CN1131872C - 修饰过的因子vii - Google Patents
修饰过的因子vii Download PDFInfo
- Publication number
- CN1131872C CN1131872C CN97195294A CN97195294A CN1131872C CN 1131872 C CN1131872 C CN 1131872C CN 97195294 A CN97195294 A CN 97195294A CN 97195294 A CN97195294 A CN 97195294A CN 1131872 C CN1131872 C CN 1131872C
- Authority
- CN
- China
- Prior art keywords
- factor
- factor vii
- factor viia
- modified
- degr
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 229940012413 factor vii Drugs 0.000 title claims abstract description 177
- 108010023321 Factor VII Proteins 0.000 title claims abstract description 174
- 102100023804 Coagulation factor VII Human genes 0.000 title claims abstract description 172
- 238000000034 method Methods 0.000 claims abstract description 60
- 230000017531 blood circulation Effects 0.000 claims abstract description 28
- 230000002107 myocardial effect Effects 0.000 claims abstract description 25
- 230000003197 catalytic effect Effects 0.000 claims abstract description 23
- 230000004048 modification Effects 0.000 claims abstract description 21
- 238000012986 modification Methods 0.000 claims abstract description 21
- 239000000203 mixture Substances 0.000 claims description 57
- 230000000302 ischemic effect Effects 0.000 claims description 36
- 108010014173 Factor X Proteins 0.000 claims description 32
- 230000008569 process Effects 0.000 claims description 25
- 150000001413 amino acids Chemical class 0.000 claims description 24
- 230000004913 activation Effects 0.000 claims description 19
- 230000010412 perfusion Effects 0.000 claims description 18
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 17
- SUNMBRGCANLOEG-UHFFFAOYSA-N monochloromethyl ketone Natural products ClCC(=O)CCl SUNMBRGCANLOEG-UHFFFAOYSA-N 0.000 claims description 16
- LDMSFLLWFAFZAF-OALUTQOASA-N (4s)-5-[[2-[[(3s)-1-chloro-6-(diaminomethylideneamino)-2-oxohexan-3-yl]amino]-2-oxoethyl]amino]-4-[[5-(dimethylamino)naphthalen-1-yl]sulfonylamino]-5-oxopentanoic acid Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(=O)(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCCN=C(N)N)C(=O)CCl LDMSFLLWFAFZAF-OALUTQOASA-N 0.000 claims description 15
- 238000006243 chemical reaction Methods 0.000 claims description 13
- 238000006555 catalytic reaction Methods 0.000 claims description 11
- 230000003680 myocardial damage Effects 0.000 claims description 10
- UFXAQJOOAQICNE-HKBOAZHASA-N (2r)-2-amino-n-[(2s)-2-[[(3s)-1-chloro-6-(diaminomethylideneamino)-2-oxohexan-3-yl]amino]-3-phenylpropanoyl]-3-phenylpropanamide Chemical compound C([C@@H](N)C(=O)NC(=O)[C@H](CC=1C=CC=CC=1)N[C@@H](CCCN=C(N)N)C(=O)CCl)C1=CC=CC=C1 UFXAQJOOAQICNE-HKBOAZHASA-N 0.000 claims description 7
- -1 organo phosphorous Chemical compound 0.000 claims description 7
- 239000000137 peptide hydrolase inhibitor Substances 0.000 claims description 6
- 238000012217 deletion Methods 0.000 claims description 5
- 230000037430 deletion Effects 0.000 claims description 5
- XYOVOXDWRFGKEX-UHFFFAOYSA-N azepine Chemical compound N1C=CC=CC=C1 XYOVOXDWRFGKEX-UHFFFAOYSA-N 0.000 claims description 4
- 238000003780 insertion Methods 0.000 claims description 4
- 230000037431 insertion Effects 0.000 claims description 4
- SFZCNBIFKDRMGX-UHFFFAOYSA-N sulfur hexafluoride Chemical compound FS(F)(F)(F)(F)F SFZCNBIFKDRMGX-UHFFFAOYSA-N 0.000 claims description 4
- 229960000909 sulfur hexafluoride Drugs 0.000 claims description 4
- 102000008847 Serpin Human genes 0.000 claims description 3
- 108050000761 Serpin Proteins 0.000 claims description 3
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 claims description 3
- 208000010125 myocardial infarction Diseases 0.000 claims description 3
- 239000003001 serine protease inhibitor Substances 0.000 claims description 3
- 229940012414 factor viia Drugs 0.000 abstract description 245
- 108010054265 Factor VIIa Proteins 0.000 abstract description 142
- 238000011282 treatment Methods 0.000 abstract description 35
- 230000023555 blood coagulation Effects 0.000 abstract description 33
- 208000007536 Thrombosis Diseases 0.000 abstract description 32
- 150000001875 compounds Chemical class 0.000 abstract description 12
- 230000000699 topical effect Effects 0.000 abstract description 12
- 230000002792 vascular Effects 0.000 abstract description 11
- 230000015572 biosynthetic process Effects 0.000 abstract description 9
- 230000010410 reperfusion Effects 0.000 abstract description 4
- 208000037891 myocardial injury Diseases 0.000 abstract description 2
- 108010000499 Thromboplastin Proteins 0.000 description 80
- 102000002262 Thromboplastin Human genes 0.000 description 80
- 210000004027 cell Anatomy 0.000 description 80
- 241001465754 Metazoa Species 0.000 description 71
- 230000000694 effects Effects 0.000 description 53
- 210000002381 plasma Anatomy 0.000 description 46
- 230000006378 damage Effects 0.000 description 44
- 210000004204 blood vessel Anatomy 0.000 description 42
- 108090000623 proteins and genes Proteins 0.000 description 42
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 33
- 241000283973 Oryctolagus cuniculus Species 0.000 description 32
- 208000027418 Wounds and injury Diseases 0.000 description 31
- 108010074860 Factor Xa Proteins 0.000 description 29
- 210000001367 artery Anatomy 0.000 description 29
- 239000000523 sample Substances 0.000 description 27
- 238000002399 angioplasty Methods 0.000 description 25
- 102000004169 proteins and genes Human genes 0.000 description 25
- 239000000243 solution Substances 0.000 description 25
- 108020004414 DNA Proteins 0.000 description 24
- 230000005764 inhibitory process Effects 0.000 description 24
- 238000002347 injection Methods 0.000 description 24
- 239000007924 injection Substances 0.000 description 24
- 238000005516 engineering process Methods 0.000 description 23
- 238000002474 experimental method Methods 0.000 description 23
- 208000014674 injury Diseases 0.000 description 23
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 23
- 206010061216 Infarction Diseases 0.000 description 22
- 230000008859 change Effects 0.000 description 22
- 230000007574 infarction Effects 0.000 description 22
- 230000014509 gene expression Effects 0.000 description 21
- 235000018102 proteins Nutrition 0.000 description 21
- 210000001772 blood platelet Anatomy 0.000 description 20
- 239000003814 drug Substances 0.000 description 20
- 239000012634 fragment Substances 0.000 description 20
- 210000001519 tissue Anatomy 0.000 description 19
- 241001504519 Papio ursinus Species 0.000 description 18
- 238000002360 preparation method Methods 0.000 description 18
- 239000000758 substrate Substances 0.000 description 18
- 238000012360 testing method Methods 0.000 description 18
- 206010053567 Coagulopathies Diseases 0.000 description 17
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 17
- 229940024606 amino acid Drugs 0.000 description 17
- 235000001014 amino acid Nutrition 0.000 description 17
- 210000004369 blood Anatomy 0.000 description 17
- 239000008280 blood Substances 0.000 description 17
- 230000035602 clotting Effects 0.000 description 17
- 229920000669 heparin Polymers 0.000 description 17
- 229960002897 heparin Drugs 0.000 description 17
- 238000013016 damping Methods 0.000 description 16
- 238000010790 dilution Methods 0.000 description 16
- 239000012895 dilution Substances 0.000 description 16
- 239000012530 fluid Substances 0.000 description 16
- 239000011780 sodium chloride Substances 0.000 description 16
- 108010094028 Prothrombin Proteins 0.000 description 13
- 102100027378 Prothrombin Human genes 0.000 description 13
- 230000004087 circulation Effects 0.000 description 13
- 230000015271 coagulation Effects 0.000 description 13
- 238000005345 coagulation Methods 0.000 description 13
- 230000008021 deposition Effects 0.000 description 13
- 238000007710 freezing Methods 0.000 description 13
- 238000005259 measurement Methods 0.000 description 13
- 229940039716 prothrombin Drugs 0.000 description 13
- 238000011160 research Methods 0.000 description 13
- 238000013171 endarterectomy Methods 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 208000037803 restenosis Diseases 0.000 description 12
- 210000002966 serum Anatomy 0.000 description 12
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 12
- 239000007983 Tris buffer Substances 0.000 description 11
- 230000002785 anti-thrombosis Effects 0.000 description 11
- 210000001715 carotid artery Anatomy 0.000 description 11
- 229940099816 human factor vii Drugs 0.000 description 11
- 239000003550 marker Substances 0.000 description 11
- 238000007789 sealing Methods 0.000 description 11
- 210000001105 femoral artery Anatomy 0.000 description 10
- 210000004165 myocardium Anatomy 0.000 description 10
- 239000012266 salt solution Substances 0.000 description 10
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 9
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 9
- 208000032843 Hemorrhage Diseases 0.000 description 9
- 108010060630 Lactoglobulins Proteins 0.000 description 9
- 239000003146 anticoagulant agent Substances 0.000 description 9
- 244000309466 calf Species 0.000 description 9
- 238000010523 cascade reaction Methods 0.000 description 9
- 210000004351 coronary vessel Anatomy 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 230000008488 polyadenylation Effects 0.000 description 9
- 230000009466 transformation Effects 0.000 description 9
- 230000009261 transgenic effect Effects 0.000 description 9
- 239000011712 vitamin K Substances 0.000 description 9
- 229940046010 vitamin k Drugs 0.000 description 9
- 241000124008 Mammalia Species 0.000 description 8
- 108091028043 Nucleic acid sequence Proteins 0.000 description 8
- 229930003448 Vitamin K Natural products 0.000 description 8
- 230000001154 acute effect Effects 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 210000003725 endotheliocyte Anatomy 0.000 description 8
- 210000005240 left ventricle Anatomy 0.000 description 8
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 8
- 239000013612 plasmid Substances 0.000 description 8
- 238000012545 processing Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 235000019168 vitamin K Nutrition 0.000 description 8
- 150000003721 vitamin K derivatives Chemical class 0.000 description 8
- 108010076282 Factor IX Proteins 0.000 description 7
- 108010011756 Milk Proteins Proteins 0.000 description 7
- 241001494479 Pecora Species 0.000 description 7
- 229940127219 anticoagulant drug Drugs 0.000 description 7
- 238000005520 cutting process Methods 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 229960004222 factor ix Drugs 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 210000005246 left atrium Anatomy 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 108700038606 rat Smooth muscle Proteins 0.000 description 7
- 102000004411 Antithrombin III Human genes 0.000 description 6
- 108090000935 Antithrombin III Proteins 0.000 description 6
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 6
- 208000001778 Coronary Occlusion Diseases 0.000 description 6
- 239000007995 HEPES buffer Substances 0.000 description 6
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 6
- 206010047249 Venous thrombosis Diseases 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 229960005348 antithrombin iii Drugs 0.000 description 6
- 210000000709 aorta Anatomy 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 239000007853 buffer solution Substances 0.000 description 6
- 230000002860 competitive effect Effects 0.000 description 6
- 239000002299 complementary DNA Substances 0.000 description 6
- 238000009826 distribution Methods 0.000 description 6
- 210000002216 heart Anatomy 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 230000037361 pathway Effects 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 230000009805 platelet accumulation Effects 0.000 description 6
- 230000008521 reorganization Effects 0.000 description 6
- 238000001890 transfection Methods 0.000 description 6
- 206010002091 Anaesthesia Diseases 0.000 description 5
- 201000001320 Atherosclerosis Diseases 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 5
- 206010011086 Coronary artery occlusion Diseases 0.000 description 5
- 206010011091 Coronary artery thrombosis Diseases 0.000 description 5
- 102000010911 Enzyme Precursors Human genes 0.000 description 5
- 108010062466 Enzyme Precursors Proteins 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 241000701109 Human adenovirus 2 Species 0.000 description 5
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical group OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 5
- 102000008192 Lactoglobulins Human genes 0.000 description 5
- 102000014171 Milk Proteins Human genes 0.000 description 5
- 108090000190 Thrombin Proteins 0.000 description 5
- 102100033571 Tissue-type plasminogen activator Human genes 0.000 description 5
- 230000037005 anaesthesia Effects 0.000 description 5
- 230000003872 anastomosis Effects 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 108091007433 antigens Proteins 0.000 description 5
- 102000036639 antigens Human genes 0.000 description 5
- 230000004872 arterial blood pressure Effects 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 208000002528 coronary thrombosis Diseases 0.000 description 5
- 230000009849 deactivation Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 230000006624 extrinsic pathway Effects 0.000 description 5
- 238000006062 fragmentation reaction Methods 0.000 description 5
- RBTKNAXYKSUFRK-UHFFFAOYSA-N heliogen blue Chemical compound [Cu].[N-]1C2=C(C=CC=C3)C3=C1N=C([N-]1)C3=CC=CC=C3C1=NC([N-]1)=C(C=CC=C3)C3=C1N=C([N-]1)C3=CC=CC=C3C1=N2 RBTKNAXYKSUFRK-UHFFFAOYSA-N 0.000 description 5
- 238000010253 intravenous injection Methods 0.000 description 5
- 210000004731 jugular vein Anatomy 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 235000013336 milk Nutrition 0.000 description 5
- 239000008267 milk Substances 0.000 description 5
- 210000004080 milk Anatomy 0.000 description 5
- 235000021239 milk protein Nutrition 0.000 description 5
- 108020004707 nucleic acids Proteins 0.000 description 5
- 102000039446 nucleic acids Human genes 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 230000003204 osmotic effect Effects 0.000 description 5
- 229920003023 plastic Polymers 0.000 description 5
- 108091033319 polynucleotide Proteins 0.000 description 5
- 102000040430 polynucleotide Human genes 0.000 description 5
- 239000002157 polynucleotide Substances 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 238000010008 shearing Methods 0.000 description 5
- JADVWWSKYZXRGX-UHFFFAOYSA-M thioflavine T Chemical compound [Cl-].C1=CC(N(C)C)=CC=C1C1=[N+](C)C2=CC=C(C)C=C2S1 JADVWWSKYZXRGX-UHFFFAOYSA-M 0.000 description 5
- 229960004072 thrombin Drugs 0.000 description 5
- 230000002537 thrombolytic effect Effects 0.000 description 5
- 230000003966 vascular damage Effects 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- 210000003462 vein Anatomy 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 5
- 229960001600 xylazine Drugs 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- 241000894006 Bacteria Species 0.000 description 4
- 102000004506 Blood Proteins Human genes 0.000 description 4
- 108010017384 Blood Proteins Proteins 0.000 description 4
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 4
- 108010073385 Fibrin Proteins 0.000 description 4
- 102000009123 Fibrin Human genes 0.000 description 4
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 4
- 208000035992 Postmortem Changes Diseases 0.000 description 4
- 108010064129 Thrombogen Proteins 0.000 description 4
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 4
- 230000035508 accumulation Effects 0.000 description 4
- 238000009825 accumulation Methods 0.000 description 4
- 206010000891 acute myocardial infarction Diseases 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- 229960004676 antithrombotic agent Drugs 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 229910001424 calcium ion Inorganic materials 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 239000000975 dye Substances 0.000 description 4
- 229950003499 fibrin Drugs 0.000 description 4
- 238000013467 fragmentation Methods 0.000 description 4
- 230000023597 hemostasis Effects 0.000 description 4
- 206010020718 hyperplasia Diseases 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 4
- 229960003299 ketamine Drugs 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 229920001184 polypeptide Polymers 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 239000006152 selective media Substances 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 230000001732 thrombotic effect Effects 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- KWPACVJPAFGBEQ-IKGGRYGDSA-N (2s)-1-[(2r)-2-amino-3-phenylpropanoyl]-n-[(3s)-1-chloro-6-(diaminomethylideneamino)-2-oxohexan-3-yl]pyrrolidine-2-carboxamide Chemical compound C([C@@H](N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)CCl)C1=CC=CC=C1 KWPACVJPAFGBEQ-IKGGRYGDSA-N 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- 102000004092 Amidohydrolases Human genes 0.000 description 3
- 108090000531 Amidohydrolases Proteins 0.000 description 3
- 206010003162 Arterial injury Diseases 0.000 description 3
- 108010081589 Becaplermin Proteins 0.000 description 3
- 241000699800 Cricetinae Species 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- DSFYPIUSAMSERP-IHRRRGAJSA-N Leu-Leu-Arg Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(O)=O)CCCN=C(N)N DSFYPIUSAMSERP-IHRRRGAJSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- 108010076504 Protein Sorting Signals Proteins 0.000 description 3
- KOSRFJWDECSPRO-UHFFFAOYSA-N alpha-L-glutamyl-L-glutamic acid Natural products OC(=O)CCC(N)C(=O)NC(CCC(O)=O)C(O)=O KOSRFJWDECSPRO-UHFFFAOYSA-N 0.000 description 3
- 238000002583 angiography Methods 0.000 description 3
- 230000002429 anti-coagulating effect Effects 0.000 description 3
- 230000000692 anti-sense effect Effects 0.000 description 3
- 108010047857 aspartylglycine Proteins 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 238000012258 culturing Methods 0.000 description 3
- 230000001186 cumulative effect Effects 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 230000003511 endothelial effect Effects 0.000 description 3
- 238000011010 flushing procedure Methods 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 108010037850 glycylvaline Proteins 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 230000012447 hatching Effects 0.000 description 3
- 230000000004 hemodynamic effect Effects 0.000 description 3
- 102000057593 human F8 Human genes 0.000 description 3
- 230000008676 import Effects 0.000 description 3
- 210000003734 kidney Anatomy 0.000 description 3
- 238000012417 linear regression Methods 0.000 description 3
- 210000001161 mammalian embryo Anatomy 0.000 description 3
- 210000005075 mammary gland Anatomy 0.000 description 3
- 239000004005 microsphere Substances 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 210000001616 monocyte Anatomy 0.000 description 3
- 238000007491 morphometric analysis Methods 0.000 description 3
- 230000014508 negative regulation of coagulation Effects 0.000 description 3
- 210000004493 neutrocyte Anatomy 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 229910052698 phosphorus Inorganic materials 0.000 description 3
- 239000002985 plastic film Substances 0.000 description 3
- 229920001155 polypropylene Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000002947 procoagulating effect Effects 0.000 description 3
- 230000000750 progressive effect Effects 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 229940047431 recombinate Drugs 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000001509 sodium citrate Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 238000007619 statistical method Methods 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 3
- 229940038773 trisodium citrate Drugs 0.000 description 3
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 2
- KKJUPNGICOCCDW-UHFFFAOYSA-N 7-N,N-Dimethylamino-1,2,3,4,5-pentathiocyclooctane Chemical group CN(C)C1CSSSSSC1 KKJUPNGICOCCDW-UHFFFAOYSA-N 0.000 description 2
- AOHKLEBWKMKITA-IHRRRGAJSA-N Arg-Phe-Ser Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CO)C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N AOHKLEBWKMKITA-IHRRRGAJSA-N 0.000 description 2
- HGKHPCFTRQDHCU-IUCAKERBSA-N Arg-Pro-Gly Chemical compound NC(N)=NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)NCC(O)=O HGKHPCFTRQDHCU-IUCAKERBSA-N 0.000 description 2
- HRGGPWBIMIQANI-GUBZILKMSA-N Asp-Gln-Leu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O HRGGPWBIMIQANI-GUBZILKMSA-N 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- KXUKWRVYDYIPSQ-CIUDSAMLSA-N Cys-Leu-Ala Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(O)=O KXUKWRVYDYIPSQ-CIUDSAMLSA-N 0.000 description 2
- HBHMVBGGHDMPBF-GARJFASQSA-N Cys-Leu-Pro Chemical compound CC(C)C[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CS)N HBHMVBGGHDMPBF-GARJFASQSA-N 0.000 description 2
- 206010051055 Deep vein thrombosis Diseases 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 241000393496 Electra Species 0.000 description 2
- 108010074864 Factor XI Proteins 0.000 description 2
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 2
- 108010049003 Fibrinogen Proteins 0.000 description 2
- 102000008946 Fibrinogen Human genes 0.000 description 2
- ZJFNRQHUIHKZJF-GUBZILKMSA-N Glu-His-Asp Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC(O)=O)C(O)=O ZJFNRQHUIHKZJF-GUBZILKMSA-N 0.000 description 2
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical group O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 241001135569 Human adenovirus 5 Species 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010021703 Indifference Diseases 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- IBMVEYRWAWIOTN-UHFFFAOYSA-N L-Leucyl-L-Arginyl-L-Proline Natural products CC(C)CC(N)C(=O)NC(CCCN=C(N)N)C(=O)N1CCCC1C(O)=O IBMVEYRWAWIOTN-UHFFFAOYSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- KAFOIVJDVSZUMD-UHFFFAOYSA-N Leu-Gln-Gln Natural products CC(C)CC(N)C(=O)NC(CCC(N)=O)C(=O)NC(CCC(N)=O)C(O)=O KAFOIVJDVSZUMD-UHFFFAOYSA-N 0.000 description 2
- VKVDRTGWLVZJOM-DCAQKATOSA-N Leu-Val-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O VKVDRTGWLVZJOM-DCAQKATOSA-N 0.000 description 2
- LBSWWNKMVPAXOI-GUBZILKMSA-N Met-Val-Ser Chemical compound CSCC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O LBSWWNKMVPAXOI-GUBZILKMSA-N 0.000 description 2
- 108010079364 N-glycylalanine Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- KWMZPPWYBVZIER-XGEHTFHBSA-N Pro-Ser-Thr Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(O)=O KWMZPPWYBVZIER-XGEHTFHBSA-N 0.000 description 2
- 101800004937 Protein C Proteins 0.000 description 2
- 102000017975 Protein C Human genes 0.000 description 2
- 229940096437 Protein S Drugs 0.000 description 2
- 208000006193 Pulmonary infarction Diseases 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 101800001700 Saposin-D Proteins 0.000 description 2
- ZIFYDQAFEMIZII-GUBZILKMSA-N Ser-Leu-Glu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O ZIFYDQAFEMIZII-GUBZILKMSA-N 0.000 description 2
- 108010023197 Streptokinase Proteins 0.000 description 2
- SVGAWGVHFIYAEE-JSGCOSHPSA-N Trp-Gly-Gln Chemical compound C1=CC=C2C(C[C@H](N)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(O)=O)=CNC2=C1 SVGAWGVHFIYAEE-JSGCOSHPSA-N 0.000 description 2
- GQVZBMROTPEPIF-SRVKXCTJSA-N Tyr-Ser-Asp Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(O)=O GQVZBMROTPEPIF-SRVKXCTJSA-N 0.000 description 2
- 208000007814 Unstable Angina Diseases 0.000 description 2
- IZFVRRYRMQFVGX-NRPADANISA-N Val-Ala-Gln Chemical compound C[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](C(C)C)N IZFVRRYRMQFVGX-NRPADANISA-N 0.000 description 2
- VHRLUTIMTDOVCG-PEDHHIEDSA-N Val-Ile-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)O)NC(=O)[C@H](C(C)C)N VHRLUTIMTDOVCG-PEDHHIEDSA-N 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 230000000735 allogeneic effect Effects 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 238000005571 anion exchange chromatography Methods 0.000 description 2
- 239000002506 anticoagulant protein Substances 0.000 description 2
- 229940058344 antitrematodals organophosphorous compound Drugs 0.000 description 2
- 239000008365 aqueous carrier Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000002146 bilateral effect Effects 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 239000012888 bovine serum Substances 0.000 description 2
- 210000002302 brachial artery Anatomy 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- XQGZSYKGWHUSDH-UHFFFAOYSA-N dazoxiben Chemical compound C1=CC(C(=O)O)=CC=C1OCCN1C=NC=C1 XQGZSYKGWHUSDH-UHFFFAOYSA-N 0.000 description 2
- 229950008000 dazoxiben Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 108010054813 diprotin B Proteins 0.000 description 2
- 238000002224 dissection Methods 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 238000004043 dyeing Methods 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 210000001671 embryonic stem cell Anatomy 0.000 description 2
- 210000003038 endothelium Anatomy 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 229940012952 fibrinogen Drugs 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 229960002989 glutamic acid Drugs 0.000 description 2
- 108010078144 glutaminyl-glycine Proteins 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- VPZXBVLAVMBEQI-UHFFFAOYSA-N glycyl-DL-alpha-alanine Natural products OC(=O)C(C)NC(=O)CN VPZXBVLAVMBEQI-UHFFFAOYSA-N 0.000 description 2
- HPAIKDPJURGQLN-UHFFFAOYSA-N glycyl-L-histidyl-L-phenylalanine Natural products C=1C=CC=CC=1CC(C(O)=O)NC(=O)C(NC(=O)CN)CC1=CN=CN1 HPAIKDPJURGQLN-UHFFFAOYSA-N 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 238000002647 laser therapy Methods 0.000 description 2
- 108010076756 leucyl-alanyl-phenylalanine Proteins 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 244000144972 livestock Species 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 230000017074 necrotic cell death Effects 0.000 description 2
- 238000011587 new zealand white rabbit Methods 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 150000002903 organophosphorus compounds Chemical class 0.000 description 2
- 229960005489 paracetamol Drugs 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 230000002980 postoperative effect Effects 0.000 description 2
- 229960000856 protein c Drugs 0.000 description 2
- 238000001742 protein purification Methods 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 230000007575 pulmonary infarction Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000003156 radioimmunoprecipitation Methods 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 239000000837 restrainer Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000012679 serum free medium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- RGHFKWPGWBFQLN-UHFFFAOYSA-M sodium;5,5-diethylpyrimidin-3-ide-2,4,6-trione Chemical compound [Na+].CCC1(CC)C([O-])=NC(=O)NC1=O RGHFKWPGWBFQLN-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 229960005202 streptokinase Drugs 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 108700004896 tripeptide FEG Proteins 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 238000007631 vascular surgery Methods 0.000 description 2
- 230000004304 visual acuity Effects 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 1
- JNTMAZFVYNDPLB-PEDHHIEDSA-N (2S,3S)-2-[[[(2S)-1-[(2S,3S)-2-amino-3-methyl-1-oxopentyl]-2-pyrrolidinyl]-oxomethyl]amino]-3-methylpentanoic acid Chemical compound CC[C@H](C)[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(O)=O JNTMAZFVYNDPLB-PEDHHIEDSA-N 0.000 description 1
- JNBCIUTTWUUDDX-LSBAASHUSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-amino-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]-4-carboxybutanoyl]amino]-4-carboxybutanoyl]amino]-4-methylpentanoic acid Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC1=CC=CC=C1 JNBCIUTTWUUDDX-LSBAASHUSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- PKDBCJSWQUOKDO-UHFFFAOYSA-M 2,3,5-triphenyltetrazolium chloride Chemical compound [Cl-].C1=CC=CC=C1C(N=[N+]1C=2C=CC=CC=2)=NN1C1=CC=CC=C1 PKDBCJSWQUOKDO-UHFFFAOYSA-M 0.000 description 1
- TYMLOMAKGOJONV-UHFFFAOYSA-N 4-nitroaniline Chemical compound NC1=CC=C([N+]([O-])=O)C=C1 TYMLOMAKGOJONV-UHFFFAOYSA-N 0.000 description 1
- 108020005029 5' Flanking Region Proteins 0.000 description 1
- WQVFQXXBNHHPLX-ZKWXMUAHSA-N Ala-Ala-His Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1cnc[nH]1)C(O)=O WQVFQXXBNHHPLX-ZKWXMUAHSA-N 0.000 description 1
- YAXNATKKPOWVCP-ZLUOBGJFSA-N Ala-Asn-Ala Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(O)=O YAXNATKKPOWVCP-ZLUOBGJFSA-N 0.000 description 1
- KIUYPHAMDKDICO-WHFBIAKZSA-N Ala-Asp-Gly Chemical compound C[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(O)=O KIUYPHAMDKDICO-WHFBIAKZSA-N 0.000 description 1
- BLGHHPHXVJWCNK-GUBZILKMSA-N Ala-Gln-Leu Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O BLGHHPHXVJWCNK-GUBZILKMSA-N 0.000 description 1
- NWVVKQZOVSTDBQ-CIUDSAMLSA-N Ala-Glu-Arg Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O NWVVKQZOVSTDBQ-CIUDSAMLSA-N 0.000 description 1
- NIZKGBJVCMRDKO-KWQFWETISA-N Ala-Gly-Tyr Chemical compound C[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 NIZKGBJVCMRDKO-KWQFWETISA-N 0.000 description 1
- OKEWAFFWMHBGPT-XPUUQOCRSA-N Ala-His-Gly Chemical compound OC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](N)C)CC1=CN=CN1 OKEWAFFWMHBGPT-XPUUQOCRSA-N 0.000 description 1
- CCDFBRZVTDDJNM-GUBZILKMSA-N Ala-Leu-Glu Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O CCDFBRZVTDDJNM-GUBZILKMSA-N 0.000 description 1
- ZBLQIYPCUWZSRZ-QEJZJMRPSA-N Ala-Phe-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](C)N)CC1=CC=CC=C1 ZBLQIYPCUWZSRZ-QEJZJMRPSA-N 0.000 description 1
- IHMCQESUJVZTKW-UBHSHLNASA-N Ala-Phe-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@H](C)N)CC1=CC=CC=C1 IHMCQESUJVZTKW-UBHSHLNASA-N 0.000 description 1
- GMGWOTQMUKYZIE-UBHSHLNASA-N Ala-Pro-Phe Chemical compound C[C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 GMGWOTQMUKYZIE-UBHSHLNASA-N 0.000 description 1
- MMLHRUJLOUSRJX-CIUDSAMLSA-N Ala-Ser-Lys Chemical compound C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCCCN MMLHRUJLOUSRJX-CIUDSAMLSA-N 0.000 description 1
- OEVCHROQUIVQFZ-YTLHQDLWSA-N Ala-Thr-Ala Chemical compound C[C@H](N)C(=O)N[C@@H]([C@H](O)C)C(=O)N[C@@H](C)C(O)=O OEVCHROQUIVQFZ-YTLHQDLWSA-N 0.000 description 1
- AAWLEICNDUHIJM-MBLNEYKQSA-N Ala-Thr-His Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)NC(=O)[C@H](C)N)O AAWLEICNDUHIJM-MBLNEYKQSA-N 0.000 description 1
- IETUUAHKCHOQHP-KZVJFYERSA-N Ala-Thr-Val Chemical compound CC(C)[C@H](NC(=O)[C@@H](NC(=O)[C@H](C)N)[C@@H](C)O)C(O)=O IETUUAHKCHOQHP-KZVJFYERSA-N 0.000 description 1
- DHONNEYAZPNGSG-UBHSHLNASA-N Ala-Val-Phe Chemical compound C[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 DHONNEYAZPNGSG-UBHSHLNASA-N 0.000 description 1
- 101710153593 Albumin A Proteins 0.000 description 1
- 206010002388 Angina unstable Diseases 0.000 description 1
- DFCIPNHFKOQAME-FXQIFTODSA-N Arg-Ala-Asn Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(O)=O DFCIPNHFKOQAME-FXQIFTODSA-N 0.000 description 1
- DBKNLHKEVPZVQC-LPEHRKFASA-N Arg-Ala-Pro Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](C)C(=O)N1CCC[C@@H]1C(O)=O DBKNLHKEVPZVQC-LPEHRKFASA-N 0.000 description 1
- XPSGESXVBSQZPL-SRVKXCTJSA-N Arg-Arg-Arg Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O XPSGESXVBSQZPL-SRVKXCTJSA-N 0.000 description 1
- RWWPBOUMKFBHAL-FXQIFTODSA-N Arg-Asn-Cys Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(O)=O RWWPBOUMKFBHAL-FXQIFTODSA-N 0.000 description 1
- BVBKBQRPOJFCQM-DCAQKATOSA-N Arg-Asn-Leu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(O)=O BVBKBQRPOJFCQM-DCAQKATOSA-N 0.000 description 1
- OANWAFQRNQEDSY-DCAQKATOSA-N Arg-Cys-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CCCN=C(N)N)N OANWAFQRNQEDSY-DCAQKATOSA-N 0.000 description 1
- NYZGVTGOMPHSJW-CIUDSAMLSA-N Arg-Glu-Cys Chemical compound C(C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CS)C(=O)O)N)CN=C(N)N NYZGVTGOMPHSJW-CIUDSAMLSA-N 0.000 description 1
- OHYQKYUTLIPFOX-ZPFDUUQYSA-N Arg-Glu-Ile Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O OHYQKYUTLIPFOX-ZPFDUUQYSA-N 0.000 description 1
- KRQSPVKUISQQFS-FJXKBIBVSA-N Arg-Gly-Thr Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCN=C(N)N KRQSPVKUISQQFS-FJXKBIBVSA-N 0.000 description 1
- LLUGJARLJCGLAR-CYDGBPFRSA-N Arg-Ile-Val Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](C(C)C)C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N LLUGJARLJCGLAR-CYDGBPFRSA-N 0.000 description 1
- JEXPNDORFYHJTM-IHRRRGAJSA-N Arg-Leu-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCCN=C(N)N JEXPNDORFYHJTM-IHRRRGAJSA-N 0.000 description 1
- ISJWBVIYRBAXEB-CIUDSAMLSA-N Arg-Ser-Glu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(O)=O ISJWBVIYRBAXEB-CIUDSAMLSA-N 0.000 description 1
- RYQSYXFGFOTJDJ-RHYQMDGZSA-N Arg-Thr-Leu Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(O)=O RYQSYXFGFOTJDJ-RHYQMDGZSA-N 0.000 description 1
- DDBMKOCQWNFDBH-RHYQMDGZSA-N Arg-Thr-Lys Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CCCN=C(N)N)N)O DDBMKOCQWNFDBH-RHYQMDGZSA-N 0.000 description 1
- MOGMYRUNTKYZFB-UNQGMJICSA-N Arg-Thr-Phe Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H]([C@H](O)C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MOGMYRUNTKYZFB-UNQGMJICSA-N 0.000 description 1
- XWGJDUSDTRPQRK-ZLUOBGJFSA-N Asn-Ala-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(N)=O XWGJDUSDTRPQRK-ZLUOBGJFSA-N 0.000 description 1
- IBLAOXSULLECQZ-IUKAMOBKSA-N Asn-Ile-Thr Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H](N)CC(N)=O IBLAOXSULLECQZ-IUKAMOBKSA-N 0.000 description 1
- HCZQKHSRYHCPSD-IUKAMOBKSA-N Asn-Thr-Ile Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O HCZQKHSRYHCPSD-IUKAMOBKSA-N 0.000 description 1
- FLJVGAFLZVBBNG-BPUTZDHNSA-N Asn-Trp-Arg Chemical compound N[C@@H](CC(=O)N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(=N)N)C(=O)O FLJVGAFLZVBBNG-BPUTZDHNSA-N 0.000 description 1
- GHWWTICYPDKPTE-NGZCFLSTSA-N Asn-Val-Pro Chemical compound CC(C)[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CC(=O)N)N GHWWTICYPDKPTE-NGZCFLSTSA-N 0.000 description 1
- XEDQMTWEYFBOIK-ACZMJKKPSA-N Asp-Ala-Glu Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(O)=O XEDQMTWEYFBOIK-ACZMJKKPSA-N 0.000 description 1
- AXXCUABIFZPKPM-BQBZGAKWSA-N Asp-Arg-Gly Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O AXXCUABIFZPKPM-BQBZGAKWSA-N 0.000 description 1
- FTNVLGCFIJEMQT-CIUDSAMLSA-N Asp-Cys-Leu Chemical compound CC(C)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CC(=O)O)N FTNVLGCFIJEMQT-CIUDSAMLSA-N 0.000 description 1
- YRBGRUOSJROZEI-NHCYSSNCSA-N Asp-His-Val Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](C(C)C)C(O)=O YRBGRUOSJROZEI-NHCYSSNCSA-N 0.000 description 1
- KTTCQQNRRLCIBC-GHCJXIJMSA-N Asp-Ile-Ala Chemical compound [H]N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O KTTCQQNRRLCIBC-GHCJXIJMSA-N 0.000 description 1
- UMHUHHJMEXNSIV-CIUDSAMLSA-N Asp-Leu-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CC(O)=O UMHUHHJMEXNSIV-CIUDSAMLSA-N 0.000 description 1
- HJCGDIGVVWETRO-ZPFDUUQYSA-N Asp-Lys-Ile Chemical compound CC[C@H](C)[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CC(O)=O)C(O)=O HJCGDIGVVWETRO-ZPFDUUQYSA-N 0.000 description 1
- FIAKNCXQFFKSSI-ZLUOBGJFSA-N Asp-Ser-Cys Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(O)=O FIAKNCXQFFKSSI-ZLUOBGJFSA-N 0.000 description 1
- HRVQDZOWMLFAOD-BIIVOSGPSA-N Asp-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)[C@H](CC(=O)O)N)C(=O)O HRVQDZOWMLFAOD-BIIVOSGPSA-N 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- 206010004053 Bacterial toxaemia Diseases 0.000 description 1
- 108010027612 Batroxobin Proteins 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- OWNRRUFOJXFKCU-UHFFFAOYSA-N Bromadiolone Chemical compound C=1C=C(C=2C=CC(Br)=CC=2)C=CC=1C(O)CC(C=1C(OC2=CC=CC=C2C=1O)=O)C1=CC=CC=C1 OWNRRUFOJXFKCU-UHFFFAOYSA-N 0.000 description 1
- 101150011672 CCL9 gene Proteins 0.000 description 1
- 101100439299 Caenorhabditis elegans cgt-3 gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 240000001432 Calendula officinalis Species 0.000 description 1
- 235000005881 Calendula officinalis Nutrition 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 102000011632 Caseins Human genes 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102100022641 Coagulation factor IX Human genes 0.000 description 1
- 102100030563 Coagulation factor XI Human genes 0.000 description 1
- 101100007328 Cocos nucifera COS-1 gene Proteins 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- PRXCTTWKGJAPMT-ZLUOBGJFSA-N Cys-Ala-Ser Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(O)=O PRXCTTWKGJAPMT-ZLUOBGJFSA-N 0.000 description 1
- QADHATDBZXHRCA-ACZMJKKPSA-N Cys-Gln-Asn Chemical compound C(CC(=O)N)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)O)NC(=O)[C@H](CS)N QADHATDBZXHRCA-ACZMJKKPSA-N 0.000 description 1
- BDWIZLQVVWQMTB-XKBZYTNZSA-N Cys-Glu-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CS)N)O BDWIZLQVVWQMTB-XKBZYTNZSA-N 0.000 description 1
- DZLQXIFVQFTFJY-BYPYZUCNSA-N Cys-Gly-Gly Chemical compound SC[C@H](N)C(=O)NCC(=O)NCC(O)=O DZLQXIFVQFTFJY-BYPYZUCNSA-N 0.000 description 1
- DZSICRGTVPDCRN-YUMQZZPRSA-N Cys-Gly-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)CNC(=O)[C@H](CS)N DZSICRGTVPDCRN-YUMQZZPRSA-N 0.000 description 1
- KPENUVBHAKRDQR-GUBZILKMSA-N Cys-His-Glu Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CCC(O)=O)C(O)=O KPENUVBHAKRDQR-GUBZILKMSA-N 0.000 description 1
- VXLXATVURDNDCG-CIUDSAMLSA-N Cys-Lys-Asp Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)NC(=O)[C@H](CS)N VXLXATVURDNDCG-CIUDSAMLSA-N 0.000 description 1
- BNCKELUXXUYRNY-GUBZILKMSA-N Cys-Lys-Glu Chemical compound C(CCN)C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](CS)N BNCKELUXXUYRNY-GUBZILKMSA-N 0.000 description 1
- CIVXDCMSSFGWAL-YUMQZZPRSA-N Cys-Lys-Gly Chemical compound C(CCN)C[C@@H](C(=O)NCC(=O)O)NC(=O)[C@H](CS)N CIVXDCMSSFGWAL-YUMQZZPRSA-N 0.000 description 1
- LHJDLVVQRJIURS-SRVKXCTJSA-N Cys-Phe-Asp Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)NC(=O)[C@H](CS)N LHJDLVVQRJIURS-SRVKXCTJSA-N 0.000 description 1
- HMWBPUDETPKSSS-DCAQKATOSA-N Cys-Pro-Lys Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CS)N)C(=O)N[C@@H](CCCCN)C(=O)O HMWBPUDETPKSSS-DCAQKATOSA-N 0.000 description 1
- DTFJUSWYECELTM-BPUTZDHNSA-N Cys-Pro-Trp Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CS)N)C(=O)N[C@@H](CC2=CNC3=CC=CC=C32)C(=O)O DTFJUSWYECELTM-BPUTZDHNSA-N 0.000 description 1
- RJPKQCFHEPPTGL-ZLUOBGJFSA-N Cys-Ser-Asp Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(O)=O RJPKQCFHEPPTGL-ZLUOBGJFSA-N 0.000 description 1
- IQXSTXKVEMRMMB-XAVMHZPKSA-N Cys-Thr-Pro Chemical compound C[C@H]([C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CS)N)O IQXSTXKVEMRMMB-XAVMHZPKSA-N 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 102000016942 Elastin Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- KVYVOGYEMPEXBT-GUBZILKMSA-N Gln-Ala-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCC(N)=O KVYVOGYEMPEXBT-GUBZILKMSA-N 0.000 description 1
- ZPDVKYLJTOFQJV-WDSKDSINSA-N Gln-Asn-Gly Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(O)=O ZPDVKYLJTOFQJV-WDSKDSINSA-N 0.000 description 1
- KYFSMWLWHYZRNW-ACZMJKKPSA-N Gln-Asp-Cys Chemical compound C(CC(=O)N)[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CS)C(=O)O)N KYFSMWLWHYZRNW-ACZMJKKPSA-N 0.000 description 1
- UZMWDBOHAOSCCH-ACZMJKKPSA-N Gln-Cys-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCC(N)=O UZMWDBOHAOSCCH-ACZMJKKPSA-N 0.000 description 1
- MFLMFRZBAJSGHK-ACZMJKKPSA-N Gln-Cys-Ser Chemical compound C(CC(=O)N)[C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CO)C(=O)O)N MFLMFRZBAJSGHK-ACZMJKKPSA-N 0.000 description 1
- RBWKVOSARCFSQQ-FXQIFTODSA-N Gln-Gln-Ser Chemical compound NC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(O)=O RBWKVOSARCFSQQ-FXQIFTODSA-N 0.000 description 1
- SNLOOPZHAQDMJG-CIUDSAMLSA-N Gln-Glu-Glu Chemical compound NC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O SNLOOPZHAQDMJG-CIUDSAMLSA-N 0.000 description 1
- FKXCBKCOSVIGCT-AVGNSLFASA-N Gln-Lys-Leu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O FKXCBKCOSVIGCT-AVGNSLFASA-N 0.000 description 1
- NYCVMJGIJYQWDO-CIUDSAMLSA-N Gln-Ser-Arg Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O NYCVMJGIJYQWDO-CIUDSAMLSA-N 0.000 description 1
- GHAXJVNBAKGWEJ-AVGNSLFASA-N Gln-Ser-Tyr Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O GHAXJVNBAKGWEJ-AVGNSLFASA-N 0.000 description 1
- YLABFXCRQQMMHS-AVGNSLFASA-N Gln-Tyr-Cys Chemical compound C1=CC(=CC=C1C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCC(=O)N)N)O YLABFXCRQQMMHS-AVGNSLFASA-N 0.000 description 1
- VEYGCDYMOXHJLS-GVXVVHGQSA-N Gln-Val-Leu Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O VEYGCDYMOXHJLS-GVXVVHGQSA-N 0.000 description 1
- FHPXTPQBODWBIY-CIUDSAMLSA-N Glu-Ala-Arg Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O FHPXTPQBODWBIY-CIUDSAMLSA-N 0.000 description 1
- XMVLTPMCUJTJQP-FXQIFTODSA-N Glu-Gln-Cys Chemical compound C(CC(=O)O)[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CS)C(=O)O)N XMVLTPMCUJTJQP-FXQIFTODSA-N 0.000 description 1
- WPLGNDORMXTMQS-FXQIFTODSA-N Glu-Gln-Ser Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(O)=O WPLGNDORMXTMQS-FXQIFTODSA-N 0.000 description 1
- HUFCEIHAFNVSNR-IHRRRGAJSA-N Glu-Gln-Tyr Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 HUFCEIHAFNVSNR-IHRRRGAJSA-N 0.000 description 1
- CGOHAEBMDSEKFB-FXQIFTODSA-N Glu-Glu-Ala Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(O)=O CGOHAEBMDSEKFB-FXQIFTODSA-N 0.000 description 1
- PXXGVUVQWQGGIG-YUMQZZPRSA-N Glu-Gly-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CCCN=C(N)N PXXGVUVQWQGGIG-YUMQZZPRSA-N 0.000 description 1
- XMPAXPSENRSOSV-RYUDHWBXSA-N Glu-Gly-Tyr Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O XMPAXPSENRSOSV-RYUDHWBXSA-N 0.000 description 1
- WTMZXOPHTIVFCP-QEWYBTABSA-N Glu-Ile-Phe Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 WTMZXOPHTIVFCP-QEWYBTABSA-N 0.000 description 1
- VSRCAOIHMGCIJK-SRVKXCTJSA-N Glu-Leu-Arg Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O VSRCAOIHMGCIJK-SRVKXCTJSA-N 0.000 description 1
- TWYFJOHWGCCRIR-DCAQKATOSA-N Glu-Pro-Arg Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(O)=O TWYFJOHWGCCRIR-DCAQKATOSA-N 0.000 description 1
- DDXZHOHEABQXSE-NKIYYHGXSA-N Glu-Thr-His Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)NC(=O)[C@H](CCC(=O)O)N)O DDXZHOHEABQXSE-NKIYYHGXSA-N 0.000 description 1
- HGJREIGJLUQBTJ-SZMVWBNQSA-N Glu-Trp-Leu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC(C)C)C(O)=O HGJREIGJLUQBTJ-SZMVWBNQSA-N 0.000 description 1
- QEJKKJNDDDPSMU-KKUMJFAQSA-N Glu-Tyr-Met Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CCSC)C(O)=O QEJKKJNDDDPSMU-KKUMJFAQSA-N 0.000 description 1
- KXRORHJIRAOQPG-SOUVJXGZSA-N Glu-Tyr-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=C(C=C2)O)NC(=O)[C@H](CCC(=O)O)N)C(=O)O KXRORHJIRAOQPG-SOUVJXGZSA-N 0.000 description 1
- QSDKBRMVXSWAQE-BFHQHQDPSA-N Gly-Ala-Thr Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)CN QSDKBRMVXSWAQE-BFHQHQDPSA-N 0.000 description 1
- OCQUNKSFDYDXBG-QXEWZRGKSA-N Gly-Arg-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)CN)CCCN=C(N)N OCQUNKSFDYDXBG-QXEWZRGKSA-N 0.000 description 1
- XEJTYSCIXKYSHR-WDSKDSINSA-N Gly-Asp-Gln Chemical compound C(CC(=O)N)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)CN XEJTYSCIXKYSHR-WDSKDSINSA-N 0.000 description 1
- QSTLUOIOYLYLLF-WDSKDSINSA-N Gly-Asp-Glu Chemical compound [H]NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O QSTLUOIOYLYLLF-WDSKDSINSA-N 0.000 description 1
- GVVKYKCOFMMTKZ-WHFBIAKZSA-N Gly-Cys-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)CN GVVKYKCOFMMTKZ-WHFBIAKZSA-N 0.000 description 1
- XXGQRGQPGFYECI-WDSKDSINSA-N Gly-Cys-Glu Chemical compound NCC(=O)N[C@@H](CS)C(=O)N[C@H](C(O)=O)CCC(O)=O XXGQRGQPGFYECI-WDSKDSINSA-N 0.000 description 1
- BYYNJRSNDARRBX-YFKPBYRVSA-N Gly-Gln-Gly Chemical compound NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(O)=O BYYNJRSNDARRBX-YFKPBYRVSA-N 0.000 description 1
- BIRKKBCSAIHDDF-WDSKDSINSA-N Gly-Glu-Cys Chemical compound NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CS)C(O)=O BIRKKBCSAIHDDF-WDSKDSINSA-N 0.000 description 1
- JUBDONGMHASUCN-IUCAKERBSA-N Gly-Glu-His Chemical compound NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](Cc1cnc[nH]1)C(O)=O JUBDONGMHASUCN-IUCAKERBSA-N 0.000 description 1
- IDOGEHIWMJMAHT-BYPYZUCNSA-N Gly-Gly-Cys Chemical compound NCC(=O)NCC(=O)N[C@@H](CS)C(O)=O IDOGEHIWMJMAHT-BYPYZUCNSA-N 0.000 description 1
- YWAQATDNEKZFFK-BYPYZUCNSA-N Gly-Gly-Ser Chemical compound NCC(=O)NCC(=O)N[C@@H](CO)C(O)=O YWAQATDNEKZFFK-BYPYZUCNSA-N 0.000 description 1
- HPAIKDPJURGQLN-KBPBESRZSA-N Gly-His-Phe Chemical compound C([C@H](NC(=O)CN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CNC=N1 HPAIKDPJURGQLN-KBPBESRZSA-N 0.000 description 1
- COVXELOAORHTND-LSJOCFKGSA-N Gly-Ile-Val Chemical compound NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(O)=O COVXELOAORHTND-LSJOCFKGSA-N 0.000 description 1
- PTIIBFKSLCYQBO-NHCYSSNCSA-N Gly-Lys-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCCCN)NC(=O)CN PTIIBFKSLCYQBO-NHCYSSNCSA-N 0.000 description 1
- OQQKUTVULYLCDG-ONGXEEELSA-N Gly-Lys-Val Chemical compound CC(C)[C@H](NC(=O)[C@H](CCCCN)NC(=O)CN)C(O)=O OQQKUTVULYLCDG-ONGXEEELSA-N 0.000 description 1
- PFMUCCYYAAFKTH-YFKPBYRVSA-N Gly-Met Chemical compound CSCC[C@@H](C(O)=O)NC(=O)CN PFMUCCYYAAFKTH-YFKPBYRVSA-N 0.000 description 1
- ZZJVYSAQQMDIRD-UWVGGRQHSA-N Gly-Pro-His Chemical compound NCC(=O)N1CCC[C@H]1C(=O)N[C@@H](Cc1cnc[nH]1)C(O)=O ZZJVYSAQQMDIRD-UWVGGRQHSA-N 0.000 description 1
- POJJAZJHBGXEGM-YUMQZZPRSA-N Gly-Ser-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CO)NC(=O)CN POJJAZJHBGXEGM-YUMQZZPRSA-N 0.000 description 1
- ZZWUYQXMIFTIIY-WEDXCCLWSA-N Gly-Thr-Leu Chemical compound [H]NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(O)=O ZZWUYQXMIFTIIY-WEDXCCLWSA-N 0.000 description 1
- TVTZEOHWHUVYCG-KYNKHSRBSA-N Gly-Thr-Thr Chemical compound [H]NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O TVTZEOHWHUVYCG-KYNKHSRBSA-N 0.000 description 1
- WSWWTQYHFCBKBT-DVJZZOLTSA-N Gly-Thr-Trp Chemical compound C[C@@H](O)[C@H](NC(=O)CN)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(O)=O WSWWTQYHFCBKBT-DVJZZOLTSA-N 0.000 description 1
- PYFIQROSWQERAS-LBPRGKRZSA-N Gly-Trp-Gly Chemical compound C1=CC=C2C(C[C@H](NC(=O)CN)C(=O)NCC(O)=O)=CNC2=C1 PYFIQROSWQERAS-LBPRGKRZSA-N 0.000 description 1
- DNAZKGFYFRGZIH-QWRGUYRKSA-N Gly-Tyr-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)CN)CC1=CC=C(O)C=C1 DNAZKGFYFRGZIH-QWRGUYRKSA-N 0.000 description 1
- SBVMXEZQJVUARN-XPUUQOCRSA-N Gly-Val-Ser Chemical compound NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(O)=O SBVMXEZQJVUARN-XPUUQOCRSA-N 0.000 description 1
- IZVICCORZOSGPT-JSGCOSHPSA-N Gly-Val-Tyr Chemical compound [H]NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(O)=O IZVICCORZOSGPT-JSGCOSHPSA-N 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 101710088172 HTH-type transcriptional regulator RipA Proteins 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- AWASVTXPTOLPPP-MBLNEYKQSA-N His-Ala-Thr Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O AWASVTXPTOLPPP-MBLNEYKQSA-N 0.000 description 1
- PDSUIXMZYNURGI-AVGNSLFASA-N His-Arg-Arg Chemical compound NC(N)=NCCC[C@@H](C(O)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CC1=CN=CN1 PDSUIXMZYNURGI-AVGNSLFASA-N 0.000 description 1
- LSQHWKPPOFDHHZ-YUMQZZPRSA-N His-Asp-Gly Chemical compound C1=C(NC=N1)C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)NCC(=O)O)N LSQHWKPPOFDHHZ-YUMQZZPRSA-N 0.000 description 1
- ZJSMFRTVYSLKQU-DJFWLOJKSA-N His-Asp-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC1=CN=CN1)N ZJSMFRTVYSLKQU-DJFWLOJKSA-N 0.000 description 1
- TVTIDSMADMIHEU-KKUMJFAQSA-N His-Cys-Phe Chemical compound N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CS)C(=O)N[C@@H](Cc1ccccc1)C(O)=O TVTIDSMADMIHEU-KKUMJFAQSA-N 0.000 description 1
- NTXIJPDAHXSHNL-ONGXEEELSA-N His-Gly-Val Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)NCC(=O)N[C@@H](C(C)C)C(O)=O NTXIJPDAHXSHNL-ONGXEEELSA-N 0.000 description 1
- QEYUCKCWTMIERU-SRVKXCTJSA-N His-Lys-Asp Chemical compound C1=C(NC=N1)C[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)O)C(=O)O)N QEYUCKCWTMIERU-SRVKXCTJSA-N 0.000 description 1
- BSVLMPMIXPQNKC-KBPBESRZSA-N His-Phe-Gly Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)NCC(O)=O BSVLMPMIXPQNKC-KBPBESRZSA-N 0.000 description 1
- FRDFAWHTPDKRHG-ULQDDVLXSA-N His-Tyr-Arg Chemical compound C([C@H](N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)C1=CN=CN1 FRDFAWHTPDKRHG-ULQDDVLXSA-N 0.000 description 1
- DRKZDEFADVYTLU-AVGNSLFASA-N His-Val-Val Chemical compound [H]N[C@@H](CC1=CNC=N1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(O)=O DRKZDEFADVYTLU-AVGNSLFASA-N 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- MKWSZEHGHSLNPF-NAKRPEOUSA-N Ile-Ala-Val Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)O)N MKWSZEHGHSLNPF-NAKRPEOUSA-N 0.000 description 1
- NCSIQAFSIPHVAN-IUKAMOBKSA-N Ile-Asn-Thr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)N NCSIQAFSIPHVAN-IUKAMOBKSA-N 0.000 description 1
- SYVMEYAPXRRXAN-MXAVVETBSA-N Ile-Cys-Phe Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)N SYVMEYAPXRRXAN-MXAVVETBSA-N 0.000 description 1
- JHCVYQKVKOLAIU-NAKRPEOUSA-N Ile-Cys-Val Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)O)N JHCVYQKVKOLAIU-NAKRPEOUSA-N 0.000 description 1
- HUORUFRRJHELPD-MNXVOIDGSA-N Ile-Leu-Glu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N HUORUFRRJHELPD-MNXVOIDGSA-N 0.000 description 1
- WLRJHVNFGAOYPS-HJPIBITLSA-N Ile-Ser-Tyr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CC=C(C=C1)O)C(=O)O)N WLRJHVNFGAOYPS-HJPIBITLSA-N 0.000 description 1
- MGUTVMBNOMJLKC-VKOGCVSHSA-N Ile-Trp-Val Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CC1=CNC2=CC=CC=C21)C(=O)N[C@@H](C(C)C)C(=O)O)N MGUTVMBNOMJLKC-VKOGCVSHSA-N 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108010065920 Insulin Lispro Proteins 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- HGCNKOLVKRAVHD-UHFFFAOYSA-N L-Met-L-Phe Natural products CSCCC(N)C(=O)NC(C(O)=O)CC1=CC=CC=C1 HGCNKOLVKRAVHD-UHFFFAOYSA-N 0.000 description 1
- RCFDOSNHHZGBOY-UHFFFAOYSA-N L-isoleucyl-L-alanine Natural products CCC(C)C(N)C(=O)NC(C)C(O)=O RCFDOSNHHZGBOY-UHFFFAOYSA-N 0.000 description 1
- SENJXOPIZNYLHU-UHFFFAOYSA-N L-leucyl-L-arginine Natural products CC(C)CC(N)C(=O)NC(C(O)=O)CCCN=C(N)N SENJXOPIZNYLHU-UHFFFAOYSA-N 0.000 description 1
- TYYLDKGBCJGJGW-UHFFFAOYSA-N L-tryptophan-L-tyrosine Natural products C=1NC2=CC=CC=C2C=1CC(N)C(=O)NC(C(O)=O)CC1=CC=C(O)C=C1 TYYLDKGBCJGJGW-UHFFFAOYSA-N 0.000 description 1
- 102000004407 Lactalbumin Human genes 0.000 description 1
- 108090000942 Lactalbumin Proteins 0.000 description 1
- XIRYQRLFHWWWTC-QEJZJMRPSA-N Leu-Ala-Phe Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 XIRYQRLFHWWWTC-QEJZJMRPSA-N 0.000 description 1
- TWQIYNGNYNJUFM-NHCYSSNCSA-N Leu-Asn-Val Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(O)=O TWQIYNGNYNJUFM-NHCYSSNCSA-N 0.000 description 1
- YKNBJXOJTURHCU-DCAQKATOSA-N Leu-Asp-Arg Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(O)=O)CCCN=C(N)N YKNBJXOJTURHCU-DCAQKATOSA-N 0.000 description 1
- IASQBRJGRVXNJI-YUMQZZPRSA-N Leu-Cys-Gly Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)NCC(O)=O IASQBRJGRVXNJI-YUMQZZPRSA-N 0.000 description 1
- KAFOIVJDVSZUMD-DCAQKATOSA-N Leu-Gln-Gln Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O KAFOIVJDVSZUMD-DCAQKATOSA-N 0.000 description 1
- DPWGZWUMUUJQDT-IUCAKERBSA-N Leu-Gln-Gly Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(O)=O DPWGZWUMUUJQDT-IUCAKERBSA-N 0.000 description 1
- GPICTNQYKHHHTH-GUBZILKMSA-N Leu-Gln-Ser Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(O)=O GPICTNQYKHHHTH-GUBZILKMSA-N 0.000 description 1
- QVFGXCVIXXBFHO-AVGNSLFASA-N Leu-Glu-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O QVFGXCVIXXBFHO-AVGNSLFASA-N 0.000 description 1
- HQUXQAMSWFIRET-AVGNSLFASA-N Leu-Glu-Lys Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CCCCN HQUXQAMSWFIRET-AVGNSLFASA-N 0.000 description 1
- HYIFFZAQXPUEAU-QWRGUYRKSA-N Leu-Gly-Leu Chemical compound CC(C)C[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC(C)C HYIFFZAQXPUEAU-QWRGUYRKSA-N 0.000 description 1
- BKTXKJMNTSMJDQ-AVGNSLFASA-N Leu-His-Gln Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)N[C@@H](CCC(=O)N)C(=O)O)N BKTXKJMNTSMJDQ-AVGNSLFASA-N 0.000 description 1
- DBSLVQBXKVKDKJ-BJDJZHNGSA-N Leu-Ile-Ala Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O DBSLVQBXKVKDKJ-BJDJZHNGSA-N 0.000 description 1
- IFMPDNRWZZEZSL-SRVKXCTJSA-N Leu-Leu-Cys Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(O)=O IFMPDNRWZZEZSL-SRVKXCTJSA-N 0.000 description 1
- YOKVEHGYYQEQOP-QWRGUYRKSA-N Leu-Leu-Gly Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O YOKVEHGYYQEQOP-QWRGUYRKSA-N 0.000 description 1
- JVTYXRRFZCEPPK-RHYQMDGZSA-N Leu-Met-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)N)O JVTYXRRFZCEPPK-RHYQMDGZSA-N 0.000 description 1
- MAXILRZVORNXBE-PMVMPFDFSA-N Leu-Phe-Trp Chemical compound C([C@H](NC(=O)[C@@H](N)CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)C1=CC=CC=C1 MAXILRZVORNXBE-PMVMPFDFSA-N 0.000 description 1
- VULJUQZPSOASBZ-SRVKXCTJSA-N Leu-Pro-Glu Chemical compound [H]N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(O)=O)C(O)=O VULJUQZPSOASBZ-SRVKXCTJSA-N 0.000 description 1
- JIHDFWWRYHSAQB-GUBZILKMSA-N Leu-Ser-Glu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@H](C(O)=O)CCC(O)=O JIHDFWWRYHSAQB-GUBZILKMSA-N 0.000 description 1
- XZNJZXJZBMBGGS-NHCYSSNCSA-N Leu-Val-Asn Chemical compound [H]N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O XZNJZXJZBMBGGS-NHCYSSNCSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- ALSRJRIWBNENFY-DCAQKATOSA-N Lys-Arg-Asn Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(O)=O ALSRJRIWBNENFY-DCAQKATOSA-N 0.000 description 1
- NQCJGQHHYZNUDK-DCAQKATOSA-N Lys-Arg-Ser Chemical compound NCCCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CO)C(O)=O)CCCN=C(N)N NQCJGQHHYZNUDK-DCAQKATOSA-N 0.000 description 1
- JBRWKVANRYPCAF-XIRDDKMYSA-N Lys-Asn-Trp Chemical compound C1=CC=C2C(=C1)C(=CN2)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCCCN)N JBRWKVANRYPCAF-XIRDDKMYSA-N 0.000 description 1
- QUYCUALODHJQLK-CIUDSAMLSA-N Lys-Asp-Asp Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O QUYCUALODHJQLK-CIUDSAMLSA-N 0.000 description 1
- NRQRKMYZONPCTM-CIUDSAMLSA-N Lys-Asp-Ser Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(O)=O NRQRKMYZONPCTM-CIUDSAMLSA-N 0.000 description 1
- PBIPLDMFHAICIP-DCAQKATOSA-N Lys-Glu-Glu Chemical compound [H]N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O PBIPLDMFHAICIP-DCAQKATOSA-N 0.000 description 1
- GPJGFSFYBJGYRX-YUMQZZPRSA-N Lys-Gly-Asp Chemical compound NCCCC[C@H](N)C(=O)NCC(=O)N[C@H](C(O)=O)CC(O)=O GPJGFSFYBJGYRX-YUMQZZPRSA-N 0.000 description 1
- LCMWVZLBCUVDAZ-IUCAKERBSA-N Lys-Gly-Glu Chemical compound [NH3+]CCCC[C@H]([NH3+])C(=O)NCC(=O)N[C@H](C([O-])=O)CCC([O-])=O LCMWVZLBCUVDAZ-IUCAKERBSA-N 0.000 description 1
- QOJDBRUCOXQSSK-AJNGGQMLSA-N Lys-Ile-Lys Chemical compound NCCCC[C@H](N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(O)=O QOJDBRUCOXQSSK-AJNGGQMLSA-N 0.000 description 1
- ORVFEGYUJITPGI-IHRRRGAJSA-N Lys-Leu-Met Chemical compound CSCC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCCCN ORVFEGYUJITPGI-IHRRRGAJSA-N 0.000 description 1
- SVSQSPICRKBMSZ-SRVKXCTJSA-N Lys-Pro-Gln Chemical compound [H]N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(O)=O SVSQSPICRKBMSZ-SRVKXCTJSA-N 0.000 description 1
- QFSYGUMEANRNJE-DCAQKATOSA-N Lys-Val-Cys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CCCCN)N QFSYGUMEANRNJE-DCAQKATOSA-N 0.000 description 1
- VKCPHIOZDWUFSW-ONGXEEELSA-N Lys-Val-Gly Chemical compound OC(=O)CNC(=O)[C@H](C(C)C)NC(=O)[C@@H](N)CCCCN VKCPHIOZDWUFSW-ONGXEEELSA-N 0.000 description 1
- 206010027336 Menstruation delayed Diseases 0.000 description 1
- AHZNUGRZHMZGFL-GUBZILKMSA-N Met-Arg-Ser Chemical compound CSCC[C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CO)C(O)=O)CCCNC(N)=N AHZNUGRZHMZGFL-GUBZILKMSA-N 0.000 description 1
- GFDBWMDLBKCLQH-IHRRRGAJSA-N Met-Phe-Cys Chemical compound CSCC[C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CS)C(=O)O)N GFDBWMDLBKCLQH-IHRRRGAJSA-N 0.000 description 1
- SPSSJSICDYYTQN-HJGDQZAQSA-N Met-Thr-Gln Chemical compound CSCC[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@H](C(O)=O)CCC(N)=O SPSSJSICDYYTQN-HJGDQZAQSA-N 0.000 description 1
- QAVZUKIPOMBLMC-AVGNSLFASA-N Met-Val-Leu Chemical compound CSCC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CC(C)C QAVZUKIPOMBLMC-AVGNSLFASA-N 0.000 description 1
- 102000003792 Metallothionein Human genes 0.000 description 1
- 108090000157 Metallothionein Proteins 0.000 description 1
- 208000034486 Multi-organ failure Diseases 0.000 description 1
- 208000010718 Multiple Organ Failure Diseases 0.000 description 1
- YBAFDPFAUTYYRW-UHFFFAOYSA-N N-L-alpha-glutamyl-L-leucine Natural products CC(C)CC(C(O)=O)NC(=O)C(N)CCC(O)=O YBAFDPFAUTYYRW-UHFFFAOYSA-N 0.000 description 1
- XZFYRXDAULDNFX-UHFFFAOYSA-N N-L-cysteinyl-L-phenylalanine Natural products SCC(N)C(=O)NC(C(O)=O)CC1=CC=CC=C1 XZFYRXDAULDNFX-UHFFFAOYSA-N 0.000 description 1
- BQVUABVGYYSDCJ-UHFFFAOYSA-N Nalpha-L-Leucyl-L-tryptophan Natural products C1=CC=C2C(CC(NC(=O)C(N)CC(C)C)C(O)=O)=CNC2=C1 BQVUABVGYYSDCJ-UHFFFAOYSA-N 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 241001561899 Otomys Species 0.000 description 1
- 241000282520 Papio Species 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- LXUJDHOKVUYHRC-KKUMJFAQSA-N Phe-Cys-Leu Chemical compound CC(C)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CC1=CC=CC=C1)N LXUJDHOKVUYHRC-KKUMJFAQSA-N 0.000 description 1
- KYYMILWEGJYPQZ-IHRRRGAJSA-N Phe-Glu-Glu Chemical compound OC(=O)CC[C@@H](C(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 KYYMILWEGJYPQZ-IHRRRGAJSA-N 0.000 description 1
- FIRWJEJVFFGXSH-RYUDHWBXSA-N Phe-Glu-Gly Chemical compound OC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 FIRWJEJVFFGXSH-RYUDHWBXSA-N 0.000 description 1
- OQTDZEJJWWAGJT-KKUMJFAQSA-N Phe-Lys-Asp Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(O)=O OQTDZEJJWWAGJT-KKUMJFAQSA-N 0.000 description 1
- BONHGTUEEPIMPM-AVGNSLFASA-N Phe-Ser-Glu Chemical compound [H]N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(O)=O BONHGTUEEPIMPM-AVGNSLFASA-N 0.000 description 1
- YCEWAVIRWNGGSS-NQCBNZPSSA-N Phe-Trp-Ile Chemical compound C([C@H](N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O)C1=CC=CC=C1 YCEWAVIRWNGGSS-NQCBNZPSSA-N 0.000 description 1
- MWQXFDIQXIXPMS-UNQGMJICSA-N Phe-Val-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC1=CC=CC=C1)N)O MWQXFDIQXIXPMS-UNQGMJICSA-N 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- 108010051508 Preconativ Proteins 0.000 description 1
- CQZNGNCAIXMAIQ-UBHSHLNASA-N Pro-Ala-Phe Chemical compound C[C@H](NC(=O)[C@@H]1CCCN1)C(=O)N[C@@H](Cc1ccccc1)C(O)=O CQZNGNCAIXMAIQ-UBHSHLNASA-N 0.000 description 1
- VCYJKOLZYPYGJV-AVGNSLFASA-N Pro-Arg-Leu Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(O)=O VCYJKOLZYPYGJV-AVGNSLFASA-N 0.000 description 1
- XWYXZPHPYKRYPA-GMOBBJLQSA-N Pro-Asn-Ile Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O XWYXZPHPYKRYPA-GMOBBJLQSA-N 0.000 description 1
- ZPPVJIJMIKTERM-YUMQZZPRSA-N Pro-Gln-Gly Chemical compound OC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@@H]1CCCN1 ZPPVJIJMIKTERM-YUMQZZPRSA-N 0.000 description 1
- AFXCXDQNRXTSBD-FJXKBIBVSA-N Pro-Gly-Thr Chemical compound [H]N1CCC[C@H]1C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(O)=O AFXCXDQNRXTSBD-FJXKBIBVSA-N 0.000 description 1
- HAEGAELAYWSUNC-WPRPVWTQSA-N Pro-Gly-Val Chemical compound [H]N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](C(C)C)C(O)=O HAEGAELAYWSUNC-WPRPVWTQSA-N 0.000 description 1
- NFLNBHLMLYALOO-DCAQKATOSA-N Pro-Leu-Cys Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@@H]1CCCN1 NFLNBHLMLYALOO-DCAQKATOSA-N 0.000 description 1
- CXGLFEOYCJFKPR-RCWTZXSCSA-N Pro-Thr-Val Chemical compound [H]N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(O)=O CXGLFEOYCJFKPR-RCWTZXSCSA-N 0.000 description 1
- GNFHQWNCSSPOBT-ULQDDVLXSA-N Pro-Trp-Gln Chemical compound C1C[C@H](NC1)C(=O)N[C@@H](CC2=CNC3=CC=CC=C32)C(=O)N[C@@H](CCC(=O)N)C(=O)O GNFHQWNCSSPOBT-ULQDDVLXSA-N 0.000 description 1
- FHJQROWZEJFZPO-SRVKXCTJSA-N Pro-Val-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H]1CCCN1 FHJQROWZEJFZPO-SRVKXCTJSA-N 0.000 description 1
- 239000004792 Prolene Substances 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 206010038563 Reocclusion Diseases 0.000 description 1
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- CWHJIJJSDGEHNS-MYLFLSLOSA-N Senegenin Chemical compound C1[C@H](O)[C@H](O)[C@@](C)(C(O)=O)[C@@H]2CC[C@@]3(C)C(CC[C@]4(CCC(C[C@H]44)(C)C)C(O)=O)=C4[C@@H](CCl)C[C@@H]3[C@]21C CWHJIJJSDGEHNS-MYLFLSLOSA-N 0.000 description 1
- QFBNNYNWKYKVJO-DCAQKATOSA-N Ser-Arg-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)CCCN=C(N)N QFBNNYNWKYKVJO-DCAQKATOSA-N 0.000 description 1
- BNFVPSRLHHPQKS-WHFBIAKZSA-N Ser-Asp-Gly Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(O)=O BNFVPSRLHHPQKS-WHFBIAKZSA-N 0.000 description 1
- SFZKGGOGCNQPJY-CIUDSAMLSA-N Ser-Asp-His Chemical compound C1=C(NC=N1)C[C@@H](C(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CO)N SFZKGGOGCNQPJY-CIUDSAMLSA-N 0.000 description 1
- BLPYXIXXCFVIIF-FXQIFTODSA-N Ser-Cys-Arg Chemical compound C(C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CO)N)CN=C(N)N BLPYXIXXCFVIIF-FXQIFTODSA-N 0.000 description 1
- KMWFXJCGRXBQAC-CIUDSAMLSA-N Ser-Cys-Lys Chemical compound C(CCN)C[C@@H](C(=O)O)NC(=O)[C@H](CS)NC(=O)[C@H](CO)N KMWFXJCGRXBQAC-CIUDSAMLSA-N 0.000 description 1
- VDVYTKZBMFADQH-AVGNSLFASA-N Ser-Gln-Tyr Chemical compound OC[C@H](N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 VDVYTKZBMFADQH-AVGNSLFASA-N 0.000 description 1
- PVDTYLHUWAEYGY-CIUDSAMLSA-N Ser-Glu-Arg Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O PVDTYLHUWAEYGY-CIUDSAMLSA-N 0.000 description 1
- YMTLKLXDFCSCNX-BYPYZUCNSA-N Ser-Gly-Gly Chemical compound OC[C@H](N)C(=O)NCC(=O)NCC(O)=O YMTLKLXDFCSCNX-BYPYZUCNSA-N 0.000 description 1
- VMLONWHIORGALA-SRVKXCTJSA-N Ser-Leu-Leu Chemical compound CC(C)C[C@@H](C([O-])=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H]([NH3+])CO VMLONWHIORGALA-SRVKXCTJSA-N 0.000 description 1
- BUYHXYIUQUBEQP-AVGNSLFASA-N Ser-Phe-Glu Chemical compound C1=CC=C(C=C1)C[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](CO)N BUYHXYIUQUBEQP-AVGNSLFASA-N 0.000 description 1
- XQAPEISNMXNKGE-FXQIFTODSA-N Ser-Pro-Cys Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CO)N)C(=O)N[C@@H](CS)C(=O)O XQAPEISNMXNKGE-FXQIFTODSA-N 0.000 description 1
- CUXJENOFJXOSOZ-BIIVOSGPSA-N Ser-Ser-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CO)NC(=O)[C@H](CO)N)C(=O)O CUXJENOFJXOSOZ-BIIVOSGPSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 108700007696 Tetrahydrofolate Dehydrogenase Proteins 0.000 description 1
- JNQZPAWOPBZGIX-RCWTZXSCSA-N Thr-Arg-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)[C@@H](C)O)CCCN=C(N)N JNQZPAWOPBZGIX-RCWTZXSCSA-N 0.000 description 1
- PAOYNIKMYOGBMR-PBCZWWQYSA-N Thr-Asn-His Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)N)O PAOYNIKMYOGBMR-PBCZWWQYSA-N 0.000 description 1
- LKEKWDJCJSPXNI-IRIUXVKKSA-N Thr-Glu-Tyr Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 LKEKWDJCJSPXNI-IRIUXVKKSA-N 0.000 description 1
- IMULJHHGAUZZFE-MBLNEYKQSA-N Thr-Gly-Ile Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(O)=O IMULJHHGAUZZFE-MBLNEYKQSA-N 0.000 description 1
- KBBRNEDOYWMIJP-KYNKHSRBSA-N Thr-Gly-Thr Chemical compound C[C@H]([C@@H](C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)O)N)O KBBRNEDOYWMIJP-KYNKHSRBSA-N 0.000 description 1
- FLPZMPOZGYPBEN-PPCPHDFISA-N Thr-Leu-Ile Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O FLPZMPOZGYPBEN-PPCPHDFISA-N 0.000 description 1
- TZJSEJOXAIWOST-RHYQMDGZSA-N Thr-Lys-Arg Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(O)=O)CCCN=C(N)N TZJSEJOXAIWOST-RHYQMDGZSA-N 0.000 description 1
- SPVHQURZJCUDQC-VOAKCMCISA-N Thr-Lys-Leu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(O)=O SPVHQURZJCUDQC-VOAKCMCISA-N 0.000 description 1
- KPMIQCXJDVKWKO-IFFSRLJSSA-N Thr-Val-Glu Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(O)=O KPMIQCXJDVKWKO-IFFSRLJSSA-N 0.000 description 1
- AKHDFZHUPGVFEJ-YEPSODPASA-N Thr-Val-Gly Chemical compound [H]N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)NCC(O)=O AKHDFZHUPGVFEJ-YEPSODPASA-N 0.000 description 1
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 1
- 208000013222 Toxemia Diseases 0.000 description 1
- 102000002070 Transferrins Human genes 0.000 description 1
- 108010015865 Transferrins Proteins 0.000 description 1
- YVXIAOOYAKBAAI-SZMVWBNQSA-N Trp-Leu-Gln Chemical compound C1=CC=C2C(C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)=CNC2=C1 YVXIAOOYAKBAAI-SZMVWBNQSA-N 0.000 description 1
- CCZXBOFIBYQLEV-IHPCNDPISA-N Trp-Leu-Leu Chemical compound CC(C)C[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)Cc1c[nH]c2ccccc12)C(O)=O CCZXBOFIBYQLEV-IHPCNDPISA-N 0.000 description 1
- DVLHKUWLNKDINO-PMVMPFDFSA-N Trp-Tyr-Leu Chemical compound [H]N[C@@H](CC1=CNC2=C1C=CC=C2)C(=O)N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(C)C)C(O)=O DVLHKUWLNKDINO-PMVMPFDFSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- JJNXZIPLIXIGBX-HJPIBITLSA-N Tyr-Ile-Cys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)N JJNXZIPLIXIGBX-HJPIBITLSA-N 0.000 description 1
- ILTXFANLDMJWPR-SIUGBPQLSA-N Tyr-Ile-Glu Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCC(=O)O)C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)N ILTXFANLDMJWPR-SIUGBPQLSA-N 0.000 description 1
- DMWNPLOERDAHSY-MEYUZBJRSA-N Tyr-Leu-Thr Chemical compound [H]N[C@@H](CC1=CC=C(O)C=C1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(O)=O DMWNPLOERDAHSY-MEYUZBJRSA-N 0.000 description 1
- VYTUETMEZZLJFU-IHRRRGAJSA-N Tyr-Pro-Cys Chemical compound C1C[C@H](N(C1)C(=O)[C@H](CC2=CC=C(C=C2)O)N)C(=O)N[C@@H](CS)C(=O)O VYTUETMEZZLJFU-IHRRRGAJSA-N 0.000 description 1
- OBKOPLHSRDATFO-XHSDSOJGSA-N Tyr-Val-Pro Chemical compound CC(C)[C@@H](C(=O)N1CCC[C@@H]1C(=O)O)NC(=O)[C@H](CC2=CC=C(C=C2)O)N OBKOPLHSRDATFO-XHSDSOJGSA-N 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 101150006005 VII gene Proteins 0.000 description 1
- ZMDCGGKHRKNWKD-LAEOZQHASA-N Val-Asn-Glu Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CCC(=O)O)C(=O)O)N ZMDCGGKHRKNWKD-LAEOZQHASA-N 0.000 description 1
- UDNYEPLJTRDMEJ-RCOVLWMOSA-N Val-Asn-Gly Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CC(=O)N)C(=O)NCC(=O)O)N UDNYEPLJTRDMEJ-RCOVLWMOSA-N 0.000 description 1
- DJEVQCWNMQOABE-RCOVLWMOSA-N Val-Gly-Asp Chemical compound CC(C)[C@@H](C(=O)NCC(=O)N[C@@H](CC(=O)O)C(=O)O)N DJEVQCWNMQOABE-RCOVLWMOSA-N 0.000 description 1
- PIFJAFRUVWZRKR-QMMMGPOBSA-N Val-Gly-Gly Chemical compound CC(C)[C@H]([NH3+])C(=O)NCC(=O)NCC([O-])=O PIFJAFRUVWZRKR-QMMMGPOBSA-N 0.000 description 1
- UMPVMAYCLYMYGA-ONGXEEELSA-N Val-Leu-Gly Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(O)=O UMPVMAYCLYMYGA-ONGXEEELSA-N 0.000 description 1
- XTDDIVQWDXMRJL-IHRRRGAJSA-N Val-Leu-His Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](CC1=CN=CN1)C(=O)O)NC(=O)[C@H](C(C)C)N XTDDIVQWDXMRJL-IHRRRGAJSA-N 0.000 description 1
- SYSWVVCYSXBVJG-RHYQMDGZSA-N Val-Leu-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)N)O SYSWVVCYSXBVJG-RHYQMDGZSA-N 0.000 description 1
- NHXZRXLFOBFMDM-AVGNSLFASA-N Val-Pro-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)C(C)C NHXZRXLFOBFMDM-AVGNSLFASA-N 0.000 description 1
- DEGUERSKQBRZMZ-FXQIFTODSA-N Val-Ser-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(O)=O DEGUERSKQBRZMZ-FXQIFTODSA-N 0.000 description 1
- JQTYTBPCSOAZHI-FXQIFTODSA-N Val-Ser-Cys Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CS)C(=O)O)N JQTYTBPCSOAZHI-FXQIFTODSA-N 0.000 description 1
- RYHUIHUOYRNNIE-NRPADANISA-N Val-Ser-Gln Chemical compound CC(C)[C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(=O)N)C(=O)O)N RYHUIHUOYRNNIE-NRPADANISA-N 0.000 description 1
- SDHZOOIGIUEPDY-JYJNAYRXSA-N Val-Ser-Trp Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@H](CO)NC(=O)[C@@H](N)C(C)C)C(O)=O)=CNC2=C1 SDHZOOIGIUEPDY-JYJNAYRXSA-N 0.000 description 1
- UQMPYVLTQCGRSK-IFFSRLJSSA-N Val-Thr-Gln Chemical compound C[C@H]([C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](C(C)C)N)O UQMPYVLTQCGRSK-IFFSRLJSSA-N 0.000 description 1
- BGTDGENDNWGMDQ-KJEVXHAQSA-N Val-Tyr-Thr Chemical compound C[C@H]([C@@H](C(=O)O)NC(=O)[C@H](CC1=CC=C(C=C1)O)NC(=O)[C@H](C(C)C)N)O BGTDGENDNWGMDQ-KJEVXHAQSA-N 0.000 description 1
- LLJLBRRXKZTTRD-GUBZILKMSA-N Val-Val-Ser Chemical compound CC(C)[C@@H](C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CO)C(=O)O)N LLJLBRRXKZTTRD-GUBZILKMSA-N 0.000 description 1
- 241000294142 Vascellum Species 0.000 description 1
- 208000024248 Vascular System injury Diseases 0.000 description 1
- 208000012339 Vascular injury Diseases 0.000 description 1
- 101710087237 Whey acidic protein Proteins 0.000 description 1
- 238000001793 Wilcoxon signed-rank test Methods 0.000 description 1
- DPDMMXDBJGCCQC-UHFFFAOYSA-N [Na].[Cl] Chemical compound [Na].[Cl] DPDMMXDBJGCCQC-UHFFFAOYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 238000001467 acupuncture Methods 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 108010070944 alanylhistidine Proteins 0.000 description 1
- 108010011559 alanylphenylalanine Proteins 0.000 description 1
- 108010050025 alpha-glutamyltryptophan Proteins 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000010100 anticoagulation Effects 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 108010013835 arginine glutamate Proteins 0.000 description 1
- 108010059459 arginyl-threonyl-phenylalanine Proteins 0.000 description 1
- 210000002565 arteriole Anatomy 0.000 description 1
- 108010092854 aspartyllysine Proteins 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940006612 barium citrate Drugs 0.000 description 1
- PAVWOHWZXOQYDB-UHFFFAOYSA-H barium(2+);2-hydroxypropane-1,2,3-tricarboxylate Chemical compound [Ba+2].[Ba+2].[Ba+2].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O PAVWOHWZXOQYDB-UHFFFAOYSA-H 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 229940088623 biologically active substance Drugs 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- UDSAIICHUKSCKT-UHFFFAOYSA-N bromophenol blue Chemical compound C1=C(Br)C(O)=C(Br)C=C1C1(C=2C=C(Br)C(O)=C(Br)C=2)C2=CC=CC=C2S(=O)(=O)O1 UDSAIICHUKSCKT-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 229940021722 caseins Drugs 0.000 description 1
- 239000012930 cell culture fluid Substances 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 210000000080 chela (arthropods) Anatomy 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 210000003109 clavicle Anatomy 0.000 description 1
- 230000001112 coagulating effect Effects 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 239000003218 coronary vasodilator agent Substances 0.000 description 1
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 1
- 150000004775 coumarins Chemical class 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 108010016616 cysteinylglycine Proteins 0.000 description 1
- 108010060199 cysteinylproline Proteins 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 102000004419 dihydrofolate reductase Human genes 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 108010054812 diprotin A Proteins 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229920002549 elastin Polymers 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 201000007386 factor VII deficiency Diseases 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 108010055341 glutamyl-glutamic acid Proteins 0.000 description 1
- 108010084264 glycyl-glycyl-cysteine Proteins 0.000 description 1
- 108010051307 glycyl-glycyl-proline Proteins 0.000 description 1
- 108010089804 glycyl-threonine Proteins 0.000 description 1
- 108010010147 glycylglutamine Proteins 0.000 description 1
- 108010015792 glycyllysine Proteins 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000021760 high fever Diseases 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 108010040030 histidinoalanine Proteins 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- YQYJSBFKSSDGFO-FWAVGLHBSA-N hygromycin A Chemical compound O[C@H]1[C@H](O)[C@H](C(=O)C)O[C@@H]1Oc1ccc(\C=C(/C)C(=O)N[C@@H]2[C@@H]([C@H]3OCO[C@H]3[C@@H](O)[C@@H]2O)O)cc1O YQYJSBFKSSDGFO-FWAVGLHBSA-N 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 238000003317 immunochromatography Methods 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003601 intercostal effect Effects 0.000 description 1
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000006623 intrinsic pathway Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002505 iron Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000013038 irreversible inhibitor Substances 0.000 description 1
- 108010000761 leucylarginine Proteins 0.000 description 1
- 108010091871 leucylmethionine Proteins 0.000 description 1
- 108010057821 leucylproline Proteins 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 210000003041 ligament Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 108010003700 lysyl aspartic acid Proteins 0.000 description 1
- 108010064235 lysylglycine Proteins 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 108010068488 methionylphenylalanine Proteins 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 238000002406 microsurgery Methods 0.000 description 1
- 210000004088 microvessel Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000013425 morphometry Methods 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 230000003387 muscular Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000001009 osteoporotic effect Effects 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 210000004681 ovum Anatomy 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000036285 pathological change Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 210000003516 pericardium Anatomy 0.000 description 1
- 238000005502 peroxidation Methods 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 108010025221 plasma protein Z Proteins 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 230000020971 positive regulation of blood coagulation Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000003805 procoagulant Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 108010029020 prolylglycine Proteins 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 235000021251 pulses Nutrition 0.000 description 1
- 238000010814 radioimmunoprecipitation assay Methods 0.000 description 1
- 108010013773 recombinant FVIIa Proteins 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 238000012882 sequential analysis Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 108010069117 seryl-lysyl-aspartic acid Proteins 0.000 description 1
- 235000020254 sheep milk Nutrition 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 238000005480 shot peening Methods 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- NGSFWBMYFKHRBD-UHFFFAOYSA-N sodium;2-hydroxypropanoic acid Chemical compound [Na+].CC(O)C(O)=O NGSFWBMYFKHRBD-UHFFFAOYSA-N 0.000 description 1
- 238000007711 solidification Methods 0.000 description 1
- 230000008023 solidification Effects 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- QTENRWWVYAAPBI-YCRXJPFRSA-N streptomycin sulfate Chemical compound OS(O)(=O)=O.OS(O)(=O)=O.OS(O)(=O)=O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O.CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](N=C(N)N)[C@H](O)[C@@H](N=C(N)N)[C@H](O)[C@H]1O QTENRWWVYAAPBI-YCRXJPFRSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- OBTWBSRJZRCYQV-UHFFFAOYSA-N sulfuryl difluoride Chemical compound FS(F)(=O)=O OBTWBSRJZRCYQV-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000009871 tenuigenin Substances 0.000 description 1
- 210000003684 theca cell Anatomy 0.000 description 1
- BHBIPLOIWQSVID-UHFFFAOYSA-N thiohypofluorous acid Chemical compound SF BHBIPLOIWQSVID-UHFFFAOYSA-N 0.000 description 1
- 108010061238 threonyl-glycine Proteins 0.000 description 1
- 230000009424 thromboembolic effect Effects 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 108010038745 tryptophylglycine Proteins 0.000 description 1
- 108010044292 tryptophyltyrosine Proteins 0.000 description 1
- 210000004231 tunica media Anatomy 0.000 description 1
- 229940072651 tylenol Drugs 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
- 238000003809 water extraction Methods 0.000 description 1
- 235000021241 α-lactalbumin Nutrition 0.000 description 1
Images

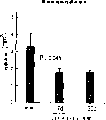
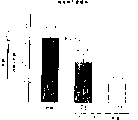


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6437—Coagulation factor VIIa (3.4.21.21)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/164—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- A61K38/166—Streptokinase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
- A61K38/482—Serine endopeptidases (3.4.21)
- A61K38/4846—Factor VII (3.4.21.21); Factor IX (3.4.21.22); Factor Xa (3.4.21.6); Factor XI (3.4.21.27); Factor XII (3.4.21.38)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
- A61K38/49—Urokinase; Tissue plasminogen activator
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21021—Coagulation factor VIIa (3.4.21.21)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Toxicology (AREA)
- Biophysics (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Materials For Medical Uses (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
因子VII的催化活性位点经修饰产生一种能有效地阻断血液凝固级联反应的化合物。这些修饰使因子VIIa基本上不能激活血浆因子X或LX。本发明涉及一种新治疗方法以及修饰过的因子VII用于预防和治疗与缺血后再灌注相关的心肌损伤,用于改善再灌注过程中局部心肌血流以及用于维持或改善病人的血管开放度的用途,以及在血管易于形成血栓处修饰过的因子VII的局部应用。
Description
发明领域
本发明涉及新的治疗方法和修饰形式的因子VII的新用途,修饰过的因子VII抑制血栓形成,维持或改善血管开放度,改善局部心肌血流和调节在缺血后再灌注过程中的心肌损伤。
发明背景
血液凝固是包含一系列不同血液成分或因子的复杂反应,最终产生血纤蛋白块。一般,血液中参与称为凝血级联反应的成分是酶原,一种能通过一种激活物(自身为活化的凝血因子)作用转变成蛋白水解酶的酶学无活性的蛋白质。已经历这种转变的凝血因子一般称为“活性因子”,并添加一个小写“a”命名(如,因子VIIa)。
需要活化的因子X(“Xa”)以转变凝血酶原为凝血酶,其再将纤维蛋白原转变成纤维蛋白,作为形成纤维蛋白块的最后阶段。有两个系统或途径促进因子X的激活。“内源性途径”指通过利用仅在血浆中存在的因子导致凝血酶形成的那些反应。一系列蛋白酶介导的反应最终产生了因子IXa,其与因子VIIIa联合,切割因子X形成Xa。在血液凝固的“外源性途径”中,同样的蛋白水解由因子VIIa及其辅因子(组织因子)进行。组织因子是一种膜结合蛋白,一般不在血浆中循环。然而,一旦血管破裂,其可在Ca2+和磷脂存在下与因子VIIa复合以催化因子X或因子IX的激活(Nemerson和Gentry,生物化学(Biochem.)25:4020-4033(1986))。虽然在止血中两种凝血途径的相对重要性不清楚,近年来,已发现因子VII和组织因子在调节血液凝固中起关键作用。
因子VII是以单链酶原形式在血浆中循环的痕量血浆糖蛋白。此酶原无催化活性(Williams等人,生物化学杂志(J.Biol.Chem.)264:7536-7543(1989);Rao等人,美国国家科学院学报(Proc.Natl.Acad.Sci),85:6687-6691(1988))。单链因子VII可以在体外通过因子Xa,因子XIIa,因子IXa或凝血酶转变成双链因子VIIa。认为因子Xa是因子VII的主要生理激活剂。象其它几种参与止血的血浆蛋白一样,因子VII的活性依赖维生素K,这是在蛋白质氨基端成簇的多谷氨酸残基的g-羧基化所需的。这些g-羧基化的谷氨酸为因子VII与磷脂的金属关联的相互作用所需。
酶原因子VII到活化的双链分子的转变通过位于大约分子中部的内肽键的切割来完成。人因子VII中,活性切割位点在Arg152-Ile153(Hagen等人,美国国家科学院院报,83:2412-2416(1986);Thim等人,生物化学(Biochem.)27:7785-7793(1988),此处引用两篇文献作为参考)。牛因子VII通过在类似的Arg152-Ile153键的切割被激活(Takeya等人,生物化学杂志(J.Biol.chem.)263:14868-14877,1988)。在组织因子、磷脂和钙离子存在下,双链因子VIIa通过有限蛋白水解迅速激活因子X或因子IX。
经常需要选择性阻断病人的凝血级联反应。例如,可以在肾透析或治疗深静脉栓塞、弥散性血管内凝血(DIC)以及其它医学疾病过程中使用如肝素、香豆素、香豆素衍生物、二氢化茚二酮衍生物等的抗凝药。例如,肝素处理或柠檬酸离子的体外处理(美国专利4,500,309)可用于透析以防止在治疗过程中的凝血。肝素也用于防止手术中病人的深静脉血栓形成。
然而,用肝素和其它抗凝药的治疗有不期望的副作用。可得的抗凝药一般地作用遍及全身,而不是特异性作用于凝血位置。例如,肝素可引起严重出血。此外,由于半衰期约80分钟,肝素被从血中迅速清除,需要频繁给药。因为肝素以抗凝血酶III(ATIII)的辅助因子起作用,而在DIC治疗中ATIII被迅速地耗贫,维持肝素的合适剂量经常是困难的,需要连续监视ATIII和肝素水平。如果ATIII极度贫乏肝素也无作用。此外,肝素的长期使用也可以增加血小板凝结且减少血小板数量,且参与骨质疏松的形成。二氢化茚二酮衍生物也可有毒副作用。
除了上述简单描述的抗凝药之外,已发现几种天然存在的蛋白质有抗凝药活性。例如,Reutelingsperger(美国专利No.4,736,018)从牛主动脉和人脐静动脉中分离了抗凝蛋白质。Maki等人(美国专利No.4,732,891)公开了人胎盘衍生的抗凝蛋白质。此外,ATIII已被提议用作治疗性抗凝药(Schipper等人,柳叶刀(Lancet)1(8069):854-856(1978);Jordan,美国专利No.4,386,025;Bock等人,美国专利No.4,517,294)。
血管壁中平滑肌细胞(SMC)的增殖是动脉粥样硬化中血管再造或应答其它血管损伤之后血管损伤形成中的重要事件。例如,动脉粥样硬化的治疗经常包括:用血管成形术、动脉内膜切除术或减少动脉粥样硬化斑切除术疏通封闭的血管,或者用旁路移植,一些通过导管插入术(血管成形术)将动脉粥样硬化斑压缩或除去,通过切开(动脉内膜切除术)从动脉壁剥离或通过天然或合成移植物分流的手术方法疏通封闭的血管。这些方法清除了血管内皮,干扰下面的内膜层,导致中层平滑肌细胞的死亡。此损伤之后的中层平滑肌细胞增生且移入内膜,这典型地在损伤后的头几周内最多六个月内发生,且当覆盖在上面的内皮层重建后停止。在人中,这些损伤由约20%细胞和80%胞外基质组成。
在约30%或更多的用血管成形术、动脉内膜切除术或旁路移植治疗的病人中,血栓和/或内膜中平滑肌细胞增生引起血管再闭塞且经常使重建手术失败。这种手术后血管的关闭称为“再狭窄”。
目前仍需要具有抗凝活性改善的物质,其能以相对低的剂量给药且不产生与传统抗凝剂相关的不期望的副作用。本发明通过提供特异性作用于损伤位置的抗凝药且进一步提供了其他相关的便利来满足这一需要。修饰过的因子VII分子给人施用治疗包括血管内凝血的各种情况特别有用。例如,虽然深静脉血栓和肺栓塞可以用传统抗凝药治疗,此处所述的修饰过的因子VII可用于在确定的如那些经历手术或有充血性心力衰竭的高危病人中防止血栓栓塞并发症。此外,修饰过的因子VII可作为组织因子介导的凝血诱导的拮抗剂,阻止凝血酶的产生和随后的纤维蛋白沉淀。同样,修饰过的因子VII可用于抑制组织因子活性,导致如对血液凝固、血栓形成或血小板沉积的抑制。
修饰过的因子VII分子特别可用于由于急性血管损伤的内膜增生或再狭窄的治疗。急性血管损伤是那些与慢性血管损伤(如动脉粥样硬化)相反的迅速发生的(即:数天至数月的)损伤。急性血管损伤常常由手术过程造成,如血管重建,其中使用了血管成形术,动脉内膜切除术,动脉粥样硬化斑切除术,血管移植物置入等技术。增生也可以作为对如:移植物置入或器官移植的延迟反应而发生。由于修饰过的因子VII比肝素更有选择性,一般地仅结合暴露于损伤位置的组织因子,且因为修饰过的因子VII不破坏其它凝血蛋白,因此当防止深静脉栓塞的预防性使用时比肝素更有效且更不易引起出血并发症。
冠状动脉疾病的最新进展包括机械性介入的使用以清除或取代病因性斑块物从而重建通过冠状动脉的足够的血流。除了各种形式的机械性介入的使用,包括气囊血管成形术、减少动脉粥样硬化斑切除术、血管斯坦特固定模(Stent)的放置、激光治疗或喷丸,这些技术的有效性仍受到治疗后六个月内约40%的再狭窄率的局限。
再狭窄被认为是由生物过程的复杂相互作用造成的,包括:血小板沉积和血栓形成,趋化因子和促分裂因子的释放以及血管平滑肌细胞迁移和增生进入扩张的动脉部分的内膜。
对机械性损伤位置的血小板积累的抑制可限制人的再狭窄速率。血小板Gp IIb/IIIa的单克隆抗体的治疗性使用可限制人的再狭窄水平(Califf等人,新英格兰医学杂志(N.Engl.J.Med.),330:956-961(1994)。该抗体可结合到血小板表面的Gp IIb/IIIa受体上因而抑制了血小板积累。此数据提示人冠状动脉中机械性损伤的位置血小板累积的抑制对发生最终治愈反应有利。当在急性血管损伤位置发生血小板积累时,这些位置的凝血酶的产生可能负责血小板的激活和其随后的累积。
如后面的实施例所示,本发明修饰过的因子VII能与细胞表面的组织因子结合。例如,DEGR-因子VIIa以与野生型因子VIIa相同或更高的亲和力与细胞表面组织因子结合。然而,DEGR-因子VIIa无酶活性,但其与组织因子结合且用作野生型因子VIIa的竞争性拮抗剂,从而抑制外源性凝血途径导致凝血酶产生的后续步骤。
修饰过的因子VII分子保持组织因子结合能力,其通过阻断凝血酶产生以及后续纤维蛋白沉积来抑制血管损伤位置的血小板积累。
由于DEGR-因子VII阻断凝血酶产生以及限制在急性血管损伤处血小板沉积的能力,保持组织因子结合活性但无因子VIIa酶活性的修饰过的因子VII分子可用于抑制血管再狭窄。
含有修饰过的因子VII的组合物当配制成药物组合物时在用于治疗病人的方法中特别有用,其中向处于不同疾病状态的个体施用这些修饰过的因子VII的组合物以治疗与凝血相关的病状。这种修饰过的因子VII分子能与组织因子结合但在凝血级联反应中只有很低的催化其它因子活化的能力,其具有较长的血浆半衰期,因而与其它抗凝药相比有相应的较长期的抗凝血活性。适于此组合物的医学适应症是那些通常用抗凝药治疗的如:深静脉血栓,肺栓塞,中风,弥散性血管内凝血(DIC),与革兰氏阴性内毒血症相关的肺和肾中纤维蛋白沉积和心肌梗死。这些组合物可用于抑制机械性血管损伤之后发生的血管再狭窄,如由气囊血管成形术,动脉内膜切除术,减少的动脉粥样硬化斑切除术,斯坦特固定模放置,激光治疗或rotablation造成的损伤,或在血管移植,斯坦特固定模,旁路移植或器官移植之后发生的损伤。这些组合物可因而用于抑制血小板沉积和相关的疾病。因此,一种抑制凝血、血管再狭窄或血小板沉积的方法,例如,包括以足有效抑制凝血、血管再狭窄或血小板沉积的量给病人施用含有修饰过的因子VII,如在其催化三联体Ser344、Asp242和His193中至少有一种氨基酸替换的组合物。该方法也可用于治疗个体中急性冠状动脉闭合(如急性心肌梗塞),其包括与组织型纤维蛋白溶酶原激活剂或链激酶联合使用修饰过的因子VII(包括DEGR-因子VII和FFR-因子VII),可加速tPA诱导的血栓溶解。修饰过的因子VII可在血栓溶解剂如组织型纤维蛋白溶酶原激活剂施用之前、同时,或紧随其后使用。
国际申请No.WO92/15686涉及修饰过的因子VIIa、多核酸和用于修饰过的因子VIIa生产的哺乳动物细胞系,以及用于抑制血液凝固的含有修饰过的因子VIIa的组合物。
国际申请No.WO94/27631涉及用于抑制血管再狭窄、组织因子活性和血小板沉积的方法。
国际申请No.WO96/12800涉及一种用于治疗急性冠状动脉闭合的方法,包括给病人施用与组织型纤溶酶原激活剂或链激酶联合使用的含有修饰过的因子VIIa的组合物。
发明概述
本发明涉及一种用于抑制病人血栓形成的方法,其包括在病人易于形成血栓的血管位置局部施用治疗有效剂量的组合物,此组合物含有在其催化中心至少有一种修饰的因子VII,此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。血栓形成的位置可与手术,显微手术、血管成形术或创伤有关。
本发明也涉及一种用于维持或改善病人中血管开放度的方法,其包括在易于减小开放度的血管位置局部施用治疗有效剂量的组合物,此组合物含有在其催化中心至少有一种修饰的因子VII,此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。血栓形成的位置可与手术、显微手术、血管成形术或创伤有关。
本发明还涉及一种预防或减少个体与缺血后再灌注相关的心肌损伤的方法,其包括给个体施用一种组合物,此组合物含有在其催化中心至少有一种修饰的药学可接受的因子VII,此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。
本发明还涉及一种改善个体在缺血后再灌注过程中局部心肌血流的方法,其包括给个体施用一种组合物,此组合物包含在其催化中心至少有一种修饰的药学可接受的因子VII此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。
在一优选的实施方案中,修饰过的因子VII包含因子VII与丝氨酸蛋白酶抑制剂的反应。在一更优选的方面,此蛋白酶抑制剂是一种有机磷化合物,一种氟化硫(Sulfanyl fluoride),一种肽卤甲基酮或一种氮杂肽。在一更为优选的方面,此蛋白酶抑制剂是一种选自Dansyl-Phe-Pro-Arg氯甲基酮,Dansyl-Glu-Gly-Arg氯甲基酮,Dansyl-Phe-Phe-Arg氯甲基酮及Phe-Phe-Arg氯甲基酮中的肽卤甲基酮,最优选的为Phe-Phe-Arg氯甲基酮。
本发明还涉及在其催化中心至少有一种修饰的因子VII用于制造用来防止或减少与缺血后再灌注相关的心肌损伤的组合物的用途,此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。本发明还涉及在其催化中心至少有一种修饰的因子VII用于制造用来改善缺血后再灌注过程中局部心肌血流的组合物的用途,此修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力。
本发明提供了抑制与缺血后再灌注相关的有害事件的方法和组合物。组织、器官或肢体的严重缺血可以由于血流减少也可以与创伤、手术操作或低血压相关。与严重缺血相关的一种并发症是在动脉系统中组织因子的上调。认为这种组织因子表达的增加刺激了促凝血反应(主要在毛细血管床),因而引发和/或维持了血管内的血栓形成。此外,通过冠状血管系统内的内皮细胞在缺血后心脏的再灌注过程中组织因子(TF)的从头合成可以导致再灌注过程中冠状动脉血流的减少,因而影响最终经历坏死的缺血性心肌层的结局。与患稳定性心绞痛的病人相比,获自患不稳定性心绞痛病人的动脉粥样硬化斑切除术标本中的组织因子抗原及促凝血活性升高。因此,认为在这些病人中不稳定心绞痛可能有大心外膜冠状动脉的亚内皮组织中组织因子暴露造成斑块破坏这一因素的参与。这将最终促进冠状动脉内血栓形成,引起冠状动脉血流确实减少。
组织因子也可以一种不同的途径影响冠状动脉血流。在缺血性组织再灌注后,可以产生闭塞性的或非闭塞性的血栓。动脉床中血栓的产生及在血栓周围血小板的沉积导致该组织中继发性缺血的产生。血栓的产生及血小板的存在于是可引起多种生物活性因子的产生和释放,包括那些从凝血途径产生的如:凝血酶和因子X以及从活化的血小板中释放的各种因子。这些因子又可引起由底部的内皮和平滑肌细胞或由相邻的单核细胞产生额外的因子,如TNF-α和IL-1。这些因子又可再激活内皮细胞导致与单核细胞和中性粒细胞结合相关的各种粘附分子的上调。一般地,与循环血接触的内皮细胞不表达明显的组织因子活性。在特定条件下,内皮细胞可通过表达组织因子样促凝血活性有效地促进凝血。特别是,外源和内源性产生的氧自由基(OFR)可刺激冠状血管内的内皮细胞合成和表达大量的组织因子。氧自由基是高活性的分子,可进攻各种细胞成分。在缺血期后血流恢复之后有大量氧自由基产生,这些氧化剂可在脂过氧化和细胞成分的其它不可逆改变之后造成一种特殊形式的再灌注介导的组织损伤。氧自由基也可急剧减低组织因子途径抑制剂(TFPI)(一种由内皮细胞合成的抑制外源性凝血途径的kunitz型蛋白质)的活性,这种氧自由基的双重效果(由内皮细胞表达组织因子和降低TFPI活性)可以将正常内皮天然的抗凝血性质转变成一种促凝血状态,因而有利于一种不希望的血管内凝血的活化。因此,在冠状循环中氧自由基介导的组织因子表达导致在缺血后再灌注过程中冠状动脉血流的明显减少。这种氧自由基介导的组织因子的表达及其带来的外源性凝血途径的活化有严重后果,因为此现象影响缺血后再灌注的病理生理特别是在经历冠状动脉血栓的急性心肌梗死病人中。
不复流现象,即在先前缺血组织中缺乏对微血管系统一致的灌注,已由Krug等人首次描述(Circ.Res.1966;19:57-62)。可以影响缺血性心肌命运的最重要的决定因素被认为是缺血过程中的侧流量、危险面积的大小以及心肌的氧需求。
在过去的十年中有大量的兴趣集中在用再灌注方案治疗急性的心肌梗死病人方面,包括冠状动脉血栓溶解,初级血管成形术或两者都有。然而,并非全部研究都证实在梗死相关的动脉复通后左心室功能的改善。目前,有相当数目的病人在梗死相关的冠状动脉床中表现出“低流量”状况。此状况至少在死亡率方面几乎完全无益。这种低流量状况被认为至少部分地是由血液缺乏再进入所有的先前缺血性心肌的血管系统的能力引起。现已令人惊讶的表明因子VIIai影响且增加在再灌注过程中局部的心肌血流。也已令人惊讶的表明因子VIIai导致不复流区域的明显减少。
单核和中性粒细胞的结合和转移以及这些细胞释放的生物活性物质(包括氧自由基的产生)可以加剧内皮细胞活化的水平及破坏程度。最终,如果这种级联反应不经抑制,就可导致全身性并发症和可能刺激多种器官衰竭。通过施用针对组织因子/因子VII结合(如:FFR-FVIIa)的特异性抑制剂封闭组织因子,从而阻断凝血外源性途径的起始,防止级联反应的起始,从而调节与缺血/再灌注相关事件的破坏程度,如,消除或减少心肌的损伤或坏死。
附图说明
图1.表示一种Ser344Ala修饰过的因子VII DNA序列的表达载体的构建。所用符号包括0-1,0-1图谱单元序列来自腺病毒5;E,是SV40的增强子;MLP,腺病毒2主要晚期启动子;SS,一套剪切位点;及pA,在晚期方向中来自SV40的聚腺苷酸化作用信号。
图2.表示与盐水处理的对照相比在动脉内膜切除的狒狒主动脉上大剂量注射DEGR-因子VIIa对血栓形成(血小板沉积)的影响。对动脉测量了60分钟以上。DEGR-因子VIIa明显地抑制了在此急性血管损伤的灵长类模型中的富含血小板的血栓的发展。
图3.表示当在恒定量的FVIIa(5nM)存在下用递增浓度的FVIIa(空心框)或是DEGR-因子VIIa(实心框)培养狒狒平滑肌细胞时所获结果。FX活化的水平随后用产色底物S-2222测定。数据以在5nm FVIIa单独存在时产生的活性的百分比用酰胺水解活性给出。
图4.显示与对照动物相比,在颈动脉动脉内膜切除术之后且用DEGR-因子VIIa处理7或30天的狒狒的内膜面积尺寸。
图5.显示在气囊损伤之后且用DEGR-因子VIIa处理的狒狒股动脉内膜面积与内膜+中层面积的比率,其中对照组包括5只血管,处理7天的检测了11只血管,处理30的检测了2只血管(n是检测的血管数)。
图6.显示用于测量梗死尺寸(IS)、不复流(NR)、危险面积(AR)、凝血酶原时间(PT)及活化的部分凝血激酶时间(aPTT)的实验方案。
图7.显示三个治疗组中以梗死的危险面积的百分比表示的再灌注期(IS)结束时梗死尺寸图,三个治疗组分别用FFR-因子VIIa,因子VIIa和盐水治疗。每条代表8只动物的±SD平均值。
图8.显示以梗死的危险面积的百分比表示的再灌注期结束时不复流(NR)的面积图(动物分别用FFR-因子VIIa,因子VIIa和盐水治疗)。每条代表8只动物的±SD平均值。
图9.显示对每只动物用多元线性回归方程计算出的预期的不复流(以左心室百分比,LV)与观察到的不复流(以LV百分比)之间的关系。
图10.显示在缺血20分钟、再灌注10分钟及2小时后用于缺血性心肌评估的局部心肌血流(RMBF)图。
图11.显示FFR-因子VIIa和因子VIIa对凝血酶原时间(PT)和活化的部分促凝血酶原激酶时间(aPTT)的影响。
具体实施方案的说明
修饰过的因子VII可为酶原的形式(即:一种单链分子)或可在其活性位点被切割。因此,“修饰过的因子VII”是指包括结合组织因子并抑制因子IX到IXa和因子X到Xa活化的修饰过的因子VII以及修饰过的因子VIIa。因子VII序列有至少一种氨基酸修饰,其中所选的修饰可显著减少活化的因子VII催化血浆因子X或IX活化的能力,因而可抑制凝血活性。修饰过的因子VII有经至少一种氨基酸替换修饰的活性位点,且其修饰过的形式能与组织因子结合。修饰过的因子VII组合物典型的是基本上纯的形式。
在优选的人和牛因子VII的实施方案中,活性位点残基Ser344被改变,用Gly,Met,Thr或更优选地用Ala取代。这种取代可单独进行或与催化三联体中在其它位点(包括His193和Asp242)的取代联合进行。
修饰过的因子VII组合物适合施用于不同的哺乳动物,特别是人,以抑制凝血级联反应。修饰过的因子VII可以与其它抗凝化合物联合或代替其它抗凝化合物施于病人。典型地,用于施于人的药物组合物含有修饰过的人因子VII蛋白质以及药学可接受的载体和缓冲剂。
因子VII在凝血级联反应中起重要作用,特别是参与了外源性途径。作为一种单链酶原蛋白质在循环血浆中存在的因子VII一旦被活化成为因子VIIa,其与组织因子及钙离子一起激活因子X成为Xa,激活因子IX成为IXa,最终形成一种纤维蛋白块。
因子VII有一个催化位点被修饰以降低因子VIIa的催化活性,同时此分子保留与组织因子结合的能力。修饰过的因子VII分子与天然的因子VII和/或因子VIIa竞争结合组织因子。结果抑制了因子X和IX的活化。
修饰过的因子VII可以由含有两种可操纵性连接的序列编码区域的一种多核苷酸分子编码,此两种序列编码区域分别编码一种前原肽和一种维生素K依赖型血浆蛋白质的gla结构域以及一种无gla结构域的因子VII蛋白质,其中一旦表达,该多核苷酸编码一种不明显激活血浆因子X或IX且能与组织因子结合的修饰过的因子VII分子。由此多核苷酸表达的修饰过的因子VII是一种生物活性的抗凝剂,即它能抑制凝血级联反应,因而抑制纤维蛋白沉积或凝块的形成。为了表达修饰过的因子VII,将此多核苷酸转染入哺乳动物细胞系,如,BHK,BHK570或293细胞系。
因子VIIa的催化活性可以通过催化中心或三联体的化学衍生抑制。通过因子VII与一种不可逆抑制剂如一种有机磷化合物,一种氟化硫(Sulfonyl fluoride),一种肽卤甲基酮或一种氮杂肽反应或通过例如酰化作用实现衍生化。优选的肽卤甲基酮包括PPACK(D-Phe-Pro-Arg氯甲基酮);(见美国专利No.4,318,904,此处引用作为参考),D-Phe-Phe-Arg和Phe-Phe-Arg氯甲基酮(FFR-cmk)以及DEGRck(Dansyl-Glu-Gly-Arg氯甲基酮)。
也可以通过氨基酸替换、插入或删除抑制因子VIIa的催化活性。在优选的实施方案中,在因子VII催化三联体的氨基酸序列中(此处限定为含有对因子VIIa催化位点有贡献的氨基酸的区域)进行氨基酸替换。一般地在或邻近构成催化位点的氨基酸上进行催化三联体的替换、插入或缺失。在人和牛因子VII蛋白中,构成催化“三联体”的氨基酸是Ser344,Asp242和His193(下标编号表明在序列中的位置)。可以用目前可能的技术包括(除了其它之外)蛋白质分离和氨基酸序列测序来测定来自其它哺乳动物种类的因子VII中的催化位点。也可以通过与其它丝氨酸蛋白酶,特别是活性位点已测定的胰凝乳蛋白酶的序列比较从而由该序列的类似活性位点残基测定催化位点(Sigler等人,分子生物学杂志(J.Mol.Biol.)35:143-164,(1968),此处引用作为参考)。
通过氨基酸替换、插入或删除以防止或是抑制因子X和/或IX由因子VIIa的活化。然而,这种修饰过的因子VII也应保持其在凝血级联反应中与真的因子VII和/或因子VIIa竞争结合组织因子的能力。可以利用如此处所述的一种凝血试验的方法或是利用如人膀胱癌细胞系J82(Sakai等人,生物化学杂志(J.Biol,chem.)264:9980-9988(1989),此处引用作为参考)的含有细胞表面组织因子的一种细胞系的竞争性结合试验的方法方便地测定这种竞争能力。
在人或牛因子VII中构成因子VII中催化位点的氨基酸,如Ser344,Asp242和His193可以被取代或删除。在本发明中优选仅改变一个氨基酸,从而减少增加此分子抗原性或是抑制其与组织因子结合的可能性,然而,可以进行两个或多个氨基酸改变(取代、添加或删除),也可以联合进行一种(些)取代、添加和删除。在一优选的关于人和牛因子VII的实施方案中,优选地用Ala替换Ser344,也可以取代以Gly,Met,Thr或其它氨基酸。优选的用Glu取代Asp以及用Lys或Arg取代His。一般地,所选的取代应尽可能少干扰蛋白质三级结构。此处引用作为参考的Dayhoft等人的模型(Atlas,蛋白质结构,1978(Atlas of Protein Structure 1978),Nat′lBiomed.Res.Found.,华盛顿特区)可用作选择其他氨基酸取代的指导。人们可以在人、牛或其它种的合适的催化位点中引入如前述的替代残基,且可以如此处所述测试所得的蛋白质催化活性抑制的水平以及所获的抗凝活性。修饰过的因子VII的催化活性将基本被抑制,一般地比相应种类的野生型因子VII的催化活性的约5%更低,更优选地比其活性的约1%更低。
可以利用重组DNA技术生产修饰过的因子VII。一般地,克隆的野生型因子VII DNA序列经过修饰以编码期望的蛋白质。再将此修饰过的序列插入一种表达载体,此载体再被转化或转染入宿主细胞。高等真核细胞,特别是培养的哺乳动物细胞是优选的宿主细胞。已知人因子VII的完整的核苷酸和氨基酸序列。见美国专利No.4,784,950,此处引用作为参考,其中描述了重组人因子VII的克隆和表达。牛因子VII序列由Takeya等人在生物化学杂志(J.Biol.Chem.)263:14868-14872(1988)中描述,此处引用作为参考。
可以通过不同的技术完成氨基酸序列的改变。可通过定点突变进行DNA序列的修饰。用来定点突变的技术为本领域周知,且由如Zoller和Smith(DNA 3:479-488,1984)所述。因此,利用因子VII的核苷酸和氨基酸序列可以引入所选择的一种(些)改变。
因子VII相应的修改包括那些用一种选自维生素K依赖型血浆蛋白质因子IX,因子X,凝血酶原,蛋白质C,蛋白质S或蛋白质Z的gla结构域取代其氨基端部分(gla结构域)的蛋白质。维生素K依赖型血浆蛋白质的gla结构域的特征为存在γ-羧基谷氨酸残基和长度一般从约30到约40个氨基酸以及在相关基因中带有相应于外显子-内含子边界位置的C末端。生产带有异源gla结构域的因子VII的方法公开于美国专利No.4,784,950,此处引用作为参考。
用于生产修饰过的因子VII的DNA序列典型地编码一种在因子VII蛋白质的氨基端的前肽原以获得正确的翻译后加工(例如:谷氨酸残基的γ-羧基化)以及由宿主分泌。前肽原可以是因子VII的或是其它如因子IX,因子X,凝血酶原,蛋白质C或蛋白质S的维生素K依赖型血浆蛋白质的。如本领域一般技术人员所知晓,在修饰过的因子VII中可进行附加的各种修饰,这些修饰不会明显损害此蛋白质作为一种抗凝药的能力。例如,在催化三联体上修饰过的因子VII也可在活化切割位点上修饰以抑制此因子VII酶原到其活化的双链形式的转变,一般地如美国专利No.5,288,629中所述,此处引用作为参考。
用于表达修饰过的因子VIIa的表达载体含有能指导一种克隆的基因或cDNA转录的启动子。优选的用于培养的哺乳动物细胞中的启动子包括病毒启动子和细胞启动子。病毒启动子包括SV40启动子(Subramani等人,分子细胞生物学(Mol.Cell.Biol.)1:854-864,1981)和CMV启动子(Boshart等人,细胞41:521-530,1985)。一种特别优选的病毒启动子是一种来自腺病毒2的主要晚期启动子(Koufman和Sharp,分子细胞生物学,2:1304-1319,1982)。细胞启动子包括鼠κ基因启动子(Bergman等人,美国国家科学院院报,81:7041-7045,1983)和鼠VH启动子(Loh等人,细胞(cell)33:85-93,1983)。一种特别优选的细胞启动子是鼠金属硫蛋白-I启动子(Palmiter等人,科学(Science)222:809-814,1983)。表达载体也可以含有一套位于启动子下游和因子VII序列本身插入位点的上游的RNA剪切位点。优选的RNA剪切位点可以获自腺病毒和/或免疫球蛋白基因。表达载体中也包含位于插入位点下游的多腺苷酸化信号。特别优选的多腺苷酸化信号包括来自SV40的早期或晚期多腺苷酸化信号(Kaufman和Sharp,出处同上),来自腺病毒5Elb区域的多腺苷酸化信号,人生长激素基因终止子(DeNoto等人,核酸研究(Nuc.Acids Res.)9:3719-3730,1981)或是来自人因子VII或牛因子VII基因的多腺苷酸化信号。表达载体也可包括一种位于启动子和RNA剪切位点之间的非编码性病毒先导序列,如腺病毒2三联基因先导以及增强子序列,如SV40增强子。
克隆的DNA序列通过例如磷酸钙介导的转染法(Wigler等人,细胞14:725-732,1978;Corsaro和Pearson,体细胞遗传学(Somatic CellGenetics)7:603-616,1981;Graham和Van der Eb,病毒学(Virology)52d:456-467,1973)或是电穿孔法(Neumann等人,EMBO.J.1:841-845,1982)导入培养的哺乳动物细胞。为了鉴别和挑选表达外源DNA的细胞,一般地将一种能赋予可选择表型(一种选择性标记)的基因与目的基因或cDNA一起导入细胞。优选的选择性标志包括对能赋予药物如新霉素、潮霉素和氨甲蝶呤的抗性的基因。选择性标记可以是一种可扩增的选择性标记。一种优选的可扩增的选择性标记是一种二氢叶酸还原酶序列。选择性标记由Thilly综述(哺乳动物细胞技术(MammalianCell Technology)Butterworth出版社,Stoneham,MA,此处引用作为参考)。选择性标记的选择在本领域普通技术人员水平之内。
选择性标记可以与在不同质粒上的目的基因同时转入细胞,也可以在同一质粒上转入。如果在同一质粒上,选择性标记和目标基因可以在不同和相同的启动子控制之下,后一情形产生一种双顺反子(dicistronic)信息。这种类型的构建物为本领域已知(例如,Levinson和Simonsen,美国专利4,713,339)。在待引入细胞的混合物中添加一个附加DNA(称作“载体DNA”)也可能是有益的。
细胞吸收该DNA之后,在合适的生长培养基上生长,典型地1-2天,以开始表达目的基因。此处所用“合适的生长培养基”指一种含有营养物和细胞生长及修饰过的因子VII表达所需的其它成份的培养基。培养基一般地包括碳源、氮源、必需氨基酸、必需糖、维生素、盐、磷脂、蛋白质和生长因子。为了产生γ-羧基化的修饰过的因子VII,培养基含有优选地从约0.1mg/ml到约5mg/ml的浓度的维生素K。再应用药物筛选用于挑选以稳定形式表达选择性标记的细胞的生长。对于已用可扩增的选择性标记转化的细胞,可增加药物浓度以挑选一种高拷贝数的克隆序列,从而增加表达水平。再筛选稳定地转染的细胞克隆中修饰过的因子VII的表达。
优选的哺乳动物细胞系包括COS-1(ATCC CRL1650),幼仓鼠肾(BHK)和293细胞系(ATCC CRL1573;Graham等人,普通病毒学杂志(J.Gen.Virol.)36:59-72,1977)。一种优选的BHK细胞系是tk-ts13BHK细胞系(Waechter和Baserga,美国国家科学院院报,79:1106-1110,1982,此处引用作为参考),此后称为BHK570细胞。BHK570细胞系已保藏在美国典型培养物保藏中心,12301 Parklawn Dr.,MD 20852,ATCC保藏号CRL 10314。tk-ts13 BHK细胞系也可从ATCC获得,其编号为CRL 1632。此外,可能用到一些其它细胞系,包括大鼠Hep I(大鼠肝细胞瘤;ATCC CRL 1600),大鼠Hep II(大鼠肝细胞瘤;ATCCCRL 1548),TCMK(ATCC CCL 139),人肺(ATCC HB8065),NCTC1469(ATCC CCL9∶1),CHO(ATCC CCL61)及DUKX细胞(Urlaub和Chasin,美国国家科学院院报77:4216-4220,1980)。
也可利用转基因动物技术生产修饰过的因子VII。优选在宿主雌性哺乳动物的乳腺内生产蛋白质。在乳腺内表达之后将目标蛋白质分泌到奶中克服了许多分离源自其它来源的蛋白质遇到的困难。奶便于收集,可大量获得且已在生化上很好地表征。此外,奶中的主要乳蛋白为高浓度的(典型的从约1到15g/L)。
从商业观点出发,显然优选的用作宿主的种类有大的产奶量。虽然可以使用较小的动物如鼠和大鼠(且是在原理的证明期优选的),优选使用家畜类哺乳动物包括,但不局限于猪、山羊、绵羊和牛。由于下列因素:在绵羊中先前的转基因的历史、奶产量、价格及用于收集绵羊奶的方便易得的设备,使绵羊成为特别优选的。关于影响所选的宿主种类的因素的比较参见WIPO公开文本WO88/00239。一般地期望选择一种已培育为乳用的宿主动物的品种,如East Friesland绵羊或是在晚些时候通过转基因品系培育以引入乳用牲畜。在任何情况下,应使用已知健康状况良好的动物。
为获得在乳腺中的表达,使用来自乳蛋白质基因的转录启动子。乳蛋白质基因包括那些编码酪蛋白(见美国专利No.5,304,489,此处引用作为参考),β-乳球蛋白,α-乳清蛋白及乳清酸性蛋白的基因。优选β-乳球蛋白(BLG)启动子。对于羊β-乳球蛋白基因,一般使用该基因序列5′侧翼的至少约406bp区域,虽然优选长达约5kbp的5′侧翼序列的更长部分,如包含5′侧翼启动子和β-乳球蛋白基因的非编码部分。见Whitelaw等人,生物化学杂志(Biochem J.)286:31-39(1992)。来自其它种类的启动子DNA的相似片段也是合适的。
β-乳球蛋白基因的其它区域也可掺入到构建体中,这可以是待表达基因的基因组区域。本领域一般认同例如,缺乏内含子的构建体与含有这种DNA序列的构建体相比表达较差(见Brinster等人,美国国家科学院院报85:836-840(1988);Palmiter等人,美国国家科学院院报88:478-482(1991);Whitelaw等人,转基因研究(Transgenic Res.)1:3-13(1991);WO 89/01343;及WO 91/02318,此处引入每一文献作为参考)。在这点上,可能情况下一般地优选使用包含编码目标蛋白质的或多肽的基因的全部或部分天然的内含子的基因组序列,因此优选还包括至少一些源自如β-乳球蛋白的基因的内含子。一种这样的区域是提供用于内含子剪切及从绵羊β-乳球蛋白基因的3′非编码区的RNA多腺苷酸化的一种DNA片段。当用于取代一种基因的天然3′非编码序列时,此种绵羊β-乳球蛋白片段可同时增强和稳定目标蛋白质或多肽的表达水平。在其它实施方案中,用源自一种乳特异性蛋白质基因的相应序列取代修饰过的因子VII起始ATG周围的区域。这种取代提供了一种推测的组织特异性起始环境以增加表达。修饰过的因子VII的整个前原序列和5′非编码区序列用例如BLG基因的那些取代是方便的,虽然也可以用较小的区域取代。
对于转基因动物中修饰过的因子VII的表达,将编码修饰过的因子VII的DNA片段可操纵地连接到其表达所必需的其他片段上以产生表达单元。这种其他片段包括上述提到的启动子,以及用于提供转录终止和mRNA多腺苷酸化的序列。表达单元也包括编码一种可操纵性地连接到编码修饰过的因子VII的片段上的分泌性信号序列的DNA片段。分泌性信号序列可以是一种天然的因子VII分泌性信号序列或可以是另一种蛋白质如乳蛋白的。见如,Von Heinje,核酸研究,14:4683-4690(1986);和Meade等人,美国专利No.4,873,316,此处引入作为参考。
用于转基因动物的表达单元的构建通过将修饰过的因子VII序列插入含有其他DNA片段的一种质粒或噬菌体载体中方便地进行,虽然表达单元实质上可以通过任何序列的连接而构建。特别方便的是提供一种含有编码一种乳蛋白的DNA片段的载体,用编码修饰过的因子VII多肽的序列取代编码此乳蛋白的序列就可构建一种包含此乳蛋白基因表达控制序列的融合基因。在任何情况下,在质粒或其它载体中表达单元的克隆有利于修饰过的因子VII序列的扩增。扩增可在细菌宿主细胞中(如E.coli)方便地进行,因此载体中典型地包括在细菌宿主细胞中有功能的复制起点和选择性标记。
再将表达单元引入所选宿主种类的受精卵(包括早期胚)。可通过几种途径,包括显微注射(如美国专利No.4,873,191),逆转录病毒转染(Jaenisch,科学240:1468-1476(1988))或利用胚胎干细胞(ES)进行定点整合(由Bradley等人,Bio/Technology 10:534-539(1992)综述)等方式中的一种完成异源DNA导入。将卵植入假妊娠的雌性动物的输卵管或子宫中以使之发育足月。在其胚系中携带导入的DNA的子代可将此DNA以正常孟德尔方式传入其子代,以使转基因种群发展。
用于生产转基因动物的一般程序为本领域已知。见如,Hogan等人,鼠胚胎操作:一种实验室手册,冷泉港实验室,1986(Manipulating theMouse Embryo:A Laboratory Manual,Cold Spring HarborLaboratory,1986);Simons 等人,Bio/Technology 6:179-183(1988);Wall等人,Biol.Reprod.32:645-651(1985);Buhler等人,Bio/Technology 8:140-143(1990);Ebert等人,Bio/Technology9:835-838(1991);Krimpenfort等人,Bio/Technology 9:844-847(1991);Wall等人,细胞生物化学杂志(J.Cell Biochem.)49:113-120(1992);美国专利No.4,873,191和4,873,316;WIPO公开文本WO88/00239,WO90/05188,WO 92/11757和GB87/00458,此处引入这些文献作为参考。用于将异源DNA序列引入到哺乳动物和其胚细胞中的技术最初是在鼠中发展的。见如,Gordon等人,美国国家科学院院报77:7380-7384,(1980);Gordon和Ruddle,科学214:1244-1246(1981);Palmiter和Brinster,细胞41:343-345(1985);Brinster等人,美国国家科学院院报,82:448-4442(1985);及Hogan等人,(出处同上)。这些技术随后被修改以适于用在较大的动物,包括家畜品种中(见如,WIPO公开文本WO88/00239,WO90/05188和WO92/11757和Simons等人,Bio/Technology 6:179-183(1988)。总之,目前最有效的用于转基因鼠或家畜产生的途径中,按照已建立的技术将数百个线性目标DNA分子注射入一个受精卵的核原(pro-nuclei)中。也可将DNA注射入受精卵的细胞质中。
也可以采用在转基因植物中的生产。表达可以是广泛的或定向于特定的器官如块茎。见Hatt,自然344:469-479(1990);Edelbaum等人,干扰素研究杂志(J.Interferon Res.)12:449-453(1992);Sijmons等人,Bio/Technology,8:217-221(1990);及欧洲专利局公开文本EP255,378。
修饰过的因子VII可以通过亲和层析在一种抗因子VII抗体柱上纯化。特别优选的是钙离子依赖型单克隆抗体的使用,如Wakabayashi等人,生物化学杂志(J.Biol.Chem.)261:11097-11108(1986)和Thim等人,生物化学(Biochem.)27:7785-7793(1988)所述,此处引入此文献作为参考。可以通过传统化学纯化手段如高效液相层析进行进一步的纯化。其它纯化方法,包括柠檬酸钡沉淀为本领域已知,可以用于纯化此处所述的新型修饰过的因子VII(一般地见,Scopes R.,蛋白质纯化(ProteinPurifcation)Springer-Verlag,N.Y.,1982)。基本上纯的修饰过的因子VII优选为至少约90~95%同质的,最优选为98~99%同质的,以用于药物用途。一旦纯化,如所期望的部分同质或达到同质这种修饰过的因子VII就可用于治疗。
修饰过的因子VII在其活化位点被切割以将其转变为双链形式。活化可以按本领域已知的方式进行,如由Osterud等人,生物化学(Biochemistry)11:2853~2857(1972);Thomas,美国专利No.4,456,591;Headner和Kisiel临床研究杂志(J.Clin.Invest.)71:1836-1841(1983);或Kisiel和Fujikawa,Behring Inst.Mitt.,73:29-42(1983)所述,此处引入作为参考。所获分子再如下述配制和施用。组合物
化合物典型地在实施干预之前24小时内及之后多达7天内施用。用于防止或减小心肌损伤的给药可通过如此处进一步详述的多种途径。化合物也可以在易于形成血栓的血管位置例如在吻合术位置或在易于减少开放度的血管位置局部施用。
在预防或治疗心肌损伤时,修饰过的因子VII的剂量对于70kg病人的作为注入和维持剂量的范围为约50mg到500mg/天,更典型的1mg到200mg/天,且更优选的从10mg到约175mg/天,取决于病人的体重和病况的严重性。
用于治疗心肌损伤的药物组合物用于在预防和/或治疗中的非肠道给药。优选地,该药物组合物是非肠道性给药,即静脉、皮下或肌内给药。用于非肠道给药的组合物包含溶于一种可接受的载体,优选地为一种水性载体中的修饰过的因子VII分子的一种溶液。可以使用多种水性载体如水、缓冲液、0.4%盐水、0.3%甘氨酸等。也可将修饰过的因子VII分子制成用于输送和定位于损伤靶点的脂质体制剂中。脂质体制剂一般地如U.S.4,837,028,U.S.4,501,728及U.S.4,975,282中所述,此处引入作为参考。可以用传统的周知的灭菌技术将组合物灭菌。所得的水溶液可以被包装使用或在无菌条件下过滤和冷冻干燥,冻干制剂可以在施用前与无菌水溶液混合。该组合物可含有近似生理条件所需的药学可接受的辅助物质,如pH调节和缓冲剂、渗透压调节剂等等,例如,乙酸钠、乳酸钠、氯化钠、氯化钾、氯化钙等。在这种制剂中修饰过的因子VII的浓度可有很大变化,即:从低于约5%,通常至少在约1%到最多达15或20%重量百分比,且主要由液体体积、粘度等以及所选的特定给药方式所决定。
因此,典型地用于静脉输注的药物组合物可由多达250ml的无菌林格溶液(Ringer′s Solution)和10mg修饰过的因子VII制成。制备非肠道给药化合物的具体方法为本领域普通技术人员已知或显而易见的,且如Remington药物科学(Remington Pharmaceutical Science)16版,Mack出版公司,Easton,PA(1982)中所详述,此处引入作为参考。
含有修饰过的因子VII分子的组合物可以在预防和/或治疗性处理中给药。在治疗性应用中,组合物如前述以足以治愈或至少部分阻止该疾病及其并发症的量施用于已患病的病人。足以达到目的量称为“治疗有效剂量”。用于此的有效剂量取决于疾病或损伤的严重性以及病人的体重和一般状况,对于70kg病人而言一般地范围从约0.05mg到约500mg修饰过的因子VII/天,常用的剂量为从约1.0mg到约200mg修饰过的因子/天。必须注意本发明的材料可以一般地应用于严重的疾病或损伤状态中,即威胁生命或可能威胁生命的情况。在这种情况下,由于所含外源物质很少且在人中修饰过的人因子VII的免疫原性普遍缺乏,可能(也可能是治疗医生期望的)施用大大过量的修饰过的因子VII组合物。
在预防性应用中,含有修饰过的因子VII的组合物施用于易于受到或处于疾病状况或损伤威胁的病人以增强病人自身的抗凝血能力。这种剂量称为“预防有效剂量”。在此用途中,精确的量同样取决于病人的健康状况和体重,对于70kg病人一般地范围从约0.05mg到约500mg,更常见的从约1.0mg到约200mg/70kg体重。
该组合物的单次或多次施用可以治疗医生所选的剂量水平和方案进行。对于救护车病人需要日常维持水平,可以用例如便携式泵系统连续注入方式完成修饰过的因子VII的施用。
修饰过的因子VII的局部运送如,在易于形成血栓的血管位置(如:吻合术处)或在易于减少开放度的血管位置局部应用修饰过的因子VII可以通过如喷雾,灌注,双气囊导管,斯坦特固定模,植入血管的移植物或斯胆特固定模,用于包被气囊导管的水凝胶或是其它已建立的方法进行。在任何情况下,药物组合物应提供足以有效治疗病人的量的修饰过的因子VII。
提供下列实施例用于阐明,而不是限制本发明。
实施例实施例1.Ser344Ala344因子VII的表达
为产生Ser344Ala因子VII活性位点突变体,用XbaI和KpnI消化质粒FVII(565+2463)/pDX(美国专利No.4,784,950,此处引入作为参考;保藏在美国典型培养物保藏中心,保藏号40205),回收所得的含有丝氨酸344编码区域的0.6kb片段。此片段如图所示克隆入XbaI,KpnI消化的M13mp19。此操作及随后的步骤一般地如下述按照标准方案进行(如Maniatis等人,分子克隆:实验室手册,冷泉港实验室出版社所述,冷泉港,N.Y.(1982),此处引入作为参考)。
利用突变的寡核苷酸ZC 1656(5′TGG GCC TCC GGC GTC CCCCTT3′)及“通用”第二引物ZC87(5′TCC CAG TCA CGA CGT3′)按照Zoller和Smith的方法在M13模板上进行突变。用激酶化的(Kinased)ZC1656筛选反应产物。挑选阳性克隆,制备模板DNA并从在1077的PstI位点到在1213的KpnI位点测序。序列分析证实存在期望的突变。突变的克隆称为1656。
用1656克隆构建一种表达载体。以约0.14kb的Pst I-KpnI片段从M13载体上分离突变的序列。此序列与来自FVII(565+2463)/pDX的1.7kb Hind III-XbaI片段,来自FVII(565+2463)pDX的0.5kb XbaI-PstI及来自FVII(565+2463)/pDX 4.3kb KpnI-HindIII片段连接,如图所示。通过用PstI消化突变体和野生型克隆及用KpnI和XbaI消化M13中插入因子VII的突变体,准备所消化DNA的Southern杂交及用放射性标记的ZC1656探查杂交证实期望的突变序列的存在。
以两种分离的(命名为#544和#545)的1656表达载体转染幼仓鼠肾细胞系BHK570(保藏在美国典型培养物保藏中心,保藏号10314)。通过以1∶10比例将铺满10cm平皿的BHK570细胞稀释入5个非选择性培养基的10cm平皿中(Dulbecco改良Eagle培养基[DMEM],含10%胎牛血清及1%PSN抗生素混合物[GIBCO生命技术,Gaithersburg,MD])。24小时后,当细胞达到20~30%铺满度时,它们将与一种编码1656突变的表达载体质粒p486(含有腺病毒5起点,SV40增强子,腺病毒2主要晚期启动子,腺病毒2三联体先导,5′和3′剪切位点,DHFRr cDNA和pML-1中的SV40多腺苷酸化信号(Lusky和Botchan,自然293:79-81,(1981))以及10mg载体DNA(超声过的鲑精DNA)共转染,如表1所示。DNA加入15ml管中,再加入0.5ml 2x Hepes(25g Hepes,40g NaCl,1.8g KCl,0.75gNa2HPO4·2H2O,5g葡萄糖用蒸镏水稀释到2.5L,pH调到pH6.95-7.0),试管中混合。通过以巴斯德吸管向DNA/Hepes溶液鼓入空气的同时加入0.5ml 0.25M CaCl2的方式沉淀每管中的DNA。振荡这些管,室温下孵育15分钟,再次振荡。用吸头将DNA混合物滴加到细胞平皿上。旋转平皿且在37℃下培养4~6小时。培养后,向每一平皿中加入2ml以Tris-盐水(0.375gKCl,0.71g Na2HPO2,8.1gNaCl,3.0gTris-HCl,0.5g蔗糖,稀释到1L,pH调到pH7.9)稀释的20%甘油。旋转平皿,室温下放置2分钟。将培养基移出平皿且换成2ml Tris-盐水。室温下放置平皿2分钟后,移去Tris-盐水,用10ml非选择性培养基替换。平皿在37℃下培养两天。
表1
转染 *
*所用DNA浓度为:克隆544:0.7mg/ml;克隆545:0.3mg/ml;p486:1.49mg/ml。
| 544 | 545 | 544对照 | 545对照 | |
| 质粒名称 | ||||
| 克隆544 | 15mml | --- | 15ml | -- |
| 克隆545 | --- | 30ml | --- | 30ml |
| p486 | 1.5ml | 1.5ml | --- | --- |
| 载体DNA | 1.6ml | 1.6ml | 1.6ml | 106ml |
培养两天后,细胞稀释于选择性培养基(DMEM,含10%透析过的胎牛血清,1%PSN抗生素混合物和150nM氨甲蝶呤)且以1∶100,1∶250和1∶500的稀释度接种于特大平皿中。平皿在37℃下培养一周。一周后,更换培养基且用选择培养基替换,检查平皿中群落的形成。
八天后,菌落形成之后从#544和#545转染的1∶500稀释的平皿中随机选择12个菌落。每个菌落接种于6孔板中的一个孔,生长于选择性培养基。七天后,平皿铺满后将每个菌落分入10cm平皿的选择性培养基中。
上述克隆及转染以表达野生型因子VII的对照细胞用35S-甲硫氨酸-半胱氨酸蛋白质标记混合物(NEN DuPont生物技术系统,Wilmington,DE)代谢性标记。菌落生长且制备用于在选择性培养基中脉冲标记实验。用磷酸缓冲盐溶液(Sigma,St.Louis,MO)冲洗细胞,在20mCi/ml35S-Cys-35S-Met中脉冲标记4小时。4小时后,收集上清液和细胞。细胞裂解基本上如Lenk和Penman所述(细胞16:289-302,1979))且用50ml Staph A(Sigma,st.Louis,Mo)预清除400ml每种裂解液。
来自代谢性标记的细胞样本通过首先与6ml抗因子VII多克隆血清孵育4小时放射免疫沉淀(RIP)。每种样品中加入60μl洗过的葡萄球菌蛋白A,4℃下轻摇样品1.5小时。离心样品,除去上清液。沉淀在0.7MRIPA缓冲液[10mM Tris,pH7.4,1%脱氧胆酸[Calbiochem公司,LaJolla,CA],1%Triton X-100,0.1%SDS,5mMEDTA,0.7M NaCl)中洗两次再在0.15M RIPA缓冲液中(10mM Tris,pH7.4,1%脱氧胆酸[Calbiochem公司,La Jolla,CA],1%Triton X-100,0.1%SDS,5mMEDTA,0.15M NaCl)洗一次。每个样品中加入100μl 1×SDS染料(50mM Tris-HCl,pH6.8,100mM二硫苏糖醇,2% SDS,0.1%溴酚蓝,10%甘油),样品煮沸5分钟,随后离心以除去蛋白A。每个样品的50μl在10%聚丙烯酰胺凝胶上电泳。结果显示,10个菌落中的9个分泌修饰过的因子VII。实施例II修饰过的因子VII的抗凝血活性
修饰过的因子VII蛋白质抑制凝血的活性在一步凝血实验中测定,利用野生型因子VII作为对照。重组蛋白质基本上按照上述方法从培养于含5mg/ml维生素K的培养基中的细胞制备。不同量的修饰过的因子VII(来自克隆544)或重组野生型因子VII用50mM Tris,pH7.5,0.1%BSA稀释到100ml。此混合物与100ml缺乏因子VII的血浆(George King生物医学公司,Overland Park,KS)和200ml促凝血酶原激酶C(Dade,Miami,FL;含有兔脑促凝血酶原激酶和11.8mM Ca++)一起孵育。在一种自动凝血计时仪(MLA Electra 800,医学实验室自动化公司,Pleasantville,NY)中进行凝血实验,利用由1∶5到1∶640收集的正常的人血浆稀释液构建的标准曲线(假定含有每毫升1个单位因子VII活性;通过从健康供体收集柠檬酸盐血清制得)将凝血时间转变成因子VII活性单位。通过此实验,修饰过的因子VII制剂未显示可检测的凝血活性。表2显示对照(未转染的)BHK细胞条件的培养基(+/-维生素K)、野生型因子VII和两株分离的表达修饰过的因子VII细胞的凝血时间的实验结果。通过在凝血时间上比对照样品的减少可见因子VII活性。
表2
| 样品 | 稀释度 | 凝血时间(秒) |
| 对照+K | 1∶5 | 33.1 |
| 1∶10 | 33.4 | |
| 对照-K | 1∶5 | 34.3 |
| 1∶10 | 33.2 | |
| 野生型因子 | 1∶20 | 19.0 |
| 1.40 | 21.5 | |
| 1∶80 | 23.3 | |
| 修饰过的因子VII(#6) | 1∶1 | 33.5 |
| 修饰过的因子VII(#10) | 1∶1 | 32.5 |
为了测定修饰过的因子VII对血浆因子底物的作用,修饰过的因子VII制剂以及重组野生型或天然的因子VII一起与因子X或因子IX孵育,通过凝血实验或聚丙烯酰胺凝胶电泳监测其活性。实施例III修饰过的因子VII结合组织因子的能力
修饰过的因子VII与野生型因子VII竞争性结合组织因子以及抑制其凝血活性的能力在一步凝血实验中在有限量的组织因子(促凝血酶原激酶)存在下测定。
在类似于实施例II所述的一步实验中确定凝血时间。在混合实验中使用有限量的组织因子、恒定量的野生型因子VII以及增加的变体因子VII的量。在含有增加的变体因子VII的量的实验中凝血时间的增加显示因子VII/VIIa凝血原活性的抑制。
在测试样品中,因子VII的量以在收集正常的血浆中测定的因子VII活性的标准曲线的百分比计算。利用在磷酸缓冲盐液(PBS)中范围从1∶5~1∶640的收集的正常血浆系列稀释液得出因子VII活性的标准曲线。为了此目的,假定正常血浆含约500ng/ml因子VII,且以此为一个活性单位。100ml因子VII缺乏的血浆,100ml血浆稀释液及200ml凝血凝酶C(Dade,Miami,FL)的混合物用于在MLA Electra800自动计时仪上测量凝血时间。为建立标准曲线,结果以每秒计的凝血时间的活性百分数(1∶5=100%活性)画图。
实验所要求的培养基含有组成少于1%血清的野生型及变体因子VII。用PBS制成稀释液以使凝血时间落入标准曲线的范围。典型的最低稀释度为1∶2。终体积为100ml。在实验中测试了命名为克隆“#10”和“#6”的两个不同的人因子VII Ser344Ala变体。列于下表的结果显示,随着因子VII变体的增加,因子VIIa的活性百分比减少。
表3:
用Ser344->Ala混合实验的结合
变体(B4A1(野生型)培养基用作在10μl/反应的100%活性)
| Ser344->Ala克隆No. | 变体在培养基中的量 | B4A1培养基的量 | BHK对照* | 因子VIIa活性的百分比 |
| #10 | 10μl | 10μl | 0 | 70 |
| #10 | 20μl | 10μl | 0 | 51 |
| #10 | 30μl | 10μl | 0 | 43 |
| #10 | 40μl | 10μl | 0 | 34 |
| #10 | 50μl | 10μl | 0 | 28 |
| #10(-K)§ | 20μl | 10μl | 0 | 78 |
| #6 | 10μl | 10μl | 0 | 74 |
| #6 | 20μl | 10μl | 0 | 56 |
| #6 | 30μl | 10μl | 0 | 46 |
| #6 | 40μl | 10μl | 0 | 41 |
| #6 | 50μl | 10μl | 0 | 32 |
| #6 | 20μl | 10μl | 0 | 85 |
| BHK对照 | 0 | 10μl | 20μl | 91 |
| BHK对照(-K) | 0 | 10μl | 20μl | 107 |
未转染条件的培养基
§除了标明“(-K)”的情况,为表达变体因子VII,
细胞在有维生素K存在下生长
这些实验表明,带有Ser344Ala取代的因子VII变体以剂量依赖型形式与天然因子VII竞争,抑制天然因子VII/VIIa的凝血原活性。因此可得出结论:Ser344Ala变体人因子VII与天然人因子VIIa竞争,从而抑制人血浆中因子X和/或IX的活性。实施例IV因子VII与PPACK的反应
重组因子VII在转染的幼仓鼠肾细胞中产生。此蛋白质的纯化和活化如Thim等人(生物化学(Biochemistry)27:7785-7793,1988),Brinkous等人(美国国家科学院院报86:1382-1386,1989)及Bjoern和Thim(Res.Discl.No.269,564,1986)所公开,此处引入作为参考。细胞培养液回收、过滤并稀释以减低盐浓度。稀释的培养液再利用含有CaCl2的洗脱缓冲液通过阴离子交换层析分离。回收因子VII流份再利用钙离子依赖抗因子VII单克隆抗体通过免疫层析进一步纯化。利用分别以CaCl2和NaCl洗脱因子VII的两步阴离子交换层析进行进一步纯化。
50mM Tris-HCl,100mM NaCl,5mM CaCl2,pH7.4中的重组因子VIIa(1mM)与20mM PPack(D-苯丙酰-脯氨酰-精氨酰氯甲基酮;Calbiochem,La Jolla,CA)孵育5,20和60分钟。再加入含有产色底物S2288(H-D-异亮氨酸-L-脯氨酰-L-精氨酸-对硝基苯胺;Kabi Vitrum AB,Molndal,瑞典)以获得2.5倍稀释液,且终浓度为0.3mM S2288。测量对硝基苯胺的产生且与利用未处理的因子VIIa为对照所获的结果比较。结果表明在这些反应条件下约60分钟后因子VIIa完全失活。实施例V DEGR-因子VIIa的产生
如实施例IV所述制备重组人因子VIIa。10mM甘氨酸缓冲液,pH8.0,10mM CaCl2,50mM NaCl中的重组人因子VIIa稀释到1.5mg/ml的浓度。加入10倍摩尔过量的溶于蒸镏水中的Dansyl-L-Glu-Gly-Arg-氯甲基酮,DEGRck,(Calbiochem,La Jolla,CA,92037)到因子VIIa中,37℃孵育2小时后,再加入10倍摩尔过量的DEGRck到混合物中,37℃下再孵育2小时。第三次加入10倍摩尔过量的DEGRck到因子VIIa中,4℃下孵育约16小时。此DEGR-因子VIIa样品在4℃下对Tris缓冲盐水(0.05M Tris-HCl,0.1M NaCl,pH7.5)充分透析以除去任何游离的DEGRck。
在因子Xa产色底物实验中测定最终DEGR-因子VIIa混合物中游离DEGRck的存在。DEGR-因子VIIa混合物与产色底物S-2222一起加入到纯化的人因子Xa中。此底物特异性地由因子Xa切割,而不被因子VIIa切割。混合物中未结合的DEGRck能与因子Xa结合,从而抑制因子Xa的产色活性。将游离DEGRck加入因子Xa的混合物中产生标准曲线,以通过因子Xa产色活性的抑制测量溶液中游离DEGRck的水平。DEGR-因子VIIa混合物的分析表明游离DEGRck∶DEGR-因子VIIa的比率在充分透析后低于0.5%,从而保证在下述不同鉴定体系中通过DEGR-因子VIIa观察到的抑制不是由于游离DEGRck的存在。实施例VI在大鼠平滑肌细胞上因子Xa的产生
通过利用特异于因子Xa的产色底物测定细胞刺激因子X转变为因子Xa的能力分析平滑肌细胞的细胞表面组织因子的存在。
大鼠血管平滑肌细胞(Clowes等人,临床医学研究(J.Clin.Invest.)93:644-651(1994))以8,000细胞/孔的密度接种于96孔培养皿(美国科学产品,Chicago,IL)的培养基中(表4)。
表4500ml Dulbecco改良Eagle培养基(DMEM);(GIBCO-BRL,Gaithersburg,MD)10%胎牛血清(Hyclone,Logan,UT)1mM丙酮酸钠(Irvine,Santa Ana,CA)0.29mg/ml L-谷氨酰胺(Hazelton,Lenexa,KS)1xPSN;(100x为5mg/ml青霉素,5mg/ml链霉素,10mg/ml新霉素)(GIBCO-BRL,Gaithersburg,MD)
37℃培养48小时后培养基换成无血清培养基(表5)
表5250ml Dulbecco改良Eagle培养基(DMEM),250ml Ham F-12培养基(Fred Hutchinson癌症研究中心,西雅图,WA)1mM丙酮酸钠0.29mg/ml L-谷氨酰胺20mM转铁蛋白(JRH,Lenexa,KS)5mM胰岛素(GIBCO-BRL)16ng硒(Aldrich,Milwaukee,WI)1mg/ml牛血清血蛋白(Sigma,St.Louis,MO)
37℃下培养细胞72小时。培养后,或加入PDGF-BB(10ng/ml)或加入10%胎牛血清到细胞中以刺激组织因子表达(Taubman等人,临床医学研究,91:547-552,1993)。监测一套平行的既无PDGF也无血清的未受刺激的细胞中的内源性活性。培养6小时后,细胞中加入重组人因子VIIa至终浓度10nM。以不加入因子VIIa的一套细胞为阴性对照。细胞在37℃培养2小时,用HEPES缓冲液(10mM HEPES,137mMNaCl,4mM KCl,5mM CaCl2,11mM葡萄糖,0.1%BSA)洗涤。洗涤后,细胞与每孔50ml的200nM血浆纯化的人因子X(补加5mM CaCl2的Tris-缓冲盐液中)培养5分钟。每孔中加入25μl 0.5M的EDTA及25ml S-2222产色底物(Kabi Pharmacia,Franklin,OH)的800mM溶液。室温下孵育平皿40分钟,用THERMOMAX微孔板读数仪(MolecularDevices,Menlo Park,CA)在405nm分析。
表6显示用因子VIIa处理过的孔与对照孔相比(无因子VIIa加入)吸光度增加。吸光度增加是孔中产生的因子Xa水平的直接测定,其随后切割产色底物,稀放出产色团。数据也表明用PDGF-BB或10%胎牛血清预处理的孔中产色活性水平比未刺激的细胞高。
表6
| 测试样品 | OD405 |
| 对照 | 0.043 |
| 内源性 | 0.247 |
| PDGF-BB | 0.360 |
| 10%FCS | 0.342 |
这些结果清楚地显示在大鼠平滑肌细胞的细胞表面上因子X到因子Xa转变是一种因子VIIa依赖性活化。实施例VII通过DEGR-因子VIIa的细胞表面产色活性的抑制
如前述将大鼠血管平滑肌细胞接种于96孔板中。如前述在无血清培养基中培养细胞72小时,添加10%胎牛血清6小时以刺激组织因子表达。刺激后,每孔中分别加入缓冲液(对照),或10nM因子VIIa,或[10nM因子VIIa+100nM DEGR-因子VIIa]。细胞在37℃培养2小时后,用HEPES缓冲液洗。洗涤后,细胞与每孔50ml的200nM因子X(补加5mMCaCl2的Tris缓冲盐溶液中)一起培养5分钟。每孔中加入25μl 0.5MEDTA和25mlS-2222(800mM)产色底物(Kabi Pharmacia)。室温下培养细胞40分钟。如前述在405nm处分析产色活性。
表7显示仅用因子VIIa处理的孔中产色活性的刺激,以及当DEGR-因子VIIa与因子VIIa共培养时刺激的抑制。这些结果显示,DEGR-因子VIIa作为因子VIIa结合的竞争性拮抗剂,抑制因子X转变为因子Xa的活性,随即抑制S-2222色原的切割。
表7
实施例VIII DEGR-因子VIIa对大鼠平滑肌细胞上细胞表面产色活性 的剂量依赖性抑制
| 测试样品 | OD405 |
| 对照 | 0.035 |
| 因子VIIa | 0.342 |
| 因子VIIa+DEGR-因子VIIa | 0.0730.73 |
大鼠血管平滑肌细胞以4,000个细胞/孔接种于96孔培养皿的补加1%胎牛血清的生长培养基中(如表4,无10%胎牛血清)。5天后除去培养基,细胞中或是仅加入浓度增加的因子VIIa,或是与增加浓度的DEGR-因子VIIa一起加入10nM因子VIIa。细胞与因子VII混合物37℃下一起培养2小时。培养后,洗涤细胞且与50ml 200nM的因子X(Tris缓冲盐水中)室温下培养5分钟。每孔中加入0.5M EDTA 25ml及800mM S-2222(Kabi Pharmacia)25ml,室温下培养平皿40分钟。如前述用微孔板读数仪在405nm处分析产色活性。
表8显示了随着加入孔中因子VIIa量的增加产色活性的剂量依赖性增加。当DEGR-因子VIIa与100nM因子VIIa混合物加入细胞时(表9),存在产色活性的剂量依赖性抑制。1∶1摩尔比的DEGR-因子VIIa∶因子VIIa抑制产色活性的约95%。这些数据表明此实验设计中DEGR-因子VIIa对培养中的平滑肌细胞的细胞表面组织因子的亲和力比天然因子VIIa明显要高。如果DEGR-因子VIIa与因子VIIa对结合组织因子有同等亲和力,则当两种分子以等摩尔比加入到细胞中所观察到的抑制水平不会如此高。表8
| 因子VIIa浓度(nM) | OD405 |
| 0.01 | 0.005 |
| 0.39 | 0.025 |
| 1.56 | 0.058 |
| 6.25 | 0.111 |
| 25.00 | 0.154 |
| 100.00 | 0.208 |
表9显示由DEGR-因子VIIa对大鼠平滑肌细胞的因子Xa产色活性的剂量依赖性抑制。DEGR-因子VIIa增加的浓度与100nM因子VIIa共培养,因子Xa产色活性用产色底物S-2222测定。
表9
实施例IX在可溶性组织因子实验中通过DEGR-因子VIIa对因子Xa 产生的抑制
| DEGR-因子VIIa浓度(nM) | OD405 |
| 0.10 | 0.208 |
| 0.39 | 0.176 |
| 1.56 | 0.116 |
| 6.25 | 0.073 |
| 25.00 | 0.026 |
| 100.00 | 0.014 |
通过产色实验建立使用纯化的重组可溶性组织因子的因子X到因子Xa的转变。组织因子从酿酒酵母(Saccharomyces cerevisiae)(Shigematsu等人,生物化学杂志(J.Biol,Chem.)267:21329-21337,1992)中表达并纯化。可溶性组织因子由W.Kisiel博士纯化并表征(新墨西哥大学)。制备含有65.9ml可溶性组织因子(2.2mM),29.0ml PCPS(1mM,Sigma,St.Louis,MO),29.5ml人因子X(4.1mM),2.77ml Hank缓冲液(25mM Tris,pH7.4,150mM NaCl,2.7mM KCl,5mM CaCl2,0.1%BSA)的反应混合物。96孔板中每孔加入40μl组织因子/因子X混合物,25ml用TBS稀释的因子VIIa及25ml用TBS稀释的DEGR-因子VIIa。也使用了40ml组织因子/因子X混合物;25ml用TBS/稀释的因子VIIa以及仅有25mlTBS的对照。10μl S-2222(4mM)产色底物加到孔中的反应混合物中且在室温培养2~10分钟。如前述在微孔板读数仪在405nm处分析结果。
利用在无DEGR-因子VIIa条件下因子VIIa浓度的增加确定因子VIIa活化因子X的标准曲线。列于表10中的结果表明,随着加入到反应混合物中因子VIIa量的测量产色活性呈剂量依赖性增加。不同量的DEGR-因子VIIa和100nM因子VIIa的同时加入导致产色活性的剂量依赖性减少(表11)。这些数据表明,DEGR-因子VIIa作为天然因子VIIa结合可溶性组织因子的竞争性拮抗剂,因而抑制因子Xa的产生,如通过针对产色底物S-2222的产色活性的降低所测。
表10
以增加浓度的因子VIIa加入到可溶性组织因子的因子Xa产色活性的刺激。用产色底物S-2222测定光密度的改变。
| 因子VIIa浓度(nM) | OD405 |
| 0.78 | 0.168 |
| 1.56 | 0.288 |
| 3.12 | 0.478 |
| 6.25 | 0.694 |
| 12.50 | 0.764 |
| 25.00 | 0.790 |
| 50.00 | 0.738 |
| 100.00 | 0.770 |
表11
通过在天然因子VIIa存在下向可溶性组织因子加入DEGR-因子VIIa测定对因子Xa产色活性的抑制。用产色底物S-2222测定光密度的改变。
实施例X通过DEGR-因子VIIa对凝血的抑制
| DEGR-因子VIIa因子(nM) | OD405 |
| 0 | 0.810 |
| 50 | 0.750 |
| 100 | 0.609 |
| 200 | 0.296 |
| 400 | 0.167 |
| 800 | 0.083 |
| 1600 | 0.055 |
监测DEGR-因子VIIa对凝血时间的作用的标准凝血实验准备如下:100ml正常狒狒血浆(用作为抗凝剂的柠檬酸钠收集)加入到以TBS(20mM Tris,pH7.4,150mM NaCl)稀释的100ml不同DEGR-因子VIIa浓度的溶液中。样品混合后在37℃短暂孵育。样品加入Electra800自动凝血计时仪(医学实验室自动化公司,Pleasantville,NY)中。孵育后,含有25mM CaCl2的200ml组织因子制剂加入到DEGR-因子VIIa制剂。组织因子制剂以源自新鲜冷冻脑组织的狒狒脑盐水提取物形式制得,用其引发狒狒血浆凝血的能力表征。选择凝血时间在约40秒钟的组织因子的浓度。
列于表12的数据表明凝血时间的剂量依赖性增加是由于DEGR-因子VIIa的添加。血浆中低至1mg/ml DEGR-因子VIIa导致凝血时间的明显增加。表12DEGR-因子VIIa导致的凝血时间的剂量依赖性增加
实施例XI DEGR-因子VIIa对血小板积累的抑制
| DEGR-因子VIIa(μg/ml血浆) | 凝血时间(秒) |
| 0 | 40.7 |
| 0.5 | 46.2 |
| 1.0 | 50.8 |
| 2.5 | 64.5 |
| 5.0 | 108.1 |
| 10.0 | 158.4 |
分析了DEGR-因子VIIa的对非人的灵长类中由于机械性损伤在动脉血栓处血小板积累的抑制能力。在狒狒中使用主动脉动脉内膜切除术模型,基本上如Lumsden等人所述(血液(blood),81:1762-1770(1993))。长度为1-2cm的狒狒主动脉部分被切下,翻转并刮擦以除去动脉的内膜及约50%的中膜。动脉回复到其正确方向,两端插管,置于狒狒的一条体外旁路中,从而通过此旁路将机械性损伤的动脉暴露于血液。在打开旁路到循环血液之前,用111In标记的自体血小板静脉注射到动物中。利用实时γ照像仪测定在损伤动脉位置累积的血小板的水平。
利用在即将打开旁路前DEGR-因子VIIa或对照盐水的大量注射进行DEGR-因子VIIa对血小板累积抑制的评价。对损伤的动脉连续测量60分钟。0.005mg/kgVIIa剂量的DEGR-因子抑制血小板累积。在以1.0mg/kg量注射时,在给药后1小时约90%的血小板累积被抑制。结果见图2。
这些数据表明,用DEGR-因子VIIa抑制组织因子可明显抑制动脉血管损伤的非人灵长类模型中富含血小板的血栓的形成。实施例XII在动脉粥样硬化兔中气囊血管成形术后DEGR-因子VIIa 抑制血管再狭窄
在新西兰白色(NZW)动脉粥样硬化兔中气囊血管成形术后评价DEGR-因子VIIa的调节损伤形成的能力。此动物模型已被很好的表征且已证明是用于评价抗血栓化合物对血管损伤形成的作用的好模型(Gimple等人,循环(Circulation)86:1536-1546(1992),及Rogosta等人,循环89:1262-1271(1994))。用于评价DEGR-因子VIIa的动物模型基本如Ragosta(出处同前)所述。
通过向兔肌内注射5mg/kg甲苯噻嗪和35mg/kg氯胺酮麻醉动物。通过在腹股沟韧带下方切开暴露近端股动脉在近端和远端结扎。用27号针头插入分离的部分。通过针刺造口。分离的片段用盐水灌满以消除残余血液,通过以80ml/min速度的空气注入干燥8分钟。空气干燥后,分离的片段再次用盐水灌满,除去结扎。用非闭塞性局部压迫维持止血。用金属钳分开此片段。在局部用1%甲苯噻嗪处理局部痉挛。手术后一天,动物用1%胆固醇和6%花生油饲养一个月直至气囊血管成形术。口服对乙酰氨基酚(Tylenol)10mg/kg 3~5天以缓解手术后疼痛。在手术后的3~5天内给1cc的Ambipen。
用于动物的测试药物输送包括初始大量注射后立即施气囊血管成形术,随后利用渗透泵通过内部颈静脉进行连续的全身性注入。注入药物持续3天。对照动物在接受肝素(150U/k,静脉注射)后进行气囊血管成形术,随后再用盐水注入。用DEGR-因子VIIa处理过的动物在接受1mg/kg大剂量注射后以50mg/kg/hr注入。
为了用于连续全身性注入的渗透泵的置入,如前述麻醉动物,在全部过程中另外肌肉内注射甲苯噻嗪和氯胺酮维持。通过颈部中线切开,用钝器解剖法分离右内部颈静脉,远端结扎。用硅化橡胶管(PE-160)导入到右内部颈静脉。制备皮下通路以使硅化橡胶管穿过。此管与渗透泵连接。渗透泵在兔背的皮下植入。右公用颈动脉用钝器解剖法分开,远端结扎。通过动脉切开术,将5F导管插入器在主动脉弓连接之前置入。抽血以确定止血参数,药物和胆固醇水平。动脉内注射20mg甲苯噻嗪。通过位于主动脉分支处上方的5F Berman导管用手注射3~4ml泛影钠-泛影葡胺进行对照主股动脉血管造影。
除去Berman导管后,在下行主动脉和位于主动脉分支上方位置中导入0.14英寸的导线。在荧光镜指导下,引入2.0到2.5mm的合适大小的气囊血管成形术导管,且在导线之前置于横跨狭窄处。用手动充气机充胀气囊到6个大气压60秒。在三次充胀之间有60秒间隔。在每个动物中的两个股动脉均进行此过程。
气囊膨胀后,撤除血管成形术导管,将Berman导管再次引入到主动脉分支上方3cm位置。在动脉内给20mg利多卡因以减少痉挛。如前述进行血管造影术后程序。在股动脉水平定出一个1cm格以计算实际直径。然后再除去导管。用3-0丝线结扎右颈动脉,逐层缝合伤口。如上述施用Ambipen和对乙酰氨基酚。
在测试化合物的即将大量注射前,注射1小时后以及在3天连续注射结束时测定血液中DEGR-因子VIIa浓度和凝血酶原时间。获得柠檬酸化的血浆1~2毫升,测定凝血酶原时间和抗原水平。
下面的标准凝固实验用于监测对照和DEGR-因子VIIa处理的动物中的凝血酶原时间。25μl测试的兔血浆(用作为抗凝剂的柠檬酸钠收集)加入到150mlTBS中(20mM Tris,pH7.4,150mM NaCl.)。混合样品且加入Electra 800自动凝血计时仪(医学实验室自动化公司,Pleasan Tville,NY)。培养后,含有25mM CaCl2的200ml促凝血酶原激酶制剂(SigmaChemical)加入到血浆制剂中。选择在对照兔血浆中约20秒产生凝血的时间的促凝血酶原激酶浓度。
用ELISA实验测定来自对照和DEGR-因子VIIa处理的兔的血浆样品中的DEGR-因子VIIa浓度。实验包括首先在0.1M碳酸盐缓冲液(pH9.6)中稀释抗人因子VII单克隆抗体(W.Kisiel博士,新墨西哥大学)到2.0mg/ml,且以100ml/孔加入到96孔板中。平板在4℃下孵育过夜,随后用洗涤缓冲液(PBS,pH7.4,含0.05%Tween20)洗两次。每孔中用200ml封闭缓冲液(PBS,pH7.4,含0.05%Tween20和1%牛血清血蛋白)封闭非特异性结合位点,37℃孵育2小时后再用洗涤缓冲液洗涤。
封闭后,范围从20到0.027ng/ml的DEGR-因子VIIa标准稀释系列与测试的兔血浆(在封闭液中1∶100~1∶4000)的稀释系列一起加入,每孔中100ml。非免疫兔血浆用作阴性对照。平皿37℃培养1小时后用洗涤缓冲液洗涤4次。
通过加入每孔100ml封闭缓冲液中1∶1000的兔抗人因子VII多克隆抗体稀释液(Kisiel博士,新墨西哥大学)检测DEGR-因子VIIa。37℃下培养平皿1小时后再用洗涤缓冲液洗5次。用每孔100毫升1∶2000稀释的山羊抗兔IgG抗体辣根过氧化物酶偶联物(Tago公司)检测特异性抗体结合。37℃下培养平皿1小时,用洗涤缓冲液洗6次。最后,加入100ml底物溶液(0.2M柠檬酸盐缓冲液中的0.42mg/ml二盐酸化邻苯二胺[OPD],pH5.0,含0.3%H2O2)。室温下1~3分钟后,通过加入100ml/孔的1N H2SO4终止颜色反应,在微孔板分光光度计上于490nm测定平板读数。通过与那些DEGR-因子VIIa标准曲线比较未知物的A490值测定血浆样品中DEGR-因子VIIa浓度。
对血浆样品的凝血酶原时间和DEGR-因子VIIa抗原水平的分析分别见表13和表14。数据代表每个单独的动物。表15显示平均凝血时间的总括。在所有情况中,DEGR-因子VIIa处理过的动物在注射后1小时时间点的凝血酶原时间延长,在注射后第3天时间点凝血酶原时间回复到接近处理前的水平。DEGR-因子VIIa抗原水平的分析也显示在1小时时间点血浆中的DEGR-因子VIIa水平较高,其血浆中范围在2-6mg/ml之间,在第3天时间点上循环水平很低。在1小时时段测得的的DEGR-因子VIIa水平与预计的凝血酶原时间增加相一致,通过在体外以DEGR-因子VIIa强化正常兔血浆测定DEGR-因子VII水平,在标准稀释促凝血酶原激酶实验中测定凝血酶原时间。
N/A=无数据
N/A=无数据表15血浆凝固时间的统计学总括不成对的t-检验X 流血前DF: 不成对的t值 概率(双尾):12 1.12 0.2852组: 数量 均值 标准离差 标准误差对照 8 24.15 1.64 0.58DEGR-VIIa 6 23.2 1.48 0.60不成对的t-检验X 血管成形术后1小时DF: 不成对的t值 概率(双尾)10 -5.44 0.0003组: 数量 均值: 标准偏差: 标准误差:对照 6 24.5 3.35 1.37DEGR-因子VIIa 6 40.8 6.53 2.67不成对的t-检验X 管成形术后3天DF: 不成对的t值 概率(双尾)13 -2.04 0.0622组: 数量 均值: 标准偏差: 标准误差:对照 8 19.54 1.53 0.54DEGR-因子VIIa 7 21.06 1.33 0.50
| 表13凝血酶原时间测定 | ||||
| 凝固时间(秒) | ||||
| 动物编号 | 处理 | 处理前 | 1小时 | 3天 |
| 73 | 对照 | 24.8 | 22.3 | 17.8 |
| 74 | 对照 | 24.8 | 27.9 | 18.6 |
| 75 | 对照 | 24.6 | N/D | 20.5 |
| 76 | 对照 | 22 | N/D | 17.9 |
| 169 | 对照 | 21.2 | 22.9 | 22 |
| 170 | 对照 | 24.9 | 23.5 | 18.6 |
| 173 | 对照 | 25.9 | 21 | 20.8 |
| 174 | 对照 | 25 | 29.4 | 20.1 |
| 77 | DEGR-FVIIa | 22.5 | 40.1 | 18.3 |
| 78 | DEGR-FVIIa | 24.3 | 34 | 20.9 |
| 80 | DEGR-FVIIa | 24.7 | 50 | 21.7 |
| 96 | DEGR-FVIIa | N/A | N/A | 21 |
| 97 | DEGR-FVIIa | 23.6 | 33.3 | 21.2 |
| 171 | DEGR-FVIIa | 20.6 | 45.8 | 21.9 |
| 172 | DEGR-FVIIa | 23.5 | 41.6 | 22.4 |
| 表14ELISA检测兔血浆中DEGR-因子VIIa | ||||
| FVIIa ELISA(ng/ml) | ||||
| 动物编号 | 处理 | 处理前 | 1小时 | 3天 |
| 73 | 对照 | 0 | 13 | 0 |
| 74 | 对照 | 36 | 14 | 4 |
| 75 | 对照 | 0 | N/A | 9 |
| 76 | 对照 | 0 | N/A | 14 |
| 169 | 对照 | 0 | 0 | 1 |
| 170 | 对照 | 0 | 0 | 0 |
| 173 | 对照 | 36 | 31 | 0 |
| 174 | 对照 | 87 | 86 | 160 |
| 77 | DEGR-FVIIa | 0 | 3,210 | 102 |
| 78 | DEGR-FVIIa | 1 | 4,950 | 7 |
| 80 | DEGR-FVIIa | 13 | 4,543 | 661 |
| 96 | DEGR-FVIIa | 65 | 4,900 | 117 |
| 97 | DEGR-FVIIa | 4 | 4,600 | 502 |
| 171 | DEGR-FVIIa | 13 | 2,145 | 212 |
| 172 | DEGR-FVIIa | 9 | 2,830 | 228 |
如前述血管成形术三周后通过左颈动脉在处死前重复一次随访血管造影术。通过一垂直的下腹部切口,分离远端主动脉,近端剥离,在主动脉分支处插入灌注管。远端主动脉用50ml盐水冲洗后再在体内用500mlHistochoice(AMRESCO,Solon,OH)溶液以120mm Hg注入超过15分钟以固定。一旦灌注开始,用过量戊巴比妥钠(3ml巴比妥钠IV,65mg/ml)处死。5cm段股动脉被对侧切割。组织保存于Histochoice溶液中用于光学显微镜观察。
为测定在气囊血管成形术位点内膜损伤的形成,将被切除的股动脉切成连续的3mm切片,包被在石蜡中,切片切自每个动脉的各种区域。切片固定于玻璃载玻片上,用苏木精和伊红及姬姆萨染色法染载玻片。用Bioquant Program进行形态测量分析以获得腔、内膜和中膜的面积测量。对来自损伤动脉的组织切片进行形态测量分析,测量总腔面积;内膜面积的测定则通过测量内弹性层之内的面积减去相应的来自每一组织切片的腔面积得到;中膜面积则通过测量外弹性层之内的面积再减去内弹性层之内的面积得到。测定对照和用DEGR-因子VIIa处理过的动物的股动脉中内膜损伤的测量表明用DEGR-因子VIIa处理过的内膜尺寸明显减少(表16)。相反,在两组中中膜面积的测量无明显差异。
| 表16.气囊血管成形术处理过的兔的内膜和中膜的测量 | ||||
| 组 | 数量 | 内膜(mm2) | 标准偏差 | 概率(双尾) |
| 对照 | 13 | 0.819 | 0.414 | 0.0138 |
| DEGR-FVIIa | 10 | 0.438 | 0.192 | |
| 组 | 数量 | 中膜(mm2) | 标准偏差 | 概率(双尾) |
| 对照 | 13 | 0.389 | 0.098 | 0.172 |
| DEGR-FVIIa | 10 | 0.329 | 0.105 | |
来自血管成形术测量的数据列于表17,如对照和DEGR-因子VIIa处理过的动物的全部三个时间点:即将施行血管成形术之前,血管成形术后立即,及血管成形术之后21天的平均腔直径(MLD)+/-标准偏差。在对照和DEGR-因子VIIa处理过的动物之间的血管成形术前或后测量的MLD无明显差异。然而观察到在用DEGR-因子VIIa处理过的动物中血管成形术之后测量的MLD明显增加。
实施例XIII DEGR-因子VIIa对狒狒平滑肌细胞(SMC)上细胞表面因子 Xa产生的抑剂
| 表17平均腔直径(MLD)的测量 | ||||
| PTCA前MLD测量 | ||||
| 组 | 数量 | 平均MLD | 标准偏差 | 概率(双尾) |
| 对照 | 13 | 1.202 | 0.24 | 0.3883 |
| DEGR-FVIIa | 10 | 1.283 | 0.19 | |
| PTCA后MLD测量 | ||||
| 组 | 数量 | 平均MLD | 标准偏差 | 概率(双尾) |
| 对照 | 13 | 1.492 | 0.551 | 0.5326 |
| DEGR-FVIIa | 10 | 1.323 | 0.725 | |
| 21天时MLD测量 | ||||
| 组 | 数量 | 平均MLD | 标准偏差 | 概率(双尾) |
| 对照 | 13 | 0.889 | 0.228 | 0.0001 |
| DEGR-FVIIa | 10 | 1.393 | 0.242 | |
基本上如前面实施例VIII所述进行细胞表面产色鉴定以测量DEGR-因子VIIa在狒狒单层平滑肌细胞(SMC)上对因子VIIa与细胞表面的组织因子结合的封闭,抑制随后因子X到因子Xa转变的功效。此方法是对如Sakai等人生物化学杂志(J.Bio.Chem.)264:9980-9980(1989)和Wildgoose等人,美国国家科学院院报87:7290-7294(1990)所述方法的改进。狒狒平滑肌细胞获自华盛顿大学,西雅图,美国,且培养自主动脉外植体。狒狒平滑肌细胞以8,000细胞/孔的浓度接种于96孔培养皿的每孔200ml的DEME培养基(补加10%胎牛血清)中,在此培养基中37℃下5%CO2中维持4天。鉴定时除去110ml培养液,每孔中加入递增浓度的因子VIIa或与DEGR-因子VIIa一起加入因子VIIa。得出因子VIIa浓度的标准曲线,范围从5nM到0.04nM。为测量DEGR-因子VIIa对因子VIIa活性的抑制活性,每测试孔中在恒量因子VIIa(5nM)存在下,加入递增浓度的DEGR-因子VIIa。因子VIIa和DEGR-因子VIIa均用HEPES缓冲液(10mM HEPES,137mM NaCl,4mM KCl,5mM CaCl2,11mM葡萄糖,0.1%BSA)稀释,且细胞中加入10ml 10x贮存液。细胞与测试化合物37℃下一起培养2小时,再用HEPES缓冲液洗3次。每孔中再加入50μl在Tris缓冲液(25mM Tris,pH7.4,150mM NaCl,2.7mM KCl,5mM CaCl2,0.1%BSA)中的200nM因子X溶液。室温下4分钟后,加入25ml 0.5M EDTA以终止因子X向因子Xa的转变。每孔中加入25μl溶于Tris缓冲液的0.8mM S-2222(一种因子Xa特异性产色底物),60分钟后在Thermomax微孔板读数仪(Molecular Devices公司,Menlo Park,CA)中405nm处读数。
图3所示结果表明,因子VIIa处理过的孔中(空心方框)酰胺水解活性呈剂量依赖性增加。吸光度的增加是孔中产生的因子Xa水平的直接测量,因子Xa随即切割产色底物。恒量因子VIIa(5nM)中加入递增水平的DEGR-因子VIIa显示随着DEGR-因子VIIa(实心方框)水平的增加,酰胺水解活性呈剂量依赖性下降。与因子VIIa等摩尔比的DEGR-因子VIIa能抑制>90%的产色活性。即使在DEGR-因子VIIa的水平低10倍时,仍有对因子Xa产色活性产生40%的抑制。这些结果支持了DEGR-因子VIIa是一种完整细胞单层平滑肌细胞表面上因子VIIa使因子X向因子Xa转变活化的极强有力的拮抗剂的结论。实施例XIV DEGR-因子VIIa对狒狒中血管血栓形成及血管损伤形成 的影响
测试了人DEGR-因子VIIa抑制在非人灵长类中组织因子(TF)和活化的因子VII(因子VIIa)调节血管损伤形成(VLF)的能力,此VLF是由机械性血管损伤诱发的。
在即将于狒狒中创造机械性血管损伤之前开始静脉注射DEGR-因子VIIa7天(5只动物)或30天(1只动物)。在第30天进行血管损伤形成测量。5只处理过的动物的结果与5只同时注入缓冲液的对照的结果比较。
测定关于实验动物的基础值:a)血小板计数,中性粒细胞计数,单核细胞计数及红细胞计数;b)血浆纤维蛋白原水平;c)血浆凝固因子VII,VIIa,X和V的活性水平以及因子VII的抗原性水平;和d)抗因子VIIa抗体水平的基线血浆样品。
在三氟溴氯乙烷麻醉及无菌操作条件下,用自体111In-血小板标记过的动物使用连续静脉内给药的tether系统接受DEGR-因子VIIa静脉内注入(初始注射1mg/kg,随后连续静脉内注入50mg/kg/hr)。动物接受颈动脉动脉内膜切除术、对侧肱动脉或对侧股动脉Fogarty气囊导管血管成形术的手术。
利用tether系统通过静脉导管连续注入以施用DEGR-因子VIIa7到30天。手术后30天用三氟溴氯乙烷麻醉动物,用含0.1%戊二醛的4%多聚甲醛在原位加压灌注固定30分钟。那时,用Harker等人,循环(Circulation)83:41-44(1991)和Hanson等人,高血压(Hypertension)18:1170-1176(1991)的方法收集血管片段(含有先前损伤过的位置)。样品在体外固定后(含0.1%戊二醛的4%多聚甲醛)低温保藏(Cryopreserved)且加工用于损伤程度的形态测量学分析。
研究了11只正常成年狒狒(Paio anubis)。其中6只动物接受DEGR-因子VIIa的注入(50mg/kg/hr),剩余5只为不接受DEGR-因子VIIa的对照动物。在采用前将动物去蠕虫且经三个月观察无疾病。全部过程经研究性动物关怀及使用委员会批准并遵守由NIH关于实验室动物的关怀及使用指南列出的操作及方法,以及动物保护法案和相关研究性政策。在用氯胺酮(10mg/kg肌内注射)和安定(0.5mg/kg静脉内注射)诱导后在三氟溴氯乙烷麻醉下进行侵入性过程。为了在随后的手术后进行的实验性操作中短期固定,使用盐酸氯胺酮(5-20mg/kg,肌内注射)。
用Hanson等人,高血压,18:I170-I176(1991)和Krupski等人,循环84:1749-1757(1991)所述的技术(此处引入作为参考)通过颈中线切口完成颈动脉动脉内膜切除术。使用动脉内膜切除术作为血管损伤模型是因为其临床上的相关性,以及已证实正常动脉的动脉内膜切除术诱导的VLF具有可重复性。简言之,从锁骨近侧到颈动脉分支远端的共用颈动脉被无包围组织的切下。大量注射硫酸盐肝素(100U/kg静脉内注射,Elkins-Simm公司,Cherry Hill,NJ)后3分钟用置于暴露血管两端的无创伤血管来交叉夹住其用颈动脉,且近侧与远端交叉夹分开1cm。再将近侧动脉段在弯钳上翻转。在获得最大翻转后,将一对聚丙烯驻留缝线(7-0)置于任一端近侧,将另一对置于暴露出腔的片段的近端。从翻转血管段分隔端的1cm处开始进行动脉内膜切除术,持续达测量长度为1cm。此步骤包括利用弯钳和手术显微镜(32x放大倍数)将正常内膜和中膜的部分厚度机械性除去。动脉内膜切除术后,血管回复到正常构象,用7-0聚丙烯缝线进行末端对末端的吻合术及在2.5倍放大倍数下连续操作将伤口逐层缝合。
为了对VLF进行形态测量分析,包埋在石蜡中且用苏木精-伊红染色相连组织成分(蚀原,弹性蛋白)的部分利用带有影像分析系统(Thomas光学测量系统,Columbus,GA)的Zeiss透视镜评价,此显像分析系统含有带高分辨率(700线)监视器的高分辨率(580线)CCD显微镜照相机,一块IBM386芯片,带有用于显像捕获和贮存的高分辨率图像分析仪的80MB计算机。用形态测量软件进行定量显像分析(Optimas,Bioscan公司,Edmonds,WA)。针对新内膜增生性损伤的总面积及相应的动脉中膜面积分析动脉截面。为进行统计学分析,利用成对和不成对数据的学生检验(双尾)比较不同的组。
结果显示在用DEGR-因子VIIa处理7天后在第30天进行研究的动物中的内膜面积与经过同样血管损伤但未接受任何DEGR-因子VIIa的对照动物相比明显下降(图4)。在用DEGR-因子VIIa处理30天且在第30天检查的动物中也发现类似结果。
利用气囊血管造影肱动脉模型的初步研究表明,用DEGR-因子VIIa治疗无可检测的益处。然而此模型已证实在狒狒中是一个组织因子起关键作用的凝血酶原模型。
狒狒中的股动脉气囊损伤研究的确表明与对照相比,DEGR-因子VIIa有统计学上明显的益处,如图5所示。实施例XV DEGR-因子VIIa对tPA诱导的血栓溶解的影响
在急性心肌梗死过程中进行性冠状动脉血栓形成主要是由与因子VIIa复合的组织因子通过外源性凝血途径调节。确定了在外源性途径中的不同位点辅助性凝血级联反应抑制对组织型纤溶酶原激活剂(TPA)血栓溶解的影响。
带有电子诱导冠状动脉血栓的正在用tPA(1mg/kg,20分钟以上)溶栓的36只狗分别被给予四种辅助性治疗的一种:9只接受30mg/kg/min的蜱抗凝固肽(TAP,一种选择性因子Xa抑制剂)90分钟。在9只狗中TF-因子VIIa复合物被重组组织因子途径抑制剂(TFPI)(100-150mg/kg/min,90分钟)抑制,在另9只狗中被DEGR-因子VIIa(1~2mg/kg次)(作为活化的因子VIIa竞争性拮抗剂)抑制。还有9只狗接受盐水对照。在溶栓后观察狗的再闭塞120分钟。这些试剂对溶栓功效的影响见下表18(数据为平均±SD)表18
*数值与盐水对照的差异为0.05显著性水平。
| 盐水 | DEGR-因子VIIa | TFPI | TAP | |
| 复流开始时间(分钟) | 32±13 | 20±7* | 21±6* | 18±10* |
| 复流持续时间(分钟) | 62±45 | 70±48 | 91±35* | 120 |
| 循环血流变化 | 70% | 89% | 56% | 0% |
| 再闭塞 | 70% | 78% | 67% | 0% |
这些数据表明,由DEGRVIIa或TFPI封闭因子Xa或TF-因子VIIa造成的外源性途径的抑制加速了t-PA诱导的血栓溶解。选择性因子Xa抑制剂在成功再灌注后维持动脉开放更为有效。实施例XVI修饰过的因子VIIa对血管内血栓形成的抑制不影响系统性 凝血
为确定与TF结合的因子VII的抑制是否会导致抗血栓形成作用,通过将外用缩窄器置于内皮损伤的免颈动脉周围(Fott′s模型)引发由于复发的血栓形成造成的循环血流变化(CFV)。利用置于缩窄器近端的Dopple流量探测器连续测定颈动脉血流。在动脉周围放置缩窄器后,6只兔中的6只形成平均频率11±2循环/小时的CFV,而在CFV最低点颈动脉血流速度平均为5±2%基线值。观察CFV30分钟后,动物接受人重组活性位点封闭的(Phe-Phe-Arg氯甲基酮)因子VIIa(因子VIIai)(0.1mg/kg/min,10分钟)的注入。因子VIIai完全消除了6只动物中6只的CFV(CFV频率=0循环/小时;P<0.05;颈动脉血流速度=106.9%基线值;p=NS,相对于基线基)。CFV抑制30分钟后,以0.1mg/kg/min的剂量注入人重组因子VIIa10分钟。因子VIIa的注入在所有动物中恢复了CFV,这表明因子VIIa与TF的结合是竞争性的。在因子VIIai注入后与基线值相比,凝血酶原时间、活化的部分促凝血酶原激酶时间、应答ADP和凝血酶的离体血小板沉积均无差异。因此,在体内引发血栓形成中,因子VII-VIIa起了重要作用。因子VIIai的施用在此模型中表现出不影响系统性凝血的强的抗血栓形成作用。实施例XVII通过局部施用修饰过的因子VIIa抑制微动脉血栓
在血管手术、微血管再造手术或再植手术中,最常见的失败原因是在吻合处的血栓。当血管经过创伤、显示出病理性改变或当使用介入性静脉移植物时闭塞性血栓形成的风险大大增加了。因而,抗血栓形成性干预经常与手术一起使用。目前可得的用于此目的的物质以非肠道途径(肝素,葡聚糖)或口服途径(ASA)施用且均带有出血副作用。此外,肝素和尤其是ASA在防止(动脉)血栓形成时仅部分有效。由于这些缺点,需要在血管手术中使用防止血栓的一种试剂,此试剂能通过可被局部运输的物质的方式在吻合术区域和血管损伤位置结合且发挥功效,避免不期望的系统性副作用。此实施例中,活性位点失活的因子VIIa在动脉损伤位点的局部施用用于产生不会诱发出血增加或其它止血缺陷的倾向的抗血栓形成作用。方法:
重为~3.5kg的瑞典鲁普兔(Swedish Loop)(雌、雄均可)用标准糖丸饲料和随意量的水喂养。用前在实验室观察至少一周无疾病。
边缘耳静脉插管,用巴比妥钠(18mg/kg)麻醉且重复注射以维持。
双侧耳引出皮瓣,制备中央动脉3cm长的片段(外径~1mm)。所有分支用10-0缝线连接和切割。操作区用等渗盐水冲洗且盖上薄塑料膜。为了保持高且恒定的血流量和防止痉挛,将动物置于热垫上且保持在约39.5℃的(正常体温约38.5℃)稍高体温下,在操作后向血管局部施用3滴10mg/ml利多卡因(再灌注后及再灌注后第30分钟测试开放度后)。
两侧血管同时置于双微血管夹中(S&T2V,S&T市场有限公司,Neuhausen,瑞士),分离两个夹子之间7mm动脉片段。进行纵向动脉切开术(7mm)后使两个夹子接近,血管复位后将血管腔翻转弄平,暴露中膜的深层。用连续10-0单线尼龙缝线闭合动脉切口(Ethilon 10-0BV-75-3,Ethicon有限公司,Edinburg,英国)。全部手术操作由一名外科医生用高质操作显微镜(Wild M-650,Leica-Heerbrugg,Heerbrugg,瑞士)进行。
通过打开血管夹使血管同时再灌注。迅速用盐水浸透过的纱布盖上,每分钟检查一次。记录动脉切开术出血完全终止的时间。
再灌注后30和120分钟,用标准显微外科空/满(empty/refill)测试评价血管开放度:用一对微弯钳轻轻地在损伤面远端堵塞血管,用另一对弯钳在下游腾空。释放第一对弯钳后,评价血管的再充满,血管分类为开放的或闭合的。闭合的血管表现为不再充满,而开放的血管表现为或快或慢的再充满,后者被称作“降低的开放度”。在最终的开放度测试后,切下血管并纵切开,移出血栓性物质并称量。化合物:
浓度为3.1mg/ml的重组化学性灭活的人因子VIIa(VIIai)或赋形剂以200ml/份贮存在编号的小瓶。实验方案:
以如下的随机双盲法处理20只兔,每只兔用作其自身对照:在如前述方法部分造成深动脉损伤后,一只耳的暴露的损伤部位用VIIai溶液(总量为0.5mg)冲洗5分钟,另一只耳用赋形剂处理。冲洗过的创面使之在另一个5分钟过程中与溶液孵育,其后所有过量的溶液用等渗盐水冲洗净。闭合动脉切口且再灌注血管。统计学方法:
利用体征(Sign)测试、血栓重量及动脉切开术出血数据等与Wilcoxon测试比较开放度结果。给出了双边(two-sided)P值。结果:
如开放率测试所得施用因子VIIai产生明显的抗血栓形成作用。在因子VIIai组中,再灌注后第30分钟血管开放度为85%,第120分钟时为75%。在赋形剂组中的相应值分别为40%和30%。此差异有统计学意义(分别为P=0.008和P=0.004)。因子VIIai组中中等血栓重量为0.3mg,在赋形剂组中为0.5mg,虽然此差异并不显著(P=0.19)。因子VIIai组中的中等动脉切开术出血时间为1.5分钟,在赋形剂组为2分钟。两组之间统计学上无差异(P=1)。
此实施例显示在动脉损伤位置局部施用因子VIIai可产生不引起出血倾向增加的抗血栓形成作用。这代表了用于预防由于手术、对血管的显微手术、血管成形术或其他损伤造成的血栓综合症治疗的高度有吸引力的模式。实施例XVIII因子VIIai的局部应用减少血栓重量并增加开放度
此实施例证实了化学性灭活的因子VIIa(因子VIIai)的局部应用减少血栓重量并增加血管开放度。
20只麻醉的兔用于此实施例。松动颈静脉并在夹子之间分离10mm片段。通过对内皮细胞的分离片段化学性(aetoxysclerol)破坏以及在该片段尾侧半限制性结扎的联合应用导入血栓。在双盲法中,一侧用0.5mg化学性灭活的因子VIIa(因子VIIai)处理,另一侧用缓冲液处理。化学性破坏后将测试物质注射入分离的片段且孵育10分钟。30和120分钟后用空/满测试控制开放度。处死后称量可能有的血栓。
| 中等血栓重量 | 血栓重量范围 | P-值 | 30分钟时开放度 | P-值 | 120分钟时开放度 | P-值 | |
| 因子VIIai缓冲液 | 0.85mg9.3mg | 0-22.3mg0-26.8mg | 0.035 | 90%55% | 0.0070 | 85%50% | 0.070 |
这些结果表明在静脉血栓模型中灭活的因子VIIa的局部应用明显减少血栓重量并提高开放度。实施例XIX因子VIIai的应用减少危险面积并改善再灌注 方法实验准备
研究了28只新西兰白兔(雌雄均有)(3.2~3.8kg)。简言之,用肌内施用氯胺酮(35mg/kg)和甲苯噻嗪(5mg/kg)的混合物麻醉动物,插管且用恒体积呼吸机通气(Harvard设备公司,Cambrige,MA)。聚丙烯导管通过左颈动脉置于主动脉及颈静脉中以分别监测动脉压力和药物的施用。通过第5个左侧肋间进行胸廓切开术并打开心包。聚丙烯导管置入左动脉心耳以用于以后的彩色微球注射。弯曲的冠状动脉的大边缘支用手术缝线圈套器将其从起始处临时性闭合约0.3cm。维持冠状动脉闭合30分钟后,将结扎松开,使再灌注5.5小时。实验过程中连续记录(Gould仪器)系统性动脉压力(Statham P23 DB压力传感器)。实验方案
在再灌注时,将动物随机分配到下列处理组中:左心房接受5ml盐水的对照组;用人重组的活性位点封闭的因子VIIa(因子VIIai,NovoNordisk,A/S,Gentofte,丹麦,1mg/kg注射入左心房)处理的组;用人重组活化的因子VIIa(FVIIai,Novo Nordisk A/S,Gentofte,丹麦,1mg/kg注射入左心房)处理的组。危险面积、梗死尺寸和不复流现象的评价
为评价在实验终止时组织灌注的分布(“不复流”现象,NR),通过左心房导管向动物注射6%的硫黄素S溶液(1mg/kg)。为获得梗死危险面积的评估,注射硫黄素后立即将冠状动脉再闭合,并通过左心房导管注射入单星蓝(E.I.DuPont;1mg/kg)溶液。立即切割心脏,将左心室从全部其他结构中切出并称重。左心室在-70℃冷冻30分钟,并以平行于房室沟方向切成8~10片。正常灌注的心肌轮廓以及危险的面积均根据单星蓝的分布描绘于一个透明的塑料片上。再在紫外光下观察心肌切片,按照硫黄素的分布正常灌注心肌的(荧光)则可明显地与缺血性心肌(无荧光)区分开来。这些区域也描绘于透明塑料片上。将切片37℃下再孵育在2%氯化三苯基四唑溶液(TTC,Sigma)中10分钟,以观察坏死的面积。再次用透明塑料片描绘正常心肌(TTC-阳性)和梗死心肌(TTC-阴性)的轮廓。
计算下列变量:1)在再灌注期终点评价梗死危险面积占左心室的百分比(单星蓝分布)(AR);2)依TTC染色标准梗死尺寸(IS)占实际发展为坏死的面积的百分比;3)再灌注期终点不接受血流的危险面积的百分数(不复流现象,NR)。局部心肌血流的测量
每处理组中的每只兔均测量局部心肌血流(RMBF)。不同颜色的塑料微球(兰、红和黄色,triton Technology,San Diego,CA)用于测量闭塞后20分钟、再灌注后10分钟和5小时的RMBF。
微球尺寸为15±1μ,悬浮于含0.01%Tween80的10%葡聚糖溶液中。在即将使用前用超声波浴超声微球5分钟,以保证充分分散。将约500,000个微球(总体积0.5~1.0ml)注射入左心室导管。在微球注射前1分钟,参比动脉血流开始减少且注射后持续1分钟。取自缺血性区域中央及取自非缺血性区域的组织样品(100~300mg)按TTC染色。按制造商指示,再通过在72℃下于4M KOH溶液中消化3小时从组织中回收微球,并通过在室温下于16M KOH溶液中消化后再经过微孔过滤,从参比血样中回收微球。按照制造商指示,从已知体积的溶剂(二甲基甲酰胺)中回收染料,用分光光度计在可见光波长测定每种染色的浓度。每种染料溶液的组合光谱通过一种矩阵转换技术分解成单组分光谱。用下式计算每个心肌样品中的血流:
RMBF=Frx Am/Ar,其中
RMBF=每分钟心肌血流的毫升(ml/min),
AM=心肌样品的吸光度,及
AR=参比血液样品的吸光度。用样品湿重除心肌血流,表示为ml/min/g。凝血研究
为测定因子VIIai和因子VIIa施用对系统性凝血的作用,在基线和药物施用后30分钟测量凝血酶原时间(PT)和活化的部分促凝血酶原激酶时间(aPTT)。在0.5ml柠檬酸钠中(3.8%)收集血液样品(4.5ml),4℃下离心(2000g)10分钟以分离血浆。从血液收集时2小时之内重复测定PT和aPTT。统计学分析
以平均值±SD表示结果。变量分析用于组间的多重比较。对于带Bonferroni修正的不成对观察用学生t-检验测试单个组之间的差异。为比较组之间的血液动力学变量及局部心肌血流,采用了带有用于重复测量的设计的双路(2-way)变量分析。结果
手术处理28只兔;在分配到处理组之前两只兔在冠状动脉闭合过程中由于室颤死亡,另外两只兔子在再灌注过程中死亡(一个为对照组,另一只在用因子VIIa处理过的组)。这些动物从后续统计分析中排除去。因此,研究中每个处理组中含有8只动物。血液动力学测量:
在所有处理组之中,冠状动脉闭塞引起心率和平均动脉压力略有下降。在实验期间的过程中三组动物在心率和平均动脉压力上未发现差异(表1)。危险面积、梗死尺寸和不复流现象的评价:
通过单星蓝在实验终点的注射所评价的由冠状动脉闭合产生的梗死危险面积在三个治疗组中相似(在对照、因子VIIai和因子VIIa处理过的动物中分别为左心室的31.6±6.3%,28.2±4.1%和29.2±5.3%,P=NS)。
在冠状动脉闭塞30分钟后和再灌注5.5小时后,对照组中向坏死发展的危险面积的值平均为59.8±12.8%(图7)。因子VIIai的施用将梗死尺寸明显减少到危险面积的28.1±11.3%,P<0.01,使用ANOVA(图1),而施用因子VIIa则与梗死尺寸明显增加到危险面积的80.1±13.1%相关(P<0.01,相对于对照和用因子VIIai处理过的兔,图7)。
在对照兔中,24.4±2.7%的危险面积显示有灌注缺陷,如在实验终止时用硫黄素S的分布所测定(不复流现象)。此不复流面积的范围分别由因子VIIai明显减少到危险面积的11.1±6.1%,由因子VII明显增加到危险面积的61.9±13.8%(P<0.01,图8)。
前面的研究表明:在缺血后再灌注过程中显示有灌注缺陷的心肌组织的量与多种参数相关;最重要的为危险面积的范围,梗死尺寸的大小及在闭塞过程中残余伴随血流的量。这些变量的控制使得对不复流现象的干预作用有更为精确的评价。在本研究中,将对照兔中不复流面积与这些参数相关联时,观察到有密切关系,其符合一多元线性回归方程:NR(占左心室(LV)的%)=-14.62+0.75(AR)+0.07(IS)+3.69(RMBF);
R2=0.98;F检验=109.3,(0.37)(0.3)(3.69),其中NR为不复流面积,AR为危险面积,RMBF为伴随血流(ml/min/g)。括号内的数字显示相关系数的标准误差。r2值为0.98的此模型归因于本研究观察的对照动物中>95%的不复流面积变量。
获自对照兔数据的多元线性回归方程中的方程相关系数用于计算每干预组中单个动物的预期不复流面积(图9)。接受因子VIIai的兔子中实际观察到的不复流的面积比应用获自多元回归分析的方程计算的预期值小。实际上,在用因子VIIai处理过的兔中,任何实际观察值小于其给定的预期不复流面积,即:此组中全部动物的值分布在获自对照动物的回归线下方(图9)。相反,用因子VIIa处理过的兔有正相反的显示,即:对于任何给定的预期不复流面积,实际观察值明显更大,此组中动物分布在对照动物的回归线上(图9)。综上,这些数据显示在因子VIIai处理过的动物中观察到的不复流现象的减少不完全是梗死面积减少造成的,这提示在缺血后再灌注过程中外源性凝血级联反应的激活对不复流现象的发生有作用。局部心肌血流的测量:
在对照动物中,贯穿研究中的非缺血性心肌的RMBF平均为1.20ml/min/克组织(数据未给出)。在同一组动物中,对缺血性心肌的RMBF在冠状动脉闭塞后20分钟、灌注后10分钟及5小时分别为0.08±0.02,1.43±0.28和0.98±0.19ml/min/克组织。与对照动物相比本研究中所用的不同药物在实验期间并不明显改变无论是正常心肌还是在缺血性心肌中的RMBF(图10)。凝血研究:
为了研究因子VIIai可能的系统性作用(可以诱发出血危险的增加),测量在第30分钟的CFV收集的及在因子VIIa和因子VIIai施用后收集的血液样品中的PT和aPTT。在30分钟CFV期终止时,PT和aPTT平均分别为8.2±0.6秒和25±3秒。在因子VIIai施用后观察到PT略微增加到10.1±0.6秒(图11)。然而,这种增加,没有达到统计学上的显著性(使用ANOVA)及带有Bonferronis修正的学生t-检验,P=0.09)。在因子VIIai施用后aPTT无明显改变(图10)。仅就因子VIIai施用后所获的值而言,因子VIIa的施用导致PT和aPTT的明显缩短(图11)。
表1:
冠状动脉闭塞-再灌注过程中的血液动力学变量
表2冠状动脉闭合及再灌注过程中局部心肌血流(ml/min/克组织)
CAO=冠状动脉闭塞;REP=再灌注实施例XIX因子VIIai的应用减少梗死尺寸及梗死的危险面积
| 心率(b/min) | |||||
| 闭塞后的时间 | 对照 | SQ29548 | 咪唑乙氧基甲酸 | R68070 | ASA |
| 0 | 175±5 | 170±4 | 178±6 | 169±5 | 1 |
| 30分钟 | 169±4 | 165±5 | 173±5 | 164±6 | 1 |
| 1小时 | 169±6 | 167±4 | 171±5 | 166±5 | 1 |
| 2小时 | 173±5 | 172±5 | 175±6 | 170±5 | 1 |
| 3小时 | 170±4 | 168±5 | 177±5 | 168±6 | 1 |
| 4小时 | 175±5 | 168±6 | 174±5 | 169±5 | 1 |
| 5小时 | 175±5 | 172±2 | 173±5 | 172±6 | 1 |
| 6小时 | 168±5 | 170±4 | 174±5 | 168±5 | 1 |
| 平均动脉压力(mm Hg) | |||||
| 闭塞后的时间 | 对照 | SQ29548 | 咪唑乙氧基甲酸 | R68070 | ASA+R68070 |
| 0 | 75±4 | 78±3 | 78±4 | 73±3 | 7 |
| 30分钟 | 69±4 | 65±4 | 71±3 | 67±4 | 6 |
| 1小时 | 69±3 | 67±4 | 71±4 | 69±4 | 6 |
| 2小时 | 73±4 | 75±4 | 75±5 | 72±4 | 7 |
| 3小时 | 74±4 | 75±3 | 77±5 | 75±3 | 7 |
| 4小时 | 75±5 | 78±4 | 76±4 | 73±4 | 7 |
| 5小时 | 73±4 | 74±5 | 78±5 | 72±4 | 7 |
| 6小时 | 73±5 | 76±4 | 74±5 | 73±5 | 7 |
| 对照 | FVIIai | FVIIa | |
| 正常心肌 | |||
| 20分钟CAO | 1.19±0.22 | 1.03±0.19 | 1.27±0.24 |
| 10分钟REP | 1.22±0.15 | 1.10±0.17 | 1.19±0.20 |
| 2小时REP | 1.17±0.18 | 1.07±0.16 | 1.22±0.19 |
| 缺血性心肌 | |||
| 20分钟CAO | 0.09±0.05 | 0.08±0.05 | 0.08±0.04 |
| 10分钟REP | 1.53±0.12 | 1.65±0.18 | 1.24±0.14 |
| 2小时REP | 0.89±0.14 | 1.23±0.15 | 0.72±0.13 |
在冠状动脉血管系统中发生在缺血后心肌再灌注过程中的组织因子暴露导致冠状动脉中血流的下降。
为鉴定组织因子暴露是否通过对凝血的活化及减少缺血后再灌注过程中冠状动脉血流来对心肌损伤有作用,将新西兰白兔先经过30分钟冠状动脉闭塞后再经过5.5小时的再灌注。再灌注时,动物随机的接受:盐水(n=8);人重组活性位点封闭的因子VIIa(因子VIIai,100μg/kg/min注入左心房10分钟,n=8)或人重组活化的因子VIIa(因子VIIa 100μg/kg/min注入左心房10分钟,n=8)。用彩色微球在缺血20分钟及再灌注10分钟和2小时后测量局部心肌血流(RMBF)。在实验终止时通过单星蓝和硫黄素的分布以及通过TTC染色测定梗死危险面积(AR),梗死尺寸(IS)和不复流面积(NR)。与对照相比,因子VIIa导致IS和NR的明显减少(分别为AR值的28.1±11.3%和11.1±6.1%,对应AR值的59.8±12.8%和24.4±8.2%P<0.01),而与对照相比,因子VIIa导致在IS和NR上的显著增加,分别为AR的80.1±13.1%和61.9±13.8%P<0.01)。在各组中血压、心率、AR及缺血20分钟的RMBF均未观察到差异。在因子VIIai处理过的动物中再灌注2小时时RMBF明显要高,而在因子VIIa处理过的兔中较低。因此,由TF介导的凝血活性对在缺血后再灌注过程中发生的心肌损伤有重要贡献。
AR=梗死危险面积;IS=梗死尺寸(IS);NR=不复流面积
| 因子VIIai(AR) | 对照(AR) | 因子VIIa(AR) | |
| IS | 28.1±11.3 | 59.8±12.8 | 80.1±13.1 |
| NR | 11.1±6.1 | 24.4±8.2 | 61.9±13.8 |
虽然上述发明已用阐明的方式和意在澄清理解的实施例进行了某种程度的详述,显然地,可以在附属的权利要求范围之内进行特定的改变和修饰。
序列表(1)一般信息:
(i)申请人:Zymo Genetics和Novo Nordisk A/S
(ii)发明名称:修饰过的因子VII
(iii)序列数目:4
(iv)相关地址:
(A)收信人:Novo Nordisk A/S,共同专利
(B)街道:Novo Alle
(C)城市:DK-2880 Bagsvaerd
(D)国家:丹麦
(v)计算机可读形式:
(A)介质类型:软盘
(B)计算机:IBM PC兼容机
(C)操作系统:PC-DOS/MS-DOS
(D)软件:PatentIn Release#1.24(vi)当前申请数据:
(A)申请号:
(B)递交日:
(C)分类,(vii)在先申请数据:
(A)申请号:08/475,845
(B)递交日:1995年6月7日
(C)分类:(2)SEQ ID 1的信息:(i)序列特征:
(A)长度:2422碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑结构:线性
(ii)分子类型:cDNA
(iii)假设:否
(iv)反义:否
(ix)特征:
(A)名称/关键词:CDS
(B)位置:28..1420
(D)其它信息:密码子起始=28
产物=因子VII
(xi)SEQ ID 1的序列描述:CCTCCCGACA ATACAGGGGC AGCACTGCAG AGATTTCATC ATG GTC TCC CAG GCC 55
Met Val Ser Gln Ala
-38 -35CTC AGG CTC CTC TGC CTT CTG CTT GGG CTT CAG GGC TGC CTG GCT GCA 103Leu Arg Leu Leu Cys Leu Leu Leu Gly Leu Gln Gly Cys Leu Ala Ala
-30 -25 -20GTC TTC GTA ACC CAG GAG GAA GCC CAC GGC GTC CTG CAC CGG CGC CGG 151Val Phe Val Thr Gln Glu Glu Ala His Gly Val Leu His Arg Arg Arg
-15 -10 -5CGC GCC AAC GCG TTC CTG GAG GAG CTG CGG CCG GGC TCC CTG GAG AGG 199Arg Ala Asn Ala Phe Leu Glu Glu Leu Arg Pro Gly Ser Leu Glu Arg
1 5 10 15GAG TGC AAG GAG GAG CAG TGC TCC TTC GAG GAG GCC CGG GAG ATC TTC 247Glu Cys Lys Glu Glu Gln Cys Ser Phe Glu Glu Ala Arg Glu Ile Phe
20 25 30AAG GAC GCG GAG AGG ACG AAG CTG TTC TGG ATT TCT TAC AGT GAT GGG 295Lys Asp Ala Glu Arg Thr Lys Leu Phe Trp Ile Ser Tyr Ser Asp Gly
35 40 45GAC CAG TGT GCC TCA AGT CCA TGC CAG AAT GGG GGC TCC TGC AAG GAC 343Asp Gln Cys Ala Ser Ser Pro Cys Gln Asn Gly Gly Ser Cys Lys Asp
50 55 60CAG CTC CAG TCC TAT ATC TGC TTC TGC CTC CCT GCC TTC GAG GGC CGG 391Gln Leu Gln Ser Tyr Ile Cys Phe Cys Leu Pro Ala Phe Glu Gly Arg
65 70 75AAC TGT GAG ACG CAC AAG GAT GAC CAG CTG ATC TGT GTG AAC GAG AAC 439Asn Cys Glu Thr His Lys Asp Asp Gln Leu Ile Cys Val Asn Glu Asn80 85 90 95GGC GGC TGT GAG CAG TAC TGC AGT GAC CAC ACG GGC ACC AAG CGC TCC 487Gly Gly Cys Glu Gln Tyr Cys Ser Asp His Thr Gly Thr Lys Arg Ser
100 105 110TGT CGG TGC CAC GAG GGG TAC TCT CTG CTG GCA GAC GGG GTG TCC TGC 535Cys Arg Cys His Glu Gly Tyr Ser Leu Leu Ala Asp Gly Val Ser Cys
115 120 125ACA CCC ACA GTT GAA TAT CCA TGT GGA AAA ATA CCT ATT CTA GAA AAA 583Thr Pro Thr Val Glu Tyr Pro Cys Gly Lys Ile Pro Ile Leu Glu Lys
130 135 140AGA AAT GCC AGC AAA CCC CAA GGC CGA ATT GTG GGG GGC AAG GTG TGC 631Arg Asn Ala Ser Lys Pro Gln Gly Arg Ile Val Gly Gly Lys Val Cys
145 150 155CCC AAA GGG GAG TGT CCA TGG CAG GTC CTG TTG TTG GTG AAT GGA GCT 679Pro Lys Gly Glu Cys Pro Trp Gln Val Leu Leu Leu Val Asn Gly Ala160 165 170 175CAG TTG TGT GGG GGG ACC CTG ATC AAC ACC ATC TGG GTG GTC TCC GCG 727Gln Leu Cys Gly Gly Thr Leu Ile Asn Thr Ile Trp Val Val Ser Ala
180 185 190GCC CAC TGT TTC GAC AAA ATC AAG AAC TGG AGG AAC CTG ATC GCG GTG 775Ala His Cys Phe Asp Lys Ile Lys Asn Trp Arg Asn Leu Ile Ala Val
195 200 205CTG GGC GAG CAC GAC CTC AGC GAG CAC GAC GGG GAT GAG CAG AGC CGG 823Leu Gly Glu His Asp Leu Ser Glu His Asp Gly Asp Glu Gln Ser Arg
210 215 220CGG GTG GCG CAG GTC ATC ATC CCC AGC ACG TAC GTC CCG GGC ACC ACC 871Arg Val Ala Gln Val Ile Ile Pro Ser Thr Tyr Val Pro Gly Thr Thr
225 230 235AAC CAC GAC ATC GCG CTG CTC CGC CTG CAC CAG CCC GTG GTC CTC ACT 919Asn His Asp Ile Ala Leu Leu Arg Leu His Gln Pro Val Val Leu Thr240 245 250 255GAC CAT GTG GTG CCC CTC TGC CTG CCC GAA CGG ACG TTC TCT GAG AGG 967Asp His Val Val Pro Leu Cys Leu Pro Glu Arg Thr Phe Ser Glu Arg
260 265 270ACG CTG GCC TTC GTG CGC TTC TCA TTG GTC AGC GGC TGG GGC CAG CTG 1015Thr Leu Ala Phe Val Arg Phe Ser Leu Val Ser Gly Trp Gly Gln Leu
275 280 285CTG GAC CGT GGC GCC ACG GCC CTG GAG CTC ATG GTC CTC AAC GTG CCC 1063Leu Asp Arg Gly Ala Thr Ala Leu Glu Leu Met Val Leu Asn Val Pro
290 295 300CGG CTG ATG ACC CAG GAC TGC CTG CAG CAG TCA CGG AAG GTG GGA GAC 1111Arg Leu Met Thr Gln Asp Cys Leu Gln Gln Ser Arg Lys Val Gly Asp
305 310 315TCC CCA AAT ATC ACG GAG TAC ATG TTC TGT GCC GGC TAC TCG GAT GGC 1159Ser Pro Asn Ile Thr Glu Tyr Met Phe Cys Ala Gly Tyr Ser Asp Gly320 325 330 335AGC AAG GAC TCC TGC AAG GGG GAC AGT GGA GGC CCA CAT GCC ACC CAC 1207Ser Lys Asp Ser Cys Lys Gly Asp Ser Gly Gly Pro His Ala Thr His
340 345 350TAC CGG GGC ACG TGG TAC CTG ACG GGC ATC GTC AGC TGG GGC CAG GGC 1255Tyr Arg Gly Thr Trp Tyr Leu Thr Gly Ile Val Ser Trp Gly Gln Gly
355 360 365TGC GCA ACC GTG GGC CAC TTT GGG GTG TAC ACC AGG GTC TCC CAG TAC 1303Cys Ala Thr Val Gly His Phe Gly Val Tyr Thr Arg Val Ser Gln Tyr
370 375 380ATC GAG TGG CTG CAA AAG CTC ATG CGC TCA GAG CCA CGC CCA GGA GTC 1351Ile Glu Trp Leu Gln Lys Leu Met Arg Ser Glu Pro Arg Pro Gly Val
385 390 395CTC CTG CGA GCC CCA TTT CCC TAG C CCAGCAGCCC TGGCCTGTGG 1396Leu Leu Arg Ala Pro Phe Pro400 405AGAGAAAGCC AAGGCTGCGT CGAACTGTCC TGGCACCAAA TCCCATATAT TCTTCTGCAG 1456TTAATGGGGT AGAGGAGGGC ATGGGAGGGA GGGAGAGGTG GGGAGGGAGA CAGAGACAGA 1516AACAGAGAGA GACAGAGACA GAGAGAGACT GAGGGAGAGA CTCTGAGGAC ATGGAGAGAG 1576ACTCAAAGAG ACTCCAAGAT TCAAAGAGAC TAATAGAGAC ACAGAGATGG AATAGAAAAG 1636ATGAGAGGCA GAGGCAGACA GGCGCTGGAC AGAGGGGCAG GGGAGTGCCA AGGTTGTCCT 1696GGAGGCAGAC AGCCCAGCTG AGCCTCCTTA CCTCCCTTCA GCCAAGCCCC ACCTGCACGT 1756GATCTGCTGG CCCTCAGGCT GCTGCTCTGC CTTCATTGCT GGAGACAGTA GAGGCATGAA 1816CACACATGGA TGCACACACA CACACGCCAA TGCACACACA CAGAGATATG CACACACACG 1876GATGCACACA CAGATGGTCA CACAGAGATA CGCAAACACA CCGATGCACA CGCACATAGA 1936GATATGCACA CACAGATGCA CACACAGATA TACACATGGA TGCACGCACA TGCCAATGCA 1996CGCACACATC AGTGCACACG GATGCACAGA GATATGCACA CACCGATGTG CGCACACACA 2056GATATGCACA CACATGGATG AGCACACACA CACCAAGTGC GCACACACAC CGATGTACAC 2116ACACAGATGC ACACACAGAT GCACACACAC CGATGCTGAC TCCATGTGTG CTGTCCTCTG 2176AAGGCGGTTG TTTAGCTCTC ACTTTTCTGG TTCTTATCCA TTATCATCTT CACTTCAGAC 2236AATTCAGAAG CATCACCATG CATGGTGGCG AATGCCCCCA AACTCTCCCC CAAATGTATT 2296TCTCCCTTCG CTGGGTGCCG GGCTGCACAG ACTATTCCCC ACCTGCTTCC CAGCTTCACA 2356ATAAACGGCT GCGTCTCCTC CGCACACCTG TGGTGCCTGC CACCCAAAAA AAAAAAAAAA 2416AAAAAA 2422(2)SEQ ID 2的信息:(i)序列特征:
(A)长度:444氨基酸
(B)类型:氨基酸
(D)拓扑结构:线性
(ii)分子类型:蛋白质
(xi)SEQ ID 2的序列描述:Met Val Ser Gln Ala Leu Arg Leu Leu Trp Leu Leu Leu Gly Leu Gln-38 -35 -30 -25Gly Cys Leu Ala Ala Val Phe Val Thr Gln Glu Glu Ala His Gly Val
-20 -15 -10Leu His Arg Arg Arg Arg Ala Asn Ala Phe Leu Glu Glu Leu Arg Pro
-5 1 5 10Gly Ser Leu Glu Arg Glu Cys Lys Glu Glu Gln Cys Ser Phe Glu Glu
15 20 25Ala Arg Glu Ile Phe Lys Asp Ala Glu Arg Thr Lys Leu Phe TrpIle
30 35 40Ser Tyr Ser Asp Gly Asp Gln Cys Ala Ser Ser Pro Cys Gln Asn Gly
45 50 55Gly Ser Cys Lys Asp Gln Leu Gln Ser Tyr Ile Cys Phe Cys Leu Pro
60 65 70Ala Phe Glu Gly Arg Asn Cys Glu Thr His Lys Asp Asp Gln Leu Ile75 80 85 90Cys Val Asn Glu Asn Gly Gly Cys Glu Gln Tyr Cys Ser Asp His Thr
95 100 105Gly Thr Lys Arg Ser Cys Arg Cys His Glu Gly Tyr Ser Leu Leu Ala
110 115 120Asp Gly Val Ser Cys Thr Pro Thr Val Glu Tyr Pro Cys Gly Lys Ile
125 130 135Pro Ile Leu Glu Lys Arg Asn Ala Ser Lys Pro Gln Gly Arg Ile Val
140 145 150Gly Gly Lys Val Cys Pro Lys Gly Glu Cys Pro Trp Gln Val Leu Leu155 160 165 170Leu Val Asn Gly Ala Gln Leu Cys Gly Gly Thr Leu Ile Asn Thr Ile
175 180 185Trp Val Val Ser Ala Ala His Cys Phe Asp Lys Ile Lys Asn Trp Arg
190 195 200Asn Leu Ile Ala Val Leu Gly Glu His Asp Leu Ser Glu His Asp Gly
205 210 215Asp Glu Gln Ser Arg Arg Val Ala Gln Val Ile Ile Pro Ser Thr Tyr
220 225 230Val Pro Gly Thr Thr Asn His Asp Ile Ala Leu Leu Arg Leu His Gln235 240 245 250Pro Val Val Leu Thr Asp His Val Val Pro Leu Cys Leu Pro Glu Arg
255 260 265Thr Phe Ser Glu Arg Thr Leu Ala Phe Val Arg Phe Ser Leu Val Ser
270 275 280Gly Trp Gly Gln Leu Leu Asp Arg Gly Ala Thr Ala Leu Glu Leu Met
285 290 295Val Leu Asn Val Pro Arg Leu Met Thr Gln Asp Cys Leu Gln Gln Ser
300 305 310Arg Lys Val Gly Asp Ser Pro Asn Ile Thr Glu Tyr Met Phe Cys Ala315 320 325 330Gly Tyr Ser Asp Gly Ser Lys Asp Ser Cys Lys Gly Asp Ser Gly Gly
335 340 345Pro His Ala Thr His Tyr Arg Gly Thr Trp Tyr Leu Thr Gly Ile Val
350 355 360Ser Trp Gly Gln Gly Cys Ala Thr Val Gly His Phe Gly Val Tyr Thr
365 370 375Arg Val Ser Gln Tyr Ile Glu Trp Leu Gln Lys Leu Met Arg Ser Glu
380 385 390Pro Arg Pro Gly Val Leu Leu Arg Ala Pro Phe Pro395 400 405(2)SEQ ID 3的信息:
(i)序列特征:
(A)长度:21碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑结构:线性
(ii)分子类型:cDNA
(iii)假设:否
(iv)反义:否
(xi)SEQ ID 3的序列描述:
TGGGCCTCCG GCGTCCCCCT T 21(2)SEQ ID 4的信息:(i)序列特征:
(A)长度:15碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑结构:线性(ii)分子类型:cDNA(iii)假设:否(iv)反义:否(xi)SEQ ID 4的序列描述:
TCCCAGTCAC GACGT 15
Claims (7)
1.在其催化中心中至少有一种修饰过的因子VII用于制造用来防止或减少与缺血后再灌注相关的心肌损伤的组合物的用途,其中的修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力,所述的修饰包括因子VII与丝氨酸蛋白酶抑制剂的反应;或者,因子VII的修饰包括在Ser、Asp和His催化三联体中至少一个氨基酸的替换、插入或删除。
2.如权利要求1的用途,其中的蛋白酶抑制剂是一种有机磷化合物,一种氟化硫,一种肽卤甲基酮或一种氮杂肽。
3.如权利要求2的用途,其中的蛋白酶抑制剂是一种选自Dansyl-Phe-Pro-Arg氯甲基酮、Dansyl-Glu-Gly-Arg氯甲基酮、Dansyl-Phe-Phe-Arg氯甲基酮和Phe-Phe-Arg氯甲基酮中的肽卤甲基酮。
4.如权利要求1的用途,其中的心肌损伤是心肌坏死。
5.在其催化中心中至少有一种修饰的因子VII用于制造用来改善在缺血后再灌注过程中局部心肌血流的组合物的用途,其中的修饰基本上抑制了修饰过的因子VII激活血浆因子X或IX的能力,所述修饰包括因子VII与丝氨酸蛋白酶抑制剂的反应;或者,因子VII的修饰包括在Ser、Asp和His催化三联体中至少一个氨基酸的替换、插入或删除。
6.如权利要求5的用途,其中的蛋白酶抑制剂是一种有机磷化合物,一种氟化硫,一种肽卤甲基酮或一种氮杂肽。
7.如权利要求6的用途,其中的蛋白酶抑制剂是一种选自Dansyl-Phe-Pr0-Arg氯甲基酮、Dansyl-Glu-Gly-Arg氯甲基酮、Dansyl-Phe-Phe-Arg氯甲基酮和Phe-Phe-Arg氯甲基酮中的肽卤甲基酮。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/660,289 US5833982A (en) | 1991-02-28 | 1996-06-07 | Modified factor VII |
| US08/660,289 | 1996-06-07 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA031066089A Division CN1515318A (zh) | 1996-06-07 | 1997-06-06 | 修饰过的因子vii |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1221427A CN1221427A (zh) | 1999-06-30 |
| CN1131872C true CN1131872C (zh) | 2003-12-24 |
Family
ID=24648892
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN97195294A Expired - Fee Related CN1131872C (zh) | 1996-06-07 | 1997-06-06 | 修饰过的因子vii |
| CNA031066089A Pending CN1515318A (zh) | 1996-06-07 | 1997-06-06 | 修饰过的因子vii |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA031066089A Pending CN1515318A (zh) | 1996-06-07 | 1997-06-06 | 修饰过的因子vii |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US5833982A (zh) |
| EP (1) | EP0910580A1 (zh) |
| JP (1) | JP2000513720A (zh) |
| KR (1) | KR20000016415A (zh) |
| CN (2) | CN1131872C (zh) |
| AU (1) | AU735012B2 (zh) |
| BR (1) | BR9709661A (zh) |
| CA (1) | CA2256761A1 (zh) |
| CZ (1) | CZ394698A3 (zh) |
| HU (1) | HUP0003077A3 (zh) |
| IL (1) | IL127099A0 (zh) |
| NO (1) | NO985668L (zh) |
| PL (1) | PL330365A1 (zh) |
| RU (1) | RU2211704C2 (zh) |
| UA (1) | UA68333C2 (zh) |
| WO (1) | WO1997047651A1 (zh) |
| ZA (1) | ZA975013B (zh) |
Families Citing this family (88)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5997864A (en) | 1995-06-07 | 1999-12-07 | Novo Nordisk A/S | Modified factor VII |
| US20040087498A1 (en) * | 1991-02-28 | 2004-05-06 | Novo Nordisk Health Care Ag | Modified factor VII |
| AU8101698A (en) * | 1997-07-18 | 1999-02-10 | Novo Nordisk A/S | Use of fviia or fviiai for the treatment of adverse conditions related to the fviia mediated intracellular signalling pathway |
| US7247708B2 (en) * | 1997-10-23 | 2007-07-24 | Regents Of The University Of Minnesota | Modified vitamin K-dependent polypeptides |
| US6693075B1 (en) * | 1997-10-23 | 2004-02-17 | Regents Of The University Of Minnesota | Modified vitamin K-dependent polypeptides |
| US6747003B1 (en) | 1997-10-23 | 2004-06-08 | Regents Of The University Of Minnesota | Modified vitamin K-dependent polypeptides |
| US6406488B1 (en) | 1998-08-27 | 2002-06-18 | Heartstent Corporation | Healing transmyocardial implant |
| EP0987274A1 (en) | 1998-09-15 | 2000-03-22 | Hoechst Marion Roussel Deutschland GmbH | Factor VIIa Inhibitors |
| EP1059302A1 (en) | 1999-06-08 | 2000-12-13 | Aventis Pharma Deutschland GmbH | Factor VIIa inhibitors |
| US6924359B1 (en) * | 1999-07-01 | 2005-08-02 | Yale University | Neovascular-targeted immunoconjugates |
| EP1095933A1 (en) | 1999-10-30 | 2001-05-02 | Aventis Pharma Deutschland GmbH | Novel N-guanidinoalkylamides, their preparation, their use, and pharmaceutical preparations comprising them |
| WO2001058935A2 (en) | 2000-02-11 | 2001-08-16 | Maxygen Aps | FACTOR VII OR VIIa-LIKE MOLECULES |
| CA2405557C (en) | 2000-04-12 | 2013-09-24 | Human Genome Sciences, Inc. | Albumin fusion proteins |
| US7220837B1 (en) | 2000-04-28 | 2007-05-22 | Regents Of The University Of Minnesota | Modified vitamin K-dependent polypeptides |
| US7812132B2 (en) | 2000-04-28 | 2010-10-12 | Regents Of The University Of Minnesota | Modified vitamin K-dependent polypeptides |
| EP1162194A1 (en) | 2000-06-06 | 2001-12-12 | Aventis Pharma Deutschland GmbH | Factor VIIa inhibitory (thio)urea derivatives, their preparation and their use |
| US6833357B2 (en) * | 2000-06-20 | 2004-12-21 | The Trustees Of The University Of Pennsylvania | Compositions and methods for modulating muscle cell and tissue contractility |
| US7160540B2 (en) * | 2000-06-30 | 2007-01-09 | Regents Of The University Of Minnesota | Methods for detecting activity of clottings factors |
| US6423826B1 (en) | 2000-06-30 | 2002-07-23 | Regents Of The University Of Minnesota | High molecular weight derivatives of vitamin K-dependent polypeptides |
| US20030211094A1 (en) * | 2001-06-26 | 2003-11-13 | Nelsestuen Gary L. | High molecular weight derivatives of vitamin k-dependent polypeptides |
| PL362320A1 (en) | 2000-12-06 | 2004-10-18 | Aventis Pharma Deutschland Gmbh | Guanidine and amidine derivatives as factor xa inhibitors |
| US6825323B2 (en) * | 2001-01-10 | 2004-11-30 | The United States Of America As Represented By The Secretary Of The Army | Compositions for treatment of hemorrhaging with activated factor VIIa in combination with fibrinogen and methods of using same |
| US7235638B2 (en) | 2001-03-22 | 2007-06-26 | Novo Nordisk Healthcare A/G | Coagulation factor VII derivatives |
| BR0208203A (pt) * | 2001-03-22 | 2005-04-19 | Novo Nordisk Healthcare Ag | Polipeptìdeo do fator vii, derivado do fator vii, composição, composição farmacêutica, construção de polinucleotìdeo, célula hospedeira eucariótica, animal transgênico, planta transgênica, e, métodos para produzir o polipeptìdeo do fator vii e um derivado do fator vii, uso de um derivado do fator vii, métodos para o tratamento de episódios de hemorragia ou distúrbios de hemorragia em um paciente ou para a intensificação do sistema hemostático normal e para inibir a formação de trobo em um paciente |
| CA2445811A1 (en) * | 2001-05-02 | 2002-11-07 | Novo Nordisk A/S | Modified fvii in treatment of ards |
| EP1270551A1 (en) | 2001-06-26 | 2003-01-02 | Aventis Pharma Deutschland GmbH | Urea derivatives with antiproteolytic activity |
| WO2003022122A2 (en) * | 2001-09-10 | 2003-03-20 | Florence Medical Ltd. | Individual ffr determination for lesions of a multi-lesioned blood vessel |
| EP2292273A3 (en) | 2001-10-10 | 2011-11-23 | BioGeneriX AG | Remodeling and glycoconjugation of peptides |
| US7214660B2 (en) | 2001-10-10 | 2007-05-08 | Neose Technologies, Inc. | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| WO2004099231A2 (en) | 2003-04-09 | 2004-11-18 | Neose Technologies, Inc. | Glycopegylation methods and proteins/peptides produced by the methods |
| US7173003B2 (en) | 2001-10-10 | 2007-02-06 | Neose Technologies, Inc. | Granulocyte colony stimulating factor: remodeling and glycoconjugation of G-CSF |
| WO2003037361A2 (en) * | 2001-11-02 | 2003-05-08 | Novo Nordisk Health Care Ag | Use of tissue factor agonist or tissue factor antagonist for treatment of conditions related to apoptosis |
| EP1314733A1 (en) | 2001-11-22 | 2003-05-28 | Aventis Pharma Deutschland GmbH | Indole-2-carboxamides as factor Xa inhibitors |
| WO2003055511A1 (en) * | 2001-12-21 | 2003-07-10 | Novo Nordisk A/S | Liquid composition of modified factor vii polypeptides |
| IL162239A0 (en) | 2001-12-21 | 2005-11-20 | Novo Nordisk Healthcare Ag | Liquid composition of factor vii polypeptides |
| KR20040065278A (ko) * | 2001-12-21 | 2004-07-21 | 노보 노르디스크 에이/에스 | 변경된 인자 ⅶ 폴리펩티드의 액체 조성물 |
| DK1499719T3 (da) * | 2002-04-30 | 2011-02-28 | Bayer Healthcare Llc | Faktor VII- eller VIIa-polypeptidvarianter |
| US20040009918A1 (en) * | 2002-05-03 | 2004-01-15 | Hanne Nedergaard | Stabilised solid compositions of modified factor VII |
| EP1503798B8 (en) * | 2002-05-03 | 2012-03-14 | Novo Nordisk Health Care AG | Stabilised solid compositions of modified factor vii |
| EP2283856B1 (en) | 2002-06-21 | 2017-09-20 | Novo Nordisk Health Care AG | Stabilised solid compositions of factor VIIa polypeptides |
| CA2502162C (en) * | 2002-09-30 | 2014-04-15 | Maxygen Holdings Ltd. | Fvii or fviia variants having increased clotting activity |
| US7358268B2 (en) | 2002-12-04 | 2008-04-15 | Sanofi-Aventis Deutschland Gmbh | Imidazole derivatives as factor Xa inhibitors |
| US7429581B2 (en) | 2002-12-23 | 2008-09-30 | Sanofi-Aventis Deutschland Gmbh | Pyrazole-derivatives as factor Xa inhibitors |
| US20040176704A1 (en) * | 2003-03-04 | 2004-09-09 | Stevens Timothy A | Collection device adapted to accept cartridge for point of care system |
| MXPA05009914A (es) * | 2003-03-18 | 2006-01-09 | Novo Nordisk Healthcare Ag | Composiciones farmaceuticas acuosas, liquidas de polipeptidos del factor vii. |
| ES2386010T3 (es) * | 2003-03-20 | 2012-08-07 | Bayer Healthcare Llc | Variantes de FVII o FVIIa |
| US7897734B2 (en) * | 2003-03-26 | 2011-03-01 | Novo Nordisk Healthcare Ag | Method for the production of proteins |
| US20040202726A1 (en) * | 2003-04-10 | 2004-10-14 | Deshay Samuel L. | Topical blood pressure composition |
| EP1479675A1 (en) | 2003-05-19 | 2004-11-24 | Aventis Pharma Deutschland GmbH | Indazole-derivatives as factor Xa inhibitors |
| EP1479680A1 (en) | 2003-05-19 | 2004-11-24 | Aventis Pharma Deutschland GmbH | Azaindole derivatives as Factor Xa inhibitors |
| US7317027B2 (en) | 2003-05-19 | 2008-01-08 | Sanofi-Aventis Deutschland Gmbh | Azaindole-derivatives as factor Xa inhibitors |
| US7223780B2 (en) | 2003-05-19 | 2007-05-29 | Sanofi-Aventis Deutschland Gmbh | Triazole-derivatives as blood clotting enzyme factor Xa inhibitors |
| US7741341B2 (en) | 2003-05-19 | 2010-06-22 | Sanofi-Aventis Deutschland Gmbh | Benzimidazole-derivatives as factor Xa inhibitors |
| EP1479677A1 (en) | 2003-05-19 | 2004-11-24 | Aventis Pharma Deutschland GmbH | New indole derivatives as factor xa inhibitors |
| CA2525224A1 (en) * | 2003-05-23 | 2004-12-02 | Michael Bech Jensen | Protein stabilization in solution |
| US7029675B1 (en) * | 2003-06-04 | 2006-04-18 | Shu-Wha Lin | Hepsin antagonist and methods of use |
| SI1644504T1 (sl) * | 2003-06-19 | 2010-06-30 | Bayer Healthcare Llc | Variante Gla domene faktorja VII ali VIIa |
| ATE547114T1 (de) | 2003-06-25 | 2012-03-15 | Novo Nordisk Healthcare Ag | Flüssige zusammensetzungen von factor vii polypeptiden |
| ES2335994T3 (es) * | 2003-07-01 | 2010-04-07 | Novo Nordisk Health Care Ag | Composicion farmaceutica liquida, acuosa de polipeptidos factor vii. |
| US9005625B2 (en) | 2003-07-25 | 2015-04-14 | Novo Nordisk A/S | Antibody toxin conjugates |
| ES2574581T3 (es) | 2003-08-14 | 2016-06-20 | Novo Nordisk Health Care Ag | Composición farmacéutica líquida acuosa de polipéptidos de tipo Factor VII |
| US20080305992A1 (en) | 2003-11-24 | 2008-12-11 | Neose Technologies, Inc. | Glycopegylated erythropoietin |
| EP2298287B1 (en) * | 2003-12-19 | 2018-04-11 | Novo Nordisk Health Care AG | Stabilised compositions of factor VII polypeptides |
| EP1568698A1 (en) | 2004-02-27 | 2005-08-31 | Aventis Pharma Deutschland GmbH | Pyrrole-derivatives as factor Xa inhibitors |
| JP2008514216A (ja) * | 2004-09-29 | 2008-05-08 | ノボ ノルディスク ヘルス ケア アクチェンゲゼルシャフト | Desgla−vii因子ポリペプチド構造の除去による、vii因子ポリペプチドの原体の精製 |
| US20080176790A1 (en) | 2004-10-29 | 2008-07-24 | Defrees Shawn | Remodeling and Glycopegylation of Fibroblast Growth Factor (Fgf) |
| US9029331B2 (en) | 2005-01-10 | 2015-05-12 | Novo Nordisk A/S | Glycopegylated granulocyte colony stimulating factor |
| EP1871795A4 (en) | 2005-04-08 | 2010-03-31 | Biogenerix Ag | COMPOSITIONS AND METHOD FOR PRODUCING GLYCOSYLATION MUTANTS OF A PROTEASE-RESISTANT HUMAN GROWTH HORMONE |
| US20070105755A1 (en) | 2005-10-26 | 2007-05-10 | Neose Technologies, Inc. | One pot desialylation and glycopegylation of therapeutic peptides |
| WO2007056191A2 (en) | 2005-11-03 | 2007-05-18 | Neose Technologies, Inc. | Nucleotide sugar purification using membranes |
| US20080242607A1 (en) | 2006-07-21 | 2008-10-02 | Neose Technologies, Inc. | Glycosylation of peptides via o-linked glycosylation sequences |
| US20100075375A1 (en) | 2006-10-03 | 2010-03-25 | Novo Nordisk A/S | Methods for the purification of polypeptide conjugates |
| DK2144923T3 (da) | 2007-04-03 | 2013-05-13 | Biogenerix Ag | Behandlingsfremgangsmåder under anvendelse af glycopegyleret g-csf |
| WO2008154639A2 (en) | 2007-06-12 | 2008-12-18 | Neose Technologies, Inc. | Improved process for the production of nucleotide sugars |
| CA2715465C (en) | 2008-02-27 | 2017-03-21 | Novo Nordisk A/S | Conjugated factor viii molecules |
| DK2379096T3 (da) | 2008-12-19 | 2019-11-25 | Baxalta GmbH | TFPI-inhibitorer og fremgangsmåder til anvendelse |
| US8450275B2 (en) | 2010-03-19 | 2013-05-28 | Baxter International Inc. | TFPI inhibitors and methods of use |
| AU2010290077C1 (en) | 2009-08-24 | 2015-12-03 | Bioverativ Therapeutics Inc. | Coagulation factor IX compositions and methods of making and using same |
| KR101294351B1 (ko) * | 2010-05-27 | 2013-08-07 | 동국대학교 산학협력단 | 뇌혈전 형광영상기술을 이용한 뇌졸중 동물모델의 뇌경색 영역의 크기 예측 방법 |
| RS63870B1 (sr) | 2012-02-15 | 2023-01-31 | Bioverativ Therapeutics Inc | Sastavi faktora viii i postupci za pravljenje i upotrebu istih |
| NZ628014A (en) | 2012-02-15 | 2016-09-30 | Biogen Ma Inc | Recombinant factor viii proteins |
| HUE046572T2 (hu) | 2012-03-21 | 2020-03-30 | Baxalta GmbH | TFPI inhibitorok és alkalmazási eljárások |
| EP3033097B1 (en) | 2013-08-14 | 2021-03-10 | Bioverativ Therapeutics Inc. | Factor viii-xten fusions and uses thereof |
| AU2016297178A1 (en) * | 2015-07-22 | 2018-01-25 | Iconic Therapeutics, Inc. | Methods for treating disorders associated with angiogenesis and neovascularization |
| TWI741992B (zh) | 2015-08-03 | 2021-10-11 | 美商百歐維拉提夫治療公司 | 因子ix融合蛋白以及其製備及使用方法 |
| WO2017121436A1 (en) | 2016-01-15 | 2017-07-20 | Rigshospitalet | Quantitative pet imaging of tissue factor expression using 18f-labled active site inhibited factor vii |
| MA46864A (fr) | 2016-11-17 | 2021-04-28 | Minerva Imaging Aps | Facteur vii inhibé par le site actif marqué par 177-lu |
| JP2021523878A (ja) | 2018-05-18 | 2021-09-09 | バイオベラティブ セラピューティクス インコーポレイテッド | 血友病aを処置する方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992015686A1 (en) * | 1991-02-28 | 1992-09-17 | Zymogenetics, Inc. | Modified factor vii |
| WO1994017631A1 (en) * | 1993-01-20 | 1994-08-04 | Rca Thomson Licensing Corporation | Digital video tape recorder for digital hdtv |
| WO1996006637A1 (en) * | 1994-08-26 | 1996-03-07 | Washington University | Method of inhibiting tissue ischemia and reperfusion injury |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4775624A (en) * | 1985-02-08 | 1988-10-04 | Eli Lilly And Company | Vectors and compounds for expression of human protein C |
| GR860984B (en) * | 1985-04-17 | 1986-08-18 | Zymogenetics Inc | Expression of factor vii and ix activities in mammalian cells |
| ATE74164T1 (de) * | 1985-04-22 | 1992-04-15 | Genetics Inst | Herstellung mit hoher leistung des aktivfaktors ix. |
| US4959318A (en) * | 1985-06-27 | 1990-09-25 | Zymogenetics, Inc. | Expression of protein C |
| US4829052A (en) * | 1986-06-11 | 1989-05-09 | Monsanto Company | Serine protease inhibitors |
| US5258288A (en) * | 1986-07-25 | 1993-11-02 | Genzyme Corporation | Vector containing DNA encoding mature human protein S |
| US4959381A (en) | 1987-02-10 | 1990-09-25 | Glaxo Group Limited | Pyridine compounds which have useful activity associated with reversible air ways obstruction |
| FR2619314B1 (fr) * | 1987-08-11 | 1990-06-15 | Transgene Sa | Analogue du facteur viii, procede de preparation et composition pharmaceutique le contenant |
| US4994371A (en) * | 1987-08-28 | 1991-02-19 | Davie Earl W | DNA preparation of Christmas factor and use of DNA sequences |
| US5023236A (en) * | 1988-04-07 | 1991-06-11 | Corvas, Inc. | Factor VII/VIIA active site inhibitors |
| US5258180A (en) * | 1988-09-02 | 1993-11-02 | Genetech, Inc. | Tissue plasminogen activator having fibrin specific properties and deletion of amino acids 466-970, compositions and methods of treatment |
| WO1990003390A1 (en) * | 1988-09-23 | 1990-04-05 | Corvas, Inc. | Peptidyl inhibitors of the initiation of coagulation |
| US5120537A (en) * | 1989-06-14 | 1992-06-09 | Oklahoma Medical Research Foundation | Factor xa based anticoagulant compositions |
| US5190919A (en) * | 1989-11-13 | 1993-03-02 | Board Of Regents, The University Of Texas System | Antihemostatic factor vii peptides |
| JP3330932B2 (ja) | 1990-01-29 | 2002-10-07 | ノボ ノルディスク ヘルス ケア アクチェンゲゼルシャフト | 抗凝固剤タンパク質 |
| US5278144A (en) * | 1990-09-04 | 1994-01-11 | Cor Therapeutics, Inc. | Antithrombosis agents |
| US5817788A (en) | 1991-02-28 | 1998-10-06 | Zymogenetics, Inc. | Modified factor VII |
| US5861374A (en) | 1991-02-28 | 1999-01-19 | Novo Nordisk A/S | Modified Factor VII |
| US5788965A (en) | 1991-02-28 | 1998-08-04 | Novo Nordisk A/S | Modified factor VII |
| US5326559A (en) * | 1991-05-16 | 1994-07-05 | Miller D Douglas | Treatment of accelerated atheosclerosis with interleukin-2 receptor targeted molecules |
| US5419760A (en) * | 1993-01-08 | 1995-05-30 | Pdt Systems, Inc. | Medicament dispensing stent for prevention of restenosis of a blood vessel |
| DK0699075T3 (da) * | 1993-05-21 | 2002-03-11 | Novo Nordisk As | Modificeret faktor VII |
-
1996
- 1996-06-07 US US08/660,289 patent/US5833982A/en not_active Expired - Fee Related
-
1997
- 1997-06-06 IL IL12709997A patent/IL127099A0/xx unknown
- 1997-06-06 ZA ZA9705013A patent/ZA975013B/xx unknown
- 1997-06-06 CN CN97195294A patent/CN1131872C/zh not_active Expired - Fee Related
- 1997-06-06 JP JP10501082A patent/JP2000513720A/ja not_active Ceased
- 1997-06-06 EP EP97925917A patent/EP0910580A1/en not_active Withdrawn
- 1997-06-06 BR BR9709661-0A patent/BR9709661A/pt not_active IP Right Cessation
- 1997-06-06 PL PL97330365A patent/PL330365A1/xx unknown
- 1997-06-06 CA CA002256761A patent/CA2256761A1/en not_active Abandoned
- 1997-06-06 UA UA98126442A patent/UA68333C2/uk unknown
- 1997-06-06 CN CNA031066089A patent/CN1515318A/zh active Pending
- 1997-06-06 AU AU30906/97A patent/AU735012B2/en not_active Ceased
- 1997-06-06 KR KR1019980709998A patent/KR20000016415A/ko not_active Application Discontinuation
- 1997-06-06 WO PCT/DK1997/000251 patent/WO1997047651A1/en not_active Application Discontinuation
- 1997-06-06 CZ CZ983946A patent/CZ394698A3/cs unknown
- 1997-06-06 HU HU0003077A patent/HUP0003077A3/hu unknown
- 1997-06-06 RU RU99100089/14A patent/RU2211704C2/ru not_active IP Right Cessation
-
1998
- 1998-11-10 US US09/189,607 patent/US6168789B1/en not_active Expired - Fee Related
- 1998-12-04 NO NO985668A patent/NO985668L/no not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992015686A1 (en) * | 1991-02-28 | 1992-09-17 | Zymogenetics, Inc. | Modified factor vii |
| WO1994017631A1 (en) * | 1993-01-20 | 1994-08-04 | Rca Thomson Licensing Corporation | Digital video tape recorder for digital hdtv |
| WO1996006637A1 (en) * | 1994-08-26 | 1996-03-07 | Washington University | Method of inhibiting tissue ischemia and reperfusion injury |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0910580A1 (en) | 1999-04-28 |
| CN1515318A (zh) | 2004-07-28 |
| UA68333C2 (en) | 2004-08-16 |
| ZA975013B (en) | 1997-12-08 |
| BR9709661A (pt) | 2000-04-25 |
| NO985668L (no) | 1999-02-04 |
| WO1997047651A1 (en) | 1997-12-18 |
| HUP0003077A2 (hu) | 2000-12-28 |
| CA2256761A1 (en) | 1997-12-18 |
| CZ394698A3 (cs) | 1999-04-14 |
| KR20000016415A (ko) | 2000-03-25 |
| HUP0003077A3 (en) | 2003-01-28 |
| PL330365A1 (en) | 1999-05-10 |
| IL127099A0 (en) | 1999-09-22 |
| US5833982A (en) | 1998-11-10 |
| RU2211704C2 (ru) | 2003-09-10 |
| JP2000513720A (ja) | 2000-10-17 |
| NO985668D0 (no) | 1998-12-04 |
| US6168789B1 (en) | 2001-01-02 |
| AU3090697A (en) | 1998-01-07 |
| AU735012B2 (en) | 2001-06-28 |
| CN1221427A (zh) | 1999-06-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1131872C (zh) | 修饰过的因子vii | |
| CN1133743C (zh) | 修饰的凝血因子vii | |
| RU2326126C2 (ru) | Полипептид фактора vii свертывания крови, его получение и применение | |
| JP4361728B2 (ja) | ヒト凝固因子vii変異型 | |
| JP4455802B2 (ja) | ヒト第vii凝固因子変異型 | |
| AU651573B2 (en) | Anticoagulant proteins | |
| EP0699075B1 (en) | MODIFIED FACTOR VII for inhibiting vascular restenosis and platelet deposition | |
| RU2325401C2 (ru) | Полипептид фактора коагуляции vii человека, его получение и применение | |
| US5817788A (en) | Modified factor VII | |
| US20100158891A1 (en) | Human Coagulation Factor VII Variants | |
| AU2001287550A1 (en) | Human coagulation factor VII variants | |
| AU2008261165A1 (en) | Human coagulation factor VII variants | |
| US6039944A (en) | Modified Factor VII | |
| US20040087498A1 (en) | Modified factor VII | |
| RU2214833C2 (ru) | Модифицированный фактор vii |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |