CN107743489B - 治疗免疫疾病、炎性疾病或癌症的7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物 - Google Patents
治疗免疫疾病、炎性疾病或癌症的7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物 Download PDFInfo
- Publication number
- CN107743489B CN107743489B CN201680017259.4A CN201680017259A CN107743489B CN 107743489 B CN107743489 B CN 107743489B CN 201680017259 A CN201680017259 A CN 201680017259A CN 107743489 B CN107743489 B CN 107743489B
- Authority
- CN
- China
- Prior art keywords
- methyl
- pyrazolo
- morpholin
- pyrimidine
- tert
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
通式(I)的化合物,其中Y表示‑CH2‑或‑CO‑;R1选自A1、A2和A3;R2表示二氧代硫代吗啉基部分B1、哌嗪基部分B2、氮杂环丁烷基部分B3或哌啶基部分B4;R3选自H、卤素和C1‑C4烷基;R4选自C1‑C4烷基、C3‑C4‑环烷基、被C1‑C4烷氧基取代的C1‑C4烷基,和CHF2;以及其药物可接受的盐。包含所述化合物的药物组合物,以及它们在治疗免疫系统疾病、炎性疾病和癌症中的用途。
Description
本发明涉及新颖的7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物、含有该衍生物的药物组合物、以及它们作为药物的用途。7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物表现出抑制PI3激酶活性的能力,并且发现可以用作药物,特别是用于治疗免疫系统疾病、炎性疾病和癌症的药物。
激酶的PI3家族负责以负责调节细胞存活、分化和迁移过程的重要信号途径之一进行的信号转导。这些激酶也负责调节免疫系统功能。 PI3K途径的异常激活已在肿瘤疾病(例如慢性淋巴细胞白血病或非霍奇金淋巴瘤)以及具有潜在的炎性过程的疾病(例如类风湿性关节炎或哮喘)中得到证实。
活性的激酶PI3由催化亚基和调节亚基组成。激酶的PI3K I型家族包括四种调节亚基:PI3K-α、PI3K-β、PI3K-γ和PI3K-δ。在所有组织中表达了同种型α和同种型β,并且它们在胚发育阶段的失活是致命的。同种型γ和同种型δ的表达受限于造血系(hematopoietic line);它们的失活是非致命的,但是导致发育生物体中出现免疫缺陷综合征。
脂质激酶PI3催化经由磷脂酰肌醇-3-磷酸酯(PIP)和磷脂酰肌醇 3,4-二磷酸酯中间体的磷脂酰肌醇磷酸化的磷脂酰肌醇(3,4,5)-三磷酸酯(PIP3)的产生。位于细胞膜的磷脂PIP3是AKT蛋白的结合位点,其磷酸化程度反映了PI3K途径的活性。
因此,PI3K的抑制是引人注目的治疗多种疾病的机制,其中细胞中AKT蛋白的磷酸化水平可发挥作用。
因为PI3Kα和PI3Kβ在广泛的组织中表达,以及它们在胚发育中的作用,非选择性的PI3K抑制剂可能具有受限的耐受性和高的毒性。
因此,需要表现出期望活性且引起不良副作用的能力受限的选择性PI3K抑制剂、特别是PI3Kδ抑制剂。
现有技术中公开了各种脂质激酶PI3抑制剂。
WO2010136491和WO2010138589公开了双环吲哚并嘧啶化合物,其表现出激酶PI3抑制活性,并且适用于治疗诸如炎性病症和免疫性病症以及癌症的疾病。
WO201110142公开了吗啉基取代的吡啶并[3,2-d]嘧啶化合物,其表现出激酶PI3抑制活性,并且适用于治疗诸如炎性病症和免疫性病症以及癌症的疾病。
本发明的目标是提供新颖的化合物,即在炎性和免疫性的疾病和病症以及癌症的治疗中具有潜在效用,表现出靶向特定PI3K同种型、特别是PI3Kδ的选择性活性的PI3K抑制剂。
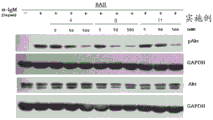
图1显示了本发明的实施例4和实施例5的化合物在不同浓度下对PI3Kδ激酶的抑制作用以及AKT蛋白质磷酸化水平。
图2显示了本发明的实施例4、8和11的化合物在不同浓度下对 PI3Kδ激酶的抑制作用以及AKT蛋白质磷酸化水平。
图3显示了本发明的实施例8和实施例50的化合物在不同浓度下对PI3Kδ激酶的抑制作用以及AKT蛋白质磷酸化水平。
图4显示了本发明的实施例8和实施例11的化合物在不同浓度下对PI3Kγ激酶的抑制作用以及AKT蛋白质磷酸化水平。
本发明涉及通式(I)的化合物

其中:
Y表示-CH2-或-CO-;
R1选自A1、A2和A3:

R2表示:
-二氧代硫代吗啉基部分B1;

-哌嗪基部分B2

其中哌嗪环的两个碳原子可以任选地通过亚甲基桥连接以形成 2,5-二氮杂双环部分,并且R5选自-SO2CH3;-C(O)-C1-C3-烷基;- C(O)-C3-C5-环烷基;被-O-C1-C3烷基取代的苯基;和-CR6R7R8,
其中R6、R7和R8独立地选自氢原子、CH3、环丙基和CONH2,条件是R6、R7和R8中的仅一个可以表示环丙基或CONH2,
或者R6、R7和R8之一表示氢原子,并且R6、R7和R8中的另外两个连接在一起形成-(CH2)2-、-(CH2)3-、-(CH2)4-或-(CH2-O-CH2)-;
-氮杂环丁烷基部分B3

其中R9选自吗啉基、2,6-二甲基吗啉基、1,1-二氧代硫代吗啉基、 4,4-二氟哌啶基和3-甲氧基氮杂环丁烷-1-基;或者
-哌啶基部分B4

其中R10选自C1-C4烷基;被OH取代的C1-C4烷基;-COO(C1-C3 烷基);-N(C1-C3烷基)2;-NHCONH-C1-C3-烷基;-NHCONH-C1-C3- 苯基;哌嗪基;和在4位被C1-C3烷基取代的哌嗪基;
R3选自H、卤素和C1-C4烷基;
R4选自C1-C4烷基、C3-C4-环烷基、被C1-C4烷氧基取代的C1-C4 烷基,和CHF2;
X1和X2具有以下含义:
(i)X1表示CH,并且X2表示CH或N;
(ii)X1表示N,并且X2表示CH,或者
(iii)X1表示CH,并且X2表示C-O-CH3;
X3、X4、X5和X6具有以下含义:
(i)X3表示N,X4表示CH,X5表示CH,并且X6表示 CH;
(ii)X3表示CH,X4表示N,X5表示CH,并且X6表示 CH;
(iii)X3表示CH,X4表示CH,X5表示N,并且X6表示 CH;
(iv)X3表示CH,X4表示CH,X5表示CH,并且X6表示 CH或CF;或者
(v)X3表示CH,X4表示CH,X5表示CF,并且X6表示 CH;
X7表示CH或N;
波浪线表示连接点;
以及其酸加成盐。
在一个实施方案中,本发明涉及式(I)的化合物,其中Y表示-CH2-。
在另一个实施方案中,本发明涉及式(I)的化合物,其中Y表示-CO-。
本发明化合物的一个组是式(I)的化合物,其中R3表示H。
本发明化合物的另一个组是式(I)的化合物,其中R3表示C1-C4 烷基。有利地,R3表示甲基。
本发明化合物的另一个组是式(I)的化合物,其中R3表示卤素原子。卤素原子包含氯原子、溴原子和氟原子,并且有利地表示氯原子。
在本发明化合物的另一个实施方案中,R2表示B1。
在本发明化合物的另一个实施方案中,R2表示B2。
本发明化合物的另一个亚组是式(I)的化合物,其中B2中的R5表示-CR6R7R8,并且R8,R6、R7和R8独立地选自氢原子和CH3,即R5表示选自CH3、CH2CH3、CH(CH3)2和C(CH3)3的C1-C4烷基。有利地, R5表示C(CH3)3(叔丁基)。
在本发明化合物的另一个实施方案中,R2表示B3。
在本发明化合物的另一个实施方案中,R2表示B4。
在本发明化合物的一个变化中,R1表示A1或A2。
在另一实施方案中,R1表示A1,其中X1表示CH并且X2表示 CH或N。
在该变体的一个亚组中,R1表示A1,其中X1表示CH,并且X2表示CH或N,并且R2表示B2或B4。特别地,X1表示CH并且X2表示CH。在所述亚组的一个实施方案中,Y表示-CH2-。有利地,B2 部分中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4部分中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A1,其中X1表示CH并且X2表示CH或N,并且R2表示B3并且Y表示-CH2-。特别地X1表示CH 并且X2表示CH。
在另一个实施方案中,R1表示A1,其中X1表示N并且X2表示 CH,并且R2表示B2或B4。在该实施方案的亚组中,Y表示-CH2-。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4 中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在另一个实施方案中,R1表示A1,其中X1表示CH并且X2表示 C-O-CH3。在该实施方案的亚组中,Y表示-CH2-。有利地,B2中的 R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
有利地,R4表示C1-C4烷基,包括甲基、乙基、正丙基、异丙基、正丁基、异丁基和叔丁基,特别是甲基、乙基或异丙基。还优选地, R4表示环丙基或环丁基。最优选地,R4为CHF2。
在本发明化合物的另一变体中,R1表示A2。
在该变体的亚组中,R1表示A2,其中X3表示N,X4表示CH, X5表示CH,并且X6表示CH,并且R2表示B2或B4。在该亚组的实施方案中,Y表示-CH2-。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A2,其中X3表示CH,X4表示N,X5表示CH,并且X6表示CH,并且R2表示B2或B4。在该亚组的实施方案中,Y表示-CH2-。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A2,其中X3表示CH,X4表示CH,X5表示N,并且X6表示CH,并且R2表示B2或B4。在该亚组的实施方案中,Y表示-CH2-。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A2,其中X3表示CH,X4表示CH,X5表示CH,并且X6表示CH或CF,特别是CH,并且R2表示B2或B4。在该亚组的实施方案中,Y表示-CH2-。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A2,其中X3表示CH,X4表示CH,X5表示CH,并且X6表示CH或CF,特别是CH,并且R2表示B2或B4。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在该变体的另一个亚组中,R1表示A2,其中X3表示CH,X4表示CH,X5表示CF,并且X6表示CH,并且R2表示B2或B4。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的 R10表示被OH取代的C1-C4烷基,特别是2-羟基丙-2-基。
在本发明化合物的另一变体中,R1表示A3部分,并且R2表示 B2或B4。有利地,B2中的R5表示C1-C4烷基,特别是叔丁基。还优选地,B4中的R10表示被OH取代的C1-C4烷基,特别是2-羟基丙 -2-基。
在本发明化合物的一个亚组中,R1表示A1或A2,并且R2表示 B2或B4。
有利地,B2中的R5表示叔丁基。
有利地,B4中的R10表示2-羟基丙-2-基。
除非另外明确限定,本文使用的术语C1-C4烷基包含甲基、乙基、正丙基、异丙基、正丁基、仲丁基和叔丁基。术语C1-C3烷基包含甲基、乙基、正丙基和异丙基。
术语C1-C4烷氧基包含甲氧基、乙氧基、正丙氧基、正丁氧基、仲丁氧基和叔丁氧基。
术语C3-C4-环烷基包含环丙基和环丁基,并且术语C3-C5-环烷基包含环丙基、环丁基和环戊基。
本发明的式(I)的化合物表现出PI3激酶抑制能力,并且发现可以用作药物,特别是用于治疗免疫系统疾病和病症、炎性疾病和病症、以及癌症。本发明化合物的具体特性是激酶PI3K delta(PI3Kδ)抑制与该激酶的其他同种型相比的高选择性。这允许期望相比作用于多种 PI3K同种型的广谱抑制剂的毒性降低。
因此本发明的另一方面是用作药物的如上文限定的式(I)的化合物。
因此本发明的另一方面是如上文限定的式(I)的化合物用于制备治疗免疫系统疾病和病症、炎性疾病和病症以及癌症的药物的用途。
本发明的另一个方面是包含如上文限定的式(I)的化合物和药物可接受的赋形剂的药物组合物。
本发明的另一个方面是治疗免疫系统疾病和病症、炎性疾病和病症、以及癌症的方法,其包括施用有效量的如上文限定的式(I)的化合物。
本发明的式(I)的化合物的酸加成盐特别包括与药物可接受的无机酸或有机酸形成的盐。优选药物可接受的酸加成盐。能够形成酸加成盐(包括与具有碱性氮原子的化合物形成的药物可接受的盐)的无机酸和有机酸,并且所述盐的制备方法是本领域熟知的。与无机酸形成的盐可以特别地包括盐酸、氢溴酸、硫酸和磷酸的盐。与有机酸形成的盐可以特别地包括甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸、萘磺酸、乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸的盐。应理解,本发明的范围也包括与不同于药物可接受的酸的其他酸形成的、但可以特别地用作本发明化合物的制备、分离和纯化的中间体的盐。
本发明的具体化合物选自在下文实施例中给出的化合物和它们的酸加成盐,特别是药物可接受的酸加成盐,包括无机酸加成盐和有机酸加成盐。
式(I)的化合物可以以在下文的本发明中间体和化合物的制备实施例中一般描述和具体描述以及在方案1、2和3中概述的方式制备。
方案1

方案2

方案3

如上述方案1所示,其中R3表示氢原子或C1-C4烷基并且其他符号具有上文所限定含义的式(I)的化合物,可以由其中Z分别表示氢原子或OH并且其他符号具有对于式(I)所限定的含义的式(II)的醛或酸获得。

其中Z表示氢原子的式(II)的化合物以还原胺化反应与式R2H的胺发生反应,以得到其中Y表示CH2并且R3表示氢原子或C1-C4烷基的式(I)的化合物。
与胺R2H的还原胺化的反应可以在以下条件中进行:温度为0℃至反应混合物中最低沸点组分的沸点,于诸如氯仿、二氯甲烷、二氧六环、二甲基甲酰胺、二甲基乙酰胺、甲苯、己烷、四氢呋喃或二甲氧基乙烷的有机溶剂中进行反应。还原剂可以为,例如,硼氢化钠、三乙酰氧基硼氢化钠或氰基氢化钠(sodium cyanohydride)。最适合的条件为,使用二氯甲烷作为溶剂,使用三乙酰氧基硼氢化钠作为还原剂,并且在室温下进行反应。
其中Z表示OH的式(II)的化合物以酰胺化反应与式R2H的胺发生反应,以得到其中Y表示-CO-并且R3表示氢原子或C1-C4烷基的式(I)的化合物。
与胺R2H的酰胺化反应可以在以下条件中进行:温度为0℃至反应混合物中最低沸点组分的沸点(回流温度),于诸如氯仿、二氯甲烷、二氧六环、二甲基甲酰胺、二甲基乙酰胺、甲苯、己烷、四氢呋喃、二甲氧基乙烷、二甲基亚砜、乙酸乙酯、乙腈或乙醚的有机溶剂中进行反应。在存在碳二亚胺、添加剂和叔胺的情况下进行反应,所述碳二亚胺例如N,N’-二异丙基碳二亚胺、N,N’-二环己基碳二亚胺、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐或1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(HATU),所述添加剂例如1-羟基苯并三唑(HOBt)或N-羟基琥珀酰亚胺,叔胺例如三乙胺(TEA)或N,N-二异丙基乙胺(DIPEA)。进行酰胺化的最适合的条件是室温,于二甲基甲酰胺中,在1-乙基-3-(3-二甲基氨基丙基)碳二亚胺 (EDCI)盐酸盐、1-羟基苯并三唑(HOBt)添加剂和三乙胺(TEA)的存在下。
可以通过将其中R3表示氢原子的式(I)的化合物卤化来制备其中 R3表示卤素原子的式(I)的化合物。
卤化反应可以在以下条件中进行:使用分子卤素或N-卤代琥珀酰亚胺作为卤化试剂,温度为0℃至混合物中最低沸点组分的沸点,于诸如氯仿、二氯甲烷、二氧六环、二甲基甲酰胺、二甲基乙酰胺、甲苯、己烷、四氢呋喃、二甲氧基乙烷、二甲基亚砜、乙酸乙酯、乙腈或乙醚的有机溶剂中进行反应。进行该反应的最适合的条件是在30℃下于二氯甲烷溶剂中使用N-卤代琥珀酰亚胺。
由其中R3表示氢原子或C1-C4烷基并且Bn表示苄基的式(III)的化合物来制备式(II)的化合物,

如方案2概述。
在制备其中R1表示A2或A3的式(II)的化合物的情况中,使式(III) 的化合物与硼酸R1B(OH)2或其酯(特别是环状酯,例如频哪醇酯)进行 Suzuki反应,以得到其中Bn为苄基保护基的式(IV)的化合物。

在Suzuki反应中,使用了钯催化剂,例如乙酸钯、四(三苯基膦基)钯(0)、双(三苯基膦基)氯化钯、三(二亚苄基丙酮)二钯(0)、1,1’-双(二苯基膦基)二茂铁二氯化钯(II)二氯甲烷加合物,使用了碱,例如磷酸钠、碳酸钠、碳酸钾、碳酸铯或乙酸钠,并且使用了有机溶剂,例如甲苯、己烷、四氢呋喃、二氧六环或1,2-二甲氧基乙烷,温度为0℃至混合物中最低沸点组分的沸点。进行该反应的最适合的条件是在回流温度下,四(三苯基膦基)钯(0)作为催化剂,2M碳酸钠水溶液作为碱,并且1,2-二甲氧基乙烷作为溶剂。
然后,通过去除保护羟基的苄基(Bn)将式(IV)的化合物脱保护,以得到式(V)的化合物。

苄基保护基的去除可以通过以下方式进行:在活性碳上的钯存在的情况下,在诸如甲醇、乙醇、二甲基甲酰胺、二氧六环、环己烷、甲苯或其混合物的有机溶剂中,在0℃至混合物中最低沸点组分的沸点的温度下,在1bar至100bar的氢气压力下,通过式(IV)的化合物的加氢来进行。进行该反应的最适合的条件是二甲基甲酰胺(DMF)和乙醇(EtOH)溶剂混合物,加入甲酸,在高温下(例如约60℃),于约1bar 的氢气压力下。
使用适合的氧化剂,通过氧化将由此得到的式(V)的化合物转化为式(II)的醛或酸。
式(V)的化合物可以通过以下方式转化为式(II)的醛:在0℃至混合物中最低沸点组分的沸点的温度下,于诸如二氯甲烷、乙腈、己烷、甲苯、二甲基甲酰胺(DMF)、二甲基乙酰胺、二甲基亚砜(DMSO)或二氧六环的溶剂中,通过使用Collins试剂、Dess-Martin试剂、重铬酸吡啶盐(PDC)、氯铬酸吡啶盐(PCC)、2-碘酰苯甲酸(IBX)、二氧化锰进行氧化,或者通过Swern反应进行氧化。进行该反应的最适合的条件是在室温下,于诸如DMF的溶剂中使用Dess-Martina试剂。
式(V)的化合物可以通过以下方式转化为式(II)的酸:在0℃至混合物中最低沸点组分的沸点的温度下,于诸如乙腈、水、甲苯、丙酮、二氧六环或四氢呋喃的溶剂中,通过使用Jones试剂、高锰酸钾、重铬酸吡啶盐(PDC)、四氧化钌、2,2,6,6-四甲基吡啶N-氧化物进行氧化。最适合的条件是在回流温度下,于水溶液中使用Jones试剂。
在制备其中R1表示A1的式(II)的化合物的情况中,首先使用式(VI) 的硝基化合物:

通过Buchwald-Hartwig反应将式(III)的化合物转化成为式(VII)的化合物

其中Bn表示苄基保护基。
在存在钯催化剂,例如乙酸钯、四(三苯基膦基)钯(0)、双(三苯基膦基)氯化钯、三(二亚苄基丙酮)二钯(0)、1,1’-双(二苯基膦基)二茂铁二氯化钯(II)二氯甲烷加合物;碱,例如磷酸钠、碳酸钠、碳酸钾、碳酸铯、乙酸钠、氢氧化钠、氢氧化钾、叔丁醇钠或叔丁醇钾;磷酸酯配体,例如三苯基膦、9,9-二甲基-4,5-双(二苯基膦)氧杂蒽、(2-联苯基) 二叔丁基膦、2-二环己基膦-2’,4’,6’-三异丙基联苯、(2-联苯基)二环己基膦;和有机溶剂,例如甲苯、己烷、四氢呋喃、二氧六环或1,2-二甲氧基乙烷的情况下,在0℃至混合物中最低沸点组分的沸点的温度下进行Buchwald-Hartwig反应。进行该反应的最适合的条件是,回流温度,存在三(二亚苄基丙酮)二钯(0)作为催化剂,9,9-二甲基-4,5-双(二苯基膦)氧杂蒽,碳酸铯,于作为溶剂的甲苯中。
然后,将式(VII)的化合物还原以得到式(VIII)的化合物

然后通过类似于针对由Z为羟基(-OH)的式(II)的化合物制备式(I) 的化合物所描述的方式,使式(VIII)的化合物进行与式R4COOH的酸的酰胺化反应,从而得到式(IX)的化合物。

通过在乙酸中加热回流,经由环化将式(IX)的化合物转化为式(X) 的化合物。

然后,通过如上文针对式(IV)的化合物中羟基的脱保护所描述的方式,将保护羟基的苄基从化合物(X)去除,从而得到式(XI)的化合物,

并且如上文针对式(V)的化合物的氧化所描述的,使用适合的氧化剂,通过氧化将由此获得的式(XI)的化合物转化为其中R1表示A1基团的式(II)的醛或酸。
式(III)的化合物可以如方案3所示制备。
根据用于类似结构的化合物的文献方法,研发了方案3中示出的中间体的制备方法。
根据方案3,与WO2011/109267所述类似,在强碱(例如氢化钠) 的存在下使苄醇与溴乙酸乙酯反应,得到2-苄氧基乙酸乙酯,然后与 WO2009/106539所述类似,在强碱(例如丁基锂)的存在下与其中R3表示氢原子或C1-C4烷基的式R3CH2CN的腈进行反应,将所述2-苄氧基乙酸乙酯转化为式(XII)的腈。与WO2009/106539所述类似,通过与肼的环化反应将式(XII)的腈转化为式(XIII)的化合物。在强碱(例如乙醇钠)的存在下通过与丙二酸二乙酯的环化反应使式(XIII)的化合物转化为式(XIV)的化合物,其通过与三氯氧磷(POCl3)的氯化进一步转化为式(XV)的化合物,然后式(XV)的化合物通过在碱(例如碳酸钠)的存在下与吗啉反应转化为式(III)的化合物。进行方案3所示反应的条件和方式在下文的制备中间体的描述中详细描述。
式R2H的中间体是可商购的已知化合物,或者可以使用已知方法通过下文的制备中间体的描述中详细示出的方式来获得。在大部分情况下,使用上文描述的含有羰基系统的化合物与适当的胺的还原胺化反应来制备中间体R2H。对于包含脲系统的化合物R2H,使用了伯胺与异氰酸酯衍生物的反应。可以在-80℃至混合物中最低沸点组分的沸腾温度的温度下,于诸如氯仿、二氯甲烷、二氧六环、二甲基甲酰胺、二甲基乙酰胺、甲苯、己烷、四氢呋喃、二甲氧基乙烷、二甲基亚砜、乙腈或乙醚的有机溶剂中进行该反应。
硼酸R1B(OH)2及其酯(例如频哪醇酯)是可商购的,或者可以使用 Miyaura反应或通过在碱或钯催化剂的存在下卤代衍生物与硼酸或其酯的反应来获得。
其中R3表示氢原子或C1-C4烷基的式R3CH2CN的腈类是可商购的已知化合物。
式(I)的化合物可以作为化合物或者以含有该化合物的药物组合物或药物制剂的形式用于本文所述的疾病和病症的治疗。通常,它们将作为含有本发明化合物或其药物可接受的盐并且结合药物可接受的载体和辅助物质的药物组合物或药物制剂来使用。
在上述病症、疾病和病况的治疗中,本发明的药物组合物可以通过任何适合的途径施用,优选口服、胃肠外或吸入途径,并且本发明的药物组合物的形式将会是根据预期施用途径而预制(destined)用于医疗的制剂。
用于口服施用的组合物可以具有固体或液体制剂的形式。固体制剂可以具有,例如,由药物可接受的非活性赋形剂通过常规方式生产的片剂或胶囊的形式,所述赋形剂例如粘合剂(例如,预凝胶化的玉米淀粉、聚乙烯吡咯烷酮或羟丙基甲基纤维素);填充剂(例如乳糖、蔗糖、羧甲基纤维素、微晶纤维素或磷酸氢钙);崩解剂(例如交联聚维酮、玉米淀粉或羟基乙酸淀粉钠);润滑剂(例如硬脂酸镁、滑石或二氧化硅);润湿剂(例如月桂基硫酸钠)。片剂可以涂布有本领域熟知的包衣,例如简单包衣、迟释/控释包衣或肠溶包衣。用于口服施用的液体制剂可以是例如,溶液、糖浆或悬浮液的形式,或者可以具有用于使用前在水或其他合适媒介物中重构的干燥固体产物的形式。这类液体制剂可以使用常规手段由药物可接受的非活性赋形剂来制备,所述赋形剂例如悬浮剂(例如山梨醇糖浆、纤维素衍生物或氢化的食用油);乳化剂(例如卵磷脂或阿拉伯胶);非水媒介物(例如扁桃油(mandelicoil)、油酯、乙醇或分馏的植物油);以及防腐剂(例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯或山梨酸)。制剂也可以包含适合的缓冲剂、调味剂、着色剂和甜味剂。
用于口服施用的制剂可以使用本领域技术人员已知的方法配制以使活性化合物受控释放。
胃肠外施用途径包括通过肌内注射和静脉内注射以及静脉输注的施用。用于胃肠外施用的组合物可以,例如,具有单位剂型的形式,例如添加有防腐剂的安瓶或多剂量容器。组合物可以具有诸如在油性或水性媒介物中的悬浮液、溶液或乳液的形式,并且可以包含诸如悬浮剂、稳定剂和/或分散剂的赋形剂。或者,活性成分可以配制为用于使用前在合适载体(例如无菌、无热源水)中重构的粉末。
用于经由吸入途径施用的组合物可以具有吸入形式,并且通过雾化施用。这类制剂包含作为气雾剂施用的活性化合物和辅助物质,所述气雾剂即悬浮在气体中的精细分散的固体或液体物质小颗粒的系统。用于雾化的辅助物质可以例如,作为等渗剂的氯化钠,作为pH调节剂和稳定剂的无机酸和氢氧化物,作为防腐剂的苯扎氯铵,作为缓冲剂的柠檬酸钠,作为表面活性剂的聚山梨醇酯80,作为共溶剂的乙醇和丙二醇,以及作为抗氧化剂的硫酸盐(VI)。通过吸入途径施用的制剂可以具有压力吸入器或干燥粉末吸入器的形式。
使用本发明化合物治疗的方法将包括向需要这类治疗的对象施用治疗有效量的本发明化合物,优选地以药物组合物的形式施用。
本发明化合物的建议剂量为每天0.1mg至约1000mg,以单剂量或多剂量。对于本领域技术人员将显而易见的是,选择获得期望生物效果所需的剂量将取决于许多因素,例如具体化合物、适应证、施用方式、患者的年龄和身体状况,并且精确剂量将由负责医生最终确定。
实施例
中间体的制备
2-苄氧基乙酸乙酯
在30分钟内,向21.8g(0.545mol)60%NaH在1000ml干燥甲苯中的悬浮液逐滴添加47ml(0.454mol)苄醇。将全部混合物在室温下搅拌4h。将悬浮液在水-冰浴中冷却,并且在45分钟内逐滴添加66ml (0.595mol)溴乙酸乙酯。将反应混合物加热至室温,并搅拌1h。将全部混合物倒入用10ml浓盐酸酸化的冰水(1200ml)。进行相分离,并且用3×乙醚萃取水相。将合并的有机相用卤水洗涤,并且经无水硫酸镁干燥。将干燥剂过滤后,在减压下蒸发有机溶剂。通过在减压下蒸馏分离残余物。蒸馏后,得到66.7g(76%)2-苄氧基乙酸乙酯,为无色液体(Tb=104-106℃/0.7tor)。1H NMR(500MHz,CDCl3)δ:7.39 -7.28(m;5H),4.63(s;2H),4.23(q;J=7.1Hz;2H),4.09(s;2H),1.28(t; J=7.1Hz;3H).MS-ESI:(m/z)计算值,对于C11H14O3[M+H]+:195.23;测定值195.1。
中间体XII-1:4-苄氧基-3-氧代丁腈
向具有氩气的(argonated)烧瓶填充750ml冷却至-78℃的干燥 THF,添加200ml(0.5mol)2.5M n-BuLi己烷溶液,然后逐滴添加28ml (0.533mol)乙腈。将全部混合物在-78℃下搅拌2h。向悬浮液逐滴添加77.7g(0.4mol)上文获得的2-苄氧基乙酸乙酯,并且在-78℃下继续搅拌1h。通过添加饱和氯化铵溶液淬灭反应。将混合物倒入冰水,并且用6M盐酸酸化。将水相用乙醚萃取。将合并的有机相用卤水洗涤,并且经无水硫酸镁干燥。将干燥剂过滤,并且在减压下蒸发溶剂。中间体XII-1不经过纯化用于下一步骤。MS-ESI:(m/z)计算值,对于 C11H11NO2[M+H]+:190.22;测定值190.1。
中间体XII-2:4-苄氧基-2-甲基-3-氧代丁腈。
由2-苄氧基乙酸乙酯和丙腈获得,与中间体XII-1类似。
中间体XIII-1:3-(苄氧基甲基)-1H-吡唑-5-胺。
向上文获得的中间体XII-1(约75.7g,0.4mol)添加500ml乙醇和 100ml(2.1mol)一水合肼。将混合物回流16h。在减压下将溶剂蒸发至干燥后,向残余物添加氯仿,并且经无水硫酸钠干燥。过滤干燥剂,并且蒸发溶剂,并且在使用100/0至95/5的乙酸乙酯/甲醇作为洗脱液的色谱柱上分离混合物。分离后,得到70.4g(87%)中间体XIII-1,为棕色油状物。1HNMR(300MHz;CDCl3)δ:7.39-7.28(m;5H);5.59(s; 1H);4.53(s;2H);4.50(s;2H)。MS-ESI:(m/z)计算值,对于C11H13N3O [M+H]+:204.25;测定值204.1。
中间体XIII-2:3-(苄氧基甲基)-4-甲基-1H-吡唑-5-胺。
由中间体XII-2获得,与对于中间体XIII-1的描述类似。
中间体XIV-1:2-(苄氧基甲基)吡唑并[1,5-a]嘧啶-5,7-二醇。
向具有由53g(0.74mol)乙醇钠和700ml乙醇得到的乙醇钠溶液的烧瓶,添加溶解于200ml乙醇的70.4g(0.35mol)中间体XIII-1和 80ml(0.53mol)丙二酸二乙酯。将反应在回流下进行24h。将反应混合物冷却至室温,然后在减压下蒸发溶剂。将残余物溶解于1200ml 水中,并且用浓盐酸酸化至pH约2。将从溶液沉淀的奶油色固体过滤、洗涤并干燥。得到79g(84%)中间体XIV-1,为奶油色固体。MS-ESI:(m/z)计算值,对于C14H13N3O3[M+Na]+:294.26;测定值294.1。
中间体XIV-2:2-(苄氧基甲基)-3-甲基吡唑并[1,5-a]嘧啶-5,7-二醇
由中间体XIII-2获得,与中间体XIV-1类似。
中间体XV-1:2-(苄氧基甲基)-5,7-三氯吡唑并[1,5-a]嘧啶
将30g(0.11mol)中间体XIV-1在270ml乙腈中的悬浮液在水- 冰浴中冷却,并且添加206ml(2.2mol)POCl3。将反应在80℃下进行 5h。将反应混合物在蒸发器中浓缩以去除乙腈和POCl3。将残余物倒入含有冰的水,并且用饱和碳酸氢钠溶液碱化至pH 5。将水相用乙酸乙酯萃取,并且分离后将有机相经无水硫酸钠干燥。将干燥剂过滤并将溶剂蒸发后,通过使用100/0至80/20的庚烷/乙酸乙酯作为洗脱液的柱色谱来纯化残余物。分离后,得到13g(38%)中间体XV-1,为淡黄色油状物。1H NMR(300MHz,CDCl3)δ:7.41-7.27(m;5H);6.96(s;1H);6.80(s;1H);4.81(s;2H);4.65(s;2H)。MS-ESI:(m/z)计算值,对于C14H11Cl2N3O[M+H]+:309.17;测定值308.0。
中间体XV-2:2-(苄氧基甲基)-5,7-二氯-3-甲基吡唑并[1,5-a]嘧啶
由中间体XIV-2获得,与对于中间体XV-1描述的工序类似。
中间体III-1:2-(苄氧基甲基)-5-氯-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶
向13g(42.3mmol)中间体XV-1溶解于450ml丙酮中的溶液添加5.38g(50.8mmol)碳酸钠和6.65ml(76.2mmol)吗啉。将反应在室温下进行1.5h。将500ml水添加至反应混合物,并且将沉淀的白色固体过滤。将固体用水和200ml水/丙酮混合物(2/1)洗涤,然后干燥。得到 14g(92%)中间体III-1,为白色固体。1H NMR(300MHz,CDCl3)δ:7.41 -7.27(m;5H);6.56(s;1H);6.06(s;1H);4.73(s;2H);4.62(s;2H);3.98 -3.90(m;4H);3.82-3.74(m;4H)。MS-ESI:(m/z)计算值,对于 C18H19ClN4O2[M+H]+:359.83;测定值359.2。
中间体III-2:2-(苄氧基甲基)-5-氯-3-甲基-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶
由中间体XV-2获得,与对于中间体III-1描述的工序类似。
中间体IV-1:2-(苄氧基甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-
a]嘧啶

向1.88g(5.24mmol)中间体III-1溶解于52ml的1,2-二甲氧基乙烷(DME)中的溶液添加1.97g(7.87mmol)吲哚-4-硼酸频哪醇酯、0.61g (0.52mmol)四(三苯基膦基)钯(0)和5.2ml的2M碳酸钠水溶液。将反应在回流下进行16h。将反应混合物冷却至室温,经 过滤,并且用乙酸乙酯洗涤固体。使用蒸发器浓缩滤液。通过使用100/0至30/70 的庚烷/乙酸乙酯作为洗脱液的柱色谱分离残余物,以得到1.91g(83%) 中间体IV-1。1H NMR(300MHz,CDCl3)δ:8.61(bs;1H);7.61(dd;J= 7.4;0.8Hz;1H);7.50-7.23(m;8H);7.13-7.07(m;1H);6.74(s;1H); 6.66(s;1H);4.81(s;2H);4.67(s;2H);4.02-3.95(m;4H);3.81-3.73(m;4H)。MS-ESI:(m/z)计算值,对于C11H13N3O[M+H]+:204.25;测定值204.1。
过滤,并且用乙酸乙酯洗涤固体。使用蒸发器浓缩滤液。通过使用100/0至30/70 的庚烷/乙酸乙酯作为洗脱液的柱色谱分离残余物,以得到1.91g(83%) 中间体IV-1。1H NMR(300MHz,CDCl3)δ:8.61(bs;1H);7.61(dd;J= 7.4;0.8Hz;1H);7.50-7.23(m;8H);7.13-7.07(m;1H);6.74(s;1H); 6.66(s;1H);4.81(s;2H);4.67(s;2H);4.02-3.95(m;4H);3.81-3.73(m;4H)。MS-ESI:(m/z)计算值,对于C11H13N3O[M+H]+:204.25;测定值204.1。
表1中示出的中间体IV-2至IV-9,由中间体III-1(R3=H)或III-2 (R3=Me)和各自的硼酸R1B(OH)2的频哪醇酯起始,并且通过与对于制备中间体IV-1所描述的类似的方式获得。
表1.


中间体V-1:[7-(吗啉-4-基)-5-(1H-吲哚-4-基)吡唑并[1,5-a]嘧啶-2-基]甲醇

向5.0g(9.1mmol)中间体IV-1在120mlDMF和60ml EtOH中的溶液添加11.3g10%Pd/C和100μl甲酸。将反应在60℃下于氢气压力下进行24h。将反应混合物冷却至室温后,在 上过滤催化,用EtOH洗涤固体,然后使用蒸发器将滤液浓缩。通过使用100/0至 0/100的庚烷/乙酸乙酯作为洗脱液的柱色谱纯化所得的混合物。分离后得到2.08g(66%)中间体V-1。1H NMR(300MHz,DMSO)δ11.36(bs; 1H);7.70-7.63(m;1H);7.59-7.52(m;1H);7.52-7.46(m;1H);7.28 -7.20(m;1H);7.14-7.09(m;1H);6.78(s;1H);6.55(s;1H);5.36(t;J =6.0Hz;1H);4.66(d;J=6.0Hz;2H);3.90-3.83(m;4H);3.83-3.75 (m;4H)。MS-ESI:(m/z)计算值,对于C19H19N5O2[M+H]+:350.39;测定值350.2。
上过滤催化,用EtOH洗涤固体,然后使用蒸发器将滤液浓缩。通过使用100/0至 0/100的庚烷/乙酸乙酯作为洗脱液的柱色谱纯化所得的混合物。分离后得到2.08g(66%)中间体V-1。1H NMR(300MHz,DMSO)δ11.36(bs; 1H);7.70-7.63(m;1H);7.59-7.52(m;1H);7.52-7.46(m;1H);7.28 -7.20(m;1H);7.14-7.09(m;1H);6.78(s;1H);6.55(s;1H);5.36(t;J =6.0Hz;1H);4.66(d;J=6.0Hz;2H);3.90-3.83(m;4H);3.83-3.75 (m;4H)。MS-ESI:(m/z)计算值,对于C19H19N5O2[M+H]+:350.39;测定值350.2。
表2中示出的中间体V-2至V-9由各自的中间体IV起始,通过与对于制备中间体V-1所描述的类似的方式获得。
表2.


中间体VII-1:2-((苄氧基)甲基)-7-(吗啉-4-基)-N-(2-硝基苯基)吡唑并[1,5-
a]嘧啶-5-胺

将1.65g(4.6mmol)中间体III-1、0.953g(6.9mmol)2-硝基苯胺、 4.49g(13.8mmol)碳酸铯、0.211g(0.23mmol)三(二亚苄基丙酮)二钯 (0)、0.266g(0.46mmol)9,9-二甲基-4,5-双(二苯基膦)氧杂蒽和46ml 干燥甲苯引入反应容器。将全部混合物用氩气保护(argonated),并且在110℃下搅拌24h。冷却至室温后,将反应混合物通过 过滤,并且用乙酸乙酯洗涤固体。使用蒸发器浓缩滤液。在使用50/50至0/100 的庚烷/乙酸乙酯系统作为洗脱液的色谱柱上分离残余物。分离后得到 1.84g(87%)中间体VII-1。MS-ESI:(m/z)计算值,对于 C24H24N6O4[M+H]+:461.49;测定值461.5。
过滤,并且用乙酸乙酯洗涤固体。使用蒸发器浓缩滤液。在使用50/50至0/100 的庚烷/乙酸乙酯系统作为洗脱液的色谱柱上分离残余物。分离后得到 1.84g(87%)中间体VII-1。MS-ESI:(m/z)计算值,对于 C24H24N6O4[M+H]+:461.49;测定值461.5。
通过与对于制备中间体VII-1所描述的类似的方式,由中间体 III-1(R3=H)或III-2(R3=烷基)和适合的式VI的硝基胺衍生物(代替 2-硝基苯胺)起始,来制备表3中示出的中间体VII-2至VII-5。
表3

| 中间体 | R<sup>2</sup> | X<sup>1</sup> | X<sup>2</sup> | MS-ESI[M+H]<sup>+</sup> |
| VII-2 | H | N | CH | 462.2 |
| VII-3 | H | CH | N | 462.2 |
| VII-4 | H | CH | C-OMe | 491.2 |
| VII-5 | Me | CH | CH | 475.2 |
中间体VIII-1:N-(2-(苄氧基甲基)-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶-5-基)
苯-1,2-二胺

将1.75g(3.8mmol)中间体VII-1和3.94g(19.0mmol)二水合二氯化锡(II)在乙醇(25ml)中的混合物在加热回流下搅拌约20h。将反应冷却至室温后,添加100ml乙酸乙酯和100ml饱和碳酸氢钠溶液。将悬浮液通过 过滤,并且分离各相。将水相用乙酸乙酯萃取两次。将合并的有机相经无水硫酸钠干燥。过滤干燥剂,并使用蒸发器浓缩溶液。使用100/0至50/50的庚烷/乙酸乙酯系统作为洗脱液,使用色谱柱(用丙胺改性的硅胶)分离混合物。分离后,得到1.30g(79%)中间体VIII-1。1H NMR(300MHz,CDCl3)δ7.41-7.27(m;5H);7.3-7.17 (m;1H);7.17-7.08(m;1H);6.87-6.75(m;2H);6.17(s;1H);5.34(s; 1H);4.68(s;2H);4.61(s;2H);3.93-3.86(m;4H);3.57-3.49(m;4H)。 MS-ESI:(m/z)计算值,对于C24H26N6O2[M+H]+:431.22;测定值431.2。
过滤,并且分离各相。将水相用乙酸乙酯萃取两次。将合并的有机相经无水硫酸钠干燥。过滤干燥剂,并使用蒸发器浓缩溶液。使用100/0至50/50的庚烷/乙酸乙酯系统作为洗脱液,使用色谱柱(用丙胺改性的硅胶)分离混合物。分离后,得到1.30g(79%)中间体VIII-1。1H NMR(300MHz,CDCl3)δ7.41-7.27(m;5H);7.3-7.17 (m;1H);7.17-7.08(m;1H);6.87-6.75(m;2H);6.17(s;1H);5.34(s; 1H);4.68(s;2H);4.61(s;2H);3.93-3.86(m;4H);3.57-3.49(m;4H)。 MS-ESI:(m/z)计算值,对于C24H26N6O2[M+H]+:431.22;测定值431.2。
表4中示出的中间体VIII-2至VIII-5(R3=H、烷基)由适合的中间体VII起始,与对于制备中间体VIII-1的描述类似地获得。
表4.

| 中间体 | R<sup>3</sup> | X<sup>1</sup> | X<sup>2</sup> | MS-ESI[M+H]<sup>+</sup> |
| VIII-2 | H | N | CH | 432.2 |
| VIII-3 | H | CH | N | 432.2 |
| VIII-4 | H | CH | C-OMe | 461.2 |
| VIII-5 | Me | CH | CH | 445.2 |
中间体IX-1:N-(2-((2-(苄氧基甲基)-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶-5-基)
氨基)苯基)丙酰胺

向0.943g(2.2mmol)中间体VIII-1溶解于50.0ml干燥DCM中的溶液添加0.364ml(0.36g,4.8mmol)丙酸、0.652g(4,8mmol)HOBt、0.920g(4.8mmol)EDCI和0.916ml(0.666g,6.6mmol)TEA。将全部混合物在室温下搅拌48h。将100ml水添加至混合物,并且分离各相。将水相用DCM萃取3次。将合并的有机相经无水硫酸钠干燥。将干燥剂过滤并将溶剂蒸发后,使用50/50至0/100的庚烷/乙酸乙酯作为洗脱液,在色谱柱上分离反应混合物。分离后,得到0.84g(79%)中间体IX-1。1HNMR(300MHz,CDCl3)δ8.43(s;1H);7.61-7.55(m;1H); 7.40-7.28(m;6H);7.17-7.11(m;2H);6.18(s;1H);5.38(s;1H);4.68 (s;2H);4.62(s;2H);3.96-3.89(m;4H);3.63-3.55(m;4H);2.32(q;J =7.6Hz;2H);1.15(t;J=7.6Hz;3H)。MS-ESI:(m/z)计算值,对于 C27H30N6O3[M+H]+:487.25;测定值487.3。
由适合的中间体VIII和酸R4COOH(代替丙酸)起始,与对于中间体IX-1的描述类似地制备表5中示出的中间体IX-2至IX-13(R3=H、烷基)。
表5.



中间体X-1:2-(苄氧基甲基)-5-(2-乙基苯并咪唑-1-基)-7-(吗啉-4-基)吡唑并
[1,5-a]嘧啶

将0.763g(1.57mmol)中间体IX-1引入烧瓶,并且溶解于200ml 冰乙酸中。将反应在回流下进行24h。将反应混合物冷却后,使用蒸发器浓缩溶液。将残余物用水稀释,然后用饱和碳酸氢钠溶液中和。将水相用乙酸乙酯萃取3次。将合并的有机级分(fraction)经硫酸钠干燥。将干燥剂过滤后,使用蒸发器蒸发溶剂。在使用100/0至0/100 的庚烷/乙酸乙酯系统作为洗脱液的色谱柱上分离反应混合物。分离后得到0.459g(62%)中间体X-1。1H NMR(300MHz,CDCl3)δ7.84-7.77 (m;1H);7.49-7.22(m;8H);6.71(s;1H);6.21(s;1H);4.80(s;2H); 4.69(s;2H);4.05-3.95(m;4H);3.91-3.82(m;4H);3.13(q;J=7.5Hz; 2H);1.42(t;J=7.5Hz;3H)。MS-ESI:(m/z)计算值,对于C27H28N6O2 [M+H]+:469.23;测定值469.2。
与对于中间体的描述类似地制备表6中示出的中间体X-2至X-13,由适合的中间体IX起始,与对于制备中间体X-1的描述类似。
表6.


中间体XI:
由适合的中间体X起始,并且使用乙酸乙酯/乙醇为1/1的混合物代替DMF和乙醇,与对于中间体V-1的描述类似地制备表7中示出的中间体XI-1至XI-13。
表7.


中间体II-1:5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-羧基醛

向0.90g(2.58mmol)中间体V-1在26ml干燥DMF中的溶液添加 1.31g(3.09mmol)Dess-Martin试剂。将全部混合物在室温下搅拌1h。将固体过滤,然后用乙酸乙酯洗涤。将得到的溶液在减压下浓缩。以 100/0至30/70的庚烷/乙酸乙酯系统在色谱柱上纯化混合物。分离后得到0.70g(78%)中间体II-1。1H NMR(300MHz,CDCl3)δ10.22(s; 1H);8.47(bs;1H);7.66-7.59(m;1H);7.57-7.50(m;1H);7.39-7.29 (m;2H);7.18-7.09(m;2H);6.83(s;1H);4.08-4.00(m;4H);3.86- 3.77(m;4H)。MS-ESI:(m/z)计算值,对于C19H17N5O2[M+H]+:348.38;测定值348.1。
由适合的中间体V起始,与对于制备中间体II-1的描述类似地制备表8中示出的中间体II-2至II-9。
表8.


中间体II-10:5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-羧酸

通过氧化中间体II-1获得。
向具有35ml 20%硫酸溶液的烧瓶中添加1.40g(4.73mmol)重铬酸钾,然后添加1.1g(3.15mmol)中间体II-1。将全部混合物回流,同时搅拌并保持在该温度持续5h。将混合物冷却至室温,并且将混合物沉淀出的固体过滤。将固体用水洗涤,然后在干燥器中经P2O5干燥。得到0.56g(49%)粗中间体II-10,并且不经纯化用于下一步骤。
由适合的中间体XI起始,与对于制备中间体II-1的描述类似地制备表9中示出的中间体II-11至II-23(R3=H、烷基)。
表9.


中间体R2H(胺)
4-(氮杂环丁烷-3-基)吗啉

步骤:1.向0.50g(2.0mmol)1-二苯甲基氮杂环丁烷-3-酮在20.0 ml干燥DCM中的溶液添加0.21ml(0.209g;2.4mmol)吗啉,然后将混合物在室温下搅拌。4h后,添加0.848g(4.0mmol)三乙酰氧基硼氢化钠,并且在室温下继续搅拌过夜。将水加入反应混合物,并且分离各相。将水相用氯仿萃取3次。合并的有机相经无水硫酸钠干燥。在过滤干燥剂并且在减压下蒸发溶剂后获得的残余物在色谱柱上纯化。将100/0/0至0/95/5的庚烷/乙酸乙酯/MeOH系统用于分离。分离后,得到0.564g的4-[1-(二苯基甲基)氮杂环丁烷-3-基]吗啉。1HNMR(300 MHz,DMSO)δ7.45-7.38(m;4H);7.30-7.22(m;4H);7.20-7.13(m; 2H);4.41(s;1H);3.58-3.49(m;4H);3.27-3.18(m;2H);2.93-2.82 (m;1H);2.80-2.71(m;2H);2.25-2.13(m;4H)。MS-ESI:(m/z)计算值,对于C20H24N2O[M+H]+:309.20;测定值309.2。
步骤2.向114mg(0.37mmol)步骤1的产物在4mlEtOH中的溶液添加114mg 10%Pd/C和10μl甲酸。将反应在室温下于氢气压力下进行48h。在 上过滤催化剂,用EtOH洗涤固体,并且将滤液在蒸发器浓缩。得到50mg(95%)4-(氮杂环丁烷-3-基)吗啉。1H NMR(300MHz,CDCl3)δ3.76-3.67(m;4H);3.55-3.43(m;2H);3.04- 2.90(m;2H);2.90-2.80(m;1H);2.39-2.23(m;4H)。MS-ESI:(m/z) 计算值,对于C7H14N2O[M+H]+:143.12;测定值143.1。
上过滤催化剂,用EtOH洗涤固体,并且将滤液在蒸发器浓缩。得到50mg(95%)4-(氮杂环丁烷-3-基)吗啉。1H NMR(300MHz,CDCl3)δ3.76-3.67(m;4H);3.55-3.43(m;2H);3.04- 2.90(m;2H);2.90-2.80(m;1H);2.39-2.23(m;4H)。MS-ESI:(m/z) 计算值,对于C7H14N2O[M+H]+:143.12;测定值143.1。
分别用2,6-二甲基吗啉、1,1-二氧代硫代吗啉、4,4-二氟哌啶或3- 甲氧基氮杂环丁烷代替吗啉,与对于制备4-(氮杂环丁烷-3-基)吗啉的描述类似地获得表10中示出的中间体R2H。
表10.

1-(氧杂环丁烷-3-基)哌嗪

步骤1.
向0.23ml(0.279g 3.9mmol)3-氧杂环丁烷酮在39.0ml干燥DCM 中的溶液添加0.60g(3.2mmol)1-Boc-哌嗪,然后将混合物在室温下搅拌。4h后,添加1.35g(6.4mmol)三乙酰氧基硼氢化钠,并且在室温下继续搅拌过夜。将水加入反应混合物,并且分离各相。将水相用氯仿萃取3次。将合并的有机相经无水硫酸钠干燥。将干燥剂过滤后,在减压下蒸发溶剂。得到0.61g粗制4-(氧杂环丁烷-3-基)哌嗪-1-羧酸叔丁酯。1H NMR(300MHz,CDCl3)δ4.68-4.52(m;4H);3.50-3.32 (m;5H);2.31-2.09(m;4H);1.43(s;9H)。MS-ESI:(m/z)计算值,对于 C12H22N2O3[M+H]+:243.17;测定值243.2。
步骤2.
向0.55g(2.8mmol)步骤1产物在28ml DCM中的溶液,添加16.8 ml三氟乙酸。将反应在室温下进行2h。加入水,并且用饱和碳酸钠溶液碱化反应混合物。分离各相,并且将水相用氯仿萃取3次。将合并的有机相经无水硫酸钠干燥。将干燥剂过滤,并且在减压下蒸发溶剂。由此得到的0.23g粗制1-(氧杂环丁烷-3-基)哌嗪未经纯化用于进一步的反应。1HNMR(300MHz,CDCl3)δ4.66-4.56(m;4H);3.66-3.56 (m;1H);3.30-3.12(m;4H);2.68-2.51(m;4H)。MS-ESI:(mIz)计算值,对于C7H14N2O[M+H]+:143.12;测定值143.1。
(1S,4S)-2-(氧杂环丁烷-3-基)-2,5-二氮杂双环[2.2.1]庚烷

由3-氧杂环丁烷酮和Boc-(1S,4S)-2,5-二氮杂双环[2.2.1]庚烷起始,与对于制备1-(氧杂环丁烷-3-基)哌嗪的描述类似地制备。
3-乙基-1-(哌啶-4-基)脲
如US2005/197333,实施例41所述进行制备。

1-苯基-3-(哌啶-4-基)脲

如US2005/197333,实施例41中对于制备3-乙基-1-(哌啶-4-基) 脲的描述类似地制备。
本发明的化合物
实施例1.2-((4-(环丙烷羰基)哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-
基)吡唑并[1,5-a]嘧啶

向124mg(0.357mmol)中间体II-1在4.0ml干燥DCM中的溶液添加69.5mg(0.428mmol)1-(环丙基羰基)哌嗪,然后在室温下搅拌。 1h后,添加151mg(0.714mmol)三乙酰氧基硼氢化钠,并且在室温下再继续搅拌1h。将水加入反应混合物,并且分离各相。将水相用氯仿萃取3次。将合并的有机相经无水硫酸钠干燥。在过滤干燥剂并且在减压下蒸发溶剂后,将残余物在色谱板上纯化。将CHCl3/MeOH为 90/10的系统用于分离。分离后,得到170mg(98%)化合物1。1HNMR (300MHz,CDCl3)δ8.55(bs;1H);7.61(dd;J=7.4;0.8Hz;1H);7.52- 7.47(m;1H);7.36-7.27(m;2H);7.13-7.08(m;1H);6.66(s;1H);6.64 (s;1H);4.03-3.95(m;4H);3.84(s;2H);3.81-3.65(m;8H);2.71- 2.53(m;4H);1.81-1.70(m;1H);1.02-0.95(m;2H);0.79-0.71(m; 2H)。MS-ESI:(m/z)计算值,对于C27H31N7O2[M+H]+:486.59;测定值486.2。
表11中示出实施例2至69的本发明化合物,其中式(I)中Y表示 -CH2-并且R3表示H或烷基,由适合的中间体II-1至II-9和II-11至 II-23以及适合的胺R2H起始,与实施例1类似地进行制备。














实施例70.2-(4-叔丁基哌嗪-1-基羰基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基) 吡唑并[1,5-a]嘧啶

向0.545g(1.50mmol)中间体II-10溶解于15ml干燥DMF中的溶液添加0.47g(3.3mmol)1-叔丁基哌嗪、0.446g(3.3mmol)HOBt、 0.633g(3.3mmol)EDCI和0.63ml(0.455g,4.5mmol)TEA。将全部混合物在室温下搅拌40h。向混合物加入50ml水,并且分离各相。将水相用DCM萃取3次。将合并的有机相经无水硫酸钠干燥。在过滤干燥剂并且蒸发溶剂后,使用100/0至0/100的庚烷/乙酸乙酯系统作为洗脱液,通过柱色谱(用丙胺改性的硅胶)分离反应混合物。分离后,得到0.263g(36%)化合物70。1H NMR(300MHz,CDCl3)δ8.90(bs;1H);7.60(dd;J=7.4;0.8Hz;1H);7.48(d;J=8.1Hz;1H);7.33-7.25 (m;2H);7.12-7.04(m;1H);6.93(s;1H);6.72(s;1H);4.01-3.93(m; 4H);3.93-3.84(m;4H);3.80-3.71(m;4H);2.78-2,55(m;4H);1.13 (s;9H)。MS-ESI:(m/z)计算值,对于C27H33N7O2[M+H]+:488.28;测定值488.3。
实施例71.5-(2-(二氟甲基)苯并咪唑-1-基)-3-氯-2-((4-叔丁基哌嗪-1-基) 甲基)-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶

向烧瓶添加50mg(0.095mmol)实施例50的化合物,并且溶解于 1.0ml DCM中,然后添加17.8mg(0.134mmol)NCS(N-氯代琥珀酰亚胺)。将全部混合物在30℃下搅拌24h,然后添加3ml焦亚硫酸钠。分离各相,并且将水相用氯仿萃取两次。将合并的有机相经无水硫酸钠干燥。在过滤干燥剂并且用蒸发器蒸发溶剂后,在使用系统CHCl3/ MeOH-90/10作为洗脱液的色谱板上分离反应混合物。分离后,得到 50mg(94%)化合物71。1H NMR(300MHz,CDCl3)δ7.97-7.89(m; 1H);7.74-7.66(m;1H);7.49-7.39(m;2H);7.37(t;J=54Hz;1H); 6.36(s;1H);4.00-3.91(m;10H);2.78-2.57(m;8H);1.07(s;9H)。 MS-ESI:(m/z)计算值,对于C279H33ClF2N8O[M+H]+:559.25;测定值 559.3。
实施例72.5-(2-(二氟甲基)苯并咪唑-1-基)-3-溴-2-((4-叔丁基哌嗪-1-基) 甲基)-7-(吗啉-4-基)吡唑并[1,5-a]嘧啶
与实施例71的化合物类似地获得,用NCB(N-溴代琥珀酰亚胺) 代替N-氯代琥珀酰亚胺。1H NMR(300MHz,CDCl3)δ7.97-7.89(m; 1H);7.74-7.66(m;1H);7.49-7.39(m;2H);7.38(t;J=54Hz;1H); 6.40(s;1H);4.01-3.92(m;10H);2.77-2.56(m;8H);1.07(s;9H)。MS-ESI:(m/z)计算值,对于C28H33BrF2N8O[M+H]+:602.20;测定值 602.2。
本发明化合物的生物活性
PI3K激酶体外抑制的测试
使用下文描述的PI3K激酶抑制实验来在体外分析本发明化合物的效果。
将测试化合物溶解于100%DMSO,并且将由此获得的溶液连续稀释于1×反应缓冲液。将重组激酶稀释于包含5×反应缓冲液、基质 (在40mM Tris缓冲液中的1mM二乙酸钠4,5-二磷酸磷脂酰肌醇(PIP2) 溶液)和水的反应混合物。向96孔板的孔中添加5μl化合物溶液和15 μl在反应混合物中的激酶溶液。为了引发测试化合物与酶的相互作用,将板在板恒温器中以适合的温度孵育10分钟,同时以600rpm进行回转式摇动。阴性对照孔含有除测试化合物和激酶之外的所有上述试剂,并且阳性对照孔含有除测试化合物之外的所有上述试剂。通过添加5 μl 150μM ATP溶液来引发酶促反应,然后将板在板恒温器中以25℃或30℃(根据测试的PI3K同种型)孵育1小时,同时以600rpm将板的内含物进行回转式摇动。在以下的表12中示出了反应条件。
表12

然后使用ADP-Glo激酶测定(Promega)对酶促反应中形成的ADP 进行检测。向96孔板的孔添加25μl ADP-Glo试剂,并且将板在板恒温器中以25℃孵育40分钟,同时以600rpm进行回转式摇动。然后向96孔板的孔中添加50μl激酶检测试剂,并且将板在板恒温器中以25℃孵育40分钟,同时以600rpm进行回转式摇动。在孵育时间后,通过Victor Light光度计(Perkin Elmer,Inc.)测量孔中的荧光强度。
根据含有不同浓度的测试化合物的孔中和对照孔中的荧光强度测量来确定IC50值。使用Graph Pad 5.03软件,使用非线性回归拟合曲线来计算这些值。使用至少4个孔的各个对照,在两个96孔板上将各种化合物测试至少四次(4个孔)。
所选的本发明化合物的针对PI3K激酶的具体同种型的抑制活性的平均结果在以下的表13中以IC50示出。在表13中,符号A、B和C具有以下含义:
A:IC50<100nM;B:100nM≤IC50<1000nM;C:IC50≥1000nM
n.d.-未测定
表13.


通过蛋白质印迹分析测试AKT蛋白质磷酸化水平
细胞中的AKT蛋白质磷酸化水平取决于PI3K途径的活性——PI3K激酶被抑制剂(测试化合物)抑制导致AKT磷酸化减少,其在蛋白质印迹法中可表现为抗pAKT抗体染色的泳道消失。激酶的抑制不影响总蛋白质水平;使用抗pAKT抗体染色是实验中的对照。各个染色的对照是甘油醛-3-磷酸脱氢酶(GAPDH)水平的测量结果,这种蛋白以恒定的量存在于细胞中。
作为由PI3Kδ测试AKT磷酸化水平的细胞模型,使用由IgM抗体刺激的RAJI细胞系(来源:伯基特淋巴瘤,供应商:ATCC)。这种抗体通过结合存在于这些细胞表面上的BCR受体而使PI3Kδ激活,并且转而增加AKT磷酸化(Winkler等人2013,Chemistry and Biology)。
作为由PI3Kγ测试AKT磷酸化水平的细胞模型,使用补体系统的 C5a组分刺激的RAW264.7细胞系(来源:Ab-MLB转化的鼠淋巴细胞系,供应商:ATCC)。这种蛋白质通过结合C5aR受体,使PI3Kγ激活,这转而导致AKT磷酸化水平提高(Winkler等人2013,Chemistryand Biology)。
将不含抑制剂的培养基中的细胞以1-1.5×106/ml的密度接种至6 孔板上。接种后直接将细胞用测试化合物处理1小时,然后刺激细胞 (Raji:抗IgM,5mg/ml,在孵育结束前15min;RAW:C5a,5ng/ml,在孵育结束前5min)。然后用含有蛋白酶抑制剂(Halt ProteaseInhibitor Cocktail,Thermo)和磷酸酶(PhosSTOP,Roche)的RIPA缓冲液 (Sigma-Aldrich)裂解细胞,并且使用BCA方法(Pierce)根据制造商的建议测定蛋白质浓度。在MiniProtean III(BioRad)设备中,使得到的裂解物以100V进行SDS-PAGE电泳2小时。电泳后,在Mini Protean III 设备中,在1小时内通过以100V的电转移技术将分离的蛋白质转移至硝化纤维膜上。根据抗体制造商的建议对所选的蛋白质进行蛋白质印迹分析。
使用的一级抗体是抗pAKT和抗AKT(Cell Signaling Technology) 以及抗GAPDH(Millipore)的。为了检测一级抗体,使用了与辣根过氧化物酶(Sigma-Aldrich)偶联的二级抗体。用LumiLight试剂(Roche)使膜结合的蛋白可视化,然后在Light Film BioMax膜(Kodak)上显影。
在使用所选的本发明化合物处理1小时的细胞中,磷酸化的AKT (pAKT)水平和总AKT水平的测定结果于附图中示出,图1至图3针对PI3Kδ,并且图4针对PI3Kγ。图3示出随着磷酸-AKT蛋白(细胞系 RAJI)的泳道强度降低而PI3Kδ活性呈剂量依赖性降低。针对RAW细胞系进行的实验(其中AKT蛋白通过激酶PI3Kγ磷酸化)未表现出磷酸化AKT量的减少,这证实了化合物对PI3Kδ激酶的选择性作用。
体内药代动力学测试
为测试本发明化合物的生物利用度,根据下文描述的方法,在大鼠上对本发明化合物的药代动力学性质进行了体内研究。
对约12周龄、体重250-300g的Wistar大鼠进行测试。以30mg/kg m.c口服给予大鼠测试化合物。在6个时间点(从施用化合物起30min、 1h、2h、4h、7h和12h)从动物收集血液样品进行分析。将血液收集至含有K2EDTA的管,并且在室温下以2000×g离心15min从而分离血清。在分析前,将收集的血清样品保持在-20℃。以5只动物为一组测试各种化合物。
通过分光光度法确定测试化合物的血清浓度。对于每一种测试化合物,测定到达最大血清浓度的时间(Tmax)、最大浓度(Cmax)和曲线下面积(AUC)。四种代表性的本发明化合物的药代动力学参数在表14 中示出:
表14.
| 实施例编号 | 4 | 8 | 11 | 50 |
| Tmax[h] | 2 | 2 | 2 | 2 |
| Cmax[ng/ml] | 600 | 200 | 300 | 900 |
| AUC[mg*h/L] | 3.58 | 1.29 | 1.25 | 5.03 |
测试化合物是可生物利用的,并且达到了这类药物通常的Tmax 和Cmax。
Claims (25)
1.通式(I)的化合物

其中:
Y表示–CH2-或-CO-;
R1选自A1和A2:

R2表示:
-哌嗪基部分B2

其中R5选自-SO2CH3;–C(O)-C1-C3-烷基;–C(O)-C3-C5-环烷基;被–O-C1-C3烷基取代的苯基;和–CR6R7R8,
其中R6、R7和R8独立地表示氢原子、CH3、环丙基或CONH2,条件是R6、R7和R8中的仅一个能够表示环丙基或CONH2,
或者R6、R7和R8之一表示氢原子,并且R6、R7和R8中的另外两个连接在一起以形成–(CH2)2–、–(CH2)3–、–(CH2)4–或–(CH2-O-CH2)-基团;
或者
-哌啶基部分B4

其中R10选自C1-C4烷基;被OH取代的C1-C4烷基;-COO(C1-C3烷基);-N(C1-C3烷基)2;-NHCONH-C1-C3-烷基;-NHCONH-苯基;哌嗪基;和在4位被C1-C3烷基取代的哌嗪基;
R3选自H、卤素和C1-C4烷基;
R4选自C1-C4烷基、C3-C4-环烷基、被C1-C4烷氧基取代的C1-C4烷基,和CHF2;
X1和X2具有以下含义:
(i)X1表示CH并且X2表示CH或N;
(ii)X1表示N并且X2表示CH,或者
(iii)X1表示CH并且X2表示C-O-CH3;
X3、X4、X5和X6具有以下含义:
(i)X3表示N,X4表示CH,X5表示CH,并且X6表示CH;
(ii)X3表示CH,X4表示N,X5表示CH,并且X6表示CH;
(iii)X3表示CH,X4表示CH,X5表示N,并且X6表示CH;
(iv)X3表示CH,X4表示CH,X5表示CH,并且X6表示CH或CF;或者
(v)X3表示CH,X4表示CH,X5表示CF,并且X6表示CH;
波浪线表示连接点;
以及其药物可接受的盐。
2.如权利要求1所述的化合物,其中Y表示–CH2-。
3.如权利要求1所述的化合物,其中Y表示-CO-。
4.如权利要求1所述的化合物,其中R3表示H。
5.如权利要求1所述的化合物,其中R3表示C1-C4烷基。
6.如权利要求1所述的化合物,其中R3表示卤素。
7.如权利要求1或4或5或6所述的化合物,其中R1表示A1并且Y表示–CH2-。
8.如权利要求7所述的化合物,其中X1表示CH并且X2表示CH或N。
9.如权利要求8所述的化合物,其中X1表示CH并且X2表示CH。
10.如权利要求9所述的化合物,其中R4表示CHF2。
11.如权利要求7所述的化合物,其中R2表示B4并且R10表示被OH取代的C1-C4烷基。
12.如权利要求11所述的化合物,其中R4表示CHF2。
13.如权利要求7所述的化合物,其中R2表示B2。
14.如权利要求13所述的化合物,其中R5表示-CR6R7R8。
15.如权利要求14所述的化合物,其中R6、R7和R8中的每一个表示CH3。
16.如权利要求1或4或5或6所述的化合物,其中R1表示A2、R2表示B2并且Y表示–CH2-。
17.如权利要求16所述的化合物,其中X3表示CH、X4表示CH、X5表示CH并且X6表示CH或CF。
18.如权利要求17所述的化合物,其中R3表示CH3-。
19.如权利要求1所定义的通式(I)化合物,其选自:
2-((4-(环丙烷羰基)哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-吲哚-4-基)-2-((4-(甲基磺酰基)哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-吲哚-4-基)-2-((4-(4-甲基哌嗪-1-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(二甲基氨基)哌啶-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-吲哚-4-基)-2-((4-(4-甲氧基苯基)哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-5-(5-氟-1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-叔丁基哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-(4-((5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-基)甲基)哌嗪-1-基)-2-甲基丙酰胺;
2-(4-((5-(5-氟-1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-基)甲基)哌嗪-1-基)-2-甲基丙酰胺;
2-((4-叔丁基哌嗪-1-基)甲基)-5-(5-氟-1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-叔丁基哌嗪-1-基)甲基)-5-(1H-吲唑-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(二甲基氨基)哌啶-1-基)甲基)-5-(5-氟-1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(5-氟-1H-吲哚-4-基)-2-((4-(4-甲基哌嗪-1-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-5-(1H-吲唑-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-氨基吡啶-5-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-氨基嘧啶-5-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-氨基嘧啶-5-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-环丙基哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-吲哚-4-基)-2-((4-甲基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-乙基哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
1-((5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-基)甲基)哌啶-4-羧酸甲酯;
2-((4-(环丙基甲基)哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-环戊基哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-叔丁基哌啶-1-基)甲基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-6-氮杂吲哚-4-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-6-氮杂吲哚-4-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-7-氮杂吲哚-4-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-7-氮杂吲哚-4-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-吲哚-4-基)-2-((4-(氧杂环丁烷-3-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(1H-5-氟吲哚-4-基)-2-((4-(氧杂环丁烷-3-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
3-乙基-1-(1-((5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-基)甲基)哌啶-4-基)脲;
1-苯基-3-(1-((5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶-2-基)甲基)哌啶-4-基)脲;
2-((4-叔丁基哌嗪-1-基)甲基)-5-(6-氟-1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-叔丁基哌嗪-1-基)甲基)-5-(1H-吲哚-4-基)-3-甲基-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-5-(1H-吲哚-4-基)-3-甲基-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-(甲基磺酰基)哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-(二甲基氨基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-(4-甲基哌嗪-1-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-(4-甲氧基苯基)哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并-[1,5-a]嘧啶;
5-(2-甲基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-甲基苯并咪唑-1-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-2-((4-(2-羟基-丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-异丙基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-异丙基苯并咪唑-1-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-环丙基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-环丙基苯并咪唑-1-基)-2-((4-(2-羟基丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基咪唑并[4,5-b]吡啶-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基咪唑并[4,5-c]吡啶-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基-5-甲氧基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-乙基-5-甲氧基苯并咪唑-1-基)-2-((4-(2-羟基-丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-二氟甲基-5-甲氧基苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-二氟甲基-5-甲氧基-苯并咪唑-1-基)-2-((4-(2-羟基-丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)咪唑并[4,5-c]吡啶-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(甲氧基甲基)-苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-2-((3-(3-甲氧基氮杂环丁烷-1-基)氮杂环丁烷-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-3-甲基-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-3-甲基-2-((4-(2-羟基-丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
2-(4-叔丁基哌嗪-1-基羰基)-5-(1H-吲哚-4-基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;
5-(2-(二氟甲基)苯并咪唑-1-基)-3-氯-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶;和
5-(2-(二氟甲基)苯并咪唑-1-基)-3-溴-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶。
20.如权利要求19所述的通式(I)化合物,其为5-(1H-6-氮杂吲哚-4-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶。
21.如权利要求19所述的通式(I)化合物,其为5-(2-(二氟甲基)苯并咪唑-1-基)-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶。
22.如权利要求19所述的通式(I)化合物,其为5-(2-(二氟甲基)苯并咪唑-1-基)-3-甲基-2-((4-叔丁基哌嗪-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶。
23.如权利要求19所述的通式(I)化合物,其为5-(2-(二氟甲基)苯并咪唑-1-基)-3-甲基-2-((4-(2-羟基-丙-2-基)哌啶-1-基)甲基)-7-(吗啉-4-基)-吡唑并[1,5-a]嘧啶。
24.药物组合物,其包含权利要求1至23中任一项所定义的式(I)的化合物和药物可接受的赋形剂。
25.权利要求1至23中任一项所定义的式(I)的化合物用于制备治疗免疫系统疾病、炎性疾病和癌症的药物的用途。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL411864 | 2015-04-02 | ||
| PL411864A PL236355B1 (pl) | 2015-04-02 | 2015-04-02 | Pochodne 7-(morfolin-4-ylo)pirazolo[1,5-α]pirymidyny jako inhibitory kinazy PI3 |
| PCT/IB2016/051792 WO2016157091A1 (en) | 2015-04-02 | 2016-03-30 | 7-(morpholin-4-yl)pyrazole[1,5-a]pyrimidine derivatives which are useful for the treatment of immune or inflammatory diseases or cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107743489A CN107743489A (zh) | 2018-02-27 |
| CN107743489B true CN107743489B (zh) | 2021-05-04 |
Family
ID=55650626
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680017259.4A Active CN107743489B (zh) | 2015-04-02 | 2016-03-30 | 治疗免疫疾病、炎性疾病或癌症的7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US10138247B2 (zh) |
| EP (1) | EP3277687B1 (zh) |
| JP (1) | JP6665201B2 (zh) |
| KR (1) | KR102559190B1 (zh) |
| CN (1) | CN107743489B (zh) |
| AU (1) | AU2016241568B2 (zh) |
| BR (1) | BR112017020131B1 (zh) |
| CA (1) | CA2978828A1 (zh) |
| DK (1) | DK3277687T3 (zh) |
| EA (1) | EA032826B1 (zh) |
| ES (1) | ES2765642T3 (zh) |
| HR (1) | HRP20192195T1 (zh) |
| HU (1) | HUE047822T2 (zh) |
| MX (1) | MX2017011423A (zh) |
| PL (2) | PL236355B1 (zh) |
| PT (1) | PT3277687T (zh) |
| SI (1) | SI3277687T1 (zh) |
| WO (1) | WO2016157091A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11834467B2 (en) | 2020-09-28 | 2023-12-05 | 1ST Biotherapeutics, Inc. | Substituted indazoles as hematopoietic progenitor kinase 1 (HPK1) inhibitors |
| CN114957261B (zh) * | 2022-05-17 | 2023-06-23 | 重庆文理学院 | 一种具有抗头颈癌作用的化合物及其制备方法和应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1205009A (zh) * | 1996-02-07 | 1999-01-13 | 詹森药业有限公司 | 吡唑并嘧啶类化合物 |
| WO2006084634A1 (en) * | 2005-02-11 | 2006-08-17 | F.Hoffmann-La Roche Ag | Pyrazolo-pyrimidine derivatives as mglur2 antagonists |
| CN101516887A (zh) * | 2006-09-20 | 2009-08-26 | 伊莱利利公司 | 作为crf1受体拮抗剂的噻唑吡唑并嘧啶类 |
| WO2010037765A2 (en) * | 2008-10-03 | 2010-04-08 | Merck Serono S.A. | 4-morpholino-pyrido[3,2-d]pyrimidines |
| CN101835779A (zh) * | 2007-10-26 | 2010-09-15 | 霍夫曼-拉罗奇有限公司 | 作为pi3激酶抑制剂的嘌呤衍生物 |
| WO2010136491A1 (en) * | 2009-05-27 | 2010-12-02 | F. Hoffmann-La Roche Ag | Bicyclic indole-pyrimidine pi3k inhibitor compounds selective for p110 delta, and methods of use |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2550729A1 (en) | 2003-12-23 | 2005-07-14 | Activbiotics, Inc. | Rifamycin analogs and uses thereof |
| MX2010009416A (es) | 2008-02-26 | 2010-09-24 | Novartis Ag | Compuestos heterociclicos como inhibidores de cxcr2. |
| JP2011521968A (ja) * | 2008-05-30 | 2011-07-28 | ジェネンテック, インコーポレイテッド | プリンpi3k阻害剤化合物および使用方法 |
| WO2010074284A1 (ja) * | 2008-12-26 | 2010-07-01 | 味の素株式会社 | ピラゾロピリミジン化合物 |
| BRPI1014572B8 (pt) * | 2009-04-16 | 2022-07-19 | Fundacion Centro Nac De Investigaciones Oncologicas Carlos Iii | Imidazopirazinas para uso como inibidores de cinase |
| CA2761445A1 (en) * | 2009-05-27 | 2010-12-02 | Genentech, Inc. | Bicyclic pyrimidine pi3k inhibitor compounds selective for p110 delta, and methods of use |
| GB0912745D0 (en) | 2009-07-22 | 2009-08-26 | Wolfson Microelectronics Plc | Improvements relating to DC-DC converters |
| CA2786294A1 (en) | 2010-02-22 | 2011-08-25 | F. Hoffmann-La Roche Ag | Pyrido[3,2-d]pyrimidine pi3k delta inhibitor compounds and methods of use |
| WO2011109267A1 (en) | 2010-03-04 | 2011-09-09 | Merck Sharp & Dohme Corp. | Inhibitors of catechol o-methyl transferase and their use in the treatment of psychotic disorders |
| US9096605B2 (en) * | 2011-08-24 | 2015-08-04 | Glaxosmithkline Llc | Pyrazolopyrimidine derivatives as PI3 kinase inhibitors |
-
2015
- 2015-04-02 PL PL411864A patent/PL236355B1/pl unknown
-
2016
- 2016-03-30 MX MX2017011423A patent/MX2017011423A/es active IP Right Grant
- 2016-03-30 DK DK16714019.3T patent/DK3277687T3/da active
- 2016-03-30 JP JP2017551627A patent/JP6665201B2/ja active Active
- 2016-03-30 US US15/562,537 patent/US10138247B2/en active Active
- 2016-03-30 HU HUE16714019A patent/HUE047822T2/hu unknown
- 2016-03-30 WO PCT/IB2016/051792 patent/WO2016157091A1/en active Application Filing
- 2016-03-30 EA EA201792087A patent/EA032826B1/ru not_active IP Right Cessation
- 2016-03-30 EP EP16714019.3A patent/EP3277687B1/en active Active
- 2016-03-30 PL PL16714019T patent/PL3277687T3/pl unknown
- 2016-03-30 AU AU2016241568A patent/AU2016241568B2/en not_active Ceased
- 2016-03-30 CA CA2978828A patent/CA2978828A1/en active Pending
- 2016-03-30 PT PT167140193T patent/PT3277687T/pt unknown
- 2016-03-30 KR KR1020177031214A patent/KR102559190B1/ko active IP Right Grant
- 2016-03-30 ES ES16714019T patent/ES2765642T3/es active Active
- 2016-03-30 SI SI201630560T patent/SI3277687T1/sl unknown
- 2016-03-30 CN CN201680017259.4A patent/CN107743489B/zh active Active
- 2016-03-30 BR BR112017020131-3A patent/BR112017020131B1/pt active IP Right Grant
-
2019
- 2019-12-06 HR HRP20192195TT patent/HRP20192195T1/hr unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1205009A (zh) * | 1996-02-07 | 1999-01-13 | 詹森药业有限公司 | 吡唑并嘧啶类化合物 |
| WO2006084634A1 (en) * | 2005-02-11 | 2006-08-17 | F.Hoffmann-La Roche Ag | Pyrazolo-pyrimidine derivatives as mglur2 antagonists |
| CN101516887A (zh) * | 2006-09-20 | 2009-08-26 | 伊莱利利公司 | 作为crf1受体拮抗剂的噻唑吡唑并嘧啶类 |
| CN101835779A (zh) * | 2007-10-26 | 2010-09-15 | 霍夫曼-拉罗奇有限公司 | 作为pi3激酶抑制剂的嘌呤衍生物 |
| WO2010037765A2 (en) * | 2008-10-03 | 2010-04-08 | Merck Serono S.A. | 4-morpholino-pyrido[3,2-d]pyrimidines |
| WO2010136491A1 (en) * | 2009-05-27 | 2010-12-02 | F. Hoffmann-La Roche Ag | Bicyclic indole-pyrimidine pi3k inhibitor compounds selective for p110 delta, and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20170132275A (ko) | 2017-12-01 |
| SI3277687T1 (sl) | 2020-02-28 |
| MX2017011423A (es) | 2018-05-17 |
| KR102559190B1 (ko) | 2023-07-25 |
| CN107743489A (zh) | 2018-02-27 |
| EP3277687B1 (en) | 2019-10-30 |
| ES2765642T3 (es) | 2020-06-10 |
| PL236355B1 (pl) | 2021-01-11 |
| AU2016241568A1 (en) | 2017-10-19 |
| JP6665201B2 (ja) | 2020-03-13 |
| EA032826B1 (ru) | 2019-07-31 |
| AU2016241568B2 (en) | 2019-09-26 |
| CA2978828A1 (en) | 2016-10-06 |
| PL411864A1 (pl) | 2016-10-10 |
| BR112017020131A2 (pt) | 2018-05-29 |
| DK3277687T3 (da) | 2019-12-16 |
| EA201792087A1 (ru) | 2018-03-30 |
| HUE047822T2 (hu) | 2020-05-28 |
| EP3277687A1 (en) | 2018-02-07 |
| WO2016157091A1 (en) | 2016-10-06 |
| US20180111939A1 (en) | 2018-04-26 |
| JP2018510192A (ja) | 2018-04-12 |
| PL3277687T3 (pl) | 2020-09-21 |
| BR112017020131B1 (pt) | 2023-02-28 |
| HRP20192195T1 (hr) | 2020-03-06 |
| US10138247B2 (en) | 2018-11-27 |
| PT3277687T (pt) | 2020-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8895544B2 (en) | Indazoles | |
| KR101994381B1 (ko) | 키나아제 억제제 | |
| AU2011217961B2 (en) | Cyclobutane and methylcyclobutane derivatives as Janus kinase inhibitors | |
| WO2009122180A1 (en) | Pyrimidine derivatives capable of inhibiting one or more kinases | |
| US9487529B2 (en) | Macrocyclic compounds as ALK, FAK and JAK2 inhibitors | |
| KR20190112000A (ko) | Rho-키나아제 억제제로서 티로신 아마이드 유도체 | |
| EA023574B1 (ru) | ПРОИЗВОДНЫЕ 6-ЦИКЛОБУТИЛ-1,5-ДИГИДРОПИРАЗОЛО[3,4-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE9A | |
| US20110257171A1 (en) | Novel imidazo[1,2-a]pyridine derivatives, method for the preparation thereof, use thereof as medicaments, pharmaceutical compositions and novel use in particular as met inhibitors | |
| AU2007316417A1 (en) | Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives and their use as protein kinase inhibitors | |
| WO2005085252A1 (en) | Imidazo ‘1,2-a’ pyrazine compounds which interact with protein kinases | |
| CA3045951A1 (en) | Aminopyrazoles as selective janus kinase inhibitors | |
| CN113631557A (zh) | Jak激酶抑制剂及其制备方法和在医药领域的应用 | |
| US20240327438A1 (en) | Prodrugs of stat3 inhibitors | |
| CN107743489B (zh) | 治疗免疫疾病、炎性疾病或癌症的7-(吗啉-4-基)吡唑并[1,5-a]嘧啶衍生物 | |
| JP6858252B2 (ja) | ラパマイシンシグナル伝達経路阻害剤のメカニズム標的、及びその治療応用 | |
| CN114671894A (zh) | 一种嘧啶并五元氮杂环类衍生物的盐、晶型及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |