CN102101847A - N-甲基-n′-(2-氯乙基)哌嗪的制备方法 - Google Patents
N-甲基-n′-(2-氯乙基)哌嗪的制备方法 Download PDFInfo
- Publication number
- CN102101847A CN102101847A CN 201010573660 CN201010573660A CN102101847A CN 102101847 A CN102101847 A CN 102101847A CN 201010573660 CN201010573660 CN 201010573660 CN 201010573660 A CN201010573660 A CN 201010573660A CN 102101847 A CN102101847 A CN 102101847A
- Authority
- CN
- China
- Prior art keywords
- piperazine
- methyl
- chloroethyl
- preparation
- bromo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- FHDYRFNCTJFNQX-UHFFFAOYSA-N 1-(2-chloroethyl)-4-methylpiperazine Chemical compound CN1CCN(CCCl)CC1 FHDYRFNCTJFNQX-UHFFFAOYSA-N 0.000 title claims abstract description 19
- 238000000034 method Methods 0.000 title abstract 3
- 238000006243 chemical reaction Methods 0.000 claims abstract description 21
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 claims abstract description 18
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 claims abstract description 18
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 claims abstract description 9
- 239000002994 raw material Substances 0.000 claims abstract description 6
- 239000003054 catalyst Substances 0.000 claims abstract description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 24
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 18
- 238000003756 stirring Methods 0.000 claims description 12
- 238000002360 preparation method Methods 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- 239000007787 solid Substances 0.000 claims description 8
- 238000000967 suction filtration Methods 0.000 claims description 8
- 239000000047 product Substances 0.000 claims description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 4
- 239000012141 concentrate Substances 0.000 claims description 4
- 238000001816 cooling Methods 0.000 claims description 4
- 239000013078 crystal Substances 0.000 claims description 4
- 238000002425 crystallisation Methods 0.000 claims description 4
- 230000008025 crystallization Effects 0.000 claims description 4
- 230000006837 decompression Effects 0.000 claims description 4
- 229960000935 dehydrated alcohol Drugs 0.000 claims description 4
- 238000004821 distillation Methods 0.000 claims description 4
- -1 filter Substances 0.000 claims description 4
- 239000000706 filtrate Substances 0.000 claims description 4
- 239000007788 liquid Substances 0.000 claims description 4
- 239000012452 mother liquor Substances 0.000 claims description 4
- 238000007789 sealing Methods 0.000 claims description 4
- 238000010025 steaming Methods 0.000 claims description 4
- 238000003786 synthesis reaction Methods 0.000 claims description 4
- 238000010792 warming Methods 0.000 claims description 4
- 230000015572 biosynthetic process Effects 0.000 claims description 3
- 239000002131 composite material Substances 0.000 claims description 2
- MAIPOMCACBNHEI-UHFFFAOYSA-N 1-(2-chloroethyl)piperazine Chemical compound ClCCN1CCNCC1 MAIPOMCACBNHEI-UHFFFAOYSA-N 0.000 claims 4
- 229940043237 diethanolamine Drugs 0.000 abstract 1
- 238000009776 industrial production Methods 0.000 abstract 1
- 238000000746 purification Methods 0.000 abstract 1
- 238000000926 separation method Methods 0.000 abstract 1
- 238000006467 substitution reaction Methods 0.000 abstract 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 14
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 10
- 239000003814 drug Substances 0.000 description 5
- 150000002431 hydrogen Chemical class 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 1
- DNXHEGUUPJUMQT-CBZIJGRNSA-N Estrone Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 DNXHEGUUPJUMQT-CBZIJGRNSA-N 0.000 description 1
- 241000532412 Vitex Species 0.000 description 1
- 235000001667 Vitex agnus castus Nutrition 0.000 description 1
- BAOWVDHYZBLYMB-UHFFFAOYSA-N [F].C1=CC=NC=C1 Chemical compound [F].C1=CC=NC=C1 BAOWVDHYZBLYMB-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000003905 agrochemical Substances 0.000 description 1
- 239000003430 antimalarial agent Substances 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 239000011280 coal tar Substances 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 229960003399 estrone Drugs 0.000 description 1
- SMANXXCATUTDDT-QPJJXVBHSA-N flunarizine Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)N1CCN(C\C=C\C=2C=CC=CC=2)CC1 SMANXXCATUTDDT-QPJJXVBHSA-N 0.000 description 1
- 229960000326 flunarizine Drugs 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000000077 insect repellent Substances 0.000 description 1
- 239000010687 lubricating oil Substances 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- ZQSQZTFHCWMNCX-UHFFFAOYSA-N oxalic acid;piperazine Chemical compound C1CNCCN1.OC(=O)C(O)=O.OC(=O)C(O)=O ZQSQZTFHCWMNCX-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- JOHZPMXAZQZXHR-UHFFFAOYSA-N pipemidic acid Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CN=C1N1CCNCC1 JOHZPMXAZQZXHR-UHFFFAOYSA-N 0.000 description 1
- 229960001732 pipemidic acid Drugs 0.000 description 1
- UCRHFBCYFMIWHC-UHFFFAOYSA-N piperaquine Chemical compound ClC1=CC=C2C(N3CCN(CC3)CCCN3CCN(CC3)C=3C4=CC=C(C=C4N=CC=3)Cl)=CC=NC2=C1 UCRHFBCYFMIWHC-UHFFFAOYSA-N 0.000 description 1
- 229950006717 piperaquine Drugs 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000007660 quinolones Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940066771 systemic antihistamines piperazine derivative Drugs 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
Landscapes
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
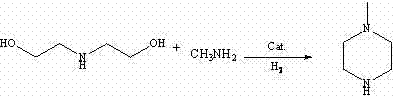

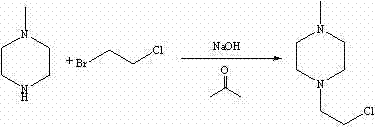
Claims (5)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010573660A CN102101847B (zh) | 2010-12-06 | 2010-12-06 | N-甲基-n′-(2-氯乙基)哌嗪的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010573660A CN102101847B (zh) | 2010-12-06 | 2010-12-06 | N-甲基-n′-(2-氯乙基)哌嗪的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102101847A true CN102101847A (zh) | 2011-06-22 |
| CN102101847B CN102101847B (zh) | 2012-09-12 |
Family
ID=44154936
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010573660A Expired - Fee Related CN102101847B (zh) | 2010-12-06 | 2010-12-06 | N-甲基-n′-(2-氯乙基)哌嗪的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102101847B (zh) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013178693A1 (de) | 2012-06-01 | 2013-12-05 | Basf Se | Verfahren zur herstellung eines mono-n-alkyl-piperazins |
| WO2013178534A1 (de) | 2012-06-01 | 2013-12-05 | Basf Se | Verfahren zur herstellung eines mono-n-alkyl-piperazins |
| US8637668B2 (en) | 2010-06-15 | 2014-01-28 | Basf Se | Process for preparing a cyclic tertiary methylamine |
| US8884015B2 (en) | 2012-06-01 | 2014-11-11 | Basf Se | Process for the preparation of a mono-N-alkypiperazine |
| WO2014184039A1 (de) | 2013-05-16 | 2014-11-20 | Basf Se | Verfahren zur herstellung von n-alkyl-piperazinen |
| US8933223B2 (en) | 2010-10-14 | 2015-01-13 | Basf Se | Process for preparing a cyclic tertiary amine |
| US8981093B2 (en) | 2012-06-06 | 2015-03-17 | Basf Se | Process for preparing piperazine |
| CN108503608A (zh) * | 2018-03-26 | 2018-09-07 | 吴彦彬 | 一种1,4-二甲基哌嗪的制备方法 |
-
2010
- 2010-12-06 CN CN201010573660A patent/CN102101847B/zh not_active Expired - Fee Related
Non-Patent Citations (2)
| Title |
|---|
| 《Eur. J. Med. Chem.》 19951231 G. Caliendo et al. Synthesis and biological activity of benzotriazole derivatives structurally related to trazodone 77-84 1-5 第30卷, * |
| 《河北工业大学学报》 20090630 郭胜辉等 CuO-NiO/Al2O3催化剂上N-甲基哌嗪合成研究 24-28 1-5 第38卷, 第3期 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8637668B2 (en) | 2010-06-15 | 2014-01-28 | Basf Se | Process for preparing a cyclic tertiary methylamine |
| US8933223B2 (en) | 2010-10-14 | 2015-01-13 | Basf Se | Process for preparing a cyclic tertiary amine |
| WO2013178693A1 (de) | 2012-06-01 | 2013-12-05 | Basf Se | Verfahren zur herstellung eines mono-n-alkyl-piperazins |
| WO2013178534A1 (de) | 2012-06-01 | 2013-12-05 | Basf Se | Verfahren zur herstellung eines mono-n-alkyl-piperazins |
| US8884015B2 (en) | 2012-06-01 | 2014-11-11 | Basf Se | Process for the preparation of a mono-N-alkypiperazine |
| US8927712B2 (en) | 2012-06-01 | 2015-01-06 | Basf Se | Process for the preparation of a mono-N-alkylpiperazine |
| US8981093B2 (en) | 2012-06-06 | 2015-03-17 | Basf Se | Process for preparing piperazine |
| WO2014184039A1 (de) | 2013-05-16 | 2014-11-20 | Basf Se | Verfahren zur herstellung von n-alkyl-piperazinen |
| CN108503608A (zh) * | 2018-03-26 | 2018-09-07 | 吴彦彬 | 一种1,4-二甲基哌嗪的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102101847B (zh) | 2012-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102101847B (zh) | N-甲基-n′-(2-氯乙基)哌嗪的制备方法 | |
| CN110467555A (zh) | 一种轴手性芳基吲哚化合物及其合成方法 | |
| CN102079737B (zh) | 一种制备芹菜素的方法 | |
| CN110627690A (zh) | 一类新型对香豆酸磺酸酯衍生物及其制备方法和应用 | |
| CN108097265A (zh) | 一种抗肿瘤靶向药物尼罗替尼芳胺中间体的制备方法 | |
| CN107098822A (zh) | 一种制备普仑司特关键中间体3‑氨基‑2‑羟基苯乙酮的制备方法 | |
| CN108863822B (zh) | 一种精制盐酸异丙肾上腺素的方法 | |
| CN107445869A (zh) | 一种盐酸二甲双胍的合成方法 | |
| CN105461580B (zh) | 一种异丙甲草胺的合成方法 | |
| CN103214534A (zh) | 一种3’-脱氧腺苷的制备方法 | |
| CN116986986A (zh) | 一种3-氧代-1-环丁烷羧酸中间体的合成方法 | |
| CN110713471B (zh) | 一种盐酸曲美他嗪的合成方法 | |
| CN101486730A (zh) | 一种磷酸肌酸钠化合物及其合成方法 | |
| CN103113408B (zh) | 一种制备磷霉素左磷右胺盐的新方法 | |
| CN112624951B (zh) | 一种氨磺必利的制备方法 | |
| CN106397188B (zh) | 一种l-菊苣酸的制备方法 | |
| CN107879979A (zh) | 一种右美托咪定的制备方法 | |
| CN112500370A (zh) | 一种路易斯碱催化合成烯酰吗啉的方法 | |
| CN108554456B (zh) | 一种稀土咪唑盐化合物作为催化剂的应用 | |
| CN105732601B (zh) | 含咪唑类功能基团的香豆素类化合物及其制备方法和应用 | |
| CN110642770B (zh) | 一种5-甲氧基吲哚的制备方法 | |
| CN109369414B (zh) | 一种制备伏虫隆中间体3,5-二氯-2,4-二氟苯胺的方法 | |
| CN109369749B (zh) | 一种落新妇甙的制备方法 | |
| CN108503580A (zh) | 一种阿哌沙班中间体的制备方法 | |
| CN115772077B (zh) | 一种由阿拉伯糖醇催化转化制备手性d-甘油酸的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: ZHOU YULIAN Free format text: FORMER OWNER: ZHANGJIAGANG TIANYOU NEW MATERIAL TECHNOLOGY CO., LTD. Effective date: 20120802 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20120802 Address after: Suzhou City, Jiangsu province 215636 Daxin Town, Zhangjiagang city along the Yangtze River Road on the north side of Taihua chemical technical room Applicant after: Zhou Yulian Address before: Zhangjiagang City Daxin Town old dam village in Suzhou City, Jiangsu province 215636 Zhangjiagang field by the new Mstar Technology Ltd Applicant before: Zhangjiagang Tianyou New Material Technology Co., Ltd. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20120912 Termination date: 20131206 |