BRPI0621886B1 - Composição de vacina contra influenza monovalente, kit, método para a produção de uma composição de vacina contra influenza para uma situação pandêmica ou uma situação pré- pandêmica, e, usos de um antígeno de vírus da influenza ou preparação antigênica do mesmo e de um adjuvante em emulsão óleo-em-água, de um antígeno de vírus da influenza pandêmica ou preparação antigênica do antígeno de hemaglutinina do vírus da influenza e de um adjuvante em emulsão óleo-em-água e de um antígeno ou preparação antigênica de uma primeira cepa de influenza - Google Patents
Composição de vacina contra influenza monovalente, kit, método para a produção de uma composição de vacina contra influenza para uma situação pandêmica ou uma situação pré- pandêmica, e, usos de um antígeno de vírus da influenza ou preparação antigênica do mesmo e de um adjuvante em emulsão óleo-em-água, de um antígeno de vírus da influenza pandêmica ou preparação antigênica do antígeno de hemaglutinina do vírus da influenza e de um adjuvante em emulsão óleo-em-água e de um antígeno ou preparação antigênica de uma primeira cepa de influenza Download PDFInfo
- Publication number
- BRPI0621886B1 BRPI0621886B1 BRPI0621886-5A BRPI0621886A BRPI0621886B1 BR PI0621886 B1 BRPI0621886 B1 BR PI0621886B1 BR PI0621886 A BRPI0621886 A BR PI0621886A BR PI0621886 B1 BRPI0621886 B1 BR PI0621886B1
- Authority
- BR
- Brazil
- Prior art keywords
- influenza
- antigen
- pandemic
- vaccine
- strain
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 313
- 206010022000 influenza Diseases 0.000 title claims abstract description 223
- 239000002671 adjuvant Substances 0.000 title claims abstract description 194
- 102000036639 antigens Human genes 0.000 title claims abstract description 188
- 108091007433 antigens Proteins 0.000 title claims abstract description 188
- 239000000427 antigen Substances 0.000 title claims abstract description 187
- 241000712461 unidentified influenza virus Species 0.000 title claims abstract description 134
- 238000002360 preparation method Methods 0.000 title claims abstract description 119
- 229960003971 influenza vaccine Drugs 0.000 title claims abstract description 79
- 239000007764 o/w emulsion Substances 0.000 title claims abstract description 61
- 238000000034 method Methods 0.000 title claims abstract description 54
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 32
- 238000002255 vaccination Methods 0.000 claims abstract description 177
- 239000000185 hemagglutinin Substances 0.000 claims abstract description 140
- 101710154606 Hemagglutinin Proteins 0.000 claims abstract description 139
- 101710093908 Outer capsid protein VP4 Proteins 0.000 claims abstract description 139
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 claims abstract description 139
- 101710176177 Protein A56 Proteins 0.000 claims abstract description 139
- 230000000890 antigenic effect Effects 0.000 claims abstract description 86
- 230000002163 immunogen Effects 0.000 claims abstract description 76
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 claims abstract description 74
- 230000028993 immune response Effects 0.000 claims abstract description 52
- 229960000984 tocofersolan Drugs 0.000 claims abstract description 30
- 229940087168 alpha tocopherol Drugs 0.000 claims abstract description 28
- 239000002076 α-tocopherol Substances 0.000 claims abstract description 28
- 235000004835 α-tocopherol Nutrition 0.000 claims abstract description 28
- -1 alpha-tocopherol Chemical compound 0.000 claims abstract description 19
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 claims abstract description 18
- 239000003995 emulsifying agent Substances 0.000 claims abstract description 17
- 229930182558 Sterol Natural products 0.000 claims abstract description 16
- 150000003432 sterols Chemical class 0.000 claims abstract description 16
- 235000003702 sterols Nutrition 0.000 claims abstract description 16
- 229930003799 tocopherol Natural products 0.000 claims abstract description 16
- 239000011732 tocopherol Substances 0.000 claims abstract description 16
- 235000010384 tocopherol Nutrition 0.000 claims abstract description 16
- 229960001295 tocopherol Drugs 0.000 claims abstract description 16
- 239000003814 drug Substances 0.000 claims abstract description 7
- 229960005486 vaccine Drugs 0.000 claims description 267
- 241000700605 Viruses Species 0.000 claims description 136
- 230000004224 protection Effects 0.000 claims description 51
- 238000012770 revaccination Methods 0.000 claims description 50
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 49
- 230000004044 response Effects 0.000 claims description 36
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 claims description 32
- 229920000053 polysorbate 80 Polymers 0.000 claims description 32
- 241000282412 Homo Species 0.000 claims description 23
- 235000013601 eggs Nutrition 0.000 claims description 23
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 claims description 17
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 claims description 17
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 claims description 16
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 claims description 16
- 238000002156 mixing Methods 0.000 claims description 16
- 229940031439 squalene Drugs 0.000 claims description 16
- 230000028996 humoral immune response Effects 0.000 claims description 13
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 10
- 230000015654 memory Effects 0.000 claims description 8
- 230000008348 humoral response Effects 0.000 claims description 7
- 230000006698 induction Effects 0.000 claims description 6
- 238000004113 cell culture Methods 0.000 claims description 5
- 229920003171 Poly (ethylene oxide) Polymers 0.000 claims description 4
- WERKSKAQRVDLDW-ANOHMWSOSA-N [(2s,3r,4r,5r)-2,3,4,5,6-pentahydroxyhexyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO WERKSKAQRVDLDW-ANOHMWSOSA-N 0.000 claims description 3
- 230000009385 viral infection Effects 0.000 claims description 2
- 238000009472 formulation Methods 0.000 abstract description 53
- 230000003053 immunization Effects 0.000 abstract description 9
- 238000002649 immunization Methods 0.000 abstract description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 8
- 230000002708 enhancing effect Effects 0.000 abstract description 5
- 210000004027 cell Anatomy 0.000 description 47
- 239000003921 oil Substances 0.000 description 46
- 235000019198 oils Nutrition 0.000 description 46
- 230000003612 virological effect Effects 0.000 description 41
- 210000002966 serum Anatomy 0.000 description 40
- 241000282339 Mustela Species 0.000 description 33
- 238000003556 assay Methods 0.000 description 32
- 230000005847 immunogenicity Effects 0.000 description 31
- 239000000839 emulsion Substances 0.000 description 29
- 102000004127 Cytokines Human genes 0.000 description 27
- 108090000695 Cytokines Proteins 0.000 description 27
- 238000010790 dilution Methods 0.000 description 26
- 239000012895 dilution Substances 0.000 description 26
- 102000005348 Neuraminidase Human genes 0.000 description 25
- 108010006232 Neuraminidase Proteins 0.000 description 25
- 230000003472 neutralizing effect Effects 0.000 description 25
- 241001465754 Metazoa Species 0.000 description 24
- 238000004448 titration Methods 0.000 description 24
- 101100525628 Picea mariana SB62 gene Proteins 0.000 description 23
- 238000012360 testing method Methods 0.000 description 23
- 238000004458 analytical method Methods 0.000 description 22
- 210000004072 lung Anatomy 0.000 description 22
- 239000002245 particle Substances 0.000 description 20
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 19
- 230000001976 improved effect Effects 0.000 description 18
- 229960004906 thiomersal Drugs 0.000 description 18
- 208000015181 infectious disease Diseases 0.000 description 17
- 210000001519 tissue Anatomy 0.000 description 17
- 108010029697 CD40 Ligand Proteins 0.000 description 16
- 102100032937 CD40 ligand Human genes 0.000 description 16
- 108010002350 Interleukin-2 Proteins 0.000 description 16
- 102000000588 Interleukin-2 Human genes 0.000 description 16
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 16
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 16
- 210000003743 erythrocyte Anatomy 0.000 description 16
- 238000002347 injection Methods 0.000 description 16
- 239000007924 injection Substances 0.000 description 16
- 238000009021 pre-vaccination Methods 0.000 description 16
- 210000002845 virion Anatomy 0.000 description 16
- 230000005875 antibody response Effects 0.000 description 15
- 230000001965 increasing effect Effects 0.000 description 15
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 14
- 229930006000 Sucrose Natural products 0.000 description 14
- 230000008901 benefit Effects 0.000 description 14
- 239000000872 buffer Substances 0.000 description 14
- 230000035931 haemagglutination Effects 0.000 description 14
- 230000005764 inhibitory process Effects 0.000 description 14
- 230000001681 protective effect Effects 0.000 description 14
- 239000005720 sucrose Substances 0.000 description 14
- 229940125575 vaccine candidate Drugs 0.000 description 14
- 230000005867 T cell response Effects 0.000 description 13
- 239000003599 detergent Substances 0.000 description 13
- 238000009826 distribution Methods 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 238000006386 neutralization reaction Methods 0.000 description 12
- 229920004890 Triton X-100 Polymers 0.000 description 11
- 238000011156 evaluation Methods 0.000 description 11
- 238000007918 intramuscular administration Methods 0.000 description 11
- 239000008363 phosphate buffer Substances 0.000 description 11
- 230000024932 T cell mediated immunity Effects 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 230000009260 cross reactivity Effects 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 239000012528 membrane Substances 0.000 description 10
- 210000004379 membrane Anatomy 0.000 description 10
- 108090000623 proteins and genes Proteins 0.000 description 10
- 206010061598 Immunodeficiency Diseases 0.000 description 9
- 239000013504 Triton X-100 Substances 0.000 description 9
- 230000036760 body temperature Effects 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000036039 immunity Effects 0.000 description 9
- 210000001806 memory b lymphocyte Anatomy 0.000 description 9
- GNENVASJJIUNER-UHFFFAOYSA-N 2,4,6-tricyclohexyloxy-1,3,5,2,4,6-trioxatriborinane Chemical compound C1CCCCC1OB1OB(OC2CCCCC2)OB(OC2CCCCC2)O1 GNENVASJJIUNER-UHFFFAOYSA-N 0.000 description 8
- 229940124873 Influenza virus vaccine Drugs 0.000 description 8
- 238000005119 centrifugation Methods 0.000 description 8
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 8
- 229960003964 deoxycholic acid Drugs 0.000 description 8
- 231100000673 dose–response relationship Toxicity 0.000 description 8
- 239000012530 fluid Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 210000004180 plasmocyte Anatomy 0.000 description 8
- 229940071643 prefilled syringe Drugs 0.000 description 8
- 239000003755 preservative agent Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 239000011123 type I (borosilicate glass) Substances 0.000 description 8
- 230000006978 adaptation Effects 0.000 description 7
- 230000000875 corresponding effect Effects 0.000 description 7
- 230000036541 health Effects 0.000 description 7
- 230000002779 inactivation Effects 0.000 description 7
- 230000001939 inductive effect Effects 0.000 description 7
- 210000004779 membrane envelope Anatomy 0.000 description 7
- 230000002335 preservative effect Effects 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 238000000108 ultra-filtration Methods 0.000 description 7
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 6
- 241000271566 Aves Species 0.000 description 6
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 230000009286 beneficial effect Effects 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 230000001186 cumulative effect Effects 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 210000000987 immune system Anatomy 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 230000003287 optical effect Effects 0.000 description 6
- 230000001105 regulatory effect Effects 0.000 description 6
- 230000004043 responsiveness Effects 0.000 description 6
- 229930182490 saponin Natural products 0.000 description 6
- 150000007949 saponins Chemical class 0.000 description 6
- 235000017709 saponins Nutrition 0.000 description 6
- 230000000405 serological effect Effects 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 229940031626 subunit vaccine Drugs 0.000 description 6
- 241000712431 Influenza A virus Species 0.000 description 5
- 208000022120 Jeavons syndrome Diseases 0.000 description 5
- 241000282341 Mustela putorius furo Species 0.000 description 5
- 102100039360 Toll-like receptor 4 Human genes 0.000 description 5
- 230000002411 adverse Effects 0.000 description 5
- 206010064097 avian influenza Diseases 0.000 description 5
- 230000001900 immune effect Effects 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000011534 incubation Methods 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 230000001717 pathogenic effect Effects 0.000 description 5
- 210000005259 peripheral blood Anatomy 0.000 description 5
- 239000011886 peripheral blood Substances 0.000 description 5
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 5
- 230000001932 seasonal effect Effects 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- 238000005199 ultracentrifugation Methods 0.000 description 5
- 239000000277 virosome Substances 0.000 description 5
- 108010042708 Acetylmuramyl-Alanyl-Isoglutamine Proteins 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 241000287828 Gallus gallus Species 0.000 description 4
- 206010060891 General symptom Diseases 0.000 description 4
- 102000003886 Glycoproteins Human genes 0.000 description 4
- 108090000288 Glycoproteins Proteins 0.000 description 4
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 description 4
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 4
- UZQJVUCHXGYFLQ-AYDHOLPZSA-N [(2s,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-4-[(2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-6-(hydroxymethyl)-4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-6-(hy Chemical compound O([C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)CC(O)[C@]1(CCC(CC14)(C)C)C(=O)O[C@H]1[C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O[C@H]4[C@@H]([C@@H](O[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O5)O)[C@H](O)[C@@H](CO)O4)O)[C@H](O)[C@@H](CO)O3)O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UZQJVUCHXGYFLQ-AYDHOLPZSA-N 0.000 description 4
- 238000013019 agitation Methods 0.000 description 4
- 229940037003 alum Drugs 0.000 description 4
- 238000013475 authorization Methods 0.000 description 4
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 4
- 210000004970 cd4 cell Anatomy 0.000 description 4
- 230000036755 cellular response Effects 0.000 description 4
- 235000013330 chicken meat Nutrition 0.000 description 4
- 235000019700 dicalcium phosphate Nutrition 0.000 description 4
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000011049 filling Methods 0.000 description 4
- 230000000951 immunodiffusion Effects 0.000 description 4
- 238000011081 inoculation Methods 0.000 description 4
- 239000002054 inoculum Substances 0.000 description 4
- 230000021633 leukocyte mediated immunity Effects 0.000 description 4
- BSOQXXWZTUDTEL-ZUYCGGNHSA-N muramyl dipeptide Chemical compound OC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O BSOQXXWZTUDTEL-ZUYCGGNHSA-N 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 4
- 229940068968 polysorbate 80 Drugs 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 210000002345 respiratory system Anatomy 0.000 description 4
- 230000003248 secreting effect Effects 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 229940031418 trivalent vaccine Drugs 0.000 description 4
- 230000007485 viral shedding Effects 0.000 description 4
- 208000017667 Chronic Disease Diseases 0.000 description 3
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 3
- 102000018697 Membrane Proteins Human genes 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 238000012408 PCR amplification Methods 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 235000014676 Phragmites communis Nutrition 0.000 description 3
- 206010035664 Pneumonia Diseases 0.000 description 3
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 230000003466 anti-cipated effect Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 238000005352 clarification Methods 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 238000011109 contamination Methods 0.000 description 3
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 210000004207 dermis Anatomy 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 208000037797 influenza A Diseases 0.000 description 3
- 208000037798 influenza B Diseases 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical class O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 3
- 229920006008 lipopolysaccharide Polymers 0.000 description 3
- 229940126601 medicinal product Drugs 0.000 description 3
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 3
- 238000002941 microtiter virus yield reduction assay Methods 0.000 description 3
- 229940035032 monophosphoryl lipid a Drugs 0.000 description 3
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 3
- 235000019796 monopotassium phosphate Nutrition 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 230000035515 penetration Effects 0.000 description 3
- 210000003800 pharynx Anatomy 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 150000003904 phospholipids Chemical class 0.000 description 3
- 239000001103 potassium chloride Substances 0.000 description 3
- 235000011164 potassium chloride Nutrition 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000011146 sterile filtration Methods 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 230000029812 viral genome replication Effects 0.000 description 3
- OILXMJHPFNGGTO-UHFFFAOYSA-N (22E)-(24xi)-24-methylcholesta-5,22-dien-3beta-ol Natural products C1C=C2CC(O)CCC2(C)C2C1C1CCC(C(C)C=CC(C)C(C)C)C1(C)CC2 OILXMJHPFNGGTO-UHFFFAOYSA-N 0.000 description 2
- HNLXNOZHXNSSPN-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OCCOCCOCCOCCOCCOCCOCCO)C=C1 HNLXNOZHXNSSPN-UHFFFAOYSA-N 0.000 description 2
- 108010068327 4-hydroxyphenylpyruvate dioxygenase Proteins 0.000 description 2
- CERZMXAJYMMUDR-QBTAGHCHSA-N 5-amino-3,5-dideoxy-D-glycero-D-galacto-non-2-ulopyranosonic acid Chemical compound N[C@@H]1[C@@H](O)CC(O)(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO CERZMXAJYMMUDR-QBTAGHCHSA-N 0.000 description 2
- OQMZNAMGEHIHNN-UHFFFAOYSA-N 7-Dehydrostigmasterol Natural products C1C(O)CCC2(C)C(CCC3(C(C(C)C=CC(CC)C(C)C)CCC33)C)C3=CC=C21 OQMZNAMGEHIHNN-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 2
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 description 2
- 239000011627 DL-alpha-tocopherol Substances 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 108060003393 Granulin Proteins 0.000 description 2
- 241000197304 H2N2 subtype Species 0.000 description 2
- 101000959820 Homo sapiens Interferon alpha-1/13 Proteins 0.000 description 2
- 241000713196 Influenza B virus Species 0.000 description 2
- 208000002979 Influenza in Birds Diseases 0.000 description 2
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 2
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 2
- 241001092142 Molina Species 0.000 description 2
- 238000011887 Necropsy Methods 0.000 description 2
- 102000011931 Nucleoproteins Human genes 0.000 description 2
- 108010061100 Nucleoproteins Proteins 0.000 description 2
- 108010058846 Ovalbumin Proteins 0.000 description 2
- 206010037660 Pyrexia Diseases 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 108020000999 Viral RNA Proteins 0.000 description 2
- 230000000240 adjuvant effect Effects 0.000 description 2
- 230000004520 agglutination Effects 0.000 description 2
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- LGJMUZUPVCAVPU-UHFFFAOYSA-N beta-Sitostanol Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(C)CCC(CC)C(C)C)C1(C)CC2 LGJMUZUPVCAVPU-UHFFFAOYSA-N 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 238000012769 bulk production Methods 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000000356 contaminant Substances 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 230000037029 cross reaction Effects 0.000 description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 229910000397 disodium phosphate Inorganic materials 0.000 description 2
- 235000019800 disodium phosphate Nutrition 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 102000013361 fetuin Human genes 0.000 description 2
- 108060002885 fetuin Proteins 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000013020 final formulation Substances 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 238000000760 immunoelectrophoresis Methods 0.000 description 2
- 229960001438 immunostimulant agent Drugs 0.000 description 2
- 239000003022 immunostimulating agent Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 229940090046 jet injector Drugs 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 231100000518 lethal Toxicity 0.000 description 2
- 230000001665 lethal effect Effects 0.000 description 2
- 229960001226 live attenuated influenza Drugs 0.000 description 2
- 229940050906 magnesium chloride hexahydrate Drugs 0.000 description 2
- DHRRIBDTHFBPNG-UHFFFAOYSA-L magnesium dichloride hexahydrate Chemical compound O.O.O.O.O.O.[Mg+2].[Cl-].[Cl-] DHRRIBDTHFBPNG-UHFFFAOYSA-L 0.000 description 2
- 210000001161 mammalian embryo Anatomy 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 229940031346 monovalent vaccine Drugs 0.000 description 2
- 229940031348 multivalent vaccine Drugs 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- CERZMXAJYMMUDR-UHFFFAOYSA-N neuraminic acid Natural products NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO CERZMXAJYMMUDR-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 229920004905 octoxynol-10 Polymers 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 229940092253 ovalbumin Drugs 0.000 description 2
- 230000002688 persistence Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 2
- 229940068196 placebo Drugs 0.000 description 2
- 239000000902 placebo Substances 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 229920002477 rna polymer Polymers 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000004062 sedimentation Methods 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 238000004513 sizing Methods 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 229940031351 tetravalent vaccine Drugs 0.000 description 2
- 229940033663 thimerosal Drugs 0.000 description 2
- 239000006163 transport media Substances 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- 239000002023 wood Substances 0.000 description 2
- RQOCXCFLRBRBCS-UHFFFAOYSA-N (22E)-cholesta-5,7,22-trien-3beta-ol Natural products C1C(O)CCC2(C)C(CCC3(C(C(C)C=CCC(C)C)CCC33)C)C3=CC=C21 RQOCXCFLRBRBCS-UHFFFAOYSA-N 0.000 description 1
- RDEIXVOBVLKYNT-VQBXQJRRSA-N (2r,3r,4r,5r)-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[(2r,3r,6s)-3-amino-6-(1-aminoethyl)oxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-5-methyl-4-(methylamino)oxane-3,5-diol;(2r,3r,4r,5r)-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[(2r,3r,6s)-3-amino-6-(aminomethyl)oxan-2-yl]o Chemical compound OS(O)(=O)=O.O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC[C@@H](CN)O2)N)[C@@H](N)C[C@H]1N.O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC[C@H](O2)C(C)N)N)[C@@H](N)C[C@H]1N.O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N RDEIXVOBVLKYNT-VQBXQJRRSA-N 0.000 description 1
- UCTWMZQNUQWSLP-VIFPVBQESA-N (R)-adrenaline Chemical compound CNC[C@H](O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-VIFPVBQESA-N 0.000 description 1
- 229930182837 (R)-adrenaline Natural products 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- YRNWIFYIFSBPAU-UHFFFAOYSA-N 4-[4-(dimethylamino)phenyl]-n,n-dimethylaniline Chemical compound C1=CC(N(C)C)=CC=C1C1=CC=C(N(C)C)C=C1 YRNWIFYIFSBPAU-UHFFFAOYSA-N 0.000 description 1
- 101710186708 Agglutinin Proteins 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 235000019737 Animal fat Nutrition 0.000 description 1
- 241000272517 Anseriformes Species 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 208000031504 Asymptomatic Infections Diseases 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 241001168968 Chroicocephalus ridibundus Species 0.000 description 1
- 241000759568 Corixa Species 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 239000003109 Disodium ethylene diamine tetraacetate Substances 0.000 description 1
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 238000011510 Elispot assay Methods 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- DNVPQKQSNYMLRS-NXVQYWJNSA-N Ergosterol Natural products CC(C)[C@@H](C)C=C[C@H](C)[C@H]1CC[C@H]2C3=CC=C4C[C@@H](O)CC[C@]4(C)[C@@H]3CC[C@]12C DNVPQKQSNYMLRS-NXVQYWJNSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010018072 General signs and symptoms Diseases 0.000 description 1
- 101150039660 HA gene Proteins 0.000 description 1
- 101710121925 Hemagglutinin glycoprotein Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 101710146024 Horcolin Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 101900159346 Influenza A virus Hemagglutinin Proteins 0.000 description 1
- 206010022004 Influenza like illness Diseases 0.000 description 1
- 241001500351 Influenzavirus A Species 0.000 description 1
- 102100034349 Integrase Human genes 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- 238000012313 Kruskal-Wallis test Methods 0.000 description 1
- 101710189395 Lectin Proteins 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 101710155913 Major envelope protein Proteins 0.000 description 1
- 101710179758 Mannose-specific lectin Proteins 0.000 description 1
- 101710150763 Mannose-specific lectin 1 Proteins 0.000 description 1
- 101710150745 Mannose-specific lectin 2 Proteins 0.000 description 1
- 101150080862 NA gene Proteins 0.000 description 1
- 238000010222 PCR analysis Methods 0.000 description 1
- 241000845082 Panama Species 0.000 description 1
- 108010046016 Peanut Agglutinin Proteins 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 206010035737 Pneumonia viral Diseases 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 235000001630 Pyrus pyrifolia var culta Nutrition 0.000 description 1
- 240000002609 Pyrus pyrifolia var. culta Species 0.000 description 1
- 241001454523 Quillaja saponaria Species 0.000 description 1
- 235000009001 Quillaja saponaria Nutrition 0.000 description 1
- 244000088415 Raphanus sativus Species 0.000 description 1
- 235000006140 Raphanus sativus var sativus Nutrition 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 235000019774 Rice Bran oil Nutrition 0.000 description 1
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 1
- 241000219287 Saponaria Species 0.000 description 1
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- 210000000447 Th1 cell Anatomy 0.000 description 1
- 241000656145 Thyrsites atun Species 0.000 description 1
- 108010060804 Toll-Like Receptor 4 Proteins 0.000 description 1
- HZYXFRGVBOPPNZ-UHFFFAOYSA-N UNPD88870 Natural products C1C=C2CC(O)CCC2(C)C2C1C1CCC(C(C)=CCC(CC)C(C)C)C1(C)CC2 HZYXFRGVBOPPNZ-UHFFFAOYSA-N 0.000 description 1
- 206010046306 Upper respiratory tract infection Diseases 0.000 description 1
- MECHNRXZTMCUDQ-UHFFFAOYSA-N Vitamin D2 Natural products C1CCC2(C)C(C(C)C=CC(C)C(C)C)CCC2C1=CC=C1CC(O)CCC1=C MECHNRXZTMCUDQ-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000008649 adaptation response Effects 0.000 description 1
- 230000004523 agglutinating effect Effects 0.000 description 1
- 239000000910 agglutinin Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- ILRRQNADMUWWFW-UHFFFAOYSA-K aluminium phosphate Chemical compound O1[Al]2OP1(=O)O2 ILRRQNADMUWWFW-UHFFFAOYSA-K 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000010775 animal oil Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000014102 antigen processing and presentation of exogenous peptide antigen via MHC class I Effects 0.000 description 1
- 230000027645 antigenic variation Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 229940076810 beta sitosterol Drugs 0.000 description 1
- VEZXCJBBBCKRPI-UHFFFAOYSA-N beta-propiolactone Chemical compound O=C1CCO1 VEZXCJBBBCKRPI-UHFFFAOYSA-N 0.000 description 1
- NJKOMDUNNDKEAI-UHFFFAOYSA-N beta-sitosterol Natural products CCC(CCC(C)C1CCC2(C)C3CC=C4CC(O)CCC4C3CCC12C)C(C)C NJKOMDUNNDKEAI-UHFFFAOYSA-N 0.000 description 1
- 239000003012 bilayer membrane Substances 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000001427 coherent effect Effects 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000012297 crystallization seed Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000011026 diafiltration Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 235000021186 dishes Nutrition 0.000 description 1
- 235000019301 disodium ethylene diamine tetraacetate Nutrition 0.000 description 1
- DGLRDKLJZLEJCY-UHFFFAOYSA-L disodium hydrogenphosphate dodecahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].OP([O-])([O-])=O DGLRDKLJZLEJCY-UHFFFAOYSA-L 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000002296 dynamic light scattering Methods 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 210000003278 egg shell Anatomy 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 229960005139 epinephrine Drugs 0.000 description 1
- 229960002061 ergocalciferol Drugs 0.000 description 1
- DNVPQKQSNYMLRS-SOWFXMKYSA-N ergosterol Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H](CC[C@]3([C@H]([C@H](C)/C=C/[C@@H](C)C(C)C)CC[C@H]33)C)C3=CC=C21 DNVPQKQSNYMLRS-SOWFXMKYSA-N 0.000 description 1
- 210000000887 face Anatomy 0.000 description 1
- 238000011832 ferret model Methods 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 235000021323 fish oil Nutrition 0.000 description 1
- 239000012537 formulation buffer Substances 0.000 description 1
- 238000003958 fumigation Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000012239 gene modification Methods 0.000 description 1
- 230000005017 genetic modification Effects 0.000 description 1
- 235000013617 genetically modified food Nutrition 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000010211 hemagglutination inhibition (HI) assay Methods 0.000 description 1
- 244000144980 herd Species 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 238000003125 immunofluorescent labeling Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000006054 immunological memory Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 229920001684 low density polyethylene Polymers 0.000 description 1
- 239000004702 low-density polyethylene Substances 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000013178 mathematical model Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- VYQNWZOUAUKGHI-UHFFFAOYSA-N monobenzone Chemical compound C1=CC(O)=CC=C1OCC1=CC=CC=C1 VYQNWZOUAUKGHI-UHFFFAOYSA-N 0.000 description 1
- 210000002850 nasal mucosa Anatomy 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 235000014571 nuts Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000003200 peritoneal cavity Anatomy 0.000 description 1
- 238000001558 permutation test Methods 0.000 description 1
- 102000013415 peroxidase activity proteins Human genes 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 230000007479 persistent immune response Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 229920002627 poly(phosphazenes) Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 229960000380 propiolactone Drugs 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000000601 reactogenic effect Effects 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 230000002787 reinforcement Effects 0.000 description 1
- 230000003014 reinforcing effect Effects 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- 239000008165 rice bran oil Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000009589 serological test Methods 0.000 description 1
- 239000010686 shark liver oil Substances 0.000 description 1
- 229940069764 shark liver oil Drugs 0.000 description 1
- KZJWDPNRJALLNS-VJSFXXLFSA-N sitosterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CC[C@@H](CC)C(C)C)[C@@]1(C)CC2 KZJWDPNRJALLNS-VJSFXXLFSA-N 0.000 description 1
- 229950005143 sitosterol Drugs 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- HCXVJBMSMIARIN-PHZDYDNGSA-N stigmasterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)/C=C/[C@@H](CC)C(C)C)[C@@]1(C)CC2 HCXVJBMSMIARIN-PHZDYDNGSA-N 0.000 description 1
- 229940032091 stigmasterol Drugs 0.000 description 1
- 235000016831 stigmasterol Nutrition 0.000 description 1
- BFDNMXAIBMJLBB-UHFFFAOYSA-N stigmasterol Natural products CCC(C=CC(C)C1CCCC2C3CC=C4CC(O)CCC4(C)C3CCC12C)C(C)C BFDNMXAIBMJLBB-UHFFFAOYSA-N 0.000 description 1
- 210000000434 stratum corneum Anatomy 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229960004793 sucrose Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- STCOOQWBFONSKY-UHFFFAOYSA-N tributyl phosphate Chemical compound CCCCOP(=O)(OCCCC)OCCCC STCOOQWBFONSKY-UHFFFAOYSA-N 0.000 description 1
- AISMNBXOJRHCIA-UHFFFAOYSA-N trimethylazanium;bromide Chemical compound Br.CN(C)C AISMNBXOJRHCIA-UHFFFAOYSA-N 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 229960000172 trivalent influenza vaccine Drugs 0.000 description 1
- 239000002691 unilamellar liposome Substances 0.000 description 1
- 239000012646 vaccine adjuvant Substances 0.000 description 1
- 229940124931 vaccine adjuvant Drugs 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 210000003501 vero cell Anatomy 0.000 description 1
- 230000007501 viral attachment Effects 0.000 description 1
- 208000009421 viral pneumonia Diseases 0.000 description 1
- 210000000605 viral structure Anatomy 0.000 description 1
- 230000001018 virulence Effects 0.000 description 1
- MECHNRXZTMCUDQ-RKHKHRCZSA-N vitamin D2 Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1\C[C@@H](O)CCC1=C MECHNRXZTMCUDQ-RKHKHRCZSA-N 0.000 description 1
- 235000001892 vitamin D2 Nutrition 0.000 description 1
- 239000011653 vitamin D2 Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 239000010497 wheat germ oil Substances 0.000 description 1
- 229940126581 whole-virion vaccine Drugs 0.000 description 1
- 150000003772 α-tocopherols Chemical class 0.000 description 1
Images















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/145—Orthomyxoviridae, e.g. influenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/525—Virus
- A61K2039/5252—Virus inactivated (killed)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55566—Emulsions, e.g. Freund's adjuvant, MF59
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16111—Influenzavirus A, i.e. influenza A virus
- C12N2760/16134—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16211—Influenzavirus B, i.e. influenza B virus
- C12N2760/16234—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
composição de vacina contra influenza monovalente, kit, método para a produção de uma composição de vacina contra influenza para uma situação pandêmica ou uma situação pre-pandêmica, e, usos de um antígeno de vírus da influenza ou preparação antigênica do mesmo e de um adjuvante em emulsão óleo-em-água, de um antígeno de vírus da influenza pandêmica ou preparação antigênica do antígeno de hemaglutinina do vírus da influenza e de um adjuvante em emulsão óleo-em-água e de um antígeno ou preparação antigênica de uma primeira cepa de influenza. a presente invenção refere-se a formulações de vacina contra influenza monovalentes e regimes de vacinação para imunização contra doença de influenza, seu uso na medicina, em particular seu uso no aumento de respostas imunes a diversos antígenos, e a métodos de preparação. em particular, a invenção refere-se a composições imunogénicas de influenza monovalentes compreendendo um antígeno de influenza ou preparação antigénica do mesmo a partir de uma cepa de vírus de influenza que está associada com um surto pandêmico ou apresentando o potencial a ser associado com um surto pandémico, em combinação com um adjuvante em emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa-tocoferol, e um agente emulsificante.
Description
[0001] A presente invenção refere-se a formulações de vacina contra influenza e regimes de vacinação para imunização contra doença de influenza, seu uso na medicina, em particular seu uso no aumento de respostas imunes a diversos antígenos, e a métodos de preparação. Em particular, a invenção refere-se a composições imunogênicas de influenza monovalentes compreendendo uma pequena quantidade de antígeno de vírus da influenza ou preparação antigênica do mesmo de uma cepa de vírus da influenza que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia, em combinação com um adjuvante de emulsão óleo-em- água.
[0002] Vírus de influenza são dos vírus mais ubíquos presentes no mundo, afetando tanto humanos como rebanhos. Influenza resulta numa carga econômica, morbidez e, até mesmo, mortalidade, que são significativos.
[0003] O vírus da influenza é um vírus envelopado com RNA com um tamanho de partículas com diâmetro de cerca de 125 nm. Ele consiste basicamente de um nucleocapsídeo interno ou núcleo de ácido ribonucleico (RNA) associado com nucleoproteína, envolvido por um envelope viral com uma estrutura de camada dupla de lipídios e glicoproteínas externas. A camada interior do envelope viral constitui-se predominantemente de proteínas de matriz, e a camada exterior consiste, em sua maior parte, de material lipídico derivado do hospedeiro. O vírus da influenza compreende dois antígenos de superfície, glicoproteínas neuraminidase (NA) e hemaglutinina (HA), que ocorrem como picos com comprimentos de 10 a 12 nm, na superfície das partículas. São estas proteínas de superfície, particularmente a hemaglutinina, que determinam a especificidade antigênica dos subtipos de influenza. Cepas de vírus são classificadas de acordo com a espécie de origem do hospedeiro, sítio geográfico e ano de isolamento, número de série, e, no caso de influenza A, por propriedades sorológicas de subtipos de HA e NA. Identificou-se 16 subtipos de HA (de H1 a H16) e nove subtipos de NA (de N1 a N9) para vírus de influenza A [Webster RG et al. "Evolution and ecology of influenza A viruses". Microbiol. Rev. 1992; 56: 152-179; Fouchier RA et al.. "Characterization of a Novel Influenza A Virus Hemagglutinin Subtype (H16) Obtained from Black-Headed Gulls". J. Virol. 2005; 79:2814-2822). Vírus de todos os subtipos de HA e NA foram recuperados de aves aquáticas, mas apenas três subtipos de HA (H1, H2, e H3) e dois subtipos de NA (N1 e N2) foram determinados como linhagens estáveis na população humana desde 1918. Apenas um subtipo de HA e um de NA são reconhecidos como vírus de influenza B.
[0004] Vírus de influenza A evoluem e sofrem cotinuamente variabilidade antigênica [Wiley D, Skehel J. "The structure and the function of the hemagglutinin membrane glycoprotein of the influenza virus". Ann. Rev. Biochem. 1987; 56:365-394]. Uma falta de verificação efetiva pela RNA polimerase viral leva a uma alta taxa de erros de transcrição que podem resultar em substituições de aminoácidos em glicoproteínas de superfície. Isto é denominado "desvio antigênico". O genoma viral segmentado permite um segundo tipo de variação antigênica. Se dois vírus de influenza infectarem simultaneamente uma célula hospedeira, redistribuição genética, denominada "desvio antigênico"pode gerar um vírus inédito com novas proteínas de superfície ou proteínas internas. Estas alterações antigênicas, tanto 'desvios' como também 'variações' são imprevisíveis e podem exercer um impacto drástico de um ponto de vista imunológico na medida em que eventualmente levam à emergência de novas cepas de influenza e que permitem ao vírus escapar do sistema imunológico causando as epidemias bem conhecidas, quase anuais. Ambas estas modificações genéticas causaram novas variantes virais responsáveis por pandemias em humanos.
[0005] HA é o antígeno mais importante na definição da especificidade sorológica das diferentes cepas de influenza. Esta proteína de 75-80 kD contém numerosas determinantes antigênicas, várias das quais encontram-se em regiões que sofrem alterações de seqüências em diferentes cepas (determinantes específicos para cepas) e outras em regiões que são comuns a muitas moléculas de HA (comuns a determinantes).
[0006] Vírus de influenza causam epidemias quase a cada inverno, com taxas de infecção para vírus de tipo A ou B de até 40% ao longo de um período de seis semanas. Infecção por influenza resulta em vários estados de doença, desde uma infecção subclínica, passando por uma infecção branda do trato respiratório superior, até uma pneumonia viral grave. Epidemias de influenza típicas causam aumentos na incidência de pneumonia e de doença do trato respiratório inferior, conforme confirmado por taxas incrementadas de hospitalização ou de mortalidade. A gravidade da doença é determinada primariamente pela idade do hospedeiro, por seu estado imunológico e pelo sítio de infecção.
[0007] Pessoas idosas, com 65 anos ou mais, são particularmente vulneráveis, contribuindo por de 80 a 90% de todas as mortes relacionadas com influenza em países desenvolvidos. Indivíduos com doenças crônicas subjacentes também experimentarão, com grande probabilidade, referidas complicações. Bebês jovens também podem sofrer doença grave. Portanto, em particular, estes grupos precisam ser protegidos. Além destes grupos 'em risco', as autoridades de saúde também estão recomendando vacinar profissionais de saúde.
[0008] A vacinação desempenha um papel crítico no controle de epidemias anuais de influenza. Vacinas contra influenza correspondentemente disponíveis são vacinas de influenza inativadas ou vivas atenuadas.
[0009] Vacinas de influenza inativadas constituem-se de três formas possíveis de preparação de antígeno: vírus inteiro inativado, subvírions em que partículas virais purificadas são disrompidas com detergentes ou outros reagentes para solubilizar o envelope lipídico (assim- chamada vacina "dividida") ou HA e NA purificada (vacina de subunidade). Estas vacinas são dadas intramuscularmente (i.m.), subcutaneamente (s.c.), ou intranasalmente (i.n.).
[00010] Vacinas de influenza para uso interpandêmico, de todos os tipos, são usualmente vacinas trivalentes. Elas contém geralmente antígenos derivados de duas cepas de vírus de influenza A e uma cepa de influenza B. Uma dose injetável de 0,5 ml convencional contém, na maior parte dos casos, (pelo menos) 15 μg de componente de antígeno de hemaglutinina de cada cepa, conforme medido por meio de imunodifusão radial simples (SRD) (J. M. Wood et al.: "An improved single radial immunodiffusion technique for the assay of influenza haemagglutinin antigen: adaptation for potency determination of inactivated whole virus and subunit vaccines" [Uma técnica de imunodifusão radial simples aperfeiçoada para o ensaio de antígeno de hemaglutinina de influenza: adaptação para determinação de potência de vírus inteiro inativado e vacinas de subunidade], J. Biol. Stand. 5 (1977) 237-247; J. M. Wood et al., "International collaborative study of single radial diffusion and Immunoelectrophoresis techniques for the assay of haemagglutinin antigen influenza virus" [Estudo colaborativo internacional sobre difusão radial simples e técnicas de imunoeletroforese para o ensaio de vírus de influenza de antígeno de hemaglutinina] J. Biol. Stand. 9 (1981 ) 317-330).
[00011] Cepas de vírus da influenza interpandêmicas a serem incorporadas em vacina de influenza a cada estação são determinadas pela Organização Mundial da Saúde em colaboração com autoridades de saúde nacionais e fabricantes de vacinas. Vacinas contra influenza interpandêmica correntemente disponíveis são consideradas seguras em todos os grupos de idade (De Donato et al. 1999, Vaccine, 17, 3094-3101). No entanto, há pouca evidência de que vacinas de influenza correntes atuam em crianças pequenas abaixo da idade de dois anos. Adicionalmente, taxas reportadas de eficácia de vacina para a prevenção de doença de influenza típica confirmada são de 23 a 72% para os idosos, sendo que são significativamente menores do que as taxas de eficácia de 60 a 90% reportadas para adultos mais jovens (Govaert, 1994, J. Am. Med. Assoc., 21, 166-1665; Gross, 1995, Ann Intern. Med. 123, 523-527). Mostrou-se que a efetividade de uma vacina de influenza correlaciona-se com titulações séricas de anticorpos de inibição de hemaglutinina (HI, hemagglutination inhibition) com relação à cepa viral, e vários estudos verificaram que adultos mais velhos apresentam menores titulações de HI após imunização contra influenza do que adultos mais jovens (Murasko, 2002, Experimental gerontology, 37, 427-439).
[00012] Uma vacina de influenza de subunidade adjuvantada com o adjuvante MF59, em forma de uma emulsão óleo-em-água encontra-se comercialmente obtenível para os idosos e para a população em risco, e demonstrou sua capacidade de induzir uma maior titulação de anticorpos do que aquela obtida com a vacina de subunidade não-adjuvantada (De Donato et al. 1999, Vaccine, 17, 3094-3101). No entanto, em uma publicação posterior, a mesma vacina não demonstrou seu perfil aperfeiçoado em comparação com uma vacina dividida não-adjuvantada (Puig-Barbera et al., 2004, Vaccine 23, 283-289).
[00013] A título de fundamentação, durante períodos inter- pandêmicos, circulam vírus de influenza que estão relacionados com aqueles da epidemia precedente. Os vírus disseminam-se entre pessoas com níveis variáveis de imunidade a partir de infecções anteriores na vida. Tal circulação, usualmente durante um período de 2 a 3 anos, promove a seleção de novas cepas que se modificaram suficientemente para causar novamente uma epidemia entre a população em geral; este processo é denominado 'desvio antigênico'. 'Variantes de desvio' podem exercer impactos diferentes em diferentes comunidades, regiões, países ou continentes a qualquer ano, embora, ao longo de vários anos, seu impacto global seja freqüentemente semelhante. Em outras palavras, uma pandemia de influenza ocorre quando aparece um novo vírus da influenza para o qual a população humana não tem imunidade. Epidemias de influenza típicas causam aumentos na incidência de pneumonia e doença do trato respiratório inferior, conforme confirmado por maiores taxas de hospitalização ou mortalidade. Os idosos ou aqueles com doenças crônicas subjacentes experimentarão mui provavelmente referidas complicações, mas bebês jovens também podem sofrer doença grave.
[00014] Em intervalos que não podem ser preditos emergem vírus de influenza inéditos com um antígeno de superfície chave, a hemaglutinina, de um tipo totalmente diferente de cepas que circularam na estação precedente. Aqui, os antígenos resultantes podem variar de 20% a 50% da proteína correspondente de cepas que circularam previamente em humanos. Isto pode resultar em vírus que escapam da 'imunidade de rebanho' e estabelecem pandemias. Este fenômeno é denominado 'desvio antigênico'. Considera-se que pelo menos as últimas pandemias passadas ocorreram quando um vírus da influenza de uma espécie diferente, como um vírus de influenza aviário ou porcino, atravessaram a barreira das espécies. Se referidos vírus possuírem o potencial de disseminar-se de humano para humano, eles podem espalhar-se pelo mundo todo dentro de poucos meses até um ano, resultando numa pandemia. Por exemplo, em 1957 (pandemia da gripa asiática), vírus do subtipo H2N2 substituíram vírus H1N1 que haviam circulado na população humana desde pelo menos 1918 quando o vírus foi isolado pela primeira vez. Os H2HA e N2NA sofreram desvio antigênico entre 1957 e 1968 até que o HA fosse substituído em 1968 (pandemia da gripe de Hong-Kong) pela emergência do subtipo de influenza H3N2, após o que o N2NA continuou a desvio juntamente com o H3HA (Nakajima et al., 1991, Epidemiol. Infect. 106, 383-395).
[00015] As características de uma cepa do vírus da influenza que lhe proporcionam o potencial de causar um surto pandêmico são: ele contém uma nova hemaglutinina em comparação com a hemaglutinina nas cepas correntemente circulantes, que podem ou não ser acompanhadas por uma alteração no subtipo neuraminidase; ele é capaz de ser transmitido horizontalmente na população humana; e ele é patogênico para humanos. Uma nova hemaglutinina pode ser uma que não foi evidente na população humana durante um período de tempo prolongado, provavelmente várias décadas, como H2. Ou ele pode ser uma hemaglutinina que não esteve circulando na população humana antes, por exemplo, H5, H9, H7 ou H6 que são encontrados em aves. Em qualquer caso a maior parte ou, pelo menos, uma grande proporção de, ou mesmo toda a população, não encontrou previamente o antígeno e é imunologicamente despreparada para o mesmo.
[00016] Realizou-se vários estudos clínicos para avaliar a segurança e a imunogenicidade em populações não-preparadas, com vacinas candidatas monovalentes contendo uma cepa pandêmica, como as cepas de H2N2 ou H9N2 não-circulantes. Estudos investigaram formulações de vírus divididos ou inteiros de várias concentrações de HA (1,9, 3,8, 3,8, 7,5 ou 15 μg HA por dose), com ou sem adjuvantação de alume. Vírus de influenza do subtipo H2N2 que circularam de 1957 até 1968 quando eles foram substituídos pelas cepas H3N2 durante a 'pandemia de Hong Kong'. Hoje em dia, indivíduos nascidos após 1968 são imunologicamente despreparados para cepas de H2N2. Estes candidatos de vacinas mostraram ser imunogênicos e bem tolerados. Resultados são reportados em Hehme, N. et al. 2002, Med. Microbiol. Immunol. 191, 203-208; em Hehme N. et al. 2004, Virus Research 103, 163-171; e dois estudos foram reportados com H5N1 (Bresson JL et al. The Lancet, 2006:367 (9523): 1657-1664; Treanor JJ et al. N. Engl. J. Med. 2006;354: 1343-1351). Outros estudos reportaram resultados com vacinas contra influenza adjuvantadas com MF59. Um estudo reportou que duas doses de uma vacina contra influenza H5N3 adjuvantada com MF59 reforçaram a imunidade contra influenza H5N1 em uma população preparada (Stephenson et al., Vaccine 2003, 21, 1687-1693) e outro estudo reportou respostas de anticorpos de reação cruzada a vírus H5N1 obtidos após três doses de vacina contra influenza H5N4 adjuvantada com MF59 (Stephenson et al., J. Infect. Diseases 2005, 191, 1210-1215).
[00017] Pessoas em risco, no caso de uma pandemia de influenza, podem ser diferentes dos grupos de risco definidos relativamente a complicações devidas a influenza sazonal. De acordo com a OMS, 50% dos casos humanos causados pela cepa H5N1 da gripe aviária ocorreram em pessoas com menos de 20 anos de vida, 90% ocorreram entre aquelas com idades <40. (OMS, registro epidemiológico semanal, 30 de junho de 2006).
[00018] Durante uma pandemia, drogas antivirais podem não ser suficientes para cobrir as necessidades, e o número de indivíduos em risco de influenza será maior do que em períodos interpandêmicos, de modo que o desenvolvimento de uma vacina apropriada com o potencial de ser produzida em grandes quantidades e com eficiente potencial de distribuição e de administração é essencial. Por estas razões, vacinas monovalentes em lugar de vacinas trivalentes estão sendo desenvolvidas para fins [de controlar] pandemias numa tentativa de reduzir o volume de vacinas, primariamente porque duas doses de vacina podem ser necessárias para se obter níveis protetores de anticorpos em recipientes imunologicamente não-tratados (Wood JM et al. Med Mircobiol Immunol. 2002; 191: 197-201. Wood JM et al. Philos Trans R Soc Lond B Biol Sci. 2001; 356:1953-1960).
[00019] Estes problemas podem ser enfrentados por meio de adjuvantação, sendo que o objetivo consiste em aumentar a imunogenicidade da vacina de forma a permitir diminuir o teor de antígeno (diminuição do antígeno) e, com isto, aumentar o número de doses de vacinas disponíveis. O uso de um adjuvante também pode superar a potencial baixa imunogenicidade do antígeno em uma população não-tratada. Exemplos do referido acima foram mostrados usando vírus H2N2 ou H9N2 inteiro inativado adjuvantado com sal de alumínio (N. Hehme et al. Virus Research 2004, 103, 163-171). Já se realizou ensaios clínicos com vacina de H5N1 de subvírion simples ou vacina de vírus H5N1 dividido adjuvantado com hidróxido de alumínio. Os resultados destes ensaios indicam que tanto vacinas de vírus H5N1 simples como também adjuvantadas são seguras até uma dose de antígeno de 90 μg (testados apenas como vacina de subvírion simples) (Bresson JL et al. The Lancet. 2006:367 (9523): 1657-1664; Treanor JJ et al. N Engl J Med. 2006; 354:1343-1351)
[00020] Portanto, necessita-se ainda de novas vacinas com uma imunogenicidade aperfeiçoada, em particular contra cepas pandêmicas pouco imunogênicas ou não imunogênicas ou para os indivíduos imunocomprometidos, como a população idosa. Necessita-se também de vacinas novas com um potencial de proteção cruzada, que poderiam ser usadas como vacinas pré-pandêmicas ou formação de estoque para preparar uma população imunologicamente não-tratada contra uma cepa de pandemia antes ou após a declaração de um pandemia. Formulação de antígeno de vacina com adjuvantes potentes é uma abordagem possível para incrementar respostas imunes a antígenos de subvírion. Proporciona-se aqui formulações adjuvantes inéditas que permitem a uma formulação diminuidora do antígeno fornecer proteção suficiente (soroconversão de sujeitos previamente soronegativos a uma titulação de HI considerada como protetora, 1:40 ou aumento de quatro vezes na titulação) de todos os grupos etários.
[00021] Em um primeiro aspecto da presente invenção proporciona-se uma composição imunogênica de influenza, em particular uma vacina, compreendendo uma pequena quantidade de antígeno de vírus da influenza ou preparação antigênica de uma cepa de vírus da influenza que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia, em combinação com um adjuvante, sendo que a pequena quantidade de antígeno não excede 15 μg de hemaglutinina (HA) por dose, e sendo que referido adjuvante é uma emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante. De maneira vantajosa, a composição de vacina é uma composição monovalente.
[00022] Em todo o documento se referirá a uma cepa pandêmica como uma cepa de influenza que está associada ou que é suscetível de estar associada com um surto de doença de influenza, como cepas de influenza pandêmica A. Cepas pandêmicas vantajosas são, embora sem limitação: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1. Outras cepas pandêmicas vantajosas em humanos são H7N3 (2 casos reportados no Canadá), H10N7 (2 casos reportados no Egito) e H5N2 (1 caso reportado no Japão).
[00023] Em outro aspecto a invenção proporciona um método para a produção de uma composição imunogênica de influenza, em particular uma vacina, para uma situação de pandemia ou uma situação pré-pandêmica, sendo que referido método compreende misturar um antígeno de vírus da influenza ou preparação antigênica do mesmo de uma cepa de vírus da influenza simples que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia, com um adjuvante de emulsão óleo-em-água como definido aqui acima, e proporcionando unidades de vacina que contêm no máximo 15 μg de antígeno de hemaglutinina de influenza por dose. O vírus da influenza pode ser derivado de ovo, derivado de planta, derivado de cultura, ou pode ser produzido de forma recombinante. De maneira vantajosa, o antígeno de vírus da influenza é derivado de ovo ou derivado de cultura de células.
[00024] Em um terceiro aspecto proporciona-se uma composição imunogênica como definida aqui para uso na medicina.
[00025] Em outro aspecto adicional, proporciona o uso de (a) uma pequena quantidade, como definido aqui, de antígeno de vírus da influenza ou preparação antigênica do mesmo de uma cepa de influenza simples associada com uma pandemia ou apresentando o potencial de estar associada com uma pandemia, e (b) um adjuvante de emulsão óleo-em-água, na fabricação de uma composição imunogênica, ou um kit, para induzir pelo menos um dentre i) uma resposta imune melhorada de células T CD4, ii) uma resposta melhorada de memória de células B, iii) uma resposta humoral melhorada, contra referido antígeno de vírus ou composição antigênica em um humano. Referida resposta imune é induzida, em particular, em um população ou indivíduo imunocomprometido, como um idoso ou um adulto em alto risco. De maneira vantajosa, a composição imunogênica é como definida aqui.
[00026] Proporciona-se também o uso de um vírus da influenza ou preparação antigênica do mesmo e um adjuvante de emulsão óleo-em-água na preparação de uma composição imunogênica como definida aqui para a vacinação de idosos humanos contra influenza.
[00027] Em uma concretização específica, a composição imunogênica é capaz de induzir tanto uma resposta imune melhorada de células T CD4 como uma resposta melhorada de células de memória B em comparação com o antígeno ou composição antigênica não-adjuvantada. Em outra concretização específica, a composição imunogênica é capaz de induzir tanto uma resposta imune melhorada de células T CD4 como também uma resposta humoral melhorada em comparação com aquela obtida com o antígeno ou composição antigênica não-adjuvantada. Em particular, referida resposta imune humoral ou proteção atende pelo menos um, vantajosamente dois, tipicamente todos os três critérios regulatórios da EU ou FDA para eficácia da vacina contra influenza. De maneira vantajosa, referida(s) resposta(s) imune(s) ou proteção é obtida após uma, vantajosamente duas, doses de vacina. Particularmente, referida(s) resposta(s) imune(s) ou proteção atendem pelo menos um, pelo menos um, vantajosamente dois, ou todos os três critérios regulatórios da EU ou FDA para eficácia da vacina contra influenza após uma dose de vacina adjuvantada. Especificamente, pelo menos um, vantajosamente dois critérios da FDA ou EU é (são) atendidos após apenas uma dose de vacina. Critérios de eficácia para a composição de acordo com a presente invenção são detalhados adicionalmente abaixo (ver Tabela 1 e abaixo sob "critérios de eficácia"). De maneira vantajosa, referida composição é administrada parenteralmente, em particular pela via intramuscular ou subcutânea.
[00028] Em uma concretização adicional, proporciona-se o uso de uma pequena quantidade de um vírus da influenza ou preparação antigênica do mesmo na fabricação de uma composição imunogênica para a revacinação de humanos previamente vacinados com uma composição imunogênica de influenza monovalente compreendendo um antígeno de influenza ou preparação antigênica do mesmo de uma cepa de vírus da influenza simples que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia, em combinação com um adjuvante de emulsão óleo-em-água como definido aqui.
[00029] Em uma concretização específica, a composição usada para a revacinação pode ser não-adjuvantada ou pode conter um adjuvante, em particular um adjuvante de emulsão óleo-em-água. Em outra concretização específica, a composição imunogênica para revacinação contém um vírus da influenza ou preparação antigênica do mesmo que compartilha epitopos de células T CD4 comuns com o vírus da influenza ou preparação antigênica viral do mesmo usada para a primeira vacinação. A composição imunogênica para uma revacinação pode conter uma quantidade clássica (i.e. cerca de 15 μg de HA) de referida cepa pandêmica variante.
[00030] De maneira vantajosa a revacinação é realizada em sujeitos que já foram vacinados na estação precedente contra influenza. De maneira vantajosa, a revacinação é realizada com uma vacina compreendendo uma cepa de influenza (p. ex., H5N1 Vietnam) que é do mesmo subtipo que aquela usado para a primeira vacinação (p. ex., H5N1 Vietnam). Em uma concretização específica, a revacinação é realizada com uma cepa desviada do mesmo subtipo, p. ex., H5N1 Indonésia. Em outra concretização, referida cepa de influenza usada para a revacinação é uma cepa desviada, i.e. é diferente daquela usada para a primeira vacinação, p. ex., apresenta um subtipo de HA ou NA diferente, como H5N2 (mesmo subtipo de HA que H5N1 mas subtipo de NA diferente) ou H7N1 (subtipo de HA diferente de H5N1, mas mesmo subtipo de NA).
[00031] De maneira vantajosa, a primeira vacinação é realizada quando da declaração de uma pandemia e re-vacinação é realizada mais tarde. Alternativamente, a primeira vacinação é parte de uma estratégia pré- pandêmica e é realizada antes da declaração de uma pandemia, como uma estratégia de preparação, permitindo desta forma que o sistema imunológico seja preparado, e sendo que a revacinação é realizada subseqüentemente. Tipicamente, a revacinação é realizada pelo menos 4 meses após a primeira vacinação, vantajosamente 6 ou de 8 a 14 meses após, vantajosamente em torno de 10 a 12 meses após, ou mesmo mais tempo. De maneira vantajosa, a revacinação um ano mais tarde ou ainda mais um ano mais tarde é capaz de reforçar a resposta imune celular e/ou de anticorpos. Isto é particularmente importante à medida que ocorrem ondas adicionais de infecção vários meses após o primeiro surto de uma pandemia. Conforme necessário, a revacinação pode ser realizada mais de uma vez.
[00032] De maneira vantajosa, referida emulsão óleo-em-água compreende um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante. Em uma outra concretização específica, referido adjuvante de emulsão óleo-em-água compreende pelo menos um óleo metabolizável numa quantidade de 0,5% a 20% do volume total, e apresenta gotículas de óleo, das quais pelo menos 70% em intensidade têm diâmetros inferiores a 1 μm. De maneira vantajosa, um referido tocoferol, como alfa tocoferol, está presente numa quantidade de 1,0% a 20%, em particular numa quantidade de 1,0% a 5% do volume total de referida composição imunogênica.
[00033] Em um aspecto adicional da presente invenção, proporciona-se o uso de um antígeno ou preparação antigênica de uma primeira cepa de influenza pandêmica na fabricação de uma composição imunogênica como definido aqui para a proteção contra infecções por influenza causadas por uma cepa de influenza variante.
[00034] Em um aspecto específico, proporciona-se um método de vacinação de uma população ou indivíduo humano imunocomprometido, como idosos ou adultos em alto risco, sendo que referido método compreende administrar a referido indivíduo ou população uma composição imunogênica de influenza compreendendo uma pequena quantidade de um antígeno de influenza ou preparação antigênica do mesmo de uma cepa de vírus da influenza pandêmica simples em combinação com um adjuvante de emulsão óleo-em-água como definido aqui.
[00035] Em outra concretização adicional, a invenção proporciona um método para revacinar humanos previamente vacinados com uma composição imunogênica de influenza monovalente compreendendo um antígeno de influenza ou preparação antigênica do mesmo de uma cepa simples de vírus da influenza pandêmica, em combinação com um adjuvante de emulsão óleo-em-água, sendo que referido método compreende administrar a referido humano uma composição imunogênica compreendendo um vírus de influenza, quer adjuvantado ou não-adjuvantado.
[00036] Em uma concretização adicional proporciona-se um método para vacinar uma população humana ou um indivíduo contra uma cepa de vírus da influenza pandêmica, seguido de revacinação de referido humano ou população contra uma cepa variante de virus da influenza, sendo que referido método compreende administrar a referido humano (i) uma primeira composição compreendendo um vírus da influenza ou preparação antigênica do mesmo de uma primeira cepa de vírus da influenza pandêmica e um adjuvante de emulsão óleo-em-água, e (ii) uma segunda composição imunogênica compreendendo uma cepa variante de vírus da influenza de referido primeira cepa de vírus da influenza. Em uma concretização específica referida cepa variante está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia. Em outra concretização específica referida cepa variante é parte de uma composição polivalente que compreende, adicionalmente a referido vírus da influenza variante pandêmica, pelo menos uma cepa variante de vírus da influenza circulante (sazonal). Em particular, referida cepa de vírus da influenza pandêmica é parte de uma composição divalente, ou trivalente, ou tetravalente compreendendo adicionalmente uma, duas ou três cepas sazonais, respectivamente.
[00037] Em todo o documento, o uso de uma pequena quantidade de antígeno de vírus da influenza pandêmico na fabricação de uma composição como definido aqui para a prevenção de infecção por influenza ou doença, e um método de tratamento de humanos usando a composição reivindicada será usado de forma intercambiável.
[00038] Outros aspectos e vantagens da presente invenção encontram-se descritos adicionalmente na descrição detalhada a seguir das concretizações preferidas dos mesmos.
[00039] Figura 1: Distribuição de tamanhos das partículas de gotículas de óleo em emulsão óleo-em-água SB62 conforme medido por meio de PCS. Figura 1A mostra medições de tamanhos do lote 1023 de SB62 com o Malvern Zetasizer 3000HS: A = diluição de 1/10000 (de Rec22 a Rec24) (Análise em Contin e modelo óptico adaptado 1,5/0,01); B = Diluição de 1/20000 (de Rec28 a Rec30) (Análise em Contin e modelo óptico adaptado 1,5/0,01). Figura 1 B mostra uma ilustração esquemática do registro 22 (parte superior) e registro 23 (parte inferior) por intensidade.
[00040] Figura 2: Experimentos com furões. Figura 2A: Teste de inibição de hemaglutinina (GMT, Hemagglutination Inhibition Test) em furões imunizados com diferentes doses de H5N1 A/Vietnam. Figura 2B: Dados médios de PCR de H5N1 (gráfico à esquerda) e dados médios de titulação viral (gráfico à esquerda) de tecidos do pulmão de furões no dia da morte ou da eutanização (dados de PCR são expressos como Unidades de Diluição de Controle (CDU, Control Diluition Units) que são determinados de uma curva padrão produzida a partir de um estoque de vírus que é diluído serialmente, sendo que cada diluição é submetida a extração com ácido nucleico e amplificação de PCR Taqman da mesma maneira que as amostras de teste. Dados de titulação viral são expressos como TCID50/g de tecido).
[00041] Figura 3: Respostas de anticorpos neutralizadores anti- A/Indonesia em furões imunizados com diferentes doses de H5N1 A/Vietnam.
[00042] Figura 4: Sinopse da fabricação de volumes de influenza monovalentes.
[00043] Figura 5: Folha de fluxo da formulação para volume final do antígeno.
[00044] Figura 6: Ensaio clínico humano com uma faixa de dosagem de antígeno de vírus dividido H5N1, adjuvantado ou não com AS03. GMT's (com 95% de CI) para anticorpo anti-HA nos momentos dos dias 0, 21 e 42.
[00045] Figura 7: Ensaio clínico humano com uma faixa de dosagem de antígeno de vírus dividido H5N1, adjuvantado ou não com AS03. Taxas de soroconversão (com 95% de CI) para anticorpo anti-HA nos dias 21 e 42 após a vacinação.
[00046] Figura 8: Ensaio clínico humano com uma faixa de dosagem do antígeno de vírus dividido H5N1, adjuvantado ou não com AS03. Taxas de soroproteção (com 95% de CI) para anticorpo anti-HA em cada um dos momentos (dia 0, dia 21 e (dia 42).
[00047] Figura 9: Ensaio clínico humano com uma faixa de dosagem de antígeno de vírus dividido H5N1, adjuvantado ou não com AS03. Fator de soroconversão (com 95% de CI) para anticorpo anti-HA após a vacinação (dia 21 e 42)
[00048] Figura 10: Titulações de neutralização (GMT: Figura 10A; taxas de soroconversão: Figura 10B) para a cepa H5N1 Vietnam. HN4 = 3,8 μg de HA não-adjuvantado; HN8 = 7,5 μg de HA não-adjuvantado; HN4AD = 3,8 μg de HA adjuvantado com AS03; HN8AD = 7,5 μg de HA adjuvantado com AS03.
[00049] Figura 11: Respostas específicas CD4 para a cepa H5N1 Vietnam. HN4 = 3,8 μg de HA não-adjuvantado; HN8 = 7,5 μg de HA não- adjuvantado; HN4AD = 3,8 μg de HA adjuvantado com AS03; HN8AD = 7,5 μg de HA adjuvantado com AS03.
[00050] Os presentes inventores verificaram que uma formulação de influenza compreendendo baixa quantidade de um vírus da influenza ou preparação antigênica do mesmo associada com uma pandemia ou suscetível de ser associada com uma pandemia, juntamente com um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante, foi capaz de melhorar a resposta imune humoral, e/ou a resposta imune de células T CD4 e/ou resposta de memória de células B contra referido antígeno ou composição antigênica em um humano ou população, em comparação com aquela obtida com o vírus não-adjuvantado ou preparação antigênica do mesmo. Eles permitirão obter proteção contra morbidez/mortalidade causada por uma cepa de influenza homóloga. As formulações adjuvantadas com um adjuvante de emulsão óleo-em-água como definido aqui serão usadas vantajosamente para induzir resposta de células T CD4 anti-influenza capazes de detecção de epitopos de influenza apresentados por moléculas de MHC de classe II. As formulações adjuvantadas com um adjuvante de emulsão óleo- em-água como definido aqui serão usadas vantajosamente para induzir uma resposta imune de reação cruzada, i.e. imunidade detectável (humoral e/ou celular) contra uma cepa variante ou contra uma faixa de cepas relacionadas. As formulações adjuvantadas serão efetivas de maneira vantajosa para objetivar o sistema imunológico humoral e/ou mediado com células para aumentar a responsividade contra cepas de influenza homólogas e desviadas (mediante vacinação e infecção). Eles também serão usados de maneira vantajosa para induzir uma estratégia de preparação cruzada, i.e. induzir memória imunológica "preparada" que facilita a resposta quando da revacinação (de uma dose) com cepa variante.
[00051] As composições de influenza pandêmicas adjuvantadas de acordo com a invenção apresentam várias vantagens: 1) Uma imunogenicidade aperfeiçoada: elas permitirão melhorar a fraca resposta imune a cepas de influenza menos imunogênicas a um nível mais elevado do que aqueles obtidos com as formulações não- adjuvantadas; 2) O uso de adjuvantes pode superar a imunogenicidade potencialmente fraca do antígeno em uma população não-tratada; 3) Elas podem levar a uma imunogenicidade melhorada em populações específicas, como nas pessoas idosas (tipicamente acima de 60 anos de vida) a níveis observados em pessoas mais jovens com idades de 18 a 60 (respostas de células T e/ou anticorpos); 4) Elas podem levar a um perfil melhorado de proteção cruzada: maior reatividade cruzada, proteção cruzada contra cepas de influenza variantes (desviadas) permitindo a estruturação de uma estratégia de preparação cruzada onde elas podem ser usadas como vacinas pré-pandêmicas permitindo adicionalmente apenas uma dose de uma vacina para pandemia necessária para incrementar a proteção contra a cepa pandêmica (circulante); 5) Ao atingirem qualquer uma ou todas estas vantagens adicionais com uma dosagem reduzida de antígeno, elas assegurarão uma maior capacidade no caso de emergência ou para a preparação visando uma situação de pandemia (diminuição do antígeno na situação pandêmica)
[00052] Outras vantagens serão aparentes a partir da descrição e da seção de exemplos abaixo. As composições para uso na presente invenção podem ser capazes de proporcionar melhor soroproteção contra influenza após revacinação, conforme avaliado pelo número de sujeitos humanos que se deparam com a influenza correlacionando-se com as proteções. Adicionalmente, a composição para uso na presente invenção também pode ser capaz de induzir uma maior resposta humoral ou resposta de memória de células B após a primeira vacinação de um sujeito humano, e uma resposta mais substancial após a revacinação, em comparação com a composição não- adjuvantada.
[00053] As composições adjuvantadas reivindicadas também podem ser capazes não só de induzir, mas também de manter níveis protetores de anticorpos contra a cepa de influenza presente na vacina, em mais indivíduos do que aqueles obtidos com a composição não-adjuvantada.
[00054] Assim, em outra concretização adicional, a composição reivindicada é capaz de assegurar uma resposta imune persistente contra doença relacionada com influenza. Em particular, por persistência compreende-se uma resposta imune de anticorpos HI que é capaz de atender critérios reguladores após pelo menos três meses, vantajosamente após pelo menos 6 meses após a vacinação. Em particular, a composição reivindicada é capaz de induzir níveis protetores de anticorpos conforme medido por meio da taxa de proteção (ver Tabela 1) em >50%, vantajosamente em >60% dos indivíduos >70% dos indivíduos, vantajosamente em >80% dos indivíduos ou vantajosamente em >90% dos indivíduos para a cepa de influenza pandêmica presente na vacina, após pelo menos três meses. Em um aspecto específico, obtém-se níveis protetores de anticorpos de >90% pelo menos 6 meses após a vacinação contra a cepa de influenza da composição de vacina.
[00055] De acordo com aspectos adicionais da presente invenção, a composição reivindicada é capaz de induzir soroproteção e soroconversão em um grau mais elevado do que aquele proporcionado pelas exigências da EU para cepas de vacinas de influenza. Isto será detalhado adicionalmente abaixo (ver tabela 1 e abaixo, sob "critérios de eficácia").
[00056] Em uma concretização, um vírus da influenza ou preparação antigênica do mesmo para uso de acordo com a presente invenção pode ser um vírus da influenza dividido ou preparação antigênica de vírus dividido do mesmo. Em uma concretização alternativa a preparação de influenza pode conter outro tipo de antígeno de influenza inativado, como vírus inteiro inativado ou HA e NA recombinante e/ou purificado (vacina de subunidade), ou um virosoma de influenza. Em uma concretização adicional, o vírus da influenza pode ser uma preparação de influenza viva atenuada.
[00057] Um vírus da influenza dividido ou preparação antigênica de vírus dividido do mesmo para uso de acordo com a presente invenção é vantajosamente uma preparação de vírus inativado em que partículas de vírus são disrompidas com detergentes ou outros reagentes para solubilizar o envelope lipídico. Vírus divididos ou preparações antigênicas de vírus divididos dos mesmos são preparadas de maneira vantajosa por meio de fragmentação de vírus de influenza inteiro, quer infeccioso ou inativado, com concentrações solubilizadoras de solventes orgânicos ou detergentes e subseqüente remoção de todo ou da maior parte do agente solubilizador e de parte ou da maior parte do material lipídico viral. Por preparação antigênica de vírus dividido do mesmo compreende-se uma preparação de vírus dividido que pode ter sido submetido a algum grau de purificação em comparação com o vírus dividido, ao mesmo tempo que conserva a maior parte das propriedades antigênicas dos componentes de vírus divididos. Por exemplo, quando produzido em ovos, o vírus dividido pode ser depletado de proteínas contaminadoras dos ovos, ou quando produzidos em cultura de células, o vírus dividido pode ser depletado de contaminantes da célula hospedeira. Uma preparação antigênica de vírus dividido pode compreender componentes antigênicos de vírus divididos de mais de uma cepa viral. Vacinas contendo vírus dividido (denominado 'vacina dividida de influenza') ou preparações antigênicas de vírus divididos geralmente contêm proteína de matriz residual e nucleoproteína e, por vezes, lipídios, e também proteínas de envelope de membrana. Referidas vacinas de vírus dividido conterão usualmente a maior parte ou todas as proteínas estruturais do vírus, embora não necessariamente nas mesmas proporções em que ocorrem no vírus inteiro.
[00058] Alternativamente, o vírus da influenza pode encontrar-se em forma de uma vacina de vírus inteiro. Isto pode provar ser uma vantagem relativamente a uma vacina de vírus dividido para uma situação de pandemia na medida em que evita a incerteza sobre se uma vacina de vírus dividido pode ser produzida com êxito para uma nova cepa de vírus da influenza. Para algumas cepas, os detergentes convencionais usados para produzir o vírus dividido podem danificar o vírus e torná-lo inútil. Embora sempre haja a possibilidade de usar diferentes detergentes e/ou desenvolver um processo diferente para produzir uma vacina dividida, isto poderia consumir tempo, que pode não estar disponível numa situação de pandemia. Adicionalmente ao maior grau de certeza obtido com uma abordagem de vírus inteiro, também há uma maior capacidade de produção de vacina do que no caso do vírus dividido por que quantidades consideráveis de antígeno são perdidas durante etapas de purificação adicionais necessárias para preparar uma vacina dividida vantajosa.
[00059] Em outra concretização, a preparação do vírus da influenza encontra-se em forma de uma vacina de influenza de subunidade purificada. Vacinas de influenza de subunidade contêm geralmente as duas principais proteínas de envelope, HA e NA, e podem apresentar uma vantagem adicional sobre todas as vacinas de vírions porque são menos reatogênicas, particularmente em vacinados jovens. Vacinas de subunidade podem ser produzidas recombinantemente ou purificadas de partículas virais disrompidas.
[00060] Em outra concretização, a preparação de vírus da influenza encontra-se em forma de um virosoma. Virosomas são vesículas esféricas, unilamelares que conservam as glicoproteínas funcionais do envelope viral HA e NA em conformação autêntica, intercalada na membrana de camada dupla de fosfolipídeos de virosomas1.
[00061] Referido vírus da influenza ou preparação antigênica do mesmo pode ser derivada de ovo ou derivada de cultura de células. Eles também podem ser produzidos em outros sistemas, como células de insetos, plantas, leveduras ou bactérias ou podem ser produzidos recombinantemente.
[00062] Por exemplo, o antígeno de vírus da influenza ou preparações antigênicas dos mesmos de acordo com a invenção pode ser derivado do método convencional do ovo embrionado, mediante desenvolvimento do vírus da influenza em ovos e purificando o fluido alantóico colhido. Ovos podem ser acumulados em grandes números em curto prazo. Alternativamente, eles podem ser derivados de qualquer um dos métodos de geração usando cultura de tecidos para desenvolver o vírus ou expressar antígenos de superfície do vírus da influenza recombinante. Substratos de células vantajosas para desenvolver os vírus incluem, por exemplo, células de rim de cão, como MDCK ou células de um clone de MDCK, células parecidas com MDCK, células de rim de macaco, como células AGMK incluindo células de Vera, linhas de células de porco vantajosas, ou qualquer outro tipo de células mamíferas vantajosas para a produção de vírus da influenza para fins de vacina. Substratos de células vantajosos também incluem células humanas, p. ex., células MRC-5 ou Per- C6. Substratos de células vantajosos não se limitam a linhas de células; por exemplo, células primárias, como fibroblastos de embrião de franco e linhas de células aviárias também são incluídas.
[00063] O antígeno de vírus da influenza ou preparação antigênica do mesmo pode ser produzido por meio de qualquer número de processos comercialmente aplicáveis, por exemplo, o processo de vírus de influenza dividido descrito na Patente dos Estados Unidos n° DD 300 833 e DD 211 444, incorporadas aqui por referência. Tradicionalmente, o vírus da influenza dividido foi produzido usando-se um tratamento com solvente/detergente, como fosfato de tri-n-butila, ou éter de dietila em combinação com Tween™ (conhecido como divisão com "Tween-éter") e este processo ainda é usado em algumas instalações de produção. Outros agentes de divisão agora empregados incluem detergentes ou enzimas proteolíticas ou sais biliares, por exemplo, desoxicolato de sódio, como descrito na patente no. DD 155 875, incorporada aqui por referência. Detergentes que podem ser usados como agentes de divisão incluem detergentes catiônicos, p. ex., brometo de cetil trimetil amônio (CTAB, cethyl trimethyl ammonium bromide), outros detergentes iônicos, p. ex., lauril sulfato, taurodesocolato, ou detergentes não- iônicos, como aqueles descritos acima incluindo Triton X-100 (por exemplo, em um processo descrito em Lina et al, 2000, Biologicals 28, 95- 103) e Triton N-101, ou combinações de quaisquer dois ou mais detergentes.
[00064] O processo de preparação para uma vacina dividida pode incluir uma quantidade de diferentes etapas de filtração e/ou outras etapas de separação, como ultracentrifugação, ultrafiltração, centrifugação zonal e cromatografia (p. ex., troca de íon) numa variedade de combinações, e opcionalmente uma etapa de inativação, p. ex., com calor, formaldeído ou β- propiolactona ou U.V. que pode ser realizada antes ou após a divisão. O processo de divisão pode ser realizado como um processo de bateladas, contínuo ou semi-contínuo. Um processo vantajosos de divisão e purificação para uma composição imunogênica dividida encontra-se descrito no WO 02/097072.
[00065] Preparações de vacina de antígeno de vacina de gripe divididas de acordo com a invenção compreendem uma quantidade residual de Tween 80 e/ou Triton X-100 remanescente do processo de produção, embora estes possam ser adicionados ou suas concentrações ajustadas após a preparação do antígeno dividido. De maneira vantajosa, tanto Tween 80 e Triton X-100 estão presentes. Faixas vantajosas para as concentrações finais destes tensoativos não-iônicos na dose de vacina são: Tween 80: de 0,01 a 1%, vantajosamente de cerca de 0,1% (v/v) Triton X-100: de 0,001 a 0,1 (% peso/volume), vantajosamente de 0,005 a 0,02% (peso/volume).
[00066] Em uma concretização específica, a concentração final para Tween 80 compreende de 0,045% - 0,09% peso/volume. Em outra concretização específica, o antígeno é proporcionado como uma mistura em concentração de 2:1, que apresenta uma concentração de Tween 80 compreendendo de 0,045% a 0,2% (peso/volume) e precisa ser diluída duas vezes até a formulação final com o adjuvantado (ou o tamponador na formulação de controle).
[00067] Em outra concretização específica, a concentração final para Triton X-100 compreende de 0,005% a 0,017% peso/volume. Em outra concretização específica, o antígeno é proporcionado como uma mistura concentrada a 2:1, que apresenta uma concentração de Triton X-100 compreendendo de 0,005% a 0,034% (peso/volume) e precisa ser diluído duas vezes na formulação final com o adjuvantado (ou o tamponador na formulação de controle).
[00068] De maneira vantajosa, a preparação de influenza é preparada na presença de baixo nível de conservante em particular tiomersal ou, vantajosamente, na ausência de tiomersal. De maneira vantajosa, a preparação de influenza resultante é estável na ausência de conservantes de organo-mercúrio, em particular a preparação não contém tiomersal residual. Em particular, a preparação de vírus da influenza compreende um antígeno de hemaglutinina estabilizado na ausência de tiomersal, ou em baixos níveis de tiomersal (geralmente de 5 μg/ml ou menos). Particularmente, a estabilização da cepa de influenza B é realizada por um derivado de alfa tocoferol, como succinato de alfa tocoferol (também conhecido como succinato de vitamina E, i.e. VES). Referidas preparações e métodos para preparar os mesmos encontram-se divulgados no WO 02/097072.
[00069] Alternativamente, particularmente para recipientes de doses múltiplas, tiomersal ou qualquer outro conservante vantajoso está presente para reduzir os riscos de contaminação. Isto é particularmente relevante para vacinas pandêmicas, projetadas para vacinar a maior quantidade possível de pessoas no menor tempo possível.
[00070] Uma composição vantajosa para re-vacinação contém três antígenos de vírions divididos inativados preparados a partir das cepas recomendadas pela OMS da estação de influenza apropriada, adicionalmente a uma cepa de influenza pandêmica.
[00071] Em uma concretização o vírus da influenza ou preparação antigênica do mesmo e o adjuvante de emulsão óleo-em-água estão contidos no mesmo recipiente. Isto é referido como "abordagem de um só frasco". De maneira vantajosa, o frasco é uma seringa pré-enchida. Em uma concretização alternativa, o vírus da influenza ou preparação antigênica do mesmo e o adjuvante de emulsão óleo-em-água estão contidos em recipientes ou frascos ou unidades separadas, e misturados pouco antes, ou após a administração ao sujeito. Isto é referido como 'abordagens de dois frascos'. A título de exemplo, quando a vacina é uma vacina de 2 componentes para um volume de dose total de dose injetada de 0,5 ml, os antígenos concentrados (por exemplo, os antígenos de vírions divididos inativados concentrados) podem ser apresentados em um frasco (330 μl) (recipiente de antígeno, como um frasco) e uma seringa pré-enchida contém o adjuvante (400 μl) (recipiente de adjuvante, como uma seringa). Tipicamente, a vacina pandêmica é uma dose injetada de 0,5 ml e frascos de doses múltiplas contêm uma mistura de frasco:frasco de 1:1 antes de se injetar no primeiro sujeito. Alternativamente, a vacina pandêmica é uma dose injetada de frasco:seringa de 1,0 ml. No momento da injeção, o conteúdo do frasco contendo os antígenos de vírions divididos inativados concentrados é removido do frasco por meio do uso da seringa contendo o adjuvante seguido de misturação cuidadosa da seringa. Antes da injeção, a agulha usada é substituída por uma agulha intramuscular e o volume é corrigido para 530 μl. Uma dose do candidato de vacina de influenza adjuvantada corresponde a 530 μl.
[00072] De maneira vantajosa, a vacina candidata de influenza pandêmica adjuvantada é uma vacina de 2 componentes consistindo de 0,5 ml de antígenos de vírions divididos inativados concentrados apresentados em um frasco de vidro de tipo I e de uma seringa de vidro de tipo I pré-enchida contendo 0,5 ml do adjuvante. Alternativamente, a vacina é uma vacina de 2 componentes apresentada em 2 frascos (um para o antígeno, um para o adjuvante, de 10 doses cada um) para mistura antes da administração ao primeiro paciente, e subseqüente armazenamento a 4°C durante um curto período de tempo (p. ex., de até uma semana) para administração subseqüente. No momento da injeção, o conteúdo da seringa pré-enchida contendo o adjuvante é injetadas no frasco que contém os antígenos de vírions divididos inativados trivalentes concentrados. Após a misturação, o conteúdo é sugado para a seringa e a agulha é substituída por uma agulha intramuscular. Uma dose da vacina candidata de influenza adjuvantada e reconstituída corresponde a 0,5 ml. Cada dose de vacina de 0,5 ml contém 1,9 μg, 3,8 μg, 7,5 μg, 15 μ ou 30 μg de hemaglutinina (HA) ou qualquer quantidade vantajosa de HA que poderia ser determinada de tal forma que a composição de vacina atenda os critérios de eficácia como definido aqui. Uma vacina dose de 1 ml (0,5 ml de adjuvante mais 0,5 ml de preparação de antígeno) também é vantajosa.
[00073] De acordo com a presente invenção, a cepa de influenza na composição imunogênica monovalente como definida aqui está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia. Referida cepa também pode ser referida como 'cepas pandêmicas'no texto abaixo. Em particular, quando a vacina é uma vacina polivalente para re-vacinação, como uma vacina divalente, ou uma vacina trivalente ou uma vacina quadrivalente, pelo menos uma cepa está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia.
[00074] Cepas vantajosas compreendem, embora sem limitação: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1. Outras cepas pandêmicas em humanos: H7N3 (2 casos reportados no Canadá), H10N7 (2 casos reportados no Egito) e H5N2 (1 caso reportado no Japão).
[00075] Referido vírus da influenza ou preparação antigênica do mesmo para re-vacinação é vantajosamente polivalente, como divalente ou trivalente ou quadrivalente ou contém ainda mais cepas de influenza. De maneira vantajosa, o vírus da influenza ou preparação antigênica do mesmo para re-vacinação é trivalente ou quadrivalente, apresentando um antígeno das três cepas de influenza diferentes, sendo que pelo menos uma cepa está associada com uma pandemia ou apresentando o potencial de estar associada com um surto pandêmico. De maneira vantajosa, a composição de re- vacinação compreende uma cepa pandêmica, que pode ser uma variante da cepa pandêmica presente na composição para a primeira vacinação, e três outras cepas, tipicamente as cepas circulantes clássicas.
[00076] Alternativamente, uma estratégia de vacina pré-pandêmica vantajosa carreia imunização periódica (como a cada 1 a 2 anos) com cepas pandêmicas de potencial diferente, com o objetivo de manter e ampliar respostas a estes vírus, ao longo do tempo. Por exemplo, em uma concretização, a primeira vacinação é preparada com a vacina monovalente adjuvantada de acordo com a reivindicada compreendendo uma cepa pandêmica, como H5N1 em um ano, seguido de uma adjuvantada compreendendo uma cepa de influenza pandêmica diferente, como H9N2, no próximo momento (p. ex., após 6 meses, um ano, ou dois anos), depois seguido de vacinação com uma composição adjuvantada compreendendo ainda outra cepa de influenza pandêmica, como H7N7, e assim por diante. Como é impossível predizer a) o timing de uma pandemia potencial, e b) a cepa pandêmica específica, esta estratégia que se baseia na composição adjuvantada acordo com a reivindicada proporcionará maior segurança para maximizar a magnitude e amplitude das respostas imunes protetoras no momento correto. Nestas estratégias, o adjuvante é vantajosamente como definido aqui.
[00077] As características de uma cepa de vírus da influenza que lhe proporcionam o potencial de causar uma pandemia ou um surto de doença de influenza associada com cepas de influenza pandêmicas são: ela contém uma nova hemaglutinina em comparação com a hemaglutinina nas cepas correntemente circulantes e, portanto, quase todas as pessoas são imunologicamente despreparadas; ela é capaz de ser transmitida horizontalmente na população humana; e ela é patogênica para humanos. Uma hemaglutinina nova pode ser uma que não tem estado evidente na população humana durante um período de tempo prolongado, provavelmente de várias décadas, como H2. Ou ela pode ser uma hemaglutinina que não tem estado circulando na população humana antes, por exemplo, H5, H9, H7 ou H6 que são encontradas em aves. Em qualquer caso, a maior parte, ou pelo menos uma grande proporção de, ou mesmo toda a população não se deparou previamente com o antígeno e é imunologicamente despreparada para o mesmo. Presentemente, o vírus de influenza A que foi identificado pela OMS como um que poderia causar potencialmente uma pandemia em humanos é o vírus da influenza aviária H5N1 altamente patogênica. Portanto, a vacina pandêmica de acordo com a invenção compreenderá vantajosamente vírus H5N1.
[00078] Determinadas partes encontram-se geralmente em um risco incrementado de tornarem-se infectadas com influenza em uma situação de pandemia. Os idosos, os cronicamente doentes e crianças pequenas são particularmente suscetíveis, mas muitos adultos jovens e pessoas aparentemente saudáveis também se encontram em risco. Para influenza H2, a parte da população nascida após 1968 encontra-se em risco incrementado. É importante que estes grupos sejam protegidos efetivamente assim que possível e de maneira simples.
[00079] Outro grupo de pessoas que se encontra em risco incrementado são os viajantes. Pessoas viajam mais hoje em dia do que anteriormente, e as regiões em que a maior parte dos vírus emerge, China e Sudeste da Ásia, tornaram-se recentemente destinos de viajem populares. Esta alteração nos padrões de viagem permite que novos vírus atinjam todo o globo em questão de semanas, ao invés de meses ou anos.
[00080] Assim, para estes grupos de pessoas há uma necessidade particular de vacinação para proteger contra influenza em uma situação de pandemia ou em uma situação pandêmica potencial. Cepas pandêmicas vantajosas são, embora sem limitação: H5N1, H9N2, H7N7, H7N1, H2N2 e H1N1. Outras cepas pandêmicas em humanos: H7N3 (2 casos reportados no Canadá), H10N7 (2 casos reportados no Egito) e H5N2 (1 caso reportado no Japão).
[00081] A composição adjuvante da invenção contém um adjuvante de emulsão óleo-em-água, vantajosamente referida emulsão compreende um óleo metabolizável numa quantidade de 0,5% a 20% do volume total, e apresentando gotículas de óleo das quais pelo menos 70% em intensidade apresentam diâmetros inferiores a 1 μm.
[00082] Para que qualquer composição óleo-em-água a ser utilizável para administração humana, a fase óleo do sistema de emulsão precisa compreender um óleo metabolizável. O significado do termo óleo metabolizável é bem conhecido na arte. Metabolizável pode ser definido como 'sendo capaz de ser transformado pelo metabolismo' (Dorland's Illustrated Medical Dictionary, W.B. Sanders Company, 25a edição (1974)). O óleo pode ser qualquer óleo vegetal, óleo de peixe, óleo animal ou óleo sintético, que não é tóxico para o recipiente e que é capaz de ser transformado pelo metabolismo. Nozes, sementes e grãos são fontes comuns de óleos vegetais. Óleos sintéticos também são parte desta invenção e podem incluir óleos comercialmente obteníveis, como NEOBEE® e outros. Um óleo metabolizável particularmente vantajoso é o esqualeno. Esqualeno (2,6,10,15,19,23-hexametil-2,6,10,14,18,22-tetracosaexaeno) é um óleo insaturado que é encontrado em grandes quantidades no óleo de fígado de tubarão, e em menores quantidades no óleo de oliva, óleo de germe de trigo, óleo de farelo de arroz, e levedura, e é um óleo particularmente vantajoso para uso nesta invenção. Esqualeno é um óleo metabolizável em virtude do fato de que é um intermediário na biossíntese do colesterol (Merck index, 10aedição, entrada n° 8619).
[00083] Emulsões óleo-em-água são bem conhecidas per se, na arte, e foram sugeridas como sendo úteis como composições de adjuvante (EP 399843; WO 95/17210).
[00084] De maneira vantajosa o óleo metabolizável está presente numa quantidade de 0,5% a 20% (concentração final) do volume total da composição imunogênica, vantajosamente numa quantidade de 1,0% a 10% do volume total, vantajosamente numa quantidade de 2,0% a 6,0% do volume total.
[00085] Em uma concretização específica, o óleo metabolizável está presente em uma quantidade final de cerca de 0,5%, 1%, 3,5% ou 5% do volume total da composição imunogênica. Em outra concretização específica, o óleo metabolizável está presente em uma quantidade final de 0,5%, 1%, 3,57% ou 5% do volume total da composição imunogênica. Uma quantidade vantajosa do esqualeno é de cerca de 10,7 mg por dose de vacina, vantajosamente de 10,4 a 11,0 mg por dose de vacina.
[00086] De maneira vantajosa os sistemas de emulsão óleo-em- água da presente invenção apresentam um pequeno tamanho das gotículas de óleo na faixa de sub-mícron. De maneira vantajosa os tamanhos das gotículas serão na faixa de 120 a 750 nm, vantajosamente tamanhos de 120 a 600 nm de diâmetro. Tipicamente, a emulsão óleo-em-água contém gotículas de óleo das quais pelo menos 70% em intensidade apresentam diâmetros inferiores a 500 nm, em particular pelo menos 80% em intensidade apresentam diâmetros inferiores a 300 nm, vantajosamente pelo menos 90% em intensidade encontram-se na faixa de 120 a 200 nm de diâmetro.
[00087] O tamanho das gotículas de óleo, i.e. o diâmetro, de acordo com a presente invenção é dado em intensidade. Há várias maneiras de determinar o diâmetro do tamanho das gotículas de óleo em intensidade. A intensidade é medida por meio do uso de um instrumento de determinação do tamanho, vantajosamente por meio de difração de luz dinâmica, como o Malvern Zetasizer 4000 ou, vantajosamente, o Malvern Zetasizer 3000HS. Um procedimento detalhado é dado no Exemplo II.2. Uma primeira possibilidade consiste em determinar o diâmetro médio z [ZAD, z average diameter] por meio de difração de luz dinâmica (PCS-Photon correlation spectroscopy [Espectroscopia de correlação de fótons]); este método proporciona adicionalmente o índice de polidispersidade (PDI, polydispersity index), e ambos, ZAD e PDI, são calculados com o algoritmo de cumulantes. Estes valores não requerem o conhecimento do índice refrativo de partículas. Um segundo meio consiste em calcular o diâmetro da gotícula de óleo por meio de determinação da distribuição total dos tamanhos das partículas por meio de outro algoritmo, quer seja o Contin, ou NNLS, ou o automático "Malvern" (o algoritmo default proporcionado pelo instrumento de dimensionamento). Na maior parte do tempo, na medida em que o índice refrativo das partículas de uma composição complexa é conhecido, considera- se somente a distribuição de intensidade, e, se necessário, a intensidade média que se origina desta distribuição.
[00088] A emulsão óleo-em-água de acordo com a invenção compreende um esterol ou um tocoferol, como alfa tocoferol. Esteróis são bem conhecidos na arte, por exemplo, colesterol é bem conhecido e é divulgado, por exemplo, no Merck Index, 11aedição, página 341, como um esterol naturalmente ocorrente encontrado na gordura animal. Outros esteróis vantajosos incluem o β-sitosterol, estigmasterol, ergosterol e ergocalciferol. Referido esterol está presente, vantajosamente, numa quantidade de 0,01% a 20% (peso/volume) do volume total da composição imunogênica, vantajosamente numa quantidade de 0,1% a 5% (peso/volume). De maneira vantajosa, quando o esterol é colesterol, ele está presente numa quantidade entre 0,02% e 0,2% (peso/volume) do volume total da composição imunogênica, tipicamente numa quantidade de 0,02% (peso/volume) em um volume de dose de vacina de 0,5 ml, ou 0,07% (peso/volume) em 0,5 ml de volume de dose de vacina ou 0,1% (peso/volume) em 0,7 ml de volume de dose de vacina.
[00089] De maneira vantajosa alfa-tocoferol ou um derivado do mesmo, como succinato de alfa-tocoferol, está presente. De maneira vantajosa alfa-tocoferol está presente numa quantidade entre 0,2% e 5,0% (v/v) do volume total da composição imunogênica, vantajosamente numa quantidade de 2,5% (v/v) em um volume de dose de vacina de 0,5 ml, ou 0,5% (v/v) em 0,5 ml de volume de dose de vacina ou 1,7-1,9% (v/v), vantajosamente 1,8% em 0,7 ml de volume de dose de vacina. A título de esclarecimento, concentrações dadas em v/v podem ser convertidas em concentração indicada em peso/volume aplicando-se o seguinte fator de conversão: uma concentração de 5% (v/v) de alfa-tocoferol é equivalente a uma concentração de 4,8% (peso/volume) de alfa-tocoferol. Uma quantidade vantajosa de alfa- tocoferol é de cerca de 11,9 mg por dose de vacina, vantajosamente de 11,6 a 12,2 mg por dose de vacina.
[00090] A emulsão óleo-em-água compreende um agente emulsificante. O agente emulsificante pode estar presente numa quantidade de 0,01 a 5,0% em peso da composição imunogênica (peso/peso), vantajosamente presente numa quantidade de 0,1 a 2,0% em peso (peso/peso). Concentrações vantajosas são de 0,5 a 1,5% em peso (peso/peso) da composição total.
[00091] O agente emulsificante pode ser, vantajosamente, monooleato de polioxietileno sorbitol (Tween 80). Em uma concretização específica, um volume de dose de vacina de 0,5 ml contém 1% (peso/peso) de Tween 80, e um volume de dose de vacina de 0,7 ml contém 0,7% (peso/peso) Tween 80. Em outra concretização específica a concentração de Tween 80 é de 0,2% (peso/peso). A quantidade vantajosa de polissorbato 80 é cerca de 4,9 mg por dose de vacina, vantajosamente de 4,6 a 5,2 mg por dose de vacina.
[00092] De maneira vantajosa uma dose de vacina compreende alfa-tocoferol numa quantidade de cerca de 11,9 mg por dose de vacina, esqualeno numa quantidade de 10,7 mg por dose de vacina, e polissorbato 80 numa quantidade de cerca de 4,9 mg por dose de vacina.
[00093] O adjuvante de emulsão óleo-em-água pode ser usado com outros adjuvantes ou imunoestimulantes e, portanto, uma concretização importante da invenção é uma formulação óleo-em-água compreendendo esqualeno ou outro óleo metabolizável, um tocoferol, como alfa tocoferol, e Tween 80. A emulsão óleo-em-água também pode conter span 85 e/ou lecitina. Tipicamente, o óleo em água compreenderá de 2 a 10% de esqualeno do volume total da composição imunogênica, de 2 a 10% de alfa tocoferol e de 0,3 a 3% de Tween 80, e pode ser produzido de acordo com o procedimento descrito no WO 95/17210. De maneira vantajosa, a relação de esqualeno:alfa tocoferol é igual ou menor que 1 porque proporciona uma emulsão mais estável. Span 85 [trioleato de polioxietileno sorbitol] (polyoxyethylene sorbitan trioleate) também pode estar presente, por exemplo, em um nível de 1%.
[00094] Na presente invenção a composição de influenza monovalente é capaz de induzir uma resposta imune melhorada de células T CD4 contra pelo menos um do(s) componente(s) de antígeno(s) ou composição antigênica, em comparação com a resposta imune de células T CD4 obtida com a composição correspondente que, não-adjuvantada, i.e. não contém qualquer adjuvante exógeno (referida aqui como 'composição simples'). Em uma concretização específica, referida resposta imune melhorada de células T CD4 é contra a cepa de influenza pandêmica.
[00095] Por 'resposta imune melhorada de células T CD4' compreende-se que uma resposta CD4 é obtida em um paciente humano após a administração da composição imunogênica adjuvantada do que aquela que é obtida após a administração da mesma composição sem adjuvante. Por exemplo, uma resposta maior de células T CD4 é obtida em um paciente humano mediante a administração de uma composição imunogênica compreendendo um vírus da influenza ou preparação antigênica do mesmo juntamente com um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um tocoferol, como alfa tocoferol, e um agente emulsificante, em comparação com a resposta induzida após a administração de uma composição imunogênica compreendendo um vírus da influenza ou preparação antigênica do mesmo que é não-adjuvantado. Referida formulação será usada vantajosamente para induzir resposta de células T CD4 antiinfluenza capaz de detecção de epitopos de influenza apresentados por moléculas MHC de classe II.
[00096] De maneira vantajosa, referida resposta imunológica induzida por uma composição de influenza dividida adjuvantada para uso na presente invenção é maior do que a resposta imunológica induzida por qualquer outra vacina contra influenza não-adjuvantada convencional, como vacina de influenza de subunidade ou vacina de vírus de influenza inteiro.
[00097] Em particular, mas não exclusivamente, referida 'resposta melhorada imune de células T CD4'é obtido em um paciente imunologicamente não-preparado, i.e. um paciente que é soronegativo para referido vírus da influenza ou antígeno. Esta soronegatividade pode ser o resultado do referido paciente apresentando um vírus ou antígeno ainda nunca deparado (assim-chamado paciente 'não-tratado'['naive']) ou, alternativamente, que falhou em responder a referido antígeno uma vez encontrado. De maneira vantajosa, referida resposta melhorada imune de células T CD4 é obtida em um sujeito imunocomprometido, como um idoso, tipicamente com pelo menos 50 anos de vida, tipicamente 65 anos de vida ou mais, ou um adulto abaixo de 65 anos de vida com uma condição de alto risco clínico (adulto em 'alto risco'), ou uma criança abaixo de dois anos.
[00098] A resposta melhorada imune de células T CD4 pode ser avaliada medindo-se o número de células que produzem qualquer uma das citocinas a seguir: • células produzindo pelo menos duas citocinas diferentes (CD40L, IL-2, IFNY, TNFα) • células produzindo pelo menos CD40L e outra citocina (IL-2, TNFα, IFNY) • células produzindo pelo menos IL-2 e outra citocina (CD40L, TNFα, IFNy) • células produzindo pelo menos IFNy e outra citocina (IL-2, TNFα, CD40L) • células produzindo pelo menos TNFα e outra citocina (IL-2, CD40L, IFNy)
[00099] Haverá uma resposta melhorada imune de células T CD4 quando células que produzem qualquer uma das citocinas acima estiverem em maior quantidade após a administração da composição adjuvantada, em comparação com a administração da composição não-adjuvantada. Tipicamente, pelo menos uma, vantajosamente duas das cinco condições mencionadas aqui, acima, serão preenchidas. Em uma concretização particular, as células que produzem todas as quatro citocinas estarão presentes numa quantidade maior no grupo adjuvantado, em comparação com o grupo não-adjuvantado.
[000100] Em uma concretização específica, uma resposta imune melhorada de células T CD4 pode ser conferida pela composição de influenza adjuvantada da presente invenção e pode ser obtida idealmente após uma única administração. A abordagem de dose simples será extremamente relevante, por exemplo, em uma situação de surto de evolução rápida. Em determinadas circunstâncias, particularmente para a população mais idosa, ou no caso de crianças jovens (abaixo de 9 anos de vida) que são vacinadas pela primeira vez contra influenza, ou no caso de uma pandemia, pode ser benéfico administrar duas doses da mesma composição para aquela estação. A segunda dose da referida mesma composição (ainda considerada como 'composição para primeira vacinação') pode ser administrada durante a resposta imune primária em andamento e é espaçada adequadamente. Tipicamente, a segunda dose da composição é dada algumas poucas semanas, ou cerca de um mês, p. ex., 2 semanas, 3 semanas, 4 semanas, 5 semanas, ou 6 semanas após a primeira dose, para auxiliar o sistema imunológico em indivíduos não- responsivos ou fracamente responsivos. Em um aspecto específico, a primeira vacinação é seguida de um curso de vacinação subseqüente de produto de vacina adjuvantada contendo uma cepa de influenza heteróloga.
[000101] Em uma concretização específica, a administração de referida composição imunogênica induz, alternativamente ou adicionalmente, uma resposta melhorada de células de memória B em pacientes administrados com a composição imunogênica adjuvantada, em comparação com a resposta de células de memória B induzida em indivíduos imunizados com a composição não-adjuvantada. Uma resposta de células de memória B melhorada deve significar uma freqüência incrementada de linfócitos B do sangue periférico capazes de diferenciação em células de plasma secretadoras de anticorpos quando do encontro com o antígeno conforme medido por meio de estímulo de diferenciação in vitro (ver seções de Exemplos, p. ex., métodos de memória de células B Elispot).
[000102] Em uma outra concretização específica adicional, a vacinação com a composição para a primeira vacinação, adjuvantada, não exerce qualquer impacto mensurável sobre a resposta CD8.
[000103] De maneira vantajosa, a composição reivindicada compreendendo um vírus da influenza ou preparação antigênica do mesmo formulada com um adjuvante de emulsão óleo-em-água, em particular um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante, será efetiva para promover respostas de células T em uma população humana imunocomprometida. De maneira vantajosa, a administração de uma dose simples da composição imunogênica para primeira vacinação, como descrito na invenção, será capaz de proporcionar melhor soro-proteção, conforme avaliado pelos correlacionamentos de proteção para vacinas de influenza, após revacinação contra influenza, do que a vacinação com uma vacina de influenza não-adjuvantada. A formulação adjuvantada de acordo com a reivindicada também induzirá uma resposta imune melhorada de células T CD4 contra vírus da influenza, em comparação com aquela obtida com a formulação não-adjuvantada. Esta propriedade pode ser associada com uma maior responsividade após vacinação ou infecção relativamente à exposição do antígeno de influenza. Adicionalmente, isto também pode ser associado com uma responsividade cruzada, i.e. uma maior capacidade de responder contra cepas de influenza variantes. Esta resposta melhorada qualitativamente e/ou quantitativamente pode ser benéfica em todas as populações no caso de pandemias, e, particularmente, em uma população humana imunocomprometida, como a população idosa (65 anos de vida e mais) e, em particular, a população idosa em alto risco. Isto também pode ser benéfico para a população de bebês (abaixo de 5 anos, vantajosamente abaixo de 2 anos de vida). Esta resposta melhorada será benéfica para o uso na preparação, p. ex., de vacina estocada contendo uma variante de desvio, antes ou no início do surto pandêmico. Isto pode resultar na redução da morbidez global e na taxa de mortalidade, e na prevenção de admissões de emergência em hospitais devido pneumonia e outras doenças parecidas com influenza. Adicionalmente isto permite induzir uma resposta de células T CD4 que é mais persistente ao longo do tempo, p. ex., ainda presente um ano após a primeira vacinação, em comparação com a resposta induzida com a formulação não-adjuvantada.
[000104] De maneira vantajosa, a resposta imune de células T CD4, como a resposta melhorada imune de células T CD4 obtida em um sujeito não-tratado, envolve a indução de uma resposta de auxiliar T CD4 de reação cruzada. Em particular, a quantidade de células T CD4 de reação cruzada é incrementada. Por resposta CD4 'de reação cruzada' compreende-se células T CD4 que objetivam epitopos compartilhados entre cepas de influenza.
[000105] Usualmente, vacinas de influenza disponíveis só são efetivas contra cepas infectantes de vírus da influenza que apresentam hemaglutinina com características antigênicas similares. Quando o vírus da influenza infectante (circulante) sofreu alterações menores (como uma mutação pontual ou um acúmulo de mutações pontuais resultando em alterações de aminoácidos no exemplo) nas glicoproteínas de superfície em particular hemaglutinina (cepa de vírus variante de desvio antigênico) a vacina ainda pode proporcionar alguma proteção, embora isto possa proporcionar apenas proteção limitada porque as variantes recentemente criadas podem escapar da imunidade induzida antes da infecção por influenza ou vacinação. Desvio antigênico é responsável por epidemias anuais que ocorrem durante períodos interpandêmicos (Wiley & Skehel, 1987, Ann. Rev. Biochem. 56, 365-394). A indução de células T CD4 de reação cruzada proporciona uma vantagem adicional para a composição da invenção, pelo fato de que também pode proporcionar proteção cruzada, em outras palavras, proteção contra infecções heterólogas, i.e. infecções causadas por uma cepa de influenza circulante que é uma variante (p. ex., um desvio) da cepa de influenza contida na composição imunogênica. Isto pode ser vantajoso quando a cepa circulante é difícil de propagar em ovos ou de produzir em cultura de células, tornando o uso de uma cepa desviada uma alternativa de trabalho. Isto também pode ser vantajoso quando o sujeito recebeu uma primeira e uma segunda vacinação com espaçamento de vários meses ou de um ano, e a cepa de influenza na composição imunogênica usada para uma segunda imunização é uma cepa de variante de desvio da cepa usada na composição usada para a primeira vacinação.
[000106] A composição imunogênica de influenza adjuvantada como definida aqui apresenta, portanto, uma maior capacidade de induzir soroproteção e células T CD4 de reação cruzada em sujeitos idosos vacinados. Esta característica pode ser associada com uma maior capacidade de responder contra uma cepa variante da cepa presente na composição imunogênica. Isto pode provar ser uma vantagem importante em uma situação de pandemia. Por exemplo, uma composição imunogênica de influenza monovalente compreendendo qualquer uma das cepa(s) H5, H2, H9, H7 ou H6 pode proporcionar uma maior capacidade de responder contra uma variante de pandemia, i.e. uma cepa de desvio de referida(s) cepa(s) pandêmica(s), seja após vacinação subseqüente com, ou após infecção por referida cepa de desvio.
[000107] Após a administração de vacina de influenza trivalente clássica (3 semanas), há um aumento substancial da freqüência de células T CD4 do sangue periférico que respondem à preparação de cepa antigênica (vírus inteiro ou antígeno dividido) que é homologia àquele presente na vacina (H3N2: A/Panamá/2007/99, H1N1: A/ New Caledonia/20/99, B: B/Shangdong/7/97) (ver Exemplo III). Um aumento comparável da freqüência pode ser observado se células T CD4 do sangue periférico forem reestimuladas com cepas de influenza classificadas como cepas desviadas (H3N2: A/Sydney/5/97, H1N1: A/Beijing/262/95, B: B/Yamanashi/166/98).
[000108] Em contraste, se células T CD4 do sangue periférico forem reestimuladas com cepas de influenza classificadas como cepa desviadas (H2N2: A/Singapura/1/57, H9N2: A/Hong Kong/1073/99) por perito no campo, não haverá aumento observável após a vacinação.
[000109] Células T CD4 que são capazes de reconhecer tanto cepas de influenza homólogas como também desviadas foram denominadas no presente documento "de reação cruzada". As composições de influenza adjuvantadas como descrito aqui têm sido capazes de apresentar reatividade cruzada hetero-subtípica, porque há reatividade cruzada que pode ser observada contra cepas de influenza desviadas. Como indicado acima, a capacidade de uma formulação de vacina para pandemia ser efetiva contra cepas pandêmicas desviadas podem provar ser uma característica importante no caso de pandemias.
[000110] Consistentemente com as observações acima, epitopos de células T CD4 compartilhadas por diferentes cepas de influenza foram identificados em humanos (Gelder C et al. 1998, Int Immunol. 10(2):211-22; Gelder CM et al. 1996 J Virol. 70(7):4787-90; Gelder CM et al. 1995 J Virol. 1995 69(12):7497-506).
[000111] Devido a suas propriedades imunogênicas, a composição reivindicada será capaz de estabelecer uma estratégia de vacinação pró-ativa contra a ameaça de uma pandemia de influenza humana, incluindo a formação de estoques de vacina pré-pandêmica de forma a melhor preparar contra o início de uma pandemia.
[000112] Especificamente, a vacina pré-pandêmica é uma que foi produzida, por exemplo, por meio do uso de engenharia genética invertida, usando-se uma cepa de H5N1 (gripe aviária) similar àquelas que circulam correntemente na população aviária. A imunidade desenvolvida em resposta à vacina pré-pandêmica permitirá ao sistema imunológico ser 'preparado' ou 'educado' atingindo um estado de prontidão e, com isto, permitindo desenvolvimento mais rápido de respostas imunes protetoras após encontrar a cepa de vírus pandêmica efetiva, levando a uma menor suscetibilidade a uma cepa pandêmica da influenza relacionada. Uma vez que uma pandemia foi declarada pela OMS, e a cepa pandêmica final foi identificada (se for uma cepa desviada), a vacina pré-pandêmica também permitirá uma resposta imune mais rápida para a vacina pandêmica quando esta última se tornar disponível.
[000113] Em uma concretização específica, a composição adjuvantada pode oferecer o benefício adicional de proporcionar melhor proteção contra cepas circulantes que sofreram uma alteração maior (como recombinação gênica, por exemplo, entre duas espécies diferentes) na hemaglutinina (desvio antigênico) contra a qual vacinas correntemente disponíveis não têm eficácia.
[000114] A composição pode compreender um adjuvante adicional, em particular, um adjuvante de ligante TRL-4, vantajosamente um derivado não-tóxico do lipídio A. Um ligante TRL-4 vantajoso é o lipídio de monofosforila 3 des-O-acilado A (3D-MPL). Outros ligante TLR-4 vantajosos são lipopolissacarídeo (LPS) e derivados, MDP (dipeptídeo de muramila) e proteína F do RSV.
[000115] Em uma concretização, a composição pode incluir adicionalmente um ligante de receptor toll-like 4 (TLR, Toll like receptor), como um derivado não-tóxico de lipídio A, particularmente lipídeo de monofosforila A ou, mais particularmente, lipídio de monofosforila 3- desacilado A (3D-MPL).
[000116] 3D-MPL é comercializado sob a marca comercial MPL® pela Corixa corporation, agora GSK (aqui MPL) e promove primariamente respostas de células T CD4+ com um fenótipo IFN-y (Th1). Ele pode ser produzido de acordo com os métodos divulgados na GB 2 220 211 A. Quimicamente, ele é uma mistura de lipídeo de monofosforila 3-desacilado A com 3, 4, 5 ou 6 cadeias aciladas. Em particular, nas composições da presente invenção usa-se 3 D-MPL de partículas pequenas. 3D-MPL de partículas pequenas tem um tamanho de partículas tal que pode ser filtrado de forma estéril através de um filtro de 0,22 μm. Referidas preparações são descritas no WO94/21292 e no Exemplo II.
[000117] 3D-MPL pode ser usado, por exemplo, numa quantidade de 1 a 100 μg (peso/volume) por dose de composição, vantajosamente numa quantidade de 10 a 50 μg (peso/volume) por dose de composição. Uma quantidade vantajosa de 3D-MPL é, por exemplo, qualquer uma de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, ou 50 μg (peso/volume) por dose de composição. De maneira vantajosa, a quantidade de 3D-MPL compreende de 25 a 75 μg (peso/volume) por dose de composição. Usualmente uma dose de composição compreenderá de cerca de 0,5 ml a cerca de 1 ml. Uma dose de vacina típica compreende de 0,5 ml, 0,6 ml, 0,7 ml, 0,8 ml, 0,9 ml ou 1 ml. Em uma concretização vantajosa, uma concentração final de 50 μg de 3D-MPL está contida por ml de composição de vacina, ou 25 μg por 0,5 ml de dose de vacina. Em outras concretizações vantajosas, uma concentração final de 35,7 μg ou 71,4 μg de 3D-MPL está contida por ml de vacina composição. Especificamente, um volume de dose de vacina de 0,5 ml contém 25 μg ou 50 μg de 3D-MPL por dose.
[000118] A dose de MPL é capaz, vantajosamente, de incrementar uma resposta imune a um antígeno em um humano. Em particular, uma quantidade vantajosa de MPL é aquela que melhora o potencial imunológico da composição, em comparação com a composição não-adjuvantada, ou, em comparação com a composição adjuvantada com outra quantidade de MPL, sendo aceitável do ponto de vista de um perfil de reatogenicidade.
[000119] Derivados sintéticos de lipídeo A são conhecidos, sendo que alguns são descritos como agonistas de TLR-4, e incluem, embora sem limitação: OM174(2-desóxi-6-o-[2-desóxi-2-[(R)-3-dodecanoiloxitetra- decanoilamino]-4-o-fosfono-β-D-glucopiranosil]-2-[(R)-3- hidroxitetradecanoilamino]-α-D-glucopiranosildi-hidrogeno fosfato), (WO 95/14026) OM 294 DP (3S,9 R)-3--[(R)-dodecanoiloxitetradecanoilamino]-4-oxo-5- aza-9(R)-[(R)-3-hidroxitetradecanoilamino]decan-1,10-diol,1,10-bis(di- hidrogeno fosfato) (WO 99/64301 e WO 00/0462) OM 197 MP-Ac DP (3S-, 9R)-3-[(R)-dodecanoiloxitetradecanoilamino]- 4-oxo-5-aza-9-[(R)-3-hidroxitetradecanoilamino]decan-1,10-diol,1-di- hidrogeno fosfato 10-(6-aminoexanoato) (WO 01/46127)
[000120] Outros ligantes de TLR-4 vantajosos são, por exemplo, lipopolissacarídeo e seus derivados, dipeptídeo de muramila (MDP) ou proteína F do vírus respiratório sincicial.
[000121] Outro imunoestimulante vantajoso para uso na presente invenção é Quil A e seus derivados. Quil A é uma preparação de saponina isolada da árvore sul-americana Quilaja Saponaria Molina e foi descrita pela primeira vez por Dalsgaard et al. em 1974 ("Saponin adjuvants", Archiv. für die gesamte Virusforschung, vol. 44, Springer Verlag, Berlim, p. 243-254) como apresentando atividade adjuvante. Fragmentos purificados de Quil A foram isolados por meio de HPLC e que conservam atividade adjuvante sem a toxicidade associada com Quil A (EP 0 362 278), por exemplo, QS7 e QS21 (também conhecido como QA7 e QA21). QS-21 é uma saponina natural derivada da casca da Quillaja saponaria Molina, que induz células T citotóxicas CD8+ (CTLs), células Th1 e uma resposta de anticorpos IgG2a predominante, e é uma saponina vantajosa no contexto da presente invenção.
[000122] Descreveu-se formulações particulares de QS21 que são particularmente vantajosas, sendo que estas formulações são particularmente vantajosas, e estas formulações compreendem adicionalmente um esterol (WO96/33739). As saponinas que fazem parte da presente invenção podem encontrar-se em forma de uma emulsão óleo-em-água (WO 95/17210).
[000123] Um aspecto da presente invenção proporciona o uso de um antígeno de influenza na fabricação de uma composição imunogênica de influenza para revacinação de humanos previamente vacinados com uma composição de influenza monovalente como reivindicado aqui, ou com referida composição de influenza monovalente compreendendo uma cepa de influenza variante, formulada com um adjuvante de emulsão óleo-em-água como definido aqui.
[000124] Tipicamente, a revacinação é realizada pelo menos 6 meses após a primeira vacinação/vacinações, vantajosamente de 8 a 14 meses após, vantajosamente em torno de 10 a 12 meses após.
[000125] A composição imunogênica para revacinação (a composição de reforço) pode conter qualquer tipo de preparação de antígeno, quer inativada, recombinante ou viva atenuada. Ela pode conter o mesmo tipo de preparação de antígeno, i.e. vírus da influenza dividido ou preparação dividida de vírus da influenza antigênica do mesmo, um vírion inteiro, um virosoma ou uma vacina de HA e NA purificada (subunidade), como a composição imunogênica usada para a primeira vacinação. Alternativamente, a composição de reforço pode conter outro tipo de antígeno de influenza, i.e. vírus da influenza dividido ou preparação dividida de vírus da influenza antigênica do mesmo, um vírion inteiro, um virosoma ou vacina de HA e NA purificada (subunidade), que não aquela usada para a primeira vacinação. De maneira vantajosa, usa-se um vírus dividido ou uma vacina de vírion inteiro.
[000126] Assim, em uma concretização, a invenção proporciona o uso de um vírus da influenza ou preparação antigênica do mesmo na fabricação de uma composição imunogênica para revacinação de humanos previamente vacinados com uma composição imunogênica pandêmica monovalente como reivindicada aqui.
[000127] A composição de reforço pode ser adjuvantada ou não- adjuvantada. Em uma concretização, a composição para re-vacinação não é adjuvantada e é uma vacina de influenza clássica contendo três antígenos de vírions divididos inativados preparados a partir de cepas recomendadas pela OMS da estação de influenza apropriada, como Fluarix™/α-Rix®/Influsplit® administrada intramuscularmente.
[000128] Em outra concretização, a composição para revacinação é adjuvantada. De maneira vantajosa, a composição de reforço compreende um adjuvante de emulsão óleo-em-água, em particular um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante. Especificamente, referido adjuvante de emulsão óleo-em-água compreende pelo menos um óleo metabolizável numa quantidade de 0,5% a 20% do volume total, e apresenta gotículas de óleo das quais pelo menos 70% em intensidade têm diâmetros inferiores a 1 μm. Alternativamente, a composição de reforço compreende um adjuvante de alume, quer hidróxido de alumínio ou fosfato de alumínio ou uma mistura de ambos.
[000129] Em uma concretização, a primeira vacinação é realizada com uma composição de influenza pandêmica como definido aqui, vantajosamente uma composição de influenza dividida, e a revacinação é realizada como a seguir.
[000130] Em uma concretização específica, a composição imunogênica para revacinação contém um vírus da influenza ou preparação antigênica do mesmo que compartilha epitopos de células T CD4 comuns com o vírus da influenza ou preparação antigênica do mesmo usado para a primeira vacinação. Um epitopo de células T CD4 comum deve significar peptídeos/seqüências/epitopos de diferentes antígenos que podem ser reconhecidos pela mesma célula CD4 (ver exemplos de epitopos descritos em: Gelder C et al. 1998, Int Immunol. 10(2):211-22; Gelder CM et al. 1996 J Virol. 70(7):4787-90; Gelder CM et al. 1995 J Virol. 1995 69(12):7497-506).
[000131] Em uma concretização de acordo com a invenção, a composição de reforço é uma composição de influenza monovalente compreendendo uma cepa de influenza que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia. Cepas vantajosas compreendem, embora sem limitação: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1. Referida cepa pode ser igual àquela, ou a uma destas, presente(s) na composição usada para a primeira vacinação. Em uma concretização alternativa, referida cepa pode ser uma cepa variante, i.e. uma cepa desviada, da cepa presente na composição usada para a primeira vacinação. Em outra concretização específica, a composição para revacinação é uma vacina de influenza polivalente. Em particular, quando a composição de reforço é uma vacina polivalente, como uma vacina divalente, trivalente ou quadrivalente, pelo menos uma cepa está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia. Em uma concretização específica, duas ou mais cepas na composição de reforço são cepas pandêmicas. Em outra concretização específica, a pelo menos uma cepa pandêmica na composição de reforço é do mesmo tipo que aquela, ou uma daquelas, presentes na composição usada para a primeira vacinação. Em uma concretização alternativa, a pelo menos uma cepa pode ser uma cepa variante, i.e. uma cepa desviada, da pelo menos uma cepa pandêmica presente na composição usada para a primeira vacinação.
[000132] Assim, em outro aspecto da presente invenção, proporciona-se o uso de um vírus da influenza ou preparação antigênica do mesmo, de uma primeira cepa de influenza pandêmica, na fabricação de uma composição imunogênica como definido aqui, para proteção contra infecções por influenza causada por uma cepa de influenza que é uma variante de referida primeira cepa de influenza.
[000133] Assim, em outro aspecto da presente invenção, proporciona-se o uso de: (a) um vírus da influenza ou preparação antigênica do mesmo, de uma primeira cepa de influenza, e (b) um adjuvante de emulsão óleo-em-água como definido aqui na fabricação de uma composição imunogênica como definido aqui, para proteção contra infecções por influenza causadas por uma cepa de influenza que é uma variante de referida primeira cepa de influenza.
[000134] A composição para revacinação pode ser adjuvantada ou não.
[000135] Tipicamente, uma composição de reforço, onde usada, é dada na próxima estação de influenza, p. ex., aproximadamente um ano após a primeira composição imunogênica. A composição de reforço também pode ser dada em anos alternados (terceira, quarta, quinta vacinação etc). A composição de reforço pode ser a mesma que a composição usada para a primeira vacinação. De maneira vantajosa, a composição de reforço contém um vírus da influenza ou preparação antigênica do mesmo que é uma cepa variante do vírus da influenza usado para a primeira vacinação. Em particular, as cepas virais de influenza ou preparação antigênica das mesmas são selecionadas de acordo com o material de referência distribuído pela Organização Mundial da Saúde, de tal forma que sejam adaptadas à cepa de influenza que está circulando no ano da revacinação. De maneira vantajosa, a primeira vacinação é realizada no momento da declaração de uma pandemia, e a revacinação é feita mais tarde. De maneira vantajosa, a revacinação é realizada com uma vacina compreendendo uma cepa de influenza (p. ex., H5N1 Vietnam) que é do mesmo subtipo que aquele usado para a primeira vacinação (p. ex., H5N1 Vietnam). Em uma concretização específica, a revacinação é realizada com uma cepa desviada do mesmo subtipo, p. ex., H5N1 Indonésia. Em outra concretização, referida cepa de influenza usada para a revacinação é uma cepa desviada, i.e. é diferente daquela usada para a primeira vacinação, p. ex., ele apresenta um subtipo diferente de HA ou NA, como H5N2 (mesmo subtipo de HA que H5N1, mas diferente do subtipo de NA) ou H7N1 (subtipo diferente de HA de H5N1, mas com o mesmo subtipo de NA).
[000136] O antígeno de influenza ou composição antigênica usada na revacinação compreende vantajosamente um adjuvante ou uma emulsão óleo-em-água, vantajosamente como descrita acima. O adjuvante pode ser um adjuvante de emulsão óleo-em-água como descrito aqui, acima, que é vantajosa, contendo opcionalmente um adjuvante adicional, como ligante de TLR-4, como 3D-MPL ou uma saponina, ou pode ser outro adjuvante vantajoso, como alume ou alternativas para alume, como polifosfazeno, por exemplo.
[000137] De maneira vantajosa, a revacinação induz qualquer um, vantajosamente dois, ou todos, dentre os seguintes: (i) uma resposta CD4 melhorada contra o vírus da influenza ou preparação antigênica do mesmo, ou (ii) uma resposta melhorada de memória de células B ou (iii) uma resposta humoral melhorada, em comparação com a resposta equivalente induzida após uma primeira vacinação com o vírus da influenza não-adjuvantado ou preparação antigênica do mesmo. De maneira vantajosa, as respostas imunológicas induzidas após a revacinação com o vírus da influenza adjuvantado ou preparação antigênica do mesmo como definido aqui, são mais substanciais do que a resposta correspondente induzida após a revacinação com a composição não-adjuvantada. De maneira vantajosa, as respostas imunológicas induzidas após a revacinação com um vírus de influenza não-adjuvantado, vantajosamente dividido, são maiores na população vacinada primeiro com a composição de influenza adjuvantada, vantajosamente dividida, do que a resposta correspondente na população vacinada primeiro com a composição de influenza não-adjuvantada, vantajosamente dividida.
[000138] Em um aspecto de acordo com a invenção, a revacinação dos sujeitos com uma composição de reforço compreendendo um vírus da influenza e um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante, como definido aqui, acima, mostrará titulações maiores de anticorpos do que os valores correspondentes no grupo de pessoas vacinado primeiro com a composição não-adjuvantada, e reforçado com a composição não-adjuvantada. O efeito do adjuvante no incremento da resposta de anticorpos à revacinação é particularmente importante na população mais idosa que, como se sabe, apresenta uma menor resposta à vacinação ou infecção por vírus de influenza. Em particular, o benefício associado com a composição adjuvantada também será marcante em termos de melhoramento da resposta de células T CD4 após a revacinação.
[000139] A composição adjuvantada da invenção será capaz de induzir uma melhor responsividade cruzada contra cepa desviada (a cepa de influenza da estação de influenza seguinte), em comparação com a proteção conferida pela vacina de controle. Referida responsividade cruzada mostrou uma maior persistência, em comparação com aquela obtida com a formulação não-adjuvantada. O efeito do adjuvante no incremento da responsividade cruzada contra cepa desviada é importante em uma situação de pandemia.
[000140] Em uma concretização adicional a invenção refere-se a um regime de vacinação em que a primeira vacinação é realizada com uma composição de influenza, vantajosamente uma composição de influenza dividida, contendo uma cepa de influenza que potencialmente poderia causar uma pandemia e a revacinação é realizada com uma composição, quer monovalente ou polivalente, compreendendo pelo menos uma cepa circulante, quer seja uma cepa de pandemia ou uma cepa clássica.
[000141] Este desvio antigênico reside principalmente em regiões do epitopo das proteínas de superfície viral hemaglutinina (HA) e neuraminidase (NA). É de conhecimento geral que qualquer diferença em epitopos de células B e CD4 entre diferentes cepas de influenza, sendo usados pelo vírus para evadir-se da resposta adaptativa do sistema imunológico do hospedeiro, desempenhará um papel importante na vacinação contra influenza.
[000142] Epitopos de células T CD4 compartilhados por diferentes cepas de influenza foram identificados em humanos (ver, por exemplo: Gelder C et al. 1998, Int Immunol. 10(2):211-22; Gelder CM et al. 1996 J Virol. 70(7):4787-90; e Gelder CM et al. 1995 J Virol. 1995 69(12):7497- 506).
[000143] Em uma concretização específica, a revacinação é realizada usando-se uma composição de reforço que contém um vírus da influenza ou preparação antigênica do mesmo que compartilha epitopos de células T CD4 comuns com o antígeno de vírus da influenza ou preparação antigênica do mesmo usado para a primeira vacinação. A invenção refere-se, portanto, ao uso da composição imunogênica compreendendo uma pandemia vírus da influenza ou preparação antigênica do mesmo e um adjuvante de emulsão óleo-em-água, em particular um adjuvante de emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, como alfa tocoferol, e um agente emulsificante, na fabricação de um componente de primeira vacinação de uma vacina de doses múltiplas, sendo que a vacina de doses múltiplas compreende adicionalmente, como uma dose de reforço, um vírus da influenza ou preparação antigênica do mesmo que compartilha epitopos de células T CD4 comuns com o antígeno de vírus da influenza pandêmica ou preparação antigênica de vírus do mesmo na dose dada na primeira vacinação.
[000144] A composição da invenção pode ser administrada por meio de qualquer via de fornecimento vantajosa, como intradérmica, mucosal p. ex., intranasal, oral, intramuscular ou subcutânea. Outras vias de fornecimento também são bem conhecidas na arte.
[000145] A via de fornecimento intramuscular é particularmente vantajosa para a composição de influenza adjuvantada. A composição de acordo com a invenção pode ser apresentada em um recipiente de monodose, ou alternativamente, um recipiente de doses múltiplas, particularmente vantajoso para uma vacina de pandemia. Neste caso, está presente tipicamente um conservante antimicrobiano, como o tiomersal, para prevenir contaminação durante o uso. Uma concentração de tiomersal de 5 μg/dose de 0,5 ml (i.e. 10 μg/ml) ou 10 μg/dose de 0,5 ml (i.e. 20 μg/ml) está presente vantajosamente. Poderia-se usar um dispositivo de fornecimento i.m., como um dispositivo de injeção de jato de líquido isento de agulha, por exemplo, o Biojector 2000 (Bioject, Portland, OR). Alternativamente, um dispositivo injetor tipo caneta, como o que é usado para fornecimento doméstico de epinefrina, poderia ser usado para permitir a auto-administração da vacina. O uso de referidos dispositivos pode ser particularmente utilizável em campanhas de imunização de grande escala, como poderia ser necessário durante uma pandemia.
[000146] O fornecimento intradérmico é outra via vantajosa. Qualquer dispositivo vantajoso pode ser usado para fornecimento intradérmico, por exemplo, dispositivos de agulha curta, como aqueles descritos nas US 4.886.499, US 5.190.521, US 5.328.483, US 5.527.288, US 4.270.537, US 5.015.235, US 5.141.496, US 5.417.662. Vacinas intradérmicas também podem ser administradas por meio de dispositivos que limitam o comprimento de penetração efetivo de uma agulha na pele, como aquele descrito no WO99/34850 e EP1092444, incorporada aqui por referência, e seus equivalentes funcionais. Também são vantajosos dispositivos de injeção a jato que fornecem vacinas líquidas na derme por meio de um injetor de jato líquido ou via uma agulha que perfura o stratum corneum e produz um jato que atinge a derme. Dispositivos de injeção por jato encontram-se descritos, por exemplo, na US 5.480.381, US 5.599.302, US 5.334.144, US 5.993.412, US 5.649.912, US 5.569.189, US 5.704.911, US 5.383.851, US 5.893.397, US 5.466.220, US 5.339.163, US 5.312.335, US 5.503.627, US 5.064.413, US 5.520.639, US 4.596.556, US 4.790.824, US 4.941.880, US 4.940.460, WO 97/37705 e WO 97/13537. Também são vantajosos dispositivos de fornecimento balísticos de pó/partículas que usam gás comprimido para acelerar a vacina em forma de pó através das camadas exteriores da pele até a derme. Adicionalmente, é possível usar seringas convencionais no método clássico de mantoux de administração intradérmica.
[000147] Outra via de administração vantajosa é a via subcutânea. É possível usar qualquer dispositivo vantajoso para fornecimento subcutâneo, por exemplo, uma agulha clássica. De maneira vantajosa, usa-se um dispositivo injetor de jato isento de agulha, como aquele publicado no WO 01/05453, WO 01/05452, WO 01/05451, WO 01/32243, WO 01/41840, WO 01/41839, WO 01/47585, WO 01/56637, WO 01/58512, WO 01/64269, WO 01/78810, WO 01/91835, WO 01/97884, WO 02/09796, WO 02/34317. De maneira vantajosa, referido dispositivo é pré-enchida com a formulação de vacina líquida.
[000148] Alternativamente, a vacina é administrada intranasalmente. Tipicamente, a vacina é administrada localmente na área naso-faríngea, vantajosamente sem ser inalada nos pulmões. É desejável usar um dispositivo de fornecimento intranasal que fornece a formulação de vacina na área naso-faríngea, sem ou substancialmente sem entrar nos pulmões.
[000149] Dispositivos vantajosos para administração intranasal das vacinas de acordo com a invenção são dispositivos de spray. Dispositivos de spray nasal comercialmente obteníveis vantajosos incluem Accuspray™ (Becton Dickinson). Nebulizadores produzem um spray muito fino que pode ser facilmente inalado nos pulmões e, portanto, não atingem eficientemente a mucosa nasal. Portanto, nebulizadores não são preferidos.
[000150] Dispositivos de spray vantajosos para uso intranasal são dispositivos para os quais o desempenho do dispositivo não depende da pressão aplicada pelo usuário. Estes dispositivos são conhecidos como dispositivos de limiar de pressão. Líquido é liberado pelo bico ejetor somente quando se aplica uma pressão de limiar. Estes dispositivos tornam mais fácil obter um spray com um tamanho regular das gotículas. Dispositivos de limiar de pressão vantajosos para uso com a presente invenção são conhecidos na arte e são descritos, por exemplo, no WO 91/13281 e EP 311 863 B e EP 516 636, incorporados aqui por referência. Referidos dispositivos são comercialmente obteníveis da Pfeiffer GmbH e também são descritos por Bommer, R. Pharmaceutical Technology Europe, setembro de 1999.
[000151] Dispositivos intranasais vantajosos produzem gotículas (medidas usando-se água como o líquido) na faixa de 1 a 200 μm, vantajosamente de 10 a 120 μm. Abaixo de 10 μm há um risco de inalação, portanto é desejável que não mais de cerca de 5% das gotículas se encontrem abaixo de 10 μm. Gotículas acima de 120 μm não se espalham tão bem quanto gotículas menores, de modo que é desejável que não mais de cerca de 5% das gotículas excedam 120 μm.
[000152] Fornecimento de dose dupla [bi-dose] é uma característica vantajosa adicional de um sistema de fornecimento intranasal para uso com as vacinas de acordo com a invenção. Dispositivos de dose dupla [bi-dose] contêm duas subdoses de uma dose de vacina simples, uma subdose para administração em cada narina. De uma maneira geral, as duas subdoses estão presentes em uma câmara simples e a construção do dispositivo permite o fornecimento eficiente de uma subdose simples a cada momento. Alternativamente, é possível usar um dispositivo de monodose para administrar as vacinas de acordo com a invenção.
[000153] Alternativamente, a via de vacinação epidérmica ou transdérmica também é contemplada na presente invenção.
[000154] Em um aspecto da presente invenção, a composição imunogênica adjuvantada para a primeira administração pode ser dada intramuscularmente, e a composição de reforço, quer adjuvantada ou não, pode ser administrada por meio de uma via diferente, por exemplo, intradérmica, subcutânea ou intranasal. Em uma concretização específica, a composição para a primeira administração contém uma quantidade de HA inferior a 15 μg para a cepa de influenza pandêmica, e a composição de reforço pode conter uma quantidade padrão de 15 μg ou, vantajosamente, uma pequena quantidade de HA, i.e. abaixo de 15 μg, que, dependendo da via de administração, pode ser dada em um volume menor.
[000155] A população-alvo a vacinar é toda a população, p. ex., adultos jovens saudáveis (p. ex., com idades de 18 a 60), idosos (tipicamente, com idades acima de 60) ou bebês/crianças. Em particular, a população-alvo pode ser imunocomprometida. Humanos imunocomprometidos são geralmente menos capazes de responder a um antígeno, em particular a um antígeno de influenza, em comparação com adultos saudáveis.
[000156] Em um aspecto de acordo com a invenção, a população- alvo é uma população que é despreparada contra a influenza, quer por ser não- tratada (como relativamente a uma cepa de pandemia), ou por ter falhado em responder previamente a infecção por influenza ou vacinação. De maneira vantajosa, a população-alvo é de pessoas idosas com, vantajosamente, idades de pelo menos 60, ou 65 anos e acima, adultos mais jovens em alto risco (i.e. entre 18 e 60 anos de vida), como pessoas que trabalham em instituições de saúde, ou aqueles adultos jovens com um fator de risco, como doença cardiovascular e pulmonar, ou diabetes. Outra população-alvo compreende todas as crianças com 6 meses de vida e acima, especialmente crianças de 6 a 23 meses de vida que experimentam uma taxa de hospitalização relativamente alta relacionada com influenza.
[000157] De maneira vantajosa, na maior parte dos casos, as composições imunogênicas de acordo com a presente invenção consistem de uma dose padrão injetável de 0,5 ml, e contém menos de 15 μg de componente de antígeno de hemaglutinina de uma cepa de influenza pandêmica, conforme medido por meio de imunodifusão radial simples (SRD, single radial immunodiffusion) (J. M. Wood et al.: J. Biol. Stand. 5 (1977) 237-247; J. M. Wood et al., J. Biol. Stand. 9 (1981) 317-330). De maneira vantajosa, o volume de dose da vacina será entre 0,5 ml e 1 ml, em particular um padrão de 0,5 ml, ou 0,7 ml de volume de dose de vacina. A adaptação ligeira do volume da dose será feita dependendo da concentração de HA na amostra volumétrica original e dependendo também da via de fornecimento com doses menores sendo dadas pela via intranasal ou intradérmica.
[000158] De maneira vantajosa, referida composição imunogênica contém uma pequena quantidade de antígeno HA - p. ex., de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 μg de HA por cepa de influenza ou que não excede 15 μg de HA por cepa. Referida baixa quantidade de quantidade de HA pode ser tão baixa quanto o for exequível na prática, desde que seja possível formular uma vacina que atenda a critérios internacionais, p. ex., EU ou FDA, quanto à eficácia, como detalhado abaixo (ver Tabela 1 e os parâmetros específicos, como indicado). Uma baixa quantidade de HA é entre de 1 a 7,5 μg de HA por cepa de influenza, vantajosamente entre de 3,5 a 5 μg, como 3,75 ou 3,8 μg de HA por cepa de influenza, tipicamente de cerca de 5 μg de HA por cepa de influenza. Outra quantidade vantajosa de HA é entre 0,1 e 5 μg de HA por cepa de influenza, vantajosamente entre 1,0 e 2 μg de HA por cepa de influenza, como 1,9 μg de HA por cepa de influenza.
[000159] De maneira vantajosa, uma dose de vacina de acordo com a invenção, em particular uma vacina com baixa quantidade de HA, pode ser proporcionada em um volume menor do que as vacinas de gripe divididas injetadas convencionais, que geralmente são em torno de 0,5, 0,7 ou 1 ml por dose. As doses de baixo volume de acordo com a invenção são, vantajosamente, abaixo de 500 μl, tipicamente abaixo de 300 μl e, vantajosamente, não acima de cerca de 200 μl ou menos por dose.
[000160] Assim, uma dose de vacina vantajosa com baixo volume de acordo com um aspecto da invenção é uma dose com uma dose baixa de antígeno em um baixo volume, p. ex., de cerca de 15 μg ou cerca de 7,5 μg de HA ou cerca de 3,0 μg de HA (por cepa) em um volume de cerca de 200 μl.
[000161] A droga contra influenza da invenção atende vantajosamente determinados critérios para vacinas. Padrões são aplicados internacionalmente para medir a eficácia de vacinas contra influenza. Variáveis sorológicas são avaliadas de acordo com critérios da Agência Européia para a Avaliação de Produtos Medicinais [European Agency for the Evaluation of Medicinal Products] para uso humano (CHMP/BWP/214/96, Committee for Proprietary Medicinal Products (CPMP). Nota para harmonização de exigências para vacinas de influenza, 1997. CHMP/BWP/214/96 circular N°96-0666:1-22) para ensaios clínicos relacionados com procedimentos de licença anuais de vacinas de influenza (Tabela 1). As exigências são diferentes para populações adultas (de 18 a 60 anos) e populações idosas (>60 anos) (Tabela 1). Para vacinas contra influenza interpandêmica, pelo menos uma das avaliações (fator de soroconversão, taxa de soroconversão, taxa de soroproteção) deveria atender as exigências Européias, para todas as cepas de influenza incluídas na vacina. A proporção de titulações iguais ou maiores a 1:40 é considerada como a mais relevante porque se espera que estas titulações se correlacionem melhor em termos de proteção [Beyer W et al. 1998. Clin Drug Invest.;15:1-12].
[000162] Como especificado na obra "Guideline on dossier structure and content for pandemic influenza vaccine marketing authorisation application" [Diretrizes para estrutura de dossiês e teor de pedido de autorização para comercialização de vacinas para influenza pandêmica] (CHMP/VEG/4717/03, 5 de abril de 2004), na ausência de critérios específicos para vacinas contra influenza derivadas de cepas não-circulantes, antecipa-se que uma vacina candidatada para pandemia deveria (pelo menos) ser capaz de elicitar suficientes respostas imunológicas para atender vantajosamente todos os padrões correntes indicados para vacinas existentes em sujeitos idosos ou adultos não preparados, após duas doses de vacina.
[000163] As composições da presente invenção atendem vantajosamente a pelo menos um destes critérios para a cepa pandêmica incluída na composição (um critério é suficiente para obter aprovação), vantajosamente pelo menos dois, ou tipicamente pelo menos todos os três critérios para proteção como indicado na Tabela 1. Tabela 1 (critérios de CHMP) * Taxa de soroconversão é definida como a proporção de sujeitos em cada grupo apresentando uma titulação pós-vacinação protetora ≥ 1:40. A taxa de soroconversão considerada é simplesmente o% de sujeitos que apresentam uma titulação de HI antes da vacinação de um aumento de pelo menos quatro vezes na quantidade de anticorpo após a vacinação. ** Fator de conversão é definido como o aumento em múltiplos das titulações geométricas médias de HI no soro (GMTs, geometric mean titres) após a vacinação, para cada cepa de vacina. Taxa de proteção é definida como a proporção de sujeitos que foram, ou soronegativos antes da vacinação e que apresentam uma titulação de HI pós- vacinação (protetora) da ordem de > 1:40, ou que foram soropositivos antes da vacinação e apresentam um aumento significativo de 4 vezes na titulação pós-vacinação; isto normalmente é aceito como indicando proteção.
* Taxa de soroconversão é definida como a proporção de sujeitos em cada grupo apresentando uma titulação pós-vacinação protetora ≥ 1:40. A taxa de soroconversão considerada é simplesmente o% de sujeitos que apresentam uma titulação de HI antes da vacinação de um aumento de pelo menos quatro vezes na quantidade de anticorpo após a vacinação. ** Fator de conversão é definido como o aumento em múltiplos das titulações geométricas médias de HI no soro (GMTs, geometric mean titres) após a vacinação, para cada cepa de vacina. Taxa de proteção é definida como a proporção de sujeitos que foram, ou soronegativos antes da vacinação e que apresentam uma titulação de HI pós- vacinação (protetora) da ordem de > 1:40, ou que foram soropositivos antes da vacinação e apresentam um aumento significativo de 4 vezes na titulação pós-vacinação; isto normalmente é aceito como indicando proteção.
[000164] Uma taxa de soroproteção de 70% é definida pela autoridade européia reguladora da saúde (CHMP - Committee for Medicinal Products for Human Use[Comitê para produtos medicinais para uso humano]) é um dos três critérios normalmente exigidos para atenderem uma vacina de influenza sazonal anual, e sendo que o CHMP também espera que se atenda uma vacina candidata para pandemia. No entanto, modelos matemáticos indicaram que uma vacina que só apresenta 30% de eficiência contra determinadas cepas desviadas também podem ser benéficas para auxiliar a reduzir a magnitude de uma pandemia (Ferguson et al, Nature 2006).
[000165] A FDA [Administração Federal para Drogas e Alimentos] publicou uma orientação obrigatória (obtenível junto ao Office of Communication, Training and Manufacturers Assistance[Escritório para comunicação, treinamento e assistência a fabricantes] (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, ou ligando para 1-800-835-4709 ou 301-827-1800, ou na internet [rede mundial de computadores] em http://www.fda.gov/cber/guidelines.htm)sobre Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines [Dados clínicos necessários para corroborar o licenciamento de vacinas para influenza pandêmica] e os critérios propostos também se baseiam nos critérios do CHMP.
[000166] Pontos de viragem apropriados incluem de maneira análoga: 1) o percentual de sujeitos que atingem uma titulação de anticorpos HI > 1:40, e 2) taxas de soroconversão, definido como um aumento de quatro vezes na titulação de anticorpos HI após a vacinação. A titulação geométrica média (GMT, geometric mean titer) deveria ser incluída nos resultados. Estes dados e os intervalos de confiança (CI) de 95% das estimativas pontuais destas avaliações deveriam ser proporcionados.
[000167] Assim, em um aspecto da invenção, proporciona-se para uma composição, método ou uso como reivindicado aqui, sendo que referida resposta imune ou proteção induzida pela administração da composição pandêmica considerada atende a todos os três critérios reguladores da EU para eficácia da vacina contra influenza. De maneira vantajosa, pelo menos um, vantajosamente dois, ou três dos critérios a seguir são atendidos para a cepa pandêmica da composição: -uma taxa de soroconversão de >50%, de >60%, de >70%, vantajosamente de >80% ou >90% na população adulta (com idades de 18 a 60), e/ou, vantajosamente, também na população de idosos (com idades >60 anos); - uma taxa de proteção de >75%, de >80%, de >85%, vantajosamente de >90% na população adulta (com idades de 18 a 60), e/ou, vantajosamente, também na população de idosos (com idades de >60 anos); - um fator de conversão de >4,0, de >5,0, de >6,0, de >7,0, de >8,0, de >9,0 ou de 10 ou acima de 10 na população adulta (com idades de 18 a 60), e/ou, vantajosamente, também na população de idosos (com idades de >60 anos).
[000168] Em uma concretização específica, a composição de acordo com a invenção atenderá tanto uma taxa de soroconversão de >60%, ou >70%, ou vantajosamente >80% e uma taxa de proteção de >75%, vantajosamente de >80% na população adulta. Em outra concretização específica, a composição de acordo com a invenção atenderá tanto um fator de conversão de >5,0, ou de >7,0 ou vantajosamente >10,0 e uma taxa de soroconversão de >60%, ou >70%, ou vantajosamente >80% na população adulta. Em outra concretização específica, a composição de acordo com a invenção atenderá tanto um fator de conversão de >5,0, ou >7,0 ou vantajosamente >10,0, e uma taxa de proteção de >75%, vantajosamente >80% na população adulta. Em outra concretização específica adicional a composição de acordo com a invenção atenderá tanto um fator de conversão de 10,0 ou acima, uma taxa de soroconversão de 80% ou acima, e uma taxa de proteção de 80% ou acima.
[000169] Em outra concretização, a vacina reivindicada atenderá uma taxa de soroproteção de pelo menos 30% contra vacinas desviadas, vantajosamente de pelo menos 40%, ou >50% ou >60% contra cepas desviadas. De maneira vantajosa, a taxa de soroproteção será de >70%, ou vantajosamente >80% contra cepas desviadas.
[000170] De maneira vantajosa, qualquer um ou todos esses critérios também são atendidos para outras populações, como em crianças e em qualquer população imunocomprometida.
[000171] De maneira vantajosa, a(s) resposta(s) vantajosa(s) é/são obtida(s) após uma dose, ou tipicamente após duas doses. É uma vantagem particular da composição reivindicada que a resposta imune é obtida após apenas uma dose de vacina adjuvantada. Assim, proporciona-se, em um aspecto da invenção, o uso de um antígeno de preparação de vírus de influenza pandêmica não-vivo, em particular uma preparação de vírus da influenza dividida, na fabricação de uma composição de vacina para uma vacinação de uma dose só contra influenza, sendo que a vacinação de uma dose só gera uma resposta imune que atende pelo menos um, vantajosamente dois ou três, exigências reguladoras internacionais para vacinas de influenza. Em outra concretização particular referida vacinação de dose única também gera, ou gera adicionalmente, resposta imune de células T CD4 e/ou uma resposta de memória de células B que é maior do que aquela obtida com a vacina não-adjuvantada. Em uma concretização particular referida resposta imune é uma resposta de anticorpos de reação cruzada ou uma resposta de células T CD4 de reação cruzada ou ambas. Em uma concretização específica, o paciente humano é imunologicamente despreparada (i.e., não apresenta imunidade preexistente) para a cepa de vacinação. Especificamente, a composição de vacina contém uma baixa quantidade de antígeno HA. Especificamente, a composição de vacina é como definida aqui. Em particular, as propriedades imunogênicas da composição de vacina são como definidas aqui. De maneira vantajosa, a vacina é administrada intramuscularmente.
[000172] Com relação à composição para revacinação, quando ela é uma composição polivalente, pelo menos dois ou todos os três critérios precisarão ser atendidos para todas as cepas, particularmente para uma nova vacina, como uma nova vacina para fornecimento através de uma via diferente. Em algumas circunstâncias, dois critérios podem ser suficientes. Por exemplo, pode ser aceitável que dois dos três critérios sejam atendidos por todas as cepas, enquanto que o terceiro critério é atendido por algumas, mas não todas as cepas (p. ex., duas das três cepas).
[000173] Em um aspecto adicional, a invenção proporciona um método de projetar uma vacina para doenças conhecidas por serem curadas ou tratadas por meio de uma ativação de células T CD4+, compreendendo 1) selecionar um antígeno contendo epitopos CD4+, e 2) combinar referido antígeno com um adjuvante de emulsão óleo-em-água como definido aqui acima, sendo que referida vacina, ao ser administrada a referido mamífero, é capaz de induzir uma resposta incrementada de células T CD4 em referido mamífero.
[000174] O ensinamento de todas as referências no presente pedido, incluindo pedidos de patentes e patentes concedidas, é incorporado aqui plenamente por referência. Qualquer pedido de patente relativamente ao qual este pedido reivindica prioridade é incorporado aqui integralmente por referência da maneira aqui descrita para publicações e referências.
[000175] Para evitar dúvidas, os termos 'compreendendo', 'compreendem' e 'compreende' usados aqui pelos inventores devem poder ser opcionalmente substituíveis pelos termos 'consistindo de', 'consistem de', e 'consiste de', respectivamente, em cada caso.
[000176] A invenção será descrita agora com referência aos exemplos não limitantes, a seguir: Exemplo I descreve métodos de leitura imunológica usados em estudos em furões e em humanos. Exemplo II descreve a preparação e caracterização da emulsão óleo-em-água e formulações adjuvantes usadas nos estudos exemplificados. Exemplo III mostra uma avaliação pré-clínica de vacinas contra influenza adjuvantadas e não-adjuvantadas em furões. Exemplo IV descreve um ensaio clínico numa população adulta com idades entre 18 e 60 anos com uma vacina contendo uma preparação de antígeno de influenza dividido de uma cepa H5N1 pandêmica e adjuvante AS03.
[000177] Indica-se abaixo métodos vantajosos que são usados rotineiramente para experimentos realizados com cepas sazonais. O leitor versado na arte compreenderá que pode ser necessário alguma adaptação ou otimização dependendo da cepa de influenza usada.
[000178] Titulações de anticorpos anti-hemaglutinina com relação aos vírus da cepa de influenza são determinadas usando-se o teste de inibição de hemaglutinação (HI). O princípio do teste de HI baseia-se na capacidade de anticorpos anti-influenza específicos inibirem a hemaglutinação de células sanguíneas vermelhas (RBC) de cavalo pela hemaglutinina (HA) do vírus da influenza. Após pré-tratamento de soros (cólera, RDE, inativação com calor,...), diluições duplas de soros são incubadas com 4 unidades de hemaglutinação da cepa de influenza. Células sanguíneas vermelhas de cavalo (adaptação: peru, ou cavalo) são então adicionadas, e classifica-se a inibição da aglutinação. As titulações são expressas como a recíproca da maior diluição de soro que inibiu completamente a hemaglutinação. Como a primeira diluição de soros foi de 1:10, um nível indetectável foi classificado como uma titulação igual a 5. Mais detalhes podem ser encontrados na seção 1.2.1. abaixo.
[000179] A temperatura corporal foi registrada por meio de sensores de temperatura (Star Oddi, Islândia) implantados no dia 14 sob anestesia com mistura de cetamina-rompun-atropina (2,5%-0,25%-0,025%) via uma pequena incisão na linha alba na cavidade peritoneal. A ferida foi fechada com pontos e inspecionada diariamente.
[000180] Esfregaços faríngeos, nasais e retais foram coletados de todos os animais. Esfregaços individuais foram colocados em 3 ml de meio de transporte de vírus (VTM, virus transport medium; PBS estéril ou solução isotônica apropriada, como BSS de Hank contendo antibióticos (100 unidades/ml de penicilina, 100 μg/ml de estreptomicina) e, ou 2% de soro bovino fetal ou 0,5% de gelatina) e armazenado a -800C até a análise. Após necrópsia cranioventral, seções craniodorsal, caudoventral e caudodorsal do pulmão direito de cada animal foram pesadas e armazenadas a -800C até a análise. Seções de pulmão foram homogeneizadas em 3 ml de VTM. Titulações virais foram determinadas com PCR TaqMan específica para H5N1 e cultura de titulação de vírus em células de rim canino Madine Darby (MDCK).
[000181] Para a PCR TaqMan, RNAs de gripe H5N1 viral foram extraídos de amostras de furão e então amplificadas por meio de um ensaio de RT-PCR quantitativo usando-se iniciadores e sondas projetados em uma região conservada do genoma da influenza. Dados foram expressos como unidades de diluição de controle (CDU, Control Diluition Units) e TCID50 por grama de tecido de pulmão ou por ml de esfregaço, respectivamente.
[000182] Unidades de Diluição de Controle (CDU, Control Diluition Units) - CDUs são determinadas a partir de uma curva padrão produzida de um estoque de vírus que é diluído serialmente, com cada diluição sendo submetida a extração com ácido nucleico e amplificação de PCR Taqman da mesma maneira que amostras de teste.
[000183] Para a cultura viral, diluições seriais de todas as amostras foram transferidas para placas de microtitulação contendo meio e células MDCK e, então, incubadas a 350C durante de 5 a 7 dias. Após a incubação, as titulações de desprendimento viral foram determinadas por meio de "Reed and Muench" e expressas como Log TCID50 por grama de tecido de pulmão ou por ml de esfregaço.
[000184] Realizou-se medições de anticorpos neutralizadores em amostras de soro congeladas e, depois, descongeladas. A neutralização de vírus por anticorpos contidos no soro é determinada em um ensaio de microneutralização. Os soros são usados sem tratamento adicional no ensaio. Cada soro é testado em triplicata. Uma quantidade padronizada de vírus é misturada com diluições seriais de soro e incubadas para permitir ligação dos anticorpos com o vírus. Adiciona-se então uma quantidade definida de células MDCK à mistura de vírus e anti-soro e isto foi incubado a 33°C. Após o período de incubação, a replicação de vírus é visualizada por meio de hemaglutinação de células sanguíneas vermelhas de galinha. A titulação de neutralização a 50% de um soro é calculada por meio do método de Reed and Muench (Am.J.Hyg. 1938, 27: 493-497).
[000185] A resposta imune foi determinada medindo-se anticorpos HI usando o método descrito pelo WHO Collaborating Centre for influenza, Centres for Disease Control, Atlanta, E.U.A. (1991).
[000186] Realizou-se medições de titulação de anticorpos em amostras de soro congeladas e, depois, descongeladas com um micrométodo padronizado e validado compreensivamente usando-se 4 unidades inibidoras de hemaglutinação (4 HIU) dos antígenos apropriados e uma suspensão de eritrócitos de 0,5% de ave (ou 0,5% de ave e cavalo para H5N1). Inibidores de soro não-específicos foram removidos com tratamento com calor e enzima destruidora de receptores. Os soros obtidos foram avaliados quanto aos níveis de anticorpos HI. Partindo de uma diluição inicial de 1:10, preparou-se uma série de diluições (por um fator de 2) até uma diluição final de 1:20480. O ponto de viragem da titulação foi considerado como a etapa de maior diluição, o que mostrou inibição completa (100%) de hemaglutinação. Todos os ensaios foram realizados em duplicata.
[000187] Glicoproteínas (hemaglutininas) encontram-se localizadas no envelope viral, e são capazes de aglutinar eritrócitos (células sanguíneas vermelhas) de muitas espécies, p. ex., frangos.
[000188] O teste de inibição de hemaglutinação é realizado em duas etapas: 1. Reação antígeno-anticorpo: o antígeno de influenza (DTA, dialysis test antigen [antígeno de teste para diálise]) reage com os anticorpos do soro do sujeito. 2. Aglutinação de antígeno excessivo: antígeno excessivo reage com células sanguíneas vermelhas adicionadas
[000189] Eritrócitos de cavalos são usados para as cepas H5N1 pandêmicas.
[000190] 0,5% (concentração final) suspensão de células sanguíneas vermelhas de cavalo em tamponador de fosfato contendo 0,5% de BSA (albumina de soro bovino, concentração final).
[000191] Esta suspensão é preparada a cada dia por meio de lavagem da célula sanguínea vermelha com o mesmo tamponador de fosfato e uma etapa de centrifugação subseqüente (10 min, 2000 rpm). Esta etapa de lavagem precisa ser repetida uma vez. Após a adição das células sanguíneas vermelhas de cavalo à mistura de reação de soros de paciente/sujeito e suspensão de vírus, as placas precisam ser incubadas à temperatura ambiente (t.a., 200C +/- 2°C) durante duas horas devido à baixa taxa de sedimentação das células sanguíneas vermelhas de cavalo.
[000192] O ensaio foi realizado placas de microtitulação revestidas com fetuína. Preparou-se um série de diluição de 2 vezes do anti-soro. Esta foi preparada e misturada com uma quantidade padronizada de virus A H3N2, H1N1 ou influenza B. O teste foi baseado na atividade biológica da neuraminidase que libera enzimaticamente ácido neuramínico da fetuína. Após clivagem do ácido neuramínico terminal a β-D-glactose-N-acetil- galactosamina foi desmascarada. Aglutinina de amendoim marcada com peroxidase de rabanete (HRP) de Arachis hypogaea, que se liga especificamente às estruturas de galactose, foi adicionada nos poços. A quantidade de aglutinina ligada pode ser detectada e quantificada em uma reação de substrato com tetrametilbenzidina (TMB). A maior diluição de anticorpos que ainda inibe a atividade de neuraminidase viral em pelo menos 50% foi indicada como a titulação de NI.
[000193] Medições de anticorpos neutralizadores foram realizadas em amostras de soro congeladas e, depois, descongeladas. A neutralização de vírus por anticorpos contidos no soro é determinada em um ensaio de microneutralização. Os soros são usados sem tratamento adicional no ensaio. Cada soro é testado em triplicata. Uma quantidade padronizada de vírus é misturada com diluições seriais de soro e incubada para permitir ligação dos anticorpos ao vírus. Adiciona-se então uma suspensão de células, contendo uma quantidade definida de células MDCK À mistura de vírus e anti-soro, e isto foi incubado a 33°C. Após o período de incubação, a replicação viral é visualizada por meio de hemaglutinação das células sanguíneas vermelhas de frango. A titulação de neutralização a 50% de um soro é calculada por meio do método de Reed and Muench (Am.J;Hyg.1938, 27: 493-497).
[000194] Células T CD4 e CD8 específicas para antígeno do sangue periférico podem ser reestimuladas in vitro para produzir IL-2, CD40L, TNF- alfa e IFN se incubadas com seu antígeno correspondente. Conseqüentemente, células T CD4 e CD8 específicas para antígeno podem ser enumeradas por meio de citometria de fluxo após marcação convencional com imunofluorescência do fenótipo celular, e também produção de citocinas intracelulares. No presente estudo, usa-se antígeno de vacina contra influenza como antígeno para reestimular células T específicas para influenza. Resultados são expressos como uma freqüência de células T CD4 ou CD8 positivas para citocina(s) na sub-população de células T CD4 ou CD8.
[000195] A tecnologia ELISPOT permite a quantificação de células B de memória específicas para um dado antígeno. Células B de memória podem ser induzidas para diferenciar-se em células de plasma in vitroapós cultivo com CpG durante 5 dias. Células de plasma específicas para antígeno geradas in vitro podem, portanto, ser enumeradas usando-se o ensaio ELISPOT. Resumidamente, células de plasma geradas in vitrosão incubadas em placas de cultura revestidas com antígeno. Células de plasma específicas para antígeno formam pontos de anticorpo/antígeno, que podem ser detectados por meio de procedimento imunoenzimático convencional. No presente estudo, usa-se cepas de vacina de influenza ou imunoglobulinas anti- humanas para revestir placas de cultura de forma a enumerar anticorpos específicos para influenza ou células de plasma secretadoras de IgG, respectivamente. Resultados são expressos como uma freqüência de células de plasma secretadoras de anticorpos específicas para influenza nas células de plasma produtoras de IgG. 1.2.6. Métodos estatísticos 1.2.6.1. Pontos de viragem primários • Percentual, intensidade e relação com a vacinação de sinais e sintomas locais e gerais solicitados durante um período de acompanhamento de 7 dias (i.e. dia da vacinação e 6 dias subseqüentes) após vacinação e global. • Percentual, intensidade e relação com a vacinação de sintomas e sinais locais e gerais não solicitados durante um período de acompanhamento de 21 dias (i.e. dia de vacinação e 20 dias subseqüentes) após vacinação e global. • Ocorrência de eventos adversos sérios durante todo o estudo.
[000196] Para a resposta imune humoral: Variáveis observadas: 1 Nos dias 0 e 21: inibição da hemaglutinação do soro (HI) e titulações de anticorpos NI, testados separadamente contra cada um dos três vírus da cepa de influenzas representadas na vacina (anticorpos anti-H1N1, anti-H3N2 & anti-B). 2 Nos dias 0 e 21: titulações de anticorpos neutralizadores, testados separadamente contra cada um dos três vírus das cepas de influenza representados na vacina
[000197] Variáveis derivadas (com intervalos de confiança de 95%): 3 titulações geométricas médias (GMTs, geometric mean titers) de anticorpos HI do soro com intervalos de confiança de 95% (95% CI) pré- e pós-vacinação 4 Taxas de soroconversão* com 95% de CI no dia 21 5 Fatores de conversão**com 95% de CI no dia 21 6 Taxas de soroproteção***com 95% de CI no dia 21 7 GMTs de anticorpos NI no soro (com intervalos de confiança de 95%) em todos os momentos. 8 Taxa de soroconversão definida como o percentual de vacinados que apresentaram um aumento de pelo menos 4 vezes nas titulações de HI no soro no dia 21, em comparação com o dia 0, para cada cepa de vacina. 9 * Fator de conversão definido como o aumento em vezes das GMTs de HI do soro no dia 21, em comparação com o dia 0, para cada cepa de vacina. 10 * Taxa de proteção definida como o percentual de vacinados com uma titulação de HI do soro = 40 após vacinação (para cada cepa de vacina) que é aceita usualmente como indicando proteção.
[000198] Nos dias 0 e 21: freqüência de células CD4/CD8 positivas para citocina por 106 em testes diferentes.
[000199] Cada teste quantifica a resposta de células T CD4/CD8 para: 11 Antígeno de peptídeo de influenza (pf) (a natureza e origem precisa destes antígenos precisa ser dada/explicada 12 Antígeno de influenza dividido (sf) 13 Antígeno de influenza inteiro (wf). Variáveis derivadas: 14 células produzindo pelo menos duas citocinas diferentes (CD40L, IL-2, IFNY, TNFα) 15 células produzindo pelo menos CD40L e outra citocina (IL-2, TNFα, IFNy) 16 células produzindo pelo menos IL-2 e outra citocina (CD40L, TNFα, IFNy) 17 células produzindo pelo menos IFNy e outra citocina (IL-2, TNFα, CD40L) 18 células produzindo pelo menos TNFα e outra citocina (IL-2, CD40L, IFNy)
[000200] A análise de imunogenicidade foi baseada na coorte vacinada total. Para cada grupo de tratamento, calculou-se os seguintes parâmetros (com intervalos de confiança de 95%): • Titulações geométricas médias (GMTs) de titulações de anticorpos HI e NI nos dias 0 e 21 • Titulações geométricas médias (GMTs) de titulações de anticorpos neutralizadores nos dias 0 e 21. • Fatores de conversão no dia 21. • Taxas de soroconversão (SC) no dia 21 definidas como o percentual de vacinados que apresentam um aumento de pelo menos 4 vezes nas titulações de HI do soro no dia 21, em comparação com o dia 0. • Taxas de proteção no dia 21 definidas como o percentual de vacinados com uma titulação de HI do soro = 1:40. • A freqüência de linfócitos T CD4/CD8 que secretam em resposta foi resumida (estatísticas descritivas) para cada grupo de vacinação, em cada momento (dia 0, dia 21) e para cada antígeno (peptídeo de influenza (pf), influenza dividido (sf) e influenza inteiro (wf)). • Estatísticas descritivas de diferença individual entre respostas em momentos (Pós-Pré) para cada grupo de vacinação e cada antígeno (pf, sf, e wf) em cada um dos 5 testes diferentes. • Um teste não-paramétrico (teste de Kruskall-Wallis) foi usado para comparar as diferenças de localização entre os 3 grupos e o valor p estatístico foi calculado para cada antígeno em cada um de 5 testes diferentes. Todos os testes de significância foram bicaudados. Valores P inferiores a, ou iguais a 0,05 foram considerados como estatisticamente significativos.
[000201] Exceto se indicado de outra forma, a emulsão óleo/água usada nos exemplos subseqüentes constitui-se de uma fase orgânica constituída de 2 óleos (alfa-tocoferol e esqualeno), e uma fase aquosa de PBS contendo Tween 80 como agente emulsificante. Exceto se indicado de outra forma, as formulações emulsão óleo-em-água adjuvante usadas nos exemplos subseqüentes foram preparadas compreendendo o seguinte componente de emulsão óleo-em-água (concentrações finais dadas): 2,5% de esqualeno (v/v), 2,5% de alfa-tocoferol (v/v), 0,9% de monooleato de polioxietileno sorbitol (v/v) (Tween 80), ver WO 95/17210. Esta emulsão, denominada AS03 nos exemplos subseqüentes, foi preparada como a seguir como um concentrado a 2:1.
[000202] Tween 80 é dissolvido em tamponador de solução salina fosfatada (PBS) dando uma solução a 2% no PBS. Para proporcionar 100 ml de emulsão de concentrado a 2:1, 5 g de DL alfatocoferol e 5 ml de esqualeno são submetidos a agitação suficiente para se obter um vórtice para misturação íntima. 90 ml de solução de PBS/Tween são adicionados e misturados intimamente. A emulsão resultante é então passada através de uma seringa e, finalmente, microfluidizada com o uso de uma máquina M110S microfluidics. As gotículas de óleo resultantes têm um tamanho de aproximadamente 120 a 180 nm (expresso como média Z medida por PCS). Os outros componentes de adjuvantes/antígeno são adicionados à emulsão em mistura simples.
[000203] A preparação da emulsão de SB62 é preparada misturando-se sob forte agitação uma fase óleo constituída de componentes hidrofóbicos (α-tocoferol e esqualeno) e uma fase aquosa contendo os componentes solúveis em água (Tween 80 e PBS mod (modificado), pH 6,8). Durante a agitação a fase óleo (1/10 do volume total) é transferida para a fase aquosa (9/10 do volume total), e a mistura é agitada durante 15 minutos à temperatura ambiente. A mistura resultante é então submetida a forças de cisalhamento, impacto e cavitação na câmara de interação de um microfluidizador (15000 psi [103500 kPa]) - 8 ciclos) para produzir gotículas da ordem de submícron (distribuição entre 100 e 200 nm). O pH resultante é entre 6,8 ± 0,1. A emulsão de SB62 é então esterilizada por meio de filtração através de uma membrana de 0,22 μm e a emulsão volumétrica estéril é armazenada sob refrigeração em recipientes Cupac a de 2 a 8°C. Injeta-se gás inerte estéril (nitrogênio ou argônio) é no volume morto do recipiente volumétrico final da emulsão de SB62 durante pelo menos 15 segundos.
[000204] A composição final da emulsão SB62 é como a seguir:
[000205] Tween 80: 1,8% (v/v) 19,4 mg/ml; Esqualeno: 5% (v/v) 42,8 mg/ml; α-tocoferol: 5% (v/v) 47,5 mg/ml; PBS-mod: NaCl 121 mM, KCl 2,38 mM, Na2HPO4 7,14 mM, KH2PO4 1,3 mM; pH 6,8 ± 0,1.
[000206] O tamanho do diâmetro das gotículas de óleo é determinado de acordo com o procedimento a seguir e nas condições experimentais a seguir. A medida dos tamanhos das gotículas é dado como uma medida da intensidade e é expressa como média z medida por meio de PCS.
[000207] Medições de tamanho foram realizadas no adjuvante de emulsão óleo-em-água: SB62 preparado de acordo com o método de escala ampliada, AS03 e AS03+MPL (50 μg/ml), sendo que estes dois últimos só são preparados imediatamente antes do uso. A composição das amostras é dada abaixo (ver seção II.2.4). Amostras foram diluídas 4000x - 8000x em PBS 7,4.
[000208] Como um controle, padrões de tamanho de partículas PL- Nanocal de 100 nm (n° de cat. 6011-1015) foi diluído em 10 mM de NaCl.
[000209] Todas as medições de tamanho foram realizadas com ambos Malvern Zetasizer 3000HS. Amostras foram medidas em uma cubeta plástica para análise Malvern numa diluição vantajosa (usualmente a uma diluição de 4000x a 20000x dependendo da concentração da amostra), e com dois modelos ópticos: - ou índice refrativo real de 0 e imaginário de 0. - ou índice refrativo das partículas real de 1.5 e imaginário de 0,01 (o modelo óptico adaptado para a emulsão, de acordo com os valores encontrados na literatura).
[000210] As condições técnicas foram: - comprimento de onda do laser: 532 nm (Zeta3000HS). - potência do laser: 50 mW (Zeta3000HS). - luz difratada detectada a 90° (Zeta3000HS). - temperatura: 25°C, - duração: determinação automática pelo programa de computador, - número: 3 medições consecutivas, - diâmetro da média z: por meio de análise de cumulantes - distribuição de tamanhos: por meio do método de Contin ou o método Automático.
[000211] O algoritmo Malvern Automático usa uma combinação de cumulantes, Contin e algoritmos de menos quadrados não-negativos (NNLS, non negative least squares).
[000212] A distribuição de intensidades pode ser convertida a distribuição de volumes graças à teoria de Mie. II.2.4. Resultados (ver Tabela 2) Análise de cumulantes (diâmetro da média Z): Tabela 2 (*) Preparado como a seguir: Água para injeção, PBS concentrado 10x, 250 μl de emulsão de SB62 e 25 μg de MPL são misturados em conjunto para se atingir um volume final de 280 μl.
(*) Preparado como a seguir: Água para injeção, PBS concentrado 10x, 250 μl de emulsão de SB62 e 25 μg de MPL são misturados em conjunto para se atingir um volume final de 280 μl.
[000213] O diâmetro médio de z (ZAD, z-average diameter) é pesado por uma quantidade de luz difratada por cada tamanho de partículas na amostra. Este valor é relacionado com uma análise monomodal da amostra e é usada principalmente para fins de reprodutibilidade.
[000214] A taxa de contagem (CR, count rate) é uma medida da luz difratada: ela corresponde a milhares de fótons por segundo.
[000215] O índice de polidispersidade (Poly) é a amplitude da distribuição. Esta é uma medida isenta de dimensão da amplitude de distribuição.
[000216] Duas outras preparações de SB62 (AS03 concentrada 2 vezes) foram preparadas e avaliadas de acordo com o procedimento explicado acima com as seguintes modificações menores:
[000217] Amostras foram medidas em uma cubeta plástica para análise de Malvern, a duas diluições determinadas para se obter valores ótimos de taxa de contagem: 10000x e 20000x para o Zetasizer 3000HS, os mesmos modelos ópticos usados no exemplo acima. Resultados são mostrados na Tabela 3. Tabela 3 "-" quando os valores obtidos não foram coerentes.
"-" quando os valores obtidos não foram coerentes.
[000218] Uma representação esquemática destes resultados é mostrada na Figura 1 para a formulação 1023. Como se pode observar, a maior parte das partículas (p. ex., pelo menos 80%) apresenta um diâmetro inferior a 300 nm em intensidade.
[000219] Formulação de SB62 foi medida em diferentes diluições com o Malvern Zetasizer 3000HS e dois modelos ópticos. O tamanho das partículas ZAD (i.e. intensidade média por análise de cumulantes) das formulações avaliadas acima foi em torno de 150-155 nm.
[000220] Quando se usa o algoritmo de cumulantes, nós não observamos qualquer influência da diluição no ZAD e polidispersidade.
[000221] Infecção por influenza no modelo de furão emula estreitamente a influenza humana, com relação a ambos, sensibilidade à infecção e a resposta clínica. O furão é extremamente sensível a infecção com ambos os vírus de influenza A e B sem adaptação prévia de cepas virais. Portanto, proporciona-se um sistema de modelo excelente para estudos de proteção conferidos por vacinas de influenza administradas.
[000222] Este estudo investigou a eficácia de vacinas divididas de H5N1 adjuvantadas com AS03 para proteger furões contra uma exposição letal com a cepa homóloga de H5N1 A/Vietnam/1194/2004 ou com uma cepa heteróloga A/Indonésia. O objetivo deste experimento consistiu em demonstrar a eficácia de uma vacina de influenza adjuvantada, em comparação com furões imunizados com PBS ou o adjuvante apenas.
[000223] 36 Furões machos adultos jovens hibridados (Mustela putorius furo) (6 furões/grupo) com idades de aproximadamente 8 meses (pesos corporais de 0,8 a 1,5 kg) foram injetados intramuscularmente nos dias 0 e 21 com uma dose humana plena (dose de vacina de 500 μl). Quatro grupos de furões (n=6) foram imunizados com quatro concentrações diferentes de A/Vietnam/1194/2004 (NIBRG-14) (15, 5,0, 1,7 e 0,6 μg de HA) em combinação com AS03 (dose humana padrão, 250 μl/dose). Dois grupos de controle consistiram de animais tratados com placebo e tratados com AS03. Soros foram coletados no dia 21 e 42 para análise de respostas sorológicas. Titulações de anticorpos para vírus homólogos foram determinadas por meio de ensaio de inibição de hemaglutinação (titulações de HI). No dia 49 todos os animais foram expostos pela via intranasal com uma dose de 106 TCID50 da cepa homotípica A/Vietnam/1194/04. Durante o curso da exposição, coletou- se esfregaços nasais, da garganta e retais para avaliar o desprendimento de vírus. Após a necrópsia, seções cranioventrais, craniodorsais, caudoventrais e caudodorsais do pulmão direito de cada animal foram pesadas e armazenadas a -800C até a análise. Titulações virais foram determinadas por meio de PCR TaqMan específica para H5N1 e cultura de titulação de vírus em células MDCK. Dados foram expressos como Unidades de Diluição de Controle (CDU) e TCID50 por grama de tecido de pulmão ou por ml de esfregaço, respectivamente. CDU são determinados a partir de uma curva padrão produzida de um estoque de vírus que é diluído serialmente, sendo que cada diluição é submetida a extração de ácido nucleico e amplificação com PCR Taqman da mesma maneira que amostras de teste. Tabela 4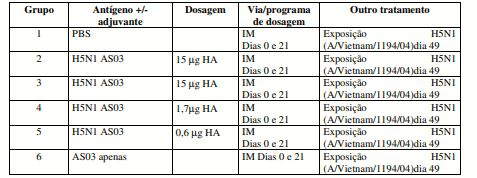
[000224] Um tamponador pré-misturado é preparado previamente no Tamponador de Volume Final (PBS pH 7,4) contendo Tiomersal, Tween 80 e Triton X100 em quantidades que consideram suas concentrações na cepa. No dia das imunizações adicionou-se 15 - 5 - 1,7 ou 0,6 μg da cepa H5N1 ao tamponador pré-misturado. Após 30 minutos de agitação, adicionou-se 250 μl de emulsão de SB62. A formulação é agitada durante 30 minutos. Injeções ocorrem dentro de uma hora após o término da formulação.
[000225] De maneira vantajosa, a formulação é preparada como a seguir. Adicionou-se Tween 80, Triton X100 e Tiomersal ao Tamponador de volume final em quantidades que consideram suas concentrações na cepa. Após 5 min de agitação, adicionou-se 15 - 5 - 1,7 ou 0,6 μg da cepa H5N1. Após 30 minutos de agitação, adicionou-se 250 μl de emulsão de SB62. A formulação é agitada durante 30 minutos. Injeções ocorrem dentro da hora seguinte ao término da formulação.
[000226] 250 μl da emulsão de SB62 é misturada com 250 μl de PBS pH 6,8, agitada durante 5 minutos e armazenada a 40C até sua administração.
[000227] De maneira vantajosa, a formulação é preparada como a seguir. 250 μl de emulsão de SB62 são misturados com 250 μl de PBS pH 6,8 e agitados durante 5 minutos. Injeções ocorrem dentro da hora seguinte ao término da formulação.
[000228] Observação: Em cada formulação adiciona-se PBS concentrado 10 vezes para atingir a isotonicidade e é concentrado 1 vez no volume final. O volume de H2O é calculado de forma a se atingir o volume objetivado. 111.2.3. Leituras (Tabela 5) Tabela 5

[000229] A Tabela 6 resume os dados de proteção, titulações de HI e carga viral em tecido do pulmão e esfregaços faríngeos obtidos em furões após exposição com uma cepa de H5N1 homóloga. Tabela 6. Proteção de furões vacinados com H5N1 adjuvantado com AS03 contra exposição com vírus de influenza H5N1 homólogo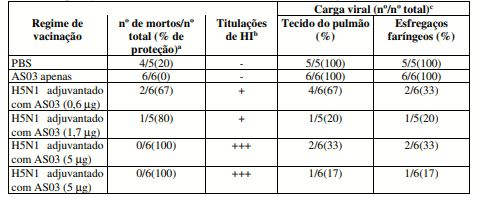 a Um animal imunizado com PBS e um imunizado com 1,7 μg de HA da vacina adjuvantada foram imunizados durante o curso da vacinação (dia 25). Não houve ligação aparente entre a vacinação e estas mortalidades. bTitulações de HI médias geométricas (D42): +++ (> 160), ++ (60-160) + (40-60), - (< 40). cNúmeros de animais com carga viral determinados por meio de cultura viral > 102 TCID50 por g de tecido ou por ml de esfregaço.
a Um animal imunizado com PBS e um imunizado com 1,7 μg de HA da vacina adjuvantada foram imunizados durante o curso da vacinação (dia 25). Não houve ligação aparente entre a vacinação e estas mortalidades. bTitulações de HI médias geométricas (D42): +++ (> 160), ++ (60-160) + (40-60), - (< 40). cNúmeros de animais com carga viral determinados por meio de cultura viral > 102 TCID50 por g de tecido ou por ml de esfregaço.
[000230] Antes da exposição com H5N1, dois animais foram eutanizados. Um animal imunizado com PBS foi eutanizado devido a perda de peso excessiva durante o curso da vacinação (dia 14). Um animal imunizado com 1,7 μg de HA da vacina adjuvantada foi eutanizado devido a perda de peso excessiva durante o curso da vacinação (dia 25). Não houve ligação aparente entre vacinação e estas mortalidades.
[000231] Exposição com A/Vietnam/1194/04 em furões vacinados com vacinas de H5N1 adjuvantadas com AS03 apresentaram uma proteção dependente da dose de antígeno ou curva de sobrevida (Tabela 6). Todos os animais imunizados com 5 ou 15 μg de HA da vacina adjuvantada com AS03 foram protegidos contra a exposição letal. É importante ressaltar que titulações de HI médicas contra vírus A/Vietnam/1194/2004 homólogo foram > 40 em todos os grupos de furões imunizados com vacinas adjuvantadas com AS03, incluindo nos grupos que receberam as menores doses, tendo-se obtido 66,67 e 80,00% de proteção contra a exposição homóloga em furões imunizados com 0,6 e 1,7 μg de vacina de H5N1 dividida adjuvantada com AS03, respectivamente. Todos os furões imunizados com PBS ou o adjuvante apenas apresentaram uma carga viral acima de 105 TCID50/g de tecido do pulmão e todos os animais desprenderam altos níveis virais na faringe durante todo o curso da infecção. Inversamente, a administração de vacinas adjuvantadas com AS03 reduziu a carga viral na maior parte dos animais até abaixo de um limiar de 102 TCID50 por grama de tecido do pulmão ou por ml de fluido de esfregaços faríngeos (Tabela 6). Apenas um ou dois furões por grupo imunizados com vacinas adjuvantadas com AS03 apresentaram cargas virais de moderadas a altas (>102 TCID50).
[000232] Uma análise estatística realizada nestes dados levou às seguintes conclusões: - todas as doses de vacina foram estatisticamente diferentes dos controles - valores P (teste exato de Fischer) compreenderam de 0,0276 para a dose mais baixa, até 0,0006 para a dose mais alta - a dose estimada para induzir 90% de proteção foi estimada neste modelo como sendo de 2,9 μg - a menor dose para induzir 100% de proteção foi estimada neste modelo como sendo entre 2,9 e 5 μg.
[000233] A resposta imune humoral à vacinação foi medida após cada imunização nos dias 21 e 42. Amostras de soro foram testadas com o teste de inibição de hemaglutinação (HI) usando-se eritrócitos de cavalo. Resultados são apresentados na Tabela 6.
[000234] Formulações divididas de H5N1 monovalente adjuvantadas com AS03 induziram as mais fortes respostas de HI à cepa homóloga, em comparação com furões imunizados com PBS ou AS03 apenas. Observou-se um efeito dose antígeno com as titulações de HI mais elevadas obtidas em furões imunizados com as doses de antígeno mais elevadas (15 ou 5 μg de HA da vacina adjuvantada), em comparação com a menor resposta imune em furões que recebem as duas doses mais baixas (1,7 ou 0,8 μg de HA da vacina adjuvantada).
[000235] Como mostrado na Tabela 6 e na Figura 2A, verificou-se uma correlação entre as titulações de HI e a proteção contra exposição de furões a H5N1 A/Vietnam. Titulações de HI superiores a 40 pareceram correlacionar-se com a proteção em furões imunizados com vacinas de H5N1 divididas adjuvantadas com AS03.
[000236] A resposta imune humoral à vacinação também foi testada por meio de ensaio de neutralização após cada imunização nos dias 21 e 42. Resultados são apresentados na Figura 3.
[000237] Formulações divididas de H5N1 monovalente adjuvantadas com AS03 induziram as mais fortes respostas de anticorpos neutralizadores à cepa heteróloga, em comparação com furões imunizados com PBS ou AS03 apenas. Observou-se um efeito dose antígeno com as titulações de HI mais altas obtidas em furões imunizados com as mais altas doses de antígeno (15 ou 5 μg de HA da vacina adjuvantada), em comparação com a menor resposta imune em furões que recebem as doses mais baixas (1,7 ou 0,8 μg de HA da vacina adjuvantada).
[000238] O desprendimento viral foi realizado por meio de cultura viral e PCR TaqMan no tecido do pulmão e em esfregaços nasais/da garganta/retais.
[000239] Como mostrado na Tabela 6, também se observou proteção em termos de redução da carga viral nos tecidos de pulmão e esfregaços faríngeos em furões imunizados com vacinas divididas de H5N1 adjuvantadas com AS03 (a maior parte dos animais abaixo de 102 TCID50 por grama de tecido ou por ml de esfregaço). Todos os furões imunizados com PBS ou o adjuvante apenas apresentaram alta carga viral em tecidos de pulmão e na faringe durante todo o curso da infecção.
[000240] Figura 2B mostra a carga viral média determinada tanto por PCR e cultura viral em cada grupo. Isto demonstrou que todos os grupos que receberam vacina adjuvantada tenderam a apresentar caras virais reduzidas relativamente aos grupos de controle que receberam PBS ou o adjuvante apenas, sendo que furões imunizados com 0,7 μg de HA da vacina adjuvantada apresentam uma redução mais modesta da carga viral. Foi possível observar pouca diferença entre furões imunizados com 1,6, 5 ou 15 μg de HA da vacina adjuvantada. Finalmente, para a carga viral nos pulmões, resultados de PCR foram consistentes com a titulação de vírus.
[000241] Além disso, o desprendimento viral em esfregaços nasais e retais, e também no plasma, foi investigado por meio de cultura viral e PCR. De uma maneira geral, a titulação viral foi menos sensível do que a análise de PCR. Esta análise mostrou a presença de vírus H5N1 nos esfregaços da cavidade nasal e retal de 2/5 furões que receberam PBS. Neste grupo, 1/5 animal também desprendeu vírus no soro. Nenhum animal imunizado com vacina dividida de H5N1 adjuvantada com AS03 desprendeu vírus em esfregaços retais ou no plasma. É interessante observar que a análise de cada um dos furões individuais mostrou que alguns furões protegidos apresentaram elevada carga viral e baixas titulações de HI, demonstrando que o mecanismo por meio do qual a proteção foi obtida pode dever-se, pelo menos em parte, a uma indução da imunidade celular (não avaliada neste experimento).
[000242] As temperaturas corporais de todos os animais foram registradas. Não se observou temperaturas corporais durante a fase de vacinação. Exposição com A/Vietnam/1194/04 induziu o início da febre com temperaturas corporais compreendendo de 400C a 420C com picos entre 12 a 24 horas após o início da exposição. Durante o curso da infecção, as temperaturas corporais diminuíram a níveis normais em animais que sobreviveram à exposição. Animais que morreram antes do dia 5 apresentaram uma rápido decréscimo da temperatura corporal temperatura corporal após o pico de febre.
[000243] Em resumo, testes sorológicos indicaram que foi possível obter titulações de HI significativamente maiores em animais imunizados com vacinas adjuvantadas, em comparação com animais imunizados com PBS ou o adjuvante apenas. Exposição com o vírus A/Vietnam/1194/04 homólogo mostrou uma curva de sobrevida dependente de antígeno-dose com carga viral reduzida no tecido do pulmão e esfregaços faríngeos dos grupos que receberam vacinas adjuvantadas, em comparação com grupos de controle. Todos os animais em grupos de controle (imunizados com PBS ou o adjuvante apenas) desprenderam vírus no trato respiratório superior com uma carga viral acima de 105 TCID50/g de pulmão, enquanto que em grupos que receberam vacinas adjuvantadas, apenas uma proporção de animais desprendeu vírus no trato respiratório superior (4/17 com de 105 a 107 TCID50/g de tecido do pulmão, maior parte abaixo de 102 TCID50 por grama de pulmão ou por ml de esfregaços faríngeos). Adicionalmente, houve titulações virais reduzidas no tecido do pulmão dos grupos que receberam vacinas adjuvantadas como, em comparação, com grupos tratados apenas com placebo e adjuvante. Esta redução foi parcialmente dependente dose de antígeno, atingindo um platô aparente a 5 μg de antígeno.
[000244] Um estudo randomizado, de fase I, cego para o observador, está sendo realizado correntemente em uma população adulta com idades de 18 a 60 anos em 2006 para avaliar a reatogenicidade e a imunogenicidade de um candidato de influenza pandêmica administrado em diferentes doses de antígeno (3,8 μg, 7,5 μg, 15 μg e 30 μg de HA) adjuvantada ou não com o adjuvante AS03. A resposta imune humoral (i.e., titulações de anticorpos anti-hemaglutinina, neutralizadores e antineuraminidase) e resposta imune mediada por célula (respostas de células T CD4 e/ou CD8) são medidas 21 dias após cada uma das duas administrações intramusculares das formulações de vacina candidata. Os grupos não- adjuvantados serviram como referência para o grupo adjuvantado respectivo que recebeu o mesmo teor de antígeno. 111.3.7. Projeto do estudo Oito grupos de 50 sujeitos cada um (planejados) receberam, em paralelo, a seguinte vacina intramuscularmente: ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus de influenza pandêmica dividido contendo 3,8 μg de HA ■ um grupo de 51 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 3,8 μg de HA e adjuvantada com AS03 ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 7,5 μg de HA ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 7,5 μg de HA e adjuvantada com AS03 ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 15 μg de HA ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 15 μg de HA e adjuvantada com AS03 ■ um grupo de 50 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 30 μg de HA ■ um grupo de 49 sujeitos recebeu duas administrações da vacina de vírus da influenza pandêmica dividido contendo 30 μg de HA e adjuvantada com AS03
[000245] O recrutamento foi realizado para assegurar que metade dos sujeitos de cada grupo apresentasse idades entre 18 e 30 anos.
[000246] Esquema de vacinação: duas injeções de vacina candidata contra influenza pandêmica no dia 0 e dia 21, coleta de amostras de sangue, análise de leitura no dia 21 e 42 (determinação de anticorpos HI, determinação de anticorpos NI, determinação de anticorpos neutralizadores, e análise de CMI), conclusão do estudo (dia 51) e fim do estudo (180 dias). IV.3. Objetivos do estudo IV.3.1. Objetivos primários - Para avaliar a resposta imune humoral induzida pelas vacinas do estudo em termos de titulações de anticorpos anti-hemaglutinina. - Para avaliar a segurança e a reatogenicidade das vacinas do estudo em termos de eventos locais solicitados e gerais adversos, eventos adversos não-solicitados e eventos adversos sérios. Para a resposta imune humoral: Variáveis observadas nos dias 0, 21, 42 e 180: - Titulações de anticorpos de inibição de hemaglutinação. Variáveis derivadas (com intervalos de confiança de 95%): - Titulações geométricas médias (GMTs) de anticorpos do soro nos dias 0, 21, 42 e 180 - Taxas de soroconversão*nos dias 21, 42 e 180 - Fatores de conversão**nos dias 21, 42 e 180 - Taxas de proteção*** nos dias 0, 21, 42 e 180 - Taxa de soroconversão para resposta de anticorpos à hemaglutinina é definida como o percentual de vacinados que apresentam, ou uma titulação de pré-vacinação < 1:10 e uma titulação pós-vacinação > 1:40 ou uma titulação pré-vacinação > 1:10 e pelo menos um aumento de quatro vezes na titulação pós-vacinação - * Fator de conversão definido como o aumento em múltiplos em GMTs de HI no soro após a vacinação, em comparação com dia 0; - ** Taxa de proteção definida como o percentual de vacinados com uma titulação de HI no soro >40 após vacinação que usualmente é aceita como indicando proteção. Para a avaliação da segurança/reatogenicidade: 1. Percentual, intensidade e relação com a vacinação de sintomas e sinais locais e gerais solicitados durante um período de acompanhamento de 7 dias (i.e., dia de vacinação e 6 dias subseqüentes) após cada vacinação e globalmente. 2. Percentual, intensidade e relação com a vacinação de sintomas e sinais locais e gerais não solicitados durante um período de acompanhamento de 21 dias após a primeira vacinação, durante um período de acompanhamento de 30 dias após a segunda vacinação e globalmente.
[000247] Ocorrência de eventos adversos sérios durante todo o estudo. IV.3.2. Objetivos secundários - Para avaliar a resposta imune humoral induzida pelas vacinas de estudo em termos de titulações de anticorpos neutralizadores no soro - Para avaliar a resposta imune mediada com células induzida pelas vacinas de estudo em termos de freqüência de linfócitos T CD4/CD8 específicos para influenza
[000248] Adicionalmente, o impacto da vacinação em células de memória B específicas para influenza usando a tecnologia Elispot será avaliado.
[000249] Variáveis observadas nos dias 0, 21, 42 e 180: titulações de anticorpos neutralizadores no soro.
[000250] Variáveis derivadas (com intervalos de confiança de 95%): - Titulações geométricas médias (GMTs) de anticorpos no soro nos dias 0, 21, 42 e 180 - Taxas de soroconversão*no dia s21, 42 e 180 - Taxa de soroconversão para resposta de anticorpos neutralizadores é definida como o percentual de vacinados com um aumento mínimo de 4 vezes na titulação após a vacinação. Para a resposta CMI: 1. Freqüência de células CD4/CD8 de citocina por 106 em testes produzindo pelo menos duas citocinas diferentes (CD40L, IL-2, TNF-α, IFN-Y) 2. Freqüência de células CD4/CD8 positivas para citocina por 106 em testes produzindo pelo menos CD40L e outra molécula-sinal (IL-2, IFN-y, TNF-α) 3. Freqüência de células CD4/CD8 positivas para citocina por 106 em testes produzindo pelo menos IL-2 e outra molécula-sinal (CD40L, IFN-y, TNF-α) 4. Freqüência de células CD4/CD8 positivas para citocina por 106 em testes produzindo pelo menos TNF-α e outra molécula-sinal (IL-2, IFN-y, CD40L)
[000251] Freqüência de células CD4/CD8 positivas para citocina por 106 em testes produzindo pelo menos IFN-y e outra molécula-sinal (CD40L, IL-2, TNF-α).
[000252] Nos dias 0, 21, 42 e 180: freqüência de células de memória B específicas para influenza por 106células no teste.
[000253] AS03 contém a emulsão de SB62 óleo-em-água, consistindo de uma fase óleo contendo DL-α-tocoferol e esqualeno, e uma fase aquosa contendo o detergente não-iônico polissorbato 80.
[000254] A substância ativa da candidata a vacina contra influenza pandêmica é um antígeno de vírus dividido inativado com formaldeído derivado da cepa de vírus de vacina A/VietNam/1194/2004 (H5N1) NIBRG- 14. A dose de antígeno HA compreende de 3,8 a 30 μg por dose.
[000255] Os volumes monovalentes de vírus divididos usados para produzir a vacina de influenza adjuvantada com AS03 são fabricados de acordo com o mesmo procedimento que o usado para a vacina contra influenza interpandêmica licenciada pela GSK Biologicals Fluarix™/α-Rix®. Para os fins deste ensaio clínico, a cepa de vírus usada para fabricar os lotes clínicos é a cepa de vacina de H5N1 A/Vietnam/1194/04 NIBRG-14 cepa de vacina de protótipo de H5N1 recombinante derivada da A/Vietnam/1194/04 altamente patogênica. Esta cepa de protótipo recombinante foi desenvolvida pela NIBSC usando engenharia genética reversa (uma referência vantajosa é Nicolson et al. 2005, Vaccine, 23, 2943-2952)). A cepa reassortant [contendo 2 ou mais porções de ácido nucleico de parentais diferentes] combina os segmentos H5 e N1 à espinha dorsal da cepa A/PR/8/34, e a H5 foi manipulada para eliminar a extensão polibásica de aminoácidos no sítio de clivagem de HA que é responsável pela alta virulência das cepas originais. Isto foi obtido transfectando-se células Vero com plasmídeos contendo o gene de HA (modificada de forma a remover os determinantes com elevada patogenicidade) e gene de NA do A/Viet Nam/1194/2004 (H5N1) isolado humano e plasmídeos contendo os genes internos de PR8. O vírus recuperado foi passado duas vezes sobre ovos e foi então denominado como o vírus de referência NIBRG-14. O caráter atenuado desta H5N1 reassortant foi documentado extensivamente em uma avaliação de segurança pré-clínica (realizada pela NIBSC), como também é feito rotineiramente para as cepas de vacina contra influenza clássicas.
[000256] A vacina candidata de influenza pandêmica adjuvantada com AS03 de acordo com a invenção é uma vacina de 2 componentes consistindo de 0,5 ml de antígenos de vírions divididos inativados concentrados apresentados em um frasco de vidro de tipo I e de uma seringa de vidro de tipo O pré-enchida contendo 0,5 ml do adjuvante AS03. No momento da injeção, o conteúdo da seringa pré-enchida contendo o adjuvante é injetado no frasco que contém os antígenos de vírion divididos inativados concentrados. Após a misturação, o conteúdo é sugado pela seringa e a agulha é substituída por uma agulha intramuscular. Uma dose da vacina candidata de influenza adjuvanta com AS03 corresponde a 1 ml. Cada dose de vacina de 1 ml contém 3,8 μg, 7,5 μg, 15 μ ou 30 μg hemaglutinina (HA) ou qualquer quantidade de HA vantajosa que poderia seria determinada para que vacina atenda os critérios de eficácia como detalhado aqui.
[000257] Alternativamente, a vacina candidata de influenza pandêmica adjuvantada com AS03 de acordo com a invenção é uma vacina de 2 componentes consistindo de 0,25 ml de antígenos de vírions divididos inativados concentrados apresentados em um frasco de vidro de tipo I e de uma seringa de vidro de tipo I pré-enchida contendo 0,25 ml do adjuvante AS03. No momento da injeção, o conteúdo da seringa pré-enchida contendo o adjuvante é injetado no frasco que contém os antígenos de vírions divididos inativados concentrados. Após a misturação, o conteúdo é sugado para o interior da seringa e a agulha é substituída por uma agulha intramuscular. Uma dose da vacina candidata contra influenza adjuvantada com AS03 corresponde a 0,5 ml. Cada dose de vacina de 0,5 ml contém 3,8 μg, 7,5 μg, 15 μ ou 30 μg hemaglutinina (HA) ou qualquer quantidade de HA vantajosa que poderia ser determinada para que a vacina atenda aos critérios de eficácia como detalhado aqui.
[000258] Os excipientes de vacina são polissorbato 80 (Tween 80), octoxinol 10 (Triton X-100), cloreto de sódio, hidrogeno fosfato de dissódio, di-hidrogeno fosfato de potássio, cloreto de potássio, hexaidrato de cloreto de magnésio e água para injeção. Adicionou-se tiomersal como um conservante antimicrobiano para prevenir a contaminação durante o uso, porque se antecipa que, quando ocorre uma pandemia, a apresentação principal será apresentada em um recipiente de doses múltiplas (frascos ou ampolas), para os quais se requer um conservante. Por esta razão, a vacina pandêmica é formulada com tiomersal a 5 μg/dose como conservante. De maneira vantajosa, a vacina pandêmica pode ser formulada com tiomersal a 10 μg/dose como conservante ou uma dose ligeiramente maior, como de até 25 μg/dose de vacina.
[000259] Os mono-volumes de vírus são preparados desenvolvendo sementes de trabalho de H5N1 em ovos de galinha embrionados. O processo de fabricação para os volumes monovalentes da cepa de H5N1 de influenza inativada dividida, ilustrada na Figura 4, é idêntica ao processo de fabricação para os volumes monovalentes de α-Rix™.
[000260] Basicamente, o processo de fabricação dos volumes monovalentes pode ser dividido em quatro partes principais: 1) Propagação das sementes de trabalho em ovos de galinha fertilizados, colhendo-se e combinando-se os fluidos alantóicos infectados de forma a se obter o "volume de vírus inteiro monovalente bruto" (etapa 1). 2) Purificação de cada cepa de vírus levando ao "volume de vírus inteiro monovalente purificado" (etapas 2-6). 3) Divisão do volume de vírus inteiro monovalente purificado com desoxicolato de sódio resultando no "volume de vírus dividido monovalente purificado" (etapas 7-8/1). 4) Inativação do volume de vírus dividido monovalente purificado em duas etapas por meio de incubação com desoxicolato de sódio e com formaldeído, seguido de ultrafiltração e filtração estéril, para se obter o "volume de vírus dividido inativado monovalente purificado", ou "volume monovalente" (etapas 8/2-9).
[000261] No dia da inoculação dos ovos embrionados, um inóculo é preparado por meio de misturação do lote de semente de vírus de trabalho com tamponador de fosfato contendo 25 μg/ml de hidrocortisona, e 0,5 mg/ml de sulfato de gentamicina. O inóculo de vírus é mantido à temperatura ambiente até à inoculação.
[000262] Usa-se ovos embrionados pré-incubados com onze dias de vida para replicação de vírus. Os ovos são transferidos para os recintos de produção após fumigação das cascas com formaldeído. Aproximadamente 120.000 ovos são inoculados com 0,2 ml do inóculo de vírus, sendo que cada um usa um dispositivo de inoculação de ovos automático. Os ovos inoculados são incubados a 34,0°C durante 72 horas.
[000263] Ao término do período de incubação, os ovos são inspecionados visualmente quanto à presença de embrião vivo e de vasos sanguíneos adequados para a idade. Os embriões são mortos por meio de resfriamento dos ovos e armazenados durante de 12 a 46 horas a de 2 a 8°C.
[000264] O fluido alantóico (aproximadamente 12 ml) dos ovos embrionados resfriados é colhido por máquinas de coleta de ovos. Os fluidos alantóicos são recolhidos em um tanque de aço inoxidável termorregulado a de 2 a 8°C. Neste estágio o produto é denominado "volume de vírus inteiro monovalente bruto". O volume de vírus inteiro monovalente bruto não é armazenado, mas imediatamente transferido à etapa de clarificação.
[000265] Todas as operações são realizadas a de 2 a 8°C, até o fluxo por meio de ultracentrifugação, que é realizada à temperatura ambiente.
[000266] O fluido alantóico coletado é clarificado por meio de centrifugação contínua com velocidade moderada. Esta etapa remove partículas grandes que poderiam ter sido coletadas durante a colheita do fluido alantóico (p. ex., partes de cascas de ovos).
[000267] Esta etapa permite clarificar adicionalmente o fluido alantóico por meio de uma precipitação de material virótico, por meio de adsorção sobre um gel de hidrogeno fosfato de cálcio dibásico. Para obter o hidrogeno fosfato de cálcio dibásico (CaHPO4) em gel, 0,5 mol/l de hidrogeno fosfato de dissódio (Na2HPO4) e 0,5 mol/l de cloreto de cálcio (CaCal2) são adicionados ao combinado de vírus clarificado para atingir uma concentração final de 1,87 g de CaHPO4 por litro.
[000268] Após sedimentação durante pelo menos 8 horas até um máximo de 36 horas, o sobrenadante é removido e o sedimento contendo o vírus de influenza é ressolubilizado por meio de adição de uma solução de EDTA dissódio a 8,7%.
[000269] O sedimento de influenza ressuspenso é filtrado através de uma membrana de filtração de 6 μm para remover as pelotas remanescentes potenciais.
[000270] O vírus da influenza é purificado adicionalmente (remoção de proteínas e fosfolipídios) e concentrado por meio de ultracentrifugação isopícnica em um gradiente de sacarose linear (de 0 a 55%) a uma taxa de fluxo de 8 a 20 litros por hora. O gradiente é formado usando-se a solução de sacarose a 55% (peso/peso) com 0,01% de timerosal, e um tamponador de fosfato pH 7,4 com 0,01% de timerosal. Isto é feito na presença de 100 ± 15 μg/ml de tiomersal para controlar a carga biológica do processo, porque a centrifugação é realizada à temperatura ambiente. Quatro frações diferentes são recuperadas medindo-se a concentração de sacarose via um refratômetro: Fração 4/1: de 55 a 47% de sacarose Fração 4/2: de 47 a 38% de sacarose Fração 4/3: de 38 a 20% de sacarose Fração 4/4: de 20 a 0% de sacarose
[000271] O limite superior da fração 4/2 é selecionado para o balanço entre um alto coeficiente de pureza HA/proteína e uma máxima recuperação de vírus inteiro. O limite entre as frações 4/2 e 4/3 é selecionado de forma a minimizar o teor de ovalbumina na fração 4/2. O limite inferior da fração 4/3 é selecionado com base no teor de HA encontrado na faixa baixa de gradiente de sacarose. Frações 4/2 e 4/3 são usadas para preparações adicionais. A maior parte dos vírus é coletada na Fração 4/2. A Fração 4/3, que contém tanto vírus como também proteínas, é purificada adicionalmente. Primeiramente, a concentração de sacarose da Fração 4/3 é reduzida abaixo de 6% (necessário para a etapa de centrifugação subseqüente) por meio de ultrafiltração. Em seguida, a Fração 4/3 é pelotizada via centrifugação para remover quaisquer contaminantes solúveis (proteínas). A pelotização é ressuspensa em tamponador de fosfato pH 7,4 e misturada intimamente para se obter uma suspensão homogênea. Os tempos de contenção são de, no máximo, 36 horas para a Fração 4/3, no máximo 60 horas para a Fração 4/2 e, no máximo, 36 horas para a Fração 4/3 purificada.
[000272] Ambas as frações, a Fração 4/3 tratada e a Fração 4/2 não- tratada, são combinadas e diluídas por meio de adição de 60 l de tamponador de fosfato pH 7,4.
[000273] Neste estágio, o combinado de material corresponde ao "volume de vírus inteiro monovalente purificado".
[000274] Fluxo através ultracentrifugação na presença de desoxicolato de sódio:
[000275] O vírus da influenza é dividido e purificado adicionalmente por meio de centrifugação através de um gradiente de sacarose linear (de 0 a 55% - formado com solução de sacarose S8a e tamponador S6a) que contém 1,5% de desoxicolato de sódio. Tween-80 está presente a 0,1% no gradiente. O vírus é processado a uma taxa de 8 litros por hora. Ao término da centrifugação, coleta-se três diferentes frações. A faixa da fração principal (Fração 7/2) é selecionada com base em validação dependente-da-cepa das condições de divisão com o objetivo de coletar uma fração que consiste de antígeno de vírus de influenza predominantemente disrompido, ao mesmo tempo em que se minimiza o mais possível de partículas de vírus inteiro possivelmente remanescentes e fosfolipídios provenientes da membrana do vírus após a divisão.
[000276] Para A/Vietnam/1194/2004 NIBRG-145 a compreende da fração 7/2 é ajustada em de 20 a 41% de sacarose. O antígeno de hemaglutinina é concentrada na Fração 7/2, que contém aproximadamente 1,2% de desoxicolato de sódio. Este material corresponde ao "volume de vírus dividido monovalente purificado".
[000277] Fração 7/2 é diluído três vezes em tamponador de fosfato S7c, que contém 0,025% de Tween-80. Em seguida, a fração 7/2 é filtrada reduzindo-se gradualmente até uma membrana de filtração de 0,45 μm, sonificada brevemente (para facilitar a filtração) e filtrada através uma membrana de 0,2 μm. Ao término da filtração, os filtros são enxaguados com tamponador de fosfato (S107c) contendo 0,025% de Tween-80. Como um resultado da filtração e enxágüe, o volume final do filtrado é 5 vezes o volume original da fração 7/2.
[000278] A solução resultante é incubada a 22 ± 2°C durante pelo menos 84 horas. Após completamento da primeira etapa de inativação, o material é diluído com tamponador de fosfato S7c para reduzir o teor total de proteína até uma concentração calculada de 500 μg/ml:
[000279] O formaldeído é adicionado a uma concentração final calculada de 100 μg/ml. A inativação ocorre em uma bolsa de polietileno de baixa densidade de uso único com volume de 100 litros a 20 ± 2°C durante pelo menos 72 horas.
[000280] O material de vírus dividido inativado é ultrafiltrado através de membranas com um valor de corte de peso molecular de 30.000 Dalton, usando consecutivamente tamponadores S7b e S1b.
[000281] Após uma redução de volume, o volume permanece constante durante a ultrafiltração (diafiltração) por meio de adição de tamponador de fosfato e tamponador de solução salina fosfatada (S1b) contendo 0,01% de Tween-80.
[000282] Durante a ultrafiltração, o teor de formaldeído, NaDoc e sacarose é reduzido.
[000283] O material é concentrado a de 15 a 25 litros e é transferido imediatamente à etapa de filtração final.
[000284] Após a ultrafiltração, o material inativado dividido é filtrado gradualmente diminuindo até uma membrana de 0,2 μm.
[000285] A filtração estéril final através de uma membrana de grau estéril de 0,22 μm é realizada em um ambiente de Classe 100. Ao término da filtração os filtros são enxaguados com solução tamponadora de solução salina fosfatada S1b, contendo 0,01% de Tween-80. Com isto, o filtrado é diluído a uma concentração de proteína inferior a 1000 μg/ml, para evitar a agregação durante o armazenamento subseqüente.
[000286] O material resultante é o "volume de vírus dividido inativado monovalente purificado" ou "volume monovalente".
[000287] Os volumes monovalentes finais de vírus de influenza H5N1 inativado dividido são armazenados a de 2 a 80C durante um máximo de 18 meses em frascos de vidro de tipo I.
[000288] A vacina candidata contra influenza pandêmica de vírus dividido inativado adjuvantada com AS03 a ser avaliada nos ensaios clínicos de fase I H5N1-007 destina-se a administração intramuscular. A vacina é uma vacina de 2 componentes que consiste de antígenos de vírion dividido inativado concentrado (H5N1) apresentados em um frasco de vidro de tipo I e do adjuvante AS03 contido em uma seringa de vidro de tipo I pré-enchida.
[000289] Uma dose de vacina contra influenza pandêmica adjuvantada com AS03 reconstituída corresponde a 1 ml. A composição é indicada na Tabela 7. Como o estudo H5N1-007 é um estudo de determinação da dose, o teor de HA por dose é diferente para cada um dos lotes clínicos a serem testados. Uma dose contém 3,8, 7,5, 15 ou 30 μg de HA. A vacina contém os seguintes resíduos do processo de fabricação da substância de droga: formaldeído, ovalbumina, sacarose, tiomersal e desoxicolato de sódio. Tabela 7 - Composição da vacina candidata contra influenza pandémica adjuvantada com AS03 reconstituída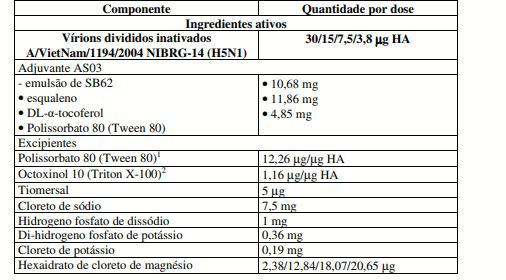
[000290] A fabricação da vacina contra influenza pandêmica adjuvantada com AS03 consiste de três etapas principais: (a) Formulação do volume final do vírus dividido (concentrada 2 x) sem adjuvante e enchimento no recipiente de antígeno (b) Preparação do adjuvante AS03 e enchimento no recipiente de adjuvante (c) Reconstituição extemporânea da vacina de vírus dividido adjuvantado com AS03 1) Formulação do volume final sem adjuvante e enchimento no recipiente de antígeno. O diagrama de fluxo da formulação é apresentado na Figura 5.
[000291] O tamanho do volume monovalente é baseado no teor de HA medido no volume monovalente antes da formulação e em um volume- alvo de 4000 ml.
[000292] O tamponador de volume final (tamponador de formulação compreendendo: Cloreto de sódio: 7,699 g/l; dodecaidrato de fosfato de dissódio: 2,600 g/l; di-hidrogeno fosfato de potássio: 0,373 g/l; cloreto de potássio: 0,2 g/l; hexaidrato de cloreto de magnésio: 0,1 g/l) e os volumes corretos de Triton X-100 (solução de Octoxinol 10 (Triton X-100) a 5%) e tiomersal (solução de consumo de Tiomersal a 0,9%) são misturados entre si sob agitação contínua. O mono-volume H5N1 é então diluído no volume resultante de tamponador-Triton X-100-tiomersal para se obter uma concentração final de 60/30/15/7,6 μg de H5N1 por ml de volume final por ml (30, 15, 7,5 ou 3,8 μg de HA/500 μl de volume final). A mistura é agitada durante de 30 a 60 minutos. O pH é verificado como sendo de 7,2 ± 0,3. Não houve necessidade de adicionar Tween 80 porque a concentração de Tween 80 (822 μg/ml) presente no mono-volume foi suficiente para atingir a concentração-alvo (12,26 μg/μg de HA).
[000293] O volume final é enchido de forma asséptica em frascos de vidro de tipo I estéreis de 3 ml (Ph. Eur.). Cada frasco contém um volume de 0,65 ml ± 0,05 ml. 2) Preparação do volume de adjuvante estéril AS03 e enchimento no recipiente de adjuvante.
[000294] O adjuvante AS03 é preparado misturando-se dois componentes: emulsão de SB62 e tamponador de fosfato.
[000295] A preparação da emulsão de SB62 é realizada misturando- se sob forte agitação de uma fase óleo constituída de componentes hidrofóbicos (α-tocoferol e esqualeno) e uma fase aquosa contendo os componentes solúveis em água (Tween 80 e tamponador de solução salina- fosfato a pH 6,8). Enquanto se agita, a fase óleo (1/10 do volume total) é transferido para a fase aquosa (9/10 do volume total), e a mistura é agitada durante 15 minutos à temperatura ambiente. A mistura resultante é então submetida a forças de cisalhamento, impacto e cavitação na câmara de interação de um microfluidizador (15000 psi [103500 kPa]) - 8 ciclos) para produzir gotículas da ordem de submícron (distribuição entre 100 e 200 nm). O pH resultante é entre 6,8 ± 0,1. A emulsão de SB62 é então esterilizada por meio de filtração através de uma membrana de 0,22 μm e a emulsão de volume estéril é armazenada refrigerada em recipientes Cupac a de 2 a 8°C. Gás inerte estéril (nitrogênio) é inundado no volume morto do recipiente de volume final da emulsão de SB62 durante pelo menos 15 segundos.
[000296] A composição final da emulsão de SB62 é como a seguir (Tabela 8): Tabela 8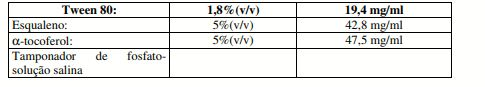

[000297] O sistema adjuvante AS03 é preparado misturando-se tamponador (PBS mod) com volume de SB62. A mistura é agitada durante de 15 a 45 minutos à temperatura ambiente, e o pH é ajustado em 6,8 ± 0,1 com NAOH (0,05 ou 0,5 M)/HCl (0,03 M ou 0,3 M). Após mais agitação durante de 15 a 20 minutos à temperatura ambiente, o pH é medido e a mistura é esterilizada por meio de filtração através de uma membrana de 0,22 μm. O adjuvante AS03 estéril é armazenado a +2-8°C até o enchimento asséptico em seringas de vidro estéreis de tipo I de 1,25 ml (Ph. Eur.). Cada seringa contém um excesso de volume de 720 μl (500 μl + 220 μl de excesso). A composição final do adjuvante AS03 é como a seguir (Tabela 9): Tabela 9 3) Reconstituição extemporânea da vacina de vírus dividido adjuvantado com AS03. No momento da injeção, o conteúdo da seringa pré- enchida contendo o adjuvante é injetado no frasco que contém os antígenos de vírions divididos inativados trivalentes concentrados. Após a misturação, o conteúdo é sugado para o interior da seringa e a agulha é substituída por uma agulha intramuscular. Uma dose da vacina candidata de influenza adjuvantada com AS03 reconstituída corresponde a 1 ml.
3) Reconstituição extemporânea da vacina de vírus dividido adjuvantado com AS03. No momento da injeção, o conteúdo da seringa pré- enchida contendo o adjuvante é injetado no frasco que contém os antígenos de vírions divididos inativados trivalentes concentrados. Após a misturação, o conteúdo é sugado para o interior da seringa e a agulha é substituída por uma agulha intramuscular. Uma dose da vacina candidata de influenza adjuvantada com AS03 reconstituída corresponde a 1 ml.
[000298] As vacinas foram administradas intramuscularmente na região deltóide do braço não-dominante. As vacinas candidatas de influenza pandêmica são vacinas de 2 componentes consistindo de antígenos apresentados em um frasco (recipiente de antígeno) e uma seringa pré-enchida contendo, ou o adjuvante (recipiente de adjuvante) ou o diluente. No momento da injeção, o conteúdo da seringa pré-enchida é injetado no frasco que contém os antígenos. Após misturação, o conteúdo é sugado para o interior da seringa. A agulha usada é substituída por uma agulha intramuscular. Uma dose da vacina corresponde a 1 ml.
[000299] Um total de 400 sujeitos foi incluído neste estudo: de 49 a 51 sujeitos em cada um dos 8 grupos. A idade média da coorte vacinada no momento da vacinação foi de 34,3 anos com um desvio padrão de 12,76 anos. A distribuição média de idades e sexo dos sujeitos em todos os 8 grupos de vacina foi similar.
[000300] A administração da vacina candidata contra influenza pandêmica adjuvantada com AS03 foi segura e clinicamente bem tolerada na população de estudo, i.e. pessoas adultas com idades entre 18 e 60 anos.
[000301] Análise da imunogenicidade foi realizada na coorte de ATP (394 sujeitos).
[000302] Para avaliar a resposta imune humoral induzida pela vacina candidata de H5N1 de influenza pandêmica adjuvantada com ASO3, calculou-se os parâmetros a seguir (com intervalos de confiança de 95%) para cada grupo de tratamento: • Titulações geométricas médias (GMTs) de titulações de anticorpos HI nos dias 0,21 e 42. • Taxas de soroconversão (SC) nos dias 21 e 42; • Fatores de conversão no dia 21 e 42; • Taxas de proteção no dia 21 e 42.
[000303] As GMTs para anticorpos HI com 95% de CI são mostradas na Tabela 10 (GMT para anticorpo anti-HI) e na Figura 6. GMTs de pré-vacinação de anticorpos para a cepa de vacinação de H5N1 encontraram-se dentro da mesma faixa nos oito grupos de estudo. Após a primeira vacinação, em todos os grupos não-adjuvantados os níveis de anticorpos anti-hemaglutinina aumentaram muito modestamente de uma maneira dependente-da-dose. Nos grupos de vacinação adjuvantados, observou-se um aumento mais preeminente dos níveis de anticorpos anti- hemaglutinina mesmo após a primeira vacinação, sendo que a GMT mais alta no grupo que recebeu a maior dose de antígeno (HN30AD). Após a segunda vacinação, as GMTs nos grupos adjuvantados aumentaram ligeiramente relativamente à primeira GMT pós-vacinação. Em comparação, observou-se GMTs significativamente mais altas após a segunda vacinação em todos os grupos adjuvantados, com um aumento dependente da dose observada do grupo de 3,8 μg ao 7,5 μg ao 15 μg. No caso do grupo de 30 μg, observou-se uma GMT menor do que para o grupo de 7,5 μg. Todos os grupos de estudo adjuvantados, incluindo a menor dose de 3,8 μg de HA, elicitaram uma resposta imune que satisfaz os critérios para licenciamento de vacinas pandêmicas com base na orientação obrigatória da FDA (março de 2006) e também nos critérios estabelecidos pela EMEA. Tabela 10 - Titulações geométricas médias (GMTs) para anticorpo anti-HA em diferentes momentos (coorte de ATP para imunogenicidade) HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 GMT = titulação geométrica média calculada em todos os sujeitos N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada 95% CI = intervalo de confiança de 95%; LL = limite inferior, UL = limite superior MIN/MAX = mínimo/máximo PRE = dose 1 de pré-vacinação PI(D21) = pós-vacinação no dia 21 PII(D42) = pós-vacinação no dia 42 fonte de dados = Apêndice da tabela IIIA
HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 GMT = titulação geométrica média calculada em todos os sujeitos N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada 95% CI = intervalo de confiança de 95%; LL = limite inferior, UL = limite superior MIN/MAX = mínimo/máximo PRE = dose 1 de pré-vacinação PI(D21) = pós-vacinação no dia 21 PII(D42) = pós-vacinação no dia 42 fonte de dados = Apêndice da tabela IIIA
[000304] Resultados são apresentados nas Tabelas 11 (fatores de conversão), 12 (taxas de soroproteção) e 13 (taxas de soroconversão).
[000305] Os fatores de conversão (Tabela 11, Figura 9) representam o aumento de múltiplo de GMTs de HI no soro para a cepa de vacina no dia 21 e 42, em comparação com dia 0. O fator de conversão após a segunda vacinação varia de 1,2 a 3,9 nos 4 grupos não-adjuvantados e de 27,9 a 60,5 nos grupos adjuvantados. Fatores de conversão nos grupos adjuvantados com AS03 são muito superiores ao aumento de 2,5 vezes da GMT requerida pelas Autoridades Européias para vacinas interpandêmicas para adultos (apresentado na Tabela 1). Correntemente, no caso de vacinas candidatadas pandêmicas aplica-se os mesmos critérios conforme utilizado para o licenciamento anual da vacina de influenza interpandêmica. É digno de nota que, exceto no caso dos grupos adjuvantados com menor concentração de antígeno, se obtém um fator de conversão de > 2,5 já após a primeira vacinação. As taxas de soroproteção (Tabela 12, Figura 8) representam a proporção de sujeitos com uma titulação de HI no soro >40 no dia 21 e 42. Antes da vacinação, verificou-se que 3 dos sujeitos (1 no grupo HN15, 1 no grupo HN8AD e 1 no grupo HN4D) apresentam níveis protetores de anticorpos para cepa de vacina H5N1 A/Vietnam/1194/2004. No caso de H5N1, obteve-se um percentual muito baixo de indivíduos soro-protegidos antes da vacinação, confirmando a observação de estudos precedentes (Bresson JL et al. The Lancet 2006:367 (9523): 1657-1664; Treanor JJ et al. N Engl J Med. 2006;354:1343-1351). No dia 21, as taxas de soroproteção nos grupos não-adjuvantados compreenderam de 0,0% a 28,6% (Tabela 12), enquanto que nos grupos adjuvantados de 26,0% a 58,3% dos sujeitos atingiram uma titulação protetora. Após a segunda dose de vacina candidata de influenza pandêmica, de 4,0 a 42,9% dos sujeitos nos grupos não- adjuvantados e de 84,0% a 95,9% nos grupos adjuvantados apresentaram um titulação igual ou maior que o limiar considerado como protetor (i.e. titulação de HI > 1:40). Conseqüentemente, até 95,9% dos sujeitos (grupo 15HNAD) que receberam uma vacina candidata pandêmica adjuvantada apresentaram uma titulação de HI no soro >40 após 2 vacinações e pareceram estar protegidos contra a cepa de vacinação de H5N1. Todas as quatro formulações candidatas excederam a taxa de soroproteção de 70% requerida na população com idades de 18 a 60 anos pelas Autoridades Européias - sendo que uma proporção substancial dos sujeitos já atingiram uma titulação protetora após a primeira dose - enquanto que vacinas candidatas não-adjuvantadas atingiram este critério. As taxas de soroconversão (Tabela 13, Figura 7) representam o percentual de vacinados que apresentaram, ou uma titulação pré-vacinação < 1:10 e uma titulação pós-vacinação > 1:40, ou uma titulação pré-vacinação > 1:10 e pelo menos um aumento de quatro vezes na titulação de pós- vacinação no dia 21 e 42, em comparação com dia 0. Após a primeira vacinação, taxas de soroconversão nos grupos não-adjuvantados compreenderam de 0,0% a 14,9% (Tabela 13). Nos grupos de estudo adjuvantados correspondentes, observou-se taxas de soroconversão entre 24,0% e 58,3% após a primeira vacinação, excedendo mesmo em 3 dos 4 grupos adjuvantados que receberam diferentes teores de antígenos as exigências da EMEA (taxa de soroconversão requerida acima de 40% na população com idades na faixa de 18 a 60 anos). Após a segunda vacinação entre 4,0% e 40,8% dos sujeitos nos grupos não-adjuvantados, mas de 82,0% a 95,9% dos sujeitos nos grupos adjuvantados, ou atingiram uma soroconversão ou um aumento de quatro vezes. Portanto, após duas vacinações, todas as quatro formulações adjuvantadas da vacina candidata atenderam o critério para licenciamento como indicado pela EMEA, mas apenas a maior dose de vacina não-adjuvantada meramente atingiu este limiar (HN30: 40,8%). Tabela 11 - Fator de soroconversão para titulação de anticorpos HAI em cada momento da pós-vacinação (coorte de ATP para imunogenicidade)
 HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg+AS03 HN15AD = H5N1 15μg+AS03 HN8AD = H5N1 7,5μg+AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados obteníveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada PRE = Pré-vacinação PI(D21) = Pós-vacinação no dia 21 PII(D42) = Pós-vacinação no dia 42 Tabela 12 - Taxas de soroproteção nos dias 0, dia 21 e dia 42 definido como o percentual de vacinados com a titulação de anti-HA no soro >1:40 (coorte de ATP para imunogenicidade)
HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg+AS03 HN15AD = H5N1 15μg+AS03 HN8AD = H5N1 7,5μg+AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados obteníveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada PRE = Pré-vacinação PI(D21) = Pós-vacinação no dia 21 PII(D42) = Pós-vacinação no dia 42 Tabela 12 - Taxas de soroproteção nos dias 0, dia 21 e dia 42 definido como o percentual de vacinados com a titulação de anti-HA no soro >1:40 (coorte de ATP para imunogenicidade)  HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada PRE = Pré-vacinação PI(D21) = Pós-vacinação no dia 21 PII(D42) = Pós-vacinação no dia 42 Tabela 13 - Taxas de soroconversão para titulação de anticorpos anti-HA em cada pós-vacinação no dia 21 e dia 42 (coorte de ATP para imunogenicidade)
HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos com titulação dentro da faixa especificada PRE = Pré-vacinação PI(D21) = Pós-vacinação no dia 21 PII(D42) = Pós-vacinação no dia 42 Tabela 13 - Taxas de soroconversão para titulação de anticorpos anti-HA em cada pós-vacinação no dia 21 e dia 42 (coorte de ATP para imunogenicidade)  HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis PI(D21) = Pós-vacinação a 21 dias PII(D42) = Pós-vacinação a 42 dias Fonte de dados = Apêndice tabela IIIA n/% = número/percentual de sujeitos com, ou uma titulação de pré-vacinação <1:10 e titulação de pós-vacinação >1:40 ou uma titulação de pré-vacinação >1:10 e um aumento mínimo de 4 vezes na titulação de pós-vacinação. 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior
HN30 = H5N1 30μg HN15 = H5N1 15μg HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03 HN15AD = H5N1 15μg + AS03 HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis PI(D21) = Pós-vacinação a 21 dias PII(D42) = Pós-vacinação a 42 dias Fonte de dados = Apêndice tabela IIIA n/% = número/percentual de sujeitos com, ou uma titulação de pré-vacinação <1:10 e titulação de pós-vacinação >1:40 ou uma titulação de pré-vacinação >1:10 e um aumento mínimo de 4 vezes na titulação de pós-vacinação. 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior
[000306] No caso de uma pandemia de influenza, grandes proporções da população serão despreparadas relativamente à cepa de influenza pandêmica e igualmente exigirão 2 doses de vacina a serem protegidas. Para reduzir o teor de antígeno na vacina pandêmica potencial e, portanto, aumentam o suprimento de vacina, emprega-se estratégias de adjuvantação após se ter mostrado que vacinas candidatas de H5N1 não- adjuvantadas (H5N1 é um candidato principal para causar a próxima pandemia de influenza) elicitam uma resposta imune mesmo após doses maiores de antígeno (Treanor JJ et al. N Engl J Med. 2006;354: 1343-1351).
[000307] Neste primeiro ensaio, aqui reportado, com uma vacina candidata contra influenza pandêmica H5N1 adjuvantada com AS03, obteve- se os seguintes resultados: • Há um benefício claro do adjuvante AS03 em comparação com as formulações de antígeno simples para todas as doses de hemaglutinina diferentes testadas. Após a segunda vacinação, houve uma superioridade clara dos grupos adjuvantados em GMTs de anticorpo HI observado: A GMT do grupo adjuvantado que recebeu a menor dose de antígeno (3,8 μg de HA) testada ainda foi 7,5 vezes maior do que a maior GMT obtida nos grupos não- adjuvantados, elicitada pela maior dose de antígeno (2 injeções a 30 μg de HA). Não houve superposição de intervalo de confiança de 95% entre qualquer um dos grupos adjuvantados com qualquer um dos grupos não- adjuvantados no dia 42. • As taxas de soroconversão no dia 42 foram de 82,0%, 90,0%, 95,9% e 85,6% para os grupos de 3,8 μg, 7,5 μg, 15 μg e 30 μg mais adjuvante, respectivamente. Isto é para todos os teores de antígeno adjuvantado com AS03 testados superiores aos 40% exigidos pelas Autoridades Européias. Apenas um dos grupos não-adjuvantados, o grupo com maior dose de antígeno (30 μg), foi meramente capaz de atingir um percentual acima do limitar determinado. • No dia 42, as taxas de soroproteção nos quatro grupos adjuvantados foram de 84,0%, 90,0%, 95,9% e 85,4% para os grupos de 3,8 μg, 7,5 μg, 15 μg e 30 μg mais adjuvante, respectivamente. O percentual requerido pela EMEA para o grupo de idade adulta abaixo de 60 anos de vida é de 70%, com isto, todos os grupos adjuvantados atenderam este critério, enquanto que nenhum dos grupos não-adjuvantados simples pôde atingir a taxa de soroproteção requerida. • Neste estudo, após duas vacinações com as diferentes formulações de vacina candidata, o fator de soroconversão foi maior que 27,9 para os quatro grupos adjuvantados, excedendo com isto amplamente a exigência determinada em 2,5. Também para os grupos não-adjuvantados, os 2 grupos que receberam as maiores doses de antígeno (15 μg e 30 μg) atenderam a exigência com 2,8 (grupo de HN15) e 3,9 (grupo HN30).
[000308] Considerando os três critérios indicados pela EMEA que também são aplicáveis para a avaliação de vacinas candidatadas de influenza pandêmica, todos os grupos adjuvantados obtidos após a segunda dose da respectiva vacina de H5N1 adjuvantada com AS03, todos os três critérios definidos para este grupo etário. Todos os grupos adjuvantados também atenderam os critérios propostos pela FDA relativos a soroconversão, soroproteção e fator de conversão, após a segunda dose.
[000309] A avaliação da imunogenicidade contra uma cepa de H5N1 antigenicamente diferente da cepa de vacina é considerada para permitir avaliação adicional do potencial de uma vacina candidata para pandemia. Realiza-se teste de reatividade cruzada nos soros de sujeitos que receberam a cepa de vacinação e avalia-se o potencial dos anticorpos induzidos pela vacina em reagir a uma cepa antigenicamente diferente. Para avaliação da reatividade cruzada selecionou-se H5N1 A/Indonésia/5/2005. H5N1 A/Indonésia pertence a Clade 2, enquanto que H5N1 A/Vietnam/1194/2004, a cepa de vacina, pertence a Clade 1, e é o 1o protótipo de cepa de vacina pandêmica de um novo grupo genético liberado pela OMS. Ambas as cepas podem ser consideradas, portanto, como antigenicamente diferentes.
[000310] Soropositivo foi definido como uma titulação de anticorpos HI >1:10. Todos os sujeitos foram soronegativos para Indonésia antes da primeira vacinação com a cepa Vietnam. Após a segunda vacinação, até 48% dos sujeitos dos grupos adjuvantados (28% do grupo de 3,8 μg, 48% do grupo de 7,5 μg, 26,5% do grupo de 15 μg, 33,3% do grupo de 30 μg) atingiram um status de soropositividade. Em comparação, não se observou qualquer soropositividade nos grupos de 3,8, 7,5 e 15 μg não-adjuvantados de forma alguma, enquanto que apenas 2% (1 sujeito) foi determinado como sendo soropositivo para H5N1 Indonésia no grupo não-adjuvantado com o maior teor de antígeno (30 μg). Tabela 14 - Taxas de soropositividade e GMTs para titulação de anticorpos HI no dia 0, dia 21 e dia 42 por grupo de vacina (coorte de ATP para imunogenicidade)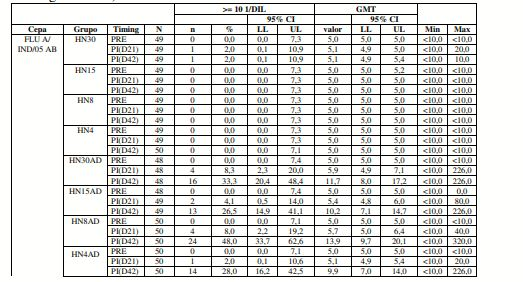 HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4=H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI >= 1:10) 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior GMT = titulação de anticorpos geométrica média Min/Max = mínimo/máximo PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42)
HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4=H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI >= 1:10) 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior GMT = titulação de anticorpos geométrica média Min/Max = mínimo/máximo PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42)
[000311] Após a segunda vacinação, até 32,0% dos sujeitos nos grupos adjuvantados foram determinados como sendo soroprotegidos contra a cepa Indonésia não contida na vacina. No grupo de 3,8μg, 7,5μg, 15μg e 30μg adjuvantado, 20,0%, 32,0%, 20,4% e 29,2% dos sujeitos apresentaram uma titulação de >1:40 após a segunda vacinação, respectivamente. Nenhum dos sujeitos nos grupos não-adjuvantados foi soroprotegido. Tabela 15 - Taxas de soroproteção (SP) para titulação de anticorpos HI no dia 0, dia 21 e dia 42 por grupo de vacina (coorte de ATP para imunogenicidade) HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4 = H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos protegidos (titulação de HI >= 1:40) n/% DESPROT = número/percentual de sujeitos desprotegidos (titulação de HI < 1:40) 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior
HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4 = H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos protegidos (titulação de HI >= 1:40) n/% DESPROT = número/percentual de sujeitos desprotegidos (titulação de HI < 1:40) 95% CI = intervalo de confiança de 95% LL = limite inferior UL = limite superior
[000312] Até 32,0% dos sujeitos nos grupos adjuvantados atingiram soroconversão contra a cepa Indonésia não contida na vacina. No grupo de 3,8μg, 7,5μg, 15μg e 30μg adjuvantado, 20,0%, 32,0%, 20,8% e 29,2% dos sujeitos soroconvertidos após a segunda vacinação, respectivamente. Para nenhum dos sujeitos nos grupos não-adjuvantados foi possível demonstrar soroconversão. Tabela 16 - Taxa de soroconversão (SC) para titulação de anticorpos HI no dia 21 e dia 42 por grupo de vacina (coorte de ATP para imunogenicidade)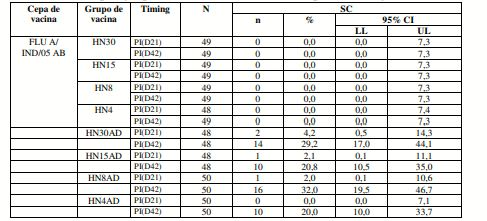 HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4=H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 PI(D21)= 21 dias após a primeira vacinação (dia 21), PII(D42)= 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual que soroconverteram no POST especificado 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior
HN30=H5N1 30μg, HN15=H5N1 15μg, HN8=H5N1 7,5μg, HN4=H5N1 3,8μg HN30AD=H5N1 30μg + AS03, HN15AD=H5N1 15μg + AS03, HN8AD=H5N1 7,5μg + AS03, HN4AD=H5N1 3,8μg + AS03 PI(D21)= 21 dias após a primeira vacinação (dia 21), PII(D42)= 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual que soroconverteram no POST especificado 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior
[000313] Fatores de soroconversão entre 2 e 2,8 foram obtidos pelos grupos adjuvantados no ensaio. Nos grupos de 3,8μg, 7,5μg, 15μg e 30μg adjuvantados, o fator de soroconversão foi de 2,0, 2,8, 2,1 e 2,3, respectivamente.
[000314] Em conclusão, após 2 doses de uma vacina candidata de vírus dividido adjuvantada com adjuvante AS03, dados de reatividade cruzada obtidos para uma cepa H5N1 de um clade diferente da cepa da vacina, que causou considerável morbidez e mortalidade em humanos na Ásia, foram positivos. Até 48% dos sujeitos apresentaram sinais de estarem preparados, e até 32% dos sujeitos se mostraram efetivamente soroprotegidos contra a cepa não-vacina. Estes resultados mostram que a adjuvantação de uma vacina para pandemia pode proporcionar reatividade cruzada contra uma variante de desvio da cepa pandêmica usada na candidata a vacina. Estes resultados estão confirmando o potencial da vacina adjuvantada de preparar e preparar de forma cruzada.
[000315] O ensaio de neutralização é um método que permite a quantificação de anticorpos que inibem a fixação, a penetração e também a propagação do vírus de influenza nas células. Embora, para o ensaio de inibição de hemaglutinina, um limiar de soroproteção esteja estabelecido, tal não é o caso neste ensaio. Alternativamente, é possível usar um aumento de quatro vezes na titulação neutralizadora para avaliar se indivíduos vacinados responderam contra a cepa de vacinação ou uma cepa heteróloga. A taxa de soroconversão é um dos parâmetros-chave da imunogenicidade usados pela CHMP/FDA para avaliar a efetividade de vacinas de influenza candidatas. Testes de amostras de soro em um ensaio de neutralização do tipo referido usando cepa desviada poderiam permitir predizer, pelo menos, a freqüência de indivíduos que foram "preparados [primed]" contra uma dada cepa diferente da cepa de vacina.
[000316] O limiar para a soropositividade é ajustado a uma titulação de >1:28, o ensaio de neutralização também é um teste altamente sensível. GMT's no dia 0 compreenderam de 14,0 a 18,1 nos grupos não-adjuvantados, e de 18,5 a 25,2 nos grupos adjuvantados. Após a segunda vacinação, titulações aumentaram para os grupos não-adjuvantados de uma maneira dependente da dose a 43,9, 61,7, 86,9 e 177,8 para os grupos de 3,8, 7,5, 15 e 30μg respectivamente. Nos grupos adjuvantados obteve-se titulações de 381,0, 421,2, 464,7 e 333,3 para os grupos de 3,8, 7,5, 15 e 30μg respectivamente, repetindo exatamente a observação feita em titulações de HI: desde o grupo de 3,8μg passando por 7,5μg ao grupo de 15μg adjuvantado observou-se um aumento dependente-da-dose na GMT (Tabela 17). Tabela 17: Taxas de soropositividade e GMTs para titulação de anticorpos neutralizadores no dia 0, dia 21 e dia 42 (coorte de ATP para imunogenicidade)
 HN30 = H5N1 30μg; HN15 = H5N1 15μg; HN8 = H5N1 7,5μg; HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03; HN15AD = H5N1 15μg + AS03; HN8AD = H5N1 7,5μg + AS03; HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI > = 1:10) 95% CI = intervalo de confiança de 95% LL = limite inferior, UL = limite superior, GMT = titulação de anticorpos média geométrica Min/Max = mínimo/máximo PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42)
HN30 = H5N1 30μg; HN15 = H5N1 15μg; HN8 = H5N1 7,5μg; HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03; HN15AD = H5N1 15μg + AS03; HN8AD = H5N1 7,5μg + AS03; HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI > = 1:10) 95% CI = intervalo de confiança de 95% LL = limite inferior, UL = limite superior, GMT = titulação de anticorpos média geométrica Min/Max = mínimo/máximo PRE = dose 1 de pré-vacinação (dia 0) PI(D21) = 21 dias após a primeira vacinação (dia 21) PII(D42) = 21 dias após a segunda vacinação (dia 42)
[000317] Como mencionado acima, usa-se um aumento de quatro vezes para determinar a soroconversão contra uma cepa de influenza. Portanto, sujeitos soropositivos no dia 0 só serão incluídos se alcançarem um aumento de quatro vezes, subtraindo desta forma um fundo potencial. Após a segunda dose, a soroconversão nos grupos não-adjuvantados pôde ser novamente observada de uma maneira dependente da dose: 20,9, 37,5, 53,5 e 76,0% dos sujeitos soroconvertidos nos grupos de 3,8, 7,5, 15 e 30μg respectivamente. Nos grupos adjuvantados, 86,0, 83,3, 86,0 e 100,0% dos sujeitos nos grupos de 3,8, 7,5, 15 e 30μg respectivamente soroconvertidos, confirmando com isto também os resultados de HI. É digno de nota que após a primeira dose de vacina adjuvantada, já de 66,7 a 88,0% dos sujeitos se apresentaram soroconvertidos nos quatro grupos adjuvantados. Tabela 18 - Taxas de soroconversão (SC) para titulação de anticorpos (de Dresden) a cada momento após a vacinação (coorte de ATP para imunogenicidade)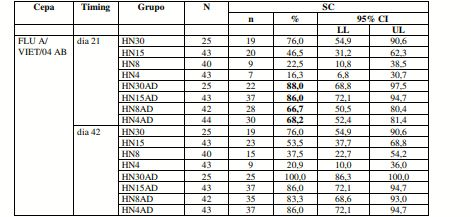 HN30 = H5N1 30μg; HN15 = H5N1 15μg; HN8 = H5N1 7,5μg; HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03; HN15AD = H5N1 15μg + AS03; HN8AD = H5N1 7,5μg + AS03; HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soroconvertidos (pelo menos um aumento de 4 vezes em POST) 95% CI = intervalo de confiança de 95%, LL = Limite inferior, UL = Limite superior
HN30 = H5N1 30μg; HN15 = H5N1 15μg; HN8 = H5N1 7,5μg; HN4 = H5N1 3,8μg HN30AD = H5N1 30μg + AS03; HN15AD = H5N1 15μg + AS03; HN8AD = H5N1 7,5μg + AS03; HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soroconvertidos (pelo menos um aumento de 4 vezes em POST) 95% CI = intervalo de confiança de 95%, LL = Limite inferior, UL = Limite superior
[000318] As titulações GMT e taxas de soroconversão para a cepa Vietnam são ilustradas em 10A e 10B respectivamente. Em conclusão, anticorpos neutralizadores medidos contra a cepa de vacina (Vietnam) confirmam os resultados obtidos pelo HI. Todos os grupos adjuvantados incluindo o grupo de menor dose de 3,8μg alcançaram uma soroconversão acima de 65% após a primeira, e acima de 80% dos sujeitos testados após a segunda dose da vacina pandêmica candidata.
[000319] Dados parciais (apenas dos grupos de 3,8μg e 7,5μg de HA adjuvantados) são apresentados aqui, abaixo. Como já discutido, devido à natureza do ensaio de neutralização que mede anticorpos que inibem a penetração na célula e a propagação célula a célula do vírus da influenza adicionalmente à inibição da fixação do vírus, a avaliação da cepa desviada permite avaliar adicionalmente o potencial da vacina em preparar também para uma cepa não-vacina.
[000320] Nos dois grupos inferiores adjuvantados, GMTs contra a cepa Indonésia alcançadas após duas doses da vacina foram de 70,6 e 73,1 para os grupos de 3,8μg e 7,5μg adjuvantados, respectivamente. Tabela 19 - Taxas de soropositividade e GMTs de titulação de anticorpos neutralizadores para a cepa Indonésia no dia 0, dia 21 e dia 42 (coorte de ATP para imunogenicidade)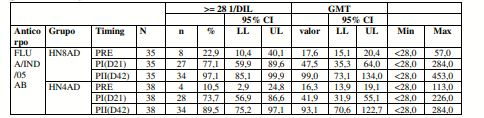 HN8AD = H5N1 7,5μg + AS03, HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI >= 1:10) 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior GMT = titulação de anticorpos geométrica média Min/Max = mínimo/máximo PRE = dose 1 pré-vacinação (dia 0), PI(D21) = 21 dias após a primeira vacinação (dia 21), PII(D42) = 21 dias após a segunda vacinação (dia 42)
HN8AD = H5N1 7,5μg + AS03, HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos soropositivos (titulação de HI >= 1:10) 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior GMT = titulação de anticorpos geométrica média Min/Max = mínimo/máximo PRE = dose 1 pré-vacinação (dia 0), PI(D21) = 21 dias após a primeira vacinação (dia 21), PII(D42) = 21 dias após a segunda vacinação (dia 42)
[000321] Ambos os grupos, de 3,8 e 7,5μg, adjuvantados receberam uma alta taxa de soroconversão contra a cepa de não-vacinação antigenicamente diferente: 84,2% dos sujeitos soroconvertidos quando testados contra a cepa A/Indonésia. Tabela 20 - Taxas de soroconversão (SC) da titulação de anticorpos neutralizadores para a cepa Indonésia em cada momento após a vacinação (coorte de ATP para imunogenicidade) HN8AD = H5N1 7,5μg + AS03, HN4AD = H5N1 3,8μg + AS03 PRE = dose 1 de pré-vacinação (dia 0), PI(D21) = 21 dias após a primeira vacinação (dia 21), PII(D42) = 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos que soroconverteram (pelo menos um aumento de 4 vezes em POST) 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior
HN8AD = H5N1 7,5μg + AS03, HN4AD = H5N1 3,8μg + AS03 PRE = dose 1 de pré-vacinação (dia 0), PI(D21) = 21 dias após a primeira vacinação (dia 21), PII(D42) = 21 dias após a segunda vacinação (dia 42) N = número de sujeitos com resultados disponíveis n/% = número/percentual de sujeitos que soroconverteram (pelo menos um aumento de 4 vezes em POST) 95% CI = intervalo de confiança de 95%, LL = limite inferior, UL = limite superior
[000322] Os dados disponíveis na reatividade cruzada no ensaio de neutralização indicam um efeito notável da vacina adjuvantada contendo uma cepa heteróloga à cepa Indonésia testada e confirmam o potencial de reação cruzada da vacina candidata.
[000323] Para avaliação da CMI, favor ver seções I.2, I.3, e IV.3.2. Um das características importantes de um adjuvante no aumento da imunogenicidade de uma vacina é a capacidade de estimular a imunidade mediada com célula, CMI. Neste ensaio, prevê-se uma avaliação de células CD4 e CD8 específicas para influenza incluindo freqüências de citocinas relacionadas com Th1 e também a avaliação da freqüência de células B de memória. Dados podem ser obtidos para as respostas de células T dos dois menores grupos de antígeno, adjuvantados ou não com AS03. Resultados de CMI são expressos como uma freqüência de células T CD4 positivas para citocina(s).
[000324] Valores médios (incluindo primeiro e terceiro quartis, ver Tabela 21) são apresentados na Figura 11. Os resultados indicaram que os grupos adjuvantados claramente induziram uma resposta CD4 muito mais forte em comparação com os grupos não-adjuvantados. Tabela 21 - Estatísticas descritivas sobre as células T CD4 positivas para freqüência (por milhão de células T CD4) em cada momento (coorte de ATP para imunogenicidade)
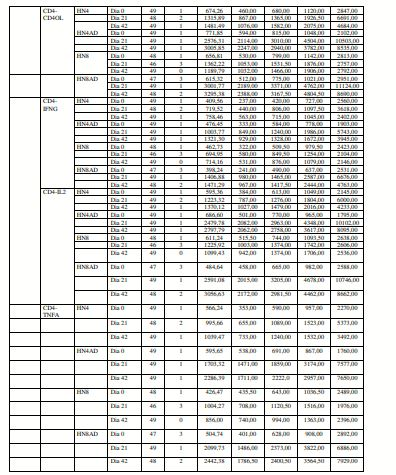 HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis; N falt. = número de sujeitos com resultados faltantes GM = média geométrica SD = Desvio padrão Q1,Q3 = primeiro e terceiro quartis MIN/MAX = mínimo/máximo
HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN8AD = H5N1 7,5μg + AS03 HN4AD = H5N1 3,8μg + AS03 N = número de sujeitos com resultados disponíveis; N falt. = número de sujeitos com resultados faltantes GM = média geométrica SD = Desvio padrão Q1,Q3 = primeiro e terceiro quartis MIN/MAX = mínimo/máximo
[000325] Na análise inferencial confirmou-se que tanto após a primeira vacinação no dia 21 (com exceção de células CD4 positivas para IFN gama) como também após a segunda vacinação no dia 42, a indução de células CD4 positivas para citocinas foi significativamente maior no grupo adjuvantado em comparação com o grupo não-adjuvantado que recebeu a mesma dose. Portanto, o efeito adjuvante observado na avaliação sorológica dos anticorpos induzidos pela vacina foi confirmado pelos resultados de CMI. De uma maneira similar, a análise mostra que o efeito sobre a CMI é claramente dependente do adjuvante, mas não da dose (comparação das doses de 3,8μg e de 7,5μg apenas), o que é consistente com os resultados de HI (ver Tabela 22). Tabela 22 - Estatísticas inferenciais (valores p de Testes de Kruskal-Wallis) relativamente às células T CD4 positivas para freqüência de citocinas em cada momento
 HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN8AD = H5N1 7,5μg +AS03 HN4AD = H5N1 3,8μg + AS03
HN8 = H5N1 7,5μg HN4 = H5N1 3,8μg HN8AD = H5N1 7,5μg +AS03 HN4AD = H5N1 3,8μg + AS03
[000326] Em conclusão, AS03 em combinação com a cepa pandêmica potencial A/Vietnam foi capaz de estimular uma resposta imune mediada com célula com as duas menores doses de antígeno testadas. Adicionalmente, a resposta observada nos grupos adjuvantados foi mais forte do que a resposta CD4 induzida pelos grupos não-adjuvantados.
[000327] O candidato predominante para a próxima pandemia de influenza é o vírus aviário H5N1, que resultou em uma alta taxa de mortalidade em casos de transmissão de ave para humano, embora a transmissão eficiente de humano para humano não tenha sido plenamente confirmada. Caso H5N1 venha a demonstrar a capacidade de disseminar-se eficientemente de pessoa para pessoa, em combinação com a rede de transporte global, o resultado pode ser, exequivelmente, um surto disseminado de influenza que afeta um elevado percentual de indivíduos, levando a maior mortalidade e morbidez em todos os países. Portanto, uma abordagem imunologicamente efetiva e diminuidora de antígeno para a vacinação precisa ser estabelecida para prevenir efeitos potencialmente devastadores de uma pandemia. Isto pode ser obtido usando-se um adjuvante vantajoso e, pela primeira vez, seria possível mostrar neste ensaio o efeito incrementador de imunogenicidade de um adjuvante inédito sobre uma vacina de H5N1 candidata.
[000328] Este estudo foi projetado para avaliar (1) a segurança e reatogenicidade em adultos saudáveis de uma vacina candidata contra influenza pandêmica adjuvantada ou não com emulsão óleo-em-água, i.e. AS03, (2) o anticorpo e respostas imunes mediadas com célula.
[000329] Dados de reatogenicidade mostram que a vacina pandêmica candidata adjuvantada induziu (independente do teor de antígeno) mais sintomas locais e gerais do que os grupos não-adjuvantados. No entanto, o perfil de segurança de todos os 4 grupos adjuvantados foi clinicamente aceitável. Não se reportou evento advento sério. A partir destes resultados, pode-se concluir que a reatogenicidade e o perfil de segurança da vacina pandêmica candidata adjuvantada com AS03 é satisfatório e clinicamente aceitável.
[000330] No que se refere à resposta imune, a vacina candidata contra influenza pandêmica adjuvantada com AS03 excedeu, com todos os teores de antígeno testados (3,8 μg, 7,5 μg, 15 μg e 30 μg de HA, H5N1 A/Vietnam/1194/2004), as exigências das Autoridade Européias para o registro anual de vacinas contra influenza de vírion dividido ("Note for Guidance on Harmonisation of Requirements for influenza Vaccines" [Nota para orientação sobre a harmonização de exigências para vacinas de influenza] para a avaliação imunológica das alterações anuais de cepa - CPMP/BWP/214/96), correntemente usado como base para a avaliação de vacinas candidatas contra influenza pandêmica (Guideline on dossier structure and content for pandemic influenza marketing authorization application [Diretriz sobre estrutura de dossiê e teor para aplicação de autorização de comercialização de influenza pandêmica], CPMP/VEG/4717/03). Os quatro diferentes teores de antígeno para uma vacina candidata de influenza pandêmica adjuvantada testados neste ensaio foram imunogênicos nos adultos saudáveis, que desenvolveram uma excelente resposta de anticorpos à hemaglutinina da influenza conforme medido com HI (Tabela 23). Tabela 23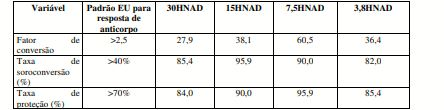
[000331] Dados que avaliam a reatividade cruzada relativamente a uma cepa antigenicamente diferente, H5N1 A/Indonésia/5/05, com o ensaio de inibição de hemaglutinina indicaram adicionalmente uma preparação cruzada [cross-priming] dos vacinados nos grupos adjuvantados contra uma cepa que sofreu variação. Medidas sorológicas foram completadas por meio de avaliação usando-se o ensaio de neutralização para cepa homóloga e heteróloga. Também por meio de ensaio de neutralização seria possível confirmar a imunogenicidade e o potencial de reação cruzada da vacina candidata.
[000332] Em resumo, 2 doses de uma vacina candidata de influenza pandêmica adjuvantada inédita induzem, na menor dose testada de 3,8 μg, uma titulação protetora contra a cepa de vacina H5N1 A/Vietnam/1194/2004 em proporção muito elevada de sujeitos, excedendo todos os critérios estabelecidos para avaliação da imunogenicidade de vacinas contra influenza. Adicionalmente, os resultados obtidos documentam a capacidade da vacina pré-pandêmica candidata de induzir imunidade contra uma cepa de variação.
[000333] Os resultados corroboram o uso da composição de vacina reivindicada para se obter soroproteção em uma situação de pandemia em que a preparação pré-pandêmica é realizada com uma vacina compreendendo uma cepa heteróloga para a cepa pandêmica circulante. Em outras palavras, a composição reivindicada pode ser usada para preparar respostas subseqüentes a cepa(s) pandêmica(s) desviadas. Os resultados corroboram o uso da composição reivindicada em um uso heterólogo e homólogo, de 1a e 2a dose, de prime-boost[preparação-reforço] de vacina contra influenza (H5N1) monovalente pandêmica adjuvantada com AS03: - preparação de pacientes com uma ou duas doses da vacina adjuvantada contendo uma cepa pandêmica (p. ex., a cepa Vietnam), a uma dose selecionada, incluindo uma baixa quantidade de HA, - seguida, vários meses depois (p. ex., 6 ou 12 meses mais tarde), de uma dose de i) ou a mesma cepa pandêmica (p. ex., cepa Vietnam, i.e. prime-boost[preparação-reforço] homólogo, ou ii) uma cepa heteróloga (p. ex., cepa Indonésia, i.e. prime-boostheterólogo), dada como um reforço. O valor desta vacina adjuvantada reside, de forma importante, numa situação em que a pandemia é declarada após indivíduos terem sido preparados antes, ou próximo do início da pandemia, uma vez ou duas vezes, ou com a mesma cepa, ou com uma cepa diferente da cepa 'pandêmica'.
Claims (42)
1. Composição de vacina contra influenza monovalente, caracterizadapelo fato de que compreende uma pequena quantidade de antígeno de vírus da influenza ou preparação antigênica de uma cepa H5, H2, H9, H7 ou H6 de vírus da influenza para uso, em combinação com um adjuvante, na proteção contra infecções por influenza causadas por uma cepa H5, H2, H9, H7 ou H6 de influenza variante, sendo que a pequena quantidade de antígeno não excede 15 μg de hemaglutinina (HA) por dose, e sendo que referido adjuvante é uma emulsão óleo-em-água compreendendo um óleo metabolizável, um esterol ou um tocoferol, e um agente emulsificante.
2. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que referido tocoferol é alfa-tocoferol.
3. Composição de acordo com a reivindicação 1 ou reivindicação 2, caracterizadapelo fato de que referido óleo metabolizável é esqualeno.
4. Composição de acordo com qualquer uma das reivindicações de 1 a 3, caracterizadapelo fato de que referido óleo metabolizável está presente em uma quantidade de 0,5% a 20% do volume total de referida composição imunogênica.
5. Composição de acordo com qualquer uma das reivindicações de 1 a 4, caracterizadapelo fato de que referido óleo metabolizável está presente em uma quantidade de 1,0% a 10% do volume total de referida composição imunogênica.
6. Composição de acordo com qualquer uma das reivindicações de 1 a 5 caracterizadapelo fato de que referido óleo metabolizável está presente em uma quantidade de 2,0% a 6,0% do volume total de referida composição imunogênica.
7. Composição de acordo com qualquer uma das reivindicações de 1 a 6, caracterizadapelo fato de que referido tocoferol ou alfa-tocoferol está presente em uma quantidade de 1,0% a 20% do volume total de referida composição imunogênica.
8. Composição de acordo com qualquer uma das reivindicações de 1 a 7, caracterizada pelo fato de que referido tocoferol ou alfa-tocoferol está presente em uma quantidade de 1,0% a 5,0% do volume total de referida composição imunogênica.
9. Composição de acordo com qualquer uma das reivindicações de 1 a 8, caracterizada pelo fato de que referido agente emulsificante é monooleato de polioxietileno sorbitol (Tween 80).
10. Composição de acordo com qualquer uma das reivindicações de 1 a 9, caracterizada pelo fato de que referido agente emulsificante está presente em uma quantidade de 0,01 a 5,0% em peso (peso/peso) de referida composição imunogênica.
11. Composição de acordo com qualquer uma das reivindicações de 1 a 10, caracterizada pelo fato de que referido agente emulsificante está presente em uma quantidade de 0,1 a 2,0% em peso (peso/peso) de referida composição imunogênica.
12. Composição de acordo com qualquer uma das reivindicações de 1 a 11, caracterizada pelo fato de que a quantidade de antígeno de HA não excede 10 μg por dose.
13. Composição de acordo com a reivindicação 12, caracterizada pelo fato de que a quantidade de antígeno de HA não excede 8 μg, ou 4 μg, ou 2 μg por dose.
14. Composição de acordo com qualquer uma das reivindicações de 11 a 13, caracterizada pelo fato de que a quantidade de antígeno de HA é entre 1 - 7,5 μg, ou de 1 - 5 μg por dose.
15. Composição de acordo com a reivindicação 14, caracterizada pelo fato de que a quantidade de antígeno de HA contém entre de 2,5 a 7,5 μg de HA por cepa.
16. Composição de acordo com qualquer uma das reivindicações de 1 a 15, caracterizada pelo fato de que referida cepa de vírus da influenza pandêmica é selecionada da lista que consiste de: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1.
17. Composição de acordo com a reivindicação 16, caracterizada pelo fato de que referida cepa de vírus da influenza pandêmica é selecionada da lista que consiste de: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1.
18. Composição de acordo com qualquer uma das reivindicações de 1 a 17, caracterizada pelo fato de que o antígeno ou composição de antígeno encontra-se na forma de: um vírus de influenza purificado, um vírus de influenza não-vivo, ou componente(s) de subunidade de vírus da influenza.
19. Composição de acordo com a reivindicação 18, caracterizada pelo fato de que referido vírus da influenza não-vivo é um vírus da influenza dividido.
20. Composição conforme reivindicada em qualquer uma das reivindicações de 1 a 19, caracterizada pelo fato de que referido antígeno de influenza ou composição antigênica é derivada de cultura de células ou produzido em ovos embrionários.
21. Composição conforme reivindicada em qualquer uma das reivindicações de 1 a 20, caracterizada pelo fato de que é para uso na medicina.
22. Kit, caracterizado pelo fato de que compreende uma unidade compreendendo uma pequena quantidade de antígeno de vírus da influenza ou preparação antigênica do mesmo e uma unidade compreendendo um adjuvante óleo-em-água como definido em qualquer uma das reivindicações de 1 a 11.
23. Kit de acordo com a reivindicação 22, caracterizado pelo fato de que referido antígeno é HA.
24. Kit de acordo com a reivindicação 23, caracterizado pelo fato de que referida quantidade de HA é como definida na reivindicação 1 ou em qualquer uma das reivindicações de 12 a 15.
25. Método para a produção de uma composição de vacina contra influenza para uma situação pandêmica ou uma situação pré- pandêmica, caracterizado pelo fato de que referido método compreende misturar um antígeno de vírus da influenza de uma cepa de vírus de influenza simples de H5, H2, H9, H7 ou H6 que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia, com um adjuvante em emulsão óleo-em-água e proporcionando unidades de vacina que contém não mais do que 15 μg de antígeno de hemaglutinina de influenza por dose.
26. Método de acordo com a reivindicação 25, caracterizado pelo fato de que o adjuvante em emulsão óleo-em-água é como definido em qualquer uma das reivindicações de 1 a 12.
27. Uso de (a) uma pequena quantidade de um antígeno de vírus da influenza ou preparação antigênica do mesmo a partir de uma cepa de influenza simples de H5, H2, H9, H7 ou H6 associada com uma pandemia ou apresentando o potencial de estar associada com uma pandemia, e (b) um adjuvante em emulsão óleo-em-água, caracterizado pelo fato de ser na fabricação de uma composição imunogênica como definida em qualquer uma das reivindicações de 1 a 21 para induzir pelo menos um de i) uma resposta imune melhorada de células T CD4, ii) uma resposta melhorada de memória de células B, contra referido vírus ou composição antigênica em um humano, iii) uma resposta humoral melhorada.
28. Uso de acordo com a reivindicação 27, caracterizado pelo fato de que referida resposta imune de células T CD4 envolve a indução de uma resposta de auxiliar T CD4 de reação cruzada ou a indução de uma resposta imune humoral de reação cruzada.
29. Uso de (a) uma pequena quantidade de um antígeno de vírus da influenza pandêmica ou preparação antigênica do antígeno de hemaglutinina do vírus da influenza de uma cepa de influenza simples associada com uma pandemia ou apresentando o potencial de estar associada com uma pandemia, e (b) um adjuvante em emulsão óleo-em-água como definido em qualquer uma das reivindicações de 1 a 11, caracterizadopelo fato de ser para a fabricação de um lote de vacina ou um kit de vacina para proteção contra infecção por vírus da influenza.
30. Uso de acordo com qualquer uma das reivindicações de 27 a 29, caracterizadopelo fato de que referida resposta imune ou proteção é obtida após uma ou duas doses de vacina.
31. Uso de acordo com qualquer uma das reivindicações de 28 a 30, caracterizadopelo fato de que referida vacina é administrada parenteralmente.
32. Método de acordo com a reivindicação 26 ou 27 ou uso de acordo com qualquer uma das reivindicações de 28 a 31, caracterizadopelo fato de que a referida quantidade de antígeno de HA é como definida em qualquer uma das reivindicações de 13 a 16.
33. Uso de um vírus da influenza ou preparação antigênica do mesmo, caracterizadopelo fato de ser para a fabricação de uma composição imunogênica para revacinação de humanos previamente vacinados com uma composição imunogênica como definida em qualquer uma das reivindicações de 1 a 20.
34. Uso de acordo com a reivindicação 33, caracterizadopelo fato de que a composição usada para a revacinação contém um adjuvante.
35. Uso de acordo com a reivindicação 34, caracterizadopelo fato de que referido adjuvante é um adjuvante em emulsão óleo-em-água.
36. Uso de acordo com qualquer uma das reivindicações de 33 a 35, caracterizado pelo fato de que referida composição imunogênica para revacinação contém um vírus da influenza ou preparação antigênica do mesmo que está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia.
37. Uso de acordo com a reivindicação 36, caracterizado pelo fato de que referida cepa pandêmica é selecionada da lista que consiste de: H5N1, H9N2, H7N7, H2N2, H7N1 e H1N1.
38. Uso de acordo com a reivindicação 36 ou reivindicação 37, caracterizado pelo fato de que referida composição imunogênica para revacinação contém um vírus da influenza ou preparação antigênica do mesmo que compartilha epitopos de células T CD4 comuns ou epitopos de células B comuns com o vírus da influenza ou preparação antigênica da mesma usada para a primeira vacinação.
39. Uso de acordo com qualquer uma das reivindicações de 33 a 38, caracterizado pelo fato de que a primeira vacinação é realizada com uma composição de influenza contendo uma cepa de influenza que poderia causar potencialmente uma pandemia e a revacinação é preparada com uma composição de influenza contendo uma cepa pandêmica circulante.
40. Uso de um antígeno ou preparação antigênica de uma primeira cepa de influenza, caracterizado pelo fato de que é na fabricação de uma composição imunogênica como definida em qualquer uma das reivindicações de 1 a 20 para proteção contra infecções por influenza causadas por uma cepa de influenza variante.
41. Uso de acordo com a reivindicação 40, caracterizado pelo fato de que a primeira cepa de influenza está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia.
42. Uso de acordo com a reivindicação 40, caracterizado pelo fato de que a cepa de influenza variante está associada com uma pandemia ou que possui o potencial de estar associada com uma pandemia.
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US83143706P | 2006-07-17 | 2006-07-17 | |
| US60/831,437 | 2006-07-17 | ||
| US60/831437 | 2006-07-17 | ||
| GB0618195A GB0618195D0 (en) | 2006-09-15 | 2006-09-15 | Influenza vaccine |
| GB0618195.2 | 2006-09-15 | ||
| GB0619090A GB0619090D0 (en) | 2006-09-27 | 2006-09-27 | Influenza vaccine |
| GB0619090.4 | 2006-09-27 | ||
| PCT/EP2006/010439 WO2008009309A1 (en) | 2006-07-17 | 2006-10-27 | Influenza vaccine |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0621886A2 BRPI0621886A2 (pt) | 2011-12-20 |
| BRPI0621886A8 BRPI0621886A8 (pt) | 2019-01-15 |
| BRPI0621886B1 true BRPI0621886B1 (pt) | 2021-09-21 |
Family
ID=41203904
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0621886-5A BRPI0621886B1 (pt) | 2006-07-17 | 2006-10-27 | Composição de vacina contra influenza monovalente, kit, método para a produção de uma composição de vacina contra influenza para uma situação pandêmica ou uma situação pré- pandêmica, e, usos de um antígeno de vírus da influenza ou preparação antigênica do mesmo e de um adjuvante em emulsão óleo-em-água, de um antígeno de vírus da influenza pandêmica ou preparação antigênica do antígeno de hemaglutinina do vírus da influenza e de um adjuvante em emulsão óleo-em-água e de um antígeno ou preparação antigênica de uma primeira cepa de influenza |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20090263422A1 (pt) |
| EP (1) | EP2422810B1 (pt) |
| KR (1) | KR20140069379A (pt) |
| CN (1) | CN105727276A (pt) |
| BR (1) | BRPI0621886B1 (pt) |
| DK (2) | DK2422810T3 (pt) |
| ES (2) | ES2469568T3 (pt) |
| PL (1) | PL2422810T3 (pt) |
| PT (2) | PT2043682E (pt) |
| SI (2) | SI2422810T1 (pt) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9923176D0 (en) * | 1999-09-30 | 1999-12-01 | Smithkline Beecham Biolog | Novel composition |
| HUE027837T2 (en) | 2005-03-23 | 2016-11-28 | Glaxosmithkline Biologicals Sa | Adjuvant use of influenza virus and oil-in-water emulsion to induce CD4 T-cell and / or enhanced B-cell cellular response |
| MX2009000660A (es) | 2006-07-17 | 2009-04-08 | Glaxosmithkline Biolog Sa | Vacuna de influenza. |
| TW200908994A (en) | 2007-04-20 | 2009-03-01 | Glaxosmithkline Biolog Sa | Vaccine |
| CN104688687A (zh) * | 2009-12-03 | 2015-06-10 | 诺华股份有限公司 | 疫苗佐剂制备中的亲水过滤 |
| KR101523215B1 (ko) | 2009-12-03 | 2015-05-27 | 노파르티스 아게 | 에멀션 미세 액화 및/또는 균질화 동안 성분들의 순환 |
| DE102009056871A1 (de) | 2009-12-03 | 2011-06-22 | Novartis AG, 4056 | Impfstoff-Adjuvantien und verbesserte Verfahren zur Herstellung derselben |
| CL2012001399A1 (es) | 2009-12-03 | 2013-03-08 | Novartis Ag | Metodo para fabricar adyuvante para vacuna (emulsion aceite/agua con escualeno, polisorbato 80 y trioleato de sorbitan), que comprende (i) formar primera emulsion en homogenizador desde un contendor a otro para formar segunda emulsion, (ii) y microfluidizar primera emulsion para formar segunda emulsion. |
| DE102009056884B4 (de) * | 2009-12-03 | 2021-03-18 | Novartis Ag | Impfstoff-Adjuvantien und verbesserte Verfahren zur Herstellung derselben |
| DE102009056883B4 (de) * | 2009-12-03 | 2012-08-16 | Novartis Ag | Impfstoff-Adjuvantien und verbesserte Verfahren zur Herstellung derselben |
| CN102869379B (zh) | 2010-04-30 | 2015-04-15 | 淡马锡生命科学研究院有限公司 | H5n1谱系的通用疫苗 |
| WO2011154976A2 (en) | 2010-06-08 | 2011-12-15 | Panacea Biotec Limited | Improved influenza vaccine |
| US9821051B1 (en) * | 2010-10-28 | 2017-11-21 | Seqirus UK Limited | Reducing hospitalization in elderly influenza vaccine recipients |
| CN106668854A (zh) * | 2016-12-23 | 2017-05-17 | 江苏中慧元通生物科技有限公司 | 一种四价亚单位流感疫苗及其制备方法 |
| GB2562241B (en) * | 2017-05-08 | 2022-04-06 | Stabilitech Biopharma Ltd | Vaccine compositions |
| JP6826203B2 (ja) * | 2017-08-28 | 2021-02-03 | 一般財団法人阪大微生物病研究会 | リアソータントインフルエンザウイルスの段階的作出方法 |
| AU2021302954A1 (en) * | 2020-06-30 | 2023-02-16 | Seqirus UK Limited | Cold filtration of oil-in-water emulsion adjuvants |
Family Cites Families (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4270537A (en) | 1979-11-19 | 1981-06-02 | Romaine Richard A | Automatic hypodermic syringe |
| DD155875A1 (de) | 1980-12-31 | 1982-07-14 | Willy Nordheim | Verfahren zur herstellung eines ballaststoffarmen inaktivierten influenzaimpfstoffes |
| DD211444A3 (de) | 1982-08-19 | 1984-07-11 | Saechsisches Serumwerk | Verfahren zur herstellung von influenza-impfstoffen |
| US4596556A (en) | 1985-03-25 | 1986-06-24 | Bioject, Inc. | Hypodermic injection apparatus |
| DD300833A7 (de) | 1985-10-28 | 1992-08-13 | Saechsische Landesgewerbefoerd | Verfahren zur herstellung von inaktivierten influenza-vollvirusimpfstoffen |
| CA1283827C (en) | 1986-12-18 | 1991-05-07 | Giorgio Cirelli | Appliance for injection of liquid formulations |
| GB8704027D0 (en) | 1987-02-20 | 1987-03-25 | Owen Mumford Ltd | Syringe needle combination |
| JP2851288B2 (ja) | 1987-06-05 | 1999-01-27 | アメリカ合衆国 | 癌診断および管理における自己分泌運動性因子 |
| US4790824A (en) | 1987-06-19 | 1988-12-13 | Bioject, Inc. | Non-invasive hypodermic injection device |
| US4941880A (en) | 1987-06-19 | 1990-07-17 | Bioject, Inc. | Pre-filled ampule and non-invasive hypodermic injection device assembly |
| US4940460A (en) | 1987-06-19 | 1990-07-10 | Bioject, Inc. | Patient-fillable and non-invasive hypodermic injection device assembly |
| DE3734306A1 (de) | 1987-10-10 | 1989-04-27 | Pfeiffer Erich Gmbh & Co Kg | Austragvorrichtung fuer fliessfaehige medien |
| US5339163A (en) | 1988-03-16 | 1994-08-16 | Canon Kabushiki Kaisha | Automatic exposure control device using plural image plane detection areas |
| US4912094B1 (en) | 1988-06-29 | 1994-02-15 | Ribi Immunochem Research Inc. | Modified lipopolysaccharides and process of preparation |
| FR2638359A1 (fr) | 1988-11-03 | 1990-05-04 | Tino Dalto | Guide de seringue avec reglage de la profondeur de penetration de l'aiguille dans la peau |
| HU212924B (en) | 1989-05-25 | 1996-12-30 | Chiron Corp | Adjuvant formulation comprising a submicron oil droplet emulsion |
| US5312335A (en) | 1989-11-09 | 1994-05-17 | Bioject Inc. | Needleless hypodermic injection device |
| US5064413A (en) | 1989-11-09 | 1991-11-12 | Bioject, Inc. | Needleless hypodermic injection device |
| DE4005528C2 (de) | 1990-02-22 | 1998-01-15 | Pfeiffer Erich Gmbh & Co Kg | Austragvorrichtung für Medien |
| US5190521A (en) | 1990-08-22 | 1993-03-02 | Tecnol Medical Products, Inc. | Apparatus and method for raising a skin wheal and anesthetizing skin |
| US5527288A (en) | 1990-12-13 | 1996-06-18 | Elan Medical Technologies Limited | Intradermal drug delivery device and method for intradermal delivery of drugs |
| GB9118204D0 (en) | 1991-08-23 | 1991-10-09 | Weston Terence E | Needle-less injector |
| SE9102652D0 (sv) | 1991-09-13 | 1991-09-13 | Kabi Pharmacia Ab | Injection needle arrangement |
| US5328483A (en) | 1992-02-27 | 1994-07-12 | Jacoby Richard M | Intradermal injection device with medication and needle guard |
| US5383851A (en) | 1992-07-24 | 1995-01-24 | Bioject Inc. | Needleless hypodermic injection device |
| US5569189A (en) | 1992-09-28 | 1996-10-29 | Equidyne Systems, Inc. | hypodermic jet injector |
| US5334144A (en) | 1992-10-30 | 1994-08-02 | Becton, Dickinson And Company | Single use disposable needleless injector |
| SG48309A1 (en) | 1993-03-23 | 1998-04-17 | Smithkline Beecham Biolog | Vaccine compositions containing 3-0 deacylated monophosphoryl lipid a |
| PT729473E (pt) | 1993-11-17 | 2001-02-28 | Deutsche Om Arzneimittel Gmbh | Dissacaridos de glucosamina metodo para a sua preparacao composicao farmaceutica contendo os mesmos e sua utilizacao |
| GB9326253D0 (en) | 1993-12-23 | 1994-02-23 | Smithkline Beecham Biolog | Vaccines |
| WO1995024176A1 (en) | 1994-03-07 | 1995-09-14 | Bioject, Inc. | Ampule filling device |
| US5466220A (en) | 1994-03-08 | 1995-11-14 | Bioject, Inc. | Drug vial mixing and transfer device |
| US5599302A (en) | 1995-01-09 | 1997-02-04 | Medi-Ject Corporation | Medical injection system and method, gas spring thereof and launching device using gas spring |
| UA56132C2 (uk) | 1995-04-25 | 2003-05-15 | Смітклайн Бічем Байолоджікалс С.А. | Композиція вакцини (варіанти), спосіб стабілізації qs21 відносно гідролізу (варіанти), спосіб приготування композиції вакцини |
| US5730723A (en) | 1995-10-10 | 1998-03-24 | Visionary Medical Products Corporation, Inc. | Gas pressured needle-less injection device and method |
| US5893397A (en) | 1996-01-12 | 1999-04-13 | Bioject Inc. | Medication vial/syringe liquid-transfer apparatus |
| GB9607549D0 (en) | 1996-04-11 | 1996-06-12 | Weston Medical Ltd | Spring-powered dispensing device |
| US5993412A (en) | 1997-05-19 | 1999-11-30 | Bioject, Inc. | Injection apparatus |
| IT1298087B1 (it) | 1998-01-08 | 1999-12-20 | Fiderm S R L | Dispositivo per il controllo della profondita' di penetrazione di un ago, in particolare applicabile ad una siringa per iniezioni |
| HUP0102332A3 (en) | 1998-06-08 | 2002-11-28 | Sca Emballage France | Fast flattening packaging |
| ATE355266T1 (de) | 1998-06-30 | 2006-03-15 | Om Pharma | Neue acylierte pseudodipeptide, verfahren zu ihrer herstellung, und diese enthaltende pharmazeutische zusammensetzungen |
| FR2796289B1 (fr) | 1999-07-16 | 2001-08-10 | Cross Site Technologies | Seringue sans aiguille avec injecteur a elements superposes |
| FR2796291B1 (fr) | 1999-07-16 | 2001-09-21 | Cross Site Technologies | Seringue sans aiguille munie d'un systeme de declenchement piezo-electrique |
| FR2796290B1 (fr) | 1999-07-16 | 2001-09-14 | Cross Site Technologies | Seringue sans aiguille fonctionnant avec un generateur d'onde de choc a travers une paroi |
| GB9923176D0 (en) * | 1999-09-30 | 1999-12-01 | Smithkline Beecham Biolog | Novel composition |
| US6494865B1 (en) | 1999-10-14 | 2002-12-17 | Becton Dickinson And Company | Intradermal delivery device including a needle assembly |
| FR2800619B1 (fr) | 1999-11-05 | 2002-02-08 | Cross Site Technologies | Seringue sans aiguille avec un moyen de poussee temporairement retenu |
| FR2802102B1 (fr) | 1999-12-08 | 2002-07-12 | Poudres & Explosifs Ste Nale | Seringue sans aiguille munie d'un tube d'ejection a section constante |
| FR2802103B1 (fr) | 1999-12-08 | 2003-10-03 | Poudres & Explosifs Ste Nale | Seringue sans aiguille fonctionnant avec entrainement du principe actif par effet tube a choc |
| WO2001046127A1 (fr) | 1999-12-22 | 2001-06-28 | Om Pharma | Pseudodipeptides acyles porteurs d'un bras auxiliaire fonctionnalise |
| FR2802820B1 (fr) | 1999-12-27 | 2002-10-18 | Poudres & Explosifs Ste Nale | Seringue sans aiguille fonctionnant par effet tube a choc, avec maintien prealable du principe actif sur le cote |
| FR2804329B1 (fr) | 2000-02-02 | 2002-12-13 | Poudres & Explosifs Ste Nale | Seringue sans aiguille munie d'un opercule contenant le principe actif |
| FR2804869B1 (fr) | 2000-02-11 | 2002-05-17 | Poudres & Explosifs Ste Nale | Seringue sans aiguille pour l'injection d'un liquide contenu dans une ampoule pre-remplie |
| FR2805749B1 (fr) | 2000-03-01 | 2002-05-17 | Poudres & Explosifs Ste Nale | Seringue sans aiguille a deux niveaux de vitesse d'injection |
| FR2807946B1 (fr) | 2000-04-19 | 2002-06-07 | Poudres & Explosifs Ste Nale | Seringue sans aiguille fonctionnant avec un chargement pyrotechnique bicomposition |
| FR2809626B1 (fr) | 2000-05-30 | 2003-03-07 | Poudres & Explosifs Ste Nale | Seringue sans aiguille avec membrane d'isolation d'un ejecteur multiconduit |
| FR2810554B1 (fr) | 2000-06-22 | 2003-05-16 | Poudres & Explosifs Ste Nale | Seringue sans aiguille munie d'un reservoir modulable |
| FR2812202B1 (fr) | 2000-07-28 | 2002-09-13 | Poudres & Explosifs Ste Nale | Seringue sans aiguille fonctionnant par mise en compression du reservoir contenant le principe actif liquide |
| FR2815544B1 (fr) | 2000-10-23 | 2003-02-14 | Poudres & Explosifs Ste Nale | Seringue sans aiguille securisee a architecture compacte |
| TWI228420B (en) | 2001-05-30 | 2005-03-01 | Smithkline Beecham Pharma Gmbh | Novel vaccine composition |
| DE202005022108U1 (de) * | 2004-03-09 | 2013-11-12 | Novartis Vaccines And Diagnostics, Inc. | Influenza-Virus-Impfstoffe |
| HUE027837T2 (en) * | 2005-03-23 | 2016-11-28 | Glaxosmithkline Biologicals Sa | Adjuvant use of influenza virus and oil-in-water emulsion to induce CD4 T-cell and / or enhanced B-cell cellular response |
| NZ567978A (en) * | 2005-11-04 | 2011-09-30 | Novartis Vaccines & Diagnostic | Influenza vaccine with reduced amount of oil-in-water emulsion as adjuvant |
| CA2628158C (en) * | 2005-11-04 | 2015-12-15 | Novartis Vaccines And Diagnostics S.R.L. | Emulsions with free aqueous-phase surfactant as adjuvants for split influenza vaccines |
| AU2006310337B9 (en) * | 2005-11-04 | 2013-11-28 | Novartis Ag | Adjuvanted influenza vaccines including cytokine-inducing agents |
| CA2628152C (en) * | 2005-11-04 | 2016-02-02 | Novartis Vaccines And Diagnostics S.R.L. | Adjuvanted vaccines with non-virion antigens prepared from influenza viruses grown in cell culture |
-
2006
- 2006-10-27 SI SI200631867T patent/SI2422810T1/sl unknown
- 2006-10-27 ES ES06806620.8T patent/ES2469568T3/es active Active
- 2006-10-27 PT PT68066208T patent/PT2043682E/pt unknown
- 2006-10-27 EP EP11190391.0A patent/EP2422810B1/en active Active
- 2006-10-27 KR KR1020147014106A patent/KR20140069379A/ko not_active IP Right Cessation
- 2006-10-27 DK DK11190391.0T patent/DK2422810T3/da active
- 2006-10-27 BR BRPI0621886-5A patent/BRPI0621886B1/pt active IP Right Grant
- 2006-10-27 DK DK06806620.8T patent/DK2043682T3/da active
- 2006-10-27 ES ES11190391.0T patent/ES2525572T3/es active Active
- 2006-10-27 SI SI200631787T patent/SI2043682T1/sl unknown
- 2006-10-27 US US12/374,346 patent/US20090263422A1/en not_active Abandoned
- 2006-10-27 PL PL11190391T patent/PL2422810T3/pl unknown
- 2006-10-27 CN CN201610112099.3A patent/CN105727276A/zh active Pending
- 2006-10-27 PT PT111903910T patent/PT2422810E/pt unknown
Also Published As
| Publication number | Publication date |
|---|---|
| PL2422810T3 (pl) | 2015-03-31 |
| PT2043682E (pt) | 2014-07-11 |
| EP2422810B1 (en) | 2014-10-22 |
| SI2422810T1 (sl) | 2015-01-30 |
| SI2043682T1 (sl) | 2014-07-31 |
| US20090263422A1 (en) | 2009-10-22 |
| BRPI0621886A2 (pt) | 2011-12-20 |
| BRPI0621886A8 (pt) | 2019-01-15 |
| DK2422810T3 (da) | 2014-11-24 |
| EP2422810A1 (en) | 2012-02-29 |
| ES2469568T3 (es) | 2014-06-18 |
| ES2525572T3 (es) | 2014-12-26 |
| CN105727276A (zh) | 2016-07-06 |
| KR20140069379A (ko) | 2014-06-09 |
| DK2043682T3 (da) | 2014-06-02 |
| PT2422810E (pt) | 2014-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11564984B2 (en) | Influenza vaccine | |
| ES2525572T3 (es) | Vacuna antigripal | |
| DK1861120T3 (en) | Use of an influenza virus and an oil-in-water emulsion adjuvant to induce CD 4 T cell and / or enhanced B memory cell response | |
| ES2377761T3 (es) | Vacuna contra la gripe de emulsión de aceite en agua | |
| US20070141078A1 (en) | Influenza vaccine | |
| US20080014217A1 (en) | Influenza vaccine | |
| US20150056249A1 (en) | Influenza vaccines | |
| US20230201329A1 (en) | Influenza vaccine | |
| CA2583555C (en) | Influenza vaccine | |
| CN101489586A (zh) | 流感疫苗 | |
| BRPI0609516B1 (pt) | uso de um vírus de influenza ou preparação antigênica do mesmo |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 27/10/2006, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |