CN111333552B - β-苯并氨基酸类化合物的合成方法 - Google Patents
β-苯并氨基酸类化合物的合成方法 Download PDFInfo
- Publication number
- CN111333552B CN111333552B CN202010274523.0A CN202010274523A CN111333552B CN 111333552 B CN111333552 B CN 111333552B CN 202010274523 A CN202010274523 A CN 202010274523A CN 111333552 B CN111333552 B CN 111333552B
- Authority
- CN
- China
- Prior art keywords
- compound
- acid
- formula
- reacting
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000001308 synthesis method Methods 0.000 title abstract description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 150
- CKLJMWTZIZZHCS-UWTATZPHSA-N D-aspartic acid Chemical compound OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 claims abstract description 61
- 229940024606 amino acid Drugs 0.000 claims abstract description 25
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 claims abstract description 23
- 229960005261 aspartic acid Drugs 0.000 claims abstract description 21
- 238000002360 preparation method Methods 0.000 claims abstract description 19
- 125000003277 amino group Chemical group 0.000 claims abstract description 12
- 125000006239 protecting group Chemical group 0.000 claims abstract description 10
- 238000005809 transesterification reaction Methods 0.000 claims abstract description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 108
- 238000006243 chemical reaction Methods 0.000 claims description 61
- -1 t-butoxycarbonyl Chemical group 0.000 claims description 34
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 24
- 229940125782 compound 2 Drugs 0.000 claims description 24
- 229940126214 compound 3 Drugs 0.000 claims description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 22
- 238000000034 method Methods 0.000 claims description 20
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 20
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 239000002904 solvent Substances 0.000 claims description 13
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 claims description 11
- 239000005695 Ammonium acetate Substances 0.000 claims description 11
- 229940043376 ammonium acetate Drugs 0.000 claims description 11
- 235000019257 ammonium acetate Nutrition 0.000 claims description 11
- 238000001816 cooling Methods 0.000 claims description 11
- 229940125904 compound 1 Drugs 0.000 claims description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 9
- UWIVVFQECQYHOB-UHFFFAOYSA-M sodium;ethanesulfinate Chemical compound [Na+].CCS([O-])=O UWIVVFQECQYHOB-UHFFFAOYSA-M 0.000 claims description 9
- 238000010438 heat treatment Methods 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 7
- BJHLFTYYTRBXRD-CYBMUJFWSA-N (3R)-3-(4-ethoxysulfonylphenyl)-3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CCOS(=O)(=O)C1=CC=C(C=C1)[C@@H](CC(=O)O)NC(=O)OC(C)(C)C BJHLFTYYTRBXRD-CYBMUJFWSA-N 0.000 claims description 5
- BJHLFTYYTRBXRD-ZDUSSCGKSA-N (3S)-3-(4-ethoxysulfonylphenyl)-3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CCOS(=O)(=O)C1=CC=C(C=C1)[C@H](CC(=O)O)NC(=O)OC(C)(C)C BJHLFTYYTRBXRD-ZDUSSCGKSA-N 0.000 claims description 5
- 229910052736 halogen Inorganic materials 0.000 claims description 5
- 238000002156 mixing Methods 0.000 claims description 5
- 238000010992 reflux Methods 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 4
- 239000000203 mixture Substances 0.000 claims description 4
- 125000004963 sulfonylalkyl group Chemical group 0.000 claims description 4
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 3
- 238000005886 esterification reaction Methods 0.000 claims description 3
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims description 3
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 claims description 3
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims description 3
- 239000011259 mixed solution Substances 0.000 claims description 3
- 238000006467 substitution reaction Methods 0.000 claims description 3
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims 9
- 125000005843 halogen group Chemical group 0.000 claims 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 235000001014 amino acid Nutrition 0.000 description 19
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 239000012065 filter cake Substances 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 150000001576 beta-amino acids Chemical class 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- 239000002994 raw material Substances 0.000 description 10
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 230000003287 optical effect Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 230000002194 synthesizing effect Effects 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- BJHLFTYYTRBXRD-UHFFFAOYSA-N 3-(4-ethoxysulfonylphenyl)-3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound C(C)(C)(C)OC(=O)NC(CC(=O)O)C1=CC=C(C=C1)S(=O)(=O)OCC BJHLFTYYTRBXRD-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 229940088710 antibiotic agent Drugs 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 150000002367 halogens Chemical group 0.000 description 3
- 229960002510 mandelic acid Drugs 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 239000011975 tartaric acid Substances 0.000 description 3
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 235000008206 alpha-amino acids Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 231100000027 toxicology Toxicity 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- UJOYFRCOTPUKAK-QMMMGPOBSA-N (S)-3-ammonio-3-phenylpropanoate Chemical group OC(=O)C[C@H](N)C1=CC=CC=C1 UJOYFRCOTPUKAK-QMMMGPOBSA-N 0.000 description 1
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000004922 2-methyl-3-pentyl group Chemical group CC(C)C(CC)* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 1
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- CWGVEMFBQJUWLU-SKTBPLDYSA-N Cc1ncsc1-c1ccc(cc1)[C@H](CC(=O)N1CCC(CC1)N1CCC(CC1)C#Cc1ccc(cc1)C(=O)NC1C(C)(C)C(Oc2ccc(C#N)c(Cl)c2)C1(C)C)NC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@@H](NC(=O)C1(F)CC1)C(C)(C)C Chemical compound Cc1ncsc1-c1ccc(cc1)[C@H](CC(=O)N1CCC(CC1)N1CCC(CC1)C#Cc1ccc(cc1)C(=O)NC1C(C)(C)C(Oc2ccc(C#N)c(Cl)c2)C1(C)C)NC(=O)[C@@H]1C[C@@H](O)CN1C(=O)[C@@H](NC(=O)C1(F)CC1)C(C)(C)C CWGVEMFBQJUWLU-SKTBPLDYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- 150000001371 alpha-amino acids Chemical class 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 125000003460 beta-lactamyl group Chemical group 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000007257 deesterification reaction Methods 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004246 ligand exchange chromatography Methods 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 238000003541 multi-stage reaction Methods 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- QOUZNKNYXDVAOK-UHFFFAOYSA-N octatrienyl-D-beta-phenylalanine Natural products CC=CC=CC=CC(=O)NC(CC(O)=O)C1=CC=CC=C1 QOUZNKNYXDVAOK-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000004537 pulping Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C315/00—Preparation of sulfones; Preparation of sulfoxides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C315/00—Preparation of sulfones; Preparation of sulfoxides
- C07C315/04—Preparation of sulfones; Preparation of sulfoxides by reactions not involving the formation of sulfone or sulfoxide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明涉及一种β‑苯并氨基酸类化合物的制备方法,包括如下步骤:(1)获取式(I)所示消旋体化合物;(2)将式(I)所示消旋体化合物与L‑天冬氨酸进行反应,得式(I‑1)所示化合物;和/或,将式(I)所示消旋体化合物与D‑天冬氨酸进行反应,得式(I‑2)所示化合物;(3)于式(I‑1)所示化合物和/或式(I‑2)所示化合物中的胺基上引入保护基团,然后进行脱酯基反应。该合成方法无需进行手性色谱拆分,即可获得不同构型的β‑苯并氨基酸类化合物,成本低,操作简单,便于工业应用,且产物手性纯度高,收率高。
Description
技术领域
本发明涉及化合物合成技术领域,特别是β-苯并氨基酸类化合物的合成方法。
背景技术
β-氨基酸是α-氨基酸增加一个亚甲基后形成的一种新类型的氨基酸。研究表明,β-多肽有着许多α-肽所不具备的生物活性,更重要的是它能在一些溶剂中形成如α-螺旋、β-折叠和β-转角等二级结构。这些重要的发现使β-氨基酸越来越受到关注。大量含天然的或非天然的β-氨基酸残基药物已经被广泛应用在医药行业中:首先是使用比较广泛的抗生素的结构中含有大量的β-氨基酸残基,如世界上最重要的一类抗生素——β-内酰胺环抗生素可以通过β-氨基酸缩合制备,抗生素Pyloricidin A和Moiramide A中存在(S)-β-苯丙氨酸残基;其次,一些药物或药物中间体也含有β-氨基酸残基,如法国的赛诺菲安万特制药公司治疗冠心病药物奥米沙班,德国的默克公司糖尿病药物西他列汀以及美国辉瑞制药公司抗艾滋病药物马拉维罗等。而且,人们研究还发现在多肽中引入部分β-氨基酸,其空间二级结构与α-氨基酸组成的多肽非常类似,但却有很多独特的性质。
然而β-氨基酸在自然界中存在较少,而且两种对映体在体内的药理作用不同,所以设法得到单一的β-氨基酸对映体就显得尤为重要。化学拆分法是研究最早的用于拆分光学异构体的方法之一,而现有的化学拆分法制备光学纯β-氨基酸的方法通常涉及多步反应,原料昂贵,操作复杂且产率较低。另有方法以醛、丙二酸和铵盐为原料,采用"一锅法"合成了β-氨基酸消旋体,然后通过应用手性配体交换色谱或者化学拆分法对所合成的氨基酸消旋体进行拆分,获得光学纯的β-氨基酸对映体。但是手性色谱柱拆分在衍生化过程中容易引入副产物,且手性柱价格昂贵,限制其工业发展。
发明内容
基于此,有必要提供一种β-苯并氨基酸类化合物的合成方法。该合成方法无需进行手性色谱拆分,即可获得不同构型的β-苯并氨基酸类化合物,成本低,操作简单,便于工业应用,且产物手性纯度高,收率高。
具体技术方案如下:
一种β-苯并氨基酸类化合物的制备方法,包括如下步骤:
(1)获取式(I)所示消旋体化合物;
(2)将式(I)所示消旋体化合物与L-天冬氨酸进行反应,得式(I-1)所示化合物;和/或,将式(I)所示消旋体化合物与D-天冬氨酸进行反应,得式(I-2)所示化合物;
(3)于式(I-1)所示化合物和/或式(I-2)所示化合物中的胺基上引入保护基团,然后进行脱酯基反应;


R1选自磺酰烷基或磺酰环烷基;
R2选自C1~C10烷基。
在其中一个实施例中,步骤(2)中,式(I)所示消旋体化合物与L-天冬氨酸的摩尔比为1:0.5~1;式(I)所示消旋体化合物与D-天冬氨酸的摩尔比为1:0.5~1。
在其中一个实施例中,步骤(2)中,所述反应采用的溶剂为甲醇和水的混合液。
在其中一个实施例中,步骤(2)中,于水和适量甲醇中混合式(I)所示消旋体化合物与L-天冬氨酸或D-天冬氨酸,升温至55~60℃溶清,然后保持温度反应0.8~1.2h;再加入甲醇,自然降温至室温,继续反应10~14h。
在其中一个实施例中,所述保护基团为叔丁氧羰基、甲氧羰基、乙氧羰基或异丙氧羰基。
在其中一个实施例中,R1选自磺酰甲基、磺酰乙基、磺酰丙基、磺酰丁基、磺酰环丙基、磺酰环戊基、磺酰环己基;
R2选自甲基、乙基、异丙基、叔丁基。
在其中一个实施例中,获取式(I)所示消旋体化合物的方法,包括如下步骤:

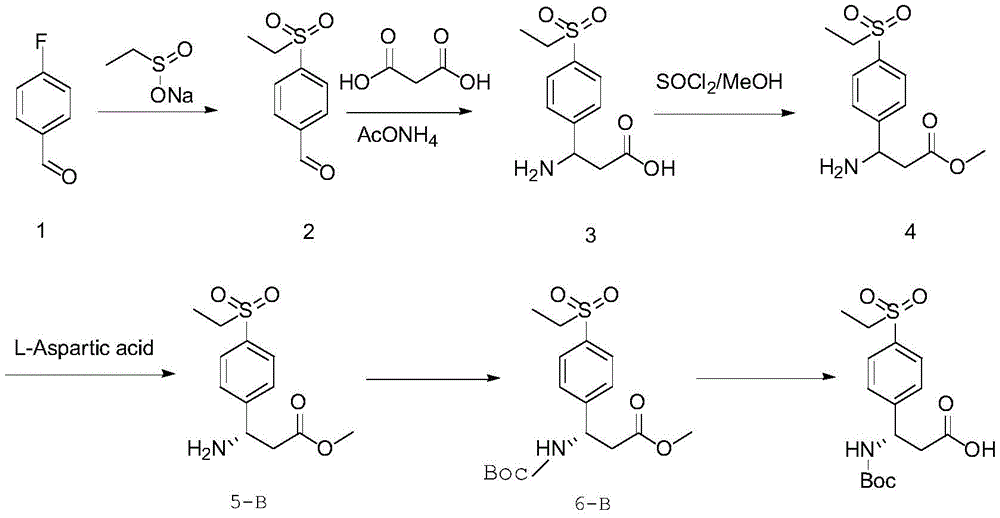
将化合物a进行取代反应,引入R1,得化合物2;X代表卤素;
将化合物b与丙二酸、醋酸铵进行反应,得化合物3;
将化合物c进行酯化反应,引入R2。
在其中一个实施例中,所述β-苯并氨基酸类化合物为(R)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:

将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与D-天冬氨酸进行反应,得化合物5-A;
于化合物5-A中的胺基上引入叔丁氧羰基(Boc)保护基团,得化合物6-A;
将化合物6-A进行脱酯基反应。
在其中一个实施例中,所述β-苯并氨基酸类化合物为(S)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:

将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与L-天冬氨酸进行反应,得化合物5-B;
于化合物5-B中的胺基上引入叔丁氧羰基(Boc)保护基团,得化合物6-B;
将化合物6-B进行脱酯基反应。
在其中一个实施例中,所述化合物2制备步骤中,所述反应的条件包括:于90~100℃反应。
在其中一个实施例中,所述化合物3制备步骤中,所述反应的条件包括:于回流条件下反应。
在其中一个实施例中,所述化合物4制备步骤中,所述反应的条件包括:先将所述化合物3和甲醇混合,控制所得混合液的温度小于20℃条件下,加入所述氯化亚砜,加完后自然升温至室温反应。
与现有技术相比较,本发明具有如下有益效果:
本发明创新地发现,对于β-苯并氨基酸类化合物的对映体的制备过程中,能够利用L-天冬氨酸/D-天冬氨酸对β-苯并氨基酸母核结构进行有效的化学拆分,有效缩短β-苯并氨基酸类化合物产品的制备步骤,操作简单、且不需要经过多次结晶就可以得到高手性纯度的产品,避免了化学拆分法中常有的产率低、操作时间长、步骤繁琐等限制其应用的问题,可直接放大应用于产业化生产,经济高效。
附图说明
图1为本发明实施例1制备的化合物2的核磁图谱;
图2为本发明实施例1制备的化合物4的HPLC监控图谱;
图3为本发明实施例1制备的化合物5-A的HPLC图谱;
图4为本发明实施例1制备的化合物5-A的核磁图谱;
图5为本发明实施例1制备的化合物5-B的HPLC图谱;
图6为本发明实施例1制备的化合物5-B的核磁图谱;
图7为本发明实施例1制备的化合物7-A的HPLC图谱;
图8为本发明实施例1制备的化合物7-A的核磁图谱;
图9为本发明实施例1制备的化合物7-B的HPLC图谱;
图10为本发明实施例1制备的化合物7-B的核磁图谱。
具体实施方式
以下结合具体实施例对本发明的β-苯并氨基酸类化合物的合成方法作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
术语“烷基”是指包含伯(正)碳原子、或仲碳原子、或叔碳原子、或季碳原子、或其组合的饱和烃。包含该术语的短语,例如,“C1~C10烷基”是指包含1~10个碳原子的烷基,每次出现时,可以互相独立地为C1烷基、C2烷基、C3烷基、C4烷基、C5烷基、C6烷基、C7烷基、C8烷基、C9烷基、C10烷基。合适的实例包括但不限于:甲基(Me、-CH3)、乙基(Et、-CH2CH3)、1-丙基(n-Pr、n-丙基、-CH2CH2CH3)、2-丙基(异丙基、i-Pr、i-丙基、-CH(CH3)2)、1-丁基(n-Bu、n-丁基、-CH2CH2CH2CH3)、2-甲基-1-丙基(i-Bu、i-丁基、-CH2CH(CH3)2)、2-丁基(s-Bu、s-丁基、-CH(CH3)CH2CH3)、2-甲基-2-丙基(叔丁基、t-Bu、t-丁基、-C(CH3)3)、1-戊基(n-戊基、-CH2CH2CH2CH2CH3)、2-戊基(-CH(CH3)CH2CH2CH3)、3-戊基(-CH(CH2CH3)2)、2-甲基-2-丁基(-C(CH3)2CH2CH3)、3-甲基-2-丁基(-CH(CH3)CH(CH3)2)、3-甲基-1-丁基(-CH2CH2CH(CH3)2)、2-甲基-1-丁基(-CH2CH(CH3)CH2CH3)、1-己基(-CH2CH2CH2CH2CH2CH3)、2-己基(-CH(CH3)CH2CH2CH2CH3)、3-己基(-CH(CH2CH3)(CH2CH2CH3))、2-甲基-2-戊基(-C(CH3)2CH2CH2CH3)、3-甲基-2-戊基(-CH(CH3)CH(CH3)CH2CH3)、4-甲基-2-戊基(-CH(CH3)CH2CH(CH3)2)、3-甲基-3-戊基(-C(CH3)(CH2CH3)2)、2-甲基-3-戊基(-CH(CH2CH3)CH(CH3)2)、2,3-二甲基-2-丁基(-C(CH3)2CH(CH3)2)、3,3-二甲基-2-丁基(-CH(CH3)C(CH3)3和辛基(-(CH2)7CH3)。
术语“环烷基”是指包含环碳原子的非芳香族烃,可以为单环烷基、或螺环烷基、或桥环烷基。包含该术语的短语,例如,“C3~C6环烷基”是指包含3~6个碳原子的环烷基,每次出现时,可以互相独立地为C3环烷基、C4环烷基、C5环烷基、C6环烷基。合适的实例包括但不限于:环丙基、环丁基、环戊基、环己基。另外,“环烷基”还可含有一个或多个双键,含有双键的环烷基的代表性实例包括环戊烯基、环己烯基、环己二烯基和环丁二烯基。
本发明的实施例提供一种β-苯并氨基酸类化合物的制备方法,包括如下步骤:
(1)获取式(I)所示消旋体化合物;
(2)将式(I)所示消旋体化合物与L-天冬氨酸进行反应,得式(I-1)所示化合物;和/或,将式(I)所示消旋体化合物与D-天冬氨酸进行反应,得式(I-2)所示化合物;
(3)于式(I-1)所示化合物和/或式(I-2)所示化合物中的胺基上引入保护基团,然后进行脱酯基反应;

R1选自磺酰烷基或磺酰环烷基;
R2选自C1~C10烷基。
可以理解地,步骤(2)中所述“和/或”表示,根据实际需求,可以采用相应构型的天冬氨酸仅制备式(I-1)所示化合物或式(I-2)所示化合物,也可以同时制备式(I-1)所示化合物和式(I-2)所示化合物两种构型的化合物。相应地,若未制备式(I-1)所示化合物或式(I-2)所示化合物,步骤(3)中进一步的保护基引入和脱酯基反应将不存在。
在其中一个具体的实施例中,步骤(2)中,式(I)所示消旋体化合物与L-天冬氨酸的摩尔比为1:0.5~1。式(I)所示消旋体化合物与D-天冬氨酸的摩尔比为1:0.5~1。合理控制消旋体化合物与L-天冬氨酸或D-天冬氨酸的摩尔比有利于异构体的拆分,提升式(I-1)所示化合物或式(I-2)所示化合物的手性纯度。具体地,式(I)所示消旋体化合物与L-天冬氨酸或D-天冬氨酸的摩尔比可以分别独立地为1:0.5、1:0.7、1:0.8、1:0.9、1:1。
作为优选地,步骤(2)中,式(I)所示消旋体化合物与L-天冬氨酸的摩尔比为1:0.7~0.9;式(I)所示消旋体化合物与D-天冬氨酸的摩尔比为1:0.7~0.9。
在其中一个具体的实施例中,步骤(2)中,所述反应采用的溶剂为甲醇和水的混合液。采用合适种类的溶剂,有利于反应速率的提升,所述溶剂中甲醇和水的配比可以任意调整,能够有效溶解反应原料即可。更为具体地,所述溶剂中甲醇和水的重量比为1:2~3。
在其中一个实施例中,步骤(2)中,所述反应的条件包括:于水和适量甲醇中混合式(I)所示消旋体化合物与L-天冬氨酸或D-天冬氨酸,升温至55~60℃溶清,然后保持温度反应0.8~1.2h;再加入甲醇,自然降温至室温,继续反应10~14h。采用合适反应温度和反应时间,有利于促进异构体的拆分,进一步提高手性纯度,同时保证反应速率。可以理解地,前述“适量甲醇”的用量是指能够使式(I)所示消旋体化合物与L-天冬氨酸或D-天冬氨酸在上述温度条件下溶清的量。反应结束后,“再加入甲醇”的目的在于尽可能将结晶盐沉淀出母液,其用量可以为式(I)所示消旋体化合物重量的2~3倍。更为具体地,“继续反应10~14h”后,还包括过滤步骤:对反应所得混合物进行过滤,滤饼用甲醇洗涤。
在其中一个具体的实施例中,所述保护基团为叔丁氧羰基、甲氧羰基、乙氧羰基或异丙氧羰基。所述保护基团的作用在于对式(I-1)或式(I-2)化合物中的胺基进行保护,以避免其对后续脱酯反应的影响。作为优选地,所述保护基团为叔丁氧羰基(Boc)。
在其中一个具体的实施例中,R1选自的磺酰烷基中的烷基部分可以为C1~C5烷基,磺酰环烷基中的环烷基部分可以为C3~C6环烷基。更为具体地,R1选自磺酰甲基、磺酰乙基、磺酰丙基、磺酰丁基、磺酰环丙基、磺酰环戊基、磺酰环己基。
在其中一个具体的实施例中,R2选自C1~C5烷基。更为具体地,R2选自甲基、乙基、异丙基、叔丁基。
在其中一个具体的实施例中,获取式(I)所示消旋体化合物的方法,包括如下步骤:

将化合物a进行取代反应,引入R1,得化合物2;X代表卤素;
将化合物b与丙二酸、醋酸铵进行反应,得化合物3;
将化合物c进行酯化反应,引入R2。
以廉价易得的对位卤素苯甲醛为起始原料,合成式(I)所示消旋体化合物,原料简单、易得,成本低。具体地,对位的卤素可以为F、Cl、Br等,优选为F。
进一步地,如上所述β-苯并氨基酸类化合物的制备方法可具体用于制备3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸,3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸是一种β-苯并氨基酸类化合物,能够作为具有一定药理活性的化学片段应用于大分子药物(如具有抗前列腺癌功效的大分子药物ARD-69)的进一步合成,而同一分子的对映异构体在毒理药理和生物活性方面具有显著差异,所以需要将R和S构型分开,以便得到药学毒理低,生物活性好的药物单体。具体地,其具有两种对映异构体,即(R)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸和(S)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸。
3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸是一种β-苯并氨基酸类化合物的两种对映异构体的现有合成路线如下:

该现有合成路线步骤较为繁琐,且需要使用到昂贵的不同种类酶手性催化剂,难以商品化,操作复杂,不利于实际生产。
本发明的实施例利用如上所述β-苯并氨基酸类化合物的制备方法进行3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸的制备,原料简单易得,成本较低,操作简单,易于放大生产。具体的技术方案如下:
在其中一个具体的实施例中,所述β-苯并氨基酸类化合物为(R)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:

将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与D-天冬氨酸进行反应,得化合物5-A;
于化合物5-A中的胺基上引入叔丁氧羰基(Boc)保护基团,得化合物6-A;
将化合物6-A进行脱酯基反应。
在其中一个实施例中,所述β-苯并氨基酸类化合物为(S)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:

将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与L-天冬氨酸进行反应,得化合物5-B;
于化合物5-B中的胺基上引入叔丁氧羰基(Boc)保护基团,得化合物6-B;
将化合物6-B进行脱酯基反应。
在其中一个具体的实施例中,所述化合物2制备步骤中,所述反应的条件包括:于90~100℃反应。更为具体地,所述化合物2制备步骤中,以溶剂溶解化合物1和乙基亚磺酸钠,然后于90~100℃反应。反应过程中可通过TLC监控反应的进程,具体地,反应完全的时间约为10~15h。所述溶剂可选用常规溶剂,能够使化合物1和乙基亚磺酸钠溶解即可,具体举例如DMSO。
在其中一个具体的实施例中,所述化合物3制备步骤中,所述反应的条件包括:于回流条件下反应。更为具体地,所述化合物2制备步骤中,以溶剂溶解化合物2和丙二酸、醋酸铵,然后升温至回流反应。反应过程中可通过TLC监控反应的进程,具体地,反应完全的时间约为10~15h。所述溶剂可选用常规溶剂,具体举例如无水乙醇。
在其中一个具体的实施例中,所述化合物4制备步骤中,所述反应的条件包括:先将所述化合物3和甲醇混合,控制所得混合液的温度小于20℃条件下,加入所述氯化亚砜,加完后自然升温至室温反应。具体地,反应过程中可通过HPLC监控反应的进程,具体地,反应完全(HPLC监控原料<5%)的时间约为10~15h。
如下为具体的实施例,如无特别说明,实施例中采用的原料均为市售获得。
实施例1
实施例中的(S)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸和(R)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸合成路线汇总如下:

合成化合物2
室温下,将DMSO(40kg)加到反应釜中搅拌,再加入化合物1(9.9kg,,1.0eq),加入乙基亚磺酸钠(10.2kg,1.1eq)搅拌均匀直至无块状物,升温至95±5℃,反应液混浊呈淡黄色,保温搅拌12h,TLC监控反应完全。将反应液冷却至室温,缓慢加入水(120kg),搅拌2h,过滤,滤饼用80kg水洗涤两次,滤饼于65℃鼓风烘干,得到化合物2,淡黄色粉末状固体(14.9kg,94%)。
1H-NMR(500MHz,CDCl3):δ10.121(s,1H),8.069(s,4H),3.171-3.127(m,2H),1.296-1.266(t,3H),图谱见图1。
合成化合物3
将无水乙醇(104kg)加入到反应釜中搅拌,再加入化合物2(14.9kg,1.0eq)、丙二酸(10.2kg,1.3eq)、醋酸铵(11.6kg,2eq),加热升温至回流搅拌12h。HPLC监控反应完全。降温冷却至室温,过滤,滤饼用无水乙醇14kg乙醇洗涤两次,滤饼于50℃鼓风烘干,得到化合物3(14.4kg,75%),白色粉末状固体。所得粗产品不经纯化直接用于下一步反应。
合成化合物4
将化合物3(14.4kg,1eq)和甲醇(72kg)加入到反应釜中,氮气保护下降温至10℃左右,缓慢滴加氯化亚砜(10.0kg,1.5eq),控制内温<20℃,体系由白色悬浊液逐渐变成浅淡黄色体系,滴加完毕后自然回温至室温,搅拌12h,HPLC监控原料<5%(图谱见图2)。于35℃下减压浓缩掉绝大部分甲醇,再加入乙酸乙酯(30kg)常温打浆1h,过滤,滤饼用乙酸乙酯(7.2kg)洗涤,滤饼用30%氨水(7.2kg)和水(17.3kg)进行溶解,再加入二氯甲烷(50.4kg)萃取,干燥后于35℃下减压浓缩得到白色固体化合物4,直接用于下一步拆分操作。
其中,HPLC监控采用的仪器及条件设置如下:
设备厂家:岛津LC-2030C;
柱子型号:CHIRALPAK AY-H;
流动相构成比例:正己烷:乙醇=1:1(体积比),再加入总流动相体积0.01%三乙胺;
时长:35分钟。
合成化合物5-A
将化合物4(7.5kg,1.0eq)、甲醇(2.25kg)、水(5.25kg)加入到反应釜中搅拌,加热升温至55~60℃溶清,再加入L-天冬氨酸(2.96kg,0.8eq),搅拌1h,体系为乳黄白色,再滴加甲醇(20.2kg),滴加完毕后自然降温至室温搅拌12h。过滤,滤饼用甲醇(3.5kg)洗涤至滤液在紫外下无荧光,HPLC检测旋光纯度使ee值≥98%(图谱见图3),得到白色粉末状固化合物5-A(4.6kg,37%(理论收率50%))。
MS(ESI,pos)m/z:271.8[m+1]。
1H-NMR(500MHz,CDCl3):δ7.866-7.849(d,2H),7.611-7.594(d,2H),4.536-4.509(m,1H),3.680(s,3H),3.134-3.089(m,2H),2.690-2.672(m,2H),1.278-1.248(t,3H),图谱见图4。
其中,HPLC检测采用的仪器及条件设置如下:
设备厂家:岛津LC-2030C;
柱子型号:CHIRALPAK AY-H;
流动相构成比例:正己烷:乙醇=1:1(体积比),再加入总流动相体积0.01%三乙胺;
时长:35分钟。
合成化合物5-B
将化合物4(5.0kg,1.0eq)、甲醇(1.5kg)、水(3.5kg)加入到反应釜中搅拌,加热升温至55~60℃溶清,再加入D-天冬氨酸(1.96kg,0.8eq),搅拌1h,体系为乳黄白色,再滴加甲醇(13.5kg),滴加完毕后自然降温至室温搅拌12h。过滤,滤饼用甲醇(3kg)次洗涤至滤液在紫外下无荧光,HPLC检测旋光纯度使ee值≥98%(图谱见图5),得到白色粉末状固化合物5-B(3.3kg,35%(理论收率50%))。
MS(ESI,pos)m/z:271.8[m+1]。
1H-NMR(500MHz,CDCl3):δ7.853-7.836(d,2H),7.573-7.557(d,2H),4.513-4.486(m,1H),3.663(s,3H),3.103-3.059(m,2H),2.655-2.639(m,2H),1.262-1.232(t,3H),图谱见图6。
其中,HPLC检测采用的仪器及条件设置如下:
设备厂家:岛津LC-2030C;
柱子型号:CHIRALPAK AY-H;
流动相构成比例:正己烷:乙醇=1:1(体积比),再加入总流动相体积0.01%三乙胺;
时长:35分钟。
化合物6-A
将化合物5-A按游离碱(2.8kg,1.0eq)、甲醇(11.2L)、水(5.4L)加入到反应釜中,再加入三乙胺(2.3kg,2.2eq),溶清,测pH>8。降温至内温10~15℃,再缓慢加入二碳酸二叔丁酯固体(5.04kg,2.2eq),控制内温<25℃,加完后常温搅拌12h,TLC监控原料反应完全。向反应釜中缓慢加入氨水(15kg,3.0eq 30%氨水+15L水),体系由白色混浊变成澄清再变成大量固体析出,加完后再搅拌1h,过滤,滤饼用水(2V×2次)洗涤干净,得到白色固体化合物6-A(1.76kg,96.2%)。
1H-NMR(500MHz,CDCl3):δ7.866-7.850(d,2H),7.508-7.491(d,2H),5.715(s,1H),5.157(s,1H),3.618(s,3H),3.115-3.070(m,2H),2.859-2.849(m,2H),1.417(s,9H),1.300-1.253(t,3H)。
化合物6-B
将化合物5-B按游离碱(4.6kg,1.0eq)、甲醇(18.4L)、水(9.2L)加入到反应釜中,再加入三乙胺(3.7kg,2.2eq),溶清,测pH>8。降温至内温10~15℃,再缓慢加入二碳酸二叔丁酯固体(6.7kg,2.2eq),控制内温<25℃,加完后常温搅拌12h,TLC监控原料反应完全。向反应釜中缓慢加入氨水(15kg,3.0eq 30%氨水+水15L),体系由白色混浊变成澄清再变成大量固体析出,加完后再搅拌1h,过滤,滤饼用水(2V×2次)洗涤干净,得到白色固体化合物6-B(2.5kg,96.0%)。
1H-NMR(500MHz,CDCl3):δ7.850-7.833(d,2H),7.495-7.478(d,2H),5.712(s,1H),5.143(s,1H),3.602(s,3H),3.100-3.056(m,2H),2.844-2.835(m,2H),1.401(s,9H),1.300-1.253(t,3H)。
化合物7-A
向反应釜中加入化合物6-A按上一步的理论值算(2.5kg,1.0eq)、甲醇(5L)、水(5L);将预先配置好的氢氧化钠溶液(341g,2.0eqNaOH+水2V)缓慢加入到反应釜中,控制内温<50℃,滴加完毕后,测pH≥13,搅拌2h体系澄清,再测pH≥13,降温至内温≤10℃,缓慢滴加2N的盐酸溶液,控制内温<20℃,调节pH为4,体系变浑浊,加入DCM(9V),再加入饱和氯化钠溶液(2V)干燥,分液取有机相再加入无水硫酸钠(1.0w)干燥,过滤,滤饼用DCM(0.5V×2次)洗涤,蒸干滤液得到化合物7-A(2.2kg,92.0%),HPLC检测旋光纯度使ee值≥98%(图谱见图7)。
MS(ESI,neg)m/z:356.1[m-1]。
1H-NMR(500MHz,CDCl3):δ9.311(s,1H),7.860-7.844(d,2H),7.512-7.496(d,2H),5.734(s,1H),5.283-5.004(m,1H),3.126-3.082(m,2H),2.869(s,2H),1.403(s,9H),1.273-1.243(m,3H),图谱见图8。
其中,HPLC检测采用的仪器及条件设置如下:
设备厂家:岛津LC-2030C;
柱子型号:CHIRALPAK AY-H;
流动相构成比例:正己烷:乙醇=1:1(体积比),再加入总流动相体积0.01%三乙胺;
时长:35分钟。
化合物7-B
向反应釜中加入化合物6-B按上一步的理论值算(1.76kg,1.0eq)、甲醇(3.5L)、水(3.5L);将预先配置好的氢氧化钠溶液(200g,2.0eqNaOH+水2V)缓慢加入到反应釜中,控制内温<50℃,滴加完毕后,测pH≥13,搅拌2h体系澄清,再测pH≥13,降温至内温≤10℃,缓慢滴加2N的盐酸溶液,控制内温<20℃,调节pH为4,体系变浑浊,加入DCM(9V),再加入饱和氯化钠溶液(2V)干燥,分液取有机相再加入无水硫酸钠(1.0w)干燥,过滤,滤饼用DCM(0.5V×2次)洗涤,蒸干滤液得到化合物7-B(1.63kg,96.0%),HPLC检测旋光纯度使ee值≥98%(图谱见图9)。
MS(ESI,neg)m/z:356.1[m-1]。
1H-NMR(500MHz,CDCl3):δ7.858-7.842(d,2H),7.514-7.497(d,2H),5.750(s,1H),5.166-54.991(m,1H),3.125-3.081(m,2H),2.861(s,2H),1.403(s,9H),1.273-1.243(m,3H),图谱见图10。
其中,HPLC检测采用的仪器及条件设置如下:
设备厂家:岛津LC-2030C;
柱子型号:CHIRALPAK AY-H;
流动相构成比例:正己烷:乙醇=1:1(体积比),再加入总流动相体积0.01%三乙胺;
时长:35分钟。
对比实验:
分别取10g(小试工艺)同一批次制备的化合物4,以D(L)-酒石酸,D(L)-扁桃酸、D(L)-樟脑磺酸或D(L)-天冬氨酸,按照同实施例1的合成步骤进行拆分,即合成化合物5A和5B,结果如下:
(1)D(L)-扁桃酸:在合成化合物5-A和5-B的步骤中,D(L)-扁桃酸无法实现化合物5-A和5-B的拆分;
(2)D(L)-酒石酸:在合成化合物5-A和5-B的步骤中,D(L)-酒石酸对化合物5-A和5-B有一定的拆分效果,但需要对结晶盐进行三到四次重结晶才能将不同异构体分离干净(即达到HPLC检测旋光纯度使ee值≥98%),导致最终拆分收率很低,拆分收率26%左右(理论值:50%),成本较高;
(3)D(L)-樟脑磺酸:在合成(即化学拆分)化合物5-A和5-B的步骤中,D(L)-樟脑磺酸对化合物5-A和5-B有一定的拆分效果,但需要对结晶盐进行三到四次重结晶才能将不同异构体分离干净(即达到HPLC检测旋光纯度使ee值≥98%),导致最终拆分收率很低,拆分收率14%左右(理论值:50%),成本较高;
(4)D(L)-天冬氨酸:在合成化合物5-A和5-B的步骤中,使用D(L)-天冬氨酸进行拆分,对第一次结晶盐进行简单的甲醇冲洗就能将异构体冲洗干净,达到HPLC检测旋光纯度ee值≥98%,避免了多次重结晶,拆分收率43%左右(理论值:50%)。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
Claims (12)
1.一种β-苯并氨基酸类化合物的制备方法,其特征在于,包括如下步骤:
(1)获取式(I)所示消旋体化合物;
(2)将式(I)所示消旋体化合物与L-天冬氨酸进行反应,得式(I-1)所示化合物;和/或,将式(I)所示消旋体化合物与D-天冬氨酸进行反应,得式(I-2)所示化合物;
(3)于式(I-1)所示化合物和/或式(I-2)所示化合物中的胺基上引入保护基团,然后进行脱酯基反应;

R1选自磺酰烷基或磺酰环烷基;
R2选自C1~C10烷基;
步骤(2)中,所述反应采用的溶剂为重量比为1:2~3的甲醇和水的混合液。
2.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,步骤(2)中,式(I)所示消旋体化合物与L-天冬氨酸的摩尔比为1:0.5~1;式(I)所示消旋体化合物与D-天冬氨酸的摩尔比为1:0.5~1。
3.根据权利要求2所述的β-苯并氨基酸类化合物的制备方法,其特征在于,步骤(2)中,式(I)所示消旋体化合物与L-天冬氨酸的摩尔比为1:0.7~0.9;式(I)所示消旋体化合物与D-天冬氨酸的摩尔比为1:0.7~0.9。
4.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,步骤(2)中,所述反应的条件包括:于水和适量甲醇中混合式(I)所示消旋体化合物与L-天冬氨酸或D-天冬氨酸,升温至55~60℃溶清,然后保持温度反应0.8~1.2h;再加入甲醇,自然降温至室温,继续反应10~14h。
5.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述保护基团为叔丁氧羰基、甲氧羰基、乙氧羰基或异丙氧羰基。
6.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,R1选自磺酰甲基、磺酰乙基、磺酰丙基、磺酰丁基、磺酰环丙基、磺酰环戊基、磺酰环己基;
R2选自甲基、乙基、异丙基、叔丁基。
7.根据权利要求1~6任一项所述的β-苯并氨基酸类化合物的制备方法,其特征在于,获取式(I)所示消旋体化合物的方法,包括如下步骤:

将化合物a进行取代反应,引入R1,得化合物2;X代表卤素;
将化合物b与丙二酸、醋酸铵进行反应,得化合物3;
将化合物c进行酯化反应,引入R2。
8.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述β-苯并氨基酸类化合物为(R)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:

将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与D-天冬氨酸进行反应,得化合物5-A;
于化合物5-A中的胺基上引入叔丁氧羰基保护基团,得化合物6-A;
将化合物6-A进行脱酯基反应。
9.根据权利要求1所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述β-苯并氨基酸类化合物为(S)-3-((叔丁氧羰基)氨基)-3-(4-(乙基磺基)苯基)丙酸;所述制备方法包括如下步骤:
将化合物1与乙基亚磺酸钠进行反应,得化合物2;
将化合物2与丙二酸、醋酸铵进行反应,得化合物3;
将化合物3与氯化亚砜、甲醇进行反应,得化合物4;
将化合物4与L-天冬氨酸进行反应,得化合物5-B;
于化合物5-B中的胺基上引入叔丁氧羰基保护基团,得化合物6-B;
将化合物6-B进行脱酯基反应。
10.根据权利要求8或9所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述化合物2制备步骤中,所述反应的条件包括:于90~100℃反应。
11.根据权利要求8或9所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述化合物3制备步骤中,所述反应的条件包括:于回流条件下反应。
12.根据权利要求8或9所述的β-苯并氨基酸类化合物的制备方法,其特征在于,所述化合物4制备步骤中,所述反应的条件包括:先将所述化合物3和甲醇混合,控制所得混合液的温度小于20℃条件下,加入所述氯化亚砜,加完后自然升温至室温反应。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010274523.0A CN111333552B (zh) | 2020-04-09 | 2020-04-09 | β-苯并氨基酸类化合物的合成方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010274523.0A CN111333552B (zh) | 2020-04-09 | 2020-04-09 | β-苯并氨基酸类化合物的合成方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111333552A CN111333552A (zh) | 2020-06-26 |
| CN111333552B true CN111333552B (zh) | 2021-07-20 |
Family
ID=71178832
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010274523.0A Active CN111333552B (zh) | 2020-04-09 | 2020-04-09 | β-苯并氨基酸类化合物的合成方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111333552B (zh) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018123918A1 (ja) * | 2016-12-26 | 2018-07-05 | 科研製薬株式会社 | ピラゾール誘導体及びそれを含有する医薬 |
| CN108409589A (zh) * | 2018-03-26 | 2018-08-17 | 爱斯特(成都)生物制药股份有限公司 | 一种带手性的β-氨基酸酯的制备方法 |
| CN109071509A (zh) * | 2016-01-29 | 2018-12-21 | 生命医药公司 | 作为ROR-γ的调节剂的苯并咪唑衍生物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9845308B2 (en) * | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| US9663516B2 (en) * | 2014-12-18 | 2017-05-30 | MyoKardia, Inc. | Bicyclic-pyrimidinedione compounds |
-
2020
- 2020-04-09 CN CN202010274523.0A patent/CN111333552B/zh active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109071509A (zh) * | 2016-01-29 | 2018-12-21 | 生命医药公司 | 作为ROR-γ的调节剂的苯并咪唑衍生物 |
| WO2018123918A1 (ja) * | 2016-12-26 | 2018-07-05 | 科研製薬株式会社 | ピラゾール誘導体及びそれを含有する医薬 |
| CN108409589A (zh) * | 2018-03-26 | 2018-08-17 | 爱斯特(成都)生物制药股份有限公司 | 一种带手性的β-氨基酸酯的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111333552A (zh) | 2020-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008062460A2 (en) | Crystalline forms of pregabalin | |
| JP5646509B2 (ja) | (1s,2r)−ミルナシプランの合成方法 | |
| JP6307087B2 (ja) | ベンズアミド化合物の合成に有用な化合物 | |
| KR20050113292A (ko) | 광학적으로 순수한 4-하이드록시-2-옥소-1-피롤리딘아세트아미드의 제조방법 | |
| CN110590635A (zh) | 左乙拉西坦及其中间体的制备方法 | |
| JP7097467B2 (ja) | ブリバラセタム中間体、その製造方法及びブリバラセタムの製造方法 | |
| EP2067768A1 (en) | A process for the preparation of (S)(+)-3-(aminomethyl)-5-methylhexanoic acid | |
| CN117886708A (zh) | 一种苯磺酸米洛巴林的合成方法 | |
| US9771317B2 (en) | Process for preparing lacosamide and related compounds | |
| JPH06263699A (ja) | L−5−(2−アセトキシ−プロピオニルアミノ)−2,4,6−トリヨード−イソフタル酸ジクロライドの製造方法 | |
| CN102267961B (zh) | 制备(3r,4r,5s)-4,5-环氧基-3-(1-乙基丙氧基)-1-环己烯-1-甲酸乙酯的方法 | |
| CN111333552B (zh) | β-苯并氨基酸类化合物的合成方法 | |
| TW201718462A (zh) | 用於製備有機化合物及中間物的方法 | |
| CA2894826C (en) | Asymmetric synthesis of a substituted pyrrolidine-2-carboxamide | |
| CN112272665A (zh) | 制备立他司特的方法 | |
| JP4190879B2 (ja) | テアニンを製造するための新規な中間体 | |
| JP2008115178A (ja) | ジフェニルアラニン−Ni(II)錯体の製造方法 | |
| JP2007510695A (ja) | ガバペンチンの調製方法 | |
| JP2010513531A (ja) | 1−アミノ、3−置換フェニルシクロペンタンカルボン酸エステルの個々の立体異性体の製造および単離方法 | |
| JP2010513531A5 (zh) | ||
| EP2805936A1 (en) | Process for preparing levomilnacipran HCL | |
| JP5344523B2 (ja) | 立体選択的にストレッカー反応を進行させ得る触媒、およびそれを用いたα−アミノニトリル誘導体を立体選択的に製造するための方法 | |
| CN112341413A (zh) | 一种用于合成布瓦西坦的中间体及其制备方法 | |
| CN114195684B (zh) | 一种氨基保护基n-取代手性氨基酸的合成方法 | |
| CA2377801A1 (en) | Process for preparing (-)-(1s,4r) n-protected 4-amino-2-cyclopentene-1-carboxylate esters |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |