CN1054847C - 氟代烷甲酰胺衍生物的生产方法 - Google Patents
氟代烷甲酰胺衍生物的生产方法 Download PDFInfo
- Publication number
- CN1054847C CN1054847C CN94108763A CN94108763A CN1054847C CN 1054847 C CN1054847 C CN 1054847C CN 94108763 A CN94108763 A CN 94108763A CN 94108763 A CN94108763 A CN 94108763A CN 1054847 C CN1054847 C CN 1054847C
- Authority
- CN
- China
- Prior art keywords
- amine
- formula
- trifluoromethyl
- carbon atom
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明公开了式(Ⅰ)氟代烷甲酰胺衍生物的制备方法,它包括:(a)在伯胺或仲胺的存在下,使式(Ⅱ)亚氨基噻唑啉与式(Ⅲ)氟代烯烃反应;(b)使所得混合物与水反应。式(Ⅰ)、式(Ⅱ)和式(Ⅲ)如下,其中各基团定义见说明书。
Description
本发明涉及氟代烷甲酰胺衍生物的生产方法。
美国专利第5312798号公开了一种除草剂二氟乙酰胺衍生物及其生产方法。但是,该方法具有需要二氟乙酸的缺陷,二氟乙酸必须从四氟乙烯经过好几个步骤才能获得,而不能进行工业规模的生产(参见例如Org.Prep.Proced.Int.19,468(1987))。因此,人们迫切需要开发一种生产氟代烷甲酰胺衍生物的有效方法。
本发明者进行了研究,以克服常规方法的缺陷。发现了生产包括上述二氟乙酰胺衍生物在内的氟代烷甲酰胺衍生物的有效方法,该方法使用易得的氟代烯烃经一步转化很方便地得到所需的化合物。
从而,本发明提供式(I)氟代烷甲酰胺衍生物的生产方法: 式中R′是卤原子、被至少一个卤原子取代的烷基或被至少一个卤原子取代的烷氧基;R2是氢原子或卤原子;R3是甲基、乙基、氯原子或溴原子;X和Y相同或不同,各自代表氢原子、氟原子、氯原子或三氟甲基,前提是X和Y不同时代表氢原子或氯原子;该方法包括下列步骤:
式中R′是卤原子、被至少一个卤原子取代的烷基或被至少一个卤原子取代的烷氧基;R2是氢原子或卤原子;R3是甲基、乙基、氯原子或溴原子;X和Y相同或不同,各自代表氢原子、氟原子、氯原子或三氟甲基,前提是X和Y不同时代表氢原子或氯原子;该方法包括下列步骤:
(a)在伯胺或仲胺化合物存在下,将式(II)亚氨基噻唑啉化合物或其盐与式(III)氟代烯烃反应;式(II)和(III)如下
XYC=CF2 (III)式中R1,R2,R3,X和Y定义同上;
(b)使所得反应混合物与水反应。
根据本发明方法,式(I)氟代烷甲酰胺可用易得的氟代烯烃一步转化生产。
在式(I)氟代烷甲酰胺衍生物中,作为R1和R2的卤原子,可以列举的有氟原子、氯原子和溴原子。R1的卤代烷基包括被至少一个卤原子取代的C1-C6烷基,如三氟甲基;卤代烷氧基包括被至少一个卤原子取代的C1-C6烷氧基,如二氟甲氧基、三氟甲氧基、1,1,2,2-四氟乙氧基和2,2,2-三氟乙氧基。氟代烷酰基XYCHC=O包括二氟乙酰基、氯氟乙酰基、2,3,3,3-四氟丙酰基和2,2-二(三氟甲基)乙酰基。
用本发明方法可制得的式(I)氟代烷甲酰胺衍生物包括:
2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(3,5-二氯苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(3-二氟甲氧基苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(3-氯苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(3-氟甲氧基苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(4-氟-3-三氟甲基苯基)-5-甲基噻唑啉,
2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-乙基噻唑啉,
2-二氟乙酰亚氨基-3-(3-氟甲基苯基)-5-氯噻唑啉,
2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-溴噻唑啉,
2-氯氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(3,5-二氯苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(3-二氟甲氧基苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(3-氯苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(3-三氟甲氧基苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(4-氟-3-三氟甲基苯基)-5-甲基噻唑啉,
2-氯氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-乙基噻唑啉,
2-氯氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-氯噻唑啉,
2-氯氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-溴噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲基苯基)-5-甲基噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3,5-二氯苯基)-5-甲基噻唑啉,
2-(3,3,3,3-四氟丙酰亚氨基)-3-(3-二氟甲氧基苯基)-5-甲基噻唑啉,
2-(3,3,3,3-四氟丙酰亚氨基)-3-(3-三氯苯基)-5-甲基噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲氧基苯基)-5-甲基噻唑啉,
2-(3,3,3,3-四氟丙酰亚氨基)-3-(4-氟-3-三氟甲基苯基)-5-甲基噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲基苯基)-5-乙基噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲基苯基)-5-氯噻唑啉,
2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲基苯基)-5-溴噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲基苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3,5-二氯苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-二氟甲氧基苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氯苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲氧基苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(4-氟-3-三氟甲基苯基)-5-甲基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲基苯基)-5-乙基噻唑啉,
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲基苯基)-5-氯噻唑啉,和
2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲基苯基)-5-溴噻唑啉。
本发明的步骤(a)在伯胺或仲胺化合物的存在下进行。本发明使用的伯胺化合物和仲胺化合物可被惰性基团取代而且只要能加成到氟代烯烃上便可,并不限于特定的化合物。
伯胺化合物和仲胺化合物分别包括一元伯胺和一元仲胺。
该一元伯胺可用式(IV)表示:
Q-NH2 (IV)式中,Q是1-20个碳原子的烷基、3-10个碳原子的烷氧基烷基、3-10个碳原子的烷硫基烷基、3-10个碳原子的环烷基、3-10个碳原子的环烷基烷基、3-10个碳原子的烯基、3-10个碳原子的炔基、7-10个碳原子的芳烷基、6-10个碳原子的杂芳烷基、5-10个碳原子的杂芳基或6-12个碳原子的芳基。
一元伯胺化合物的优选实例有甲胺、乙胺、正丙胺、异丙胺、正丁胺、异丁胺、仲丁胺、叔丁胺、正戊胺、正己胺、正辛胺、2-乙基己胺、十二胺、十四胺、十八胺、二十胺、2-甲氧基乙胺、2-乙氧基乙胺、3-甲氧基丙胺、3-乙氧基丙胺、2-甲硫基乙胺、环丙胺、环戊胺或环己胺、环己基甲胺、烯丙胺、炔丙胺、苄胺、苯乙胺、3-苯基-1-丙胺、4-苯基-1-丁胺、2-氨甲基吡啶、氨基吡啶、苯胺和萘胺。
所述一元仲胺可用式(V)表示:
Q-NH-Q (V)式中,两Q相同或不同,各自定义同上,两Q基团可一起形成-(CH2)4-基团、-(CH2)5-基团或-(CH2)2-O-(CH2)2-基团,其中各亚烷基可被至少一个C1-C3烷基取代。
所述一元仲胺的优选实例有二甲胺、二乙胺、二正丙胺、二丙异胺、二正丁胺、二异丁胺、二戊胺、二己胺、二烯丙胺、二环己胺、N-乙基甲胺、N-甲基丙胺、N-甲基异丙胺、N-甲基丁胺、N-甲基己胺、N-甲基环己胺、N-乙基丙胺,N-乙基异丙胺,N-乙基丁胺,N-乙基己胺、N-乙基环己胺、N-甲基苄胺、N-乙基苄胺、二苄胺、N-甲基苯胺、N-乙基苯胺和N-丙基苯胺;和环胺如吡咯烷、哌啶、2-甲基哌啶和吗啉。
此外,伯胺化合物和仲胺化合物分别包括多元伯胺化合物和多元仲胺化合物。
所述多元伯胺化合物和多元仲胺化合物可用式(VI)表示:
Q2N-Z-N-L2 (VI)式中,Z是亚苯基、-(CH2)2-基团或-(CH2)3-基团;Q相同或不同,各自定义如上,L与Q相同;两个Q基团和两个L基团均可形成-(CH2)2-NH-(CH2)2-基团,其中各亚烷基可以被至少一个C1-C3烷基取代;并且Q和L中至少有一个是氢原子。
该多元伯胺化合物或多元仲胺化合物的优选实例有乙二胺衍生物,例如:乙二胺、N-甲基乙二胺、N,N-二甲基乙二胺、N,N′-二甲基乙二胺、N,N,N′-三甲基乙二胺、N-乙基乙二胺、N,N-二乙基乙二胺、N,N′-二乙基乙二胺、N,N,N′-三乙基乙二胺、N,N-二甲基-N′-乙基乙二胺、N,N-二乙基-N′-甲基乙二胺、N-丙基乙二胺;丙二胺衍生物,例如:N-甲基-1,3-丙二胺、N,N-二甲基-1,3-丙二胺,N,N′-二甲基-1,3-丙二胺,N,N-二乙基-1,3-丙二胺、N,N′-二乙基-1,3-丙二胺、N,N-二丁基-1,3-丙二胺和N,N,N′-三甲基-1,3-丙二胺;哌嗪、N-甲基哌嗪、2-甲基哌嗪、1-(2-氨乙基)哌啶、4-(2-氨乙基)吗啉、1-(2-氨乙基)吡咯烷、1-(2-氨乙基)哌嗪、2-(2-氨乙基)吡啶、4-(3-氨丙基)吗啉或1-(3-氨丙基)-2-甲基哌啶;和芳族多元胺如苯二胺。
此外,未包括在上述式(VI)中的乙醛合氨和1,3-二(4-哌啶基)丙烷也是多元仲胺的例子。
式(III)氟代烯烃包括三氟乙烯、四氟乙烯、六氟丙烯、八氟丁烯和一氯三氟乙烯。这些氟代烯烃市场有售。
在本发明中,必要时可以使用碱,对该碱并无特别限定,只要它能捕获反应生成的氟化氢便可。伯胺或仲胺可直接用作碱。其它可用的碱包括无机碱如碱金属氢氧化物和碱土金属氢氧化物(例如:氢氧化钠、氢氧化钾)、碱金属碳酸盐和碱土金属碳酸盐(例如:碳酸钠、碳酸钾)和碱金属碳酸氢盐,和有机碱如三乙胺、N,N-二甲胺、N,N-二乙胺、吡啶和喹啉。
必要时步骤(a)可在溶剂存在下进行。所用的溶剂是疏质子极性溶剂如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮或二甲亚砜;烷基腈溶剂如乙腈或丙腈;醚溶剂如四氢呋喃、二甲氧基乙烷、二甘醇二甲醚或三甘醇二甲醚;囟代溶剂如氯仿或二氯乙烷;或芳香烃溶剂如苯、甲苯或一氯代苯,或其混合物。但并不限于上述溶剂。
步骤(a)的反应温度为0-200℃,优选20-150℃或溶剂的沸点。就反应物的量而言,通常对于1mol上述式(II)亚氨基咪唑啉化合物使用1-10mol伯胺化合物或仲胺化合物;必要时加入的碱的量通常为1-10mol;并且通常在步骤(a)中使用1mol至摩尔过量的氟代烯烃。反应可在大气压力或在1-10kg/cm2(表压)的压力下进行。
在本发明中,步骤(a)通常这样进行:将式(III)氟代烯烃加到式(II)亚氨基噻唑啉化合物和伯胺化合物或仲胺化合物的溶液中;或者分下面两步进行:
(i)使氟代烯烃与伯胺化合物或仲胺化合物反应;然后
(ii)使所得反应混合物与式(II)亚氨基噻唑啉化合物反应。
然后,使上述第(ii)步所得的反应混合物与水反应得所需化合物。因为步骤(b)通常在步骤(a)之后进行,所以步骤(b)可以在与步骤(a)所用溶剂相同的溶剂存在下进行;然而,步骤(b)使用的溶剂可任意地从步骤(a)可用的溶剂中选择。
在步骤(b)中,反应温度通常在0-100℃或溶剂的沸点进行。对于1mol式(II)亚氨基噻唑啉化合物,水的用量通常为1mol至摩尔过量。
在步骤(b)完成后,用常规后处理方法如用有机溶剂提取的方法和/或浓缩的方法得到本发明化合物。必要时,可使所得化合物经重结晶和柱色谱操作进一步纯化。
式(II)亚氨基噻唑啉化合物是本发明的原料,它可通过日本专利申请第325259/1992或美国专利第5312798号说明书所述的方法制得。
在式(II)亚氨基噻唑啉化合物中,作为卤原子,可以列举出氟原子、氯原子和溴原子。卤代烷基包括被至少一个囱原子取代的C1-C6烷基,如三氟甲基;卤代烷氧基包括被至少一个卤原子取代的C1-C6烷氧基,如二氟甲氧基、三氟甲氧基、1,1,2,2-四氟乙氧基和2,2,2-三氟乙氧基。
式(II)亚氨基咪唑化合物包括2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉、2-亚氨基-3-(3,5-二氯苯基)-5-甲基噻唑啉、2-亚氨基-3-(3-二氟甲氧基苯基)-5-甲基噻唑啉、2-亚氨基-3-(3-氯苯基)-5-甲基噻唑啉、2-亚氨基-3-(3-三氟甲氧基苯基)-5-甲基噻唑啉、2-亚氨基-3-(4-氟-3-三氟甲基苯基)-5-甲基噻唑啉、2-亚氨基-3-(3-三氟甲基苯基)-5-乙基噻唑啉、2-亚氨基-3-(3-三氟甲基苯基)-5-氯噻唑啉和2-亚氨基-3-(3-三氟甲基苯基)-5-溴噻唑啉。此外,在该反应中还可使用无机酸(如盐酸)或有机酸与上述化合物的盐。
下面用实施例进一步说明本发明,但它们并不构成对本发明范围的限定。
实施例1
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.77g,3.0mmol)、二乙胺(1.06g,14.5mmol)和三乙胺(0.91g,9.0mmol)的N,N-二甲基甲酰胺(15ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌17小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙酸乙酯提取。乙酸乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱(洗脱剂,正己烷∶乙酸乙酯=2∶1),得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.82g,2.4mmol,收率:81%)。
m.p.,123.9℃;
1H NMR(CDCl3,TMS)δ(ppm):2.39(s,3H),5.89(t,1H,J=54.9Hz),6.97(s,1H),7.6-7.8(m,4H).
实施例2
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.52g,2.0mmol)、哌啶(0.51g,6.0mmol)和三乙胺(0.61g,6.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌9小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙酸乙酯提取。乙酸乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.61g,1.8mmol,收率:91%)。
实施例3
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.52g,2.0mmol)、哌啶(0.51g,6.0mmol)和三乙胺(0.61g,6.0mmol)的甲苯(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌15小时。冷却至环境温度后,将反应混合物倒入冰水中,用甲苯提取。甲苯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.48g,1.4mmol,收率:71%)。
实施例4
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.52g,2.0mmol)、正丁胺(0.44g,6.0mmol)和三乙胺(0.61g,6.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌13小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚层依次用5%盐酸、水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.50g,1.5mmol,收率:74%)。
实施例5
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.52g,2.0mmol)、二乙胺(0.44g,6.0mmol)和三乙胺(0.61g,6.0mmol)的N,N-二甲基甲酰胺(15ml)溶液投入反应烧瓶中,将四氟乙烯(0.70g,7.0mmol)从气袋引到烧瓶中,在50℃和烧瓶压力下反应21小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙酸乙酯提取。乙酸乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.44g,1.3mmol,收率:65%)。
实施例6
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉盐酸盐(1.47g,5.0mmol)、哌啶(0.64g,7.5mmol)和三乙胺(1.77g,1.75mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌13小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.56g,4.6mmol,收率:93%)。
实施例7
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.77g,3.0mmol)、二乙胺(1.32g,18.0mmol)和碳酸钾(1.04g,7.5mmol)的N,N-二甲基甲酰胺(15ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌32小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙酸乙酯提取。乙酸乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.53g,1.6mmol,收率:53%)。
实施例8
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和哌啶(0.64g,7.5mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌21小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.24g,3.7mmol,收率:74%)。回收未反应的2-氨基-3-(3-氟甲基苯基)-5-甲基噻唑啉(0.30g)部分。
实施例9
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和3-(N,N-二甲氨基)丙胺(0.77g,7.5mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.3升/小时)反应,同时于50℃剧烈搅拌17小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,(洗脱剂,正己烷∶乙酸乙酯=2∶1),得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.59g,1.8mmol,收率:35%)。回收未反应的2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.50g)部分。
实施例10
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和哌嗪(0.65g,7.5mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌9小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.24g,3.7mmol,收率:74%)。
实施例11
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉盐酸盐(1.47g,5.0mmol)、哌嗪(0.64g,7.5mmol)和三乙胺(1.52g,15.0mmol)的二甘醇二甲醚(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌24小时。冷却至环境温度后,将反应混合物倒入冰水中,用乙醚提取。乙醚乙酯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.01g,3.0mmol,收率:60%)。
实施例12
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和1-(2-氨乙基)哌嗪(0.78g,6.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌32小时。冷却至环境温度后,将反应混合物倒入饱和碳酸氢钠水溶液中,用乙醚提取。乙醚层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.18g,3.5mmol,收率:70%)。回收未反应的2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.12g)部分。
实施例13
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和哌嗪(0.52g,6.0mmol)的甲苯(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于80℃剧烈搅拌48小时。冷却至环境温度后,将反应混合物倒入饱和碳酸氢钠水溶液中,用甲苯提取。甲苯层依次用水和盐水洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,3.8mmol,收率:77%)。
实施例14
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉盐酸盐(1.47g,5.0mmol)和吗啉(1.57g,18.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于60℃剧烈搅拌2小时。冷却至环境温度后,将反应混合物倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取两次(100ml×2)。甲苯层用5%盐酸洗涤两次(50ml×2)、再依次用水(70ml)和盐水(70ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.47g,4.4mmol,收率:87%)。
实施例15
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和吗啉(0.96g,11.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时)反应,同时于50℃剧烈搅拌13小时。冷却至环境温度后,将反应混合物倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取两次(100ml×2)。甲苯层用5%盐酸洗涤两次(50ml×2)、再依次用水(50ml)和盐水(50ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.64g,4.9mmol,收率:98%)。
实施例16
将哌啶(0.57g,6.7mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时),于50℃剧烈搅拌反应10小时。冷却至环境温度后,向反应混合物中加2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉盐酸盐(1.29g,5.0mmol),于室温反应过夜,然后于50℃反应8小时,冷却至环境温度后,将反应溶液倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取两次(100ml×2)。甲苯层用5%盐酸洗涤两次(50ml×2)、再依次用水(70ml)和盐水(70ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.83g,2.5mmol,收率:49%)。
将洗涤后分出的盐酸水溶液层中和,然后在冰水冷却下用32%氢氧化钠碱化,然后该溶液用乙醚提取两次(100ml×2)。乙醚层用无水硫硫镁干燥,减压蒸去溶剂。这样便回收了2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.62g)。
实施例17
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.64g,2.5mmol)和吗啉(0.48g,5.5mmol)的乙腈(5ml)溶液投入反应烧瓶中,加入四氟乙烧(约0.7升/小时),在50℃剧烈搅拌反应13小时,冷却至环境温度后,将反应溶液倒入饱和碳酸氢钠水溶液(50ml)中,用甲苯提取两次(50ml×2)。甲苯层用5%盐酸洗涤两次(30ml×2),再依次用水(50ml)和盐水(50ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.64g,1.9mmol,收率:76%)。
实施例18
将哌啶(1.70g,20.0mmol)的乙腈(10ml)溶液投入反应烧瓶中,加入四氟乙烯(约0.7升/小时),于室温剧烈搅拌反应7.5小时。冷却至环境温度后,向反应混合物中加2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉盐酸盐(5.89g,20.0mmol)和乙腈(25ml),于室温反应过夜。冷却至环境温度后,将反应溶液倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取三次(50ml×3)。甲苯层用5%盐酸洗涤两次(30ml×2)、再依次用水(100ml)和盐水(100ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(5.33g,15.9mmol,收率:79%)。
将洗涤后分出的盐酸水溶液层中和,然后在冰水冷却下用32%氢氧化钠碱化,然后该溶液用乙醚提取两次(100ml×2)。乙醚层用无水硫硫镁干燥,减压蒸去溶剂。这样便回收了2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.75g)。
实施例19
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(2.28g,8.8mmol)和哌啶(0.98g,11.5mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烧(约0.7升/小时),在室温剧烈搅拌反应60小时,冷却至环境温度后,将反应溶液倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取两次(10ml×2)。合并的甲苯层用5%盐酸洗涤两次(50ml×2)再依次用水(70ml)和盐水(70ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(2.32g,6.9mmol,收率:78%)。
将洗涤后分出的盐酸水溶液层中和,然后在冰水冷却下用32%氢氧化钠碱化,然后该溶液用乙醚提取两次(100ml×2)。乙醚层用无水硫硫镁干燥,减压蒸去溶剂。这样便回收了2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.34g)。
实施例20
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.47g,5.0mmol)、三乙胺(2ml)和N-甲基苯胺(1.29g,12.0mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入四氟乙烧(约0.7升/小时)反应,同时于80℃剧烈搅拌30小时,冷却至环境温度后,将反应混合物倒入饱和碳酸氢钠水溶液(100ml)中,用甲苯提取两次(10ml×2)。合并的甲苯层用10%盐酸洗涤两次(50ml×2),再依次用水(70ml)和盐水(70ml)洗涤,用无水硫酸镁干燥。蒸除溶剂,残留物经硅胶柱色谱,得2-二氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.45g,1.34mmol,收率:27%)。
将洗涤后分出的盐酸水溶液层中和,然后在冰水冷却下用32%氢氧化钠碱化,然后该溶液用乙醚提取两次(100ml×2)。乙醚层用无水硫硫镁干燥,减压蒸去溶剂。这样便回收了2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(0.50g)。
实施例21
将2-亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和二乙胺(0.51g,7.0mmol)的N,N-二甲酰胺(10ml)溶液投入反应烧瓶中,将六氟丙烯从气袋引到烧瓶中,在室温和烧瓶压力下剧烈搅拌反应3.5小时。反应完全后,类似于上述方法对反应混合物进行后处理,得2-(2,3,3,3-四氟丙酰亚氨基)-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.48g,3.8mmol,收率77%)。
m.p.,162.0℃;
1H NMR(CDCl3,内标.TMS)δ(ppm):2.40(s,3H),5.10(dq,1H,J=47.1,7.7Hz),6.98(s,1H),7.6-7.9(m,4H).
实施例22
在本例中,按照与实施例15相同的方法,只是用三氟乙烯代替四氟乙烯,制得2-氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉。
实施例23
在本例中,按照与实施例15相同的方法,只是用八氟异丁烯代替四氟乙烯,制得2-(2,2-二(三氟甲基)乙酰亚氨基)-3-(3-三氟甲基苯基)-5-甲基噻唑啉。
实施例24
将2-亚氨基-3-(3-氟甲基苯基)-5-甲基噻唑啉(1.29g,5.0mmol)和二乙胺(1.21g,16.5mmol)的N,N-二甲基甲酰胺(10ml)溶液投入反应烧瓶中,加入一氯三氟乙烯(约0.7升/小时),在室温下剧烈搅拌反应6小时。反应完全后,类似于上述方法对反应混合物进行后处理,得2-氯氟乙酰亚氨基-3-(3-三氟甲基苯基)-5-甲基噻唑啉(1.19g,3.4mmol,收率67%)。
m.p.,155.4℃;
1H NMR(CDCl3,内标TMS)δ(ppm):2.40(s,3H),6.34(d,1H,J=51.4Hz),6.98(s,1H),7.6-7.9(m,4H).
Claims (9)
1.式(I)氟代烷甲酰胺衍生物的生产方法: 式中R′是卤原子、被至少一个卤原子取代的烷基或被至少一个卤原子取代的烷氧基;R2是氢原子或卤原子;R3是甲基、乙基、氯原子或溴原子;X和Y相同或不同,各自代表氢原子、氟原子、氯原子或三氟甲基,前提是X和Y不同时代表氢原子或氯原子;该方法包括下列步骤:
式中R′是卤原子、被至少一个卤原子取代的烷基或被至少一个卤原子取代的烷氧基;R2是氢原子或卤原子;R3是甲基、乙基、氯原子或溴原子;X和Y相同或不同,各自代表氢原子、氟原子、氯原子或三氟甲基,前提是X和Y不同时代表氢原子或氯原子;该方法包括下列步骤:
(a)在伯胺或仲胺化合物存在下,将式(II)亚氨基噻唑啉化合物或其盐与式(III)氟代烯烃反应;式(II)和(III)如下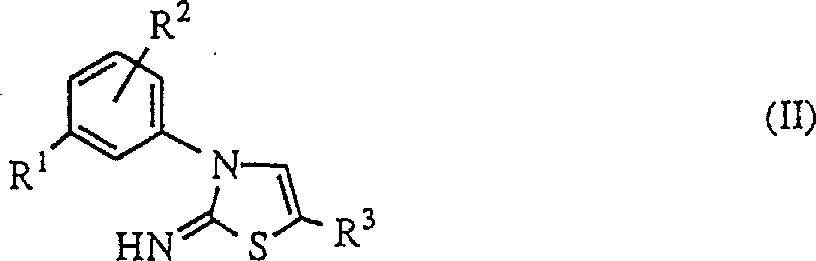
XYC=CF2 (III)式中R1,R2,R3,X和Y定义同上;
(b)使所得反应混合物与水反应。
2.按照权利要求1的方法,其中反应在碱的存在下进行。
3.按照权利要求1的方法,其中步骤(a)是这样进行的:先使氟代烯烃III与伯胺化合物或仲胺化合物反应,然后再将所得反应混合物与亚氨基噻唑啉化合物II反应。
4.按照权利要求1的方法,其中所述伯胺化合物可用式(I)表示:
Q-NH2 (IV)式中,Q是1-20个碳原子的烷基、3-10个碳原子的烷氧基烷基、3-10个碳原子的烷硫基烷基、3-10个碳原子的环烷基、3-10个碳原子的环烷基烷基、3-10个碳原子的烯基、3-10个碳原子的炔基、7-10个碳原子的芳烷基、6-10个碳原子的杂芳烷基、5-10个碳原子的杂芳基或6-12个碳原子的芳基;所述仲胺化合物可用式(V)表示:
Q-NH-Q (V)式中,两Q相同或不同,各自定义同上,两Q基团可一起形成-(CH2)4-基团、-(CH2)5-基团或-(CH2)2-O-(CH2)2-基团,其中各亚烷基可被至少一个C1-C3烷基取代;或者所述伯胺化合物或仲胺化合物可用下式表示:
Q2N-Z-N-L2式中,Z是亚苯基、-(CH2)2-基团或-(CH2)2-基团;Q的定义同上,L的定义与Q相同;两个Q基团和两个L基团均可形成-(CH2)2-NH-(CH2)2-基团,其中各亚烷基可以被至少一个C1-C3烷基取代;并且Q和L中至少有一个是氢原子。
5.按照权利要求1的方法,其中伯胺化合物选自甲胺、乙胺、正丙胺、异丙胺、正丁胺、异丁胺、仲丁胺、叔丁胺、正戊胺、正己胺、正辛胺、2-乙基己胺、十二胺、十四胺、十八胺、二十胺、2-甲氧基乙胺、2-乙氧基乙胺、3-甲氧基丙胺、3-乙氧基丙胺、2-甲硫基乙胺、环丙胺、环戊胺或环己胺、环己基甲胺、烯丙胺、炔丙胺、苄胺、苯乙胺、3-苯基-1-丙胺、4-苯基-1-丁胺、苯胺、2-氨甲基吡啶、氨基吡啶、和萘胺;仲胺化合物选自:二甲胺、二乙胺、二正丙胺、二丙异胺、二正丁胺、二异丁胺、二戊胺、二己胺、二烯丙胺、二环己胺、N-乙基甲胺、N-甲基丙胺、N-甲基异丙胺、N-甲基丁胺、N-甲基己胺、N-甲基环己胺、N-乙基丙胺、N-乙基异丙胺、N-乙基丁胺、N-乙基己胺、N-乙基环己胺、N-甲基苄胺、N-乙基苄胺、二苄胺、N-甲基苯胺、N-乙基苯胺、N-丙基苯胺、吡咯烷、哌啶、2-甲基哌啶和吗啉。
6.按照权利要求1的方法,其中伯胺化合物选自:乙二胺、N-甲基乙二胺、N,N-二甲基乙二胺、N,N′-二甲基乙二胺、N,N,N′-三甲基乙二胺、N-乙基乙二胺、N,N-二乙基乙二胺、N,N′-二乙基乙二胺、N,N,N′-三乙基乙二胺、N,N-二甲基-N′-乙基乙二胺、N,N-二乙基-N′-甲基乙二胺、N-丙基乙二胺;N-甲基-1,3-丙二胺、N,N-二甲基-1,3-丙二胺,N,N′-二甲基-1,3-丙二胺,N,N-二乙基-1,3-丙二胺、N,N′-二乙基-1,3-丙二胺、N,N-二丁基-1,3-丙二胺和N,N,N′-三甲基-1,3-丙二胺、哌嗪、N-甲基哌嗪、2-甲基哌嗪、1-(2-氨乙基)哌啶、4-(2-氨乙基)吗啉、1-(2-氨乙基)吡咯烷、1-(2-氨乙基)哌嗪、2-(2-氨乙基)吡啶、4-(3-氨丙基)吗啉、1-(3-氨丙基)-2-甲基哌啶、苯二胺、乙醛合氨和1,3-二(4-哌啶基)丙烷。
7.按照权利要求1的方法,其中仲胺化合物选自二乙胺、哌啶、吡咯烷、哌嗪和吗啉。
8.按照权利要求1的方法,其中氟代烯烃是三氟乙烯、四氟乙烯、六氟丙烯、八氟异丁烯或一氯三氟乙烯。
9.按照权利要求8的方法,其中氟代烯烃是四氟乙烯。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP187966/93 | 1993-07-29 | ||
| JP18796693 | 1993-07-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1105026A CN1105026A (zh) | 1995-07-12 |
| CN1054847C true CN1054847C (zh) | 2000-07-26 |
Family
ID=16215274
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94108763A Expired - Fee Related CN1054847C (zh) | 1993-07-29 | 1994-07-29 | 氟代烷甲酰胺衍生物的生产方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5484933A (zh) |
| EP (1) | EP0636616B1 (zh) |
| KR (1) | KR950003279A (zh) |
| CN (1) | CN1054847C (zh) |
| CA (1) | CA2129213A1 (zh) |
| DE (1) | DE69421337T2 (zh) |
| TW (1) | TW299329B (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI353991B (en) | 2003-05-06 | 2011-12-11 | Syntonix Pharmaceuticals Inc | Immunoglobulin chimeric monomer-dimer hybrids |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4567175A (en) * | 1983-06-03 | 1986-01-28 | Tanabe Seiyaku Co., Ltd. | 8-Chloro-1,5-benzothiazepine derivatives |
| US4590188A (en) * | 1984-02-18 | 1986-05-20 | Tanabe Seiyaku Co., Ltd. | 1,5-benzothiazepine derivatives and their pharmaceutical use |
| EP0529482A1 (en) * | 1991-08-23 | 1993-03-03 | Sumitomo Chemical Company, Limited | Iminothiazolines, their production and use as herbicides, and intermediates for their production |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5244863A (en) * | 1990-03-12 | 1993-09-14 | Sumitomo Chemical Company, Limited | Iminothiazolines, their production and use as herbicides, and intermediates for their production |
| CA2077737A1 (en) * | 1991-09-11 | 1993-03-12 | Shinichi Kawamura | Herbicidal composition |
-
1994
- 1994-07-26 KR KR1019940018120A patent/KR950003279A/ko active IP Right Grant
- 1994-07-28 TW TW083106891A patent/TW299329B/zh active
- 1994-07-29 DE DE69421337T patent/DE69421337T2/de not_active Expired - Fee Related
- 1994-07-29 CN CN94108763A patent/CN1054847C/zh not_active Expired - Fee Related
- 1994-07-29 US US08/282,562 patent/US5484933A/en not_active Expired - Fee Related
- 1994-07-29 EP EP94111882A patent/EP0636616B1/en not_active Expired - Lifetime
- 1994-07-29 CA CA002129213A patent/CA2129213A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4567175A (en) * | 1983-06-03 | 1986-01-28 | Tanabe Seiyaku Co., Ltd. | 8-Chloro-1,5-benzothiazepine derivatives |
| US4590188A (en) * | 1984-02-18 | 1986-05-20 | Tanabe Seiyaku Co., Ltd. | 1,5-benzothiazepine derivatives and their pharmaceutical use |
| EP0529482A1 (en) * | 1991-08-23 | 1993-03-03 | Sumitomo Chemical Company, Limited | Iminothiazolines, their production and use as herbicides, and intermediates for their production |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2129213A1 (en) | 1995-01-30 |
| DE69421337T2 (de) | 2000-03-23 |
| EP0636616B1 (en) | 1999-10-27 |
| US5484933A (en) | 1996-01-16 |
| CN1105026A (zh) | 1995-07-12 |
| DE69421337D1 (de) | 1999-12-02 |
| EP0636616A1 (en) | 1995-02-01 |
| TW299329B (zh) | 1997-03-01 |
| KR950003279A (ko) | 1995-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1089346C (zh) | 含氟弹性体 | |
| CN1295200C (zh) | 2-丁基-3-(4-[3-(二丁基氨基)丙氧基]苯甲酰基)-5-硝基苯并呋喃盐酸盐及其制备 | |
| CN1037680C (zh) | 制备7-取代-9-[(取代甘氨酰)氨基]-6-去甲基-6-去氧四环素的新方法 | |
| CN1097054C (zh) | 三环类吡唑衍生物及其组合物 | |
| CN1239487C (zh) | 制备奎替阿平的中间体及所述中间体的制备方法 | |
| CN1315320A (zh) | 作为抗糖尿病药的醚和酰胺类化合物及制备方法 | |
| CN1930164A (zh) | 制备n-([1,2,4]三唑并嘧啶-2-基)芳基磺酰胺的改进方法 | |
| CN1875006A (zh) | 氟甲基取代的杂环的制备方法 | |
| CN1109049A (zh) | 1,2-二酰基-2-(叔烷基)肼的制备方法 | |
| CN1072652C (zh) | 取代的苯甲酰氨基噻唑衍生物以及含有它们的药物 | |
| CN1054847C (zh) | 氟代烷甲酰胺衍生物的生产方法 | |
| CN1052979C (zh) | 恩丹西酮的制备方法 | |
| CN1247562C (zh) | 制备螺环季酮酸衍生物的方法 | |
| CN1445258A (zh) | 电荷传输聚合物 | |
| CN1205204C (zh) | 3-羟甲基-苯并[b]噻吩衍生物及其制备方法 | |
| CN1255404C (zh) | 制备取代的咪唑并吡啶化合物的方法 | |
| CN1616384A (zh) | 二氟烷基芳香族化合物 | |
| CN1037175C (zh) | 新的卤代甲基苯甲酰氰类化合物及其制备方法 | |
| CN1742002A (zh) | 新的哌嗪衍生物和它们作为合成中间体的应用 | |
| CN1138027A (zh) | 2-(2,6-二卤代苯氨基)苯乙酰氧乙酸衍生物的硝酸酯及其制备方法 | |
| CN1594287A (zh) | 3-羟基丙烯腈金属盐的制备方法 | |
| CN1220676C (zh) | 酸酐的制造方法 | |
| CN1447800A (zh) | 制备噻唑衍生物的催化方法 | |
| CN1974553A (zh) | 罗匹尼罗及其衍生物的制备方法 | |
| CN1325381A (zh) | 酮亚胺的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |