CN101516886B - 噻吩吡唑并嘧啶化合物 - Google Patents
噻吩吡唑并嘧啶化合物 Download PDFInfo
- Publication number
- CN101516886B CN101516886B CN2007800349472A CN200780034947A CN101516886B CN 101516886 B CN101516886 B CN 101516886B CN 2007800349472 A CN2007800349472 A CN 2007800349472A CN 200780034947 A CN200780034947 A CN 200780034947A CN 101516886 B CN101516886 B CN 101516886B
- Authority
- CN
- China
- Prior art keywords
- hexane
- thiophene
- methyl
- dimethyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 Cc1c(*)cc(*)[s]1 Chemical compound Cc1c(*)cc(*)[s]1 0.000 description 7
- KQJDDNCSNLUGAK-UHFFFAOYSA-N CCC(CC)c([n]1nc2C)cc(C)nc1c2I Chemical compound CCC(CC)c([n]1nc2C)cc(C)nc1c2I KQJDDNCSNLUGAK-UHFFFAOYSA-N 0.000 description 1
- YTETWZNORTXMFJ-UHFFFAOYSA-N CCC(CC)c1cc(C)nc2cc(C)n[n]12 Chemical compound CCC(CC)c1cc(C)nc2cc(C)n[n]12 YTETWZNORTXMFJ-UHFFFAOYSA-N 0.000 description 1
- FRAKGJKEMLMXMD-UHFFFAOYSA-N Cc1ncn[n]1C Chemical compound Cc1ncn[n]1C FRAKGJKEMLMXMD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Addiction (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Psychiatry (AREA)
- Diabetes (AREA)
- Gynecology & Obstetrics (AREA)
- Immunology (AREA)
- Heart & Thoracic Surgery (AREA)
- Obesity (AREA)
- Child & Adolescent Psychology (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Pregnancy & Childbirth (AREA)
- Cardiology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明涉及式(I)化合物
Description
发明领域
本发明涉及新噻吩吡唑并嘧啶化合物、其药物组合物及其作为CRF1受体拮抗剂在治疗精神病学和神经内分泌障碍(psychiatric andneuroendocrine disorders)、神经疾病和代谢综合征中的用途。
发明背景
促肾上腺皮质激素释放因子(CRF)是41氨基酸肽,它是垂体前叶腺阿黑皮素原(POMC)源性肽分泌的主要生理调节剂。除了它在垂体腺的内分泌作用之外,CRF的免疫组织化学定位已证明:该激素在中枢神经系统中具有广泛的下丘脑外分布,并产生各种各样自主的、电生理和行为效应,这与脑中的神经递质或神经调节剂作用一致。还有证据表明:CRF在整合免疫系统对生理、心理和免疫应激物的应答中起重要作用。
CRF已牵涉精神病学障碍和神经疾病,包括抑郁和焦虑,以及下列病症:阿尔茨海默病、亨廷顿舞蹈病、进行性核上性麻痹、肌萎缩侧索硬化、帕金森病、癫痫、偏头痛、酒精和精神活性物质滥用及相关的戒断症状、肥胖症、代谢综合征、先天性肾上腺增生症、库欣病、高血压、卒中、肠易激综合征、应激诱导的胃溃疡、经前期综合征、性功能障碍、早产、炎性疾病、变态反应、多发性硬化、内脏痛、睡眠障碍、垂体肿瘤或异位垂体源性肿瘤(ectopic pituitary-derived tumor)、慢性疲劳综合征和纤维肌瘤。
已经鉴定了CRF受体亚型CRF1和CRF2,它们在脑内非均相分布,因此提示有潜在的功能多样性。例如,广泛分布的脑CRF1受体强烈地牵涉于伴随暴露于环境应激物的情绪性。重要的是,CRF1而不是CRF2受 体显示介导选择致焦虑样行为(select anxiogenic like behavior)。更离散的中膈/下丘脑分布和替代内源性配体的可用性表明CRF2受体的不同功能作用。例如,据报道:对CRF2受体相对于CRF1受体具有优先亲和力的新型CRF家族神经肽可抑制食欲,而不产生用选择性CRF1激动作用所观察到的行为激活特征。在其他的情况下,CRF2激动作用产生与所报道的CRF1拮抗剂或CRF1基因缺失的那些作用类似的作用。例如,尽管CRF2激动剂已被推荐为减肥药,但是CRF1拮抗剂也可以是重要的肥胖症疗法。
某些可用作CRF拮抗剂的吡咯并[2,3-d]嘧啶、吡咯并[3,2-d]嘧啶、吡唑并[1,5-a]嘧啶、1,2,3-三唑并[4,5-b]吡啶和吡唑并[1,5-a]-1,3,5-三嗪在WO94/13676、WO 97/29109、WO 98/08847和WO 98/03510中描述。
本发明提供了可用作CRF1受体拮抗剂的新型噻吩吡唑并嘧啶。鉴于上述,期望发现新的有效和选择性的CRF1拮抗剂,它作为潜在有价值的治疗剂用于治疗精神病学和神经内分泌障碍、神经疾病和代谢综合征。进一步,由于大部分市售CNS和心血管药物显示不期望的生物利用度特性,所以还期望发现相对于已知CRF拮抗剂如CP154526和NBI30775而言具有优异生物利用度特性的新化合物。
发明概述
在一个实施方案中,本发明涉及式I化合物或其药学上可接受的盐,

其中:
R1和R2独立地是氢或C1-C3烷基;
R3是 
R4是Cl或甲基;
R5是氢、Br、硝基、甲氧基、甲氧基甲基、二甲氨基、乙氧羰基、乙酰氨基、乙酰氧基、 
在另一个实施方案中,本发明涉及治疗患者的抑郁或重度抑郁症、焦虑、酒精或精神活性物质滥用、肥胖症、高血压、代谢综合征、肠应激综合征、癫痫、卒中、睡眠障碍、变态反应、偏头痛、经前期综合征(PMS)、不育症、性功能障碍、先天性肾上腺增生症、库欣病、早产、应激诱导的胃溃疡、炎性疾病、垂体肿瘤或异位垂体源性肿瘤、慢性疲劳综合征、纤维肌瘤、内脏痛或多发性硬化的方法,包括:对有需要的患者施用治疗有效量的式I化合物或其药学上可接受的盐。
在另一个实施方案中,本发明涉及式I化合物或其药学上可接受的盐在制备药物中的用途,所述药物用于治疗抑郁或重度抑郁症、焦虑、酒精或精神活性物质滥用、肥胖症、高血压、代谢综合征、肠易激综合征、癫痫、卒中、睡眠障碍、变态反应、偏头痛、经前期综合征(PMS)、不育症、性功能障碍、先天性肾上腺增生症、库欣病、早产、应激诱导的胃溃疡、炎性疾病、垂体肿瘤或异位垂体源性肿瘤、慢性疲劳综合征、纤维肌瘤、内脏痛或多发性硬化。
在另一个实施方案中,本发明涉及用作药物的式I化合物或其药学上可接受的盐。
发明详述
如上所用且贯穿整个本发明说明书的下列术语,除非另有说明,具有下列含义:
“烷基”表示饱和脂肪族烃基,其可以是直链或支链的,在链中具有1至5个碳原子。
“药学上可接受的赋形剂”是指增加制剂特性的药学上可接受的制剂 载体、溶液或添加剂。这种赋形剂必须与制剂的其他成分相容,且对其受者无害,为技术人员众所周知(例如参见Remingtons PharmaceuticalSciences,第19版,Mack Publishing Company,1995)。
“药学上可接受的盐”是指本发明化合物的相对无毒的、无机和有机酸加成盐和碱加成盐。这些盐可在最后分离和纯化化合物时原位制备。特别地,酸加成盐可通过分别将经纯化的化合物以其游离碱形式与合适有机或无机酸反应并分离由此生成的盐而制备(例如参见RemingtonsPharmaceutical Sciences,第19版,Mack Publishing Company,1995)。
“治疗有效量”或“有效量”是指本发明式I化合物或含有本发明式I化合物的药物组合物的量,所述量诱发研究人员、兽医、医学博士或其他临床医生正寻求的组织、系统、动物或人的生物或医学反应或者对所述组织、系统、动物或人的期望治疗作用。
术语“治疗(treatment)”、“治疗(treat)”、“治疗(treating)”等表示包括延缓和逆转病症的进展。这些术语还包括减轻、改善、降低、消除或减少障碍或病症的一种或多种症状,即使并未实际消除所述障碍或病症并且即使并未延缓或逆转所述障碍或病症本身的进展。术语“治疗”及类似术语还包括防止(例如预防性)和姑息治疗。疾病的防止表现为延长或延迟疾病症状的发生。
分子结构中的符号“——”指示具体取代基的连接位置。
当任何变量在任意组分或式I中出现多于一次时,它每次出现的定义独立于它每个其他次出现的定义。同时,只有这种组合形成稳定的化合物,取代基和/或变量的组合才是允许的。在选择本发明化合物时,本领域技术人员应认识到:各种取代基即R1、R2等的选择应符合化学结构连接性众所周知的原则。
根据贯穿本公开文本所用的标准命名法,先描述指定侧链的末端部分,继之以相邻的官能团直至连接的点。例如,芳基羰基氨基烷基取代基等于芳基-C(O)-NH-烷基-。
本发明包括特定类的发明,如下述:
(a)式I化合物或其药学上可接受的盐,其中R1和R2独立地是乙基或正丙基;
(b)式I化合物或其药学上可接受的盐,其中R3是 

(c)式I化合物或其药学上可接受的盐,其中R5是甲氧基、甲氧基甲基、二甲氨基或 
(d)式I化合物或其药学上可接受的盐用于治疗抑郁或焦虑的用途;
(e)式I化合物或其药学上可接受的盐用于治疗酒精或精神活性物质滥用的用途;
(f)显示CRF1结合的Ki值≤500nM的式I化合物或其药学上可接受的盐;
(g)显示CRF1结合的Ki值≤50nM的式I化合物或其药学上可接受的盐;
(h)显示CRF1结合的Ki值≤5nM的式I化合物或其药学上可接受的盐;
(i)显示CRF1结合的Ki值≤500nM并且相对于CRF2与CRF1选择性结合(即较低Ki)的式I化合物或其药学上可接受的盐;
(j)显示CRF1结合的Ki值≤50nM并且相对于CRF2与CRF1选择性结合(即较低Ki)的式I化合物或其药学上可接受的盐;和
(k)显示CRF1结合的Ki值≤5nM并且相对于CRF2与CRF1选择性结合(即较低Ki)的式I化合物或其药学上可接受的盐;
(l)生物利用度特性优于已知CRF拮抗剂(例如CP154526和NBI30775)的式I化合物或其药学上可接受的盐。
本发明的化合物优选配制成经各种途径施用的药物组合物。优选地, 这些组合物用于口服施用。这些药物组合物及用于制备它们的方法为本领域所众所周知(例如参见Remington:The Science and Practice of Pharmacy,A.Gennaro等人编,第19版,Mack Publishing Co.,1995)。
式I化合物通常在宽剂量范围内有效。例如,每天剂量通常落在约0.0001至约30mg/kg体重的范围内。在某些情况下,低于前述范围下限的剂量水平可能超出足够量,而在其他情况下仍可使用更大的剂量却不会引起任何有害副作用,因此上述剂量范围并不旨在以任何方式限制本发明的范围。应理解,实际所施用的化合物的量将由医生根据有关情况确定,包括待治疗的病症、所选的施用途径、所施用的实际化合物、个别患者的年龄、重量和反应及患者症状的严重度。
式I化合物是CRF-1拮抗剂,因此可用于治疗通过减少CRF1受体刺激可治疗的病症。促肾上腺皮质激素释放因子(CRF)是41氨基酸肽,它是垂体前叶腺的阿黑皮素原(POMC)源性肽分泌的主要生理调节剂[J.Rivier等人,Proc.Natl.Acad.Sci(USA)80:4851(1983);W.Vale等人,Science213:1394(1981)],它已与许多种医学病症有联系。例如,除了它在垂体腺的内分泌作用之外,CRF的免疫组织化学定位已证明所述激素在中枢神经系统中具有广泛的下丘脑外分布并产生各种各样自主的、电生理和行为效应,这与脑中神经递质或神经调节剂的作用一致[W.Vale等人,Rec.Prog.Horm.Res.39:245(1983);G.F.Koob,Persp.Behav.Med.2:39(1985);E.B.De Souza等人,J.Neurosci.5:3189(1985)]。还有证据显示:CRF在整合免疫系统对生理、心理和免疫应激物的应答中起重要作用[例如参见J.E.Blalock,Physiological Reviews 69:1(1989);J.E.Morley,Life Sci.41:527(1987)]。
CRF涉及精神病学障碍和神经疾病,包括抑郁和焦虑[D.M.Nielsen,Life Sci.78:909-919;H.E.Kunzel等人,J.Psychiatr.Res.37:525-533;D.R.Gehlert等人,Eur.J.Pharmacol.509:145-153]。还假定CRF作用于阿尔茨海默病、亨廷顿舞蹈病、进行性核上性麻痹和肌萎缩侧索硬化的病因学和病理生理学中,由于它们涉及中枢神经系统中的CRF神经元功能障碍[综 述参见E.B.De Souze,Hosp.Practice 23:59(1988)]。已显示CRF的长期施用产生多巴胺系统的损害,这表明在帕金森病中起作用[E.Izzo等人,Pharmacol.Biochem.Behav.81:701-708(2005)]。CRF所涉及的其他神经障碍包括癫痫[T.Z.Baram等人,Brain Res.770:89-95(1997)]和偏头痛[T.C.Theoharides等人,Endocrinology 136:5745-5750(1995)]。CRF已牵涉酒精和精神活性物质滥用及相关的戒断症状[D.H.Overstreet等人,Pharmacol.Biochem.Behav.77:405-413;Y.Shaham等人,Psychopharmacology(Berl)137:184-190]。此外,有证据表明CRF在各种内分泌障碍和心血管疾病如肥胖症[E.Timofeeva和D.Richard,Neuroendocrinology 66:327-340(1997)]、代谢性综合征[A.M.Ward等人,Metabolism 53:720-726(2004)]、先天性肾上腺增生症[D.P.Merke和G.B.Cutler Jr.,Endocrinol.Metab.Clin.North Am.30:121-135(2001)]、库欣病[M.Labeur等人,Curr.DrugTargets Immune Endocr.Metabol.Disord.4:335-342(2004)]、高血压[R.J.Briscoe,等人,Brain Res.881:204-207(2000)]和卒中[S.L.Stevens等人,J.Cereb.Blood Flow Metab.23:1151-1159(2003)]中起作用。已显示胃紊乱如肠易激综合征[Y.Tache等人,Eur J.Surg.Suppl:16-22(2002)]和应激诱导的胃溃疡[K.E.Gabry等人,Mol.Psychiatry 7:474-483,433(2002)]与CRF有关。此外,显示CRF在人类女性健康的各种领域如经前期综合征[F.Facchinetti等人,Psychosom.Med.56:418-422(1994)]、不育症[L.Ghizzoni等人,Endocrinology 138:4806-4811(1997)]、性功能障碍[J.E.Jones等人,Am.J.Physiol.Regul.Integr.Comp.Physiol.283:R591-597(2002)]和早产[P.D.Wadhwa等人,Am.J.Obstet.Gynecol.191:1063-1069(2004)]中起作用。还有证据显示CRF在免疫系统中起主要作用,显示用于治疗炎性疾病[A.Gravanis和A.N.Margioris,Currr.Med.Chem.12:1503-1512(2005)]、变态反应[L.K.Singh等人.,Brain Behav.Immun.13:225-239(1999)]、多发性硬化和其他自身免疫性疾病[C.Benou等人,J.Immunol.174:5407-5413(2005)]的治疗可能性。除前述之外,CRF已牵涉内脏痛[M.Nijsen等人,Neurogastroenterol.Motil.17:423-432(2005)]、睡眠障碍[T.M. Buckley和A.F.Schatzberg,J.Clin.Endocrinol.Metab.90:3106-3114(2005)]、垂体肿瘤或异位垂体源性肿瘤[K.D.Dieterich等人,J.Clin.Endocrinol.Metab.83:3327-3331(1998)]、慢性疲劳综合征和纤维肌痛[G.Neeck和L.J.Crofford,Rheum.Dis.Clin.North Am.26:989-1002(2000)]。
已鉴定CRF受体亚型CRF1和CRF2,且它们在脑内非均相分布[D.T.Chalmers等人,TIPS 17:166-72(1996)],因此提示有潜在的功能多样性[S.C.Heinrichs等人,Regul.Peptides 71:15(1997)]。例如,广泛分布的脑CRF1受体强烈牵涉于伴随暴露于环境应激物的情绪性[G.Liebsch等人,Regul.Peptides 59:229-39(1995);D.W.Schulz,PNAS 93:10477-82(1996)]。重要的是,CRF1而不是CRF2受体显示介导选择致焦虑样行为[Heinrichs等人,1997]。更离散的中膈/下丘脑分布[D.T.Chalmers等人,J.Neurosci.15(10):6340-50(1995)]和替代内源性配体的可用性[J.Vaughan等人,Nature 378:287-92(1995)]表明CRF2受体的不同功能作用[Heinrichs等人,1997]。例如,据报道:相对于CRF1受体对CRF2具有优先亲和力的新型CRF家族神经肽可抑制食欲,而不产生用选择性CRF1激动作用所观察到的行为激活特征(H.Tezval等人,PNAS 101(25):9468-9473(2004)]。在一些情况下,CRF2激动作用产生与所报道的CRF1拮抗剂或CRF1基因缺失的那些作用类似的作用[S.C.Heinrichs,Trends in PharmacologicalSciences 20(8):311-5(1999)]。例如,尽管CRF2激动剂已被推荐为减肥药,但是CRF1拮抗剂也可以是重要的肥胖症疗法[C.Contoreggi等人,Neuroendocrinology 80(2):111-23(2004)]。
制备本发明化合物
本发明的所有化合物可例如通过按照下述流程中所列出的合成途径以化学方法制备。然而,下列详述并不旨在以任何方法限制本发明的范围。例如,所述每个途径的特定合成步骤可以以不同方式组合或与不同流程的步骤结合来制备另外的式(I)化合物。每步骤的产物可通过常规方法回收, 包括萃取、蒸发、沉淀、色谱法、过滤、研磨、结晶等。除非另有指出,在下述流程中所有取代基如先前定义,且合适的试剂为本领域众所周知和理解。
流程1

式(I)化合物的制备可根据流程1所述的反应进行。合适的式(I)化合物是其中R1、R2、R3、R4和R5如对式I所定义的化合物。
在步骤1中,在回流乙酸中将乙酰乙酸乙酯和5-甲基-2H-吡唑-3-基胺缩合,形成2,5-二甲基-4H-吡唑并[1,5-a]嘧啶-7-酮(1)。
随后在步骤2中,在惰性溶剂如甲苯中、在溶剂的回流温度、使用氯氧化磷(phosphorous oxychloride)和二甲基苯胺将式(1)的吡唑并[1,5-a]嘧啶-7-酮转化成7-氯-2,5-二甲基-吡唑并[1,5-a]嘧啶。
在流程1步骤3中,在惰性溶剂如甲苯中、在回流温度将式(3)的Grignard试剂(X=Cl或Br)与式(2)的氯化物反应,提供式(4)的7-烷基吡唑并嘧啶。作为替代选择,如步骤4所示,式(4)的7-烷基吡唑并嘧啶通过二酮与5-甲基-2H-吡唑-3-基胺的缩合来获得。该反应在60至80℃的温度、用催化量的哌啶在乙醇中进行(Novinson,T.等人J.Med.Chem.1975,18, 460)。当R1=H时,获得区域异构体的混合物,例如当R2=丙基时,它们在步骤5的碘化作用后分离。
在步骤5中,在乙腈中使用过量的N-碘代琥珀酰亚胺将式(4)的吡唑并嘧啶官能化成式(5)的碘代吡唑并嘧啶。
在流程1步骤6中,在Negishi交叉偶联反应中将式(5)的碘代吡唑并嘧啶与杂环锌卤化物(R3ZnX)(X=Cl或Br)反应。杂环锌卤化物可使用本领域技术人员众所周知的方法来产生。例如,有机锌试剂通过将杂环溴化物与锌金属反应,或者使用非卤代杂环与正丁基锂、仲丁基锂或叔丁基锂形成杂环锂试剂、然后用ZnCl2进行锂-锌交换而产生。在钯催化剂如二氯[1,1’-双(二苯基膦基)二茂铁]钯(II)二氯甲烷存在下、在惰性溶剂如THF中、在回流温度将有机锌试剂与式(5)的碘代吡唑并嘧啶偶联约12至36小时,提供式(I)的吡唑并嘧啶。
作为替代选择,在步骤7中,杂环代硼酸(R3B(OH)2)用于与式(5)的碘代化合物的Suzuki偶联反应。对于这种偶联工艺,有很多技术人员可用的反应条件。优选的条件使用甲苯/乙醇/2M碳酸钠的溶剂混合物与钯催化剂如双(三叔丁基膦)钯(0)在60℃至回流温度进行约12至36小时。
容易理解的是,杂芳基代硼酸可使用本领域众所周知的工艺、通过类似于本文所述的那些方法来制备。例如,噻吩和苯并噻吩可使用N-溴代琥珀酰亚胺来溴化,接着使用卤素金属交换用烷基锂试剂转化成代硼酸,然后用硼酸三甲酯处理,经处理水解。
本领域技术人员应理解,如式(Ic)化合物中的噻吩并吡啶通过将市售可得的噻吩并[3,2-b]吡啶-7-醇转化成溴化物、随后卤素金属交换并用质子源如甲醇/水淬灭而容易获得。然后用溴在3位溴化、随后与甲基代硼酸Suzuki偶联,提供3-甲基-噻吩并[3,2-b]吡啶,后者如上所述使用ZnCl2与式(5)的碘代吡唑并嘧啶偶联。
流程2

式(7)、(8)或(9)化合物的形成可根据流程2所述的反应进行。合适的式(7)、(8)或(9)化合物是其中R1、R2和R4如对式I所定义且“Het”如所示定义的化合物。
在流程2步骤1中,式(6)的噻吩用N-溴代琥珀酰亚胺在惰性溶剂如二氯甲烷或氯仿中溴化。
在步骤2中,式(7)的溴代噻吩与杂环锌试剂偶联,提供式(8)的噻吩杂环。杂环锌试剂从溴代杂环和锌金属形成,并与式(7)的溴代噻吩原位反应。该反应在惰性溶剂如THF中、在溶剂的回流温度进行。作为替代选择,杂环锂试剂可例如使用正丁基锂、仲丁基锂或叔丁基锂与1-甲基-1,2,4-三唑形成,随后用ZnCl2进行锂-锌交换,获得杂环锌试剂。
在流程2步骤3中,将式(6)的噻吩硝化,得到式(9)的硝基噻吩。优选的条件使用含有70%硝酸的二氯甲烷和TFA的溶剂混合物。本领域技术人员应理解,式(9)的硝基噻吩可用作进一步官能化的中间体。例如,硝基可被还原,酰化得到乙酰胺,或者被烷基化得到二烷基胺。
同时应认识到,制备式(8)化合物所需的步骤可以以任何次序进行,由此2-杂环噻吩可使用本领域已知方法来制备,接着官能化成溴代噻吩或噻吩基代硼酸,然后与式(5)的碘代吡唑并嘧啶偶联。
流程3

式(10)化合物的生成可根据流程3所述的反应进行。合适的式(10)化合物是其中R1、R2和R4如对式I所定义且R5a=-OCH3、-OC(O)CH3、-CH2OCH3或-CO2CH2CH3的化合物。
容易理解,式(7)的溴代噻吩代表有用的中间体,技术人员可将它容易地转化成多种取代的噻吩。例如,形成噻吩基格氏试剂后用氧处理(Hurd,C.D.和Kreuz,K.JACS 1950,72,5543),提供2-羟基噻吩,其被酰化,提供乙酰氧基衍生物。所述格氏试剂还可用亲电试剂如氰基甲酸乙酯处理,提供乙氧羰基取代的噻吩。用正丁基锂、仲丁基锂或叔丁基锂的卤素-锂交换可提供噻吩基锂试剂,它随后可与亲电试剂如卤代烃类如碘代甲基甲醚反应。此外,用氧化铜、碘化钠和甲醇钠处理提供2-甲氧基噻吩。
如本文所用,“TLC”是指薄层色谱法;“HPLC”是指高效液相色谱法;“δ”是指相对于四甲基硅烷低场的百万分率;“THF”是指四氢呋喃;“EtOAc”是指乙酸乙酯;“HOAc”是指乙酸;“MeOH”是指甲醇;“DME”是指二甲氧基乙烷。
实施例
无需进一步阐述,认为本领域技术人员可使用前面描述最大限度地实施本发明。提供下列制备和实施例以更详细地描述本发明。它们旨在示例而非以任何方式限制本发明。试剂和起始原料为本领域普通技术人员容易获得或容易合成的。实施例1-27提供了代表性化合物并示例了其制备。实施例A-C示例了可用于测定本发明化合物生物特性的各种生物测定法。本领域技术人员将敏捷地认识到实施例中所述工艺的适当变化。本发明化合物的名称由ChemDraw  7.0.1版提供。
7.0.1版提供。
制备1
2,5-二甲基-4H-吡唑并[1,5-a]嘧啶-7-酮
逐滴加入乙酰乙酸乙酯(128g,0.98mol)至5-甲基-2H-吡唑-3-基胺(100g,0.95mol)的乙酸溶液(500mL),使温度保持在25-28℃。回流加热混合物10小时,然后冷却至室温。将反应物加至冷却至5℃的叔丁基甲醚(5L),使温度保持在低于10℃。在5℃搅拌1小时,过滤。将所生成的物质真空干燥过夜,提供白色固体(158g,96%)。
制备2
7-氯-2,5-二甲基-吡唑并[1,5-a]嘧啶
将N,N-二甲基苯胺(9.7mL,76.7mmol)加至2,5-二甲基-4H-吡唑并[1,5-a]嘧啶-7-酮(10.0g,61.3mmol)在甲苯(150mL)中的悬液。逐滴加入三氯氧化磷(11.2mL,122.6mmol)至此白色悬液。在惰性气氛下回流3小时,冷却至室温,使用减压浓缩反应物成褐色油。将所述油溶解于乙酸乙酯(250mL)中,用1N NaOH碱化。分离,用另外的乙酸乙酯(2×100mL)萃取碱性水相。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩,得到棕色固体。使用快速色谱法纯化该物质,用80%己烷/20%(30%THF/己烷)至0%己烷/100%(30%THF/己烷)以20%增量的阶段梯度洗脱,提供淡绿色固体(6.65g,59%)。ES/MS m/z(35Cl)182.3(M+1)+。
制备3
7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶

在惰性气氛下将催化量的碘和  镁(1.0M,在THF中,52mL,52mmol)装入配置有回流冷凝器的烘干烧瓶中。加热至45℃,将3-溴代戊烷(5.3mL,42.9mmol)加至反应物。当格氏反应开始时温度达到顶峰。在50℃搅拌反应物另外4小时,冷却至室温。使金属镁沉降,在正氩压下用套管引流(canulate off)格氏试剂到含有无水甲苯(50mL)中的7-氯-2,5-二甲基吡唑并[1,5-a]嘧啶(5.19g,28.6mmol)的烧瓶中。在惰性气氛下加热至回流 达48小时。将反应物冷却至室温,用水淬灭。用乙酸乙酯(150mL)稀释反应物,加入水(50mL)。分离,用乙酸乙酯(75mL)萃取水相。合并有机相,经无水硫酸镁干燥,减压浓缩。使用快速色谱法纯化,用80%戊烷/20%(30%THF/戊烷)至0%戊烷/100%(30%THF/戊烷)以20%增量的阶段梯度洗脱,得到黄色油(2.59g,42%)。ES/MS m/z 218.1(M+1)+。
镁(1.0M,在THF中,52mL,52mmol)装入配置有回流冷凝器的烘干烧瓶中。加热至45℃,将3-溴代戊烷(5.3mL,42.9mmol)加至反应物。当格氏反应开始时温度达到顶峰。在50℃搅拌反应物另外4小时,冷却至室温。使金属镁沉降,在正氩压下用套管引流(canulate off)格氏试剂到含有无水甲苯(50mL)中的7-氯-2,5-二甲基吡唑并[1,5-a]嘧啶(5.19g,28.6mmol)的烧瓶中。在惰性气氛下加热至回流 达48小时。将反应物冷却至室温,用水淬灭。用乙酸乙酯(150mL)稀释反应物,加入水(50mL)。分离,用乙酸乙酯(75mL)萃取水相。合并有机相,经无水硫酸镁干燥,减压浓缩。使用快速色谱法纯化,用80%戊烷/20%(30%THF/戊烷)至0%戊烷/100%(30%THF/戊烷)以20%增量的阶段梯度洗脱,得到黄色油(2.59g,42%)。ES/MS m/z 218.1(M+1)+。
使用商业可得的格氏试剂或如上所述制备格氏试剂,基本上如制备3中所述制备下列化合物。
| 制备 编号 | 化学名称 | 物理数据: NMR或MS(m/z) |
| 4 | 7-(1-丙基-丁基)-2,5-二甲基 -吡唑并[1,5-a]嘧啶 | 246.3(M+1)+ |
| 5 | 7-异丙基-2,5-二甲基-吡唑 并[1,5a]嘧啶 | 1H NMR(400MHz,CDCl3):6.41(s, 1H),6.30(s,1H),3.81-3.77(m,1H), 2.52(s,3H),2.47(s,3H),1.37(d,J= 7.0Hz,6H) |
制备6
7-丁基-2,5-二甲基-吡唑并[1,5-a]嘧啶
将5-甲基-2H-吡唑-3-基胺(217mg,2.17mmol)、壬烷-2,4-二酮(339mg,2.39mmol)和哌啶(1滴)加至乙醇(10mL),在80℃加热过夜。冷却至室温,浓缩至干。经硅胶柱色谱法(0-20%乙酸乙酯/己烷)纯化,获得两种异构体的混合物(2g)。
制备7
7-(1-乙基-丙基)-3-碘代-2,5-二甲基-吡唑并[1,5-a]嘧啶

将7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(2.14g,9.84mmol)溶解于无水乙腈(25mL)中,以10分钟的间隔加入6份(每份0.5g)的N-碘代 琥珀酰亚胺(3.0g,13.3mmol)。搅拌反应物4小时。蒸发(Strip off)乙腈,用二氯甲烷(100mL)稀释所生成的油。用饱和氯化铵溶液(2×50mL)洗涤橙色溶液。收集有机相,经无水硫酸镁干燥,过滤,减压浓缩以获得暗红色油。使用快速色谱法纯化,用100%戊烷/0%(20%乙酸乙酯/戊烷)至0%戊烷/100%(20%乙酸乙酯/戊烷)以50%增量的阶段梯度洗脱,得到橙色油(3.28g,97%)。1H NMR(400MHz,CDCl3):6.44(s,1H),3.59(m,1H),2.61(s,3H),2.49(s,3H),1.86-1.76(m,4H),0.85(t,J=7.5Hz,6H)。
基本上如制备7中所述制备下列化合物。
| 制备 编号 | 化学名称 | 物理数据: NMR或MS(m/z) |
| 8 | 7-(1-丙基-丁基)-3-碘代-2,5- 二甲基-吡唑并[1,5-a]嘧啶 | 1H NMR(400MHz,CDCl3):6.42 (s,1H),3.74-3.70(m,1H),2.58(s, 3H),2.46(s,3H),1.74-1.68(m, 4H),1.28-1.14(m,4H),0.84(t,J= 7.0Hz,6H). |
| 9 | 7-异丙基-3-碘代-2,5-二甲基- 吡唑并[1,5a]嘧啶 | 316.0(M+1)+ |
| 10* | 7-丁基-3-碘代-2,5-二甲基-吡 唑并[1,5-a]嘧啶 | 330(M+1)+ |
*处理:用Na2S2O3水溶液洗涤有机物。对制备6的异构体混合物进行反应。在硅胶上分离两种异构体(0-20%EtOAc/己烷)。
实施例1
3-(3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶

将 锌(5g/100mL THF,20mL,15.0mmol)悬浮于无水 THF(10mL)中,加入2-溴代-3-氯噻吩(1.98g,10.0mmol)。在惰性气氛下使混合物在油浴(85℃)中回流3小时。将反应物冷却至室温,离心剩余的锌金属。在正氩压下套管引流试剂溶液至新容器中,加入7-(1-乙基-丙基)-3-碘代-2,5-二甲基-吡唑并[1,5-a]嘧啶(1.72g,5.0mmol)。通过正氩压使溶液脱气10-15分钟,加入二氯[1,1’-双(二苯基-膦基)二茂铁]钯(II)二氯甲烷(0.225g,0.275mmol)。在惰性气氛下于回流温度搅拌过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分,合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(15%乙酸乙酯/己烷)至0%己烷/100%(15%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到黄色固体(1.35g,40%)。ES/MS m/z(35Cl)334.4(M+1)+。
基本上如实施例1中所述制备下列化合物。
| 实施例 编号 | 化学名称 | MS(m/z) |
| 2 | 3-(3-氯-噻吩-2-基)-7-(1-丙基-丁基)-2,5-二甲 基-吡唑并[1,5-a]嘧啶 | (35Cl)362.0 (M+1)+ |
| 3 | 3-(3-氯-噻吩-2-基)-7-异丙基-2,5-二甲基-吡唑 并[1,5-a]嘧啶 | (35Cl)306.0 (M+1)+ |
实施例4
3-(3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将3-甲基-2-噻吩基溴化锌(11.0mL,5.5mmol)、无水THF(6.0mL)和7-(1-乙基-丙基)-3-碘代-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.536g,1.56mmol)装入烘干烧瓶。用正氩压脱气10-15分钟,加入二氯[1,1’-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.12g,0.15mmol)。在惰性气氛下在油浴(100℃)中回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%戊烷 /0%(25%乙酸乙酯/戊烷)至0%戊烷/100%(25%乙酸乙酯/戊烷)以10%增量的阶段梯度洗脱,得到白色泡沫。用己烷研磨,过滤。(0.432g,8%)。1HNMR(400MHz,CDCl3):7.26(d,J=4.8,MHz,1H),6.96(d,J=5.3Hz,1H),6.42(s,1H),3.64-3.60(m,1H),2.53(s,3H),2.43(s,3H),2.14(s,3H),1.87-1.79(m,4H),0.88(t,J=7.0Hz,6H)。
实施例5
3-(5-溴代-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶

将3-(3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(1.16g,3.5mmol)溶解于二氯甲烷(15mL),以一个等分试样加入N-溴代琥珀酰亚胺(0.69g,3.85mmol)。在惰性气氛下搅拌2小时,使用TLC确认反应完全。用二氯甲烷(50mL)稀释反应物,用水(75mL)、盐水(50mL)洗涤,经无水硫酸镁干燥,过滤,减压浓缩,得到黄色固体(1.56g,定量收率)。1HNMR(400MHz,DMSO):7.39(s,1H),6.88(s,1H),3.48-3.44(m,1H),2.45(s,3H),2.39(s,3H),1.80-1.73(m,4H),0.76(t,J=7.0Hz,6H)。
基本上如实施例5中所述制备下列化合物。
| 实施例 编号 | 化学名称 | MS(m/z) |
| 6 | 3-(5-溴-3-氯-噻吩-2-基)-7-(1-丙基-丁基)-2,5- 二甲基-吡唑并[1,5-a]嘧啶 | (35Cl81Br) 441.7(M+) |
| 7* | 3-(5-溴-3-甲基-噻吩-2-基)-7-(1-乙基-丙 基)-2,5-二甲基-吡唑并[1,5-a]嘧啶 | (81Br)393.8 (M+1)+ |
| 8* | 3-(5-溴-3-氯-噻吩-2-基)-7-异丙基-2,5-二甲基- 吡唑并[1,5-a]嘧啶 | (35Cl81Br) 385.0(M+) |
*通过用己烷/乙酸乙酯洗脱的柱色谱法纯化。
实施例9
3-[3-氯-5-(2-甲基-2H-[1,2,4]三唑-3-基)-噻吩-2-基]-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶

将  锌(5g/100mL THF,5.5mL,4.2mmol)悬浮于无水THF(5mL),加入1-甲基-5-溴-1,2,4-三唑(0.4g,2.5mmol)。在惰性气氛下在油浴(85℃)中回流3小时。将反应物冷却至室温,离心剩余的锌金属。在正氩压下引流试剂溶液至新容器中,加入3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.64g,1.45mmol)。通过正氩压将溶液脱气10-15分钟,加入二氯[1,1-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.136g,0.167mmol)。在惰性气氛下回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(30%THF/己烷)至0%己烷/100%(30%THF/己烷)以10%增量的阶段梯度洗脱,得到黄色固体(0.298g,50%)。ES/MS m/z(35Cl)415.0(M+1)+。
锌(5g/100mL THF,5.5mL,4.2mmol)悬浮于无水THF(5mL),加入1-甲基-5-溴-1,2,4-三唑(0.4g,2.5mmol)。在惰性气氛下在油浴(85℃)中回流3小时。将反应物冷却至室温,离心剩余的锌金属。在正氩压下引流试剂溶液至新容器中,加入3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.64g,1.45mmol)。通过正氩压将溶液脱气10-15分钟,加入二氯[1,1-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.136g,0.167mmol)。在惰性气氛下回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(30%THF/己烷)至0%己烷/100%(30%THF/己烷)以10%增量的阶段梯度洗脱,得到黄色固体(0.298g,50%)。ES/MS m/z(35Cl)415.0(M+1)+。
使用市售可得的6-甲基-2-吡啶基溴化锌,基本上如实施例9中所述制备下列化合物。

实施例11
3-(3-氯-5-甲氧基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.46g,1.1mmol)、氧化铜(0.045g,0.56mmol)、碘化钠(0.020g,0.11mmol)、25%甲醇钠/甲醇溶液(10mL)和无水甲醇(10mL)装入烘干烧瓶中。在惰性气氛下将反应物在油浴(90℃)中回流整个周末。用冰水淬灭反应物,用乙醚(3×100mL)萃取。合并有机相部分,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(35%乙酸乙酯/己烷)至0%己烷/100%(35%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到白色泡沫(0.086g,21%)。ES/MS m/z(35Cl) 363.7(M+1)+。
实施例12
乙酸4-氯-5-[7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶-3-基]噻吩-2-基酯
将异丙基溴(0.25mL,2.7mmol)加至3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.74,1.8mmol)在  镁(1.0M,在THF中,2.7mL,2.7mmol)和无水THF(3mL)中的悬液中。在惰性气氛下回流1小时,将反应物冷却至室温。在正压下于室温将氧吹至放热反应中达90分钟。用饱和氯化铵(30mL)淬灭反应,用乙酸乙酯(2×100mL)萃取,减压浓缩。将油溶解于乙醚(150mL)中,用0.1N氢氧化钠(3×75mL)洗涤,用乙醚(50mL)反萃取碱性水相。用饱和氯化铵酸化水相,用二氯甲烷(4×75mL)萃取。合并二氯甲烷部分,经无水硫酸镁干燥,过滤,减压浓缩。将粗油(0.138g,0.39mmol)溶解于二氯甲烷(2mL)和三乙胺(0.23mL,1.6mmol)和乙酰氯(0.034mL,0.47mmol)中。在惰性气氛下搅拌1小时,用二氯甲烷(50mL)稀释,用水(50mL)洗涤。经无水硫酸镁干燥有机相,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(25%乙酸乙酯/己烷)至0%己烷/100%(25%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到白色泡沫状物(0.084g,21%)。ES/MS m/z (35Cl)392.0(M+1)+。
镁(1.0M,在THF中,2.7mL,2.7mmol)和无水THF(3mL)中的悬液中。在惰性气氛下回流1小时,将反应物冷却至室温。在正压下于室温将氧吹至放热反应中达90分钟。用饱和氯化铵(30mL)淬灭反应,用乙酸乙酯(2×100mL)萃取,减压浓缩。将油溶解于乙醚(150mL)中,用0.1N氢氧化钠(3×75mL)洗涤,用乙醚(50mL)反萃取碱性水相。用饱和氯化铵酸化水相,用二氯甲烷(4×75mL)萃取。合并二氯甲烷部分,经无水硫酸镁干燥,过滤,减压浓缩。将粗油(0.138g,0.39mmol)溶解于二氯甲烷(2mL)和三乙胺(0.23mL,1.6mmol)和乙酰氯(0.034mL,0.47mmol)中。在惰性气氛下搅拌1小时,用二氯甲烷(50mL)稀释,用水(50mL)洗涤。经无水硫酸镁干燥有机相,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(25%乙酸乙酯/己烷)至0%己烷/100%(25%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到白色泡沫状物(0.084g,21%)。ES/MS m/z (35Cl)392.0(M+1)+。
实施例13
3-(3-氯-5-甲氧基甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a] 嘧啶(0.41,1.0mmol)和无水THF(5mL)装入烘干烧瓶。在惰性气氛下冷冻至-70℃,加入正丁基锂(2.5M己烷溶液,0.42mL,1.05mmol)。黄色反应物转变成暗红色。加入碘代甲基甲醚(0.09mL,1.10mmol),使反应物温至室温。用乙酸乙酯(100mL)稀释反应混合物,用水(75mL)和盐水(75mL)洗涤。经无水硫酸镁干燥有机部分,减压浓缩至橙色油。使用快速色谱法纯化所得残渣,用100%己烷/0%(20%乙酸乙酯/己烷)至0%己烷/100%(20%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到黄色固体(0.107g,28%)。ES/MS m/z(35Cl)378.3(M+1)+。
实施例14
4-氯-5-[7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶-3-基]-噻吩-2-甲酸乙酯
在惰性气氛下将3-(5-溴-3-氯-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基吡唑并[1,5-a]嘧啶(0.62g,1.5mmol)在无水THF(5mL)中的混合物冷冻至0℃,加入乙基氯化镁(2M THF溶液,0.83mL,1.65mmol)。将反应物搅拌5分钟,温至室温,搅拌另外的15分钟,然后将反应温度降低至0℃。加入在无水THF(1mL)中稀释的氰基甲酸乙酯(0.16mL,1.58mmol)。使反应物温至室温,搅拌1小时。用饱和碳酸氢钠(20mL)淬灭反应,用乙醚(2×75mL)萃取。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化,用100%己烷/0%(20%乙酸乙酯/己烷)至0%己烷/100%(20%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱。真空干燥所得物质,提供白色固体(0.114g,19%)。ES/MS m/z(35Cl)406.3(M+1)+。
制备11
2-(5-溴-4-甲基-噻吩-2-基)-6-甲基-吡啶
将2.0M二异丙氨基锂(9.76mL,19.52mmol)加至2-溴-3-甲基-噻吩(2.0mL,17.75mmol)和THF(30mL)的-78℃溶液。45分钟后加入ZnCl2(0.5M,在THF中,39.0mL,19.50mmol),搅拌溶液30分钟。加入2-溴-6-甲基-吡啶(2.4mL,21.29mmol)和Pd(PPh3)4(0.50g,0.44mmol)。将反应物温至环境温度,搅拌2小时。用饱和NH4Cl溶液(20mL)洗涤反应物。 用CH2Cl2(30mL)洗涤水层。合并有机层,用饱和NH4Cl溶液(20mL)洗涤,经Na2SO4干燥,过滤,浓缩。通过硅胶柱色谱法(10%-20%EtOAc/己烷梯度)纯化所得残渣,提供标题化合物(2.34g,49%)。LC-ES/MS m/z(79Br/81Br)267.7/269.5(M+H)+。
制备12
3-甲基-5-(6-甲基-吡啶-2-基)-噻吩-2-代硼酸
在氮气氛下将2-(5-溴-4-甲基-噻吩-2-基)-6-甲基-吡啶(37.6g,0.14mol)和硼酸三异丙基酯(34.2g,0.182mol,42.3mL)溶解于甲苯(100mL)和THF(160mL)。将溶液冷却至-40℃。使用添加漏斗历经40分钟缓慢加入正丁基锂(2.5M,在己烷中,70mL,0.175mol)。溶液中的温度从-40℃转变至-35℃。添加结束后,在-40℃将混合物搅拌2小时。将反应物温至0℃,加入THF(40mL),然后加入2N HCl水溶液(120mL),形成白色固体。加入1N NaOH直至pH=7且所有盐都溶解。分离有机层,用二乙醚萃取水层(3×)。合并有机层,经MgSO4干燥,过滤,浓缩。将THF加至残渣,然后加入己烷。过滤所得的黄色沉淀,重复沉淀步骤数次,提供标题化合物(19.7g,60%)。1H NMR(CD3OD):δ2.47(bs,3H),2.61(s,3H),7.20(d,J=7.7Hz,1H),7.60(bs,1H),7.64(bd,J=7.7Hz,1H),7.75(t,J=7.7Hz,1H)。
实施例15
7-(1-乙基-丙基-2,5-二甲基-3-[3-甲基-5-(6-甲基-吡啶-2-基)噻吩-2-基]-吡唑并[1,5-a]嘧啶

将2-(5-代硼酸-4-甲基-噻吩-2-基)-6-甲基-吡啶(0.29g,1.24mmol)、无水甲苯(4mL)、无水乙醇(1mL)、2M碳酸钠(1.1mL,2.2mmol)和7-(1-乙基-丙 基)-3-碘-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.25g,0.73mmol)装入烘干烧瓶。用正氩压将混合物脱气30分钟,加入双(三叔丁基膦)钯(0)(0.10g,0.086mmol)。在惰性气氛下使反应物在油浴(100℃)中回流过夜。将反应物冷却至室温,用乙酸乙酯(75mL)和水(25mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(25%乙酸乙酯/己烷)至0%己烷/100%(25%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到灰白色泡沫状物(0.215g,73%)。ES/MS m/z 405.4(M+1)+。
制备13
7-溴-噻吩并[3,2-b]吡啶
在110℃将噻吩并[3,2-b]吡啶-7-醇(5.00g,33mmol)和三溴氧化磷(50g,174mmol)一起加热3小时。将热反应混合物倾入冰和5N NaOH的混合物中,用CH2Cl2萃取。用Na2SO4干燥有机相,蒸发。使用硅胶色谱柱(己烷∶EtOAC=3∶1)纯化所得粗品,得到标题化合物(4.19g,59%)。ES/MS m/z(81Br)215(M+)。
制备14
噻吩并[3,2-b]吡啶
将7-溴-噻吩并[3,2-b]吡啶(3.69g,17mmol)溶解于干燥THF(20mL),冷却至-78℃。将正丁基锂(1.6M,在己烷中,21.2mL,34mmol)缓慢加至-78℃混合物,在-78℃搅拌20分钟。加入MeOH/H2O=1/1(20mL),在室温搅拌1小时。用CH2Cl2萃取反应混合物。用饱和NaCl溶液洗涤有机部分,经Na2SO4干燥,蒸发。使用硅胶色谱法纯化所得残渣,用100%己烷至己烷∶乙酸乙酯=10∶1洗脱,得到标题化合物(1.44g,62%)。ES/MS m/z 136(M+1)+。
制备15
3-溴-噻吩并[3,2-b]吡啶
将噻吩并[3,2-b]吡啶(3.45g,25.6mmol)、碳酸氢钠(2.15g,25.6mmol)、K2HPO4(6.69g,38.4mmol)和MgSO4(4.01g,33.3mmol)合并于 CHCl3(60mL)中。在回流下搅拌混合物,缓慢加入Br2(1.57mL,30.7mmol)。在回流下将反应混合物搅拌过夜。加入更多的溴(0.7mL),在回流下将反应物搅拌4小时。冷却至室温,加入水,用CHCl3萃取。用饱和Na2S2O3溶液和饱和盐水洗涤有机部分。经Na2SO4干燥,蒸发。将物质从己烷/CH2Cl2中重结晶,获得标题化合物(3.94g,72%)。ES/MS m/z(81Br)215(M+)。
制备16
3-甲基-噻吩并[3,2-b]吡啶
准备三个含有DME/水/EtOH=7/3/1(4mL)中的3-溴-噻吩并[3,2-b]吡啶(214mg,1.0mmol)和甲基代硼酸(180mg,3.0mmol)的可微波反应小瓶。加入2M Na2CO3(1.5mL,3.0mmol),吹入氮气15分钟。加入Pd(PPh3)4(58mg,0.05mmol),密封小瓶。在130℃于微波中加热小瓶30分钟。合并所有反应混合物,加入水和CH2Cl2,萃取。分离CH2Cl2层,经Na2SO4干燥,过滤,蒸发。使用硅胶色谱法纯化所得物质,用己烷∶乙酸乙酯∶2M NH3的MeOH溶液=20∶4∶1洗脱,得到标题化合物(193mg,43%)。ES/MS m/z150(M+1)+。
实施例16
7-(1-乙基-丙基)-2,5-二甲基-3-(3-甲基-噻吩并[3,2-b]吡啶-2-基)-吡唑并[1,5-a]嘧啶

将3-甲基噻吩并[3,2-b]吡啶(0.213g,1.42mmol)和无水THF(5mL)装入烘干schrenk烧瓶,在惰性气氛下冷却至-78℃。加入正丁基锂(2.5M,在己烷中,0.72mL,1.78mmol),在降低的温度搅拌30分钟。以一个等分试样加入无水氯化锌(0.58g,4.26mmol),在降低的温度搅拌15分钟。使反应物温至室温,搅拌另外的30分钟。用无水THF(5mL)稀释反应物,加入7-(1- 乙基-丙基)-3-碘-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.406g,0.08mmol)。用正氩压脱气15分钟,加入二氯[1,1’-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.093g,0.114mmol)。在惰性气氛下在油浴(90℃)中使反应物回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化,用100%己烷/0%(15%乙酸乙酯/己烷)至0%己烷/100%(15%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到白色泡沫状物(0.059g,14%)。ES/MS m/z 365.0(M+1)+。
实施例17
3-[3-甲基-5-(2-甲基-2H-[1,2,4]三唑-3-基)-噻吩-2-基]-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将1-甲基-1,2,4-三唑(0.320,3.81mmol)、无水THF(10mL)装入烘干烧瓶,在惰性气氛下冷冻至-78℃。加入正丁基锂(2.5M,在己烷中,1.52mL,3.81mmol),在降低的温度搅拌30分钟。以一个等分试样加入无水氯化锌(1.06g,7.75mmol),在降低的温度搅拌15分钟。使反应物温至室温,搅拌另外的30分钟。将无水THF(5mL)中的3-(5-溴-3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.50g,1.27mmol)加至反应物。用正氩压脱气15分钟,加入二氯[1,1’-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.147g,0.127mmol)。在惰性气氛下将反应物在油浴(90℃)中回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化,用100%己烷/0%(40%THF/己烷)至0%己烷/100%(40%THF/己烷)以10%增量的阶段梯度洗脱。用己烷/乙醚(3∶1)研磨所得物质,得到白色固体(0.371g,74%)。ES/MS m/z 395.0(M+1)+。
实施例18
3-(5-硝基-3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基吡唑并[1,5-a]嘧啶
将3-(3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.419g,1.33mmol)溶解于二氯甲烷(2.5mL)中,在惰性气氛下搅拌,在冰浴 中冷冻至0℃,加入三氟乙酸(5mL)。将浓(70%)硝酸(0.132g,1.47mmol)逐滴加至反应物中。溶液的颜色从黄色转变成暗绿色。在惰性气氛下同时在冰浴中搅拌1小时,使用TLC确认反应完全。用二氯甲烷(80mL)稀释反应物,用饱和碳酸氢钠淬灭。分离,用二氯甲烷(50mL)萃取碱性水相。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(30%乙酸乙酯/己烷)至0%己烷/100%(30%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到黄色固体(0.363g,76%)。1HNMR(400MHz,CDCl3):δ7.81(s,1H),6.51(s,1H),3.62-3.58(m,1H),2.56(s,3H),2.48(s,3H),2.18(s,3H),1.88-1.70(m,4H),0.88(t,J=7.5Hz,6H)。
实施例19
N-{5-[7-(1-乙基-丙基)-2,5-二甲基吡唑并[1,5-a]嘧啶-3-基]-4-甲基-噻吩-2-基}乙酰胺
将3-(5-硝基-3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶(0.33g,0.92mmol)溶解于含有10%披钯碳(0.10g)的THF(10mL)中,用真空/氢气流吹扫脱气(3×),在氢气氛(55psi)下于室温搅拌3小时。使用TLC确认反应完全。用THF(50mL)稀释反应物,经硅藻土过滤,然后浓缩至粗橙色油(0.328g)。将油溶解于二氯甲烷(4mL)和1.0MNaOH(1mL)中。将乙酰氯(0.038mL,0.52mmol)加至反应物,于室温在密封的反应容器中搅拌整个周末。用二氯甲烷(100mL)稀释反应物,用水洗涤。收集有机相,经无水硫酸镁干燥,过滤,在减压下浓缩至红色油。使用快速色谱法纯化所述油,用100%己烷/0%(15%乙酸乙酯/10%7N氨化甲醇/75%己烷)至0%己烷/100%(15%乙酸乙酯/10%7N氨化甲醇/75%己烷)以10%增量的阶段梯度洗脱,得到褐色泡沫状物(0.048g,26%)。ES/MS m/z371.0(M+1)+。
实施例20
3-[3-氯-5-(2-甲基-2H-[1,2,4]三唑-3-基)-噻吩-2-基]-7-(1-丙基-丁基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将1-甲基-1,2,4-三唑(0.120,1.43mmol)和无水THF(5mL)装入烘干烧瓶,在惰性气氛下冷冻至-78℃。加入正丁基锂(2.5M,在己烷中,0.60mL,1.43mmol),在降低的温度搅拌30分钟。以一个等分试样加入无水氯化锌(0.400g,2.90mmol),在降低的温度搅拌15分钟。使反应物温至室温,搅拌另外的30分钟。将无水THF(5mL)中的3-(5-溴-3-氯-噻吩-2-基)-7-(1-丙基-丁基)-2,5-二甲基-吡唑并[1,5a]嘧啶(0.500g,1.27mmol)加至反应物。用正氩压脱气15分钟,加入四(三苯基膦)钯(0)(0.060g,0.052mmol)。在惰性气氛下于油浴(90℃)中回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(25%THF/己烷)至0%己烷/100%(25%THF/己烷)以20%增量的阶段梯度洗脱,得到黄色固体(0.140g,66%)。ES/MS m/z(35Cl)441.7(M+1)+。
基本上如实施例20中所述制备下列化合物。
| 实施例 编号 | 化学名称 | MS (m/z) |
| 21 | 3-[3-氯-5-(2-甲基-2H-[1,2,4]三唑-3-基)-噻吩-2- 基]-7-异丙基-2,5-二甲基-吡唑并[1,5a]嘧啶 | 387.0 (M+1)+ |
制备17
2-溴-5-氟-3-甲基苯并[b]噻吩
用N-溴代琥珀酰亚胺(56.32g,0.318mol,1.05当量)处理5-氟-3-甲基苯并[b]噻吩(50.32g,0.303mol)在乙腈(350mL)中的机械搅拌的溶液。起初的 吸热使反应温度下降至17℃。随后的放热使反应温度历经10分钟增加至40℃,此时通过应用冰水浴使反应物冷却至18-20℃。在室温搅拌反应物另外的35分钟,用水(350mL)缓慢稀释所得浆液。搅拌浆液10分钟。将产物过滤,用50∶50 乙腈∶水(100mL)洗涤,干燥,得到无色结晶固体(65.56g,88%)。
实施例22
3-(5-氟-3-甲基-苯并[b]噻吩-2-基)-7-异丙基-2,5-二甲基吡唑并[1,5-a]嘧啶

将2-溴-5-氟-3-甲基-苯并[b]噻吩(0.490,2.00mmol)和无水THF(5mL)装入烘干烧瓶,在惰性气氛下冷冻至-50℃。逐滴加入正丁基锂(2.5M,在己烷中,0.80mL,2.0mmol),搅拌30分钟。以一个等分试样加入无水氯化锌(0.550g,4.00mmol),在降低的温度搅拌15分钟。使反应物温至室温,搅拌另外的30分钟。加入无水THF(5mL)中的7-异丙基-3-碘-2,5-二甲基-吡唑并[1,5a]嘧啶(0.316g,1.00mmol)。用正氩压脱气15分钟,加入二氯[1,1’-双(二苯基膦基)二茂铁]钯(II)二氯甲烷(0.082g,0.1mmol)。在惰性气氛下于油浴(90℃)中回流过夜。将反应物冷却至室温,用饱和氯化铵淬灭,用乙酸乙酯(75mL)稀释。分离,用乙酸乙酯(50mL)萃取水相部分。合并有机相,经无水硫酸镁干燥,过滤,减压浓缩。使用快速色谱法纯化所得残渣,用100%己烷/0%(20%乙酸乙酯/己烷)至0%己烷/100%(20%乙酸乙酯/己烷)以20%增量的阶段梯度洗脱,获得灰白色泡沫(0.206g,58%)。ES/MS m/z354.0(M+1)+。
基本上如实施例22中所述制备下列化合物。
| 实施例 编号 | 化学名称 | MS(m/z) |
| 23 | 7-(1-乙基-丙基)-3-(5-氟-3-甲基-苯并[b]噻吩 -2-基)-2,5-二甲基-吡唑并[1,5-a]嘧啶 | (APCI)382.2 (M+1)+ |
| 24 | 7-(1-丙基-丁基)-3-(5-氟-3-甲基-苯并[b]噻吩 -2-基)-2,5-二甲基-吡唑并[1,5-a]嘧啶 | (APCI)410.0 (M+1)+ |
制备18
5-氟-3-甲基-苯并[b]噻吩-2-基代硼酸
在干燥烧瓶中合并5-氟-3-甲基-苯并[b]噻吩(312mg,1.88mmol)与干THF(4mL)。冷却至-78℃,加入正丁基锂(1.6N,在己烷中,1.18mL,1.90mmol)。在-78℃搅拌1.5小时,然后加入硼酸三甲酯(0.23mL,2.02mmol)。搅拌3小时,将冷浴升至-20℃。加入5N盐酸直至反应物为酸性(pH=4)。用水稀释,用乙酸乙酯萃取(3×)。合并有机相,经硫酸钠干燥,过滤,蒸发。在二氯甲烷中研磨,得到白色固体(258mg,65%)。ES/MSm/z 209(M-1)-。
实施例25
7-丁基-3-(5-氟-3-甲基-苯并[b]噻吩-2-基)-2,5-二甲基-吡唑并[1,5-a]嘧啶
将四(三苯基膦)钯(0)(31mg,0.03mmol)和7-丁基-3-碘-2,5-二甲基-吡唑并[1,5-a]嘧啶(174mg,0.53mmol)加至经脱气的无水THF(10mL)。搅拌混合物10分钟。加入2-代硼酸-5-氟-3-甲基-苯并[b]噻吩(111mg,0.53mmol)和碳酸钠水溶液(112mg,1.06mmol,5mL水中)。将混合物加热至70℃达24小时。冷却至室温。用乙醚稀释,然后用水和盐水洗涤。经硫酸镁干燥有机相,过滤,浓缩。经HPLC纯化残渣,用乙腈/水/TFA洗脱,得到标题化合物(55mg,28%)。ES/MS m/z 368(M+1)+。
制备19
4-氯-噻吩-2-甲腈
将22-L反应烧瓶装备冷却浴、空气搅拌器、气体添加管和温度计探针。 用氮净化烧瓶,然后装入AlCl3(1025g,7.69mol)和CHCl3(6.6L,16.5vol.)。将混合物冷却至0-5℃,使用添加漏斗历经10-15分钟逐滴加入2-噻吩甲腈(400g,3.66mol),同时将温度保持在≤10℃。在≤10℃向混合物表面下充气Cl2气体(300g,4.23mol,1.16当量)1.25小时。通过GC监控反应的进程。将反应混合物的等分试样淬灭入6N HCl中,用EtOAc萃取,经Na2SO4干燥,过滤,注射滤液。
当通过GC分析反应完全(当根据GC的面积%(起始原料∶产物∶二氯化物)的比率约为(1∶5.8∶1)时,则认为反应完全)时,使用添加漏斗历经1.5小时逐滴加入6N HCl(8.0L),同时保持温度为≤20℃。添加HCl极其放热,放出气体。将反应物移至分液漏斗,分层。用CHCl3(4.0L)萃取水层,合并氯仿层,用水(6.0L)洗涤。经Na2SO4干燥有机部分,过滤,真空浓缩,得到淡黄色的半固体(575g,109%)。GC(60℃至280℃温度梯度)面积%分析显示有约68%产物(t保留时间=6.5分钟),主要杂质是未反应的起始原料(t保留时间=5.1分钟)和二氯化产物(t保留时间=7.4分钟)。GC法:柱:DB1;T注射=300℃;T初始=60℃,t=2.0分钟;T最终=280℃,速率=18℃/分钟。
制备20
4-氯-2-噻吩甲酰胺
将12-L反应烧瓶装备冷却浴、空气搅拌器和温度计探针,装入KOH(288.6g,5.143mol)和水(6.04L)以形成放热至约31℃的溶液。将溶液冷却至约28℃,将4-氯-2-噻吩甲腈(671.3g,4.675mol)装入混合物(少量固体未溶解)。加入EtOH(675mL),此时发生逐渐放热,持续1-1.5小时直至约38℃。在环境温度将反应物搅拌过夜。真空过滤反应混合物,用水洗涤,干燥,得到粗产物。将该固体溶解于EtOAc(10.0L),用Na2SO4和活性炭处理1-2小时,然后过滤,用EtOAc洗涤。于45℃经旋转蒸发器浓缩滤液直至固体开始沉淀。释放真空,使温度增至60-65℃以再溶解固体。在60℃边搅拌边缓慢加入庚烷(3.5L)以沉淀固体。在60℃搅拌15-20分钟,然后将混合物冷却至30-40℃,过滤。用庚烷洗涤固体(2×0.75L),干燥,得到标题化合物,为白色固体(235.4g,31%)。从滤液获得第二批产物(67.8g, 9%)。
制备21
4-氯-N-二甲氨基亚甲基-2-噻吩甲酰胺
将5-L反应烧瓶装备上加热套、空气搅拌器、Dean-Stark装置和温度计探针。装入4-氯-2-噻吩甲酰胺(300g,1.856mol)和二甲基甲酰胺缩二甲醇(872mL)以形成浆液。将混合物逐渐加热至96℃同时收集馏出物(大部分是MeOH)。去除加热套,冷却至≤25℃。使用添加漏斗加入水(3.0L),将温度保持在≤35℃。用EtOAc(3.0L,1.5L)萃取反应混合物。合并有机物,用水(1.5L)洗涤。经Na2SO4干燥有机相,过滤,真空浓缩,得到粗产物(400g)。
在50-60℃将所述产物溶解于EtOAc(320mL,0.8vol)。缓慢加入庚烷(1700mL,4.25vol.),同时逐渐增加温度至70℃。将晶种加至混浊溶液以启动沉淀。将所得混合物在室温搅拌过夜,然后过滤,用庚烷洗涤。干燥固体,得到标题化合物,为白色固体(329.8g,82%)。
制备22
5-(4-氯-噻吩-2-基)-1-甲基-1H-[1,2,4]三唑
将3-L反应烧瓶装备冷却浴、空气搅拌器和温度计探针,装入4-氯-N-二甲氨基亚甲基-2-噻吩甲酰胺(155g,0.715mol)和HOAc(1500mL)以形成溶液。使用冰水冷却浴以使温度保持在≤30℃,使用添加漏斗历经15-20分钟逐滴加入甲基肼(33.2g,0.721mol)以形成浅黄色浆液。将反应物逐渐加热至90℃,在90℃保持30分钟。经GC分析混合物,然后使反应物冷却至约70℃,浓缩至粘稠的油/浆液。缓慢加入水(1.67L)以沉淀固体,将混合物冷却至<30℃,过滤,用水(1.67L)洗涤。将湿固体(125.8g)溶解于温热的叔丁基甲醚(1.64L),经Na2SO4干燥,过滤,浓缩至干,提供标题化合物,为淡黄色固体(85.8g,60%)。
制备23
5-(5-溴-4-氯-噻吩-2-基)-1-甲基-1H-[1,2,4]三唑
将3-L反应烧瓶装备冷却浴、空气搅拌器和温度计探针,装入5-(4-氯-噻吩-2-基)-1-甲基-1H-[1,2,4]三唑(105.3g,0.527mol)、乙腈(1053mL)和 HOAc(105mL)以形成溶液。在30-60分钟内分批加入N-溴代琥珀酰亚胺(103.2g,0.580mol)同时将温度保持在≤31℃。搅拌1小时,此时GC分析显示反应完全。将反应混合物倾入水(2.1L,20体积)中,搅拌30分钟,过滤,用水洗涤(2×1L)。于45℃在真空烘箱中将产物干燥过夜,得到标题化合物,为淡黄色固体(123.0g,84%)。
实施例26
7-丁基-3-[3-氯-5-(2-甲基-2H-[1,2,4]三唑-3-基)-噻吩-2-基]-2,5-二甲基-吡唑并[1,5-a]嘧啶

将5-(5-溴-4-氯-噻吩-2-基)-1-甲基-1H-[1,2,4]三唑(169mg,0.61mmol)加至无水THF(5mL)。冷却至-78℃。加入叔丁基锂(0.81mL,1.37mmol,1.7M,在戊烷中)。搅拌45分钟,然后缓慢加入氯化锌(1.5mL,1.76mmol,0.5M,在THF中)。搅拌5分钟,然后加热至室温,搅拌15分钟。加入双(三叔丁基膦)钯(0)(62mg,0.12mmol)和7-丁基-3-碘-2,5-二甲基-吡唑并[1,5-a]嘧啶(200mg,0.61mmol)。加热至回流过夜。将反应物经硅藻土过滤,浓缩至干。使用硅胶柱色谱法纯化,用0-50%乙酸乙酯/己烷洗脱,得到标题化合物(36mg,15%)。ES/MS m/z(35Cl)401(M+1)+。
实施例27
{5-[7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5-a]嘧啶-3-基]-4-甲基-噻吩-2-基}-二甲基-胺盐酸盐
将3-(5-硝基-3-甲基-噻吩-2-基)-7-(1-乙基-丙基)-2,5-二甲基-吡唑并[1,5a]嘧啶(0.36g,1.0mmol)溶解于含有10%披钯碳(0.20g)的THF(5mL)中,用真空/氢气流吹扫脱气(3×),在氢气氛(55psi)下于室温搅拌3小时。使用TLC确认反应完全。用THF(50mL)稀释反应物,经硅藻土过滤。以60% 油悬液形式加入氢化钠(0.10g,2.25mmol),在室温搅拌20分钟。将碘甲烷(0.140mL,2.25mmol)加至反应物,于50℃在密封的反应容器中搅拌过夜。用水淬灭反应物,用乙酸乙酯(100mL)稀释。用水、饱和氯化铵和盐水洗涤有机部分。经无水硫酸镁干燥,过滤,减压浓缩至油。使用快速色谱法纯化所得残渣,用100%己烷/0%(25%乙酸乙酯/己烷)至0%己烷/100%(25%乙酸乙酯/己烷)以10%增量的阶段梯度洗脱,得到泡沫(0.145g,41%)。将产物(0.13g,0.365mmol)溶解于甲醇(3mL)中,加入4.0M HCl-二噁烷(3mL)。将反应物搅拌30分钟,用正氩压泄放(blow down)反应物。在真空烘箱中干燥,提供白色固体(0.125g)。ES/MS m/z 357.2(M+1)+。
实施例A
使用离体结合的体内功效评价
为了评价体内功效,使用离体结合评价本发明化合物。使用如D.R.Gehlert等人,EJP 509:145-153(2005)中提供的方法,经由口服途径向大鼠施用化合物。然后如Gehlert等人所述,离体评价125I-蛙皮降压肽与小脑的结合。例如,实施例20在10mg/kg时提供74%抑制。
实施例B
CRF1滤膜结合试验
通过使用由Stanford许可的Phoenix逆转录病毒表达系统,克服了基于质粒的人CRF1表达在生成具有足以开展结合试验的受体密度的重组细胞系方面的局限性。稳定的HEK-hCRF1细胞系用于制备膜,且结合反应物(200μl)如下建立:50μl 125I-蛙皮降压肽(最终0.2nM)、50μl化合物和100μlCRF1膜(25μg/反应)。将反应物在室温孵化2小时,然后经预处理的FBMillipore玻璃纤维滤板(96孔)过滤而终止。将所述板用冰冷的试验缓冲液(50mM tris、12.5mM NaCl、1mM EDTA、10mM MgCl2、0.05%BSA,pH 7.2)洗涤两次,风干过夜,用100μl Microscint 40在MicroBeta计数器中计数。非特异性结合(NSB)在0.5μM未标记的蛙皮降压肽存在下测定。通常进行一式三份测定,中值数据点由Graph Pad Prism绘图。
使用此试验,所实施的本发明化合物(除了实施例2、3、6、7和8,它们用作其他实施例的中间体且未进行测试)抑制125I-蛙皮降压肽(4nM)在转瓶(roller)/贴壁细胞中的结合,Ki(抑制常数)≤500nM。例如,实施例20显示Ki为4.4nM。
实施例C
CRF2滤膜结合试验
通过使用由Stanford许可的Phoenix逆转录病毒表达系统,克服了基于质粒的人CRF2表达在生成具有足以开展结合试验的受体密度的重组细胞系方面的局限性。稳定的HEK-hCRF2细胞系用于制备膜,且结合反应物(200μl)如下建立:50μl 125I-蛙皮降压肽(终浓度0.2nM)、50μl化合物和100μl CRF2膜(25μg/反应)。将反应物在室温孵化2小时,然后经预处理的FB Millipore玻璃纤维滤板(96孔)过滤而终止。将所述板用冰冷的试验缓冲液(50mM tris、12.5mM NaCl、1mM EDTA、10mM MgCl2、0.05%BSA,pH 7.2)洗涤两次,风干过夜,用100μl Microscint 40在MicroBeta计数器中计数。非特异性结合(NSB)在0.5μM未标记的蛙皮降压肽存在下测定。作为替代选择,使用闪烁亲近测定法评价化合物。此测定如下建立:50ul 125I-蛙皮降压肽(0.2nM终浓度)、50μl化合物或未标记的蛙皮降压肽(NSB)和100μl含有250μg麦胚凝集素(WGA)SPA珠和CRF2膜(1.5μg/反应)。将各板在室温孵化4-5小时,然后以200Xg离心10分钟。使用Wallac Trilux闪烁计数器评价结合的放射性。结合通常进行一式三份测定,中值数据点由Graph Pad Prism绘图。化合物起初以固定浓度筛选,如果记录充分的活性,那么随后产生浓度-反应曲线。
本发明化合物优选显示对CRF2受体的弱亲和力(相对于CRF1)。例如,实施例20在50μM浓度时显示39%抑制。
实施例D
生物利用度和药动学特性
分布容积(Vdist)将体内药物的量与血液或血浆中药物的浓度相联系。分布容积是指以与血液或血浆中相同的浓度包含体内药物总量所需的液 体体积:Vdist=体内药物的量/血液或血浆中药物的浓度(Goodman和Gillman,s)。对于10mg剂量和10mg/L的血浆浓度,分布容积为1升。分布容积反映药物存在于血管外组织中的程度。大的分布容积反映相较于血浆蛋白结合,化合物结合组织成分的趋向。在临床环境下,Vdist可用于测定达到稳态浓度的负荷剂量(loading dose)。
为了测试分布容积,对雄性Sprague Dawley大鼠(N=3)施予单次1mg/kg静脉内剂量的化合物给药后0.08至24小时的各时间点收集多个血浆样品。血浆样品经LC/MS/MS分析以测定血浆浓度。进行血浆药动学计算,以测定包括Vdist和血浆清除率(Clp)的药动学参数。
与其他CRF拮抗剂如CP154526(Schulz等人,Proc.Natl.Acad.Sci.(USA),93:10477(1996))和NBI30775(Chen等人,Drug DevelopmentResearch,65:216(2005))比较,本发明化合物优选具有优良的生物利用度特性。
Claims (7)
1.式I的化合物或其药学上可接受的盐,
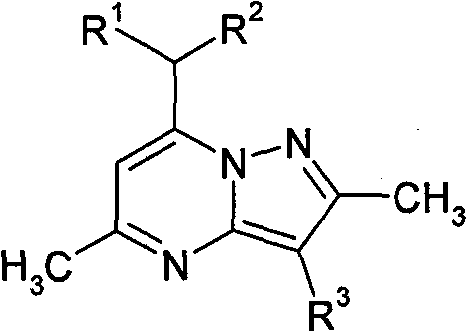
其中:
R1和R2独立地是氢或C1-C3烷基;
R3是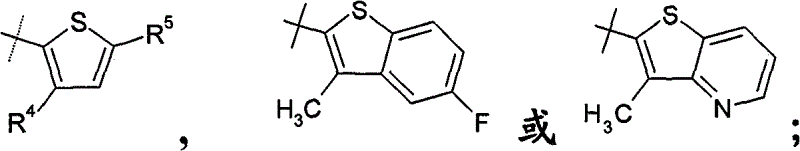
R4是Cl或甲基;
R5是氢、Br、硝基、甲氧基、甲氧基甲基、二甲氨基、乙氧羰基、乙酰氨基、乙酰氧基、
2.权利要求1的化合物或其药学上可接受的盐,其中R1和R2独立地是乙基或正丙基。
3.权利要求1或2的化合物或其药学上可接受的盐,其中R3是
4.根据权利要求1至2任一项的化合物,其中R5是甲氧基、甲氧基甲基、二甲氨基或
5.药物组合物,其包含:根据权利要求1至4任一项的化合物或其药学上可接受的盐和药学上可接受的赋形剂。
6.根据权利要求1至4任一项的化合物或其药学上可接受的盐在制备用于治疗焦虑或抑郁的药物中的用途。
7.根据权利要求1至4任一项的化合物或其药学上可接受的盐在制备用于治疗酒精或精神活性物质滥用的药物中的用途。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82627306P | 2006-09-20 | 2006-09-20 | |
| US60/826,273 | 2006-09-20 | ||
| PCT/US2007/078352 WO2008036541A1 (en) | 2006-09-20 | 2007-09-13 | Thiophene pyrazolopyrimidine compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101516886A CN101516886A (zh) | 2009-08-26 |
| CN101516886B true CN101516886B (zh) | 2012-02-29 |
Family
ID=38988344
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007800349472A Expired - Fee Related CN101516886B (zh) | 2006-09-20 | 2007-09-13 | 噻吩吡唑并嘧啶化合物 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8110580B2 (zh) |
| EP (1) | EP2076515B1 (zh) |
| JP (1) | JP5184536B2 (zh) |
| KR (1) | KR101088196B1 (zh) |
| CN (1) | CN101516886B (zh) |
| AU (1) | AU2007297481B2 (zh) |
| BR (1) | BRPI0717070A2 (zh) |
| CA (1) | CA2662000C (zh) |
| EA (1) | EA016056B1 (zh) |
| ES (1) | ES2428543T3 (zh) |
| MX (1) | MX2009003098A (zh) |
| WO (1) | WO2008036541A1 (zh) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2025674A1 (de) | 2007-08-15 | 2009-02-18 | sanofi-aventis | Substituierte Tetrahydronaphthaline, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| US9029411B2 (en) | 2008-01-25 | 2015-05-12 | Millennium Pharmaceuticals, Inc. | Thiophenes and uses thereof |
| WO2010090716A1 (en) | 2009-01-30 | 2010-08-12 | Millennium Pharmaceuticals, Inc. | Heteroaryls and their use as pi3k inhibitors |
| US8796314B2 (en) | 2009-01-30 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9090601B2 (en) | 2009-01-30 | 2015-07-28 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
| WO2012021611A1 (en) | 2010-08-11 | 2012-02-16 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8859768B2 (en) | 2010-08-11 | 2014-10-14 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| TW201217365A (en) | 2010-08-11 | 2012-05-01 | Millennium Pharm Inc | Heteroaryls and uses thereof |
| TW201307309A (zh) | 2010-10-13 | 2013-02-16 | Millennium Pharm Inc | 雜芳基化合物及其用途 |
| EP2729455B1 (en) | 2011-07-05 | 2016-09-14 | Lupin Limited | Biaryl derivatives as nachr modulators |
| EP2567959B1 (en) | 2011-09-12 | 2014-04-16 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| RS65839B1 (sr) | 2014-01-21 | 2024-09-30 | Neurocrine Biosciences Inc | Crf1 receptor antagonisti za tretman kongenitalne adrenalne hiperplazije |
| JP6364967B2 (ja) * | 2014-05-30 | 2018-08-01 | 東ソー株式会社 | ジチエノベンゾジチオフェンの製造方法 |
| EP3238676B1 (en) | 2016-04-29 | 2019-01-02 | The Procter and Gamble Company | Absorbent core with profiled distribution of absorbent material |
| WO2019036503A1 (en) | 2017-08-14 | 2019-02-21 | Spruce Biosciences, Inc. | CORTICOTROPIN RELEASE FACTOR RECEPTOR ANTAGONISTS |
| JP7285222B2 (ja) * | 2017-08-14 | 2023-06-01 | スプルース バイオサイエンシズ インコーポレイテッド | 副腎皮質刺激ホルモン放出因子受容体拮抗薬 |
| EP4643950A3 (en) | 2018-04-27 | 2026-01-14 | Spruce Biosciences, Inc. | Methods for treating testicular and ovarian adrenal rest tumors |
| US20220023266A1 (en) | 2018-12-07 | 2022-01-27 | Neurocrine Biosciences, Inc. | Crf1 receptor antagonist, pharmaceutical formulations and solid forms thereof for the treatment of congenital adrenal hyperplasia |
| JP7623365B2 (ja) | 2019-09-27 | 2025-01-28 | ニューロクライン バイオサイエンシーズ,インコーポレイテッド | Crf受容体アンタゴニストおよび使用方法 |
| TW202134241A (zh) * | 2019-12-04 | 2021-09-16 | 美商紐羅克里生物科學有限公司 | Crf受體拮抗劑及使用方法 |
| KR20240023691A (ko) | 2020-08-12 | 2024-02-22 | 스프루스 바이오사이언시스 인코포레이티드 | 다낭성 난소 증후군을 치료하기 위한 방법 및 조성물 |
| US11708372B2 (en) | 2021-11-19 | 2023-07-25 | Spruce Biosciences, Inc. | Crystalline composition of tildacerfont and methods of use and preparation thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997029109A1 (en) * | 1996-02-07 | 1997-08-14 | Janssen Pharmaceutica N.V. | Pyrazolopyrimidines as crf receptor antagonists |
| WO2001023388A2 (en) * | 1999-09-30 | 2001-04-05 | Neurogen Corporation | AMINO SUBSTITUTED PYRAZOLO[1,5,-a]-1,5-PYRIMIDINES AND PYRAZOLO[1,5-a]-1,3,5-TRIAZINES |
| WO2005063755A1 (en) * | 2003-12-22 | 2005-07-14 | Sb Pharmco Puerto Rico Inc | Crf receptor antagonists and methods relating thereto |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR19990067704A (ko) | 1992-12-17 | 1999-08-25 | 디. 제이. 우드, 스피겔 알렌 제이 | 부신피질자극호르몬 유리인자 길항제로서의 피롤로피리미딘을함유하는 약학 조성물 |
| HU229024B1 (en) | 1996-07-24 | 2013-07-29 | Bristol Myers Squibb Pharma Co | Azolo-pyridimidines, pharmaceutical compositions containing the same and use thereof |
| JP3621706B2 (ja) | 1996-08-28 | 2005-02-16 | ファイザー・インク | 置換された6,5―ヘテロ―二環式誘導体 |
| EP1869049B1 (en) * | 2005-03-21 | 2009-03-04 | Eli Lilly And Company | Imidazopyridazine compounds |
-
2007
- 2007-09-13 JP JP2009529307A patent/JP5184536B2/ja not_active Expired - Fee Related
- 2007-09-13 KR KR1020097005694A patent/KR101088196B1/ko not_active Expired - Fee Related
- 2007-09-13 EA EA200970302A patent/EA016056B1/ru not_active IP Right Cessation
- 2007-09-13 WO PCT/US2007/078352 patent/WO2008036541A1/en not_active Ceased
- 2007-09-13 AU AU2007297481A patent/AU2007297481B2/en not_active Ceased
- 2007-09-13 ES ES07814842T patent/ES2428543T3/es active Active
- 2007-09-13 US US12/438,953 patent/US8110580B2/en active Active
- 2007-09-13 BR BRPI0717070-0A2A patent/BRPI0717070A2/pt not_active IP Right Cessation
- 2007-09-13 CN CN2007800349472A patent/CN101516886B/zh not_active Expired - Fee Related
- 2007-09-13 CA CA2662000A patent/CA2662000C/en not_active Expired - Fee Related
- 2007-09-13 MX MX2009003098A patent/MX2009003098A/es active IP Right Grant
- 2007-09-13 EP EP07814842.6A patent/EP2076515B1/en not_active Not-in-force
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997029109A1 (en) * | 1996-02-07 | 1997-08-14 | Janssen Pharmaceutica N.V. | Pyrazolopyrimidines as crf receptor antagonists |
| WO2001023388A2 (en) * | 1999-09-30 | 2001-04-05 | Neurogen Corporation | AMINO SUBSTITUTED PYRAZOLO[1,5,-a]-1,5-PYRIMIDINES AND PYRAZOLO[1,5-a]-1,3,5-TRIAZINES |
| WO2005063755A1 (en) * | 2003-12-22 | 2005-07-14 | Sb Pharmco Puerto Rico Inc | Crf receptor antagonists and methods relating thereto |
Non-Patent Citations (2)
| Title |
|---|
| MAJO V.J.,et al.Facile Palladium-Catalyzed Synthesis of 3-Arylpyrazolo-[1,5a]pyrimidines.《ADVANCED SYNTHESIS AND CATALYSIS》.2003,第345卷(第5期),620-624. * |
| MAJOV.J.,etal.FacilePalladium-CatalyzedSynthesisof3-Arylpyrazolo-[1 5a]pyrimidines.《ADVANCED SYNTHESIS AND CATALYSIS》.2003 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2010504343A (ja) | 2010-02-12 |
| MX2009003098A (es) | 2009-04-01 |
| US8110580B2 (en) | 2012-02-07 |
| ES2428543T3 (es) | 2013-11-08 |
| CN101516886A (zh) | 2009-08-26 |
| AU2007297481A1 (en) | 2008-03-27 |
| KR101088196B1 (ko) | 2011-11-30 |
| EA200970302A1 (ru) | 2009-10-30 |
| CA2662000C (en) | 2013-01-08 |
| CA2662000A1 (en) | 2008-03-27 |
| KR20090053921A (ko) | 2009-05-28 |
| EP2076515A1 (en) | 2009-07-08 |
| JP5184536B2 (ja) | 2013-04-17 |
| BRPI0717070A2 (pt) | 2013-09-24 |
| WO2008036541A1 (en) | 2008-03-27 |
| EA016056B1 (ru) | 2012-01-30 |
| AU2007297481B2 (en) | 2012-03-08 |
| EP2076515B1 (en) | 2013-07-17 |
| US20100022560A1 (en) | 2010-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101516886B (zh) | 噻吩吡唑并嘧啶化合物 | |
| CN101516887B (zh) | 作为crf1受体拮抗剂的噻唑吡唑并嘧啶类 | |
| AU2009211004C1 (en) | Novel pyrazolo [3, 4 -d] pyrimidine derivatives as anti -cancer agents | |
| EP1451161B1 (en) | Imidazopyridines, pyrimidines and triazines for enhancing cognition as gaba-a alpha 5 receptor subtype ligands | |
| CA2272705C (en) | Fused bicyclic pyrimidine derivatives | |
| CN101479276B (zh) | 药用化合物 | |
| US20120202799A1 (en) | Condensed Azepine Derivatives As Bromodomain Inhibitors | |
| CN106458996A (zh) | 作为p97复合物的抑制剂的稠合嘧啶 | |
| CN105102458A (zh) | 作为自分泌运动因子抑制剂的二氢吡啶并嘧啶化合物 | |
| TW202045509A (zh) | 7H-吡咯并[2,3-d]嘧啶-4-胺衍生物 | |
| EP1421087A1 (en) | Diazacycloalkanes as oxytocin agonists | |
| CA2020297A1 (en) | 4h-indolo [1,2-d](1,2,4]triazolo[4,3-a](1,4)benzodiazepines, a process for their preparation and their use as medicaments | |
| AU2002318005B2 (en) | Diazacycloalkanes as oxytocin agonists | |
| CA2151242A1 (en) | Novel triazoloquinazolines, their preparation and use | |
| AU2002318005A1 (en) | Diazacycloalkanes as oxytocin agonists | |
| HK1132505B (zh) | 作為crf1受體拮抗劑的噻唑吡唑並嘧啶 | |
| CA2204030A1 (en) | Oxazolyl- and thiazolylimidazo-benzo- and thienodiazepines and their use as medicaments |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20120229 Termination date: 20160913 |