑制浓度 | |
| 化合物 | MIC(μg/mL) |
| 红霉素AAFGJ | 0.030.31>100>100 |
| 表3肠胃动素受体去敏作用 | ||
| 化合物 | 暴露给化合物后的活性(保留的原始活性的%) | |
| 1μM化合物 | 10μM化合物 | |
| F | 100 | 100 |
| G | 100 | 100 |
| J | 88 | 88 |


Claims (20)





Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US40734502P | 2002-08-29 | 2002-08-29 | |
| US60/407,345 | 2002-08-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1665799A true CN1665799A (zh) | 2005-09-07 |
| CN100363359C CN100363359C (zh) | 2008-01-23 |
Family
ID=31978464
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB038150530A Expired - Fee Related CN100363359C (zh) | 2002-08-29 | 2003-08-26 | 运动内酯化合物 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US6946482B2 (zh) |
| EP (1) | EP1532131B1 (zh) |
| JP (1) | JP4796300B2 (zh) |
| KR (1) | KR101051613B1 (zh) |
| CN (1) | CN100363359C (zh) |
| AT (1) | ATE417856T1 (zh) |
| AU (1) | AU2003273254B2 (zh) |
| CA (1) | CA2492846C (zh) |
| CY (1) | CY1108808T1 (zh) |
| DE (1) | DE60325377D1 (zh) |
| DK (1) | DK1532131T3 (zh) |
| ES (1) | ES2316805T3 (zh) |
| PT (1) | PT1532131E (zh) |
| SI (1) | SI1532131T1 (zh) |
| WO (1) | WO2004019879A2 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016173308A1 (zh) * | 2015-04-28 | 2016-11-03 | 天津国际生物医药联合研究院 | 大环内酯类衍生物及其用途 |
| CN106478541A (zh) * | 2016-10-17 | 2017-03-08 | 南开大学 | 大环内酯类衍生物及其用途 |
| CN106478542A (zh) * | 2016-10-17 | 2017-03-08 | 南开大学 | 一种大环内脂类衍生物盐、其制备方法及用途 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8063021B2 (en) | 2002-01-17 | 2011-11-22 | Kosan Biosciences Incorporated | Ketolide anti-infective compounds |
| US7407941B2 (en) * | 2003-08-26 | 2008-08-05 | Pfizer, Inc. | N-desmethyl-N-substituted-11-deoxyerythromycin compounds |
| GB0327720D0 (en) * | 2003-11-28 | 2003-12-31 | Biotica Tech Ltd | Erythromycins and process for their preparation |
| US7211568B2 (en) * | 2003-12-18 | 2007-05-01 | Kosan Biosciences Incorporated | 9-Desoxoerythromycin compounds as prokinetic agents |
| US7468428B2 (en) * | 2004-03-17 | 2008-12-23 | App Pharmaceuticals, Llc | Lyophilized azithromycin formulation |
| US20060116336A1 (en) * | 2004-03-17 | 2006-06-01 | American Pharmaceutical Partners, Inc. | Lyophilized azithromycin formulation |
| US7582611B2 (en) | 2005-05-24 | 2009-09-01 | Pfizer Inc. | Motilide compounds |
| UA87569C2 (ru) * | 2005-05-24 | 2009-07-27 | Пфайзер Инк. | Соединения для лечения расстройств желудочно-кишечной моторики |
| US20070135362A1 (en) * | 2005-12-08 | 2007-06-14 | Yaoquan Liu | Method for demethylating the 3'-dimethylamino group of erythromycin compounds |
| EP2054429B1 (en) | 2006-09-11 | 2013-11-06 | Tranzyme Pharma, Inc. | Macrocyclic antagonists of the motilin receptor for treatment of gastrointestinal dysmotility disorders |
| BRPI0719565A2 (pt) * | 2006-12-05 | 2013-12-10 | Pfizer | Polimorfos de motilídeo |
| US20080287371A1 (en) * | 2007-05-17 | 2008-11-20 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor for modulation of the migrating motor complex |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US391594A (en) * | 1888-10-23 | Arc-extinguisher for electric switches or cut-outs | ||
| US4382085A (en) | 1982-03-01 | 1983-05-03 | Pfizer Inc. | 4"-Epi erythromycin A and derivatives thereof as useful antibacterial agents |
| US4382086A (en) | 1982-03-01 | 1983-05-03 | Pfizer Inc. | 9-Dihydro-11,12-ketal derivatives of erythromycin A and epi-erythromycin A |
| PL136968B1 (en) | 1983-02-10 | 1986-04-30 | Tarchominskie Zaklad Farma | Process for preparing cyclic 11,12-carbonate of erythromycin a |
| JPS60218321A (ja) * | 1984-04-13 | 1985-11-01 | Satoshi Omura | 消化管収縮運動促進剤 |
| EP0184921A3 (en) * | 1984-12-08 | 1986-10-29 | Beecham Group Plc | Erythromycin derivatives |
| GB8608080D0 (en) | 1986-04-02 | 1986-05-08 | Fujisawa Pharmaceutical Co | Solid dispersion composition |
| US4742049A (en) * | 1986-06-04 | 1988-05-03 | Abbott Laboratories | Semisynthetic erythromycin antibiotics |
| US4857641A (en) | 1987-08-14 | 1989-08-15 | Pfizer Inc. | C.12 modified erythromycin A derivatives |
| ATE128714T1 (de) | 1989-03-28 | 1995-10-15 | Abbott Lab | Erythromycinderivate. |
| US5190871A (en) | 1989-06-12 | 1993-03-02 | Eli Lilly And Company | Use of the site-specific integrating function of phage φC31 |
| US5824513A (en) | 1991-01-17 | 1998-10-20 | Abbott Laboratories | Recombinant DNA method for producing erythromycin analogs |
| US5523418A (en) | 1991-04-09 | 1996-06-04 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| AU663089B2 (en) | 1991-04-09 | 1995-09-28 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| DK0623021T3 (da) | 1992-01-21 | 1999-02-08 | Abbott Lab | 4"-deoxyerythromycinderivater |
| MY113693A (en) | 1992-05-26 | 2002-05-31 | Chugai Pharmaceutical Co Ltd | Erythromycin derivatives having an enterokinesis stimulating action |
| US6066721A (en) | 1995-07-06 | 2000-05-23 | Stanford University | Method to produce novel polyketides |
| US5712146A (en) | 1993-09-20 | 1998-01-27 | The Leland Stanford Junior University | Recombinant combinatorial genetic library for the production of novel polyketides |
| EP1493814A1 (en) | 1995-07-06 | 2005-01-05 | The Leland Stanford Junior University | Cell-free synthesis of polyketides |
| US6077943A (en) | 1996-03-01 | 2000-06-20 | Takeda Chemical Industries, Ltd. | Method of producing erythromycin derivative |
| WO1997035590A1 (en) * | 1996-03-25 | 1997-10-02 | Platt Chris E | 10-aza-9-deoxo-11-deoxy-erythromycin a and derivatives combined with sulfisoxazole |
| US5712253A (en) | 1996-06-18 | 1998-01-27 | Abbott Laboratories | Macrocyclic 13-membered ring derivatives of erythromycins A and B |
| US6271255B1 (en) | 1996-07-05 | 2001-08-07 | Biotica Technology Limited | Erythromycins and process for their preparation |
| US5922849A (en) | 1996-11-22 | 1999-07-13 | Abbott Laboratories | Process for preparation of N-demethyl-4"-deoxy-erthromycins A and B |
| WO1998040392A1 (fr) * | 1997-03-10 | 1998-09-17 | Taisho Pharmaceutical Co., Ltd. | Derives d'erythromycine a |
| US6169168B1 (en) | 1997-10-29 | 2001-01-02 | Taisho Pharmaceuticals Co., Ltd. | Erythromycin A derivatives |
| WO1999021867A1 (fr) | 1997-10-29 | 1999-05-06 | Taisho Pharmaceutical Co., Ltd. | Derives d'erythromycine a |
| US6084079A (en) | 1998-05-15 | 2000-07-04 | Keyes; Robert F. | Process for preparing N-demethyl-N-alkyl erythromycin derivatives |
| AU1447700A (en) | 1998-10-28 | 2000-05-15 | Kosan Biosciences, Inc. | Library of novel "unnatural" natural products |
| EP1124969A2 (en) | 1998-10-29 | 2001-08-22 | Kosan Biosciences, Inc. | Recombinant oleandolide polyketide synthase |
| JP2002535387A (ja) | 1999-01-27 | 2002-10-22 | コーサン バイオサイエンシーズ, インコーポレイテッド | オリゴケチドの合成 |
| CN1241931C (zh) | 1999-04-16 | 2006-02-15 | 高山生物科学股份有限公司 | 大环内酯抗感染药物 |
| US6524841B1 (en) | 1999-10-08 | 2003-02-25 | Kosan Biosciences, Inc. | Recombinant megalomicin biosynthetic genes and uses thereof |
| IL148934A0 (en) | 1999-10-27 | 2002-09-12 | Kosan Biosciences Inc | Heterologous production of polyketides |
| CA2399198A1 (en) | 2000-02-18 | 2001-08-23 | Christopher Carreras | Motilide compounds |
| JP2005529579A (ja) | 2000-05-02 | 2005-10-06 | コーサン バイオサイエンシーズ, インコーポレイテッド | ポリケチドの生合成のための過剰産生宿主 |
| CA2429709A1 (en) * | 2000-12-01 | 2002-07-04 | Gary Ashley | Motilide compounds |
| US20020094962A1 (en) | 2000-12-01 | 2002-07-18 | Gary Ashley | Motilide compounds |
| CA2437576A1 (en) | 2001-02-15 | 2002-08-22 | Kosan Biosciences, Inc. | Method for evaluating therapeutic efficacy |
| CA2451391A1 (en) * | 2001-07-03 | 2003-01-16 | Chiron Corporation | C12 modified erythromycin macrolides and ketolides having antibacterial activity |
| ITMI20021726A1 (it) | 2002-08-01 | 2004-02-02 | Zambon Spa | Macrolidi ad attivita' antiinfiammatoria. |
-
2003
- 2003-08-26 KR KR1020047021161A patent/KR101051613B1/ko not_active IP Right Cessation
- 2003-08-26 SI SI200331492T patent/SI1532131T1/sl unknown
- 2003-08-26 JP JP2004531645A patent/JP4796300B2/ja not_active Expired - Fee Related
- 2003-08-26 DK DK03755757T patent/DK1532131T3/da active
- 2003-08-26 PT PT03755757T patent/PT1532131E/pt unknown
- 2003-08-26 US US10/648,946 patent/US6946482B2/en not_active Expired - Lifetime
- 2003-08-26 DE DE60325377T patent/DE60325377D1/de not_active Expired - Lifetime
- 2003-08-26 CN CNB038150530A patent/CN100363359C/zh not_active Expired - Fee Related
- 2003-08-26 AU AU2003273254A patent/AU2003273254B2/en not_active Ceased
- 2003-08-26 CA CA2492846A patent/CA2492846C/en not_active Expired - Fee Related
- 2003-08-26 AT AT03755757T patent/ATE417856T1/de active
- 2003-08-26 WO PCT/US2003/026991 patent/WO2004019879A2/en active Application Filing
- 2003-08-26 EP EP03755757A patent/EP1532131B1/en not_active Expired - Lifetime
- 2003-08-26 ES ES03755757T patent/ES2316805T3/es not_active Expired - Lifetime
-
2009
- 2009-02-13 CY CY20091100165T patent/CY1108808T1/el unknown
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016173308A1 (zh) * | 2015-04-28 | 2016-11-03 | 天津国际生物医药联合研究院 | 大环内酯类衍生物及其用途 |
| CN106138038A (zh) * | 2015-04-28 | 2016-11-23 | 天津国际生物医药联合研究院 | 大环内酯类衍生物及其用途 |
| CN106138038B (zh) * | 2015-04-28 | 2021-04-20 | 天津国际生物医药联合研究院 | 大环内酯类衍生物及其用途 |
| CN106478541A (zh) * | 2016-10-17 | 2017-03-08 | 南开大学 | 大环内酯类衍生物及其用途 |
| CN106478542A (zh) * | 2016-10-17 | 2017-03-08 | 南开大学 | 一种大环内脂类衍生物盐、其制备方法及用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1532131A2 (en) | 2005-05-25 |
| CA2492846A1 (en) | 2004-03-11 |
| WO2004019879A3 (en) | 2004-06-03 |
| EP1532131A4 (en) | 2007-05-30 |
| JP4796300B2 (ja) | 2011-10-19 |
| EP1532131B1 (en) | 2008-12-17 |
| AU2003273254B2 (en) | 2008-10-30 |
| PT1532131E (pt) | 2009-02-18 |
| ATE417856T1 (de) | 2009-01-15 |
| CA2492846C (en) | 2012-09-25 |
| DK1532131T3 (da) | 2009-02-09 |
| CN100363359C (zh) | 2008-01-23 |
| ES2316805T3 (es) | 2009-04-16 |
| KR20050038594A (ko) | 2005-04-27 |
| DE60325377D1 (de) | 2009-01-29 |
| US6946482B2 (en) | 2005-09-20 |
| SI1532131T1 (sl) | 2009-04-30 |
| WO2004019879A8 (en) | 2004-07-29 |
| US20040138150A1 (en) | 2004-07-15 |
| AU2003273254A1 (en) | 2004-03-19 |
| JP2005537317A (ja) | 2005-12-08 |
| WO2004019879A2 (en) | 2004-03-11 |
| CY1108808T1 (el) | 2014-04-09 |
| KR101051613B1 (ko) | 2011-07-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1665799A (zh) | 运动内酯化合物 | |
| KR100851418B1 (ko) | 에포틸론 및 에포틸론 유도체의 생산을 위한 재조합 방법 및 물질 | |
| EA001744B1 (ru) | Новые эритромицины и способы их получения | |
| CN1284081A (zh) | 新的大环内酯类化合物 | |
| KR20010021933A (ko) | 신규한 폴리케타이드의 제조방법 | |
| EP1303615A2 (en) | Fermentation process for epothilones | |
| CN1351608A (zh) | 13-甲基红霉素衍生物 | |
| EP1307579A2 (en) | Bio-intermediates for use in the chemical synthesis of polyketides | |
| CN1326460A (zh) | 十三元氮杂大环内酯类化合物及其作为抗生剂的应用 | |
| AU2001238476A1 (en) | Motocide compounds | |
| CN1225096A (zh) | 溴代台勾霉素化合物 | |
| CN103102396B (zh) | 新型硫链丝菌素类似物及其制法和用途 | |
| US6939861B2 (en) | Amido macrolides | |
| CN105777870A (zh) | 新型硫链丝菌素类似物及其制法和用途 | |
| CA2533583C (en) | N-desmethyl-n-substituted-11-deoxyerythromycin compounds | |
| WO2003061671A1 (en) | Amido macrolides | |
| US20090275594A1 (en) | 3-hydrazone piperazinyl rifamycin derivatives useful as antimicrobial agents | |
| AU2001279025B2 (en) | Fermentation process for epothilones | |
| AU2001279025A1 (en) | Fermentation process for epothilones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: PHISAI CO., LTD. Free format text: FORMER OWNER: KOSAN BIOSCIENCES INC. Effective date: 20070413 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20070413 Address after: American New York Applicant after: Pfizer Inc. Address before: American California Applicant before: Kosan Biosciences, Inc. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20080123 Termination date: 20150826 |
|
| EXPY | Termination of patent right or utility model |