WO2023090361A1 - 改変d領域を含むヒト免疫グロブリン重鎖遺伝子座を有する哺乳動物人工染色体ベクター、及びそのベクターを保持する細胞又は非ヒト動物 - Google Patents
改変d領域を含むヒト免疫グロブリン重鎖遺伝子座を有する哺乳動物人工染色体ベクター、及びそのベクターを保持する細胞又は非ヒト動物 Download PDFInfo
- Publication number
- WO2023090361A1 WO2023090361A1 PCT/JP2022/042560 JP2022042560W WO2023090361A1 WO 2023090361 A1 WO2023090361 A1 WO 2023090361A1 JP 2022042560 W JP2022042560 W JP 2022042560W WO 2023090361 A1 WO2023090361 A1 WO 2023090361A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- human
- region
- sequence
- heavy chain
- immunoglobulin heavy
- Prior art date
Links
- 239000013598 vector Substances 0.000 title claims abstract description 205
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 title claims abstract description 194
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 title claims abstract description 185
- 230000002759 chromosomal effect Effects 0.000 title abstract description 11
- 238000000034 method Methods 0.000 claims abstract description 93
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 claims abstract description 41
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 claims abstract description 40
- 238000004519 manufacturing process Methods 0.000 claims abstract description 20
- 210000004962 mammalian cell Anatomy 0.000 claims abstract description 13
- 210000004027 cell Anatomy 0.000 claims description 264
- 108090000623 proteins and genes Proteins 0.000 claims description 138
- 238000005215 recombination Methods 0.000 claims description 133
- 230000006798 recombination Effects 0.000 claims description 132
- 108700026244 Open Reading Frames Proteins 0.000 claims description 126
- 241000283690 Bos taurus Species 0.000 claims description 75
- 239000002773 nucleotide Substances 0.000 claims description 74
- 125000003729 nucleotide group Chemical group 0.000 claims description 74
- 210000000723 mammalian artificial chromosome Anatomy 0.000 claims description 71
- 108091007433 antigens Proteins 0.000 claims description 66
- 102000036639 antigens Human genes 0.000 claims description 66
- 239000000427 antigen Substances 0.000 claims description 64
- 102000004190 Enzymes Human genes 0.000 claims description 50
- 108090000790 Enzymes Proteins 0.000 claims description 50
- 210000000349 chromosome Anatomy 0.000 claims description 50
- 241001465754 Metazoa Species 0.000 claims description 35
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 33
- 210000004507 artificial chromosome Anatomy 0.000 claims description 32
- 238000012217 deletion Methods 0.000 claims description 32
- 230000037430 deletion Effects 0.000 claims description 32
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 30
- 241000283984 Rodentia Species 0.000 claims description 28
- 108060003951 Immunoglobulin Proteins 0.000 claims description 25
- 108010052160 Site-specific recombinase Proteins 0.000 claims description 25
- 150000001413 amino acids Chemical class 0.000 claims description 25
- 102000018358 immunoglobulin Human genes 0.000 claims description 25
- 230000003053 immunization Effects 0.000 claims description 24
- 241000282693 Cercopithecidae Species 0.000 claims description 20
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 claims description 18
- 238000012986 modification Methods 0.000 claims description 18
- 230000004048 modification Effects 0.000 claims description 18
- 238000010362 genome editing Methods 0.000 claims description 15
- 241000124008 Mammalia Species 0.000 claims description 14
- 241001494479 Pecora Species 0.000 claims description 10
- 238000005516 engineering process Methods 0.000 claims description 10
- 239000003550 marker Substances 0.000 claims description 10
- 241000251730 Chondrichthyes Species 0.000 claims description 8
- 241000283973 Oryctolagus cuniculus Species 0.000 claims description 8
- 210000004408 hybridoma Anatomy 0.000 claims description 8
- 241000283073 Equus caballus Species 0.000 claims description 7
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 7
- 210000004989 spleen cell Anatomy 0.000 claims description 7
- 102100029567 Immunoglobulin kappa light chain Human genes 0.000 claims description 6
- 101710189008 Immunoglobulin kappa light chain Proteins 0.000 claims description 6
- 238000012258 culturing Methods 0.000 claims description 6
- 210000001165 lymph node Anatomy 0.000 claims description 6
- 102000039446 nucleic acids Human genes 0.000 claims description 6
- 108020004707 nucleic acids Proteins 0.000 claims description 6
- 150000007523 nucleic acids Chemical class 0.000 claims description 6
- 102000004169 proteins and genes Human genes 0.000 claims description 5
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 4
- 238000012270 DNA recombination Methods 0.000 claims description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 210000004369 blood Anatomy 0.000 claims description 4
- 239000008280 blood Substances 0.000 claims description 4
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 4
- 238000002823 phage display Methods 0.000 claims description 4
- 210000005260 human cell Anatomy 0.000 claims description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 156
- 239000012634 fragment Substances 0.000 description 119
- 238000004458 analytical method Methods 0.000 description 76
- 108020004414 DNA Proteins 0.000 description 75
- 241000699670 Mus sp. Species 0.000 description 55
- 238000010222 PCR analysis Methods 0.000 description 46
- 108010091086 Recombinases Proteins 0.000 description 44
- 102000018120 Recombinases Human genes 0.000 description 43
- 239000002609 medium Substances 0.000 description 38
- 239000013612 plasmid Substances 0.000 description 36
- 229930189065 blasticidin Natural products 0.000 description 34
- 210000003917 human chromosome Anatomy 0.000 description 34
- 108020005004 Guide RNA Proteins 0.000 description 33
- 239000002299 complementary DNA Substances 0.000 description 30
- 230000014509 gene expression Effects 0.000 description 30
- 108091008146 restriction endonucleases Proteins 0.000 description 29
- 210000000952 spleen Anatomy 0.000 description 26
- 241000700159 Rattus Species 0.000 description 25
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 24
- 108091033409 CRISPR Proteins 0.000 description 23
- 101000607560 Homo sapiens Ubiquitin-conjugating enzyme E2 variant 3 Proteins 0.000 description 23
- 102100039936 Ubiquitin-conjugating enzyme E2 variant 3 Human genes 0.000 description 23
- 238000003780 insertion Methods 0.000 description 23
- 230000037431 insertion Effects 0.000 description 23
- 238000002965 ELISA Methods 0.000 description 22
- 239000013604 expression vector Substances 0.000 description 22
- 108091035539 telomere Proteins 0.000 description 22
- 102000055501 telomere Human genes 0.000 description 22
- 239000005090 green fluorescent protein Substances 0.000 description 21
- 210000003411 telomere Anatomy 0.000 description 21
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 19
- 238000002649 immunization Methods 0.000 description 18
- 238000004520 electroporation Methods 0.000 description 17
- 210000002257 embryonic structure Anatomy 0.000 description 17
- 102000012960 Immunoglobulin kappa-Chains Human genes 0.000 description 16
- 108010090227 Immunoglobulin kappa-Chains Proteins 0.000 description 16
- 238000012790 confirmation Methods 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 13
- 238000003776 cleavage reaction Methods 0.000 description 13
- 238000010586 diagram Methods 0.000 description 13
- 230000007017 scission Effects 0.000 description 13
- 102100035102 E3 ubiquitin-protein ligase MYCBP2 Human genes 0.000 description 12
- 239000003814 drug Substances 0.000 description 12
- 239000000523 sample Substances 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 101100384865 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cot-1 gene Proteins 0.000 description 11
- 238000002744 homologous recombination Methods 0.000 description 11
- 230000006801 homologous recombination Effects 0.000 description 11
- 238000012546 transfer Methods 0.000 description 11
- 241000282412 Homo Species 0.000 description 10
- 102100034349 Integrase Human genes 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- 102000040430 polynucleotide Human genes 0.000 description 10
- 108091033319 polynucleotide Proteins 0.000 description 10
- 239000002157 polynucleotide Substances 0.000 description 10
- 108010061833 Integrases Proteins 0.000 description 9
- 108010006785 Taq Polymerase Proteins 0.000 description 9
- 101150046339 fur gene Proteins 0.000 description 9
- 210000000415 mammalian chromosome Anatomy 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- 238000012408 PCR amplification Methods 0.000 description 8
- 235000010724 Wisteria floribunda Nutrition 0.000 description 8
- 230000028993 immune response Effects 0.000 description 8
- 230000016784 immunoglobulin production Effects 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 230000000717 retained effect Effects 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 239000006228 supernatant Substances 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- 238000011144 upstream manufacturing Methods 0.000 description 8
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 7
- 241000725303 Human immunodeficiency virus Species 0.000 description 7
- 241000282567 Macaca fascicularis Species 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 238000003163 cell fusion method Methods 0.000 description 7
- 210000002950 fibroblast Anatomy 0.000 description 7
- 239000007758 minimum essential medium Substances 0.000 description 7
- 210000000130 stem cell Anatomy 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 210000002230 centromere Anatomy 0.000 description 6
- 238000010276 construction Methods 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- 108010048367 enhanced green fluorescent protein Proteins 0.000 description 6
- 238000010195 expression analysis Methods 0.000 description 6
- 238000010363 gene targeting Methods 0.000 description 6
- 239000013642 negative control Substances 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 230000036961 partial effect Effects 0.000 description 6
- 210000005259 peripheral blood Anatomy 0.000 description 6
- 239000011886 peripheral blood Substances 0.000 description 6
- 230000008707 rearrangement Effects 0.000 description 6
- 238000010354 CRISPR gene editing Methods 0.000 description 5
- 206010059866 Drug resistance Diseases 0.000 description 5
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 5
- 238000007792 addition Methods 0.000 description 5
- 238000000246 agarose gel electrophoresis Methods 0.000 description 5
- 210000002459 blastocyst Anatomy 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 210000004602 germ cell Anatomy 0.000 description 5
- 230000014759 maintenance of location Effects 0.000 description 5
- 210000001161 mammalian embryo Anatomy 0.000 description 5
- 230000013011 mating Effects 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 239000012460 protein solution Substances 0.000 description 5
- 238000012163 sequencing technique Methods 0.000 description 5
- 238000004904 shortening Methods 0.000 description 5
- 210000001082 somatic cell Anatomy 0.000 description 5
- 241000894007 species Species 0.000 description 5
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 4
- 108700028369 Alleles Proteins 0.000 description 4
- 241000725101 Clea Species 0.000 description 4
- 102000018251 Hypoxanthine Phosphoribosyltransferase Human genes 0.000 description 4
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- 230000005856 abnormality Effects 0.000 description 4
- 239000011543 agarose gel Substances 0.000 description 4
- 229960000723 ampicillin Drugs 0.000 description 4
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 4
- 210000004102 animal cell Anatomy 0.000 description 4
- 238000000137 annealing Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 230000001747 exhibiting effect Effects 0.000 description 4
- 238000000684 flow cytometry Methods 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 238000010899 nucleation Methods 0.000 description 4
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 238000012216 screening Methods 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 230000005945 translocation Effects 0.000 description 4
- CXNPLSGKWMLZPZ-GIFSMMMISA-N (2r,3r,6s)-3-[[(3s)-3-amino-5-[carbamimidoyl(methyl)amino]pentanoyl]amino]-6-(4-amino-2-oxopyrimidin-1-yl)-3,6-dihydro-2h-pyran-2-carboxylic acid Chemical compound O1[C@@H](C(O)=O)[C@H](NC(=O)C[C@@H](N)CCN(C)C(N)=N)C=C[C@H]1N1C(=O)N=C(N)C=C1 CXNPLSGKWMLZPZ-GIFSMMMISA-N 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 241000700198 Cavia Species 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- 108010051219 Cre recombinase Proteins 0.000 description 3
- 241000699800 Cricetinae Species 0.000 description 3
- 241000283086 Equidae Species 0.000 description 3
- 108010046276 FLP recombinase Proteins 0.000 description 3
- 108700021430 Kruppel-Like Factor 4 Proteins 0.000 description 3
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 3
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 238000011530 RNeasy Mini Kit Methods 0.000 description 3
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 3
- 238000012300 Sequence Analysis Methods 0.000 description 3
- 102000004142 Trypsin Human genes 0.000 description 3
- 108090000631 Trypsin Proteins 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- CXNPLSGKWMLZPZ-UHFFFAOYSA-N blasticidin-S Natural products O1C(C(O)=O)C(NC(=O)CC(N)CCN(C)C(N)=N)C=CC1N1C(=O)N=C(N)C=C1 CXNPLSGKWMLZPZ-UHFFFAOYSA-N 0.000 description 3
- 230000024245 cell differentiation Effects 0.000 description 3
- 239000002771 cell marker Substances 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 238000004925 denaturation Methods 0.000 description 3
- 230000036425 denaturation Effects 0.000 description 3
- 230000005782 double-strand break Effects 0.000 description 3
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 3
- 229960005542 ethidium bromide Drugs 0.000 description 3
- 230000001605 fetal effect Effects 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000003472 neutralizing effect Effects 0.000 description 3
- 230000001717 pathogenic effect Effects 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 210000001988 somatic stem cell Anatomy 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- 244000063299 Bacillus subtilis Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 241000282832 Camelidae Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 101710098119 Chaperonin GroEL 2 Proteins 0.000 description 2
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 2
- 241000711573 Coronaviridae Species 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 108091029865 Exogenous DNA Proteins 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 101000686179 Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Protein RecA Proteins 0.000 description 2
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- 229930193140 Neomycin Natural products 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 239000012124 Opti-MEM Substances 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 241000282579 Pan Species 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 108700008625 Reporter Genes Proteins 0.000 description 2
- 101150086694 SLC22A3 gene Proteins 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 2
- 101150117115 V gene Proteins 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 101150063416 add gene Proteins 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000007910 cell fusion Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 230000002998 immunogenetic effect Effects 0.000 description 2
- 239000003547 immunosorbent Substances 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 101150111214 lin-28 gene Proteins 0.000 description 2
- 238000001638 lipofection Methods 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 229960004927 neomycin Drugs 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 244000045947 parasite Species 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 229920002523 polyethylene Glycol 1000 Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 230000008672 reprogramming Effects 0.000 description 2
- ZFLJHSQHILSNCM-UHFFFAOYSA-N reversine Chemical compound C1CCCCC1NC1=NC(NC=2C=CC(=CC=2)N2CCOCC2)=NC2=C1N=CN2 ZFLJHSQHILSNCM-UHFFFAOYSA-N 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 108091005957 yellow fluorescent proteins Proteins 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 241000203069 Archaea Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000025721 COVID-19 Diseases 0.000 description 1
- 101100290380 Caenorhabditis elegans cel-1 gene Proteins 0.000 description 1
- 241000204900 Carcharhinus leucas Species 0.000 description 1
- 241000904011 Carcharhinus perezii Species 0.000 description 1
- 241000722713 Carcharodon carcharias Species 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 101150097493 D gene Proteins 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 241000702191 Escherichia virus P1 Species 0.000 description 1
- 101000834253 Gallus gallus Actin, cytoplasmic 1 Proteins 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 101000843762 Homo sapiens Immunoglobulin heavy diversity 1-1 Proteins 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108700029228 Immunoglobulin Heavy Chain Genes Proteins 0.000 description 1
- 102100030644 Immunoglobulin heavy diversity 1-1 Human genes 0.000 description 1
- 108091029795 Intergenic region Proteins 0.000 description 1
- 101150008942 J gene Proteins 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 241000127282 Middle East respiratory syndrome-related coronavirus Species 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108010047620 Phytohemagglutinins Proteins 0.000 description 1
- 208000020584 Polyploidy Diseases 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 108020005091 Replication Origin Proteins 0.000 description 1
- 241000315672 SARS coronavirus Species 0.000 description 1
- 108091005609 SARS-CoV-2 Spike Subunit S1 Proteins 0.000 description 1
- 241000235343 Saccharomycetales Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 101800000904 Spike protein S1 Proteins 0.000 description 1
- 101000910035 Streptococcus pyogenes serotype M1 CRISPR-associated endonuclease Cas9/Csn1 Proteins 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 238000010459 TALEN Methods 0.000 description 1
- 208000035199 Tetraploidy Diseases 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 1
- 241000223104 Trypanosoma Species 0.000 description 1
- 210000001766 X chromosome Anatomy 0.000 description 1
- 210000002593 Y chromosome Anatomy 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000002266 amputation Methods 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 239000007640 basal medium Substances 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 210000004958 brain cell Anatomy 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 108091036078 conserved sequence Proteins 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- JVHIPYJQMFNCEK-UHFFFAOYSA-N cytochalasin Natural products N1C(=O)C2(C(C=CC(C)CC(C)CC=C3)OC(C)=O)C3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 JVHIPYJQMFNCEK-UHFFFAOYSA-N 0.000 description 1
- ZMAODHOXRBLOQO-UHFFFAOYSA-N cytochalasin-A Natural products N1C(=O)C23OC(=O)C=CC(=O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 ZMAODHOXRBLOQO-UHFFFAOYSA-N 0.000 description 1
- 238000004163 cytometry Methods 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 210000001842 enterocyte Anatomy 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 108091006047 fluorescent proteins Proteins 0.000 description 1
- 102000034287 fluorescent proteins Human genes 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 210000002064 heart cell Anatomy 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 210000004754 hybrid cell Anatomy 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 229940027941 immunoglobulin g Drugs 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229920012128 methyl methacrylate acrylonitrile butadiene styrene Polymers 0.000 description 1
- 210000000472 morula Anatomy 0.000 description 1
- 239000012120 mounting media Substances 0.000 description 1
- 210000005088 multinucleated cell Anatomy 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 210000004681 ovum Anatomy 0.000 description 1
- 230000001885 phytohemagglutinin Effects 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000012846 protein folding Effects 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 108010054624 red fluorescent protein Proteins 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 238000010079 rubber tapping Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 210000002363 skeletal muscle cell Anatomy 0.000 description 1
- 210000004927 skin cell Anatomy 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 210000004988 splenocyte Anatomy 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 210000004340 zona pellucida Anatomy 0.000 description 1
Images












Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New breeds of animals
- A01K67/027—New breeds of vertebrates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/06—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies from serum
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
- C12N5/12—Fused cells, e.g. hybridomas
- C12N5/16—Animal cells
Definitions
- the present invention relates to mammalian artificial chromosome vectors comprising human immunoglobulin heavy chain loci and human immunoglobulin light chain loci with altered human immunoglobulin heavy chain D regions.
- the present invention also relates to mammalian cells or non-human animals comprising said mammalian artificial chromosome vector.
- the present invention further relates to a method for modifying the D region in the production of the mammalian artificial chromosome vector.
- Human antibodies are used as therapeutic agents for tumors and autoimmune diseases because they are not recognized as foreign substances by the human body. Such antibodies can be produced by using phage display technology or human antibody-producing mice.
- Human antibody-producing mice are Trans-chromosomic (TC) mice carrying mouse artificial chromosome vectors containing full-length human immunoglobulin heavy chain and light chain loci (Patent Document 1, Patent Document 2, Non-Patent Document 1).
- Non-Patent Document 3 discloses a platform for achieving further expanded human antibody diversity.
- An object of the present invention is to provide means for achieving expansion of human antibody diversity in human antibody-producing non-human animals.
- the present inventors focused on the finding that cattle efficiently produce broadly neutralizing antibodies against HIV (MJ Burke et al., Viruses 2020, 12, 473; doi : 10.3390/v12040473), found a way to expand the diversity of human antibodies and/or emerge human antibodies with longer than normal antibody heavy chain CDR3s.
- the present invention includes the features shown below.
- the human-derived genomic sequence from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region has the following (1) and (2), or (1) and (3), a mammalian artificial chromosome vector characterized in that it is replaced with a modified sequence of the D region consisting of a combination of the D region
- the modified sequence is (1) the human-derived genomic sequence between the open reading frames (ORF) from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, or the ORF except between D3-9 and D3-10
- the human-derived genomic sequence between comprises a sequence truncated to a length of 49 bp or more from each VDJ recombination sequence end of the inter-ORF region flanked by the VDJ recombination sequences, and (2) instead of the ORF sequences from D1-1 to D1-26 of
- the ORF sequence from B1-1 to B9-4 of the bovine-derived immunoglobulin heavy chain locus D region has a nucleotide sequence of SEQ ID NOs: 127 to 149 or has 90% or more identity with the nucleotide sequence.
- the ORF sequence from 1S5 to 1S35 of the monkey-derived immunoglobulin heavy chain locus D region comprises a nucleotide sequence of SEQ ID NOS: 150 to 175 or a nucleotide sequence having 90% or more identity with the nucleotide sequence.
- a non-human comprising the mammalian artificial chromosome vector according to any one of [1] to [10] and having disrupted endogenous immunoglobulin heavy chain, ⁇ light chain and ⁇ light chain genes or loci Mammal.
- a non-human animal comprising human immunoglobulin heavy chain and light chain loci, wherein the human-derived genomic sequence from D1-1 to D1-26 of the D region of the human immunoglobulin heavy chain locus is: (1) and (2), or (1) and (3), is replaced with a modified sequence of the D region, wherein the modified sequence of the D region is (1) the human-derived genomic sequence between the open reading frames (ORF) from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, or the ORF except between D3-9 and D3-10
- the human-derived genomic sequence between comprises a sequence truncated to a length of 49 bp or more from each VDJ recombination sequence end of the inter-ORF region flanked by the VDJ recombination sequences, and (2) instead of the ORF sequences from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, from B1-1 to B9-4 of the bovine-derived immunoglobulin heavy chain locus D region
- the non-human animal according to any one of [14] to [17] is immunized with a target antigen, and spleen cells, lymph node cells or B cells from the non-human animal producing an antibody that binds to the target antigen , fusing the spleen cells, lymph node cells or B cells with myeloma cells to form hybridomas, culturing the hybridomas to obtain monoclonal antibodies that bind to the target antigen, How to make.
- a mammalian artificial chromosome vector comprising a first site-specific recombinase recognition site, a promoter and a second site-specific recombinase recognition site.
- the mammalian artificial chromosome vector according to [23] wherein the deleted D region is a region from D1-1 to D1-26.
- the human-derived genomic sequence of the D region of the human immunoglobulin heavy chain locus or its human minimalization i.e., the inter-open reading frame (ORF) region.
- Antibody diversity by replacing the ORF (gene) sequence of the D region sequence with an ORF sequence of a D region derived from an animal such as a bovine or a monkey, or a modified ORF sequence of a human D region sequence can be extended and human antibodies can be generated that preferably contain a CDRH3 of 15 to 50 or more amino acids in length.
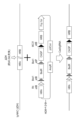
- FIG. 2 is a diagram relating to Example 1;
- FIG. 2A shows CHO cells (cell clone TF7-B10) in which one copy of Ig-NAC ( ⁇ DH) is maintained independently of the host mouse chromosome.
- Ig-NAC( ⁇ DH) arrowheads represent human chromosomal regions (red) and arrows represent artificial chromosomal regions (green).
- FIG. 2B shows that Ig-NAC ( ⁇ DH) contains human immunoglobulin (Ig) heavy chains (arrowheads or green) and kappa light chains (arrowheads or red).
- FIG. 2 shows the results of Example 1; This figure shows restriction enzyme AfiIII in plasmid pRP[Exp]-CAG>mCherry (synthesized by Vector Builder) having CAGP (CAG promoter; conjugate of cytomegalovirus enhancer and chicken ⁇ -actin promoter).
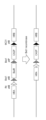
- FIG. 10 is a diagram related to Example 2;
- plasmid CAG- ⁇ C31-HRB-Fcy::fur was cleaved with restriction enzymes NotI (NEB) and SpeI (NEB), and a plasmid HRA-derived fragment ("A") was inserted upstream of CAGP to produce HDR. It indicates that the vector was obtained.
- BsdR represents the blasticidin resistance gene
- CAGP the CAG promoter
- R4 attB, Bxb1 attB and Bxb1 attP the recombinase recognition site
- AmpR the ampicillin resistance gene
- pUCori the pUC replication origin.
- FIG. 10 is a diagram related to Example 2; This figure shows that the HDR vector was inserted near the DH region deletion site ( ⁇ DH) of Ig-NAC ( ⁇ DH) in CHO cells by genome editing (Cas9/gRNA).
- FIG. 10 is a diagram related to Example 2; This figure shows removal of the blasticidin resistance gene from the HDR vector by site-specific recombination by introducing Bxb1 recombinase into cells.
- FIG. 10 is a diagram relating to Example 3; This figure shows that each genomic sequence between ORFs D1-1 to D1-26 of the human immunoglobulin heavy chain gene D region is shortened to a size of approximately 200 bp, minimizing the D region.
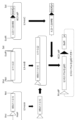
- FIG. 11 is a diagram relating to Example 6; This figure shows the procedure for generating a human minimalized IgHD vector from human miniA, human miniB and human miniC.
- human miniA is from R4 attP and downstream of HRA to D fragment 3-10
- human miniB is from D fragment 3-10 to 3-22
- human miniC is from D fragment 3-22 to HRB.
- ⁇ C31 attP and blasticidin resistance gene (BSDR) respectively.
- FIG. 11 is a diagram relating to Example 6; This figure shows that site-specific recombination between ⁇ C31 attB in the HDR vector with the blasticidin resistance gene removed shown in FIG. Generation of HDR vector with IgHD inserted.
- FIG. 11 is a diagram relating to Example 6; This figure shows that site-specific recombination between ⁇ C31 attB in the HDR vector with the blasticidin resistance gene removed shown in FIG. Generation of HDR vector with IgHD inserted.
- FIG. 11 is a diagram relating to Example 6; This figure shows removal of unnecessary sequences such as the CAG promoter and the blasticidin resistance gene from the human minimalized IgHD-inserted HDR vector generated in FIG. 9 by introducing R4 recombinase into the cells. .
- FIG. 11 is a diagram relating to Example 6; This figure shows human minimalized IgHD (a total of 23 pieces from D2-2 to D5-24 in the figure) in a vector (Fig. 8) containing the human minimalized immunoglobulin heavy chain locus D region, and bovine human IgHD. (A total of 23 from B1-1 to B9-4 in the figure) are used to construct vectors.
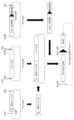
- FIG. 11 is a diagram relating to Example 9; This figure shows the procedure for constructing a bovinized human IgHD vector carrying bovine miniA, B, and C.
- bovine human IgHD was divided into three parts in the same manner as in Example 5, and bovine miniA, bovine miniB, and bovine miniC, which were the three-part plasmid DNAs, were chemically synthesized (the synthesis was entrusted to Eurofins Genomics).
- FIG. 9 shows the procedure for constructing a bovinized human IgHD vector carrying bovine miniA, B, and C.
- bovine human IgHD was divided into three parts in the same manner as in Example 5, and bovine miniA, bovine miniB, and bovine miniC, which were the three-part plasmid DNAs, were chemically synthesized (the synthesis was entrusted to Eurofins Genomics).
- FIG. 11 is a diagram relating to Example 9; This figure shows the procedure for constructing a bovinized human IgHD
- Ig immunoglobulin
- Example 14 shows the results of Example 14;
- This figure shows antigen (SARS -Using a 96-well plate on which the CoV2 spike protein S1 receptor binding site (RBD) was immobilized and an anti-human IgG Fc antibody, the indicated 1/1,000 to 1/1,000,000-fold dilution of the anti-
- the results of ELISA performed on serum are shown. At this time, it was confirmed that an immune response was generated by continuous booster immunization, and the antibody titer against RBD (vertical axis, OD at 450 nm) increased.
- b1-b5 represent boosters 1-5.
- Example 11 shows the results of Example 18; This figure shows the structure of the simianized human IgHD region obtained by replacing the human minimalized IgHD region (approximately 43 kb) with the cynomolgus monkey D fragment (approximately 44 kb).
- FIG. 20 is a diagram relating to Example 19; This figure shows 26 modified long DH sequences to replace D fragments 1-1 to 1-26 of the human long minimalized IgHD region.
- FIG. 29 is a diagram relating to Example 29;
- the present invention in a first aspect, in a mammalian artificial chromosome vector comprising human immunoglobulin heavy chain and light chain loci, D1-1 to D1 of the human immunoglobulin heavy chain locus D region A mammal characterized in that the human-derived genomic sequence up to -26 is replaced with a modified sequence of the D region consisting of a combination of (1) and (2) or (1) and (3) below
- An animal artificial chromosome vector wherein the modified sequence of the D region is (1) human-derived genomic sequence between ORFs from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, or human-derived genome between the above ORFs excluding between D3-9 and D3-10 the sequence comprises a truncated sequence of 49 bp or more from each VDJ recombination sequence end of the inter-ORF region flanked by the VDJ recombination sequences; and (2) instead of the OR
- Human immunoglobulin heavy chain locus D region herein refers to all ORFs (also referred to as “genes” (or polypeptide-encoding regions) or “D fragments”), VDJ recombination sequences, and all It refers to the entirety including the inter-ORF (ie, between genes) regions of the .
- VDJ recombination sequences are sequences flanking the upstream and downstream of the V, D and J regions of human immunoglobulin heavy and light chain loci, the heavy chain, light chain kappa and light chain lambda genes It contains conserved sequences of 7, 23, 9 and/or 12 bases in different locations depending on the locus.
- the recombinant sequence contains 28 bp (ie, 9 bp, 12 bp (spacer), 7 bp) of bases.
- Mammalian artificial chromosome vectors are, but not limited to, artificial chromosome vectors made from mammalian chromosomes such as humans, rodents (e.g., mice, rats, hamsters, guinea pigs, etc.), more specifically in the genomic sequence from the long arm and short arm (if any) comprising the centromere and telomere from said animal and having, for example, 99.5% to 100%, preferably 100%, of the genes removed
- rodents e.g., mice, rats, hamsters, guinea pigs, etc.
- an artificially engineered chromosomal vector containing a human immunoglobulin heavy chain locus comprising an altered antibody heavy chain D region and, optionally, a human immunoglobulin light chain locus.
- telomeres in the present specification are natural telomeres of the same or different species, or artificial telomeres.
- homologous means an animal of the same species as the mammal from which the chromosome fragment of the artificial chromosome vector is derived, while “heterologous” means a mammal other than the animal (which includes humans).
- An artificial telomere sequence refers to an artificially produced sequence having a telomere function, such as the (TTAGGG)n sequence (n means repeat).
- telomere sequence into an artificial chromosome can be performed by a method including telomere truncation (cleavage by artificially inserting a telomere sequence) as described in, for example, International Publication WO00/10383. Telomere truncation can be used for chromosome shortening in the creation of artificial chromosomes of the present invention.
- a human antibody heavy chain variable region consists of framework (FR) 1, CDRH1, FR2, CDRH2, FR3, CDRH3, FR4, and part of the constant region from the N-terminal side.
- FR1, CDRH1, FR2, CDRH2 and FR3 are encoded primarily by V region nucleotide sequences of the VDJ sequence formed by recombination (or rearrangement) of the human immunoglobulin heavy chain locus.
- CDRH3 and FR4 are mainly encoded by the D region nucleotide sequence and J region nucleotide sequence of the above VDJ sequence, respectively.
- the D region is modified in order to produce a human antibody containing an antibody heavy chain CDR3 with a length of 18 or more amino acids.
- the human antibody light chain variable region consists of FR1, CDRL1, FR2, CDRL2, FR3, CDRL3, FR4, and part of the constant region, and FR1, CDRL1, FR2, CDRL2, FR3 and CDRL3 (part) It is encoded mainly by the V region nucleotide sequence of the VJ sequence formed by recombination (or rearrangement) of the human immunoglobulin light chain locus, and CDRH3 (part) and FR4 are mainly encoded by the VJ sequence is encoded by the J region nucleotide sequence of
- the present invention expands the diversity of human antibodies, preferably The D region genomic sequence of the human antibody heavy chain locus is modified so that an antibody containing a longer (eg, 18 or more) amino acid length of CDRH3 than usual in a human antibody can be obtained.
- modified sequences of the human D region are described below.
- An example of a modified sequence is that the genomic sequence of the inter-ORF (also referred to as "inter-genic") region of the human immunoglobulin heavy chain locus D region is flanked by VDJ recombination sequences at the ends of each VDJ recombination sequence of the inter-ORF region.
- a modified D region genomic sequence comprising a sequence truncated to a length of 49 bp or more from the be.
- the D region (also referred to as the “IgHD” region) of the human immunoglobulin heavy chain locus is present on human chromosome 14 (14q32.33) and is 5′ ⁇ 3′ in order, for example, D1-1 (position 23599). Accession No. X97051), D2-2 (Positions 35049-37615; Accession No. J00232), D3-3 (Positions 37616-39425; Accession No. X13972), D4-4 (Positions 39426-40474; Accession No. X13972), D5-5 (Positions 40475-41880; Accession No. X13972), D6-6 (Positions 41881-43056; Accession No.
- X13972 D1-7 (Positions 43057-44649; Accession No. X13972), D2-8 (Positions 44650-47262) Accession No. X13972), D3-9 (Positions 47263-48619; Accession No. X13972), D3-10 (Positions 48620-49158; Accession No. X13972), D4-11 (Positions 49159-50078; Accession No.
- the ORF sequences D1-1 to D1-26 of the human immunoglobulin heavy chain (IgH) D region are, for example, the nucleotide sequences of SEQ ID NOS: 101-126 below. Also, an example of the size between ORFs after shortening is shown between each sequence.
- SEQ ID NO: 101 (IGHD1-1): ggtacaactggaacgac (Size between ORFs of D1-1 and D2-2 + VDJ recombination sequence (28bp ⁇ 2): 256bp)
- SEQ ID NO: 102 (IGHD2-2): aggatatgtagtagtaccagctgctatgcc (Size between ORFs of D2-2 and D3-3 + VDJ recombination sequence (28bp x 2): 256bp)
- SEQ ID NO: 103 (IGHD3-3): gtattacgatttttggagtggttattatacc (Size between ORFs of D3-3 and D4-4 + VDJ recombination sequence (28bp x 2): 256bp)
- SEQ ID NO: 104 (IGHD4-4): tgactacagtaactac (Size between ORFs of D4-4 and D5-5 +
- the modified sequence contains the modified ORF of the human IgHD region, and each human-derived genomic sequence of the inter-ORF region (that is, intergenic region) is flanked by the VDJ-recombined sequence. to a length of 49 bp or more, such as from about 100 bp to about 500 bp, from about 150 bp to about 300 bp, or from about 200 bp to about 250 bp.
- the length of the modified human IgHD region after minimization is preferably about 10 kb or less so that it can be inserted into a (conventional) plasmid vector. For example, when shortening to about 200 bp, it is minimized to a length of about 8 kb.
- the above shortening (or minimization) method is not particularly limited. can be deleted.
- D1-1 to D1-26 of the human D region is divided into several (eg, 3 to 5) segments, and each segment is minimized. They can be ligated side by side to create the human minimalized D region of interest ( Figure 8).
- the nucleotide sequence of the D region which includes the ORF sequences from D1-1 to D1-26 of the human minimalized D region, the VDJ recombination sequence (28 bp ⁇ 2) and the sequence between the minimalized ORFs, is, for example, the nucleotide sequence of SEQ ID NO: 1 below.
- nucleotide sequence containing an addition.
- a human minimalized D region sequence such as SEQ ID NO: 1 can be used when replacing the human IgHD region ORF with a heterologous ORF.
- Another modified sequence includes the genomic sequence of the immunoglobulin heavy chain locus D region derived from a non-human animal capable of producing an antibody with an antibody heavy chain CDR3 amino acid length of 18 or more, or a modified sequence thereof.
- Such non-human animals are, for example, animals such as cows, monkeys (eg, cynomolgus monkeys), sheep, horses, rabbits, birds, sharks (eg, bull sharks, reef sharks, and white sharks).
- Information retrieval from, for example, the International ImmunoGeneTics information system (IMGT) can confirm that at least the above animal species are species with D segments longer than the longest D segment in humans (37 base pairs).
- the bovine immunoglobulin heavy chain locus D region (21q24), for example, IGHD B1-1, B2-1, B3-1, B4-1, B9-1, B1-2, B2-2, B5-2, B8-2, B6-2, B1-3, B2-3, B3-3, B7-3, B5-3, B6-3, B1-4, B2-4, B3- 4, B7-4, B5-4, B6-4 and B9-4.
- the ORF sequence from B1-1 to B9-4 of the bovine immunoglobulin heavy chain locus D region can be replaced to include
- the nucleotide sequence of the bovine human minimalized D region thus obtained is, for example, the nucleotide sequence of SEQ ID NO: 2 below, or 90% or more, 95% or more, 97% or more, 98% or more, or It includes nucleotide sequences that have 99% or more identity, or nucleotide sequences that contain deletions, substitutions or additions of one or several nucleotides (or bases) in the nucleotide sequence.
- the ORF sequences from B1-1 to B9-4 of the bovine IgHD region are, for example, the nucleotide sequences of SEQ ID NOs: 127-149 below.
- Cynomolgus monkey immunoglobulin heavy chain locus D region (Chr7q; G.-Y. Yu et al., Immunogenetics 2016; 68: 417-428) is 5′ ⁇ 3′ in order, for example, IGHD 1S5, 2S11, 3S6 , 4S24,5S8,6S4,1S10,2S17,3S18,2S34,5S14,5S31,6S3,1S27,2S22,4S36,4S30,5S37,6S20,1S33,2S28,6S32,3S12,6S38,6S26, and 1S39 about 44kb ( Figure 15).
- the ORF sequences from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region are replaced with the ORF sequences from 1S5 to 1S35 of the monkey immunoglobulin heavy chain locus D region.
- the ORF sequences from 1S5 to 1S35 of the cynomolgus monkey D region are, for example, the nucleotide sequences of SEQ ID NOs: 150-175 below.
- SEQ ID NO: 150 (1S5): ggtataactggaactac SEQ ID NO: 151 (2S11): agaatattgtagtagtacttactgctcctcc SEQ ID NO: 152 (3S6): gtattacgaggatgattacggttactattacacccacagcgt SEQ ID NO: 153 (4S24): tgactacggtagcagctac SEQ ID NO: 154 (5S8): gtggatacagctacagttac SEQ ID NO: 155 (6S4): gggtatagcagcggctggtac SEQ ID NO: 156 (1S10): ggtatagctggaacgac SEQ ID NO: 157 (2S17
- another modified sequence is created based on a human antibody heavy chain CDR3 sequence of 18 amino acids or more in place of the ORFs from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, It contains 26 modified ORF sequences from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region.
- Such a modified D region ORF is, for example, a nucleotide sequence shown in SEQ ID NOs: 75 to 100 as shown in FIG.
- a nucleotide sequence having an identity of 99% or more, or a nucleotide sequence containing deletion, substitution or addition of one or several nucleotides (or bases) in the nucleotide sequence, D1-1 of the human IgHD region to D1-26 can be replaced with the corresponding 26 modified ORF sequences described above.
- the nucleotide sequence of the human minimalized D region so replaced is, for example, the nucleotide sequence of SEQ ID NO: 3, or 90% or more, 95% or more, 97% or more, 98% or more, or 99% or more of said nucleotide sequence. It includes nucleotide sequences with identity or nucleotide sequences containing deletions, substitutions or additions of one or several nucleotides (or bases) in said nucleotide sequences.
- a method for determining a modified ORF sequence based on a human antibody heavy chain CDR3 sequence can include, for example, the steps shown below.
- the upstream from the sequence "WG" on the N-terminal side of VH is defined as CDR3 (M. Chiu et al. ., Antibodies 2019;8(4):55; doi.org/10.3390/antib8040055).
- CDR3 M. Chiu et al. ., Antibodies 2019;8(4):55; doi.org/10.3390/antib8040055.
- known sequences with CDR3 of 18 amino acids or more are searched.
- the nucleotide sequence of CDR3 is extracted.
- synonymous (no amino acid changes) substitution mutations are introduced into the somatic mutation hotspot sequence to the extent possible.
- Identity in the present specification can be determined using a protein or gene search system such as BLAST or FASTA with or without introducing gaps (Zheng Zhang et al., J. Comput.Biol.2000;7:203-214;Altschul, SF et al., Journal of Molecular Biology1990;215:403-410;Pearson, WR et al., Proc.Natl.Acad Sci USA, 1988;85:2444-2448).
- the D region of the human immunoglobulin heavy chain locus can be modified, for example, by methods comprising the following steps.
- a mammalian artificial chromosome vector [Ig-NAC]) containing human immunoglobulin heavy and light chain loci is provided.
- Ig-NAC has a structure that includes a human immunoglobulin heavy chain locus and a human immunoglobulin light chain ⁇ (or ⁇ ) locus from the telomere side to the centromere side (eg, Japanese Patent No. 6868250).
- Ig-NAC is capable of stable replication and distribution as a chromosome independent of the original chromosome of the cell to be introduced.
- examples of this vector are rodent artificial chromosome vectors (eg mouse or rat artificial chromosome vectors). Production of rodent artificial chromosome vectors is described, for example, in Japanese Patent No. 6775224 and Japanese Patent No. 6868250 by the present applicant.
- a mammalian artificial chromosome vector may be produced, for example, by telomere truncation of a mammalian chromosome, comprising the centromere of the animal, a long arm segment near the centromere and, if present in the animal, a short arm segment, and a telomere .
- the mouse-derived chromosome fragment is any chromosome of mouse 1 to 19, X and Y chromosomes, preferably any fragment of chromosome 1 to 19 (at least the number of all endogenous genes in the long arm 99.5%, preferably nearly 100% deleted long arm fragment), the fragment includes a long arm fragment in which the long arm distal is deleted from the mouse chromosome long arm site near the centromere .
- Mouse chromosome sequence information is available from DDBJ/EMBL/GenBank, Santa Cruz Biotechnology, Inc.; Available from Chromosome Databases such as.
- the long arm fragment is non-limiting, for example, AC121307, AC161799 etc.
- the long arm region distal to the position is deleted consists of fragments.
- the long arm fragment consists of, but not limited to, a long arm fragment in which a region distal to Gm35974 has been deleted.
- it consists of a mouse chromosome 10-derived long arm fragment obtained by deleting the distal long arm from the gene Gm8155, which is the chromosomal region of the mouse chromosome 10 long arm.
- mouse artificial chromosome vectors include mouse artificial chromosomes contained in deposited cell line DT40 (10MAC) T5-26 (international deposit number NITE BP-02656), or deposited cell line DT40 (16MAC) T1-14 (international deposit number NITE BP-02657) includes mouse artificial chromosomes.
- the mammalian artificial chromosome vector of the present invention is, for example, the following steps (a) to (c): (a) obtaining cells that retain mammalian chromosomes; (b) deleting the mouse chromosome long-arm distal so that it does not contain the majority (99.5%-100%, preferably 100%) of the endogenous gene(s), and (c) long-arm proximal can be made by a method comprising the step of inserting one or more DNA sequence insertion sites into
- the order of steps (b) and (c) may be reversed.
- a mammalian artificial chromosome vector of the present invention To produce a mammalian artificial chromosome vector of the present invention, first, cells that retain mammalian chromosomes are produced. For example, mammalian fibroblasts carrying mammalian chromosomes labeled with drug resistance genes (e.g., blasticidin S resistance gene (BSr)) and mouse A9 cells (ATCC VA20110-2209 ) by cell fusion and transferring the chromosome from a mouse A9 hybrid cell carrying a mammalian chromosome labeled with a drug resistance gene into a cell with a high rate of homologous recombination.
- mammalian fibroblasts can be obtained based on the method described in the literature.
- mouse fibroblasts can be established from C57B6 mice available from CLEA Japan.
- Chicken DT40 cells Dieken et al., Nature Genetics, 12:174-182, 1996), for example, can be used as cells with a high homologous recombination rate.
- the transfer can be performed by a known chromosomal transfer method, for example, the micronuclear cell fusion method (Koi et al., Jpn. J. Cancer Res., 80:413-418, 1973).
- ⁇ Step (1b)> In cells that retain a single chromosome from a mammal, the long arm distal of the mammalian chromosome and, if present in the animal, the short arm distal are deleted. At this time, the important thing is to delete (or remove or delete) most of the endogenous genes present on the long and short arms and construct an artificial chromosome that retains the mammalian centromere. This is at least 99.5%, preferably at least 99.7%, more preferably at least 99.8%, most preferably 99.9-100% of the total endogenous gene(s) present on the long and short arms Most preferably, the cutting position is determined so as to delete (or remove or delete) 100%.
- telomere truncation Specifically, in cells that retain mammalian chromosomes, a targeting vector that retains the telomere sequence is constructed, and a clone in which an artificial or natural telomere sequence is inserted at a desired position on the chromosome by homologous recombination is obtained. gives deletion mutants by telomere truncation.
- the desired position is the cutting position of the distal long arm to be deleted, and the telomere sequence is substituted and inserted at this position by homologous recombination to delete the distal long arm.
- Such positions can be appropriately set by designing the target sequence when constructing the targeting vector.
- a target sequence is designed based on the DNA sequence of the long arm of a mammalian chromosome, and set so that telomere truncation occurs on the telomere side of the target sequence. This results in a mammalian chromosomal fragment in which most of the endogenous gene has been deleted. Telomere truncation can be similarly performed for other chromosomes. The same is true for amputation of the distal short arm.
- a recognition site for a site-specific recombination enzyme can preferably be inserted. That is, certain enzymes are known to recognize a specific recognition site and specifically cause recombination of DNA at the recognition site, and in the mammalian artificial chromosome vector in the present invention, such an enzyme and its A system of recognition sites for enzymes can be used to insert and load the desired human immunoglobulin heavy chain or locus and/or human immunoglobulin light chain gene or locus sequences.
- a Cre enzyme derived from bacteriophage P1 and a loxP sequence system as its recognition site Cre/loxP system; B.
- a known method such as a homologous recombination method can be used to insert the recognition site for such a site-specific recombination enzyme, and the insertion position and number are appropriately set within the proximal long arm and proximal short arm. can do.
- One type of recognition site or different types of recognition sites can be inserted into the mammalian artificial chromosome vector. Insertion of the target gene or locus by setting the recognition site, that is, the human immunoglobulin heavy chain gene or locus, the human immunoglobulin light chain ⁇ gene or locus, or the human immunoglobulin light chain ⁇ gene or locus Since the position can be specified, the insertion position is constant and free from unintended position effects.
- a reporter gene may preferably be inserted into the mammalian artificial chromosome vector, in addition to the sequence of the above gene or locus of interest.
- reporter genes include, but are not limited to, fluorescent protein (e.g., green fluorescent protein (GFP or EGFP), yellow fluorescent protein (YFP), etc.) gene, tag protein-encoding DNA, ⁇ -galactosidase gene, luciferase gene and the like.
- the mammalian artificial chromosome vector may further contain a selectable marker gene.
- Selectable markers are useful in selecting cells transformed with the vector.
- selectable marker genes include positive selectable marker genes and negative selectable marker genes, or both.
- Positive selectable marker genes include drug resistance genes such as neomycin resistance gene, ampicillin resistance gene, blasticidin S (BS) resistance gene, puromycin resistance gene, geneticin (G418) resistance gene, hygromycin resistance gene, etc.
- Negative selectable marker genes include, for example, herpes simplex thymidine kinase (HSV-TK) gene, diphtheria toxin A fragment (DT-A) gene and the like. HSV-TK is commonly used in combination with ganciclovir or acyclovir.
- human immunoglobulin gene or locus means a human immunoglobulin heavy chain gene or locus from human chromosome 14, a human immunoglobulin light chain from human chromosome 2, unless otherwise specified. It refers to the kappa gene or locus and/or the human immunoglobulin light chain lambda gene or locus from human chromosome 22.
- the human immunoglobulin gene or locus is, for example, human chromosome 14 immunoglobulin heavy locus (human) NC — 000014.9 ((base number 105586437..106879844) or (base number 105264221..107043718)), human immunoglobulin kappa locus (human) NC — 000002.12 ((base number 88857361..90235368) or (base number 88560086..90265666)) of chromosome 2, and immunoglobulin lambda locus of human chromosome 22 (human) NC_000022.11 (( (base numbers 22026076..22922913) or (base numbers 21620362..23823654)).
- the human immunoglobulin heavy chain gene or locus is about 1.3 Mb base length, the human immunoglobulin light chain ⁇ gene or locus is about 1.4 Mb base length, and the human immunoglobulin light chain ⁇ gene. Alternatively, the locus is about 0.9 Mb base long.
- the mouse antibody heavy chain gene or locus is on mouse chromosome 12
- the mouse antibody light chain ⁇ gene or locus is on mouse chromosome 6,
- the mouse antibody light chain ⁇ gene or The locus is on mouse chromosome 16.
- the mouse antibody heavy chain gene or locus is Chromosome 12, NC_000078.6 (113258768..116009954, complement)
- the mouse antibody light chain ⁇ gene or locus is Chromosome 6, NC_000072.6 (67555636 ..70726754)
- the mouse antibody light chain ⁇ gene or locus is represented by the base sequence described in Chromosome 16, NC — 000082.6 (19026858..19260844, complement).
- the rat antibody heavy chain gene or locus is on rat chromosome 6
- the rat antibody light chain ⁇ gene or locus is on rat chromosome 4
- the rat antibody light chain ⁇ gene or locus is on rat chromosome 4.
- the locus is on rat chromosome 11.
- the nucleotide sequences of these genes or loci are available from NCBI in the United States (GenBank, etc.), published literature, and the like.
- ⁇ Second step> In a mammalian artificial chromosome vector (Ig-NAC) containing the human immunoglobulin (heavy chain and / or light chain) locus prepared in the first step, from the human immunoglobulin heavy chain locus, for example, genome editing (J M. Chylinski et al., Science 2012;337:816-821) and site-specific recombination techniques (e.g., use of site-specific recombination enzymes) to combine the V and J regions of the human heavy chain locus.
- the D region present between is deleted to create 'Ig-NAC( ⁇ DH)'.
- Genome editing in this specification is a technology for editing and genetically modifying genomic DNA using artificial cleavage enzymes such as TALEN (TALE nuclease), CRISPR-Cas system, etc. Any technique of genome editing can be used in the method of the invention, but preferably the CRISPR-Cas system is used.
- TALEN TALE nuclease
- CRISPR-Cas system CRISPR-Cas system
- the CRISPR/Cas9 system was discovered from the adaptive immune system against viruses and plasmids of bacteria and archaea, but the vector construction is relatively simple and it is possible to modify multiple genes at the same time (Jineket et al. al., Science, 17, 337(6096): 816-821, 2012; Sander et al., Nature biotechnology, 32(4): 347-355, 2014).
- This system includes a Cas9 protein and a guide RNA (gRNA) with an approximately 20 base pair target sequence.
- the gRNA recognizes the PAM sequence near the target sequence and specifically binds to the target genomic DNA, and the Cas9 protein induces a double-strand break upstream of the 5' side of the PAM sequence. do.
- the site of the human immunoglobulin heavy chain locus targeted by the guide RNA is, but not limited to, the 5' upstream site of the human D1-1 fragment, and the target sequence of the guide RNA is, for example, the 5' -AGATCCTCCATGCGTGCTGTGGG-3' (SEQ ID NO: 4).
- Site-specific recombination enzymes in this specification are enzymes that specifically recombine with the target DNA sequence at the recognition sites of these enzymes. Site-specific recombination can be performed by using such a recombination enzyme and its recognition site.
- site-specific recombinases are Cre integrase (also called Cre recombinase), Flp recombinase, ⁇ C31 integrase, R4 integrase, TP901-1 integrase, Bxb1 integrase, and the like.
- the recognition sites of the enzyme include, for example, loxP (Cre recombinase recognition site), FRT (Flp recombinase recognition site), ⁇ C31attB and ⁇ C31attP ( ⁇ C31 recombinase recognition site), R4attB and R4attP (R4 recombinase recognition site), TP901-1attB and TP901-1attP (TP901-1 recombinase recognition site), or Bxb1attB and Bxb1attP (Bxb1 recombinase recognition site).
- acceptor site for introducing a synthetic D region polynucleotide is inserted into the site from which the D region of the human immunoglobulin heavy chain locus has been deleted.
- acceptor sites are cassettes containing recognition sites for site-specific recombination enzymes.
- An example of the method for inserting the acceptor site is described in ⁇ Third Step> below.
- the site-specific recombination enzymes and their recognition sites are, for example, the above Cre recombinase and loxP system, Flp recombinase and FRT system, ⁇ C31 recombinase and ⁇ C31attB and ⁇ C31attP systems, and the like.
- a site-specific recombinase eg, Cre
- Cre can catalyze site-specific recombination reactions between recognition site sequences (eg, between loxPs).
- ⁇ Third step> A synthetic D region polynucleotide is inserted into the acceptor site by a method such as site-specific recombination technology or genome editing technology.
- the synthetic D region polynucleotide is a human-derived genomic sequence from D1-1 to D1-26 of the D region of the human immunoglobulin heavy chain locus, as described in Section 1 above, which includes (1) and ( 2), or a modified D region substituted with a modified sequence of the D region consisting of a combination of (1) and (3), wherein the modified sequence is (1) human-derived genomic sequence between the open reading frames (ORF) from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, or the ORF except between D3-9 and D3-10
- the human-derived genomic sequence between comprises a sequence truncated to a length of 49 bp or more from each VDJ recombination sequence end of the inter-ORF region flanked by the VDJ recombination sequences, and (2) Instead of the ORF sequence from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, the bovine-derived immunoglobulin heavy chain locus D region from B1-1
- the method of replacing the human immunoglobulin heavy chain D region in the production of the mammalian artificial chromosome vector with its modified sequence comprises the following steps: can include (1′) providing a mammalian artificial chromosome vector containing human immunoglobulin heavy and light chain loci; (2 ') step (1 ') to delete the human immunoglobulin heavy chain D region from the mammalian artificial chromosome vector, (3') a cassette containing a first site-specific recombination enzyme recognition site, a promoter and a second site-specific recombination enzyme recognition site at the deletion site of the D region of the vector obtained in step (2'); insert, (4') modification of the human immunoglobulin heavy chain D region using a second site-specific recombinase at the second site-specific recombinase recognition site of the cassette inserted in step (3') inserting a cassette comprising a
- the modified sequence of the human immunoglobulin heavy chain D region, the first site-specific recombinant enzyme recognition site, the second site-specific recombinant enzyme recognition site, the first site-specific recombinant enzyme, and the second Site-specific recombination enzymes are not limited, but for example those described above can be used.
- the promoter is not limited, but for example, the CAG promoter can be used.
- step (2 ') in order to delete the human immunoglobulin heavy chain D region from the mammalian artificial chromosome vector of step (1 '), for example, the above genome editing, using techniques such as site-specific recombination can be done.
- the polynucleotide and a site-specific recombination enzyme recognition site at its 5′ end A vector containing a containing cassette (for example, the sequence of ⁇ C31-BsdR-oriC-R4) is prepared, and a site-specific recombination reaction is performed between the vector and the Ig-NAC ( ⁇ DH) to generate Ig-NAC ( ⁇ DH ) can be inserted into the human heavy chain D region cleavage site (FIGS. 5, 6, 9 and 10).
- Ig-NAC human immunoglobulin heavy chain V region partial sequence and human immunoglobulin heavy chain J region (and optionally , C region) to create a vector containing a site-specific recombination enzyme recognition site between the partial sequence, genome editing (Cas9 / gRNA) between this vector and Ig-NAC ( ⁇ DH)
- Ig-NAC Insert the site-specific recombinase recognition site into the human heavy chain D region cleavage site of ( ⁇ DH), act another site-specific recombinase, and human heavy chain D region of the Ig-NAC ( ⁇ DH)
- An Ig-NAC is generated that contains a site-specific recombinase recognition site at the cleavage site.
- a vector containing the polynucleotide and a site-specific recombination enzyme recognition site at its 5' end is prepared, and a site-specific recombination enzyme (for example, ⁇ C31 recombinase) is acted on, and another site-specific recombinase (eg, Bxb1 recombinase or R4 recombinase) is acted on to the human heavy chain D region cleavage site of the above Ig-NAC ( ⁇ DH) synthetic D region polynucleotide
- ⁇ DH human heavy chain D region cleavage site of the above Ig-NAC
- the invention still further provides, in another aspect, a mammalian artificial chromosome vector comprising human immunoglobulin heavy and light chain loci, wherein said human immunoglobulin heavy chain loci lack a D region,
- a mammalian artificial chromosome vector comprising a first site-specific recombinase recognition site, a promoter and a second site-specific recombinase recognition site in place of the deleted D region.
- the above vector may further contain a third site-specific recombination enzyme recognition site.
- mammalian artificial chromosome vector is the vector produced in the above step (3') (e.g., Fig. 9).
- the deleted D region is the region from D1-1 to D1-26, or the region from D1-1 to D7-27.
- the first site-specific recombination enzyme recognition site and the second site-specific recombination enzyme recognition site are not limited, but those described above, for example, can be used.
- the promoter is not limited, but for example, the CAG promoter can be used.
- the invention provides a mammalian cell comprising the mammalian artificial chromosome vector described in Section 1 above.
- Mammalian cells By introducing a human immunoglobulin locus in which the antibody heavy chain D region has been modified ("modified Ig-NAC" in Section 1) into mammalian-derived cells via the mammalian artificial chromosome vector of the present invention, Mammalian cells can be engineered that are capable of producing human antibodies.
- modified Ig-NAC modified immunoglobulin locus in which the antibody heavy chain D region has been modified
- the mammalian artificial chromosome vector of the present invention can be transferred or introduced into any mammalian cell.
- Techniques therefor include, for example, the micronucleus cell fusion method, lipofection, calcium phosphate method, microinjection, electroporation and the like, and the preferred method is the micronucleus cell fusion method.
- micronucleus cell fusion method cells having the ability to form micronuclei containing the mammalian artificial chromosome vector of the present invention (e.g., mouse A9 cells) are combined with other desired cells by micronucleus fusion. It is a method of transfecting into cells.
- Cells having the ability to form micronuclei are treated with a polyploid-inducing agent (e.g., colcemid, colchicine, etc.) to form micronucleated multinucleated cells, and treated with cytochalasin to form micronuclei. can perform cell fusion with cells of a polyploid-inducing agent (e.g., colcemid, colchicine, etc.) to form micronucleated multinucleated cells, and treated with cytochalasin to form micronuclei. can perform cell fusion with cells of
- the above-mentioned vector-introducible cells are mammalian cells such as rodent cells and human cells. It includes cells, stem cells such as somatic stem cells, somatic cells, fetal cells, adult cells, normal cells, primary cultured cells, passaged cells, established cells, and the like.
- Stem cells include, for example, embryonic stem (ES) cells, embryonic germ (EG) cells, somatic stem cells such as mesenchymal stem cells, induced pluripotent stem (iPS) cells, nuclear transfer cloned embryo-derived embryonic stem ( ntES) cells and the like.
- Preferred cells are e.g. rodents such as mice, rats, hamsters (e.g.
- Chinese hamsters guinea pigs, primates such as humans, monkeys, chimpanzees, ungulates such as cows, pigs, sheep, goats, horses and camels.
- somatic cells stem cells, progenitor cells, and non-human germline cells from mammals such as mammals.
- the vectors are more stably retained in rodent cells or tissues into which the vectors of the present invention have been introduced.
- Non-limiting examples of somatic cells include hepatocytes, enterocytes, kidney cells, splenocytes, lung cells, heart cells, skeletal muscle cells, brain cells, skin cells, bone marrow cells, fibroblasts, and the like.
- ES cells are stem cells with pluripotency and semi-permanent proliferative ability established from the inner cell mass of the blastocyst of a fertilized egg derived from mammals (MJ Evans and MH Kaufman (1981) Nature 292: 154-156; JA Thomson et al. (1999) Science 282: 1145-1147; JA Thomson et al. 7844-7848; JA Thomson et al. (1996) Biol.Reprod.55:254-259; JA Thomson and VS Marshall (1998) Curr. -165).
- Rat ES cells as well as mouse ES cells (MJ Evans and MH Kaufman, Nature 1981; 292(5819): 154-156), can be obtained from rat blastocyst stage embryos or 8-cell stage embryos. It is a cell line with pluripotency and self-renewal ability, which is established from the inner cell mass of the embryo.
- rat blastocysts with lysed zona pellucida are cultured on mouse embryonic fibroblast (MEFF) feeders with medium containing leukemia inhibitory factor (LIF) and 7-10 days later, blastocysts
- LIF leukemia inhibitory factor
- the outgrowth formed from the cells is dispersed, transferred onto a MEF feeder and cultured, and after about 7 days, ES cells emerge.
- rat ES cells see, for example, K.K. Kawaharada et al. , World J Stem Cells 2015;7(7):1054-1063.
- iPS cells are formed by introducing a specific reprogramming factor (DNA or protein) into somatic cells (including somatic stem cells), culturing them in an appropriate medium, and subculturing them to form colonies in about 3 to 5 weeks. can be generated.
- Reprogramming factors are, for example, the combination consisting of Oct3/4, Sox2, Klf4 and c-Myc; the combination consisting of Oct3/4, Sox2 and Klf4; the combination consisting of Oct4, Sox2, Nanog and Lin28; , Klf4, c-Myc, Nanog and Lin28 are known (K. Takahashi and S. Yamanaka, Cell 126: 663-676 (2006); WO2007/069666; M.
- mitomycin C-treated mouse fetal fibroblast cell lines eg, STO
- vector-introduced somatic cells eg, about 10 4 to 10 5 cells
- Feeder cells are not necessarily required (Takahashi, K. et al., Cell 131:861-872 (2007)).
- the basal medium is, for example, Dulbecco's Modified Eagle Medium (DMEM), Ham's F-12 medium, mixed medium thereof, or the like, and the ES cell medium is mouse ES cell medium, primate ES cell medium (Reprocell), or the like. can be used.
- Non-human animal in the third aspect of the present invention, a non-human animal comprising human immunoglobulin heavy chain and light chain loci, the D region of the human immunoglobulin heavy chain locus from D1-1 to D1- Up to 26 human-derived genomic sequences are replaced with a modified sequence of the D region consisting of a combination of (1) and (2) or (1) and (3) below, and the modified sequence of the D region but, (1) human-derived genomic sequence between D1-1 to D1-26 open reading frames (ORF) of the human immunoglobulin heavy chain locus D region, or the ORF except between D3-9 and D3-10
- the human-derived genomic sequence between comprises a sequence truncated to a length of 49 bp or more from each VDJ recombination sequence end of the inter-ORF region flanked by the VDJ recombination sequences, and (2) Instead of the ORF sequence from D1-1 to D1-26 of the human immunoglobulin heavy chain locus D region, the bovine-derived immunoglobulin heavy
- the invention comprises a mammalian artificial chromosome vector as described in Section 1 above, and wherein the endogenous immunoglobulin heavy chain, kappa light chain and lambda light chain genes or loci are disrupted.
- non-human animals refers to animals other than humans, including primates such as monkeys and chimpanzees, rodents such as mice, rats, hamsters and guinea pigs, cattle, pigs, sheep, goats, horses and camels. including, but not limited to, mammals such as ungulates such as hoofed animals.
- Non-human animals For example, by techniques such as mammalian artificial chromosome vector-mediated technology, gene targeting technology, genome editing technology, and / or microinjection technology, pluripotent cells such as the above ES cells and iPS cells, or germ cells, eggs Human immunoglobulins can be produced by introducing a human immunoglobulin locus (“modified Ig-NAC”) in which the human immunoglobulin heavy chain D region is modified into totipotent cells such as mother cells and spermatogonia. Non-human animals can be produced that do.
- modified Ig-NAC human immunoglobulin locus
- Non-human animals can be produced that do.
- the endogenous loci corresponding to the human immunoglobulin heavy and light chain kappa, lambda loci are disrupted (or knocked out) and, if desired, the non-human animal
- the human immunoglobulin light chain ⁇ locus from human chromosome 22 can be inserted (or knocked in). Techniques such as gene targeting, genome editing, etc. can be used for the method of disruption.
- a targeting vector is constructed to disrupt the endogenous locus, or to replace/insert (or knock-in) the exogenous DNA in place of the endogenous locus.
- a vector for disrupting an endogenous locus and inserting foreign DNA comprises a 5′ upstream sequence (about 2.5 kb or more) and a 3′ downstream sequence (about 2.5 kb or more) of the endogenous locus, Between these sequences can be included, for example, an exogenous drug resistance gene sequence, or an exogenous DNA sequence as described above to replace the endogenous locus to be disrupted.
- Drug resistance genes include, for example, neomycin resistance genes and ampicillin resistance genes.
- the CRISPR / Cas9 system uses Cas9 protein and a guide RNA (gRNA) having a target sequence of about 20 base pairs to co-express them in cells, so that gRNA is near the target sequence and binds specifically to target genomic DNA, Cas9 protein can induce double-strand breaks 5′ upstream of the PAM sequence.
- gRNA guide RNA
- the antibody heavy chain and light chain loci of non-human animals can be cleaved and removed, or the human immunoglobulin heavy chain D region derived from human chromosome 14 can be added to the cleaved site.
- a vector containing foreign DNA corresponding to a modified human immunoglobulin heavy chain locus, a human immunoglobulin light chain ⁇ locus derived from human chromosome 2, and a human immunoglobulin light chain ⁇ locus derived from human chromosome 22 can be used to insert (or knock-in) the foreign DNA.
- a vector containing the foreign DNA of interest is introduced into ES cells or iPS cells derived from a non-human animal to disrupt the endogenous locus derived from the animal, and the DNA sequence of interest is inserted into the genome. be able to.
- the insertion of the foreign DNA of interest can be confirmed by identifying the amplified product by PCR using forward and reverse primers prepared based on the inserted sequence.
- a chimeric non-human animal can be produced by microinjecting ES cells or iPS cells into which the foreign DNA sequence has been inserted into an early embryo of a foster parent non-human animal.
- the non-human animal in which the endogenous gene locus has been disrupted is, for example, a chimeric non-human animal containing a human immunoglobulin locus in which the human immunoglobulin heavy chain D region has been modified or its progeny animal, and a cluster of the corresponding endogenous gene. It can be produced by mating animals in which the endogenous gene is heterozygously deleted, which are obtained by mating chimeric animals or progeny animals in which the entire gene is deleted, and further mating the animals.
- non-human animals of the present invention include a human immunoglobulin heavy chain locus with a modified human immunoglobulin heavy chain D region derived from human chromosome 14 and a human immunoglobulin light chain ⁇ locus derived from human chromosome 2.
- a non-human animal comprising a chromosome-derived human immunoglobulin light chain ⁇ locus and a human immunoglobulin light chain ⁇ locus derived from human chromosome 22, or a non-human animal comprising the artificial chromosome vector.
- Non-human animals are preferably rodents such as mice and rats containing the mouse artificial chromosome vector.
- Animal cells e.g., chicken B cell line DT40
- a human immunoglobulin light chain kappa locus from human chromosome 2 modified by introduction of site-specific recombinase recognition sites (e.g., loxP or FRT), and sites
- site-specific recombinase recognition sites e.g., loxP or FRT
- Each animal cell e.g., DT40
- a human immunoglobulin light chain ⁇ locus derived from human chromosome 22 modified by introducing a specific recombination enzyme recognition site e.g., loxP or FRT
- MAC mouse artificial chromosome
- CHO Chinese hamster ovary cells
- a site-specific recombination enzyme e.g., Cre or Flp
- human Rodent cells containing a MAC vector containing a kappa immunoglobulin light chain locus as well as rodent cells containing a MAC vector containing a human immunoglobulin light chain lambda locus can be generated.
- human immunoglobulin Animal cells containing a human immunoglobulin heavy chain locus in which the heavy chain D region is modified are fused with rodent cells (e.g., CHO) containing a MAC vector by a cell fusion method to generate a human immunoglobulin heavy chain D region.
- rodent cells e.g., CHO
- Rodent cells can be generated that contain MAC vectors containing modified human immunoglobulin heavy chain loci ("modified Ig-NAC" above).
- a rodent cell containing a MAC vector comprising a human immunoglobulin heavy chain locus in which the human immunoglobulin heavy chain D region is modified and a human immunoglobulin light chain ⁇ locus, and the human immunoglobulin heavy chain D region
- Each of the rodent cells containing the MAC vector containing the modified human immunoglobulin heavy chain locus and the human immunoglobulin light chain ⁇ gene or locus derived from human chromosome 22, by the micronuclear cell fusion method A human immunoglobulin heavy chain locus in which the human immunoglobulin heavy chain D region derived from human chromosome 14 is fused with a human animal (e.g., mouse or rat) pluripotent stem cell (e.g., ES cell or iPS cell) and a non-human animal (e.g., mouse or rat) pluripotent stem cell containing a MAC vector containing a human immunoglobulin light chain kappa locus from human chromosome 2, and
- Generating a non-human animal (e.g., mouse or rat) pluripotent stem cell containing a MAC vector containing a human immunoglobulin heavy chain locus and a human immunoglobulin light chain ⁇ locus derived from human chromosome 22. can be done.
- Non-human animal comprising a MAC vector comprising a human immunoglobulin heavy chain locus and a human immunoglobulin light chain ⁇ locus derived from human chromosome 2, wherein the human immunoglobulin heavy chain D region derived from human chromosome 14 above is modified (e.g., mouse or rat) pluripotent stem cells, and a human immunoglobulin heavy chain locus with an altered human immunoglobulin heavy chain D region from human chromosome 14 and a human immunoglobulin light chain ⁇ from human chromosome 22
- Each non-human animal (e.g., mouse or rat) pluripotent stem cell containing a MAC vector containing the gene locus is transformed into an early embryo (e.g., 8-cell stage embryo or blastocyst stage) of a non-human animal that is B cell-deficient or non-deficient.
- the chimeric animal When the chimeric animal retains endogenous B cells, the chimeric animal is further an allogeneic non-human animal lacking mouse antibody heavy chain, light chain ⁇ and light chain ⁇ loci (or lacking B cells). (eg mice or rats).
- the human immunoglobulin heavy chain locus in which the human immunoglobulin heavy chain D region derived from human chromosome 14 is modified and the human immunoglobulin light chain ⁇ derived from human chromosome 2 A MAC vector containing the locus, or a human immunoglobulin heavy chain locus with a modified human immunoglobulin heavy chain D region derived from human chromosome 14 and a human immunoglobulin light chain ⁇ locus derived from human chromosome 22.
- Non-human animals can be generated that contain each of the above MAC vectors.
- Non-human animals eg, mice or rats
- mice or rats can be generated that contain MAC vectors containing the light chain ⁇ locus.
- all endogenous antibody genes or loci corresponding to human immunoglobulin heavy chain genes or loci and human immunoglobulin light chain ⁇ and ⁇ genes or loci are disrupted. or preferably knocked out, thereby allowing only human antibodies to be produced in said non-human animal.
- the present invention further provides for producing human antibodies using the non-human animals described in Section 3 comprising a mammalian artificial chromosome vector comprising human immunoglobulin heavy and light chain loci, and producing the human antibodies.
- a method for producing a human antibody is provided, comprising recovering.
- a "human antibody” as used herein may be any class or subclass of human immunoglobulin (Ig).
- Such classes include IgG, IgA, IgM, IgD and IgE, and subclasses include IgG1, IgG2, IgG3, IgG4, IgA1 and IgA2.
- IgG chains are called ⁇ chains, ⁇ 1, ⁇ 2, ⁇ 3 and ⁇ 4 chains corresponding to IgG1-IgG4, IgA, IgM, IgD and IgE are referred to as ⁇ -chains ( ⁇ 1 and ⁇ 2), ⁇ -chains, ⁇ -chains and ⁇ -chains respectively.
- All antibody light chains have ⁇ and ⁇ chains, and it is known that ⁇ chain gene rearrangement occurs when ⁇ chain gene rearrangement is unsuccessful in the process of immunoglobulin gene rearrangement.
- the human immunoglobulin heavy chain locus is VH1, VH2 . .
- V variable region genes including VHm (where m is, for example, 38 to 46), DH1, DH2, . . D (diversity) region genes including DHn (where n is 23), JH1, JH2. .
- the D region gene of the human immunoglobulin heavy chain locus is a synthetic D region described as a modified sequence consisting of a combination of (1) and (2) or (1) and (3) in Section 1 above Including substitutions by nucleotide sequence.
- the antibody produced through the rearrangement of the human immunoglobulin gene is difficult to consistently produce with conventional human antibodies, such as 18 to 50 amino acids or longer Human antibodies can be produced that contain ultralong CDRH3. Human antibodies containing such ultra-long-chain CDRH3 are difficult to produce by conventional monoclonal antibody-producing methods, since they occur at a low frequency in humans.
- a human antibody molecule consists of two human antibody heavy chains and two human antibody light chains, each heavy chain and each light chain being linked by two disulfide bonds, and two heavy chains (C) It has a structure linked by two disulfide bonds in the region.
- each of the variable (V) regions of the heavy and light chains of an antibody molecule has three regions with particularly large mutations (hypervariable regions), which are called complementarity determining regions (CDRs).
- the hypervariable regions of the heavy chain variable region are called CDRH1, CDRH2 and CDRH3 from the N-terminal side, while the hypervariable regions of the light chain variable region are called CDRL1, CDRL2 and CDRL3 from the N-terminal side.
- the present invention expands the diversity of human antibodies by rearranging the human immunoglobulin genes.
- the human antibody containing ultralong CDRH3 produced by the non-human animal of the present invention has a structure consisting of a long ( ⁇ -ribbon) Stalk and a Knob, and the Knob has a loop and multiple disulfide bonds.
- Antibodies that can be obtained by the method of the present invention are ultralong antibodies with a length of 18 or more, 20 or more, 25 or more, 30 or more, 40 or more, or 50 or more amino acids in the antibody heavy chain CDR3. Since it is a human antibody containing CDRH3, it can be used against pathogenic viruses such as human immunodeficiency virus (HIV), influenza virus, coronaviruses (eg, SARS-CoV, MERS-CoV), RS (Respiratory syncytial) virus, and other pathogens ( For example, it is considered to be effective for diseases including infections caused by malaria parasites, trypanosoma parasites).
- the human antibodies are therapeutically useful because they allow a "hook" type interaction that binds to the receptor cavity of the virus or pathogen at the protruding CDRH3 region.
- the antibodies of the present invention can be made into cocktails of multiple types of antibodies, and can be used not only for treatment but also for enhancing preventive effects.
- Specific antibody production methods include the following methods.
- the human antibody-producing non-human animal (eg, mouse or rat) of the present invention can be immunized with a target antigen such as an HIV envelope protein to produce a polyclonal antibody that specifically binds to the target antigen.
- a target antigen such as an HIV envelope protein
- the above animal is immunized with a target antigen
- the B lymphocytes of the animal individual with an increased antigen-specific antibody titer are used to fuse with myeloma cells to produce hybridomas (immortalized B cells), and the human in the hybridoma supernatant is Broadly neutralizing human monoclonal antibodies can be selected by screening monoclonal antibodies by ELISA analysis for evaluation of antigen-binding ability and, if necessary, evaluation of infection with HIV or the like.
- the present invention provides, in a fourth aspect, the production of an antibody, comprising immunizing the non-human animal according to Section 3 above with a target antigen and obtaining an antibody that binds to the target antigen from the blood of the non-human animal.
- a fabrication method is provided.
- the invention further comprises immunizing a non-human animal according to Section 3 above with a target antigen, and extracting spleen cells or lymph node cells from said non-human animal producing antibodies that bind to said target antigen. obtaining, fusing the spleen cells or lymph node cells with myeloma cells to form hybridomas, culturing the hybridomas, and obtaining monoclonal antibodies that bind to the target antigen. do.
- the invention further comprises immunizing a non-human animal according to Section 3 above with a target antigen and obtaining B cells from said non-human animal producing antibodies that bind to said target antigen;
- a nucleic acid encoding a protein consisting of an antibody heavy chain and light chain, or a variable region thereof, is obtained from the B cell, and an antibody that binds to the target antigen by DNA recombination technology or phage display technology using the nucleic acid (e.g., , recombinant antibodies, monoclonal antibodies, single chain antibodies (scFv), etc.).
- the above nucleic acids include DNA (including cDNA) or RNA (including mRNA).
- the above scFv is a recombinant antibody fragment produced by binding an antibody heavy chain variable region polypeptide and an antibody light chain variable region polypeptide via a linker.
- variable regions of human antibodies are expressed on the surface of phages as, for example, single-chain antibodies (scFv) or Fabs, and phages that bind to antigens are selected (Nature Biotechnology, 2005; 23(9): 1105- 1116).
- scFv single-chain antibodies
- Fabs Fabs
- phages that bind to antigens are selected (Nature Biotechnology, 2005; 23(9): 1105- 1116).
- a human antibody can be obtained by constructing an expression vector having the sequence and introducing it into an appropriate host for expression (WO92/01047, WO92/ 20791, WO93/06213, WO93/11236, WO93/19172, WO95/01438, WO95/15388, Annu.Rev.Immunol, 1994;12:433-455, Nature Biotechnology, 2005; 05-1116) .
- Antibodies prepared by the above method are prepared by filling blood (antiserum) or cell supernatant containing the antibody with a carrier bound to an antigenic substance or an immunoglobulin G-binding carrier (e.g., agarose gel, silica gel, etc.). It can be applied to a column and then recovered and purified by a technique such as affinity chromatography, which involves eluting the human antibody bound to the carrier from the carrier.
- a carrier bound to an antigenic substance or an immunoglobulin G-binding carrier e.g., agarose gel, silica gel, etc.
- Ig-NAC ( ⁇ DH) construction with deleted human IgHD region In order to construct Ig-NAC ( ⁇ DH) with deleted human IgHD region, CRISPR / Cas9 gRNA was designed, evaluated, selected, DH region removal was performed by two-site cleavage in CHO cells retaining Ig-NAC, and Ig-NAC ( ⁇ DH) retaining CHO cells were obtained by screening.
- Plasmids expressing candidate gRNAs designed based on Cas9 and gRNA-expressing all-in-one vector eSpCas9 (1.1) were constructed.
- CHO cells bearing Ig-NAC were transfected with the all-in-one vector with Lipofectamine LTX (Invitrogen) according to the manufacturer's protocol.
- DNA was extracted from the transfected cells using the PureGene kit (Qiagen), the cleavage activity was confirmed with the Cel-1 assay kit (IDT), and vectors with the following gRNA sequences exhibiting high activity were selected.
- gRNA Up3 5'-AGATCCTCCATGCGTGCTGT (GGG)-3' (SEQ ID NO: 4) (Here, the gRNA sequence is a sequence of 20 bases excluding the PAM sequence (GGG).)
- gRNA Down4 5'-CTGCGGCATGAACCCAATGC(AGG)-3' (SEQ ID NO: 5) (Here, the gRNA sequence is a sequence of 20 bases excluding the PAM sequence (AGG).)
- the DH region was deleted in each combination of gRNA Up3 (“5′guide3” in the figure) and Down4 (“5′guide4” in the figure) (FIG. 1).
- the all-in-one vector for each gRNA and the CMV-tdTomato expression vector were introduced into Ig-NAC-retaining CHO cells (Patent No. 6868250) using Lipofectamine LTX (Invitrogen) according to the manufacturer's protocol. Cells were obtained by sorting.
- the EGFP signal indicates retention of Ig-NAC, and the tdTomato signal indicates lipofection.
- the co-positive cells were further cultured, and EGFP-positive and tdTomato-negative cells were collected in a 384-well plate by single-cell sorting 8 days after the first sorting.
- the grown clones were further scaled up and screened as described below.
- junction primer F1 5'-TCCCACGGCCCAAGGAAGACAAGACACA-3' (SEQ ID NO: 6)
- Junction primer R1 5'-TCGAACACGCTCCTAGCATTGCACAGCC-3' (SEQ ID NO: 7)
- Deletion confirmation primer F1 5'-ACACCTGTCTCCCGGGTTGTG-3' (SEQ ID NO: 8)
- Deletion confirmation primer R1 5'-AAGAAACGCAGGACGGTGGA-3' (SEQ ID NO: 9)
- Defect confirmation primer F2 5'-CAGCAGCCACTCTGATCCCA-3' (SEQ ID NO: 10)
- Deletion confirmation primer R2 5'-CTGGTGGACTCTACGGCGAA-3' (SEQ ID NO: 11)
- Deletion confirmation primer F3 5'-
- KOD FX (TOYOBO) was used as the enzyme, and the reaction conditions were 35 cycles of 98°C for 15 seconds, 60°C for 30 seconds, and 68°C for 90 seconds. Out of 381 clones obtained from all combinations, removal of the D region was confirmed in 35 clones. After that, PCR for confirming the presence of antibody gene regions other than DH on Ig-NAC was performed by the method described in Japanese Patent No. 6868250) to select candidate Ig-NAC ( ⁇ DH) clones.
- Ig-NAC ( ⁇ DH) is maintained independently without being integrated into the host chromosome, in order to confirm that it maintains the expected structure, in Japanese Patent No. 6868250
- FISH analysis FISH analysis by the described method
- CHO cells in which one copy of Ig-NAC ( ⁇ DH) was retained independently of the host chromosome were obtained (FIG. 2), and the cell clone was TF7-B10. named.
- Example 2 Insertion of acceptor site for introducing synthetic D region into Ig-NAC ( ⁇ DH) Genome editing and homologous recombination were performed on the DH region deleted in Ig-NAC ( ⁇ DH) prepared in Example 1. This allows introduction of a chemically synthesized DH region by inserting an acceptor site. Therefore, the sequences before and after the DH region deletion site of Ig-NAC ( ⁇ DH) (V region side: HRA, J fragment side: HRB) and Bxb1 attB, Bxb1 attP, ⁇ C31 attB, R4 attB (Patent No.
- Bxb1 attB (SEQ ID NO: 18) TGGCCGTGGCCGTGCTCGTCCTCGTCGGCCGGCTTGTCGACGACGGCGGTCTCCGTCGTCAGGATCATCCGGGGCCAC
- Bxb1 attP (SEQ ID NO: 19) TATGGCCGTGATGACCTGTGTCTTCGTGGTTTGTCTGGTCAACCACCGCGGTCTCAGTGGTGTACGGTACAAACCCA
- ⁇ C31 attB (SEQ ID NO: 20) GATGTAGGTCACGGTCTCGAAGCCGCGGTGCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTACTCCACCTCACCCATCTGGTCCATCATGATGAACGGGTCGAGGTGGCGGTAGTTGATCCCGGCGAACGCGCGCGCGGCGCACCGGGAAGCCCTCGCCCTCGAAACCGCTGGGCGCGGTGGTCACGGTGAGCACGGGACGTGCGACGGCGTCGGCGGGTGCGGATACGGGGCAGCGTCAGCGGGTT CTCGACGGTCACGGCGGGCAT
- R4 attB (SEQ ID NO:21) GCGCCCAAGTTGCCCATGACCATGCCGAAGCAGTGGTAGAAGGGCACCGGCAGACAC
- Fcy fur gene into CAG promoter-carrying vector
- Insertion of HDR vector into Ig-NAC ( ⁇ DH)-carrying CHO cells selectively kills cells in which insertion by random integration other than the desired homologous recombination has occurred
- a negative selectable marker, the Fcy::fur gene was inserted to allow The Fcy::fur gene was obtained by PCR amplification from pSelect-zeo-Fcy::fur plasmid (Invivogen) DNA using the following primers and conditions.
- Fcy-fur-F1 5'-CATGAATCTAGAAGTGCAGGTGCCAGAACATT-3' (SEQ ID NO: 22)
- fur-BspH1-R1 5'-CATGAATCATGACCCCCTGAACCTGAAACATA-3' (SEQ ID NO: 23)
- Taq DNA polymerase uses KODone (TOYOBO), reaction conditions: 98° C., 10 seconds; 60° C., 5 seconds; 35 cycles of PCR were performed at °C for 13 seconds.
- the DNA fragment of the Fcy::fur gene was purified by agarose gel electrophoresis, and both ends were cleaved with restriction enzymes BspHI (NEB) and XbaI (NEB). Plasmid DNA containing CAG promoter pRP[Exp]-CAG>mCherry (synthesized by Vector Builder), inserting the above Fcy::fur gene into restriction enzymes AfiIII (NEB) and XbaI (NEB) cleavage sites to create a plasmid CAG-Fcy::fur was obtained (Fig. 3).
- Plasmid CAG-Fcy::fur was cleaved with restriction enzyme XbaI, and a pHRB-derived fragment cleaved at both ends with XbaI was inserted between the CAG promoter and Fcy::fur gene to obtain plasmid CAG- ⁇ C31-HRB-Fcy::fur. was obtained (Fig. 3).
- HDR vector plasmid 2 Preparation of HDR vector plasmid 2 above. Plasmid DNA for introducing HRA and site-specific recombination enzyme site R4 attB, Bxb1 attB, Bxb1 attP, blasticidin resistance gene into plasmid CAG- ⁇ C31-HRB-Fcy::fur prepared in , pHRA is Eurofin Synthesized by Genomics. This plasmid was cleaved with restriction enzyme NotI and restriction enzyme SpeI.
- CRISPR/Cas9 allows efficient homologous recombination by causing two DNA double strand breaks at the target site.
- Guide RNAs, gRNA-A1 and gRNA-B1 were designed at two locations (targets) near the DH region deletion site on Ig-NAC ( ⁇ DH).
- gRNA-A1 GGGCCCAGGCGCCGTTTAAT AGG (SEQ ID NO: 24) (Here, the gRNA sequence is a sequence of 20 bases excluding the PAM sequence (AGG).)
- gRNA-B1 TGCCGCAGAGTGCCAGGTGCAGG (SEQ ID NO: 25) (Here, the gRNA sequence is a sequence of 20 bases excluding the PAM sequence (AGG).)
- NGG recognized by Cas9 was removed, and oligo DNAs added with a restriction enzyme BbsI recognition sequence were synthesized (Eurofins Genomics). Further, after annealing these oligo DNAs, the plasmid DNA cleaved with the restriction enzyme BbsI (NEB) was cloned into the Cas9 expression vector (px330, addgene Plasmid #42230) cleaved with the restriction enzyme BbsI. The presence or absence of insertion of oligo DNA was confirmed by performing PCR on the resulting E. coli colonies using the following primers and conditions. U6F was used as the forward primer.
- BbsI restriction enzyme
- gRNA-A1R was used to confirm gRNA-A1
- gRNA-B1R was used to confirm gRNA-B1.
- U6F GAGGGCCTATTTCCCATGATTCC (SEQ ID NO: 26)
- gRNA-A1F CACCGGGGCCCAGGGCGCCGTTTAAT (SEQ ID NO: 27)
- gRNA-A1R AAACATTAAACGGCGCCTGGGCCCC (SEQ ID NO: 28)
- gRNA-B1F CACCGTGCCGCAGAGTGCCAGGGTGC (SEQ ID NO: 29)
- gRNA-B1R AAACGCACCTGGCACTCTGCGGCAC (SEQ ID NO: 30)
- Taq DNA polymerase uses (EmeraldAmp PCR Master Mix, Takara Bio), reaction conditions: 98°C, 2 minutes, once ⁇ 98°C, 10 seconds; 60°C, seconds; 68°C, 13 seconds, 35 cycles of PCR did
- the target band was obtained around 330 bp by 2% agarose gel electrophoresis. Colonies were cultured in liquid LB medium and ampicillin 100 ⁇ L/mL, and plasmid DNA (Nucleospin Plasmid Transfection-grade, Takara Bio) was used. Cas9-gRNA expression vector A1, B1) was obtained.
- HDR Vector Insertion into Ig-NAC( ⁇ DH) By genome editing using Cas9/gRNA, the HDR vector was inserted near the DH region deletion site of Ig-NAC( ⁇ DH) in CHO cells (FIG. 5).
- the HDR vector and the Cas9-gRNA expression vector A1, B1 were introduced by electroporation using the CHO cell clone TF7B10 carrying the Ig-NAC ( ⁇ DH) prepared in Example 1.
- CHO cells TF7B10 were trypsinized, suspended in OptiMEM (Thermo Fisher Scientific) to 1 ⁇ 10 7 cells/mL, and then 5 ⁇ g of HDR vector, 2.5 ⁇ g of gRNA-A1, 2.5 ⁇ g of gRNA- Electroporation was performed at 150 V for 7.5 ms using NEPA21 (Nepagene) in the presence of B1.
- Cells were seeded in three 96-well plates after electroporation, and after 48 to 72 hours, 800 ⁇ g / mL G418 (Fuji Film Wako Pure Chemical) and 1600 ⁇ g / mL blasticidin (Fuji Film Wako Pure Chemical), 10% FBS ( SIGMA), cultured in F12 medium containing 1600 ⁇ g / mL blasticidin, 200 ⁇ M 5FC (Tokyo Kasei Kogyo), 10% FBS after 84 to 96 hours from the start of culture, and cultured by replacing the medium with F12 medium containing drug resistant clones. obtained.
- PCR analysis Cultivate the isolated drug-resistant clones, extract genomic DNA from the cells using Cellysis (Qiagen), use this genomic DNA as a template, three primers specific for homologous recombination (below), and Ig-NAC One strain (TF7B10-74) was selected from which a band specific for homologous recombination was obtained by PCR analysis using 12 types of Ig-NAC-specific primers prepared above (Japanese Patent No. 6868250). Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification.
- Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C. for 2 minutes, denatures at 98 ° C.
- HRAprimerF1 5'-GAACTGACCGTCTGCTCCA-3' (SEQ ID NO: 31)
- Bxb1attBF1 5'-TGCCGAAGCAGTGGTAGAAG-3' (SEQ ID NO: 32)
- ⁇ C31F1 5′-GGGCAACGTGCTGGTTATTG-3′
- HRAprimerR1 5'-GTGCCCTTCTACCACTGCTTC-3'
- Bxb1attPR1 5'-AGAGTGAAGCAGAACGTGGG-3'
- HRBR1 5'-TGCTTTTTCGCAACCATGGG-3' (SEQ ID NO: 36)
- the horizontal column indicates the clone name
- the vertical column indicates the primers and elongation time used in PCR. ⁇ indicates positive.
- Example 3 Removal of blasticidin-resistant gene by site-specific recombination using Bxb1 recombinase
- Bxb1 recombinase In order to determine the presence or absence of cydin resistance, it is necessary to remove the blasticidin resistance gene inserted by homologous recombination.
- the blasticidin resistance gene As shown in the HDR vector, the blasticidin resistance gene is located between Bxb1 attB and Bxb1 attP, and introduction of Bxb1 recombinase into the cell causes site-specific recombination between Bxb1 attB-attP, Ablation of the blasticidin resistance gene is possible (Fig. 6).
- Bxb1 recombinase gene transfer to remove blasticidin-resistant gene The Bxb1 recombinase gene expression vector (Japanese Patent No. 6868250) is obtained in Example 2, and the acceptor site-inserted Ig-NAC ( ⁇ DH) is retained. It was introduced into CHO cells TF7B10-74, which are living in the same region, by electroporation. TF7B10-74 cells were trypsinized, suspended in Opti MEM to 2 ⁇ 10 6 cells/mL, added with 10 ⁇ g of Bxb1 recombinase gene expression vector, and incubated with NEPA21 (Neppagene) at 150 V, 7.5 ms. electroporation was performed.
- NEPA21 Neppagene
- Cells were seeded in one 10 cm dish after electroporation. After 5 days, the cells were trypsinized and seeded at 150 cells per 384-well plate. After seeding on a 384-well plate, the cells were cultured in F12 medium containing 800 ⁇ g/mL G418 and 10% FBS for 9 days, and single clones were picked up.
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, two specific primers after Bxb1 site-specific recombination, and on Ig-NAC PCR analysis was performed using 12 kinds of Ig-NAC-specific primers (Japanese Patent No. 6868250) prepared in 1998. Among the two primers specific for Bxb1 site-specific recombination, primerHRAF1-Bxb1attPR2 shifts the band from around 2800 bp to around 1500 bp after site-specific recombination. Two strains (clone D1, J23) were selected that gave specific bands after Bxb1 site-specific recombination.
- Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C. for 2 minutes, denatures at 98 ° C. for 10 seconds, anneals at 60 ° C. for 5 seconds, extends at 68 ° C., 5 seconds if the length of the amplified fragment is 500 bp or less , 35 cycles of 100 sec/kb for ⁇ 500 bp.
- HRAprimerF1 5'-GAACTGACCGTCTGCTCCA-3' (SEQ ID NO: 31)
- Bxb1attPR2 5'-GGGGCGTACTTGGCATATGA-3' (SEQ ID NO: 37)
- Fluorescence in situ hybridization (FISH) FISH analysis was performed according to the method described in (Japanese Patent No. 6868250) using a human gene-specific probe human cot1 (invitrogen) and a mouse gene-specific probe mouse cot1 (invitrogen).
- Table 2 shows the results of the above PCR analysis, blasticidin resistance confirmation, and FISH analysis.
- the rows indicate the clone names, and the columns indicate the primers and extension times used in PCR.
- ⁇ indicates positive for Bxb1 recombination, and x indicates negative for Bxb1 recombination.
- Example 4 Introduction of Ig-NAC ( ⁇ DH/Bsr-) into CHO cell line HPRT(-) by micronucleation method (MMCT) Ig whose blasticidin resistance gene removal by Bxb1 recombinase was confirmed in Example 3
- the TF7B10-74-D1 clone carrying -NAC ( ⁇ DH/Bsr-) was found to be tetraploid while independently carrying Ig-NAC by FISH analysis (Table 2). Since this cell is important as a platform cell for introducing chemically synthesized DNA, Ig-NAC ( ⁇ DH/Bsr-) was transfected into the normal diploid CHO cell line HPRT(-) by MMCT.
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, PCR was performed using the same primers as in the PCR analysis performed in Example 2. . The band of interest could be confirmed in 14 of the 24 cell clones picked up.
- FISH analysis 6 cells were arbitrarily selected from 14 cell clones in which the band of interest was confirmed by PCR analysis, and FISH analysis was performed. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250). Table 3 shows the results of PCR analysis and FISH analysis for the 6 cell clones subjected to FISH analysis. In Table 3, the rows indicate the clone names, and the columns indicate the primers and extension times used in PCR. ⁇ indicates positive for Bxb1 recombination. In the table, Ig-NACx1 indicates that 1 copy of Ig-NAC is detected per cell, and Ig-NACx2 indicates that 2 copies of Ig-NAC are detected per cell.
- Example 5 Insertion of chemically synthesized DNA containing human minimalized IgHD by site-specific recombination using ⁇ C31 recombinase
- the human IgHD region is as large as approximately 40.2 kb, most of which is composed of fragments other than the D fragment encoding the antibody. is an array. For example, there is about 2500 bp of non-antibody coding region between the first and second D fragments. On the other hand, the length between the 9th D fragment and the 10th D fragment is as short as about 100 bp.
- plasmid DNA human miniA, human miniB, and human miniC were chemically synthesized by dividing the vector sequence into three (Eurofins Genomics).
- human miniA is R4 attP and downstream from HRA to D fragment 3-10
- human miniB is D fragment 4-11 to 3-22
- human miniC is D fragment 4-23 to HRB and ⁇ C31 attP , which has a blasticidin-resistant gene.
- the human minimalized IgHD vector carrying this human miniA, B, and C is described in 1 below. and 2. (FIG. 8).
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, ⁇ C31 site-specific recombination followed by one specific primer and human minimalized IgHD PCR analysis was performed using 2 types of primers prepared inside and 12 types of Ig-NAC-specific primers prepared on Ig-NAC. Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification.
- Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C. for 2 minutes, denatures at 98 ° C. for 10 seconds, anneals at 60 ° C.
- BsdR1 5'-ATGCAGATCGAGAAGCACCT-3' (SEQ ID NO: 38) 2-8to3-9F: 5'-AACGCACCAGACCAGCAGAC-3' (SEQ ID NO: 39) 4-11to5-12R: 5'-GACGTGGGCCTAGAGGCT-3' (SEQ ID NO: 40) 3-22to4-23F: 5'-CAGGCGGGGAAGATTCAGAA-3' (SEQ ID NO: 41) 4-23to5-24R: 5'-TAGAGAGCCTGGGCCTAGAG-3' (SEQ ID NO: 42)
- FISH analysis FISH analysis was performed on 3 clones for which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250). Table 4 shows the results of PCR analysis and FISH analysis for the three cell clones subjected to FISH analysis.
- Ig-NACx1 indicates that 1 copy of Ig-NAC is detected per cell
- Ig-NACx2 indicates that 2 copies of Ig-NAC are detected per cell.
- Example 6 Partial sequence removal by site-specific recombination using R4 recombinase Among the sequences introduced on Ig-NAC ( ⁇ DH), the CAG promoter and the blasticidin resistance gene are not necessary for antibody gene expression. is an array of These sequences are flanked by R4 attB and attP and can be removed by site-specific recombination between R4 attB-attP by introducing R4 recombinase into cells (Fig. 10).
- R4 recombinase gene expression vector introduction R4 recombinase gene expression vector (Patent No. 6868250) is confirmed to be inserted into a human minimalized IgHD vector, and CHO cells 1-10 with a high retention rate of one Ig-NAC It was introduced into pEFI-8 (Example 5) by electroporation. 1-10 pEFI-8 cells were trypsinized, suspended in Opti MEM to 3 ⁇ 10 6 cells/mL, added with 10 ⁇ g of R4 recombinase gene expression vector, and incubated at 150 V, 7 using NEPA21 (Nepagene). Electroporation was performed for 0.5 ms.
- Cells were seeded in one 10 cm dish after pulsing. After 24-48 hours, the cells were trypsinized and seeded at 150 cells per 384-well plate. Two weeks after seeding on a 384-well plate, the cells were cultured in F12 medium containing 800 ⁇ g/mL G418 and 10% FBS, and 24 single clones were picked up.
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, one specific primer after R4 site-specific recombination, and human minimalized IgHD PCR analysis was performed using 2 types of primers prepared inside and 12 types of Ig-NAC-specific primers prepared on Ig-NAC. PCR was performed using primers specific for the R4 site-specific recombination, and 6 strains were selected that gave specific bands after the R4 site-specific recombination. Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification. Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C.
- R4attLF2 5'-CCCTCAAGGTGTGAACAGGG-3' (SEQ ID NO: 43)
- R4attLR2 5'-CTGGAGGGAGGCATGTTCTG-3' (SEQ ID NO: 44)
- FISH analysis was performed on 6 cell clones in which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250). Table 5 shows the results of PCR analysis and FISH analysis for 6 cell clones subjected to FISH analysis.
- the horizontal column indicates the clone name, and the vertical column indicates the primers and extension time used in PCR. ⁇ indicates positive.
- Ig-NACx1 indicates that Ig-NAC is detected at 1 copy per cell.
- Example 7 Introduction of human minimalized IgHD-loaded Ig-NAC by micronucleation method (MMCT) into mouse ES cell line
- MMCT micronucleation method
- mouse ES cells HKD31 6TG- in which both alleles of the endogenous immunoglobulin heavy chain and ⁇ chain loci have been disrupted 9 strain (XO) (Patent No. 4082740) was used.
- the HKD31 6TG-9 strain was cultured using mitomycin C-treated fetal mouse fibroblast cell lines as feeder cells, and 15% FBS and 1% nucleoside, penicillin streptomycin, penicillin streptomycin, and 1% ES cell culture medium were placed on the feeder cell layer.
- the cells were cultured at 37 degrees using Dulbecco's Modified Eagle's Medium (DMEM) containing Non-Essential Amino Acids, L-glutamine solution, 2-mercaptoethanol and LIF.
- DMEM Dulbecco's Modified Eagle's Medium
- 400 nM paclitaxel and 500 nM reversine were added to approximately 4 ⁇ 10 7 1-10 pEF1-K5 and 1-10 pEF1-M24 to prepare micronuclei.
- the total amount of micronuclei obtained was suspended in 5 mL of DMEM and centrifuged to precipitate.
- Mouse ES cells HKD31 6TG-9 strain of 5 ⁇ 10 6 to 4 ⁇ 10 6 were dispersed with trypsin, washed 3 times with DMEM, and added to 5 mL. After being suspended in DMEM, it was added onto the precipitate of micronuclei. The supernatant was removed by centrifugation at 1200 rpm for 5 minutes.
- the precipitate was loosened well by tapping, and 0.5 mL of PEG solution (2.5 g PEG 1000 ⁇ Fuji Film Wako Pure Chemical>, 3 mL DMEM ⁇ Fuji Film Wako Pure Chemical>, 0.5 mL DMSO ⁇ Fuji Film Wako Pure Chemical>) was added. In addition, it was shaken in a water bath at 37°C, and after 90 seconds, 13 mL of DMEM was slowly added, centrifuged at 1200 rpm for 5 minutes, the supernatant was removed, the precipitate was suspended in 30 mL of ES cell medium, and feeder cells were seeded in advance. Three 10 cm dishes were seeded. After 48 hours, the medium was replaced with an ES cell medium supplemented with 350 ⁇ g/mL G418, and then the medium was replaced with the same composition once every two days. 14 drug-resistant colonies that emerged after 1 week to 10 days were picked up.
- PEG solution 2.5 g PEG 1000 ⁇ Fu
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, PCR was performed using the same primers as in the PCR analysis performed in Example 5. . The target band could be confirmed in 5 of the 14 picked up.
- Example 8 Preparation of chimeric mice from mouse ES cell lines containing human minimalized IgHD-containing Ig-NAC Chimeric mice were prepared from human minimalized IgHD-containing Ig-NAC mouse ES cells in order to confirm human antibody production. .
- mice are produced according to the method of Gene Targeting, Experimental Medicine, 1995.
- the host is MCH (ICR) (white, purchased from Clea Japan), or HKLD mice exhibiting antibody gene deficiency (Igh/Ig ⁇ KO) or low expression (Igl1 low).
- Cell stage embryos were used. It is possible to determine whether or not the pups born as a result of transplanting the injected embryos into foster mothers are chimeras based on their coat color. By transplanting the embryos into foster mothers, chimeric mice (having a dark brown coat color) can be obtained.
- a total of 211 ICR embryos were injected with 4 clones of mouse ES cell lines harboring human minimalized Ig-NAC and transplanted into 6 foster mothers. As a result, a total of 5 GFP-positive chimeric mice were obtained. As a result of judging the chimera rate by coat color, the chimera rate was 10 to 40% for 5 chimeric mice.
- Mouse ES cell line 2 clones were injected into 465 HKLD embryos and transplanted into 27 foster mothers, resulting in 14 GFP-positive individuals. As a result of judging the chimera rate by coat color, the chimera rate was 5 to 50% for 14 chimeric mice.
- bovine D fragment 8-2 the position of human D fragment 3-10 was replaced with bovine D fragment 8-2.
- the order of the bovine D fragment was not changed. Since bovine has fewer D fragments than humans, the three human D fragments that were not replaced were deleted along with 100 bp before and after. Thus, bovinized human IgHD was designed as shown in (Fig. 11).
- bovine human IgHD was divided into three parts in the same manner as in Example 5, and the plasmid DNA bovine miniA (HRA) and the bovine D fragments B1-1 to B8 corresponding to the regions of human D fragments 1-1 to 3-10 were obtained.
- bovine miniB including bovine D fragments B6-2 to B5-4 corresponding to the regions of human D fragments 4-11 to 3-22
- bovine miniC human D fragments 4-23 to 1- A bovine D fragment B6-4 to B9-4 corresponding to the region of 26, containing HRB, ⁇ C31 attP, and the blasticidin resistance gene
- the bovinized human minimalized IgHD vector carrying bovine miniA, B, and C was prepared by the following procedure 1. 2. (Fig. 12).
- bovine miniC fragment into R4 attP-HRA/1-1_3-22 Human miniC was cleaved with restriction enzymes EcoRI and SalI.
- a bovinized human IgHD vector was constructed by inserting the fragment human miniC into R4 attP-HRA/1-1 — 3-22 which was similarly cut with restriction enzymes EcoRI and SalI.
- Bovinized Human IgHD Vector by ⁇ C31 Recombinase It was carried out in the same manner as in Example 5. 1-10 cells (Example 4) were trypsinized and suspended in Opti MEM to 3 ⁇ 10 6 cells/mL, then 1 ⁇ g of ⁇ C31 recombinase gene expression vector ⁇ C31pEF1 (Patent No. 6868250) and 3 ⁇ g of A bovine human IgHD vector was added and electroporation was performed at 150 V for 7.5 ms using NEPA21 (Nepagene). Two 384-well plates were seeded after pulsing. After 48 to 72 hours, the cells were cultured by replacing the medium with F12 medium containing 800 ⁇ g/mL G418, 800 ⁇ g/mL blasticidin and 10% FBS. After 13 days, 16 plants resistant to blasticidin were picked up.
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and PCR analysis was performed using this genomic DNA as a template and the same primers as in Example 5.
- FISH analysis was performed on 4 clones arbitrarily selected from 16 cell clones in which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250).
- Table 7 shows the results of PCR analysis and FISH analysis for the four cell clones subjected to FISH analysis.
- the rows indicate the clone names, and the columns indicate the primers and extension times used in PCR. ⁇ indicates positive.
- Ig-NACx1 indicates that 1 copy of Ig-NAC is detected per cell
- Ig-NACx2 indicates that 2 copies of Ig-NAC are detected per cell.
- Example 10 Removal of partial sequence by site-specific recombination using R4 recombinase 1.
- Introduction of R4 Recombinase Gene Expression Vector It was carried out in the same manner as in Example 6.
- 1-10 C4 cells (Example 9) in which introduction of the bovine human IgHD vector was confirmed were trypsinized, suspended in Opti MEM at 3 ⁇ 10 6 cells/mL, and then subjected to 10 ⁇ g of R4 recombinase gene expression. The vector was added and electroporation was performed at 150 V, 7.5 ms using NEPA21 (Nepagene). Cells were seeded in one 10 cm dish after pulsing.
- the cells were trypsinized and seeded at 150 cells per 384-well plate. After seeding in a 384-well plate, the cells were cultured in F12 medium containing 800 ⁇ g/mL G418 and 10% FBS for 17 days, and 22 single clones were picked up.
- PCR analysis The isolated cells are cultured, genomic DNA is extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, one specific primer after R4 site-specific recombination as in Example 6 , and 2 primers made in human minimalized IgHD and 12 Ig-NAC specific primers made on Ig-NAC. PCR was performed using primers specific for the R4 site-specific recombination, and 3 clones were selected which gave specific bands after the R4 site-specific recombination.
- FISH analysis FISH analysis was performed on 3 clones for which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250). Table 8 shows the results of PCR analysis and FISH analysis for the three cell clones subjected to FISH analysis. In Table 8, the rows indicate the clone names, and the columns indicate the primers and extension times used in PCR. ⁇ indicates positive. In the table, Ig-NACx1 indicates that Ig-NAC is detected at 1 copy per cell.
- bovine minimalized IgHD bovine human minimalized IgHD
- MMCT micronucleation method
- Mouse ES cell line HKD31 6TG-9 (XO) in which both alleles of the endogenous immunoglobulin heavy chain and ⁇ chain loci have been disrupted was used as the chromosome recipient cell (Japanese Patent No. 4082740). Micronuclei were formed in the same manner as in Example 7, fused with mouse ES cell HKD31 6TG-9 strain (XO), and the cells were seeded on three 10 cm dishes. After 48 hours, the medium was replaced with an ES cell medium supplemented with 350 ⁇ g/mL G418, and then the medium was replaced with the same composition once every two days. Twenty-three drug-resistant colonies that appeared after 1 week to 10 days were picked up.
- PCR analysis The isolated cells were cultured, genomic DNA was extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, PCR was performed using the same primers as in the PCR analysis performed in Example 6. . The desired band could be confirmed in 14 of the 23 picked up.
- Example 12 Production of chimeric mice from mouse ES cell lines containing Ig-NAC containing minimalized bovine IgHD In order to confirm human antibody production, chimeric mice were produced from mouse ES cells containing Ig-NAC containing minimalized bovine IgHD.
- mice are produced according to the method of Gene Targeting, Experimental Medicine, 1995.
- the host is MCH (ICR) (white, purchased from Clea Japan), or HKLD mice exhibiting antibody gene deficiency (Igh/Ig ⁇ KO) or low expression (Igl1 low).
- Cell stage embryos were used. It is possible to determine whether or not the pups born as a result of transplanting the injected embryos into foster mothers are chimeras based on their coat color. By transplanting the embryos into foster mothers, chimeric mice (having a dark brown coat color) can be obtained.
- mice harboring Ig-NAC containing bovine minimalized IgHD Eight clones of mouse ES cells harboring Ig-NAC containing bovine minimalized IgHD were injected into a total of 591 ICR embryos and transplanted into 32 foster mothers. As a result, 18 GFP-positive individuals were obtained. As a result, the chimera rate was 5-60% for 18 chimeric mice. In addition, 5 clones were injected into a total of 380 HKLD embryos and transplanted into 21 foster mothers, resulting in 13 GFP-positive individuals. As a result of judging the chimera rate by coat color, the chimera rate was 5 to 80% for 13 chimeric mice.
- Example 13 Human minimalized chimeric mouse analysis (B cell analysis, ELISA, cDNA preparation) B cell differentiation analysis, plasma antibody concentration measurement ELISA analysis, and gene sequence analysis by cDNA recovery from spleen can be performed to evaluate whether human antibodies are being produced from human minimalized chimeric mice.
- B-cell differentiation analysis If cells positive for both EGFP and B220, which is a B-cell marker, can be detected, B-cell differentiation due to the contribution of antibody production is indicated, and this is an indirect evaluation of the functional expression of the antibody.
- Peripheral blood samples obtained from chimeric mice were subjected to flow cytometry analysis of B220 by the method described in Japanese Patent No. 6868250. As a result, not only GFP-positive cell populations but also GFP and B220 co-positive populations were confirmed. Therefore, it was shown that a functional antibody was expressed. The highest GFP positive rate per individual was obtained in 28.12% of individuals, and the highest GFP and B220 copositive rate per individual was 2.30%.
- RNA derived from human antibody-producing mouse spleen is synthesized from RNA derived from human antibody-producing mouse spleen, and human antibody gene variable region cloning and base sequencing are performed. Analysis and evaluation were carried out in the same manner as in Japanese Patent No. 6868250. After collection, the spleens were stored overnight at 4°C in RNAlater (Ambion), then removed from the solution and stored at -80°C. 1 mL of ISOGEN (Nippon Gene) is added to the pressure tube containing the zirconia beads, and 50 mg of tissue is added.
- ISOGEN Natural Gene
- Bovine minimalized chimeric mouse analysis B cell analysis, ELISA, cDNA preparation
- B cell differentiation analysis, plasma antibody concentration measurement ELISA analysis, and gene sequence analysis by cDNA recovery from spleen can be performed as an evaluation of whether human antibodies are being produced from bovine minimalized chimeric mice.
- B-cell differentiation analysis If cells positive for both EGFP and B220, which is a B-cell marker, can be detected, B-cell differentiation due to the contribution of antibody production will be indicated, and this will be an indirect evaluation of the functional expression of the antibody.
- Peripheral blood samples obtained from chimeric mice were subjected to flow cytometry analysis of B220 by the method described in Japanese Patent No. 6868250. As a result, not only GFP-positive cell populations but also GFP and B220 co-positive populations were confirmed. Therefore, it was shown that a functional antibody was expressed. The highest GFP positive rate per individual was obtained in 40.38% of individuals, and the highest GFP and B220 copositive rate per individual was 3.23%.
- RNA derived from human antibody-producing mouse spleen is synthesized from RNA derived from human antibody-producing mouse spleen, and human antibody gene variable region cloning and base sequencing are performed. Analysis and evaluation were performed in the same manner as in Japanese Patent No. 6868250. After collection, the spleens were stored overnight at 4°C in RNAlater (Ambion), then removed from the solution and stored at -80°C. 1 mL of ISOGEN (Nippon Gene) is added to the pressure tube containing the zirconia beads, and 50 mg of tissue is added.
- ISOGEN Natural Gene
- Human minimalized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse repertoire analysis 1. Human antibody gene heavy chain region cloning and base sequencing from human minimalized ( ⁇ IgH / ⁇ Ig ⁇ -ES) chimeric mouse spleen cDNA cDNA derived from chimeric mouse spleen retaining human minimalized IgHD was subjected to PCR using the following primers, and human antibody The gene heavy chain variable region was amplified. As a negative control, spleen-derived cDNA obtained from non-chimeric mouse ICR was used.
- HIGMEX1-2 5'-CCAAGCTTCAGGAGAAAGTGATGGAGTC-3' (SEQ ID NO: 45)
- VH1/5BACK 5'-CAGGTTGCAGCTGCAGCAGTCTGG-3'
- VH4BACK 5'-CAGGTGCAGCTGCAGGAGTCGGG-3'
- VH3BACK 5'- GAGGTGCAGCTGCAGGAGTCTGG-3' (SEQ ID NO: 48)
- PCR was performed under the conditions of 35 cycles of 98°C for 10 seconds, 59°C for 5 seconds, and 68°C for 5 seconds using a combination of constant region x variable region (three types). All amplified products were detected by ethidium bromide staining after 1.5% agarose gel electrophoresis. As a result, an amplification product was detected around the expected 470 bp in all combinations. On the other hand, no specific amplification product was detected at the same position in all of the negative controls. These amplified products were extracted from an agarose gel and treated with restriction enzymes using HindIII and PstI. This was cloned into the HindIII, PstI sites of the pUC119 (TAKARA) vector.
- the VH1 family-derived clones, the VH4 family-derived clones, and the VH3 family-derived clones were sequenced to determine the nucleotide sequences of the amplified products.
- Example 16 Repertoire analysis of bovine minimalized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mice Using cDNA derived from bovine minimalized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse spleen, PCR was performed by the method shown in Example 15, A human antibody gene heavy chain variable region was amplified. As a negative control, spleen-derived cDNA obtained from non-chimeric mouse ICR was used. The results demonstrate the use of the bovine D fragment in the chimeric mouse spleen Ig ⁇ -cDNA.
- bovine D fragment-specific primers were designed and used.
- cowF1 5'-ATTATCTGCAGAAGTCTGACCCGCACACAGG-3' (SEQ ID NO: 49)
- cowF2 5'-ATTATCTGCAGTGTGGAGCTGGCCAATGCAT-3' (SEQ ID NO: 50)
- cowF3 5'-ATTATCTGCAGATGGTTATGGTTATGGTTATGGTTATGG-3' (SEQ ID NO: 51)
- cowR1 5'-GTCGGAAGCTTATAACCACAACCATAACCAT-3' (SEQ ID NO: 52)
- the expected length (forward primer and HIGMEX1-2 prepared in the fragment: around 250 bp, reverse primer prepared in the fragment and the variable region primer used in Example 15: around 400 bp) amplified product. was detected.
- no specific amplification product was detected at the same position in all of the negative controls.
- These amplified products were extracted from an agarose gel and treated with restriction enzymes using HindIII and PstI. This was cloned into the HindIII, PstI sites of the pUC119 (TAKARA) vector. The plasmid into which the amplified product was inserted was sequenced to determine the base sequence.
- clone 1 D fragment (bovine D1-3): TGTGGAGCTGGCCA (SEQ ID NO: 53) J fragment (human J3): ATGCTTTTGATATCTGGGGCCAAGGGACAATGGTCACCGTCTCTTCAG (SEQ ID NO: 54) clone 2 D fragment (bovine 6-3 or 6-4): ATGGTTATGGTTATGGTTATGGTTGTGGTTATGGTTATGGTTATAC (SEQ ID NO: 55) J fragment (human J1): CTTCCAGCACTGGGGCCAGGGCACCCTGGTCACCGTCTCCTCAG (SEQ ID NO: 56) clone 3 V fragment (human V3-23): GGGGGAGGCTTGGTACAGCCTGGGGGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTAGCAGCTATGCCATGAGCTGGGTCCGCCAGGCTCCAGGGAAGGGGCTGGAGTGGGTTCTCAGCTATTAGTGGTAGTGGTGGTAGCACATACTACGCAGACTCCGTGAAGGGCC
- the amino acid sequence derived from the clone 2 cDNA sequence is the following sequence: It was confirmed that the length of CDRH3 was 18 amino acids or more (underlined number of amino acids) or longer chain CDR3.
- Example 17 Human minimalized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse immune response An antigen protein was administered to human minimalized chimeric mice, and enzyme-linked immunosorbent assay (Enzyme) was performed using serum or plasma samples collected after administration. - Linked Immuno Sorbent Assay (hereinafter referred to as ELISA) can be performed to evaluate whether immune responses and antigen-specific antibodies are induced.
- Enzyme enzyme-linked immunosorbent assay
- ELISA Linked Immuno Sorbent Assay
- Antiserum ELISA Plasma is prepared from peripheral blood 3 days after each immunization. In order to evaluate the titer of antigen-specific IgG in plasma, antiserum ELISA is performed using a 96-well plate on which the antigen is immobilized and an anti-human IgG Fc antibody.
- Bovine minimalized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse immune response Receptor binding domain (hereinafter referred to as RBD) of SARS-CoV2 spike protein S1 was administered to bovine minimized chimeric mice as an antigen and administered.
- ELISA was performed using serum or plasma samples collected later to assess the induction of immune responses and antigen-specific antibodies.
- Antiserum ELISA Plasma was prepared from peripheral blood 3 days after each immunization. In order to evaluate the titer of antigen-specific IgG in plasma, antiserum ELISA was performed using a 96-well plate on which the antigen was immobilized and an anti-human IgG Fc antibody. As a result, it was confirmed that continuous booster immunization resulted in an immune response and an increase in the antibody titer against RBD (Fig. 14).
- OGAB Organic Gene Assembly in Bacillus subtilis
- simian IgHD vector by ⁇ C31 recombinase It was carried out in the same manner as in Example 5. 1-10 cells were trypsinized and suspended in Opti MEM to 3 ⁇ 10 6 cells/mL, then 1 ⁇ g of ⁇ C31 recombinase gene expression vector ⁇ C31pEF1 (Patent No. 6868250) and 3 ⁇ g of simian IgHD vector were added. In addition, electroporation was performed at 150 V for 7.5 ms using NEPA21 (Nepagene). Cells were seeded in two 384-well plates after pulsing. After 48 hours, the medium was replaced with F12 medium containing 800 ⁇ g/mL G418, 800 ⁇ g/mL blasticidin and 10% FBS, and cultured. After 12 days, 17 plants resistant to blasticidin were picked up.
- PCR analysis The isolated cells were cultured, and genomic DNA was extracted from the cells using Cellysis (Qiagen). In addition to 12 types of Ig-NAC-specific primers prepared on Ig-NAC, PCR analysis was performed using one type of simian IgHD-specific primer shown below. Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification. Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C. for 2 minutes, denatures at 98 ° C. for 10 seconds, anneals at 60 ° C.
- KODone TOYOBO
- the following five types of primers were prepared and long PCR was performed. Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification.
- Taq polymerase was PrimeSTAR TM GXL Premix Fast (Takara Bio Inc.) and the following PCR was performed by a step-down method.
- PACF1 5'- CGGTGTGCGGTTGTATGCCTGCTGT-3' (SEQ ID NO: 64) 3-3to4-4R: 5'-GGCCAGGCAGGATGATGGACTCCCA-3' (SEQ ID NO: 65) 3-3F: 5'- CGGTTACTATTACACCCCACAGCGTC-3' (SEQ ID NO: 66) 3-10F2: 5'-TTGTAGTGGTGGTGTCTGCTACACC-3' (SEQ ID NO: 67) 3-16R: 5'-GTAGTTACTGTATTCACACAGTGAC-3' (SEQ ID NO: 68) F37: 5'-CTGCAGGCCCTGTCCTCTTC-3' (SEQ ID NO: 69) 4-23R: 5'- ATAGTAATACCACTGTGGGAGGGCC-3' (SEQ ID NO: 70)
- FISH analysis was performed on one cell clone in which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250).
- Table X shows the results of PCR analysis and FISH analysis in one cell clone subjected to FISH analysis.
- the rows indicate the clone names, and the columns indicate the primers and extension times used in PCR. ⁇ indicates positive.
- Ig-NACx1 indicates that Ig-NAC is detected at 1 copy per cell.
- Example 20 Partial sequence removal (simianization) by site-specific recombination using R4 recombinase 1.
- Introduction of R4 Recombinase Gene Expression Vector It was carried out in the same manner as in Example 6.
- a cell clone in which introduction of the simianized human IgHD vector was confirmed (Example 19) was treated with trypsin, suspended in OptiMEM to a concentration of 3 ⁇ 10 6 cells/mL, and then 10 ⁇ g of the R4 recombinase gene expression vector was added.
- NEPA21 Nappagene was used to perform electroporation at 150 V for 7.5 ms.
- One 10 cm dish was seeded after pulsing.
- the cells were trypsinized and seeded at 150 cells per 384-well plate. Seventeen days after seeding on the 384-well plate, the medium was replaced with F12 medium containing 800 ⁇ g/mL G418 and 10% FBS, cultured, and single clones were picked up.
- PCR analysis Culturing the isolated cells, extracting genomic DNA from the cells using Cellysis (Qiagen), using this genomic DNA as a template, one specific primer after R4 site-specific recombination as in Example 6 , and 2 primers prepared in simian IgHD, and 12 Ig-NAC-specific primers prepared on Ig-NAC were used for PCR analysis. PCR was performed using specific primers after R4 site-specific recombination, and clones (1-10 M9-I9) were selected that gave a specific band after R4 site-specific recombination.
- FISH analysis was performed on the clone (1-10 M9-I9) for which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250).
- Example 21 Introduction of simian IgHD-loaded Ig-NAC into mouse ES cell line by micronucleation method (MMCT) 1. Transfer of Ig-NAC loaded with simian IgHD into mouse ES cells using MMCT method Clones in which R4 site-specific recombination was confirmed by PCR and FISH analysis in Example 10 are used as chromosome donor cells. Mouse ES cell line HKD31 6TG-9 (XO) in which both alleles of the endogenous immunoglobulin heavy chain and ⁇ chain loci are disrupted is used as the chromosome recipient cell (Patent No. 4082740).
- XO XO
- Micronuclei are formed in the same manner as in Example 7, fused with mouse ES cell HKD31 6TG-9 strain (XO), and seeded on three 10 cm dishes. After 48 hours, the medium is replaced with an ES cell medium supplemented with 350 ⁇ g/mL G418, and then the medium is replaced with the same composition once every two days. Pick up drug-resistant colonies that appear after 1 week to 10 days.
- PCR analysis The isolated cells are cultured, genomic DNA is extracted from the cells using Cellysis (Qiagen), and using this genomic DNA as a template, PCR is performed using the same primers as in the PCR analysis performed in Example 6. .
- mice were generated from mouse ES cell lines containing simian IgHD-containing Ig-NAC. make. Chimeric mice are produced according to the procedure of Gene Targeting, Experimental Medicine, 1995, using mouse ES cells retaining Ig-NAC containing simian IgHD.
- the host is MCH (ICR) (white, purchased from Clea Japan) or HKLD mice exhibiting antibody gene deficiency (Igh/Ig ⁇ KO) or low expression (Igl1 low). Morula and 8 cells obtained by mating. Stage embryos are used.
- Example 23 Monkey IgHD chimeric mouse analysis (B cell analysis, ELISA, cDNA preparation) B cell differentiation analysis, plasma antibody concentration measurement ELISA analysis, and gene sequence analysis by recovering cDNA from spleen can be performed to evaluate whether monkey IgHD chimeric mice produce monkey IgHD human antibodies.
- B-cell differentiation analysis If cells positive for both EGFP and B220, which is a B-cell marker, can be detected, B-cell differentiation due to the contribution of antibody production will be indicated, and this will be an indirect evaluation of the functional expression of the antibody.
- Peripheral blood samples obtained from chimeric mice were subjected to flow cytometry analysis of B220 by the method described in Japanese Patent No. 6868250. As a result, not only GFP-positive cell populations but also GFP and B220 co-positive populations were confirmed. This indicates that a functional antibody is expressed.
- RNA derived from human antibody-producing mouse spleen is synthesized from RNA derived from human antibody-producing mouse spleen, and human antibody gene variable region cloning and base sequencing are performed. Analysis and evaluation are performed in the same manner as in Japanese Patent No. 6868250. After collection, the spleens were stored overnight at 4°C in RNAlater (Ambion), then removed from the solution and stored at -80°C. 1 mL of ISOGEN (Nippon Gene) is added to the pressure tube containing the zirconia beads, and 50 mg of tissue is added.
- ISOGEN Natural Gene
- RNA After crushing with the device (about 30 to 45 seconds) and confirming that the tissue was crushed, after spin down, 1 mL of supernatant was collected, 200 ⁇ L of chloroform was added, and RNeasy Mini kit was used according to the manufacturer's protocol. Extract the RNA.
- Example 24 Monkeyized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse repertoire analysis 1.
- Human antibody gene heavy chain region cloning and base sequencing from simian ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse spleen cDNA cDNA derived from chimeric mouse spleen retaining simian IgHD was subjected to PCR using the following primers to determine the human antibody gene weight. Amplify the chain variable region. Spleen-derived cDNA obtained from non-chimeric mouse ICR is used as a negative control.
- Constant region HIGMEX1-2: 5'-CCAAGCTTCAGGAGAAAGTGATGGAGTC-3' (SEQ ID NO: 71)
- VH1/5BACK 5'-CAGGTGCAGCTGCAGCAGTCTGG-3'
- VH4BACK 5'-CAGGTGCAGCTGCAGGAGTCGGG-3'
- VH3BACK 5'-GAGGTGCAGCTGCAGGAGTCTGG-3' (SEQ ID NO: 74)
- PCR is performed under the conditions of 35 cycles of 98°C for 10 seconds, 59°C for 5 seconds, and 68°C for 5 seconds using a combination of constant region x variable region (three types). All amplified products are detected by ethidium bromide staining after 1.5% agarose gel electrophoresis. After the amplified product is extracted from an agarose gel, it is treated with restriction enzymes using HindIII and PstI. This is cloned into the HindIII, PstI sites of the pUC119 (TAKARA) vector. The plasmid into which the amplification product has been inserted is sequenced to determine the base sequence of the amplification product.
- Example 25 Monkeyized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mouse immune response An antigen protein was administered to monkeyized ( ⁇ IgH/ ⁇ Ig ⁇ -ES) chimeric mice, and enzyme-linked immunosorbent assay (Enzyme) was performed using serum or plasma samples.
- enzyme-linked immunosorbent assay (hereinafter referred to as ELISA) can be performed to evaluate whether immune responses and antigen-specific antibodies are induced.
- an antigen protein solution of 1 mg/mg was prepared with PBS containing 0.4 M arginine, Freund's Complete adjuvant (Sigma F5881) was used for initial immunization, and Sigma Adjuvant System TM (SAS TM ) (Sigma S6322) was used for booster immunization. ) to prepare the challenge solution. 200 ⁇ L for initial immunization (100 ⁇ g antigen, intraperitoneal), 100 ⁇ L for booster (50 ⁇ g antigen, intraperitoneal, every 2 weeks), and 100 ⁇ L for final immunization (50 ⁇ g antigen, 1 mg/mg antigen protein solution, intravenous injection) to 6-week-old chimeric mice. ).
- Antiserum ELISA Plasma is prepared from peripheral blood 3 days after each immunization. In order to evaluate the titer of antigen-specific IgG in plasma, antiserum ELISA is performed using a 96-well plate on which the antigen is immobilized and an anti-human IgG Fc antibody.
- Example 26 Establishment of a mouse strain that retains human minimalized IgHD-loaded Ig-NAC and has disrupted endogenous immunoglobulin heavy chain and ⁇ chain loci.
- mouse ES cells retaining Ig-NAC loaded with human minimalized IgHD produced as a chimeric mouse produced as shown in Example 8, the endogenous immunoglobulin heavy chain and ⁇ chain were disrupted mouse strain
- a mouse strain harboring Ig-NAC loaded with human minimalized IgHD and in which the endogenous immunoglobulin heavy and kappa chain loci were disrupted was established by crossing with .
- Example 27 Establishment of a mouse strain that retains bovine minimalized IgHD-loaded Ig-NAC and in which the endogenous immunoglobulin heavy chain and ⁇ chain loci are disrupted
- mouse ES cell TT2F XO
- mouse ES cells that retain bovine minimalized IgHD-loaded Ig-NAC prepared as wild-type mice or chimeric mice prepared as shown in Example 12 endogenous immunoglobulin heavy chains and ⁇ chains are destroyed.
- a mouse strain was established in which the endogenous immunoglobulin heavy chain and ⁇ chain loci were disrupted while retaining the bovine minimalized IgHD-loaded Ig-NAC.
- the resulting mouse individual (#241) which retains the bovine minimalized IgHD-loaded Ig-NAC and has the endogenous immunoglobulin heavy chain and ⁇ chain loci disrupted, was subjected to flow cytometry by the method described in Example 13. A metric analysis was performed. As a result, the GFP positive rate of peripheral blood mononuclear cells was 99.09%, and the GFP and B220 co-positive rate of peripheral blood mononuclear cells was 6.26%. It was comparable to mouse individuals that retained wild-type Ig-NAC and had the endogenous immunoglobulin heavy chain and ⁇ chain loci disrupted (Japanese Patent No. 6868250).
- Example 28 Establishment of a mouse strain that retains simian IgHD-loaded Ig-NAC and has disrupted endogenous immunoglobulin heavy chain and ⁇ chain loci
- a mouse strain that retains simian IgHD-loaded Ig-NAC and has disrupted endogenous immunoglobulin heavy chain and ⁇ chain loci
- monkeys A mouse strain can be established that carries an IgHD-loaded Ig-NAC with disruption of the endogenous immunoglobulin heavy and kappa chain loci.
- Example 29 Insertion of chemically synthesized DNA containing human long chain minimalized IgHD by site-specific recombination using ⁇ C31 recombinase D fragments 1-1 to 1-26 of human minimalized IgHD designed in Example 5
- Human long-chain minimalized IgHD vectors substituted with 26 modified long-chain DH sequences shown in FIG. and 2. It was prepared by the procedure of.
- Human long DH sequences were designed based on long (18 amino acids or more) CDR3 sequences found in human antibody cDNA sequences registered with NCBI.
- Human long-chain miniA, human long-chain miniB, and human long-chain miniC vectors were synthesized by Eurofins Genomics in the same manner as in Example 5.
- Human long chain miniA Fragment into Human Long Chain MiniB Human long chain miniA was cleaved with restriction enzymes XhoI and NotI.
- human long chain miniA/B vector was constructed by inserting long chain miniA into human long chain miniB cleaved with restriction enzymes XhoI and NotI.
- Human long chain miniC was cleaved with restriction enzymes EcoRI and SalI.
- a human long chain minimalized IgHD vector was constructed by inserting a human long chain miniC fragment into a human long chain miniA/B vector cleaved with restriction enzymes EcoRI and SalI.
- PCR analysis In the same manner as in Example 5, 1 primer specific after ⁇ C31 site-specific recombination, and 2 primers generated in human long chain minimalized IgHD, Ig-NAC specific generated on Ig-NAC 3. using 12 different primers; PCR analysis of the clones obtained in . Approximately 0.1 ⁇ g of genomic DNA was used for PCR amplification.
- Taq polymerase uses KODone (TOYOBO), performs one cycle of 98 ° C. for 2 minutes, denatures at 98 ° C. for 10 seconds, anneals at 60 ° C. for 5 seconds, elongation at 68 ° C., 5 seconds if the length of the amplified fragment is 500 bp or less , 35 cycles of 100 sec/length (bp) for ⁇ 500 bp.
- FISH analysis was performed on the clone (1-10 HL15) for which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250).
- Example 30 Partial sequence removal by site-specific recombination using R4 recombinase Among the sequences introduced onto Ig-NAC ( ⁇ DH), the CAG promoter and the blasticidin resistance gene are not necessary for antibody gene expression. is an array of These sequences are flanked by R4 attB and R4 attP and can be removed by site-specific recombination between R4 attB-attP by introducing R4 recombinase into cells.
- R4 recombinase gene expression vector introduction As in Example 6, the R4 recombinase gene expression vector (Patent No. 6868250) was confirmed to contain a human long-chain minimalized IgHD vector, and one Ig-NAC was retained. A high percentage CHO cell clone (1-10 HL15) was transfected by electroporation. The resulting G418-resistant clones were picked.
- PCR analysis As in Example 6, 1. PCR analysis was performed using the genomic DNA of the G418-resistant clone obtained in 2. above as a template, and clones (1-10 HL15-28, 1-10 HL15-43) that gave specific bands after R4 site-specific recombination were selected.
- FISH analysis was performed on cell clones (1-10 HL15-28, 1-10 HL15-43) in which the band of interest was confirmed by PCR analysis. FISH analysis was performed using a mouse gene-specific probe mouse cot1 (invitrogen) according to the method described in (Japanese Patent No. 6868250).
- Example 31 Introduction of human long-chain minimalized IgHD-loaded Ig-NAC into mouse ES cell line by micronucleation method (MMCT) In Example 30, PCR and FISH analysis confirmed that R4 site-specific recombination occurred Using the obtained clones as chromosome donor cells, human long-chain minimalized IgHD-containing Ig-NAC transfer is performed into mouse ES cells using the MMCT method.
- MMCT micronucleation method
- PCR Analysis Similar to Example 7, the isolated cells are cultured and genomic DNA is extracted from the cells using Celllysis (Qiagen). PCR is performed using this genomic DNA as a template, and clones from which the band of interest is detected are selected.
- Example 32 Generation of chimeric mice from mouse ES cell lines harboring human long-chain minimalized IgHD-containing Ig-NAC Chimeric mice are generated from minimized IgHD-containing Ig-NAC mouse ES cells.
- Mouse ES cell line clones harboring human long-chain minimalized HD-containing Ig-NAC are injected into ICR embryos and implanted into foster mothers.
- GFP-positive chimeric mice are obtained.
- the obtained chimeric mice are analyzed by the methods described in Examples 13, 14, 15 and the like. The results demonstrate the use of the long human D fragment in the chimeric mouse spleen Ig ⁇ -cDNA.
- Example 33 Construction of synthetic D region-introduced acceptor-bearing Ig-NAC with deletion of human IgHD7-27 26 fragments have been deleted and 7-27, which lies far from the D region and near the J region, has not been deleted.
- design evaluate and select CRISPR / Cas9 gRNA at two locations before and after D fragment 7-27.
- human IgHD7-27 is deleted by cleaving the Ig-NAC-holding CHO cells 1-10 (Example 4) equipped with the introduction platform of chemically synthesized DNA by genome editing using the above gRNA and Cas9.
- the synthesized D region-introduced acceptor-retaining Ig-NAC is obtained.
- the present invention expands the diversity of human antibodies. Preferably, it enables the production of human antibodies comprising CDRH3 of 18 to 50 or more amino acids.
- SEQ ID NO: 1 Nucleotide sequence of human minimalized D region
- SEQ ID NO: 2 Nucleotide sequence of human minimalized D region replaced with bovine ORF737. . B1-1 to replace 767 D2-2 1024. . B2-1 to replace 1039 D3-3 1296. . B3-1 to replace 1331 D4-4 1588. . B4-1 to replace 1630 D5-5 1887. . B9-1 to replace 1900 D6-6 2157. . B1-2 to replace 2187 D1-7 2444. . B2-2 to replace 2459 D2-8 2715. . B5-2 to replace 2756 D3-9 2910. . B8-2 to replace 3057 D3-10 3374. . B6-2 to replace 3431 D4-11 3688. .
- SEQ ID NOS: 26-52 Primers SEQ ID NO: 61: Amino Acid Sequences of Peptides Containing Long CDRH3
- SEQ ID NOS: 62-74 Primers SEQ ID NOS: 75: Substituted for Nucleotide Sequences of Human DH Numbers 1-1 Modified Long DH Sequence
- SEQ ID NO:76 Modified Long DH Sequence Substituted for the Nucleotide Sequence of Human DH Numbers 2-2
- SEQ ID NO:78 Modified Long DH Sequence Substituted for the Nucleotide Sequence of Human DH Numbers 4-4
- SEQ ID NO:79 Modified Long DH Substituted for the Nucleotide Sequence of Human DH Numbers 5-5
- SEQ ID NO:80 modified long DH sequence substitute
Abstract
ヒト抗体産生非ヒト動物において、ヒト抗体多様性の拡張を達成するための手段を提供することである。 ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座及びヒト免疫グロブリン軽鎖遺伝子座を含む哺乳動物人工染色体ベクター、この哺乳動物人工染色体ベクターを含む哺乳動物細胞又は非ヒト動物、並びに、この人工染色体ベクターの製造においてヒト免疫グロブリン重鎖D領域をその改変配列で置換する方法。
Description
本発明は、ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座及びヒト免疫グロブリン軽鎖遺伝子座を含む哺乳動物人工染色体ベクターに関する。
本発明はまた、上記哺乳動物人工染色体ベクターを含む哺乳動物細胞又は非ヒト動物に関する。
本発明はさらに、上記哺乳動物人工染色体ベクターの製造における上記D領域の改変方法に関する。
本発明はまた、上記哺乳動物人工染色体ベクターを含む哺乳動物細胞又は非ヒト動物に関する。
本発明はさらに、上記哺乳動物人工染色体ベクターの製造における上記D領域の改変方法に関する。
ヒト抗体は、人体では異物として認識されないため腫瘍や自己免疫疾患などの治療薬として使用されている。このような抗体は、ファージディスプレイ技術やヒト抗体産生マウスの使用によって作製可能である。ヒト抗体産生マウスは、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座全長を含むマウス人工染色体ベクターを保持するTrans-chromosomic(TC)マウスである(特許文献1、特許文献2、非特許文献1)。
例えばヒト免疫不全ウイルス(HIV)、新型コロナウイルス(COVID-19)、インフルエンザウイルスなどのウイルスによる感染症の受動免疫のためにヒト抗体を使用するとき、ウイルス標的抗原のマスキング、或いはウイルス標的抗原が迅速に変異を起こすなどの理由のために中和抗体はしばしば有効性を欠くことがよく知られている。変異に対しては、防御可能な新たな抗体の作製が必要になるため、感染症対策には多大の時間と費用が必要となる。また、病原性ウイルスの標的抗原のマスキングに対しては、免疫系を活性化するための手法や、抗ウイルス剤などの治療剤の開発などを対策として講ずる必要があるため、迅速に感染症を抑制することは難しいと思われる。
このような状況において、ヒトにおいて抗体ができにくい標的抗原に対してはヒト抗体産生マウスの利用価値は限定的であるため、例えばヒト抗体多様性(human antibody repertoire)を拡張して当該標的抗原に対する抗体取得を容易にする技術の開発が望まれているし、また、さらに拡張されたヒト抗体多様性を達成するためのプラットフォームの構築が提案されている(非特許文献3)。
K.Tomizuka et al.,Proc.Natl.Acad.Sci.USA,2000;97(2):922-927
M.A.Alfaleh et al.,Front.Immunol.2020;11:Article 1986,https://doi.org/10.3389/fimmu.2020.01986
A.R.Rees,MABS 2020,12(1),e1729683
本発明の課題は、ヒト抗体産生非ヒト動物において、ヒト抗体多様性の拡張を達成するための手段を提供することである。
この関連でY.Diら(Immunology August 2021;00:1-14,doi:10.1111/imm.13407)は、ゲノム編集を用いてウシDH遺伝子領域をマウスゲノム上のDH遺伝子領域と組み換えたB-DHマウスを作製し、抗原(OVA,BSA,EWL,KLH)で免疫し、超長鎖(ultralong)CDRH3を含む抗体の産生を調べたが、超長鎖CDRH3はかなり低率で観察されたにすぎなかったと報告している。
この関連でY.Diら(Immunology August 2021;00:1-14,doi:10.1111/imm.13407)は、ゲノム編集を用いてウシDH遺伝子領域をマウスゲノム上のDH遺伝子領域と組み換えたB-DHマウスを作製し、抗原(OVA,BSA,EWL,KLH)で免疫し、超長鎖(ultralong)CDRH3を含む抗体の産生を調べたが、超長鎖CDRH3はかなり低率で観察されたにすぎなかったと報告している。
本発明者らは、上記課題を解決するためにウシがHIVに対する広域中和抗体を効率的に産生するという知見に注目し(M.J.Burke et al.,Viruses 2020,12,473;doi:10.3390/v12040473)、ヒト抗体の多様性を拡張するための手法、及び/又は、抗体重鎖CDR3が通常より長いヒト抗体を出現する手法を見出した。
すなわち、本発明は、以下に示す特徴を包含する。
[1] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターにおいて、前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されていることを特徴とする哺乳動物人工染色体ベクターであって、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
前記哺乳動物人工染色体ベクター。
[2] 前記(1)のヒト動物免疫グロブリン重鎖遺伝子座D領域のORF間のヒト由来ゲノム配列が100~500bpの長さに短縮された配列を含む、[1]に記載の哺乳動物人工染色体ベクター。
[3] 前記ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列が、配列番号127~149のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[4] 前記サル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列が、配列番号150~175のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[5] 前記(3)の26種の改変ORF配列が、配列番号75~100のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[6] 前記D領域の改変配列が、配列番号2又は配列番号3のヌクレオチド配列、或いは該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]~[5]のいずれかに記載の哺乳動物人工染色体ベクター。
[7] 前記ヒト免疫グロブリン重鎖遺伝子座のD領域がさらにD7-27を欠失している、[1]~[6]のいずれかに記載の哺乳動物人工染色体ベクター。
[8] 前記ベクターが、齧歯類人工染色体ベクターである、[1]~[7]のいずれかに記載の哺乳動物人工染色体ベクター。
[9] 前記齧歯類人工染色体ベクターが、マウス人工染色体ベクターである、[8]に記載の哺乳動物人工染色体ベクター。
[10] 前記軽鎖遺伝子座が、κ軽鎖遺伝子座及び/又はλ軽鎖遺伝子座である、[1]~[9]のいずれかに記載の哺乳動物人工染色体ベクター。
[11] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターを含む哺乳動物細胞。
[12] 前記細胞が、齧歯類細胞又はヒト細胞である、[11]に記載の哺乳動物細胞。
[13] 前記細胞が、哺乳動物由来のiPS細胞及びES細胞からなる群から選択される分化多能性幹細胞である、[11]に記載の哺乳動物細胞。
[14] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターを含み、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている非ヒト哺乳動物。
[15] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む非ヒト動物であって、前記ヒト免疫グロブリン重鎖遺伝子座のD領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されており、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ことを特徴とし、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている、前記非ヒト動物。
[16] 前記動物が、齧歯類である、[14]又は[15]に記載の非ヒト動物。
[17] 前記齧歯類が、マウス又はラットである、[16]に記載の非ヒト動物。
[18] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該非ヒト動物の血液から該標的抗原と結合する抗体を取得することを含む、抗体の作製方法。
[19] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物から脾臓細胞、リンパ節細胞又はB細胞を取得し、前記脾臓細胞、リンパ節細胞又はB細胞とミエローマ細胞を融合してハイブリドーマを形成し、該ハイブリドーマを培養して、前記標的抗原と結合するモノクローナル抗体を取得することを含む、抗体の作製方法。
[20] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物からB細胞を取得し、前記B細胞から抗体重鎖及び軽鎖、或いはそれらの可変領域、からなるタンパク質をコードする核酸を取得し、該核酸を用いてDNA組み換え技術又はファージディスプレイ技術によって前記標的抗原と結合する抗体を作製することを含む、抗体の作製方法。
[21] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターの製造において、ヒト免疫グロブリン重鎖D領域をその改変配列で置換する方法であって、
(1’)ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを準備する、
(2’)工程(1’)の前記哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除する、
(3’)工程(2’)で得られたベクターの前記D領域の削除部位に、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、
(4’)工程(3’)で挿入された前記カセットの第2の部位特異的組み換え酵素認識部位に、第2の部位特異的組み換え酵素を用いて、前記ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、選択マーカー及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、並びに、
(5’)第1の部位特異的組み換え酵素を用いる部位特異的組み換えによって、第1の部位特異的組み換え酵素認識部位、前記ヒト免疫グロブリン重鎖D領域の改変配列及び第2の部位特異的組み換え酵素認識部位を含むカセットが前記D領域の削除部位に挿入された前記人工染色体ベクターを得る、
ことを含む前記方法。
[22] 前記ヒト免疫グロブリン重鎖D領域の削除が、ゲノム編集によって行われる、[21]に記載の方法。
[23] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターであって、前記ヒト免疫グロブリン重鎖遺伝子座がD領域を欠失しており、該欠失されたD領域に代えて、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含む、哺乳動物人工染色体ベクター。
[24] 前記欠失されたD領域が、D1-1からD1-26までの領域である、[23]に記載の哺乳動物人工染色体ベクター。
[25] 前記欠失されたD領域が、D1-1からD7-27までの領域である、[23]に記載の哺乳動物人工染色体ベクター。
本明細書は本願の優先権の基礎となる日本国特許出願番号2021-186124号の開示内容を包含する。
[1] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターにおいて、前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されていることを特徴とする哺乳動物人工染色体ベクターであって、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
前記哺乳動物人工染色体ベクター。
[2] 前記(1)のヒト動物免疫グロブリン重鎖遺伝子座D領域のORF間のヒト由来ゲノム配列が100~500bpの長さに短縮された配列を含む、[1]に記載の哺乳動物人工染色体ベクター。
[3] 前記ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列が、配列番号127~149のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[4] 前記サル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列が、配列番号150~175のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[5] 前記(3)の26種の改変ORF配列が、配列番号75~100のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]又は[2]に記載の哺乳動物人工染色体ベクター。
[6] 前記D領域の改変配列が、配列番号2又は配列番号3のヌクレオチド配列、或いは該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、[1]~[5]のいずれかに記載の哺乳動物人工染色体ベクター。
[7] 前記ヒト免疫グロブリン重鎖遺伝子座のD領域がさらにD7-27を欠失している、[1]~[6]のいずれかに記載の哺乳動物人工染色体ベクター。
[8] 前記ベクターが、齧歯類人工染色体ベクターである、[1]~[7]のいずれかに記載の哺乳動物人工染色体ベクター。
[9] 前記齧歯類人工染色体ベクターが、マウス人工染色体ベクターである、[8]に記載の哺乳動物人工染色体ベクター。
[10] 前記軽鎖遺伝子座が、κ軽鎖遺伝子座及び/又はλ軽鎖遺伝子座である、[1]~[9]のいずれかに記載の哺乳動物人工染色体ベクター。
[11] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターを含む哺乳動物細胞。
[12] 前記細胞が、齧歯類細胞又はヒト細胞である、[11]に記載の哺乳動物細胞。
[13] 前記細胞が、哺乳動物由来のiPS細胞及びES細胞からなる群から選択される分化多能性幹細胞である、[11]に記載の哺乳動物細胞。
[14] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターを含み、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている非ヒト哺乳動物。
[15] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む非ヒト動物であって、前記ヒト免疫グロブリン重鎖遺伝子座のD領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されており、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ことを特徴とし、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている、前記非ヒト動物。
[16] 前記動物が、齧歯類である、[14]又は[15]に記載の非ヒト動物。
[17] 前記齧歯類が、マウス又はラットである、[16]に記載の非ヒト動物。
[18] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該非ヒト動物の血液から該標的抗原と結合する抗体を取得することを含む、抗体の作製方法。
[19] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物から脾臓細胞、リンパ節細胞又はB細胞を取得し、前記脾臓細胞、リンパ節細胞又はB細胞とミエローマ細胞を融合してハイブリドーマを形成し、該ハイブリドーマを培養して、前記標的抗原と結合するモノクローナル抗体を取得することを含む、抗体の作製方法。
[20] [14]~[17]のいずれかに記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物からB細胞を取得し、前記B細胞から抗体重鎖及び軽鎖、或いはそれらの可変領域、からなるタンパク質をコードする核酸を取得し、該核酸を用いてDNA組み換え技術又はファージディスプレイ技術によって前記標的抗原と結合する抗体を作製することを含む、抗体の作製方法。
[21] [1]~[10]のいずれかに記載の哺乳動物人工染色体ベクターの製造において、ヒト免疫グロブリン重鎖D領域をその改変配列で置換する方法であって、
(1’)ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを準備する、
(2’)工程(1’)の前記哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除する、
(3’)工程(2’)で得られたベクターの前記D領域の削除部位に、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、
(4’)工程(3’)で挿入された前記カセットの第2の部位特異的組み換え酵素認識部位に、第2の部位特異的組み換え酵素を用いて、前記ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、選択マーカー及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、並びに、
(5’)第1の部位特異的組み換え酵素を用いる部位特異的組み換えによって、第1の部位特異的組み換え酵素認識部位、前記ヒト免疫グロブリン重鎖D領域の改変配列及び第2の部位特異的組み換え酵素認識部位を含むカセットが前記D領域の削除部位に挿入された前記人工染色体ベクターを得る、
ことを含む前記方法。
[22] 前記ヒト免疫グロブリン重鎖D領域の削除が、ゲノム編集によって行われる、[21]に記載の方法。
[23] ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターであって、前記ヒト免疫グロブリン重鎖遺伝子座がD領域を欠失しており、該欠失されたD領域に代えて、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含む、哺乳動物人工染色体ベクター。
[24] 前記欠失されたD領域が、D1-1からD1-26までの領域である、[23]に記載の哺乳動物人工染色体ベクター。
[25] 前記欠失されたD領域が、D1-1からD7-27までの領域である、[23]に記載の哺乳動物人工染色体ベクター。
本明細書は本願の優先権の基礎となる日本国特許出願番号2021-186124号の開示内容を包含する。
本発明によって、ヒト抗体産生非ヒト動物(例えば、齧歯類)において、ヒト免疫グロブリン重鎖遺伝子座のD領域のヒト由来ゲノム配列又はそのヒトミニマル化(すなわち、オープンリーディングフレーム(ORF)間領域の短縮化を含む)D領域配列のORF(遺伝子)配列を、例えばウシやサルなどの動物由来のD領域のORF配列又はヒトD領域配列の改変ORF配列と置換することにより、抗体の多様性を拡張することができるし、また好ましくは15~50若しくはそれ以上のアミノ酸長のCDRH3を含むヒト抗体を出現することが可能である。
本発明をさらに詳細に説明する。
1.哺乳動物人工染色体ベクター
本発明は、第一の態様において、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターにおいて、上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されていることを特徴とする哺乳動物人工染色体ベクターであって、前記D領域の改変配列が、
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く上記ORF間のヒト由来ゲノム配列が、VDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
前記哺乳動物人工染色体ベクターを提供する。
1.哺乳動物人工染色体ベクター
本発明は、第一の態様において、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターにおいて、上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されていることを特徴とする哺乳動物人工染色体ベクターであって、前記D領域の改変配列が、
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く上記ORF間のヒト由来ゲノム配列が、VDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
前記哺乳動物人工染色体ベクターを提供する。
本明細書中の「ヒト免疫グロブリン重鎖遺伝子座D領域」は、全てのORF(「遺伝子」(若しくはポリペプチドをコードする領域)又は「D断片」とも称する。)、VDJ組み換え配列、及び全てのORF間(すなわち、遺伝子と遺伝子の間の)領域を含む全体を指す。
本明細書中「VDJ組み換え配列」は、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座のV、D及びJ領域の上流及び下流に隣接する配列であり、重鎖、軽鎖κ及び軽鎖λ遺伝子座に応じて異なる配置で7、23、9及び/又は12塩基からなる保存された配列を含む。ヒト免疫グロブリン重鎖遺伝子座のD領域の場合、組み換え配列は28bp(すなわち、9bp、12bp(スペーサー)、7bp)の塩基を含む。
哺乳動物人工染色体ベクターは、非限定的に、例えばヒト、齧歯類(例えば、マウス、ラット、ハムスター、モルモットなど)などの哺乳動物の染色体から作製された人工染色体ベクターであり、より具体的には、該動物由来のセントロメア、及びテロメアを含み、かつ遺伝子類の例えば99.5%~100%、好ましくは100%、が除去された長腕及び(もしあれば)短腕由来のゲノム配列中に、改変された抗体重鎖D領域を含むヒト免疫グロブリン重鎖遺伝子座及び、場合により、ヒト免疫グロブリン軽鎖遺伝子座を含む、人工的に作製された染色体ベクターである。
本明細書中の「テロメア」は、同種又は異種の天然テロメア、或いは、人工テロメアである。ここで、同種とは、人工染色体ベクターの染色体断片が由来する哺乳動物と同種の動物を意味し、一方、異種とは、該動物以外の哺乳動物(これには、ヒトを含む)を意味する。また、人工テロメアの配列は、(TTAGGG)n配列(nは、繰り返しを意味する。)などの人工的に作製されたテロメア機能を有する配列を指す。人工染色体へのテロメア配列の導入は、例えば国際公開WO00/10383に記載されるようなテロメアトランケーション(テロメア配列を人工的に挿入することにより切断を行う。)を含む方法によって行うことができる。テロメアトランケーションは、本発明の人工染色体の作製において染色体の短縮のために使用することができる。
[ヒト免疫グロブリン重鎖遺伝子座のD領域の改変]
以下に、上記(1)と、上記(2)又は(3)の組み合わせからなる改変配列について説明する。
本発明の人工染色体ベクターに含まれるヒト免疫グロブリン(単に「ヒト抗体」とも称する。)重鎖遺伝子座は、V領域、D領域、J領域及びC領域においてD領域のゲノム配列のみが改変されている。ここで、D領域のヒト由来ゲノム配列の改変は、ヒトD領域のORF(若しくは、遺伝子)配列の改変又はORF間領域の短縮化(「ミニマル化」とも称する。)を含み、さらに、上記ヒトD領域のヒト由来ゲノム配列、又はヒトD領域のミニマル化配列、中のORF配列を別の動物種由来のD領域ORF配列、或いは18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列、と置換する。
以下に、上記(1)と、上記(2)又は(3)の組み合わせからなる改変配列について説明する。
本発明の人工染色体ベクターに含まれるヒト免疫グロブリン(単に「ヒト抗体」とも称する。)重鎖遺伝子座は、V領域、D領域、J領域及びC領域においてD領域のゲノム配列のみが改変されている。ここで、D領域のヒト由来ゲノム配列の改変は、ヒトD領域のORF(若しくは、遺伝子)配列の改変又はORF間領域の短縮化(「ミニマル化」とも称する。)を含み、さらに、上記ヒトD領域のヒト由来ゲノム配列、又はヒトD領域のミニマル化配列、中のORF配列を別の動物種由来のD領域ORF配列、或いは18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列、と置換する。
一般に、ヒト抗体重鎖可変領域は、N末端側からフレームワーク(FR)1、CDRH1、FR2、CDRH2、FR3、CDRH3、FR4、及び定常領域の一部からなる。FR1、CDRH1、FR2、CDRH2及びFR3は、ヒト免疫グロブリン重鎖遺伝子座の組み換え(又は、再編成)によって形成されるVDJ配列の主にV領域ヌクレオチド配列によってコードされている。また、CDRH3及びFR4はそれぞれ、主に上記VDJ配列のD領域ヌクレオチド配列及びJ領域ヌクレオチド配列によってコードされている。本発明ではアミノ酸長18以上の抗体重鎖CDR3を含むヒト抗体を産生するために、D領域の改変が行われる。
一方、ヒト抗体軽鎖可変領域は、FR1、CDRL1、FR2、CDRL2、FR3、CDRL3、FR4、及び定常領域の一部からなり、FR1、CDRL1、FR2、CDRL2、FR3及びCDRL3(一部)は、ヒト免疫グロブリン軽鎖遺伝子座の組み換え(又は、再編成)によって形成されるVJ配列の主にV領域ヌクレオチド配列によってコードされており、また、CDRH3(一部)及びFR4は、主に上記VJ配列のJ領域ヌクレオチド配列によってコードされている。
また、一般に、標的抗原が特異的に結合する抗体領域は、抗体重鎖超可変領域のCDRH3ドメインであることが知られているため、本発明では、ヒト抗体の多様性を拡張し、好ましくはヒト抗体において通常より長い(例えば、18以上の)アミノ酸長のCDRH3を含む抗体を得ることができるように、上記ヒト抗体重鎖遺伝子座のD領域ゲノム配列を改変する。
以下では、ヒトD領域の改変配列について説明する。
改変配列の例は、上記ヒト免疫グロブリン重鎖遺伝子座D領域のORF間(又は「遺伝子間」とも称する。)領域のゲノム配列が、VDJ組み換え配列が隣接するORF間領域の各VDJ組み換え配列末端から49bp以上に短縮された配列を含む、例えば約100~約500bp、約150bp~約300bp、又は約200bp~約250bpなどの長さに短縮された配列を含む、改変されたD領域ゲノム配列である。
改変配列の例は、上記ヒト免疫グロブリン重鎖遺伝子座D領域のORF間(又は「遺伝子間」とも称する。)領域のゲノム配列が、VDJ組み換え配列が隣接するORF間領域の各VDJ組み換え配列末端から49bp以上に短縮された配列を含む、例えば約100~約500bp、約150bp~約300bp、又は約200bp~約250bpなどの長さに短縮された配列を含む、改変されたD領域ゲノム配列である。
ヒト免疫グロブリン重鎖遺伝子座のD領域(「IgHD」領域ともいう。)は、ヒト14番染色体(14q32.33)に存在し、5’→3’の順に、例えば、D1-1(位置23599-35048;アクッセション番号X97051)、D2-2(位置35049-37615;アクッセション番号J00232)、D3-3(位置37616-39425;アクッセション番号X13972)、D4-4(位置39426-40474;アクッセション番号X13972)、D5-5(位置40475-41880;アクッセション番号X13972)、D6-6(位置41881-43056;アクッセション番号X13972)、D1-7(位置43057-44649;アクッセション番号X13972)、D2-8(位置44650-47262;アクッセション番号X13972)、D3-9(位置47263-48619;アクッセション番号X13972)、D3-10(位置48620-49158;アクッセション番号X13972)、D4-11(位置49159-50078;アクッセション番号X13972)、D5-12(位置50079-51316;アクッセション番号X13972)、D6-13(位置51317-52324;アクッセション番号X13972)、D1-14(位置52325-53909;アクッセション番号X13972)、D2-15(位置53910-56408;アクッセション番号J00234)、D3-16(位置5640-58143;アクッセション番号X97051)、D4-17(位置58144-59187;アクッセション番号X97051)、D5-18(位置59188-60591;アクッセション番号X97051)、D6-19(位置60592-61769;アクッセション番号X97051)、IGHD1-20(位置61770;アクッセション番号X97051)、D2-21(位置61770-65916;アクッセション番号X97051)、D3-22(位置65917-67762;アクッセション番号X93616)、D4-23(位置67763-68826;アクッセション番号X97051)、D5-24(位置68827-70492;アクッセション番号X97051)、D6-25(位置70493-71926;アクッセション番号X97051)、及びD1-26(位置719267-79765;アクッセション番号X97051)の約40kbを超えるヌクレオチド配列、並びにその他の配列(D7-27)を含む(M.Ruiz et al.Exp.Clin.Immunogenet.16,173-184(1999))。或いは、ヒト天然IgHD領域の配列は、NG_001019.6(ヌクレオチド番号(位置):960181~1000622(40442bp))にも開示されている。
ヒト免疫グロブリン重鎖(IgH)D領域のD1-1からD1-26までのORF配列は、例えば、以下の配列番号101~126のヌクレオチド配列である。また、短縮化後のORF間のサイズの例を各配列間に示した。
配列番号101(IGHD1-1):
ggtacaactggaacgac
(D1-1とD2-2のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号102(IGHD2-2):
aggatattgtagtagtaccagctgctatgcc
(D2-2とD3-3のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号103(IGHD3-3):
gtattacgatttttggagtggttattatacc
(D3-3とD4-4のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号104(IGHD4-4):
tgactacagtaactac
(D4-4とD5-5のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号105(IGHD5-5):
gtggatacagctatggttac
(D5-5とD6-6のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号106(IGHD6-6):
gagtatagcagctcgtcc
(D6-6とD1-7のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号107(IGHD1-7):
ggtataactggaactac
(D1-7とD2-8のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号108(IGHD2-8):
aggatattgtactaatggtgtatgctatacc
(D2-8とD3-9のORF間のサイズ+VDJ組み換え配列(28bp×2):255bp)
配列番号109(IGHD3-9):
gtattacgatattttgactggttattataac
(D3-9とD3-10のORF間のサイズ+VDJ組み換え配列(28bp×2):153bp)
配列番号110(IGHD3-10):
gtattactatggttcggggagttattataac
(D3-10とD4-11のORF間のサイズ+VDJ組み換え配列(28bp×2):316bp)
配列番号111(IGHD4-11):
tgactacagtaactac
(D4-11とD5-12のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号112(IGHD5-12):
gtggatatagtggctacgattac
(D5-12とD6-13のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号113(IGHD6-13):
gggtatagcagcagctggtac
(D6-13とD1-14のORF間のサイズ+VDJ組み換え配列(28bp×2):259bp)
配列番号114(IGHD1-14):
ggtataaccggaaccac
(D1-14とD2-15のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号115(IGHD2-15):
aggatattgtagtggtggtagctgctactcc
(D2-15とD3-16のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号116(IGHD3-16):
gtattatgattacgtttgggggagttatgcttatacc
(D3-16とD4-17のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号117(IGHD4-17):
tgactacggtgactac
(D4-17とD5-18のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号118(IGHD5-18):
gtggatacagctatggttac
(D5-18とD6-19のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号119(IGHD6-19):
gggtatagcagtggctggtac
(D6-19とD1-20のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号120(IGHD1-20):
ggtataactggaacgac
(D1-20とD2-21のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号121(IGHD2-21):
agcatattgtggtggtgattgctattcc
(D2-21とD3-22のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号122(IGHD3-22):
gtattactatgatagtagtggttattactac
(D3-22とD4-23のORF間のサイズ+VDJ組み換え配列(28bp×2):316bp)
配列番号123(IGHD4-23):
tgactacggtggtaactcc
(D4-23とD5-24のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号124(IGHD5-24):
gtagagatggctacaattac
(D5-24とD6-25のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号125(IGHD6-25):
gggtatagcagcggctac
(D6-25とD1-26のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号126(IGHD1-26):
ggtatagtgggagctactac
配列番号101(IGHD1-1):
ggtacaactggaacgac
(D1-1とD2-2のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号102(IGHD2-2):
aggatattgtagtagtaccagctgctatgcc
(D2-2とD3-3のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号103(IGHD3-3):
gtattacgatttttggagtggttattatacc
(D3-3とD4-4のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号104(IGHD4-4):
tgactacagtaactac
(D4-4とD5-5のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号105(IGHD5-5):
gtggatacagctatggttac
(D5-5とD6-6のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号106(IGHD6-6):
gagtatagcagctcgtcc
(D6-6とD1-7のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号107(IGHD1-7):
ggtataactggaactac
(D1-7とD2-8のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号108(IGHD2-8):
aggatattgtactaatggtgtatgctatacc
(D2-8とD3-9のORF間のサイズ+VDJ組み換え配列(28bp×2):255bp)
配列番号109(IGHD3-9):
gtattacgatattttgactggttattataac
(D3-9とD3-10のORF間のサイズ+VDJ組み換え配列(28bp×2):153bp)
配列番号110(IGHD3-10):
gtattactatggttcggggagttattataac
(D3-10とD4-11のORF間のサイズ+VDJ組み換え配列(28bp×2):316bp)
配列番号111(IGHD4-11):
tgactacagtaactac
(D4-11とD5-12のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号112(IGHD5-12):
gtggatatagtggctacgattac
(D5-12とD6-13のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号113(IGHD6-13):
gggtatagcagcagctggtac
(D6-13とD1-14のORF間のサイズ+VDJ組み換え配列(28bp×2):259bp)
配列番号114(IGHD1-14):
ggtataaccggaaccac
(D1-14とD2-15のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号115(IGHD2-15):
aggatattgtagtggtggtagctgctactcc
(D2-15とD3-16のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号116(IGHD3-16):
gtattatgattacgtttgggggagttatgcttatacc
(D3-16とD4-17のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号117(IGHD4-17):
tgactacggtgactac
(D4-17とD5-18のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号118(IGHD5-18):
gtggatacagctatggttac
(D5-18とD6-19のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号119(IGHD6-19):
gggtatagcagtggctggtac
(D6-19とD1-20のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号120(IGHD1-20):
ggtataactggaacgac
(D1-20とD2-21のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号121(IGHD2-21):
agcatattgtggtggtgattgctattcc
(D2-21とD3-22のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号122(IGHD3-22):
gtattactatgatagtagtggttattactac
(D3-22とD4-23のORF間のサイズ+VDJ組み換え配列(28bp×2):316bp)
配列番号123(IGHD4-23):
tgactacggtggtaactcc
(D4-23とD5-24のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号124(IGHD5-24):
gtagagatggctacaattac
(D5-24とD6-25のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号125(IGHD6-25):
gggtatagcagcggctac
(D6-25とD1-26のORF間のサイズ+VDJ組み換え配列(28bp×2):256bp)
配列番号126(IGHD1-26):
ggtatagtgggagctactac
上記改変配列は、上記ヒトIgHD領域の改変ORFを含み、かつ、ORF間領域(即ち、遺伝子間領域)の各ヒト由来ゲノム配列を、VDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上、例えば、約100bp~約500bp、約150bp~約300bp、又は約200bp~約250bp、の長さになるように短縮(若しくは、ミニマル化)することを含む。ミニマル化後の改変されたヒトIgHD領域の長さは、(通常の)プラスミドベクターに挿入可能なように約10kb以下であることが好ましい。例えば約200bpに短縮するときには、約8kbの長さにミニマル化される。
上記の短縮(若しくは、ミニマル化)の方法は、特に制限はないが、例えば、ORF間領域中、各ORFに隣接するVDJ組み換え配列末端から49bp以上は残し、ORF間領域の配列中央部から均等に削除してもよい。また、ミニマル化は、例えばヒトD領域のD1-1~D1-26を数個(例えば3~5個)のセグメントに分け、各セグメントのミニマル化を行い、得られたミニマル化セグメントを5’側から順番に連結して目的のヒトミニマル化D領域を作製することができる(図8)。ヒトミニマル化D領域のD1-1からD1-26までのORF配列、VDJ組み換え配列(28bp×2)及びミニマル化ORF間配列を含むD領域のヌクレオチド配列は、例えば下記の配列番号1のヌクレオチド配列、又は該配列と90%以上、95%以上、97%以上、98%以上、若しくは99%以上の同一性、又は該ヌクレオチド配列において1若しくは数個のヌクレオチド(若しくは塩基)の欠失、置換若しくは付加を含むヌクレオチド配列を有するものである。
配列番号1:




本明細書中で使用する「数個」とは、2~10の整数をいう。
配列番号1のようなヒトミニマル化D領域の配列は、ヒトIgHD領域ORFを異種のORFと置換するときに使用することができる。
本発明に関わる上記ヒトD領域のミニマル化によって、例えばキメラマウス脾臓Igμ-cDNAにおけるヒトミニマル化D断片の使用頻度はn=27(野生型の場合n=15)に高まる(図13)。このことは、ヒト免疫グロブリン重鎖D領域のORF間領域を短縮化しても、抗体の多様性に影響がないことを示している。
別の改変配列は、抗体重鎖CDR3のアミノ酸長が18以上となる抗体を産生することができる非ヒト動物由来の免疫グロブリン重鎖遺伝子座D領域のゲノム配列又はその改変配列を含む。
このような非ヒト動物は、例えばウシ、サル(例えば、カニクイザル)、ヒツジ、ウマ、ウサギ、トリ、サメ(例えばオオメジロザメ、メジロザメ及びコモリザメ)などの動物である。例えばthe international ImMunoGeneTics information system(IMGT)からの情報検索により、少なくとも上記の動物種が、ヒトで最長のD断片(37塩基対)より長いD断片をもつ種であることを確認することができる。以下では、ウシとサルについて具体的に説明するが、同様の手法によって、他の動物種についても上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて他の動物種由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含むように置換することができる。
ウシ免疫グロブリン重鎖遺伝子座D領域(21q24)は、5’→3’の順に、例えば、IGHDのB1-1、B2-1、B3-1、B4-1、B9-1、B1-2、B2-2、B5-2、B8-2、B6-2、B1-3、B2-3、B3-3、B7-3、B5-3、B6-3、B1-4、B2-4、B3-4、B7-4、B5-4、B6-4及びB9-4のヌクレオチド配列を含む。
上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列を含むように置換することができる。このようにして得られるウシ化ヒトミニマル化D領域のヌクレオチド配列は、例えば、下記の配列番号2のヌクレオチド配列、又は該配列と90%以上、95%以上、97%以上、98%以上、若しくは99%以上の同一性、又は該ヌクレオチド配列において1若しくは数個のヌクレオチド(若しくは塩基)の欠失、置換若しくは付加を含むヌクレオチド配列を有するヌクレオチド配列を含む。
配列番号2:




上記ウシIgHD領域のB1-1からB9-4までのORF配列は、例えば以下の配列番号127~149のヌクレオチド配列である。
配列番号127(B1-1):
agaataccgtgatgatggttactgctacacc
配列番号128(B1-2):
agaatatcgtgatgatggttactgctacacc
配列番号129(B1-3):
agactatcgtgatgatggttactgctacacccacagtgactcaggccctgacataaagtctgacccgcacacaggtgtggagctggccaatgcatccccaggggcactgggctcccaag
配列番号130(B1-4):
agaatatcgtgatgatggttactgctacacc
配列番号131(B2-1):
ttactatagtgaccac
配列番号132(B2-2):
ttactatagtgaccac
配列番号133(B2-3):
ttactatagtgaccac
配列番号134(B2-4):
ttactatagtgaccac
配列番号135(B3-1):
gtattgtggtagctattgtggtagttattatggtac
配列番号136(B3-3):
gtattgtggtagctattgtggtagttattatggtac
配列番号137(B3-4):
gtattgtggtagctattgtggtagttattatggtac
配列番号138(B4-1):
gtagttatagtggttatggttatggttatagttatggttatac
配列番号139(B5-2):
atgatacgataggtgtggttgtagttattgtagtgttgctac
配列番号140(B5-3):
atgatacgataggtgtggttttagttattgtagtgttgctac
配列番号141(B5-4):
atgatacgataggtgtggttttagttattgtagtgttgctac
配列番号142(B6-2):
gtagttgttatagtggttatggttatggttgtggttatggttatggttatgattatac
配列番号143(B6-3):
gtagttgttatagtggttatggttatggttatggttgtggttatggttatggttatac
配列番号144(B6-4):
gtagttgttatagtggttatggttatggttatggttgtggttatggttatggttatac
配列番号145(B7-3):
gtagttatggtggttatggttatggtggttatggttgttatggttatggttatggttatggttatac
配列番号146(B7-4):
gtagttatggtggttatggttatggtggttatggttgttatggttatggttatggttatggttatggttatac
配列番号147(B8-2):
gtagttgtcctgatggttatagttatggttatggttgtggttatggttatggttgtagtggttatgattgttatggttatggtggttatggtggttatggtggttatggttatagtagttatagttatagttatacttacgaatatac
配列番号148(B9-1):
gaactcggtggggc
配列番号149(B9-4):
gaactcggtggggc
配列番号127(B1-1):
agaataccgtgatgatggttactgctacacc
配列番号128(B1-2):
agaatatcgtgatgatggttactgctacacc
配列番号129(B1-3):
agactatcgtgatgatggttactgctacacccacagtgactcaggccctgacataaagtctgacccgcacacaggtgtggagctggccaatgcatccccaggggcactgggctcccaag
配列番号130(B1-4):
agaatatcgtgatgatggttactgctacacc
配列番号131(B2-1):
ttactatagtgaccac
配列番号132(B2-2):
ttactatagtgaccac
配列番号133(B2-3):
ttactatagtgaccac
配列番号134(B2-4):
ttactatagtgaccac
配列番号135(B3-1):
gtattgtggtagctattgtggtagttattatggtac
配列番号136(B3-3):
gtattgtggtagctattgtggtagttattatggtac
配列番号137(B3-4):
gtattgtggtagctattgtggtagttattatggtac
配列番号138(B4-1):
gtagttatagtggttatggttatggttatagttatggttatac
配列番号139(B5-2):
atgatacgataggtgtggttgtagttattgtagtgttgctac
配列番号140(B5-3):
atgatacgataggtgtggttttagttattgtagtgttgctac
配列番号141(B5-4):
atgatacgataggtgtggttttagttattgtagtgttgctac
配列番号142(B6-2):
gtagttgttatagtggttatggttatggttgtggttatggttatggttatgattatac
配列番号143(B6-3):
gtagttgttatagtggttatggttatggttatggttgtggttatggttatggttatac
配列番号144(B6-4):
gtagttgttatagtggttatggttatggttatggttgtggttatggttatggttatac
配列番号145(B7-3):
gtagttatggtggttatggttatggtggttatggttgttatggttatggttatggttatggttatac
配列番号146(B7-4):
gtagttatggtggttatggttatggtggttatggttgttatggttatggttatggttatggttatggttatac
配列番号147(B8-2):
gtagttgtcctgatggttatagttatggttatggttgtggttatggttatggttgtagtggttatgattgttatggttatggtggttatggtggttatggtggttatggttatagtagttatagttatagttatacttacgaatatac
配列番号148(B9-1):
gaactcggtggggc
配列番号149(B9-4):
gaactcggtggggc
カニクイザル免疫グロブリン重鎖遺伝子座D領域(Chr7q;G.-Y.Yu et al.,Immunogenetics 2016;68:417-428)は、5’→3’の順に、例えば、IGHDの1S5,2S11,3S6,4S24,5S8,6S4,1S10,2S17,3S18,2S34,5S14,5S31,6S3,1S27,2S22,4S36,4S30,5S37,6S20,1S33,2S28,6S32,3S12,6S38,6S26,及び1S39の約44kbのヌクレオチド配列を含む(図15)。
上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、サル免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列を含むように置換する。
上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、サル免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列を含むように置換する。
カニクイザルD領域の1S5から1S35までのORF配列は、例えば以下の配列番号150~175のヌクレオチド配列である。
配列番号150(1S5):
ggtataactggaactac
配列番号151(2S11):
agaatattgtagtagtacttactgctcctcc
配列番号152(3S6):
gtattacgaggatgattacggttactattacacccacagcgt
配列番号153(4S24):
tgactacggtagcagctac
配列番号154(5S8):
gtggatacagctacagttac
配列番号155(6S4):
gggtatagcagcggctggtac
配列番号156(1S10):
ggtatagctggaacgac
配列番号157(2S17):
agaatactgtactggtagtggttgctatgcc
配列番号158(3S18):
gtactggggtgattattatgac
配列番号159(2S34):
agcatattgtagtggtggtgtctgctacacc
配列番号160(5S14):
gtggatacagctacagttaccacagttttgccacc
配列番号161(5S31):
gtggatatagctacggttac
配列番号162(6S9):
gggtatagcagctggtcc
配列番号163(1S27):
ggtataactggaatgac
配列番号164(2S22):
agaatattgtagtggtatttactgctatgcc
配列番号165(4S36):
tgaatacagtaactac
配列番号166(4S30):
tgactacggtaactac
配列番号167(5S37):
ggggatacagtgggtacagttac
配列番号168(6S20):
gggtatagcggcagctggaac
配列番号169(1S33):
ggaacacctggaacgac
配列番号170(2S28):
agcacactgtagtgatagtggctgctcctcc
配列番号171(6S32):
gggtatagcggtggctggtcc
配列番号172(3S12):
gtattactatagtggtagttattactaccacagtgt
配列番号173(6S38):
gggtatagcagcagcta
配列番号174(6S26):
gggtatagcagcggctggtcc
配列番号175(1S39):
ggtatagtgggaactacaac
配列番号150(1S5):
ggtataactggaactac
配列番号151(2S11):
agaatattgtagtagtacttactgctcctcc
配列番号152(3S6):
gtattacgaggatgattacggttactattacacccacagcgt
配列番号153(4S24):
tgactacggtagcagctac
配列番号154(5S8):
gtggatacagctacagttac
配列番号155(6S4):
gggtatagcagcggctggtac
配列番号156(1S10):
ggtatagctggaacgac
配列番号157(2S17):
agaatactgtactggtagtggttgctatgcc
配列番号158(3S18):
gtactggggtgattattatgac
配列番号159(2S34):
agcatattgtagtggtggtgtctgctacacc
配列番号160(5S14):
gtggatacagctacagttaccacagttttgccacc
配列番号161(5S31):
gtggatatagctacggttac
配列番号162(6S9):
gggtatagcagctggtcc
配列番号163(1S27):
ggtataactggaatgac
配列番号164(2S22):
agaatattgtagtggtatttactgctatgcc
配列番号165(4S36):
tgaatacagtaactac
配列番号166(4S30):
tgactacggtaactac
配列番号167(5S37):
ggggatacagtgggtacagttac
配列番号168(6S20):
gggtatagcggcagctggaac
配列番号169(1S33):
ggaacacctggaacgac
配列番号170(2S28):
agcacactgtagtgatagtggctgctcctcc
配列番号171(6S32):
gggtatagcggtggctggtcc
配列番号172(3S12):
gtattactatagtggtagttattactaccacagtgt
配列番号173(6S38):
gggtatagcagcagcta
配列番号174(6S26):
gggtatagcagcggctggtcc
配列番号175(1S39):
ggtatagtgggaactacaac
或いは、別の改変配列は、上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORFに代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む。
ヒト抗体のなかには頻度は低いが長鎖CDRH3配列の存在が知られている(L.Yu and Y.Guan,Frontiers in Immunology,2014,24,doi:10.3389/fimmu.2014.00250)。
このような改変D領域ORFは、例えば図16に示されるような配列番号75~100に示されるヌクレオチド配列、又は該ヌクレオチド配列と90%以上、95%以上、97%以上、98%以上、若しくは99%以上の同一性を有するヌクレオチド配列、又は該ヌクレオチド配列において1若しくは数個のヌクレオチド(若しくは塩基)の欠失、置換若しくは付加を含むヌクレオチド配列を含むものであり、ヒトIgHD領域のD1-1からD1-26までのORFを、対応する26種類の上記改変ORF配列と置換することができる。そのように置換したヒトミニマル化D領域のヌクレオチド配列は、例えば、配列番号3のヌクレオチド配列、又は該ヌクレオチド配列と90%以上、95%以上、97%以上、98%以上、若しくは99%以上の同一性を有するヌクレオチド配列、又は該ヌクレオチド配列において1若しくは数個のヌクレオチド(若しくは塩基)の欠失、置換若しくは付加を含むヌクレオチド配列を含むものである。
配列番号3:




ヒト抗体重鎖CDR3配列に基づいて改変ORF配列を決定する方法は、例えば以下に示すような工程を含むことができる。
この例では、ヒト抗体重鎖アミノ酸配列のVHのC末端側の配列「CAR」/「CAK」以降、VHのN末端側の配列「WG」より上流をCDR3と定義する(M.Chiu et al.,Antibodies 2019;8(4):55;doi.org/10.3390/antib8040055)。
第1工程では、CDR3が18アミノ酸以上となる公知の配列を検索する。
第2工程では、CDR3の塩基配列を抽出する。
第3工程では、体細胞突然変異ホットスポット配列に、同義(アミノ酸が変わらない)置換変異を可能な範囲で導入する。
この例では、ヒト抗体重鎖アミノ酸配列のVHのC末端側の配列「CAR」/「CAK」以降、VHのN末端側の配列「WG」より上流をCDR3と定義する(M.Chiu et al.,Antibodies 2019;8(4):55;doi.org/10.3390/antib8040055)。
第1工程では、CDR3が18アミノ酸以上となる公知の配列を検索する。
第2工程では、CDR3の塩基配列を抽出する。
第3工程では、体細胞突然変異ホットスポット配列に、同義(アミノ酸が変わらない)置換変異を可能な範囲で導入する。
本明細書中の「同一性」は、BLASTやFASTAによるタンパク質又は遺伝子の検索システムを用いて、ギャップを導入して、又はギャップを導入しないで、決定することができる(Zheng Zhang et al.,J.Comput.Biol.2000;7:203-214;Altschul,S.F.et al.,Journal of Molecular Biology1990;215:403-410;Pearson,W.R.et al.,Proc.Natl.Acad.Sci.U.S.A.、1988;85:2444-2448)。
[ヒト免疫グロブリン重鎖遺伝子座のD領域の改変方法]
ヒト免疫グロブリン重鎖遺伝子座のD領域は、例えば次のような工程を含む方法によって改変することができる。
ヒト免疫グロブリン重鎖遺伝子座のD領域は、例えば次のような工程を含む方法によって改変することができる。
<第1工程>
ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳類人工染色体ベクター([Ig-NAC])を準備する。
ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳類人工染色体ベクター([Ig-NAC])を準備する。
Ig-NACは、テロメア側からセントロメア側に向かってヒト免疫グロブリン重鎖遺伝子座、ヒト免疫グロブリン軽鎖κ(若しくはλ)遺伝子座を含む構造を有する(例えば特許第6868250号公報)。
Ig-NACは、導入すべき細胞本来の染色体から独立した染色体として安定に複製及び分配が可能である。このベクターの例は、齧歯類人工染色体ベクター(例えば、マウス又はラット人工染色体ベクター)である。齧歯類人工染色体ベクターの作製は、例えば、本出願人による特許第6775224号公報、特許第6868250号公報などに記載されている。
具体的な作製法は以下の通りである。
哺乳動物人工染色体ベクターは、例えば哺乳動物染色体のテロメアトランケーションによって作製されてもよい、該動物のセントロメア、セントロメア近傍の長腕断片及び、もしその動物に存在すれば、短腕断片、及びテロメアを含む。例えば、マウス由来の染色体断片は、マウスの1~19番、X及びY染色体のうちの任意の染色体、好ましくは1番~19番染色体のいずれかの断片(長腕の全内在遺伝子数の少なくとも99.5%、好ましくはほぼ100%が削除された長腕断片)であり、該断片には、セントロメア近傍のマウス染色体長腕の部位から長腕遠位が削除された長腕断片が含まれる。マウスの染色体の配列情報は、DDBJ/EMBL/GenBank、Santa Cruz Biotechnology,Inc.などのChromosome Databasesから入手可能である。より具体的には、例えばマウス15番染色体断片由来の人工染色体ベクターの場合、上記長腕断片は、非限定的に例えば、AC121307、AC161799などの位置よりも遠位の領域が削除された長腕断片からなる。或いは、マウス16番染色体断片由来の人工染色体ベクターの場合、前記長腕断片は、非限定的に例えばGm35974より遠位の領域が削除された長腕断片からなる。或いは、マウス10番染色体長腕の染色体部位である遺伝子Gm8155から長腕遠位を削除したマウス10番染色体由来の長腕断片からなる。
哺乳動物人工染色体ベクターは、例えば哺乳動物染色体のテロメアトランケーションによって作製されてもよい、該動物のセントロメア、セントロメア近傍の長腕断片及び、もしその動物に存在すれば、短腕断片、及びテロメアを含む。例えば、マウス由来の染色体断片は、マウスの1~19番、X及びY染色体のうちの任意の染色体、好ましくは1番~19番染色体のいずれかの断片(長腕の全内在遺伝子数の少なくとも99.5%、好ましくはほぼ100%が削除された長腕断片)であり、該断片には、セントロメア近傍のマウス染色体長腕の部位から長腕遠位が削除された長腕断片が含まれる。マウスの染色体の配列情報は、DDBJ/EMBL/GenBank、Santa Cruz Biotechnology,Inc.などのChromosome Databasesから入手可能である。より具体的には、例えばマウス15番染色体断片由来の人工染色体ベクターの場合、上記長腕断片は、非限定的に例えば、AC121307、AC161799などの位置よりも遠位の領域が削除された長腕断片からなる。或いは、マウス16番染色体断片由来の人工染色体ベクターの場合、前記長腕断片は、非限定的に例えばGm35974より遠位の領域が削除された長腕断片からなる。或いは、マウス10番染色体長腕の染色体部位である遺伝子Gm8155から長腕遠位を削除したマウス10番染色体由来の長腕断片からなる。
マウス人工染色体ベクターの例は、寄託細胞株DT40(10MAC)T5-26(国際寄託番号NITE BP-02656)に含まれるマウス人工染色体、或いは、寄託細胞株DT40(16MAC)T1-14(国際寄託番号NITE BP-02657)に含まれるマウス人工染色体を包含する。
本発明の哺乳動物人工染色体ベクターは、例えば以下の工程(a)~(c):
(a)哺乳動物染色体を保持する細胞を得る工程、
(b)内在遺伝子(数)の大部分(99.5%~100%、好ましくは100%)を含まないようにマウス染色体の長腕遠位を削除する工程、及び
(c)長腕近位に1つ以上のDNA配列挿入部位を挿入する工程
を含む方法によって作製することができる。ここで、工程(b)及び(c)の順序は逆であってもよい。
(a)哺乳動物染色体を保持する細胞を得る工程、
(b)内在遺伝子(数)の大部分(99.5%~100%、好ましくは100%)を含まないようにマウス染色体の長腕遠位を削除する工程、及び
(c)長腕近位に1つ以上のDNA配列挿入部位を挿入する工程
を含む方法によって作製することができる。ここで、工程(b)及び(c)の順序は逆であってもよい。
以下、各工程について説明する。
<工程(1a)>
本発明の哺乳動物人工染色体ベクターを作製するには、まず、哺乳動物染色体を保持する細胞を作製する。例えば、薬剤耐性遺伝子(例えば、blasticidin S registance gene(BSr))で標識された哺乳動物染色体を保持する哺乳動物線維芽細胞とG418耐性遺伝子であるneo遺伝子を導入したマウスA9細胞(ATCC VA20110-2209)とを細胞融合し、薬剤耐性遺伝子で標識された哺乳動物染色体を保持するマウスA9雑種細胞から、その染色体を相同組み換え率の高い細胞に移入することにより作製することができる。哺乳動物線維芽細胞は、文献記載の方法に基づいて入手することが可能であり、例えば、マウス線維芽細胞は日本クレアより入手可能なC57B6系統のマウスより樹立可能である。相同組み換え率の高い細胞としては、例えば、ニワトリDT40細胞(Dieken et al.,Nature Genetics,12:174-182,1996)を利用できる。さらにまた、上記移入は、公知の染色体移入法、例えば、微小核細胞融合法(Koi et al.,Jpn.J.Cancer Res.,80:413-418,1973)によって行うことができる。
<工程(1a)>
本発明の哺乳動物人工染色体ベクターを作製するには、まず、哺乳動物染色体を保持する細胞を作製する。例えば、薬剤耐性遺伝子(例えば、blasticidin S registance gene(BSr))で標識された哺乳動物染色体を保持する哺乳動物線維芽細胞とG418耐性遺伝子であるneo遺伝子を導入したマウスA9細胞(ATCC VA20110-2209)とを細胞融合し、薬剤耐性遺伝子で標識された哺乳動物染色体を保持するマウスA9雑種細胞から、その染色体を相同組み換え率の高い細胞に移入することにより作製することができる。哺乳動物線維芽細胞は、文献記載の方法に基づいて入手することが可能であり、例えば、マウス線維芽細胞は日本クレアより入手可能なC57B6系統のマウスより樹立可能である。相同組み換え率の高い細胞としては、例えば、ニワトリDT40細胞(Dieken et al.,Nature Genetics,12:174-182,1996)を利用できる。さらにまた、上記移入は、公知の染色体移入法、例えば、微小核細胞融合法(Koi et al.,Jpn.J.Cancer Res.,80:413-418,1973)によって行うことができる。
<工程(1b)>
哺乳動物由来の単一の染色体を保持する細胞において、該哺乳動物染色体の長腕遠位及び、もし動物に存在すれば、短腕遠位を削除する。このとき、重要なことは、長腕及び短腕上に存在する内在遺伝子の大部分を削除(又は、除去若しくは欠失)し、哺乳動物セントロメアを保持する人工染色体を構築することである。これは長腕及び短腕上に存在する全内在遺伝子(数)の少なくとも99.5%、好ましくは少なくとも99.7%、より好ましくは少なくとも99.8%、最も好ましくは99.9~100%、さらに最も好ましくは100%を削除(又は、除去若しくは欠失)するように切断位置を決定することである。そうすることによって、人工染色体が導入された、哺乳動物由来の、好ましくはマウス、ラットなどのげっ歯類由来の、細胞、組織又は個体において安定かつ高保持率で保持される。上記内在遺伝子の削除は、例えば、テロメアトランケーションにより行うことができる。具体的には、哺乳動物染色体を保持する細胞において、テロメア配列を保持するターゲティングベクターを構築し、相同組み換えにより染色体上の所望の位置に人工若しくは天然テロメア配列が挿入されたクローンを取得し、これによってテロメアトランケーションにより欠失変異体が得られる。例えば、所望の位置(又は、部位)が削除すべき長腕遠位の切断位置であり、この位置にテロメア配列が相同組み換えにより置換、挿入されて長腕遠位が削除される。このような位置は、ターゲティングベクターを構築する際の標的配列の設計により、適宜設定できる。例えば、哺乳動物染色体長腕のDNA配列に基づいて標的配列を設計し、その標的配列よりもテロメア側でテロメアトランケーションが起こるように設定される。これにより、内在遺伝子の大部分が削除された哺乳動物染色体断片が得られる。他の染色体の場合にも同様にテロメアトランケーションを実施できる。短腕遠位の切断の場合も同様である。
哺乳動物由来の単一の染色体を保持する細胞において、該哺乳動物染色体の長腕遠位及び、もし動物に存在すれば、短腕遠位を削除する。このとき、重要なことは、長腕及び短腕上に存在する内在遺伝子の大部分を削除(又は、除去若しくは欠失)し、哺乳動物セントロメアを保持する人工染色体を構築することである。これは長腕及び短腕上に存在する全内在遺伝子(数)の少なくとも99.5%、好ましくは少なくとも99.7%、より好ましくは少なくとも99.8%、最も好ましくは99.9~100%、さらに最も好ましくは100%を削除(又は、除去若しくは欠失)するように切断位置を決定することである。そうすることによって、人工染色体が導入された、哺乳動物由来の、好ましくはマウス、ラットなどのげっ歯類由来の、細胞、組織又は個体において安定かつ高保持率で保持される。上記内在遺伝子の削除は、例えば、テロメアトランケーションにより行うことができる。具体的には、哺乳動物染色体を保持する細胞において、テロメア配列を保持するターゲティングベクターを構築し、相同組み換えにより染色体上の所望の位置に人工若しくは天然テロメア配列が挿入されたクローンを取得し、これによってテロメアトランケーションにより欠失変異体が得られる。例えば、所望の位置(又は、部位)が削除すべき長腕遠位の切断位置であり、この位置にテロメア配列が相同組み換えにより置換、挿入されて長腕遠位が削除される。このような位置は、ターゲティングベクターを構築する際の標的配列の設計により、適宜設定できる。例えば、哺乳動物染色体長腕のDNA配列に基づいて標的配列を設計し、その標的配列よりもテロメア側でテロメアトランケーションが起こるように設定される。これにより、内在遺伝子の大部分が削除された哺乳動物染色体断片が得られる。他の染色体の場合にも同様にテロメアトランケーションを実施できる。短腕遠位の切断の場合も同様である。
<工程(1c)>
DNA配列挿入部位として、好ましくは部位特異的組み換え酵素の認識部位を挿入することができる。すなわち、ある種の酵素が特定の認識部位を認識して特異的にその認識部位でDNAの組み換えを起こす現象が知られており、本発明における哺乳動物人工染色体ベクターでは、このような酵素とその酵素の認識部位からなる系を利用して、目的とするヒト免疫グロブリン重鎖若しくは遺伝子座、及び/又は、ヒト免疫グロブリン軽鎖遺伝子又は遺伝子座の配列を挿入、搭載できる。このような系として、例えば、バクテリオファージP1由来のCre酵素と、その認識部位であるloxP配列の系(Cre/loxP系;B.Sauer in Methods of Enzymology;1993,225:890-900)や、出芽酵母由来のFlp酵素と、その認識部位であるFRT(Flp Recombination Target)配列の系(Flp/FRT系)や、ストレプトミセスファージ由来のφC31インテグレースと、その認識部位であるφC31attB/attP配列の系、R4インテグレースと、その認識部位であるR4attB/attP配列の系、TP901-1インテグレースと、その認識部位であるTP901-1attB/attP配列の系、Bxb1インテグレースと、その認識部位であるBxb1attB/attP配列の系、などを挙げることができるが、DNA配列挿入部位として機能しうるのであれば、上記の系に限定されないものとする。
DNA配列挿入部位として、好ましくは部位特異的組み換え酵素の認識部位を挿入することができる。すなわち、ある種の酵素が特定の認識部位を認識して特異的にその認識部位でDNAの組み換えを起こす現象が知られており、本発明における哺乳動物人工染色体ベクターでは、このような酵素とその酵素の認識部位からなる系を利用して、目的とするヒト免疫グロブリン重鎖若しくは遺伝子座、及び/又は、ヒト免疫グロブリン軽鎖遺伝子又は遺伝子座の配列を挿入、搭載できる。このような系として、例えば、バクテリオファージP1由来のCre酵素と、その認識部位であるloxP配列の系(Cre/loxP系;B.Sauer in Methods of Enzymology;1993,225:890-900)や、出芽酵母由来のFlp酵素と、その認識部位であるFRT(Flp Recombination Target)配列の系(Flp/FRT系)や、ストレプトミセスファージ由来のφC31インテグレースと、その認識部位であるφC31attB/attP配列の系、R4インテグレースと、その認識部位であるR4attB/attP配列の系、TP901-1インテグレースと、その認識部位であるTP901-1attB/attP配列の系、Bxb1インテグレースと、その認識部位であるBxb1attB/attP配列の系、などを挙げることができるが、DNA配列挿入部位として機能しうるのであれば、上記の系に限定されないものとする。
このような部位特異的組み換え酵素の認識部位の挿入のためには、公知の方法、例えば、相同組み換え法が利用でき、挿入位置及び数は、長腕近位及び短腕近位内に適宜設定することができる。
哺乳動物人工染色体ベクターには、1つの種類の認識部位又は異なる種類の認識部位を挿入することができる。認識部位の設定により、目的とする遺伝子若しくは遺伝子座、すなわち、ヒト免疫グロブリン重鎖遺伝子若しくは遺伝子座、ヒト免疫グロブリン軽鎖κ遺伝子若しくは遺伝子座、又はヒト免疫グロブリン軽鎖λ遺伝子若しくは遺伝子座の挿入位置を特定することができるので、挿入位置が一定となり、想定外の位置効果(position effect)を受けることもなくなる。
哺乳動物人工染色体ベクターには、好ましくは、目的とする上記遺伝子又は遺伝子座の配列の他に、レポーター遺伝子を挿入してもよい。レポーター遺伝子としては、特に限定するものではないが、例えば、蛍光タンパク質(例えば、緑色蛍光タンパク質(GFP又はEGFP)、黄色蛍光タンパク質(YFP)、など)遺伝子、タグタンパク質コードDNA、β-ガラクトシダーゼ遺伝子、ルシフェラーゼ遺伝子などが挙げられる。
哺乳動物人工染色体ベクターにはさらに、選択マーカー遺伝子を含んでもよい。選択マーカーは、該ベクターで形質転換された細胞を選別する際に有効である。選択マーカー遺伝子としては、ポジティブ選択マーカー遺伝子及びネガティブ選択マーカー遺伝子のいずれか、又はその両方が例示される。ポジティブ選択マーカー遺伝子には、薬剤耐性遺伝子、例えばネオマイシン耐性遺伝子、アンピシリン耐性遺伝子、ブラストサイジンS(BS)耐性遺伝子、ピューロマイシン耐性遺伝子、ジェネティシン(G418)耐性遺伝子、ハイグロマイシン耐性遺伝子などが含まれる。また、ネガティブ選択マーカー遺伝子には、例えば単純ヘルペスチミジンキナーゼ(HSV-TK)遺伝子、ジフテリアトキシンA断片(DT-A)遺伝子などが包含される。一般に、HSV-TKは、ガンシクロビル又はアシクロビルと組み合わせて使用される。
本明細書中で使用する「ヒト免疫グロブリン遺伝子若しくは遺伝子座」は、特に断らない限り、ヒト14番染色体由来のヒト免疫グロブリン重鎖遺伝子若しくは遺伝子座、ヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子若しくは遺伝子座、及び/又はヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子若しくは遺伝子座を指す。具体的には、ヒト免疫グロブリン遺伝子若しくは遺伝子座は、例えばヒト14番染色体のimmunoglobulin heavy locus(human)NC_000014.9((塩基番号105586437..106879844)或いは(塩基番号105264221..107043718))、ヒト2番染色体のimmunoglobulin kappa locus (human)NC_000002.12((塩基番号88857361..90235368)或いは(塩基番号88560086..90265666))、及びヒト22番染色体のimmunoglobulin lambda locus(human)NC_000022.11((塩基番号22026076..22922913)或いは(塩基番号21620362..23823654))に記載される塩基配列よって表される。ヒト免疫グロブリン重鎖遺伝子若しくは遺伝子座は、約1,3Mbの塩基長であり、ヒト免疫グロブリン軽鎖κ遺伝子若しくは遺伝子座は、約1.4Mbの塩基長であり、ヒト免疫グロブリン軽鎖λ遺伝子若しくは遺伝子座は、約0.9Mbの塩基長である。
因みに、マウスの抗体重鎖遺伝子若しくは遺伝子座は、マウス第12番染色体上にあり、マウスの抗体軽鎖κ遺伝子若しくは遺伝子座は、マウス第6番染色体上にあり、マウス抗体軽鎖λ遺伝子若しくは遺伝子座は、マウス第16番染色体上にある。具体的には、マウス抗体重鎖遺伝子若しくは遺伝子座は、例えばChromosome 12,NC_000078.6(113258768..116009954,complement)、マウス抗体軽鎖κ遺伝子若しくは遺伝子座は、Chromosome 6,NC_000072.6(67555636..70726754)、マウス抗体軽鎖λ遺伝子若しくは遺伝子座は、Chromosome 16,NC_000082.6(19026858..19260844,complement)に記載される塩基配列よって表される。
また、ラットの抗体重鎖遺伝子若しくは遺伝子座は、ラット第6番染色体上にあり、ラットの抗体軽鎖κ遺伝子若しくは遺伝子座は、ラット第4番染色体上にあり、ラット抗体軽鎖λ遺伝子若しくは遺伝子座は、ラット第11番染色体上にある。同様に、これらの遺伝子若しくは遺伝子座の塩基配列は、米国NCBI(GenBankなど)、公知文献などから入手可能である。
<第2工程>
上記第1工程で作製されたヒト免疫グロブリン(重鎖及び/又は軽鎖)遺伝子座を含む哺乳動物人工染色体ベクター(Ig-NAC)において、ヒト免疫グロブリン重鎖遺伝子座から、例えばゲノム編集(J.M.Chylinski et al.,Science 2012;337:816-821)や部位特異的組み換え技術(例えば、部位特異的組み換え酵素の使用)などの方法により、ヒト重鎖遺伝子座のV領域とJ領域の間に存在するD領域を削除して「Ig-NAC(ΔDH)」を作製する。
上記第1工程で作製されたヒト免疫グロブリン(重鎖及び/又は軽鎖)遺伝子座を含む哺乳動物人工染色体ベクター(Ig-NAC)において、ヒト免疫グロブリン重鎖遺伝子座から、例えばゲノム編集(J.M.Chylinski et al.,Science 2012;337:816-821)や部位特異的組み換え技術(例えば、部位特異的組み換え酵素の使用)などの方法により、ヒト重鎖遺伝子座のV領域とJ領域の間に存在するD領域を削除して「Ig-NAC(ΔDH)」を作製する。
本明細書中の「ゲノム編集」は、例えばTALEN(TALEヌクレアーゼ)などの人工切断酵素、CRISPR-Casシステムなどを使用してゲノムDNAの編集や遺伝子改変を行う技術である。本発明の方法では、ゲノム編集の任意の技術を使用することができるが、好ましくはCRISPR-Casシステムの使用である。
特にCRISPR/Cas9システムは、細菌や古細菌のウイルスやプラスミドに対する適応免疫機構から発見されたが、ベクターの構築が比較的簡便であり、複数の遺伝子を同時に改変することが可能である(Jinek et al.,Science,17,337(6096):816-821,2012;Sander et al.,Nature biotechnology,32(4):347-355,2014)。このシステムは、Cas9タンパク質と約20塩基対標的配列をもつガイドRNA(guide RNA;gRNA)を含む。細胞内でこれらを共発現させることにより、gRNAが標的配列近傍のPAM配列を認識して標的ゲノムDNAと特異的に結合し、Cas9タンパク質がPAM配列の5’側上流で二本鎖切断を誘導する。現在最も使用されているCas9タンパク質はSpCas9型であり、PAM配列がNGG(N=C,G,A若しくはT)であることから変異導入位置の制約が少ない。本発明では、ガイドRNAが標的とするヒト免疫グロブリン重鎖遺伝子座の部位は、非限定的に、例えばヒトD1-1断片の5’上流部位であり、ガイドRNAの標的配列は、例えば5’-AGATCCTCCATGCGTGCTGTGGG-3’(配列番号4)である。
本明細書中の「部位特異的組み換え酵素」とは、これら酵素の認識部位で特異的に目的のDNA配列と組み換えを起こすための酵素である。このような組み換え酵素とその認識部位とを利用することによって部位特異的組み換えを行うことができる。部位特異的組み換え酵素の例は、Creインテグラーゼ(Creリコンビナーゼとも称する。)、Flpリコンビナーゼ、φC31インテグラーゼ、R4インテグラーゼ、TP901-1インテグラーゼ、Bxb1インテグラーゼなどである。また、該酵素の認識部位には、例えばloxP(Creリコンビナーゼ認識部位)、FRT(Flpリコンビナーゼ認識部位)、φC31attB及びφC31attP(φC31リコンビナーゼ認識部位)、R4attB及びR4attP(R4リコンビナーゼ認識部位)、TP901-1attB及びTP901-1attP(TP901-1リコンビナーゼ認識部位)、或いはBxb1attB及びBxb1attP(Bxb1リコンビナーゼ認識部位)などが含まれる。
ヒト免疫グロブリン重鎖遺伝子座のD領域を削除した部位に、合成D領域ポリヌクレオチドを導入するためのアクセプター部位を挿入する。アクセプター部位の例は、部位特異的組み換え酵素の認識部位を含むカセットである。該アクセプター部位の挿入方法の例は、下記の<第3工程>に記載されている。
ここで、部位特異的組み換え酵素とその認識部位は、例えば上記のCreリコンビナーゼとloxPの系、FlpリコンビナーゼとFRTの系、φC31リコンビナーゼとφC31attB及びφC31attPの系などである。部位特異的組み換え酵素(例えばCre)は、認識部位配列間(例えばloxP間)で部位特異的組み換え反応を触媒することができる。
<第3工程>
部位特異的組み換え技術、ゲノム編集技術などの方法によって、上記アクセプター部位に合成D領域ポリヌクレオチドを挿入する。
部位特異的組み換え技術、ゲノム編集技術などの方法によって、上記アクセプター部位に合成D領域ポリヌクレオチドを挿入する。
合成D領域ポリヌクレオチドは、上記セクション1に記載したような、上記ヒト免疫グロブリン重鎖遺伝子座のD領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されている改変D領域であり、上記改変配列が、
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く上記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメなどの脊椎動物由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ように改変された配列である。
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く上記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメなどの脊椎動物由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ように改変された配列である。
一実施形態において、上記の哺乳動物人工染色体ベクターの製造においてヒト免疫グロブリン重鎖D領域をその改変配列(上記「合成D領域ポリヌクレオチド」と同義である。)で置換する方法は、下記の工程を含むことができる。
(1’)ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを準備する、
(2’)工程(1’)の前記哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除する、
(3’)工程(2’)で得られたベクターの前記D領域の削除部位に、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、
(4’)工程(3’)で挿入された前記カセットの第2の部位特異的組み換え酵素認識部位に、第2の部位特異的組み換え酵素を用いて、前記ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、選択マーカー及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、並びに、
(5’)第1の部位特異的組み換え酵素を用いる部位特異的組み換えによって、第1の部位特異的組み換え酵素認識部位、前記ヒト免疫グロブリン重鎖D領域の改変配列及び第2の部位特異的組み換え酵素認識部位を含むカセットが前記D領域の削除部位に挿入された前記人工染色体ベクターを得る。
(1’)ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを準備する、
(2’)工程(1’)の前記哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除する、
(3’)工程(2’)で得られたベクターの前記D領域の削除部位に、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、
(4’)工程(3’)で挿入された前記カセットの第2の部位特異的組み換え酵素認識部位に、第2の部位特異的組み換え酵素を用いて、前記ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、選択マーカー及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、並びに、
(5’)第1の部位特異的組み換え酵素を用いる部位特異的組み換えによって、第1の部位特異的組み換え酵素認識部位、前記ヒト免疫グロブリン重鎖D領域の改変配列及び第2の部位特異的組み換え酵素認識部位を含むカセットが前記D領域の削除部位に挿入された前記人工染色体ベクターを得る。
上記工程における、ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、第2の部位特異的組み換え酵素認識部位、第1の部位特異的組み換え酵素、及び第2の部位特異的組み換え酵素は、制限はないが、例えば上に説明したものを使用できる。また、プロモーターも制限はないが、例えばCAGプロモーターなどを使用できる。
工程(2’)において、工程(1’)の哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除するために、例えば、上記のゲノム編集、部位特異的組み換えなどの技術を使用することができる。
具体的には、上記Ig-NAC(ΔDH)のヒト重鎖D領域切断部位に上記合成D領域ポリヌクレオチドを挿入するために、該ポリヌクレオチドと、その5’末端に部位特異的組み換え酵素認識部位含有カセット(例えば、φC31-BsdR-oriC-R4の配列)とを含むベクターを作製し、該ベクターと上記Ig-NAC(ΔDH)との間で部位特異的組み換え反応を行ってIg-NAC(ΔDH)のヒト重鎖D領域切断部位に合成D領域ポリヌクレオチドを挿入することができる(図5、6、9及び10)。
或いは、Ig-NAC(ΔDH)のヒト重鎖D領域切断部位に合成D領域ポリヌクレオチドを挿入するために、ヒト免疫グロブリン重鎖V領域部分配列とヒト免疫グロブリン重鎖J領域(及び、場合により、C領域)の部分配列との間に部位特異的組み換え酵素認識部位を含むベクターを作製し、このベクターとIg-NAC(ΔDH)の間でゲノム編集(Cas9/gRNA)を行ってIg-NAC(ΔDH)のヒト重鎖D領域切断部位に上記部位特異的組み換え酵素認識部位を挿入し、さらに別の部位特異的組み換え酵素を作用して、上記Ig-NAC(ΔDH)のヒト重鎖D領域切断部位に部位特異的組み換え酵素認識部位を含むIg-NACを作製する。さらに、このIg-NACに上記合成D領域ポリヌクレオチドを挿入するために、該ポリヌクレオチドと、その5’末端に部位特異的組み換え酵素認識部位とを含むベクターを作製し、部位特異的組み換え酵素(例えばφC31リコンビナーゼ)を作用し、さらに別の部位特異的組み換え酵素(例えばBxb1リコンビナーゼ又はR4リコンビナーゼ)を作用して、上記Ig-NAC(ΔDH)のヒト重鎖D領域切断部位に合成D領域ポリヌクレオチドを含む目的の改変Ig-NACを作製することができる(図10)。
本発明はさらに、別の態様において、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターであって、前記ヒト免疫グロブリン重鎖遺伝子座がD領域を欠失しており、該欠失されたD領域に代えて、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含む、哺乳動物人工染色体ベクターを提供する。
上記ベクターにはさらに、第3の部位特異的組み換え酵素認識部位を含んでもよい。
上記哺乳動物人工染色体ベクターの例は、上記工程(3’)で作製されるベクターである(例えば図9)。
上記欠失されたD領域が、D1-1からD1-26までの領域であるか、或いは、D1-1からD7-27までの領域である。
上記ベクターにおいて、第1の部位特異的組み換え酵素認識部位及び第2の部位特異的組み換え酵素認識部位は、制限はないが、例えば上に説明したものを使用できる。また、プロモーターも制限はないが、例えばCAGプロモーターなどを使用できる。
2.哺乳動物細胞
本発明は、第二の態様において、上記セクション1に記載した哺乳動物人工染色体ベクターを含む哺乳動物細胞を提供する。
本発明は、第二の態様において、上記セクション1に記載した哺乳動物人工染色体ベクターを含む哺乳動物細胞を提供する。
本発明の哺乳動物人工染色体ベクターを介して哺乳動物由来の細胞類に、抗体重鎖D領域が改変されたヒト免疫グロブリン遺伝子座(セクション1の「改変Ig-NAC」)を導入することによって、ヒト抗体を産生可能にする哺乳動物細胞を作製することができる。
本発明の哺乳動物人工染色体ベクターは、任意の哺乳動物細胞に移入又は導入することができる。そのための手法には、例えば、微小核細胞融合法、リポフェクション、リン酸カルシウム法、マイクロインジェクション、エレクトロポレーションなどが含まれるが、好ましい手法は微小核細胞融合法である。
微小核細胞融合法は、本発明の哺乳動物人工染色体ベクターを含有する微小核形成能を有する細胞(例えばマウスA9細胞)と、所望の他の細胞との微小核融合によって上記ベクターを上記他の細胞に移入する方法である。微小核形成能を有する細胞は、倍数体誘発剤(例えばコルセミド、コルヒチンなど)で処理して微小核多核細胞を形成し、サイトカラシン処理により微小核体を形成する処理を行ったのちに、所望の細胞との細胞融合を行うことができる。
上記のベクター導入可能な細胞は、齧歯類細胞、ヒト細胞などの哺乳動物細胞であり、例えば卵母細胞、精子細胞などの生殖系列細胞、胚性幹(ES)細胞、精子幹(GS)細胞、体性幹細胞などの幹細胞、体細胞、胎児細胞、成体細胞、正常細胞、初代培養細胞、継代細胞、株化細胞などを包含する。幹細胞には、例えば胚性幹(ES)細胞、胚性生殖(EG)細胞、間葉系幹細胞などの体性幹細胞、人工多能性幹(iPS)細胞、核移植クローン胚由来胚性幹(ntES)細胞などが含まれる。好ましい細胞は、例えばマウス、ラット、ハムスター(例えば、チャイニーズハムスター)、モルモットなどの齧歯類、ヒト、サル、チンパンジーなどの霊長類、ウシ、ブタ、ヒツジ、ヤギ、ウマ、ラクダなどの有蹄類などの哺乳動物由来の体細胞、幹細胞、前駆細胞、及び非ヒト生殖系列細胞からなる群から選択される。細胞が齧歯類由来の細胞である場合、本発明のベクターが導入された齧歯類の細胞や組織において、ベクターがより安定に保持される。
体細胞の例は、非限定的に、肝細胞、腸細胞、腎細胞、脾細胞、肺細胞、心臓細胞、骨格筋細胞、脳細胞、皮膚細胞、骨髄細胞、線維芽細胞などである。
ES細胞は、哺乳動物由来の受精卵の胚盤胞の内部細胞塊から樹立された分化多能性と半永久的増殖能とを備えた幹細胞である(M.J.Evans and M.H.Kaufman(1981)Nature 292:154-156;J.A.Thomson et al.(1999)Science 282:1145-1147;J.A.Thomson et al.(1995)Proc.Natl.Acad.Sci.USA 92:7844-7848;J.A.Thomson et al.(1996)Biol.Reprod.55:254-259;J.A.Thomson and V.S.Marshall(1998)Curr.Top.Dev.Biol.38:133-165)。
ラットのES細胞は、マウスES細胞の場合と同様に(M.J.Evans and M.H.Kaufman,Nature 1981;292(5819):154-156)、ラット胚盤胞期胚又は8細胞期胚の内部細胞塊から樹立される、多分化能と自己複製能をもつ細胞株である。例えば、卵透明帯を溶解したラット胚盤胞を、白血病抑制因子(LIF)を含有する培地を用いてマウス胚性線維芽細胞(MEFF)フィーダー上で培養し、7~10日後に胚盤胞から形成されるアウトグロウス(outgrowth)を分散し、これをMEFフィーダー上に移して培養し、約7日後、ES細胞が出現する。ラットES細胞の作製については、例えばK.Kawaharada et al.,World J Stem Cells 2015;7(7):1054-1063に記載されている。
iPS細胞は、体細胞(体性幹細胞を含む)に、ある特定のリプログラミング因子(DNA又はタンパク質)を導入し、適当な培地にて培養、継代培養することによって約3~5週間でコロニーを生成することができる。リプログラミング因子は、例えばOct3/4、Sox2、Klf4及びc-Mycからなる組み合わせ;Oct3/4、Sox2及びKlf4からなる組み合わせ;Oct4、Sox2、Nanog及びLin28からなる組み合わせ;或いは、Oct3/4、Sox2、Klf4、c-Myc、Nanog及びLin28からなる組み合わせなどが知られている(K.Takahashi and S.Yamanaka,Cell 126:663-676(2006);WO2007/069666;M.Nakagawa et al.,Nat.Biotechnol.26:101-106(2008);K.Takahashi et al.,Cell 131:861-872(2007);J.Yu et al.,Science 318:1917-1920(2007);J.Liao et al.,Cell Res.18,600-603(2008))。培養例は、マイトマイシンC処理したマウス胎仔線維芽細胞株(例えばSTO)をフィーダー細胞とし、このフィーダー細胞層上でES細胞用培地を用いて、ベクター導入体細胞(例えば約104~105細胞/cm2)を約37℃の温度で培養することを含む。フィーダー細胞は必ずしも必要ではない(Takahashi,K.et al.,Cell 131:861-872(2007))。基本培地は、例えばダルベッコ改変イーグル培地(DMEM)、ハムF-12培地、それらの混合培地などであり、ES細胞用培地は、マウスES細胞用培地、霊長類ES細胞用培地(リプロセル社)などを使用することができる。
3.非ヒト動物
本発明は、第三の態様において、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む非ヒト動物であって、上記ヒト免疫グロブリン重鎖遺伝子座のD領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されており、上記D領域の改変配列が、
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメなどの脊椎動物由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ことを特徴とし、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている、非ヒト動物を提供する。
本発明は、第三の態様において、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む非ヒト動物であって、上記ヒト免疫グロブリン重鎖遺伝子座のD領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されており、上記D領域の改変配列が、
(1)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメなどの脊椎動物由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)上記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ことを特徴とし、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている、非ヒト動物を提供する。
本発明は、上記非ヒト動物の別の態様において、上記セクション1に記載した哺乳動物人工染色体ベクターを含み、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている非ヒト動物を提供する。
本明細書中の「非ヒト動物」という用語は、ヒトを除く、サル、チンパンジーなどの霊長類、マウス、ラット、ハムスター、モルモットなどの齧歯類、ウシ、ブタ、ヒツジ、ヤギ、ウマ、ラクダ科動物などの有蹄類などの哺乳動物を含むが、これらに限定されない。
[非ヒト動物の作製]
例えば哺乳動物人工染色体ベクターを介する技術、ジーンターゲティング技術、ゲノム編集技術、及び/又は顕微注入技術などの手法によって、上記ES細胞やiPS細胞などの分化多能性細胞に、或いは、生殖細胞、卵母細胞、精原細胞などの全能性細胞に、ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン遺伝子座(「改変Ig-NAC」)を導入することによって、ヒト免疫グロブリンを産生可能にする非ヒト動物を作製することができる。
例えば哺乳動物人工染色体ベクターを介する技術、ジーンターゲティング技術、ゲノム編集技術、及び/又は顕微注入技術などの手法によって、上記ES細胞やiPS細胞などの分化多能性細胞に、或いは、生殖細胞、卵母細胞、精原細胞などの全能性細胞に、ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン遺伝子座(「改変Ig-NAC」)を導入することによって、ヒト免疫グロブリンを産生可能にする非ヒト動物を作製することができる。
そのような非ヒト動物では、ヒト免疫グロブリン重鎖及び軽鎖κ、λ遺伝子座に対応する内在遺伝子座が破壊(若しくは、ノックアウト)されていること、並びに、必要に応じて、非ヒト動物のゲノム上の破壊された内在遺伝子座に、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座、ヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座、及びヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座に相当する各外来DNAを挿入(若しくは、ノックイン)することができる。破壊の方法には、例えばジーンターゲティング法、ゲノム編集などの技術を使用することができる。
ジーンターゲティング法では、上記内在遺伝子座を破壊するための、或いは、さらに上記内在遺伝子座に代えて上記外来DNAを置換/挿入(若しくは、ノックイン)するための、ターゲティングベクターを構築する。内在遺伝子座を破壊する、及び外来DNAを挿入するためのベクターは、内在遺伝子座の5’側上流配列(約2.5kb以上)及び3’側下流配列(約2.5kb以上)を含み、これらの配列間に例えば外来性薬剤耐性遺伝子配列、又は、破壊すべき内在遺伝子座を置換するための上記外来DNA配列を含むことができる。薬剤耐性遺伝子は、例えばネオマイシン耐性遺伝子、アンピシリン耐性遺伝子などである。
ゲノム編集では、上記の通り、例えばCRISPR/Cas9システムは、Cas9タンパク質と約20塩基対標的配列をもつガイドRNA(gRNA)を用いて細胞内でこれらを共発現させることにより、gRNAが標的配列近傍のPAM配列を認識して標的ゲノムDNAと特異的に結合し、Cas9タンパク質がPAM配列の5’側上流で二本鎖切断を誘導することができる。この技術を用いることによって、非ヒト動物の抗体重鎖及び軽鎖遺伝子座を切断除去することができるし、或いは、さらに上記切断部位に、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座、ヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座、及びヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座に相当する各外来DNAを含むベクターを用いて該外来DNAを挿入(若しくは、ノックイン)することができる。
上記の目的の外来DNAを含むベクターを、非ヒト動物由来のES細胞又はiPS細胞に導入し、該動物由来の上記内在遺伝子座を破壊し、並びに、目的の上記DNA配列をゲノム上に挿入することができる。目的の外来DNAが挿入されたことの確認は、挿入配列に基づいて作製されたフォワード及びリバースプライマーを使用するPCR法による増幅産物の同定によって行うことができる。上記外来DNA配列が挿入されたES細胞又はiPS細胞を仮親非ヒト動物の初期胚に顕微注入し、キメラ非ヒト動物を作製することができる。
上記内在遺伝子座が破壊された非ヒト動物は、例えば、上記ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン遺伝子座を含むキメラ非ヒト動物若しくはその子孫動物と、対応する内在遺伝子をクラスターごと欠失させたキメラ動物若しくは子孫動物とを交配させて得られた該内在遺伝子がヘテロに欠失した動物同士をさらに交配させることによって作出することができる。
本発明の非ヒト動物の例は、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含む非ヒト動物又は上記人工染色体ベクターを含む非ヒト動物、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含む非ヒト動物又は上記人工染色体ベクターを含む非ヒト動物、或いは、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座、ヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座、及びヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含む非ヒト動物又は上記人工染色体ベクターを含む非ヒト動物である。
非ヒト動物は、好ましくは、上記マウス人工染色体ベクターを含む、マウスやラットなどの齧歯類である。
以下において、マウス人工染色体(MAC)ベクターを利用する非ヒト動物の作製例について説明する。
部位特異的組み換え酵素の認識部位(例えばloxP又はFRT)を導入して改変されたヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含む動物細胞(例えばニワトリB細胞株DT40)、及び部位特異的組み換え酵素の認識部位(例えばloxP又はFRT)を導入して改変されたヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含む動物細胞(例えばDT40)のそれぞれを、細胞融合法によりマウス人工染色体(MAC)ベクターを含む齧歯類細胞(例えばチャイニーズハムスター卵巣細胞(CHO))に移入したのち、部位特異的組み換え酵素(例えばCre又はFlp)の作用又は該酵素の発現誘導により、ヒト免疫グロブリン軽鎖κ遺伝子座を含むMACベクターを含む齧歯類細胞、並びにヒト免疫グロブリン軽鎖λ遺伝子座を含むMACベクターを含む齧歯類細胞を作製することができる。
部位特異的組み換え酵素の認識部位(例えばloxP又はFRT)を導入して改変されたヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含む動物細胞(例えばニワトリB細胞株DT40)、及び部位特異的組み換え酵素の認識部位(例えばloxP又はFRT)を導入して改変されたヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含む動物細胞(例えばDT40)のそれぞれを、細胞融合法によりマウス人工染色体(MAC)ベクターを含む齧歯類細胞(例えばチャイニーズハムスター卵巣細胞(CHO))に移入したのち、部位特異的組み換え酵素(例えばCre又はFlp)の作用又は該酵素の発現誘導により、ヒト免疫グロブリン軽鎖κ遺伝子座を含むMACベクターを含む齧歯類細胞、並びにヒト免疫グロブリン軽鎖λ遺伝子座を含むMACベクターを含む齧歯類細胞を作製することができる。
一方、動物細胞(例えばDT40)に保持されるヒト14番染色体上のヒト免疫グロブリン重鎖遺伝子座の近傍に部位特異的組み換え酵素の認識部位(例えばloxP又はFRT)を導入したのち、ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座を含む動物細胞を、細胞融合法によりMACベクターを含む齧歯類細胞(例えばCHO)と融合して、ヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座を含むMACベクター(上記の「改変Ig-NAC」)を含む齧歯類細胞を作製することができる。
上記のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト免疫グロブリン軽鎖κ遺伝座を含むMACベクターを含む齧歯類細胞、並びに上記のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子若しくは遺伝子座を含むMACベクターを含む齧歯類細胞のそれぞれを、微小核細胞融合法により、非ヒト動物(例えばマウス又はラット)分化多能性幹細胞(例えばES細胞又はiPS細胞)と融合して、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含むMACベクターを含む非ヒト動物(例えばマウス又はラット)分化多能性幹細胞、並びにヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含むMACベクターを含む非ヒト動物(例えばマウス又はラット)分化多能性幹細胞を作製することができる。
上記のヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含むMACベクターを含む非ヒト動物(例えばマウス又はラット)分化多能性幹細胞、並びにヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含むMACベクターを含む非ヒト動物(例えばマウス又はラット)分化多能性幹細胞のそれぞれを、B細胞欠損又は非欠損の非ヒト動物の初期胚(例えば8細胞期胚又は胚盤胞期胚)に移植して、上記MACベクターのそれぞれを含むキメラ動物を作製する。該キメラ動物が内在性B細胞を保持する場合には、さらにキメラ動物を、マウス抗体重鎖、軽鎖κ及び軽鎖λ遺伝子座を欠損する(或いは、B細胞を欠損する)同種非ヒト動物(例えばマウス又はラット)と交配する。その後、目的の非ヒト動物を選別することにより、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト2番染色体由来のヒト免疫グロブリン軽鎖κ遺伝子座を含むMACベクター、或いは、ヒト14番染色体由来のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座とヒト22番染色体由来のヒト免疫グロブリン軽鎖λ遺伝子座を含む上記MACベクターのそれぞれを含む非ヒト動物を作製することができる。
或いはさらに、上記の2種の異なる子孫動物同士の交配により、上記のヒト免疫グロブリン重鎖D領域が改変されたヒト免疫グロブリン重鎖遺伝子座、ヒト免疫グロブリン軽鎖κ遺伝子座、及びヒト免疫グロブリン軽鎖λ遺伝子座を含むMACベクターを含む非ヒト動物(例えばマウス又はラット)を作製することができる。
上記の方法によって作製される非ヒト動物においては、ヒト免疫グロブリン重鎖遺伝子若しくは遺伝子座、並びに、ヒト免疫グロブリン軽鎖κ及びλ遺伝子若しくは遺伝子座に対応する全ての内在抗体遺伝子若しくは遺伝子座が破壊若しくはノックアウトされていることが好ましく、これによって該非ヒト動物においてヒト抗体のみを産生可能となる。
4.ヒト抗体の作製方法
本発明はさらに、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを含むセクション3に記載の非ヒト動物を用いてヒト抗体を産生し、該ヒト抗体を回収することを含む、ヒト抗体の製造方法を提供する。
本発明はさらに、ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを含むセクション3に記載の非ヒト動物を用いてヒト抗体を産生し、該ヒト抗体を回収することを含む、ヒト抗体の製造方法を提供する。
本明細書における「ヒト抗体」は、ヒト免疫グロブリン(Ig)のいずれのクラス及びサブクラスでもよい。そのようなクラスには、IgG、IgA、IgM、IgD及びIgEが含まれ、サブクラスには、IgG1、IgG2、IgG3、IgG4、IgA1及びIgA2が含まれる。これらのクラス及びサブクラスは、重鎖の違いによって分けることが可能であり、IgG鎖は、γ鎖と称し、IgG1~IgG4に対応してγ1、γ2、γ3及びγ4鎖と称し、IgA、IgM、IgD、IgEはそれぞれα鎖(α1及びα2)、μ鎖、δ鎖、ε鎖と称する。いずれの抗体の軽鎖にもκ鎖とλ鎖があり、免疫グロブリン遺伝子の再編成の過程でκ鎖遺伝子の再編成が不成功に終わるとλ鎖遺伝子の再編成が起こることが知られている。
また、ヒト免疫グロブリン重鎖遺伝子座は、5’から3’に向けて、VH1,VH2..VHm(ここで、mは、例えば38~46である。)を含むV(variable)領域遺伝子、DH1,DH2..DHn(ここで、nは、23である。)を含むD(diversity)領域遺伝子、JH1,JH2..JHr(ここで、rは、6である。)を含むJ(joining)領域遺伝子、Cμ,Cδ,Cγ3,Cγ1,Cα1,Cγ2,Cγ4,Cε,Cα2を含むC(constant)領域遺伝子を含むが、本発明では、ヒト免疫グロブリン重鎖遺伝子座のD領域遺伝子は、上記セクション1の(1)及び(2)、又は(1)及び(3)の組み合わせからなる改変配列として記載の合成D領域ヌクレオチド配列による置換を含む。
上記非ヒト動物では、上記のヒト免疫グロブリン遺伝子の再編成を介して産生される抗体が、従来のヒト抗体では定常的に作製が困難であった例えばアミノ酸数18~50若しくはそれ以上の超長鎖(ultralong)CDRH3を含むヒト抗体が産生されうる。このような超長鎖CDRH3を含むヒト抗体はヒトでの出現頻度が低いため、通常のモノクローナル抗体作製法では作製困難である。
ヒト抗体分子は、2本のヒト抗体重鎖と2本のヒト抗体軽鎖からなり、各重鎖と各軽鎖は2つのジスルフィド結合によって結合されており、並びに、2本の重鎖は定常(C)領域で2つのジスルフィド結合によって結合された構造を有する。また、抗体分子の重鎖及び軽鎖の可変(V)領域にはそれぞれ、とりわけ変異の大きい部分が3箇所(超可変領域)あり、相補性決定領域(complementarity determining region(CDR))と呼ばれており、重鎖可変領域の超可変領域はN末端側からCDRH1、CDRH2及びCDRH3と呼ばれ、一方、軽鎖可変領域の超可変領域はN末端側からCDRL1、CDRL2及びCDRL3と呼ばれている。このCDR領域の配列の違いにより抗体の抗原に対する結合特性が変化する。本発明により、ヒト免疫グロブリン遺伝子類の再編成によるヒト抗体の多様性が拡張される。
本発明の非ヒト動物が産生する超長鎖(ultralong)CDRH3を含むヒト抗体は、長い(β-ribbon)StalkとKnobからなる構造を有しており、Knobにはループと複数のジスルフィド結合が存在する(J.Dong et al.,Frontiers in Immunology,2019;10:Article 558;J.K.Haakenson et al.,Frontiers in Immunology 2018;9:Article1 262)。
本発明の非ヒト動物が産生する超長鎖(ultralong)CDRH3を含むヒト抗体は、長い(β-ribbon)StalkとKnobからなる構造を有しており、Knobにはループと複数のジスルフィド結合が存在する(J.Dong et al.,Frontiers in Immunology,2019;10:Article 558;J.K.Haakenson et al.,Frontiers in Immunology 2018;9:Article1 262)。
本発明の方法によって得ることが可能である抗体は、抗体重鎖CDR3のアミノ酸長が18以上、20以上、25以上、30以上、40以上、又は50以上となるような超長鎖(ultralong)CDRH3を含むヒト抗体であるため、例えばヒト免疫不全ウイルス(HIV)、インフルエンザウイルス、コロナウイルス(例えばSARS-CoV、MERS-CoV)、RS(Respiratory syncytial)ウイルスなどの病原性ウイルスや他の病原体(例えばマラリア原虫、トリパノソーマ原虫)を原因とする感染症を含む疾病に有効であると考えられる。上記ヒト抗体は、突出したCDRH3領域で上記ウイルスや病原体の受容体のくぼみに結合する「カギ」型の相互作用を可能とするため治療上有用である。
本発明の抗体は、複数種の抗体カクテルとすることも可能であり、治療だけでなく予防効果を増強するために使用することもできる。
具体的な抗体作製法は、以下の方法を包含する。
例えば、本発明の上記ヒト抗体産生非ヒト動物(例えばマウス又はラット)にHIVエンベロープタンパク質などの標的抗原を免疫して該標的抗原と特異的に結合するポリクローナル抗体を作製することができる。
或いは、上記動物に標的抗原を免疫し、抗原特異的抗体価が上昇した動物個体のBリンパ球を用いてミエローマ細胞と融合しハイブリドーマ(不死化B細胞)を作製し、ハイブリドーマ上清中のヒトモノクローナル抗体について、抗原結合能評価のためのELISA解析、必要であればHIVなどの感染評価により選抜を行い、広域中和抗体ヒトモノクローナル抗体を選択することができる。
すなわち、本発明は、第四の態様において、上記セクション3に記載の非ヒト動物に標的抗原を免疫し、該非ヒト動物の血液から該標的抗原と結合する抗体を取得することを含む、抗体の作製方法を提供する。
或いは、別の態様において、本発明はさらに、上記セクション3に記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物から脾臓細胞又はリンパ節細胞を取得し、前記脾臓細胞又はリンパ節細胞とミエローマ細胞を融合してハイブリドーマを形成し、該ハイブリドーマを培養して、前記標的抗原と結合するモノクローナル抗体を取得することを含む、抗体の作製方法を提供する。
或いは、さらに別の態様において、本発明はさらに、上記セクション3に記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物からB細胞を取得し、前記B細胞から抗体重鎖及び軽鎖、或いはそれらの可変領域、からなるタンパク質をコードする核酸を取得し、該核酸を用いてDNA組み換え技術又はファージディスプレイ技術によって前記標的抗原と結合する抗体(例えば、組み換え抗体、モノクローナル抗体、一本鎖抗体(scFv)など)を作製することを含む、抗体の作製方法を提供する。
上記核酸は、DNA(cDNAを含む)又はRNA(mRNAを含む)を含む。
また、上記scFvは、抗体重鎖可変領域ポリペプチドと抗体軽鎖可変領域ポリペプチドとをリンカーを介して結合して作製された組み換え抗体フラグメントである。
ファージディスプレイ法では、ヒト抗体の可変領域を例えば一本鎖抗体(scFv)又はFabとしてファージ表面に発現させて、抗原に結合するファージを選択する(Nature Biotechnology,2005;23(9):1105-1116)。抗原に結合することで選択されたファージの遺伝子を解析することによって、抗原に結合するヒト抗体の可変領域をコードするDNA配列を決定することができる。抗原に結合するscFvのDNA配列が明らかになれば、当該配列を有する発現ベクターを作製し、適当な宿主に導入して発現させることによりヒト抗体を取得することができる(WO92/01047、WO92/20791、WO93/06213、WO93/11236、WO93/19172、WO95/01438、WO95/15388、Annu.Rev.Immunol,1994;12:433-455、Nature Biotechnology,2005;23(9):1105-1116)。
上記方法によって作製される抗体は、該抗体を含む血液(抗血清)や細胞上清などを、抗原物質を結合した担体や免疫グロブリンG結合性担体(例えばアガロースゲル、シリカゲル、など)を充填したカラムにアプライし、ついで、該担体に結合したヒト抗体を担体から溶出することを含むアフィニティクロマトグラフィーなどの手法によって回収、精製することができる。
下記の実施例を参照しながら、本発明をさらに具体的に説明するが本発明の技術的範囲はそれらの実施例によって制限されないものとする。
[実施例1]ヒトIgHD領域を削除したIg-NAC(ΔDH)構築
ヒトIgHD領域を削除したIg-NAC(ΔDH)を構築するために、DH領域を挟む2カ所(上流、下流)にCRISPR/Cas9のgRNAを設計、評価、選定し、Ig-NACを保持するCHO細胞内での2ヵ所切断によりDH領域除去を実施、スクリーニングによりIg-NAC(ΔDH)保持CHO細胞を取得した。
ヒトIgHD領域を削除したIg-NAC(ΔDH)を構築するために、DH領域を挟む2カ所(上流、下流)にCRISPR/Cas9のgRNAを設計、評価、選定し、Ig-NACを保持するCHO細胞内での2ヵ所切断によりDH領域除去を実施、スクリーニングによりIg-NAC(ΔDH)保持CHO細胞を取得した。
1.gRNA発現ベクターの構築及び活性評価による選定
Cas9とgRNAを発現するオールインワンベクターeSpCas9(1.1)(Addgene Plasmid #71814)をベースとして設計した候補gRNAを発現するプラスミドを構築した。Ig-NACを保持するCHO細胞に、メーカーのプロトコルに従ってLipofectamine LTX(Invitrogen)でオールインワンベクターを導入した。導入した細胞からPureGene kit(Qiagen)を用いてDNAを抽出しCel-1 assay kit(IDT)で切断活性を確認し、活性の高かった以下のgRNA配列のベクターを選択した。
gRNA Up3:5’-AGATCCTCCATGCGTGCTGT (GGG)-3’(配列番号4)
(ここでgRNA配列は、PAM配列(GGG)を除く20塩基の配列である。)
gRNA Down4:5’-CTGCGGCATGAACCCAATGC (AGG)-3’(配列番号5)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
Cas9とgRNAを発現するオールインワンベクターeSpCas9(1.1)(Addgene Plasmid #71814)をベースとして設計した候補gRNAを発現するプラスミドを構築した。Ig-NACを保持するCHO細胞に、メーカーのプロトコルに従ってLipofectamine LTX(Invitrogen)でオールインワンベクターを導入した。導入した細胞からPureGene kit(Qiagen)を用いてDNAを抽出しCel-1 assay kit(IDT)で切断活性を確認し、活性の高かった以下のgRNA配列のベクターを選択した。
gRNA Up3:5’-AGATCCTCCATGCGTGCTGT (GGG)-3’(配列番号4)
(ここでgRNA配列は、PAM配列(GGG)を除く20塩基の配列である。)
gRNA Down4:5’-CTGCGGCATGAACCCAATGC (AGG)-3’(配列番号5)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
2.DH領域を削除したクローンの取得
gRNA Up3(図中、「5’guide3」)とDown4(図中、「5’guide4」)の各組み合わせでDH領域の削除を実施した(図1)。各gRNAのオールインワンベクター及びCMV-tdTomato発現ベクターを、メーカーのプロトコルに従ってLipofectamine LTX(Invitrogen)により、Ig-NAC保持CHO細胞(特許第6868250号公報)へ導入し、3日後にEGFP/tdTomato共陽性の細胞をソーティングにより取得した。なお、EGFPのシグナルはIg-NACを保持していることを示し、tdTomatoのシグナルはリポフェクションを受けたことを示す。共陽性細胞をさらに培養し、一回目のソーティングから8日後にEGFP陽性かつtdTomato陰性の細胞をシングルセルソーティングにより384ウェルプレートに回収した。増殖してきたクローンについてさらにスケールアップを行い、以下に示すようにスクリーニングを行った。
gRNA Up3(図中、「5’guide3」)とDown4(図中、「5’guide4」)の各組み合わせでDH領域の削除を実施した(図1)。各gRNAのオールインワンベクター及びCMV-tdTomato発現ベクターを、メーカーのプロトコルに従ってLipofectamine LTX(Invitrogen)により、Ig-NAC保持CHO細胞(特許第6868250号公報)へ導入し、3日後にEGFP/tdTomato共陽性の細胞をソーティングにより取得した。なお、EGFPのシグナルはIg-NACを保持していることを示し、tdTomatoのシグナルはリポフェクションを受けたことを示す。共陽性細胞をさらに培養し、一回目のソーティングから8日後にEGFP陽性かつtdTomato陰性の細胞をシングルセルソーティングにより384ウェルプレートに回収した。増殖してきたクローンについてさらにスケールアップを行い、以下に示すようにスクリーニングを行った。
3.DH領域を削除したクローンのスクリーニング
上記で取得した細胞について、PureGene kit(Qiagen)を用いて抽出したDNAを鋳型に、以下のジャンクション確認、DH領域削除確認プライマーにてPCRスクリーニングを行った。
JunctionプライマーF1:5’-TCCCACGGCCCAAGGAAGACAAGACACA-3’(配列番号6)
JunctionプライマーR1:5’-TCGAACACGCTCCTAGCATTGCACAGCC-3’(配列番号7)
欠損確認プライマーF1:5’-ACACCTGTCTCCGGGTTGTG-3’(配列番号8)
欠損確認プライマーR1:5’-AAGAAACGCAGGACGGTGGA-3’(配列番号9)
欠損確認プライマーF2:5’-CAGCAGCCACTCTGATCCCA-3’(配列番号10)
欠損確認プライマーR2:5’-CTGGTGGACTCTACGGCGAA-3’(配列番号11)
欠損確認プライマーF3:5’-GGGACCCTCAAGGTGTGAAC-3’(配列番号12)
欠損確認プライマーR3:5’-AGGGTGCTTGGGTCCTGTTA-3’(配列番号13)
欠損確認プライマーF4:5’-ACAAGCCAGGGAGCTGTTTC-3’(配列番号14)
欠損確認プライマーR4:5’-TCAAGAAATCCGAGGCGACA-3’(配列番号15)
上記で取得した細胞について、PureGene kit(Qiagen)を用いて抽出したDNAを鋳型に、以下のジャンクション確認、DH領域削除確認プライマーにてPCRスクリーニングを行った。
JunctionプライマーF1:5’-TCCCACGGCCCAAGGAAGACAAGACACA-3’(配列番号6)
JunctionプライマーR1:5’-TCGAACACGCTCCTAGCATTGCACAGCC-3’(配列番号7)
欠損確認プライマーF1:5’-ACACCTGTCTCCGGGTTGTG-3’(配列番号8)
欠損確認プライマーR1:5’-AAGAAACGCAGGACGGTGGA-3’(配列番号9)
欠損確認プライマーF2:5’-CAGCAGCCACTCTGATCCCA-3’(配列番号10)
欠損確認プライマーR2:5’-CTGGTGGACTCTACGGCGAA-3’(配列番号11)
欠損確認プライマーF3:5’-GGGACCCTCAAGGTGTGAAC-3’(配列番号12)
欠損確認プライマーR3:5’-AGGGTGCTTGGGTCCTGTTA-3’(配列番号13)
欠損確認プライマーF4:5’-ACAAGCCAGGGAGCTGTTTC-3’(配列番号14)
欠損確認プライマーR4:5’-TCAAGAAATCCGAGGCGACA-3’(配列番号15)
酵素はKOD FX(TOYOBO)を用い、反応条件は98℃15秒、60℃30秒、68℃90秒を35サイクル行った。全ての組み合わせで得られたクローン計381クローンのうち、35クローンでD領域の除去を確認した。その後Ig-NAC上のDH以外の抗体遺伝子領域の存在確認PCRについては、特許第6868250号公報)に記載の方法で行い、候補Ig-NAC(ΔDH)クローンを選定した。
4.Ig-NAC(ΔDH)の構造確認
Ig-NAC(ΔDH)が宿主染色体に組み込まれず独立して維持されていること、期待された構造を保っていることを確認するために特許第6868250号公報に記載の方法でFISH解析を行った結果、宿主染色体とは独立して1コピーのIg-NAC(ΔDH)が保持されたCHO細胞の取得を確認し(図2)、この細胞クローンをTF7-B10と名付けた。
Ig-NAC(ΔDH)が宿主染色体に組み込まれず独立して維持されていること、期待された構造を保っていることを確認するために特許第6868250号公報に記載の方法でFISH解析を行った結果、宿主染色体とは独立して1コピーのIg-NAC(ΔDH)が保持されたCHO細胞の取得を確認し(図2)、この細胞クローンをTF7-B10と名付けた。
[実施例2]Ig-NAC(ΔDH)に合成D領域を導入するためのアクセプター部位の挿入
実施例1で作製したIg-NAC(ΔDH)において削除されたDH領域に、ゲノム編集及び相同組み換えを利用して、アクセプター部位を挿入することにより、化学合成したDH領域を導入することを可能とする。そのために、Ig-NAC(ΔDH)のDH領域削除部位の前後の配列(V領域側:HRA、J断片側:HRB)及び、Bxb1 attB、Bxb1 attP、ΦC31 attB、R4 attB(特許第6868250号公報)(それぞれの配列は下記の通り)、CAGプロモーター(pRP[Exp]-CAG>mCherry、Vector Builder社にて合成)、ブラストサイジン耐性遺伝子(富士フィルム和光純薬)、Fcy::fur遺伝子(pSELECT-zeo-Fcy::fur、Invivogen)が図4の通り配置されたHDRベクターを以下に示す手順で作製した。
実施例1で作製したIg-NAC(ΔDH)において削除されたDH領域に、ゲノム編集及び相同組み換えを利用して、アクセプター部位を挿入することにより、化学合成したDH領域を導入することを可能とする。そのために、Ig-NAC(ΔDH)のDH領域削除部位の前後の配列(V領域側:HRA、J断片側:HRB)及び、Bxb1 attB、Bxb1 attP、ΦC31 attB、R4 attB(特許第6868250号公報)(それぞれの配列は下記の通り)、CAGプロモーター(pRP[Exp]-CAG>mCherry、Vector Builder社にて合成)、ブラストサイジン耐性遺伝子(富士フィルム和光純薬)、Fcy::fur遺伝子(pSELECT-zeo-Fcy::fur、Invivogen)が図4の通り配置されたHDRベクターを以下に示す手順で作製した。
HRA:(配列番号16)
GACCTGAGAGCCGCTCCCTGAAGTGTCCCCATTGGGAAGGATGGGGCCTGTGTCTCCAGGCTCTGGGAGGACAGAATCCTGACCTCAACAGTGGCCGGCACGGACACAACTGGCCCCATCCCGGGGACGCTGACCAGCGCTGGGCAACTTTTCCCTTCCCCGACGACTGAGCCCCGAGCACCCTCCCTGCTCCCCTACCACCTCCCTTTACAAGGCTGTGGCCTCTGCACAGATGATAATGGAGCTTGGCTCATTCCCCTAGAGTCGGTAGGGAGTTAAGGACAAAACTCAGTTTCCTCCACCTGAACTCAAGTCTGCCTATGTTTACCTAATCACACCTGGTGGACAGTTTGGACAAACTTGCACACTCAGAGACACAGACACTTCTAGAAATCATTATCTCCCTGCCCCGGGGACCCCACTCCAGCAGAAGTCTGCTAGGCACTGGCCTGGGCCCTCCTGCTGTCCTAGGAGGCTGCTGACCTCCTGCCTGGCTCCTGTCCCCAGGTCCAGAGTCAGAGCAGACTCCAGGGACGCTGCAGGCTAGGAAGCCGCCCCCTCCAGGCGAGGGTCTAGTGCAGGTGCCCAGGACAAGAAAGATTGTGAATGCAGGAATGACTGGGCCACACCCCTCCCGTGCACGCCCCCTCCTGCCCTGCACCCCACAGCCCAGCCCCCCGTGCTGGATGCCCCCCCACAGCAGAGGTGCTGTTCTGTGATCCCCTGGGAAAGACGCCCTCAACCTCCACCCTGTCCCACGGCCCAAGGAAGACAAGACACAGGCCCTCTCCTCACAGTCTCCCCACCTGGCTCCTGCTGGGACCCTCAAGGTGTGAACAGGGAGGATGGTTGTCTGGGTGGCCCCTAGGAGCCCAGATCTTCACTCCACAGACCCCAACCCAAGCACCCCCTTCTGCAGGGCCCAGCTCATCCCCCTCCTCCTCCCTCTGCTCTCCTCT
GACCTGAGAGCCGCTCCCTGAAGTGTCCCCATTGGGAAGGATGGGGCCTGTGTCTCCAGGCTCTGGGAGGACAGAATCCTGACCTCAACAGTGGCCGGCACGGACACAACTGGCCCCATCCCGGGGACGCTGACCAGCGCTGGGCAACTTTTCCCTTCCCCGACGACTGAGCCCCGAGCACCCTCCCTGCTCCCCTACCACCTCCCTTTACAAGGCTGTGGCCTCTGCACAGATGATAATGGAGCTTGGCTCATTCCCCTAGAGTCGGTAGGGAGTTAAGGACAAAACTCAGTTTCCTCCACCTGAACTCAAGTCTGCCTATGTTTACCTAATCACACCTGGTGGACAGTTTGGACAAACTTGCACACTCAGAGACACAGACACTTCTAGAAATCATTATCTCCCTGCCCCGGGGACCCCACTCCAGCAGAAGTCTGCTAGGCACTGGCCTGGGCCCTCCTGCTGTCCTAGGAGGCTGCTGACCTCCTGCCTGGCTCCTGTCCCCAGGTCCAGAGTCAGAGCAGACTCCAGGGACGCTGCAGGCTAGGAAGCCGCCCCCTCCAGGCGAGGGTCTAGTGCAGGTGCCCAGGACAAGAAAGATTGTGAATGCAGGAATGACTGGGCCACACCCCTCCCGTGCACGCCCCCTCCTGCCCTGCACCCCACAGCCCAGCCCCCCGTGCTGGATGCCCCCCCACAGCAGAGGTGCTGTTCTGTGATCCCCTGGGAAAGACGCCCTCAACCTCCACCCTGTCCCACGGCCCAAGGAAGACAAGACACAGGCCCTCTCCTCACAGTCTCCCCACCTGGCTCCTGCTGGGACCCTCAAGGTGTGAACAGGGAGGATGGTTGTCTGGGTGGCCCCTAGGAGCCCAGATCTTCACTCCACAGACCCCAACCCAAGCACCCCCTTCTGCAGGGCCCAGCTCATCCCCCTCCTCCTCCCTCTGCTCTCCTCT
HRB:(配列番号17)
CTCACTGAAGGCAGCCTCAGGGTGCCCAGGGGCAGGCAGGGTGGGGGTGAGGCTTCCAGCTCCAACCGCTCCACTAGCCGAGACTAAGGAAGTGAGAGGCAGCCAGAAATCCAGACCATTCCATAGCAAATGGATTTCATTAAAGTTACCAGACTTCAGTGTAAGTAACATGAGCCCCATGCACAACAATCCCTTATGAAGGGGAAGTCAGTGTCGCCTCGGATTTCTTGAAAAACACAAAAACTTATCAATGCCTGTAAAAGTCTGTTGGAAAGAAAATATGATTCAAGAATGTTATGCCCAACAAAGCTGGCATATTTTCTACCCGGACACACTCAGGGAATGTGGTCCCTTGAGTGCTTCTCTCACTGCGTAAATCCTACGTGGTGTTTAAGCATATTCATAAATGTGTATGTCTATTTTTATGTGTAAGATGGTTCATTTTTATTTTATTTATTCAATATGTACAATAAAGAATATTGACAAATAGGCTGGACATGGTGGCTCCCACCTGTAATCCCAGCCCTTTGGGAGGCCGAGGCGGGCAGATCACCTGAGGTCTGGAGTTCGAGACCAGCCTGGCCAACATGATGAAAACCCATCTCTACTAAAAATACAAAGATTAGCCAGGCATGGTGGTGCATGCCTGTAATCCCAGCCACTCAGGAGGCTGAGACAGGAGAAATGCGTGAACCCGGAAGGCGGAGGTTGCAGTGAGCCGAGATCACACCACTGCACTCCAGCCTGGCGACAGAGCAAGATTCCATCTCAAAAAAAAAAAGACAAAGAAATTTGTTTTTTTGAATAAAGACAAATTTCATCACACGAAGATAAAGATGCAAAGCTCCAGACAGGAAGGCACGGACAGCACAGTGAAGCCCGGAGCGGGCGCTGGGGGGCCAGGGGCATGGCGGGGGTGCCAGCGTCTCTCGGTGCCTACCATGGCCACTCCAGCCTGTGTTCTCACGAGGATGGCTGTGT
CTCACTGAAGGCAGCCTCAGGGTGCCCAGGGGCAGGCAGGGTGGGGGTGAGGCTTCCAGCTCCAACCGCTCCACTAGCCGAGACTAAGGAAGTGAGAGGCAGCCAGAAATCCAGACCATTCCATAGCAAATGGATTTCATTAAAGTTACCAGACTTCAGTGTAAGTAACATGAGCCCCATGCACAACAATCCCTTATGAAGGGGAAGTCAGTGTCGCCTCGGATTTCTTGAAAAACACAAAAACTTATCAATGCCTGTAAAAGTCTGTTGGAAAGAAAATATGATTCAAGAATGTTATGCCCAACAAAGCTGGCATATTTTCTACCCGGACACACTCAGGGAATGTGGTCCCTTGAGTGCTTCTCTCACTGCGTAAATCCTACGTGGTGTTTAAGCATATTCATAAATGTGTATGTCTATTTTTATGTGTAAGATGGTTCATTTTTATTTTATTTATTCAATATGTACAATAAAGAATATTGACAAATAGGCTGGACATGGTGGCTCCCACCTGTAATCCCAGCCCTTTGGGAGGCCGAGGCGGGCAGATCACCTGAGGTCTGGAGTTCGAGACCAGCCTGGCCAACATGATGAAAACCCATCTCTACTAAAAATACAAAGATTAGCCAGGCATGGTGGTGCATGCCTGTAATCCCAGCCACTCAGGAGGCTGAGACAGGAGAAATGCGTGAACCCGGAAGGCGGAGGTTGCAGTGAGCCGAGATCACACCACTGCACTCCAGCCTGGCGACAGAGCAAGATTCCATCTCAAAAAAAAAAAGACAAAGAAATTTGTTTTTTTGAATAAAGACAAATTTCATCACACGAAGATAAAGATGCAAAGCTCCAGACAGGAAGGCACGGACAGCACAGTGAAGCCCGGAGCGGGCGCTGGGGGGCCAGGGGCATGGCGGGGGTGCCAGCGTCTCTCGGTGCCTACCATGGCCACTCCAGCCTGTGTTCTCACGAGGATGGCTGTGT
Bxb1 attB:(配列番号18)
TGGCCGTGGCCGTGCTCGTCCTCGTCGGCCGGCTTGTCGACGACGGCGGTCTCCGTCGTCAGGATCATCCGGGCCAC
TGGCCGTGGCCGTGCTCGTCCTCGTCGGCCGGCTTGTCGACGACGGCGGTCTCCGTCGTCAGGATCATCCGGGCCAC
Bxb1 attP:(配列番号19)
TATGGCCGTGATGACCTGTGTCTTCGTGGTTTGTCTGGTCAACCACCGCGGTCTCAGTGGTGTACGGTACAAACCCA
TATGGCCGTGATGACCTGTGTCTTCGTGGTTTGTCTGGTCAACCACCGCGGTCTCAGTGGTGTACGGTACAAACCCA
ΦC31 attB:(配列番号20)
GATGTAGGTCACGGTCTCGAAGCCGCGGTGCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTACTCCACCTCACCCATCTGGTCCATCATGATGAACGGGTCGAGGTGGCGGTAGTTGATCCCGGCGAACGCGCGGCGCACCGGGAAGCCCTCGCCCTCGAAACCGCTGGGCGCGGTGGTCACGGTGAGCACGGGACGTGCGACGGCGTCGGCGGGTGCGGATACGCGGGGCAGCGTCAGCGGGTTCTCGACGGTCACGGCGGGCAT
GATGTAGGTCACGGTCTCGAAGCCGCGGTGCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTACTCCACCTCACCCATCTGGTCCATCATGATGAACGGGTCGAGGTGGCGGTAGTTGATCCCGGCGAACGCGCGGCGCACCGGGAAGCCCTCGCCCTCGAAACCGCTGGGCGCGGTGGTCACGGTGAGCACGGGACGTGCGACGGCGTCGGCGGGTGCGGATACGCGGGGCAGCGTCAGCGGGTTCTCGACGGTCACGGCGGGCAT
R4 attB:(配列番号21)
GCGCCCAAGTTGCCCATGACCATGCCGAAGCAGTGGTAGAAGGGCACCGGCAGACAC
GCGCCCAAGTTGCCCATGACCATGCCGAAGCAGTGGTAGAAGGGCACCGGCAGACAC
1.CAGプロモーター保持ベクターへのFcy::fur遺伝子の挿入
HDRベクターをIg-NAC(ΔDH)保持CHO細胞へ挿入した際に、目的の相同組み換え以外のランダムインテグレーションによる挿入が起きた細胞を選択的に死滅させるために、ネガティブ選択マーカーであるFcy::fur遺伝子を挿入した。pSelect-zeo-Fcy::furプラスミド(Invivogen)DNAから下記プライマー、及び条件を用いてFcy::fur遺伝子をPCR増幅して得た。
Fcy-fur-F1:5’-CATGAATCTAGAAGTGCAGGTGCCAGAACATT-3’(配列番号22)
fur-BspH1-R1:5’-CATGAATCATGACCCCCTGAACCTGAAACATA-3’(配列番号23)
HDRベクターをIg-NAC(ΔDH)保持CHO細胞へ挿入した際に、目的の相同組み換え以外のランダムインテグレーションによる挿入が起きた細胞を選択的に死滅させるために、ネガティブ選択マーカーであるFcy::fur遺伝子を挿入した。pSelect-zeo-Fcy::furプラスミド(Invivogen)DNAから下記プライマー、及び条件を用いてFcy::fur遺伝子をPCR増幅して得た。
Fcy-fur-F1:5’-CATGAATCTAGAAGTGCAGGTGCCAGAACATT-3’(配列番号22)
fur-BspH1-R1:5’-CATGAATCATGACCCCCTGAACCTGAAACATA-3’(配列番号23)
100ng/μLのpSelect-zeo-Fcy::furプラスミドDNA(0.1μL)をテンプレートとして、TaqDNAポリメラーゼはKODone(TOYOBO)を使用し、反応条件:98℃,10秒;60℃,5秒;68℃,13秒で35サイクルのPCRを行った。
PCR後Fcy::fur遺伝子をアガロースゲル電気泳動によりDNA断片を精製したのち、制限酵素BspHI(NEB)及びXbaI(NEB)で両端を切断した。CAGプロモーターが入ったプラスミドDNA pRP[Exp]-CAG>mCherry(Vector Builder社にて合成)中の制限酵素AfiIII(NEB)及びXbaI(NEB)切断部位へ上記Fcy::fur遺伝子を挿入してプラスミドCAG-Fcy::furを得た(図3)。
2.CAGプロモーターとFcy::fur遺伝子間にΦC31attB及びHRBを挿入したベクタープラスミドCAG-ΦC31-HRB-Fcy::furの作製
上記1.で作製したプラスミドCAG-Fcy::furに化学合成DNAを導入する際に使用する、ΦC31 attB及びHRBが入ったプラスミドDNA、pHRBはユーロフィンジェノミクス株式会社にて合成した。このプラスミドを制限酵素XbaIで切断した。プラスミドCAG-Fcy::furを制限酵素XbaIで切断し、CAGプロモーターとFcy::fur遺伝子間に、XbaIで両端を切断したpHRB由来断片を挿入してプラスミドCAG-ΦC31-HRB-Fcy::furを得た(図3)。
上記1.で作製したプラスミドCAG-Fcy::furに化学合成DNAを導入する際に使用する、ΦC31 attB及びHRBが入ったプラスミドDNA、pHRBはユーロフィンジェノミクス株式会社にて合成した。このプラスミドを制限酵素XbaIで切断した。プラスミドCAG-Fcy::furを制限酵素XbaIで切断し、CAGプロモーターとFcy::fur遺伝子間に、XbaIで両端を切断したpHRB由来断片を挿入してプラスミドCAG-ΦC31-HRB-Fcy::furを得た(図3)。
3.HDRベクタープラスミドの作製
上記2.で作製したプラスミドCAG-ΦC31-HRB-Fcy::furに、HRA及び部位特異的組み換え酵素サイトR4 attB、Bxb1 attB、Bxb1 attP、ブラストサイジン耐性遺伝子を導入するためのプラスミドDNA、pHRAはユーロフィンジェノミクス社にて合成した。このプラスミドを制限酵素NotI及び制限酵素SpeIで切断した。プラスミドCAG-ΦC31-HRB-Fcy::furを制限酵素NotI及び制限酵素SpeIで切断し、CAGの上流側にプラスミドHRA由来の2437bp断片を挿入してHDRベクターを得た(図4)。
上記2.で作製したプラスミドCAG-ΦC31-HRB-Fcy::furに、HRA及び部位特異的組み換え酵素サイトR4 attB、Bxb1 attB、Bxb1 attP、ブラストサイジン耐性遺伝子を導入するためのプラスミドDNA、pHRAはユーロフィンジェノミクス社にて合成した。このプラスミドを制限酵素NotI及び制限酵素SpeIで切断した。プラスミドCAG-ΦC31-HRB-Fcy::furを制限酵素NotI及び制限酵素SpeIで切断し、CAGの上流側にプラスミドHRA由来の2437bp断片を挿入してHDRベクターを得た(図4)。
4.CRISPR/Cas9によるIg-NAC(ΔDH)の切断
CRISPR/Cas9により、標的部位において2箇所のDNA二重鎖切断を起こすことで、効率的な相同組み換えが可能となる。Ig-NAC(ΔDH)上のDH領域削除部位付近の2箇所(標的)にガイドRNA、gRNA-A1及びgRNA-B1を設計した。
gRNA-A1:GGGCCCAGGCGCCGTTTAAT AGG (配列番号24)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
gRNA-B1:TGCCGCAGAGTGCCAGGTGC AGG (配列番号25)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
CRISPR/Cas9により、標的部位において2箇所のDNA二重鎖切断を起こすことで、効率的な相同組み換えが可能となる。Ig-NAC(ΔDH)上のDH領域削除部位付近の2箇所(標的)にガイドRNA、gRNA-A1及びgRNA-B1を設計した。
gRNA-A1:GGGCCCAGGCGCCGTTTAAT AGG (配列番号24)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
gRNA-B1:TGCCGCAGAGTGCCAGGTGC AGG (配列番号25)
(ここでgRNA配列は、PAM配列(AGG)を除く20塩基の配列である。)
これらのgRNAからCas9が認識するNGGを除き、制限酵素BbsI認識配列を付加したオリゴDNAをそれぞれ合成した(ユーロフィンジェノミクス社)。さらにこれらのオリゴDNAをアニーリング後、制限酵素BbsI(NEB)で切断したプラスミドDNAを制限酵素BbsIで切断したCas9発現ベクター(px330、addgene Plasmid #42230)にクローニングした。得られた大腸菌コロニーを下記プライマー、及び条件を用いてPCRすることにより、オリゴDNAの挿入の有無を確認した。フォワードプライマーはU6Fを使用した。リバースプライマーはgRNA-A1確認用としてgRNA-A1R、gRNA-B1確認用としてgRNA-B1Rを使用した。
U6F:GAGGGCCTATTTCCCATGATTCC (配列番号26)
gRNA-A1F:CACCGGGGCCCAGGCGCCGTTTAAT (配列番号27)
gRNA-A1R:AAACATTAAACGGCGCCTGGGCCCC (配列番号28)
gRNA-B1F:CACCGTGCCGCAGAGTGCCAGGTGC (配列番号29)
gRNA-B1R:AAACGCACCTGGCACTCTGCGGCAC (配列番号30)
U6F:GAGGGCCTATTTCCCATGATTCC (配列番号26)
gRNA-A1F:CACCGGGGCCCAGGCGCCGTTTAAT (配列番号27)
gRNA-A1R:AAACATTAAACGGCGCCTGGGCCCC (配列番号28)
gRNA-B1F:CACCGTGCCGCAGAGTGCCAGGTGC (配列番号29)
gRNA-B1R:AAACGCACCTGGCACTCTGCGGCAC (配列番号30)
TaqDNAポリメラーゼは(EmeraldAmp PCR Master Mix、タカラバイオ)を使用し、反応条件:98℃,2分,1回→98℃,10秒;60℃,秒;68℃,13秒で35回サイクルのPCRを行った。
PCR後2%アガロースゲル電気泳動により、330bp付近に目的のバンドが得られたコロニーを液体LB培地及びアンピシリン100μL/mLで培養し、(Nucleospin Plasmid Transfection-grade、タカラバイオ)を用いてプラスミドDNA(Cas9-gRNA発現ベクターA1、B1)を得た。
5.Ig-NAC(ΔDH)へのHDRベクター挿入
Cas9/gRNAを使用するゲノム編集により、HDRベクターをCHO細胞中のIg-NAC(ΔDH)のDH領域削除部位付近に挿入した(図5)。HDRベクターとCas9-gRNA発現ベクターA1、B1を、実施例1で作製したIg-NAC(ΔDH)を保持するCHO細胞クローンTF7B10を用いてエレクトロポレーション法を実施して導入した。CHO細胞TF7B10をトリプシン処理し、1×107個/mLになるようにOptiMEM(Thermo Fisher Scientific)で懸濁してから、5μg のHDRベクター、2.5μg のgRNA-A1、2.5μgのgRNA-B1存在下でNEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞をエレクトロポレーション後96穴プレート3枚へ播種し、48~72時間後に800μg/mL G418(富士フィルム和光純薬)と1600μg/mLブラストサイジン(富士フィルム和光純薬)、10%FBS(SIGMA)を含むF12培地で培養し、培養開始84~96時間後に1600μg/mLブラストサイジン、200μM 5FC(東京化成工業)、10%FBSを含むF12培地で培地交換して培養し、薬剤耐性クローンを取得した。
Cas9/gRNAを使用するゲノム編集により、HDRベクターをCHO細胞中のIg-NAC(ΔDH)のDH領域削除部位付近に挿入した(図5)。HDRベクターとCas9-gRNA発現ベクターA1、B1を、実施例1で作製したIg-NAC(ΔDH)を保持するCHO細胞クローンTF7B10を用いてエレクトロポレーション法を実施して導入した。CHO細胞TF7B10をトリプシン処理し、1×107個/mLになるようにOptiMEM(Thermo Fisher Scientific)で懸濁してから、5μg のHDRベクター、2.5μg のgRNA-A1、2.5μgのgRNA-B1存在下でNEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞をエレクトロポレーション後96穴プレート3枚へ播種し、48~72時間後に800μg/mL G418(富士フィルム和光純薬)と1600μg/mLブラストサイジン(富士フィルム和光純薬)、10%FBS(SIGMA)を含むF12培地で培養し、培養開始84~96時間後に1600μg/mLブラストサイジン、200μM 5FC(東京化成工業)、10%FBSを含むF12培地で培地交換して培養し、薬剤耐性クローンを取得した。
6.PCR解析
単離した薬剤耐性クローンを培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、相同組み換え特異的なプライマー3種(下記)、及びIg-NAC上に作製したIg-NAC特異的なプライマー12種(特許第6868250号公報)を用いてPCR解析により、相同組み換え特異的なバンドが得られた1株(TF7B10-74)を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
HRAprimerF1:5’-GAACTGACCGTCTGCTCCA-3’(配列番号31)
Bxb1attBF1:5’-TGCCGAAGCAGTGGTAGAAG-3’(配列番号32)
ΦC31F1:5’-GGGCAACGTGCTGGTTATTG-3’(配列番号33)
HRAprimerR1:5’-GTGCCCTTCTACCACTGCTTC-3’(配列番号34)
Bxb1attPR1:5’-AGAGTGAAGCAGAACGTGGG-3’(配列番号35)
HRBR1:5’-TGCTTTTTCGCAACCATGGG-3’(配列番号36)
単離した薬剤耐性クローンを培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、相同組み換え特異的なプライマー3種(下記)、及びIg-NAC上に作製したIg-NAC特異的なプライマー12種(特許第6868250号公報)を用いてPCR解析により、相同組み換え特異的なバンドが得られた1株(TF7B10-74)を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
HRAprimerF1:5’-GAACTGACCGTCTGCTCCA-3’(配列番号31)
Bxb1attBF1:5’-TGCCGAAGCAGTGGTAGAAG-3’(配列番号32)
ΦC31F1:5’-GGGCAACGTGCTGGTTATTG-3’(配列番号33)
HRAprimerR1:5’-GTGCCCTTCTACCACTGCTTC-3’(配列番号34)
Bxb1attPR1:5’-AGAGTGAAGCAGAACGTGGG-3’(配列番号35)
HRBR1:5’-TGCTTTTTCGCAACCATGGG-3’(配列番号36)
表1において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。

[実施例3]Bxb1リコンビナーゼを用いた部位特異的組み換えによるブラストサイジン耐性遺伝子の除去
化学合成DNAをIg-NAC(ΔDH)上に導入する際に、目的部位で導入されているか否かをブラストサイジン耐性の有無で判断するため、相同組み換えによって挿入したブラストサイジン耐性遺伝子を除去する必要がある。HDRベクターに示している通り、ブラストサイジン耐性遺伝子はBxb1 attB及びBxb1 attPの間に位置しており、Bxb1リコンビナーゼを細胞に導入することによってBxb1 attB-attP間で部位特異的組み換え反応が起こり、ブラストサイジン耐性遺伝子の除去が可能である(図6)。
化学合成DNAをIg-NAC(ΔDH)上に導入する際に、目的部位で導入されているか否かをブラストサイジン耐性の有無で判断するため、相同組み換えによって挿入したブラストサイジン耐性遺伝子を除去する必要がある。HDRベクターに示している通り、ブラストサイジン耐性遺伝子はBxb1 attB及びBxb1 attPの間に位置しており、Bxb1リコンビナーゼを細胞に導入することによってBxb1 attB-attP間で部位特異的組み換え反応が起こり、ブラストサイジン耐性遺伝子の除去が可能である(図6)。
1.Bxb1リコンビナーゼ遺伝子導入によるブラストサイジン耐性遺伝子の除去
方法は、Bxb1リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を実施例2で取得されたアクセプター部位挿入済みのIg-NAC(ΔDH)を保持しているCHO細胞TF7B10-74にエレクトロポレーション法により導入した。TF7B10-74細胞をトリプシン処理し、2×106個/mLとなるようにOpti MEMに懸濁してから10μgのBxb1リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はエレクトロポレーション後10cmディッシュ1枚に播種した。5日後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後9日間800μg/mL G418、10%FBSを含むF12培地で培養し、シングルクローンをピックアップした。
方法は、Bxb1リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を実施例2で取得されたアクセプター部位挿入済みのIg-NAC(ΔDH)を保持しているCHO細胞TF7B10-74にエレクトロポレーション法により導入した。TF7B10-74細胞をトリプシン処理し、2×106個/mLとなるようにOpti MEMに懸濁してから10μgのBxb1リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はエレクトロポレーション後10cmディッシュ1枚に播種した。5日後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後9日間800μg/mL G418、10%FBSを含むF12培地で培養し、シングルクローンをピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、Bxb1部位特異的組み換え後特異的なプライマー2種、及びIg-NAC上に作製したIg-NAC特異的なプライマー12種(特許第6868250号公報)を用いてPCR解析を行った。Bxb1部位特異的組み換え後特異的なプライマー2種のうち、primerHRAF1-Bxb1attPR2では、部位特異的組み換え後2800bp付近から1500bp付近にバンドがシフトする。Bxb1部位特異的組み換え後特異的なバンドが得られた2株(クローンD1、J23)を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
HRAprimerF1:5’-GAACTGACCGTCTGCTCCA-3’(配列番号31)
Bxb1attPR2:5’-GGGGCGTACTTGGCATATGA-3’(配列番号37)
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、Bxb1部位特異的組み換え後特異的なプライマー2種、及びIg-NAC上に作製したIg-NAC特異的なプライマー12種(特許第6868250号公報)を用いてPCR解析を行った。Bxb1部位特異的組み換え後特異的なプライマー2種のうち、primerHRAF1-Bxb1attPR2では、部位特異的組み換え後2800bp付近から1500bp付近にバンドがシフトする。Bxb1部位特異的組み換え後特異的なバンドが得られた2株(クローンD1、J23)を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
HRAprimerF1:5’-GAACTGACCGTCTGCTCCA-3’(配列番号31)
Bxb1attPR2:5’-GGGGCGTACTTGGCATATGA-3’(配列番号37)
3.ブラストサイジン耐性確認
Bxb1部位特異的組み換え後特異的なバンドが得られた2株(クローンD1、J23)に対して、800μg/mLG418と1600μg/mLブラストサイジン、10%FBSを含むF12培地で培養し、ブラストサイジン耐性の有無を確認した。2株中1株(クローンD1:TF7B10-74-D1)がブラストサイジン存在下で死滅した。
Bxb1部位特異的組み換え後特異的なバンドが得られた2株(クローンD1、J23)に対して、800μg/mLG418と1600μg/mLブラストサイジン、10%FBSを含むF12培地で培養し、ブラストサイジン耐性の有無を確認した。2株中1株(クローンD1:TF7B10-74-D1)がブラストサイジン存在下で死滅した。
4.フルオレッセンスインサイチューハイブリダイゼーション(FISH)
FISH解析は(特許第6868250号公報)に記された方法に従い、ヒト遺伝子特異的プローブhuman cot1(invitrogen)及びマウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
FISH解析は(特許第6868250号公報)に記された方法に従い、ヒト遺伝子特異的プローブhuman cot1(invitrogen)及びマウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
上述のPCR解析、ブラストサイジン耐性確認、FISH解析の結果を表2に示した。表2において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○はBxb1組み換え陽性を、×はBxb1組み換え陰性を示した。
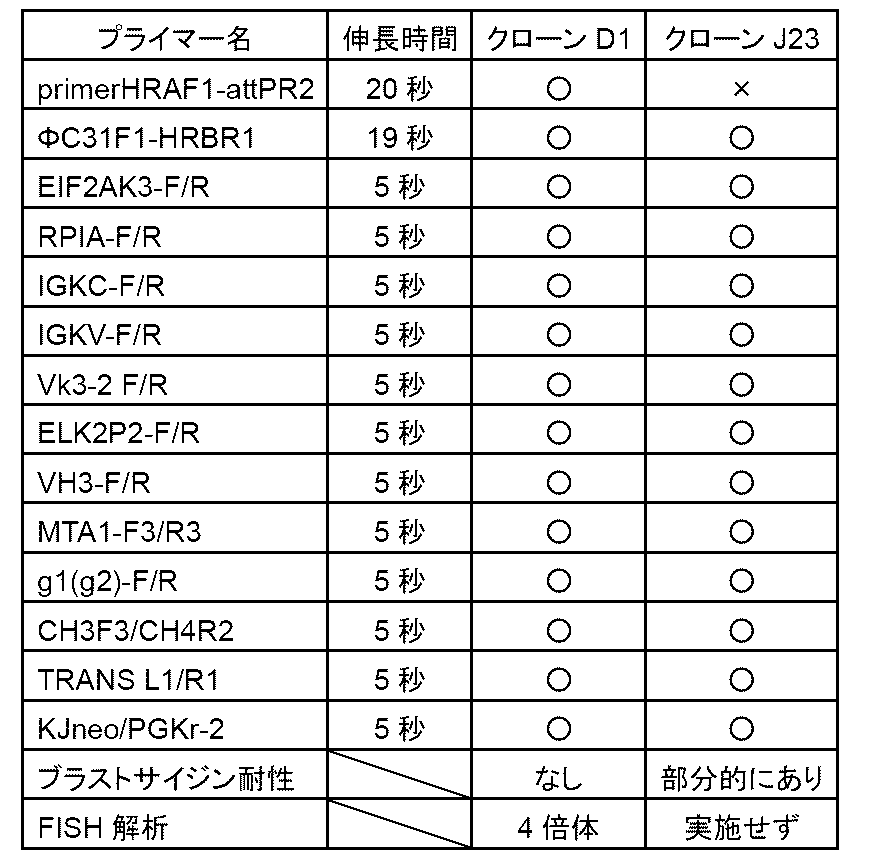
[実施例4]CHO細胞HPRT(-)株への微小核形成法(MMCT)によるIg-NAC(ΔDH/Bsr-)導入
実施例3でBxb1リコンビナーゼによるブラストサイジン耐性遺伝子除去が確認されたIg-NAC(ΔDH/Bsr-)を保持するTF7B10-74-D1クローンは、FISH解析によりIg-NACが独立保持されている一方で4倍体であることが分かった(表2)。この細胞は、化学合成DNAの導入プラットフォーム細胞として重要であるため、MMCTによって、正常な2倍体であるCHO細胞HPRT(-)株にIg-NAC(ΔDH/Bsr-)を移入した。
実施例3でBxb1リコンビナーゼによるブラストサイジン耐性遺伝子除去が確認されたIg-NAC(ΔDH/Bsr-)を保持するTF7B10-74-D1クローンは、FISH解析によりIg-NACが独立保持されている一方で4倍体であることが分かった(表2)。この細胞は、化学合成DNAの導入プラットフォーム細胞として重要であるため、MMCTによって、正常な2倍体であるCHO細胞HPRT(-)株にIg-NAC(ΔDH/Bsr-)を移入した。
1.MMCTによるIg-NAC(ΔDH/Bsr-)のCHO細胞HPRT(-)株への移入
400nMのパクリタクセル及び500nMのリバーシンを約2×108個のTF7B10-74-D1から微小核を調製した。得られた微小核を4mLの1×フィトヘマグルチニン(PHA)-P(富士フィルム和光純薬)で懸濁した。約2×107個のレシピエントであるCHO細胞HPRT(-)株をトリプシン処理後DMEMで3回洗浄後、微小核/pHAP懸濁液で置換し、37℃15分静置した。上清を除去し、PEG溶液(2.5g PEG1000<富士フィルム和光純薬>、3mLのDMEM<富士フィルム和光純薬>、0.5mLのDMSO<富士フィルム和光純薬>)を2mL加えて室温で1分30秒静置後、10mLのDMEMをゆっくり加えた。この後DMEMで3回洗浄し、37℃で温めた10%FBSを含むF12培地10mLで懸濁し、10cmディッシュで培養した。細胞は、24時間後にパッセージを行い、48時間後に96穴プレート48枚に800μg/mLG418、10%FBSを含むF12培地で培地交換して培養した。約2週間後に出現した薬剤耐性コロニー24個をピックアップした。
400nMのパクリタクセル及び500nMのリバーシンを約2×108個のTF7B10-74-D1から微小核を調製した。得られた微小核を4mLの1×フィトヘマグルチニン(PHA)-P(富士フィルム和光純薬)で懸濁した。約2×107個のレシピエントであるCHO細胞HPRT(-)株をトリプシン処理後DMEMで3回洗浄後、微小核/pHAP懸濁液で置換し、37℃15分静置した。上清を除去し、PEG溶液(2.5g PEG1000<富士フィルム和光純薬>、3mLのDMEM<富士フィルム和光純薬>、0.5mLのDMSO<富士フィルム和光純薬>)を2mL加えて室温で1分30秒静置後、10mLのDMEMをゆっくり加えた。この後DMEMで3回洗浄し、37℃で温めた10%FBSを含むF12培地10mLで懸濁し、10cmディッシュで培養した。細胞は、24時間後にパッセージを行い、48時間後に96穴プレート48枚に800μg/mLG418、10%FBSを含むF12培地で培地交換して培養した。約2週間後に出現した薬剤耐性コロニー24個をピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例2で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした24個中14個の細胞クローンで目的のバンドを確認することができた。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例2で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした24個中14個の細胞クローンで目的のバンドを確認することができた。
3.FISH解析
PCR解析で目的のバンドが確認された14個の細胞クローンから任意に6個選択し、FISH解析を実施した。FISH解析は(特許第6868250号公報)に記載された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った6個の細胞クローンにおけるPCR解析、FISH解析の結果を表3に示した。表3において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○はBxb1組み換え陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出、Ig-NACx2はIg-NACが細胞あたり2コピー検出されることを示す。
PCR解析で目的のバンドが確認された14個の細胞クローンから任意に6個選択し、FISH解析を実施した。FISH解析は(特許第6868250号公報)に記載された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った6個の細胞クローンにおけるPCR解析、FISH解析の結果を表3に示した。表3において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○はBxb1組み換え陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出、Ig-NACx2はIg-NACが細胞あたり2コピー検出されることを示す。

[実施例5]ΦC31リコンビナーゼを用いた部位特異的組み換えによるヒトミニマル化IgHDを含む化学合成DNAの挿入
ヒトIgHD領域は、約40.2kbと大きいが、そのほとんどは抗体をコードするD断片以外の配列である。例えばD断片の1個目及び2個目の間には非抗体コード領域が約2500bpある。一方で9個目のD断片と10個目の間は約100bpと短い。今回デザインしたヒトミニマル化IgHDではD断片1-1から6-26の前後にある非抗体コード領域100bpのみを残し、約7.8kbと大幅にDNAサイズを小さくした。ただし、断片間が100bpよりも短い場合は削除や付加を行っていない。また、D領域のみの改変を行うために実施例1で削除したD領域の削除部位に塩基の重複や削除なくD領域が隙間なく繋がるように合成DNAのデザインを行った(図7)。つまり、HRAより下流からD断片1-1までと、D断片6-26からHRBまでの配列はミニマル化せずに保持した。さらにΦC31 attPを保持する化学合成DNAを作製することにより、実施例2で導入したアクセプター部位にあるΦC31 attBとの間で部位特異的組み換えを起こすことが可能である。
ヒトIgHD領域は、約40.2kbと大きいが、そのほとんどは抗体をコードするD断片以外の配列である。例えばD断片の1個目及び2個目の間には非抗体コード領域が約2500bpある。一方で9個目のD断片と10個目の間は約100bpと短い。今回デザインしたヒトミニマル化IgHDではD断片1-1から6-26の前後にある非抗体コード領域100bpのみを残し、約7.8kbと大幅にDNAサイズを小さくした。ただし、断片間が100bpよりも短い場合は削除や付加を行っていない。また、D領域のみの改変を行うために実施例1で削除したD領域の削除部位に塩基の重複や削除なくD領域が隙間なく繋がるように合成DNAのデザインを行った(図7)。つまり、HRAより下流からD断片1-1までと、D断片6-26からHRBまでの配列はミニマル化せずに保持した。さらにΦC31 attPを保持する化学合成DNAを作製することにより、実施例2で導入したアクセプター部位にあるΦC31 attBとの間で部位特異的組み換えを起こすことが可能である。
以下、化学合成DNA組み換え部位であるΦC31 attP及びR4 attP、ベクター導入の有無を確認するためのブラストサイジン耐性遺伝子、ヒトミニマル化IgHDを保持するベクターの作製について記す。まずベクター配列を3分割したプラスミドDNAヒト miniA、ヒトminiB、ヒトminiCをそれぞれ化学合成した(ユーロフィンジェノミクス社)。ここで、ヒトminiAはR4 attP及びHRAより下流からD断片3-10までを、ヒトminiBはD断片4-11より3-22までを、ヒトminiCはD断片4-23よりHRBまで及びΦC31 attP、ブラストサイジン耐性遺伝子をもつ。このヒトminiA,B,Cを保持するヒトミニマル化IgHDベクターを以下1.及び2.の手順(図8)で作製した。
1.ヒトminiBへのヒトminiA断片の挿入
ヒトminiAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したヒトminiBにminiAを挿入することにより、R4 attP-HRA/1-1_3-22を得た。
ヒトminiAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したヒトminiBにminiAを挿入することにより、R4 attP-HRA/1-1_3-22を得た。
2.R4 attP-HRA/1-1_3-22へのヒトminiC断片の挿入
ヒトminiCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したR4 attP-HRA/1-1_3-22にヒトminiC断片を挿入することにより、ヒトミニマル化IgHDベクターを構築した。
ヒトminiCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したR4 attP-HRA/1-1_3-22にヒトminiC断片を挿入することにより、ヒトミニマル化IgHDベクターを構築した。
3.ΦC31リコンビナーゼによるヒトミニマル化IgHDベクターの導入
実施例4で構築した化学合成DNAの導入プラットフォーム細胞にあるΦC31 attBとヒトミニマル化IgHDベクター上にあるΦC31 attPとの間で部位特異的組み換えを起こすことが可能である(図9)。方法としては、ΦC31リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)及びヒトミニマル化IgHDベクターを、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)にエレクトロポレーション法により導入した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのヒトミニマル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後384穴プレート2枚に播種した。細胞は48~72時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。11日後にブラストサイジン耐性があった24クローンをピックアップした。
実施例4で構築した化学合成DNAの導入プラットフォーム細胞にあるΦC31 attBとヒトミニマル化IgHDベクター上にあるΦC31 attPとの間で部位特異的組み換えを起こすことが可能である(図9)。方法としては、ΦC31リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)及びヒトミニマル化IgHDベクターを、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)にエレクトロポレーション法により導入した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのヒトミニマル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後384穴プレート2枚に播種した。細胞は48~72時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。11日後にブラストサイジン耐性があった24クローンをピックアップした。
4.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、ΦC31部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
BsdR1:5’ - ATGCAGATCGAGAAGCACCT - 3’(配列番号38)
2-8to3-9F:5’ - AACGCACCAGACCAGCAGAC - 3’(配列番号39)
4-11to5-12R:5’ -GACGTGGGGCCTAGAGGCT - 3’(配列番号40)
3-22to4-23F:5’ - CAGGCGGGGAAGATTCAGAA- 3’(配列番号41)
4-23to5-24R:5’ - TAGAGAGCCTGGGCCTAGAG- 3’(配列番号42)
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、ΦC31部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
BsdR1:5’ - ATGCAGATCGAGAAGCACCT - 3’(配列番号38)
2-8to3-9F:5’ - AACGCACCAGACCAGCAGAC - 3’(配列番号39)
4-11to5-12R:5’ -GACGTGGGGCCTAGAGGCT - 3’(配列番号40)
3-22to4-23F:5’ - CAGGCGGGGAAGATTCAGAA- 3’(配列番号41)
4-23to5-24R:5’ - TAGAGAGCCTGGGCCTAGAG- 3’(配列番号42)
5.FISH解析
PCR解析で目的のバンドが確認された3クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った3個の細胞クローンにおけるPCR解析、FISH解析の結果を表4に記した。
PCR解析で目的のバンドが確認された3クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った3個の細胞クローンにおけるPCR解析、FISH解析の結果を表4に記した。
表4において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出、Ig-NACx2はIg-NACが細胞あたり2コピー検出されることを示す。
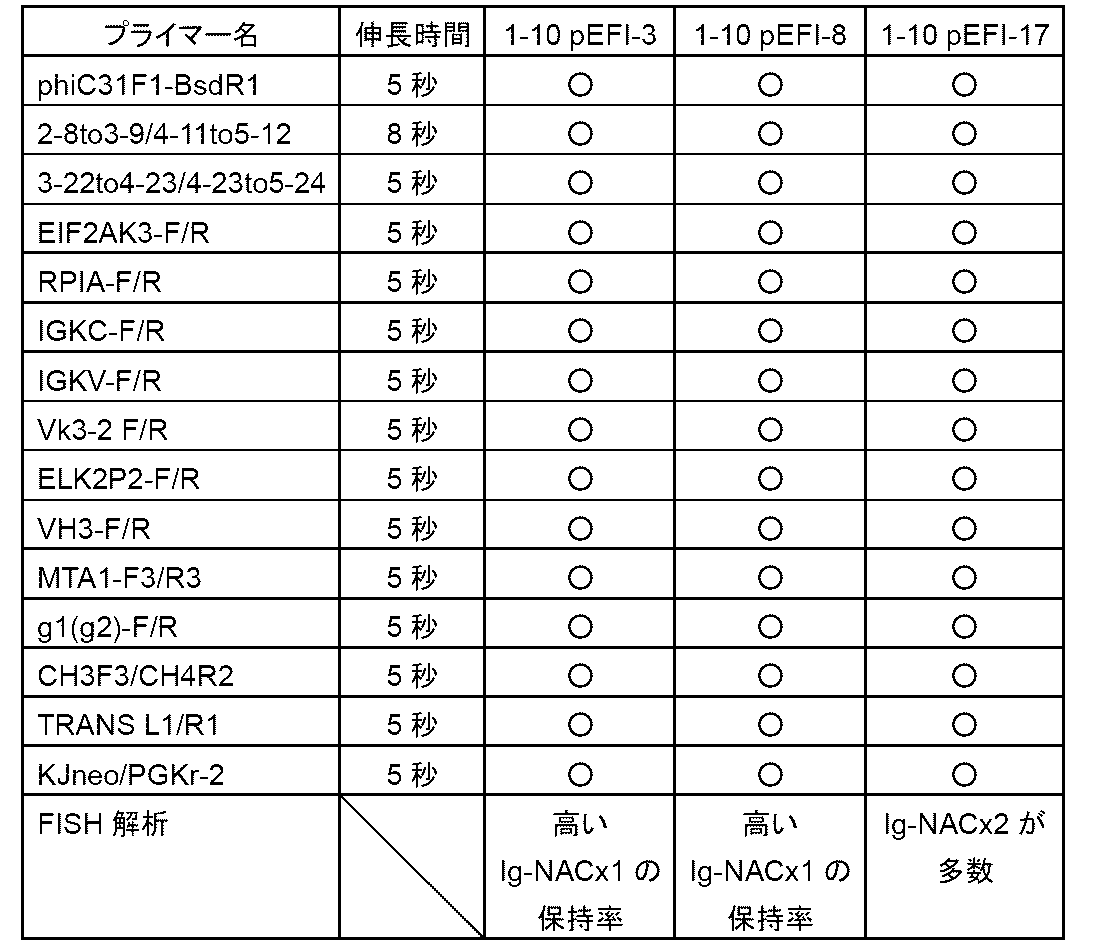
[実施例6]R4リコンビナーゼを用いた部位特異的組み換えによる一部配列除去
Ig-NAC(ΔDH)上に導入した配列の内、CAGプロモーターやブラストサイジン耐性遺伝子などは抗体遺伝子の発現に不要となる配列である。これらの配列は、R4 attB及びattPに挟まれており、R4リコンビナーゼを細胞に導入することによってR4 attB-attP間で部位特異的組み換え反応が起こり、除去することが可能である(図10)。
Ig-NAC(ΔDH)上に導入した配列の内、CAGプロモーターやブラストサイジン耐性遺伝子などは抗体遺伝子の発現に不要となる配列である。これらの配列は、R4 attB及びattPに挟まれており、R4リコンビナーゼを細胞に導入することによってR4 attB-attP間で部位特異的組み換え反応が起こり、除去することが可能である(図10)。
1.R4リコンビナーゼ遺伝子発現ベクター導入
R4リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を、ヒトミニマル化IgHDベクターの挿入が確認されており、かつ1本のIg-NAC保持率が高かったCHO細胞1-10 pEFI-8(実施例5)にエレクトロポレーション法により導入した。1-10 pEFI-8細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はパルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後2週間800μg/mLG418、10%FBSを含むF12培地で培養し、シングルクローンを24個ピックアップした。
R4リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を、ヒトミニマル化IgHDベクターの挿入が確認されており、かつ1本のIg-NAC保持率が高かったCHO細胞1-10 pEFI-8(実施例5)にエレクトロポレーション法により導入した。1-10 pEFI-8細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はパルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後2週間800μg/mLG418、10%FBSを含むF12培地で培養し、シングルクローンを24個ピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、R4部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRしたところ、R4部位特異的組み換え後特異的なバンドが得られた6株を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
R4attLF2:5’ -CCCTCAAGGTGTGAACAGGG- 3’(配列番号43)
R4attLR2:5’ -CTGGAGGGAGGCATGTTCTG- 3’(配列番号44)
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、R4部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRしたところ、R4部位特異的組み換え後特異的なバンドが得られた6株を選定した。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
R4attLF2:5’ -CCCTCAAGGTGTGAACAGGG- 3’(配列番号43)
R4attLR2:5’ -CTGGAGGGAGGCATGTTCTG- 3’(配列番号44)
3.FISH解析
PCR解析で目的のバンドが確認された6個の細胞クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った6個の細胞クローンにおけるPCR解析、FISH解析の結果を表5に示した。
PCR解析で目的のバンドが確認された6個の細胞クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った6個の細胞クローンにおけるPCR解析、FISH解析の結果を表5に示した。
表5において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。
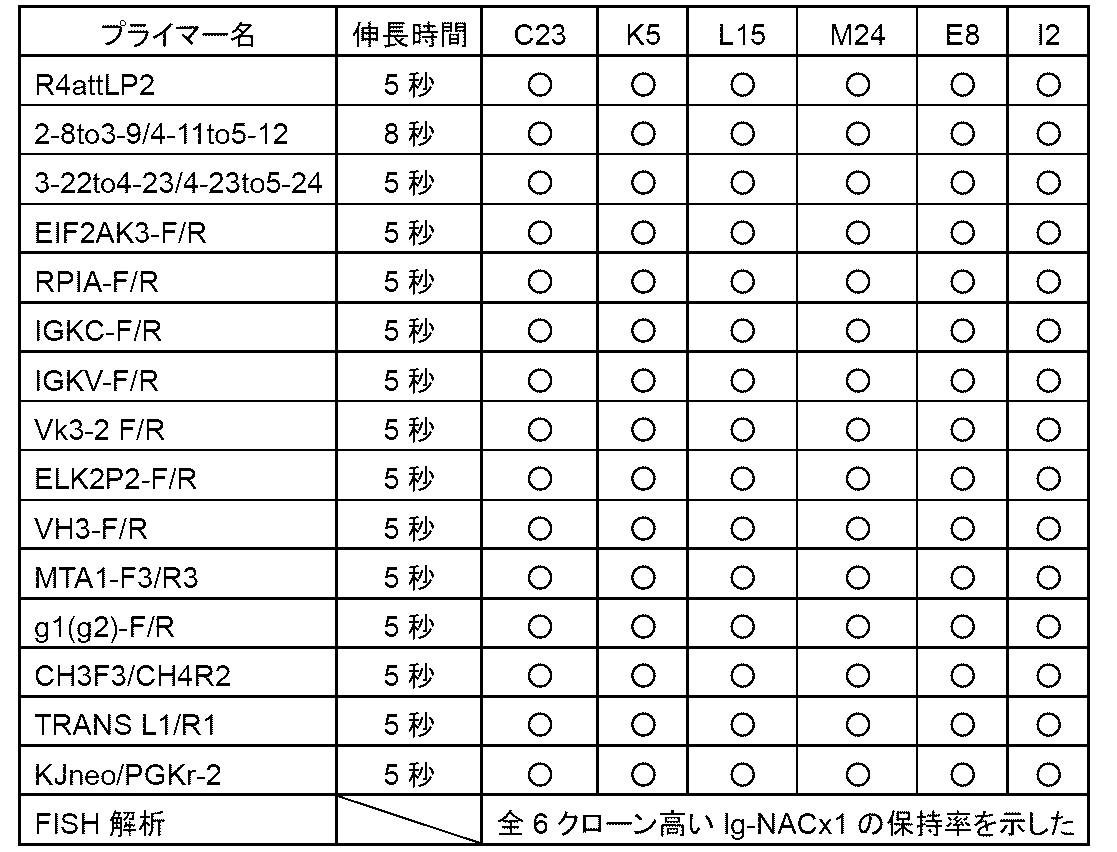
[実施例7]マウスES細胞株への微小核形成法(MMCT)によるヒトミニマル化IgHD搭載Ig-NAC導入
実施例6でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認された6クローンから任意に選択した2クローン:K5(1-10 pEF1-K5)及びM24(1-10 pEF1-M24)を染色体供与細胞として用い、MMCT法を用いてマウスES細胞へのヒトミニマル化IgHD搭載Ig-NAC移入を行った。
実施例6でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認された6クローンから任意に選択した2クローン:K5(1-10 pEF1-K5)及びM24(1-10 pEF1-M24)を染色体供与細胞として用い、MMCT法を用いてマウスES細胞へのヒトミニマル化IgHD搭載Ig-NAC移入を行った。
1.MMCT法を用いたマウスES細胞へのヒトミニマル化IgHD搭載Ig-NAC移入
染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号)を用いた。HKD31 6TG-9株の培養法はマイトマイシンC処理したマウス胎仔線維芽細胞株をフィーダー細胞とし、このフィーダー細胞層上でES細胞用培地である15%のFBS及び1%のヌクレオサイド、ペニシリンストレプトマイシン、Non-Essential Amino Acids、L-グルタミンソリューション、2-メルカプトエタノール、LIFを含むダルベッコ改変イーグル培地(DMEM)を用いて37度で培養した。まず、400nMのパクリタクセル及び500nMのリバーシンを約4×107個の1-10 pEF1-K5及び1-10 pEF1-M24に加え、微小核を調製した。得られた微小核全量を5mLのDMEMに懸濁、遠心により沈殿にした。5×106~4×106個(MMCT2日前に10cmディッシュ1/2を10cmx1に播種)のマウスES細胞HKD31 6TG-9株をトリプシンで分散させた後、DMEMで3回洗浄し、5mLのDMEMに懸濁したのち、微小核の沈澱上に加えた。1200rpm、5分間遠心して上清を除いた。沈殿をタッピングによりよくほぐし、PEG溶液(2.5g PEG1000<富士フィルム和光純薬>、3mLのDMEM<富士フィルム和光純薬>、0.5mLのDMSO<富士フィルム和光純薬>)0.5mLを加えて37度のウォーターバスで振り、90秒後に13mLのDMEMをゆっくりと加え、1200rpm、5分間遠心して上清を除き、沈殿を30mLのES細胞用培地に懸濁し、予めフィーダー細胞を播いた10cmディッシュ3枚に播種した。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行った。1週間~10日後に出現した薬剤耐性コロニー14個をピックアップした。
染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号)を用いた。HKD31 6TG-9株の培養法はマイトマイシンC処理したマウス胎仔線維芽細胞株をフィーダー細胞とし、このフィーダー細胞層上でES細胞用培地である15%のFBS及び1%のヌクレオサイド、ペニシリンストレプトマイシン、Non-Essential Amino Acids、L-グルタミンソリューション、2-メルカプトエタノール、LIFを含むダルベッコ改変イーグル培地(DMEM)を用いて37度で培養した。まず、400nMのパクリタクセル及び500nMのリバーシンを約4×107個の1-10 pEF1-K5及び1-10 pEF1-M24に加え、微小核を調製した。得られた微小核全量を5mLのDMEMに懸濁、遠心により沈殿にした。5×106~4×106個(MMCT2日前に10cmディッシュ1/2を10cmx1に播種)のマウスES細胞HKD31 6TG-9株をトリプシンで分散させた後、DMEMで3回洗浄し、5mLのDMEMに懸濁したのち、微小核の沈澱上に加えた。1200rpm、5分間遠心して上清を除いた。沈殿をタッピングによりよくほぐし、PEG溶液(2.5g PEG1000<富士フィルム和光純薬>、3mLのDMEM<富士フィルム和光純薬>、0.5mLのDMSO<富士フィルム和光純薬>)0.5mLを加えて37度のウォーターバスで振り、90秒後に13mLのDMEMをゆっくりと加え、1200rpm、5分間遠心して上清を除き、沈殿を30mLのES細胞用培地に懸濁し、予めフィーダー細胞を播いた10cmディッシュ3枚に播種した。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行った。1週間~10日後に出現した薬剤耐性コロニー14個をピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした14個中5個で目的のバンドを確認することができた。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした14個中5個で目的のバンドを確認することができた。
3.核型解析
6wellで培養したPCR陽性クローンに関して0.05μg/mLのMAS(フナコシ)を加えて1時間30分後トリプシン処理を行う。1500rpm、5minで遠心後、0.074Mの塩化カリウム5mLで懸濁し、20分間静置した。カルノア処理を行った後に、標本を作成した。この標本に50μLの封入剤(SlowfadeTM Gold antifade with DAPI)でDAPI染色した。その後、マウス染色体の核型異常がないか確認した。このPCR解析、核型解析の結果を表6に記した。
表6において、クローンごとのPCR及び核型解析の結果を示す。横列にクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。
6wellで培養したPCR陽性クローンに関して0.05μg/mLのMAS(フナコシ)を加えて1時間30分後トリプシン処理を行う。1500rpm、5minで遠心後、0.074Mの塩化カリウム5mLで懸濁し、20分間静置した。カルノア処理を行った後に、標本を作成した。この標本に50μLの封入剤(SlowfadeTM Gold antifade with DAPI)でDAPI染色した。その後、マウス染色体の核型異常がないか確認した。このPCR解析、核型解析の結果を表6に記した。
表6において、クローンごとのPCR及び核型解析の結果を示す。横列にクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。
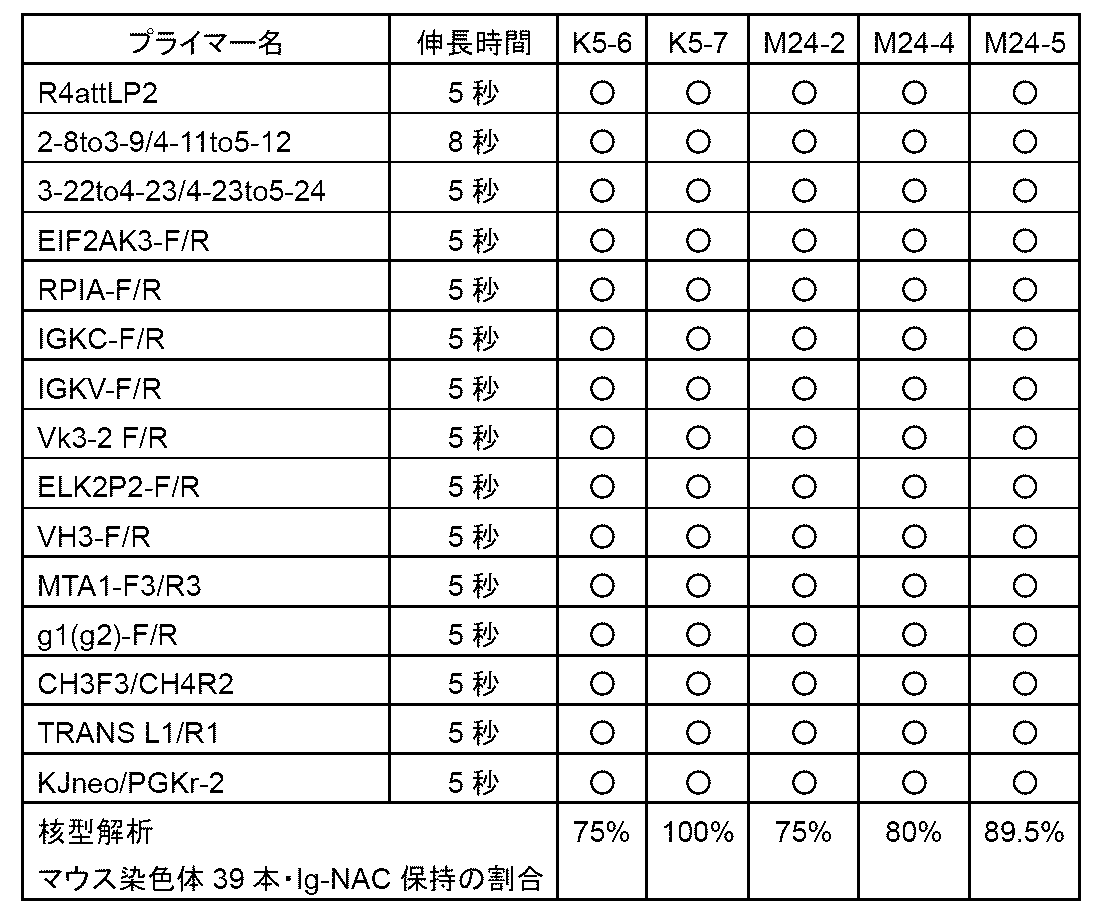
[実施例8]ヒトミニマル化IgHD含有Ig-NACを保持するマウスES細胞株からのキメラマウス作製
ヒト抗体産生を確認するためにヒトミニマル化IgHD含有Ig-NACマウスES細胞からキメラマウスを作製した。
ヒト抗体産生を確認するためにヒトミニマル化IgHD含有Ig-NACマウスES細胞からキメラマウスを作製した。
実施例7で得られたヒトミニマル化IgHD含有Ig-NACを保持するマウスES細胞を用いて、ジーンターゲティング、実験医学、1995の手法に従い、キメラマウスを作製する。宿主としてはMCH(ICR)(白色、日本クレア社より購入)、若しくは抗体遺伝子の欠損(Igh/Igκ KO)若しくは低発現(Igl1 low)を呈するHKLDマウスの雌雄交配により得られる桑実胚及び8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。胚を仮親に移植することで、キメラマウス(毛色に濃茶色の部分の認められる)が得られる。
ヒトミニマル化含有Ig-NACを保持するマウスES細胞株4クローンを計211のICR胚にインジェクションし、仮親6匹に移植した。その結果計5匹のGFP陽性キメラマウスが得られた。毛色によるキメラ率判定の結果、キメラ率は、5匹のキメラマウスについて10~40%であった。マウスES細胞株2クローンについては465個のHKLD胚にインジェクションを実施し27匹の仮親に移植した結果、14匹のGFP陽性個体が得られた。毛色によるキメラ率判定の結果、キメラ率は、14匹のキメラマウスについて5~50%であった。
[実施例9]ΦC31リコンビナーゼを用いた部位特異的組み換えによるウシ化ヒトIgHDを含む化学合成DNAの挿入
ウシにおいては、40~70アミノ酸長と非常に長いCDRH3をもつ抗体が約10%の頻度で観察される(Wangら、Cell、153:1379-93、2013)。長いCDRH3は抗体分子表面から突き出た形を取り、タンパク質の折り畳みで隠されていたエピトープなど、通常の抗体の結合が困難な部位にも結合できる。この特徴をヒト抗体に取り入れるため、ヒトミニマル化DHにおいて抗体のコード領域であるヒトD断片配列をウシD断片配列に置換することを行った。この時、ヒトD断片3-10の位置にウシD断片8-2が該当するように置換した。ただし、ウシD断片の順序は変えていない。ウシのD断片数はヒトよりも少ないため、置き換えなかった3つのヒトD断片は前後100bpと共に削除した。このようにして(図11)のようにウシ化ヒトIgHDをデザインした。
ウシにおいては、40~70アミノ酸長と非常に長いCDRH3をもつ抗体が約10%の頻度で観察される(Wangら、Cell、153:1379-93、2013)。長いCDRH3は抗体分子表面から突き出た形を取り、タンパク質の折り畳みで隠されていたエピトープなど、通常の抗体の結合が困難な部位にも結合できる。この特徴をヒト抗体に取り入れるため、ヒトミニマル化DHにおいて抗体のコード領域であるヒトD断片配列をウシD断片配列に置換することを行った。この時、ヒトD断片3-10の位置にウシD断片8-2が該当するように置換した。ただし、ウシD断片の順序は変えていない。ウシのD断片数はヒトよりも少ないため、置き換えなかった3つのヒトD断片は前後100bpと共に削除した。このようにして(図11)のようにウシ化ヒトIgHDをデザインした。
このウシ化ヒトIgHDを実施例5と同様に3分割し、3分割したプラスミドDNAウシminiA(HRA、及びヒトD断片1-1から3-10の領域に対応するウシD断片B1-1からB8-2を含む)、ウシminiB(ヒトD断片4-11から3-22の領域に対応するウシD断片B6-2からB5-4を含む)、ウシminiC(ヒトD断片4-23から1-26の領域に対応するウシD断片B6-4からB9-4、HRB、ΦC31 attP、及びブラストサイジン耐性遺伝子を含む)を化学合成した(ユーロフィンジェノミクス)。このウシminiA,B,Cを保持するウシ化ヒトミニマル化IgHDベクターを以下の手順1.、2.の通りで作製した(図12)。
1.ウシminiBへのウシminiA断片の挿入
ウシminiAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したウシminiBにminiAを挿入することにより、R4 attP-HRA/1-1_3-22(HRA、及びウシD断片B1-1からB5-4を含む)を得た。
ウシminiAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したウシminiBにminiAを挿入することにより、R4 attP-HRA/1-1_3-22(HRA、及びウシD断片B1-1からB5-4を含む)を得た。
2.R4 attP-HRA/1-1_3-22へのウシminiC断片の挿入
ヒトminiCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したR4 attP-HRA/1-1_3-22に断片ヒトminiCを挿入することにより、ウシ化ヒトIgHDベクターを構築した。
ヒトminiCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したR4 attP-HRA/1-1_3-22に断片ヒトminiCを挿入することにより、ウシ化ヒトIgHDベクターを構築した。
3.ΦC31リコンビナーゼによるウシ化ヒトIgHDベクターの導入
実施例5の方法と同様に実施した。1-10細胞(実施例4)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのウシ化ヒトIgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後384穴プレート2枚に播種した。細胞は48~72時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。13日後にブラストサイジン耐性があった16個ピックアップした。
実施例5の方法と同様に実施した。1-10細胞(実施例4)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのウシ化ヒトIgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後384穴プレート2枚に播種した。細胞は48~72時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。13日後にブラストサイジン耐性があった16個ピックアップした。
4.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5と同じプライマーを用いてPCR解析を実施した。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5と同じプライマーを用いてPCR解析を実施した。
5.FISH解析
PCR解析で目的のバンドが確認された16個の細胞クローンから任意に選択した4クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
PCR解析で目的のバンドが確認された16個の細胞クローンから任意に選択した4クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
FISH解析を行った4個の細胞クローンにおけるPCR解析、FISH解析の結果を表7に記した。
表7において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出、Ig-NACx2はIg-NACが細胞あたり2コピー検出されることを示す。
表7において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出、Ig-NACx2はIg-NACが細胞あたり2コピー検出されることを示す。

[実施例10]R4 リコンビナーゼを用いた部位特異的組み換えによる一部配列除去
1.R4リコンビナーゼ遺伝子発現ベクター導入
実施例6と同様の方法で実施した。ウシ化ヒトIgHDベクターの導入が確認された1-10 C4細胞(実施例9)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞は、パルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後17日間800μg/mL G418、10%FBSを含むF12培地で培養し、シングルクローンを22個ピックアップした。
1.R4リコンビナーゼ遺伝子発現ベクター導入
実施例6と同様の方法で実施した。ウシ化ヒトIgHDベクターの導入が確認された1-10 C4細胞(実施例9)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞は、パルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後17日間800μg/mL G418、10%FBSを含むF12培地で培養し、シングルクローンを22個ピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6と同様のR4部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRしたところ、R4部位特異的組み換え後特異的なバンドが得られた3クローンを選定した。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6と同様のR4部位特異的組み換え後特異的なプライマー1種、及びヒトミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRしたところ、R4部位特異的組み換え後特異的なバンドが得られた3クローンを選定した。
3.FISH解析
PCR解析で目的のバンドが確認された3クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った3個の細胞クローンにおけるPCR解析、FISH解析の結果を表8に示した。表8において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。
PCR解析で目的のバンドが確認された3クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。FISH解析を行った3個の細胞クローンにおけるPCR解析、FISH解析の結果を表8に示した。表8において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。
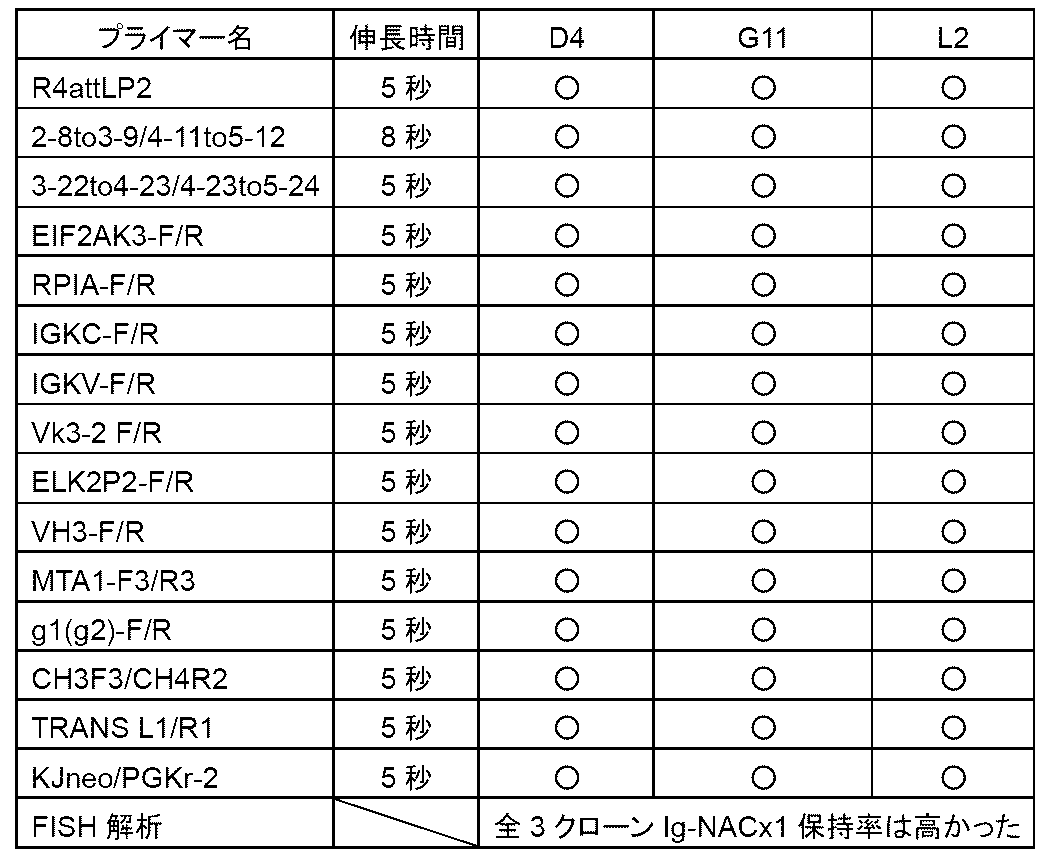
[実施例11]マウスES細胞株への微小核形成法(MMCT)によるウシミニマル化IgHD(ウシ化ヒトミニマル化IgHD)含有Ig-NAC導入
1.MMCT法を用いたマウスES細胞へのウシミニマル化IgHD搭載Ig-NAC移入
実施例10でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認された3クローンから任意に選択した2クローン:G11(1-10 C4-G11)及びL2(1-10 C4-L2)を染色体供与細胞として用いた。染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号公報)を用いた。実施例7と同様の方法で微小核を形成し、マウスES細胞HKD31 6TG-9株(XO)と融合し、細胞を10cmディッシュ3枚に播種した。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行った。1週間~10日後に出現した薬剤耐性コロニー23個をピックアップした。
1.MMCT法を用いたマウスES細胞へのウシミニマル化IgHD搭載Ig-NAC移入
実施例10でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認された3クローンから任意に選択した2クローン:G11(1-10 C4-G11)及びL2(1-10 C4-L2)を染色体供与細胞として用いた。染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号公報)を用いた。実施例7と同様の方法で微小核を形成し、マウスES細胞HKD31 6TG-9株(XO)と融合し、細胞を10cmディッシュ3枚に播種した。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行った。1週間~10日後に出現した薬剤耐性コロニー23個をピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした23個中14個で目的のバンドを確認することができた。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6で行ったPCR解析と同じプライマーを用いてPCRを実施した。ピックアップした23個中14個で目的のバンドを確認することができた。
3.核型解析
実施例7と同様の方法を用いて、マウス染色体の核型異常がないか確認した。このPCR解析、核型解析の結果を表9に記した。核型解析をしたもののうち、マウス染色体が39本かつIg-NACx1の保持率が高く、転座がない9クローンが得られた。表9において、クローンごとのPCR及び核型解析の結果を示す。横列にクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、「転」はロバートソン転座が見られるものである。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。
実施例7と同様の方法を用いて、マウス染色体の核型異常がないか確認した。このPCR解析、核型解析の結果を表9に記した。核型解析をしたもののうち、マウス染色体が39本かつIg-NACx1の保持率が高く、転座がない9クローンが得られた。表9において、クローンごとのPCR及び核型解析の結果を示す。横列にクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、「転」はロバートソン転座が見られるものである。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。

[実施例12]ウシミニマル化IgHD含有Ig-NACを保持するマウスES細胞株からのキメラマウス作製
ヒト抗体産生を確認するためにウシミニマル化IgHD含有Ig-NAC保持マウスES細胞からキメラマウスを作製した。
ヒト抗体産生を確認するためにウシミニマル化IgHD含有Ig-NAC保持マウスES細胞からキメラマウスを作製した。
得られたウシミニマル化IgHD含有Ig-NACを保持するマウスES細胞を用いて、ジーンターゲティング、実験医学、1995の手法に従い、キメラマウスを作製する。宿主としてはMCH(ICR)(白色、日本クレア社より購入)、若しくは抗体遺伝子の欠損(Igh/Igκ KO)若しくは低発現(Igl1 low)を呈するHKLDマウスの雌雄交配により得られる桑実胚及び8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。胚を仮親に移植することで、キメラマウス(毛色に濃茶色の部分の認められる)が得られる。
ウシミニマル化IgHD含有Ig-NACを保持するマウスES細胞8クローンを計591個のICR胚にインジェクションし、32匹の仮親へ移植した結果、18匹のGFP陽性個体が得られ、毛色によるキメラ率判定の結果、キメラ率は、18匹のキメラマウスについて5~60%であった。また、5クローンを計380個のHKLD胚にインジェクションを実施し、21匹の仮親に移植した結果、13匹のGFP陽性個体が得られた。毛色によるキメラ率の判定の結果、キメラ率は、13匹のキメラマウスについて5~80%であった。
[実施例13]ヒトミニマル化キメラマウス解析(B細胞解析、ELISA、cDNA調製)
ヒトミニマル化キメラマウスからヒト抗体が産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
ヒトミニマル化キメラマウスからヒト抗体が産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
1.B細胞分化解析
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されたため、機能性抗体が発現していることが示された。個体ごとのGFP陽性率は最高で28.12%の個体が得られ、個体ごとのGFP及びB220共陽性率は2.30%が最高であった。
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されたため、機能性抗体が発現していることが示された。個体ごとのGFP陽性率は最高で28.12%の個体が得られ、個体ごとのGFP及びB220共陽性率は2.30%が最高であった。
2.ELISAによる抗体発現解析
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
3.ヒト抗体の発現解析及び配列同定
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価した。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出した。
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価した。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出した。
[実施例14]ウシミニマル化キメラマウス解析(B細胞解析、ELISA、cDNA調製)
ウシミニマル化キメラマウスからヒト抗体が産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
ウシミニマル化キメラマウスからヒト抗体が産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
1.B細胞分化解析
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されたため、機能性抗体が発現していることが示された。個体ごとのGFP陽性率は最高で40.38%の個体が得られ、個体ごとのGFP及びB220共陽性率は3.23%が最高であった。
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されたため、機能性抗体が発現していることが示された。個体ごとのGFP陽性率は最高で40.38%の個体が得られ、個体ごとのGFP及びB220共陽性率は3.23%が最高であった。
2.ELISAによる抗体発現解析
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
3.ヒト抗体の発現解析及び配列同定
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価した。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出した。
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価した。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出した。
[実施例15]ヒトミニマル化(ΔIgH/ΔIgκ-ES)キメラマウスレパトワ解析
1.ヒトミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体遺伝子重鎖領域クローニングと塩基配列決定
ヒトミニマル化IgHDを保持するキメラマウス脾臓由来cDNAについて下記プライマーによりPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅した。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いた。
定常領域用:
HIGMEX1-2:5’ - CCAAGCTTCAGGAGAAAGTGATGGAGTC - 3’(配列番号45)
可変領域用:
VH1/5BACK:5’ - CAGGTGCAGCTGCAGCAGTCTGG - 3’(配列番号46)
VH4BACK:5’ - CAGGTGCAGCTGCAGGAGTCGGG - 3’(配列番号47)
VH3BACK:5’ - GAGGTGCAGCTGCAGGAGTCTGG - 3’(配列番号48)
1.ヒトミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体遺伝子重鎖領域クローニングと塩基配列決定
ヒトミニマル化IgHDを保持するキメラマウス脾臓由来cDNAについて下記プライマーによりPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅した。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いた。
定常領域用:
HIGMEX1-2:5’ - CCAAGCTTCAGGAGAAAGTGATGGAGTC - 3’(配列番号45)
可変領域用:
VH1/5BACK:5’ - CAGGTGCAGCTGCAGCAGTCTGG - 3’(配列番号46)
VH4BACK:5’ - CAGGTGCAGCTGCAGGAGTCGGG - 3’(配列番号47)
VH3BACK:5’ - GAGGTGCAGCTGCAGGAGTCTGG - 3’(配列番号48)
PCRは、定常領域用×可変領域用(3種)の組み合わせで98℃10秒、59℃5秒、68℃5秒を35サイクルの条件で行った。全ての増幅産物は1.5%アガロースゲル電気泳動後、臭化エチジウム染色することにより検出した。その結果、全て組み合わせにおいて、期待される470bp付近に増幅産物が検出された。一方、陰性コントロールでは全てにおいて同じ位置に特異的増幅産物は検出されなかった。これらの増幅産物は、アガロースゲルより抽出した後、HindIII及びPstIを用いた制限酵素処理を行った。これをpUC119(TAKARA)ベクターのHindIII,PstI部位にクローニングした。増幅産物の挿入されたプラスミドのうちVH1ファミリー由来クローン、VH4ファミリー由来クローン、VH3ファミリー由来クローンに対してそれぞれシーケンスを実施し、増幅産物の塩基配列を決定した。
2.ヒトミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体遺伝子重鎖可変領域塩基配列の解析
上記1.において決定した塩基配列について、各クローンにおいて使用されている既知の生殖系列V遺伝子(「V断片」という)、D遺伝子(「D断片」という)、J遺伝子(「J断片」という)の特定を行った。そのうち由来するD断片を特定できた27クローンの結果を表10に示す。また野生型Ig-NACを保持するヒト抗体産生マウス(特許第6868250号公報)より取得した脾臓由来cDNAを用いて同様の解析を行った15クローンの結果を表11に示す。
上記1.において決定した塩基配列について、各クローンにおいて使用されている既知の生殖系列V遺伝子(「V断片」という)、D遺伝子(「D断片」という)、J遺伝子(「J断片」という)の特定を行った。そのうち由来するD断片を特定できた27クローンの結果を表10に示す。また野生型Ig-NACを保持するヒト抗体産生マウス(特許第6868250号公報)より取得した脾臓由来cDNAを用いて同様の解析を行った15クローンの結果を表11に示す。
その結果、キメラマウス脾臓Igμ-cDNAにおけるD断片の使用は、ヒトミニマル型の27クローンにおいて13種のD断片が使用されているのに対して、野生型ヒト抗体産生マウスの15クローンにおいては6種であり、またそのうちD断片3-10を使用しているクローンが7クローンと偏りが観察された(図13)。


[実施例16]ウシミニマル化(ΔIgH/ΔIgκ-ES)キメラマウスレパトワ解析
ウシミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAを用いて、実施例15に示された方法でPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅した。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いた。その結果、キメラマウス脾臓Igμ-cDNAにおけるウシD断片の使用が示される。
ウシミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAを用いて、実施例15に示された方法でPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅した。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いた。その結果、キメラマウス脾臓Igμ-cDNAにおけるウシD断片の使用が示される。
またウシD断片の使用を確認するため、以下に示すウシD断片特異的なプライマーを設計し用いた。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いた。
cowF1:5’-ATTATCTGCAGAAGTCTGACCCGCACACAGG - 3’(配列番号49)
cowF2:5’-ATTATCTGCAGTGTGGAGCTGGCCAATGCAT - 3’(配列番号50)
cowF3:5’-ATTATCTGCAGATGGTTATGGTTATGGTTATGG - 3’(配列番号51)
cowR1:5’-GTCGGAAGCTTATAACCACAACCATAACCAT - 3’(配列番号52)
cowF1:5’-ATTATCTGCAGAAGTCTGACCCGCACACAGG - 3’(配列番号49)
cowF2:5’-ATTATCTGCAGTGTGGAGCTGGCCAATGCAT - 3’(配列番号50)
cowF3:5’-ATTATCTGCAGATGGTTATGGTTATGGTTATGG - 3’(配列番号51)
cowR1:5’-GTCGGAAGCTTATAACCACAACCATAACCAT - 3’(配列番号52)
断片内に作製したフォワードプライマー(cowF1、cowF2、cowF3、)と実施例15で用いたHIGMEX1-2(定常領域用のプライマー)、断片内に作製したリバースプライマー(cowR1)と実施例15で用いた可変領域用プライマー(VH1/5BACK、VH4BACK、VH3BACK)の組み合わせで98℃10秒、59℃5秒、68℃5秒を35サイクルの条件で行った。全ての増幅産物は1.5%アガロースゲル電気泳動後、臭化エチジウム染色することにより検出した。その結果、期待される長さ(断片内に作製したフォワードプライマーとHIGMEX1-2:250bp付近、断片内に作製したリバースプライマーと実施例15で用いた可変領域用プライマー:400bp付近)の増幅産物が検出された。一方、陰性コントロールでは全てにおいて同じ位置に特異的増幅産物は検出されなかった。これらの増幅産物は、アガロースゲルより抽出した後、HindIII及びPstIを用いた制限酵素処理を行った。これをpUC119(TAKARA)ベクターのHindIII,PstI部位にクローニングした。増幅産物の挿入されたプラスミドのシーケンスを実施し、塩基配列を決定した。
上記において決定した塩基配列について、各クローンにおいて使用されている既知の生殖系列D断片、及びV断片或いはJ断片の特定を行った。そのうち由来するウシD断片が特定された4クローンの塩基配列を以下に示す。
クローン1
D断片(ウシD1-3):
TGTGGAGCTGGCCA(配列番号53)
J断片(ヒトJ3):
ATGCTTTTGATATCTGGGGCCAAGGGACAATGGTCACCGTCTCTTCAG(配列番号54)
クローン2
D断片(ウシ6-3或いは6-4):
ATGGTTATGGTTATGGTTATGGTTGTGGTTATGGTTATGGTTATAC(配列番号55)
J断片(ヒトJ1):
CTTCCAGCACTGGGGCCAGGGCACCCTGGTCACCGTCTCCTCAG(配列番号56)
クローン3
V断片(ヒトV3-23):
GGGGGAGGCTTGGTACAGCCTGGGGGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTAGCAGCTATGCCATGAGCTGGGTCCGCCAGGCTCCAGGGAAGGGGCTGGAGTGGGTCTCAGCTATTAGTGGTAGTGGTGGTAGCACATACTACGCAGACTCCGTGAAGGGCCGGTTCACCATCTCCAGAGACAATTCCAAGAACACGCTGTATCTGCAAATGAACAGCCTGAGAGCCGAGGACACGGCCGTATATTACTGTGCGAAAGA(配列番号57)
D断片(ウシ6-3或いは6-4):
TAGTTGTTATAGTGGTTATGGTTATGGTTATGGTTGTGGTTAT(配列番号58)
クローン4
V断片(ヒトV2-5):
AGGAGTCTGGTCCTACGCTGGTGAAACCCACACAGACCCTCACGCTGACCTGCACCTTCTCTGGGTTCTCACTCAGCACTAGTGGAGTGGGTGTGGGCTGGATCCGTCAGCCCCCAGGAAAGGCCCTGGAGTGGCTTGCATTCATTTATTGGGATGATGATAAGCGCTACAGCCCATCTCTGAAGAGCAGGCTCACCATCACCAAGGACACCTCCAAAAACCAGGTGGTCCTTACAATGACCAACATGGACCCTGTGGACACAGCCACATATTACTGTGCACACAG(配列番号59)
D断片(ウシ6-3或いは6-4):
TTGTTATAGTGGTTATGGTTATGGTTATGGTTGTGGTTAT(配列番号60)
D断片(ウシD1-3):
TGTGGAGCTGGCCA(配列番号53)
J断片(ヒトJ3):
ATGCTTTTGATATCTGGGGCCAAGGGACAATGGTCACCGTCTCTTCAG(配列番号54)
クローン2
D断片(ウシ6-3或いは6-4):
ATGGTTATGGTTATGGTTATGGTTGTGGTTATGGTTATGGTTATAC(配列番号55)
J断片(ヒトJ1):
CTTCCAGCACTGGGGCCAGGGCACCCTGGTCACCGTCTCCTCAG(配列番号56)
クローン3
V断片(ヒトV3-23):
GGGGGAGGCTTGGTACAGCCTGGGGGGTCCCTGAGACTCTCCTGTGCAGCCTCTGGATTCACCTTTAGCAGCTATGCCATGAGCTGGGTCCGCCAGGCTCCAGGGAAGGGGCTGGAGTGGGTCTCAGCTATTAGTGGTAGTGGTGGTAGCACATACTACGCAGACTCCGTGAAGGGCCGGTTCACCATCTCCAGAGACAATTCCAAGAACACGCTGTATCTGCAAATGAACAGCCTGAGAGCCGAGGACACGGCCGTATATTACTGTGCGAAAGA(配列番号57)
D断片(ウシ6-3或いは6-4):
TAGTTGTTATAGTGGTTATGGTTATGGTTATGGTTGTGGTTAT(配列番号58)
クローン4
V断片(ヒトV2-5):
AGGAGTCTGGTCCTACGCTGGTGAAACCCACACAGACCCTCACGCTGACCTGCACCTTCTCTGGGTTCTCACTCAGCACTAGTGGAGTGGGTGTGGGCTGGATCCGTCAGCCCCCAGGAAAGGCCCTGGAGTGGCTTGCATTCATTTATTGGGATGATGATAAGCGCTACAGCCCATCTCTGAAGAGCAGGCTCACCATCACCAAGGACACCTCCAAAAACCAGGTGGTCCTTACAATGACCAACATGGACCCTGTGGACACAGCCACATATTACTGTGCACACAG(配列番号59)
D断片(ウシ6-3或いは6-4):
TTGTTATAGTGGTTATGGTTATGGTTATGGTTGTGGTTAT(配列番号60)
以上の結果、キメラマウス脾臓においてウシD断片と、ヒトV断片或いはJ断片が連結したcDNAが検出された。例えばクローン2のcDNA配列より導き出されるアミノ酸配列は、下記の配列:
 の通りであり、CDRH3の長さは18アミノ酸(下線部のアミノ酸数)以上の長鎖CDR3であることが確認された。
の通りであり、CDRH3の長さは18アミノ酸(下線部のアミノ酸数)以上の長鎖CDR3であることが確認された。
[実施例17]ヒトミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス免疫応答
ヒトミニマル化キメラマウスに抗原タンパク質を投与し、投与後に採取した血清或いは血漿サンプルを用いて酵素結合免疫吸着測定法(Enzyme-Linked Immuno Sorbent Assay:以下、ELISA)を行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価することができる。
ヒトミニマル化キメラマウスに抗原タンパク質を投与し、投与後に採取した血清或いは血漿サンプルを用いて酵素結合免疫吸着測定法(Enzyme-Linked Immuno Sorbent Assay:以下、ELISA)を行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価することができる。
1.抗原の投与
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)と、追加免疫ではSigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと)、最終免疫では100μL(抗原50μg、1mg/mg抗原タンパク質溶液、静注)を投与する。
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)と、追加免疫ではSigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと)、最終免疫では100μL(抗原50μg、1mg/mg抗原タンパク質溶液、静注)を投与する。
2.抗血清ELISA
各免疫投与3日後に末梢血から血漿を調製する。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行う。
各免疫投与3日後に末梢血から血漿を調製する。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行う。
[実施例18]ウシミニマル化(ΔIgH/ΔIgκ-ES)キメラマウス免疫応答
ウシミニマル化キメラマウスにSARS-CoV2スパイクタンパク質S1の受容体結合部位(Receptor Binding Domain:以下、RBD)を抗原として投与し、投与後に採取した血清或いは血漿サンプルを用いてELISAを行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価した。
ウシミニマル化キメラマウスにSARS-CoV2スパイクタンパク質S1の受容体結合部位(Receptor Binding Domain:以下、RBD)を抗原として投与し、投与後に採取した血清或いは血漿サンプルを用いてELISAを行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価した。
1.抗原の投与
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)と、追加免疫ではSigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと5回)を投与した。
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)と、追加免疫ではSigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと5回)を投与した。
2.抗血清ELISA
各免疫投与3日後に末梢血から血漿を調製した。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行った。その結果、継続的な追加免疫により免疫応答し、RBDに対する抗体力価が上昇することが確認された(図14)。
各免疫投与3日後に末梢血から血漿を調製した。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行った。その結果、継続的な追加免疫により免疫応答し、RBDに対する抗体力価が上昇することが確認された(図14)。
[実施例19]ΦC31リコンビナーゼを用いた部位特異的組み換えによるサル化IgHDを含む化学合成DNA(シンプロジェン)の挿入
ヒトと進化的に近縁であり、抗体遺伝子配列が解明されているカニクイザルD断片(各D断片の塩基配列は、http://www.imgt.org/に記載されている。)をヒトD断片と置換し、D断片以外の領域は全てヒト抗体D領域遺伝子配列を維持した約40kbpのサル化ヒトIgHDをデザインした(図15)。
ヒトと進化的に近縁であり、抗体遺伝子配列が解明されているカニクイザルD断片(各D断片の塩基配列は、http://www.imgt.org/に記載されている。)をヒトD断片と置換し、D断片以外の領域は全てヒト抗体D領域遺伝子配列を維持した約40kbpのサル化ヒトIgHDをデザインした(図15)。
1.サル化IgHDを含む化学合成DNAの作製
置換するヒトのD断片数は26、一方でカニクイザルのD断片数は40であるため、カニクイザルのD断片同士で相互に類似している配列を除外し、ヒトIgHDと比較し変化の大きいものを優先して、以下の表12に示す26種に絞り込んだ。また、4つのヒトD断片(D4-11、D1-14、D4-23、D5-24)の前後に存在するVDJ組み換え配列の変異を正常型に変換した。
置換するヒトのD断片数は26、一方でカニクイザルのD断片数は40であるため、カニクイザルのD断片同士で相互に類似している配列を除外し、ヒトIgHDと比較し変化の大きいものを優先して、以下の表12に示す26種に絞り込んだ。また、4つのヒトD断片(D4-11、D1-14、D4-23、D5-24)の前後に存在するVDJ組み換え配列の変異を正常型に変換した。

この26種のカニクイザルD断片を保持するサル化IgHDに加え、化学合成DNA組み換え部位であるΦC31 attP及びR4 attP、ベクター導入の有無を確認するためのブラストサイジン耐性遺伝子をもつベクターをシンプロジェン株式会社の枯草菌を用いたOGAB(Ordered Gene Assembly in Bacillus subtilis)法により化学合成した。
2.ΦC31リコンビナーゼによるサル化IgHDベクターの導入
実施例5の方法と同様に実施した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのサル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はパルス後に384穴プレート2枚に播種した。48時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。12日後にブラストサイジン耐性があった17個ピックアップした。
実施例5の方法と同様に実施した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのサル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。細胞はパルス後に384穴プレート2枚に播種した。48時間後に800μg/mL G418、800μg/mLブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。12日後にブラストサイジン耐性があった17個ピックアップした。
3.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5と同じΦC31部位特異的組み換え後特異的なプライマー1種及びIg-NAC上に作製したIg-NAC特異的なプライマー12種に加えて、以下に示すサル化IgHD特異的なプライマー1種を用いてPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
3-10monkeyF:5’-CACCCACAGTGTCACAGAGT- 3’(配列番号62)
4-11monkeyR:5’-TCACTGTGGGTGGCAAAACT- 3’(配列番号63)
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例5と同じΦC31部位特異的組み換え後特異的なプライマー1種及びIg-NAC上に作製したIg-NAC特異的なプライマー12種に加えて、以下に示すサル化IgHD特異的なプライマー1種を用いてPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/kbのサイクルを35サイクル行った。
3-10monkeyF:5’-CACCCACAGTGTCACAGAGT- 3’(配列番号62)
4-11monkeyR:5’-TCACTGTGGGTGGCAAAACT- 3’(配列番号63)
さらに、サル化IgHD全体がクローンに保持されているか否かを確認するために、以下に示す5種のプライマーを作製し、ロングPCRを行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはPrimeSTARTM GXL Premix Fast(タカラバイオ株式会社)を用いてステップダウン方式により以下のようなPCRを行った。
98℃2分1サイクル→変性98℃10秒、アニーリング及び伸長は72℃で、増幅断片の長さ10kbp以下の場合、5秒/長さ(kb)、10kbを超える場合、10秒/kb 5サイクル→変性98℃10秒、アニーリング及び伸長は70℃で、増幅断片の長さ10kbp以下の場合、5秒/長さ(kb)、10kbを超える場合、10秒/kb 5サイクル→変性98℃10秒、アニーリング及び伸長は68℃で、増幅断片の長さ10kbp以下の場合、5秒/長さkb、10kbを超える場合、10秒/kbのサイクルを30サイクル行った。
PACF1:5’- CGGTGTGCGGTTGTATGCCTGCTGT - 3’(配列番号64)
3-3to4-4R:5’- GGCCAGGCAGGATGATGGACTCCCA - 3’(配列番号65)
3-3F:5’- CGGTTACTATTACACCCACAGCGTC - 3’(配列番号66)
3-10F2:5’- TTGTAGTGGTGGTGTCTGCTACACC - 3’(配列番号67)
3-16R:5’- GTAGTTACTGTATTCACACAGTGAC - 3’(配列番号68)
F37:5’- CTGCAGGCCCTGTCCTCTTC - 3’(配列番号69)
4-23R:5’- ATAGTAATACCACTGTGGGAGGGCC - 3’(配列番号70)
PACF1:5’- CGGTGTGCGGTTGTATGCCTGCTGT - 3’(配列番号64)
3-3to4-4R:5’- GGCCAGGCAGGATGATGGACTCCCA - 3’(配列番号65)
3-3F:5’- CGGTTACTATTACACCCACAGCGTC - 3’(配列番号66)
3-10F2:5’- TTGTAGTGGTGGTGTCTGCTACACC - 3’(配列番号67)
3-16R:5’- GTAGTTACTGTATTCACACAGTGAC - 3’(配列番号68)
F37:5’- CTGCAGGCCCTGTCCTCTTC - 3’(配列番号69)
4-23R:5’- ATAGTAATACCACTGTGGGAGGGCC - 3’(配列番号70)
4.FISH解析
PCR解析で目的のバンドが確認された1個の細胞クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
PCR解析で目的のバンドが確認された1個の細胞クローンに対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
FISH解析を行った1個の細胞クローンにおけるPCR解析、FISH解析の結果を表Xに記した。
表13において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。
表13において、横列がクローン名、縦列にPCRで使用したプライマー及び伸長時間を示す。○は陽性を示した。表中、Ig-NACx1はIg-NACが細胞あたり1コピー検出されることを示す。

[実施例20]R4リコンビナーゼを用いた部位特異的組み換えによる一部配列除去(サル化)
1.R4リコンビナーゼ遺伝子発現ベクター導入
実施例6と同様の方法で実施した。サル化ヒトIgHDベクターの導入が確認された細胞クローン(実施例19)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後17日後に、800μg/mLG418、10%FBSを含むF12培地で培地交換して培養し、シングルクローンをピックアップした。
1.R4リコンビナーゼ遺伝子発現ベクター導入
実施例6と同様の方法で実施した。サル化ヒトIgHDベクターの導入が確認された細胞クローン(実施例19)をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから10μgのR4リコンビナーゼ遺伝子発現ベクターを加え、NEPA21(ネッパジーン)を用いて150V,7.5msでエレクトロポレーションを行った。パルス後10cmディッシュ1枚に播種した。24~48時間後に細胞をトリプシン処理し、384穴プレート1枚あたり150個になるように細胞を播種した。384穴プレート播種後17日後に、800μg/mLG418、10%FBSを含むF12培地で培地交換して培養し、シングルクローンをピックアップした。
2.PCR解析
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6と同様のR4部位特異的組み換え後特異的なプライマー1種、及びサル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRを実施し、R4部位特異的組み換え後特異的なバンドが得られるクローン(1-10 M9-I9)を選定した。
単離した細胞を培養し、細胞からCellysis(キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6と同様のR4部位特異的組み換え後特異的なプライマー1種、及びサル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いてPCR解析を行った。R4部位特異的組み換え後特異的なプライマーを用いてPCRを実施し、R4部位特異的組み換え後特異的なバンドが得られるクローン(1-10 M9-I9)を選定した。
3.FISH解析
PCR解析で目的のバンドが確認されたクローン(1-10 M9-I9)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
PCR解析で目的のバンドが確認されたクローン(1-10 M9-I9)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
[実施例21]マウスES細胞株への微小核形成法(MMCT)によるサル化IgHD搭載Ig-NAC導入
1.MMCT法を用いたマウスES細胞へのサル化IgHD搭載Ig-NAC移入
実施例10でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認されたクローンを染色体供与細胞として用いる。染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号)を用いる。実施例7と同様の方法で微小核を形成し、マウスES細胞HKD31 6TG-9株(XO)と融合し、10cmdish3枚に播種する。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行う。1週間~10日後に出現する薬剤耐性コロニーをピックアップする。
1.MMCT法を用いたマウスES細胞へのサル化IgHD搭載Ig-NAC移入
実施例10でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認されたクローンを染色体供与細胞として用いる。染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号)を用いる。実施例7と同様の方法で微小核を形成し、マウスES細胞HKD31 6TG-9株(XO)と融合し、10cmdish3枚に播種する。48時間後から350μg/mLのG418を加えたES細胞用培地と交換し、この後、2日に1回同じ組成で培地交換を行う。1週間~10日後に出現する薬剤耐性コロニーをピックアップする。
2.PCR解析
単離した細胞を培養し、細胞からCellysis (キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6で行ったPCR解析と同じプライマーを用いてPCRを実施する。
単離した細胞を培養し、細胞からCellysis (キアゲン)を用いてゲノムDNAを抽出し、このゲノムDNAを鋳型として、実施例6で行ったPCR解析と同じプライマーを用いてPCRを実施する。
3.核型解析
実施例7と同様の方法を用いて、マウス染色体の核型異常がないか確認する。核型解析をしたもののうち、マウス染色体が39本かつIg-NACx1の保持率が高く、転座がないクローンを選抜する。
実施例7と同様の方法を用いて、マウス染色体の核型異常がないか確認する。核型解析をしたもののうち、マウス染色体が39本かつIg-NACx1の保持率が高く、転座がないクローンを選抜する。
[実施例22]サル化IgHD含有Ig-NACを保持するマウスES細胞株からのキメラマウス作製
ヒト抗体産生を確認するためにサル化IgHD含有Ig-NACを保持するマウスES細胞株からキメラマウスを作製する。得られるサル化IgHD含有Ig-NACを保持するマウスES細胞を用いて、ジーンターゲティング、実験医学、1995の手法に従い、キメラマウスを作製する。宿主としてはMCH(ICR)(白色、日本クレア社より購入)若しくは抗体遺伝子の欠損(Igh/Igκ KO)若しくは低発現(Igl1 low)を呈するHKLDマウスの雌雄交配により得られる桑実胚及び8細胞期胚を用いる。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。胚を仮親に移植することで、キメラマウス(毛色に濃茶色の部分の認められる)が得られる。
ヒト抗体産生を確認するためにサル化IgHD含有Ig-NACを保持するマウスES細胞株からキメラマウスを作製する。得られるサル化IgHD含有Ig-NACを保持するマウスES細胞を用いて、ジーンターゲティング、実験医学、1995の手法に従い、キメラマウスを作製する。宿主としてはMCH(ICR)(白色、日本クレア社より購入)若しくは抗体遺伝子の欠損(Igh/Igκ KO)若しくは低発現(Igl1 low)を呈するHKLDマウスの雌雄交配により得られる桑実胚及び8細胞期胚を用いる。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。胚を仮親に移植することで、キメラマウス(毛色に濃茶色の部分の認められる)が得られる。
[実施例23]サル化IgHDキメラマウス解析(B細胞解析、ELISA、cDNA調製)
サル化IgHDキメラマウスからサル化IgHDヒト抗体が産生産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
サル化IgHDキメラマウスからサル化IgHDヒト抗体が産生産生されているかの評価として、B細胞分化解析、血漿中抗体濃度測定ELISA解析、脾臓からのcDNA回収による遺伝子配列解析を行うことができる。
1.B細胞分化解析
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されれば、機能性抗体が発現していることが示される。
EGFP及びB細胞マーカーであるB220が共に陽性である細胞が検出できれば、抗体産生寄与によるB細胞分化が示され、抗体の機能性発現の間接的評価になる。キメラマウスから取得した末梢血をサンプルとして特許第6868250号公報に記載の方法でB220のフローサイトメトリー解析を行った結果、GFP陽性の細胞集団のみならず、GFP及びB220共陽性の集団が確認されれば、機能性抗体が発現していることが示される。
2.ELISAによる抗体発現解析
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
キメラマウスから得られた血漿を用いて特許第6868250号公報に記載の方法で、マウスの抗体発現の有無確認も含め、マウス抗体(mγ、mμ、mκ、mλ)、ヒト抗体(hγ、hμ、hκ、hλ、hγ1、hγ2、hγ3、hγ4、hα、hε、hδ)の血漿中の濃度を測定する。
3.ヒト抗体の発現解析及び配列同定
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価する。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)し、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出する。
ヒト抗体産生マウス脾臓由来RNAからcDNAを合成し、ヒト抗体遺伝子可変領域クローニングと塩基配列決定を行う。特許第6868250号公報と同様に実施することで解析、評価する。脾臓は回収後RNAlater(Ambion)で一晩4℃保存した後、溶液から取り出し-80℃で保管した。ジルコニアビーズの入った耐圧チューブに1mLのISOGEN(ニッポンジーン)を添加し、組織50mgを加える。装置で破砕(30~45秒程度)し、組織が破砕されたことを確認した後、スピンダウン後、1mLの上清を分取し、200μLのクロロフォルムを加え、RNeasy Mini kitを用いメーカープロトコルに従ってRNAを抽出する。
[実施例24]サル化(ΔIgH/ΔIgκ-ES)キメラマウスレパトワ解析
1.サル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体遺伝子重鎖領域クローニングと塩基配列決定
サル化IgHDを保持するキメラマウス脾臓由来cDNAについて下記プライマーによりPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅する。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いる。
定常領域:
HIGMEX1-2:5’-CCAAGCTTCAGGAGAAAGTGATGGAGTC-3’(配列番号71)
可変領域用:
VH1/5BACK:5’-CAGGTGCAGCTGCAGCAGTCTGG-3’(配列番号72)
VH4BACK:5’-CAGGTGCAGCTGCAGGAGTCGGG-3’(配列番号73)
VH3BACK:5’-GAGGTGCAGCTGCAGGAGTCTGG-3’(配列番号74)
1.サル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体遺伝子重鎖領域クローニングと塩基配列決定
サル化IgHDを保持するキメラマウス脾臓由来cDNAについて下記プライマーによりPCRを行い、ヒト抗体遺伝子重鎖可変領域を増幅する。陰性コントロールとして非キメラマウスICRより取得した脾臓由来cDNAを用いる。
定常領域:
HIGMEX1-2:5’-CCAAGCTTCAGGAGAAAGTGATGGAGTC-3’(配列番号71)
可変領域用:
VH1/5BACK:5’-CAGGTGCAGCTGCAGCAGTCTGG-3’(配列番号72)
VH4BACK:5’-CAGGTGCAGCTGCAGGAGTCGGG-3’(配列番号73)
VH3BACK:5’-GAGGTGCAGCTGCAGGAGTCTGG-3’(配列番号74)
PCRは定常領域用×可変領域用(3種)の組み合わせで98℃10秒、59℃5秒、68℃5秒を35サイクルの条件で行う。全ての増幅産物は1.5%アガロースゲル電気泳動後、臭化エチジウム染色することにより検出する。増幅産物は、アガロースゲルより抽出した後、HindIII及びPstIを用いた制限酵素処理を行う。これをpUC119(TAKARA)ベクターのHindIII,PstI部位にクローニングする。増幅産物の挿入されたプラスミドに対してシーケンスを実施し、増幅産物の塩基配列を決定する。
2.サル化(ΔIgH/ΔIgκ-ES)キメラマウス脾臓由来cDNAからのヒト抗体 遺伝子重鎖可変領域塩基配列の解析
1.において決定した塩基配列について、各クローンにおいて使用されている既知の生殖系列V遺伝子断片、D断片、J断片の特定を行う。その結果、VDJ組み換えによりサルDH配列を含むヒト抗体遺伝子重鎖可変領域cDNAの存在が確認される。
1.において決定した塩基配列について、各クローンにおいて使用されている既知の生殖系列V遺伝子断片、D断片、J断片の特定を行う。その結果、VDJ組み換えによりサルDH配列を含むヒト抗体遺伝子重鎖可変領域cDNAの存在が確認される。
[実施例25]サル化(ΔIgH/ΔIgκ-ES)キメラマウス免疫応答
サル化(ΔIgH/ΔIgκ-ES)キメラマウスに抗原タンパク質を投与し血清或いは血漿サンプルを用いて酵素結合免疫吸着測定法(Enzyme-Linked Immuno Sorbent Assay:以下、ELISA)を行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価することができる。
サル化(ΔIgH/ΔIgκ-ES)キメラマウスに抗原タンパク質を投与し血清或いは血漿サンプルを用いて酵素結合免疫吸着測定法(Enzyme-Linked Immuno Sorbent Assay:以下、ELISA)を行うことで、免疫応答及び抗原特異的抗体が誘導されるか評価することができる。
1.抗原の投与
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)、追加免疫では、Sigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと)、最終免疫では100μL(抗原50μg、1mg/mg抗原タンパク質溶液、静注)を投与する。
0.4Mアルギニンを含むPBSにより1mg/mgの抗原タンパク質溶液を調製し、初回免疫ではFreund’s Complete adjuvant(Sigma F5881)、追加免疫では、Sigma Adjuvant SystemTM(SASTM)(Sigma S6322)と1:1で混合することで抗原投与液を準備する。6週齢のキメラマウスに初回免疫では200μL(抗原100μg、腹腔)、追加免疫では100μL(抗原50μg、腹腔、2週間ごと)、最終免疫では100μL(抗原50μg、1mg/mg抗原タンパク質溶液、静注)を投与する。
2.抗血清ELISA
各免疫投与3日後に末梢血から血漿を調製する。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行う。
各免疫投与3日後に末梢血から血漿を調製する。血漿中の抗原特異的IgGの力価を評価するため、抗原を固相化した96穴プレートと抗ヒトIgG Fc抗体を用いて抗血清ELISAを行う。
[実施例26]ヒトミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統の樹立
実施例7でマウスES細胞TT2F(XO)を受容細胞として作製されたヒトミニマル化IgHD搭載Ig-NACを保持するマウスES細胞を用いて、実施例8で示した通りに作製されたキメラマウスを内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、ヒトミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立した。得られた、ヒトミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(#200)について、実施例13で示した通りの方法でフローサイトメトリー解析を行った。その結果、末梢血単核球細胞のGFP陽性率は96.97%、末梢血単核球細胞のGFP及びB220共陽性率は18.78%であり、この値は、ヒトミニマル化IgHDの代わりに野生型Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(特許第6868250号公報)と同等であった。
実施例7でマウスES細胞TT2F(XO)を受容細胞として作製されたヒトミニマル化IgHD搭載Ig-NACを保持するマウスES細胞を用いて、実施例8で示した通りに作製されたキメラマウスを内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、ヒトミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立した。得られた、ヒトミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(#200)について、実施例13で示した通りの方法でフローサイトメトリー解析を行った。その結果、末梢血単核球細胞のGFP陽性率は96.97%、末梢血単核球細胞のGFP及びB220共陽性率は18.78%であり、この値は、ヒトミニマル化IgHDの代わりに野生型Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(特許第6868250号公報)と同等であった。
[実施例27]ウシミニマル化IgHD搭載Ig-NACを保持し、かつ内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統の樹立
実施例11でマウスES細胞TT2F(XO)を受容細胞として作製されたウシミニマル化IgHD搭載Ig-NACを保持するマウスES細胞を用いて、実施例12で示した通りに作製されたキメラマウスを野生型マウス或いは内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、ウシミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立した。得られた、ウシミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(#241)について、実施例13で示した通りの方法でフローサイトメトリー解析を行った。その結果、末梢血単核球細胞のGFP陽性率は99.09%、末梢血単核球細胞のGFP及びB220共陽性率は6.26%であり、この値は、ウシミニマル化IgHDの代わりに野生型Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(特許第6868250号公報)と同等であった。
実施例11でマウスES細胞TT2F(XO)を受容細胞として作製されたウシミニマル化IgHD搭載Ig-NACを保持するマウスES細胞を用いて、実施例12で示した通りに作製されたキメラマウスを野生型マウス或いは内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、ウシミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立した。得られた、ウシミニマル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(#241)について、実施例13で示した通りの方法でフローサイトメトリー解析を行った。その結果、末梢血単核球細胞のGFP陽性率は99.09%、末梢血単核球細胞のGFP及びB220共陽性率は6.26%であり、この値は、ウシミニマル化IgHDの代わりに野生型Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス個体(特許第6868250号公報)と同等であった。
[実施例28]サル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統の樹立
実施例22で作製されるキメラマウス、或いは実施例21でマウスES細胞TT2F(XO)を受容細胞として実施例22で示した通りに作製されるキメラマウスを野生型マウス或いは内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、サル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立できる。
実施例22で作製されるキメラマウス、或いは実施例21でマウスES細胞TT2F(XO)を受容細胞として実施例22で示した通りに作製されるキメラマウスを野生型マウス或いは内因性免疫グロブリン重鎖、κ鎖を破壊したマウス系統と交配することで、サル化IgHD搭載Ig-NACを保持し、内因性免疫グロブリン重鎖及びκ鎖遺伝子座が破壊されたマウス系統を樹立できる。
[実施例29]ΦC31リコンビナーゼを用いた部位特異的組み換えによるヒト長鎖ミニマル化IgHDを含む化学合成DNAの挿入
実施例5で設計したヒトミニマル化IgHDのD断片1-1から1-26を、図16に示す26種の改変長鎖DH配列と置換したヒト長鎖ミニマル化IgHDベクターを実施例5と同様に以下の1.及び2.の手順で作製した。ヒト長鎖DH配列は、NCBIに登録されているヒト抗体cDNA配列の中に見出される、長い(18アミノ酸以上)CDR3配列にもとづき設計した。ヒト長鎖miniA、ヒト長鎖miniB、ヒト長鎖miniCの各ベクターは実施例5同様に、ユーロフィンジェノミクス社にて合成されたものを用いた。
実施例5で設計したヒトミニマル化IgHDのD断片1-1から1-26を、図16に示す26種の改変長鎖DH配列と置換したヒト長鎖ミニマル化IgHDベクターを実施例5と同様に以下の1.及び2.の手順で作製した。ヒト長鎖DH配列は、NCBIに登録されているヒト抗体cDNA配列の中に見出される、長い(18アミノ酸以上)CDR3配列にもとづき設計した。ヒト長鎖miniA、ヒト長鎖miniB、ヒト長鎖miniCの各ベクターは実施例5同様に、ユーロフィンジェノミクス社にて合成されたものを用いた。
1.ヒト長鎖miniBへのヒト長鎖miniA断片の挿入
ヒト長鎖miniAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したヒト長鎖miniBに長鎖miniAを挿入することにより、ヒト長鎖miniA/Bベクターを構築した。
ヒト長鎖miniAを制限酵素XhoI及びNotIで切断した。同様に制限酵素XhoI及びNotIで切断したヒト長鎖miniBに長鎖miniAを挿入することにより、ヒト長鎖miniA/Bベクターを構築した。
2.ヒト長鎖miniA/Bベクターへのヒト長鎖miniC断片の挿入
ヒト長鎖miniCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したヒト長鎖miniA/Bベクターにヒト長鎖miniC断片を挿入することにより、ヒト長鎖ミニマル化IgHDベクターを構築した。
ヒト長鎖miniCを制限酵素EcoRI及びSalIで切断した。同様に制限酵素EcoRI及びSalIで切断したヒト長鎖miniA/Bベクターにヒト長鎖miniC断片を挿入することにより、ヒト長鎖ミニマル化IgHDベクターを構築した。
3.ΦC31リコンビナーゼによるヒト長鎖ミニマル化IgHDベクターの導入
実施例5と同様の方法で、ΦC31リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)及びヒト長鎖ミニマル化IgHDベクターを、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)にエレクトロポレーション法により導入した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのヒト長鎖ミニマル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V、7.5msでエレクトロポレーションを行った。細胞はパルス後に384穴プレート2枚に播種した。48~72時間後に800μg/mLのG418、800μg/mLのブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。11日後にブラストサイジン耐性があったクローンをピックアップした。
実施例5と同様の方法で、ΦC31リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)及びヒト長鎖ミニマル化IgHDベクターを、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)にエレクトロポレーション法により導入した。1-10細胞をトリプシン処理し、3×106個/mLとなるようにOpti MEMに懸濁してから1μgのΦC31リコンビナーゼ遺伝子発現ベクターΦC31pEF1(特許第6868250号公報)及び3μgのヒト長鎖ミニマル化IgHDベクターを加え、NEPA21(ネッパジーン)を用いて150V、7.5msでエレクトロポレーションを行った。細胞はパルス後に384穴プレート2枚に播種した。48~72時間後に800μg/mLのG418、800μg/mLのブラストサイジン、10%FBSを含むF12培地で培地交換して培養した。11日後にブラストサイジン耐性があったクローンをピックアップした。
4.PCR解析
実施例5と同様の方法で、ΦC31部位特異的組み換え後特異的なプライマー1種、及びヒト長鎖ミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いて3.で得られたクローンのPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/長さ(bp)のサイクルを35サイクル行った。
実施例5と同様の方法で、ΦC31部位特異的組み換え後特異的なプライマー1種、及びヒト長鎖ミニマル化IgHD内に作製したプライマー2種、Ig-NAC上に作製したIg-NAC特異的なプライマー12種を用いて3.で得られたクローンのPCR解析を行った。PCR増幅には約0.1μgのゲノムDNAを使用した。TaqポリメラーゼはKODone(TOYOBO)を用い、98℃2分を1サイクル行ったのち、変性98℃10秒、アニーリング60℃5秒、伸長は68℃で、増幅断片の長さ500bp以下の場合5秒、500bp以上の場合100秒/長さ(bp)のサイクルを35サイクル行った。
5.FISH解析
PCR解析で目的のバンドが確認されたクローン(1-10 HL15)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
PCR解析で目的のバンドが確認されたクローン(1-10 HL15)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
[実施例30]R4リコンビナーゼを用いた部位特異的組み換えによる一部配列除去
Ig-NAC(ΔDH)上に導入した配列の内、CAGプロモーターやブラストサイジン耐性遺伝子などは抗体遺伝子の発現に不要となる配列である。これらの配列は、R4 attB及びR4 attPに挟まれており、R4リコンビナーゼを細胞に導入することによってR4 attB-attP間で部位特異的組み換え反応が起こり、除去することが可能である。
Ig-NAC(ΔDH)上に導入した配列の内、CAGプロモーターやブラストサイジン耐性遺伝子などは抗体遺伝子の発現に不要となる配列である。これらの配列は、R4 attB及びR4 attPに挟まれており、R4リコンビナーゼを細胞に導入することによってR4 attB-attP間で部位特異的組み換え反応が起こり、除去することが可能である。
1.R4リコンビナーゼ遺伝子発現ベクター導入
実施例6と同様に、R4リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を、ヒト長鎖ミニマル化IgHDベクターの挿入が確認されており、かつ1本のIg-NAC保持率が高いCHO細胞クローン(1-10 HL15)に、エレクトロポレーション法により導入した。得られるG418耐性クローンをピックアップした。
実施例6と同様に、R4リコンビナーゼ遺伝子発現ベクター(特許第6868250号公報)を、ヒト長鎖ミニマル化IgHDベクターの挿入が確認されており、かつ1本のIg-NAC保持率が高いCHO細胞クローン(1-10 HL15)に、エレクトロポレーション法により導入した。得られるG418耐性クローンをピックアップした。
2.PCR解析
実施例6と同様に、1.で得られるG418耐性クローンのゲノムDNAを鋳型としてPCR解析を行い、R4部位特異的組み換え後特異的なバンドが得られるクローン(1-10 HL15-28、1-10 HL15-43)を選定した。
実施例6と同様に、1.で得られるG418耐性クローンのゲノムDNAを鋳型としてPCR解析を行い、R4部位特異的組み換え後特異的なバンドが得られるクローン(1-10 HL15-28、1-10 HL15-43)を選定した。
3.FISH解析
PCR解析で目的のバンドが確認される細胞クローン(1-10 HL15-28、1-10 HL15-43)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
PCR解析で目的のバンドが確認される細胞クローン(1-10 HL15-28、1-10 HL15-43)に対してFISH解析を実施した。FISH解析は(特許第6868250号公報)に記された方法に従い、マウス遺伝子特異的プローブmouse cot1(invitrogen)を用いて行った。
[実施例31]マウスES細胞株への微小核形成法(MMCT)によるヒト長鎖ミニマル化IgHD搭載Ig-NAC導入
実施例30でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認されたクローンを染色体供与細胞として用い、MMCT法を用いてマウスES細胞へのヒト長鎖ミニマル化IgHD含有Ig-NAC移入を行う。
実施例30でPCR及びFISH解析によりR4部位特異的組み換えが起きたことが確認されたクローンを染色体供与細胞として用い、MMCT法を用いてマウスES細胞へのヒト長鎖ミニマル化IgHD含有Ig-NAC移入を行う。
1.MMCT法を用いたマウスES細胞へのヒト長鎖ミニマル化IgHD含有Ig-NACの移入
実施例7と同様に、染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号公報)を用いて微小核形成法(MMCT)によるヒト長鎖ミニマル化IgHD含有Ig-NAC導入を行い、出現した薬剤耐性コロニーをピックアップする。
実施例7と同様に、染色体受容細胞としては、内因性免疫グロブリン重鎖、κ鎖遺伝子座の両アレルが破壊されているマウスES細胞HKD31 6TG-9株(XO)(特許第4082740号公報)を用いて微小核形成法(MMCT)によるヒト長鎖ミニマル化IgHD含有Ig-NAC導入を行い、出現した薬剤耐性コロニーをピックアップする。
2.PCR解析
実施例7と同様に、単離した細胞を培養し、細胞からCelllysis(キアゲン)を用いてゲノムDNAを抽出する。このゲノムDNAを鋳型としてPCRを実施し、目的のバンドが検出されるクローンを選択する。
実施例7と同様に、単離した細胞を培養し、細胞からCelllysis(キアゲン)を用いてゲノムDNAを抽出する。このゲノムDNAを鋳型としてPCRを実施し、目的のバンドが検出されるクローンを選択する。
3.核型解析
実施例7と同様に染色体標本を作成した。この標本をDAPI染色し、マウス染色体の核型異常がないクローンを選択する。
実施例7と同様に染色体標本を作成した。この標本をDAPI染色し、マウス染色体の核型異常がないクローンを選択する。
[実施例32]ヒト長鎖ミニマル化IgHD含有Ig-NACを保持するマウスES細胞株からのキメラマウス作製
実施例8と同様に、ヒト抗体産生を確認するために実施例31で得られたヒトミニマル化IgHD含有Ig-NACマウスES細胞からキメラマウスを作製する。
ヒト長鎖ミニマル化HD含有Ig-NACを保持するマウスES細胞株クローンをICR胚にインジェクションを実施し、仮親に移植する。その結果GFP陽性キメラマウスが得られる。
得られたキメラマウスを実施例13、実施例14、実施例15などに記載された方法により解析する。その結果、キメラマウス脾臓Igμ-cDNAにおける、長鎖ヒトD断片の使用が示される。
実施例8と同様に、ヒト抗体産生を確認するために実施例31で得られたヒトミニマル化IgHD含有Ig-NACマウスES細胞からキメラマウスを作製する。
ヒト長鎖ミニマル化HD含有Ig-NACを保持するマウスES細胞株クローンをICR胚にインジェクションを実施し、仮親に移植する。その結果GFP陽性キメラマウスが得られる。
得られたキメラマウスを実施例13、実施例14、実施例15などに記載された方法により解析する。その結果、キメラマウス脾臓Igμ-cDNAにおける、長鎖ヒトD断片の使用が示される。
[実施例33]ヒトIgHD7-27を削除した合成D領域導入アクセプター保持Ig-NACの構築
ヒトIgHD領域を削除したIg-NAC(ΔDH)では、ヒトD断片1-1から1-26までの合計26断片を削除しており、D領域から離れて存在しJ領域の近くにある7-27は削除していない。D断片7-27が残存していることへの影響をなくすため、D断片7-27の前後2カ所にCRISPR/Cas9のgRNAを設計、評価、選定する。その後、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)に対して、上記gRNA及びCas9を用いたゲノム編集によって2ヵ所切断することにより、ヒトIgHD7-27を削除した合成D領域導入アクセプター保持Ig-NACを取得する。
ヒトIgHD領域を削除したIg-NAC(ΔDH)では、ヒトD断片1-1から1-26までの合計26断片を削除しており、D領域から離れて存在しJ領域の近くにある7-27は削除していない。D断片7-27が残存していることへの影響をなくすため、D断片7-27の前後2カ所にCRISPR/Cas9のgRNAを設計、評価、選定する。その後、化学合成DNAの導入プラットフォーム搭載Ig-NAC保持CHO細胞1-10(実施例4)に対して、上記gRNA及びCas9を用いたゲノム編集によって2ヵ所切断することにより、ヒトIgHD7-27を削除した合成D領域導入アクセプター保持Ig-NACを取得する。
本発明により、ヒト抗体の多様性が拡張される。好ましくは18~50若しくはそれ以上のアミノ酸数のCDRH3を含むヒト抗体を作製することを可能にする。
配列番号1:ヒトミニマル化D領域のヌクレオチド
配列番号2:ウシORFで置換されたヒトミニマル化D領域のヌクレオチド配列
737..767 D2-2に代わるB1-1
1024..1039 D3-3に代わるB2-1
1296..1331 D4-4に代わるB3-1
1588..1630 D5-5に代わるB4-1
1887..1900 D6-6に代わるB9-1
2157..2187 D1-7に代わるB1-2
2444..2459 D2-8に代わるB2-2
2715..2756 D3-9に代わるB5-2
2910..3057 D3-10に代わるB8-2
3374..3431 D4-11に代わるB6-2
3688..3806 D5-12に代わるB1-3
4063..4078 D6-13に代わるB2-3
4338..4373 D1-14に代わるB3-3
4630..4696 D2-15に代わるB7-3
4953..4994 D3-16に代わるB5-3
5251..5308 D4-17に代わるB6-3
5565..5595 D5-18に代わるB1-4
5852..5867 D6-19に代わるB2-4
6124..6159 D1-20に代わるB3-4
6416..6488 D1-21に代わるB7-4
6745..6785 D3-22に代わるB5-4
7103..7160 D4-23に代わるB6-4
7417..7430 D5-24に代わるB9-4
配列番号3:改変ORFで置換されたヒトミニマル化D領域のヌクレオチド配列
647..690 改変D1-1
947..990 改変D2-2
1247..1290 改変D3-3
1547..1590 改変D4-4
1847..1890 改変D5-5
2147..2190 改変D6-6
2447..2508 改変D1-7
2765..2808 改変DL2-8
3064..3149 改変D3-9
3303..3385 改変D3-10
3702..3760 改変D4-11
4017..4078 改変D5-12
4335..4384 改変D6-13
4644..4711 改変D1-14
4968..5035 改変D2-15
5292..5365 改変D3-16
5622..5695 改変D4-17
5952..6016 改変D5-18
6273..6334 改変D6-19
6591..6652 改変D1-20
6909..6961 改変D2-21
7218..7264 改変D3-22
7581..7642 改変D4-23
7899..7960 改変D5-24
8217..8263 改変D6-25
8520..8578 改変D1-26
配列番号4:ガイドRNAであるgRNA Up3の標的ヌクレオチド配列
21..23: PAM配列
配列番号5:ガイドRNAであるgRNA Down4の標的ヌクレオチド配列
21..23: PAM配列
配列番号6~15:プライマー
配列番号18:Bxb1 attBのヌクレオチド配列
配列番号19:Bxb1 attPのヌクレオチド配列
配列番号20:ΦC31 attBのヌクレオチド配列
配列番号21:R4 attBのヌクレオチド配列
配列番号22~23:プライマー
配列番号24:ガイドRNAであるgRNA-A1の標的ヌクレオチド配列
21..23: PAM配列
配列番号25:ガイドRNAであるgRNA-B1の標的ヌクレオチド配列
21..23: PAM配列
配列番号26~52:プライマー
配列番号61:長鎖CDRH3を含むペプチドのアミノ酸配列
配列番号62~74:プライマー
配列番号75:ヒトDH番号1-1のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号76:ヒトDH番号2-2のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号77:ヒトDH番号3-3のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号78:ヒトDH番号4-4のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号79:ヒトDH番号5-5のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号80:ヒトDH番号6-6のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号81:ヒトDH番号1-7のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号82:ヒトDH番号2-8のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号83:ヒトDH番号3-9のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号84:ヒトDH番号3-10のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号85:ヒトDH番号4-11のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号86:ヒトDH番号5-12のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号87:ヒトDH番号6-13のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号88:ヒトDH番号1-14のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号89:ヒトDH番号2-15のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号90:ヒトDH番号3-16のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号91:ヒトDH番号4-17のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号92:ヒトDH番号5-18のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号93:ヒトDH番号6-19のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号94:ヒトDH番号1-20のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号95:ヒトDH番号2-21のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号96:ヒトDH番号3-22のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号97:ヒトDH番号4-23のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号98:ヒトDH番号5-24のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号99:ヒトDH番号6-25のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号100:ヒトDH番号1-26のヌクレオチド配列に代えて置換された改変長鎖DH配列
本明細書で引用した全ての刊行物、特許及び特許出願はそのまま引用により本明細書に組み入れられるものとする。
配列番号2:ウシORFで置換されたヒトミニマル化D領域のヌクレオチド配列
737..767 D2-2に代わるB1-1
1024..1039 D3-3に代わるB2-1
1296..1331 D4-4に代わるB3-1
1588..1630 D5-5に代わるB4-1
1887..1900 D6-6に代わるB9-1
2157..2187 D1-7に代わるB1-2
2444..2459 D2-8に代わるB2-2
2715..2756 D3-9に代わるB5-2
2910..3057 D3-10に代わるB8-2
3374..3431 D4-11に代わるB6-2
3688..3806 D5-12に代わるB1-3
4063..4078 D6-13に代わるB2-3
4338..4373 D1-14に代わるB3-3
4630..4696 D2-15に代わるB7-3
4953..4994 D3-16に代わるB5-3
5251..5308 D4-17に代わるB6-3
5565..5595 D5-18に代わるB1-4
5852..5867 D6-19に代わるB2-4
6124..6159 D1-20に代わるB3-4
6416..6488 D1-21に代わるB7-4
6745..6785 D3-22に代わるB5-4
7103..7160 D4-23に代わるB6-4
7417..7430 D5-24に代わるB9-4
配列番号3:改変ORFで置換されたヒトミニマル化D領域のヌクレオチド配列
647..690 改変D1-1
947..990 改変D2-2
1247..1290 改変D3-3
1547..1590 改変D4-4
1847..1890 改変D5-5
2147..2190 改変D6-6
2447..2508 改変D1-7
2765..2808 改変DL2-8
3064..3149 改変D3-9
3303..3385 改変D3-10
3702..3760 改変D4-11
4017..4078 改変D5-12
4335..4384 改変D6-13
4644..4711 改変D1-14
4968..5035 改変D2-15
5292..5365 改変D3-16
5622..5695 改変D4-17
5952..6016 改変D5-18
6273..6334 改変D6-19
6591..6652 改変D1-20
6909..6961 改変D2-21
7218..7264 改変D3-22
7581..7642 改変D4-23
7899..7960 改変D5-24
8217..8263 改変D6-25
8520..8578 改変D1-26
配列番号4:ガイドRNAであるgRNA Up3の標的ヌクレオチド配列
21..23: PAM配列
配列番号5:ガイドRNAであるgRNA Down4の標的ヌクレオチド配列
21..23: PAM配列
配列番号6~15:プライマー
配列番号18:Bxb1 attBのヌクレオチド配列
配列番号19:Bxb1 attPのヌクレオチド配列
配列番号20:ΦC31 attBのヌクレオチド配列
配列番号21:R4 attBのヌクレオチド配列
配列番号22~23:プライマー
配列番号24:ガイドRNAであるgRNA-A1の標的ヌクレオチド配列
21..23: PAM配列
配列番号25:ガイドRNAであるgRNA-B1の標的ヌクレオチド配列
21..23: PAM配列
配列番号26~52:プライマー
配列番号61:長鎖CDRH3を含むペプチドのアミノ酸配列
配列番号62~74:プライマー
配列番号75:ヒトDH番号1-1のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号76:ヒトDH番号2-2のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号77:ヒトDH番号3-3のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号78:ヒトDH番号4-4のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号79:ヒトDH番号5-5のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号80:ヒトDH番号6-6のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号81:ヒトDH番号1-7のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号82:ヒトDH番号2-8のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号83:ヒトDH番号3-9のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号84:ヒトDH番号3-10のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号85:ヒトDH番号4-11のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号86:ヒトDH番号5-12のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号87:ヒトDH番号6-13のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号88:ヒトDH番号1-14のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号89:ヒトDH番号2-15のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号90:ヒトDH番号3-16のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号91:ヒトDH番号4-17のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号92:ヒトDH番号5-18のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号93:ヒトDH番号6-19のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号94:ヒトDH番号1-20のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号95:ヒトDH番号2-21のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号96:ヒトDH番号3-22のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号97:ヒトDH番号4-23のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号98:ヒトDH番号5-24のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号99:ヒトDH番号6-25のヌクレオチド配列に代えて置換された改変長鎖DH配列
配列番号100:ヒトDH番号1-26のヌクレオチド配列に代えて置換された改変長鎖DH配列
本明細書で引用した全ての刊行物、特許及び特許出願はそのまま引用により本明細書に組み入れられるものとする。
Claims (25)
- ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターにおいて、前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されていることを特徴とする哺乳動物人工染色体ベクターであって、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
前記哺乳動物人工染色体ベクター。 - 前記(1)のヒト動物免疫グロブリン重鎖遺伝子座D領域のORF間のヒト由来ゲノム配列が100~500bpの長さに短縮された配列を含む、請求項1に記載の哺乳動物人工染色体ベクター。
- 前記ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列が、配列番号127~149のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、請求項1又は2に記載の哺乳動物人工染色体ベクター。
- 前記サル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列が、配列番号150~175のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、請求項1又は2に記載の哺乳動物人工染色体ベクター。
- 前記(3)の26種の改変ORF配列が、配列番号75~100のヌクレオチド配列又は該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、請求項1又は2に記載の哺乳動物人工染色体ベクター。
- 前記D領域の改変配列が、配列番号2又は配列番号3のヌクレオチド配列、或いは該ヌクレオチド配列と90%以上の同一性を有するヌクレオチド配列を含む、請求項1~5のいずれか1項に記載の哺乳動物人工染色体ベクター。
- 前記ヒト免疫グロブリン重鎖遺伝子座のD領域がさらにD7-27を欠失している、請求項1~6のいずれか1項に記載の哺乳動物人工染色体ベクター。
- 前記ベクターが、齧歯類人工染色体ベクターである、請求項1~7のいずれか1項に記載の哺乳動物人工染色体ベクター。
- 前記齧歯類人工染色体ベクターが、マウス人工染色体ベクターである、請求項8に記載の哺乳動物人工染色体ベクター。
- 前記軽鎖遺伝子座が、κ軽鎖遺伝子座及び/又はλ軽鎖遺伝子座である、請求項1~9のいずれか1項に記載の哺乳動物人工染色体ベクター。
- 請求項1~10のいずれか1項に記載の哺乳動物人工染色体ベクターを含む哺乳動物細胞。
- 前記細胞が、齧歯類細胞又はヒト細胞である、請求項11に記載の哺乳動物細胞。
- 前記細胞が、哺乳動物由来のiPS細胞及びES細胞からなる群から選択される分化多能性幹細胞である、請求項11に記載の哺乳動物細胞。
- 請求項1~10のいずれか1項に記載の哺乳動物人工染色体ベクターを含み、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている非ヒト哺乳動物。
- ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む非ヒト動物であって、前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのヒト由来ゲノム配列が、下記の(1)及び(2)、又は(1)及び(3)、の組み合わせからなる該D領域の改変配列と置換されており、前記D領域の改変配列が、
(1)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのオープンリーディングフレーム(ORF)間のヒト由来ゲノム配列、又は、D3-9とD3-10間を除く前記ORF間のヒト由来ゲノム配列がVDJ組み換え配列が隣接したORF間領域の各VDJ組み換え配列末端から49bp以上の長さに短縮された配列を含み、並びに、
(2)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、ウシ由来の免疫グロブリン重鎖遺伝子座D領域のB1-1からB9-4までのORF配列、又はサル由来の免疫グロブリン重鎖遺伝子座D領域の1S5から1S35までのORF配列、又はヒツジ、ウマ、ウサギ、トリ若しくはサメ由来の免疫グロブリン重鎖遺伝子座D領域のORF配列を含む、或いは、
(3)前記ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までのORF配列に代えて、18アミノ酸以上のヒト抗体重鎖CDR3配列に基づいて作製された、ヒト免疫グロブリン重鎖遺伝子座D領域のD1-1からD1-26までの26種の改変ORF配列を含む、
ことを特徴とし、かつ内在性免疫グロブリン重鎖、κ軽鎖及びλ軽鎖遺伝子若しくは遺伝子座が破壊されている、前記非ヒト動物。 - 前記動物が、齧歯類である、請求項14又は15に記載の非ヒト動物。
- 前記齧歯類が、マウス又はラットである、請求項16に記載の非ヒト動物。
- 請求項14~17のいずれか1項に記載の非ヒト動物に標的抗原を免疫し、該非ヒト動物の血液から該標的抗原と結合する抗体を取得することを含む、抗体の作製方法。
- 請求項14~17のいずれか1項に記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物から脾臓細胞、リンパ節細胞又はB細胞を取得し、前記脾臓細胞、リンパ節細胞又はB細胞とミエローマ細胞を融合してハイブリドーマを形成し、該ハイブリドーマを培養して、前記標的抗原と結合するモノクローナル抗体を取得することを含む、抗体の作製方法。
- 請求項14~17のいずれか1項に記載の非ヒト動物に標的抗原を免疫し、該標的抗原と結合する抗体を産生する前記非ヒト動物からB細胞を取得し、前記B細胞から抗体重鎖及び軽鎖、或いはそれらの可変領域、からなるタンパク質をコードする核酸を取得し、該核酸を用いてDNA組み換え技術又はファージディスプレイ技術によって前記標的抗原と結合する抗体を作製することを含む、抗体の作製方法。
- 請求項1~10のいずれか1項に記載の哺乳動物人工染色体ベクターの製造において、ヒト免疫グロブリン重鎖D領域をその改変配列で置換する方法であって、
(1’)ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターを準備する、
(2’)工程(1’)の前記哺乳動物人工染色体ベクターからヒト免疫グロブリン重鎖D領域を削除する、
(3’)工程(2’)で得られたベクターの前記D領域の削除部位に、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、
(4’)工程(3’)で挿入された前記カセットの第2の部位特異的組み換え酵素認識部位に、第2の部位特異的組み換え酵素を用いて、前記ヒト免疫グロブリン重鎖D領域の改変配列、第1の部位特異的組み換え酵素認識部位、選択マーカー及び第2の部位特異的組み換え酵素認識部位を含むカセットを挿入する、並びに、
(5’)第1の部位特異的組み換え酵素を用いる部位特異的組み換えによって、第1の部位特異的組み換え酵素認識部位、前記ヒト免疫グロブリン重鎖D領域の改変配列及び第2の部位特異的組み換え酵素認識部位を含むカセットが前記D領域の削除部位に挿入された前記人工染色体ベクターを得る、
ことを含む前記方法。 - 前記ヒト免疫グロブリン重鎖D領域の削除が、ゲノム編集によって行われる、請求項21に記載の方法。
- ヒト免疫グロブリン重鎖及び軽鎖遺伝子座を含む哺乳動物人工染色体ベクターであって、前記ヒト免疫グロブリン重鎖遺伝子座がD領域を欠失しており、該欠失されたD領域に代えて、第1の部位特異的組み換え酵素認識部位、プロモーター及び第2の部位特異的組み換え酵素認識部位を含む、哺乳動物人工染色体ベクター。
- 前記欠失されたD領域が、D1-1からD1-26までの領域である、請求項23に記載の哺乳動物人工染色体ベクター。
- 前記欠失されたD領域が、D1-1からD7-27までの領域である、請求項23に記載の哺乳動物人工染色体ベクター。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-186124 | 2021-11-16 | ||
| JP2021186124 | 2021-11-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023090361A1 true WO2023090361A1 (ja) | 2023-05-25 |
Family
ID=86397075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/042560 WO2023090361A1 (ja) | 2021-11-16 | 2022-11-16 | 改変d領域を含むヒト免疫グロブリン重鎖遺伝子座を有する哺乳動物人工染色体ベクター、及びそのベクターを保持する細胞又は非ヒト動物 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023090361A1 (ja) |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| WO2000010383A1 (en) | 1998-08-21 | 2000-03-02 | Kirin Beer Kabushiki Kaisha | Method for modifying chromosomes |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| JP4082740B2 (ja) | 1997-02-28 | 2008-04-30 | キリンファーマ株式会社 | 内因性遺伝子が破壊されている分化多能性保持細胞 |
| JP4318736B2 (ja) | 1995-08-29 | 2009-08-26 | 協和発酵キリン株式会社 | ヒト抗体遺伝子を発現する非ヒト動物とその利用 |
| JP2014529998A (ja) * | 2011-09-19 | 2014-11-17 | カイマブ・リミテッド | ヒトへの使用に合わせて作製された抗体、可変ドメインおよび鎖 |
| WO2018079857A1 (ja) * | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| JP6775224B2 (ja) | 2018-03-16 | 2020-10-28 | 国立大学法人鳥取大学 | マウス人工染色体ベクター及びその使用 |
| WO2021003152A1 (en) * | 2019-07-01 | 2021-01-07 | Trianni, Inc. | Transgenic mammals and methods of use thereof |
| JP2021186124A (ja) | 2020-05-27 | 2021-12-13 | 株式会社三洋物産 | 遊技機 |
-
2022
- 2022-11-16 WO PCT/JP2022/042560 patent/WO2023090361A1/ja active Application Filing
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| JP4318736B2 (ja) | 1995-08-29 | 2009-08-26 | 協和発酵キリン株式会社 | ヒト抗体遺伝子を発現する非ヒト動物とその利用 |
| JP4082740B2 (ja) | 1997-02-28 | 2008-04-30 | キリンファーマ株式会社 | 内因性遺伝子が破壊されている分化多能性保持細胞 |
| WO2000010383A1 (en) | 1998-08-21 | 2000-03-02 | Kirin Beer Kabushiki Kaisha | Method for modifying chromosomes |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| JP2014529998A (ja) * | 2011-09-19 | 2014-11-17 | カイマブ・リミテッド | ヒトへの使用に合わせて作製された抗体、可変ドメインおよび鎖 |
| WO2018079857A1 (ja) * | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| JP6868250B2 (ja) | 2016-10-31 | 2021-05-12 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| JP6775224B2 (ja) | 2018-03-16 | 2020-10-28 | 国立大学法人鳥取大学 | マウス人工染色体ベクター及びその使用 |
| WO2021003152A1 (en) * | 2019-07-01 | 2021-01-07 | Trianni, Inc. | Transgenic mammals and methods of use thereof |
| JP2021186124A (ja) | 2020-05-27 | 2021-12-13 | 株式会社三洋物産 | 遊技機 |
Non-Patent Citations (33)
| Title |
|---|
| A. R. REES, MABS, vol. 12, no. 1, 2020, pages e1729683 |
| ALTSCHUL, S. F. ET AL., JOURNAL OF MOLECULAR BIOLOGY, vol. 215, 1990, pages 403 - 410 |
| ANNU. REV. IMMUNOL, vol. 12, 1994, pages 433 - 455 |
| B. SAUER, METHODS OF ENZYMOLOGY, vol. 225, 1993, pages 890 - 900 |
| DIEKEN ET AL., NATURE GENETICS, vol. 12, 1996, pages 174 - 182 |
| G.-Y. YU ET AL., IMMUNOGENETICS, vol. 68, 2016, pages 417 - 428 |
| J. A. THOMSON ET AL., BIOL. REPROD., vol. 55, 1996, pages 254 - 259 |
| J. A. THOMSON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 7844 - 7848 |
| J. A. THOMSON ET AL., SCIENCE, vol. 282, 1999, pages 1145 - 1147 |
| J. A. THOMSONV. S. MARSHALL, CURR. TOP. DEV. BIOL., vol. 38, 1998, pages 133 - 165 |
| J. DONG ET AL., FRONTIERS IN IMMUNOLOGY, vol. 10, 2019 |
| J. K. HAAKENSON ET AL., FRONTIERS IN IMMUNOLOGY, vol. 9, 2018 |
| J. LIAO ET AL., CELL RES, vol. 18, 2008, pages 600 - 603 |
| J. YU ET AL., SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| J.M. CHYLINSKI ET AL., SCIENCE, vol. 337, no. 6096, 2012, pages 816 - 821 |
| K. KAWAHARADA ET AL., WORLD J. STEM CELLS, vol. 7, no. 7, 2015, pages 1054 - 1063 |
| K. TAKAHASHIS. YAMANAKA, CELL, vol. 126, 2006, pages 663 - 676 |
| K. TOMIZUKA ET AL., PROC. NATL. ACAD. SCI. USA, vol. 97, no. 2, 2000, pages 922 - 927 |
| KOI ET AL., JPN. J. CANCER RES., vol. 80, 1973, pages 413 - 418 |
| KOTI, M. ; KATAEVA, G. ; KAUSHIK, A.K.: "Novel atypical nucleotide insertions specifically at V"H-D"H junction generate exceptionally long CDR3H in cattle antibodies", MOLECULAR IMMUNOLOGY, PERGAMON, GB, vol. 47, no. 11-12, 1 July 2010 (2010-07-01), GB , pages 2119 - 2128, XP027072110, ISSN: 0161-5890, DOI: 10.1016/j.molimm.2010.02.014 * |
| M. A. ALFALEH ET AL., FRONT. IMMUNOL., vol. 11, 2020, Retrieved from the Internet <URL:https://doi.org/10.3389/fimmu.2020.01986> |
| M. CHIU ET AL., ANTIBODIES, vol. 8, no. 4, 2019, pages 55 |
| M. J. EVANSM. H. KAUFMAN, NATURE, vol. 292, no. 5819, 1981, pages 154 - 156 |
| M. NAKAGAWA ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 101 - 106 |
| M. RUIZ ET AL., EXP. CLIN. IMMUNOGENET., vol. 16, 1999, pages 173 - 184 |
| M.J. BURKE ET AL., VIRUSES, vol. 12, 2020, pages 473 |
| NATURE BIOTECHNOLOGY, vol. 23, no. 9, 2005, pages 1105 - 1116 |
| PEARSON, W. R. ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 85, 1988, pages 2444 - 2448 |
| SANDER ET AL., NATURE BIOTECHNOLOGY, vol. 32, no. 4, 2014, pages 347 - 355 |
| TAKAHASHI, K ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| WANG ET AL., CELL, vol. 153, 2013, pages 1379 - 93 |
| Y. DI ET AL., IMMUNOLOGY, vol. 00, August 2021 (2021-08-01), pages 1 - 14 |
| ZHENG ZHANG ET AL., J.COMPUT. BIOL., vol. 7, 2000, pages 203 - 214 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6479024B2 (ja) | 抗体親和性の最適化のための高スループットマウスモデル | |
| US7105348B2 (en) | Methods of modifying eukaryotic cells | |
| JP6641379B2 (ja) | トランスジェニックマウス | |
| JP4115281B2 (ja) | ヒト抗体λ軽鎖遺伝子を含むヒト人工染色体、および子孫伝達可能な該ヒト人工染色体を含む非ヒト動物 | |
| JP5557217B2 (ja) | マウス人工染色体ベクター | |
| JP6868250B2 (ja) | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 | |
| US20220369609A1 (en) | Transgenic mammals and methods of use thereof | |
| US20210000087A1 (en) | Transgenic mammals and methods of use thereof | |
| JP6775224B2 (ja) | マウス人工染色体ベクター及びその使用 | |
| KR20180038554A (ko) | 공통 경쇄를 갖는 항체 생산을 위한 유전자도입 동물 | |
| WO2023090361A1 (ja) | 改変d領域を含むヒト免疫グロブリン重鎖遺伝子座を有する哺乳動物人工染色体ベクター、及びそのベクターを保持する細胞又は非ヒト動物 | |
| KR20230147213A (ko) | 면역글로불린 중쇄 암호화 서열에서 dh-dh 재배열 가능한 비인간 동물 | |
| US11946063B2 (en) | Long germline DH genes and long HCDR3 antibodies | |
| Price | MUrine tools to catch high-affinity plasma cells (MUTCHAP) | |
| WO2022235759A1 (en) | Transgenic rodents expressing chimeric equine-rodent antibodies and methods of use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22895642 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023562377 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022895642 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022895642 Country of ref document: EP Effective date: 20240319 |