CN116178307A - 1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 - Google Patents
1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 Download PDFInfo
- Publication number
- CN116178307A CN116178307A CN202310030742.8A CN202310030742A CN116178307A CN 116178307 A CN116178307 A CN 116178307A CN 202310030742 A CN202310030742 A CN 202310030742A CN 116178307 A CN116178307 A CN 116178307A
- Authority
- CN
- China
- Prior art keywords
- dichlorophenyl
- piperazine
- reaction
- ethyl acetate
- piperazine hydrochloride
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- CYQFNNSFAGXCEC-UHFFFAOYSA-N 1-(2,3-dichlorophenyl)piperazine;hydrochloride Chemical compound [Cl-].ClC1=CC=CC(N2CC[NH2+]CC2)=C1Cl CYQFNNSFAGXCEC-UHFFFAOYSA-N 0.000 title claims abstract description 47
- 238000001308 synthesis method Methods 0.000 title claims abstract description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims abstract description 67
- 238000006243 chemical reaction Methods 0.000 claims abstract description 67
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims abstract description 50
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims abstract description 44
- UDQMXYJSNNCRAS-UHFFFAOYSA-N 2,3-dichlorophenylpiperazine Chemical compound ClC1=CC=CC(N2CCNCC2)=C1Cl UDQMXYJSNNCRAS-UHFFFAOYSA-N 0.000 claims abstract description 36
- -1 2-chloro-6-fluorotrichlorobenzyl Chemical group 0.000 claims abstract description 33
- 238000000034 method Methods 0.000 claims abstract description 28
- 229910000027 potassium carbonate Inorganic materials 0.000 claims abstract description 21
- DZGCGKFAPXFTNM-UHFFFAOYSA-N ethanol;hydron;chloride Chemical compound Cl.CCO DZGCGKFAPXFTNM-UHFFFAOYSA-N 0.000 claims abstract description 19
- 239000003960 organic solvent Substances 0.000 claims abstract description 14
- 238000004064 recycling Methods 0.000 claims abstract description 12
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 11
- 238000003786 synthesis reaction Methods 0.000 claims abstract description 11
- 239000007858 starting material Substances 0.000 claims abstract description 9
- NPXCSDPOOVOVDQ-UHFFFAOYSA-N 1,2-dichloro-3-fluorobenzene Chemical compound FC1=CC=CC(Cl)=C1Cl NPXCSDPOOVOVDQ-UHFFFAOYSA-N 0.000 claims abstract description 8
- 238000005660 chlorination reaction Methods 0.000 claims abstract description 7
- 238000006482 condensation reaction Methods 0.000 claims abstract description 6
- 230000017858 demethylation Effects 0.000 claims abstract description 4
- 238000010520 demethylation reaction Methods 0.000 claims abstract description 4
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 105
- 238000005406 washing Methods 0.000 claims description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 29
- 239000000463 material Substances 0.000 claims description 25
- 239000012074 organic phase Substances 0.000 claims description 23
- 239000000243 solution Substances 0.000 claims description 23
- 239000012065 filter cake Substances 0.000 claims description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 19
- 239000007788 liquid Substances 0.000 claims description 19
- 239000000047 product Substances 0.000 claims description 16
- 239000012043 crude product Substances 0.000 claims description 15
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 claims description 14
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 12
- 229910000041 hydrogen chloride Inorganic materials 0.000 claims description 12
- 238000003756 stirring Methods 0.000 claims description 12
- 238000004821 distillation Methods 0.000 claims description 11
- 239000000203 mixture Substances 0.000 claims description 11
- 239000006227 byproduct Substances 0.000 claims description 10
- 239000003054 catalyst Substances 0.000 claims description 10
- 238000000605 extraction Methods 0.000 claims description 10
- 238000004817 gas chromatography Methods 0.000 claims description 9
- 239000002910 solid waste Substances 0.000 claims description 9
- 230000002194 synthesizing effect Effects 0.000 claims description 9
- 238000001816 cooling Methods 0.000 claims description 8
- 238000002425 crystallisation Methods 0.000 claims description 8
- 230000008025 crystallization Effects 0.000 claims description 8
- 238000001035 drying Methods 0.000 claims description 8
- 238000010438 heat treatment Methods 0.000 claims description 8
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 claims description 8
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 7
- 239000007864 aqueous solution Substances 0.000 claims description 7
- 239000000460 chlorine Substances 0.000 claims description 7
- 229910052801 chlorine Inorganic materials 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 238000012544 monitoring process Methods 0.000 claims description 6
- 239000002904 solvent Substances 0.000 claims description 6
- 238000001914 filtration Methods 0.000 claims description 5
- 239000012452 mother liquor Substances 0.000 claims description 5
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical group ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 claims description 5
- 238000000967 suction filtration Methods 0.000 claims description 5
- 238000003810 ethyl acetate extraction Methods 0.000 claims description 4
- 238000005119 centrifugation Methods 0.000 claims description 3
- 238000010494 dissociation reaction Methods 0.000 claims description 3
- 230000005593 dissociations Effects 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 2
- 239000002585 base Substances 0.000 claims description 2
- 238000002156 mixing Methods 0.000 claims description 2
- 239000002994 raw material Substances 0.000 abstract description 12
- 238000000746 purification Methods 0.000 abstract description 9
- 230000008901 benefit Effects 0.000 abstract description 3
- 239000003814 drug Substances 0.000 abstract description 3
- 239000012847 fine chemical Substances 0.000 abstract description 3
- 239000002699 waste material Substances 0.000 abstract description 3
- 238000005516 engineering process Methods 0.000 abstract description 2
- 238000009835 boiling Methods 0.000 description 14
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 description 10
- 229960004372 aripiprazole Drugs 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 9
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 8
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 8
- 201000000980 schizophrenia Diseases 0.000 description 8
- 239000012535 impurity Substances 0.000 description 7
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 5
- 239000002920 hazardous waste Substances 0.000 description 5
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 238000007599 discharging Methods 0.000 description 4
- 229960003638 dopamine Drugs 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000011698 potassium fluoride Substances 0.000 description 4
- 235000003270 potassium fluoride Nutrition 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000006096 absorbing agent Substances 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 239000001103 potassium chloride Substances 0.000 description 3
- 235000011164 potassium chloride Nutrition 0.000 description 3
- 238000011085 pressure filtration Methods 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 230000008016 vaporization Effects 0.000 description 3
- HVKCZUVMQPUWSX-UHFFFAOYSA-N 1-bromo-2,3-dichlorobenzene Chemical compound ClC1=CC=CC(Br)=C1Cl HVKCZUVMQPUWSX-UHFFFAOYSA-N 0.000 description 2
- BRPSAOUFIJSKOT-UHFFFAOYSA-N 2,3-dichloroaniline Chemical compound NC1=CC=CC(Cl)=C1Cl BRPSAOUFIJSKOT-UHFFFAOYSA-N 0.000 description 2
- OACPOWYLLGHGCR-UHFFFAOYSA-N 2-chloro-6-fluorobenzaldehyde Chemical compound FC1=CC=CC(Cl)=C1C=O OACPOWYLLGHGCR-UHFFFAOYSA-N 0.000 description 2
- XNTIGDVFBDJLTQ-UHFFFAOYSA-N 2-chloro-6-fluorobenzoic acid Chemical compound OC(=O)C1=C(F)C=CC=C1Cl XNTIGDVFBDJLTQ-UHFFFAOYSA-N 0.000 description 2
- 102000049773 5-HT2A Serotonin Receptor Human genes 0.000 description 2
- 102000017911 HTR1A Human genes 0.000 description 2
- 101150015707 HTR1A gene Proteins 0.000 description 2
- 208000019022 Mood disease Diseases 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 239000003693 atypical antipsychotic agent Substances 0.000 description 2
- 229940127236 atypical antipsychotics Drugs 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- 235000019441 ethanol Nutrition 0.000 description 2
- 238000009776 industrial production Methods 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- 229940054010 other antipsychotics in atc Drugs 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000003444 phase transfer catalyst Substances 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical class C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- KMAQZIILEGKYQZ-UHFFFAOYSA-N 1-chloro-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(Cl)=C1 KMAQZIILEGKYQZ-UHFFFAOYSA-N 0.000 description 1
- YMDZDFSUDFLGMX-UHFFFAOYSA-N 2-chloro-n-(2-chloroethyl)ethanamine;hydron;chloride Chemical compound [Cl-].ClCC[NH2+]CCCl YMDZDFSUDFLGMX-UHFFFAOYSA-N 0.000 description 1
- 108010072564 5-HT2A Serotonin Receptor Proteins 0.000 description 1
- 102000040125 5-hydroxytryptamine receptor family Human genes 0.000 description 1
- 108091032151 5-hydroxytryptamine receptor family Proteins 0.000 description 1
- URHLNHVYMNBPEO-UHFFFAOYSA-N 7-(4-bromobutoxy)-3,4-dihydro-1h-quinolin-2-one Chemical compound C1CC(=O)NC2=CC(OCCCCBr)=CC=C21 URHLNHVYMNBPEO-UHFFFAOYSA-N 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 101150049660 DRD2 gene Proteins 0.000 description 1
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 1
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 1
- 108050004812 Dopamine receptor Proteins 0.000 description 1
- 102000015554 Dopamine receptor Human genes 0.000 description 1
- 101150104779 HTR2A gene Proteins 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 102000003834 Histamine H1 Receptors Human genes 0.000 description 1
- 108090000110 Histamine H1 Receptors Proteins 0.000 description 1
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 230000002932 anti-schizophrenic effect Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000001335 demethylating effect Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000010413 mother solution Substances 0.000 description 1
- 231100000957 no side effect Toxicity 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000036632 reaction speed Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 238000009834 vaporization Methods 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/06—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals
- C07D295/073—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals with the ring nitrogen atoms and the substituents separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明属于精细化工领域,涉及精细医药中间体的合成和提纯技术。本发明公开了一种1‑(2,3‑二氯苯基)哌嗪盐酸盐的合成方法:以2‑氯‑6‑氟三氯苄为起始原料,通过氯化脱甲基得到中间体2,3‑二氯氟苯;中间体2,3‑二氯氟苯和无水哌嗪在碳酸钾及有机溶剂DMSO体系中进行缩合反应,得到1‑(2,3‑二氯苯基)哌嗪;1‑(2,3‑二氯苯基)哌嗪和氯化氢乙醇溶液成盐,得到1‑(2,3‑二氯苯基)哌嗪盐酸盐。本发明1‑(2,3‑二氯苯基)哌嗪盐酸盐的合成和提纯方法,原料成本低,反应步骤少,产品收率高,并可实现循环经济和不新增三废排放。
Description
技术领域
本发明属于精细化工领域,涉及精细医药中间体的合成和提纯技术,特别是涉及一种1-(2,3-二氯苯基)哌嗪盐酸盐合成方法。
背景技术
日本大冢公司开发的阿立哌唑是一种多巴胺系统稳定剂,临床主治各种急慢性精神分裂症和情感障碍,其化学结构如下:

阿立哌唑为喹啉酮衍生物,属第三代非典型抗精神病药物,对精神分裂症阳性和阴性症状均有显著疗效。与其它抗精神病药物拮抗多巴胺受体不同,阿立哌唑不但能部分激动多巴胺D2受体,使多巴胺信号达到稳定正常水平,而且也是5-HT受体的部分激动剂。
阿立哌唑是一种多巴胺系统稳定剂,对精神分裂症阳性和阴性症状均有显著疗效,用于治疗精神分裂症,能够显著改善这类精神分裂症状,但却没有其他抗精神病药常见的一些副作用,例如体重的增加和非自主性肌肉活动等,但可能会导致头痛、焦虑及失眠等症状。其为喹啉酮衍生物,属第三代非典型抗精神病药物。阿立哌唑与多巴胺D2、D3、5-HT1A和5-HT2A受体有很高的亲和力,与D4、5-HT2c、5-HT7、a1、H1受体及5-HT重吸收位点具有中度亲和力,它通过对D2和5-HT1A受体的部分激动作用及对5-HT2A受体的拮抗作用来产生抗精神分裂症作用。
阿立哌唑自发明至今,对于它的合成许多科学家都做的非常深入,经文献查证,目前主要的合成路线如下所示:

目前阿立哌唑最经典、最经济的合成路线是:7-(4-溴丁氧基)-3,4二氢喹诺酮和碘化钠在乙腈中回流,加入1-(2,3二氯苯基)哌嗪和三乙胺回流,蒸出溶剂,得到的物质溶于氯仿,用水洗,无水硫酸镁干燥,用乙醇重结晶即可。
目前已知阿立哌唑合成都需要以1-(2,3二氯苯基)哌嗪为主要中间体,1-(2,3-二氯苯基)哌嗪或其盐酸盐是合成阿立哌唑的主要原料。1-(2,3-二氯苯基)哌嗪的合成有许多种方法,有由于多种路线回收率低,而且使用的原材料成本高昂,不适合工业化生产。
据文献报道,目前主要的合成路线由以下几种:
路线一:据文献《化学试剂》2006,28(8):507-509《1-(2,3-二氯苯基)哌嗪的合成及其工艺研究》报道,由间硝基氯苯和哌嗪在碳酸钾和正丁醇中回流,制备得到邻氯硝基苯哌嗪,然后经酯化和还原,最后通过重氮化反应和脱酯反应制得目标化合物。其第一步收率84%,第二步收率98%,第三步收率83%,第四步收率78%,总收率53.3%。这也是目前生产上所采用的路线。
路线一的反应式如下:

路线二:采用2,3-二氯溴苯为原料,哌嗪在铜为催化剂下反应制备而得目标化合物,该路线步骤少而简单,但由于所用原材料2,3-二氯溴苯价格高昂,而且整个反应收率不到20%,因此不适合于产业化。
路线二的反应式如下:

路线三:采用二乙醇胺为原料,以氯仿为溶剂,和二氯亚砜反应得到1、4-二氯二乙胺盐酸盐,在有氢氧化钾水溶液中和2,3-二氯苯胺反应得到1-(2,3-二氯苯基)哌嗪盐酸盐,第一步收率78%,第二步收率38.7%,参考文献见《精细化工中间体》2008,37(7):18-20《1-(2,3-二氯苯基)哌嗪的合成新方法》。
路线三的反应式如下:

路线四:第四种合成路线是以2,3-二氯苯胺为原料,和双-(2-氯乙基)胺盐酸盐在相转移催化剂下进行环合反应,收率65.6%,含量99.67%(参考文献见:中国发明专利CN201210286068.1一种1-(2,3-二氯苯基)哌嗪盐酸盐的制备)。该路线合成路线短,但需要使用相转移催化剂,从生产成本来说不适合工业化生产。
路线四的反应式如下:

2-氯-6-氟三氯苄是生产2-氯-6-氟苯甲醛生产的副产物,含量≥95%,2-氯-6-氟二氯苄含量≤1.5%,水分≤0.05%,其它最大未知单杂小于2.0%;虽然2-氯-6-氟三氯苄可用于制备2-氯-6-氟苯甲酸,但2-氯-6-氟苯甲酸还没有规模化的市场需求,需求量极低。除此之外,2-氯-6-氟三氯苄目前也缺乏其它商业化的用途。
发明内容
本发明要解决的技术问题是提供一种1-(2,3-二氯苯基)哌嗪盐酸盐合成和提纯方法。
为了解决上述技术问题,本发明提供一种1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法:
以2-氯-6-氟三氯苄为起始原料,通过氯化脱甲基(深度氯化脱甲基)得到中间体2,3-二氯氟苯;中间体2,3-二氯氟苯和无水哌嗪在碳酸钾及有机溶剂DMSO体系中进行缩合反应,得到1-(2,3-二氯苯基)哌嗪;1-(2,3-二氯苯基)哌嗪和氯化氢乙醇溶液进行成盐,得到1-(2,3-二氯苯基)哌嗪盐酸盐产品。
步骤1:

步骤2:
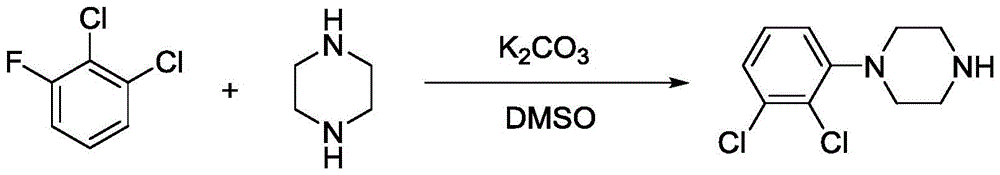
步骤3:

作为本发明的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法的改进,依次包括以下步骤:
1)、将作为起始原料的2-氯-6-氟三氯苄加热至融化,然后投入反应釜中;
2)、向反应釜内投入催化剂,然后升温至140~150℃,再通入氯气(液氯通过汽化所得)进行光氯化反应,通过气相色谱对反应进程进行监控,当反应釜内物料中2-氯-6-氟三氯苄含量≤1.0%后停止反应,得反应后物料Ⅰ;
催化剂的重量是作为起始原料的2-氯-6-氟三氯苄的0.09~0.11%(优选0.1%);
上述%均为重量%;
氯气:2-氯-6-氟三氯苄=1:3.45~3.55(优选1:3.5)的重量比;
光氯化反应时采用的是主要波长为365nm的紫外光;
说明:光氯化反应过程中产生的氯化氢进入氯化氢吸收器遇水后最终形成作为副产的盐酸;
3)、将步骤2)所得的反应后物料Ⅰ通过减压(-0.095MPa左右的压力)精馏,收集塔顶馏份,得作为中间体的2,3-二氯氟苯(纯度大于99.5%)和作为副产品的四氯化碳;
说明:
收集66-70℃的塔顶馏份,为2,3-二氯氟苯;
收集60-65℃的塔顶馏份,为四氯化碳;
4)、将步骤3)所得2,3-二氯氟苯以及碳酸钾、无水哌嗪和作为有机溶剂的DMSO投入反应釜(缩合反应釜),升温至120~130℃反应,并通过气相色谱对反应进程进行监控,当2,3-二氯氟苯剩余至≤1.0%,停止反应,得反应后物料Ⅱ;
2,3-二氯氟苯:碳酸钾:无水哌嗪=1.5~1.7:1:1.5~1.7重量比;
2,3-二氯氟苯:DMSO=1:1~2.5重量比;
5)、于抽滤桶内,将步骤4)所得反应后物料Ⅱ降温至60±10℃,加入作为有机溶剂的乙酸乙酯,充分搅拌,压滤(放料压滤),滤饼用乙酸乙酯进行洗涤,收集有机相(压滤所得滤液+洗涤后所得的洗涤液)和固体废渣;
其中固体废渣主要是氟化钾和碳酸钾,作为危废进行处置;
作为有机溶剂的乙酸乙酯:步骤4)所得反应后物料Ⅱ=0.3-0.4:1的重量比
洗涤滤饼用的乙酸乙酯:步骤4)所得反应后物料Ⅱ=0.1-0.2:1的重量比;
6)、将步骤5)所得有机相(压滤所得滤液+洗涤后所得的洗涤液)进行减压(-0.095MPa左右的压力)蒸馏,并根据沸点的差异性,分别回收乙酸乙酯、DMSO与哌嗪的混合物以及1-(2,3-二氯苯基)哌嗪粗品;
说明:
收集沸点为55-65℃的成分,为乙酸乙酯;
收集沸点为66-160℃的成分,为DMSO与哌嗪的混合物;
收集沸点为161-220℃的成分,为1-(2,3-二氯苯基)哌嗪粗品;
回收的DMSO与哌嗪混合物返回至步骤4)进行循环套用,回收的乙酸乙酯返回至步骤5)进行循环套用;因此,此两者不需要进行精馏提纯;
7)、将步骤6)所得1-(2,3-二氯苯基)哌嗪粗品投入反应釜,先加入作为溶剂的无水乙醇充分搅拌至溶解(全部溶解),再加入氯化氢乙醇溶液进行析晶,直至pH为5-6时停止滴加氯化氢乙醇溶液,离心,并用无水乙醇对滤饼进行洗涤(洗涤两次),得1-(2,3-二氯苯基)哌嗪盐酸盐湿品,干燥(通过烘干)后,得1-(2,3-二氯苯基)哌嗪盐酸盐产品。
氯化氢乙醇溶液中,氯化氢的质量浓度为16-20%;
作为溶剂的无水乙醇:1-(2,3-二氯苯基)哌嗪粗品=2.9-3.5:1的重量比;
用于洗涤滤饼的无水乙醇:1-(2,3-二氯苯基)哌嗪粗品=0.9~1.1:1的重量比。
作为本发明的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法的进一步改进,还包括以下步骤:
8)、步骤7)离心所得母液以及无水乙醇对滤饼进行洗涤所得的洗涤液合并后,通过常压精馏,回收无水乙醇返回步骤7)进行循环套用;
9)、精馏后釜底液通过碱游离和乙酸乙酯萃取,将1-(2,3-二氯苯基)哌嗪进行回收,萃取所得有机相返回步骤6),跟步骤5)所得有机相进行混合,通过减压蒸馏,回收乙酸乙酯和1-(2,3-二氯苯基)哌嗪粗品;
碱游离和乙酸乙酯萃取为:往精馏釜底液中加入20%的碳酸钾水溶液,搅拌反应1±0.2h后,再加入乙酸乙酯进行萃取,其中碳酸钾水溶液与精馏后釜底液的用量比关系为1:1.0-2.0(质量比);乙酸乙酯与精馏后釜底液的用量比关系为1.0-2.0:1(质量比)。
作为本发明的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法的进一步改进:催化剂为三氯化磷。
本发明针对现有的技术问题,开发出一条新的合成路线,以2-氯-6-氟三氯苄为主要起始原料,通过氯化脱甲基、缩合、成盐重结晶等步骤,得到1-(2,3-二氯苯基)哌嗪盐酸盐产品。
本发明的1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法具有工艺简单、产品收率高和质量优等特点,而且还可以显著降低1-(2,3-二氯苯基)哌嗪盐酸盐的生产成本,具有很好的经济和社会效益。因此,本发明给1-(2,3-二氯苯基)哌嗪盐酸盐合成增加了一个降低原料成本和更加简洁的合成路线。本发明所得的1-(2,3-二氯苯基)哌嗪盐酸盐后续按照常规方式可用于治疗各种急慢性精神分裂症和情感障碍的药物阿立哌唑的合成。
本发明1-(2,3-二氯苯基)哌嗪盐酸盐的合成和提纯方法,原料成本低,反应步骤少,产品收率高,并可实现循环经济和不新增三废排放。该方法以副产2-氯-6-氟三氯苄为起始原料。本发明针对以副产2-氯-6-氟三氯苄为原料所合成产物的成份复杂、杂质含量高的特点,采用通过精馏+成盐重结晶的工艺路线,对反应产物进行提纯,得高纯度1-(2,3-二氯苯基)哌嗪盐酸盐。
附图说明
下面结合附图对本发明的具体实施方式作进一步详细说明。
图1为本发明的工艺流程图。
具体实施方式
下面结合具体实施例对发明作进一步说明,但不作为对发明的限制,凡基于本发明所述的技术均属于本发明的组成部分。
本发明所述的原材料:
2-氯-6-氟三氯苄,是生产2-氯-6-氟苯甲醛的副产物,通过减压精馏所得;含量95%。
无水乙醇,工业级,市售;
乙酸乙酯,工业级,市售;
DMSO(二甲基亚砜),工业级,市售;
液氯,工业级,市售;
三氯化磷,工业级,市售;
碳酸钾,工业级,市售;
无水哌嗪,工业级,市售;
氯化氢乙醇,自制,氯化氢含量18%;
室温,一般指15-25℃。
实施例1、
1)、将608公斤2-氯-6-氟三氯苄(含量95%)加热(约55-65℃)至融化,然后投入反应釜(光氯化反应器)中。
2)、向步骤1)所述的反应釜中投入0.6公斤催化剂三氯化磷,然后升温至140~150℃,再通入174公斤氯气(液氯经过汽化所得)进行光氯化反应,光氯化反应时采用的是主要波长为365nm的紫外光,功率为1KW的高压汞灯3支。
通过气相色谱对反应进程进行监控,当反应釜内物料中2-氯-6-氟三氯苄含量0.82%(质量%)时停止反应,在上述光氯化反应条件下,反应时间约为24小时,得767公斤反应后物料Ⅰ;
光氯化反应过程中产生的氯化氢气体被排放至氯化氢吸收器内,与水混合后,形成作为副产的盐酸。
3)、将步骤2)所得的反应后物料Ⅰ送入减压精馏塔进行减压(-0.095MPa左右的压力)精馏,收集塔顶馏份,具体为:
收集66-70℃的塔顶馏份,得382公斤中间体2,3-二氯氟苯(纯度99.6%,折合收率为94.22%);
收集60-65℃的塔顶馏份,得355公斤作为副产品的四氯化碳。
4)、将步骤3)所得382公斤中间体2,3-二氯氟苯以及240公斤碳酸钾、398公斤无水哌嗪和750公斤有机溶剂DMSO投入缩合反应釜,升温至120-130℃反应,并通过气相色谱对反应进程进行监控,当2,3-二氯氟苯剩余至投料量的0.48%(重量%),停止反应,得1720公斤反应后物料Ⅱ;反应产生的约50公斤二氧化碳成为尾气。
此步骤4)的反应时间约为24小时。
5)、将步骤4)所得反应后物料Ⅱ降温至60℃左右后进入抽滤桶内,加入700公斤乙酸乙酯,充分搅拌,并降到室温,放料压滤,分别得滤饼和滤液;滤饼用300公斤乙酸乙酯分两次进行洗涤,将洗涤后所得的洗涤液与滤液合并,共收集2495公斤有机相;洗涤后的滤饼为固体废渣,重225公斤;
固体废渣主要是氟化钾和碳酸钾,作为危废进行处置。
6)、将步骤5)所得有机相送入蒸馏釜进行减压(-0.095MPa的压力)蒸馏,并根据沸点的差异性,分别收集,具体为:
收集沸点为55-65℃的成分,回收得到987公斤乙酸乙酯;
收集沸点为66-160℃的成分,回收得到950公斤DMSO与哌嗪混合物(其中DMSO占78.95%、哌嗪占21.05%);此%为重量%;
收集沸点为161-220℃的成分,回收得到507公斤1-(2,3-二氯苯基)哌嗪粗品,
还有51公斤的蒸馏釜底残液作为危废处置。
上述回收的DMSO与哌嗪混合物返回至步骤4)进行循环套用,因此不需要进行精馏提纯;
同理,上述回收的乙酸乙酯返回至步骤5)进行循环套用,因此不需要进行精馏提纯;
7)、将步骤6)所得1-(2,3-二氯苯基)哌嗪粗品507公斤投入反应釜(成盐析晶釜),先加入1500公斤无水乙醇充分搅拌下使得粗品全部溶解,再加入氯化氢乙醇溶液进行析晶,直至pH为5-6,停止加入氯化氢乙醇溶液(共加入490公斤氯化氢乙醇溶液,氯化氢含量18%),离心,并用500公斤无水乙醇分两次对滤饼进行洗涤,得1-(2,3-二氯苯基)哌嗪盐酸盐湿品,60-70℃烘干120分钟后得1-(2,3-二氯苯基)哌嗪盐酸盐产品530公斤。1-(2,3-二氯苯基)哌嗪盐酸盐含量为99.89%,最大单杂为0.082%;
离心所得滤液与两次洗涤所得洗涤液进行合并,共计2448公斤作为母液;
说明:1-(2,3-二氯苯基)哌嗪盐酸盐湿品烘干过程中,少量的乙醇被挥发了。
8)、步骤7)所得母液进入精馏塔,通过常压精馏,回收2380公斤无水乙醇(返回步骤7)进行循环套用;所得的精馏釜底液主要成分为1-(2,3-二氯苯基)哌嗪盐酸盐和焦油为杂质。
9)、将步骤8)所得精馏釜底液(68公斤)送入游离萃取釜内,再加入90公斤20%的碳酸钾溶液进行游离,即,1-(2,3-二氯苯基)哌嗪盐酸盐和碳酸钾进行反应,得到氯化钾和1-(2,3-二氯苯基)哌嗪。因为成盐后不溶于有机溶剂,因此需要游离后通过有机溶剂萃取。
而后再加入100公斤乙酸乙酯进行萃取,分别形成位于上层的有机相和下层无机相(主要为含氯化钾和碳酸钾的水溶液);将萃取所得有机相150公斤返回步骤6),跟步骤5)所得有机相进行混合,一起通过减压蒸馏,回收乙酸乙酯和1-(2,3-二氯苯基)哌嗪粗品。
实施例1的成品收率80.76%,此数据未包括步骤9)精馏釜底液游离萃取所得1-(2,3-二氯苯基)哌嗪,如果算上此,则收率可达89.63%。
成品收率=1-(2,3-二氯苯基)哌嗪盐酸盐的摩尔量/2-氯-6-氟三氯苄的摩尔量。
实施例2、
1)~3)、同实施例1的步骤1)~步骤3)。
4)、将步骤3)所得382公斤中间体2,3-二氯氟苯、240公斤碳酸钾、198公斤无水哌嗪、200公斤有机溶剂DMSO和实施例1步骤6)蒸馏所得950公斤DMSO与哌嗪混合物(其中DMSO占78.95%、哌嗪21.05%)投入反应釜,升温至120-130℃反应,并通过气相色谱对反应进程进行监控,当2,3-二氯氟苯剩余至投料量的0.67%,停止反应,得1920公斤反应后物料Ⅱ;反应产生的50公斤二氧化碳成为尾气。
5)、将步骤4)所得反应后物料Ⅱ降温至60℃左右,加入实施例1步骤6)精馏回收所得700公斤乙酸乙酯,充分搅拌,并降到室温,放料压滤,滤饼用实施例1步骤6)精馏回收所得287公斤乙酸乙酯和13公斤新鲜的乙酸乙酯分两次进行洗涤,将洗涤后所得的洗涤液与滤液合并,共收集2695公斤有机相和225公斤固体废渣;
其中固体废渣主要是氟化钾和碳酸钾,作为危废进行处置。
6)、将步骤5)所得有机相和实施例1步骤8)所得萃取有机相150公斤混合物送入蒸馏釜进行减压(-0.095MPa的压力)蒸馏,并根据沸点的差异性,分别收集,具体为:
收集沸点为55-65℃的成分,回收得到1085公斤乙酸乙酯;
收集沸点为66-160℃的成分,回收得到950公斤DMSO与哌嗪混合物(其中DMSO占78.84%、哌嗪占21.16%);此%为重量%;
收集沸点为161-220℃的成分,回收得到555公斤1-(2,3-二氯苯基)哌嗪粗品,
还有53公斤的蒸馏釜底残液作为危废处置。
上述回收的DMSO与哌嗪混合物返回至步骤4)进行循环套用,因此不需要进行精馏提纯;
同理,上述回收的乙酸乙酯返回至步骤5)进行循环套用,因此不需要进行精馏提纯;
7)、将步骤6)所得1-(2,3-二氯苯基)哌嗪粗品555公斤投入反应釜(成盐析晶釜),先加入先加入实施例1步骤8)精馏回收的1500公斤无水乙醇充分搅拌下使得粗品全部溶解,再加入氯化氢乙醇溶液进行析晶,直至pH为5-6,停止滴加氯化氢乙醇溶液(共加535公斤氯化氢乙醇溶液,氯化氢含量18%),离心,并用500公斤无水乙醇分两次对滤饼进行洗涤,得1-(2,3-二氯苯基)哌嗪盐酸盐湿品,60-70℃烘干120分钟后得1-(2,3-二氯苯基)哌嗪盐酸盐产品582公斤。产品1-(2,3-二氯苯基)哌嗪盐酸盐含量为99.92%,最大单杂为0.067%;
抽滤所得滤液与两次洗涤所得洗涤液进行合并,共计2482公斤作为母液。
8)、步骤7)所得母液,通过常压精馏,回收2410公斤无水乙醇(返回步骤7)进行循环套用;所得的精馏釜底液主要成分为1-(2,3-二氯苯基)哌嗪盐酸盐和焦油为杂质。
9)、将步骤8)所得精馏釜底液(72公斤)送入游离萃取釜内,再加入90公斤20%的碳酸钾溶液进行游离,再加入100公斤乙酸乙酯进行萃取,分别形成位于上层的有机相和下层无机相(主要要含氯化钾和碳酸钾的水溶液);取萃取所得有机相150公斤返回步骤6),跟步骤5)所得有机相进行混合,一起通过减压蒸馏,回收乙酸乙酯和1-(2,3-二氯苯基)哌嗪粗品。
实施例2的成品收率88.72%,此数据未包括步骤9)精馏釜底液游离萃取所得1-(2,3-二氯苯基)哌嗪。
对比例1、
步骤1)~步骤3)同实施例1;
4)、将实施例1步骤4)中的“DMSO”改成正丁醇,重量保持不变,仍然为750公斤;其余等同于实施例1的步骤4);具体如下:
将实施例1步骤3)所得382公斤中间体2,3-二氯氟苯以及240公斤碳酸钾、398公斤无水哌嗪和750公斤有机溶剂正丁醇投入缩合反应釜,升温至120-130℃反应,并通过气相色谱对反应进程进行监控,反应24h时,2,3-二氯氟苯剩余投料量的70.52%(重量%),继续反应24h,2,3-二氯氟苯剩余投料量的63.16%(重量%),停止反应,得反应后物料Ⅱ;
即,通过48h反应,原料转化率仅为36.84%。
5)、将步骤4)所得反应后物料Ⅱ降温至60℃左右后进入抽滤桶内,加入700公斤乙酸乙酯,充分搅拌,并降到室温,放料压滤,分别得滤饼和滤液;滤饼用300公斤乙酸乙酯分两次进行洗涤,将洗涤后所得的洗涤液与滤液合并,收集有机相;洗涤后的滤饼为固体废渣,其中固体废渣主要是氟化钾和碳酸钾,作为危废进行处置。
6)、将步骤5)所得有机相送入蒸馏釜进行减压(-0.095MPa的压力)蒸馏,并根据沸点的差异性,分别收集,收集沸点为160-220℃的成分,回收得到124公斤1-(2,3-二氯苯基)哌嗪粗品。
7)、将步骤6)所得1-(2,3-二氯苯基)哌嗪粗品投入反应釜(成盐析晶釜),先加入370公斤无水乙醇充分搅拌下使得粗品全部溶解,再加入氯化氢乙醇溶液进行析晶,直至pH为5-6,停止滴加氯化氢乙醇溶液(共滴加120公斤氯化氢乙醇溶液,氯化氢含量18%),离心,并用125公斤无水乙醇分两次对滤饼进行洗涤,得1-(2,3-二氯苯基)哌嗪盐酸盐湿品,60-70℃烘干120分钟后得1-(2,3-二氯苯基)哌嗪盐酸盐产品102公斤。产品1-(2,3-二氯苯基)哌嗪盐酸盐含量为97.26%,最大单杂为2.43%;
对比例1的成品收率15.55%,而且质量未达到规定的要求。
对比例2、
步骤1)同实施例1;
2)、将实施例1步骤2)中的催化剂由“三氯化磷”改成“二氯亚砜”,用量保持不变,仍然为0.6公斤;其余等同于实施例1的步骤4),具体如下:
向步骤1)所述的反应釜中投入0.6公斤催化剂二氯亚砜,然后升温至140~150℃,再通入280公斤氯气(液氯经过汽化所得)进行光氯化反应,光氯化反应时采用的是主要波长为365nm的紫外光,功率为1KW的高压汞灯3支。
通过气相色谱对反应进程进行监控,当反应釜内物料中2-氯-6-氟三氯苄含量0.96%时停止反应,在上述光氯化反应条件下,反应时间约为72小时,得765公斤反应后物料Ⅰ;
光氯化反应过程中产生的氯化氢气体被排放至氯化氢吸收器内,与水混合后,形成作为副产的盐酸。
此案例显示:反应速度很慢,反应周期从24h延长到72h,并且收率明显下降,主要是焦油量增多,氯气消耗量增加;
3)、将步骤2)所得的反应后物料Ⅰ送入减压精馏塔进行减压(-0.095MPa左右的压力)精馏,收集塔顶馏份,具体为:
收集66-70℃的塔顶馏份,得342公斤中间体2,3-二氯氟苯(纯度99.4%,折合收率为87.44%),远低于实施例1的收率。而且氯气消耗量增加50%左右。
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
Claims (7)
1.1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法,其特征在于:
以2-氯-6-氟三氯苄为起始原料,通过氯化脱甲基得到中间体2,3-二氯氟苯;中间体2,3-二氯氟苯和无水哌嗪在碳酸钾及有机溶剂DMSO体系中进行缩合反应,得到1-(2,3-二氯苯基)哌嗪;1-(2,3-二氯苯基)哌嗪和氯化氢乙醇溶液成盐,得到1-(2,3-二氯苯基)哌嗪盐酸盐。
2.根据权利要求1所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于依次包括以下步骤:
1)、将作为起始原料的2-氯-6-氟三氯苄加热至融化,然后投入反应釜中;
2)、向反应釜内投入催化剂,然后升温至140~150℃,再通入氯气进行光氯化反应,通过气相色谱对反应进程进行监控,当反应釜内物料中2-氯-6-氟三氯苄含量≤1.0%后停止反应,得反应后物料Ⅰ;
催化剂的重量是作为起始原料的2-氯-6-氟三氯苄的0.09~0.11%;
氯气:2-氯-6-氟三氯苄=1:3.45~3.55的重量比;
光氯化反应时采用的是主要波长为365nm的紫外光;
3)、将步骤2)所得的反应后物料Ⅰ通过减压精馏,收集塔顶馏份,得作为中间体的2,3-二氯氟苯和作为副产品的四氯化碳;
4)、将步骤3)所得的2,3-二氯氟苯以及碳酸钾、无水哌嗪和作为有机溶剂的DMSO投入反应釜,升温至120~130℃反应,并通过气相色谱对反应进程进行监控,当2,3-二氯氟苯剩余至≤1.0%,停止反应,得反应后物料Ⅱ;
2,3-二氯氟苯:碳酸钾:无水哌嗪=1.5~1.7:1:1.5~1.7重量比;
2,3-二氯氟苯:DMSO=1:1~2.5重量比;
5)、于抽滤桶内,将步骤4)所得反应后物料Ⅱ降温至60±10℃,加入作为有机溶剂的乙酸乙酯,充分搅拌,压滤,滤饼用乙酸乙酯进行洗涤,收集有机相和固体废渣;
6)、将步骤5)所得有机相进行减压蒸馏,分别回收乙酸乙酯、DMSO与哌嗪的混合物以及1-(2,3-二氯苯基)哌嗪粗品;
7)、将步骤6)所得1-(2,3-二氯苯基)哌嗪粗品投入反应釜,先加入作为溶剂的无水乙醇充分搅拌至溶解,再加入氯化氢乙醇溶液进行析晶,直至pH为5-6时停止滴加氯化氢乙醇溶液,离心,并用无水乙醇对滤饼进行洗涤,得1-(2,3-二氯苯基)哌嗪盐酸盐湿品,干燥,得1-(2,3-二氯苯基)哌嗪盐酸盐。
3.根据权利要求2所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于还包括以下步骤:
8)、步骤7)离心所得母液以及无水乙醇对滤饼进行洗涤所得的洗涤液合并后,通过常压精馏,回收无水乙醇返回步骤7)进行循环套用;
9)、精馏后釜底液通过碱游离和乙酸乙酯萃取,将1-(2,3-二氯苯基)哌嗪进行回收,萃取所得有机相返回步骤6),跟步骤5)所得有机相进行混合,通过减压蒸馏,回收乙酸乙酯和1-(2,3-二氯苯基)哌嗪粗品;
碱游离和乙酸乙酯萃取为:往精馏釜底液中加入20%的碳酸钾水溶液,搅拌反应1±0.2h后,再加入乙酸乙酯进行萃取,其中碳酸钾水溶液与精馏后釜底液的用量比关系为1:1.0~2.0;乙酸乙酯与精馏后釜底液的用量比关系为1.0~2.0:1。
4.根据权利要求2或3所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于:
催化剂为三氯化磷。
5.根据权利要求4所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于:
所述步骤5)中:
作为有机溶剂的乙酸乙酯:步骤4)所得反应后物料Ⅱ=0.3-0.4:1的重量比;
洗涤滤饼用的乙酸乙酯:步骤4)所得反应后物料Ⅱ=0.1-0.2:1的重量比。
6.根据权利要求5所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于:
所述步骤6)中:
回收的DMSO与哌嗪混合物返回至步骤4)进行循环套用,回收的乙酸乙酯返回至步骤5)进行循环套用。
7.根据权利要求6所述的1-(2,3-二氯苯基)哌嗪盐酸盐合成方法,其特征在于:
所述步骤7)中:
氯化氢乙醇溶液中,氯化氢的质量浓度为16-20%;
作为溶剂的无水乙醇:1-(2,3-二氯苯基)哌嗪粗品=2.9-3.5:1的重量比;
用于洗涤滤饼的无水乙醇:1-(2,3-二氯苯基)哌嗪粗品=0.9~1.1:1的重量比。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202310030742.8A CN116178307B (zh) | 2023-01-10 | 2023-01-10 | 1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202310030742.8A CN116178307B (zh) | 2023-01-10 | 2023-01-10 | 1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN116178307A true CN116178307A (zh) | 2023-05-30 |
| CN116178307B CN116178307B (zh) | 2024-05-14 |
Family
ID=86435868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202310030742.8A Active CN116178307B (zh) | 2023-01-10 | 2023-01-10 | 1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN116178307B (zh) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115340494A (zh) * | 2022-07-27 | 2022-11-15 | 安徽修一制药有限公司 | 一种高纯度阿立哌唑的合成方法 |
-
2023
- 2023-01-10 CN CN202310030742.8A patent/CN116178307B/zh active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115340494A (zh) * | 2022-07-27 | 2022-11-15 | 安徽修一制药有限公司 | 一种高纯度阿立哌唑的合成方法 |
Non-Patent Citations (1)
| Title |
|---|
| CHRISTIAN BORCH JACOBSEN, MORTEN MELDAL, AND FREDERIK DINESS: "Mechanism and Scope of Base-Controlled Catalyst-Free N-Arylation of Amines with Unactivated Fluorobenzenes", CHEM. EUR. J., vol. 2017, no. 23, 13 December 2016 (2016-12-13), pages 846 - 851 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116178307B (zh) | 2024-05-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1762556A1 (en) | Process for producing dichloropropanol from glycerol | |
| CN101070298B (zh) | 一种ε-己内酰胺的提纯精制方法 | |
| JP2012051914A (ja) | グリセロールからジクロロプロパノールを製造するための方法であって、該グリセロールが最終的にバイオディーゼルの製造における動物性脂肪の転化から生じる方法 | |
| US20080249324A1 (en) | Process For Preparing 1,3-Dibromoacetone, 1-3-Dichloroacetone and Epichlorohydrin | |
| CN105348172A (zh) | (s)-1-(4-甲氧基-3-乙氧基)苯基-2-甲磺酰基乙胺的制备及阿普斯特的制备方法 | |
| CN107915631B (zh) | 一种连续合成4-氯乙酰乙酸甲酯的后处理方法 | |
| CN111393403A (zh) | 一种碳酸亚乙烯酯的制备方法 | |
| CN114539048B (zh) | 一种卡龙酸酐中间体及卡龙酸酐的制备方法 | |
| CN109053627B (zh) | 头孢曲松钠二氯甲烷母液中2-巯基苯并噻唑、三乙胺和二氯甲烷的综合回收方法 | |
| CN116178307B (zh) | 1-(2,3-二氯苯基)哌嗪盐酸盐的合成方法 | |
| CN1356903A (zh) | 含哌嗪环化合物的新型合成和结晶方法 | |
| CN106631823B (zh) | 一种氯卡色林中间体的制备方法 | |
| CN108299149B (zh) | 高纯度oled中间体1-溴芘的合成法 | |
| CN108947919A (zh) | 一种抗痛风药Lesinurad的新型制备方法及其关键中间体 | |
| CN113773322B (zh) | 一种Filgotinib的制备方法 | |
| CN102702060A (zh) | 一种维那卡兰中间体的拆分母液中副产物的外消旋化回收方法 | |
| CN107162889A (zh) | 一种2‑叔戊基蒽醌制备与提纯的方法 | |
| CN115124466A (zh) | 一种四氢罂粟碱盐酸盐的合成方法 | |
| CN111269149B (zh) | 一种5-(3,3-二甲基胍基)-2-氧代戊酸的生产工艺 | |
| CN104016945B (zh) | 一种半富马酸喹硫平的制备方法 | |
| CN113354528A (zh) | 一种苯乙酸生产方法 | |
| CN107915632B (zh) | 一种连续合成4-氯-3-氧代-丁酸乙酯的后处理方法 | |
| CN112645901A (zh) | 一种1-环己基哌嗪的制备方法 | |
| CN111303045A (zh) | 2-乙氧基-4,6-二氟嘧啶的生产工艺 | |
| CN111170871A (zh) | 一种6-溴-2,4-二硝基苯胺合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |