[001] Este pedido reivindica prioridade do Pedido Provisório U.S. No 61/017.418, depositado em 28 de dezembro de 2007, e Pedido Provisório U.S. No 61/017.881 depositado em 31 de dezembro de 2007, cada um dos quais é aqui incorporado por referência na sua totalidade.
CAMPO DA INVENÇÃO
[002] No geral, a invenção diz respeito às formulações de VWF recombinante e métodos para fabricar uma composição que compreenda VWF recombinante.
FUNDAMENTOS DA INVENÇÃO
[003] O fator de von Willebrand (VWF) é uma glicoproteína que circula no plasma como uma série de multímeros variando no tamanho de cerca de 500 a 20.000 kD. As formas multiméricas de VWF são compostas de subunidades de polipeptídeo de 250 kD ligadas entre si pelas ligações de dissulfeto. O VWF medeia a adesão de plaqueta inicial ao sub-endotélio da parede de vaso danificada. Apenas os multímeros maiores exibem atividade hemostática. É assumido que as células endoteliais secretam formas poliméricas grandes de VWF e aquelas formas de VWF que têm um peso molecular baixo (VWF de peso molecular baixo) surge da clivagem proteolítica. Os multímeros tendo massas moleculares grandes são armazenados nos corpos de Weibel-Pallade de células endoteliais e liberados na estimulação.
[004] O VWF é sintetizado pelas células endoteliais e megacariócitos como prepró-VWF que consiste em uma extensão grande de domínios repetidos. Na clivagem do peptídeo de sinal, pró-VWF dimeriza através das ligações de dissulfeto na sua região de terminal C. Os dímeros servem como protômeros para a multimerização, que é controlada pelas ligações de dissulfeto entre os terminais de extremidade livre. A montagem para multímeros é seguida pela remoção proteolítica da sequência de propeptídeo (Leyte et al., Biochem. J. 274 (1991), 257-261).
[005] O produto da tradução primária prognosticado a partir do cDNA clonado do VWF é um polipeptídeo precursor de 2813 resíduos (prepró-VWF). O prepró-VWF consiste em um peptídeo de sinal de 22 aminoácido e um propeptídeo de 741 aminoácidos, com a VWF madura que compreende 2050 aminoácidos (Ruggeri Z. A., e Ware, J., FASEB J., 308-316 (1993)).
[006] Defeitos no VWF são a causa da doença de von Willebrand (VWD), que é caracterizada por um fenótipo de hemorragia mais ou menos pronunciado. A VWD tipo 3 é a forma mais grave em que o VWF é completamente perdido, e a VWD tipo 1 diz respeito a uma perda quantitativa de VWF e o seu fenótipo pode ser muito brando. A VWD tipo 2 diz respeito a defeitos qualitativos de VWF e pode ser tão grave quanto a VWD tipo 3. A VWD tipo 2 tem muitas sub formas, algumas sendo associadas com a perda ou a diminuição de multímeros de peso molecular alto. A síndrome de von Willebrand tipo 2a (VWS-2A) é caracterizada por uma perda de multímeros tanto intermediários quanto grandes. A VWS-2B é caracterizada por uma perda de multímeros de peso molecular mais alto. Outras doenças e distúrbios relacionados com o VWF são conhecidos na técnica.
[007] A Patente US. Nos 6.531.577, 7.166.709, e Pedido de Patente Européia No 04380188.5, descrevem formulações de VWF derivadas do plasma. Entretanto, além de problemas de quantidade e pureza com VWF derivado de plasma, existe também um risco de patógenos transportados pelo sangue (por exemplo, vírus e a doença de Creutzfeldt-Jakob variante (vCJD).
[008] Assim existe uma necessidade na técnica para desenvolver uma formulação farmacêutica estável que compreende VWF recombinante.
SUMÁRIO DA INVENÇÃO
[009] A presente invenção fornece formulações úteis para as composições que compreendem VWF recombinante, resultando em uma composição farmacêutica altamente estável. A composição farmacêutica estável é útil como um agente terapêutico no tratamento de indivíduos que sofrem de distúrbios ou condições que possam se beneficiar da administração de VWF recombinante.
[010] Em uma forma de realização, a invenção fornece uma formulação farmacêutica líquida estável de um Fator de von Willebrand recombinante (rVWF) que compreende: (a) um rVWF; (b) um agente de tamponação; (c) um ou mais sais; (d) opcionalmente um agente de estabilização; e (e) opcionalmente um tensoativo; em que o rVWF compreende um polipeptídeo selecionado do Grupo que consiste de: a) a sequência de aminoácidos apresentada na SEQ ID NO: 3; b) um análogo biologicamente ativo, fragmento ou variante de a); c) um polipeptídeo codificado pelo polinucleotídeo apresentado na SEQ ID NO: 1; d) um análogo, fragmento ou variante biologicamente ativos de c); e e) um polipeptídeo codificado por um polinucleotídeo que hibridiza ao polinucleotídeo apresentado na SEQ ID NO: 1 sob condições de hibridização moderadamente severas; em que o tampão é compreendido de um agente de tamponação de pH em uma faixa de cerca de 0,1 mM a cerca de 500 mM e em que o pH está em uma faixa de cerca de 2,0 a cerca de 12,0; em que o sal está a uma concentração de cerca de 1 a 500 mM; em que o agente de estabilização está a uma concentração de cerca de 0,1 a 1000 mM; e em que o tensoativo está a uma concentração de cerca de 0,01 g/L a 0,5 g/L.
[011] Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que o rVWF compreende a sequência de aminoácidos apresentada na SEQ ID NO: 3. Em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o agente de tamponação é selecionado do grupo que consiste em citrato de sódio, glicina, histidina, Tris e combinações destes agentes. Já em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o agente de tamponação é citrato. Ainda em uma outra forma de realização da invenção, a formulação anteriormente mencionada é fornecida em que o pH está na faixa de 6,0 a 8,0, ou 6,5 a 7,3. Em uma forma de realização realizada, a formulação anteriormente mencionada é fornecida em que o pH é 7,0. Em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o agente de tamponação é citrato e o pH é 7,0.
[012] Ainda em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o sal é selecionado do grupo que consiste de cloreto de cálcio, cloreto de sódio e cloreto de magnésio. Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que o sal está em uma faixa de concentração de 0,5 a 300 mM. Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que o sal é cloreto de cálcio a uma concentração de 10 mM.
[013] Em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o rVWF compreende a sequência de aminoácidos apresentada na SEQ ID NO: 3; em que o agente de tamponação é citrato e o pH é 7,0; e em que o sal é cloreto de cálcio a uma concentração de 10 mM. Ainda em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o rVWF compreende a sequência de aminoácidos apresentada na SEQ ID NO: 3; em que o agente de tamponação é citrato de sódio e o pH é 7,0; e em que o sal é cloreto de cálcio a uma concentração de 10 mM e NaCl a uma concentração de 100 mM.
[014] Outras formulações também são consideradas pela presente invenção. Por exemplo, em uma forma de realização, uma formulação anteriormente mencionada é fornecida em que o um ou mais agentes de tamponação é histidina e Tris em uma concentração de 3,3 mM cada um. Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que o pH é 7,0. Já em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o primeiro sal é cloreto de sódio em uma concentração de 30 mM e o segundo sal é cloreto de cálcio em uma concentração de 0,56 mM.
[015] Ainda em uma outra forma de realização da invenção, uma formulação anteriormente mencionada é fornecida em que o agente de estabilização é selecionado do Grupo que consiste em manitol, lactose, sorbitol, xilitol, sacarose, trealose, manose, maltose, lactose, glicose, rafinose, celobiose, gentiobiose, isomaltose, arabinose, glicosamina, frutose e combinações destes agentes de estabilização. Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que os agentes de estabilização são trealose em uma concentração de 7,8 mM e manitol em uma concentração de 58,6 mM.
[016] Em uma outra forma de realização, uma formulação anteriormente mencionada é fornecida em que o tensoativo é selecionado do grupo que consiste em digitonina, Triton X-100, Triton X-114, TWEEN-20, TWEEN-80 e combinações destes tensoativos. Em uma outra forma de realização, a formulação anteriormente mencionada é fornecida em que o tensoativo é TWEEN-80 a 0,03 g/L.
[017] Em uma forma de realização da invenção, uma formulação anteriormente mencionada é fornecida em que o rVWF compreende a sequência de aminoácidos apresentada na SEQ ID NO: 3; em que os agentes de tamponação são histidina em uma concentração de 3,3 mM e Tris em uma concentração de 3,3 mM no pH 7,0; em que o primeiro sal é cloreto de sódio em uma concentração de 30 mM e o segundo sal é cloreto de cálcio em uma concentração de 0,56 mM; em que os agentes de estabilização são trealose em uma concentração de 7,8 mM de manitol em uma concentração de 58,6 mM; e em que o tensoativo é TWEEN-80 a 0,03 g/L.
DESCRIÇÃO RESUMIDA DAS FIGURAS
[018] A Figura 1 mostra que rVWF não é estável em tampão Advate depois de 26 semanas, devido à presença de glutationa.
[019] A Figura 2 mostra que rVWF é estável em tampão Advate 1:3 por até 12 semanas a 4°C.
[020] A Figura 3 mostra que a estabilidade de uma formulação com base em citrato é melhor do que a formulação em tampão Advate 1:3 contendo 0,1 M de glutationa.
[021] A Figura 4 mostra que a concentração de rVWF é estável acima de 26 semanas em tampão Advate.
[022] A Figura 5 mostra que a concentração de rVWF é estável com o tempo em tampão Advate 1:3.
[023] A Figura 6 mostra que a concentração de rVWF é estável com o tempo em tampão com base em citrato.
[024] A Figura 7 mostra que a maioria dos excipientes aumentam a temperatura de desdobramento de rVWF em cerca de 1 ou 2 °C.
[025] A Figura 8 mostra que CaCl2 10 mM, aumenta a temperatura de desdobramento de rVWF em cerca de 8°C a cerca de 67°C.
[026] A Figura 9 mostra que o efeito de CaCl2 é similar no pH 7,3 e pH 6,5.
DESCRIÇÃO DETALHADA DA INVENÇÃO
Definição de termos
[027] A menos que de outro modo definido, todos os termos técnicos e científicos aqui usados têm o mesmo significado como habitualmente entendido por uma pessoa de habilidade comum na técnica à qual esta invenção pertence. As seguintes referências provêem uma pessoa de habilidade com uma definição geral de muitos dos termos usados nesta invenção: Singleton, et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY (2a ed. 1994); THE CAMBRIDGE DICTIONARY OF SCIENCE AND TECHNOLOGY (Walker ed., 1988); THE GLOSSARY OF GENETICS, 5a ED., R. Rieger, et al. (eds.), Springer Verlag (1991); e Hale e Marham, THE HARPER COLLINS DICTIONARY OF BIOLOGY (1991).
[028] Cada publicação, pedido de patente, patente, e outras referências aqui citadas é incorporada por referência na sua totalidade até o grau em que a mesma não seja incompatível com a presente divulgação.
[029] É aqui mencionado que, como usado neste relatório descritivo e nas reivindicações anexas, as formas singulares “um,” “uma,” “o”, e “a” incluem referência plural a menos que o contexto claramente dite de outro modo.
[030] Como aqui usado, os seguintes termos têm os significados atribuídos aos mesmos a menos que de outro modo especificado.
[031] O termo “que compreende,” com respeito a um composto de peptídeo, significa que um composto pode incluir aminoácidos adicionais em cada um ou em ambos os terminais amino e carbóxi da dada sequência. Naturalmente, estes aminoácidos adicionais não devem significantemente interferir com a atividade do composto. Com respeito a uma composição da presente invenção, o termo “que compreende” significa que uma composição pode incluir componentes adicionais. Estes componentes adicionais não devem interferir significantemente com a atividade da composição.
[032] O termo “farmacologicamente ativo” significa que uma substância assim descrita é determinada ter atividade que afeta um parâmetro médico (por exemplo, mas não limitado à pressão sanguínea, contagem de célula sanguínea, nível de colesterol) ou estado de doença (por exemplo, mas não limitado a câncer, distúrbios autoimunes).
[033] Como aqui usado os termos “expressa,” “expressando” e “expressão” significam permitir ou fazer com que a informação em uma sequência de gene ou DNA se torne manifesta, por exemplo, produzindo uma proteína pela ativação das funções celulares envolvidas na transcrição e tradução de uma sequência de gene ou DNA correspondente. Uma sequência de DNA é expressa em ou por uma célula para formar um “produto de expressão” tal como uma proteína. O produto de expressão por si só, por exemplo a proteína resultante, também pode ser dito ser “expressado.” Um produto de expressão pode ser caracterizado como intracelular, extracelular ou secretado. O termo “intracelular” significa dentro de uma célula. O termo “extracelular” significa fora de uma célula, tal como uma proteína de transmembrana. Uma substância é “secretada” por uma célula se a mesma aparece em medição significante fora da célula, de algum lugar na ou dentro da célula.
[034] Como aqui usado um “polipeptídeo” refere-se a um composto polimérico de resíduos de aminoácido, variantes estruturais, variantes estruturais que ocorrem naturalmente relacionadas, e análogos que não ocorrem naturalmente sintéticos destes ligados por intermédio de ligações peptídicas. Os polipeptídeos sintéticos podem ser preparados, por exemplo, usando um sintetizador de polipeptídeo automatizado. O termo “proteína” tipicamente refere-se a polipeptídeos grandes. O termo “peptídeo” tipicamente referem-se aos polipeptídeos curtos.
[035] Como aqui usado um “fragmento” de um polipeptídeo é intencionado a referir-se a qualquer porção de um polipeptídeo ou proteína menor do que o polipeptídeo de tamanho natural ou produto de expressão de proteína.
[036] Como aqui usado um “análogo” refere-se a qualquer um de dois ou mais polipeptídeos substancialmente similares em estrutura e tendo a mesma atividade biológica, mas podem ter graus variáveis de atividade, à molécula inteira, ou a um fragmento desta. Análogos diferem na composição de suas sequências de aminoácidos com base em uma ou mais mutações envolvendo a substituição de um ou mais aminoácidos por outros aminoácidos. As substituições podem ser conservativas ou não conservativas com base na relacionabilidade físico-química ou funcional do aminoácido que está sendo substituído e o aminoácido que o substitui.
[037] Como aqui usado uma “variante” refere-se a um polipeptídeo, proteína ou análogo destes que é modificado para compreender porções químicas adicionais não normalmente uma parte da molécula. Tais porções podem modular a solubilidade, absorção, meia-vida biológica, da molécula etc. As porções alternativamente podem diminuir a toxicidade da molécula e eliminar ou atenuar qualquer efeito colateral indesejado da molécula, etc. As porções capazes de mediar tais efeitos são divulgadas em Remington’s Pharmaceutical Sciences (1980). Os procedimentos para a ligação de tais porções a uma molécula são bem conhecidos na técnica. Por exemplo, a variante pode ser um fator de coagulação sanguínea tendo uma modificação química que confere uma meia-vida mais longa in vivo para a proteína. Em vários aspectos, polipeptídeos são modificados pela glicosilação, peguilação e/ou polissialilação.
VWF Recombinante
[038] As sequências de polinucleotídeo e aminoácido de prepró-VWF são apresentadas na SEQ ID NO: 1 e SEQ ID NO: 2, respectivamente, e são disponíveis nos Acessos do GenBank Nos NM_000552 e NP_000543, respectivamente. A sequência de aminoácidos que corresponde à proteína de VWF madura é apresentada na SEQ ID NO: 3 (correspondente aos aminoácidos 764 a 2813 da sequência de aminoácidos de prepró-VWF de tamanho natural).
[039] Uma forma de rVWF útil tem pelo menos a propriedade de estabilização in vivo, por exemplo ligação, de pelo menos uma molécula do Fator VIII (FVIII) e tendo opcionalmente um padrão de glicosilação que é farmacologicamente aceitável. Os seus exemplos específicos incluem VWF sem domínio A2 resistente assim à proteólise (Lankhof et al., Thromb. Haemost. 77: 1008-1013, 1997), e o fragmento VWF de Val 449 a Asn 730 incluindo o domínio que liga a glicoproteína 1b e os sítios de ligação para colágeno e heparina (Pietu et al., Biochem. Biophys. Res. Commun. 164: 1339-1347, 1989). A determinação da capacidade de um VWF para estabilizar pelo menos uma molécula de FVIII pode ser realizada em mamíferos deficientes em VWF de acordo com métodos conhecidos no estado da técnica.
[040] A rVWF da presente invenção pode ser produzida por qualquer método conhecido na técnica. Um exemplo específico é divulgado na WO86/06096 publicada em 23 de outubro de 1986 e Pedido de Patente U.S. No 07/559.509, depositado em 23 de julho de 1990, que é aqui incorporado por referência com respeito aos métodos de produzir VWF recombinante. Assim, métodos são conhecidos na técnica para (i) a produção de DNA recombinante pela engenharia genética, por exemplo via transcrição reversa de RNA e/ou amplificação de DNA, (ii) introduzir DNA recombinante dentro de células procarióticas ou eucarióticas pela transfecção, por exemplo via eletroporação ou microinjeção, (iii) cultivar as ditas células transformadas, por exemplo em uma maneira contínua ou escalonada, (iv) expressar VWF, por exemplo constitutivamente ou na indução, e (v) isolar o dito VWF, por exemplo do meio de cultura ou pela colheita das células transformadas, de modo a (vi) obter rVWF purificado, por exemplo via cromatografia de troca aniônica ou cromatografia de afinidade. Um VWF recombinante pode ser fabricado em células hospedeiras transformadas usando técnicas de DNA recombinante bem conhecidas no ramo. Por exemplo, sequências que codificam o polipeptídeo podem ser excisadas de DNA usando enzimas de restrição adequadas.
[041] Alternativamente, a molécula de DNA pode ser sintetizada usando as técnicas de síntese química, tais como o método de fosforamidato. Também, uma combinação destas técnicas pode ser usada.
[042] A invenção também fornece vetores que codificam polipeptídeos da invenção em um hospedeiro apropriado. O vetor compreende o polinucleotídeo que codifica o polipeptídeo operativamente ligado às sequências de controle de expressão apropriadas. Os métodos de efetuar esta ligação operativa, antes ou depois do polinucleotídeo ser inserido dentro do vetor, são bem conhecidos. As sequências de controle de expressão incluem promotores, ativadores, realçadores, operadores, sítios de ligação ribossômica, sinais de início, sinais de parada, sinais de encapuzamento, sinais de poliadenilação, e outros sinais envolvidos com o controle de transcrição ou tradução. O vetor resultante tendo o polinucleotídeo nele é usado para transformar um hospedeiro apropriado. Esta transformação pode ser realizada usando métodos bem conhecidos na técnica.
[043] Qualquer uma de um grande número de células hospedeiras disponíveis e bem conhecidas pode ser usada na prática desta invenção. A seleção de um hospedeiro particular é dependente de vários fatores reconhecidos pela técnica, incluindo, por exemplo, compatibilidade com o vetor de expressão escolhido, toxicidade dos peptídeos codificados pela molécula de DNA, taxa de transformação, facilidade de recuperação dos peptídeos, características de expressão, biossegurança e custos. Um equilíbrio destes fatores deve ocorrer com o entendimento de que nem todas as células hospedeiras são igualmente eficazes para a expressão de uma sequência de DNA particular. Dentro destas diretrizes gerais, as células hospedeiras microbianas úteis incluem células de bactérias, levedura e outros fungos, insetos, plantas, mamífero (incluindo o ser humano) em cultura, ou outros hospedeiros conhecidos na técnica.
[044] Em seguida, o hospedeiro transformado é cultivado e purificado. As células hospedeiras podem ser cultivadas sob condições de fermentação convencionais de modo que os compostos desejados sejam expressos. Tais condições de fermentação são bem conhecidos na técnica. Finalmente, os polipeptídeos são purificados da cultura pelos métodos bem conhecidos na técnica.
[045] Dependendo da célula hospedeira utilizada para expressar um composto da invenção, grupos de carboidrato (oligossacarídeo) podem ser convenientemente ligados aos sítios que são conhecidos como sendo sítios de glicosilação em proteínas. No geral, os oligossacarídeos ligados em O são ligados aos resíduos serina (Ser) ou treonina (Thr), enquanto que os oligossacarídeos ligados em N são ligados aos resíduos de asparagina (Asn) quando eles são parte da sequência Asn-X-Ser/Thr, onde X pode ser qualquer aminoácido exceto prolina. X é preferivelmente um dos 19 aminoácidos que ocorrem naturalmente não contando prolina. As estruturas de oligossacarídeos ligados em N e ligados em O e os resíduos de açúcar encontrados em cada tipo são diferentes. Um tipo de açúcar que é habitualmente encontrado em ambos é o ácido N-acetilneuramínico (aludido como ácido siálico). O ácido siálico é usualmente o resíduo terminal dos oligossacarídeos tanto ligado em N quanto ligado em O e, em virtude da sua carga negativa, pode conferir propriedades ácidas ao composto glicosilado. Tal(is) sítio(s) pode(m) ser incorporado(s) no ligante dos compostos desta invenção e são preferivelmente glicosilados por uma célula durante a produção recombinante dos compostos de polipeptídeo (por exemplo, em células de mamífero tais como CHO, BHK, COS). Entretanto, tais sítios podem ser glicosilados ainda pelos procedimentos sintéticos ou semi- sintéticos conhecidos na técnica.
[046] Alternativamente, os compostos podem ser fabricados pelos métodos sintéticos. Por exemplo, as técnicas de síntese de fase sólida podem ser usadas. As técnicas adequadas são bem conhecidas no ramo, e incluem aquelas descritas em Merrifield (1973), Chem. Polypeptides, pp. 335-61 (Katsoyannis e Panayotis eds.); Merrifield (1963), J. Am. Chem. Soc. 85: 2149; Davis et al. (1985), Biochem. Intl. 10: 394-414; Stewart e Young (1969), Solid Phase Peptide Synthesis; Pat. U.S. No 3.941.763; Finn et al. (1976), The Proteins (3a ed.) 2: 105-253; e Erickson et al. (1976), The Proteins (3a ed.) 2: 257-527. A síntese de fase sólida é a técnica preferida de fabricar peptídeos individuais visto que é o método mais eficaz em custo de fabricar peptídeos pequenos. Fragmentos, variantes e análogos de VWF
[047] Os métodos para preparar fragmentos, variantes ou análogos de polipeptídeo são bem conhecidos na técnica.
[048] Os fragmentos de um polipeptídeo são preparados usando, sem limitação, a clivagem enzimática (por exemplo, tripsina, quimiotripsina) e também usando meios recombinantes para gerar um fragmento de polipeptídeo tendo uma sequência de aminoácidos específica. Os fragmentos de polipeptídeo podem ser gerados que compreendem uma região da proteína tendo uma atividade particular, tal como um domínio de multimerização ou qualquer outro domínio de VWF identificável conhecido na técnica.
[049] Os métodos de fabricar análogos de polipeptídeo também são bem conhecidos. Os análogos de sequência de aminoácidos de um polipeptídeo podem ser análogos substitucionais, insercionais, de adição ou deleção. Os análogos de deleção, incluindo fragmentos de um polipeptídeo, carecem de um ou mais resíduos da proteína nativa que não são essenciais para a função ou atividade imunogênica. Os análogos insercionais envolvem a adição, por exemplo, de aminoácido(s) em um ponto não terminal no polipeptídeo. Este análogo pode incluir a inserção de um epítopo imunorreativo ou simplesmente um resíduo único. Os análogos adicionais, incluindo fragmentos de um polipeptídeo, incluem a adição de um ou mais aminoácidos em cada um de ambos os terminais de uma proteína e incluem, por exemplo, proteínas de fusão.
[050] Os análogos substitucionais tipicamente trocam um aminoácido do tipo selvagem por um outro em um ou mais sítios dentro da proteína, e podem ser planejados para modular uma ou mais propriedades do polipeptídeo sem a perda de outras funções ou propriedades. Em um aspecto, as substituições são substituições conservativas. Por “substituição de aminoácido conservativa” é intencionado a substituição de um aminoácido com um aminoácido tendo uma cadeia lateral de um caráter químico similar. Os aminoácidos similares para fabricar substituições conservativas incluem aqueles tendo uma cadeia lateral ácida (ácido glutâmico, ácido aspártico); uma cadeia lateral básica (arginina, lisina, histidina); uma cadeia lateral de amida polar (glutamina, asparagina); uma cadeia lateral hidrofóbica, alifática (leucina, isoleucina, valina, alanina, glicina); uma cadeia lateral aromática (fenilalanina, triptofano, tirosina); uma cadeia lateral pequena (glicina, alanina, serina, treonina, metionina); ou uma cadeia lateral de hidroxila alifática (serina, treonina).
[051] Os análogos podem ser substancialmente homólogos ou substancialmente idênticos ao VWF recombinante a partir dos quais os mesmos são derivados. Os análogos preferidos são aqueles que retêm pelo menos alguma da atividade biológica do polipeptídeo do tipo selvagem, por exemplo atividade de coagulação sanguínea.
[052] As variantes de polipeptídeo consideradas incluem polipeptídeos quimicamente modificados por técnicas tais como ubiquitinação, glicosilação, incluindo polissialação, conjugação aos agentes terapêuticos ou de diagnóstico, rotulação, ligação de polímero covalente tal como peguilação (derivação com polietileno glicol), introdução de ligações não hidrolisáveis, e inserção ou substituição pela síntese química de aminoácidos tais como ornitina, que normalmente não ocorrem em proteínas humanas. As variantes retêm as mesmas ou essencialmente as mesmas propriedades de ligação de moléculas não modificadas da invenção. Tal modificação química pode incluir a ligação direta ou indireta (por exemplo, via um ligante) de um agente ao polipeptídeo de VWF. No caso de ligação indireta, é considerado que o ligante pode ser hidrolisável ou não hidrolisável.
[053] Preparar análogos de polipeptídeo peguilados no geral compreenderá as etapas de (a) reagir o polipeptídeo com polietileno glicol (tal como um éster reativo ou derivado de aldeído de PEG) sob condições por meio das quais a ligação do polipeptídeo de construção torna-se ligado a um ou mais grupos PEG, e (b) obter o(s) produto(s) de reação. No geral, as condições de reação ótimas para as reações de acilação serão determinadas com base nos parâmetros conhecidos e o resultado desejado. Por exemplo, quanto maior a razão de PEG:proteína, maior a porcentagem de produto poli-peguilado. Em algumas formas de realização, a construção de ligação terá uma única porção de PEG no terminal N. Polietileno glicol (PEG) pode ser ligado ao fator de coagulação sanguínea para fornecer uma meia-vida mais longa in vivo. O grupo PEG pode ser de qualquer peso molecular conveniente e pode ser linear ou ramificado. O peso molecular médio do PEG varia de cerca de 2 quiloDalton (“kD”) a cerca de 100 kDa, de cerca de 5 kDa a cerca de 50 kDa, ou de cerca de 5 kDa a cerca de 10 kDa. Os grupos PEG são ligados ao fator de coagulação sanguínea via acilação ou alquilação redutiva através de um grupo reativo natural ou engendrado na porção de PEG (por exemplo, um grupo aldeído, amino, tiol ou éster) a um grupo reativo no fator de coagulação sanguínea (por exemplo, um grupo aldeído, amino ou éster) ou por qualquer outra técnica conhecida no ramo.
[054] Os métodos para preparar polipeptídeo polissialilado são descritos na Publicação de Patente dos Estados Unidos 20060160948, Fernandes et Gregoriadis; Biochim. Biophys. Acta 1341: 26-34, 1997, e Saenko et al., Haemophilia 12: 42-51, 2006. Em resumo, uma solução de ácido colomínico contendo 0,1 M de NaIO4 é agitada no escuro na temperatura ambiente para oxidar a CA. A solução de CA ativada é dialisada contra, por exemplo, tampão 0,05 M de fosfato de sódio, pH 7,2 no escuro e esta solução foi adicionada a uma solução de rVWF e incubadas por 18 h na temperatura ambiente no escuro sob agitação suave. Os reagentes livres podem ser depois separados do conjugado de rVWF-ácido polissiálico pela ultrafiltração/diafiltração. A conjugação de rVWF com ácido polissiálico também pode ser obtida usando glutaraldeído como reagente de reticulação (Migneault et al., Biotechniques 37: 790-796, 2004).
[055] É além disso considerado que um polipeptídeo da invenção possa ser uma proteína de fusão com um segundo agente que é um polipeptídeo. Em uma forma de realização, o segundo agente que é um polipeptídeo, sem limitação, é uma enzima, um fator de crescimento, um anticorpo, uma citocina, uma quimiocina, um receptor de superfície celular, o domínio extracelular de um receptor de superfície celular, uma molécula de adesão celular, ou fragmento ou domínio ativo de uma proteína descrita acima. Em uma forma de realização realizada, o segundo agente é um fator de coagulação sanguínea tal como Fator VIII, Fator VII, Fator IX. A proteína de fusão considerada é fabricada pelas técnicas químicas ou recombinantes bem conhecidas no ramo.
[056] Também é considerado que os polipeptídeos prepró-VWF e pró- VWF podem fornecer um benefício terapêutico nas formulações da presente invenção. Por exemplo, a Patente US No 7.005.502 descreve uma preparação farmacêutica que compreende quantidades substanciais de pró-VWF que induz a geração de trombina in vitro. Além dos fragmentos, variantes, ou análogos recombinantes, biologicamente ativos da VWF madura, que ocorrem naturalmente, a presente invenção considera o uso de fragmentos, variantes, ou análogos recombinantes biologicamente ativos dos polipeptídeos prepró- VWF (apresentado na SEQ ID NO: 2) ou pró-VWF (resíduos de aminoácido de 23 a 764 da SEQ ID NO: 2) nas formulações aqui descritas.
[057] Polinucleotídeos que codificam fragmentos, variantes e análogos podem ser facilmente gerados por um técnico de habilidade para codificar fragmentos, variantes, ou análogos biologicamente ativos da molécula que ocorre naturalmente que possui a mesma atividade biológica ou similar para a molécula que ocorre naturalmente. Estes polinucleotídeos podem ser preparados usando técnicas de PCR, digestão/ligação de molécula que codifica DNA, e outros. Assim, uma pessoa de habilidade na técnica será capaz de gerar mudanças de base únicas no filamento de DNA para resultar em um códon alterado e uma mutação de sentido errado, usando qualquer método conhecido na técnica, incluindo, mas não limitado à mutagênese específica de sítio. Como aqui usado, a frase “condições de hibridização moderadamente severas” significa, por exemplo, hibridização a 42 °C em 50 % de formamida e lavando a 60 °C em 0,1 x SSC, 0,1 % de SDS. É entendido por aqueles de habilidade na técnica que a variação nestas condições ocorre com base no comprimento e teor de base de nucleotídeo GC das sequências a serem hibridizadas. As fórmulas padrão na técnica são apropriadas para determinar as condições de hibridização exatas. Ver Sambrook et al., 9.47-9.51 em Molecular Cloning, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, Nova Iorque (1989).
Formulações e excipientes no geral,
[058] Os excipientes são aditivos que são incluídos em uma formulação porque eles comunicam ou realçam a estabilidade e liberação de um produto medicamentoso. Independente da razão para a sua inclusão, os excipientes são um componente integral de um produto medicamentoso e, portanto, necessitam ser seguros e bem tolerados pelos pacientes. Para os medicamentos de proteína, a escolha de excipientes é particularmente importante porque eles podem afetar tanto a eficácia quanto a imunogenicidade do medicamento. Consequentemente, as formulações de proteína precisam ser desenvolvidas com a seleção apropriada de excipientes que produzem estabilidade, segurança, e comerciabilidade adequadas.
[059] O desafio principal no desenvolvimento de formulações para as proteínas terapêuticas é a estabilização do produto contra os estresses de fabricação, remessa e armazenagem. O papel dos excipientes de formulação é fornecer a estabilização contra estes estresses. Os excipientes também podem ser utilizados para reduzir a viscosidade de formulações de proteína de alta concentração de modo a permitir a sua liberação e realçar a conveniência do paciente. No geral, os excipientes podem ser classificados com base nos mecanismos pelos quais eles estabilizam proteínas contra vários estresses químicos e físicos. Alguns excipientes são usados para aliviar os efeitos de um estresse específico ou para regular uma susceptibilidade particular de uma proteína específica. Outros excipientes têm efeitos mais gerais sobre as estabilidades físicas e covalentes de proteínas. Os excipientes descritos são organizados pelo seu tipo químico ou seu papel funcional nas formulações. Descrições resumidas dos modos de estabilização são fornecidas quando do debate de cada tipo de excipiente.
[060] Dadas as divulgações e orientação aqui fornecidas, aqueles de habilidade na técnica saberão qual quantidade ou faixa de excipiente podem ser incluídos em qualquer formulação particular para se obter uma formulação biofarmacêutica da invenção que promove a retenção na estabilidade do produto biofarmacêutico (por exemplo, um polipeptídeo). Por exemplo, a quantidade e tipo de um sal a ser incluído em uma formulação biofarmacêutica da invenção podem ser selecionados com base na osmolalidade desejada (isto é, isotônica, hipotônica ou hipertônica) da solução final assim como as quantidades e osmolalidade de outros componentes a serem incluídos na formulação. Similarmente, pela exemplificação com referência ao tipo de poliol ou açúcar incluídos em uma formulação, a quantidade de um tal excipiente dependerá da sua osmolalidade.
[061] Por via de exemplo, a inclusão de cerca de 5% de sorbitol pode alcançar isotonicidade, enquanto cerca de 9% de um excipiente de sacarose são necessários para alcançar isotonicidade. A seleção da quantidade ou faixa de concentrações de um ou mais excipientes que podem ser incluídos dentro de uma formulação biofarmacêutica da invenção foi exemplificado acima pela referência aos sais, polióis e açúcares. Entretanto, aqueles habilitados na técnica entenderão que as considerações aqui descritas e outras exemplificadas pela referência aos excipientes específicos são igualmente aplicáveis a todos os tipos e combinações de excipientes incluindo, por exemplo, sais, aminoácidos, outros agentes de tonicidade, tensoativos, estabilizantes, agentes de volume, crioprotetores, lioprotetores, antioxidantes, íons metálicos, agentes queladores e/ou conservantes.
[062] Além disso, onde um excipiente particular é relatado em concentração molar, aqueles habilitados na técnica reconhecerão que a porcentagem equivalente (%) p/v (por exemplo, (gramas de substância em uma amostra de solução/ml de solução) X 100 %) da solução também é considerada.
[063] Naturalmente, uma pessoa tendo habilidade comum na técnica reconhecerá que as concentrações dos excipientes aqui descritos compartilham uma interdependência dentro de uma formulação particular. Por via de exemplo, a concentração de um agente de volume pode ser reduzida onde, por exemplo, existe uma concentração de polipeptídeo alta ou onde, por exemplo, existe uma concentração alta de agente de estabilização. Além disso, uma pessoa tendo habilidade comum na técnica reconheceria que, de modo a manter a isotonicidade de uma formulação particular em que não existe nenhum agente de volume, a concentração de um agente de estabilização pode ser ajustada consequentemente (isto é, uma quantidade “tonificante” de estabilizador pode ser usada). Os excipientes comuns são conhecidos na técnica e podem ser encontrados em Powell et al., Compendium of Excipients fir Parenteral Formulations (1998), PDA J. Pharm. Sci. Technology, 52: 238-311.
Tampões e agentes de tamponação
[064] A estabilidade de uma formulação de polipeptídeo farmacologicamente ativa é usualmente observada ser máxima em uma faixa de pH estreita. Esta faixa de pH de estabilidade ótima precisa ser identificada inicialmente durante os estudos de pré-formulação. Vários métodos, tal como estudos de estabilidade acelerada e estudos de triagem calorimétrica, foram demonstrados serem úteis nesta tentativa (Remmele R. L. Jr., et al., Biochemistry, 38(16): 5241-7 (1999)). Uma vez que uma formulação é finalizada, o produto medicamentoso deve ser fabricado e mantido por toda a sua vida de prateleira. Consequentemente, os agentes de tamponação são quase sempre utilizados para controlar o pH na formulação.
[065] Os ácidos orgânicos, fosfatos e Tris foram utilizados rotineiramente como tampões em formulações de proteína. A capacidade de tamponamento das espécies de tamponamento é máxima em um pH igual ao pKa e diminui conforme o pH aumenta ou diminui sempre a partir deste valor. Noventa porcento da capacidade de tamponamento existe dentro de uma unidade de pH do seu pKa. A capacidade de tamponamento também aumenta proporcionalmente com o aumento da concentração de tampão.
[066] Vários fatores precisam ser considerados quando da escolha de um tampão. Primeiro e o principal, a espécie de tampão e a sua concentração precisam ser definidos com base no seu pKa e a formulação de pH desejada. Igualmente importante é garantir que o tampão seja compatível com o polipeptídeo e outros excipientes de formulação, e não catalisam quaisquer reações de degradação. Um terceiro aspecto importante a ser considerado é a sensação de pungência e irritação que o tampão pode induzir na administração. Por exemplo, citrato é conhecido causar pungência na injeção (Laursen T, et al., Basic Clin Pharmacol Toxicol., 98(2): 218-21 (2006)). O potencial para pungência e irritação é maior para os medicamentos que são administrados por intermédio das vias subcutânea (SC) ou intramuscular (IM), onde a solução de medicamento permanece no sítio por um período relativamente mais longo de tempo do que quando administrado pela via IV onde a formulação é diluída rapidamente no sangue na administração. Para as formulações que são administradas pela infusão IV direta, a quantidade total de tampão (e qualquer outro componente de formulação) necessita ser monitorada. Deve-se ter particularmente cuidado a cerca dos íons potássio administrados na forma do tampão de fosfato de potássio, que pode induzir efeitos cardiovasculares em um paciente (Hollander-Rodriguez JC, et al., Am. Fain. Physician., 73(2): 283-90 (2006)).
[067] O sistema de tampão presente nas composições é selecionado para ser fisiologicamente compatível e para manter um pH desejado da formulação farmacêutica. Em uma forma de realização, o pH da solução está entre pH 2,0 e pH 12,0. Por exemplo, o pH da solução pode ser 2,0, 2,3, 2,5, 2,7, 3,0, 3,3, 3,5, 3,7, 4,0, 4,3, 4,5, 4,7, 5,0, 5,3, 5,5, 5,7, 6,0, 6,3, 6,5, 6,7, 7,0, 7,3, 7,5, 7,7, 8,0, 8,3, 8,5, 8,7, 9,0, 9,3, 9,5, 9,7, 10,0, 10,3, 10,5, 10,7, 11,0, 11,3, 11,5, 11,7, ou 12,0.
[068] O composto de tamponamento de pH pode estar presente em qualquer quantidade adequada para manter o pH da formulação em um nível predeterminado. Em uma forma de realização, a concentração do tamponamento de pH está entre 0,1 mM e 500 mM (1 M). Por exemplo, é considerado que o agente de tamponação de pH seja de pelo menos 0,1, 0,5, 0,7, 0,8 0,9, 1,0, 1,2, 1,5, 1,7, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 500 mM.
[069] Os agentes de tamponação de pH exemplares usados para tamponar a formulação como aqui apresentada incluem, mas não são limitados a glicina, histidina, glutamato, succinato, fosfato, acetato, citrato, Tris e aminoácidos ou misturas de aminoácidos, incluindo, mas não limitados a aspartato, histidina e glicina.
Sais
[070] Sais são frequentemente adicionados para aumentar a concentração iônica da formulação, que pode ser importante para a solubilidade, estabilidade física, e isotonicidade da proteína. Os sais podem afetar a estabilidade física de proteínas em uma variedade de modos. Íons podem estabilizar o estado nativo das proteínas pela ligação aos resíduos carregados na superfície da proteína. Alternativamente, os sais podem estabilizar o estado desnaturado pela ligação aos grupos de peptídeo ao longo da cadeia principal de proteína (-CONH-). Os sais também podem estabilizar a conformação da proteína nativa pela proteção das interações eletrostáticas repulsivas entre resíduos dentro de uma molécula de proteína. Os sais nas formulações de proteína também podem proteger das interações eletrostáticas atrativas entre as moléculas de proteína que podem levar à agregação e insolubilidade da proteína. Nas formulações fornecidas, a concentração de sal está entre 0,1, 1, 10, 20, 30, 40, 50, 80, 100, 120, 150, 200, 300, e 500 mM.
Estabilizadores e agentes de volume
[071] Nas presentes formulações farmacêuticas, um estabilizador (ou uma combinação de estabilizadores) pode ser adicionado para prevenir ou reduzir a agregação e degradação química induzidas pela armazenagem. Uma solução nebulosa ou turva na reconstituição indica que a proteína precipitou ou pelo menos agregou. O termo “estabilizador” significa um excipiente capaz de impedir a agregação ou outra degradação física, assim como a degradação química (por exemplo, autólise, desamidação, oxidação, etc.) em um estado aquoso. Os estabilizadores que são convencionalmente utilizados nas composições farmacêuticas incluem, mas não são limitados à, sacarose, trealose, manose, maltose, lactose, glicose, rafinose, celobiose, gentiobiose, isomaltose, arabinose, glicosamina, frutose, manitol, sorbitol, glicina, arginina HCl, compostos de poli-hidróxi, incluindo polissacarídeos tais como dextrano, amido, hidroxietil amido, ciclodextrinas, N-metil pirolideno, celulose e ácido hialurônico, cloreto de sódio, [Carpenter et al., Develop. Biol. Standard 74: 225, (1991)]. Nas presentes formulações, o estabilizante é incorporado em uma concentração de cerca de 0,1, 0,5, 0,7, 0,8 0,9, 1,0, 1,2, 1,5, 1,7, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 500, 700, 900, ou 1000 mM.
[072] Se desejado, as formulações também incluem quantidades apropriadas de agentes reguladores do volume e osmolaridade. Os agentes de volume incluem, por exemplo, manitol, glicina, sacarose, polímeros tais como dextrano, polivinilpirrolidona, carboximetil celulose, lactose, sorbitol, trealose ou xilitol. Em uma forma de realização, o agente de volume é manitol. O agente de volume é incorporado em uma concentração de cerca de 0,1, 0,5, 0,7, 0,8 0,9, 1,0, 1,2, 1,5, 1,7, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 500, 700, 900, ou 1000 mM.
Tensoativos
[073] As moléculas de proteína têm uma alta propensão para interagir com superfícies que as tornam suscetíveis à absorção e desnaturação nas interfaces ar-líquido, frasco-líquido e líquido-líquido (óleo de silicona). Este caminho de degradação foi observado ser inversamente dependente da concentração de proteína e resulta na formação de agregados de proteína solúveis e insolúveis ou a perda de proteína da solução via absorção pelas superfícies. Além da absorção pela superfície do recipiente, a degradação induzida pela superfície é exacerbada com a agitação física, como seria experienciado durante a remessa e manuseio do produto.
[074] Tensoativos são habitualmente usados apenas em formulações de proteína para prevenir a degradação induzida pela superfície. Os tensoativos são moléculas anfipáticas com a capacidade de competir com as proteínas quanto as posições interfaciais. As porções hidrofóbicas das moléculas de tensoativo ocupam posições interfaciais (por exemplo, ar/líquido), enquanto que as porções hidrofílicas das moléculas permanecem orientadas contra o solvente volumoso. Em concentrações suficientes (tipicamente em torno da concentração micelar crítica do detergente), uma camada de superfície de moléculas de tensoativo serve para impedir as moléculas de proteína de absorver na interface. Com isso, a degradação induzida pela superfície é minimizada. Os tensoativos mais habitualmente usados são ésteres de ácido graxo de polietoxilatos de sorbitano, isto é, polissorbato 20 e polissorbato 80. Os dois diferem apenas no comprimento da cadeia alifática que comunica caráter hidrofóbico às moléculas, C-12 e C-18, respectivamente. Consequentemente, polissorbato-80 é mais ativo na superfície e tem uma concentração micelar crítica mais baixa do que o polissorbato-20.
[075] Detergentes também podem afetar a estabilidade conformacional termodinâmica de proteínas. Aqui mais uma vez, os efeitos de um dado excipiente detergente serão específicos da proteína. Por exemplo, polissorbatos foram mostrados reduzir a estabilidade de algumas proteínas e aumentar a estabilidade de outras. A desestabilização pelos detergentes de proteínas pode ser racionalizada em termos das caudas hidrofóbicas das moléculas de detergente que podem engrenar na ligação específica com estados de proteína parcial ou totalmente desdobrados. Estes tipos de interações podem causar uma mudança no equilíbrio conformacional contra os estados de proteína mais expandidos (isto é aumentar a exposição de porções hidrofóbicas da molécula de proteína em complemento à ligação de polissorbato). Alternativamente, se o estado nativo da proteína exibe alguma superfície hidrofóbica, a ligação de detergente ao estado nativo pode estabilizar aquela conformação.
[076] Um outro aspecto de polissorbatos é que eles são inerentemente susceptíveis à degradação oxidativa. Frequentemente, como matérias primas, eles contêm quantidades suficientes de peróxidos para causar a oxidação das cadeias laterais de resíduo de proteína, especialmente metionina. O potencial para o dano oxidativo que surge da adição de estabilizador enfatiza o ponto que as concentrações eficazes mais baixas de excipientes devam ser usadas em formulações. Para os tensoativos, a concentração eficaz para uma dada proteína dependerá do mecanismo de estabilização. Foi postulado que se o mecanismo de estabilização de tensoativo está relacionado com a prevenção da desnaturação da superfície a concentração eficaz estará em torno da concentração micelar crítica do detergente. Ao contrário, se o mecanismo de estabilização está associado com interações de proteína-detergente específicas, a concentração de tensoativo eficaz estará relacionada com a concentração de proteína e a estequiometria da interação (Randolph T. W., et al., Pharm Biotechnol., 13: 159-75 (2002)).
[077] Os tensoativos também podem ser adicionados em quantidades apropriadas para prevenir o fenômeno de agregação relacionada com a superfície durante o congelamento e secagem [Chang, B, J. Pharm. Sci. 85: 1325, (1996)]. Os tensoativos exemplares incluem tensoativos aniônicos, catiônicos, não iônicos, zwitteriônicos, e anfotéricos incluindo tensoativos derivados dos aminoácidos que ocorre naturalmente. Os tensoativos aniônicos incluem, mas não são limitados a lauril sulfato de sódio, dioctil sulfossuccinato de sódio e dioctil sulfonato de sódio, ácido quenodesoxicólico, sal sódico de N- lauroilsarcosina, dodecil sulfato de lítio, sal sódico do ácido 1-octanossulfônico, colato de sódio hidratado, desoxicolato de sódio, e sal sódico do ácido glicodesoxicólico. Os tensoativos catiônicos incluem, mas não são limitados a cloreto de benzalcônio ou cloreto de benzetônio, cloreto de cetilpiridínio monoidratado, e brometo de hexadeciltrimetilamônio. Os tensoativos zwitteriônicos incluem, mas não são limitados a CHAPS, CHAPSO, SB3-10, e SB3-12. Os tensoativos não iônicos incluem, mas não são limitados a digitonina, Triton X-100, Triton X-114, TWEEN-20, e TWEEN-80. Os tensoativos também incluem, mas não são limitados a lauromacrogol 400, estearato de polioxil 40, óleo de mamona polioxietileno hidrogenado 10, 40, 50 e 60, monoestearato de glicerol, polissorbato 40, 60, 65 e 80, lecitina de soja e outros fosfolipídeos tais como dioleil fosfatidil colina (DOPC), dimiristoilfosfatidil glicerol (DMPG), dimiristoilfosfatidil colina (DMPC), e (dioleil fosfatidil glicerol) DOPG; éster do ácido graxo de sacarose, metil celulose e carboximetil celulose. As composições que compreendem estes tensoativos, individualmente ou como uma mistura em razões diferentes, são, portanto, ainda fornecidas. Nas presentes formulações, o tensoativo é incorporado em uma concentração de cerca de 0,01 a cerca de 0,5 g/l.
Outros componentes excipientes comuns Aminoácidos
[078] Os aminoácidos têm encontrado uso versátil nas formulações de proteína como tampões, agentes de volume, estabilizantes e antioxidantes. A histidina e o ácido glutâmico são utilizados para tamponar formulações de proteína na faixa de pH de 5,5 a 6,5 e 4,0 a 5,5 respectivamente. O grupo imidazol da histidina tem um pKa = 6,0 e o grupo carboxila de cadeia lateral de ácido glutâmico tem um pKa de 4,3 que torna estes aminoácidos adequados para o tamponamento nas suas respectivas faixas de pH. O ácido glutâmico é particularmente útil em tais casos (por exemplo, Stemgen®). A histidina é habitualmente encontrada em formulações de proteína comercializadas (por exemplo, Xolar®, Herceptin®, Recombinate®),e este aminoácido fornece uma alternativa ao citrato, um tampão conhecido ser pungente na injeção. De maneira interessante, a histidina também foi relatada ter um efeito estabilizante, como observado nas formulações com ABX-IL8 (um anticorpo de IgG2), com respeito à agregação quando usado em concentrações altas nas apresentações tanto líquidas quanto liofilizadas (Chen B, et al., Pharm Res., 20(12): 1952-60 (2003)). A histidina (até 60 mM) também foi observada reduzir a viscosidade de uma formulação de alta concentração deste anticorpo. Entretanto, no mesmo estudo, os autores observaram agregação aumentada e descoloração nas formulações contendo histidina durante estudos de congelamento-descongelamento do anticorpo em recipientes de aço inoxidável. Os autores atribuíram isto a um efeito de íons ferro lixiviados da corrosão de recipientes de aço. Um outro aviso de prudência com a histidina é que a mesma sofre foto-oxidação na presença de íons metálicos (Tomita M, et al., Biochemistry, 8(12): 5149-60 (1969)). O uso de metionina como um antioxidante em formulações parece promissor; a mesma foi observada ser eficaz contra vários estresses oxidativos (Lam XM, et al., J Pharm Sci., 86(11): 1250-5 (1997)).
[079] Os aminoácidos glicina, prolina, serina e alanina foram mostrados estabilizar proteínas pelo mecanismo de exclusão preferencial. A glicina também é um agente de volume habitualmente usado em formulações liofilizadas (por exemplo, Neumega ®, Genotropin®, Humatrope®). A arginina foi mostrada ser um agente eficaz na inibição da agregação e foi usada em formulações tanto líquidas quanto liofilizadas (por exemplo, Activase®, Avonex®, Enbrel® líquidos). Além disso, a eficiência realçada de redobra de certas proteínas na presença de arginina foi atribuída à sua supressão da reação de agregação competitiva durante a redobra.
Antioxidantes
[080] A oxidação de resíduos de proteína surge de várias fontes diferentes. Além da adição de antioxidantes específicos, a prevenção de dano oxidativo à proteína envolve o controle cuidadoso de vários fatores por todo o processo de fabricação e armazenagem do produto tal como oxigênio atmosférico, temperatura, exposição á luz, e contaminação química. Os antioxidantes farmacêuticos mais habitualmente usados são agentes redutores, descontaminantes de oxigênio/radical livre, ou agentes queladores. Os antioxidantes nas formulações terapêuticas de proteína são solúveis em água e permanecem ativos por toda a vida de prateleira do produto. Os agentes redutores e descontaminantes de oxigênio/ radical livre funcionam pela remoção de espécies de oxigênio ativo em solução. Os agentes de quelação tais como EDTA são eficazes pela ligação de contaminantes de metal traço que promovem a formação de radical livre. Por exemplo, EDTA foi utilizado na formulação líquida de fator de crescimento de fibroblasto ácido para inibir a oxidação catalisada por íon metálico de resíduos de cisteína. EDTA foi usado em produtos comercializados como Kineret® e Ontak®.
[081] Além da eficácia de vários excipientes para prevenir a oxidação da proteína, o potencial dos próprios antioxidantes para induzir outras mudanças covalentes ou físicas para a proteína é de interesse. Por exemplo, os agentes redutores podem causar o rompimento das ligações intramoleculares de dissulfeto, que pode levar ao embaralhamento de dissulfeto. Na presença de íons metálicos de transição, ácido ascórbico e EDTA foram mostrados promover a oxidação da metionina em várias proteínas e peptídeos (Akers MJ, e Defelippis MR. Peptides and Proteins as Parenteral Solutions. Em: Pharmaceutic Formulation Development of Peptides and Proteins. Sven Frokjaer, Lars Hovgaard, editores. Pharmaceutic Science. Taylor and Francis, UK (1999)); Fransson J. R., J. Pharm. Sci. 86(9): 4046-1050 (1997); Yin J, et al., Pharm Res., 21(12): 2377-83 (2004)). Tiossulfato de sódio foi relatado reduzir os níveis de oxidação da metionina induzidos por luz e temperatura em rhuMab HER2; entretanto, a formação de um aduto de tiossulfato-proteína foi também relatada neste estudo (Lam XM, Yang JY, et al., J Pharm Sci. 86(11): 1250-5 (1997)). A seleção de um antioxidante apropriado é feita de acordo com os estresses e sensibilidades específicos da proteína.
Íons metálicos
[082] No geral, íons metálicos de transição são indesejados nas formulações de proteína porque eles podem catalisar reações de degradação física e química em proteínas. Entretanto, íons metálicos específicos são incluídos nas formulações quando eles são cofatores para proteínas e em formulações em suspensão de proteínas onde eles formam complexos de coordenação (por exemplo, suspensão em zinco de insulina). Recentemente, o uso de íons de magnésio (10 a 120 mM) foi proposto inibir a isomerização de ácido aspártico para ácido isoaspártico (WO 2004039337).
[083] Dois exemplos onde íons metálicos conferem estabilidade ou atividade aumentada em proteínas são desoxirribonuclease humana (rhDNase, Pulmozyme®), e Fator VIII. No caso de rhDNase, íons Ca+2 (até 100 mM) aumentaram a estabilidade da enzima através de um sítio de ligação específico (Chen B, et al., J Pharm Sci., 88(4): 477-82 (1999)). De fato, a remoção de íons cálcio da solução com EGTA causou um aumento na desamidação e agregação. Entretanto, este efeito foi observado apenas com íons Ca+2; outros cátions bivalentes Mg+2, Mn+2 e Zn+2 foram observados desestabilizar a rhDNase. Efeitos similares foram observados no Fator VIII. Os íons Ca+2 e Sr+2 estabilizaram a proteína enquanto outros como Mg+2, Mn+2 e Zn+2, Cu+2 e Fe+2 desestabilizaram a enzima (Fatouros, A., et al., Mt. J. Pharm., 155, 121-131 (1997). Em um estudo separado com Fator VIII, um aumento significante na taxa de agregação foi observado na presença de íons Al+3 (Derrick TS, et al., J. Pharm. Sci., 93(10): 2549-57 (2004)). Os autores mencionam que outros excipientes como os sais de tampão são frequentemente contaminados com íons Al+3 e ilustram a necessidade para usar excipientes de qualidade apropriada em produtos formulados.
Conservantes
[084] Conservantes são necessários quando do desenvolvimento de formulações parenterais de uso múltiplo que envolvem mais do que uma extração do mesmo recipiente. A sua função primária é inibir o crescimento microbiano e garantir a esterilidade do produto por toda a vida de prateleira ou período de uso do produto medicamentoso. Os conservantes habitualmente usados incluem álcool benzílico, fenol e m-cresol. Embora os conservantes tenham uma longa história de uso, o desenvolvimento de formulações de proteína que incluem conservantes pode ser desafiador. Os conservantes quase sempre têm um efeito desestabilizante (agregação) sobre as proteínas, e isto tem se tornado um fator principal na limitação do seu uso em formulações de proteína de dose múltipla (Roy S, et al., J Pharm Sci., 94(2): 382-96 (2005)).
[085] Até agora, a maioria dos medicamentos de proteína tem sido formulada apenas para uso único. Entretanto, quando formulações de dose múltipla são possíveis, elas têm a vantagem adicional de permitir a conveniência do paciente, e comerciabilidade aumentada. Um bom exemplo é aquele do hormônio do crescimento humano (hGH) onde o desenvolvimento de formulações preservadas tem levado à comercialização de apresentações de caneta de injeção mais conveniente, de uso múltiplo. Pelo menos quatro de tais dispositivos de caneta contendo formulações preservadas de hGH estão correntemente disponíveis no mercado. Norditropin® (líquido, Novo Nordisk), Nutropin AQ® (líquido, Genentech) & Genotropin (liofilizado - cartucho de câmara dupla, Pharmacia & Upjohn) contêm fenol enquanto Somatrope® (Eli Lilly) é formulado com m-cresol.
[086] Diversos aspectos precisam ser considerados durante o desenvolvimento da formulação de formas de dosagem preservadas. A concentração de conservante eficaz no produto medicamentoso deve ser otimizada. Isto requer testar um dado conservante na forma de dosagem com faixas de concentração que conferem eficácia antimicrobiana sem comprometer a estabilidade da proteína. Por exemplo, três conservantes foram triados com êxito no desenvolvimento de uma formulação líquida para o receptor de interleucina-1 (Tipo I), usando a calorimetria por varredura diferencial (DSC). Os conservantes foram ordenados em série com base no seu impacto sobre a estabilidade nas concentrações habitualmente usadas em produtos comercializados (Remmele RL Jr., et al., Pharm Res., 15(2): 200-8 (1998)).
[087] Alguns conservantes podem causar reações no local da injeção, que é um outro fator que precisa consideração quando da escolha de um conservante. Em testes clínicos que focalizam sobre a avaliação de conservantes e tampões em Norditropina, a percepção de dor foi observada ser mais baixa em formulações contendo fenol e álcool benzílico quando comparadas com uma formulação contendo m-cresol (Kappelgaard A. M., Harm Res. 62 Supl 3: 98-103 (2004)). De maneira interessante, entre os conservantes habitualmente usados, o álcool benzílico possui propriedades anestésicas (Minogue SC, e Sun DA., Anesth Analg., 100(3): 683-6 (2005)).
Liofilização
[088] Também é considerado que as formulações que compreendem um polipeptídeo VWF da invenção podem ser liofilizadas antes da administração. A liofilização é realizada usando técnicas comuns na técnica e devem ser otimizados para a composição que é desenvolvida [Tang et al., Pharm Res. 21: 191-200, (2004) e Chang et al., Pharm Res. 13: 243-9 (1996)].
[089] Um ciclo de liofilização é, em um aspecto, composto de três etapas: congelamento, secagem primária, e secagem secundária [A. P. Mackenzie, Phil Trans R Soc Londres, Ser B, Biol 278: 167 (1977)]. Na etapa de congelamento, a solução é esfriada para iniciar a formação de gelo. Além disso, esta etapa induz a cristalização do agente de volume. O gelo sublima no estágio de secagem primária, que é conduzido pela redução da pressão da câmara abaixo da pressão de vapor do gelo, usando um vácuo e introduzindo calor para promover a sublimação. Finalmente, a água absorvida ou ligada é removida no estágio de secagem secundário sob a pressão da câmara reduzida e em uma temperatura de prateleira elevada. O processo produz um material conhecido como uma torta liofilizada. Depois disso a torta pode ser reconstituída com água estéril ou diluente adequado para injeção.
[090] O ciclo liofilização não apenas determina o estado físico final dos excipientes, mas também afeta outros parâmetros tais como tempo de reconstituição, aparência, estabilidade e teor de umidade final. A estrutura da composição no estado congelado processa-se através de diversas transições (por exemplo, transições vítreas, umectações e cristalizações) que ocorrem em temperaturas específicas e podem ser usadas para entender e otimizar o processo de liofilização. A temperatura de transição vítrea (Tg e/ou Tg’) pode fornecer informação a cerca do estado físico de um soluto e pode ser determinada pela calorimetria por varredura diferencial (DSC). A Tg e Tg’ são um parâmetro importante que deve ser levado em consideração quando do planejamento do ciclo de liofilização. Por exemplo, a Tg’ é importante para a secagem primária. Além disso, no estado seco, a temperatura de transição vítrea fornece informação sobre a temperatura de armazenagem do produto final.
Métodos de Preparação
[091] A presente invenção considera ainda métodos para a preparação de formulações farmacêuticas. Uma variedade de carregadores aquosos, por exemplo, água estéril para injeção, água com conservantes para uso em dose múltipla, ou água com quantidades apropriadas de tensoativos (por exemplo, polissorbato-20), 0,4 % de solução salina, 0,3 % de glicina, ou suspensões aquosas podem conter o composto ativo em mistura com excipientes adequados para a fabricação de suspensões aquosas. Em vários aspectos, tais excipientes são agentes de suspensão, por exemplo carboximetilcelulose sódica, metilcelulose, hidroxipropilmetilcelulose, alginato de sódio, polivinilpirrolidona, goma de tragacanto e goma acácia; agentes de dispersão ou umectantes podem ser um fosfatídeo que ocorre naturalmente, por exemplo lecitina, ou produtos de condensação de um óxido de alquileno com ácidos graxos, por exemplo estearato de polioxietileno, ou produtos de condensação de óxido de etileno com álcoois alifáticos de cadeia longa, por exemplo heptadecaetil-eneoxicetanol, ou produtos de condensação de óxido de etileno com ésteres parciais derivados de ácidos graxos e um hexitol tal como monooleato de polioxietileno sorbitol, ou produtos de condensação de óxido de etileno com ésteres parciais derivados de ácidos graxos e anidridos de hexitol, por exemplo monooleato de polietileno sorbitano. As suspensões aquosas também podem conter um ou mais conservantes, por exemplo p- hidroxibenzoato de etila, ou n-propila.
Administração
[092] Para administrar composições aos seres humanos ou animais de teste, em um aspecto, as composições compreendem um ou mais carregadores farmaceuticamente aceitáveis. As frases “farmaceuticamente” ou “farmacologicamente” aceitáveis referem-se às entidades moleculares e composições que são estáveis, inibem a degradação da proteína tal como produtos de agregação e clivagem, e, além disso, não produzem reações alérgicas, ou outras reações adversas quando administradas usando vias bem conhecidas na técnica, como descrito abaixo. “Carregadores farmaceuticamente aceitáveis” incluem quaisquer e todos os solventes, meios de dispersão, revestimento, agentes antibacterianos e antifúngicos, agentes isotônicos e retardadores de absorção clinicamente úteis e outros, incluindo aqueles agentes divulgados acima.
[093] As formulações farmacêuticas podem ser administradas oral, tópica, transdérmica, parenteralmente, pela pulverização de inalação, vaginal, retalmente, ou pela injeção intracraniana. O termo parenteral como aqui usado inclui injeções subcutâneas, intravenosas, intramusculares, injeção intracistérnica, ou técnicas de infusão. A administração pela injeção intravenosa, intradérmica, intramuscular, intramamária, intraperitoneal, intratecal, retrobulbar, intrapulmonar e ou implantação cirúrgica em um sítio particular também é considerada. No geral, as composições são essencialmente isentas de pirógenos, assim como outras impurezas que poderiam ser nocivas para o receptor.
[094] Administrações únicas ou múltiplas das composições podem ser realizadas com os níveis de dose e padrão sendo selecionado pelo médico tratante. Para a prevenção ou tratamento de doença, a dosagem apropriada dependerá do tipo de doença a ser tratada, como definido acima, a severidade e curso da doença, seja o medicamento administrado com propósitos preventivos ou terapêuticos, terapia prévia, a história clínica do paciente e resposta ao medicamento, e a discrição do médico atendente.
Kits
[095] Como um aspecto adicional, a invenção inclui kits que compreendem uma ou mais formulações farmacêuticas embaladas em uma maneira que facilite o seu uso para a administração aos pacientes. Em uma forma de realização, um tal kit inclui a formulação farmacêutica aqui descrita (por exemplo, uma composição que compreenda uma proteína ou peptídeo terapêuticos), embalados em um recipiente tal como uma garrafa ou frasco selados, com um rótulo afixado ao recipiente ou incluído na embalagem que descreva o uso do composto ou composição na prática do método. Em uma forma de realização, a formulação farmacêutica é embalada no recipiente tal que a quantidade de topo livre no recipiente (por exemplo, a quantidade de ar entre a formulação líquida e o topo do recipiente) seja muito pequena. Preferivelmente, a quantidade de topo livre é negligenciável (isto é, quase nenhuma). Em uma forma de realização, o kit contém um primeiro recipiente tendo uma composição terapêutica de proteína ou peptídeo e um segundo recipiente tendo uma solução de reconstituição fisiologicamente aceitável para a composição. Em um aspecto, a formulação farmacêutica é embalada em uma forma de dosagem unitária. O kit pode incluir ainda um dispositivo adequado para administrar a formulação farmacêutica de acordo com uma via específica de administração. Preferivelmente, o kit contém um rótulo que descreve o uso das formulações farmacêuticas.
Dosagens
[096] O regime de dosagem envolvido em um método para tratar uma condição aqui descrita será determinado pelo médico atendente, considerando vários fatores que modificam a ação de medicamentos, por exemplo a idade, condição, peso corporal, sexo e dieta do paciente, a severidade de qualquer infecção, tempo de administração e outros fatores clínicos. Por via de exemplo, uma dose típica de um VWF recombinante da presente invenção é de aproximadamente 50 U/kg, igual a 500 μg/kg.
[097] As formulações da invenção podem ser administradas por um bolo inicial seguido por uma infusão contínua para manter níveis circulantes terapêuticos de produto medicamentoso. Como um outro exemplo, o composto inventivo pode ser administrado como uma dose de uma vez. Aqueles de habilidade comum na técnica facilmente otimizarão dosagens e regimes de administração eficazes como determinado pela boa prática médica e a condição clínica do paciente individual. A frequência de dosagem dependerá dos parâmetros farmacocinéticos dos agentes e da via de administração. A formulação farmacêutica ótima será determinada por uma pessoa habilitada na técnica dependendo da via de administração e da dosagem desejada. Ver por exemplo, Remington’s Pharmaceutical Sciences, 18a Ed. (1990, Mack Publishing Co., Easton, PA 18042) páginas 1435 a 1712, a divulgação da qual é por meio deste incorporada por referência. Tais formulações podem influenciar o estado físico, estabilidade, taxa de liberação in vivo, e taxa de depuração in vivo dos agentes administrados. Dependendo da via de administração, uma dose adequada pode ser calculada de acordo com o peso corporal, área de superfície corporal ou tamanho do órgão. As dosagens apropriadas podem ser averiguadas através do uso de ensaios estabelecidos para determinar as dosagens de nível sanguíneo em conjunção com dados de dose-resposta apropriados. O regime de dosagem final será determinado pelo médico atendente, considerando vários fatores que modificam a ação dos medicamentos, por exemplo a atividade específica do medicamento, a severidade do dano e a responsividade do paciente, a idade, condição, peso corporal, sexo e dieta do paciente, a severidade de qualquer infecção, tempo de administração e outros fatores clínicos. Conforme estudos são conduzidos, outras informações emergirão com respeito aos níveis de dosagem apropriados e duração de tratamento para várias doenças e condições.
[098] Os seguintes exemplos não são intencionados a serem limitantes, mas apenas exemplares das formas de realização específicas da invenção.
Exemplo 1
Experimentos de agitação
[099] De modo a determinar a quantidade de precipitação de rVWF em várias formulações, a recuperação percentual de rVWF a seguir da agitação turbulenta foi testada sob uma variedade de condições.
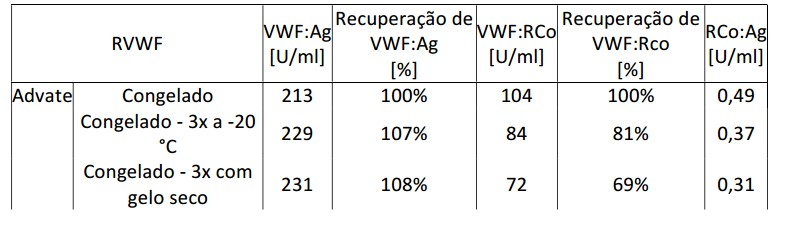
[0100] rVWF em tampão Advate (90 mM de NaCl, 1,68 mM de CaCl2, 10 mM de L-histidina, 10 mM de tris, 0,26 mM de glutationa, 23,4 mM de trealose, 175,7 mM de manitol, e 0,1 g/L de TWEEN-80, pH 7,0) ou tampão Advate 1:3 (tampão Advate diluído 3 vezes em água) foi submetido à agitação turbulenta em um agitador na temperatura ambiente (RT) por 0 min, 1 min, 2,5 horas, ou 4 dias, e a recuperação percentual do rVWF foi medida em relação ao material de partida antes de agitar. Como mostrado na Tabela 1, perdas de cerca de 40 a 80% foram observadas no tampão Advate enquanto perdas de cerca de 20 a 30% foram observadas no tampão Advate 1:3. O antígeno de VWF VWF:Ag corresponde à quantidade de VWF que pode ser detectada em um ELISA específico de VWF usando anticorpo policlonal anti-VWF, enquanto que VWF:RCo corresponde à quantidade de VWF que causa aglutinação de plaquetas estabilizadas na presença de ristocetina. Em ambos os casos plasma de referência humano calibrado contra o padrão de WHO real foi usado como padrão (1 ml de plasma de referência usualmente contém 1U de VWF). Tabela 1. Influência do tempo de agitação turbulenta sobre a recuperação de rVWF

[0101] O efeito de congelamento/descongelamento e liofilização foi também testado nos experimentos de agitação. O congelamento foi realizado a -20°C em um ambiente frio a -20°C ou sobre gelo seco, o descongelamento em ambos os casos até a temperatura ambiente e ambos iniciados a partir das formulações líquidas. Como para a liofilização, as amostras de VWF formuladas aqui descritas foram congeladas dentro de um liofilizador em escala piloto a <-40°C e foram liofilizadas usando um programa lyo padrão. A agitação foi realizada diretamente com as formulações líquidas (2 ml em frascos de 5 ml). Como mostrado na Tabela 2, a recuperação percentual de rVWF foi mais alta em tampão Advate 1:3 comparada com tampão Advate. Tabela 2.
[0102] A recuperação percentual também foi medida nos experimentos de agitação com rVWF sendo armazenado em seringas com topo livre e sem topo livre. De maneira interessante, quando rVWF é armazenado em seringas sem topo livre e agitado como descrito acima, nenhuma precipitação de rVWF foi observada. Ao contrário, quando rVWF é armazenado em seringas com topo livre, alguma precipitação foi observada.
[0103] Em resumo, a agitação turbulenta resultou em pelo menos 30% de perda de rVWF em tampão Advate ou tampão Advate 1:3, com tampão Advate mostrando perdas mais altas de recuperação comparado ao tampão Advate 1:3. De maneira interessante, os mesmos precipitados observados nos experimentos de agitação turbulenta não foram observados quando rVWF foi armazenado e transportado ~5000 km em um automóvel (representando a agitação esperada durante o transporte). A precipitação de rVWF pôde ser eliminada pela armazenagem em seringas sem topo livre.
Exemplo 2
Estabilidade de VWF recombinante
[0104] A estabilidade de rVWF foi testada avaliando-se o nível de atividade de rVWF presente em várias formulações.
[0105] Como mostrado na Figura 1, rVWF não é estável em tampão Advate depois de 26 semanas devido à presença de 0,3 mM de glutationa. Como mostrado na Figura 2, entretanto, rVWF é mais estável em tampão Advate 1:3 (por exemplo, por até 12 semanas a 4°C).
[0106] Como mostrado na Figura 3, a estabilidade de uma formulação com base em citrato (15 mM de citrato de sódio, 10 mM de CaCl2, 100 mM de NaCl, pH 7,0) é melhor do que a formulação de tampão Advate 1:3 contendo 0,1 M de glutationa.
[0107] Do mesmo modo, a concentração de rVWF foi medida com o tempo em vários tampões. Como mostrado na Figura 4, Figura 5 e Figura 6, a concentração de rVWF é estável com o tempo em tampão Advate, tampão Advate 1:3, e tampão com base em citrato, respectivamente.
Exemplo 4
Caracterização das formulações líquidas
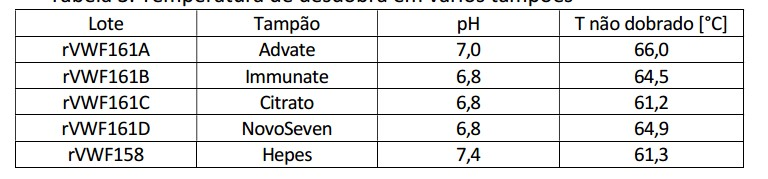
[0108] A calorimetria por varredura diferencial (DSC) foi usada para avaliar o grau de desdobra de proteína (rVWF) em vários tampões. Como mostrado na Tabela 3, tampão Advate pH 7,0 é o ideal para a estabilização.
[0109] A DSC é uma técnica termoanalítica em que as diferenças na quantidade de calor requerida para aumentar a temperatura de uma amostra e referências são medidas como uma função da temperatura. O resultado de um experimento de DSC é uma curva de fluxo de calor versus temperatura ou versus tempo.
[0110] O Calorímetro por Varredura Diferencial pode varrer através de uma faixa de temperaturas enquanto aquece e esfria e o mesmo determina uma transição de fase, isto é a fusão, cristalização, ou transição vítrea, medindo-se a quantidade de calor necessário para atingir uma temperatura ajustada. O calorímetro foi calibrado com um conjunto de metais puros (zinco, índio e estanho) que têm uma capacidade calorífica conhecida, Cp e ponto de fusão, Tm. O respectivo tampão de referência foi colocado no capilar de referência e a amostra de rVWF foi colocada no capilar de amostra do instrumento. Tabela 3. Temperatura de desdobra em vários tampões
[0111] Tampão componentes e concentrações: A) Advate: 5,26 g/l de NaCl 0,248 g/l de CaCl2 32 g/l de D-manitol 8 g/1 de Trealose 1,56 g/l de Histidina 1,2 g/l de Tris 0,08 g/l de vermelho de glutadiona, pH = 7,0 B) Immunate: 5,25 g/l de Glicina 2,2 g/l de NaCl 5,25 g/l de NaCit3 5,25 g/l de Lisina-HCl 0,62 g/l de CaCl2 pH = 6,8 C) Citrato: 3 g/l de Glicina 2,92 g/l de NaCl 2,5 g/l de NaCit3 30 g/l de D-Manitol 10 g/l de Trealose pH = 6,8 D) Novo Seven: 0,75 g/l de Glicina 2,92 g/l de NaCl 1,47 g/l de CaCl2 30 g/l de D-Manitol: pH = 6,8 rVWF158: 20 mM de Hepes, 150 mM de NaCl, 5 g/l de sacarose, pH 7,4
[0112] Além disso, como mostrado na Figura 7, a maior parte dos excipientes de formulação aumentam a temperatura de desdobra em cerca de 1 a 2°C. A Figura 8 mostra que 10 mM de CaCl2 aumenta a temperatura de desdobra em -8°C a -67°C, uma temperatura de desdobra que também pode ser atingida pelo tampão Advate. Este efeito do CaCl2 é similar no pH 7,3 e 6,5, como mostrado na Figura 9. Finalmente, o efeito da trealose e sacarose foram analisados sobre a temperatura de desdobra. Comparado apenas ao citrato, nem a trealose nem a sacarose aumentaram a temperatura de desdobra de rVWF. Um resumo dos dados da temperatura de desdobra (Tmax) para rVWF na presença de vários excipientes é apresentada na Tabela 4. Tabela 4.
[0113] Além dos vários tampões, a DSC foi usada para avaliar a temperatura de desdobra de rVWF em vários valores de pH em tampão Advate. Os resultados são mostrados na Tabela 5, abaixo. O tampão Advate pH 7,0 é o ideal para a estabilização (isto é, temperatura de desdobra mais alta; Pico 1) de rVWF. Tabela 5.
[0114] O espectro de fluorescência de rVWF em tampão Advate e tampão Advate 1:3 foi avaliado depois da armazenagem em várias temperaturas para várias dimensões de tempo. Nenhuma (ou apenas levemente) mudança no espectro de fluorescência foi observado depois da armazenagem a 40°C de 0 a 28 dias em tampões Advate ou Advate 1:3. Nenhuma diferença foi observada em outras temperaturas.
[0115] Do mesmo modo, a degradação de rVWF foi avaliada usando filtração em gel (Superose 6). Embora alguma degradação fosse observada depois de 26 semanas a 4°C em tampão Advate, quase nenhuma degradação de rVWF em tampão Advate 1:3 foi observada depois de 26 semanas a 4°C. A 40°C, glutationa aumentou a quantidade de degradação com o tempo (embora em um grau mais lento em tampão Advate 1:3).
[0116] Com base nos Exemplos acima, o tampão Advate 1:3 oferece uma vantagem com respeito ao congelamento/descongelamento e recuperação depois da liofilização quando comparado com o tampão Advate não diluído. Além disso, o tampão Advate 1:3 pode estabilizar (por exemplo, manter a atividade biológica) a atividade de rVWF durante a incubação a 40°C melhor que o tampão Advate. rVWF em tampão Advate 1:3 é estável por 4 semanas de incubação a 4°C. Finalmente, a DSC tem demonstrado que p pH 7,0 é ideal para prevenir a degradação de rVWF (isto é, mostrou a temperatura de desdobra mais alta).
[0117] Assim, em vista dos dados aqui apresentados, uma formulação foi proposta para rVWF incluindo 15 mM de citrato (ou glicina ou histidina), 10 mM de CaCl2, pH 6,5 a 7,3, ajustada para a osmolaridade desejada pelo NaCl. Por exemplo, em uma forma de realização, a fórmula com base em citrato é 15 mM de citrato de sódio, 10 mM de CaCl2, 100 mM de NaCl, pH 7,0.
[0118] Alternativamente, um tampão Advate ou Advate 1:3, sem glutationa, também são considerados: Advate: 90 mM de NaCl, 1,68 mM de CaCl2, 10 mM de L-histidina, 10 mM de Tris, 0,26 mM de glutationa, 23,4 mM de trealose, 175,7 mM de manitol, e 0,1 g/L de TWEEN-80, pH 7,0; Advate 1:3: 30 mM de NaCl, 0,56 mM de CaCl2, 3,3 mM de L-histidina, 3,3 mM de tris, 7,8 mM de trealose, 58,6 mM de manitol, e 0,03 g/L de TWEEN-80, pH 7,0.