CN103153287A - (R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺的药物组合物及其施用 - Google Patents
(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺的药物组合物及其施用 Download PDFInfo
- Publication number
- CN103153287A CN103153287A CN2011800511228A CN201180051122A CN103153287A CN 103153287 A CN103153287 A CN 103153287A CN 2011800511228 A CN2011800511228 A CN 2011800511228A CN 201180051122 A CN201180051122 A CN 201180051122A CN 103153287 A CN103153287 A CN 103153287A
- Authority
- CN
- China
- Prior art keywords
- compound
- tablet
- weight
- approximately
- amount
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2077—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/18—Drugs for disorders of the endocrine system of the parathyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Abstract
一种药物组合物,其包含:化合物1(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺和至少一种选自填充剂、稀释剂、崩解剂、表面活性剂、助流剂和润滑剂的赋形剂,所述组合物适合口服施用给有此需要的患者以治疗CFTR介导的疾病,诸如囊性纤维化。还公开了治疗有此需要的患者的方法,所述方法包括:施用化合物1的药物组合物。
Description
相关申请的交叉引用
本申请要求2010年8月23日提交的美国临时专利申请序列号61/375,976和2011年7月11日提交的美国临时专利申请序列号61/506,220的优先权,这两篇申请的完整内容通过引用并入本文。
技术领域
本发明涉及包含(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺(化合物1)的药物组合物、用于制备这类组合物的方法、和用于施用包含所述化合物的药物组合物的方法。
背景技术
CFTR是cAMP/ATP-介导的阴离子通道,其在多种细胞类型(包括吸收上皮细胞和分泌上皮细胞)中表达,在所述细胞处它调整通过膜的阴离子流,以及其它离子通道和蛋白的活性。在上皮细胞中,CFTR的正常功能对整个机体(包括呼吸和消化组织)中电解质转运的维持是关键性的。CFTR由大约1480个氨基酸组成,其编码由串联重复的跨膜结构域组成的蛋白,每个跨膜结构域包含6个跨膜螺旋和1个核苷酸结合结构域。两个跨膜结构域通过调节通道活性和细胞运输的具有多个磷酸化位点的大的、极性的、调节性的(R)-结构域连接。
编码CFTR的基因已被鉴定和测序(参见Gregory,R.J.等人(1990)Nature 347:382-386;Rich,D.P.等人(1990)Nature 347:358-362),(Riordan,J.R.等人(1989)Science 245:1066-1073)。该基因的缺陷导致CFTR中的突变,该突变引起囊性纤维化(下称“CF”),囊性纤维化是人类最常见的致命性遗传疾病。在美国大约每2500个婴儿中有大约1个受囊性纤维化影响。在全部美国人口中,多达1千万人携带有单拷贝的所述缺陷基因,而没有明显的疾病效应。相反,带有两个拷贝的CF相关基因的个体遭受CF的虚弱和致命效应(包括慢性肺病)的痛苦。
在患有囊性纤维化的患者中,在呼吸上皮细胞中内源性表达的CFTR中的突变引起顶部阴离子分泌(apical anion secretion)减少,导致离子和流体转运失衡。所引起的阴离子转 运减少促进肺中的粘液蓄积增加,以及伴随的微生物感染,其最终导致CF患者死亡。除了呼吸疾病以外,CF患者通常还遭受胃肠问题和胰腺功能不全的痛苦,其如果不经治疗,会导致死亡。另外,大多数患有囊性纤维化的男性不能生育,而患有囊性纤维化的女性生育力下降。与两个拷贝的CF相关基因的严重效应相反,带有单拷贝的CF相关基因的个体表现出对霍乱和因腹泻所致脱水的抗性的增加-也许这解释了人群中相对高频率的CF基因的原因。
CF染色体的CFTR基因的序列分析已经揭示多种致病性突变(Cutting,G.R.等人(1990)Nature 346:366-369;Dean,M.等人(1990)Cell 61:863:870;和Kerem,B-S.等人(1989)Science245:1073-1080;Kerem,B-S等人(1990)Proc.Natl.Acad.Sci.USA 87:8447-8451)。迄今为止,如在科学和医学文献中所报道的,已鉴定了CF基因中超过1000个致病性突变。最普遍的突变是CFTR氨基酸序列的508位苯丙氨酸的缺失,通常将其称为ΔF508-CFTR。这种突变发生在大约70%的囊性纤维化病例中,并与严重的疾病相关联。其它突变包括R117H和G551D。
ΔF508-CFTR中508位残基的缺失阻止了初生蛋白的正确折叠。这导致该突变蛋白不能从ER中出来并转运至质膜。其结果是,膜中存在的通道数量远少于在表达野生型CFTR的细胞中所观察到的数量。除了运输受损外,这种突变导致缺陷性通道门控。合起来,膜中通道数量的减少和缺陷性门控导致通过上皮的阴离子转运减少,从而导致缺陷性离子和流体转运。(Quinton,P.M.(1990),FASEB J.4:2709-2727)。然而,研究表明,膜中ΔF508-CFTR数量的减少是功能性的,尽管其比野生型CFTR少。(Dalemans等人(1991),Nature Lond.354:526-528;Denning等人,同上;Pasyk和Foskett(1995),J.Cell.Biochem.270:12347-50)。除ΔF508-CFTR之外,CFTR中导致缺陷性运输、合成和/或通道门控的其它引起疾病的突变可被上调或下调以改变阴离子分泌,并且改变疾病进程和/或严重程度。
虽然除转运阴离子之外CFTR还转运多种分子,但显然这种作用(阴离子转运)代表了转运离子和水跨越上皮的重要机理中的-种要素。其它要素包括上皮Na+通道、ENaC、Na+/2Cl-/K+协同转运蛋白、Na+-K+-ATP酶泵和基底外侧膜K+通道,其负责将氯离子(chloride)摄入细胞。
这些要素经由它们的选择性表达和细胞内定位,共同发挥作用以实现通过上皮的定向转运。氯离子吸收通过存在于顶端膜上的ENaC和CFTR以及在细胞基底外侧表面上表达的Na+-K+-ATP酶泵和Cl-通道的协调活性而发生。氯离子从腔侧的次级主动转运导致细胞内氯离子的蓄积,然后所述氯离子能够经由Cl-通道被动离开细胞,导致向量转运。基底外侧 表面上的Na+/2Cl-/K+协同转运蛋白、Na+-K+-ATP酶泵和基底外侧膜K+通道以及位于腔侧的CFTR的排列,经由位于腔侧的CFTR协调氯离子的分泌。因为水本身可能从不主动转运,所以它跨过上皮的流动依赖于由钠和氯离子大量流动所产生的微小跨上皮渗透梯度。
如以上所讨论的,据信ΔF508-CFTR中第508残基的缺失阻止了初生蛋白正确地折叠,导致这种突变体蛋白不能从ER中出来并转运到质膜。结果,存在于质膜的成熟蛋白的量不足,并且上皮组织内的氯离子转运显著降低。事实上已显示,这种由ER机构进行的ATP结合盒(ABC)转运蛋白的缺陷性内质网(ER)加工的细胞现象不仅是CF疾病的潜在基础,而且是大量其它单独和遗传疾病的潜在基础。ER机构可能发生机能障碍的两种方式为:通过失去与蛋白的ER输出的偶联导致降解,或者通过这些有缺陷的/错误的折叠的蛋白的ER累积[Aridor M,等人,Nature Med.,5(7),第745-751页(1999);Shastry,B.S.,等人,Neurochem.International,43,第1-7页(2003);Rutishauser,J.,等人,Swiss Med Wkly,132,第211-222页(2002);Morello,JP等人,TIPS,21,第466-469页(2000);Bross P.等人,Human Mut.,14,第186-198页(1999)]。
在国际PCT公开WO 2010053471和WO 2010054138(所述公开通过引用整体并入本文)中,将(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺公开为CFTR活性的调节剂,并因而公开为诸如囊性纤维化等CFTR介导的疾病的有用治疗。在2010年3月25日提交的美国临时专利申请序列号61/317,376、2010年4月1日提交的美国临时专利申请序列号61/319,953、2010年4月7日提交的美国临时专利申请序列号61/321,561和2010年4月7日提交的美国临时专利申请序列号61/321,636(它们都通过引用整体并入本文)中,公开了(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺的形式A和无定形形式。但是,仍然需要可容易地制备的且适合用作治疗剂的包含化合物1的药物组合物。
发明内容
本发明涉及药物组合物、药物制剂和固体剂型,其包含具有下式结构的(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺(化合物1):

在一个方面,本发明的特征在于用于口服施用的片剂,其包含:a)化合物1;b)填充剂;c)稀释剂;d)崩解剂;e)润滑剂;和f)助流剂。
在某些实施方案中,化合物1呈基本上无定形的形式(化合物1无定形形式)。在其它实施方案中,化合物1呈基本上结晶的固体形式。在一个实施方案中,化合物1呈基本上结晶的形式A(化合物1形式A)。在其它实施方案中,化合物1呈固体(即,无定形的和结晶的)形式的混合物。
在一个实施方案中,化合物1或化合物1无定形形式以在约1mg至约250mg范围内的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以在约10mg至约250mg范围内的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以在约25mg至约250mg范围内的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以约50mg至约200mg的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以约10mg的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以约50mg的量存在于所述片剂中。在一个实施方案中,化合物1或化合物1无定形形式以约100mg的量存在于所述片剂中。
在一个实施方案中,按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约1重量%至约80重量%范围内。在一个实施方案中,按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约4重量%至约50重量%范围内。在一个实施方案中,按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约10重量%至约50重量%范围内。在一个实施方案中,按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约20重量%至约30重量%范围内。在一个实施方案中,所述片剂中的化合物1或化合物1无定形形式的量是片剂的约5重量%。在一个实施方案中,所述片剂中的化合物1或化合物1无定形形式的量是片剂的约25重量%。
在一个实施方案中,所述填充剂选自:纤维素、改性纤维素、羧甲基纤维素钠、乙基纤维素、羟甲基纤维素、羟丙基纤维素、乙酸纤维素、微晶纤维素、磷酸氢钙、蔗糖、乳糖、玉米淀粉、马铃薯淀粉或它们的任意组合。在一个实施方案中,所述填充剂是微晶纤维素(MCC),且以在约10重量%至约90重量%(按片剂的重量计)范围内的量存在于所述片 剂中。在一个实施方案中,所述填充剂是微晶纤维素(MCC),且以在约10重量%至约45重量%(按片剂的重量计)范围内的量存在于所述片剂中。
在一个实施方案中,所述稀释剂选自:乳糖一水合物、甘露醇、山梨醇、纤维素、磷酸钙、淀粉、糖或它们的任意组合。在一个实施方案中,所述稀释剂是乳糖一水合物,且以在约10重量%至约90重量%(按片剂的重量计)范围内的量存在于所述片剂中。在一个实施方案中,所述稀释剂是乳糖一水合物,且以在约10重量%至约45重量%(按片剂的重量计)范围内的量存在于所述片剂中。
在一个实施方案中,所述崩解剂选自:琼脂、藻胶、碳酸钙、羧甲基纤维素、纤维素、羟丙基纤维素、低取代的羟丙基纤维素、粘土、交联羧甲纤维素钠、交聚维酮、树胶、硅酸镁铝、甲基纤维素、波拉克林钾、海藻酸钠、淀粉羟乙酸钠、玉米淀粉、马铃薯淀粉、木薯淀粉或它们的任意组合。在一个实施方案中,所述崩解剂是交联羧甲纤维素钠,且以占片剂重量的6重量%或更小的浓度存在于所述片剂中。
在一个实施方案中,所述润滑剂选自:硬脂酸镁、硬脂酸钙、硬脂酸锌、硬脂酸钠、硬脂酸、硬脂酸铝、亮氨酸、山嵛酸甘油酯、氢化植物油、硬脂酰富马酸钠或它们的任意组合。在一个实施方案中,所述润滑剂是硬脂酸镁,且具有小于2重量%(按片剂的重量计)的浓度。
在一个实施方案中,所述助流剂选自:胶体二氧化硅、滑石、玉米淀粉或它们的组合。在一个实施方案中,所述助流剂是胶体二氧化硅,且具有占片剂重量的3重量%或更小的浓度。
在一个实施方案中,所述片剂另外包含着色剂。
在一个方面,本发明的特征在于包含多个颗粒的片剂,所述组合物包含:a)化合物1无定形形式,其量为约10重量%至约50重量%(按所述组合物的重量计);b)填充剂,其量为约10重量%至约30重量%(按所述组合物的重量计);c)稀释剂,其量为约10重量%至约30重量%(按所述组合物的重量计);d)崩解剂,其量为约1重量%至约5重量%(按所述组合物的重量计);e)润滑剂,其量为约0.3重量%至约3重量%(按所述组合物的重量计);和f)助流剂,其量为约0.3重量%至约3重量%(按所述组合物的重量计)。
在一个实施方案中,化合物1是化合物1无定形形式,且是在喷雾干燥的分散体中。在一个实施方案中,所述喷雾干燥的分散体包含聚合物。在一个实施方案中,所述聚合物是羟丙甲基纤维素(HPMC)。在一个实施方案中,所述聚合物是醋酸羟丙甲基纤维素琥珀酸酯(HPMCAS)。
在一个实施方案中,所述聚合物以20重量%至70重量%的量存在。在一个实施方案中,所述聚合物以30重量%至60重量%的量存在。在一个实施方案中,所述聚合物以约49.5重量%的量存在。
在一个实施方案中,所述片剂另外包含表面活性剂。在一个实施方案中,所述表面活性剂是月桂基硫酸钠。在一个实施方案中,所述表面活性剂以0.1重量%至5重量%的量存在。在一个实施方案中,所述表面活性剂以约0.5重量%的量存在。
在另一个方面,本发明的特征在于具有表1所示配方的片剂。
表1.

在另一个方面,本发明的特征在于具有表2所示配方的片剂。
表2.

在另一个方面,本发明的特征在于具有表3所示配方的片剂。
表3.

在另一个方面,本发明提供了一种片剂形式的药物组合物,其包含化合物1和一种或多种药学上可接受的赋形剂,例如,填充剂、崩解剂、表面活性剂、稀释剂、助流剂和润滑剂和它们的任意组合,其中所述片剂具有在约30分钟内至少约50%的溶出度。在另一个实施方案中,所述溶出速率是在约30分钟内至少约75%。在另一个实施方案中,所述溶出速率是在约30分钟内至少约90%。
在另一个方面,本发明提供了一种片剂形式的药物组合物,其包含粉末掺合物或颗粒,所述粉末掺合物或颗粒包含化合物1和一种或多种药学上可接受的赋形剂,例如,填充剂、崩解剂、表面活性剂、稀释剂、助流剂和润滑剂,其中所述片剂具有至少约5kP(kP=kilo Ponds;1kP=约9.8N)的硬度。在另一个实施方案中,所述片剂具有在400转以后小于1.0%的目标脆碎度。
在另一个方面,本发明提供了如本文所述的片剂,其另外包含其它治疗剂。在一个实施方案中,所述其它治疗剂是溶粘蛋白剂、支气管扩张剂、抗生素、抗感染剂、抗炎剂、除了化合物1以外的CFTR调节剂或营养剂。在某些实施方案中,所述其它治疗剂是N-(5-羟基-2,4-二叔丁基-苯基)-4-氧代-1H-喹啉-3-甲酰胺。
在一个方面,本发明的特征在于一种施用片剂的方法,所述方法包括:每天至少1次地给患者口服施用片剂,所述片剂包含:a)约25-200mg化合物1无定形形式;b)填充剂;c)稀释剂;d)崩解剂;e)表面活性剂;f)助流剂;和g)润滑剂。在一个实施方案中,所述片剂包含约2.5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约10mg化合物1无定形形式。在一个实施方案中,所述片剂包含约25mg化合物1无定形形式。在一个实施方案中,所述片剂包含约50mg化合物1无定形形式。在一个实施方案中,所述片剂包含约100mg化合物1无定形形式。在一个实施方案中,所述片剂包含约150mg化合物1无定形形式。在一个实施方案中,所述片剂包含约200mg化合物1无定形形式。
在一个方面,本发明的特征在于一种施用片剂的方法,所述方法包括:每天2次地给患者口服施用片剂,所述片剂包含:a)约2.5-200mg化合物1无定形形式;b)填充剂;c)稀释剂;d)崩解剂;e)表面活性剂;f)助流剂;和g)润滑剂。在一个实施方案中,所述片剂包含约2.5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约10mg化合物1无定形形式。在一个实施方案中,所述片剂包含约25mg化合物1无定形形式。在一个实施方案中,所述片剂包含约50mg化合物1无定形形式。在一个实施方案中,所述片剂包含约100mg化合物1无定形形式。在一个实施方案中,所述片剂包含约150mg化合物1无定形形式。在一个实施方案中,所述片剂包含约200mg化合物1无定形形式。
在一个方面,本发明的特征在于一种施用片剂的方法,所述方法包括:每12小时1次地给患者口服施用片剂,所述片剂包含:a)约2.5-200mg化合物1无定形形式;b)a填充剂;c)稀释剂;d)崩解剂;e)表面活性剂;f)助流剂;和g)润滑剂。在一个实施方案中,所述片剂包含约2.5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约5mg化合物1无定形形式。在一个实施方案中,所述片剂包含约10mg化合物1无定形形式。在一个实施方案中,所述片剂包含约25mg化合物1无定形形式。在一个实施方案中,所述片剂包含约50mg化合物1无定形形式。在一个实施方案中,所述片剂包含约100mg化合物1无定形形式。在一个实施方案中,所述片剂包含约200mg化合物1无定形形式。
在一个方面,本发明的特征在于一种治疗受试者的疾病或减轻其严重程度的方法,所述方法包括:给所述受试者施用本发明的片剂,其中所述疾病选自:囊性纤维化、哮喘、吸烟诱发的慢性阻塞性肺病、慢性支气管炎、鼻鼻窦炎、便秘、胰腺炎、胰腺功能不全、由先天性输精管两侧缺失(CBAVD)导致的男性不育、轻度肺病、自发性胰腺炎、变应性支气管肺曲菌病(ABPA)、肝病、遗传性肺气肿、遗传性血色素沉着病、凝血-纤维蛋白溶解缺陷、蛋白质C缺乏、1型遗传性血管水肿、脂质加工缺陷、家族性高胆固醇血症、1型乳糜微粒血症、无β脂蛋白血症、溶酶体贮积病、I-细胞病/假性赫尔勒病(pseudo-Hurler)、粘多糖贮积症、桑德霍夫病/泰-萨病、II型克-纳二氏综合征(Crigler-Najjar type II)、多内分泌腺病/高胰岛素血症、糖尿病、拉伦侏儒症、髓过氧化物酶缺乏、原发性甲状旁腺功能减退、黑素瘤、1型Glycanosis CDG病、先天性甲状腺功能亢进、成骨不全、遗传性低纤维蛋白原血症、ACT缺乏症、尿崩症(DI)、神经生长性(neurophyseal)尿崩症、肾源性尿崩症、夏-马-图三氏综合征、佩-梅病、神经变性疾病、阿尔茨海默病、帕金森病、肌萎缩性侧索硬化、进行性核上性麻痹、皮克病、几种多聚谷氨酰胺神经障碍、亨廷顿舞蹈病、I型脊髓小脑共济失调、脊髓延髓肌肉萎缩症、齿状核红核苍白球丘脑下部核萎缩、肌强直性营养不良、海绵样脑病、遗传性克雅病(由于朊病毒蛋白加工缺陷所致)、法布里病、Straussler-Scheinker综合征、慢性阻塞性肺病(COPD)、干眼病、斯耶格伦病、骨质疏松症、骨质减少、戈汉综合征、氯离子通道病、先天性肌强直病(Thomson和Becker形式)、III型巴特综合征、登特病、惊跳症(hyperekplexia)、癫痫、惊跳症、溶酶体贮积病、Angelman综合征、原发性纤毛运动障碍(PCD)、纤毛的结构和/或功能的遗传性疾病、具有内脏逆位的PCD(也称作卡塔格纳综合征)、没有内脏逆位的PCD、或纤毛发育不良。
在一个实施方案中,所述疾病是囊性纤维化、肺气肿、慢性阻塞性肺病或骨质疏松症。在一个实施方案中,所述疾病是囊性纤维化。
在一个实施方案中,所述受试者具有含(CFTR)ΔF508突变的囊性纤维化跨膜受体(CFTR)。在一个实施方案中,所述受试者具有含R117H突变的囊性纤维化跨膜受体(CFTR)。在一个实施方案中,所述受试者具有含G551D突变的囊性纤维化跨膜受体(CFTR)。
在一个实施方案中,所述方法包括:施用其它治疗剂。在一个实施方案中,所述其它治疗剂是溶粘蛋白剂、支气管扩张剂、抗生素、抗感染剂、抗炎剂、除了化合物1以外的CFTR调节剂或营养剂。在某些实施方案中,所述其它治疗剂是N-(5-羟基-2,4-二叔丁基-苯基)-4-氧代-1H-喹啉-3-甲酰胺。
附图说明
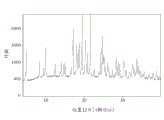
图1是通过喷雾干燥方法制备的化合物1无定形形式的X-射线粉末衍射图样。
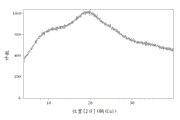
图2是通过喷雾干燥方法制备的化合物1无定形形式的调制式差示扫描量热法(MDSC)示踪图。
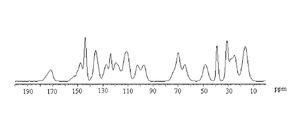
图3是化合物1无定形形式的固态13C NMR谱(15.0kHz自旋)。
图4是化合物1无定形形式的固态19F NMR谱(12.5kHz自旋)。
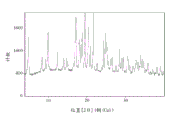
图5是通过旋转蒸发方法制备的化合物1无定形形式的X-射线粉末衍射图样。
图6是通过旋转蒸发方法制备的化合物1无定形形式的调制式差示扫描量热法(MDSC)示踪图。
图7是用DCM作为溶剂,通过料浆技术(2周)制备的化合物1形式A的实际X-射线粉末衍射图样。
图8是从化合物1形式A的单晶计算出的X-射线粉末衍射图样。
图9是化合物1形式A的差示扫描量热法(DSC)示踪图。
图10是自乙腈通过快速蒸发法制备的化合物1形式A的实际X-射线粉末衍射图样。
图11是使用EtOAc和庚烷,通过反溶剂法制备的化合物1形式A的实际X-射线粉末衍射图样。
图12是化合物1形式A基于单晶X-射线分析的构象图。
图13是化合物1形式A的固态13C NMR谱(15.0kHz自旋)。
图14是化合物1形式A的固态19F NMR谱(12.5kHz自旋)。
具体实施方式
定义
本文使用的术语“CFTR”意指囊性纤维化跨膜传导调节因子或其具有调节因子活性的突变,包括但不限于ΔF508CFTR和G551D CFTR (对于CFTR突变参见例如http://www.genet.sickkids.on.ca/cftr/)。
本文使用的术语“无定形的”表示由无序排列的分子组成并且不具有可辨别的晶格的固体形式。
本文使用的“结晶的”表示其中结构单元以固定的几何模式或晶格排列,使得结晶的固体具有刚性长程序的化合物或组合物。构成晶体结构的结构单元可为原子、分子或离子。结晶的固体显示确定的熔点。
本文使用的术语“调节”意指增加或减少例如活性达可测量的量。
本文使用的术语“化学稳定的”意指固体形式的化合物1在经受指定条件时不分解为一种或多种不同的化合物,例如经受40℃/75%相对湿度达特定的时间段,例如1天、2天、3天、1周、2周或者更长。在某些实施方案中,固体形式的化合物1中有少于25%分解,在某些实施方案中,少于约20%,少于约15%,少于约10%,少于约5%,少于约3%,少于约1%,少于约0.5%的所述形式的化合物1在所指定的条件下分解。在某些实施方案中,没有可测量的量的固体形式的化合物1分解。
本文使用的术语“物理稳定的”意指化合物1的固体形式在经受特定条件时不变成为化合物1的一种或多种不同物理形式(例如如经XRPD、DSC等测量的不同固体形式),例如经受40℃/75%相对湿度达特定的时间段,例如1天、2天、3天、1周、2周或者更长。在某些实施方案中,少于25%的化合物1的固体形式在经受指定条件时变成为一种或多种不同的物理形式。在某些实施方案中,少于约20%,少于约15%,少于约10%,少于约5%,少于约3%,少于约1%,少于约0.5%的化合物1的固体形式在经受指定条件时变成为化合物1的一种或多种不同物理形式。在某些实施方案中,没有可测量的量的化合物1的固体形式变成为化合物1的一种或多种物理上不同的固体形式。
术语“基本上不含”(如在短语“基本上不含形式X”中),当关于化合物1的指定固体形式(例如本文描述的无定形或结晶形式)时,意指存在少于20%(按重量计)的指定形式(诸指定形式)或共结晶剂(co-form)(诸共结晶剂)(例如化合物1的结晶或无定形形式),更优选地存在少于10%(按重量计)的指定形式(诸指定形式),更优选地存在少于5%(按重量计)的指定形式(诸指定形式),和最优选地存在少于1%(按重量计)的指定形式(诸指定形式)。
本文使用的“分散体”表示其中一种物质(分散相)以离散单元分布于全部第二种物质(连续相或媒介物)中的分散系统。分散相的大小可显著变化(例如纳米尺寸的胶体粒子,至大小为多个微米)。通常,分散相可为固体、液体或气体。在固体分散体的情况下,分散和连续相两者均为固体。在药学应用中,固体分散体可包括在无定形聚合物(连续相)中的结晶药物(分散相)或者在无定形聚合物(连续相)中的无定形药物(分散相)。在某些实施方案中,无定形固体分散体包括构成分散相的聚合物和构成连续相的药物。在某些实施方案中,分散体包括无定形化合物1或基本上无定形的化合物1。
术语“固体无定形分散体”一般指两种或更多种组分(通常为药物和聚合物)的固体分散体,但是可能含有其它组分诸如表面活性剂或其它药用赋形剂,其中化合物1为无定 形或基本上无定形(例如基本上不含结晶化合物1),并且无定形药物的物理稳定性和/或溶出度和/或溶解度被其它组分增强。
缩写“MTBE”和“DCM”分别代表甲基叔丁基醚和二氯甲烷。
缩写“XRPD”代表X-射线粉末衍射。
缩写“DSC”代表差示扫描量热法。
缩写“TGA”代表热重量分析法。
本文使用的术语“活性药物成分”或“API”表示生物活性化合物。示例性的API包括(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺(化合物1)。
当在本文中用于表示(R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺(化合物1)时,术语“固体形式”和有关的术语表示,包含化合物1的固体形式例如无定形粉末或晶体等,其主要不是呈液体或气体状态。
本文使用的术语“基本上无定形”是指在其分子位置中几乎不具有或没有长程序的固体材料。例如,基本上无定形的材料的结晶度小于约15%(例如,小于约10%的结晶度或小于约5%的结晶度)。还应当指出,术语“基本上无定形”包括描述词“无定形”,后者表示结晶度为零(0%)的物质。
本文使用的术语“基本上结晶的”(如在短语基本上结晶的化合物1形式A中)是指在其分子位置中主要具有长程序的固体材料。例如,基本上结晶的材料具有超过约85%的结晶度(例如,超过约90%的结晶度或超过约95%的结晶度)。还应当指出,术语“基本上结晶的”包括描述词“结晶的”,后者表示具有100%结晶度的物质。
当用于描述物质、组分、产物或形式时,术语“结晶的”和本文使用的有关术语是指,通过X-射线衍射测得,所述物质、组分或产物是基本上结晶的(参见,例如,Remington:The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins,Baltimore,Md.(2003);The United States Pharmacopeia(美国药典),第23版,1843-1844(1995))。
本文使用的术语“组合物”通常表示2种或更多种组分的组合物,通常是一种或多种药物(例如,一种药物(例如,化合物1无定形形式))和一种或多种药用赋形剂的组合物。
本文使用的术语“固体剂型”通常表示这样的药物组合物:当以口服给药模式使用时,其包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在这样的固体剂型中,将活性化合物与至少一种惰性的药学上可接受的赋形剂或载体相混合。
本文使用的“赋形剂”包括在药物组合物中的功能性的和非功能性的成分。
本文使用的“崩解剂”是水合药物组合物并辅助片剂分散的赋形剂。本文使用的“稀释剂”或“填充剂”是增加药物组合物的体积(bulkiness)的赋形剂。
本文使用的“表面活性剂”是赋予药物组合物增加的溶解度和/或可润湿能力的赋形剂。
本文使用的“粘合剂”是赋予药物组合物增加的粘结力或拉伸强度(例如,硬度)的赋形剂。
本文使用的“助流剂”是赋予药物组合物增加的流动性质的赋形剂。
本文使用的“着色剂”是赋予药物组合物希望的颜色的赋形剂。着色剂的实例包括可商购的色素,诸如FD&C Blue #1铝色淀(FD&C Blue #1 Aluminum Lake)、FD&C Blue#2、其它FD&C Blue的颜料、二氧化钛、氧化铁和/或它们的组合。在一个实施方案中,本发明提供的药物组合物为紫色。
本文使用的“润滑剂”是加入到压制成片剂的药物组合物中的赋形剂。润滑剂有助于将颗粒压紧成片剂和药物组合物的片剂从模压机射出。
本文使用的“立方厘米”和“cc”可互换地用于表示体积单位。应当指出,1cc=1mL。
本文使用的“kiloPond”和“kP”可互换地使用以及表示力的量度,其中1kP=大约9.8牛顿。
本文使用的“脆碎度”是指即使有外部压力的情况下片剂也能保持完整且保持其形式的性质。使用方程式1表示的数学表达式,可以对脆碎度进行定量:

其中W0是片剂的原始重量,Wf是片剂穿过脆碎度测定器以后的最终重量。使用标准的美国药典测试仪器来测量脆碎度,所述仪器翻滚实验片剂100或400转。本发明的某些片剂具有小于5.0%的脆碎度。在另一个实施方案中,所述脆碎度小于2.0%。在另一个实施方案中,在400转以后的目标脆碎度小于1.0%。
本文使用的“平均粒径”是使用诸如激光散射、图像分析或筛析等技术测得的平均粒径。在一个实施方案中,用于制备本发明提供的药物组合物的颗粒具有小于1.0mm的平均粒径。
本文使用的“堆密度”是指物质颗粒质量除以颗粒所占总体积的值。总体积包括颗 粒体积、颗粒间空体积和内部孔体积。堆密度不是物质的固有性质;它可以随加工物质的方法而变化。在一个实施方案中,用于制备本发明提供的药物组合物的颗粒具有约0.5-0.7g/cc的堆密度。
本发明的药物化合物的有效量或“治疗有效量”可以随诸如下述因素而变化:受试者的疾病状况、年龄和体重,以及本发明的化合物在受试者引起所需应答的能力。可以调节剂量方案,以提供最适治疗应答。有效量也是这样的量:其中本发明的化合物的治疗有益作用超过任何有毒或有害作用(例如,副作用)。
如本文使用的,其除非另外指出,术语化合物的“治疗有效量”和“有效量”是指这样量:其足以在疾病或障碍的治疗或控制中提供治疗益处,或延迟或最小化与疾病或障碍有关的一种或多种症状。化合物的“治疗有效量”和“有效量”是指治疗剂的这样的量:其单独地或与一种或多种其它药剂组合地,在疾病或障碍的治疗或控制中提供治疗益处。术语“治疗有效量”和“有效量”可以包括这样的量:其改善总体治疗,减少或避免疾病或障碍的症状或原因,或增强其它治疗剂的治疗效果。
如在在短语“基本上纯的化合物1无定形形式”中使用的,“基本上纯的”是指大于约90%纯度。在另一个实施方案中,基本上纯的表示大于约95%纯度。在另一个实施方案中,基本上纯的表示大于约98%纯度。在另一个实施方案中,基本上纯的表示大于约99%纯度。
就化合物1(即,化合物1无定形形式或化合物1形式A)而言,当与组合物或剂型中的成分的剂量、量或重量百分数结合使用时,术语“约”和“大约”意指由本领域普通技术人员公认提供相当于自指定剂量、量或重量百分数得到的药理作用的剂量、量或重量百分数。具体地讲,术语“约”或“大约”意指如由本领域普通技术人员测定的对特定值可接受的误差,其部分地取决于所述数值被如何测量或测定。在某些实施方案中,术语“约”或“大约”意指在1、2、3或4个标准偏差内。在某些实施方案中,术语“约”或“大约”意指在给定值或范围的30%、25%、20%、15%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%或0.05%内。
除非另外指出,本文描绘的结构也意指包括所述结构的所有异构体(例如对映、非对映和几何(或构象))形式,例如对每一个不对称中心的R和S构型,(Z)和(E)双键异构体以及(Z)和(E)构象异构体。因此,本发明化合物的单一立体化学异构体以及对映异构体、非对映异构体和几何(或构象)异构体的混合物处于本发明的范围内。本文包括化合物1的所有互变异构形式。例如,化合物1可作为互变异构体存在,本文包括其两者:
另外,除非另外指出,本文描绘的结构也意指包括仅在存在富含一种或多种同位素富集的原子方面不同的化合物。例如,其中一个或多个氢原子用氘或氚替代,或者一个或多个碳原子用富含13C-或14C-的碳替代的化合物1处于本发明的范围内。这样的化合物例如用作分析工具、生物测定中的探针或具有改善的治疗状况的化合物。
药物组合物
本发明提供了包含化合物1无定形形式或化合物1形式A的药物组合物、药物制剂和固体剂型诸如片剂。在该方面的某些实施方案中,存在于药物组合物中的化合物1的量是2.5mg、5mg、10mg、25mg、50mg、75mg、100mg、125mg、150mg或200mg。在该方面的某些实施方案中,存在于药物组合物中的化合物1的重量/重量相对百分比是10-50%。在这些和其它实施方案中,化合物1作为基本上纯的化合物1无定形形式存在。“基本上纯的”是指大于90%纯的;优选地大于95%纯的;更优选地大于99.5%纯的(即,不与化合物1的晶型混合)。
因而,在一个方面,本发明提供了一种药物组合物,其包含:
a.化合物1无定形形式;
b.填充剂;
c.崩解剂;
d.稀释剂;
e.润滑剂;和
g.助流剂。
在该方面的一个实施方案中,所述药物组合物包含2.5mg化合物1无定形形式。在该方面的一个实施方案中,所述药物组合物包含5mg化合物1无定形形式。在该方面的一个实施方案中,所述药物组合物包含10mg化合物1无定形形式。在该方面的一个实施方案中,所述药物组合物包含25mg化合物1无定形形式。在该方面的另一个实施方案中,所述药物组合物包含50mg化合物1无定形形式。在该方面的另一个实施方案中,所述药物组合物包含100mg化合物1无定形形式。在该方面的另一个实施方案中,所述药物组合物包含125mg化合物1无定形形式。在该方面的另一个实施方案中,所述药物组合物包含150mg 化合物1无定形形式。在该方面的另一个实施方案中,所述药物组合物包含200mg化合物1无定形形式。
在某些实施方案中,所述药物组合物包含化合物1无定形形式,其中按组合物的重量计,化合物1无定形形式以下述量存在:至少4重量%(例如,至少5重量%、至少10重量%、至少20重量%、至少30重量%、至少40重量%、至少50重量%或至少60重量%)。
在某些实施方案中,所述药物组合物包含化合物1无定形形式、填充剂、稀释剂、崩解剂、助流剂和润滑剂。在该实施方案中,所述组合物包含约4重量%至约50重量%(例如,约10-45重量%)的化合物1无定形形式(按组合物的重量计),且更通常地,20重量%至约40重量%(例如,约25-30重量%)的化合物1无定形形式(按组合物的重量计)。
在某些实施方案中,所述药物组合物包含化合物1无定形形式、填充剂、稀释剂、崩解剂、助流剂和润滑剂。在该实施方案中,所述组合物包含约4重量%至约50重量%(例如,约10-45重量%)的化合物1无定形形式(按组合物的重量计),且更通常地,20重量%至约40重量%(例如,约25-30重量%)的化合物1无定形形式(按组合物的重量计)。
所述组合物中的化合物1无定形形式的浓度取决于几种因素,诸如提供所需量的化合物1无定形形式需要的药物组合物的量和药物组合物的期望的溶出度特性。
在另一个实施方案中,所述药物组合物包含这样的化合物1:其中通过光散射测得(例如,使用可从英格兰的Malvern Instruments得到的Malvern Mastersizer),处于它的固体形式的化合物1具有0.1微米至10微米的平均粒径。在另一个实施方案中,化合物1的粒度是1微米至5微米。在另一个实施方案中,化合物1具有2.0微米的粒度D50。
如指出的,除了化合物1无定形形式以外,在本发明的某些实施方案中,作为口服制剂的药物组合物也包含一种或多种赋形剂诸如填充剂、崩解剂、表面活性剂、稀释剂、助流剂、润滑剂、着色剂或芳香剂和它们的任意组合。
适用于本发明的填充剂与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、硬度、化学稳定性、物理稳定性或生物活性。示例性的填充剂包括:纤维素、改性纤维素、(例如羧甲基纤维素钠、乙基纤维素、羟甲基纤维素、羟丙基纤维素)、乙酸纤维素、微晶纤维素、磷酸钙、磷酸氢钙、淀粉(例如玉米淀粉、马铃薯淀粉)、糖(例如,山梨醇)乳糖、蔗糖等)或它们的任意组合。
因而,在一个实施方案中,所述药物组合物包含至少一种填充剂,按组合物的重量计,其量为至少5重量%(例如,至少约20重量%、至少约30重量%或至少约40重量%)。例如,所述药物组合物包含占所述组合物重量的约10重量%至约60重量%(例如,约10重 量%至约55重量%、约15重量%至约30重量%、或约20重量%至约25重量%)的填充剂。在另一个实施例中,所述药物组合物包含占所述组合物重量的至少约20重量%(例如,至少20重量%或至少20重量%)的微晶纤维素,例如MCC Avicel PH102。
适用于本发明的崩解剂会增强所述药物组合物的分布,且与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的化学稳定性、物理稳定性、硬度或生物活性。示例性的崩解剂包括:交联羧甲纤维素钠、淀粉羟乙酸钠或它们的组合。
因而,在一个实施方案中,所述药物组合物包含崩解剂,其量为所述组合物重量的约10重量%或更小(例如,约7重量%或更小、约6重量%或更小、或约5重量%或更小)。例如,所述药物组合物包含占所述组合物重量的约1重量%至约10重量%(例如,约1.5重量%至约7.5重量%、或约2.5重量%至约6重量%)的崩解剂。在某些实施例中,所述药物组合物包含占所述组合物重量的约0.1%至约10重量%(例如,约0.5重量%至约7.5重量%、或约1.5重量%至约6重量%)的崩解剂。在其它实施例中,所述药物组合物包含占所述组合物重量的约0.5%至约10重量%(例如,约1.5重量%至约7.5重量%、或约2.5重量%至约6重量%)的崩解剂。
适用于本发明的表面活性剂会增加所述药物组合物的润湿性,且与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的化学稳定性、物理稳定性、硬度或生物活性。示例性的表面活性剂包括:月桂基硫酸钠(SLS)、硬脂酰富马酸钠(SSF)、聚氧乙烯20脱水山梨糖醇单油酸酯(例如,吐温TM)、它们的任意组合等。
因而,在一个实施方案中,所述药物组合物包含表面活性剂,其量为所述组合物重量的约10重量%或更小(例如,约5重量%或更小、约2重量%或更小、约1重量%或更小、约0.8重量%或更小、或约0.6重量%或更小)。例如,所述药物组合物包含占所述组合物重量的约10重量%至约0.1重量%(例如,约5重量%至约0.2重量%、或约2重量%至约0.3重量%)的表面活性剂。在另一个实施例中,所述药物组合物包含占所述组合物重量的约10重量%至约0.1重量%(例如,约5重量%至约0.2重量%、或约2重量%至约0.3重量%)的月桂基硫酸钠。
适用于本发明的稀释剂可以给制剂增加必要的体积,以制备具有期望的大小的片剂,且通常与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、硬度、化学稳定性、物理稳定性或生物活性。示例性的稀释剂包括:糖,例如糖果剂的糖、可压缩的糖、葡聚糖结合剂、糊精、葡萄糖、乳糖、乳糖一水合物、甘露醇、山梨醇、纤维素和改性纤维素,例如纤维素粉末、滑石粉、磷酸钙、淀粉或它们的任意组合。
因而,在一个实施方案中,所述药物组合物包含稀释剂,其量为所述组合物重量的40重量%或更小(例如,35重量%或更小、30重量%或更小、或25重量%或更小、或20重量%或更小、或15重量%或更小、或10重量%或更小)。例如,所述药物组合物包含占所述组合物重量的约40重量%至约1重量%(例如,约35重量%至约5重量%、或约30重量%至约7重量%、约25重量%至约15重量%)的稀释剂。在另一个实施例中,所述药物组合物包含占所述组合物重量的40重量%或更小(例如,35重量%或更小、或25重量%或更小)的乳糖一水合物。在另一个实施例中,所述药物组合物包含占所述组合物重量的约35重量%至约1重量%(例如,约30重量%至约5重量%、或约25重量%至约10重量%)的乳糖一水合物。
适用于本发明的助流剂会增加所述药物组合物的流动性质,且与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、硬度、化学稳定性、物理稳定性或生物活性。示例性的助流剂包括:胶体二氧化硅、滑石或它们的组合。
因而,在一个实施方案中,所述药物组合物包含助流剂,其量为所述组合物重量的2重量%或更小(例如,1.75重量%、1.25重量%或更小、或1.00重量%或更小)。例如,所述药物组合物包含占所述组合物重量的约2重量%至约0.05重量%(例如,约1.5重量%至约0.07重量%、或约1.0重量%至约0.09重量%)的助流剂。在另一个实施例中,所述药物组合物包含占所述组合物重量的2重量%或更小(例如,1.75重量%、1.25重量%或更小、或1.00重量%或更小)的胶体二氧化硅。在另一个实施例中,所述药物组合物包含占所述组合物重量的约2重量%至约0.05重量%(例如,约1.5重量%至约0.07重量%、或约1.0重量%至约0.09重量%)的胶体二氧化硅。
在某些实施方案中,所述药物组合物可以包括口服固体药物剂型,其可以包含润滑剂,所述润滑剂可以防止造粒珠混合物(granulate-bead admixture)附着于表面(例如,搅拌钵、压模和/或冲压机的表面)。润滑剂还可以减小造粒机内的颗粒间摩擦,并改善压制的药物组合物在模压机中的压制和射出。所述润滑剂还与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、硬度或生物活性。示例性的润滑剂包括:硬脂酸镁、硬脂酸钙、硬脂酸锌、硬脂酸钠、硬脂酸、硬脂酸铝、亮氨酸、山嵛酸甘油酯、氢化植物油或它们的任意组合。在一个实施方案中,所述药物组合物包含润滑剂,其量为所述组合物重量的5重量%或更小(例如,4.75重量%、4.0重量%或更小、或3.00重量%或更小、或2.0重量%或更小)。例如,所述药物组合物包含占所述组合物重量的约5重量%至约0.10重量%(例如,约4.5重量%至约0.5重量%、或约3重量%至约0.5重量%)的润滑剂。在另一个 实施例中,所述药物组合物包含占所述组合物重量的5重量%或更小(例如,4.0重量%或更小、3.0重量%或更小、或2.0重量%或更小、或1.0重量%或更小)的硬脂酸镁。在另一个实施例中,所述药物组合物包含占所述组合物重量的约5重量%至约0.10重量%(例如,约4.5重量%至约0.15重量%、或约3.0重量%至约0.50重量%)的硬脂酸镁。
本发明的药物组合物可以任选地包含一种或多种着色剂、矫味剂和/或芳香剂,以增强所述组合物的视觉吸引力、味觉和/或气味。合适的着色剂、矫味剂或芳香剂与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、化学稳定性、物理稳定性、硬度或生物活性。在一个实施方案中,所述药物组合物包含着色剂、矫味剂和/或芳香剂。在一个实施方案中,本发明提供的药物组合物为紫色。
在某些实施方案中,所述药物组合物包括或可以制成片剂,可以用着色剂包衣所述片剂,并任选地使用合适的墨水用图形(logo)、其它图像和/或文字进行标记。在其它实施方案中,所述药物组合物包括或可以制成片剂,可以用着色剂包衣所述片剂,上蜡,并任选地使用合适的墨水用图形、其它图像和/或文字进行标记。合适的着色剂和墨水与所述药物组合物的成分相容,即,它们基本上不减小所述药物组合物的溶解度、化学稳定性、物理稳定性、硬度或生物活性。所述合适的着色剂和墨水可以是任意颜色,且是基于水的或基于溶剂的。在一个实施方案中,用着色剂包衣由所述药物组合物制成的片剂,然后使用合适的墨水用图形、其它图像和/或文字进行标记。例如,包含本文所述药物组合物的片剂可以被约3重量%(例如,小于约6重量%、或小于约4重量%)的包含着色剂的薄膜包衣进行包衣。可以用图形和文字标记所述着色的片剂,所述图形和文字使用合适的墨水指示片剂中活性成分的强度。在另一个实施例中,包含本文所述药物组合物的片剂可以被约3重量%(例如,小于约6重量%或小于约4重量%)的包含着色剂的薄膜包衣进行包衣。
在另一个实施方案中,用着色剂包衣由所述药物组合物制成的片剂,上蜡,然后使用合适的墨水用图形、其它图像和/或文字进行标记。例如,包含本文所述药物组合物的片剂可以被约3重量%(例如,小于约6重量%、或小于约4重量%)的包含着色剂的薄膜包衣进行包衣。可以用巴西棕榈蜡粉末将着色的片剂上蜡,所述巴西棕榈蜡粉末的重量占起始片芯重量的约0.01%w/w。可以用图形和文字标记所述上蜡的片剂,所述图形和文字使用合适的墨水指示片剂中活性成分的强度。在另一个实施例中,包含本文所述药物组合物的片剂可以被约3重量%(例如,小于约6重量%、或小于约4重量%)的包含着色剂的薄膜包衣进行包衣。可以用巴西棕榈蜡粉末将着色的片剂上蜡,所述巴西棕榈蜡粉末的重量占起始片芯重量的约0.01%w/w。可以用图形和文字标记所述上蜡的片剂,所述图形和文字使用药品级墨水 诸如黑色墨水(例如,  S-1-17823,一种基于溶剂的墨水,可从宾夕法尼亚州的Colorcon,Inc.of West Point商购获得)指示片剂中活性成分的强度。
S-1-17823,一种基于溶剂的墨水,可从宾夕法尼亚州的Colorcon,Inc.of West Point商购获得)指示片剂中活性成分的强度。
一种示例性的药物组合物包含占所述组合物重量的约4重量%至约70重量%(例如,约10重量%至约60重量%、约15重量%至约50重量%、或约25重量%至约50重量%、或约20重量%至约70重量%、或约30重量%至约70重量%、或约40重量%至约70重量%、或约50重量%至约70重量%)的化合物1无定形形式。前述组合物还可以包括一种或多种药学上可接受的赋形剂,例如,约20重量%至约50重量%的填充剂;约1重量%至约5重量%的崩解剂;约2重量%至约0.25重量%的表面活性剂;约1重量%至约30重量%的稀释剂;约2重量%至约0.05重量%的助流剂;和约5重量%至约0.1重量%的润滑剂。或者,所述药物组合物包含这样的组合物,其含有:约15重量%至约70重量%(例如,约20重量%至约60重量%、约25重量%至约55重量%、或约30重量%至约50重量%)的化合物1无定形形式(按组合物的重量计);和一种或多种赋形剂,例如,约20重量%至约50重量%的填充剂;约1重量%至约5重量%的崩解剂;约2重量%至约0.25重量%的表面活性剂;约1重量%至约30重量%的稀释剂;约2重量%至约0.05重量%的助流剂;和约5重量%至约0.1重量%的润滑剂。
另一种示例性的药物组合物包含:约4重量%至约70重量%(例如,约10重量%至约60重量%、约15重量%至约50重量%、或约25重量%至约50重量%、或约20重量%至约70重量%、或约30重量%至约70重量%、或约40重量%至约70重量%、或约50重量%至约70重量%)的化合物1无定形形式(按组合物的重量计)和一种或多种赋形剂,例如,约20重量%至约50重量%的填充剂;约1重量%至约5重量%的崩解剂;约2重量%至约0.25重量%的表面活性剂;约1重量%至约30重量%的稀释剂;约2重量%至约0.05重量%的助流剂;和约2重量%至约0.1重量%的润滑剂。
在一个实施方案中,本发明是干燥的掺合物或颗粒状药物组合物,其包含:
a.约25重量%的化合物1无定形形式(按组合物的重量计);
b.约22.5重量%的微晶纤维素(按组合物的重量计);
c.约22.5重量%的乳糖一水合物(按组合物的重量计);
d.约3重量%的交联羧甲纤维素钠(按组合物的重量计);
e.约0.25重量%的月桂基硫酸钠(按组合物的重量计);
f.约0.5重量%的硬脂酸镁(按组合物的重量计);和
g.约1.25重量%的胶体二氧化硅(按组合物的重量计)。
在一个实施方案中,本发明是干燥的掺合物或颗粒状药物组合物,其包含:
a.约25重量%的化合物1无定形形式(按组合物的重量计);
b.约22.5重量%的微晶纤维素(按组合物的重量计);
c.约22.5重量%的乳糖一水合物(按组合物的重量计);
d.约3重量%的交联羧甲纤维素钠(按组合物的重量计);
e.约0.25重量%的月桂基硫酸钠(按组合物的重量计);
f.约0.5重量%的硬脂酸镁(按组合物的重量计);
g.约1.25重量%的胶体二氧化硅(按组合物的重量计);和
h.约25重量%的聚合物。
在一个实施方案中,本发明是干燥的掺合物或颗粒状药物组合物,其包含:
a.约5重量%的化合物1无定形形式(按组合物的重量计);
b.约42.9重量%的微晶纤维素(按组合物的重量计);
c.约42.9重量%的乳糖一水合物(按组合物的重量计);
d.约3重量%的交联羧甲纤维素钠(按组合物的重量计);
e.约0.5重量%的硬脂酸镁(按组合物的重量计);
g.约1.25重量%的胶体二氧化硅(按组合物的重量计);和
h.约5重量%的聚合物。
在另一个实施方案中,所述聚合物是HPMCAS。
本发明的药物组合物可以加工成片剂形式、胶囊剂形式、药袋形式、锭剂形式或适合口服施用的其它固体形式。因而,在某些实施方案中,所述药物组合物是片剂形式。
在本发明的另一种药物口服制剂中,具有5-21kP±20%的初始硬度的成形的药物片剂组合物包含:约25重量%的化合物1无定形形式;约22.5重量%的微晶纤维素(按组合物的重量计);约22.5重量%的乳糖一水合物(按组合物的重量计);约3重量%的交联羧甲纤维素钠(按组合物的重量计);约0.25重量%的月桂基硫酸钠(按组合物的重量计);约0.5重量%的硬脂酸镁(按组合物的重量计);和约1.25重量%的胶体二氧化硅(按组合物的重量计)。其中所述成形药物片剂中的化合物1无定形形式的量是在每片约25mg至约200mg(例如,50mg或75mg或100mg或150mg或200mg)化合物1无定形形式的范围内。
在某些实施方案中,所述成形药物片剂含有约10mg化合物1无定形形式。在某些实施方案中,所述成形药物片剂含有约50mg化合物1无定形形式。在某些实施方案中,所 述成形药物片剂含有约100mg化合物1无定形形式。
本发明的另一个方面提供了一种由片剂或胶囊剂组成的药物制剂,所述片剂或胶囊剂包括化合物1无定形形式和其它赋形剂(例如,填充剂、崩解剂、表面活性剂、助流剂、着色剂、润滑剂或它们的任意组合),它们中的每一种如上所述和在下面的实施例中所述,其中所述片剂具有在约30分钟内至少约50%(例如,至少约60%、至少约70%、至少约80%、至少约90%、或至少约99%)的溶出度。在一个实施例中,所述药物组合物由片剂组成,所述片剂包括25mg至200mg(例如,25mg或50mg或75mg或100mg或150mg或200mg)的量的化合物1无定形形式和一种或多种赋形剂(例如,填充剂、崩解剂、表面活性剂、助流剂、着色剂、润滑剂或它们的任意组合),它们中的每一种如上所述和在下面的实施例中所述,其中所述片剂具有在约30分钟内约50%至约100%(例如,约55%至约95%、或约60%至约90%)的溶出度。
在一个实施方案中,所述片剂包含组合物,所述组合物包含:至少约10mg(例如,至少约25mg、至少约30mg、至少约40mg或至少约50mg)的化合物1无定形形式;和一种或多种选自填充剂、稀释剂、崩解剂、表面活性剂、助流剂和润滑剂的赋形剂。在另一个实施方案中,所述片剂包含包含组合物,所述组合物包含:至少约10mg(例如,至少约25mg、至少约30mg、至少约40mg、至少约50mg、至少约100mg或至少150mg)的化合物1无定形形式和一种或多种选自填充剂、稀释剂、崩解剂、表面活性剂、助流剂和润滑剂的赋形剂。
可以使用标准的美国药典II型仪器来测量溶出度,所述仪器采用溶解在900mL去离子水(用50mM磷酸二氢钾在pH 6.8缓冲)中的0.1%CTAB作为溶出介质,其在约37℃的温度以约50-75rpm搅拌。在所述仪器的每个试验管中测试单个实验片剂。还可以使用标准的美国药典II型仪器来测量溶出度,所述仪器采用溶解在900mL 50mM磷酸钠缓冲液(pH6.8)中的0.7%月桂基硫酸钠作为溶出介质,其在约37℃的温度以约65rpm搅拌。在所述仪器的每个试验管中测试单个实验片剂。还可以使用标准的美国药典II型仪器来测量溶出度,所述仪器采用溶解在900mL 50mM磷酸钠缓冲液(pH 6.8)中的0.5%月桂基硫酸钠作为溶出介质,其在约37℃的温度以约65rpm搅拌。在所述仪器的每个试验管中测试单个实验片剂。
制备化合物1无定形形式和化合物1形式A的方法
化合物1为起点,并且在一个实施方案中,化合物1可按照方案1-4,通过使酰氯部分与胺部分偶联来制备。
方案1.酰氯部分的合成。

方案2.酰氯部分的合成——替代合成。
方案3.胺部分的合成。

方案4.化合物1的形成。
制备化合物1无定形形式的方法
从化合物1或甚至化合物1的晶型开始,通过旋转蒸发方法或通过喷雾干燥方法可制备化合物1无定形形式。
将化合物1溶解于合适的溶剂如甲醇中,并旋转蒸发甲醇,剩下泡沫,产生化合物1 无定形形式。在某些实施方案中,将温水浴用于加快蒸发。
也可使用喷雾干燥方法自化合物1制备化合物1无定形形式。喷雾干燥为一种将液体进料转化为干燥颗粒形式的方法。任选地,二级干燥过程诸如流化床干燥或真空干燥可用于把残留的溶剂减少至药学上可接受的水平。通常,喷雾干燥包括使高度分散的液体混悬液或溶液与足够体积的热空气接触,以产生蒸发和干燥液滴。要喷雾干燥的制剂可为任何溶液、粗粒混悬液、浆料、胶态分散体或糊剂,它们可使用所选择的喷雾干燥设备雾化。在标准程序下,制剂被喷雾成温热过滤的空气流,其蒸发溶剂并将干燥的产物传送至收集器(例如旋风分离器)。所消耗的空气然后与溶剂一起排出,或者备选地所消耗的空气被送往冷凝器以捕获和潜在地回收溶剂。可商购得到的设备类型可用于实施喷雾干燥。例如,商业上的喷雾干燥器由Buchi Ltd.And Niro制造(例如由Niro制造的PSD line喷雾干燥器)(参见US2004/0105820;US 2003/0144257)。
喷雾干燥通常采用约3%-约30%重量,例如约4%-约20%重量,优选地为至少约10%的固体负荷的材料(即药物和赋形剂)。通常,固体负荷的上限由所生成溶液的粘度(例如泵送能力)和溶液中组分的溶解度来控制。一般地,溶液的粘度可决定所生成的粉末产品中的粒子大小。
用于喷雾干燥的技术和方法可见于Perry’s Chemical Engineering Handbook,第6版,R.H.Perry,D.W.Green & J.O.Maloney编辑),McGraw-Hill book co.(1984);和Marshall“Atomization and Spray-Drying”50,Chem.Eng.Prog.Monogr.Series 2(1954)。通常,喷雾干燥采用以下入口温度进行:约60℃-约200℃,例如约95℃-约185℃、约110℃-约182℃、约96℃-约180℃,例如约145℃。喷雾干燥通常采用以下出口温度进行:约30℃-约90℃,例如约40℃-约80℃、约45℃-约80℃,例如约75℃。雾化流速通常为约4kg/h-约12kg/h,例如约4.3kg/h-约10.5kg/h,例如约6kg/h或约10.5kg/h。进料流速通常为约3kg/h-约10kg/h,例如约3.5kg/h-约9.0kg/h,例如约8kg/h或约7.1kg/h。雾化比通常为约0.3-1.7,例如约0.5-1.5,例如约0.8或约1.5。
去除溶剂可需要随后的干燥步骤,诸如盘式干燥、流化床干燥(例如约室温-约100℃)、真空干燥、微波干燥、滚桶干燥或双锥形真空干燥(例如约室温-约200℃)。
在一个实施方案中,固体分散体被流化床干燥。
在一种方法中,溶剂包括挥发性溶剂,例如具有小于约100℃的沸点的溶剂。在某些实施方案中,溶剂包括溶剂的混合物,例如挥发性溶剂的混合物或者挥发性与非挥发性溶剂的混合物。当使用溶剂的混合物时,这种混合物可包含一种或多种非挥发性溶剂,例如,其 中非挥发性溶剂以少于约15%,例如少于约12%、少于约10%、少于约8%、少于约5%、少于约3%或少于约2%存在于混合物中。
优选的溶剂为其中化合物1具有至少约10mg/ml(例如至少约15mg/ml、20mg/ml、25mg/ml、30mg/ml、35mg/ml、40mg/ml、45mg/ml、50mg/ml或者更大)的溶解度的那些溶剂。更优选的溶剂包括其中化合物1具有至少约20mg/ml的溶解度的那些溶剂。
可被试验的示例性溶剂包括丙酮、环己烷、二氯甲烷、N,N-二甲基乙酰胺(DMA)、N,N-二甲基甲酰胺(DMF)、1,3-二甲基-2-咪唑啉酮(DMI)、二甲亚砜(DMSO)、二噁烷、乙酸乙酯、乙醚、冰醋酸(HAc)、甲乙酮(MEK)、N-甲基-2-吡咯烷酮(NMP)、甲基叔丁基醚(MTBE)、四氢呋喃(THF)、戊烷、乙腈、甲醇、乙醇、异丙醇、乙酸异丙酯和甲苯。示例性的共溶剂包括丙酮/DMSO、丙酮/DMF、丙酮/水、MEK/水、THF/水、二噁烷/水。在双溶剂系统中,溶剂可以约0.1%-约99.9%存在。在一些优选的实施方案中,水为与丙酮的共溶剂,其中水以约0.1%-约15%,例如约9%-约11%,例如约10%存在。在一些优选的实施方案中,水为与MEK的共溶剂,其中水以约0.1%-约15%,例如约9%-约11%,例如约10%存在。在某些实施方案中,溶剂溶液包含3种溶剂。例如,丙酮和水可与第三种溶剂诸如DMA、DMF、DMI、DMSO或HAc混合。在其中无定形的化合物1为固体无定形分散体的组分的情况下,优选的溶剂溶解化合物1和聚合物两者。合适的溶剂包括以上描述的那些溶剂,例如MEK、丙酮、水、甲醇,及其混合物。
可改变粒度和干燥温度范围以制备最佳的固体分散体。如由熟练的实践者应意识到的那样,粒度小将导致改善的溶剂的去除。然而申请人已经发现,较小的粒子可导致粒子蓬松,其在一些情况下对于下游加工诸如压片而言不能提供最佳的固体分散体。在较高温度下,可发生化合物1的结晶或化学降解。在较低温度下,不能去除足够量的溶剂。本文的方法提供最佳粒度和最佳干燥温度。
通常,粒度使得D10(μm)小于约5,例如小于约4.5、小于约4.0或小于约3.5,D50(μm)通常小于约17,例如小于约16、小于约15、小于约14、小于约13,且D90(μm)通常小于约175,例如小于约170、小于约170、小于约150、小于约125、小于约100、小于约90、小于约80、小于约70、小于约60或小于约50。通常喷雾干燥的粒子的堆密度为约0.08g/cc-约0.20g/cc,例如约0.10-约0.15g/cc,例如约0.11g/cc或约0.14g/cc。喷雾干燥的粒子的振实密度通常为:对于轻敲10次,在约0.08g/cc-约0.20g/cc,例如约0.10-约0.15g/cc的范围内,例如为约0.11g/cc或约0.14g/cc;对于轻敲500次,在0.10g/cc-约0.25g/cc,例如约0.11-约0.21g/cc的范围内,例如为约0.15g/cc、约0.19g/cc或约0.21g/cc;对 于轻敲1250次,在0.15g/cc-约0.27g/cc,例如约0.18-约0.24g/cc的范围内,例如为约0.18g/cc、约0.19g/cc、约0.20g/cc或约0.24g/cc;和对于轻敲2500次,在0.15g/cc-约0.27g/cc,例如约0.18-约0.24g/cc的范围内,例如为约0.18g/cc、约0.21g/cc、约0.23g/cc或约0.24g/cc。
聚合物
包含化合物1无定形形式和聚合物(或固态载体)的固体分散体也包括在本文中。例如,化合物1作为固体无定形分散体的组分,作为无定形化合物存在。这种固体无定形分散体通常包含化合物1和聚合物。示例性的聚合物包括纤维素类聚合物诸如HPMC或HPMCAS和含有吡咯烷酮的聚合物诸如PVP/VA。在某些实施方案中,这种固体无定形分散体包含一种或多种另外的赋形剂,诸如表面活性剂。
在一个实施方案中,聚合物能够溶解于水性介质中。聚合物的溶解度可为不依赖于pH的或pH依赖性的。后者包括一种或多种肠溶性聚合物。术语“肠溶性聚合物”表示相对于胃的更酸性的环境,优先溶于肠的较低酸性环境的聚合物,例如不溶于酸性的水性介质但是当pH高于5-6时可溶的聚合物。合适的聚合物应为化学和生物学上惰性的。为了改善固体分散体的物理稳定性,聚合物的玻璃化转变温度(Tg)应尽可能地高。例如,优选的聚合物具有至少等于或大于药物(即化合物1)的玻璃化转变温度的玻璃化转变温度。其它优选的聚合物具有在药物(即化合物1)的约10-约15℃范围内的玻璃化转变温度。聚合物的合适的玻璃化转变温度的实例包括至少约90℃、至少约95℃、至少约100℃、至少约105℃、至少约110℃、至少约115℃、至少约120℃、至少约125℃、至少约130℃、至少约135℃、至少约140℃、至少约145℃、至少约150℃、至少约155℃、至少约160℃、至少约165℃、至少约170℃、或至少约175℃(如在干燥条件下测量的那样)。不希望受到理论的束缚,据信根本的机制是具有较高Tg的聚合物通常在室温具有较低分子可动性,这可能是稳定无定形固体分散体的物理稳定性的关键因素。
另外,聚合物的吸湿性应低至例如小于约10%。为了在该应用中进行比较的目的,聚合物或组合物的吸湿性在约60%相对湿度下表征。在一些优选的实施方案中,聚合物具有小于约10%的吸水率,例如小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%或小于约2%的吸水率。吸湿性也可影响固体分散体的物理稳定性。通常,聚合物中吸附的水分可大大地降低聚合物以及生成的固体分散体的Tg,这将进一步降低以上描述的固体分散体的物理稳定性。
在一个实施方案中,聚合物为一种或多种水溶性的聚合物或部分水溶性的聚合物。 水溶性或部分水溶性的聚合物包括但不限于纤维素衍生物(例如羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC))或乙基纤维素;聚乙烯吡咯烷酮(PVP);聚乙二醇(PEG);聚乙烯醇(PVA);丙烯酸酯,诸如聚甲基丙烯酸酯(例如  );环糊精(例如β-环糊精)及其共聚物和衍生物,包括例如PVP-VA(聚乙烯吡咯烷酮-醋酸乙烯酯)。
);环糊精(例如β-环糊精)及其共聚物和衍生物,包括例如PVP-VA(聚乙烯吡咯烷酮-醋酸乙烯酯)。
在某些实施方案中,聚合物为羟丙基甲基纤维素(HPMC),诸如HPMC E50、HPMCE15或HPMC60SH50)。
如本文讨论的那样,聚合物可为pH依赖性的肠溶性聚合物。这样的pH依赖性肠溶性聚合物包括但不限于纤维素衍生物(例如醋酸邻苯二甲酸纤维素(CAP))、羟丙基甲基纤维素邻苯二甲酸酯(HPMCP)、醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)、羧甲基纤维素(CMC)或其盐(例如钠盐诸如(CMC-Na));乙酸-1,2,4-苯三酸纤维素(CAT)、醋酸邻苯二甲酸羟丙基纤维素(HPCAP)、醋酸邻苯二甲酸羟丙基甲基纤维素(HPMCAP)和醋酸邻苯二甲酸甲基纤维素(MCAP)或聚甲基丙烯酸酯(例如  E)。在某些实施方案中,聚合物为醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)。在某些实施方案中,聚合物为醋酸羟丙基甲基纤维素琥珀酸酯HG级(HPMCAS-HG)。
E)。在某些实施方案中,聚合物为醋酸羟丙基甲基纤维素琥珀酸酯(HPMCAS)。在某些实施方案中,聚合物为醋酸羟丙基甲基纤维素琥珀酸酯HG级(HPMCAS-HG)。
在又一个实施方案中,聚合物为聚乙烯吡咯烷酮共聚物,例如乙烯吡咯烷酮/醋酸乙烯酯共聚物(PVP/VA)。
在其中化合物1与聚合物,例如与HPMC、HPMCAS或PVP/VA聚合物形成固体分散体的实施方案中,聚合物相对于固体分散体的总重量的量在约0.1重量%-99重量%的范围内。除非另外指定,如所描述的药物、聚合物及其它赋形剂在分散体中的百分数以重量百分数给出。聚合物的量一般地为至少约20%,并且优选地为至少约30%,例如至少约35%、至少约40%、至少约45%或约50%(例如49.5%)。所述量一般地为约99%或者更少,并且优选地为约80%或者更少,例如约75%或者更少、约70%或者更少、约65%或者更少、约60%或者更少,或者约55%或者更少。在一个实施方案中,聚合物以分散体的总重量的最多约50%(并且甚至更具体地讲,在约40%-50%之间,诸如约49%、约49.5%或约50%)的量存在。HPMC和HPMCAS可以多种级别得自ShinEtsu,例如HPMCAS可以多种级别得到,包括AS-LF、AS-MF、AS-HF、AS-LG、AS-MG、AS-HG。这些级别中的每一种随着乙酸酯和琥珀酸酯的取代百分数而变化。
在某些实施方案中,化合物1和聚合物以大致相等的量存在,例如聚合物和药物中的每一种组成分散体的重量百分数的约一半。例如,聚合物以约49.5%存在和药物以约50%存在。
在某些实施方案中,所合并的化合物1和聚合物代表在喷雾干燥之前的非固体分散体的1%-20%w/w总固体含量。在某些实施方案中,所合并的化合物1和聚合物代表在喷雾干燥之前的非固体分散体的5%-15%w/w总固体含量。在某些实施方案中,所合并的化合物1和聚合物代表在喷雾干燥之前的非固体分散体的约11%w/w总固体含量。
在某些实施方案中,所述分散体还包含其它次要成分,诸如表面活性剂(例如SLS)。在某些实施方案中,表面活性剂以少于分散体的约10%存在,例如少于约9%,少于约8%,少于约7%,少于约6%,少于约5%,少于约4%,少于约3%,少于约2%,约1%或约0.5%存在。
在包含聚合物的实施方案中,聚合物应以有效稳定固体分散体的量存在。稳定包括抑制或防止化合物1的结晶。这样的稳定将抑制化合物1从无定形形式转化为结晶形式。例如,聚合物将会防止至少一部分(例如约5%、约10%、约15%、约20%、约25%、约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%或者更多)的化合物1从无定形形式转化为结晶形式。稳定化作用可例如通过测量固体分散体的玻璃化转变温度、测量无定形材料的弛豫率(rate of relaxation)或者通过测量化合物1的溶解度或生物利用度来测量。
用于与化合物1组合,例如形成固体分散体诸如无定形固体分散体的合适的聚合物应具有一种或多种以下性质:
聚合物的玻璃化转变温度应具有不低于比化合物1的玻璃化转变温度低约10-15℃的温度。优选地,聚合物的玻璃化转变温度大于化合物1的玻璃化转变温度,并且通常比药品期望的储存温度高至少50℃。例如,至少约100℃、至少约105℃、至少约105℃、至少约110℃、至少约120℃、至少约130℃、至少约140℃、至少约150℃、至少约160℃、至少约160℃或者更大。
聚合物应为相对非吸湿性的。例如,当在标准条件下储存时,聚合物应吸附少于约10%的水,例如少于约9%、少于约8%、少于约7%,少于约6%,或少于约5%,少于约4%,或少于约3%的水。优选地,当在标准条件下储存时,聚合物应基本上不含吸附水。
聚合物在适合于喷雾干燥法的溶剂中,相对于化合物1应具有相似或更好的溶解度。在优选的实施方案中,聚合物将溶解于一种或多种与化合物1相同的溶剂或溶剂系统中。优选的是聚合物可溶于至少一种含有非羟基溶剂的溶剂诸如二氯甲烷、丙酮或其组合中。
当与化合物1合并时(例如在固体分散体或在液体混悬液中),相对于化合物1在不 存在聚合物时的溶解度,或相对于化合物1在与参考聚合物合并时的溶解度,聚合物应增大化合物1在水性和生理相对介质中的溶解度。例如,聚合物可以通过减少从固体无定形分散体或从液体混悬液转化为结晶化合物1的无定形化合物1的量来增大无定形化合物1的溶解度。
聚合物应减小无定形物质的弛豫率。
聚合物应增大化合物1的物理和/或化学稳定性。
聚合物应改善化合物1的可制造性。
聚合物应改善化合物1的处理、给药或储存性质中的一种或多种。
聚合物不应与其它药用组分例如赋形剂不利地相互作用。
候选聚合物(或其它组分)的适用性可使用本文描述的形成无定形组合物的喷雾干燥法(或其它方法)进行测试。候选组合物可就稳定性、耐晶体形成性或其它性质进行比较,并且可以与参考制剂例如纯的无定形化合物1或结晶化合物1的制剂进行比较。例如,候选组合物可被测试以确定其是否抑制出现溶剂介导的结晶的时间,或者抑制在受控条件下在给定的时间转化百分率达至少50%、75%、100%或110%并且参考制剂或者候选组合物可被测试以确定相对于结晶化合物1其是否改善生物利用度或溶解度。
表面活性剂
固体分散体或其它组合物可包含表面活性剂。表面活性剂或表面活性剂混合物通常将减小固体分散体与水性介质之间的界面张力。合适的表面活性剂或表面活性剂混合物也可增强化合物1自固体分散体的水溶解度和生物利用度。连同本发明一起使用的表面活性剂包括但不限于去水山梨糖醇脂肪酸酯(例如司盘类  )、聚氧乙烯去水山梨糖醇脂肪酸酯(例如吐温类
)、聚氧乙烯去水山梨糖醇脂肪酸酯(例如吐温类  )、月桂基硫酸钠(SLS)、十二烷基苯磺酸钠(SDBS)、磺基丁二酸钠二辛酯(多库酯钠)、二氧胆酸钠盐(DOSS)、去水山梨糖醇单硬脂酸酯、去水山梨糖醇三硬脂酸酯、十六烷基三甲基溴化铵(HTAB)、N-月桂酰肌氨酸钠、油酸钠、肉豆蔻酸钠、硬脂酸钠、棕榈酸钠、月桂酸聚乙二醇甘油酯(Gelucire 44/14)、乙二胺四乙酸(EDTA)、维生素E d-α生育酚基聚乙二醇1000琥珀酸酯(TPGS)、卵磷脂、MW 677-692、谷氨酸单钠一水合物、辛酸癸酸聚乙二醇甘油酯(Labrasol)、PEG 8辛酸/癸酸甘油酯类、二乙二醇单乙基醚(Transcutol)、二乙二醇单乙醚、聚乙二醇硬脂酸酯15(Solutol HS-15)、聚乙二醇/羟基硬脂酸酯、牛磺胆酸、普朗尼克(Pluronic)F68、普朗尼克F108和普朗尼克F127(或任何其它聚氧乙烯-聚氧丙烯共聚物
)、月桂基硫酸钠(SLS)、十二烷基苯磺酸钠(SDBS)、磺基丁二酸钠二辛酯(多库酯钠)、二氧胆酸钠盐(DOSS)、去水山梨糖醇单硬脂酸酯、去水山梨糖醇三硬脂酸酯、十六烷基三甲基溴化铵(HTAB)、N-月桂酰肌氨酸钠、油酸钠、肉豆蔻酸钠、硬脂酸钠、棕榈酸钠、月桂酸聚乙二醇甘油酯(Gelucire 44/14)、乙二胺四乙酸(EDTA)、维生素E d-α生育酚基聚乙二醇1000琥珀酸酯(TPGS)、卵磷脂、MW 677-692、谷氨酸单钠一水合物、辛酸癸酸聚乙二醇甘油酯(Labrasol)、PEG 8辛酸/癸酸甘油酯类、二乙二醇单乙基醚(Transcutol)、二乙二醇单乙醚、聚乙二醇硬脂酸酯15(Solutol HS-15)、聚乙二醇/羟基硬脂酸酯、牛磺胆酸、普朗尼克(Pluronic)F68、普朗尼克F108和普朗尼克F127(或任何其它聚氧乙烯-聚氧丙烯共聚物  或饱和的聚乙醇酸化甘油酯
或饱和的聚乙醇酸化甘油酯  )。可连同本发明一起使用的这类表面活性剂的具体实例包括但不限于司盘65、司盘25、吐温20、Capryol 90、普朗尼克 F108、月桂基硫酸钠(SLS)、维生素E TPGS、普朗尼克类和共聚物。SLS通常为优选的。
)。可连同本发明一起使用的这类表面活性剂的具体实例包括但不限于司盘65、司盘25、吐温20、Capryol 90、普朗尼克 F108、月桂基硫酸钠(SLS)、维生素E TPGS、普朗尼克类和共聚物。SLS通常为优选的。
表面活性剂(例如SLS)相对于固体分散体的总重量的量可在0.1-15%之间。优选地,该量为约0.5%-约10%,更优选地为约0.5%-约5%,例如约0.5-4%、约0.5-3%、约0.5-2%、约0.5-1%或约0.5%。
在某些实施方案中,表面活性剂相对于固体分散体的总重量的量为至少约0.1%,优选地为约0.5%。在这些实施方案中,表面活性剂将以不多于约15%,和优选地不多于约12%、约11%、约10%、约9%、约8%、约7%、约6%、约5%、约4%、约3%、约2%或约1%的量存在。其中表面活性剂以约0.5重量%的量存在的实施方案为优选的。
可以与针对测试聚合物所描述的类似的方式测试候选表面活性剂(或其它组分)用于本发明的适用性。
形成化合物1形式A的方法
在一个实施方案中,化合物1形式A通过将化合物1在合适的溶剂中浆化有效量的时间进行制备。在另一个实施方案中,合适的溶剂为乙酸乙酯、二氯甲烷、MTBE、乙酸异丙酯、各种比例的水/乙醇溶液、各种比例的水/乙腈溶液、各种比例的水/甲醇溶液或各种比例的水/异丙醇溶液。例如,各种比例的水/乙醇溶液包括:水/乙醇1∶9(体积/体积)、水/乙醇1∶1(体积/体积)和水/乙醇9∶1(体积/体积)。各种比例的水/乙腈溶液包括水/乙腈1∶9(体积/体积)、水/乙腈1∶1(体积/体积)和水/乙腈9∶1(体积/体积)。各种比例的水/甲醇溶液包括水/甲醇1∶9(体积/体积)、水/甲醇1∶1(体积/体积)和水/甲醇9∶1(体积/体积)。各种比例的水/异丙醇溶液包括水/异丙醇1∶9(体积/体积)、水/异丙醇1∶1(体积/体积)和水/异丙醇9∶1(体积/体积)。
通常,把约40mg的化合物1用约1.5ml的合适溶剂(目标浓度为26.7mg/ml)在室温浆化有效量的时间。在某些实施方案中,有效量的时间为约24小时至约2周。在某些实施方案中,有效量的时间为约24小时至约1周。在某些实施方案中,有效量的时间为约24小时至约72小时。然后收集固体。
在另一个实施方案中,化合物1形式A通过将化合物1溶解于合适的溶剂中,然后蒸发溶剂进行制备。在一个实施方案中,合适的溶剂为其中化合物1具有大于20mg/ml的溶解度的溶剂。例如,这些溶剂包括乙腈、甲醇、乙醇、异丙醇、丙酮等。
通常,将化合物1溶解于合适的溶剂中,过滤,然后放置缓慢蒸发或快速蒸发。缓慢蒸发的实例为用其中具有一个戳洞的石蜡封口膜覆盖包含化合物1溶液的容器(诸如小瓶)。快速蒸发的实例为无覆盖地放置包含化合物1溶液的容器(诸如小瓶)。然后收集固体。
另一方面,本发明的特征在于制备化合物1形式A的方法,所述方法包括将化合物1溶解于第一溶剂中,并加入化合物1在其中具有不佳溶解度(溶解度<1mg/ml)的第二溶剂。例如,所述第一溶剂可为化合物1在其中具有大于20mg/ml溶解度的溶剂,例如乙酸乙酯、乙醇、异丙醇或丙酮。所述第二溶剂可为例如庚烷或水。
通常,将化合物1溶解于第一溶剂中并过滤,以除去任何晶种。在搅拌的同时缓慢加入第二溶剂。固体沉淀并经过滤收集。
制备药物组合物的方法
本发明的剂量单位形式可以通过在压力下压缩或压制混合物或组合物(例如,粉末或颗粒)以形成稳定的三维形状(例如,片剂)来制备。本文使用的“片剂”包括所有形状和大小的压制的药物剂量单位形式(不论是包衣或者是未包衣的)。
本文使用的表述“剂量单位形式”是指适合于待治疗患者的药剂的物理上离散的单位。通常,压缩的混合物的密度大于压缩之前的该混合物的密度。本发明的剂量单位形式可以具有几乎任何形状,包括凹和/或凸状表面、圆形或楔形角和圆形至直线性的形状。在某些实施方案中,本发明的压制剂型包括具有平表面的圆形片剂。本发明的固体药物剂型可以通过本领域普通技术人员已知的、形成压制固体药物剂型的任何压缩和压制方法来制备。在具体实施方案中,本文提供的制剂可以使用药物制剂领域技术人员已知的常规方法来制备,例如,在相关的教科书中所描述的方法。参见,例如,Remington:The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins,Baltimore,Md.(2003);Ansel等人,Pharmaceutical Dosage Forms And Drug Delivery Systems,第7版,Lippincott Williams & Wilkins,(1999);The Handbook of Pharmaceutical Excipients,第4版,Rowe等编,American Pharmaceuticals Associaion(2003);Gibson,Pharmaceutical Preformulation And Formulation,CRC Press(2001),这些参考文献特此通过引用整体并入本文。
制粒和压制
在某些实施方案中,可以对包含活性剂化合物1无定形形式和包括的药学上可接受的赋形剂(例如填充剂、稀释剂、崩解剂、表面活性剂、助流剂、润滑剂或它们的任意组合)的固体形式(包括粉末)进行干法制粒过程。干法制粒过程导致粉末聚集成为具有适合于进一步加工的大小的大颗粒。干法制粒可以提高混合物的流动性,从而能够制备符合质量变化或装量差异要求的片剂。
本文所描述的制剂可以使用一个或多个混合和干法制粒步骤来制备。混合和制粒步骤的顺序和数量似乎不是关键性的。然而,可以对赋形剂和化合物1中的至少一种进行干法 制粒或湿式高剪切制粒,而后压制成为片剂。在片剂压制之前,使化合物1无定形形式和赋形剂结合在一起进行的干法制粒似乎意外地是在本发明组合物和制剂的组分之间提供紧密物理接触的简单、廉价和有效方法,并由此产生具有良好稳定性的片剂制剂。干法制粒可以通过机械方法进行,与本文还涉及的湿式制粒方法相反,这种方法将能量转移至混合物,而不使用任何液体物质(不是水溶液形式、基于有机溶质的溶液形式或其混合物形式)。通常,机械方法需要压缩,例如碾压所提供的压缩。干法制粒的另一个方法的例子是击压法(slugging)。
在某些实施方案中,碾压是包括高强度机械压缩一种或多种物质的制粒方法。在某些实施方案中,在2个反向旋转辊之间,将包含粉末混合物的药物组合物压缩,即碾压,以产生固体薄片,随后将其在筛网中粉碎,从而形成颗粒物质。在这种颗粒物质中,可以获得组分之间的紧密机械接触。碾压设备的例子是  Gerteis 3W-Polygran(得自Gerteis Maschinen+Processengineering AG)。
Gerteis 3W-Polygran(得自Gerteis Maschinen+Processengineering AG)。
在某些实施方案中,可以在不使用任何液体物质(不是水溶液形式、基于有机溶质的溶液形式或其混合物形式)的条件下,进行按照本发明的片剂压制,即,干法制粒方法。在典型的实施方案中,得到的核或片剂具有在1至15kP范围内的压缩强度;例如,1.5至12.5kP,优选在2至10kP范围内的压缩强度。
制备方法概述
在某些实施方案中,按照本文给定的配方称量组分。接下来,将所有颗粒内的组分过筛,并很好地混合。可以用合适的润滑剂(例如,硬脂酸镁)润滑组分。下一步可以包括压缩/击压粉末混合物和筛过的组分。接下来,将压缩或击压过的掺合物碾磨成颗粒,并过筛,以获得所需要的大小。接下来,进一步用例如硬脂酸镁润滑颗粒。接下来,可以在合适的冲头上压制本发明的颗粒组合物以形成按照本发明的各种药物制剂。任选地,可以用薄膜、着色剂或其它包衣将片剂包衣。
本发明的另一个方面提供了制备药物组合物的方法,该方法包括:提供组合物的混合物,其中组合物包含化合物1无定形形式和一种或多种选自下列的赋形剂:填充剂、稀释剂、助流剂、表面活性剂、润滑剂、崩解剂,并将该组合物压制成为片剂,这种片剂具有在约30分钟内至少约50%的溶出度。
在另一个实施方案中,进行湿式制粒方法,以由粉末组分和液体组分的混合物得到本发明的药物制剂。例如,按照本文给定的配方称量包含组合物(所述组合物包含化合物1无定形形式和一种或多种选自下列的赋形剂:填充剂、稀释剂、助流剂、表面活性剂、润滑 剂、崩解剂)的混合物的药物组合物。接下来,将所有颗粒内的组分过筛,并在高剪切或低剪切造粒机中使用水或水与表面活性剂或水与粘合剂或水与表面活性剂和粘合剂混合,以使该粉末掺合物形成颗粒。也可以使用非水流体,其可以与或不与表面活性剂和/或粘合剂一起使用,以使粉末掺合物形成颗粒。接下来,可以任选使用合适的碾磨机来碾磨湿润的颗粒。接下来,水可以任选地通过以任何合适的方式干燥组分从而从混合物中除去。接下来,可以任选将干燥的颗粒碾磨至所需大小。接下来,可以通过掺合的方式加入颗粒外赋形剂(例如填充剂、稀释剂和崩解剂)。接下来,可以用硬脂酸镁和崩解剂(例如,交联羧甲纤维素钠)进一步润滑定径的颗粒。接下来,可以将本发明的颗粒组合物筛分足够的时间,以获得合适的粒径,而后在合适的冲头上压制成按照本发明的各种药物制剂。任选地,可以用薄膜、着色剂或其它包衣将片剂包衣。
这种示例性混合物的每种组分如上所述和下面实施例所述。此外,该混合物可以包含任选的添加剂,例如上面描述的和下面实施例描述的一种或多种着色剂、一种或多种调味剂和/或一种或多种芳香剂。在某些实施方案中,上面和下面实施例也提供了该混合物中的这些组分(和任何任选的添加剂)的每种组分的相对浓度(例如,重量%)。可以顺序地、或以任何组合加入形式提供组成该混合物的组分;并且可以以任何顺序提供组分或组分的组合。在一个实施方案中,润滑剂是最后加入到该混合物中的组分。
在另一个实施方案中,该混合物包含下述物质的组合物:化合物1无定形形式,和任何一种或多种选自助流剂、表面活性剂、稀释剂、润滑剂、崩解剂和填充剂的赋形剂,其中以粉末形式提供这些组分当中的每种组分(例如,提供平均直径(利用光散射测定)为250μm或更小(例如,150μm或更小、100μm或更小、50μm或更小、45μm或更小、40μm或更小、或35μm或更小)的颗粒)。例如,该混合物包含化合物1无定形形式、稀释剂、助流剂、表面活性剂、润滑剂、崩解剂和填充剂的组合物,其中以粉末形式提供这些组分当中的每种组分(例如,提供平均直径(利用光散射测定)为250μm或更小(例如,150μm或更小、100μm或更小、50μm或更小、45μm或更小、40μm或更小、或35μm或更小)的颗粒)。在另一个实施例中,该混合物包含化合物1无定形形式、稀释剂、表面活性剂、润滑剂、崩解剂和填充剂的组合物,其中以粉末形式提供这些组分当中的每种组分(例如,提供平均直径(利用光散射测定)为250μm或更小(例如,150μm或更小、100μm或更小、50μm或更小、45μm或更小、40μm或更小、或35μm或更小)的颗粒)。
在另一个实施方案中,该混合物包含化合物1无定形形式与下列的任何组合的组合 物:助流剂、稀释剂、表面活性剂、润滑剂、崩解剂和填充剂,其中这些组分当中的每种组分基本上不含水。每种组分包含占所述组分重量的小于5重量%(例如、小于2重量%、小于1重量%、小于0.75重量%、小于0.5重量%或小于0.25重量%)的水。例如,该混合物包含化合物1无定形形式、稀释剂、助流剂、表面活性剂、润滑剂、崩解剂和填充剂的组合物,其中这些组分当中的每种组分基本上不含水。在某些实施方案中,每种组分包含占所述组分重量的小于5重量%(例如,小于2重量%、小于1重量%、小于0.75重量%、小于0.5重量%、或小于0.25重量%)的水。
在另一个实施方案中,将该混合物压制成片剂是如下实现的:用混合物填充模板(form)(例如,模具),并对混合物施加压力。这可以使用模压机或其它类似的装置来实现。在某些实施方案中,可以首先将化合物1无定形形式和赋形剂的混合物加工成颗粒形式。然后,可以按照药物领域已知的方法筛分颗粒并压制成片剂或进行配制以用于包封。还应该注意,在每次压制期间,对模板中的混合物施加压力可以使用相同压力重复进行,或在挤压期间使用不同压力来进行。在另一个实施例中,可以使用施加足够压力的模压机来压制粉末化的组分或颗粒的混合物,以形成在大约30分钟时溶解大约50%或溶解更多的片剂(例如,在大约30分钟时溶解大约55%或更多,或者在大约30分钟时溶解大约60%或更多)。例如,使用模压机来压制该混合物,以产生至少大约5kP(至少大约5.5kP,至少大约6kP,至少大约7kP,至少大约10kP或至少15kP)的片剂硬度。在有些情况下,压缩该混合物,以产生大约5和20kP之间的片剂硬度。
在某些实施方案中,包含本文所描述药物组合物的片剂可以用占片剂重量的大约3.0重量%的薄膜包衣进行包衣,其中薄膜包衣包含着色剂。在某些情况下,用于包衣片剂的着色剂混悬液或溶液包含占该着色剂混悬液或溶液重量的大约20%w/w的固体。在更进一步情况下,可以用图形、其它图像或文字给包衣片剂做标记。
在另一个实施方案中,制备药物组合物的方法包括:提供固体形式的混合物,例如,粉末和/或液体组分的混合物,该混合物包含化合物1无定形形式和一种或多种选自下列的赋形剂:助流剂、稀释剂、表面活性剂、润滑剂、崩解剂和填充剂;将该混合物混合,直到混合物基本上均匀为止,并将该混合物压制或压缩成颗粒形式。然后,如上面所描述或下面实施例所描述,可以将包含化合物1无定形形式的颗粒组合物压制成片剂,或配制到胶囊剂中。可替换地,制备药物组合物的方法包括:提供化合物1无定形形式和一种或多种下列赋形剂的混合物:助流剂、稀释剂、表面活性剂、润滑剂、崩解剂和填充剂;将该混合物混合,直到混合物基本上均匀为止,并使用辊压机,使用下面实施例列出的干法制粒组合 物,将该混合物压制/压缩成颗粒形式,或者可替换地,使用下面实施例列出的高剪切湿式颗粒压缩方法,将该混合物压制/压缩成颗粒。药物制剂,例如,本文所描述的片剂,可以使用颗粒来制备,这种颗粒除了结合所选择的本文描述的赋形剂之外,还结合了化合物1无定形形式。
在某些实施方案中,使用手工搅拌、混合器、搅拌机、其任何联用形式等等,通过搅拌、掺和、摇动等等来将混合物混合。当顺序地加入组分或组分的组合物时,混合可以在顺序加入之间进行、可以在连续加入组分的整个过程中进行混合、在加入所有组分或组分的组合物之后进行混合或其任何组合形式。将混合物混合,直到它具有基本上均匀的组成时为止。
在一个实施方案中,本发明的药物组合物可以根据下述流程图来制备:

在另一个实施方案中,本发明的药物组合物可以根据下述流程图来制备:

在另一个实施方案中,本发明的药物组合物可以根据下述流程图来制备:

在另一个实施方案中,化合物1无定形形式是聚合物和表面活性剂的混合物的50重量%,使用的胶体二氧化硅助流剂的商标是Cabot M5P,使用的交联羧甲纤维素钠崩解剂的商标是AcDiSol,使用的微晶纤维素填充剂的商标是Avicel PH101,使用的乳糖一水合物稀释剂的商标是Foremost 310。在另一个实施方案中,所述化合物1无定形形式聚合物是羟丙甲基纤维素(HPMC),所述表面活性剂是月桂基硫酸钠。在另一个实施方案中,所述化合物1无定形形式聚合物是醋酸羟丙甲基纤维素琥珀酸酯(HPMCAS)。在另一个实施方案中,所述化合物1无定形形式聚合物是醋酸羟丙甲基纤维素琥珀酸酯-高级(HPMCAS-HG)。
在不同的实施方案中,可以与化合物1无定形形式一起配制第二治疗剂,以形成单一或单次剂量形式,例如,片剂或胶囊剂。
按照United States Pharmacopoeia 29(United States Pharmacopeial Convention,Inc.,Rockville,Md.,2005(“USP”))中的Test 711“Dissolution”,可以对上面制备的剂型进行体外溶解评价,以测定活性物质从该剂型中释放的速率。通过例如高效液相色谱(HPLC)的技术,方便地测定活性物质的含量和杂质水平。
在某些实施方案中,本发明包括使用包装材料,例如,高密度聚乙烯(HDPE)、低密度聚乙烯(LDPE)和/或聚丙烯和/或玻璃的容器和密封盒(closures),玻璃纸薄片,铝箔袋和 由铝或高密度聚氯乙烯(PVC)组成,任选包括干燥剂、聚乙烯(PE)、聚偏二氯乙烯(PVDC)、PVC/PE/PVDC等等的泡罩或条板。在使用药物领域通常利用的化学或物理杀菌技术将包装和其内含物进行合适的杀菌之后,这些包装材料可以用于以无菌方式保存各种药物组合物和制剂。
施用药物组合物的方法
在一个方面,可以每天或大约每24小时给患者施用本发明的药物组合物一次。可替换地,可以每天两次或大约每12小时1次地给患者施用本发明的药物组合物。以口服制剂形式施用这些药物组合物,所述口服制剂包含约2.5mg、5mg、10mg、25mg、50mg、100mg、125mg、150mg或200mg化合物1无定形形式。在该方面,除了化合物1无定形形式之外,药物组合物还包含填充剂、稀释剂、崩解剂、表面活性剂、助流剂和润滑剂。
还可以理解,本发明的化合物和药学上可接受的组合物和制剂可以在联合治疗中使用;也就是说,化合物1无定形形式和其药学上可接受的组合物可以与一种或多种其它所需要的疗法或医疗过程同时施用、在其之前或在其之后施用。用于联合方案的疗法(治疗或过程)的具体组合将考虑目标疗法和/或过程的相容性以及所要达到的目标治疗效果。将理解,所使用的疗法对于相同病症可以达到目标效果(例如,本发明的化合物可以与另一种治疗相同病症所使用的药剂同时施用),或它们可以达到不同效果(例如,控制任何副作用)。本文使用的“其它治疗剂”(其通常用于治疗或预防具体疾病(例如,CFTR介导的疾病)或病症)被称为“适合于所治疗的疾病或病症的治疗剂”。
在一个实施方案中,其它治疗剂选自:溶粘蛋白剂、支气管扩张剂、抗生素、抗感染剂、消炎剂、除了本发明的化合物1以外的CFTR调节剂或营养剂。
在一个实施方案中,所述其它治疗剂为抗生素。用于本文的示例性抗生素包括妥布霉素,包括妥布霉素吸入性粉末(TIP);阿奇霉素;氨曲南(aztreonam),包括氨曲南的雾化形式;阿米卡星,包括其脂质体剂型;环丙沙星,包括其适合于经吸入给药的制剂;左氧氟沙星,包括其气雾剂;和两种抗生素例如磷霉素与妥布霉素的组合。
在另一个实施方案中,所述其它药剂为粘液溶解药(mucolyte)。用于本文的示例性粘液溶解药(mucolytes)包括 
在另一个实施方案中,所述其它药剂为支气管扩张剂。示例性的支气管扩张剂包括沙丁胺醇、硫酸奥西那林(metaprotenerol sulfate)、醋酸吡布特罗、沙美特罗或tetrabuline sulfate。
在另一个实施方案中,所述其它药剂有效恢复肺部气道表面液体。这类药剂改善盐 进出细胞的运动,使得肺部气道中的粘液被更多水合,并因此更易于清除。示例性的这类药剂包括高渗盐水、地纽福索钠(denufosol tetrasodium)([[(3S,5R)-5-(4-氨基-2-氧代嘧啶-1-基)-3-羟基氧杂戊环-2-基(hydroxyoxolan-2-yl)]甲氧基-羟基磷酰基][[[(2R,3S,4R,5R)-5-(2,4-二氧代嘧啶-1-基)-3,4-二羟基氧杂戊环-2-基]甲氧基-羟基磷酰基]氧基-羟基磷酰基]磷酸氢盐)或bronchitol(甘露醇的吸入剂型)。
在另一个实施方案中,所述其它药剂为抗炎剂,即可减少肺部炎症的药剂。用于本文的示例性的这类药剂包括布洛芬、二十二碳六烯酸(DHA)、西地那非、吸入性谷胱甘肽、吡格列酮、羟氯喹或斯伐他汀。
在另一个实施方案中,所述其它药剂为除了化合物1以外的CFTR调节剂,即具有调节CFTR活性作用的药剂。示例性的这类药剂包括阿他卢仑(  3-[5-(2-氟苯基)-1,2,4-噁二唑-3-基]苯甲酸)、西那普肽、兰考韦泰、地来司他(一种重组人中性粒细胞弹性蛋白酶抑制剂)和考前列酮(7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-二氟-3-甲基戊基]-2-羟基-6-氧代八氢环戊二烯并[b]吡喃-5-基}庚酸)。
3-[5-(2-氟苯基)-1,2,4-噁二唑-3-基]苯甲酸)、西那普肽、兰考韦泰、地来司他(一种重组人中性粒细胞弹性蛋白酶抑制剂)和考前列酮(7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-二氟-3-甲基戊基]-2-羟基-6-氧代八氢环戊二烯并[b]吡喃-5-基}庚酸)。
在另一个实施方案中,所述其它药剂为营养剂。示例性的营养剂包括胰脂肪酶(胰酶替代品),包括 或  (以前的
(以前的  )、
)、  或谷胱甘肽吸入。在一个实施方案中,所述其它营养剂为胰脂肪酶。
或谷胱甘肽吸入。在一个实施方案中,所述其它营养剂为胰脂肪酶。
在另一个实施方案中,所述其它药剂为选自以下的化合物:庆大霉素、姜黄素、环磷酰胺、4-苯基丁酸酯、美格鲁特、非洛地平、尼莫地平、Philoxin B、染料木黄酮(geniestein)、芹菜配基、cAMP/cGMP调节剂例如咯利普兰、西地那非、米力农、他达拉非、氨力农、异丙肾上腺素、沙丁胺醇和沙美特罗、脱氧精胍菌素、HSP 90抑制剂、HSP70抑制剂、蛋白酶体抑制剂例如环氧霉素、乳胞素等。
在其它实施方案中,所述其它药剂为在WO 2004028480、WO 2004110352、WO2005094374、WO 2005120497或WO 2006101740中公开的化合物。
在另一个实施方案中,所述其它药剂为呈现CFTR调节活性的苯并[c]喹嗪鎓衍生物或呈现CFTR调节活性的苯并吡喃衍生物。
在另一个实施方案中,所述其它药剂为在美国专利号7,202,262、美国专利号6,992,096、US20060148864、US20060148863、US20060035943、US20050164973、W02006110483、WO2006044456、WO2006044682、WO2006044505、WO2006044503、WO2006044502或WO2004091502中公开的化合物。在另一个实施方案中,所述其它药剂为 在WO2004080972、WO2004111014、WO2005035514、WO2005049018、WO2006099256、WO2006127588或WO2007044560中公开的化合物。在另一个实施方案中,所述其它药剂为N-(5-羟基-2,4-二叔丁基-苯基)-4-氧代-1H-喹啉-3-甲酰胺。
在一个实施方案中,可以将100mg化合物1施用给有此需要的受试者,然后共同施用150mg N-(5-羟基-2,4-二叔丁基-苯基)-4-氧代-1H-喹啉-3-甲酰胺(化合物2)。在另一个实施方案中,可以将100mg化合物1施用给有此需要的受试者,然后共同施用250mg化合物2。在这些实施方案中,通过施用一个或多个本发明的片剂,可以实现所述剂量的量。化合物2可以作为包含化合物2和药学上可接受的载体的药物组合物施用。施用的持续时间可以持续至实现疾病的改善,或直到受试者的医师通知,例如施用的持续时间可以是小于1周、1周、2周、3周、或1个月或更久。在共同施用阶段之前,可以存在仅单独施用化合物1的阶段。例如,可以施用100mg化合物1达2周,然后共同施用150mg或250mg化合物2达另外1周。
在一个实施方案中,可以给有此需要的受试者每天1次地施用100mg化合物1,然后每天1次地共同施用150mg化合物2。在另一个实施方案中,可以给有此需要的受试者每天1次地施用100mg化合物1,然后每天1次地共同施用250mg化合物2。在这些实施方案中,通过施用一个或多个本发明的片剂,可以实现所述剂量的量。化合物2可以作为包含化合物2和药学上可接受的载体的药物组合物施用。施用的持续时间可以持续至实现疾病的改善,或直到受试者的医师通知,例如施用的持续时间可以是小于1周、1周、2周、3周、或1个月或更久。在共同施用阶段之前,可以存在仅单独施用化合物1的阶段。例如,可以施用100mg化合物1达2周,然后共同施用150mg或250mg化合物2达另外1周。
在一个实施方案中,可以给有此需要的受试者每天1次地施用100mg化合物1,然后每12小时地共同施用150mg化合物2。在另一个实施方案中,可以给有此需要的受试者每天1次地施用100mg化合物1,然后每12小时地共同施用250mg化合物2。在这些实施方案中,通过施用一个或多个本发明的片剂,可以实现所述剂量的量。化合物2可以作为包含化合物2和药学上可接受的载体的药物组合物施用。施用的持续时间可以持续至实现疾病的改善,或直到受试者的医师通知,例如施用的持续时间可以是小于1周、1周、2周、3周、或1个月或更久。在共同施用阶段之前,可以存在仅单独施用化合物1的阶段。例如,可以施用100mg化合物1达2周,然后共同施用150mg或250mg化合物2达另外1周。
这些组合用于治疗本文描述的疾病,包括囊性纤维化。这些组合也用于本文描述的试剂盒。
存在于本发明组合物中的其它治疗剂的量将不多于将以包含所述治疗剂作为唯一活性药剂的组合物通常施用的量。优选地,目前公开的组合物中的其它治疗剂的量将在通常存在于包含所述药剂作为唯一治疗活性药剂的组合物中的量的约50%-100%范围内。
药物组合物的治疗用途
在某些实施方案中,包含化合物1无定形形式和任选的其它药剂的药学上可接受的组合物可用于治疗患者的囊性纤维化或减轻其严重程度,所述患者在呼吸道和非呼吸道上皮的顶端膜(apical membrane)中呈现残留的CFTR活性。使用本领域已知的方法,可容易地检测在上皮表面存在残留的CFTR活性,所述方法例如标准的电生理学、生物化学或组织化学技术。这类方法使用体内或离体电生理技术测量汗液或唾液的Cl-浓度,或者监测细胞表面密度的离体生化或组织化学技术鉴定CFTR活性。使用这类方法,在杂合或同型结合多种不同突变的患者,包括在同型结合或杂合最常见的突变ΔF508及其它突变诸如G551D突变或R117H突变的患者,可容易地检测残留的CFTR活性。
在一个实施方案中,本文描述的化合物1无定形形式或其药学上可接受的组合物可用于治疗患者的囊性纤维化或减轻其严重程度,所述患者的囊性纤维化处于呈现残留CFTR活性的某些基因型例如III类突变(调节或门控受损)、IV类突变(转运改变)或V类突变(合成减少)的范围内(Lee R.Choo-Kang,Pamela L.,Zeitlin,Type I,II,III,IV,and V cystic fibrosisTansmembrane Conductance Regulator Defects and Opportunities of Therapy;Current Opinion in Pulmonary Medicine 6:521-529,2000)。呈现残留CFTR活性的其它患者基因型包括对这些类型之一同型结合或与包括I类突变、II类突变或缺乏分类的突变的任何其它类型突变杂合的患者。
在一个实施方案中,本文描述的化合物1无定形形式或其药学上可接受的组合物可用于治疗患者的囊性纤维化或减轻其严重程度,所述患者的囊性纤维化处于某些临床表型的范围内,例如通常与上皮的顶端膜中的残留CFTR活性的量有关的中度至轻度临床表型。这种表型包括呈现胰腺功能不全的患者或诊断出患有自发性胰腺炎和先天性输精管两侧缺失或轻度肺疾病的患者。
所需要的确切的量将在受试者之间变化,这取决于受试者的物种、年龄和一般条件、感染的严重性、具体药剂、其给药方式等。本发明的化合物优选地以便于施用和剂量一致的剂量单位形式配制。本文使用的表述“剂量单位形式”表示适合于待治疗的患者的药剂的物理离散单位。然而应该理解,本发明化合物和组合物总每天用法将由主治医生在合理的医学判断范围内确定。对于任何具体患者或生物体的具体有效剂量水平将依多种因素而定, 这些因素包括被治疗的障碍和障碍的严重性;所采用的具体化合物的活性;所采用的具体组合物;患者的年龄、体重、一般健康状况、性别和饮食;所采用的具体化合物的给药时间、给药途径和排泄速率;治疗持续时间;与所采用的具体化合物组合或同时使用的药物以及医学领域熟知的类似因素。本文使用的术语“患者”意指动物,优选地为哺乳动物,且最优选地为人。
实施例
方法和材料
调制式差示扫描量热法(MDSC)和差示扫描量热法(DSC)
调制式差示扫描量热法(MDSC)用于测试化合物的无定形形式和喷雾干燥的分散体的玻璃化转变温度。差示扫描量热法(DSC)用于测定结晶材料的熔点和在不同的多晶形物之间进行辨别。数据使用TA DSC Q2000差示扫描热量计(TA Instruments,New Castle,DE)收集。这种仪器用铟校正。将约1-5mg样品称重至用有一个洞的盖子卷曲的铝密封的锅中。对于MDSC以2℃/分钟的加热速率,伴随每60秒调整+/-1℃,从-20℃-220℃扫描样品。对于DSC以10℃/分钟的加热速率,从25℃-220℃扫描样品。数据通过Thermal Advantage Q SeriesTM软件(版本:2.7.0.380)收集和通过Universal Analysis软件(版本:4.4A,build:4.4.0.5)(TA Instruments,New Castle,DE)分析。
XRPD(X-射线粉末衍射)
X-射线粉末衍射用于表征迄今产生的各批样品的物理形式和表征所鉴定的不同多晶形物。化合物的XRPD数据在PANalytical X’pert Pro Powder X-ray Diffractometer(Almelo,the Netherlands)上收集。XRPD图样在室温用铜辐射(1.54060A)记录。X-射线使用Cu密封管,在45kV,40mA下,用Nickel Kβ(镍Kβ)抑制滤波器产生。入射光束由可变发散狭缝组成,以确保在样品上和在衍射光侧为恒定的发光波长;使用具有以扫描模式测量的2.12°2θ的有效长度的快速线性固态探测器。将粉末样品填充在零背景硅固定装置的缩进区并进行旋转以获得更好的统计数据。在4-40°2θ测量对称扫描,步长为0.017°且扫描阶跃时间为15.5秒。数据收集软件为X’pert Data Collector(版本2.2e)。数据分析软件为X’pert Data Viewer(版本1.2d)或者X’pert Highscore(版本:2.2c)。
热重量分析(TGA)
TGA用于研究所表征的各批样品中的残留溶剂的存在,并且鉴定样品发生分解的温度。TGA数据在TA Q500 Thermogravimetric Analyzer(热重量分析仪)(TA Instruments,New Castle,DE)上收集。以10℃/分钟的加热速率,从25℃-300℃扫描重约2-5mg的样品。数据通过 Thermal Advantage Q SeriesTM软件(版本2.5.0.255)收集和通过Universal Analysis软件(版本4.4A,build 4.4.0.5)(TA Instruments,New Castle,DE)分析。
化合物1形式A单晶结构测定
衍射数据在配备有密封管Cu Kα源和Apex II CCD检测器的Bruker Apex II衍射仪上获得。结构使用SHELX程序(Sheldrick,G.M.,Acta Cryst.,(2008)A64,112-122)解析和精修。基于强度统计和系统消光解析和精修C2空间群的结构。使用类似的衍射测定绝对构型。Flack参数精确至0.00(18)表明所述模型代表正确的对映体[(R)]。
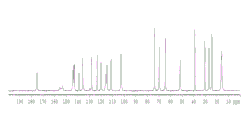
固态NMR
固态NMR在配备有Bruker-Biospin 4mm HFX探针的Bruker-Biospin 400MHz宽孔光谱仪上进行。样品被填充至4mm ZrO2转子中并在Magic Angle Spinning(MAS)条件下旋转,旋转速度为12.5kHz。质子弛豫时间首先使用1H MAS T1饱和恢复松弛实验测量,以建立13C交叉极化(CP)MAS试验的适当的再循环延迟。碳CPMAS实验的CP接触时间设为2ms。采用具有线性斜坡电压的CP质子脉冲(从50%至100%)。Hartmann-Hahn匹配基于外参比样品(甘氨酸)进行优化。氟MAS谱伴随质子去耦进行记录。为获得13C和19F两者,使用TPPM15去耦序列,场强为约100kHz。
试剂和化合物

在化合物的名称可能未正确描述所述化合物的结构的本申请的任何地方,结构替代名称并以结构为主。
化合物1的合成
酰氯部分
(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-1-乙酸乙酯-乙腈的合成
用氮气吹扫反应器,并加入900mL甲苯。通过氮气吹扫来使溶剂脱气,吹扫不少于16小时。然后向该反应器中加入Na3PO4(155.7g,949.5mmol),而后加入双(二亚苄基丙酮)钯(0) (7.28g,12.66mmol)。在23℃,历时10分钟从氮气吹扫的加料漏斗中加入10%w/w叔丁基膦在己烷(51.23g,25.32mmol)中的溶液。将该混合物搅拌50分钟,此时,历时1分钟加入5-溴-2,2-二氟-1,3-苯并间二氧杂环戊烯(75g,316.5mmol)。额外搅拌50分钟之后,历时5分钟向该混合物中加入氰基乙酸乙酯(71.6g,633.0mmol),而后加入一份水(4.5mL)。历时40分钟将该混合物加热至70℃,每1-2小时用HPLC分析反应物至产物的转化率(%)。观察到完全转化之后(在5-8小时之后,通常为100%转化率),将该混合物冷却至20-25℃,通过西莱特垫过滤。用甲苯(2X 450mL)冲洗西莱特垫,并在60-65℃将合并的有机物在真空下浓缩至300mL。向浓缩物中加入225mL DMSO,在70-80℃真空浓缩,直到溶剂停止蒸馏为止。将该溶液冷却至20-25℃,并在步骤2的制备中用DMSO稀释至900mL。1H NMR(500MHz,CDCl3)δ7.16-7.10(m,2H),7.03(d,J=8.2Hz,1H),4.63(s,1H),4.19(m,2H),1.23(t,J=7.1Hz,3H)。
(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈的合成.

历时20分钟向上述(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-1-乙基乙酸酯-乙腈的DMSO溶液中加入3N HCl(617.3mL,1.85mol),同时保持内部温度<40℃。然后历时1小时将该混合物加热到75℃,每1-2小时用HPLC分析转化率(%)。当观察到转化率>99%时(通常在5-6小时之后),将该反应物冷却至20-25℃,用MTBE(2X 525mL)萃取,在萃取期间,用足够的时间完成相分离。将合并的有机萃取物用5%NaCl(2X 375mL)洗涤。然后将该溶液转入适合于1.5-2.5Torr真空蒸馏的装置(其配备有冷却的接收烧瓶)中。将该溶液在<60℃真空浓缩,除去溶剂。然后,在125-130℃(烘箱温度)和1.5-2.0Torr,从得到的油中蒸馏出(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈。从5-溴-2,2-二氟-1,3-苯并间二氧杂环戊烯中分离出(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈,为清澈的油,产率66%(2步),HPLC纯度为91.5%AUC(相当于95%的w/w测定)。1H NMR(500MHz,DMSO)δ7.44(br s,1H),7.43(d,J=8.4Hz,1H),7.22(dd,J=8.2,1.8Hz,1H),4.07(s,2H)。
(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲腈的合成.

通过氮气吹扫,将50%w/w NaOH储备溶液脱气不少于16h。类似地,将适当量的MTBE脱气数小时。向用氮气吹扫过的反应器中加入脱气的MTBE(143mL),随后加入(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈(40.95g,207.7mmol)和四丁基溴化铵(2.25g,10.38mmol)。记录混合物的体积,并通过氮气吹扫将该混合物脱气30min。加入足够的脱气的MTBE,以使混合物恢复至脱气前的起始体积。在23.0℃,历时10min向搅拌的混合物中加入脱气的50%w/w NaOH(143mL),随后历时30min加入1-溴-2-氯乙烷(44.7g,311.6mmol)。以1h间隔,用HPLC分析反应的%转化率。在取样之前,停止搅拌,并进行相分离。将顶部有机相取样用于分析。当观察到%转化率>99%时(通常在2.5-3h以后),将所述反应混合物冷却至10℃,并以维持温度<25℃的速率加入水(461mL)。调节温度至20-25℃,并分离相。注意:应当允许足够的时间以完成相分离。用MTBE(123mL)萃取水相,并用1N HCl(163mL)和5%NaCl(163mL)洗涤合并的有机相。在真空下在40-50℃,将(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲腈在MTBE中的溶液浓缩至164mL。给溶液加入乙醇(256mL),并在真空下在50-60℃再次浓缩至164mL。加入乙醇(256mL),并在真空下在50-60℃将混合物浓缩至164mL。将得到的混合物冷却至20-25℃,并用乙醇稀释至266mL,用于下一步制备。1H NMR(500MHz,DMSO)δ7.43(d,J=8.4Hz,1H),7.40(d,J=1.9Hz,1H),7.30(dd,J=8.4,1.9Hz,1H),1.75(m,2H),1.53(m,2H)。
1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸的合成.

历时20min,向得自前一步的(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲腈在乙醇中的溶液中加入6N NaOH(277mL),并历时45min加热至77-78℃的内部温度。在16h以后,通过HPLC监测反应进程。注意:监测(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲腈和由(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲腈的部分水解产生的伯酰胺的消耗。当观察到%转化率>99%时(在16h以后通常100%转化率),将所述反应混合物冷却至25℃,并加入乙醇(41mL)和DCM(164mL)。将溶液冷却至10℃,并以维持温度<25℃的速率 加入6N HCl(290mL)。温热至20-25℃以后,使相发生分离。收集底部有机相,并用DCM(164mL)反萃取顶部水相。注意:由于高无机盐浓度,水相在萃取之前和之后稍微混浊。合并有机物,并在真空下浓缩至164mL。加入甲苯(328mL),并在70-75℃将混合物浓缩至164mL。将混合物冷却至45℃,加入MTBE(364mL),并在60℃搅拌20min。将溶液冷却至25℃,并精密过滤(polish filtered),以除去残余的无机盐。使用MTBE(123mL)冲洗反应器和收集的固体。将合并的有机物转移至干净的反应器,用于下一步制备。
1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸的分离。

在真空下,将得自前一步的1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸溶液浓缩至164mL,加入甲苯(328mL),并在70-75℃浓缩至164mL。然后将混合物加热至100-105℃,得到均匀的溶液。在该温度搅拌30min以后,历时2小时将溶液冷却至5℃,并在5℃维持3小时。然后将混合物过滤,并用冷的1∶1甲苯/正庚烷(2X 123mL)洗涤反应器和收集的固体。将所述物质在真空下在55℃干燥17小时,得到1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸,为灰白色结晶固体。从(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈分离出1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸,产率为79%(3步,包括分离),HPLC纯度为99.0%AUC。ESI-MS m/z计算值242.04,实测值241.58(M+1)+;1HNMR(500MHz,DMSO)δ12.40(s,1H),7.40(d,J=1.6Hz,1H),7.30(d,J=8.3Hz,1H),7.17(dd,J=8.3,1.7Hz,1H),1.46(m,2H),1.17(m,2H)。
酰氯部分的替代合成
(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-甲醇的合成。

将可商购得到的2,2-二氟-1,3-苯并间二氧杂环戊烯-5-甲酸(1.0当量)在甲苯(10体积)中浆化。经加料漏斗以保持温度为15-25℃的速率加入  (2当量)。在加入结束时,使温度升至40℃保持2小时,然后经加料漏斗小心加入10%(w/w)NaOH水溶液(4.0当量),期间保持温度在40-50℃。在搅拌另外30分钟后,在40℃分离各层。把有机相冷却至20℃,然后用水(2x1.5体积)洗涤,干燥(Na2SO4),过滤并浓缩,得到粗品(2,2-二氟-1,3-苯并间二 氧杂环戊烯-5-基)-甲醇,其直接用于下一步。
(2当量)。在加入结束时,使温度升至40℃保持2小时,然后经加料漏斗小心加入10%(w/w)NaOH水溶液(4.0当量),期间保持温度在40-50℃。在搅拌另外30分钟后,在40℃分离各层。把有机相冷却至20℃,然后用水(2x1.5体积)洗涤,干燥(Na2SO4),过滤并浓缩,得到粗品(2,2-二氟-1,3-苯并间二 氧杂环戊烯-5-基)-甲醇,其直接用于下一步。
5-氯甲基-2,2-二氟-1,3-苯并间二氧杂环戊烯的合成.
把(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-甲醇(1.0当量)溶于MTBE(5体积)中。加入催化量的DMAP(1mol%),并经加料漏斗加入SOCl2(1.2当量)。以维持反应器中的温度为15-25℃的速率加入SOCl2。使温度升至30℃保持1小时,然后冷却至20℃,然后经加料漏斗加入水(4体积),期间保持温度在低于30℃。在搅拌另外30分钟之后,分离各层。搅拌有机层,并加入10%(w/v)NaOH水溶液(4.4体积)。在搅拌15-20分钟之后,分离各层。然后干燥(Na2SO4)有机相,过滤并浓缩,得到粗品5-氯甲基-2,2-二氟-1,3-苯并间二氧杂环戊烯,其直接用于下一步。
(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈的合成.

将5-氯甲基-2,2-二氟-1,3-苯并间二氧杂环戊烯(1当量)在DMSO(1.25体积)中的溶液加入到NaCN(1.4当量)在DMSO(3体积)中的料浆中,期间保持温度在30-40℃之间。搅拌混合物1小时,然后加入水(6体积),随后加入MTBE(4体积)。在搅拌30分钟之后,分离各层。用MTBE(1.8体积)萃取水层。用水(1.8体积)洗涤合并的有机层,干燥(Na2SO4),过滤并浓缩,得到粗品(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-乙腈(95%),其直接用于下一步。
剩余的步骤与上述的酸部分的合成相同。
胺部分
2-溴-5-氟-4-硝基苯胺的合成.
向烧瓶中加入3-氟-4-硝基苯胺(1.0当量),然后加入乙酸乙酯(10体积),并搅拌以使全部固体溶解。分批加入N-溴代琥珀酰亚胺(1.0当量)以保持内部温度为22℃。在反应结束时,在旋转蒸发仪上真空浓缩反应混合物。使残余物在蒸馏水(5体积)中浆化,以溶解并除去琥珀酰亚胺(琥珀酰亚胺也可经水后处理程序去除)。倾析水,并把固体在2-丙醇(5体积)中浆化过夜。过滤生成的料浆,并用2-丙醇洗涤湿滤饼,伴随N2流,在50℃于真空烘箱中干燥过夜,直到达到恒重。分离黄棕色固体(50%产率,97.5%AUC)。其它杂质为溴代-位置异构体(regioisomer)(1.4%AUC)和二溴代-加合物(1.1%AUC)。1H NMR(500MHz,DMSO)δ8.19(1H,d,J=8.1Hz),7.06(br.s,2H),6.64(d,1H,J=14.3Hz)。
苄基羟乙酸化-4-铵-2-溴-5-氟苯胺甲苯磺酸盐的合成.

向在N2下彻底干燥的烧瓶中加入以下物质:活化的粉状4A分子筛(50重量%,基于2-溴-5-氟-4-硝基苯胺)、2-溴-5-氟-4-硝基苯胺(1.0当量)、高氯酸锌二水合物(20mol%)和甲苯(8体积)。在室温搅拌混合物NMT 30分钟。最后,以稳流加入在甲苯(2体积)中的(R)-苄基缩水甘油醚(2.0当量)。把反应物加热至80℃(内部温度),并搅拌约7小时或者直到2-溴-5-氟-4-硝基苯胺为<5%AUC。
把反应物冷却至室温,并加入西莱特(50重量%),随后加入乙酸乙酯(10体积)。过滤生成的混合物以去除西莱特和分子筛,并用乙酸乙酯(2体积)洗涤。用氯化铵溶液(4体积,20%w/v)洗涤滤液。用碳酸氢钠溶液(4体积x2.5%w/v)洗涤有机层。在旋转蒸发仪上真空浓缩有机层。把生成的料浆溶于乙酸异丙酯(10体积)中,并把该溶液转移至Buchi氢化器。
向氢化器中加入5重量%Pt(S)/C(1.5mol%),并在N2下在30℃(内部温度)搅拌混合物。先后用N2和氢气吹洗反应物。把氢化器压力调节至1巴氢气,并快速搅拌混合物(>1200rpm)。在反应结束时,通过西莱特垫过滤催化剂,并用二氯甲烷(10体积)洗涤。真空浓缩滤液。用二氯甲烷(2体积)去除任何剩余的乙酸异丙酯,并在旋转蒸发仪上浓缩至干。
把生成的残余物溶于二氯甲烷(10体积)中。加入对甲苯磺酸一水合物(12当量)并搅拌过夜。过滤产物,用二氯甲烷(2体积)洗涤,并抽吸干燥。把湿滤饼转移至干燥盘,并置 于真空烘箱中,伴随N2流于45℃干燥,直到达到恒重。分离到苄基羟乙酸化-4-铵-2-溴-5-氟苯胺甲苯磺酸盐,为灰白色固体。
手性纯度测定为>97%ee。
(3-氯-3-甲基丁-1-炔基)三甲基硅烷的合成.

把炔丙醇(1.0当量)加入到容器中。加入盐酸水溶液(37%,3.75体积)并开始搅拌。在固体醇溶解期间,观察到适度的吸热(5-6℃)。把生成的混合物搅拌过夜(16小时),缓慢变为深红色。向30L夹套容器中加入水(5体积),然后冷却至10℃。把反应混合物经真空缓慢转移至水中,同时保持混合物的内部温度低于25℃。加入己烷(3体积),并把生成的混合物搅拌0.5小时。使这些相沉降,排出水相(pH<1),并弃去。使用旋转蒸发器真空浓缩有机相,得到为红色油的产物。
(4-(苄氧基)-3,3-二甲基丁-1-炔基)三甲基硅烷的合成.

方法A
该部分中的所有当量和体积描述词都基于250g反应物。把镁屑(69.5g,2.86mol,2.0当量)加入到3L的4颈反应器中,并在氮气下用磁力搅拌器搅拌0.5小时。把反应器浸没在冰水浴中。把炔丙基氯(250g,1.43mol,1.0当量)在THF(1.8L,7.2体积)中的溶液在搅拌下缓慢加入到反应器中,直到观察到初始放热(约10℃)。使用1H-NMR光谱法经IPC确认格氏试剂形成。一旦放热减弱,缓慢加入溶液的剩余部分,期间保持批次温度<15℃。加入需要约3.5小时。把生成的深绿色混合物倾析到2L带盖的瓶中。
该部分中的所有当量和体积描述词都基于500g反应物。向22L反应器中加入苄基氯甲基醚(95%,375g,2.31mol,0.8当量)在THF(1.5L,3体积)中的溶液。在冰水浴中冷却反应器。把如上所述制备的2批格氏试剂合并,然后经加料漏斗缓慢加入到苄基氯甲基醚溶液中,期间保持批温度低于25℃。加入需要1.5小时。把反应混合物搅拌过夜(16小时)。
该部分中的所有当量和体积描述词都基于1kg反应物。在30L夹套反应器中制备15%氯化铵溶液(1.5kg在8.5kg水中,10体积)。使溶液冷却至5℃。将如上所述制备的2批格氏反应混合物合并,然后经集管容器(header vessel)转移至氯化铵溶液中。在该淬灭中观察 到放热,淬灭以例如保持内部温度低于25℃的速率进行。一旦转移完成,将容器夹套温度设定为25℃。加入己烷(8L,8体积),并搅拌混合物0.5小时。在使这些相沉降之后,排出水相(pH 9)并弃去。用水(2L,2体积)洗涤剩余的有机相。使用22L旋转蒸发器真空浓缩有机相,得到为橙色油的粗品产物。
方法B
把镁屑(106g,4.35mol,1.0当量)加入到22L反应器中,然后悬浮于THF(760mL,1体积)中。用冰水浴冷却容器,以使得批温度达到2℃。向反应器中缓慢加入炔丙基氯(760g,4.35mol,1.0当量)在THF(4.5L,6体积)中的溶液。在加入100mL之后,停止加入,并搅拌混合物直到观察到13℃放热(exotherm),表明格氏试剂开始形成。一旦放热减弱,缓慢加入另外500mL炔丙基氯溶液,期间保持批次温度<20℃。使用1H-NMR光谱法经IPC确认格氏试剂形成。缓慢加入剩余的炔丙基氯溶液,期间保持批次温度<20℃。加入需要约1.5小时。把生成的深绿色溶液搅拌0.5小时。使用1H-NMR光谱法经IPC确认格氏试剂形成。向反应器加料漏斗中加入纯净的苄基氯甲基醚,然后滴加到反应器中,期间保持批次温度低于25℃。加入需要1.0小时。把反应混合物搅拌过夜。使用与方法A相同的程序和相对量的材料进行水后处理和浓缩,得到为橙色油的产物。
4-苄氧基-3,3-二甲基丁-1-炔的合成.
向30L夹套反应器中加入甲醇(6体积),然后冷却至5℃。向反应器中加入氢氧化钾(85%,1.3当量)。当氢氧化钾溶解时,观察到15-20℃放热。把夹套温度设定为25℃。加入4-苄氧基-3,3-二甲基-1-三甲基甲硅烷基丁-1-炔(1.0当量)在甲醇(2体积)中的溶液,并搅拌生成的混合物,直到经HPLC监测反应完成。在25℃的典型反应时间为3-4小时。用水(8体积)稀释反应混合物,然后搅拌0.5小时。加入己烷(6体积),并把生成的混合物搅拌0.5小时。使各相沉降,然后排出水相(pH 10-11)并弃去。先后用KOH(85%,0.4当量)在水中的溶液(8体积)和水(8体积)洗涤有机相。然后使用旋转蒸发器浓缩有机相,得到为橙黄色油的标题物料。该物料的典型纯度在80%范围内,主要存在单一杂质。1H NMR(400MHz,C6D6)δ7.28(d,2H,J=7.4Hz),7.18(t,2H,J=7.2Hz),7.10(d,1H,J=7.2Hz),4.35(s,2H),3.24(s,2H),1.91(s,1H),1.25(s,6H)。
N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚的合成.
方法A
苄基羟乙酸化4-氨基-2-(4-苄氧基-3,3-二甲基丁-1-炔基)-5-氟苯胺的合成.

如下使苄基羟乙酸化-4-铵-2-溴-5-氟苯胺甲苯磺酸盐成游离碱(freebased):在EtOAc(5体积)和饱和NaHCO3溶液(5体积)中搅拌所述固体,直到得到澄清的有机层。分离生成的各层,先后用饱和NaHCO3溶液(5体积)和盐水洗涤有机层,并真空浓缩,得到为油的苄基羟乙酸化-4-铵-2-溴-5-氟苯胺甲苯磺酸盐。
然后,向烧瓶中加入苄基羟乙酸化-4-铵-2-溴-5-氟苯胺甲苯磺酸盐(游离碱,1.0当量)、Pd(OAc)(4.0mol%)、dppb(6.0mol%)和粉状K2CO3(3.0当量),并在室温与乙腈(6体积)一起搅拌。通过用N2通风口鼓泡,将生成的反应混合物脱气约30分钟。然后以快速流加入溶解于乙腈(2体积)中的4-苄氧基-3,3-二甲基丁-1-炔(11当量),加热至80℃并搅拌,直到实现4-铵-2-溴-5-氟苯胺甲苯磺酸盐的完全消耗。把反应料浆冷却至室温,通过西莱特垫过滤,并用乙腈(2体积)洗涤。真空浓缩滤液,并将残余物再溶解于EtOAc(6体积)中。有机层用NH4Cl溶液(20%w/v,4体积)和盐水(6体积)洗涤两次。浓缩生成的有机层,得到棕色的油,并原样用于下一步反应。
N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚的合成.

在室温,将苄基羟乙酸化-4-氨基-2-(4-苄氧基-3,3-二甲基丁-1-炔基)-5-氟苯胺的粗品油溶解于乙腈(6体积)中,并加入(MeCN)2PdCl2(15mol%)。生成的混合物使用N2通风口脱气约30分钟。然后把反应混合物在80℃于N2覆盖下搅拌过夜。使反应混合物冷却至室温,通过西莱特垫过滤,并用乙腈(1体积)洗涤滤饼。真空浓缩生成的滤液,并再溶解于EtOAc(5体积)中。加入Deloxane-II THP(5重量%,基于N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙 基)-6-氟吲哚的理论产率),并在室温搅拌过夜。然后通过硅胶(silica)垫(深度2.5英寸,直径6英寸滤器)过滤混合物,并用EtOAc(4体积)洗涤。把滤液浓缩为深棕色残余物,并原样用于下一步反应。
粗品N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚的再纯化:
把粗品N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚溶解于二氯甲烷(约1.5体积)中,并通过硅胶垫过滤,最初使用30%EtOAc/庚烷,其中弃去杂质。然后用50%EtOAc/庚烷洗涤硅胶垫,以分离N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚,直到在滤液中观察到淡淡的颜色。真空浓缩该滤液,得到棕色的油,其在室温放置结晶。1H NMR(400MHz,DMSO)δ7.38-7.34(m,4H),7.32-7.23(m,6H),7.21(d,1H,J=12.8Hz),6.77(d,1H,J=9.0Hz),6.06(s,1H),5.13(d,1H,J=4.9Hz),4.54(s,2H),4.46(br.s,2H),4.45(s,2H),4.33(d,1H,J=12.4Hz),4.09-4.04(m,2H),3.63(d,1H,J=9.2Hz),3.56(d,1H,J=9.2Hz),3.49(dd,1H,J=9.8,4.4Hz),3.43(dd,1H,J=9.8,5.7Hz),1.40(s,6H)。
N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚的合成.
方法B

将乙酸钯(33g,0.04当量)、dppb(94g,0.06当量)和碳酸钾(1.5kg,3.0当量)装入反应器中。将游离碱化的油苄基羟乙酸化-4-铵-2-溴-5-氟苯胺(1.5kg,1.0当量)溶解在乙腈(8.2L,4.1体积)中,然后加入反应器中。用氮气将混合物吹扫不少于1h。将4-苄氧基-3,3-二甲基丁-1-炔(70%,1.1kg,1.05当量)在乙腈中的溶液加入混合物中,然后用氮气吹扫不少于1h。将混合物加热至80℃,然后搅拌过夜。通过HPLC进行IPC,并在16h以后确定反应结束。将混合物冷却至环境温度,然后通过西莱特垫(228g)过滤。用乙腈(2x2L,2体积)洗涤反应器和西莱特垫。在22L旋转蒸发器上浓缩合并的各相,直到已经收集8L溶剂,剩下在7L(3.5体积)乙腈中的粗产物。
将二-乙腈二氯化钯(144g,0.15当量)加入反应器中。将粗溶液转移回反应器中,并用乙腈(4L,2体积)洗涤旋转蒸发器玻壳(roto-vap bulb)。用氮气将合并的溶液吹扫不少于1 h。将所述反应混合物加热至80℃保持不少于16h。通过HPLC进行的过程控制显示原料的完全消耗。通过西莱特(300g)过滤所述反应混合物。用乙腈(3L,1.5体积)洗涤反应器和滤饼。通过旋转蒸发,将合并的滤液浓缩为油。将所述油溶解在乙酸乙酯(8.8L,4.4体积)中。用20%氯化铵(5L,2.5体积)洗涤溶液,随后用5%盐水(5L,2.5体积)洗涤。将硅胶(3.5kg,1.8重量当量)加入有机相中,将其搅拌过夜。加入Deloxan THP II金属清除剂(358g)和庚烷(17.6L),并将得到的混合物搅拌不少于3h。通过烧结玻璃漏斗,过滤混合物。用在庚烷中的30%乙酸乙酯(25L)洗涤滤饼。在减压下浓缩合并的滤液,得到N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚,为棕色糊状物(1.4kg)。
化合物1的合成
苄基保护的化合物1的合成.

将1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸(1.3当量)在甲苯(2.5体积,基于1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸)中浆化,并把混合物加热至60℃。经加料漏斗加入SOCl2(1.7当量)。把生成的混合物搅拌2小时。使用旋转蒸发仪蒸除甲苯和过量的SOCl2。加入另外的甲苯(2.5体积,基于1-(2,2-二氟-1,3-苯并间二氧杂环戊烯-5-基)-环丙烷甲酸)并再次蒸馏。将粗品酰氯溶于二氯甲烷(2体积)中,并经加料漏斗加入到N-苄基羟乙酸化-5-氨基-2-(2-苄氧基-1,1-二甲基乙基)-6-氟吲哚(1.0当量)和三乙胺(2.0当量)在二氯甲烷(7体积)中的混合物中,同时保持0-3℃(内部温度)。生成的混合物在0℃搅拌4小时,然 后温热至室温过夜。向反应混合物中加入蒸馏水(5体积),搅拌不少于30分钟并分离各层。先后用20重量%K2CO3(4体积x2)和盐水洗液(4体积)洗涤有机相并浓缩,得到为稠的棕色油的粗品苄基保护的化合物1,其使用硅胶垫过滤以进一步纯化。
硅胶垫过滤:将粗品苄基保护的化合物1在活性炭Darco-G(10重量%,基于苄基保护的化合物1的理论产率)存在下溶于乙酸乙酯(3体积)中,并在室温搅拌过夜。向该混合物中加入庚烷(3体积)并通过硅胶垫(粗品苄基保护的化合物1的2x重量)过滤。用乙酸乙酯/庚烷(1∶1,6体积)洗涤硅胶垫或直到在滤液没有检测到什么颜色。真空浓缩滤液,得到为粘稠棕红色油的苄基保护的化合物1,并直接用于下一步。
再次纯化:把苄基保护的化合物1再次溶于二氯甲烷(1体积,基于苄基保护的化合物1的理论产率)中并加载到硅胶垫(粗品苄基保护的化合物1的2x重量)上。用二氯甲烷(2体积,基于苄基保护的化合物1的理论产率)洗涤硅胶垫并弃去滤液。用30%乙酸乙酯/庚烷(5体积)洗涤硅胶垫并真空浓缩滤液,得到为粘稠橙红色油的苄基保护的化合物1,并直接用于下一步。
化合物1的合成

方法A
把20L高压釜用氮气冲洗3次,然后加入披钯炭(Evonik E 101 NN/W,5%Pd,60%湿的,200g,0.075mol,0.04当量)。高压釜然后用氮气冲洗3次。经抽吸向高压釜加入粗品苄基保护的化合物1(1.3kg,约1.9mol)在THF(8L,6体积)中的溶液。把容器加盖并然后用氮气冲洗3次。伴随温和搅拌下,把容器用氢气吹洗3次,经用氮气稀释排放至大气。把高压釜用氢气增压至3巴,并把搅拌速率增加至800rpm。观察到快速吸氢(溶解)。一旦吸收减弱,把容器加热至50℃。
为了安全目的,在每一个工作日结束时关闭恒温器。把容器用氢气增压至4巴并然后与氢气发生器专用液罐分离。
在2整天的反应之后,向混合物中加入更多的Pd/C(60g,0.023mol,0.01当量)。这通过用氮气冲洗3次并然后通过固体加料口加入催化剂实现。如之前那样重新开始反应。在4整天之后,根据HPLC认为反应完成:不仅起始原料而且相应于单-苄基化中间体的峰消失。
通过西莱特垫过滤反应混合物。用THF(2L,1.5体积)洗涤容器和滤饼。然后用水润湿西莱特垫并适当地弃去滤饼。使用旋转蒸发器浓缩合并的滤液和THF洗液,得到为黑油的粗品产物,1kg。
在以下纯化中的当量和体积基于1kg粗品物料。把粗品黑油溶于1∶1乙酸乙酯-庚烷中。把混合物加到烧结漏斗中,已经用1∶1乙酸乙酯-庚烷饱和的硅胶垫(1.5kg,1.5wt.当量)。硅胶垫首先用1∶1乙酸乙酯-庚烷(6L,6体积)冲洗,然后用纯的乙酸乙酯(14L,14体积)冲洗。以4个部分收集洗脱液,所述部分经HPLC分析。
在以下纯化中的当量和体积基于0.6kg粗品物料。经旋转蒸发浓缩部分3,得到棕色泡沫(600g),然后再溶解于MTBE(1.8L,3体积)中。把深棕色的溶液在环境温度搅拌过夜,期间发生结晶。加入庚烷(55mL,0.1体积)并把混合物搅拌过夜。使用布氏漏斗过滤混合物并用3∶1 MTBE-庚烷(900mL,1.5体积)洗涤滤饼。把滤饼风干1小时,然后在环境温度真空干燥16小时,得到253g为灰白色固体的化合物1。
用于以下纯化的当量和体积基于1.4kg粗品物料。将来自以上硅胶过滤的部分2和3以及来自先前反应的物料合并并浓缩,得到1.4kg黑油。把混合物再次进行以上描述的硅胶过滤(1.5kg硅胶,先后用3.5L,2.3体积的1∶1乙酸乙酯-庚烷和9L,6体积的纯乙酸乙酯洗脱),浓缩得到褐色泡沫状固体(390g)。
用于以下纯化的当量和体积基于390g粗品物料。褐色固体不溶于MTBE,因此溶于甲醇(1.2L,3体积)中。使用配备有长程蒸馏头的4L莫顿反应器,把混合物蒸馏至2体积。加入MTBE(1.2L,3体积)并把混合物蒸馏回至2体积。加入第2份MTBE(1.6L,4体积)并把混合物蒸馏回至2体积。加入第3份MTBE(1.2L,3体积)并把混合物蒸馏回至3体积。经GC分析蒸馏物显示其包含约6%甲醇。把恒温器设为48℃(低于MTBE-甲醇共沸混合物的沸腾温度,其沸腾温度为52℃)。使混合物经2小时冷却至20℃,期间发生相对快速结晶。在把混合物搅拌2小时之后,加入庚烷(20mL,0.05体积)并把混合物搅拌过夜(16小时)。使用布氏漏斗过滤混合物并用3∶1 MTBE-庚烷(800mL,2体积)洗涤滤饼。把滤饼风干1 小时,然后在环境温度真空干燥16小时,得到130g为灰白色固体的化合物1。
方法B
把苄基保护的化合物1溶解在THF(3体积)中,然后蒸发(stripped)至干燥,以去除任何残留溶剂。把苄基保护的化合物1再溶解于THF(4体积)中,并加入到含有5重量%Pd/C(2.5mol%,60%湿的,Degussa E5E101NN/W)的氢化器中。把反应物的内部温度调节至50℃,并先后用N2(x5)和氢气(x3)冲洗。把氢化器压力调节至3巴氢气并快速搅拌(>1100rpm)混合物。在反应结束时,通过西莱特垫过滤催化剂并用THF(1体积)洗涤。真空浓缩滤液,得到棕色泡沫残余物。将生成的残余物溶解于MTBE(5体积)中,并加入0.5N HCl溶液(2体积)和蒸馏水(1体积)。把混合物搅拌不少于30分钟并分离生成的各层。先后用10重量%K2CO3溶液(2体积x2)和盐水洗液洗涤有机相。把有机层加入到含有硅胶(25重量%)、Deloxan-THPII(5重量%,75%湿的)和Na2SO4的烧瓶中并搅拌过夜。通过西莱特垫过滤生成的混合物并用10%THF/MTBE(3体积)洗涤。真空浓缩滤液,得到为淡褐色泡沫的粗品化合物1。
自母液回收化合物1:选项A.
硅胶垫过滤:真空浓缩母液,得到棕色泡沫,将其溶于二氯甲烷(2体积)中,并通过硅胶垫(粗品化合物1的3x重量)过滤。用乙酸乙酯/庚烷(1∶1,13体积)洗涤硅胶垫并弃去滤液。硅胶垫用10%THF/乙酸乙酯(10体积)洗涤并真空浓缩滤液,得到为淡褐色泡沫的化合物1。按照以上结晶程序分离剩余的化合物1。
自母液回收化合物1:选项B.
硅胶柱色谱法:在硅胶上层析(50%乙酸乙酯/己烷-100%乙酸乙酯)之后,分离为淡褐色泡沫的期望的化合物。按照以上结晶程序分离剩余的化合物1。
化合物1的额外重结晶
将固体化合物1(1.35kg)悬浮于IPA(5.4L,4体积)中,然后加热至82℃。在完全溶解后(目测),缓慢地加入庚烷(540mL,0.4体积)。将混合物冷却至58℃。然后将混合物缓慢地冷却至51℃,在此期间发生结晶。关闭热源,让重结晶混合物自然地冷却过夜。使用台式布氏漏斗,过滤该混合物,并用IPA(2.7L,2体积)洗涤滤饼。在通气下在所述漏斗中干燥滤饼8h,然后在真空中在45-50℃烘干过夜,得到1.02kg重结晶的化合物1。
化合物1也可通过在公开的美国专利申请US20090131492(其通过引用并入本文)中公开的几种合成途径之一制备。
下面表4列举了化合物1的分析数据。
表4.

化合物1无定形形式的合成
喷雾干燥法
将9.95g醋酸羟丙甲基纤维素琥珀酸酯HG级(HPMCAS-HG)与50mg月桂基硫酸钠(SLS)一起称重至500ml烧杯中。把MeOH(200ml)与所述固体混合。把物料搅拌4小时。为了确保最大溶解,在搅拌2小时后,把溶液超声处理5分钟,然后继续搅拌剩余的2小时。非常细(fin)的HPMCAS混悬液仍保持在溶液中。然而,在倾斜容器之后,视觉观察确定没有胶粘的部分残留在容器壁上或粘于底部。
把化合物1(10g)倾入到500ml烧杯中,并继续搅拌该系统。使用以下参数,喷雾干燥溶液:
配方描述:化合物1形式A/HPMCAS/SLS(50/49.5/0.5)
B小型喷雾干燥仪

回收大约16g化合物1无定形形式(80%产率)。经XRPD(图1)和DSC(图2)确认化合物1无定形形式。
化合物1无定形形式的固态13C NMR谱显示在图3中。表5提供相关峰的化学位移。
表5.

化合物1无定形形式的固态19F NMR谱显示在图4中。具有星号的峰意指旋转边带。表6提供相关峰的化学位移。
表6.

旋转蒸发法
把化合物1(约10g)溶解于180ml MeOH中,并在50℃浴中旋转蒸发为泡沫。XRPD(图5)和DSC(图6)确认化合物1的无定形形式。
化合物1形式A的合成
浆化方法
对于EtOAc、MTBE、乙酸异丙酯或DCM,把约40mg的化合物1与1-2ml任何一种上述溶剂一起加入到小瓶中。把料浆在室温搅拌24小时至2周,并通过离心(带有滤器)混悬液,收集化合物1形式A。图7公开了通过该方法用DCM作为溶剂得到的化合物1形式A的实际XRPD图样。表7列出了图7的峰。
表7.
从化合物1形式A的单晶结构计算出的X-射线衍射图样显示在图8中。表8列出了图8的计算峰。
表8.

化合物1形式A的DSC示踪图显示在图9中。化合物1形式A的熔点发生在约172-178℃。
对于EtOH/水溶液,把约40mg的化合物1加入到3个单独的小瓶中。在第一个小瓶中加入1.35ml的EtOH和0.15ml水。在第二个小瓶中加入0.75ml的EtOH和0.75ml水。在第三个小瓶中加入0.15ml的EtOH和1.35ml水。所有3个小瓶在室温搅拌24小时。然后分别离心(带有滤器)每一混悬液,以收集化合物1形式A。
对于异丙醇/水溶液,把约40mg的化合物1加入到3个单独的小瓶中。在第一个小瓶中加入1.35ml的异丙醇和0.15ml水。在第二个小瓶中加入0.75ml的异丙醇和0.75ml水。在第三个小瓶中加入0.15ml的异丙醇和1.35ml水。所有3个小瓶在室温搅拌24小时。然后分别离心(带有滤器)每一混悬液,以收集化合物1形式A。
对于甲醇/水溶液,把约40mg的化合物1加入到小瓶中。加入0.5ml甲醇和1ml水并在室温搅拌混悬液24小时。把混悬液离心(带有滤器),以收集化合物1形式A。对于乙腈,把约50mg的化合物1与2.0ml乙腈一起加入到小瓶中。在室温搅拌混悬液24小时并经离心(带有滤器)收集化合物1形式A。
对于乙腈/水溶液,把约50mg的化合物1溶解于2.5ml乙腈中,在声处理之后得到澄清的溶液。过滤溶液并吸取1ml到小瓶中。加入2.25ml水,得到浑浊的混悬液。在室温搅拌混悬液24小时并经离心(带有滤器)收集化合物1形式A。
慢蒸发法
把约55mg的化合物1溶解于0.5ml丙酮中,在声处理之后得到澄清的溶液。过滤溶液并吸取0.2ml到小瓶中。小瓶用其中具有一个戳洞的石蜡封口膜覆盖并放置。经过滤收集重结晶的化合物1形式A。
快蒸发法
对于异丙醇,把约43mg的化合物1溶解于2.1ml异丙醇中,在声处理之后得到澄清的溶液。把溶液过滤到小瓶中并无盖放置。经过滤收集重结晶的化合物1形式A。
对于甲醇,把约58mg的化合物1溶解于0.5ml甲醇中,在声处理之后得到澄清的溶液。过滤溶液,吸取0.2ml到无盖小瓶中并放置。经过滤收集重结晶的化合物1形式A。
对于乙腈,把约51mg的化合物1溶解于2.5ml乙腈中,在声处理之后得到澄清的溶液。过滤溶液,吸取一半溶液到无盖小瓶中并放置。经过滤收集重结晶的化合物1形式A。图10公开了经该方法制备的化合物1形式A的XRPD图样。
反溶剂法
对于EtOAc/庚烷,把约30mg的化合物1溶解于1.5ml的EtOAc中,在声处理之后得到澄清的溶液。过滤溶液并在缓慢搅拌的同时向过滤的溶液中加入2.0ml庚烷。把溶液搅拌另外10分钟并放置。经过滤收集重结晶的化合物1形式A。图11公开了经该方法制备的化合物1形式A的XRPD图样。
对于异丙醇/水,把约21mg的化合物1溶解于1.0ml异丙醇中,在声处理之后得到澄清的溶液。过滤溶液,得到0.8ml溶液。在缓慢搅拌的同时加入1.8ml水。加入另外0.2ml水,得到浑浊的混悬液。将搅拌停止5分钟,得到澄清的溶液。把溶液搅拌另外2分钟并放置。经过滤收集重结晶的化合物1形式A。
对于乙醇/水,把约40mg的化合物1溶解于1.0ml乙醇中,在声处理之后得到澄清的溶液。过滤溶液并加入1.0ml水。把溶液在室温搅拌1天。经过滤收集重结晶的化合物1形式A。
对于丙酮/水,把约55mg的化合物1溶解于0.5ml丙酮中,在声处理之后得到澄清的溶液。过滤溶液并吸取0.2ml到小瓶中。加入1.5ml水,然后加入另外0.5ml水,得到浑浊的混悬液。把混悬液在室温搅拌1天。经过滤收集化合物1形式A。
以下表9概述形成化合物1形式A的各种技术。
表9.


得到化合物1形式A的单晶数据,其提供了关于晶体结构的另外细节,包括晶格大小和堆积。
晶体制备
通过自浓的甲醇溶液(10mg/ml)慢蒸发得到化合物1形式A的晶体。选择具有0.20×0.05×0.05mm尺寸的化合物1形式A的无色晶体,用矿物油清洁,安装于MicroMount并聚集在Bruker APEXII衍射仪上。得到在倒易空间中分开的3批40个框架,其提供了取向矩阵和初始晶胞参数。得到最后的晶胞参数并基于完整的数据集精修。
实验
对  分辨率使用0.5°步长,每一个框架曝光30秒,得到倒易空间的衍射数据集。数据在室温[295(2)K]下收集。强度的集成(Integration of intensities)和晶胞参数的精修使用APEXII软件实现。在数据收集后观察晶体显示没有分解的迹象。
分辨率使用0.5°步长,每一个框架曝光30秒,得到倒易空间的衍射数据集。数据在室温[295(2)K]下收集。强度的集成(Integration of intensities)和晶胞参数的精修使用APEXII软件实现。在数据收集后观察晶体显示没有分解的迹象。
表10.化合物1形式A的晶体数据.

几何学:使用全协方差矩阵评价所有esds(除了两个l.s.平面之间的双面角的esd)。在距离、角度和扭转角的esds中单独考虑晶胞esds;晶胞参数的esds之间的相关性仅在它们被晶体对称性限定时使用。晶胞esds的近似(各向同性)处理用于评价涉及l.s.平面的esds。
表11.化合物1形式A晶体的数据收集参数.

数据收集:Apex II;晶胞精修:Apex II;数据简化:Apex II;用于解析结构的程序:SHELXS97(Sheldrick,1990);用于精修结构的程序:SHELXL97(Sheldrick,1997);分子图形学:汞(Mercury);用于制作出版材料的软件:publCIF。
表12.化合物1形式A晶体的精修参数.

精修:对ALL反射的F2精修。加权R-因子wR和拟合优度S基于F2,常规R-因子R基于F,且F对负F2设为零。F2的阈值表达>2σ(F2)仅用于计算R-因子(gt)等,并且与对精修的反射选择不相关。基于F2的R-因子在统计学上约为基于F的两倍那样大,并且基于ALL数据的R-因子将甚至更大。
基于单晶X-射线分析的化合物1形式A的构象图显示在图12中。晶体结构显示分子的致密堆积。化合物1形式A为单斜的C2空间群,具有以下晶胞尺寸: 

 β=95.867(6)°,γ=90°。
β=95.867(6)°,γ=90°。
化合物1形式A的固态13C NMR谱显示在图13中。表13提供相关峰的化学位移。
表13.

化合物1形式A的固态19F NMR谱显示在图14中。具有星号的峰意指旋转边带。表14提供相关峰的化学位移。
表14.

包含化合物1的示例性口服药物制剂
用在表15-17中列出的组分和量,制备片剂。
表15.

表16.


表17.
从碾压颗粒组合物形成片剂
设备/工艺
设备

筛分/称重
合并作为固体喷雾干燥分散体的化合物1无定形形式和Cabot M5P,并穿过20号网筛筛分,并在32RPM在2-L Turbula T2F振荡器混合器中掺合10分钟。
颗粒内掺合
加入AcDiSol、Avicel PH101和Foremost 310,并掺合另外15分钟。然后使掺合物穿过Quadro Comill 197(筛:0.032”R;叶轮:1607;RPM:1000RPM)。穿过20号网筛手工筛分硬脂酸镁,其量为上述掺合物的量(体积)的2-3倍。在32RPM在Turbula混合器中将得到的混合物掺合4分钟。
碾压
在Korsch XL100旋转式压片机(重力供料架1/2”直径、圆形、扁平面刻印)中将上述掺合物击压至约0.72-0.77固相分数。通过测量重量、高度,并使用在形成过程中确定的物料的真实密度,计算固相分数。对于旋转式压片机击压工艺,压缩力将随模具的填充体积和最终的半 制品的重量而变化。用研钵和研棒将半制品轻轻破碎成大约1/4英寸的块。使破碎的半制品穿过Quadro Comill 197(筛:0.079”G;叶轮:1607;RPM:1000)。
颗粒外掺合
穿过20号网筛手工筛分颗粒外Cabot M5P,其量为上述掺合物的量(体积)的2-3倍。将该颗粒外Cabot M5P预掺合物加入主掺合物中,并在32RPM在2-L Turbula T2F振荡器混合器中掺合15分钟。穿过20号网筛手工筛分颗粒外硬脂酸镁,其量为上述掺合物的量(体积)的2-3倍。将该颗粒外硬脂酸镁预掺合物加入主掺合物中,并在32RPM在Turbular混合器中掺合4分钟。
压制
使用具有重力供料架和0.289”x 0.5879”改进椭圆形刻印的Korsch XL 100,将片剂压制成14.5±3.5kp的目标硬度。
薄膜包衣
使用锅包衣机,例如O’Hara Labcoat,可以给片剂包被薄膜。
印刷
用例如Hartnett Delta印刷机,可以给薄膜包衣片剂在片剂的一面或双面上印刷花押字。
定量施用方案
在另一个方面,本发明涉及治疗受试者的CFTR介导的疾病的方法,该方法包括:给有此需要的受试者施用有效量的本发明提供的药物组合物。在另一个实施方案中,每两周给受试者施用一次该药物组合物。在另一个实施方案中,每周给受试者施用一次该药物组合物。在另一个实施方案中,每三天给受试者施用一次该药物组合物。在另一个实施方案中,每天给受试者施用一次该药物组合物。在一个实施方案中,当药物组合物是根据表1、2或3的片剂时,每天施用一次。
测定
用于检测和测量化合物的ΔF508-CFTR校正性质的测定
用于测定化合物的ΔF508-CFTR调节性质的膜电位光学方法
光学膜电位测定采用Gonzalez和Tsien(参见Gonzalez,J.E.和R.Y.Tsien(1995)“Voltage sensing by fluorescence resonance energy transfer in single cells”Biophys J 69(4):1272-80,和Gonzalez,J.E.and R.Y.Tsien(1997)“Improved indicators of cell membrane potential that use fluorescence resonance energy transfer”Chem Biol 4(4):269-77)描述的电压敏感的FRET传感器,结合用于测量荧光变化的仪器操作例如电压/离子探针读出仪(Voltage/Ion Probe Reader) (VIPR)(参见Gonzalez,J.E.,K.Oades,等人(1999)“Cell-based assays and instrumentation for screening ion-channel targets”Drug Discov Today 4(9):431-439)。
这些电压敏感测定基于膜可溶性电压敏感染料DiSBAC2(3)和附着于质膜出口小叶并起FRET供体作用的荧光磷脂CC2-DMPE之间的荧光共振能量转移(FRET)的变化。膜电位(Vm)的变化引起荷负电的DiSBAC2(3)跨质膜重新分布和来自CC2-DMPE的能量转移的量相应地发生变化。荧光发射的变化使用VIPRTM II监测,后者为集成的液体处理器和荧光检测器,被设计为在96-或384-孔微量滴定板中进行基于细胞的筛选。
1.校正化合物的鉴定
为了鉴定校正与ΔF508-CFTR相关的运输缺陷的小分子,开发了单独添加HTS检测格式。在受试化合物存在或不存在(阴性对照)下,把细胞在无血清培养基中,于37℃温育16小时。作为阳性对照,把在384-孔板中铺展的细胞于27℃温育16小时,以“温度校正”ΔF508-CFTR。细胞随后用Krebs Ringer溶液冲洗3X,并加载电压敏感染料。为了激活ΔF508-CFTR,向每孔中与不含Cl-的培养基一起加入10μM毛喉素和CFTR增效剂、染料木黄酮(20μM)。加入不含Cl-的培养基促进应答于ΔF508-CFTR激活的Cl-外流,并使用基于FRET的电压传感器染料光学监测生成的膜去极化。
2.增效剂化合物的鉴定
为了鉴定ΔF508-CFTR的增效剂,开发了双重添加HTS检测格式。在第一次添加期间,向每孔中加入不含Cl-的培养基(与或不与受试化合物一起)。22秒后,第二次加入含有2-10μM毛喉素的不含Cl-的培养基以激活ΔF508-CFTR。在两次添加之后细胞外Cl-浓度为28mM,这促进应答于ΔF508-CFTR激活的Cl-外流,并使用基于FRET的电压传感器染料光学监测生成的膜去极化。
3.溶液
浴液#1:(以mM表示)NaCl 160、KCl 4.5、CaCl22、MgCl2 1、HERES 10,pH 7.4含NaOH。
不含氯离子的浴液:浴液#1中的氯化物盐用葡糖酸盐替代。
CC2-DMPE:制备为在DMSO中的10mM储备溶液,并储存于-20℃。
DiSBAC2(3):制备为在DMSO中的10mM储备溶液,并储存于-20℃。
4.细胞培养
稳定表达ΔF508-CFTR的NIH3T3小鼠成纤维细胞用于膜电位的光学测量。在175cm2培养瓶中,于5%CO2和90%湿度下,在37℃把细胞保持在用2mM谷氨酰胺、10%胎牛血清、1X NEAA、β-ME、1X青霉素/链霉素和25mM HERES补充的Dulbecco氏改良的伊格尔培 养基中。对所有的光学测定,把细胞以30,000/孔接种在384-孔基质胶包被的平板中,并在37℃培养2小时,然后在27℃培养24小时,用于增效剂测定。对于校正测定,于27℃或37℃,在化合物存在和不存在下把细胞培养16-24小时。
用于测定化合物的ΔF508-CFTR调节性质的电生理学测定
1.尤斯室(Ussing chamber)测定
在表达ΔF508-CFTR的极化上皮细胞上进行尤斯室(Using chamber)实验,以进一步表征在光学测定中鉴定的ΔF508-CFTR调节剂。在Costar Snapwell细胞培养池(cell culture inserts)上生长的FRTΔF508-CFTR上皮细胞被安装在尤斯室(Physiologic Instruments,Inc.,San Diego,CA)中,并采用电压钳系统(Department of Bioengineering,University of Iowa,IA和PhysiologicInstruments,Inc.,San Diego,CA)使单层持续短路。通过应用2-mV脉冲测量跨上皮膜电阻。在这些条件下,FRT上皮表现出4KΩ/cm2或者更高的电阻。把溶液维持在27℃,并用空气鼓泡。电极偏移电位和流体电阻采用无细胞池校正。在这些条件下,电流反映了Cl-穿过在顶端膜中表达的ΔF508-CFTR的流动。采用MP100A-CE界面和AcqKnowledge软件(v3.2.6;BIOPAC Systems,Santa Barbara,CA),数字地获得ISC。
2.校正化合物的鉴定
典型方案使用基底外侧至顶端膜Cl-浓度梯度。为建立这种梯度,将正常的林格氏液用于基底外侧膜,而顶端NaCl用等摩尔的葡萄糖酸钠(用NaOH滴定至pH 7.4)替代以给出跨上皮的大的Cl-浓度梯度。所有实验用完整的单层进行。为了充分激活ΔF508-CFTR,应用毛喉素(10μM)和PDE抑制剂IBMX(100μM),随后加入CFTR增效剂染料木黄酮(50μM)。
如在其它细胞类型中观察到的那样,在低温温育稳定地表达ΔF508-CFTR的FRT细胞会增加CFTR在质膜中的功能密度。为了测定校正化合物的活性,把细胞在37℃用10μM受试化合物温育24小时,并随后冲洗3X,然后记录。把在化合物处理过的细胞中的cAMP-和染料木黄酮-介导的ISC归一化为27℃和37℃对照组,并表示为活性百分比。与37℃对照组相比,用校正化合物预温育细胞会显著增加cAMP和染料木黄酮介导的ISC。
3.增效剂化合物的鉴定
典型方案使用基底外侧至顶端膜Cl-浓度梯度。为建立这种梯度,将正常的林格氏液用于基底外侧膜,并用制霉菌素(360μg/ml)透化处理,而顶端NaCl用等摩尔的葡萄糖酸钠(用NaOH滴定至pH 7.4)替代,以给出跨上皮的大的Cl-浓度梯度。在制霉菌素透化处理后30分钟,进行所有实验。把毛喉素(10μM)和所有受试化合物加入到细胞培养池的两侧。将假定的ΔF508-CFTR增效剂的效力与已知增效剂染料木黄酮的效力相比较。
4.溶液
基底外侧溶液(以mM表示):NaCl(135)、CaCl2(1.2)、MgCl2(1.2)、K2HPO4(2.4)、KHPO4(0.6)、N-2-羟基乙基哌嗪-N’-2-乙磺酸(HEPES)(10)和葡萄糖(10)。溶液用NaOH滴定至pH7.4。
顶端溶液(以mM表示):与基底外侧溶液相同,但用葡萄糖酸钠(135)替代NaCl。
5.细胞培养
将表达ΔF508-CFTR(FRTΔF508-CFTR)的Fisher大鼠上皮(FRT)细胞用于自我们的光学测定鉴定的假定ΔF508-CFTR调节剂的尤斯室实验。在Costar Snapwell细胞培养池中培养细胞,并于37℃和5%CO2下,在用5%胎牛血清、100U/ml青霉素和100μg/ml链霉素补充的Coon氏改良的Ham氏F-12培养基中培养5天。在用于表征化合物的增效剂活性之前,于27℃把细胞温育16-48小时以校正ΔF508-CFTR。为了测定校正化合物的活性,于27℃或37℃,在化合物存在或不存在下把细胞温育24小时。
6.全细胞记录
采用穿孔膜片、全细胞记录监测温度和受试化合物校正的稳定表达ΔF508-CFTR的NIH3T3细胞中的宏观ΔF508-CFTR电流(IΔF508)。简言之,采用Axopatch 200B膜片钳放大器(Axon Instruments Inc.,Foster City,CA)在室温下进行IΔF508的电压钳记录。所有记录在10kHz的采样频率和1kHz低通滤波下获得。当充满胞内溶液时吸量管具有5-6MΩ的电阻。在这些记录条件下,计算的室温下Cl-的逆转电位(ECl)为-28mV。所有记录具有>20GΩ的密封电阻和<15MΩ串联电阻。采用配备有连同Clampex 8的Digidata 1320A/D界面的PC(Axon Instruments Inc.)进行脉冲产生、数据采集和分析。浴含有<250μl的盐水并采用重力驱动的灌注系统以2ml/分钟的速率持续灌注。
7.校正化合物的鉴定
为了测定校正化合物增加功能性ΔF508-CFTR在质膜中的密度的活性,我们使用以上描述的穿孔膜片记录技术测量用校正化合物处理24-hr后的电流密度。为了充分激活ΔF508-CFTR,把10μM毛喉素和20μM染料木黄酮加至细胞。在我们的记录条件下,在27℃温育24小时后的电流密度高于在37℃温育24小时后观察到的电流密度。这些结果与已知的低温温育对质膜中ΔF508-CFTR的密度的作用相吻合。为了测定校正化合物对CFTR电流密度的作用,于37℃把细胞与10μM的受试化合物温育24小时,并把电流密度与27℃和37℃对照组相比较(%活性)。记录之前,用胞外记录培养液冲洗细胞3X以除去任何残留的受试化合物。与37℃对照组相比较,用10μM的校正化合物预温育显著增加cAMP和染料木黄酮 依赖的电流。
8.增效剂化合物的鉴定
也采用穿孔膜片记录技术研究ΔF508-CFTR增效剂在稳定表达ΔF508-CFTR的NIH3T3细胞中增加宏观ΔF508-CFTR Cl-流(IΔF508)的能力。自光学测定鉴定的增效剂诱发了与光学测定中观察到的类似的效力和有效性的IΔF508的剂量依赖性增加。在检测的所有细胞中,在使用增效剂之前和期间的反转电位为约-30mV,其为计算的ECl(-28mV)。
9.溶液
胞内溶液(以mM表示):Cs-天冬氨酸(90)、CsCl(50)、MgCl2(1)、HEPES(10)和240μg/ml两性霉素-B(用CsOH把pH调至7.35)。
胞外溶液(以mM表示):N-甲基-D-葡萄糖胺(NMDG)-Cl(150)、MgCl2(2)、CaCl2(2)、HEPES(10)(用HCl把pH调至7.35)。
10.细胞培养
稳定表达ΔF508-CFTR的NIH3T3小鼠成纤维细胞用于全细胞记录。在175cm2培养瓶中,于5%CO2和90%湿度下,在37℃把细胞保持在用2mM谷氨酰胺、10%胎牛血清、1XNEAA、β-ME、1X青霉素/链霉素和25mM HEPES补充的Dulbecco氏改良的伊格尔培养基中。对全细胞记录,把2500-5000个细胞接种到聚-L-赖氨酸包被的玻璃盖玻片上并在用前于27℃培养24-48小时以测定增效剂的活性;并且于37℃在校正化合物存在或不存在下温育以用于测量校正剂的活性。
11.单通道记录
采用切除的内面外向式膜片观察NIH3T3细胞中稳定表达的温度校正的ΔF508-CFTR的单通道活性和增效剂化合物的活性。简言之,采用Axopatch 200B膜片钳放大器(Axon Instruments Inc.)在室温下进行单通道活性的电压钳记录。所有记录在10kHz的采样频率和400Hz低通滤波下获得。膜片钳(Patch pipettes)自Corning Kovar Sealing #7052玻璃(World Precision Instruments,Inc.,Sarasota,FL)制作并具有5-6MΩ的电阻(当充满胞外溶液时)。通过加入1mM Mg-ATP和75nM的cAMP依赖的蛋白激酶、催化亚基(PKA;Promega Corp.Madison,WI),在切除后激活ΔF508-CFTR。在通道活性稳定后,采用重力驱动的微灌注术系统灌注膜片。邻近膜片放置流入物,导致1-2秒内溶液完全交换。为在快速灌注期间维持ΔF508-CFTR活性,把非特异性磷酸酶抑制剂F-(10mM NaF)加入到浴液中。在这些记录条件下,通道活性在整个膜片记录期间保持恒定(最高达60分钟)。通过自胞内溶液移动至胞外溶液的正电荷产生的电流(阴离子以相反方向移动)作为正电流显示。钳制电位(pipette potential)(Vp)保持在80mV。
自包含≤2个活性通道的膜片分析通道活性。在实验过程中同时开口的最大数量决定了活性通道的数量。为了测定单通道电流幅度,自120秒的ΔF508-CFTR活性记录的数据于100Hz被“离线(off-line)”过滤,然后被用于构建用多高斯函数拟合的所有点振幅直方图(采用Bio-Patch Analysis软件(Bio-Logic Comp.法国))。自120秒的通道活性测定总微观电流和开放概率(Po)。采用Bio-Patch软件或自关系式Po=I/i(N)确定Po,其中I=平均电流,i=单通道电流幅值,且N=膜片中活性通道的数量。
12.溶液
胞外溶液(以mM表示):NMDG(150)、天冬氨酸(150)、CaCl2(5)、MgCl2(2)和HEPES(10)(用Tris碱把pH调至7.35)。
胞内溶液(以mM表示):NMDG-Cl(150)、MgCl2(2)、EGTA(5)、TES(10)和Tris碱(14)(用HCl把pH调至7.35)。
13.细胞培养
稳定表达ΔF508-CFTR的NIH3T3小鼠成纤维细胞用于离体膜膜片钳记录。在175cm2培养瓶中,于5%CO2和90%湿度下,在37℃把细胞维持在用2mM谷氨酰胺、10%胎牛血清、1X NEAA、β-ME、1X青霉素/链霉素和25mM HEPES补充的Dulbecco氏改良的伊格尔培养基中。对单通道记录,把2500-5000个细胞接种到聚-L-赖氨酸包被的玻璃盖玻片上并在用前于27℃培养24-48小时。
采用以上描述的程序,已经测量了化合物1的活性即EC50,并显示在表18中。
表18.

其它实施方案
在本公开内容中提及的所有出版物和专利都通过引用并入本文,其程度如同具体地且单独地指出每篇单独的出版物或专利申请通过引用并入。如果通过引用并入的任何专利或出版物的术语的含义与在本公开内容中使用的术语的含义相矛盾,意图以本公开内容中的术语的含义为准。此外,前面的讨论仅仅公开和描述了本发明的示例性的实施方案。本领域技术人员从这样的讨论中和从附图和权利要求书中会容易地认识到,在不脱离下述权利要求书所定义的 本发明的精神和范围内,可以对其做出各种不同的变化、修改和变动。
Claims (59)
1.一种用于口服施用的片剂,其包含:
a. 化合物1;
b. 填充剂;
c. 稀释剂;
d. 崩解剂;
e. 润滑剂;和
f. 助流剂。
2.根据权利要求1所述的片剂,其中化合物1是化合物1无定形形式的形式。
3.根据权利要求1或2所述的片剂,其中化合物1或化合物1无定形形式以在约1 mg至约250 mg范围内的量存在于所述片剂中。
4.根据权利要求1-3中任一项所述的片剂,其中化合物1或化合物1无定形形式以在约10 mg至约250 mg范围内的量存在于所述片剂中。
5.根据权利要求1-4中任一项所述的片剂,其中化合物1或化合物1无定形形式以在约25 mg至约250 mg范围内的量存在于所述片剂中。
6.根据权利要求1-5中任一项所述的片剂,其中化合物1或化合物1无定形形式以约50 mg至约200 mg的量存在于所述片剂中。
7.根据权利要求1-4中任一项所述的片剂,其中化合物1或化合物1无定形形式以约10 mg的量存在于所述片剂中。
8.根据权利要求1-6中任一项所述的片剂,其中化合物1或化合物1无定形形式以约50 mg的量存在于所述片剂中。
9.根据权利要求1-6中任一项所述的片剂,其中化合物1或化合物1无定形形式以约100 mg的量存在于所述片剂中。
10.根据权利要求1-4中任一项所述的片剂,其中按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约1重量%至约80重量%范围内。
11.根据权利要求1-5中任一项所述的片剂,其中按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约10重量%至约50重量%范围内。
12.根据权利要求1-5中任一项所述的片剂,其中按片剂的重量计,所述片剂中的化合物1或化合物1无定形形式的量是在约20重量%至约30重量%范围内。
13.根据权利要求1-4中任一项所述的片剂,其中所述片剂中的化合物1或化合物1无定形形式的量是所述片剂的约4重量%。
14.根据权利要求1-6中任一项所述的片剂,其中所述片剂中的化合物1或化合物1无定形形式的量是所述片剂的约25重量%。
15.根据权利要求1-14中任一项所述的片剂,其中所述填充剂选自:纤维素、改性纤维素、羧甲基纤维素钠、乙基纤维素、羟甲基纤维素、羟丙基纤维素、乙酸纤维素、微晶纤维素、磷酸氢钙、蔗糖、乳糖、玉米淀粉、马铃薯淀粉或它们的任意组合。
16.根据权利要求1-15中任一项所述的片剂,其中所述填充剂是微晶纤维素(MCC),且按片剂的重量计,其以在约10重量%至约90重量%范围内的量存在于所述片剂中。
17.根据权利要求1-16中任一项所述的片剂,其中所述稀释剂选自:乳糖一水合物、甘露醇、山梨醇、纤维素、磷酸钙、淀粉、糖或它们的任意组合。
18.根据权利要求1-17中任一项所述的片剂,其中所述稀释剂是乳糖一水合物,且按片剂的重量计,其以在约10重量%至约90重量%范围内的量存在于所述片剂中。
19.根据权利要求1-18中任一项所述的片剂,其中所述崩解剂选自:琼脂、藻胶、碳酸钙、羧甲基纤维素、纤维素、羟丙基纤维素、低取代的羟丙基纤维素、粘土、交联羧甲纤维素钠、交聚维酮、树胶、硅酸镁铝、甲基纤维素、波拉克林钾、海藻酸钠、淀粉羟乙酸钠、玉米淀粉、马铃薯淀粉、木薯淀粉或它们的任意组合。
20.根据权利要求1-19中任一项所述的片剂,其中所述崩解剂是交联羧甲纤维素钠,且按片剂的重量计,其以6重量%或更小的浓度存在于所述片剂中。
21.根据权利要求1-20中任一项所述的片剂,其中所述润滑剂选自:硬脂酸镁、硬脂酸钙、硬脂酸锌、硬脂酸钠、硬脂酸、硬脂酸铝、亮氨酸、山嵛酸甘油酯、氢化植物油、硬脂酰富马酸钠或它们的任意组合。
22.根据权利要求1-21中任一项所述的片剂,其中所述润滑剂是硬脂酸镁,且按片剂的重量计,其具有小于2重量%的浓度。
23.根据权利要求1-22中任一项所述的片剂,其中所述助流剂选自:胶体二氧化硅、滑石、玉米淀粉或它们的组合。
24.根据权利要求1-23中任一项所述的片剂,其中所述助流剂是胶体二氧化硅,且按片剂的重量计,其具有3重量%或更小的浓度。
25.根据权利要求1-24中任一项所述的片剂,其中所述片剂另外包含着色剂。
26.一种包含多个颗粒的片剂,所述组合物包含:
a. 化合物1无定形形式,按所述组合物的重量计,其量为约4重量%至约50重量%;
b. 填充剂,按所述组合物的重量计,其量为约10重量%至约45重量%;
c. 稀释剂,按所述组合物的重量计,其量为约10重量%至约45重量%;
d. 崩解剂,按所述组合物的重量计,其量为约1重量%至约5重量%;
e. 润滑剂,按所述组合物的重量计,其量为约0.3重量%至约3重量%;和
f. 助流剂,按所述组合物的重量计,其量为约0.3重量%至约3重量%。
27.根据权利要求1-26中任一项所述的片剂,其中化合物1是化合物1无定形形式,且是在喷雾干燥的分散体中。
28.根据权利要求26所述的片剂,其中所述喷雾干燥的分散体包含聚合物。
29.根据权利要求28所述的片剂,其中所述聚合物是羟丙甲基纤维素(HPMC)。
30.根据权利要求28或29所述的片剂,其中所述聚合物以20重量%至70重量%的量存在。
31.根据权利要求28-30中任一项所述的片剂,其中所述聚合物以30重量%至60重量%的量存在。
32.根据权利要求28-31中任一项所述的片剂,其中所述聚合物以约49.5重量%的量存在。
33.根据权利要求27-32中任一项所述的片剂,所述片剂另外包含表面活性剂。
34.根据权利要求33所述的片剂,其中所述表面活性剂是月桂基硫酸钠。
35.根据权利要求33或34所述的片剂,其中所述表面活性剂以0.1重量%至5重量%的量存在。
36.根据权利要求33-35中任一项所述的片剂,其中所述表面活性剂以约0.5重量%的量存在。
37.一种具有下表所示配方的片剂:
。
38.一种具有下表所示配方的片剂:
。
39.一种具有下表所示配方的片剂:
。
40.一种施用片剂的方法,所述方法包括:每天至少1次地给患者口服施用片剂,所述片剂包含:
a. 约1-200 mg化合物1无定形形式;
b. 填充剂;
c. 稀释剂;
d. 崩解剂;
e. 表面活性剂;
f. 助流剂;和
g. 润滑剂。
41.根据权利要求40所述的方法,其中所述片剂包含约10 mg化合物1无定形形式。
42.根据权利要求40所述的方法,其中所述片剂包含约50 mg化合物1无定形形式。
43.根据权利要求40所述的方法,其中所述片剂包含约100 mg化合物1无定形形式。
44.一种施用片剂的方法,所述方法包括:每天2次地给患者口服施用片剂,所述片剂包含:
a. 约1-200 mg化合物1无定形形式;
b. 填充剂;
c. 稀释剂;
d. 崩解剂;
e. 表面活性剂;
f. 助流剂;和
g. 润滑剂。
45.根据权利要求44所述的方法,其中所述片剂包含约10 mg化合物1无定形形式。
46.根据权利要求44所述的方法,其中所述片剂包含约50 mg化合物1无定形形式。
47.根据权利要求44所述的方法,其中所述片剂包含约100 mg化合物1无定形形式。
48.一种施用片剂的方法,所述方法包括:每12小时1次地给患者口服施用片剂,所述片剂包含:
a. 约1-200 mg化合物1无定形形式;
b. 填充剂;
c. 稀释剂;
d. 崩解剂;
e. 表面活性剂;
f. 助流剂;和
g. 润滑剂。
49.根据权利要求48所述的方法,其中所述片剂包含约10 mg化合物1无定形形式。
50.根据权利要求48所述的方法,其中所述片剂包含约50 mg化合物1无定形形式。
51.根据权利要求48所述的方法,其中所述片剂包含约100 mg化合物1无定形形式。
52.一种治疗受试者的疾病或减轻其严重程度的方法,所述方法包括:给所述受试者施用根据权利要求1-39中任一项所述的片剂或药物组合物,其中所述疾病选自:囊性纤维化、哮喘、吸烟诱发的慢性阻塞性肺病、慢性支气管炎、鼻鼻窦炎、便秘、胰腺炎、胰腺功能不全、由先天性输精管两侧缺失(CBAVD)导致的男性不育、轻度肺病、自发性胰腺炎、变应性支气管肺曲菌病(ABPA)、肝病、遗传性肺气肿、遗传性血色素沉着病、凝血-纤维蛋白溶解缺陷、蛋白质C缺乏、1型遗传性血管水肿、脂质加工缺陷、家族性高胆固醇血症、1型乳糜微粒血症、无β脂蛋白血症、溶酶体贮积病、I-细胞病/假性赫尔勒病、粘多糖贮积症、桑德霍夫病/泰-萨病、II型克-纳二氏综合征、多内分泌腺病/高胰岛素血症、糖尿病、拉伦侏儒症、髓过氧化物酶缺乏、原发性甲状旁腺功能减退、黑素瘤、1型聚糖病CDG、先天性甲状腺功能亢进、成骨不全、遗传性低纤维蛋白原血症、ACT缺乏症、尿崩症(DI)、神经生长性尿崩症、肾源性尿崩症、夏-马-图三氏综合征、佩-梅病、神经变性疾病、阿尔茨海默病、帕金森病、肌萎缩性侧索硬化、进行性核上性麻痹、皮克病、几种多聚谷氨酰胺神经障碍、亨廷顿舞蹈病、I型脊髓小脑共济失调、脊髓延髓肌肉萎缩症、齿状核红核苍白球丘脑下部核萎缩、肌强直性营养不良、海绵样脑病、由朊病毒蛋白加工缺陷所致的遗传性克雅病、法布里病、Straussler-Scheinker综合征、慢性阻塞性肺病、干眼病、斯耶格伦病、骨质疏松症、骨质减少、戈汉综合征、氯离子通道病、先天性肌强直病(Thomson和Becker形式)、III型巴特综合征、登特病、惊跳症、癫痫、惊跳症、溶酶体贮积病、Angelman综合征、原发性纤毛运动障碍(PCD)、纤毛的结构和/或功能的遗传性疾病、具有内脏逆位的PCD (也称作卡塔格纳综合征)、没有内脏逆位的PCD或纤毛发育不良。
53.根据权利要求52所述的方法,其中所述疾病是囊性纤维化、肺气肿、干眼病、慢性阻塞性肺病或骨质疏松症。
54.根据权利要求52所述的方法,其中所述疾病是囊性纤维化。
55.根据权利要求52-54中任一项所述的方法,其中所述受试者具有含ΔF508突变的囊性纤维化跨膜受体(CFTR)。
56.根据权利要求52-55中任一项所述的方法,其中所述受试者具有含R117H突变的囊性纤维化跨膜受体(CFTR)。
57.根据权利要求52-56中任一项所述的方法,其中所述受试者具有含G551D突变的囊性纤维化跨膜受体(CFTR)。
58.根据权利要求52-57中任一项所述的方法,其中所述方法包括:施用其它治疗剂。
59.根据权利要求58所述的方法,其中所述其它治疗剂是溶粘蛋白剂、支气管扩张剂、抗生素、抗感染剂、抗炎剂、除了化合物1以外的CFTR调节剂或营养剂。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37597610P | 2010-08-23 | 2010-08-23 | |
| US61/375,976 | 2010-08-23 | ||
| US201161506220P | 2011-07-11 | 2011-07-11 | |
| US61/506,220 | 2011-07-11 | ||
| PCT/US2011/048565 WO2012027247A2 (en) | 2010-08-23 | 2011-08-22 | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103153287A true CN103153287A (zh) | 2013-06-12 |
Family
ID=45594557
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2011800511228A Pending CN103153287A (zh) | 2010-08-23 | 2011-08-22 | (R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺的药物组合物及其施用 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20120046330A1 (zh) |
| EP (1) | EP2608779A2 (zh) |
| JP (1) | JP2013536231A (zh) |
| CN (1) | CN103153287A (zh) |
| AR (1) | AR084473A1 (zh) |
| AU (2) | AU2011293658B2 (zh) |
| BR (1) | BR112013004443A8 (zh) |
| CA (1) | CA2808501A1 (zh) |
| MX (1) | MX2013002035A (zh) |
| NZ (1) | NZ606805A (zh) |
| RU (1) | RU2013112937A (zh) |
| TW (2) | TWI619515B (zh) |
| WO (1) | WO2012027247A2 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106163300A (zh) * | 2013-12-20 | 2016-11-23 | 株式会社大赛璐 | 营养组合物 |
| CN110840847A (zh) * | 2014-04-15 | 2020-02-28 | 沃泰克斯药物股份有限公司 | 用于治疗囊性纤维化跨膜传导调节因子介导的疾病的药物组合物 |
| CN111818918A (zh) * | 2018-02-05 | 2020-10-23 | 弗特克斯药品有限公司 | 用于治疗囊性纤维化的药物组合物 |
Families Citing this family (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| RU2006111093A (ru) | 2003-09-06 | 2007-10-27 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | Модуляторы атр-связывающих кассетных транспортеров |
| US7977322B2 (en) | 2004-08-20 | 2011-07-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| LT2489659T (lt) | 2004-06-24 | 2018-03-26 | Vertex Pharmaceuticals Incorporated | Atp rišančios kasetės transporterių moduliatoriai |
| MX2008002019A (es) | 2005-08-11 | 2008-04-16 | Vertex Pharma | Moduladores del regulador de conductancia transmembrana de fibrosis quistica. |
| SI2404919T1 (sl) | 2005-11-08 | 2013-12-31 | Vertex Pharmaceuticals Incorporated | Heterocikliäśna spojina uporabna kot modulator za prenaĺ alce z atp-vezavno kaseto |
| RS60205B1 (sr) | 2005-12-28 | 2020-06-30 | Vertex Pharma | Farmaceutske kompozicije amorfnog oblika n-[2,4-bis(1,1-dimetiletil)-5-hidroksifenil]-1,4-dihidro-4-oksohinolin-3-karboksamida |
| CA2635760C (en) | 2005-12-28 | 2014-07-15 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| MX2008012945A (es) | 2006-04-07 | 2009-01-15 | Vertex Pharma | Moduladores de transportadores de cartucho de union a adenosina-trifosfato celular (atp). |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US8563573B2 (en) | 2007-11-02 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| CA2686838C (en) | 2007-05-09 | 2017-03-14 | Vertex Pharmaceuticals Incorporated | Modulators of cftr |
| NZ583420A (en) | 2007-08-24 | 2012-06-29 | Vertex Pharma | Isothiazolopyridinones useful for the treatment of (inter alia) cystic fibrosis |
| NZ585980A (en) * | 2007-11-16 | 2012-04-27 | Vertex Pharma | Isoquinoline modulators of atp-binding cassette transporters |
| CA2708146A1 (en) * | 2007-12-07 | 2009-06-18 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| PT2639222T (pt) | 2007-12-07 | 2016-11-01 | Vertex Pharma | Processo de produção de ácidos cicloalquilcarboxamido-piridina-benzoicos |
| CA2706920C (en) | 2007-12-07 | 2018-02-13 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| US20100036130A1 (en) | 2007-12-07 | 2010-02-11 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| ES2647531T3 (es) | 2008-02-28 | 2017-12-22 | Vertex Pharmaceuticals Incorporated | Derivados de heteroarilo como moduladores de CFTR |
| CA2718310C (en) | 2008-03-31 | 2018-08-07 | Vertex Pharmaceuticals Incorporated | Pyridyl derivatives as cftr modulators |
| CN102164587A (zh) | 2008-09-29 | 2011-08-24 | 沃泰克斯药物股份有限公司 | 3-(6-(1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)环丙烷甲酰氨基)-3-甲基吡啶-2-基)苯甲酸的剂量单元 |
| PT2349263E (pt) * | 2008-10-23 | 2014-07-28 | Vertex Pharma | Moduladores de regulador da condutância transmembranar da fibrose cística |
| KR101955863B1 (ko) | 2009-03-20 | 2019-03-07 | 버텍스 파마슈티칼스 인코포레이티드 | 낭성 섬유증 막횡단 전도도 조절자의 조정자의 제조 방법 |
| NZ700556A (en) * | 2010-03-25 | 2016-04-29 | Vertex Pharma | Solid forms of (r)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| US8802868B2 (en) | 2010-03-25 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxo1-5-yl)-N-(1-(2,3-dihydroxypropyl-6-fluoro-2-(1-hydroxy-2-methylpropan2-yl)-1H-Indol-5-yl)-Cyclopropanecarboxamide |
| HRP20211752T1 (hr) | 2010-04-07 | 2022-02-18 | Vertex Pharmaceuticals Incorporated | Farmaceutski pripravci 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksamido)-3-metilpiridin-2-il)benzojeve kiseline i njihova primjena |
| TWI549950B (zh) | 2010-04-07 | 2016-09-21 | 維泰克斯製藥公司 | 3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之固體形式 |
| SG10201505700QA (en) * | 2010-04-22 | 2015-08-28 | Vertex Pharma | Process of producing cycloalkylcarboxamido-indole compounds |
| US8563593B2 (en) | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| MX357328B (es) | 2011-11-08 | 2018-07-05 | Vertex Pharma | Moduladores de trasportadores de casete enlazante de atp. |
| NZ629199A (en) | 2012-02-27 | 2017-01-27 | Vertex Pharma | Pharmaceutical compositions comprising a solid dispersion of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| US8674108B2 (en) | 2012-04-20 | 2014-03-18 | Vertex Pharmaceuticals Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethy)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| AU2013270681A1 (en) * | 2012-06-08 | 2014-12-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of CFTR -mediated disorders |
| US9012496B2 (en) | 2012-07-16 | 2015-04-21 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide and administration thereof |
| WO2014042945A1 (en) * | 2012-09-11 | 2014-03-20 | Bend Research, Inc. | Methods for making pharmaceutical solid dosage forms of spray-dried dispersions |
| US9944700B2 (en) | 2013-03-13 | 2018-04-17 | Novartis Ag | Notch2 binding molecules for treating respiratory diseases |
| PL3068392T3 (pl) | 2013-11-12 | 2021-07-19 | Vertex Pharmaceuticals Incorporated | Proces wytwarzania kompozycji farmaceutycznych do leczenia chorób, w których pośredniczy cftr |
| EP3900726A1 (en) * | 2014-01-09 | 2021-10-27 | Verastem, Inc. | Compositions and methods for treatment of abnormal cell growth |
| EP2932966A1 (en) | 2014-04-16 | 2015-10-21 | Novartis AG | Gamma secretase inhibitors for treating respiratory diseases |
| EP3798214B1 (en) | 2014-10-06 | 2022-09-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| RU2749213C2 (ru) | 2014-10-07 | 2021-06-07 | Вертекс Фармасьютикалз Инкорпорейтед | Сокристаллы модуляторов регулятора трансмембранной проводимости при кистозном фиброзе |
| JP6494757B2 (ja) | 2014-11-18 | 2019-04-03 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | ハイスループット試験高速液体クロマトグラフィーを行うプロセス |
| US10167278B2 (en) | 2014-12-31 | 2019-01-01 | Auspex Pharmaceuticals, Inc. | Cyclopropanecarboxamide modulators of cystic fibrosis transmembrane conductance regulator |
| US10196384B2 (en) | 2015-03-31 | 2019-02-05 | Vertex Pharmaceuticals (Europe) Limited | Deuterated CFTR modulators |
| WO2017180794A1 (en) | 2016-04-13 | 2017-10-19 | Skyline Antiinfectives, Inc. | Deuterated o-sulfated beta-lactam hydroxamic acids and deuterated n-sulfated beta-lactams |
| EP3509570A1 (en) * | 2016-09-07 | 2019-07-17 | Celgene Corporation | Tablet compositions |
| ES2900263T3 (es) | 2016-09-30 | 2022-03-16 | Vertex Pharma | Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador |
| JOP20190125B1 (ar) | 2016-12-09 | 2022-03-14 | Vertex Pharma | مُعدِّل لمنظم موصلية التليف الكيسي عبر الغشاء، وتركيبات صيدلانية، وطرق للعلاج، وعملية لتصنيع المُعدِّل |
| CA3066084A1 (en) | 2017-06-08 | 2018-12-13 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| EP3654969A1 (en) | 2017-07-17 | 2020-05-27 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| TWI799435B (zh) | 2017-08-02 | 2023-04-21 | 美商維泰克斯製藥公司 | 製備化合物之製程 |
| CA3078893A1 (en) | 2017-10-19 | 2019-04-25 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of cftr modulators |
| EP3720849A2 (en) | 2017-12-08 | 2020-10-14 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| PT3752510T (pt) | 2018-02-15 | 2023-03-15 | Vertex Pharma | Macrociclos como moduladores do regulador de condutância de transmembrana da fibrose cística, suas composições farmacêuticas, seu uso no tratamento da fibrose cística e processos para produzi-los |
| EP3774825A1 (en) | 2018-04-13 | 2021-02-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| CA3067611A1 (en) | 2019-01-15 | 2020-07-15 | Apotex Inc. | Processes for the preparation of tezacaftor and intermediates thereof |
| TW202115092A (zh) | 2019-08-14 | 2021-04-16 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
| EP4013760A1 (en) | 2019-08-14 | 2022-06-22 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| TW202120517A (zh) | 2019-08-14 | 2021-06-01 | 美商維泰克斯製藥公司 | 製備cftr調節劑之方法 |
| CN111346062A (zh) * | 2020-03-11 | 2020-06-30 | 左人杰 | 一种以复合钙盐增加主药稳定性的口崩片及其制备方法 |
| WO2022013360A1 (en) * | 2020-07-17 | 2022-01-20 | Synthon B.V. | Pharmaceutical composition comprising ivacaftor |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007079139A2 (en) * | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2010053471A1 (en) * | 2008-11-06 | 2010-05-14 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020017295A1 (en) | 2000-07-07 | 2002-02-14 | Weers Jeffry G. | Phospholipid-based powders for inhalation |
| US6777400B2 (en) | 2000-08-05 | 2004-08-17 | Smithkline Beecham Corporation | Anti-inflammatory androstane derivative compositions |
| JP4977319B2 (ja) | 2002-09-30 | 2012-07-18 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 嚢胞性線維症膜コンダクタンス制御因子蛋白質阻害薬およびその使用方法 |
| WO2004080972A1 (en) | 2003-03-12 | 2004-09-23 | Vertex Pharmaceuticals Incorporated | Pirazole modulators of atp-binding cassette transporters |
| PT3889142T (pt) | 2003-04-11 | 2022-08-22 | Ptc Therapeutics Inc | Compostos de ácido 1,2,4-oxodiazolobenzoico e sua utilização para a supressão sem sentido e o tratamento de doenças |
| WO2004110352A2 (en) | 2003-05-16 | 2004-12-23 | The Regents Of The University Of California | Compounds having activity in increasing ion transport by mutant-cftr and uses thereof |
| WO2004111014A1 (en) | 2003-06-06 | 2004-12-23 | Vertex Pharmaceuticals Incorporated | Pyrimidine derivatives as modulators of atp-binding cassette transporters |
| JP2007509044A (ja) | 2003-10-08 | 2007-04-12 | バーテックス ファーマシューティカルズ インコーポレイテッド | シクロアルキルピラニル基を含むatp結合カセットトランスポーターのモジュレータ |
| US7407976B2 (en) | 2003-11-14 | 2008-08-05 | Vertex Pharmaceuticals Incorporated | Thiazoles and oxazoles useful as modulators of ATP-Binding Cassette transporters |
| US7414037B2 (en) | 2004-03-30 | 2008-08-19 | The Regents Of The University Of California | Hydrazide-containing CFTR inhibitor compounds and uses thereof |
| US20050222271A1 (en) * | 2004-03-31 | 2005-10-06 | Le Huang | Novel amorphous form of memantine hydrochloride |
| EP1765347A4 (en) | 2004-06-04 | 2008-10-01 | Univ California | COMPOUNDS WITH ION TRANSPORTER HEALING EFFECT BY MUTANT CFTR AND ITS USE |
| JP2008515985A (ja) | 2004-10-13 | 2008-05-15 | ピーティーシー セラピューティクス,インコーポレーテッド | ナンセンス抑制の化合物とその使用方法 |
| EP2363128B1 (en) | 2005-03-11 | 2016-02-17 | Vertex Pharmaceuticals Incorporated | Indole modulators of ATP-binding cassette transporters |
| CN101605543A (zh) | 2005-03-18 | 2009-12-16 | 加利福尼亚大学董事会 | 具有校正突变-cftr加工活性的化合物及其应用 |
| US8716321B2 (en) | 2005-04-08 | 2014-05-06 | Ptc Therapeutics, Inc. | Methods for dosing an orally active 1,2,4-oxadiazole |
| AU2006251624A1 (en) | 2005-05-24 | 2006-11-30 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding cassette transporters |
| WO2007044560A2 (en) | 2005-10-06 | 2007-04-19 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| MX2008012945A (es) * | 2006-04-07 | 2009-01-15 | Vertex Pharma | Moduladores de transportadores de cartucho de union a adenosina-trifosfato celular (atp). |
| CN102164587A (zh) * | 2008-09-29 | 2011-08-24 | 沃泰克斯药物股份有限公司 | 3-(6-(1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)环丙烷甲酰氨基)-3-甲基吡啶-2-基)苯甲酸的剂量单元 |
| UA104876C2 (uk) | 2008-11-06 | 2014-03-25 | Вертекс Фармасьютікалз Інкорпорейтед | Модулятори atф-зв'язувальних касетних транспортерів |
| CA2796642A1 (en) * | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
-
2011
- 2011-08-22 BR BR112013004443A patent/BR112013004443A8/pt not_active Application Discontinuation
- 2011-08-22 US US13/214,419 patent/US20120046330A1/en not_active Abandoned
- 2011-08-22 RU RU2013112937/15A patent/RU2013112937A/ru not_active Application Discontinuation
- 2011-08-22 NZ NZ60680511A patent/NZ606805A/en not_active IP Right Cessation
- 2011-08-22 AU AU2011293658A patent/AU2011293658B2/en not_active Ceased
- 2011-08-22 MX MX2013002035A patent/MX2013002035A/es not_active Application Discontinuation
- 2011-08-22 CA CA2808501A patent/CA2808501A1/en not_active Abandoned
- 2011-08-22 CN CN2011800511228A patent/CN103153287A/zh active Pending
- 2011-08-22 EP EP11751753.2A patent/EP2608779A2/en not_active Withdrawn
- 2011-08-22 WO PCT/US2011/048565 patent/WO2012027247A2/en active Application Filing
- 2011-08-22 JP JP2013526057A patent/JP2013536231A/ja active Pending
- 2011-08-23 TW TW103137082A patent/TWI619515B/zh active
- 2011-08-23 TW TW100130218A patent/TW201238611A/zh unknown
- 2011-08-23 AR ARP110103065 patent/AR084473A1/es not_active Application Discontinuation
-
2015
- 2015-06-03 AU AU2015202975A patent/AU2015202975B2/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007079139A2 (en) * | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2010053471A1 (en) * | 2008-11-06 | 2010-05-14 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
Non-Patent Citations (1)
| Title |
|---|
| 崔福德等: "《药剂学》", 31 August 2002, article "固体分散技术", pages: 443 - 446 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106163300A (zh) * | 2013-12-20 | 2016-11-23 | 株式会社大赛璐 | 营养组合物 |
| CN106163300B (zh) * | 2013-12-20 | 2020-03-13 | 株式会社大赛璐 | 营养组合物 |
| US10869883B2 (en) | 2013-12-20 | 2020-12-22 | Daicel Corporation | Nutrient composition having lipid metabolism-improving action |
| CN110840847A (zh) * | 2014-04-15 | 2020-02-28 | 沃泰克斯药物股份有限公司 | 用于治疗囊性纤维化跨膜传导调节因子介导的疾病的药物组合物 |
| CN110840847B (zh) * | 2014-04-15 | 2022-07-29 | 沃泰克斯药物股份有限公司 | 用于治疗囊性纤维化跨膜传导调节因子介导的疾病的药物组合物 |
| CN111818918A (zh) * | 2018-02-05 | 2020-10-23 | 弗特克斯药品有限公司 | 用于治疗囊性纤维化的药物组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2011293658B2 (en) | 2015-03-05 |
| US20120046330A1 (en) | 2012-02-23 |
| MX2013002035A (es) | 2013-03-25 |
| RU2013112937A (ru) | 2014-09-27 |
| AU2011293658A1 (en) | 2013-04-04 |
| WO2012027247A2 (en) | 2012-03-01 |
| AR084473A1 (es) | 2013-05-22 |
| WO2012027247A3 (en) | 2012-05-24 |
| TWI619515B (zh) | 2018-04-01 |
| CA2808501A1 (en) | 2012-03-01 |
| EP2608779A2 (en) | 2013-07-03 |
| BR112013004443A2 (pt) | 2016-06-07 |
| AU2015202975B2 (en) | 2016-05-12 |
| JP2013536231A (ja) | 2013-09-19 |
| NZ606805A (en) | 2015-03-27 |
| BR112013004443A8 (pt) | 2018-01-02 |
| TW201503911A (zh) | 2015-02-01 |
| AU2015202975A1 (en) | 2015-06-18 |
| TW201238611A (en) | 2012-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103153287A (zh) | (R)-1-(2,2-二氟苯并[d][1,3]间二氧杂环戊烯-5-基)-N-(1-(2,3-二羟基丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1H-吲哚-5-基)环丙烷甲酰胺的药物组合物及其施用 | |
| US10058546B2 (en) | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxo1-5-y1)-N-(1-(2,3-dihydroxypropy1)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-y1)-1H-indol-5-y1) cyclopropanecarbox-amide and administration thereof | |
| JP7003085B2 (ja) | Cftrが媒介する疾患の処置のための医薬組成物 | |
| CN109081804B (zh) | 环丙烷甲酰胺的固体形式 | |
| AU2011237601B2 (en) | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyriodin-2-yl)benzoic acid and administration thereof | |
| CN104168890A (zh) | 3-(6-(1-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)环丙烷甲酰胺基)-3-甲基吡啶-2-基)苯甲酸的制剂 | |
| AU2016213697B2 (en) | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid and administration thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1186120 Country of ref document: HK |
|
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20130612 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1186120 Country of ref document: HK |