WO2003061671A1 - Amido macrolides - Google Patents
Amido macrolides Download PDFInfo
- Publication number
- WO2003061671A1 WO2003061671A1 PCT/US2003/001398 US0301398W WO03061671A1 WO 2003061671 A1 WO2003061671 A1 WO 2003061671A1 US 0301398 W US0301398 W US 0301398W WO 03061671 A1 WO03061671 A1 WO 03061671A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- compound
- formula
- compounds
- Prior art date
Links
- 239000003120 macrolide antibiotic agent Substances 0.000 title abstract description 10
- 229940041033 macrolides Drugs 0.000 title description 6
- 125000003368 amide group Chemical group 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 254
- 238000000034 method Methods 0.000 claims description 122
- 125000003545 alkoxy group Chemical group 0.000 claims description 39
- 125000000217 alkyl group Chemical group 0.000 claims description 34
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 29
- 125000003342 alkenyl group Chemical group 0.000 claims description 27
- 125000000304 alkynyl group Chemical group 0.000 claims description 27
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 26
- 125000003118 aryl group Chemical group 0.000 claims description 26
- 125000005018 aryl alkenyl group Chemical group 0.000 claims description 21
- 125000005015 aryl alkynyl group Chemical group 0.000 claims description 21
- 150000003839 salts Chemical class 0.000 claims description 18
- 241000894006 Bacteria Species 0.000 claims description 13
- 125000003302 alkenyloxy group Chemical group 0.000 claims description 12
- 125000005133 alkynyloxy group Chemical group 0.000 claims description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 11
- 208000035143 Bacterial infection Diseases 0.000 claims description 10
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 10
- 201000010099 disease Diseases 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 241000606768 Haemophilus influenzae Species 0.000 claims description 7
- 230000030135 gastric motility Effects 0.000 claims description 7
- 239000008194 pharmaceutical composition Substances 0.000 claims description 7
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 208000021302 gastroesophageal reflux disease Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 5
- 229940047650 haemophilus influenzae Drugs 0.000 claims description 5
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 3
- 206010001076 Acute sinusitis Diseases 0.000 claims description 3
- 206010006458 Bronchitis chronic Diseases 0.000 claims description 3
- 206010021518 Impaired gastric emptying Diseases 0.000 claims description 3
- 206010024971 Lower respiratory tract infections Diseases 0.000 claims description 3
- 201000009906 Meningitis Diseases 0.000 claims description 3
- 241000588655 Moraxella catarrhalis Species 0.000 claims description 3
- 206010029803 Nosocomial infection Diseases 0.000 claims description 3
- 201000007100 Pharyngitis Diseases 0.000 claims description 3
- 206010035664 Pneumonia Diseases 0.000 claims description 3
- 206010054048 Postoperative ileus Diseases 0.000 claims description 3
- 206010057190 Respiratory tract infections Diseases 0.000 claims description 3
- 206010062255 Soft tissue infection Diseases 0.000 claims description 3
- 241000191967 Staphylococcus aureus Species 0.000 claims description 3
- 241000194017 Streptococcus Species 0.000 claims description 3
- 206010046306 Upper respiratory tract infection Diseases 0.000 claims description 3
- 230000001154 acute effect Effects 0.000 claims description 3
- 206010006451 bronchitis Diseases 0.000 claims description 3
- 208000007451 chronic bronchitis Diseases 0.000 claims description 3
- 206010012601 diabetes mellitus Diseases 0.000 claims description 3
- 208000001288 gastroparesis Diseases 0.000 claims description 3
- 206010040872 skin infection Diseases 0.000 claims description 3
- 206010044008 tonsillitis Diseases 0.000 claims description 3
- 241001148471 unidentified anaerobic bacterium Species 0.000 claims description 3
- 206010060968 Arthritis infective Diseases 0.000 claims description 2
- 241000192125 Firmicutes Species 0.000 claims description 2
- 206010031252 Osteomyelitis Diseases 0.000 claims description 2
- 241000193998 Streptococcus pneumoniae Species 0.000 claims description 2
- 229940031000 streptococcus pneumoniae Drugs 0.000 claims description 2
- 125000002861 (C1-C4) alkanoyl group Chemical group 0.000 claims 2
- 125000001589 carboacyl group Chemical group 0.000 claims 2
- 241000193996 Streptococcus pyogenes Species 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 119
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 119
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 111
- 239000000243 solution Substances 0.000 description 110
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 102
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Natural products O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 75
- 229960003276 erythromycin Drugs 0.000 description 72
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 71
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 70
- 239000000203 mixture Substances 0.000 description 70
- -1 hemisuccinates Chemical class 0.000 description 66
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 60
- 229930006677 Erythromycin A Natural products 0.000 description 58
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 55
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 43
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 41
- 235000019439 ethyl acetate Nutrition 0.000 description 41
- 239000000047 product Substances 0.000 description 39
- 229910001868 water Inorganic materials 0.000 description 39
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 38
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 38
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 36
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 35
- 239000012267 brine Substances 0.000 description 29
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 29
- 239000007787 solid Substances 0.000 description 27
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 26
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 23
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 23
- 238000002360 preparation method Methods 0.000 description 23
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 23
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 19
- 230000015572 biosynthetic process Effects 0.000 description 19
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 19
- 229930001119 polyketide Natural products 0.000 description 19
- 238000010898 silica gel chromatography Methods 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 18
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 18
- 239000000284 extract Substances 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 17
- 125000000830 polyketide group Chemical group 0.000 description 17
- 0 CC(CC(*)C12)OC1*2=C([C@](C)[C@@](C(C)C(O[C@](CCNC(Cc1c[n]c2ccccc12)=*)[C@](C)(C([C@@](C)C([C@](C)C1)=*)O)O)=O)OC(CC2(C)OC)OC(C)C2O)[C@]1(C)O Chemical compound CC(CC(*)C12)OC1*2=C([C@](C)[C@@](C(C)C(O[C@](CCNC(Cc1c[n]c2ccccc12)=*)[C@](C)(C([C@@](C)C([C@](C)C1)=*)O)O)=O)OC(CC2(C)OC)OC(C)C2O)[C@]1(C)O 0.000 description 16
- 229960000583 acetic acid Drugs 0.000 description 16
- 239000002253 acid Substances 0.000 description 16
- 235000017557 sodium bicarbonate Nutrition 0.000 description 16
- KDCIHNCMPUBDKT-UHFFFAOYSA-N hexane;propan-2-one Chemical compound CC(C)=O.CCCCCC KDCIHNCMPUBDKT-UHFFFAOYSA-N 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 14
- 239000007832 Na2SO4 Substances 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 14
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 14
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 14
- 229910052938 sodium sulfate Inorganic materials 0.000 description 14
- 108010030975 Polyketide Synthases Proteins 0.000 description 13
- 239000012074 organic phase Substances 0.000 description 13
- 239000011347 resin Substances 0.000 description 12
- 229920005989 resin Polymers 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 11
- 238000004519 manufacturing process Methods 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- 150000007970 thio esters Chemical class 0.000 description 11
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- 235000011054 acetic acid Nutrition 0.000 description 10
- 125000002252 acyl group Chemical group 0.000 description 10
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 10
- 238000000855 fermentation Methods 0.000 description 10
- 230000004151 fermentation Effects 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 235000011152 sodium sulphate Nutrition 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 9
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 9
- ZFBRGCCVTUPRFQ-UHFFFAOYSA-N erythronolide-B Natural products CCC1OC(=O)C(C)C(O)C(C)C(O)C(C)(O)CC(C)C(=O)C(C)C(O)C1C ZFBRGCCVTUPRFQ-UHFFFAOYSA-N 0.000 description 9
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 9
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- 150000007513 acids Chemical class 0.000 description 8
- 239000007822 coupling agent Substances 0.000 description 8
- 229930193775 erythronolide Natural products 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000002609 medium Substances 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- 229910052763 palladium Inorganic materials 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- IIIRAXWPUAYFPM-UHFFFAOYSA-N 2-[3-(furan-2-yl)phenyl]acetic acid Chemical compound OC(=O)CC1=CC=CC(C=2OC=CC=2)=C1 IIIRAXWPUAYFPM-UHFFFAOYSA-N 0.000 description 7
- FFDNRFMIOVQZMT-UHFFFAOYSA-N 3-chloropropanal Chemical compound ClCCC=O FFDNRFMIOVQZMT-UHFFFAOYSA-N 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- 238000007865 diluting Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 239000003835 ketolide antibiotic agent Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- HQZOLNNEQAKEHT-UHFFFAOYSA-N (3R,4S,5R,6S,7S,9R,11R,12S,13R,14R)-14-ethyl-4,6,12-trihydroxy-3,5,7,9,11,13-hexamethyloxacyclotetradecane-2,10-dione Natural products CCC1OC(=O)C(C)C(O)C(C)C(O)C(C)CC(C)C(=O)C(C)C(O)C1C HQZOLNNEQAKEHT-UHFFFAOYSA-N 0.000 description 6
- HRNGDAQBEIFYGL-UHFFFAOYSA-N 3,4-dihydroxy-4-tetradeca-3,6-dienoyloxybutanoic acid Chemical compound CCCCCCCC=CCC=CCC(=O)OC(O)C(O)CC(O)=O HRNGDAQBEIFYGL-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- QLNAVQRIWDRPHA-UHFFFAOYSA-N iminophosphane Chemical compound P=N QLNAVQRIWDRPHA-UHFFFAOYSA-N 0.000 description 6
- CDDKNECSGAGQBS-UHFFFAOYSA-N methyl 2-[3-(furan-2-yl)phenyl]acetate Chemical compound COC(=O)CC1=CC=CC(C=2OC=CC=2)=C1 CDDKNECSGAGQBS-UHFFFAOYSA-N 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 5
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 5
- 241000187559 Saccharopolyspora erythraea Species 0.000 description 5
- 241000187432 Streptomyces coelicolor Species 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 150000001540 azides Chemical class 0.000 description 5
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 5
- 238000005119 centrifugation Methods 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 229940102396 methyl bromide Drugs 0.000 description 5
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 5
- 150000002923 oximes Chemical class 0.000 description 5
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 5
- 238000000638 solvent extraction Methods 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- NOGFHTGYPKWWRX-UHFFFAOYSA-N 2,2,6,6-tetramethyloxan-4-one Chemical compound CC1(C)CC(=O)CC(C)(C)O1 NOGFHTGYPKWWRX-UHFFFAOYSA-N 0.000 description 4
- HSJKGGMUJITCBW-UHFFFAOYSA-N 3-hydroxybutanal Chemical compound CC(O)CC=O HSJKGGMUJITCBW-UHFFFAOYSA-N 0.000 description 4
- YHVUVJYEERGYNU-UHFFFAOYSA-N 4',8-Di-Me ether-5,7,8-Trihydroxy-3-(4-hydroxybenzyl)-4-chromanone Natural products COC1(C)CC(O)OC(C)C1O YHVUVJYEERGYNU-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 229920001353 Dextrin Polymers 0.000 description 4
- 239000004375 Dextrin Substances 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 238000013019 agitation Methods 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- AJSDVNKVGFVAQU-BIIVOSGPSA-N cladinose Chemical compound O=CC[C@@](C)(OC)[C@@H](O)[C@H](C)O AJSDVNKVGFVAQU-BIIVOSGPSA-N 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- 235000019425 dextrin Nutrition 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- 125000001475 halogen functional group Chemical group 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 238000006140 methanolysis reaction Methods 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 150000003881 polyketide derivatives Chemical class 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 238000011218 seed culture Methods 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 230000002194 synthesizing effect Effects 0.000 description 4
- PLNTYOACSMHWBN-UHFFFAOYSA-N 1,1-di(propan-2-yloxy)cyclohexane Chemical compound CC(C)OC1(OC(C)C)CCCCC1 PLNTYOACSMHWBN-UHFFFAOYSA-N 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 3
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 3
- YKFRUJSEPGHZFJ-UHFFFAOYSA-N N-trimethylsilylimidazole Chemical compound C[Si](C)(C)N1C=CN=C1 YKFRUJSEPGHZFJ-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- YFFOFFWSBYZSOI-UHFFFAOYSA-N Narbonolid Natural products CCC1OC(=O)C(C)C(=O)C(C)C(O)C(C)CC(C)C(=O)C=CC1C YFFOFFWSBYZSOI-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- NSFFHOGKXHRQEW-UHFFFAOYSA-N Thiostrepton B Natural products N1C(=O)C(C)NC(=O)C(=C)NC(=O)C(C)NC(=O)C(C(C)CC)NC(C(C2=N3)O)C=CC2=C(C(C)O)C=C3C(=O)OC(C)C(C=2SC=C(N=2)C2N=3)NC(=O)C(N=4)=CSC=4C(C(C)(O)C(C)O)NC(=O)C(N=4)CSC=4C(=CC)NC(=O)C(C(C)O)NC(=O)C(N=4)=CSC=4C21CCC=3C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-UHFFFAOYSA-N 0.000 description 3
- 239000003570 air Substances 0.000 description 3
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 210000004671 cell-free system Anatomy 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 108091008053 gene clusters Proteins 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 238000007327 hydrogenolysis reaction Methods 0.000 description 3
- 230000033444 hydroxylation Effects 0.000 description 3
- 238000005805 hydroxylation reaction Methods 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 3
- YFFOFFWSBYZSOI-HQWJGCFGSA-N narbonolide Chemical compound CC[C@H]1OC(=O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)C[C@@H](C)C(=O)\C=C\[C@H]1C YFFOFFWSBYZSOI-HQWJGCFGSA-N 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 229930188070 thiostrepton Natural products 0.000 description 3
- NSFFHOGKXHRQEW-AIHSUZKVSA-N thiostrepton Chemical compound C([C@]12C=3SC=C(N=3)C(=O)N[C@H](C(=O)NC(/C=3SC[C@@H](N=3)C(=O)N[C@H](C=3SC=C(N=3)C(=O)N[C@H](C=3SC=C(N=3)[C@H]1N=1)[C@@H](C)OC(=O)C3=CC(=C4C=C[C@H]([C@@H](C4=N3)O)N[C@H](C(N[C@@H](C)C(=O)NC(=C)C(=O)N[C@@H](C)C(=O)N2)=O)[C@@H](C)CC)[C@H](C)O)[C@](C)(O)[C@@H](C)O)=C\C)[C@@H](C)O)CC=1C1=NC(C(=O)NC(=C)C(=O)NC(=C)C(N)=O)=CS1 NSFFHOGKXHRQEW-AIHSUZKVSA-N 0.000 description 3
- 229940063214 thiostrepton Drugs 0.000 description 3
- NSFFHOGKXHRQEW-OFMUQYBVSA-N thiostrepton A Natural products CC[C@H](C)[C@@H]1N[C@@H]2C=Cc3c(cc(nc3[C@H]2O)C(=O)O[C@H](C)[C@@H]4NC(=O)c5csc(n5)[C@@H](NC(=O)[C@H]6CSC(=N6)C(=CC)NC(=O)[C@@H](NC(=O)c7csc(n7)[C@]8(CCC(=N[C@@H]8c9csc4n9)c%10nc(cs%10)C(=O)NC(=C)C(=O)NC(=C)C(=O)N)NC(=O)[C@H](C)NC(=O)C(=C)NC(=O)[C@H](C)NC1=O)[C@@H](C)O)[C@](C)(O)[C@@H](C)O)[C@H](C)O NSFFHOGKXHRQEW-OFMUQYBVSA-N 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- 125000006737 (C6-C20) arylalkyl group Chemical group 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- WZOFMKRJXYOGCK-UHFFFAOYSA-N 2-(4-pyridin-3-ylimidazol-1-yl)acetic acid Chemical compound OC(=O)CN1C=NC(C=2C=NC=CC=2)=C1 WZOFMKRJXYOGCK-UHFFFAOYSA-N 0.000 description 2
- TZKFFLQSOIKYTC-UHFFFAOYSA-N 2-(furan-2-yl)-2-phenylacetic acid Chemical compound C=1C=CC=CC=1C(C(=O)O)C1=CC=CO1 TZKFFLQSOIKYTC-UHFFFAOYSA-N 0.000 description 2
- QSKPIOLLBIHNAC-UHFFFAOYSA-N 2-chloro-acetaldehyde Chemical compound ClCC=O QSKPIOLLBIHNAC-UHFFFAOYSA-N 0.000 description 2
- GQOJGBVTUSRBPQ-UHFFFAOYSA-N 2-quinolin-3-ylacetic acid Chemical compound C1=CC=CC2=CC(CC(=O)O)=CN=C21 GQOJGBVTUSRBPQ-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- VBWYZPGRKYRKNV-UHFFFAOYSA-N 3-propanoyl-1,3-benzoxazol-2-one Chemical compound C1=CC=C2OC(=O)N(C(=O)CC)C2=C1 VBWYZPGRKYRKNV-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 2
- HQZOLNNEQAKEHT-IBBGRPSASA-N 6-deoxyerythronolide B Chemical compound CC[C@H]1OC(=O)[C@H](C)[C@@H](O)[C@H](C)[C@@H](O)[C@@H](C)C[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@H]1C HQZOLNNEQAKEHT-IBBGRPSASA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 238000006237 Beckmann rearrangement reaction Methods 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- 238000005675 Corey-Kim oxidation reaction Methods 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- LLQPHQFNMLZJMP-UHFFFAOYSA-N Fentrazamide Chemical compound N1=NN(C=2C(=CC=CC=2)Cl)C(=O)N1C(=O)N(CC)C1CCCCC1 LLQPHQFNMLZJMP-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 102000008109 Mixed Function Oxygenases Human genes 0.000 description 2
- 108010074633 Mixed Function Oxygenases Proteins 0.000 description 2
- YQLFLCVNXSPEKQ-UHFFFAOYSA-N Mycarose Natural products CC1OC(O)CC(C)(O)C1O YQLFLCVNXSPEKQ-UHFFFAOYSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 238000006069 Suzuki reaction reaction Methods 0.000 description 2
- 240000008042 Zea mays Species 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- 150000001241 acetals Chemical class 0.000 description 2
- 239000003377 acid catalyst Substances 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 150000001350 alkyl halides Chemical class 0.000 description 2
- 150000008052 alkyl sulfonates Chemical class 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 2
- 235000011130 ammonium sulphate Nutrition 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 150000001543 aryl boronic acids Chemical class 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 2
- 229960004099 azithromycin Drugs 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 230000001851 biosynthetic effect Effects 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- XNRHTMDHGDWBGP-UHFFFAOYSA-N carbamic acid;hydrochloride Chemical compound Cl.NC(O)=O XNRHTMDHGDWBGP-UHFFFAOYSA-N 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 description 2
- 238000007360 debenzoylation reaction Methods 0.000 description 2
- 125000004663 dialkyl amino group Chemical group 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 101150117988 eryK gene Proteins 0.000 description 2
- VRZVPALEJCLXPR-UHFFFAOYSA-N ethyl 4-methylbenzenesulfonate Chemical compound CCOS(=O)(=O)C1=CC=C(C)C=C1 VRZVPALEJCLXPR-UHFFFAOYSA-N 0.000 description 2
- UVECLJDRPFNRRQ-UHFFFAOYSA-N ethyl trifluoromethanesulfonate Chemical compound CCOS(=O)(=O)C(F)(F)F UVECLJDRPFNRRQ-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 235000013312 flour Nutrition 0.000 description 2
- 238000003682 fluorination reaction Methods 0.000 description 2
- 238000005187 foaming Methods 0.000 description 2
- PZJSZBJLOWMDRG-UHFFFAOYSA-N furan-2-ylboronic acid Chemical compound OB(O)C1=CC=CO1 PZJSZBJLOWMDRG-UHFFFAOYSA-N 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000000640 hydroxylating effect Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- SEOVTRFCIGRIMH-UHFFFAOYSA-N indole-3-acetic acid Chemical compound C1=CC=C2C(CC(=O)O)=CNC2=C1 SEOVTRFCIGRIMH-UHFFFAOYSA-N 0.000 description 2
- KMAKOBLIOCQGJP-UHFFFAOYSA-N indole-3-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CNC2=C1 KMAKOBLIOCQGJP-UHFFFAOYSA-N 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical group 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 2
- 230000001035 methylating effect Effects 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 238000007069 methylation reaction Methods 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- JYAQWANEOPJVEY-LYFYHCNISA-N mycarose Chemical compound C[C@H](O)[C@H](O)[C@](C)(O)CC=O JYAQWANEOPJVEY-LYFYHCNISA-N 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 2
- PFDLUBNRHMFBGI-HRVFELILSA-N oleandolide Chemical compound O=C1[C@H](C)[C@@H](O)[C@@H](C)[C@@H](C)OC(=O)[C@H](C)[C@@H](O)[C@H](C)[C@@H](O)[C@@H](C)C[C@]11OC1 PFDLUBNRHMFBGI-HRVFELILSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- 150000004714 phosphonium salts Chemical class 0.000 description 2
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 2
- 238000012809 post-inoculation Methods 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- VQMSRUREDGBWKT-UHFFFAOYSA-N quinoline-4-carboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=NC2=C1 VQMSRUREDGBWKT-UHFFFAOYSA-N 0.000 description 2
- 238000010188 recombinant method Methods 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- GRZHHTYDZVRPIC-UHFFFAOYSA-N (benzyloxy)acetic acid Chemical compound OC(=O)COCC1=CC=CC=C1 GRZHHTYDZVRPIC-UHFFFAOYSA-N 0.000 description 1
- SCZNXLWKYFICFV-UHFFFAOYSA-N 1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound C1CCCNN2CCCC=C21 SCZNXLWKYFICFV-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 1
- JJJRVOAWTBMQAR-UHFFFAOYSA-N 2-(2-thiophen-2-ylphenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC=C1C1=CC=CS1 JJJRVOAWTBMQAR-UHFFFAOYSA-N 0.000 description 1
- KYNNBXCGXUOREX-UHFFFAOYSA-N 2-(3-bromophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1 KYNNBXCGXUOREX-UHFFFAOYSA-N 0.000 description 1
- MMAHKERVPDCCJG-UHFFFAOYSA-N 2-(3-pyridin-4-ylphenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(C=2C=CN=CC=2)=C1 MMAHKERVPDCCJG-UHFFFAOYSA-N 0.000 description 1
- XDUGISXCDRTJLK-UHFFFAOYSA-N 2-(3-thiophen-3-ylphenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(C2=CSC=C2)=C1 XDUGISXCDRTJLK-UHFFFAOYSA-N 0.000 description 1
- KPZPARSXURXOHY-UHFFFAOYSA-N 2-(4-methyl-5-phenyl-1,3-oxazol-2-yl)acetic acid Chemical compound N1=C(CC(O)=O)OC(C=2C=CC=CC=2)=C1C KPZPARSXURXOHY-UHFFFAOYSA-N 0.000 description 1
- QQKKTOPRRGBBCT-UHFFFAOYSA-N 2-(5-chloro-1-benzothiophen-3-yl)acetic acid Chemical compound C1=C(Cl)C=C2C(CC(=O)O)=CSC2=C1 QQKKTOPRRGBBCT-UHFFFAOYSA-N 0.000 description 1
- QCXJFLREQGIACT-UHFFFAOYSA-N 2-(6-methoxy-1-benzofuran-3-yl)acetic acid Chemical compound COC1=CC=C2C(CC(O)=O)=COC2=C1 QCXJFLREQGIACT-UHFFFAOYSA-N 0.000 description 1
- QRUHYAWZHFTNEA-UHFFFAOYSA-N 2-(furan-2-yl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1C1=CC=CO1 QRUHYAWZHFTNEA-UHFFFAOYSA-N 0.000 description 1
- AWVWPEMJYQLBGQ-UHFFFAOYSA-N 2-[2-(furan-2-yl)phenyl]acetic acid Chemical compound OC(=O)CC1=CC=CC=C1C1=CC=CO1 AWVWPEMJYQLBGQ-UHFFFAOYSA-N 0.000 description 1
- URHUEWWRTFKARN-UHFFFAOYSA-N 2-[3-(furan-3-yl)phenyl]acetic acid Chemical compound OC(=O)CC1=CC=CC(C2=COC=C2)=C1 URHUEWWRTFKARN-UHFFFAOYSA-N 0.000 description 1
- LDEMYLVVZJLDFL-UHFFFAOYSA-N 2-[4-(4-chlorophenyl)phenyl]acetic acid Chemical compound C1=CC(CC(=O)O)=CC=C1C1=CC=C(Cl)C=C1 LDEMYLVVZJLDFL-UHFFFAOYSA-N 0.000 description 1
- JQMFQLVAJGZSQS-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JQMFQLVAJGZSQS-UHFFFAOYSA-N 0.000 description 1
- WAKFRZBXTKUFIW-UHFFFAOYSA-N 2-bromo-2-phenylacetic acid Chemical compound OC(=O)C(Br)C1=CC=CC=C1 WAKFRZBXTKUFIW-UHFFFAOYSA-N 0.000 description 1
- PZGXJYSPQYRCBB-UHFFFAOYSA-N 2-chlorobutanal Chemical group CCC(Cl)C=O PZGXJYSPQYRCBB-UHFFFAOYSA-N 0.000 description 1
- LADJHQSUVNTXES-UHFFFAOYSA-N 2-oxo-2-(3,4,5-trimethoxyphenyl)acetic acid Chemical compound COC1=CC(C(=O)C(O)=O)=CC(OC)=C1OC LADJHQSUVNTXES-UHFFFAOYSA-N 0.000 description 1
- MBXSRGURWIZVDM-UHFFFAOYSA-N 2-phenyl-2-(1h-pyrrol-2-yl)acetic acid Chemical compound C=1C=CC=CC=1C(C(=O)O)C1=CC=CN1 MBXSRGURWIZVDM-UHFFFAOYSA-N 0.000 description 1
- QIMHOHNDXHYFIP-UHFFFAOYSA-N 2-phenyl-2-thiophen-2-ylacetic acid Chemical compound C=1C=CC=CC=1C(C(=O)O)C1=CC=CS1 QIMHOHNDXHYFIP-UHFFFAOYSA-N 0.000 description 1
- HZKNCFYAVVPIGV-UHFFFAOYSA-N 2-quinolin-3-ylpropanoic acid Chemical compound C1=CC=CC2=CC(C(C(O)=O)C)=CN=C21 HZKNCFYAVVPIGV-UHFFFAOYSA-N 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- YFOKBFRTGLSZLU-UHFFFAOYSA-N 3-(1h-imidazol-5-yl)pyridine Chemical compound N1C=NC=C1C1=CC=CN=C1 YFOKBFRTGLSZLU-UHFFFAOYSA-N 0.000 description 1
- GYSMCWFKLRPSTA-UHFFFAOYSA-N 3-(5-pyridin-2-ylthiophen-2-yl)propanoic acid Chemical compound S1C(CCC(=O)O)=CC=C1C1=CC=CC=N1 GYSMCWFKLRPSTA-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- ZGIKWINFUGEQEO-UHFFFAOYSA-N 3-bromoquinoline Chemical compound C1=CC=CC2=CC(Br)=CN=C21 ZGIKWINFUGEQEO-UHFFFAOYSA-N 0.000 description 1
- FIMRRWLTRBEAOM-UHFFFAOYSA-N 4-(4-chlorophenyl)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(Cl)C=C1 FIMRRWLTRBEAOM-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- DOQLCJMCQWQQHK-UHFFFAOYSA-N 4-chlorobutanal Chemical compound ClCCCC=O DOQLCJMCQWQQHK-UHFFFAOYSA-N 0.000 description 1
- BYHDDXPKOZIZRV-UHFFFAOYSA-N 5-phenylpentanoic acid Chemical compound OC(=O)CCCCC1=CC=CC=C1 BYHDDXPKOZIZRV-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- VTIKDEXOEJDMJP-UHFFFAOYSA-N Actinorhodine Natural products CC1OC(CC(=O)O)CC2=C1C(=O)c3c(O)c(cc(O)c3C2=O)c4cc(O)c5C(=O)C6=C(C(C)OC(CC(=O)O)C6)C(=O)c5c4O VTIKDEXOEJDMJP-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 101100313763 Arabidopsis thaliana TIM22-2 gene Proteins 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010005940 Bone and joint infections Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 238000009631 Broth culture Methods 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- IFTVVFCNXZPXHE-LOJMCQKSSA-N C[C@@H](C[C@@H](C)C(O[C@H](CCl)C[C@@H]([C@@H](C)C([C@H](C)C1)=O)O)=O)C[C@@H]1O Chemical compound C[C@@H](C[C@@H](C)C(O[C@H](CCl)C[C@@H]([C@@H](C)C([C@H](C)C1)=O)O)=O)C[C@@H]1O IFTVVFCNXZPXHE-LOJMCQKSSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- ZOYWWAGVGBSJDL-UHFFFAOYSA-N D-desosamine Natural products CC1CC(N(C)C)C(O)C(O)O1 ZOYWWAGVGBSJDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 241000606790 Haemophilus Species 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930190833 Megalomicin Natural products 0.000 description 1
- LRWRQTMTYVZKQW-WWDNQWNISA-N Megalomicin A Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)O[C@@H]1O[C@@H](C)[C@H](O)[C@@H](C1)N(C)C)(C)O)CC)[C@H]1C[C@@](C)(O)[C@@H](O)[C@H](C)O1 LRWRQTMTYVZKQW-WWDNQWNISA-N 0.000 description 1
- 241000276489 Merlangius merlangus Species 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000218923 Micromonospora megalomicea Species 0.000 description 1
- 102000006833 Multifunctional Enzymes Human genes 0.000 description 1
- 108010047290 Multifunctional Enzymes Proteins 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 125000004633 N-oxo-pyridyl group Chemical group 0.000 description 1
- GDIHDSKOVXEJQQ-UHFFFAOYSA-N NC(O)=O.NC(O)=O.NC(O)=O.N Chemical compound NC(O)=O.NC(O)=O.NC(O)=O.N GDIHDSKOVXEJQQ-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 101100168477 Saccharopolyspora erythraea (strain ATCC 11635 / DSM 40517 / JCM 4748 / NBRC 13426 / NCIMB 8594 / NRRL 2338) eryF gene Proteins 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 235000019764 Soybean Meal Nutrition 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 241000187398 Streptomyces lividans Species 0.000 description 1
- 241000946831 Streptomyces narbonensis Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 238000006859 Swern oxidation reaction Methods 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 125000005236 alkanoylamino group Chemical group 0.000 description 1
- 125000004947 alkyl aryl amino group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 238000009635 antibiotic susceptibility testing Methods 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 125000001691 aryl alkyl amino group Chemical group 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 150000001499 aryl bromides Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- JPNZKPRONVOMLL-UHFFFAOYSA-N azane;octadecanoic acid Chemical class [NH4+].CCCCCCCCCCCCCCCCCC([O-])=O JPNZKPRONVOMLL-UHFFFAOYSA-N 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004602 benzodiazinyl group Chemical group N1=NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 150000001559 benzoic acids Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004600 benzothiopyranyl group Chemical group S1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 238000006480 benzoylation reaction Methods 0.000 description 1
- 125000004599 benzpyrazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000005841 biaryl group Chemical group 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- QRZAKQDHEVVFRX-UHFFFAOYSA-N biphenyl-4-ylacetic acid Chemical compound C1=CC(CC(=O)O)=CC=C1C1=CC=CC=C1 QRZAKQDHEVVFRX-UHFFFAOYSA-N 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 238000002815 broth microdilution Methods 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000007036 catalytic synthesis reaction Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000004617 chromonyl group Chemical group O1C(=CC(C2=CC=CC=C12)=O)* 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 125000000332 coumarinyl group Chemical group O1C(=O)C(=CC2=CC=CC=C12)* 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000005366 cycloalkylthio group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000003386 deoximation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- VTJCSBJRQLZNHE-CSMHCCOUSA-N desosamine Chemical compound C[C@@H](O)C[C@H](N(C)C)[C@@H](O)C=O VTJCSBJRQLZNHE-CSMHCCOUSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 125000004986 diarylamino group Chemical group 0.000 description 1
- NFKGQHYUYGYHIS-UHFFFAOYSA-N dibutyl propanedioate Chemical compound CCCCOC(=O)CC(=O)OCCCC NFKGQHYUYGYHIS-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 101150042354 eryF gene Proteins 0.000 description 1
- ZFBRGCCVTUPRFQ-HWRKYNCUSA-N erythronolide B Chemical class CC[C@H]1OC(=O)[C@H](C)[C@@H](O)[C@H](C)[C@@H](O)[C@](C)(O)C[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@H]1C ZFBRGCCVTUPRFQ-HWRKYNCUSA-N 0.000 description 1
- DNHFCULEKISSPX-UHFFFAOYSA-N ethyl 6-chloro-5-fluoro-4-oxo-1h-quinazoline-2-carboxylate Chemical compound C1=C(Cl)C(F)=C2C(=O)NC(C(=O)OCC)=NC2=C1 DNHFCULEKISSPX-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000012025 fluorinating agent Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000001279 glycosylating effect Effects 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000004476 heterocycloamino group Chemical group 0.000 description 1
- 125000004470 heterocyclooxy group Chemical group 0.000 description 1
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical class C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- IENZCGNHSIMFJE-UHFFFAOYSA-N indole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NC=CC2=C1 IENZCGNHSIMFJE-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 150000007931 macrolactones Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229950005761 megalomicin Drugs 0.000 description 1
- 238000003328 mesylation reaction Methods 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 229940087646 methanolamine Drugs 0.000 description 1
- 239000002032 methanolic fraction Substances 0.000 description 1
- NHFBYYMNJUMVOT-UHFFFAOYSA-N methyl 2-bromo-2-phenylacetate Chemical compound COC(=O)C(Br)C1=CC=CC=C1 NHFBYYMNJUMVOT-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- WNZWVZCUFRJIBG-UHFFFAOYSA-N n-(2-sulfanylethyl)propanamide Chemical compound CCC(=O)NCCS WNZWVZCUFRJIBG-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000006225 natural substrate Substances 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- ODUCDPQEXGNKDN-UHFFFAOYSA-N nitroxyl Chemical compound O=N ODUCDPQEXGNKDN-UHFFFAOYSA-N 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000005188 oxoalkyl group Chemical group 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229960003424 phenylacetic acid Drugs 0.000 description 1
- 239000003279 phenylacetic acid Substances 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- CUQOHAYJWVTKDE-UHFFFAOYSA-N potassium;butan-1-olate Chemical compound [K+].CCCC[O-] CUQOHAYJWVTKDE-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- QAQREVBBADEHPA-IEXPHMLFSA-N propionyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 QAQREVBBADEHPA-IEXPHMLFSA-N 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- NRTYMEPCRDJMPZ-UHFFFAOYSA-N pyridine;2,2,2-trifluoroacetic acid Chemical compound C1=CC=NC=C1.OC(=O)C(F)(F)F NRTYMEPCRDJMPZ-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- DJXNJVFEFSWHLY-UHFFFAOYSA-N quinoline-3-carboxylic acid Chemical compound C1=CC=CC2=CC(C(=O)O)=CN=C21 DJXNJVFEFSWHLY-UHFFFAOYSA-N 0.000 description 1
- VXGYRCVTBHVXMZ-UHFFFAOYSA-N quinoline-6-carboxylic acid Chemical compound N1=CC=CC2=CC(C(=O)O)=CC=C21 VXGYRCVTBHVXMZ-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 208000020029 respiratory tract infectious disease Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 239000011369 resultant mixture Substances 0.000 description 1
- YJYCYAISSKFKFN-UHFFFAOYSA-N s-[2-(propanoylamino)ethyl] propanethioate Chemical compound CCC(=O)NCCSC(=O)CC YJYCYAISSKFKFN-UHFFFAOYSA-N 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000004455 soybean meal Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- SWQDRYKDDGFPLL-UHFFFAOYSA-N tert-butyl prop-2-enyl carbonate Chemical compound CC(C)(C)OC(=O)OCC=C SWQDRYKDDGFPLL-UHFFFAOYSA-N 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 238000006227 trimethylsilylation reaction Methods 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- Erythromycins are macrolide antibiotics, glycosylated polyketides originally discovered in 1952 in the metabolic products of a strain of Streptomyces erythreus, now classified as Saccharopolyspora erythraea.
- the antibiotic occurs in various forms, designated A, B, C, and D. Since their discovery, many have worked to prepare derivatives of the molecule to improve or modify its properties. The focus of much of this work involved chemical modification of the naturally produced erythromycin molecule. For example, clarithromycin is a semi-synthetic antibiotic that is made by chemically methylating the hydroxyl group at C-6.
- Azalides such as azithromycin
- erythromycin derivatives where the C-9 ketone has been replaced with a N-CH 2 unit through a Beckmann rearrangement. See, for example, O'Connell et al., "Azalides and methods of making same," U.S. Patent No. 6,270,768, incorporated herein by reference.
- Ketolides are erythromycin derivatives where the C-3 cladinose sugar is chemically removed and the resulting free hydroxyl group converted into a keto group.
- Phan et ah "2-Halo-6-O-substituted ketolide derivatives ”
- U.S. Patent No. 6,124,269 describes ketolides with a cyclic carbamate group at C-ll and C-12 and an O-alkylaryl group at C-6.
- Agouridas et al., "Erythromycin compounds” U.S. Patent No.
- 5,635,485 also describes ketolides with a cyclic carbamate group at C-ll and C-12 but which have a -OMe group at C-6 and an alkylaryl group at the carbamate nitrogen.
- the complexity of the macrolide molecule has limited medicinal chemistry efforts to produce derivatives of the naturally occurring erythromycins and their precursors.
- PKS's modular polyketide synthases
- PKS's are multifunctional enzymes that catalyze the formation of the polyketide chains through repeated reactions between acylthioesters.
- the Sac. erythraea PKS known as 6-deoxyerythronolide B synthase (DEBS)
- DEBS 6-deoxyerythronolide B synthase
- DEBS produces the polyketide macrolactone 6-deoxyerythronolide B, which is processed by additional tailoring enzymes present in Sac. erythraea to make erythromycins A-D.
- the collective assembly of the PKS gene and the genes for the tailoring enzymes are referred to as the biosynthetic gene cluster.
- the organization of the gene cluster is described in Summers et al., "Polyketide-associated sugar biosynthesis genes," U.S. Patent No. 5,998,194, incorporated herein by reference. Recombinant methods using vectors encoding a variety of PKS's, including the PKS from Sac.
- Khosla et al. "Recombinant production of novel polyketides," U.S. Patent Nos. 5,672,491, 5,830,750, 5,962,290, 6,022,731, and 6,077,696; Khosla et al, "Recombinant combinatorial genetic library for the production of novel polyketides," U.S. Patent No. 5,712,146; Khosla et al., “Method to produce novel polyketides," U.S. Patent Nos. 6,066,721, 6,221,641, and 6,261,816; Barr et al., "Production of polyketides in bacteria and yeast," U.S. Patent Nos.
- the present invention fulfills this need by providing novel erythromycins, ketolides, and azalides.
- the invention provides various compounds and pharmaceutically acceptable salts thereof.
- the invention provides compounds and pharmaceutically acceptable salts thereof of the formula I
- R 1 is C Cg substituted or unsubstituted alkyl, C 2 -C 8 substituted or unsubstituted alkenyl, C 2 -C 8 substituted or unsubstituted alkynyl, C -C 15 substituted or unsubstituted aryl, C 5 -C 2 o substituted or unsubstituted arylalkyl, C 5 -C 20 substituted or unsubstituted biarylalkyl, C 5 -C 2 o substituted or unsubstituted arylalkenyl, or C 5 -C o substituted or unsubstituted arylalkynyl;
- R 2 is H, C ⁇ -C substituted or unsubstituted alkyl, C 2 -C 4 substituted or unsubstituted alkenyl, or C 2 -C substituted or unsubstituted alkynyl;
- R 3 is H, C ⁇ -C alkanoyl, or benzoyl;
- R 4 is H or OH
- R 5 is H, OH, C ⁇ -C 4 alkoxy, C 2 -C 4 alkenyloxy, or C 2 -C 4 alkynyloxy;
- the invention provides compounds of formula I where
- R 2 is H and m is 0.
- the invention provides compounds of formula I where R 2 is H and m is 1.
- the invention provides compounds of formula I where R 2 is H and m is 2.
- the invention provides compounds of formula I which have any of the following structures:
- the invention provides compounds of formula II and pharmaceutically acceptable salts thereof
- R is H, C ⁇ -C 4 alkanoyl, or benzoyl
- R 4 is H or OH
- R 5 is H, -C 4 alkoxy, C 2 -C 4 alkenyloxy, or C 2 -C 4 alkynyloxy
- the invention provides compounds of formula II where m is 0. In other embodiments, the invention provides compounds of formula II where m is 1. In yet other embodiments, the invention provides compounds of formula II where m is 2.
- the invention provides compounds of formula II which have any of the following structures:
- the invention provides compounds of formula III and pharmaceutically acceptable salts thereof
- R 1 is C 1 -C 8 substituted or unsubstituted alkyl, C 2 -C 8 substituted or unsubstituted alkenyl, C 2 -C 8 substituted or unsubstituted alkynyl, C 4 -C 15 substituted or unsubstituted aryl, C 5 -C 20 substituted or unsubstituted arylalkyl, C 5 -C 20 substituted or unsubstituted biarylalkyl, C 5 -C 20 substituted or unsubstituted arylalkenyl, or C 5 -C o substituted or unsubstituted arylalkynyl;
- R 2 is H, C C substituted or unsubstituted alkyl, C 2 -C 4 substituted or unsubstituted alkenyl, or C 2 -C 4 substituted or unsubstituted alkynyl;
- R 3 is H, -C 4 alkanoyl, or benzoyl;
- R 4 is H or OH
- R 5 is H, C 1 -C 4 alkoxy, C 2 -C alkenyloxy, or C 2 -C 4 alkynyloxy;
- the invention provides compounds of formula III in which m is 0.
- the invention provides compounds of formula III in which m is 1.
- the invention provides compounds of formula II in which m is 2.
- the invention provides compounds of formula III where R 2 is H and m is 0.
- the invention provides compounds of formula IH where R 2 is H and m is 1.
- the invention provides compounds of formula III where R 2 is H and m is 2.
- the invention provides compounds of formula III in which R 1 is a C -C 15 substituted or unsubstituted aryl, C 5 -C 2 o substituted or unsubstituted arylalkyl, C 5 -C 0 substituted or unsubstituted biarylalkyl, C 5 -C 20 substituted or unsubstituted arylalkenyl, or C 5 -C o substituted or unsubstituted arylalkynyl; and R 2 is H.
- the invention provides compounds of formula IN and pharmaceutically acceptable salts thereof
- R 1 is C C 8 substituted or unsubstituted alkyl, C 2 -C 8 substituted or unsubstituted alkenyl, C 2 -C 8 substituted or unsubstituted alkynyl, C 4 -C ⁇ 5 substituted or unsubstituted aryl, C 5 -C 0 substituted or unsubstituted arylalkyl, C 5 -C o substituted or unsubstituted biarylalkyl, C 5 -C 20 substituted or unsubstituted arylalkenyl, or C 5 -C 2 o substituted or unsubstituted arylalkynyl;
- R 2 is H, C ⁇ -C 4 substituted or unsubstituted alkyl, C 2 -C 4 substituted or unsubstituted alkenyl, or C 2 -C substituted or unsubstituted alkynyl;
- R 3 is H, C ⁇ -C 4 alkanoyl, or benzoyl
- R 5 is H, C ⁇ -C 4 alkoxy, C 2 -C alkenyloxy, or C -C 4 alkynyloxy;
- R 8 is H or F;
- the invention provides compounds of formula IV in which R 2 is H.
- the invention provides compounds of formula IV in which R 1 is C 6 -Ci 5 substituted or unsubstituted aryl, Cs-C 20 substituted or unsubstituted arylalkyl, C 5 -C 20 substituted or unsubstituted biarylalkyl, C 5 -C 2 o substituted or unsubstituted arylalkenyl, or C 5 -C 2 o substituted or unsubstituted arylalkynyl; and R 2 is H.
- the invention provides compounds of formula IV in which R 1 is C 6 -C 15 substituted or unsubstituted aryl, C 5 -C 2 o substituted or unsubstituted arylalkyl, C 5 -C 2 o substituted or unsubstituted biarylalkyl, C 5 -C 20 substituted or unsubstituted arylalkenyl, or C 5 -C 2 o substituted or unsubstituted arylalkynyl; R 2 is H; and X is NH.
- the invention provides compounds of formula IV which have any of the following structures:
- the invention provides compounds of formulas I, III, or IV in which R 1 is a group having any of the following formulas:
- the invention provides compounds of the formula V and pharmaceutically acceptable salts thereof
- R 1 is -C 8 substituted or unsubstituted alkyl, C 2 -C 8 substituted or unsubstituted alkenyl, C 2 -C 8 substituted or unsubstituted alkynyl, C 4 -C 15 substituted or unsubstituted aryl, C 5 -C 20 substituted or unsubstituted arylalkyl, C 5 - 0 substituted or unsubstituted biarylalkyl, C 5 -C 2 o substituted or unsubstituted arylalkenyl, or C 5 -C o substituted or unsubstituted arylalkynyl;
- R 2 is H, -C 4 substituted or unsubstituted alkyl, C 2 -C substituted or unsubstituted alkenyl, or C 2 -C substituted or unsubstituted alkynyl;
- R 4 is H or OH;
- R 6 is OH or OMe;
- the invention provides pharmaceutical formulations and medicaments.
- Such pharmaceutical formulations and medicaments include any of the compounds of the invention and a pharmaceutically acceptable carrier.
- the pharmaceutical formulation or medicament includes any compound of formulae I - V and a pharmaceutically acceptable carrier.
- the invention provides a method of treating a bacterial infection with compounds of the formulae I - V.
- a method of treating a bacterial infection includes administering a compound of formulae I-V to a patient in need thereof.
- the bacterial infection results from Gram positive bacteria, Gram negative bacteria or anaerobic bacteria.
- the bacteria are Staphylococcus aureus, Streptococcus epidermidis, Strep, pneumoniae, Strep, pyogenes, enterococci, Moraxella catarrhalis or Haemophilus influenzae.
- the bacterial infection is community-acquired pneumonia, acute exacerbated chronic bronchitis, acute sinusitis, tonsillitis/pharyngitis, upper respiratory tract infection, lower respiratory tract infection, skin infection, soft tissue infection, meningitis, hospital-acquired infection, bone infection or joint infection.
- the invention provides a method of treating a gastric motility disease.
- the method includes treating a patient with gastric motility disease with a compound of formula I.
- the gastric motility disease is gastro-esophageal reflux disease (GERD), post-operative ileus, diabetes, or gastroparesis.
- Figure 1 shows a variety of 15-amidoerythromycins prepared according to the methods of the invention.
- Figure 2 shows some embodiments of additional 15-amidoerythromycins that can be prepared according to the methods of the invention.
- Figure 3 shows a synthetic procedure that may be used to prepare certain macrolides according to the invention.
- R 1 is as defined in the specification and can include any of the groups shown in Figures 1 and 2.
- the present invention relates to erythromycin derivatives, intermediates thereto, and methods for their use in the treatment of disease.
- inventive compounds possess antibacterial activity against Gram positive, Gram negative, and anaerobic bacteria, and are useful as broad-spectrum antibacterial agents for the treatment of bacterial infections in humans and animals. These compounds are effective against diverse strains including but not limited to Staphylococcus aureus, Streptococcus epiderniidis, Strep, pneumoniae, Strep, pyogenes, enterococci, Moraxella catarrhalis and Haemophilus influenzae.
- Exemplary infections that may be treated include community-acquired pneumonia, acute exacerbated chronic bronchitis, acute sinusitis, tonsillitis/pharyngitis, upper and lower respiratory tract infections, skin and soft tissue infections, meningitis, hospital-acquired infections, and bone and joint infections.
- Certain of the inventive compounds also possess prokinetic activity and are useful in the treatment of diseases of gastric motility, including but not limited to gastro-esophageal reflux disease (GERD), post-operative ileus, diabetes, and gastroparesis.
- GSD gastro-esophageal reflux disease
- crystalline forms for the compounds may exist as polymorphs and as such are intended to be included in the present invention.
- some of the compounds may form solvates with water (i.e., hydrates) or common organic solvents, and such solvates are also encompassed within the scope of this invention.
- the salts of the compounds of this invention refer to nontoxic "pharmaceutically acceptable salts.”
- Other salts may, however, be useful in the preparation of compounds according to this invention or of their pharmaceutically acceptable salts.
- Suitable pharmaceutically acceptable salts of the compounds include acid addition salts which may, for example, be formed by mixing a solution of the compound with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid, glucoheptonic acid, lactobionic acid, and dodecylsulfonic acid.
- a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid, glucoheptonic acid, lactobionic acid, and dode
- the present invention includes within its scope prodrugs of the compounds of this invention.
- prodrugs will be functional derivatives of the compounds that are readily convertible in vivo into the required compound.
- examples of prodrugs of the inventive compounds include but are not limited to 2 '-O-esters such as acetates, propionates, hemisuccinates, stearates, and the like.
- the term "administering" shall encompass the treatment of the various disorders described with the compound specifically disclosed or with a compound which may not be specifically disclosed, but which converts to the specified compound in vivo after administration to the patient. Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
- substituents and substitution patterns on the compounds of this invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art as well as those methods set forth herein.
- substituents include alkyl, alkenyl, alkynyl, aryl, halo, trifluoromethoxy, trifluoromethyl, hydroxy, alkoxy, cycloalkyloxy, heterocyclooxy, alkanoyl, alkanoyloxy, amino, alkylamino, aralkylamino, cycloalkylamino, heterocycloamino, dialkylamino, alkanoylamino, thio, alkylthio, cycloalkylthio, heterocyclothio, ureido, nitro, cyano, carboxy, caroboxylalkyl, carbamyl, alkoxycarbonyl, alkylthiono, arylthiono, alkylsulfonyl, sulfonamindo, aryloxy, and the like, in addition to those otherwise specified herein.
- the substituent may be further substituted, for example, by halo, hydroxy, al
- arylalkylcarboxamido refers to a group of the formula
- terapéuticaally effective amount means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disease or disorder being treated.
- alkyl refers to straight or branched chain hydrocarbons, optionally substituted.
- alkenyl refers to a straight or branched chain hydrocarbon with at least one carbon-carbon double bond.
- Alkynyl refers to a straight or branched chain hydrocarbon with at least one carbon-carbon triple bound.
- halogen refers to fluorine, chlorine, bromine and iodine.
- cycloalkyl refers to optionally substituted, saturated cyclic hydrocarbon ring systems, preferably containing 1 to 3 rings and 3 to 7 carbons per ring which may be further fused with an unsaturated C 3 -C carbocyclic ring.
- Exemplary groups include cyclopropyl, cyclobutyl, cyclopentyl, cydohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, and adamantyl.
- Exemplary substituents include one or more alkyl groups or one or more groups described above as alkyl substituents.
- aryl refers to aromatic monocyclic, fused bicyclic, or fused polycyclic hydrocarbon or heterocyclic groups having 1 to 20 carbon atoms in the ring portions, such as phenyl, naphthyl, pyrrolyl, indolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadazolyl, isothiazolyl, furyl, thienyl, oxadiazolyl, pyridinyl, N-oxo-pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, tetrazinyl, triazinyl, triazolyl, benzothiazolyl, benzoxazolyl, benzothienyl, quinolinyl, quinolinyl-
- N-oxide isoquinolinyl, benzimidazolyl, benzofuryl, chromonyl, coumarinyl, cinnolinyl, quinoxalinyl, indazolyl, benzisothiazolyl, benzisoxazolyl, benzodiazinyl, benzofurazanyl, benzothiopyranyl, benzpyrazolyl, indolinyl, isochromanyl, isoindolinyl, naphthyridinyl, phthalazinyl, purinyl, quinazolinyl, and the like.
- Aryl groups may be substituted.
- biasing refers to a combination of two bonded, nonfused aryl groups as defined above.
- exemplary biaryl groups include biphenyl, furylphenyl, phenylfuryl, thienylphenyl, phenylthienyl, pyridylphenyl, phenylpyridyl, furylpyridyl, pyridylfuryl, pyrrolylpyridyl, pyridylthienyl, isoxazolylthienyl, isoxazolylphenyl, and the like.
- the above defined groups may be substituted by one or more substituents.
- substituents include but are not limited to alkyl, alkenyl, alkynyl, aryl, halo, haloalkyl, hydroxy, alkoxy, haloalkoxy, cycloalkoxy, oxo, aryloxy, alkanoyl, alkanoyloxy, amino, alkylamino, dialkylamino, arylamino, diarylamino, alkylarylamino, sulfonamido, carbonyl, carboxyl, alkoxycarbonyl, amidocarbonyl, oxoalkyl, cyano, nitro, carbamyl, guanidine, amidino, sulfonyl, and the like.
- R 1 is Q-Q substituted or unsubstituted alkyl, C 2 -C 8 substituted or unsubstituted alkenyl, C 2 -C 8 substituted or unsubstituted alkynyl, C4-Q5 substituted or unsubstituted aryl, C 5 -C o substituted or unsubstituted arylalkyl, C 5 -C 20 substituted or unsubstituted biarylalkyl, C 5 -C 2 o substituted or unsubstituted arylalkenyl, or C 5 -C 20 substituted or unsubstituted arylalkynyl;
- R 2 is H, C 1 -C 4 substituted or unsubstituted alkyl, C 2 -C 4 substituted or unsubstituted alkenyl, or Q-Q substituted or unsubstituted alkynyl;
- R 3 is H, Q-Q alkanoyl,
- R 1 is Q-Q 5 substituted or unsubstituted arylalkyl, C 6 -Q 5 substituted or unsubstituted biarylalkyl, Q-Q 5 substituted or unsubstituted arylalkenyl, or C 6 -C 15 substituted or unsubstituted arylalkynyl.
- compounds of formula (I) are provided wherein R 1 is Q-Q 5 substituted or unsubstituted aryl, Q-Qo substituted or unsubstituted arylalkyl, or Q-Qosubstituted or unsubstituted biarylalkyl; and R 2 is H.
- the azidoerythromycin is treated with an acylating reagent, for example acetic anhydride, propionic anhydride, benzoic anhydride, and the like, at ambient temperature in an inert solvent such as ethyl acetate, dichloromethane, or acetonitrile, to produce the 2'-O-acyl azidoerythromycin.
- an acylating reagent for example acetic anhydride, propionic anhydride, benzoic anhydride, and the like
- an inert solvent such as ethyl acetate, dichloromethane, or acetonitrile
- the azide is then reduced using a phosphine such as trimethylphosphine or triphenylphosphine in a solvent such as tetrahydrofuran (THF) or a mixture of THF and dichloromethane, and the intermediate phosphinimine is reacted with a carboxylic acid, a carbodiimide such as l-[3-(dimethylamino)propyl]-2-ethylcarbodiimide hydrochloride (EDCI) or dicyclohexyl-carbodiimide, and a coupling adjuvant such as 1-hydroxybenzotriazole (HOBt), N-hydroxysuccinimide (HOSu), or l-hydroxy-7- azabenzotriazole (HABt), to produce the 2'-O-acyl-amidoerythromycin.
- a phosphine such as trimethylphosphine or triphenylphosphine in a solvent such as tetrahydr
- the azidoerythromycin is treated with trimethylphosphine, and the resulting phosphinimine is reacted with a carboxylic acid and a coupling agent such as hexafluorophosphate (HBTU), O-(7-azabenzotriazol-l-yl)-N,N,N',N'- tetramethyluronium hexafluorophosphate (HATU), or a similar coupling agent which incorporates the coupling agent and adjuvant into one molecule.
- HBTU hexafluorophosphate
- HATU O-(7-azabenzotriazol-l-yl)-N,N,N',N'- tetramethyluronium hexafluorophosphate
- the carboxylic acids used in the preparation of the inventive compounds include alkanoic acids, alkenoic acids, alkynoic acids, N-protected amino acids and/or peptides such as N-BOC, N-Cbz, and N-FMOC amino acids and/or peptides, benzoic acids, heterocyclic carboxylic acids, biaryl carboxylic acids, arylacetic acids, biarylacetic acids, and the like, each of which may be substituted by a variety of groups.
- the carboxylic acids used in the preparation of the inventive compounds are biarylacetic acids.
- One means of preparing these biarylacetic acids is via a Suzuki coupling as illustrated in Scheme 2.
- a haloarylacetic ester is coupled with an arylboronic acid in the presence of a palladium catalyst and base to provide the biarylacetic ester.
- the ester is subsequently saponified to provide the biarylacetic acid.
- Suitable palladium catalysts include tetrakis(triphenylphosphine)palladium, palladium on carbon with triphenylphosphine, and similar sources of palladium(O).
- the 2'-O-acyl azidoerythromycin is reacted with trimethylphosphine to generate the phosphinimine, which is then alkylated by reaction with an alkyl halide or alkyl sulfonate, R 2 X.
- Suitable alkylating agents include but are not limited to methyl iodide, methyl bromide, methyl triflate, ethyl tosylate, ethyl triflate, ethyl iodide, allyl bromide, and propargyl bromide.
- the resulting phosphonium salt is hydrolyzed, and the product amine is acylated with the carboxylic acid, R !
- the method comprises the steps of (1) chemically synthesizing a racemic chlorinated diketide thioester; (2) growing a culture of a first organism in the presence of the racemic chlorinated diketide thioester so as to produce a chlorinated erythronolide; (3) partially purifying the chlorinated erythronolide; (4) chemically converting the chlorinated erythronolide into the azido erythronolide; (5) growing a culture of a second organism in the presence of the azido erythronolide, so as to produce an azidoerythromycin; and (6) purifying the azidoerythromycin.
- a method for preparing 15- azidoerythromycin comprises the steps of (1) chemically synthesizing racemic (2S* 5R*)-5-chloro-3-hydroxy-2-methylpentanoyl N- propionylcysteamine thioester; (2) growing a culture of a first organism in the presence of racemic (2S* 3R*)-5-chloro-3-hydroxy-2-methylpentanoyl N- propionylcysteamine thioester so as to produce 15-chloro-6-deoxyerythronolide B; (3) partially purifying the 15-chloro-6-deoxy erythronolide B so produced; (4) chemically converting the 15-chloro-6-deoxyerythronolide B into 15-azido-6-deoxyerythronolide B; (5) growing a culture of a second organism in the presence of the 15-azido-6- deoxyerythronolide B, so as to produce
- a method for preparing 14-azido-14- desmethylerythromycin comprises the steps of (1) chemically synthesizing racemic (2S* JR*)-4-chloro-3-hydroxy-2-methylbutanoyl N- propionylcysteamine thioester; (2) growing a culture of a first organism in the presence of racemic (2S* 3R*)-4-chloro-3-hydroxy-2-methylbutanoyl N- propionylcysteamine thioester so as to produce 15 -chloro-6-deoxy erythronolide B; (3) partially purifying the 14-chloro-14-desmethyl-6-deoxyerythronolide B so produced; (4) chemically converting the 14-chloro-14-desmethyl-6-deoxyerythronolide B into 14-azido-14-desmethyl-6-deoxy erythronolide B; (5) growing a culture of a second organism in the presence of the 14-azi
- a method for preparing 15- (azidomethyl)-erythromycin comprises the steps of (1) chemically synthesizing racemic (2S* 3R*)-4-chloro-3-hydroxy-2-methylhexanoyl N- propionylcysteamine thioester; (2) growing a culture of a first organism in the presence of racemic (2S*, 5R*)-4-chloro-3-hydroxy-2-methylhexanoyl N- propionylcysteamine thioester so as to produce 15-(chloromethyl)-6- deoxyerythronolide B; (3) partially purifying the 15-(chloromethyl)-6- deoxyerythronolide B so produced; (4) chemically converting the 15-(chloromethyl)- 6-deoxyerythronolide B into 15-(azidomethyl)-6-deoxyerythronolide B; (5) growing a culture of a second organism in the presence of the 15-(a
- the thioester so produced is (2S* 3R*)-5-chloro-3-hydroxy-2-methylpentanoate N- propionylcysteamine thioester.
- the aldehyde used is chloroacetaldehyde
- the thioester so produced is (2S* 3R*)-4-chloro-3-hydroxy-2-methylbutanoate N- propionylcysteamine thioester.
- the aldehyde used is 4-chlorobutyraldehyde
- the thioester so produced is (2S* 3R*)-6-chloro-3-hydroxy-2-methylhexanoate N- propionylcysteamine thioester.
- the preparation of 13-substituted 6-deoxyerythronolide B compounds is described in Khosla et al, U.S. Patent Nos. 6,080,555, 6,274,560, 6,066,721, and 6,261,816; and Ashley et al, U.S. Patent 6,492,562; each of which is incorporated herein by reference.
- 15-chloro-6-deoxyerythronolide B is prepared by a method in which racemic (2S* 3R*)-5-chloro-3-hydroxy-2- methylpentanoyl N-propionylcysteamine thioester is provided to a 6- deoxyerythronolide B synthase ("DEBS") that is unable to act on its natural substrate, propionyl CoA, due to a mutation in the ketosynthase domain of module 1 of DEBS.
- DEBS 6- deoxyerythronolide B synthase
- This recombinant DEBS can be expressed in the natural host that normally produces erythromycin, Saccharopolyspora erythraea, or the entire PKS gene cluster can be inserted by plasmid in a suitable host such as S. coelicolor (see e.g., Jacobsen et al, Science 277: 367-369 (1997), incorporated herein by reference) or S. lividans which has been modified to delete its endogenous actinorhodin polyketide synthesis mechanism.
- the host is S. coelicolor CH999/pJRJ2, which expresses a mutant 6-DEB synthase having an inactivated module 1 ketosynthase.
- a cell free system as described in Khosla et al, "Synthesis of polyketides from diketides," U.S. Patent No. 6,080,555; Khosla et al, “Cell-free synthesis of polyketides,” U.S. Patent 6,274,560; and PCT Publication No. WO 97/02358, each of which is incorporated herein by reference, may also be employed by producing the relevant PKS proteins recombinantly and effecting their secretion or lysing the cells containing them.
- a typical cell-free system would include the appropriate PKS, NADPH and an appropriate buffer and substrates required for the catalytic synthesis of polyketides.
- the appropriate thioester diketide substrates are provided to PKS enzymes other than the 6-dEB synthase of Saccharopolyspora erythraea.
- PKS enzymes include the 6-dEB synthase of Micromonospora megalomicea and its KS1° derivative described in McDaniel &
- the appropriate thioester diketide substrates are provided to any of the above PKS enzymes in a host cell that is capable of performing one or more of the post-PKS hydroxylation and/or glycosylation steps leading to the erythromycins.
- a chlorinated erythronolide B is prepared by providing a chlorinated thioester diketide to a strain comprising both DEBS and a 6- hydroxylase, for example the eryF hydroxylase from Sac. erythraea.
- the production of 14-chloro-14-desmethylerythronolide B is described in Example 72 below.
- 15-chloro-6-deoxyerythronolide B The preparation of 15-chloro-6-deoxyerythronolide B is detailed in Example 2 below. Using the same methods but starting with the appropriate diketide thioesters described above, 14-chloro-14-desmethyl-6-deoxy erythronolide B and 15- (chloromethyl)-6-deoxyerythronolide B are prepared as described below in Examples 71 and 73.
- the chlorinated erythronolide so produced is reacted with sodium azide and sodium iodide in DMSO so as to produce the azido erythronolide by displacement of the chloride with azide. This is detailed in the Examples below.
- 15-azido-6-deoxyerythronolide B is prepared from 15- chloro-6-deoxyerythronolide B (Example 3)
- 14-azido-14-desmethyl-6- deoxyerythronolide B is prepared from 14-chloro-14-desmethyl-6-deoxyerythronolide B (Example 74)
- 15-(azidomethyl)- 6-deoxyerythronolide B is prepared from 15- (chloromethyl)- 6-deoxyerythronolide B (Example 75)
- 15-azido-erythronolide B is prepared from 15-chloro-erythronolide B
- 14-azido-14-desmethyl-erythronolide B is prepared from 14-chloro-14-desmethyl-erythronolide B
- 15-(azidomethyl)- erythronolide B is prepared from 15-(chloromethyl)-erythronolide B.
- the azidoerythronolide prepared according to one of the above methods is then added to a culture of a bioconversion organism capable of glycosylating at the C-3 and C-5 positions, and optionally hydroxylating at C-6, hydroxylating at the C-12 position and/or methylating a 3-O- mycarosyl group, depending on the strain and fermentation conditions employed.
- the bioconversion organism is Saccharopolyspora erythraea K39-14V, a strain in which the module 1 ketosynthase of the native DEBS genes has been inactivated as described in Santi et al., "Hosts for the biosynthesis of polyketides," PCT publication WO 01/83803, incorporated herein by reference.
- K39- 14V contains the enzyme activities needed to convert an erythronolide into the erythromycin A derivative, i.e., hydroxylation at C-6, addition of mycarose to the 3- OH, addition of desosamine to the 5-OH, hydroxylation at C-12, and methylation of the mycarose.
- K39-14V is supplied with 15- azido-6-deoxyerythronolide B under fermentation conditions wherein 15- azidoerythromycin A is the predominant product. This is detailed below in Example 4.
- K39-14V is supplied with 15-azido-6- deoxyerythronolide B under fermentation conditions wherein 15-azidoerythromycin B is the predominant product, for example under conditions of limited oxygen.
- K39-14V is supplied with 14-azido-14- desmethyl-6-deoxyerythronolide B under fermentation conditions wherem 14-azido- 14-desmethylerythromycin A is produced.
- K39-14V is supplied with 14-azido-14-desmethyl-6-deoxyerythronolide B under fermentation conditions wherein 14-azido-14-desmethylerythromycin B is produced (Example 76).
- K39-14V is supplied with 15- (azidomethyl)-6-deoxyerythronolide B under fermentation conditions wherein 15- (azidomethyl)-erythromycin A is produced.
- K39-14V is supplied with 15 -(azidomethyl)-6-deoxy erythronolide B under fermentation conditions wherein 15-(azidomethyl)-erythromycin B is produced (Example 77).
- a bioconversion strain that lacks an active 12-hydroxylase, encoded by the eryK gene or a homologue, is used to convert the azido-6-deoxyerythromycin into the azidoerythromycin B.
- the azidoerythromycin B compounds are produced using a mutant of K39-14V wherein the eryK gene encoding the 12-hydroxylase has been inactivated or deleted.
- the resulting azidoerythromycins are isolated as described for 15- azidoerythromycin A in Example 4 below.
- the cells are removed from the fermentation by centrifugation, and the broth is extracted to remove the azidoerythromycins.
- the extraction is performed using absorption onto a solid resin, such as XAD or HP20.
- the azidoerythromycins are subsequently eluted from the resin with an organic solvent such as methanol, ethanol, acetone, acetonitrile, or ethyl acetate.
- the extraction is performed by mixing the broth with an immiscible organic solvent in which the azidoerythromycin is soluble, such as ethyl acetate, dichloromethane, or chloroform. After concentration of the extract, the residue is subjected to chromatography to provide purified azidoerythromycins.
- an immiscible organic solvent in which the azidoerythromycin is soluble such as ethyl acetate, dichloromethane, or chloroform.
- these compounds are prepared from azidoerythromycins as illustrated in Scheme 4 and exemplified in Example 80.
- the azidoerythromycin is first treated with hydroxylamine, typically in the presence of an acid catalyst such as acetic acid and in a mixture of water and isopropanol as solvent, to produce the 9-oxime.
- This procedure is exemplified in detail with 15- azidoerythromycin A in Example 48 below.
- the 9-oxime is protected, for example using an acetal such as 1,1-diisopropoxycyclohexane (DIPCH) or 2,2- dimethoxypropane in the presence of a mild acid catalyst such as pyridinium p- toluenesulfonate (PPTS).
- DIPCH 1,1-diisopropoxycyclohexane
- PPTS pyridinium p- toluenesulfonate
- Other protecting groups such as trialkylsilyl ethers, can also be used to protect the 9-oxime.
- the 2'- and 4" -OH groups are next protected, for example as trimethylsilyl ethers by treatment with a mixture of trimethylsilylimidazole and chlorotrimethylsilane, as described below in Example 50.
- This process is exemplified below in Example 51.
- a palladium catalyst for example palladium acetate and triphenylphosphine
- the 9-oxime and 2'- and 4"-OH groups are deprotected by treatment with acetic acid in acetonitrile.
- R 3 , R 4 , and m are as defined above;
- R 5 is H, Q-Q alkoxy, Q-Q alkenyloxy, or Q-Q alkynyloxy; and
- R 8 is H or F.
- the cladinose is removed from the compound of formula (I) by treatment with a mild acid in the presence of water, for example using PPTS in a mixture of acetone and water as exemplified below in Example 54, or using aqueous HCl.
- a mild acid for example using PPTS in a mixture of acetone and water as exemplified below in Example 54, or using aqueous HCl.
- the 3-OH is oxidized to the ketone (Example 56).
- Suitable oxidation methods include but are not limited to the Corey-Kim (N- chlorosuccinimide, dimethylsulfide, triethylamine), Swern (oxalyl chloride, DMSO, triethylamine), Moffat (dicyclohexylcarbodiimide, DMSO), and modified Pfizer- Moffat (l-[3-(dimethylamino)propyl]-3-ethylcarbodiimide, pyridinium trifluoroacetate, DMSO) conditions.
- NFBS N- fluorobenzenesulfonimide
- R 1 , R 2 , R 3 , R 4 , R 8 , and m are as defined above and R 5 is H, Q-Q alkoxy, Q- Q alkenyloxy, or Q-Q alkynyloxy.
- compounds of formula (III) are provided wherein R 1 is C 6 -C 15 substituted or unsubstituted arylalkyl, Q-Q5 substituted or unsubstituted biarylalkyl, Q-Q 5 substituted or unsubstituted arylalkenyl, or Q-Q 5 substituted or unsubstituted arylalkynyl.
- R 1 is C 6 -C 15 substituted or unsubstituted arylalkyl, Q-Q5 substituted or unsubstituted biarylalkyl, Q-Q 5 substituted or unsubstituted arylalkenyl, or Q-Q 5 substituted or unsubstituted arylalkynyl.
- R 2 is
- R 5 is alkoxy are provided.
- R 1 is Q-Q 5 substituted or unsubstituted aryl, Q-Qosubstituted or unsubstituted arylalkyl, or Q-Qosubstituted or unsubstituted biarylalkyl;
- R 2 is H; and
- R 5 is alkoxy.
- the compound of formula (II) is treated with trimethylphosphine, and the resulting phosphinimine is reacted with a carboxylic acid, a coupling agent such as a carbodiimide, and a coupling adjuvant such as HOBt, HABt, or HOSu as discussed above and exemplified below in Example 65.
- a coupling agent such as a carbodiimide
- a coupling adjuvant such as HOBt, HABt, or HOSu as discussed above and exemplified below in Example 65.
- the compound of formula (II) is treated with trimethylphosphine, and the resulting phosphinimine is reacted with a carboxylic acid and a coupling agent such as O-benzotriazol-l-yl-N,N,N',N'- tetramethyluronium hexafluorophosphate (HBTU), O-(7-azabenzotriazol-l-yl)- N,N,N',N' -tetramethyluronium hexafluorophosphate (HATU), or a similar coupling agent which incorporates the coupling agent and adjuvant into one molecule.
- HBTU O-benzotriazol-l-yl-N,N,N',N'- tetramethyluronium hexafluorophosphate
- HATU O-(7-azabenzotriazol-l-yl)- N,N,N',N' -tetramethyluronium hexaflu
- R benzoyl
- Suitable alkylating agents include but are not limited to methyl iodide, methyl bromide, methyl triflate, ethyl tosylate, ethyl triflate, ethyl iodide, allyl bromide, and propargyl bromide.
- compounds of formula (IV) wherein R 1 , R 2 , R 3 , R 8 , and m are as defined above and R 5 is H, Q-Q alkoxy, Q-Q alkenyloxy, or Q-Q alkynyloxy, and X is O or NR 7 , wherein R 7 is H, Q-Q alkyl, or C 6 -C 20 arylalkyl.
- R 1 is C 6 -Q 5 substituted or unsubstituted arylalkyl, Q-Q 5 substituted or unsubstituted biarylalkyl, C 6 -C 15 substituted or unsubstituted arylalkenyl, or Q-Q 5 substituted or unsubstituted arylalkynyl.
- R 1 is Q-Q 5 substituted or unsubstituted aryl, Q-C 20 substituted or unsubstituted arylalkyl, Q-Qo substituted or unsubstituted biarylalkyl, Q-C 20 substituted or unsubstituted arylalkenyl, or Q-Qo substituted or unsubstituted arylalkynyl; and R 2 is H.
- R 1 is Q-Q 5 substituted or unsubstituted aryl, Q-C 20 substituted or unsubstituted arylalkyl, Q-Qn substituted or unsubstituted biarylalkyl, Q-C 20 substituted or unsubstituted arylalkenyl, or Q-Qo substituted or unsubstituted arylalkynyl;
- R 2 is H; and
- X is NH.
- Example 57 A representative procedure for mesylation is given in Example 57.
- the 11-O-mesylate is treated with 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU) to produce the 10,11-anhydrocompound, as detailed in Example 58.
- DBU 1,8- diazabicyclo[5.4.0]undec-7-ene
- a representative procedure for fluorination is given in Example 60.
- 1,1- carbonyldiimidazole and a strong base for example sodium bis(trimethylsilyl)amide (NaHMDS) in an inert solvent such as tetrahydrofuran at a temperature between - 10°C and 30 °C
- an amine for example ammonia
- potassium tert-butoxide provides the 11,12-cyclic carbamate.
- the cladinose is removed, for example by treatment with aqueous acid, and the resulting 3-OH is subjected to hydrogenolysis to reduce the azide to the amine.
- Hydrogenolysis is performed in a solvent such as methanol in the presence of a metal catalyst, such as palladium or palladium supported on an inert material such as charcoal, and an acid, such as HCl, acetic acid, or similar, under a moderate pressure of hydrogen gas, such as that provided by a hydrogen balloon.
- the acid may be generated in situ, for example by addition of chlorotrimethylsilane to methanol.
- the resulting ammonium salt is condensed with a carboxylic acid R ⁇ OOH as described above.
- the 3-OH is oxidized to the 3-oxo group, for example using the Dess-Martin periodinane, Swern oxidation, Corey-Kim oxidation, or similar.
- the 2' -protecting group is removed by methanolysis. This procedure is exemplified in detail in Example 81 below.
- a representative procedure for debenzoylation is given in Example 62.
- R 1 , R 2 , R 4 , R 6 and m are as defined above; and R 9 is H or Q-Q alkyl.
- compounds of formula (V) are provided wherein R 1 is Q-Q 5 substituted or unsubstituted arylalkyl, C 6 -C 15 substituted or unsubstituted biarylalkyl, Q-Q 5 substituted or unsubstituted arylalkenyl, or Q-Q 5 substituted or unsubstituted arylalkynyl.
- R 1 is Q-Q 5 substituted or unsubstituted aryl, Q-Qo substituted or unsubstituted arylalkyl, or Q-Qo substituted or unsubstituted biarylalkyl.
- the oximinoether is then reduced, for example using sodium borohydride, under conditions wherein the azide group remains intact.
- the resulting azalide is then alkylated, for example methylated using formaldehyde and formic acid as described for the production of azithromycin in Kobrehel & Djokic, U.S. Patent No. 4,517,359, incorporated herein by reference, to produce the azidoazalide.
- the azidoazalide is converted into an embodiment of the compounds of formula (V) as illustrated in Scheme 12.
- the 2'-OH is protected, for example as the acetate or benzoate, and the azide is reduced by treatment with a phosphine, for example trimethylphosphine or triphenylphosphine, and the amide is formed by condensation with carboxylic acid R ! COOH as described above.
- the 2' -protecting group is removed by methanolysis.
- the azidoazalide is converted into an embodiment of the compounds of formula (V) by protecting the 2'-OH as the acetate or benzoate, reducing the azide to the amine by hydrogenolysis and forming the amide by condensation of the aminoazalide with R ! COOH as described above.
- This invention further provides a method of treating bacterial infections, or enhancing the activity of other anti-bacterial agents, in warm-blooded animals, which comprises administering to the animals a compound of the invention alone or in admixture with a diluent or in the form of a medicament according to the invention.
- the compounds When the compounds are employed for the above utility, they may be combined with one or more pharmaceutically acceptable carriers, e.g., solvents, diluents, and the like, and may be administered orally in such forms as tablets, capsules, dispersible powders, granules, or suspensions containing for example, from about 0.5% to 5% of suspending agent, syrups containing, for example, from about 10% to 50% of sugar, and elixers containing, for example, from about 20% to 50% ethanol, and the like, or parenterally in the form of sterile injectable solutions or suspensions containing from about 0.5% to 5% suspending agent in an isotonic medium.
- These pharmaceutical preparations may contain, for example, from about 0.5% up to about 90% of the active ingredient in combination with the carrier, more usually between 5% and 60% by weight.
- compositions for topical application may take the form of liquids, creams or gels, containing a therapeutically effective concentration of a compound of the invention admixed with a dermatologically acceptable carrier.
- any of the usual pharmaceutical media may be employed.
- Solid carriers include starch, lactose, dicalcium phosphate, microcrystalline cellulose, sucrose, and kaolin, while liquid carriers include sterile water, polyethylene glycols, non-ionic surfactants and edible oils such as corn, peanut and sesame oils, as are appropriate to the nature of the active ingredient and the particular form of administration.
- Adjuvants customarily employed in the preparation of pharmaceutical compositions may be advantageously included, such as flavoring agents, coloring agents, preserving agents, and antioxidants, for example, vitamin E, ascorbic acid, BHT and BHA.
- the active compounds may also be administered parenterally or intraperitoneally. Solutions or suspensions of these active compounds as a free base or pharmacologically acceptable salt can be prepared in water suitably mixed with a surfactant such as hydroxypropyl-cellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations may contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol, and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration and the severity of the condition being treated. However, in general, satisfactory results are obtained when the compounds of the invention are administered at a daily dosage of from about 0.1 mg/kg to about 400 mg/kg of animal body weight, preferably given once a day, or in divided doses two to four times a day, or in sustained release form. For most large mammals, including humans, the total daily dosage is from about 0.07 g to 7.0 g, preferably from about 100 mg to 1000 mg.
- Dosage forms suitable for internal use comprise from about 100 mg to 500 mg of the active compound in intimate admixture with a solid or liquid pharmaceutically acceptable carrier. This dosage regiment may be adjusted to provide the optimal therapeutic response. For example, several divided doses may be administered daily or the dose may be proportionally reduced as indicated by the exigencies of the therapeutic situation.
- compositions and medicaments are carried out by any method known in the art, for example, by mixing the active ingredient(s) with the diluent(s) to form a pharmaceutical composition (e.g., a granulate) and then forming the composition into the medicament (e.g., tablets).
- a pharmaceutical composition e.g., a granulate
- the medicament e.g., tablets
- a seed culture of Streptomyces coelicolor CH999/pJRJ2 was made by inoculating 1 mL of frozen mycelium into a 250 mL baffled flask containing 50 mL of FKA medium (corn starch, 45 g L; corn steep liquor, 10 g/L; dried, inactivated brewers yeast, 10 g/L; and CaCO 3 , 1 g/L), 0.050 mL of 50 mg/mL thiostrepton in DMSO (filter sterilized), and 0.500 mL of 50% Antifoam B.
- the flask was incubated at 30 °C with shaking at 175 rpm for 48 hours (Innova floor shaker).
- the culture was transferred into a 2.8-L baffled flask containing 500 mL of FKA medium, 0.500 mL of 50 mg/mL thiostrepton in DMSO, and 5 mL of 50% Antifoam B, and the flask was incubated at 30 °C with shaking at 175 rpm for 48 hours.
- a 10-L stirred tank bioreactor (B. Braun) was autoclaved, filled with 5 L of sterile FKA medium and 5 mL of 50 mg/mL thiostrepton in DMSO, and then inoculated with 500 mL (10%) of seed culture.
- the bioreactor was run for 24 hours at 30 °C with stirring at 600 rpm, sparged with air at 1.33 LPM, and the pH was maintained at 6.50 by automated addition of 2.5 N NaOH and 2.5 N H 2 SO 4 .
- Three liters of the above culture was used to inoculate a 100-L bioreactor containing 55 L of sterile FKA medium with 2 g/L Tastone 310 added prior to sterilization.
- the fermentor agitation rate was set at a tip speed of 2.5 m s, the temperature was maintained at 30°Q the pH was controlled at pH 6.5 by automated addition of 2.5 N NaOH and 2.5 N H 2 SO 4 , and the airflow was set at 0.4 vvm.
- Foaming was controlled by automated addition of 50% Antifoam B.
- the dissolved oxygen was maintained at > 50% air saturation by cascade control using the agitation rate (tip speed of 2.5-3.0 m/s) and back pressure (0.1-0.4 bar) in that order.
- the 15-chloro-6-deoxyerythronolide B was isolated by solid phase extraction.
- the broth was clarified by centrifugation and loaded onto a column containing HP-20 resin (Rohm and Haas) at a concentration of 1 L resin/20 g 15-chloro-6- deoxyerythronolide B.
- the column was then equilibrated with 5 column volumes of water at a flow rate of 2-4 mL/cm 2 -min.
- the loaded resin was washed with 2 column volumes of water followed by 2 column volumes of 30% methanol in water.
- the 15- chloro-6-deoxyerythronolide B was eluted from the resin with methanol.
- the fractions containing 15-chloro-6-deoxy-erythronolide B were identified by HPLC with ELSD detection.
- the methanol fractions containing 15-chloro-6-deoxyerythronolide B were pooled, and the volatiles were removed under reduced pressure.
- the dried solids were extracted with 3-5 L of methanol and filtered to yield a solution containing 6-10 mg/mL 15-chloro-6-deoxyerythronolide B, which was diluted with an equal volume of water.
- This solution was loaded onto a column of HP20SS (1 L resin/20 g of 15- chloro-6-deoxyerythronolide B), which was then washed with 2 column volumes of 50% aqueous methanol.
- the 15-chloro-6-deoxyerythronolide B was then eluted with 70% methanol in water, and the fractions were analyzed by HPLC.
- a seed culture of Saccharopolyspora erythraea K39-14V was made by inoculating a 1 mL aliquot of frozen mycelium into each of three 250 mL baffled flasks containing 50 mL of VI medium (com starch, 16 g/L; com dextrin, 10 g/L; soya meal flour, 15 g/L; com steep liquor, 5 g/L; soy bean oil, 6 g/L; sodium chloride, 2.5 g/L; ammonium sulfate, 1 g/L; and CaCO , 4 g/L) and 0.100 mL of Antifoam B.
- VI medium com starch, 16 g/L; com dextrin, 10 g/L; soya meal flour, 15 g/L; com steep liquor, 5 g/L; soy bean oil, 6 g/L; sodium chloride, 2.5 g/L; ammonium sulfate
- the flasks were incubated at 34 °C with shaking at 175 rpm for 48 hours (Innova floor shaker). Each culture was transferred into a 2.8-L baffled flask containing 500 mL of VI medium and 1 mL of Antifoam B, and the flasks were incubated at 34 °C with shaking at 175 rpm for 48 hours.
- the fermentor agitation rate was set at a tip speed of 2-4 m/s, the pH was controlled at pH 7.0 by automated addition of 2.5 N NaOH and 2.5 N H 2 SO 4 , the temperature was maintained at 34°C, and the airflow was set at 0.15 vvm. Foaming was controlled by automated addition of 50% Antifoam B.
- Each fermentor was inoculated with a 500- mL seed culture prepared above. During the fermentation, the dissolved oxygen was maintained at > 80% air saturation by cascade control using agitation rate (tip speed of 2-4 m/s)airflow (0.15-0.5 vvm), and oxygen enrichment in that order.
- the 15-azido-erythromycin A was isolated by solid phase extraction.
- the broth was adjusted to pH 9 using 2.5 N NaOH, clarified by centrifugation, and loaded onto a column containing HP-20 resin (Rohm and Haas) at a concentration of 1 L resin/20 g 15-azidoerythromycin A.
- the column was then equilibrated with 5 column volumes of water at a flow rate of 2-4 mL/cm 2 -min.
- the loaded resin was washed with 2 column volumes of water.
- the 15-azido-erythromycin A was eluted from the resin with 5 column volumes of methanol.
- the fractions containing 15-azidoerythromycin A were identified by HPLC, pooled, and the volatiles were removed under reduced pressure.
- the dried solids were mixed with 800 mL of acetone and 3.2-L of hexane for 20 minutes. The mixture was then filtered using a #4 Whatman filter paper. The solids were extracted twice in this manner, and the filtrates were combined and evaporated.
- the crude product was dissolved in methanol and diluted with an equal volume of water. This solution was loaded onto a column of HP20SS (1 L resin 20 g of 15-azido-erythromycin A), which was then washed successively with 1 column volume of 50% aqueous methanol, 3 column volumes of 3:2 methanol/water, 3 column volumes of 7:3 methanol/water, 10 column volumes of 4:1 methanol/water, and finally 5 column volumes of 100% methanol. The fractions were analyzed by HPLC. Product-containing fractions were pooled and evaporated to dryness to yield 13 g of 15-azidoerythromycin A of 95% purity.
- the solution is stirred at room temperature for 45 minutes before transferring to a solution of the carboxylic acid (0.092 mmol, 1.5 eq), l-[(3-(dimethylamino)propyl]-l-ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes. The resulting solution is stirred at room temperature for 3 to 14 hours before partitioning between ethyl acetate (10 ml) and NaHCO3 (10 ml).
- aqueous phase is extracted with ethyl acetate (3 x 10 ml) and the combined organics further washed with brine (25 ml) before drying (Na2SO4), filtering, and concentrating under reduced pressure.
- the residue is dissolved in methanol (2 ml) and stirred at 50 °C for 14 hours before concentrating under reduced pressure.
- This compound was prepared according to the method of Example 6 using 4- phenylbutyric acid.
- This compound was prepared according to the method of Example 6 using benzyloxyacetic acid.
- This compound was prepared according to the method of Example 6 using (3,4,5-trimethoxybenzoyl)formic acid.
- This compound was prepared according to the method of Example 6 using (4- methyl-5-phenyloxazol-2-yl)acetic acid.
- This compound was prepared according to the method of Example 6 using quinoline-3-carboxylic acid.
- This compound was prepared according to the method of Example 6 using pyridine-3-carboxylic acid.
- This compound was prepared according to the method of Example 6 using indole-3 -carboxylic acid.
- This compound was prepared according to the method of Example 6 using 4- biphenylacetic acid.
- This compound was prepared according to the method of Example 6 using 3- (3-furyl)phenylacetic acid.
- This compound was prepared according to the method of Example 6 using 3- (3-thienyl)phenylacetic acid.
- This compound was prepared according to the method of Example 6 using 2- (2-thienyl)phenylacetic acid.
- This compound is prepared according to the method of Example 6 using 3-(4- pyridyl)phenylacetic acid.
- This compound is prepared according to the method of Example 6 using 3-(2- furyl)pyridyl-5-acetic acid.
- This compound is prepared according to the method of Example 6 using 3-(2- thienyl)-pyridyl-5-acetic acid.
- This compound is prepared according to the method of Example 6 using 3-(2- pyrrolyl)-pyridyl-5 -acetic acid.
- This compound is prepared according to the method of Example 6 using 5- phenylthienyl-2-acetic acid.
- This compound is prepared according to the method of Example 6 using 5-(2- pyridyl)-thienyl-2-acetic acid.
- This compound is prepared according to the method of Example 6 using 5-(3- isoxazolyl)-thienyl-2-acetic acid.
- This compound is prepared according to the method of Example 6 using 3-(5- (2-pyridyl)thien-2-yl)propionic acid.
- IPCH ketal (4.2 g), 2 M methyl bromide in ether (3.9 mL), 7 mL of THF, and 7 mL of DMSO was cooled on ice.
- N-Chlorosuccinimide (0.191 g, 1.427 mmol, 1.5 eq) was added to a solution of dimethyl sulfide (0.085 ml, 1.712 mmol, 1.8 eq) and dichloromethane (5 ml) at -15 °C.
- the solution was stirred at -15 °C for 15 minutes before adding a solution of 1,1-carbonyldiimidazole (0.167 g, 1.032 mmol, 3.0 eq) in tetrahydrofuran (3.0 ml) dropwise.
- the solution was stirred at -15 °C for 15 minutes before warming to room temperature over a period of 40 minutes. Sat. aq. NaHCO3 (25 ml) was added followed by ethyl acetate (50 ml), and the solution was partitioned. The organics were further washed with sat. aq.
- the solution is stirred at room temperature for 45 minutes before transferring to a solution of the carboxylic acid (0.092 mmol, 1.5 eq), l-[(3-(dimethylamino)propyl]-l- ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1- hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes. The resulting solution is stirred at room temperature for 14 hours before partitioning between ethyl acetate (10 ml) and NaHCO 3 (10 ml).
- the aqueous phase is extracted with ethyl acetate (3 10 ml) and the combined organics further washed with brine (25 ml) before drying (Na 2 SO 4 ), filtering, and concentrating under reduced pressure.
- the product is purified by silica gel chromatography using acetone/hexane + 1% Et 3 N.
- the solution is stirred at room temperature for 45 minutes before transferring to a solution of the carboxylic acid (0.092 mmol, 1.5 eq), l-[(3- (dimethylamino)propyl]-l-ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes. The resulting solution is stirred at room temperature for 14 hours before partitioning between ethyl acetate (10 ml) and NaHCO3 (10 ml).
- the aqueous phase is extracted with ethyl acetate (3 x 10 ml) and the combined organics further washed with brine (25 ml) before drying (Na2SO4), filtering, and concentrating under reduced pressure.
- the product is purified by silica gel chromatography using acetone/hexane + 1% Et 3 N.
- the solution is stirred at room temperature for 45 minutes before transferring to a solution of the carboxylic acid (0.092 mmol, 1.5 eq), l-[(3-(dimethylamino)propyl]-l-ethylcarbodiimide hydrochloride (0.019 g, 0.099 mmol, 1.6 eq) and 1-hydroxybenzotriazole (0.017 g, 0.124 mmol, 2.0 eq) in dichloromethane or tetrahydrofuran (1.0 ml) also stirred at room temperature for 45 minutes. The resulting solution is stirred at room temperature for 14 hours before partitioning between ethyl acetate (10 ml) and NaHCO3 (10 ml).
- the aqueous phase is extracted with ethyl acetate (3 x 10 ml) and the combined organics further washed with brine (25 ml) before drying (Na2SO4), filtering, and concentrating under reduced pressure.
- the product is purified by silica gel chromatography using acetone/hexane + 1% Et 3 N.
- step (b) Oxime protection.
- the crude oxime from step (a) above (34.0 g, 42.5 mmol) was suspended in 120 ml of CH 2 C1 2 and treated with 1,1 -diisopropoxycyclohexane (51.0 ml, 246.8 mmol, 6 eq.) and pyridinium p-toluenesulfonate (24.8 g, 98.8 mmol, 2 eq.) for 15 hours at ambient temperature.
- the mixture was diluted with 600 ml of CH C1 2 , and then washed sequentially with saturated NaHCO , water, and brine.
- the organic phase was dried with MgSO 4 , filtered, and evaporated to yield brown syrup.
- the temperature of the mixture was lowered to -15 C, and liquid NH3 was added in by condensation of anhydrous ammonia gas using a dry ice condenser.
- the mixture was capped and stirred for 2h at -15 C.
- Liquid NH3 was dripped in above o mixture again before the mixture was capped, and stirred for another 2h at 0 C.
- the mixture was capped and stirred at 0 C for another lh after liquid NH3 was dripped in the mixture for the third time.
- 15 ml of a 1 N solution of potassium tert-butoxide in THF 15 mmol, 1.2 eq.
- step (c) Cladinose removal.
- the crude product from step (b) was dissolved in 120 ml of EtOH and 120 ml of 2N aqueous HCl, and the resulting solution was heated at
- EXAMPLE 82 2'-O-benzoyl-6-O-methyl-3-descladinosyl-ll-amino-ll-deoxy-3-15- aminoerythromycin A 11,12-cyclic carbamate hydrochloride 400 mg of 10% activated palladium on carbon was weighed out into a round bottom flask under nitrogen, and 50 ml of methanol was added in slowly followed by sequential addition of 400 mg of 2'-O-benzoyl-6-O-methyl-3-descladinosyl-ll- amino-ll-deoxy-3-15-azidoerythromycin A 11,12-cyclic carbamate (0.53 mmol) (Example 81) and 0.13 mL (2 eq.) of trimethylsilyl chloride.
- step (b) The compound from step (b) above was dissolved in 2 ml of methanol and heated at 70 C under stirring overnight. Evaporation of the methanol under vacuum gave a yellow solid, which was purified by flash column to provide final 15- amidoketolide.
- step (e) The compound from step (b) above was dissolved in 2 ml of methanol and heated at 70 C under stirring overnight. Evaporation of the methanol under vacuum gave a yellow solid, which was purified by flash column to provide final 15- amidoketolide.
- This compound was prepared according to the methods of Example 83, using
- This compound was prepared according to the methods of Example 83, using 2-([l ,2,4]-tetrazol-l-yl)pyrid-5-ylacetic acid.
- step (c) 3-(2-furanyl)phenylacetic acid.
- a solution of the methyl 3-(2- furanyl)phenylacetate from step (b) in 5 ml of methanol was added 10 ml of IN NaOH aqueous solution, the resulting solution was stirred overnight before diluting with 40 of ml IN NaOH aqueous solution and washing with 2 X 30 ml CH 2 C1 2 .
- the aqueous solution was then acidified with 2N HCl, and extracted with 3 X 30 ml of CH2CI 2 .
- the organic extracts were combined and were dried with MgSO 4 , filtered, and evaporated to give 0.32 g of the product as a golden-colored solid.
- Compounds prepared according to this general procedure include: (a) 3-quinolylacetic acid, using 3-bromoquinoline; and (b) 2-([l,2,4]-triazol-l-yl) ⁇ yrid-5-yl-acetic acid, using 2-([l,2,4]-tetrazol-l- yl)- 5-bromo-pyridine.
- the resulting solution was heated at 70 C for 30 min before it was diluted with 20 ml of water, and extracted with 3 X 30 ml of ethyl acetate. The organic extracts were combined and washed sequentially with water, and brine. The organic phase was dried with MgSO 4 , filtered, and evaporated to give 60 mg of a slight yellow solid.
- MICs Minimum inhibitory concentrations
- NCCLS broth microdilution procedure for susceptibility testing for bacteria that grow aerobically (National Committee for Clinical Laboratory Standards, 1997. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 4 th ed. Approved standard.
- Stock solutions were prepared on the day of the test and appropriate aliquots were added to cation adjusted Mueller-Hinton broth (CAMHB) or Haemophilus test media. Two-fold serial dilutions were prepared and added to wells in microtiter plates. Final test concentrations ranged from 16 to 0.015 ug/ml. Broth cultures of bacteria inoculated from growth on overnight plates for all test bacteria except Streptococcus pneumoniae and Haemophilus influenzae were incubated at 35°C and then adjusted to the Kirby Bauer standard and diluted in
- CAMHB to achieve a final inoculum concentration of approximately 5xl0 5 CFU/ml.
- Inocula for S. pneumoniae and H. influenzae were prepared by directly suspending colonies from an overnight plate, adjusting the turbidity and diluting as above.
- S. pneumoniae media was supplemented with 2.5% lysed horse blood. All plates were incubated in ambient air at 35°C for 20-24 h for S. pneumoniae and Haemophilus influenzae and 16-20 h for all other bacteria. The MIC endpoints were determined by reading the lowest concentration of test compound that completely inhibited the growth of the test bacteria. Results for compounds described in the above Examples are listed in Table 1.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Description

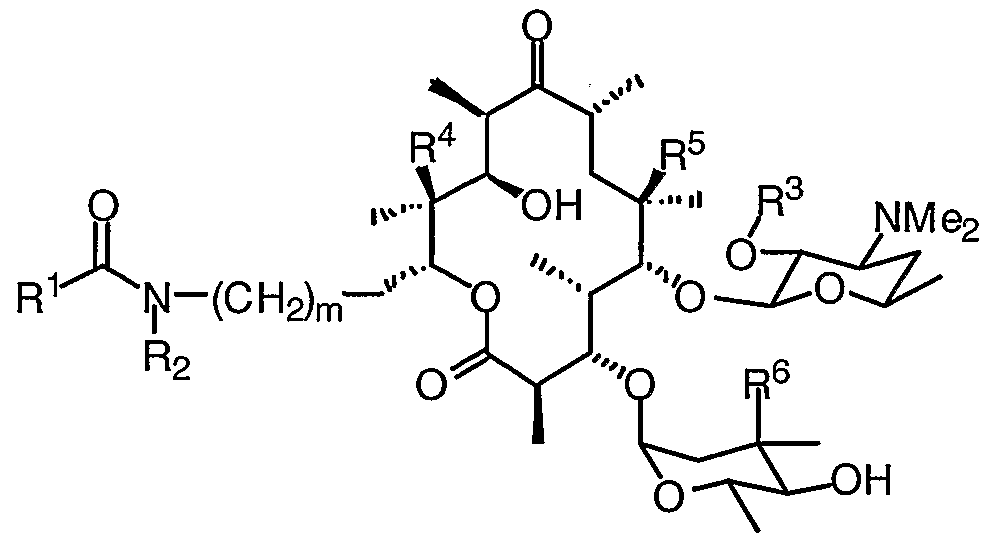
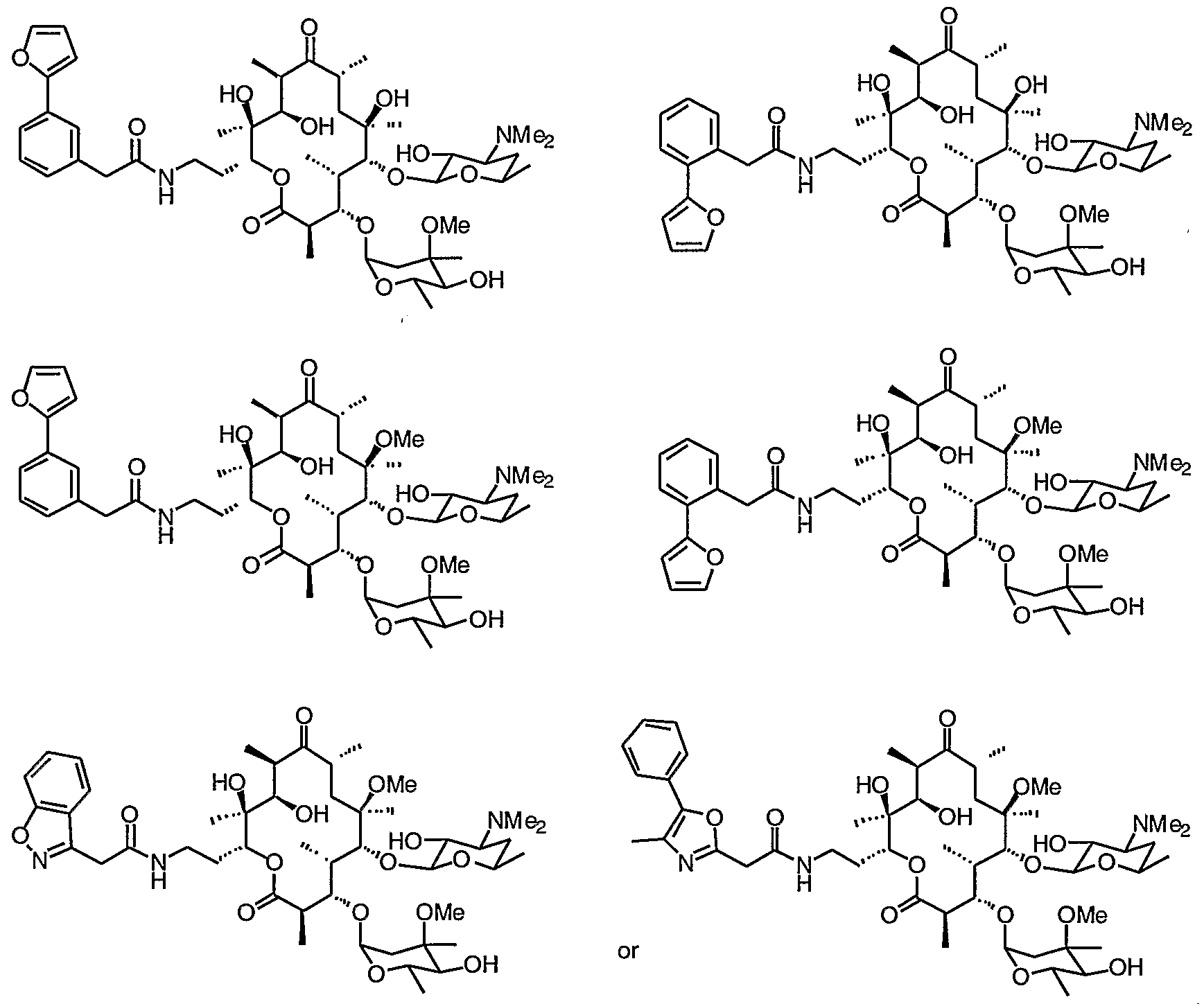



















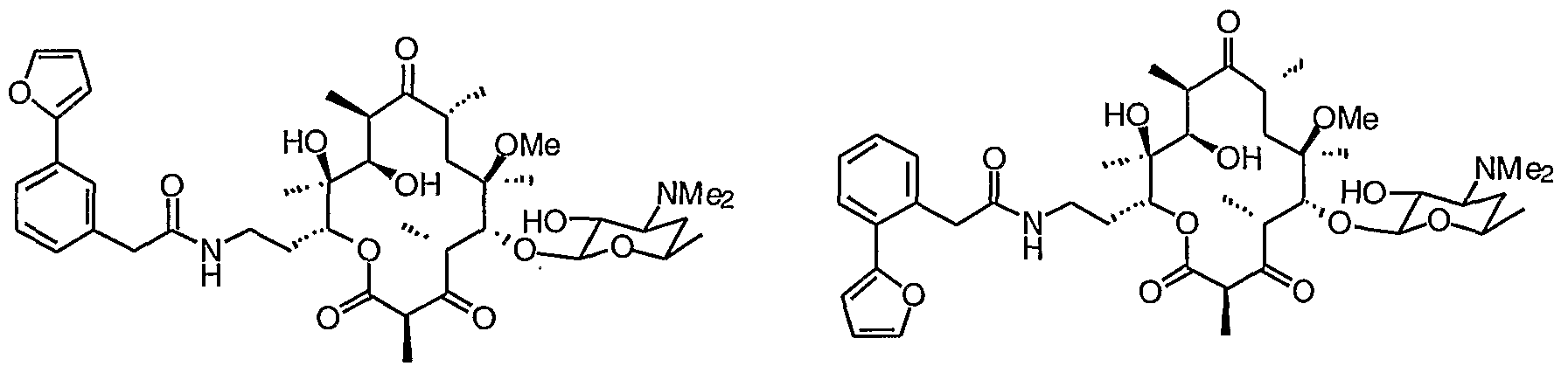















































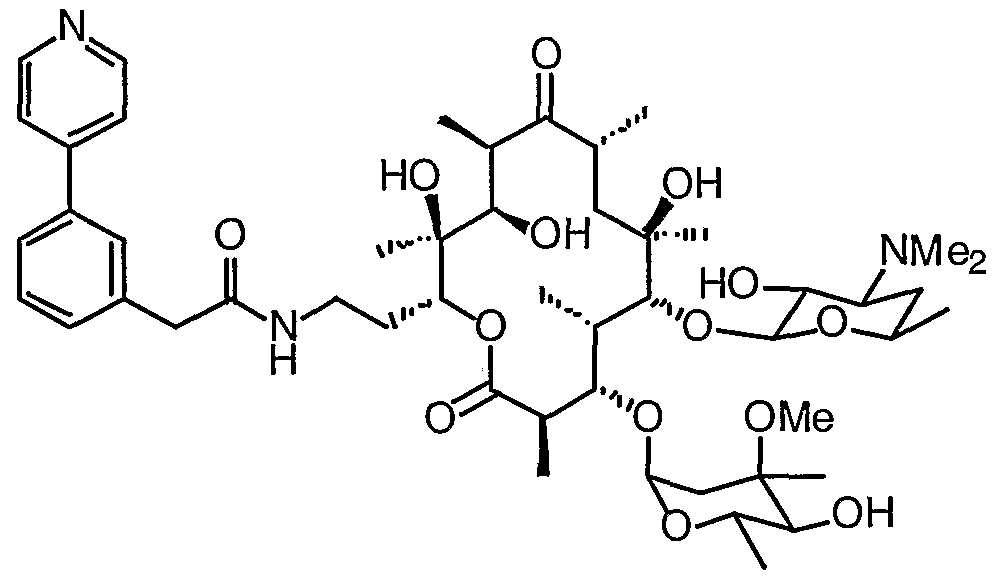


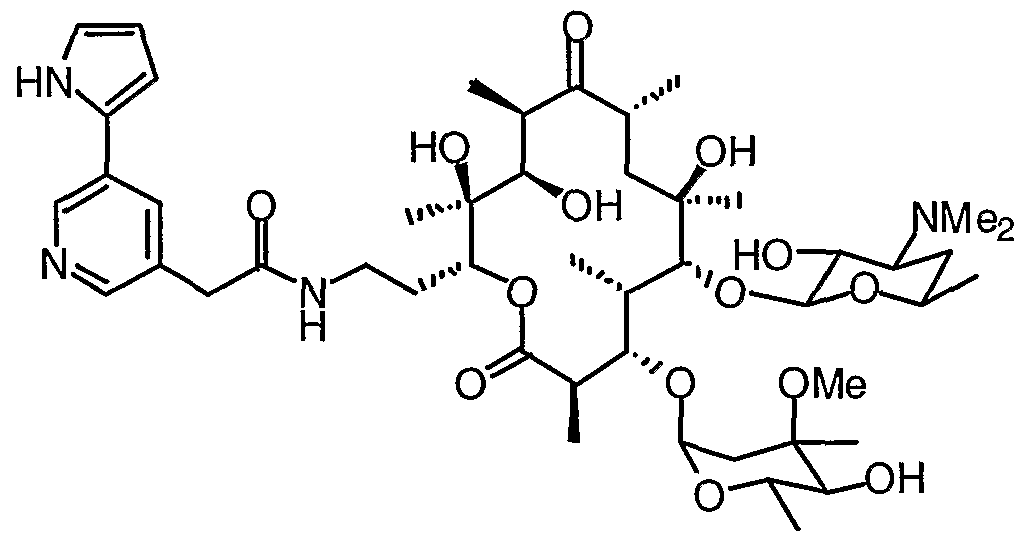





































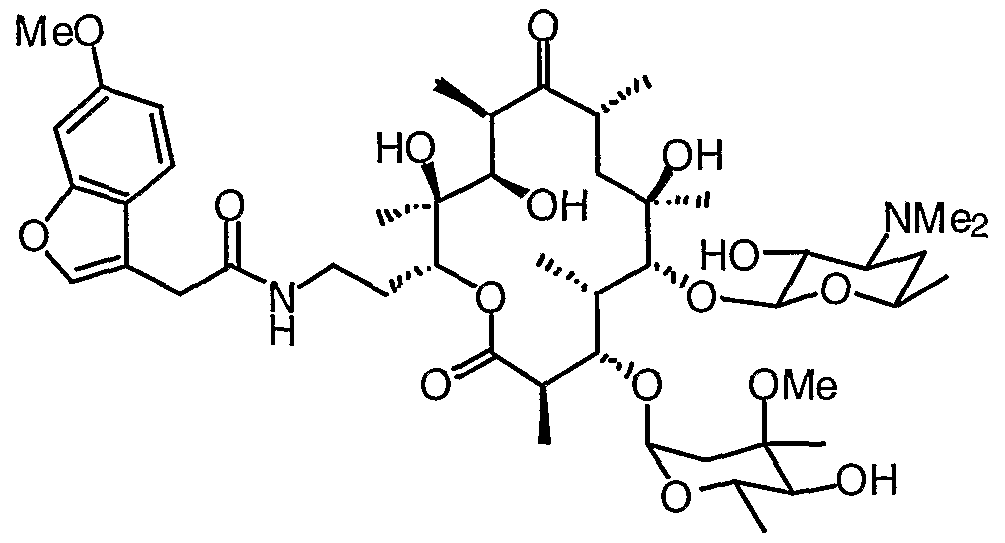


Claims












Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003561615A JP2005538927A (en) | 2002-01-17 | 2003-01-17 | Amide macrolide |
| CA002471383A CA2471383A1 (en) | 2002-01-17 | 2003-01-17 | Amido macrolides |
| EP03713257A EP1471923A4 (en) | 2002-01-17 | 2003-01-17 | Amido macrolides |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35015302P | 2002-01-17 | 2002-01-17 | |
| US60/350,153 | 2002-01-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003061671A1 true WO2003061671A1 (en) | 2003-07-31 |
Family
ID=27613367
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/001398 WO2003061671A1 (en) | 2002-01-17 | 2003-01-17 | Amido macrolides |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1471923A4 (en) |
| JP (1) | JP2005538927A (en) |
| CA (1) | CA2471383A1 (en) |
| WO (1) | WO2003061671A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008530035A (en) * | 2005-02-09 | 2008-08-07 | バジリア ファルマスーチカ アーゲー | New macro ride |
| CN101973979A (en) * | 2010-10-27 | 2011-02-16 | 浙江大学 | Antibacterial compound and application thereof |
| US8063021B2 (en) | 2002-01-17 | 2011-11-22 | Kosan Biosciences Incorporated | Ketolide anti-infective compounds |
| CN102707059A (en) * | 2012-05-31 | 2012-10-03 | 华中农业大学 | Colloidal gold test paper strip for residue detection of tylosin and tilmicosin, method for using colloidal gold test paper strip and application of colloidal gold test paper strip |
| EP3290427A1 (en) | 2005-08-24 | 2018-03-07 | Melinta Therapeutics, Inc. | Triazole compounds and methods of making and using the same |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6100240A (en) * | 1998-10-09 | 2000-08-08 | Pfizer Inc | Macrolide derivatives |
-
2003
- 2003-01-17 EP EP03713257A patent/EP1471923A4/en not_active Withdrawn
- 2003-01-17 CA CA002471383A patent/CA2471383A1/en not_active Abandoned
- 2003-01-17 WO PCT/US2003/001398 patent/WO2003061671A1/en not_active Application Discontinuation
- 2003-01-17 JP JP2003561615A patent/JP2005538927A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6100240A (en) * | 1998-10-09 | 2000-08-08 | Pfizer Inc | Macrolide derivatives |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1471923A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8063021B2 (en) | 2002-01-17 | 2011-11-22 | Kosan Biosciences Incorporated | Ketolide anti-infective compounds |
| JP2008530035A (en) * | 2005-02-09 | 2008-08-07 | バジリア ファルマスーチカ アーゲー | New macro ride |
| EP3290427A1 (en) | 2005-08-24 | 2018-03-07 | Melinta Therapeutics, Inc. | Triazole compounds and methods of making and using the same |
| CN101973979A (en) * | 2010-10-27 | 2011-02-16 | 浙江大学 | Antibacterial compound and application thereof |
| CN101973979B (en) * | 2010-10-27 | 2013-04-17 | 浙江大学 | Antibacterial compound and application thereof |
| CN102707059A (en) * | 2012-05-31 | 2012-10-03 | 华中农业大学 | Colloidal gold test paper strip for residue detection of tylosin and tilmicosin, method for using colloidal gold test paper strip and application of colloidal gold test paper strip |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1471923A4 (en) | 2006-01-11 |
| EP1471923A1 (en) | 2004-11-03 |
| JP2005538927A (en) | 2005-12-22 |
| CA2471383A1 (en) | 2003-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6458771B1 (en) | Ketolide antibacterials | |
| USRE39836E1 (en) | Macrolide antinfective agents | |
| EP1642901B1 (en) | Novel erythromycin derivatives | |
| AU731301B2 (en) | Erythromycins and process for their preparation | |
| US7015203B2 (en) | Macrolides | |
| US6590083B1 (en) | Ketolide antibacterials | |
| US6939861B2 (en) | Amido macrolides | |
| US6794366B2 (en) | Macrolide antiinfective agents | |
| WO2003061671A1 (en) | Amido macrolides | |
| JP2005531603A (en) | 3-Descradinosyl-6-O-carbamoyl and 6-O-carbonoyl macrolide antibacterial agents | |
| EP1171448A2 (en) | Macrolide antiinfective agents | |
| MXPA00006604A (en) | Novel macrolides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2471383 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003561615 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003713257 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003713257 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003713257 Country of ref document: EP |