WO2024111552A1 - 経口固形製剤 - Google Patents
経口固形製剤 Download PDFInfo
- Publication number
- WO2024111552A1 WO2024111552A1 PCT/JP2023/041643 JP2023041643W WO2024111552A1 WO 2024111552 A1 WO2024111552 A1 WO 2024111552A1 JP 2023041643 W JP2023041643 W JP 2023041643W WO 2024111552 A1 WO2024111552 A1 WO 2024111552A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oral solid
- dissolution test
- phenylpyridin
- tetrahydronaphthalen
- hydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
Definitions
- the present invention relates to an oral solid formulation containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof.
- TrkA Tropomyosin receptor kinase A
- Patent Document 1 discloses that a pharmaceutical composition of compound (I) can be appropriately combined with any pharma- ceutically acceptable additive (e.g., excipient, binder, lubricant, disintegrant, surfactant, flow enhancer, coating agent, plasticizer, masking agent, colorant, flavoring agent, preservative, isotonicity agent, pH adjuster, stabilizer, dispersant, antioxidant, buffer, preservative, fragrance, solubilizer, absorption enhancer, gelling agent, suspending agent, emulsifier, commonly used suitable additive, or solvent) to form various dosage forms (e.g., tablets, capsules, granules, powder, pills, aerosols, inhalants, ointments, patches, suppositories, injections, lozenges, liquids, spirits, suspensions, extracts, elixirs, etc.).
- any pharma- ceutically acceptable additive e.g., excipient, binder, lubricant, disintegrant, surfact
- An object of the present invention is to provide an oral solid preparation containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea (compound (I)) or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient.
- Another object of the present invention is to provide an oral solid preparation capable of suppressing a rapid increase in the blood concentration of an active ingredient compound at an early stage after administration of the preparation to a subject.
- compound (I) which exhibits large fluctuations in blood concentration, i.e., which shows a rapid increase in blood concentration at an early stage after administration and then disappears quickly by the time of the next administration, there was concern that a temporarily high blood concentration of compound (I) may lead to adverse effects such as the manifestation of toxicity. Under such circumstances, it has become urgent to provide a preparation containing compound (I) or a pharma- ceutical acceptable salt or solvate thereof as an active ingredient, which can suppress the manifestation of toxicity.
- the present inventors have conducted intensive studies to achieve the above-mentioned object, and as a result, have designed a preparation in which a core containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea (compound (I)) or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient is coated with a specific coating agent, and have found an oral solid preparation which, when the preparation produced based on the design is administered to a subject, makes it possible to suppress a rapid increase in the blood concentration of compound (I) at an early stage after administration.
- a solid oral preparation having excellent sustained blood concentration of compound (I) can be obtained by using a coating agent that starts to dissolve at a specific pH.
- an oral solid preparation which contains compound (I) and is expected to be highly safe
- an oral solid preparation which, when administered to a subject, maintains the blood concentration of compound (I) for a long period of time, particularly an oral solid preparation which is expected to have a long-lasting medicinal effect when taken once or twice a day, and have completed the present invention.
- compound (I) contained in the oral solid formulation of the present invention has TrkA inhibitory activity
- administration of the formulation can have an effect of improving diseases in which TrkA is involved (e.g., pain, pruritus, etc.).
- the present invention is an oral solid formulation containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof, as shown in the following embodiments, and more specifically, may be as follows: [1] to [21-3].
- An oral solid formulation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt thereof, or a solvate thereof as an active ingredient, the formulation comprising: An oral solid formulation comprising: (1) a core containing the active ingredient; and (2) a coating layer covering the core, the coating layer containing at least one coating agent selected from, for example, hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the coating agent for covering the core is preferably at least one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, and hypromellose acetate succinate; more preferably one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, and hypromellose acetate succinate.
- the coating agent for covering the core is preferably at least one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose; more preferably one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the methacrylic acid copolymer is, for example, methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, dry methacrylic acid copolymer LD, etc.; preferably, the methacrylic acid copolymer is at least one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD; more preferably, it is one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD.
- at least one additive selected from the group consisting of plasticizers, excipients, binders, disintegrants, lubricants, surfactants, pH regulators, solubilizers, stabilizers, antioxidants, adsorbents, flavorings, sweeteners, colorants, flavorings, flow agents, lubricants, and coating agents other than the coating agent according to the above aspect [1] (including a second coating agent).
- the plasticizer is, for example, at least one selected from triethyl citrate, tributyl citrate, Macrogol (registered trademark) (polyethylene glycol), propylene glycol, sodium lauryl sulfate ethanol, triacetin, polysorbate 80, and the like; preferably at least one selected from the group consisting of triethyl citrate, Macrogol (registered trademark) (polyethylene glycol), propylene glycol, triacetin, and polysorbate 80; more preferably triethyl citrate.
- the second coating agent is, for example, at least one selected from titanium oxide, talc, crystalline cellulose, calcium carbonate, shellac, etc.; preferably, at least one selected from the group consisting of titanium oxide, talc, crystalline cellulose, calcium carbonate, and shellac; more preferably, titanium oxide and/or talc.
- the oral solid formulation according to any one of the above aspects [3] to [3-2], wherein the excipient is at least one selected from, for example, glucose, lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose, sucrose, white sugar, mannitol (D-mannitol), mannitol, xylitol, sorbitol (D-sorbitol), crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch, dibasic calcium phosphate dihydrate, isomalt, anhydrous calcium phosphate, anhydrous calcium hydrogen phosphate, corn starch, pregelatinized starch, partially pregelatinized starch, light anhydrous silicic acid, titanium oxide, etc.
- the excipient is at least one selected from, for example, glucose, lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose
- the excipient is preferably at least one selected from the group consisting of lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose, white sugar, mannitol (D-mannitol), crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch, anhydrous calcium hydrogen phosphate, corn starch, pregelatinized starch, and partially pregelatinized starch; more preferably at least one selected from the group consisting of crystalline cellulose, mannitol (D-mannitol), and granulated lactose; even more preferably one selected from the group consisting of mannitol, crystalline cellulose, a mixture of mannitol and crystalline cellulose, and a mixture of crystalline cellulose and granulated lactose.
- lactose monohydrate, spray-dried monohydrate, anhydrous, etc.
- granulated lactose white sugar,
- the fluidizing agent is at least one selected from the group consisting of silicon dioxide (light anhydrous silicic acid, silica gel), talc, hydrous silicon dioxide, magnesium aluminometasilicate, and calcium hydrogen phosphate granules.
- the fluidizing agent is preferably at least one selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; more preferably, it is at least one selected from the group consisting of light anhydrous silicic acid, a mixture of light anhydrous silicic acid and talc, talc, and magnesium aluminometasilicate.
- the oral solid formulation according to any one of the above aspects [3] to [3-2], wherein the disintegrant is at least one selected from, for example, starches (corn starch, potato starch, starch, pregelatinized starch, partially pregelatinized starch), sodium carboxymethyl starch, carmellose (carboxymethylcellulose-CMC), carmellose calcium, croscarmellose sodium, crospovidone, sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, polyvinylpyrrolidone (PVPP), methylcellulose, microcrystalline cellulose, crystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose (L-HPC), sodium alginate, corn starch, sodium carboxymethyl starch, etc.
- starches corn starch, potato starch, starch, pregelatinized starch, partially pregelatinized starch
- sodium carboxymethyl starch carmellose (carboxymethylcellulose-CMC), carmellose calcium, croscarmellose sodium
- the disintegrant is preferably at least one selected from the group consisting of sodium carboxymethyl starch, carmellose (carboxymethylcellulose-CMC), croscarmellose sodium, crospovidone, sodium starch glycolate, calcium carboxymethylcellulose, polyvinylpyrrolidone (PVPP), microcrystalline cellulose (crystalline cellulose), lower alkyl-substituted hydroxypropylcellulose (L-HPC), and sodium carboxymethyl starch; more preferably at least one selected from sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- PVPP polyvinylpyrrolidone
- L-HPC lower alkyl-substituted hydroxypropylcellulose
- sodium carboxymethyl starch more preferably at least one selected from sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- the oral solid formulation according to any one of aspects [3] to [3-2], wherein the lubricant is at least one selected from, for example, magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, carboxymethylcellulose, stearic acid, sodium lauryl sulfate, carnauba wax, sucrose fatty acid esters, polyethylene glycol, glycerin, stearic acid monoglyceride, castor oil, hydrogenated castor oil, etc.
- the lubricant is at least one selected from, for example, magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, carboxymethylcellulose, stearic acid, sodium lauryl sulfate, carnauba wax, sucrose fatty acid
- the lubricant is preferably at least one selected from the group consisting of magnesium stearate, calcium stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, stearic acid, sodium lauryl sulfate, and carnauba wax; more preferably at least one selected from magnesium stearate and sodium stearyl fumarate; even more preferably magnesium stearate or sodium stearyl fumarate.
- [8A] An oral solid formulation according to any one of the above aspects [1] to [7-1], in which the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation is, for example, about 10.0% or more, about 13.0% or more, about 15.0% or more, about 18.0% or more, or about 20.0% or more, when a dissolution test is performed using the second dissolution test fluid according to the paddle method of the dissolution test method described in the Japanese Pharmacopoeia, 18th Edition (excluding oral solid formulations in which the coating agent is methacrylic acid copolymer S) 1 hour after the start of the dissolution test.
- An oral solid formulation according to any one of the above aspects [1] to [7-1], in which the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation is, for example, about 10.0% or more, about 13.0% or more, or about 15.0% or more, 1 hour or 6 hours after the start of the dissolution test, when the dissolution test is performed using the second dissolution test fluid according to the paddle method of the dissolution test method described in the Japanese Pharmacopoeia, 18th Edition.
- the oral solid formulation according to any one of the above aspects [1] to [9-1], wherein the coating layer begins to dissolve in a solution having a pH in the range of about 5.0 to about 7.0, and the active ingredient in the formulation is dissolved.
- the pH at which the coating layer starts to dissolve is, for example, in a range having an upper and/or lower limit of a pH value of about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0 (for example, in a range of about 5.0 to about 7.0, about 5.0 to about 6.5, about 5.0 to about 6.0, about 5.0 to about 5.5, about 5.5 to about 7.0, about 5.5 to about 6.5, about 5.5 to about 6.0, about 6.0 to about 7.0, about 6.0 to about 6.5, about 6.5 to about 7.0), preferably about 5.0 to about 7.0; more preferably about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0.
- An oral solid formulation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt thereof, or a solvate thereof as an active ingredient, the formulation comprising: (1) A core containing the following components (a) to (e): (a) the active ingredient; (b) at least one excipient selected from the group consisting of microcrystalline cellulose, granulated lactose, and microcrystalline cellulose; (c) at least one fluidizing agent selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; (d) at least one disintegrant selected from croscarmellose sodium and sodium starch glycolate; and (e) at least one lubricant selected from magnesium stearate and sodium stearyl fumarate, and (2) An oral solid formulation
- the coating agent for covering the core is preferably one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the methacrylic acid copolymer is, for example, methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, dry methacrylic acid copolymer LD, etc.
- the methacrylic acid copolymer is, for example, at least one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD; more preferably, it is one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD.
- the oral solid formulation according to any one of the above aspects [11] to [11-3] (excluding oral solid formulations in which the coating agent is methacrylic acid copolymer S) characterized in that, when a dissolution test is performed using the second fluid for dissolution test in accordance with the paddle method of dissolution test described in the Japanese Pharmacopoeia, 18th Edition, the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation 1 hour after the start of the dissolution test is about 13.0% or more.
- the pH at which the coating layer starts to dissolve is, for example, in a range having an upper and/or lower limit of a pH value of about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0 (for example, in a range of about 5.0 to about 7.0, about 5.0 to about 6.5, about 5.0 to about 6.0, about 5.0 to about 5.5, about 5.5 to about 7.0, about 5.5 to about 6.5, about 5.5 to about 6.0, about 6.0 to about 7.0, about 6.0 to about 6.5, about 6.5 to about 7.0), preferably about 5.0 to about 7.0; more preferably about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0.
- An oral solid preparation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient, the preparation comprising: (1) A core containing the following components (a) to (e): (a) the active ingredient; (b) at least one excipient selected from the group consisting of microcrystalline cellulose, granulated lactose, and microcrystalline cellulose; (c) at least one fluidizing agent selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; (d) at least one disintegrant selected from croscarmellose sodium and sodium starch glycolate; and (e) at least one lubricant selected from magnesium stearate and sodium stearyl fumarate, and (2) An oral solid formulation having a
- the coating agent for coating the core is, for example, at least one coating agent selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the coating agent for coating the core is preferably one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the methacrylic acid copolymer is, for example, methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, dry methacrylic acid copolymer LD, etc.
- the methacrylic acid copolymer is, for example, at least one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD; more preferably, it is one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD.
- [11A-7] The oral solid formulation according to any one of the above aspects [11A] to [11A-6] (excluding oral solid formulations in which the coating agent is methacrylic acid copolymer S) characterized in that, when a dissolution test is conducted using the second fluid for dissolution test in accordance with the paddle method of dissolution test described in the Japanese Pharmacopoeia, 18th Edition, the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation one hour after the start of the dissolution test is about 13.0% or more.
- [11A-7A] An oral solid formulation according to any one of the above aspects [11A] to [11A-6], wherein the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation 1 hour after the start of the dissolution test is, for example, about 10.0% or more, about 13.0% or more, about 15.0% or more, about 18.0% or more, or about 20.0% or more, when a dissolution test is performed using the second fluid for dissolution test in accordance with the paddle method of dissolution test described in the Japanese Pharmacopoeia, 18th Edition (excluding oral solid formulations in which the coating agent is methacrylic acid copolymer S).
- [11A-8] The oral solid formulation according to any one of the above aspects [11A] to [11A-7C], wherein when a dissolution test is performed using a first dissolution test fluid in accordance with the paddle method of the dissolution test method described in the Japanese Pharmacopoeia, 18th Edition, the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation is about 2.5% or less 1 hour after the start of the dissolution test.
- the dissolution rate is preferably about 2.0% or less; more preferably about 1.0% or less; and even more preferably about 0.5% or less.
- An oral solid preparation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient, the preparation comprising: (1) A core comprising the following components (a) and (b): (a) the active ingredient; (b) at least one additive selected from the group consisting of excipients, flow agents, disintegrants, and lubricants; and (2) A coating layer covering the core, the coating layer containing at least one coating agent that causes the coating layer of the formulation to begin to dissolve in a solution having a pH in the range of about 5.0 to about 7.0.
- the excipient is preferably at least one component selected from the group consisting of lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose, sucrose, mannitol (D-mannitol), crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch, anhydrous calcium hydrogen phosphate, corn starch, pregelatinized starch, and partially pregelatinized starch; more preferably at least one component selected from the group consisting of crystalline cellulose, mannitol (D-mannitol), and granulated lactose; even more preferably one selected from the group consisting of mannitol, crystalline cellulose, a mixture of mannitol and crystalline cellulose, and a mixture of crystalline cellulose and granulated lactose.
- lactose monohydrate, spray-dried monohydrate, anhydrous, etc.
- granulated lactose sucrose
- the fluidizing agent is preferably at least one selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; more preferably, it is one selected from the group consisting of light anhydrous silicic acid, a mixture of light anhydrous silicic acid and talc, talc, and magnesium aluminometasilicate.
- the disintegrant is preferably at least one selected from the group consisting of sodium carboxymethyl starch, carmellose (carboxymethylcellulose-CMC), croscarmellose sodium, crospovidone, sodium starch glycolate, calcium carboxymethylcellulose, polyvinylpyrrolidone (PVPP), microcrystalline cellulose (crystalline cellulose), lower alkyl-substituted hydroxypropylcellulose (L-HPC), and sodium carboxymethyl starch; more preferably at least one selected from sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- PVPP polyvinylpyrrolidone
- L-HPC lower alkyl-substituted hydroxypropylcellulose
- sodium carboxymethyl starch more preferably at least one selected from sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- the lubricant is preferably at least one selected from the group consisting of magnesium stearate, calcium stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, stearic acid, sodium lauryl sulfate, and carnauba wax; more preferably at least one selected from magnesium stearate and sodium stearyl fumarate; even more preferably magnesium stearate or sodium stearyl fumarate.
- the coating agent for coating the core is at least one coating agent selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the coating agent for coating the core is preferably one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the methacrylic acid copolymer is, for example, methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, dry methacrylic acid copolymer LD, etc., and preferably, the methacrylic acid copolymer is at least one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD; more preferably, it is one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S, and dry methacrylic acid copolymer LD.
- the oral solid formulation according to any one of the above aspects [11B] to [11B-10] (excluding oral solid formulations in which the coating agent is methacrylic acid copolymer S) characterized in that, when a dissolution test is conducted using the second fluid for dissolution test in accordance with the paddle method of dissolution test described in the Japanese Pharmacopoeia, 18th Edition, the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation one hour after the start of the dissolution test is about 13.0% or more.
- [11B-12] The oral solid formulation according to any one of the above aspects [11B] to [11B-11C], wherein when a dissolution test is performed using a first fluid for dissolution test in accordance with the paddle method of dissolution test described in the Japanese Pharmacopoeia, 18th Edition, the dissolution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea contained in the formulation is about 2.5% or less 1 hour after the start of the dissolution test.
- the dissolution rate is preferably about 2.0% or less; more preferably about 1.0% or less; and even more preferably about 0.5% or less.
- [12-2A] The oral solid formulation according to any one of the above aspects [1] to [11B-13], which contains 50 mg of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea, or a pharma- ceutically acceptable salt thereof, or a solvate thereof, characterized in that, 12 hours or 24 hours after the initial administration of the formulation to a subject, the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the plasma of the subject is about 90 nM or more, about 95 nM or more, about 100 nM or more, about 105 nM or more, or about 110 nM or more.
- [12-3A] The oral solid formulation according to any one of the above aspects [1] to [11B-13], which contains 150 mg of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea, or a pharma- ceutically acceptable salt thereof, or a solvate thereof, characterized in that, 12 hours or 24 hours after the initial administration of the formulation to a subject, the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the plasma of the subject is about 90 nM or more, about 95 nM or more, about 100 nM or more, about 105 nM or more, or about 110 nM or more. [12-4] In the above embodiments [12] to [12-3
- the oral solid formulation according to any one of aspects [1] to [14], wherein the content of the second coating agent that may be further contained in the coating layer is, for example, in the range of 0.0 to about 25.0% by mass, preferably in the range of about 1.0 to about 20.0% by mass, and more preferably in the range of about 1.0 to about 6.0% by mass, based on the total mass of the oral solid formulation.
- [20] The oral solid formulation according to any one of the above aspects [1] to [19], wherein the release of the active ingredient is intestinal immediate release.
- [20-1] The oral solid formulation according to any one of the above aspects [1] to [19], wherein the release of the active ingredient is in the form of sustained release or insolubility in the stomach.
- [20-2] The oral solid preparation according to any one of the above aspects [1] to [19], wherein the release of the active ingredient is a sustained release in the stomach or an insoluble and rapid release in the intestine.
- the disease in which TrkA is involved is at least one or more diseases selected from the group consisting of pain, cancer, inflammation/inflammatory disease, allergic disease, skin disease, pruritus, neurodegenerative disease, infectious disease, Sjögren's syndrome, endometriosis, renal disease, osteoporosis, and the like.
- the disease involving TrkA is preferably at least one disease selected from pain (pain such as nociceptive pain typified by osteoarthritis, rheumatoid arthritis, bone fracture, interstitial cystitis, chronic pancreatitis, pain associated with prostatitis, chronic lower back pain, diabetic peripheral neuropathy pain, postoperative pain, pelvic pain, cancer pain, etc., neuropathic pain, acute pain, chronic pain, inflammatory pain, etc.) and pruritus (generalized pruritus, localized pruritus, generalized pruritus, etc.).
- pain such as nociceptive pain typified by osteoarthritis, rheumatoid arthritis, bone fracture, interstitial cystitis, chronic pancreatitis, pain associated with prostatitis, chronic lower back pain, diabetic peripheral neuropathy pain, postoperative pain, pelvic pain, cancer pain, etc., neuropathic pain, acute pain, chronic pain, inflammatory pain, etc.
- pruritus generallyized pruri
- the disease involving TrkA is osteoarthritis pain, arthralgia, neuropathic pain, postoperative pain, lower back pain, diabetic neuropathy, intraoperative pain, cancer pain, chemotherapy-induced pain, headache (including cluster headache, tension headache, and migraine pain), trigeminal neuralgia, herpes zoster pain, postherpetic neuralgia, carpal tunnel syndrome, inflammatory pain, pain from rheumatoid arthritis, colitis, pain from interstitial cystitis, visceral pain, pain from kidney stones, pain from gallstones, sore throat, fibromyalgia, chronic pain syndrome, thalamic pain, and the like.
- Pain syndromes pain from stroke, phantom limb pain, sunburn, radiculopathy, complex regional pain syndrome, HIV sensory neuropathy, central neuropathic pain syndrome, multiple sclerosis pain, Parkinson's disease pain, spinal cord injury pain, period pain, toothache, pain from bone metastases, pain from endometriosis, pain from uterine fibroids, nociceptive pain, hyperalgesia, temporomandibular joint pain, neuroblastoma, ovarian cancer, uterine cancer, glioblastoma multiforme, cervical cancer, pancreatic cancer, colon cancer, rectal cancer, prostate cancer, melanoma, myeloma, thyroid cancer, lung cancer (small cell lung Cancer, non-small cell lung cancer), brain tumor, esophageal cancer, kidney cancer, osteoma, blood cancer (chronic myeloid leukemia, acute lymphoblastic leukemia, Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ALL), acute myeloid leukemia (A
- the present invention has the following effects when an oral solid formulation containing compound (I) is administered to a subject: (1) a rapid increase in the blood concentration of compound (I) can be controlled, and (2) the blood concentration of compound (I) can be maintained for a long period of time.
- 1 shows the result data of the dissolution test of Test Example 1.
- 1 shows the result data of the dissolution test of Test Example 2.
- 1 shows the result data of the dissolution test of Test Example 3.
- 1 shows the result data of the dissolution test in Test Example 4.
- 1 shows the result data of the dissolution test of Test Example 5.
- 1 shows the result data of the dissolution test of Test Example 6.
- 1 shows the result data of the dissolution test of Test Example 7.
- 1 shows the result data of the dissolution test of Test Example 8.
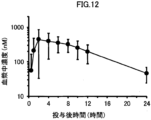
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Comparative Example 1) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 1-1) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 1-2) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 2-1) was orally administered to beagle dogs.
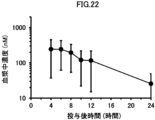
- 2 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 2-2) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 2-3) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 2-4) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 2-5) was orally administered to beagle dogs.
- 1 shows the result data of the dissolution test of Test Example 9.
- 1 shows the result data of the dissolution test of Test Example 10.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 5-1) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 5-2-1) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 5-3) was orally administered to beagle dogs.
- 1 is a graph showing the change over time in the plasma concentration of compound (I) when a solid oral preparation produced according to the formulation of (Example 5-4) was orally administered to beagle dogs.
- the present invention relates to an oral solid preparation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient.
- an oral solid preparation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient.
- the term "core” refers to a solid, preferably a tablet or its central part, formed by compressing the active ingredient 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof together with a pharma- ceutically acceptable additive.
- the shape and size of the core are not particularly limited, but it is preferable that the core surface has a curved surface that can be coated with a coating agent.
- the shape of the solid preparation having a coating layer obtained by coating the core with a coating agent may be, for example, a shape that is easy to handle as a preparation and easy for the subject to take, such as a circle, a triangle, an ellipse, etc.
- the method for producing the core containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as an active ingredient is not particularly limited, but it is possible to select a production method that is generally used for producing the core or tablet of a pharmaceutical preparation, such as direct compression, dry granulation, wet granulation, or fluidized bed granulation.
- additives there are no particular limitations on pharma- ceutically acceptable additives as long as they are used in formulation, but additives described in reference books for formulation research, such as "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021" (the contents of this reference book are incorporated herein by reference), can be used.
- additives such as excipients, disintegrants, flow agents, lubricants, binders, stabilizers, and colorants can be used as components used to maintain the manufacturability of the formulation, the appropriate hardness, disintegrability, dissolution, and other functions of the formulation.
- the additive contained in the core of the present invention is at least one selected from the group consisting of excipients, flow agents, disintegrants, and lubricants, and is preferably an excipient, flow agent, disintegrant, and lubricant.
- the core of the present invention may further contain additives such as binders, stabilizers, surfactants, pH regulators, solubilizers, antioxidants, adsorbents, flavorings, sweeteners, colorants, and fragrances.
- the term "coating layer” refers to a layer that contains a coating agent (including a second coating agent) described below (e.g., hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, aminoalkyl methacrylate copolymer, cellulose acetate phthalate, carboxymethylethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, titanium oxide, talc, etc.) used to coat the core and is laminated or coated on the surface of the core.
- a coating agent including a second coating agent described below (e.g., hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, aminoalkyl methacrylate copolymer, cellulose acetate phthalate, carboxymethylethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, titanium oxide, talc, etc.) used to coat the core and is laminated
- the surface of the core of the present invention is covered over its entire surface with the above-mentioned coating layer.
- the oral solid formulation of the present invention has a property of dissolving when it comes into contact with, for example, a solution having a pH range of about 5.0 to about 7.0, or a range having a pH value of about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0 as the upper and/or lower limit (e.g., about 5.0 to about 7.0, about 5.0 to about 6.5, about 5.0 to about 6.0, about 5.0 to about 5.5, about 5.5 to about 7.0, about 5.5 to about 6.5, about 5.5 to about 6.0, about 6.0 to about 7.0, about 6.0 to about 6.5, about 6.5 to about 7.0), preferably a solution having a pH range of about 5.0 to about 7.0, more preferably a solution having a pH of about 5.0, about 5.5, about 6.0, about 6.5, or about 7.0, and releasing the active ingredient contained in
- the coating layer may further contain additives such as plasticizers, excipients, binders, disintegrants, lubricants, surfactants, pH regulators, solubilizers, stabilizers, antioxidants, adsorbents, flavorings, sweeteners, colorants, flavorings, flow agents, and lubricants.
- additives such as plasticizers, excipients, binders, disintegrants, lubricants, surfactants, pH regulators, solubilizers, stabilizers, antioxidants, adsorbents, flavorings, sweeteners, colorants, flavorings, flow agents, and lubricants.
- Additives that can be additionally contained in the core or coating layer can be appropriately selected from additives listed in reference books for formulation research, such as the Pharmaceutical Additives Dictionary 2021, edited by the Japan Pharmaceutical Additives Association, February 2021.
- the additives that can be additionally contained in the core or coating layer exemplified here can be added to either the core or the coating layer.
- one compound may play multiple roles (e.g., excipient and binder, etc.).
- solution includes liquids such as aqueous solutions and body fluids.
- a solution with a pH of about 1 to about 2 corresponds to the pH of gastric fluid, and a solution with a pH of about 4 to about 8 corresponds to the pH of intestinal fluid.
- Coating can be performed using any coating method commonly used in the technical field. Specific examples include pan coating, fluidized bed coating, aerated dry pan coating, rolling coating, and spray coating. Coating can be performed using a coating device appropriate for each coating method.
- the surface of the core can be coated with a coating liquid made by mixing the above-mentioned coating agents with additives such as colorants, flavorings, sweeteners, and fragrances as necessary.
- the ratio of solvents (water, organic solvents, etc.) used when producing the coating agent is not particularly limited, but can be used in any ratio.
- the type of organic solvent is not particularly limited, and examples that can be used include alcohol solvents (e.g., methanol, ethanol, isopropyl alcohol, etc.), ketones (e.g., acetone, etc.), halogenated hydrocarbons (e.g., chloroform, dichloromethane, etc.), etc.
- oral solid preparation means a solid preparation type for oral administration.
- examples of the oral solid preparation include tablets, powders, granules, capsules, lozenges, pills, etc.
- the oral solid preparation of the present invention is preferably a tablet.
- the tablet is not particularly limited, but examples of the tablet include orally disintegrating tablets, chewable tablets, effervescent tablets, dispersible tablets, dissolving tablets, sugar-coated tablets, coated tablets, and other types of tablets.
- the oral solid preparation of the present invention is preferably a coated tablet.
- the oral solid preparation of the present invention is preferably an oral solid preparation (tablet) having a uniform content of the active ingredient (e.g., 5 mg, 10 mg, 20 mg, 30 mg, 50 mg, 100 mg, 150 mg, etc.) prepared by blending 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof as the active ingredient with a pharmaceutical carrier (additives such as excipients, disintegrants, flow agents, lubricants, etc.), granulating, tableting, and coating with a coating agent (which may contain a plasticizer, etc.).
- a pharmaceutical carrier additive such as excipients, disintegrants, flow agents, lubricants, etc.
- a coating agent which may contain a plasticizer, etc.
- the pharma- ceutically acceptable salts of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea are not particularly limited as long as they are pharma-ceutically acceptable salts, and examples thereof include mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, and phosphoric acid; aliphatic monocarboxylic acids such as formic acid, acetic acid, propionic acid, butyric acid, valeric acid, enanthic acid, capric acid, myristic acid, palmitic acid, stearic acid, lactic acid, sorbic acid, and mandelic acid; aromatic monocarboxylic acids such as benzoic acid and salicylic acid; oxalic acid, malocarboxylic acid, and the like.
- mineral acids such as hydrochloric acid
- the salt examples include organic carboxylic acids such as aliphatic dicarboxylic acids such as carboxylic acid, succinic acid, fumaric acid, maleic acid, malic acid, tartaric acid, and aliphatic tricarboxylic acids such as citric acid; organic sulfonic acids such as aliphatic sulfonic acids such as methanesulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, and aromatic sulfonic acids such as benzenesulfonic acid and p-toluenesulfonic acid; inorganic base addition salts with metals such as alkali metals or alkaline earth metals such as sodium, potassium, magnesium, and calcium; and organic base addition salts such as methylamine, ethylamine, ethanolamine, pyridine, lysine, arginine, and ornithine. These salts can be obtained by a conventional method, for example, by mixing an equivalent amount of
- solvate refers to a molecular complex containing 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea and one or more pharma- ceutically acceptable solvent molecules (e.g., water, ethanol, etc.). When the solvent molecule is water, it is specifically called a "hydrate".
- solvent molecules e.g., water, ethanol, etc.
- the content of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt or solvate thereof used in the present invention is, for example, in the range of about 5.0 to about 70.0% by mass, preferably about 10.0 to about 50.0% by mass, and more preferably about 10.0 to about 30.0% by mass, based on the total mass of the oral solid formulation.
- an “excipient” refers to an additive described in a reference book for formulation research, such as "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021," and is a component added for the purpose of achieving a certain size and concentration in the formulation of tablets, etc.
- excipients include, but are not limited to, glucose, lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose, sucrose, white sugar, mannitol (D-mannitol), mannitol, xylitol, sorbitol (D-sorbitol), crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch, dibasic calcium phosphate dihydrate, isomalt, anhydrous calcium phosphate, anhydrous calcium hydrogen phosphate, corn starch, pregelatinized starch, partially pregelatinized starch, light anhydrous silicic acid, titanium oxide, and mixtures thereof.
- Preferred excipients are at least one selected from the group consisting of lactose (monohydrate, spray-dried monohydrate, anhydrous, etc.), granulated lactose, sucrose, mannitol (D-mannitol), crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch, anhydrous calcium hydrogen phosphate, corn starch, pregelatinized starch, and partially pregelatinized starch; more preferably, at least one selected from the group consisting of mannitol, crystalline cellulose, and granulated lactose; even more preferably, one selected from the group consisting of mannitol, crystalline cellulose, a mixture of mannitol and crystalline cellulose, and a mixture of crystalline cellulose and granulated lactose.
- lactose monohydrate, spray-dried monohydrate, anhydrous, etc.
- granulated lactose sucrose, mannitol (
- the content of the excipient used in the present invention is, for example, in the range of about 20.0 to about 90.0% by mass, preferably in the range of about 30.0 to about 80.0% by mass, and more preferably in the range of about 40.0 to about 75.0% by mass, based on the total mass of the oral solid formulation.
- the term "disintegrant” refers to an additive described in a reference book for formulation research, such as "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021," which is added for the purpose of disintegrating the formulation (tablet) by absorbing moisture in the body and expanding, etc., and facilitating the release of the active ingredient.
- Disintegrants are not particularly limited, but examples thereof include starches (corn starch, potato starch, starch, pregelatinized starch, partially pregelatinized starch), sodium carboxymethyl starch, carmellose (carboxymethylcellulose-CMC), carmellose calcium, croscarmellose sodium, crospovidone, sodium starch glycolate, sodium carboxymethylcellulose, calcium carboxymethylcellulose, polyvinylpyrrolidone (PVPP), methylcellulose, microcrystalline cellulose (crystalline cellulose), lower alkyl-substituted hydroxypropylcellulose (L-HPC), sodium alginate, corn starch, sodium carboxymethyl starch, and mixtures thereof.
- starches corn starch, potato starch, starch, pregelatinized starch, partially pregelatinized starch
- sodium carboxymethyl starch carmellose (carboxymethylcellulose-CMC), carmellose calcium, croscarmellose sodium, crospovidone, sodium starch glycolate, sodium carboxymethylcellulose, calcium carboxymethylcellulose
- Preferred disintegrants are at least one selected from the group consisting of sodium carboxymethyl starch, carmellose (carboxymethylcellulose-CMC), croscarmellose sodium, crospovidone, sodium starch glycolate, calcium carboxymethylcellulose, polyvinylpyrrolidone (PVP), microcrystalline cellulose (crystalline cellulose), lower alkyl-substituted hydroxypropyl cellulose (L-HPC) and sodium carboxymethyl starch, more preferably at least one selected from the group consisting of sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- PVP polyvinylpyrrolidone
- L-HPC lower alkyl-substituted hydroxypropyl cellulose
- sodium carboxymethyl starch more preferably at least one selected from the group consisting of sodium croscarmellose and sodium starch glycolate; even more preferably, sodium croscarmellose or sodium starch glycolate.
- the content of the disintegrant used in the present invention is, for example, in the range of 0.0 to about 30.0% by mass, preferably in the range of about 1.0 to about 20.0% by mass, and more preferably in the range of about 2.0 to about 8.0% by mass, based on the total mass of the oral solid formulation.
- fluidizer refers to an additive described in reference books for formulation research, such as the Pharmaceutical Additives Dictionary 2021, edited by the Japan Pharmaceutical Additives Association, February 2021, and is a component added for the purpose of improving the fluidity of powder before tableting.
- Fluidizers are not particularly limited, but examples include silicon dioxide (light anhydrous silicic acid, silica gel), talc, hydrous silicon dioxide, magnesium aluminometasilicate, calcium hydrogen phosphate granules, and mixtures thereof.
- a preferred fluidizer is at least one selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; more preferably, one selected from the group consisting of light anhydrous silicic acid, a mixture of light anhydrous silicic acid and talc, and talc and magnesium aluminometasilicate.
- the content of the fluidizing agent used in the present invention is, for example, in the range of 0.0 to about 15.0% by weight, preferably in the range of about 0.1 to about 10.0% by weight, and more preferably in the range of about 0.5 to about 5.0% by weight, based on the total weight of the oral solid dosage form.
- a "lubricant” is an additive described in a reference book for formulation research, such as "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021," and is a component that is suitably added in the process of producing cores by tableting in order to eliminate adhesion of active ingredients to the compression device, insufficient hardness of the core (core defect) caused by compression force, etc.
- lubricants include, but are not limited to, magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate (also known as sodium stearyl fumarate), a mixture of magnesium stearate and sodium lauryl sulfate, talc, carboxymethylcellulose, stearic acid, sodium lauryl sulfate, carnauba wax, sucrose fatty acid esters, polyethylene glycol, glycerin, stearate monoglyceride, castor oil, hydrogenated castor oil, and mixtures thereof.
- Preferred lubricants are at least one selected from the group consisting of magnesium stearate, calcium stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, stearic acid, sodium lauryl sulfate, and carnauba wax; more preferably, at least one selected from the group consisting of magnesium stearate and sodium stearyl fumarate; even more preferably, magnesium stearate or sodium stearyl fumarate.
- the content of the lubricant used in the present invention is, for example, in the range of 0.0 to about 10.0% by mass, preferably in the range of about 0.1 to about 5.0% by mass, and more preferably in the range of about 0.5 to about 3.0% by mass, based on the total mass of the oral solid formulation.
- a "plasticizer” is an additive described in a reference book for formulation research, such as "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021," and is a component added for the purpose of interacting with the polymer used to make the polymer have a flexible structure, thereby enabling a constant release of the active ingredient from the formulation.
- the plasticizer is not particularly limited, but examples thereof include triethyl citrate, tributyl citrate, macrogol (polyethylene glycol), propylene glycol, sodium lauryl sulfate ethanol, triacetin, polysorbate 80, and mixtures thereof.
- a preferred plasticizer is at least one selected from the group consisting of triethyl citrate, macrogol (polyethylene glycol), propylene glycol, triacetin, and polysorbate 80; more preferably, triethyl citrate.
- the content of the plasticizer used in the present invention is, for example, in the range of 0.0 to about 10.0% by mass, preferably in the range of about 0.1 to about 5.0% by mass, and more preferably in the range of about 0.5 to about 3.0% by mass, based on the total mass of the oral solid formulation.
- coating agent refers to an additive described in reference books for formulation research, such as the "Dictionary of Pharmaceutical Additives 2021, edited by the Japan Pharmaceutical Additives Association, February 2021," and is a component that can be added for the purpose of coating the surface of a solid preparation.
- the coating agent is not particularly limited, but a fat-soluble coating is preferred, and examples thereof include hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, aminoalkyl methacrylate copolymer, cellulose acetate phthalate, carboxymethylethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, ethylcellulose, titanium oxide, talc, etc., or mixtures thereof.
- examples of coating agents that can be contained in the coating layer include hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, carboxymethylethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, and mixtures thereof.
- a preferred coating agent contained in the coating layer is at least one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose; more preferably, it is one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- the second coating agent that can be contained in the coating layer can be, for example, titanium oxide, talc, crystalline cellulose, calcium carbonate, shellac, a mixture thereof, etc.
- a preferred second coating agent is at least one selected from the group consisting of titanium oxide, talc, crystalline cellulose, calcium carbonate, and shellac; more preferably, titanium oxide and/or talc.
- the content of the coating agent in the coating layer used in the present invention is, for example, in the range of about 1.0 to about 25.0 mass%, preferably in the range of about 2.0 to about 20.0 mass%, and more preferably in the range of about 3.5 to about 15.0 mass%.
- the content of the second coating agent used in the present invention is, for example, in the range of 0.0 to about 25.0% by mass, and preferably in the range of about 1.0 to about 20.0% by mass, based on the total mass of the oral solid dosage form.
- an oral solid preparation prepared using one selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose as the coating agent contained in the coating layer is a preparation that is substantially insoluble in an acidic range, but is at least partially soluble in a weakly acidic or basic range.
- the acidic range indicates a pH of about 0.5 to about 4.5, preferably about 1.0 to about 2.0
- the weakly acidic or basic range indicates a pH of about 5.0 to 9.0, preferably about 5.0 to about 6.5 or about 5.0 to about 7.0.
- the pH of the solution at which the coating layer of the oral solid formulation begins to dissolve and the active ingredient in the formulation is dissolved is sometimes referred to as the "dissolution pH.”
- the dissolution pH can be, for example, in the range of about pH 4.0 to about 8.0.
- the dissolution pH of the oral solid preparation is, for example, in the range of about 5.0 to about 7.0, about 5.0 to about 6.5, about 5.0 to about 6.0, about 5.0 to about 5.5, about 5.5 to about 7.0, about 5.5 to about 6.5, about 5.5 to about 6.0, about 6.0 to about 7.0, about 6.0 to about 6.5, and about 6.5 to about 7.0.
- a method for setting the dissolution pH to a range of, for example, about 5.0 to about 7.0 includes using a commercially available coating agent. By using, for example, one or a combination of the coating agents listed in Table 1 as the coating agent contained in the coating layer, an oral solid preparation that allows for control of the dissolution pH can be produced.
- dissolution pH it is also possible to adjust the dissolution pH by combining multiple coating agents.
- the methods for adjusting the dissolution pH are not limited to these.
- the methacrylic acid copolymer includes methacrylic acid copolymer LD (e.g., Eudragit L30D-55 (molecular weight: up to 320,000 g/mol)), methacrylic acid copolymer L (e.g., Eudragit L100 (molecular weight: up to 125,000 g/mol)), methacrylic acid copolymer S (e.g., Eudragit S100 (molecular weight: up to 125,000 g/mol)), and the like, and also includes dried methacrylic acid copolymer LD obtained by drying methacrylic acid copolymer LD (e.g., Eudragit L100-55 (molecular weight: up to 320,000 g/mol)), and the like.
- methacrylic acid copolymer LD e.g., Eudragit L30D-55 (molecular weight: up to 320,000 g/mol
- methacrylic acid copolymer L e.g., Eudragit
- the methacrylic acid copolymer LD is a copolymer of methacrylic acid and ethyl acrylate (ratio 1:1) (structural formula below): [wherein n1, x1, and y1 are arbitrary numbers determined according to the molecular weight], and methacrylic acid copolymer LD usually has the form of an emulsion obtained by dissolving methacrylic acid and ethyl acrylate in an aqueous solution of polysorbate 80 and sodium lauryl sulfate. It usually dissolves in a solution of pH 5.5 or more.
- Methacrylic acid copolymer L is a copolymer of methacrylic acid and methyl methacrylate (ratio 1:1) (structural formula below): [wherein n2, x2, and y2 are arbitrary numbers determined depending on the molecular weight], and is usually in the form of a powder that dissolves in a solution with a pH of 6.0 or higher.
- Methacrylic acid copolymer S is a copolymer of methacrylic acid and methyl methacrylate (ratio 1:2) (structural formula below): [wherein n3, x3, and y3 are arbitrary numbers determined depending on the molecular weight], and it usually has the form of a powder that dissolves in a solution with a pH of 7.0 or higher.
- the amount of coating agent used can also be adjusted as appropriate.
- the amount of coating agent (dried methacrylic acid copolymer LD) used for the tablets (oral solid preparations) prepared in Examples 3-1 and 3-2 described below is twice and three times the amount of the coating agent used in Example 2-1, respectively.
- the amount of coating agent (methacrylic acid copolymer L) used for the tablets (oral solid preparations) prepared in Example 5-2-2 described below is approximately 0.7 times the amount of the coating agent used in Example 5-2-1.
- the dissolution rate of the active ingredient contained in the oral solid preparation can be measured by, for example, the rotating basket method or the paddle method described in the 18th Revised Edition of the Japanese Pharmacopoeia.
- the in vitro dissolution rate (dissolution rate) of the active ingredient contained in the oral solid preparation can be measured, for example, based on the dissolution test method described in the Japanese Pharmacopoeia, 18th Edition. This test method measures the time and rate of dissolution of the active ingredient in a test solution in vitro.
- the test solution used for the measurement includes, but is not limited to, dissolution test solution 1 (a liquid with a pH of about 1.2 simulating gastric fluid, obtained by dissolving 2.0 g of sodium chloride in 7.0 mL of hydrochloric acid and water to make 1000 mL, also called JP1 solution) or dissolution test solution 2 (a liquid with a pH of about 6.8 simulating intestinal fluid, obtained by adding 1 volume of water to 1 volume of phosphate buffer with a pH of 6.8, also called JP2 solution).
- Measurement methods include, for example, the rotating basket method and paddle method of the dissolution test methods described in the Japanese Pharmacopoeia, 18th Edition.
- the dissolution rate of the active ingredient (compound (I)) after 1 hour is, for example, about 2.5% or less.
- the dissolution rate of the active ingredient (compound (I)) after 1 hour from the start of the dissolution test is preferably about 2.0% or less, more preferably about 1.0% or less, and even more preferably about 0.5% or less.
- the dissolution rate of the active ingredient (compound (I)) one hour after the start of the dissolution test is, for example, about 10.0% or more, about 13.0% or more, about 15.0% or more, about 18.0% or more, about 20.0% or more, etc.
- the dissolution rate of the active ingredient (compound (I)) 6 hours after the start of the dissolution test can be, for example, about 10.0% or more, about 13.0% or more, or about 15.0% or more, etc.
- the "Japanese Pharmacopoeia dissolution test first fluid” may be referred to as the “dissolution test first fluid.”
- the "Japanese Pharmacopoeia dissolution test second fluid” may be referred to as the “dissolution test second fluid.”
- the pH in the digestive tract is in the stomach in the range of about 1 to about 2, in the small intestine in the range of about 4 to about 7, and in the large intestine in the range of about 7 to about 8, with the pH increasing as it progresses to the lower digestive tract.
- release refers to the amount and release rate of the active ingredient contained in an oral solid preparation.
- the release rate refers to the amount of active ingredient released from the preparation within a certain period of time.
- intestinal rapid release means that when a dissolution test of an oral solid preparation is conducted using the second dissolution test fluid in accordance with the dissolution test method described in the 18th Edition of the Japanese Pharmacopoeia, the active ingredient (compound (I)) contained in the preparation exhibits a dissolution rate of about 13.0% or more relative to the total mass of the active ingredient (compound (I)) originally contained in the preparation one hour after the start of the dissolution test.
- sustained release or insoluble in the stomach means that when a dissolution test is conducted using the first dissolution test fluid in accordance with the dissolution test method described in the Japanese Pharmacopoeia, 18th Edition, the active ingredient (compound (I)) contained in the formulation exhibits a dissolution rate of about 2.5% or less relative to the total mass of the active ingredient (compound (I)) originally contained in the formulation 1 hour after the start of the dissolution test. In particular, when the dissolution rate is about 0.5% or less, the formulation is insoluble.
- the word "approximately” when used, it may include values up to ⁇ 20% of the numerical value, and preferably up to ⁇ 10% of the numerical value.
- diseases in which TrkA is involved include pain, cancer, inflammation/inflammatory diseases, allergic diseases, skin diseases, pruritus, neurodegenerative diseases, infectious diseases, Sjogren's syndrome, endometriosis, renal diseases, osteoporosis, and the like.
- the pain may be treated with osteoarthritis pain, arthralgia, neuropathic pain, post-operative pain, low back pain, diabetic neuropathy, intraoperative pain, cancer pain, chemotherapy-induced pain, headache (including cluster headache, tension headache, migraine pain), trigeminal neuralgia, shingles pain, post-herpetic neuralgia, carpal tunnel syndrome, inflammatory pain, pain from rheumatoid arthritis, colitis, interstitial cystitis pain, visceral pain, pain from kidney stones, pain from gallstones, sore throat, fibromyalgia, chronic pain syndromes, thalamic pain syndromes, pain from stroke, Phantom limb pain, sunburn, radiculopathy, complex regional pain syndrome, HIV sensory neuropathy, central neuropathic pain syndrome, multiple sclerosis pain, Parkinson's disease pain, spinal cord injury pain, menstrual pain, toothache, pain from bone metastases, pain from endometriosis, pain from uterine fibroids, nociceptive pain, hyperalges
- the oral solid formulation of the present invention can be used for subjects in need of prevention and/or treatment of diseases involving TrkA.
- the subjects are humans or non-human mammals (dogs, cats, rats, mice, monkeys, cows, horses, pigs, sheep, etc.).
- the content of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea (compound (I)) or a pharma- ceutically acceptable salt or solvate thereof contained in the oral solid preparation is not particularly limited as long as it is an amount that is effective in preventing and/or treating a disease involving TrkA, and examples of the amount of compound (I) that may be included include 5 mg, 10 mg, 20 mg, 30 mg, 50 mg, 100 mg, 150 mg, and the like, as well as amounts within ranges with these as upper or lower limits.
- a pharma- ceutically acceptable salt of compound (I), a solvate of compound (I), or a solvate of a pharma-ceutically acceptable salt of compound (I) is included, the content of each of them is, for example, the equivalent amount of the salt, solvate, or solvate of the salt corresponding to the amount of compound (I).
- the concentration of compound (I) in the subject's plasma 12 hours or 24 hours after the initial administration is about 90 nM or more, about 95 nM or more, about 100 nM or more, about 105 nM or more, or about 110 nM or more.
- the oral solid formulation of the present invention may also be a formulation in which cracking of the formulation is prevented.
- cracks this phenomenon may also be called crazing, lamination, capping, etc.
- the formulation of Example 1-2 containing 50 mg of compound (I) did not generate cracks during the manufacturing process, but when a formulation containing 150 mg of compound (I) was manufactured using that formulation (Reference Example 1), cracks were confirmed during the core manufacturing stage. The cracks were thought to be caused by insufficient binding strength of the drug powder and poor releasability from the mortar.
- Example 2-1 the excipient in the formulation of Example 2-1 was changed to a mixture of mannitol and crystalline cellulose to increase the binding strength of the drug powder, and the flow agent was changed to a mixture of light anhydrous silicic acid and talc to improve releasability. This made it possible to mold tablets or cores with a hardness that did not cause any cracks, even when the compound (I) content in the formulation was increased to a maximum of 150 mg.
- the method for producing the oral solid formulation of the present invention is described below, but includes known methods including steps such as grinding, mixing, granulation, drying, sieving, granulation, molding (tabletting), coating, and crystallization.
- the grinding step is not particularly limited in terms of the equipment or means, so long as it is a method that can normally pharmaceutical grind the drug and suitable additives.
- grinding equipment include impact grinders, hammer mills, ball mills, jet grinders, colloid mills, etc.
- the grinding conditions are not particularly limited as long as they are appropriately selected.
- the mixing step of each component subsequent to grinding is not particularly limited in terms of the equipment or means, so long as it is a method that can normally pharmaceutical uniformly mix each component.
- Granulation may be used as a method for producing the oral solid formulation of the present invention.
- the equipment or means there are no particular limitations on the equipment or means, so long as the method can granulate the drug and appropriate pharmaceutical additives in a normal pharmaceutical manner.
- Examples of granulation methods and equipment used in wet granulation, which uses a solvent such as water or a binding liquid in which an appropriate amount of binder is dispersed or dissolved in water include, but are not limited to, high-speed stirring granulation, crushing (pulverization) granulation, fluidized bed granulation, extrusion granulation, rolling granulation, spray granulation, and equipment used in these methods.
- a wet granulation method using a non-aqueous solvent or a dry granulation method that does not use a solvent can also be selected.
- equipment In the drying process, there are no particular limitations on the equipment or means used, so long as the method is generally suitable for pharmaceutical drying.
- equipment include, but are not limited to, a forced-air dryer, a reduced-pressure dryer, a vacuum dryer, a fluidized-bed granulation dryer, etc.
- the equipment or means there are no particular limitations on the equipment or means, so long as the method is capable of sieving or sizing in a pharmaceutical manner.
- equipment include, but are not limited to, a sieve, a Comil, a power mill, etc.
- the apparatus and means are not particularly limited as long as they are a method for molding the formulation or core of the present invention.
- a method for molding the formulation or core of the present invention there may be mentioned, without being limited to, a method in which a drug and appropriate pharmaceutical additives are mixed and then directly compressed to form a pharmaceutical composition without carrying out a granulation/drying step, a method in which a pharmaceutical composition is formed by granulation and drying and then compressed to form a pharmaceutical composition, and a method in which a pharmaceutical composition (e.g., plain tablet or core) is formed by granulation and further mixing with a lubricant and then compressed to form a pharmaceutical composition, etc.
- tableting machines include rotary tableting machines, single punch tableting machines, and oil presses.
- the tableting conditions such as the tableting pressure are not particularly limited as long as the cores can be formed and the cores are not damaged during the manufacturing process.
- the hitting pressure (main pressure) when compressed and molded using a tabletop rotary tableting machine (PICCOLA NOVA, manufactured by RIVA S.A.) is 0.5 to 20.0 kN, preferably 1.0 to 10.0 kN, and more preferably 3.0 to 8.0 kN.
- the upper and lower limits can be combined arbitrarily as desired.
- the hardness of the core is not particularly limited as long as it is hard enough not to break during the coating process.
- the hardness of the compressed product measured using a tabletop rotary tablet press (PICCOLA NOVA, manufactured by RIVA S.A.) is 30 N or more, preferably 40 N or more, and more preferably 50 N or more.
- a coating can be applied to the surface of the cores after tableting, if desired.
- the method is not particularly limited as long as it is a method commonly used in pharmaceutical coating, such as pan coating and dip coating.
- the coating agent may be added in an appropriate amount, either alone or in combination of two or more.
- the coating rate is not particularly limited as long as it forms a film on the core. For example, it is 1% by mass to 20% by mass, preferably 2% by mass to 18% by mass, more preferably 3% by mass to 16% by mass, based on the total mass of the tablet.
- the coating agent may be dried by any method that is generally suitable for pharmaceutical drying and is not particularly limited.
- the additives used in this example are those listed in Table 2.
- Example 1-1 A solid preparation was prepared using 220 g of the preparation of Comparative Example 1 as a core according to the recipe in Table 3 below. Specifically, the core was placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, and talc were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 1-1.
- PRC-GTX-mini film coating machine
- talc a solution in which Eudragit L100-55, triethyl citrate, and talc were dispersed or dissolved in absolute ethanol and purified water
- 762.5 g of mannitol and 44.00 g of croscarmellose sodium were added to the mixture obtained, and mixed by hand using a polyethylene bag, and then sieved to break up the aggregates, and 2.00 g of magnesium stearate was added and mixed by hand to obtain a mixed powder for tableting.
- the mixed powder for tableting obtained was compressed using a benchtop rotary tableting machine (PICCOLA NOVA, manufactured by RIVA S.A.) to obtain cores.
- the obtained cores (219.8 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, and talc were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 1-2.
- PRC-GTX-mini film coating machine
- talc a solution in which Eudragit L100-55, triethyl citrate, and talc were dispersed or dissolved in absolute ethanol and purified water
- Dissolution test (1) For each of the preparations produced in Comparative Example 1, Example 1-1, and Example 1-2, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method of the Japanese Pharmacopoeia, 18th Edition, dissolution test first fluid (JP1 fluid) as the dissolution test fluid, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series
- the UV method was performed using an ultraviolet-visible spectrophotometer (V-660, manufactured by JASCO Corporation) (wavelength: 210 nm).
- V-660 ultraviolet-visible spectrophotometer
- the elution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea is shown in Table 4 and FIG. 1.
- Dissolution test (2) For each of the preparations produced in Comparative Example 1, Example 1-1, and Example 1-2, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method of the Japanese Pharmacopoeia, 18th Edition, dissolution test second fluid (JP2 fluid) as the dissolution test medium, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- JP2 fluid dissolution test second fluid
- the UV method was performed using an ultraviolet-visible spectrophotometer (V-660, manufactured by JASCO Corporation) (wavelength: 210 nm).
- V-660 ultraviolet-visible spectrophotometer
- the elution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea is shown in Table 5 and FIG. 2.
- the obtained cores (500.10 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 2-1.
- PRC-GTX-mini film coating machine
- a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 2-1.
- 2120.00 g of mannitol, 16.50 g of talc, 132.00 g of croscarmellose sodium, and 165.00 g of crystalline cellulose were added to the mixture obtained, and mixed by hand using a polyethylene bag, and then sieved to break up the aggregates, and 33.00 g of magnesium stearate was added and mixed by hand to obtain a mixed powder for tableting.
- the mixed powder for tableting obtained was compressed using a tabletop rotary tableting machine (PICCOLA NOVA, manufactured by RIVA S.A.) to obtain cores.
- the obtained cores (600.56 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain a formulation (tablet) of Example 2-2.
- PRC-GTX-mini film coating machine
- a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain a formulation (tablet) of Example 2-2.
- Dissolution test (3) For each of the preparations (tablets) produced in Examples 2-1, 2-3, 2-4, and 2-5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method of the 18th revised Japanese Pharmacopoeia, using the first fluid of the dissolution test method (JP1 fluid) as the dissolution test fluid, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- JP1 fluid the first fluid of the dissolution test method
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the dissolution test solution was measured using high performance liquid chromatography (HPLC), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the dissolution rate.
- HPLC high performance liquid chromatography
- HPLC method measurements were performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- Dissolution test (4) For each of the preparations (tablets) produced in Examples 2-1, 2-3, 2-4, and 2-5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the second fluid for the dissolution test (JP2 fluid) as the dissolution test medium, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the elution test solution was measured using high performance liquid chromatography (HPLC method), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the elution rate.
- the HPLC method was performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- the obtained cores (600.1 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparations (tablets) of Examples 3-1 and 3-2.
- Dissolution test (5) For each of the preparations (tablets) produced in Examples 3-1 and 3-2, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the first fluid of the dissolution test method (JP1 fluid) as the dissolution test fluid, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- JP1 fluid the first fluid of the dissolution test method
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the dissolution test solution was measured using high performance liquid chromatography (HPLC), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the dissolution rate.
- HPLC high performance liquid chromatography
- HPLC method measurements were performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- Dissolution test (6) For each of the preparations (tablets) produced in Examples 3-1 and 3-2, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the second fluid for the dissolution test (JP2 fluid) as the dissolution test medium, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the elution test solution was measured using high performance liquid chromatography (HPLC method), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the elution rate.
- the HPLC method was performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- the obtained cores (130.0 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-1.
- PRC-GTX-mini film coating machine
- a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-1.
- the obtained cores (150.1 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-2.
- the obtained cores (140.1 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-3.
- PRC-GTX-mini film coating machine
- a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-3.
- the obtained cores (150.0 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the formulation (tablet) of Example 4-4.
- mannitol 1.10 g of talc, 8.8 g of croscarmellose sodium, and 11.0 g of crystalline cellulose were added to the mixture obtained, and mixed by hand using a polyethylene bag, and then sieved to break up the aggregates, and 4.4 g of sodium stearyl fumarate (sodium stearyl fumarate) was added and mixed by hand to obtain a mixed powder for tableting.
- the mixed powder for tableting obtained was compressed using a tabletop rotary tableting machine (PICCOLA NOVA, manufactured by RIVA S.A.) to obtain cores.
- the obtained cores (150.1 g) were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-5.
- PRC-GTX-mini film coating machine
- a solution in which Eudragit L100-55, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 4-5.
- Dissolution test (7) For each of the preparations (tablets) produced in Examples 4-1, 4-2, 4-3, 4-4, and 4-5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the first fluid of the dissolution test method (JP1 fluid) as the dissolution test fluid, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the dissolution test solution was measured using high performance liquid chromatography (HPLC), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the dissolution rate.
- HPLC high performance liquid chromatography
- HPLC method measurements were performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- Dissolution test (8) For each of the preparations (tablets) produced in Examples 4-1, 4-2, 4-3, 4-4, and 4-5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the second fluid for the dissolution test (JP2 fluid) as the dissolution test medium, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the elution test solution was measured using high performance liquid chromatography (HPLC method), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the elution rate.
- the HPLC method was performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- PK studies Pharmacokinetic (PK) tests using the tablets (oral solid preparations) produced in the above Examples 1-1, 1-2, 2-1, 2-2, 2-3, 2-4, 2-5 and Comparative Example 1 will be described below.
- the vertical axis of the graphs in Figures 9 to 16 represents the plasma concentration (blood concentration) (nM) of compound (I), and the horizontal axis represents the time (hours) after administration of compound (I).
- Comparative Example PK1 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12, 14, 22 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, manufactured by Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- a standard plasma sample was prepared by serially diluting a 1 mg/mL solution of compound (I) with plasma, and the plasma concentration of compound (I) was calculated from the calibration curve, and the plasma concentration transition data shown in FIG. 9 was obtained.
- the oral solid preparation manufactured by the formulation of (Comparative Example PK1) was orally administered, the peak plasma concentration (662 nM) was reached 2 hours after administration due to good absorbability, and then rapidly decreased from the plasma, and most of the compound (I) in the plasma disappeared 24 hours after administration (plasma concentration 12 hours after administration: 91.6 nM, plasma concentration 24 hours after administration: 18.6 nM).
- the peak plasma concentration of this preparation is thought to increase in a dose-dependent manner, and it was predicted to show a peak plasma concentration of about 1410 nM at a dose of 50 mg/body weight and about 4240 nM at a dose of 150 mg/body weight.
- Example PK1-1 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12, 14, 22 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 10. As shown in FIG. 10, when the oral solid preparation produced by the formulation of (Example 1-1) was orally administered, the maximum plasma concentration (374 nM) was reached 4 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 114 nM, plasma concentration 24 hours after administration: 116 nM).
- Example PK1-2 (1) Administration of test preparations and blood sampling
- blood samples (0.5 mL) were collected from the brachial cephalic vein of the beagle dogs at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 11. As shown in FIG. 11, when the oral solid preparation produced by the formulation of (Example 1-2) was orally administered, the maximum plasma concentration (634 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 202 nM, plasma concentration 24 hours after administration: 581 nM).
- Example PK2-1 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 12. As shown in FIG. 12, when the oral solid preparation produced by the formulation of (Example 2-1) was orally administered, the maximum plasma concentration (440 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 195 nM, plasma concentration 24 hours after administration: 46.3 nM).
- Example PK2-2 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the content of compound (I) in this preparation was 6.1 times higher than that of the oral solid preparation manufactured by the formulation of Comparative Example 1, but the maximum plasma concentration after administration was almost the same at 1.1 times, indicating that the oral solid preparation manufactured by the formulation of Example 2-2 suppressed a rapid increase in plasma concentration.
- Example PK2-3 (1) Administration of test preparations and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 14. As shown in FIG. 14, when the oral solid preparation produced by the formulation of (Example 2-3) was orally administered, the maximum plasma concentration (664 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 184 nM, plasma concentration 24 hours after administration: 552 nM).
- Example PK2-4 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 15. As shown in FIG. 15, when the oral solid preparation produced by the formulation of (Example 2-4) was orally administered, the maximum plasma concentration (577 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 189 nM, plasma concentration 24 hours after administration: 41.7 nM).
- Example PK2-5) (1) Administration of test preparations and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Comparative Example PK1 (2) to obtain the plasma concentration time course data shown in FIG. 16. As shown in FIG. 16, when the oral solid preparation produced by the formulation of (Example 2-5) was orally administered, the maximum plasma concentration (397 nM) was reached 4 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 135 nM, plasma concentration 24 hours after administration: 38.2 nM).
- ⁇ Production of the formulation (tablets) of Example 5-1> 200 g of the cores obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which carboxymethylethylcellulose, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 5-1.
- a film coating machine PRC-GTX-mini, manufactured by Powrex
- Example 5-2-1> 200 g of the core obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> was placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which methacrylic acid copolymer L, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 5-2-1.
- PRC-GTX-mini manufactured by Powrex
- ⁇ Production of the formulation (tablets) of Example 5-2-2> 200 g of the core obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> was placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which methacrylic acid copolymer L, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 5-2-2.
- PRC-GTX-mini manufactured by Powrex
- Example 5-3 ⁇ Production of the formulation (tablets) of Example 5-3>
- the cores (200 g) obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which cellulose acetate phthalate, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain the preparation (tablet) of Example 5-3.
- PRC-GTX-mini film coating machine
- ⁇ Production of the formulation (tablets) of Example 5-4> 200 g of the core obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> was placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which methacrylic acid copolymer S, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and acetone, to obtain the preparation (tablet) of Example 5-4.
- a film coating machine PRC-GTX-mini, manufactured by Powrex
- ⁇ Production of the formulation (tablets) of Reference Example 5> 200 g of the cores obtained in the above ⁇ Production of Cores of Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5> were placed in a film coating machine (PRC-GTX-mini, manufactured by Powrex) and film-coated with a solution in which aminoalkyl methacrylate copolymer E, triethyl citrate, talc, and titanium oxide were dispersed or dissolved in absolute ethanol and purified water, to obtain a preparation (tablet) of Reference Example 5.
- PRC-GTX-mini manufactured by Powrex
- Dissolution test 9 For each of the preparations (tablets) produced in Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4, and Reference Example 5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, dissolution test fluid as dissolution test fluid 1 (JP1 fluid), and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the dissolution test solution was measured using high performance liquid chromatography (HPLC), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the dissolution rate.
- HPLC high performance liquid chromatography
- HPLC method measurements were performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- ⁇ Test Example 10 Dissolution test (10) For each of the preparations (tablets) produced in Examples 5-1, 5-2-1, 5-2-2, 5-3, 5-4 and Reference Example 5, a dissolution test was carried out using a dissolution tester (NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.) under the conditions of the paddle method of the dissolution test method in the 18th revised Japanese Pharmacopoeia, using the second fluid of the dissolution test method (JP2 fluid) as the dissolution test medium, and a paddle rotation speed of 50 rpm.
- a dissolution tester NTR-8000AC series, NTR-8400AC series, NTR-8600AS series, manufactured by Toyama Sangyo Co., Ltd.
- JP2 fluid the second fluid of the dissolution test method
- the peak area of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the elution test solution was measured using high performance liquid chromatography (HPLC method), and the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea was calculated from the obtained peak area to calculate the elution rate.
- the HPLC method was performed using an HPLC system (manufactured by Shimadzu Corporation) (wavelength: 210 nm).
- the column used in the HPLC method was a ZORBAX SB-Aq, with an inner diameter of 4.6 mm, a length of 150 mm, and a particle size of 3.5 ⁇ m (manufactured by Agilent), and was maintained at 40° C.
- the elution rate of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea is shown in Table 17 and FIG.
- PK studies The pharmacokinetic (PK) test using the tablets (solid oral preparations) produced in the above Examples 5-1, 5-2-1, 5-3 and 5-4 will be described below.
- the vertical axis of the graphs in Figures 19 to 22 represents the plasma concentration (nM) of compound (I), and the horizontal axis represents the time after administration (hours) of compound (I).
- Example PK5-1 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, manufactured by Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- a standard plasma sample was prepared by serially diluting a 1 mg/mL solution of compound (I) with plasma, and the plasma concentration of compound (I) was calculated from the calibration curve, and the plasma concentration transition data shown in FIG. 19 was obtained.
- the maximum plasma concentration (491 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 303 nM, plasma concentration 24 hours after administration: 61.5 nM).
- Example PK5-2-1 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- Example PK5-1 (2) Measurement of Plasma Concentration of Compound (I) The plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Example PK5-1 (2) to obtain the plasma concentration time course data shown in FIG. 20. As shown in FIG. 20, when the oral solid preparation produced by the formulation of (Example 5-2-1) was orally administered, the maximum plasma concentration (581 nM) was reached 6 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 163 nM, plasma concentration 24 hours after administration: 26.7 nM).
- Example PK5-3 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- Example 5-3 The plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Example PK5-1 (2) to obtain the plasma concentration time course data shown in FIG. 21.
- the maximum plasma concentration (381 nM) was reached 2 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 98.6 nM, plasma concentration 24 hours after administration: 17.9 nM).
- Example PK5-4 (1) Administration of test preparation and blood sampling
- blood was collected from the brachial cephalic vein of the beagle dogs (0.5 mL) at 0.5, 1, 2, 4, 6, 8, 10, 12 and 24 hours.
- the collected blood was centrifuged (micro high-speed refrigerated centrifuge MX-201, manufactured by Tommy Seiko Co., Ltd., 10,000 ⁇ g, 5 minutes, 4° C.) to collect plasma.
- Example 5-4 Measurement of Plasma Concentration of Compound (I)
- the plasma concentration of Compound (I) was calculated in the same manner as in the concentration measurement in Example PK5-1 (2) to obtain the plasma concentration time course data shown in FIG. 22.
- the maximum plasma concentration (243 nM) was reached 4 hours after administration, and the plasma concentration of compound (I) was maintained at a high level even after 12 hours after administration (plasma concentration 12 hours after administration: 116 nM, plasma concentration 24 hours after administration: 25.6 nM).
- An oral solid formulation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt thereof, or a solvate thereof as an active ingredient, the formulation comprising: (1) a core containing the active ingredient; and (2) a coating layer covering the core, the coating layer containing at least one coating agent selected from the group consisting of hypromellose phthalate, methacrylic acid copolymer, hypromellose acetate succinate, cellulose acetate phthalate, and carboxymethylethylcellulose.
- a solid oral preparation comprising: [2] The oral solid formulation according to [1], wherein the methacrylic acid copolymer is at least one selected from the group consisting of methacrylic acid copolymer LD, methacrylic acid copolymer L, methacrylic acid copolymer S and dry methacrylic acid copolymer LD. [3] The oral solid formulation according to [1] or [2], wherein the coating layer further contains at least one plasticizer selected from the group consisting of triethyl citrate, tributyl citrate, polyethylene glycol, propylene glycol, sodium lauryl sulfate, ethanol, triacetin, and polysorbate 80.
- the excipient is at least one selected from the group consisting of glucose, lactose, granulated lactose, sucrose, white sugar, mannitol, mannitol, xylitol, sorbitol, crystalline cellulose, microcrystalline cellulose, silicic acid, starch, corn starch, potato starch,
- the disintegrant is at least one selected from the group consisting of starches, sodium carboxymethyl starch, carmellose, carmellose calcium, croscarmellose sodium, crospovidone, sodium starch glycolate, sodium carboxymethylcellulose, calcium carboxymethylcellulose, polyvinylpyrrolidone, methylcellulose, microcrystalline cellulose, crystalline cellulose, lower alkyl-substituted hydroxypropylcellulose, sodium alginate, corn starch, and sodium
- the lubricant is at least one selected from the group consisting of magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, a mixture of magnesium stearate and sodium lauryl sulfate, talc, carboxymethylcellulose, stearic acid, sodium lauryl sulfate, carnauba wax, sucrose fatty acid esters, polyethylene glycol, glycerin, stearic acid monoglyceride, castor oil, and hydrogenated castor oil.
- An oral solid formulation comprising 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea or a pharma- ceutically acceptable salt thereof, or a solvate thereof as an active ingredient, the formulation comprising: (1) A core containing the following components (a) to (e): (a) the active ingredient; (b) at least one excipient selected from the group consisting of microcrystalline cellulose, granulated lactose, and microcrystalline cellulose; (c) at least one fluidizing agent selected from the group consisting of light anhydrous silicic acid, talc, and magnesium aluminometasilicate; (d) at least one disintegrant selected from croscarmellose sodium and sodium starch glycolate; and (e) at least one lubricant selected from magnesium stearate and sodium stearyl fumarate, and (2) A coating layer that
- a solid oral preparation comprising: [15] The oral solid formulation according to any one of [1] to [14], which contains 50 mg of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea, or an equivalent amount of a pharma- ceutically acceptable salt thereof, or an equivalent amount of a solvate thereof, wherein the amount or equivalent amount is such that the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the plasma of a subject is 90 nM or more 12 hours after the initial administration of the formulation to the subject.
- [16] The oral solid formulation according to any one of [1] to [14], which contains 50 mg of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea, or an equivalent amount of a pharma- ceutically acceptable salt thereof, or an equivalent amount of a solvate thereof, wherein the amount or equivalent amount is such that the concentration of 1-((1R,2R)-2-hydroxy-4,4-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-3-(2-phenylpyridin-3-yl)urea in the plasma of a subject is 90 nM or higher either 12 hours or 24 hours after the initial administration of the formulation to the subject.
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Dermatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202380078556.XA CN120225199A (zh) | 2022-11-21 | 2023-11-20 | 口服固体制剂 |
| EP23894559.6A EP4623916A1 (en) | 2022-11-21 | 2023-11-20 | Oral solid formulation |
| JP2024560142A JPWO2024111552A1 (https=) | 2022-11-21 | 2023-11-20 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022185858 | 2022-11-21 | ||
| JP2022-185858 | 2022-11-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024111552A1 true WO2024111552A1 (ja) | 2024-05-30 |
Family
ID=91195626
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/041643 Ceased WO2024111552A1 (ja) | 2022-11-21 | 2023-11-20 | 経口固形製剤 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4623916A1 (https=) |
| JP (1) | JPWO2024111552A1 (https=) |
| CN (1) | CN120225199A (https=) |
| WO (1) | WO2024111552A1 (https=) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002179571A (ja) * | 2000-12-11 | 2002-06-26 | Nikken Chem Co Ltd | 小型徐放性錠剤 |
| WO2002087549A1 (fr) * | 2001-04-25 | 2002-11-07 | Taisho Pharmaceutical Co., Ltd. | Comprimes a liberation prolongee du type a unites multiples |
| JP2004525162A (ja) * | 2001-04-10 | 2004-08-19 | サン・ファーマシューティカル・インダストリーズ・リミテッド | 時限パルス放出組成物 |
| WO2009063916A1 (ja) * | 2007-11-16 | 2009-05-22 | Asahi Kasei Chemicals Corporation | 水系フィルムコーティング液、および、フィルムコーティング顆粒、ならびに、これを用いた錠剤 |
| JP2014172855A (ja) * | 2013-03-08 | 2014-09-22 | Kyorin Pharmaceutical Co Ltd | 口腔内速崩壊性錠剤 |
| JP2017222645A (ja) * | 2016-06-10 | 2017-12-21 | 大原薬品工業株式会社 | 腸溶性被覆顆粒の耐酸性が改善された圧縮成形錠剤 |
| WO2018199166A1 (ja) | 2017-04-27 | 2018-11-01 | 持田製薬株式会社 | 新規テトラヒドロナフチルウレア誘導体 |
| JP2018193360A (ja) * | 2017-05-18 | 2018-12-06 | エルメッド エーザイ株式会社 | 湿製錠剤の製造方法及び湿製錠剤の品質向上方法 |
| JP2019523258A (ja) * | 2016-07-25 | 2019-08-22 | ハンミ ファーム. シーオー., エルティーディー. | エソメプラゾール、又は薬学的に許容可能なその塩を含むpH依存性薬物放出特性が改善した剤形 |
| WO2019189717A1 (ja) | 2018-03-30 | 2019-10-03 | 持田製薬株式会社 | テトラヒドロナフチルウレア誘導体の製造方法 |
-
2023
- 2023-11-20 JP JP2024560142A patent/JPWO2024111552A1/ja active Pending
- 2023-11-20 WO PCT/JP2023/041643 patent/WO2024111552A1/ja not_active Ceased
- 2023-11-20 EP EP23894559.6A patent/EP4623916A1/en active Pending
- 2023-11-20 CN CN202380078556.XA patent/CN120225199A/zh active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002179571A (ja) * | 2000-12-11 | 2002-06-26 | Nikken Chem Co Ltd | 小型徐放性錠剤 |
| JP2004525162A (ja) * | 2001-04-10 | 2004-08-19 | サン・ファーマシューティカル・インダストリーズ・リミテッド | 時限パルス放出組成物 |
| WO2002087549A1 (fr) * | 2001-04-25 | 2002-11-07 | Taisho Pharmaceutical Co., Ltd. | Comprimes a liberation prolongee du type a unites multiples |
| WO2009063916A1 (ja) * | 2007-11-16 | 2009-05-22 | Asahi Kasei Chemicals Corporation | 水系フィルムコーティング液、および、フィルムコーティング顆粒、ならびに、これを用いた錠剤 |
| JP2014172855A (ja) * | 2013-03-08 | 2014-09-22 | Kyorin Pharmaceutical Co Ltd | 口腔内速崩壊性錠剤 |
| JP2017222645A (ja) * | 2016-06-10 | 2017-12-21 | 大原薬品工業株式会社 | 腸溶性被覆顆粒の耐酸性が改善された圧縮成形錠剤 |
| JP2019523258A (ja) * | 2016-07-25 | 2019-08-22 | ハンミ ファーム. シーオー., エルティーディー. | エソメプラゾール、又は薬学的に許容可能なその塩を含むpH依存性薬物放出特性が改善した剤形 |
| WO2018199166A1 (ja) | 2017-04-27 | 2018-11-01 | 持田製薬株式会社 | 新規テトラヒドロナフチルウレア誘導体 |
| JP2018193360A (ja) * | 2017-05-18 | 2018-12-06 | エルメッド エーザイ株式会社 | 湿製錠剤の製造方法及び湿製錠剤の品質向上方法 |
| WO2019189717A1 (ja) | 2018-03-30 | 2019-10-03 | 持田製薬株式会社 | テトラヒドロナフチルウレア誘導体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4623916A1 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4623916A1 (en) | 2025-10-01 |
| CN120225199A (zh) | 2025-06-27 |
| JPWO2024111552A1 (https=) | 2024-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2748686T3 (es) | Dispersión sólida amorfa que comprende taxano, comprimido que comprende la misma y método de preparación de los mismos | |
| TWI540128B (zh) | 醫藥組合物 | |
| JP7156945B2 (ja) | 有機化合物のガレヌス製剤 | |
| JP5479909B2 (ja) | 新規製剤 | |
| TWI790364B (zh) | 包含烷基硫酸鈉之醫藥組合物 | |
| JP2023513444A (ja) | ダサチニブの非晶質固体分散体及びその使用 | |
| KR20200138005A (ko) | 디메틸푸마르산염을 함유한 장용성 정제 | |
| CN103505453B (zh) | 一种奥利司他口服固体制剂及其制备方法 | |
| WO2024111552A1 (ja) | 経口固形製剤 | |
| ES2992303T3 (es) | Preparación administrada de modo oral que contiene solifenacina y tamsulosina | |
| WO2020048449A1 (zh) | 包含1,3,5-三嗪衍生物或其盐的固体药物组合物 | |
| JP2025526729A (ja) | ナポラフェニブを含む非晶質固体分散体 | |
| JP5106119B2 (ja) | シクロオキシゲナーゼ−2阻害剤を含む経口投与用の薬剤、およびその調製方法 | |
| WO2025144120A1 (en) | Multi-layer pharmaceutical formulation of mirabegron and solifenacin succinate | |
| HK40114583A (zh) | Eed抑制剂固体分散体、包含其的口服制剂及其制备方法 | |
| TW201338780A (zh) | 藥物組成及其製備方法 | |
| NZ723991B2 (en) | Amorphous solid dispersion comprising taxane, tablet comprising the same, and method for preparing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23894559 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380078556.X Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024560142 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023894559 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380078556.X Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2023894559 Country of ref document: EP Effective date: 20250623 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023894559 Country of ref document: EP |