WO2023016484A1 - 磺酰胺衍生物、其制备方法及其在医药上的应用 - Google Patents
磺酰胺衍生物、其制备方法及其在医药上的应用 Download PDFInfo
- Publication number
- WO2023016484A1 WO2023016484A1 PCT/CN2022/111395 CN2022111395W WO2023016484A1 WO 2023016484 A1 WO2023016484 A1 WO 2023016484A1 CN 2022111395 W CN2022111395 W CN 2022111395W WO 2023016484 A1 WO2023016484 A1 WO 2023016484A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- cancer
- group
- alkyl
- pharmaceutically acceptable
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 27
- HFFXLYHRNRKAPM-UHFFFAOYSA-N 2,4,5-trichloro-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C(=CC(Cl)=C(Cl)C=2)Cl)=N1 HFFXLYHRNRKAPM-UHFFFAOYSA-N 0.000 title description 3
- 239000003814 drug Substances 0.000 claims abstract description 36
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 23
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 18
- 201000011510 cancer Diseases 0.000 claims abstract description 15
- 238000011282 treatment Methods 0.000 claims abstract description 6
- 230000002265 prevention Effects 0.000 claims abstract description 3
- 150000001875 compounds Chemical class 0.000 claims description 247
- -1 cycloalkane radical Chemical class 0.000 claims description 172
- 125000000217 alkyl group Chemical group 0.000 claims description 154
- 150000003839 salts Chemical class 0.000 claims description 152
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 136
- 125000003545 alkoxy group Chemical group 0.000 claims description 124
- 125000000623 heterocyclic group Chemical group 0.000 claims description 118
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 111
- 229910052736 halogen Inorganic materials 0.000 claims description 98
- 150000002367 halogens Chemical class 0.000 claims description 98
- 125000003118 aryl group Chemical group 0.000 claims description 78
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 78
- 125000001072 heteroaryl group Chemical group 0.000 claims description 65
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims description 40
- 101000944179 Homo sapiens Histone acetyltransferase KAT6A Proteins 0.000 claims description 39
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 38
- 102100033071 Histone acetyltransferase KAT6A Human genes 0.000 claims description 38
- 206010006187 Breast cancer Diseases 0.000 claims description 34
- 208000026310 Breast neoplasm Diseases 0.000 claims description 34
- 125000003342 alkenyl group Chemical group 0.000 claims description 34
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 34
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 34
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 32
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 32
- 125000000304 alkynyl group Chemical group 0.000 claims description 31
- 125000001188 haloalkyl group Chemical group 0.000 claims description 31
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 31
- 239000000126 substance Substances 0.000 claims description 30
- 125000005844 heterocyclyloxy group Chemical group 0.000 claims description 29
- 238000000034 method Methods 0.000 claims description 29
- 125000004043 oxo group Chemical group O=* 0.000 claims description 29
- 125000001424 substituent group Chemical group 0.000 claims description 29
- 125000004432 carbon atom Chemical group C* 0.000 claims description 22
- 206010060862 Prostate cancer Diseases 0.000 claims description 21
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 21
- 101000944174 Homo sapiens Histone acetyltransferase KAT6B Proteins 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 19
- 102100033070 Histone acetyltransferase KAT6B Human genes 0.000 claims description 18
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 16
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 16
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 15
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 14
- 229910020008 S(O) Inorganic materials 0.000 claims description 14
- 201000005202 lung cancer Diseases 0.000 claims description 14
- 208000020816 lung neoplasm Diseases 0.000 claims description 14
- 206010018338 Glioma Diseases 0.000 claims description 13
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 13
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 13
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 11
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 claims description 11
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 claims description 11
- 125000002947 alkylene group Chemical group 0.000 claims description 11
- 230000002401 inhibitory effect Effects 0.000 claims description 11
- 208000032612 Glial tumor Diseases 0.000 claims description 10
- 125000003341 7 membered heterocyclic group Chemical group 0.000 claims description 9
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 9
- 206010033128 Ovarian cancer Diseases 0.000 claims description 9
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 9
- 201000007270 liver cancer Diseases 0.000 claims description 9
- 208000014018 liver neoplasm Diseases 0.000 claims description 9
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 9
- 206010005003 Bladder cancer Diseases 0.000 claims description 8
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 8
- 206010014733 Endometrial cancer Diseases 0.000 claims description 8
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 8
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 8
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 8
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 8
- 201000007455 central nervous system cancer Diseases 0.000 claims description 8
- 208000025997 central nervous system neoplasm Diseases 0.000 claims description 8
- 201000010881 cervical cancer Diseases 0.000 claims description 8
- 125000004474 heteroalkylene group Chemical group 0.000 claims description 8
- 208000032839 leukemia Diseases 0.000 claims description 8
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 8
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 8
- 201000002528 pancreatic cancer Diseases 0.000 claims description 8
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 8
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 8
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 8
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 7
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 6
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 5
- 125000003277 amino group Chemical group 0.000 claims description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 5
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 4
- 125000006164 6-membered heteroaryl group Chemical group 0.000 claims description 4
- 206010061424 Anal cancer Diseases 0.000 claims description 4
- 208000007860 Anus Neoplasms Diseases 0.000 claims description 4
- 206010005949 Bone cancer Diseases 0.000 claims description 4
- 208000018084 Bone neoplasm Diseases 0.000 claims description 4
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 4
- 206010007953 Central nervous system lymphoma Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 4
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 4
- 201000001342 Fallopian tube cancer Diseases 0.000 claims description 4
- 208000013452 Fallopian tube neoplasm Diseases 0.000 claims description 4
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 claims description 4
- 208000032271 Malignant tumor of penis Diseases 0.000 claims description 4
- 208000000821 Parathyroid Neoplasms Diseases 0.000 claims description 4
- 208000002471 Penile Neoplasms Diseases 0.000 claims description 4
- 206010034299 Penile cancer Diseases 0.000 claims description 4
- 208000007913 Pituitary Neoplasms Diseases 0.000 claims description 4
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 4
- 206010039491 Sarcoma Diseases 0.000 claims description 4
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 4
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 4
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 4
- 206010057644 Testis cancer Diseases 0.000 claims description 4
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 4
- 208000023915 Ureteral Neoplasms Diseases 0.000 claims description 4
- 206010046392 Ureteric cancer Diseases 0.000 claims description 4
- 206010046431 Urethral cancer Diseases 0.000 claims description 4
- 206010046458 Urethral neoplasms Diseases 0.000 claims description 4
- 206010047741 Vulval cancer Diseases 0.000 claims description 4
- 208000004354 Vulvar Neoplasms Diseases 0.000 claims description 4
- 201000005188 adrenal gland cancer Diseases 0.000 claims description 4
- 208000024447 adrenal gland neoplasm Diseases 0.000 claims description 4
- 201000011165 anus cancer Diseases 0.000 claims description 4
- 210000003169 central nervous system Anatomy 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- 201000004101 esophageal cancer Diseases 0.000 claims description 4
- 206010017758 gastric cancer Diseases 0.000 claims description 4
- 201000010536 head and neck cancer Diseases 0.000 claims description 4
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 4
- 201000002313 intestinal cancer Diseases 0.000 claims description 4
- 208000020984 malignant renal pelvis neoplasm Diseases 0.000 claims description 4
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 208000016800 primary central nervous system lymphoma Diseases 0.000 claims description 4
- 201000007444 renal pelvis carcinoma Diseases 0.000 claims description 4
- 201000000849 skin cancer Diseases 0.000 claims description 4
- 206010062261 spinal cord neoplasm Diseases 0.000 claims description 4
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 4
- 201000011549 stomach cancer Diseases 0.000 claims description 4
- 201000003120 testicular cancer Diseases 0.000 claims description 4
- 201000002510 thyroid cancer Diseases 0.000 claims description 4
- 206010046885 vaginal cancer Diseases 0.000 claims description 4
- 208000013139 vaginal neoplasm Diseases 0.000 claims description 4
- 201000005102 vulva cancer Diseases 0.000 claims description 4
- 208000017604 Hodgkin disease Diseases 0.000 claims description 3
- 208000021519 Hodgkin lymphoma Diseases 0.000 claims description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 claims description 3
- 206010027406 Mesothelioma Diseases 0.000 claims description 3
- 201000005746 Pituitary adenoma Diseases 0.000 claims description 3
- 206010061538 Pituitary tumour benign Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 claims description 3
- 208000021310 pituitary gland adenoma Diseases 0.000 claims description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 2
- 208000002495 Uterine Neoplasms Diseases 0.000 claims 1
- 208000029824 high grade glioma Diseases 0.000 claims 1
- 201000011614 malignant glioma Diseases 0.000 claims 1
- 206010046766 uterine cancer Diseases 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 8
- 108010033293 Lysine Acetyltransferases Proteins 0.000 abstract description 3
- 102000007077 Lysine Acetyltransferases Human genes 0.000 abstract description 3
- 229940124530 sulfonamide Drugs 0.000 abstract 1
- 150000003456 sulfonamides Chemical class 0.000 abstract 1
- 229940124597 therapeutic agent Drugs 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 219
- 238000006243 chemical reaction Methods 0.000 description 178
- 239000000047 product Substances 0.000 description 94
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Natural products CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 89
- 230000002829 reductive effect Effects 0.000 description 83
- 238000004949 mass spectrometry Methods 0.000 description 71
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 62
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 61
- 238000005481 NMR spectroscopy Methods 0.000 description 60
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 57
- 239000012071 phase Substances 0.000 description 57
- 239000003480 eluent Substances 0.000 description 46
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 46
- 238000010898 silica gel chromatography Methods 0.000 description 46
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 44
- 239000000706 filtrate Substances 0.000 description 43
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 229910052757 nitrogen Inorganic materials 0.000 description 39
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 36
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 34
- 210000004027 cell Anatomy 0.000 description 34
- 238000004128 high performance liquid chromatography Methods 0.000 description 33
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 32
- 229940079593 drug Drugs 0.000 description 29
- 239000000203 mixture Substances 0.000 description 27
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 26
- 125000006413 ring segment Chemical group 0.000 description 25
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 24
- 239000012074 organic phase Substances 0.000 description 23
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 22
- 238000012360 testing method Methods 0.000 description 22
- 239000000872 buffer Substances 0.000 description 21
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 20
- 229910052805 deuterium Inorganic materials 0.000 description 19
- 238000001514 detection method Methods 0.000 description 18
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 17
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 17
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 17
- 101710203703 Peptidyl-lysine N-acetyltransferase YjaB Proteins 0.000 description 17
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 17
- 239000001099 ammonium carbonate Substances 0.000 description 17
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 17
- 239000003153 chemical reaction reagent Substances 0.000 description 16
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 14
- 238000003016 alphascreen Methods 0.000 description 14
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 125000002619 bicyclic group Chemical group 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 108090000623 proteins and genes Proteins 0.000 description 12
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 12
- VPMIAOSOTOODMY-KJAPKAAFSA-N (4r)-6-[(e)-2-[6-tert-butyl-4-(4-fluorophenyl)-2-propan-2-ylpyridin-3-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1/C=C/C=1C(C(C)C)=NC(C(C)(C)C)=CC=1C1=CC=C(F)C=C1 VPMIAOSOTOODMY-KJAPKAAFSA-N 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical group CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 11
- 206010041067 Small cell lung cancer Diseases 0.000 description 11
- 230000014509 gene expression Effects 0.000 description 11
- 125000005842 heteroatom Chemical group 0.000 description 11
- 229910052717 sulfur Inorganic materials 0.000 description 11
- VCUKKMIXURRDKL-UHFFFAOYSA-N 9-(dimethylamino)-3-(4-ethylphenyl)pyrido[1,2]thieno[3,4-d]pyrimidin-4-one Chemical compound C1=CC(CC)=CC=C1N1C(=O)C(SC=2C3=C(N(C)C)C=CN=2)=C3N=C1 VCUKKMIXURRDKL-UHFFFAOYSA-N 0.000 description 10
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 10
- 125000004429 atom Chemical group 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 239000011324 bead Substances 0.000 description 10
- 229910052739 hydrogen Inorganic materials 0.000 description 10
- 239000001257 hydrogen Substances 0.000 description 10
- 239000002609 medium Substances 0.000 description 10
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 10
- 125000003367 polycyclic group Chemical group 0.000 description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 description 10
- 239000011593 sulfur Chemical group 0.000 description 10
- 241000699670 Mus sp. Species 0.000 description 9
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- 239000001301 oxygen Substances 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 8
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 8
- 125000004070 6 membered heterocyclic group Chemical group 0.000 description 8
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 8
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 8
- 108010007005 Estrogen Receptor alpha Proteins 0.000 description 8
- 102000007594 Estrogen Receptor alpha Human genes 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 235000011114 ammonium hydroxide Nutrition 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 238000012054 celltiter-glo Methods 0.000 description 8
- 229910052731 fluorine Inorganic materials 0.000 description 8
- 210000002381 plasma Anatomy 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- 239000004472 Lysine Substances 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 229910052794 bromium Inorganic materials 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 239000006285 cell suspension Substances 0.000 description 7
- 238000010790 dilution Methods 0.000 description 7
- 239000012895 dilution Substances 0.000 description 7
- 239000012065 filter cake Substances 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 238000004809 thin layer chromatography Methods 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- HWWYDZCSSYKIAD-UHFFFAOYSA-N 3,5-dimethylpyridine Chemical compound CC1=CN=CC(C)=C1 HWWYDZCSSYKIAD-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 6
- 229920001213 Polysorbate 20 Polymers 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 6
- RRUDCFGSUDOHDG-UHFFFAOYSA-N acetohydroxamic acid Chemical compound CC(O)=NO RRUDCFGSUDOHDG-UHFFFAOYSA-N 0.000 description 6
- 229960001171 acetohydroxamic acid Drugs 0.000 description 6
- ZSLZBFCDCINBPY-ZSJPKINUSA-N acetyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 ZSLZBFCDCINBPY-ZSJPKINUSA-N 0.000 description 6
- ADFWQBGTDJIESE-UHFFFAOYSA-N anacardic acid 15:0 Natural products CCCCCCCCCCCCCCCC1=CC=CC(O)=C1C(O)=O ADFWQBGTDJIESE-UHFFFAOYSA-N 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 125000001153 fluoro group Chemical group F* 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 239000012046 mixed solvent Substances 0.000 description 6
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 6
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 125000005330 8 membered heterocyclic group Chemical group 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 102220497176 Small vasohibin-binding protein_T47D_mutation Human genes 0.000 description 5
- 108010090804 Streptavidin Proteins 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000000903 blocking effect Effects 0.000 description 5
- 229940098773 bovine serum albumin Drugs 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 239000001569 carbon dioxide Substances 0.000 description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 239000012448 Lithium borohydride Substances 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 4
- 230000021736 acetylation Effects 0.000 description 4
- 238000006640 acetylation reaction Methods 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 150000001335 aliphatic alkanes Chemical class 0.000 description 4
- KAOMOVYHGLSFHQ-UTOQUPLUSA-N anacardic acid Chemical compound CCC\C=C/C\C=C/CCCCCCCC1=CC=CC(O)=C1C(O)=O KAOMOVYHGLSFHQ-UTOQUPLUSA-N 0.000 description 4
- 235000014398 anacardic acid Nutrition 0.000 description 4
- 239000007900 aqueous suspension Substances 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- 230000005757 colony formation Effects 0.000 description 4
- 229940125782 compound 2 Drugs 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000005984 hydrogenation reaction Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000010253 intravenous injection Methods 0.000 description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 125000003226 pyrazolyl group Chemical group 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 150000003457 sulfones Chemical class 0.000 description 4
- 150000003462 sulfoxides Chemical class 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000011534 wash buffer Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 3
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- MWVKLRSIDOXBSE-UHFFFAOYSA-N 5-(1-piperidin-4-ylpyrazol-4-yl)-3-(6-pyrrolidin-1-yl-1,3-benzoxazol-2-yl)pyridin-2-amine Chemical compound NC1=NC=C(C2=CN(N=C2)C2CCNCC2)C=C1C(OC1=C2)=NC1=CC=C2N1CCCC1 MWVKLRSIDOXBSE-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical group [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- 241000662429 Fenerbahce Species 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 102100033069 Histone acetyltransferase KAT8 Human genes 0.000 description 3
- 101000944170 Homo sapiens Histone acetyltransferase KAT8 Proteins 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical class [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 3
- 102000004142 Trypsin Human genes 0.000 description 3
- 108090000631 Trypsin Proteins 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 238000004296 chiral HPLC Methods 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 235000003599 food sweetener Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical class IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 230000001394 metastastic effect Effects 0.000 description 3
- 206010061289 metastatic neoplasm Diseases 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 239000004530 micro-emulsion Substances 0.000 description 3
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- HPJGEESDHAUUQR-SKGSPYGFSA-N (2s)-2-[[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-1-[(2s)-5-(diaminomethylideneamino)-2-[[(2s)-2-[[(2s)-3-naphthalen-2-yl-2-(3-pyridin-3-ylpropanoylamino)propanoyl]amino]-3-phenylpropanoyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]amino]buta Chemical compound NC(=O)C[C@@H](C(N)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=C2C=CC=CC2=CC=1)NC(=O)CCC=1C=NC=CC=1)CC1=CC=CC=C1 HPJGEESDHAUUQR-SKGSPYGFSA-N 0.000 description 2
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 2
- JNPGUXGVLNJQSQ-BGGMYYEUSA-M (e,3r,5s)-7-[4-(4-fluorophenyl)-1,2-di(propan-2-yl)pyrrol-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)N1C(C(C)C)=C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)C(C=2C=CC(F)=CC=2)=C1 JNPGUXGVLNJQSQ-BGGMYYEUSA-M 0.000 description 2
- DPRJPRMZJGWLHY-HNGSOEQISA-N (e,3r,5s)-7-[5-(4-fluorophenyl)-3-propan-2-yl-1-pyrazin-2-ylpyrazol-4-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound OC(=O)C[C@H](O)C[C@H](O)/C=C/C=1C(C(C)C)=NN(C=2N=CC=NC=2)C=1C1=CC=C(F)C=C1 DPRJPRMZJGWLHY-HNGSOEQISA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- PRDFBSVERLRRMY-UHFFFAOYSA-N 2'-(4-ethoxyphenyl)-5-(4-methylpiperazin-1-yl)-2,5'-bibenzimidazole Chemical compound C1=CC(OCC)=CC=C1C1=NC2=CC=C(C=3NC4=CC(=CC=C4N=3)N3CCN(C)CC3)C=C2N1 PRDFBSVERLRRMY-UHFFFAOYSA-N 0.000 description 2
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 2
- HMNKTRSOROOSPP-UHFFFAOYSA-N 3-Ethylphenol Chemical compound CCC1=CC=CC(O)=C1 HMNKTRSOROOSPP-UHFFFAOYSA-N 0.000 description 2
- VLJSLTNSFSOYQR-UHFFFAOYSA-N 3-propan-2-ylphenol Chemical compound CC(C)C1=CC=CC(O)=C1 VLJSLTNSFSOYQR-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 239000012103 Alexa Fluor 488 Substances 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 102100034535 Histone H3.1 Human genes 0.000 description 2
- 101710195388 Histone H3.1 Proteins 0.000 description 2
- 101710195387 Histone H3.2 Proteins 0.000 description 2
- 102100033068 Histone acetyltransferase KAT7 Human genes 0.000 description 2
- 101000944166 Homo sapiens Histone acetyltransferase KAT7 Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- 108010058846 Ovalbumin Proteins 0.000 description 2
- 241001614181 Phera Species 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000002583 anti-histone Effects 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical class [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 235000008504 concentrate Nutrition 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 230000003054 hormonal effect Effects 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- HBEDNENASUYMPO-LJQANCHMSA-N n-hydroxy-4-[[(2r)-3-oxo-2-(thiophen-2-ylmethyl)-2,4-dihydroquinoxalin-1-yl]methyl]benzamide Chemical compound C1=CC(C(=O)NO)=CC=C1CN1C2=CC=CC=C2NC(=O)[C@H]1CC1=CC=CS1 HBEDNENASUYMPO-LJQANCHMSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 229940092253 ovalbumin Drugs 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- SFLXUZPXEWWQNH-UHFFFAOYSA-K tetrabutylazanium;tribromide Chemical compound [Br-].[Br-].[Br-].CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC SFLXUZPXEWWQNH-UHFFFAOYSA-K 0.000 description 2
- 229960005371 tolbutamide Drugs 0.000 description 2
- 201000011294 ureter cancer Diseases 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 1
- LAJAFFLJAJMYLK-CVOKMOJFSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[[(7s)-4-methoxy-7-morpholin-4-yl-6,7,8,9-tetrahydro-5h-benzo[7]annulen-3-yl]amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound N1([C@H]2CCC3=CC=C(C(=C3CC2)OC)NC=2N=C(C(=CN=2)Cl)N[C@H]2[C@H]([C@@]3([H])C[C@@]2(C=C3)[H])C(N)=O)CCOCC1 LAJAFFLJAJMYLK-CVOKMOJFSA-N 0.000 description 1
- RNKOUSCCPHSCFE-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanol Chemical compound COC1=CC=C(CO)C(OC)=C1 RNKOUSCCPHSCFE-UHFFFAOYSA-N 0.000 description 1
- SHAHPWSYJFYMRX-GDLCADMTSA-N (2S)-2-(4-{[(1R,2S)-2-hydroxycyclopentyl]methyl}phenyl)propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C[C@@H]1[C@@H](O)CCC1 SHAHPWSYJFYMRX-GDLCADMTSA-N 0.000 description 1
- PHDIJLFSKNMCMI-ITGJKDDRSA-N (3R,4S,5R,6R)-6-(hydroxymethyl)-4-(8-quinolin-6-yloxyoctoxy)oxane-2,3,5-triol Chemical compound OC[C@@H]1[C@H]([C@@H]([C@H](C(O1)O)O)OCCCCCCCCOC=1C=C2C=CC=NC2=CC=1)O PHDIJLFSKNMCMI-ITGJKDDRSA-N 0.000 description 1
- NUJWKQSEJDYCDB-GNRVTEMESA-N (3s)-1-[(1s,2r,4r)-4-[methyl(propan-2-yl)amino]-2-propylcyclohexyl]-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-2-one Chemical compound CCC[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 NUJWKQSEJDYCDB-GNRVTEMESA-N 0.000 description 1
- PNHBRYIAJCYNDA-VQCQRNETSA-N (4r)-6-[2-[2-ethyl-4-(4-fluorophenyl)-6-phenylpyridin-3-yl]ethyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1CCC=1C(CC)=NC(C=2C=CC=CC=2)=CC=1C1=CC=C(F)C=C1 PNHBRYIAJCYNDA-VQCQRNETSA-N 0.000 description 1
- FRJJJAKBRKABFA-TYFAACHXSA-N (4r,6s)-6-[(e)-2-[6-chloro-4-(4-fluorophenyl)-2-propan-2-ylquinolin-3-yl]ethenyl]-4-hydroxyoxan-2-one Chemical compound C(\[C@H]1OC(=O)C[C@H](O)C1)=C/C=1C(C(C)C)=NC2=CC=C(Cl)C=C2C=1C1=CC=C(F)C=C1 FRJJJAKBRKABFA-TYFAACHXSA-N 0.000 description 1
- QOLHWXNSCZGWHK-BWBORTOCSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-(11-phenoxyundecylcarbamoyloxy)-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@@H]([C@@H](OC(=O)NCCCCCCCCCCCOC=3C=CC=CC=3)C(O1)(C(O)=O)C(O)(C(O2)C(O)=O)C(O)=O)O)C1=CC=CC=C1 QOLHWXNSCZGWHK-BWBORTOCSA-N 0.000 description 1
- ZRYMMWAJAFUANM-INIZCTEOSA-N (7s)-3-fluoro-4-[3-(8-fluoro-1-methyl-2,4-dioxoquinazolin-3-yl)-2-methylphenyl]-7-(2-hydroxypropan-2-yl)-6,7,8,9-tetrahydro-5h-carbazole-1-carboxamide Chemical compound C1[C@@H](C(C)(C)O)CCC2=C1NC1=C2C(C2=C(C(=CC=C2)N2C(C3=CC=CC(F)=C3N(C)C2=O)=O)C)=C(F)C=C1C(N)=O ZRYMMWAJAFUANM-INIZCTEOSA-N 0.000 description 1
- QRDAPCMJAOQZSU-KQQUZDAGSA-N (e)-3-[4-[(e)-3-(3-fluorophenyl)-3-oxoprop-1-enyl]-1-methylpyrrol-2-yl]-n-hydroxyprop-2-enamide Chemical compound C1=C(\C=C\C(=O)NO)N(C)C=C1\C=C\C(=O)C1=CC=CC(F)=C1 QRDAPCMJAOQZSU-KQQUZDAGSA-N 0.000 description 1
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 1
- VAVHMEQFYYBAPR-ITWZMISCSA-N (e,3r,5s)-7-[4-(4-fluorophenyl)-1-phenyl-2-propan-2-ylpyrrol-3-yl]-3,5-dihydroxyhept-6-enoic acid Chemical compound CC(C)C1=C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)C(C=2C=CC(F)=CC=2)=CN1C1=CC=CC=C1 VAVHMEQFYYBAPR-ITWZMISCSA-N 0.000 description 1
- 125000005943 1,2,3,6-tetrahydropyridyl group Chemical group 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- FJZNNKJZHQFMCK-LRDDRELGSA-N 1-[(3S,4R)-4-(2,6-difluoro-4-methoxyphenyl)-2-oxopyrrolidin-3-yl]-3-phenylurea Chemical compound C1(=CC(=CC(=C1[C@H]1[C@@H](C(=O)NC1)NC(=O)NC1=CC=CC=C1)F)OC)F FJZNNKJZHQFMCK-LRDDRELGSA-N 0.000 description 1
- OJGXNQVFIXZTSA-UHFFFAOYSA-N 1-bromo-2-ethyl-4-methoxybenzene Chemical compound CCC1=CC(OC)=CC=C1Br OJGXNQVFIXZTSA-UHFFFAOYSA-N 0.000 description 1
- FDHRABWQLONJBS-UHFFFAOYSA-N 1-bromo-2-methyl-4-(2,2,2-trifluoroethoxy)benzene Chemical compound CC1=CC(OCC(F)(F)F)=CC=C1Br FDHRABWQLONJBS-UHFFFAOYSA-N 0.000 description 1
- ITADIVACIVJFJF-UHFFFAOYSA-N 1-bromo-4-methoxy-2-propan-2-ylbenzene Chemical compound COC1=CC=C(Br)C(C(C)C)=C1 ITADIVACIVJFJF-UHFFFAOYSA-N 0.000 description 1
- OKSRAPDSZKICKZ-UHFFFAOYSA-N 1-methoxy-3-(trifluoromethoxy)benzene Chemical compound COC1=CC=CC(OC(F)(F)F)=C1 OKSRAPDSZKICKZ-UHFFFAOYSA-N 0.000 description 1
- YKYWUHHZZRBGMG-JWTNVVGKSA-N 1-methyl-2-[[(1r,5s)-6-[[5-(trifluoromethyl)pyridin-2-yl]methoxymethyl]-3-azabicyclo[3.1.0]hexan-3-yl]methyl]benzimidazole Chemical compound C1([C@@H]2CN(C[C@@H]21)CC=1N(C2=CC=CC=C2N=1)C)COCC1=CC=C(C(F)(F)F)C=N1 YKYWUHHZZRBGMG-JWTNVVGKSA-N 0.000 description 1
- WLPACIZIAIUYDM-UHFFFAOYSA-N 1-methylsulfonylpyrazole Chemical compound CS(=O)(=O)N1C=CC=N1 WLPACIZIAIUYDM-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- UNECAYDZDLGSLP-UHFFFAOYSA-N 2,3-dihydro-1-benzofuran-7-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC2=C1OCC2 UNECAYDZDLGSLP-UHFFFAOYSA-N 0.000 description 1
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003764 2,4-dimethylpentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- KLVZDQNSNTUNGU-UHFFFAOYSA-N 2,6-dimethoxybenzenesulfonyl chloride Chemical compound COC1=CC=CC(OC)=C1S(Cl)(=O)=O KLVZDQNSNTUNGU-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- JGMXNNSYEFOBHQ-OWOJBTEDSA-N 2-[(e)-4-morpholin-4-ylbut-2-enyl]-1,1-dioxothieno[3,2-e]thiazine-6-sulfonamide Chemical compound O=S1(=O)C=2SC(S(=O)(=O)N)=CC=2C=CN1C\C=C\CN1CCOCC1 JGMXNNSYEFOBHQ-OWOJBTEDSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- ZPXCHEDAXFJYBZ-UHFFFAOYSA-N 2-bromo-1-methoxy-3-methylbenzene Chemical compound COC1=CC=CC(C)=C1Br ZPXCHEDAXFJYBZ-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- CSDSSGBPEUDDEE-UHFFFAOYSA-N 2-formylpyridine Chemical compound O=CC1=CC=CC=N1 CSDSSGBPEUDDEE-UHFFFAOYSA-N 0.000 description 1
- BEDNMYGUVYQPTP-UHFFFAOYSA-N 2-methoxy-4-methylbenzenesulfonyl chloride Chemical compound COC1=CC(C)=CC=C1S(Cl)(=O)=O BEDNMYGUVYQPTP-UHFFFAOYSA-N 0.000 description 1
- FMOOXIZNAXBKGD-UHFFFAOYSA-N 2-methoxy-6-methylbenzenesulfonyl chloride Chemical compound COC1=CC=CC(C)=C1S(Cl)(=O)=O FMOOXIZNAXBKGD-UHFFFAOYSA-N 0.000 description 1
- GYOBZOBUOMDRRN-UHFFFAOYSA-N 2-methoxybenzenesulfonyl chloride Chemical compound COC1=CC=CC=C1S(Cl)(=O)=O GYOBZOBUOMDRRN-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- 125000004336 3,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- YLLRPQWLASQXSI-UHFFFAOYSA-N 3-(4b,8a,9,9a-tetrahydro-4aH-pyrido[2,3-b]indol-4-ylamino)phenol Chemical compound Oc1cccc(NC2=CC=NC3NC4C=CC=CC4C23)c1 YLLRPQWLASQXSI-UHFFFAOYSA-N 0.000 description 1
- MWDVCHRYCKXEBY-LBPRGKRZSA-N 3-chloro-n-[2-oxo-2-[[(1s)-1-phenylethyl]amino]ethyl]benzamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)CNC(=O)C1=CC=CC(Cl)=C1 MWDVCHRYCKXEBY-LBPRGKRZSA-N 0.000 description 1
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- XDCOYBQVEVSNNB-UHFFFAOYSA-N 4-[(7-naphthalen-2-yl-1-benzothiophen-2-yl)methylamino]butanoic acid Chemical compound OC(=O)CCCNCc1cc2cccc(-c3ccc4ccccc4c3)c2s1 XDCOYBQVEVSNNB-UHFFFAOYSA-N 0.000 description 1
- TXEBWPPWSVMYOA-UHFFFAOYSA-N 4-[3-[(1-amino-2-chloroethyl)amino]propyl]-1-[[3-(2-chlorophenyl)phenyl]methyl]-5-hydroxyimidazolidin-2-one Chemical compound NC(CCl)NCCCC1NC(=O)N(Cc2cccc(c2)-c2ccccc2Cl)C1O TXEBWPPWSVMYOA-UHFFFAOYSA-N 0.000 description 1
- TZHQWUAOIWRFSW-UHFFFAOYSA-N 4-bromo-2,6-difluorobenzonitrile Chemical compound FC1=CC(Br)=CC(F)=C1C#N TZHQWUAOIWRFSW-UHFFFAOYSA-N 0.000 description 1
- ARDAEBQYIBXEMA-UHFFFAOYSA-N 4-bromo-2-fluoro-6-hydroxybenzonitrile Chemical compound OC1=CC(Br)=CC(F)=C1C#N ARDAEBQYIBXEMA-UHFFFAOYSA-N 0.000 description 1
- CAMRWHABCOLYHJ-UHFFFAOYSA-N 4-bromo-3-ethylphenol Chemical compound CCC1=CC(O)=CC=C1Br CAMRWHABCOLYHJ-UHFFFAOYSA-N 0.000 description 1
- FJFASOHNLDGZGR-UHFFFAOYSA-N 4-bromo-3-methoxy-n,n-dimethylaniline Chemical compound COC1=CC(N(C)C)=CC=C1Br FJFASOHNLDGZGR-UHFFFAOYSA-N 0.000 description 1
- RUTNWXBHRAIQSP-UHFFFAOYSA-N 4-bromo-3-methoxyaniline Chemical compound COC1=CC(N)=CC=C1Br RUTNWXBHRAIQSP-UHFFFAOYSA-N 0.000 description 1
- QVCXGNUBIRQHII-UHFFFAOYSA-N 4-bromo-3-propan-2-ylphenol Chemical compound CC(C)C1=CC(O)=CC=C1Br QVCXGNUBIRQHII-UHFFFAOYSA-N 0.000 description 1
- DVDBJVCALYSLBY-UHFFFAOYSA-N 4-bromo-6-fluoro-2-hydroxy-3-(3-hydroxypropyl)benzonitrile Chemical compound BrC1=C(C(=C(C#N)C(=C1)F)O)CCCO DVDBJVCALYSLBY-UHFFFAOYSA-N 0.000 description 1
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 1
- QZRFQULIACMIQI-UHFFFAOYSA-N 5-bromo-7-fluoro-3,4-dihydro-2H-chromene-8-carbonitrile Chemical compound BrC1=C2CCCOC2=C(C(=C1)F)C#N QZRFQULIACMIQI-UHFFFAOYSA-N 0.000 description 1
- HIHOEGPXVVKJPP-JTQLQIEISA-N 5-fluoro-2-[[(1s)-1-(5-fluoropyridin-2-yl)ethyl]amino]-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyridine-3-carbonitrile Chemical compound N([C@@H](C)C=1N=CC(F)=CC=1)C(C(=CC=1F)C#N)=NC=1NC=1C=C(C)NN=1 HIHOEGPXVVKJPP-JTQLQIEISA-N 0.000 description 1
- LUEBMKPHZDSPFH-UHFFFAOYSA-N 5-fluoro-2-methoxybenzenesulfonyl chloride Chemical compound COC1=CC=C(F)C=C1S(Cl)(=O)=O LUEBMKPHZDSPFH-UHFFFAOYSA-N 0.000 description 1
- BEUYIWOWEBGZHX-UHFFFAOYSA-N 5-methoxy-2,3-dihydro-1-benzofuran Chemical compound COC1=CC=C2OCCC2=C1 BEUYIWOWEBGZHX-UHFFFAOYSA-N 0.000 description 1
- RRELDGDKULRRDM-UHFFFAOYSA-N 6-[2-chloro-4-nitro-5-(oxan-4-yloxy)anilino]-3,4-dihydro-1H-quinolin-2-one Chemical compound [O-][N+](=O)c1cc(Cl)c(Nc2ccc3NC(=O)CCc3c2)cc1OC1CCOCC1 RRELDGDKULRRDM-UHFFFAOYSA-N 0.000 description 1
- IGHGEZPZOVFQLI-UHFFFAOYSA-N 6-bromo-5-methoxy-2,3-dihydro-1-benzofuran Chemical compound C1=C(Br)C(OC)=CC2=C1OCC2 IGHGEZPZOVFQLI-UHFFFAOYSA-N 0.000 description 1
- SJOLEHYHMOBNIB-UHFFFAOYSA-N 6-methoxy-2,3-dihydro-1h-indene-5-sulfonyl chloride Chemical compound C1=C(S(Cl)(=O)=O)C(OC)=CC2=C1CCC2 SJOLEHYHMOBNIB-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 241000143437 Aciculosporium take Species 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- DGJMHKMYSDYOFP-MRXNPFEDSA-N C=CC(N(CCC1)C[C@@H]1N1N=C(C2=CN(CC(C3=CC=CC=C3)(F)F)N=N2)C2=C(N)N=CN=C12)=O Chemical compound C=CC(N(CCC1)C[C@@H]1N1N=C(C2=CN(CC(C3=CC=CC=C3)(F)F)N=N2)C2=C(N)N=CN=C12)=O DGJMHKMYSDYOFP-MRXNPFEDSA-N 0.000 description 1
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 1
- WUZBOJXXYMKMMF-UHFFFAOYSA-N COC1=CC2=NC=3N(C(N(C(C=3N2C=C1)=O)CCC)=O)CCCCNC(=O)C1=CC=C(C=C1)S(=O)(=O)F Chemical compound COC1=CC2=NC=3N(C(N(C(C=3N2C=C1)=O)CCC)=O)CCCCNC(=O)C1=CC=C(C=C1)S(=O)(=O)F WUZBOJXXYMKMMF-UHFFFAOYSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 101000936911 Chionoecetes opilio Sarcoplasmic/endoplasmic reticulum calcium ATPase Proteins 0.000 description 1
- CYSWUSAYJNCAKA-FYJFLYSWSA-N ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O Chemical compound ClC1=C(C=CC=2N=C(SC=21)OCC)OC1=CC=C(C=N1)/C=C/[C@H](C)NC(C)=O CYSWUSAYJNCAKA-FYJFLYSWSA-N 0.000 description 1
- QBXVXKRWOVBUDB-GRKNLSHJSA-N ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C Chemical compound ClC=1C(=CC(=C(CN2[C@H](C[C@H](C2)O)C(=O)O)C1)OCC1=CC(=CC=C1)C#N)OCC1=C(C(=CC=C1)C1=CC2=C(OCCO2)C=C1)C QBXVXKRWOVBUDB-GRKNLSHJSA-N 0.000 description 1
- 229940126279 Compound 14f Drugs 0.000 description 1
- 229940126559 Compound 4e Drugs 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 101100411591 Dictyostelium discoideum rab8B gene Proteins 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical group [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 101150076104 EAT2 gene Proteins 0.000 description 1
- 101150064205 ESR1 gene Proteins 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 101000882584 Homo sapiens Estrogen receptor Proteins 0.000 description 1
- 101100019690 Homo sapiens KAT6B gene Proteins 0.000 description 1
- 101000801643 Homo sapiens Retinal-specific phospholipid-transporting ATPase ABCA4 Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 108010075710 Lysine Acetyltransferase 5 Proteins 0.000 description 1
- 102000011785 Lysine Acetyltransferase 5 Human genes 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- TZCCKCLHNUSAMQ-DUGSHLAESA-N NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N Chemical compound NC(=O)C[C@H](NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](Cc2ccc(F)cc2)NC(=O)[C@H](Cc3c[nH]c4ccccc34)NC(=O)Cc5cccs5)C(=O)N TZCCKCLHNUSAMQ-DUGSHLAESA-N 0.000 description 1
- 206010029098 Neoplasm skin Diseases 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 238000003559 RNA-seq method Methods 0.000 description 1
- 102100033617 Retinal-specific phospholipid-transporting ATPase ABCA4 Human genes 0.000 description 1
- 229940125907 SJ995973 Drugs 0.000 description 1
- 101100148749 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SAS2 gene Proteins 0.000 description 1
- 101100148751 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SAS3 gene Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 241000872198 Serjania polyphylla Species 0.000 description 1
- KZVWEOXAPZXAFB-BQFCYCMXSA-N Temocaprilat Chemical compound C([C@H](N[C@H]1CS[C@@H](CN(C1=O)CC(=O)O)C=1SC=CC=1)C(O)=O)CC1=CC=CC=C1 KZVWEOXAPZXAFB-BQFCYCMXSA-N 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical class [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- NELWQUQCCZMRPB-UBPLGANQSA-N [(2r,3r,4r,5r)-4-acetyloxy-5-(4-amino-5-ethenyl-2-oxopyrimidin-1-yl)-2-methyloxolan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](C)O[C@H]1N1C(=O)N=C(N)C(C=C)=C1 NELWQUQCCZMRPB-UBPLGANQSA-N 0.000 description 1
- BEXZJJQVPWJPOA-VOTSOKGWSA-N [(e)-hept-2-enyl] 6-methyl-4-(4-nitrophenyl)-2-oxo-3,4-dihydro-1h-pyrimidine-5-carboxylate Chemical compound CCCC\C=C\COC(=O)C1=C(C)NC(=O)NC1C1=CC=C([N+]([O-])=O)C=C1 BEXZJJQVPWJPOA-VOTSOKGWSA-N 0.000 description 1
- HGDWHTASNMRJMP-UHFFFAOYSA-N [1-(hydroxyamino)-1-oxo-5-(3-phenoxyphenyl)pentan-2-yl]phosphonic acid Chemical compound ONC(=O)C(P(O)(O)=O)CCCC1=CC=CC(OC=2C=CC=CC=2)=C1 HGDWHTASNMRJMP-UHFFFAOYSA-N 0.000 description 1
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 1
- AJRQIXBBIDPNGK-BVSLBCMMSA-N benzyl n-[(2s)-1-[[(2s)-1-(1,3-benzothiazol-2-yl)-1-oxo-3-[(3s)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]carbamate Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](C[C@H]1C(NCC1)=O)C(=O)C=1SC2=CC=CC=C2N=1)C(=O)OCC1=CC=CC=C1 AJRQIXBBIDPNGK-BVSLBCMMSA-N 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- UORVGPXVDQYIDP-BJUDXGSMSA-N borane Chemical class [10BH3] UORVGPXVDQYIDP-BJUDXGSMSA-N 0.000 description 1
- 208000024119 breast tumor luminal A or B Diseases 0.000 description 1
- CVDGHGWEHQIJTE-UHFFFAOYSA-N bromo(bromomethoxy)methane Chemical compound BrCOCBr CVDGHGWEHQIJTE-UHFFFAOYSA-N 0.000 description 1
- JAMFGQBENKSWOF-UHFFFAOYSA-N bromo(methoxy)methane Chemical compound COCBr JAMFGQBENKSWOF-UHFFFAOYSA-N 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000030570 cellular localization Effects 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 210000001726 chromosome structure Anatomy 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940127206 compound 14d Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125872 compound 4d Drugs 0.000 description 1
- 229940126115 compound 4f Drugs 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- CHVJITGCYZJHLR-UHFFFAOYSA-N cyclohepta-1,3,5-triene Chemical compound C1C=CC=CC=C1 CHVJITGCYZJHLR-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- VRLDVERQJMEPIF-UHFFFAOYSA-N dbdmh Chemical compound CC1(C)N(Br)C(=O)N(Br)C1=O VRLDVERQJMEPIF-UHFFFAOYSA-N 0.000 description 1
- 150000001975 deuterium Chemical group 0.000 description 1
- RAFNCPHFRHZCPS-UHFFFAOYSA-N di(imidazol-1-yl)methanethione Chemical compound C1=CN=CN1C(=S)N1C=CN=C1 RAFNCPHFRHZCPS-UHFFFAOYSA-N 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- AVAACINZEOAHHE-VFZPANTDSA-N doripenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](CNS(N)(=O)=O)C1 AVAACINZEOAHHE-VFZPANTDSA-N 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 201000007281 estrogen-receptor positive breast cancer Diseases 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- YWKVMGDEOUPQGN-UHFFFAOYSA-N ethyl 2-[4-[2-(3-hydroxy-1-azabicyclo[2.2.2]octan-3-yl)ethynyl]phenyl]acetate Chemical compound C1=CC(CC(=O)OCC)=CC=C1C#CC1(O)C(CC2)CCN2C1 YWKVMGDEOUPQGN-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 102000055652 human KAT6A Human genes 0.000 description 1
- 102000050265 human KAT6B Human genes 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 238000003125 immunofluorescent labeling Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical class CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- JFOZKMSJYSPYLN-QHCPKHFHSA-N lifitegrast Chemical compound CS(=O)(=O)C1=CC=CC(C[C@H](NC(=O)C=2C(=C3CCN(CC3=CC=2Cl)C(=O)C=2C=C3OC=CC3=CC=2)Cl)C(O)=O)=C1 JFOZKMSJYSPYLN-QHCPKHFHSA-N 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- UIUXUFNYAYAMOE-UHFFFAOYSA-N methylsilane Chemical compound [SiH3]C UIUXUFNYAYAMOE-UHFFFAOYSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- AFDQGRURHDVABZ-UHFFFAOYSA-N n,n-dimethylformamide;sulfur trioxide Chemical compound O=S(=O)=O.CN(C)C=O AFDQGRURHDVABZ-UHFFFAOYSA-N 0.000 description 1
- QAPTWHXHEYAIKG-RCOXNQKVSA-N n-[(1r,2s,5r)-5-(tert-butylamino)-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](NC(C)(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 QAPTWHXHEYAIKG-RCOXNQKVSA-N 0.000 description 1
- NFPUARSXIMWASK-GOEBONIOSA-N n-[(5r,7s)-2-(3-chlorophenyl)-1-oxa-3-azaspiro[4.5]dec-2-en-7-yl]acetamide Chemical compound C1[C@@H](NC(=O)C)CCC[C@@]11OC(C=2C=C(Cl)C=CC=2)=NC1 NFPUARSXIMWASK-GOEBONIOSA-N 0.000 description 1
- PHVXTQIROLEEDB-UHFFFAOYSA-N n-[2-(2-chlorophenyl)ethyl]-4-[[3-(2-methylphenyl)piperidin-1-yl]methyl]-n-pyrrolidin-3-ylbenzamide Chemical compound CC1=CC=CC=C1C1CN(CC=2C=CC(=CC=2)C(=O)N(CCC=2C(=CC=CC=2)Cl)C2CNCC2)CCC1 PHVXTQIROLEEDB-UHFFFAOYSA-N 0.000 description 1
- XZMHJYWMCRQSSI-UHFFFAOYSA-N n-[5-[2-(3-acetylanilino)-1,3-thiazol-4-yl]-4-methyl-1,3-thiazol-2-yl]benzamide Chemical compound CC(=O)C1=CC=CC(NC=2SC=C(N=2)C2=C(N=C(NC(=O)C=3C=CC=CC=3)S2)C)=C1 XZMHJYWMCRQSSI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 238000005935 nucleophilic addition reaction Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000011369 optimal treatment Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- RPGWZZNNEUHDAQ-UHFFFAOYSA-N phenylphosphine Chemical compound PC1=CC=CC=C1 RPGWZZNNEUHDAQ-UHFFFAOYSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 238000010837 poor prognosis Methods 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940125944 selective estrogen receptor degrader Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 230000009211 stress pathway Effects 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical compound [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- FRACPXUHUTXLCX-BELIEFIBSA-N tert-butyl N-{1-[(1S)-1-{[(1R,2S)-1-(benzylcarbamoyl)-1-hydroxy-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]carbamoyl}-2-cyclopropylethyl]-2-oxopyridin-3-yl}carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CN(C1=O)[C@@H](CC2CC2)C(=O)N[C@@H](C[C@@H]3CCNC3=O)[C@H](C(=O)NCC4=CC=CC=C4)O FRACPXUHUTXLCX-BELIEFIBSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 230000001875 tumorinhibitory effect Effects 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- BFDBKMOZYNOTPK-UHFFFAOYSA-N vonoprazan Chemical compound C=1C=CN=CC=1S(=O)(=O)N1C=C(CNC)C=C1C1=CC=CC=C1F BFDBKMOZYNOTPK-UHFFFAOYSA-N 0.000 description 1
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/424—Oxazoles condensed with heterocyclic ring systems, e.g. clavulanic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the disclosure belongs to the field of medicine, and relates to a sulfonamide derivative, its preparation method and its application in medicine.
- the present disclosure relates to a sulfonamide derivative represented by general formula (I), its preparation method and a pharmaceutical composition containing the derivative, and its use as a KAT inhibitor in the preparation of a drug for treating and/or preventing cancer use.
- Lysine acetyltransferases are a class of enzymes that can catalyze the transfer of acetyl groups from acetyl-CoA to lysine ⁇ -amino groups in protein substrates.
- the acetylation of lysine can affect the function of proteins, thus playing an important regulatory role in chromosome structure, gene transcription regulation, DNA binding ability, enzyme activity and stability, protein interaction and intracellular localization.
- KATs are divided into several subfamilies, among which MYST (MOZ, YBF2/SAS3, SAS2, TIP60) is the largest one, including KAT5 (TIP60), KAT6A (MOZ; MYST3), KAT6B (MORF; MYST4), KAT7 (HBO; MYST2 ) and KAT8 (MOF; MYST1).
- KAT6A/B as the main member of the MYST family, plays a vital role in development, maintenance of stem cells in the hematopoietic and immune systems, and tumor development and drug resistance.
- KAT6A and KAT6B were amplified in a variety of tumors.
- KAT6A is located in the amplicon region of chromosome 8p11-p12, which is amplified in 10-15% of breast cancers, and its copy number is positively correlated with mRNA expression and associated with poor prognosis.
- Both KAT6A and KAT6B were significantly highly expressed in breast cancer.
- Further subtype analysis showed that there was a certain correlation between the high expression of KAT6A/B and the expression level of ER ⁇ , revealing that KAT6A/B may be a potential target of ER + /HER2 - breast cancer.
- KAT6A/B selective inhibitor CTx-648 showed antitumor activity in ER+ breast cancer both in vitro and in vivo, and the expression level of KAT6A was correlated with the sensitivity of CTx-648. In ER + breast cancer cells with high KAT6A expression, CTx-648 can downregulate ER ⁇ expression, and H3K23Ac can be used as a pharmacodynamic biomarker of KAT6 inhibitors.
- KAT6A/B inhibitors have clinical development value as single agents or in combination with existing ER + /HER2 - breast cancer therapies such as fulvestrant, CDK4/6 inhibitors, and even SERD and SERCA. of.
- KAT6A/B inhibitors have potential application prospects in glioma, B cell lymphoma, liver cancer, ovarian cancer and other tumor types, and can be used as indication expansion.
- Published KAT6 inhibitor patent applications include WO2016198507A1, WO2019243491A1, WO2019043139A1, WO2019108824A1, WO2020216701A1, WO2020002587A1, WO2020254946A1 and WO2020254989A1, etc.
- Ring A is selected from cycloalkyl, heterocyclyl, aryl and heteroaryl;
- Ring B is cycloalkyl or heterocyclyl
- L is a chemical bond, an alkylene group or a heteroalkylene group; wherein each of the alkylene or heteroalkylene groups is independently selected from the group consisting of hydroxyl, halogen, alkyl, haloalkyl, hydroxyalkyl, alkoxy and one or more substituents in haloalkoxy;
- Each R 1 , each R 2 , and R 3 are the same or different, and each independently selected from a hydrogen atom, halogen, cyano, nitro, oxo, alkenyl, alkynyl, alkyl, alkoxy, cycloalkane radical, heterocyclyl, aryl, heteroaryl, -OR 5 , -C(O)R 6 , -C(O)OR 6 , -OC(O)R 6 , -NHC(O)OR 6 , - NR 7 R 8 , -C(O)NR 7 R 8 , -S(O) r R 6 and -S(O) r NR 7 R 8 ; the alkenyl, alkynyl, alkyl, alkoxy radical, cycloalkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxyl, halogen, cyano, amino, nitro, oxo, alken
- R4 is a hydrogen atom
- Ring C is selected from cycloalkyl, heterocyclyl, aryl and heteroaryl;
- R is selected from hydrogen atom, hydroxyl, halogen, alkyl, haloalkyl, hydroxyalkyl, alkoxy, haloalkoxy and cycloalkyl;
- Each R 4a is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, cyano, nitro, oxo, alkenyl, alkynyl, alkyl, haloalkyl, hydroxyalkyl, alkoxy, Haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy, heterocyclyloxy and -NR 9 R 10 ;
- R 5 and R 6 are the same or different, and are each independently selected from a hydrogen atom, alkenyl, alkynyl, alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl; wherein the alkenyl, alkynyl radical, alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxyl, halogen, cyano, amino, nitro, oxo, alkenyl, alkynyl, alkane radical, haloalkyl, hydroxyalkyl, alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy and One or more substituents in heterocyclyloxy are substituted;
- R 7 , R 8 , R 9 , and R 10 are the same or different, and are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocyclic group, an aryl group, and a heteroaryl group; wherein the alkyl group, ring Alkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxy, halo, cyano, amino, nitro, oxo, alkenyl, alkynyl, alkyl, haloalkyl, hydroxy Alkyl, alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy and heterocyclyloxy substituted by one or more substituents;
- R 7 and R 8 form a heterocyclic group together with the connected N atom
- R 9 and R 10 form a heterocyclic group together with the connected N atom
- the heterocyclic group is optionally selected from from hydroxy, halogen, cyano, amino, nitro, oxo, alkenyl, alkynyl, alkyl, haloalkyl, hydroxyalkyl, alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl
- p 0, 1, 2, 3 or 4;
- q 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- r 0, 1 or 2.
- Ring A is selected from cycloalkyl, heterocyclyl, aryl and heteroaryl;
- Ring B is selected from cycloalkyl or heterocyclyl
- L is a chemical bond, an alkylene group or a heteroalkylene group; wherein each of the alkylene or heteroalkylene groups is independently selected from the group consisting of hydroxyl, halogen, alkyl, haloalkyl, hydroxyalkyl, alkoxy and one or more substituents in haloalkoxy;
- Each R 1 , each R 2 , and R 3 are the same or different, and each independently selected from a hydrogen atom, halogen, cyano, nitro, oxo, alkenyl, alkynyl, alkyl, alkoxy, cycloalkane radical, heterocyclyl, aryl, heteroaryl, -OR 5 , -C(O)R 6 , -C(O)OR 6 , -OC(O)R 6 , -NHC(O)OR 6 , - NR 7 R 8 , -C(O)NR 7 R 8 , -S(O) r R 6 and -S(O) r NR 7 R 8 ; the alkenyl, alkynyl, alkyl, alkoxy radical, cycloalkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxyl, halogen, cyano, amino, nitro, oxo, alken
- R4 is a hydrogen atom
- Ring C is aryl or heteroaryl
- R is selected from hydrogen atom, hydroxyl, halogen, alkyl, haloalkyl, hydroxyalkyl, alkoxy, haloalkoxy and cycloalkyl;
- Each R 4a is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, cyano, nitro, oxo, alkenyl, alkynyl, alkyl, haloalkyl, hydroxyalkyl, alkoxy, Haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy, heterocyclyloxy and -NR 9 R 10 ;
- R 5 and R 6 are the same or different, and are each independently selected from a hydrogen atom, alkenyl, alkynyl, alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl; wherein the alkenyl, alkynyl radical, alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxyl, halogen, cyano, amino, nitro, oxo, alkenyl, alkynyl, alkane radical, haloalkyl, hydroxyalkyl, alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy and One or more substituents in heterocyclyloxy are substituted;
- R 7 , R 8 , R 9 , and R 10 are the same or different, and are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocyclic group, an aryl group, and a heteroaryl group; wherein the alkyl group, ring Alkyl, heterocyclyl, aryl and heteroaryl are each independently selected from the group consisting of hydroxy, halo, cyano, amino, nitro, oxo, alkenyl, alkynyl, alkyl, haloalkyl, hydroxy Alkyl, alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy and heterocyclyloxy substituted by one or more substituents;
- R 7 and R 8 form a heterocyclic group together with the connected N atom
- R 9 and R 10 form a heterocyclic group together with the connected N atom
- the heterocyclic group is optionally selected from Hydroxy, Halo, Cyano, Amino, Nitro, Oxo, Alkenyl, Alkynyl, Alkyl, Haloalkyl, Hydroxyalkyl, Alkoxy, Haloalkoxy, Cycloalkyl, Heterocyclyl, Aryl , heteroaryl, cycloalkylalkyl, heterocyclylalkyl, cycloalkyloxy and heterocyclyloxy are substituted by one or more substituents;
- p 0, 1, 2, 3 or 4;
- q 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- n 0, 1, 2, 3 or 4;
- r 0, 1 or 2.
- the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof wherein R 3 is selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1- 6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and 3 to 8 membered cycloalkyl; preferably, R 3 is a hydrogen atom.
- the compound represented by general formula (I) or a pharmaceutically acceptable salt thereof wherein ring C is selected from 3 to 8 membered cycloalkyl, 3 to 8 membered heterocyclyl, 6 to 10-membered aryl and 5- to 10-membered heteroaryl; preferably, ring C is 5- to 10-membered heteroaryl or 6- to 10-membered aryl; more preferably, ring C is 5- or 6-membered heteroaryl ; Most preferably, ring C is pyrazolyl.
- the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof wherein ring C is a 5-membered or 6-membered heterocyclic group, a 5-membered or 6-membered heteroaryl group;
- ring C is selected from pyrazolyl, pyridyl, furyl and tetrahydrofuryl; more preferably, ring C is pyrazolyl.
- the compound represented by the general formula (I) or a pharmaceutically acceptable salt thereof wherein R 0 is selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1- 6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and 3 to 8 membered cycloalkyl; preferably, R is selected from hydrogen atom, hydroxyl, halogen and C 1-6 alkyl; preferably, R 0 is selected from a hydrogen atom, hydroxyl and Cl; more preferably, R 0 is a hydrogen atom.
- m is 0 or 1, preferably 1.
- R4 is R 4a and n are as defined in general formula (I);
- R4 is
- the compound represented by general formula (I) or a pharmaceutically acceptable salt thereof is a compound represented by general formula (II) or a pharmaceutically acceptable salt thereof:
- Ring C is a 5- to 10-membered heteroaryl group, preferably a 5-membered or 6-membered heteroaryl group;
- Ring A, ring B, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (I).
- R4 is R 4a and n are as defined in general formula (I);
- R 4 is R 4a and n are as defined in general formula (I);
- R4 is
- R 4 is
- the compound represented by general formula (I) or a pharmaceutically acceptable salt thereof is a compound represented by general formula (Ii) or a pharmaceutically acceptable salt thereof:
- Ring C is a 3- to 8-membered heterocyclic group or a 5- to 10-membered heteroaryl group, preferably a 5- or 6-membered heterocyclic group, a 5- or 6-membered heteroaryl group;
- Ring A, ring B, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (I).
- the compound represented by general formula (I), general formula (II) or general formula (Ii) or a pharmaceutically acceptable salt thereof wherein ring A is a 6- to 10-membered aryl group Or 5 to 10 membered heteroaryl, the 6 to 10 membered aryl is preferably phenyl or naphthyl; the 5 to 10 membered heteroaryl is preferably pyridyl, quinolinyl and benzoxazolyl.
- the compound represented by general formula (I), general formula (II) or general formula (Ii) or a pharmaceutically acceptable salt thereof wherein ring A is a 6- to 10-membered aryl group Or 5 to 10 membered heteroaryl, the 6 to 10 membered aryl is preferably selected from phenyl, naphthyl, The 5- to 10-membered heteroaryl is preferably pyridyl, quinolinyl and benzoxazolyl.
- Ring B is a 4 to 7 membered heterocyclyl or a 4 to 7 membered cycloalkyl
- ring B is a 4- to 7-membered heterocyclic group
- Ring B is a 4- to 7-membered heterocyclic group, wherein the 4- to 7-membered heterocyclic group contains 1-3 oxygen atoms;
- ring B is a 5-membered or 6-membered heterocyclic group, wherein the 5-membered or 6-membered heterocyclic group contains 1 or 2 oxygen atoms.
- the compound represented by general formula (I), general formula (II) or general formula (Ii) or a pharmaceutically acceptable salt thereof, wherein ring B is selected from preferably R 2 can be substituted at any substitutable position on ring B.
- ring B is selected from More preferably, Ring B is R 2 can be substituted at any substitutable position on ring B.
- the compound represented by general formula (I) or (II) or a pharmaceutically acceptable salt thereof is a compound represented by general formula (III) or a pharmaceutically acceptable salt thereof the salt:
- Each R a , R b , R c and R d are the same or different, and each independently selected from a hydrogen atom, a hydroxyl group, a halogen, a cyano group, an amino group, an alkenyl group, an alkynyl group, an alkyl group, a haloalkyl group, a hydroxyalkyl group, Alkoxy, haloalkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, cycloalkyloxy and heterocyclyloxy;
- s 0, 1, 2 or 3;
- L, R 1 , R 4a , p and n are as defined in the general formula (I).
- each Rc and Rd are the same or different, and are each independently selected from hydrogen atom, halogen, C1-6 alkyl and C1-6 alkoxy;
- each R c and R d are the same or different, and each independently is a hydrogen atom or a halogen;
- each Rc and Rd are the same or different, and are each independently a hydrogen atom or a fluorine atom;
- both R c and R d are hydrogen atoms.
- the compound represented by general formula (I), general formula (II) or general formula (III) or a pharmaceutically acceptable salt thereof is represented by general formula (IV) Compound or its pharmaceutically acceptable salt:
- L, X, R 1 , R 4a , p, n and s are as defined in general formula (III).
- each R a and R b are the same or different, and are each independently selected from a hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy , C 1-6 haloalkoxy, 3 to 8 membered cycloalkyl, 3 to 8 membered heterocyclyl, 3 to 8 membered cycloalkyloxy and 3 to 8 membered heterocyclyloxy;
- each R a and R b are the same or different, and are each independently selected from a hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy ;
- each R a and R b are the same or different, and are each independently selected from a hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6
- the compound represented by general formula (III) or general formula (IV) or a pharmaceutically acceptable salt thereof wherein X is O or CH 2 ; preferably, X is CH 2 .
- the compound represented by general formula (III) or a pharmaceutically acceptable salt thereof wherein R c and R d are the same or different, and each independently is a hydrogen atom or a halogen; and/ or X is O or CH2 ; and/or s is 1 or 2.
- each R 1 is the same or different, and each independently selected from a hydrogen atom, a hydroxyl group, a halogen, a cyano group, an amino group, a C 1-6 alkyl group, a C 1-6 haloalkyl group, a C 1-6 hydroxyalkyl group, C 1-6 alkoxy, C 1-6 haloalkoxy, oxo, 3 to 8 membered cycloalkyl, C 1-6 alkoxy C 1-6 alkyl, -OR 5 , -C(O )R 6 , -C(O)OR 6 , -C(O)NR 7 R 8 and -S(O) r R 6 , r, R 5 , R 6 , R 7 and R 8 such as general formula (I) defined in; preferably
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkane Oxygen, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl, -C(O)OCH 3 and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each are independently a hydrogen atom or a C 1-6 alkyl group;
- each R 1 is the same or different, and each independently selected from hydrogen atom, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently is a hydrogen atom or a C 1-6 alkyl group;
- each R 1 is the same or different, and is independently C 1-6 alkoxy.
- each R 1 is the same or different, and each independently is methoxy.
- each R 1 is the same or different, and each independently selected from a hydrogen atom, a fluorine atom, a chlorine atom, a methyl group, an ethyl group, an isopropyl group, a methoxy group, an ethoxy group, a trifluoromethoxy group, a methylamino, dimethylamino and Preferably, R 1 is methoxy.
- each R 2 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, cyano, amino, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 Haloalkoxy, 3 to 8 membered cycloalkyl, 3 to 8 membered heterocyclyl, oxo, 3 to 8 membered cycloalkyloxy and 3 to 8 membered heterocyclyloxy; preferably, each R The same or different, and each independently selected from a hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy and oxo ; Further preferably, each R 2 is the same or different, and is independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C
- each R 4a is the same or different, and each independently selected from a hydrogen atom, hydroxyl, halogen, cyano, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1 -6 alkoxy, C 1-6 haloalkoxy, -NR 9 R 10 , 3 to 8 membered cycloalkyl, 3 to 8 membered heterocyclyl, 3 to 8 membered cycloalkyloxy and 3 to 8 membered Heterocyclyloxy, R 9 and R 10 are as defined in the general formula (I); preferably, each R 4a is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl , C 1-6 hydroxyalkyl and C 1-6 alkoxy
- the compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III) or general formula (IV) or its pharmaceutically acceptable Salt wherein L is a chemical bond or a C 1-6 alkylene group; preferably selected from chemical bond, -CH 2 - or -CH 2 CH 2 -, more preferably chemical bond and -CH 2 -, most preferably chemical bond.
- the compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III) or general formula (IV) or its pharmaceutically acceptable Salt where n is 1 or 2.
- the compound represented by general formula (III) or general formula (IV) or a pharmaceutically acceptable salt thereof wherein each R 4a is the same or different, and each independently selected from halogen, Hydroxy, C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy, and n is 1 or 2, or R 4a is a hydrogen atom, and n is 3.
- the compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III) or general formula (IV) or its pharmaceutically acceptable Salt wherein R 5 and R 6 are the same or different, and are each independently selected from a hydrogen atom, C 1-6 alkyl, 3 to 8 membered cycloalkyl, 3 to 12 membered heterocyclic group, 6 to 10 membered aryl group and 5 to 10 membered heteroaryl; wherein said C 1-6 alkyl, 3 to 8 membered cycloalkyl, 3 to 12 membered heterocyclic group, 6 to 10 membered aryl and 5 to 10 membered heteroaryl Each independently optionally selected from hydroxyl, halogen, cyano, amino, nitro, oxo, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 One or more substituents in alkoxy, C 1-6 haloalkoxy
- R 5 and R 6 are the same or different, and are independently selected from C 1-6 alkyl, C 1-6 haloalkyl and 3-8 membered cycloalkyl.
- the compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III) or general formula (IV) or its pharmaceutically acceptable Salt wherein R 7 and R 8 are the same or different, and each independently selected from hydrogen atom, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, 3 to 8 membered cycloalkyl and a 3- to 12-membered heterocyclic group; preferably, R 7 and R 8 are the same or different, and each independently is a hydrogen atom or a C 1-6 alkyl group; more preferably, R 7 and R 8 are hydrogen atoms.
- the compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III) or general formula (IV) or its pharmaceutically acceptable Salt wherein R 9 and R 10 are the same or different, and are each independently selected from a hydrogen atom, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, 3 to 8 membered cycloalkyl and 3- to 12-membered heterocyclyl; preferably, R9 and R10 are hydrogen atoms.
- the compound represented by general formula (I), general formula (II) and general formula (Ii) or a pharmaceutically acceptable salt thereof, wherein for R 1a and R 1b are the same or different, and each independently selected from a hydrogen atom, hydroxyl, halogen, cyano, amino, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, oxo, 3 to 8 membered cycloalkyl, C 1-6 alkoxy C 1-6 alkyl, -OR 5 , -C(O) R 6 , -C(O)OR 6 , -C(O)NR 7 R 8 and -S(O) r R 6 , r, R 5 , R 6 , R 7 and R 8 are as in general formula (I) defined; preferably, R 1a and R 1b are the same or different, and each independently selected from hydrogen atom,
- the compound represented by general formula (I), general formula (II) and general formula (Ii) or a pharmaceutically acceptable salt thereof, wherein for R 1a and R 1b are the same or different, and each independently selected from a hydrogen atom, hydroxyl, halogen, cyano, amino, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, oxo, 3 to 8 membered cycloalkyl, C 1-6 alkoxy C 1-6 alkyl, -OR 5 , -C(O) R 6 , -C(O)OR 6 , -C(O)NR 7 R 8 and -S(O) r R 6 , r, R 5 , R 6 , R 7 and R 8 are as in general formula (I) defined; preferably, R 1a and R 1b are the same or different, and each independently selected from hydrogen atom,
- the compound represented by general formula (III) or a pharmaceutically acceptable salt thereof wherein for Preferably, selected from More preferably, for
- the compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof wherein selected from Preferably, selected from More preferably, for
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl and -C(O)OCH 3 ;
- p is 1, 2 or 3;
- each R 2 is the same or different, and each independently selected from a hydrogen atom, halogen, C 1-6 alkyl and C 1-6 alkoxy;
- q is 1;
- R 3 is a hydrogen atom;
- R 4 is Each R 4a is the same or different, and each independently selected from a hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy;
- n is 1 or 2;
- ring C is a 5- to 10-member
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl , -C(O)OCH 3 and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently is a hydrogen atom or a C 1-6 alkyl group;
- p is 0, 1, 2, 3 or 4 ;
- Each R 2 is the same or different, and each independently selected from a hydrogen atom, halogen, C 1-6 alkyl and C 1-6 alkoxy; q is 0, 1, 2, 3 or 4;
- R 3 is hydrogen atom;
- R 4 is Each R 4a is the same or different, and each independently selected from hydrogen atom, hydroxyl,
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl and -C(O)OCH 3 ;
- p is 1, 2 or 3;
- each R 2 is the same or different, and each independently selected from a hydrogen atom, halogen, C 1-6 alkyl and C 1-6 alkoxy;
- q is 1;
- each R 4a is the same or different, and each independently selected from a hydrogen atom, halogen, hydroxyl, C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy;
- n is 1 or 2;
- Ring A is a 6- to 10-membered aryl group;
- Ring B is a 4-
- each R 1 is the same or different, and each independently selected from hydrogen atom, halogen, C 1- 6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently represents a hydrogen atom or C 1-6 alkyl;
- p is 0, 1, 2 or 3;
- each R 2 is the same or different, and each independently is a hydrogen atom or a halogen;
- q is 0, 1 or 2;
- each R 4a is the same or different, and Each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy;
- n is 0, 1, 2 or 3;
- ring A is 6 to 10-membered aryl;
- Ring B is a 4- to 7-membered heterocyclic group;
- each R 1 is the same or different, and each independently selected from hydrogen atom, halogen, C 1- 6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently represents a hydrogen atom or C 1-6 alkyl;
- p is 0, 1, 2 or 3;
- each R 2 is the same or different, and each independently is a hydrogen atom or a halogen;
- q is 0, 1 or 2;
- each R 4a is the same or different, and Each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy;
- n is 0, 1, 2 or 3;
- ring A is 6 to 10-membered aryl;
- ring B is a 4- to 7-membered heterocyclic group;
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl and -C(O)OCH 3 ;
- p is 1, 2 or 3;
- X is O or CH 2 ;
- R c and R d are both hydrogen atoms;
- each R 4a is the same or different, and each independently selected from halogen, hydroxyl , C 1-6 alkyl, C 1-6 hydroxyalkyl and C 1-6 alkoxy, and n is 1 or 2, or R 4a are all hydrogen atoms, and n is 3;
- s is 1 or 2;
- L is a chemical bond or -CH 2 -.
- each R 1 is the same or different, and each independently selected from hydrogen atom, halogen, C 1- 6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently represents a hydrogen atom or C 1-6 alkyl;
- p is 0, 1, 2 or 3;
- X is O or CH 2 ;
- each R c and R d are the same or different, and each independently is a hydrogen atom or a halogen;
- R 4a is a hydrogen atom ;
- n is 3;
- s is 1 or 2; and
- L is a chemical bond.
- the compound represented by general formula (III) or a pharmaceutically acceptable salt thereof wherein each R 1 is the same or different, and each independently selected as C 1-6 alkoxy; p is 2; X is O or CH 2 ; each R c and R d is the same or different, and each independently is a hydrogen atom or a fluorine atom; R 4a is a hydrogen atom; n is 3; s is 1 or 2; L is a chemical bond.
- the compound represented by general formula (III) or a pharmaceutically acceptable salt thereof wherein each R 1 is the same or different, and each independently selected as C 1-6 alkoxy; p is 1 or 2; X is CH 2 ; each of R c and R d is the same or different, and each independently is a hydrogen atom or a fluorine atom; n is 0; s is 1 or 2; and L is a chemical bond.
- each R 1 is the same or different, and each independently selected from hydrogen atom, hydroxyl, halogen, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 hydroxyalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy, C 1-6 alkoxy C 1-6 alkyl and -C(O)OCH 3 ;
- p is 1, 2 or 3;
- X is O or CH 2 ;
- each R 4a is the same or different, and each independently selected from halogen, hydroxyl, C 1-6 alkyl, C 1 -6 hydroxyalkyl and C 1-6 alkoxy, and n is 1 or 2, or R 4a are both hydrogen atoms, and n is 3;
- s is 1 or 2; and L is a chemical bond or -CH 2 -.
- each R 1 is the same or different, and each independently selected from hydrogen atom, halogen, C 1- 6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, C 1-6 haloalkoxy and -NR 7 R 8 ;
- R 7 and R 8 are the same or different, and each independently represents a hydrogen atom or C 1-6 alkyl; p is 0, 1, 2 or 3; X is O or CH 2 ; R 4a are all hydrogen atoms; n is 3; s is 1 or 2;
- the compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof wherein each R 1 is the same or different, and each independently is C 1-6 alkoxy; p is 1 or 2; X is CH 2 ; R 4a are all hydrogen atoms; n is 3; s is 1 or 2; and L is a chemical bond.
- the compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof wherein each R 1 is the same or different, and each independently is C 1-6 alkoxy; p is 2; X is O or CH 2 ; R 4a are all hydrogen atoms; n is 3; s is 1 or 2; and L is a chemical bond.
- the compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof wherein each R 1 is the same or different, and each independently is C 1-6 alkoxy; p is 1 or 2 ; X is CH2; n is 0; s is 1 or 2; and L is a chemical bond.
- Typical compounds of the present disclosure include, but are not limited to:
- Ring B, R 2 , R 3 , R 4 and q are as defined in the general formula (I).
- Ring B, ring C, R 2 , R 4a , q and n are as defined in the general formula (II).
- Ring B, ring C, R 2 , R 4a , q and n are as defined in the general formula (Ii).
- X is halogen , preferably bromine
- Ring A, ring B, R 1 , R 2 , R 3 , L, p and q are as defined in the general formula (I).
- X is halogen , preferably bromine
- Ring A, ring B, R 1 , R 2 , L, p and q are as defined in the general formula (II).
- Another aspect of the present disclosure relates to a compound or a salt thereof represented by general formula (IIIA'):
- X is halogen , preferably bromine
- R 1 , R c , R d , s, p, X and L are as defined in general formula (III).
- X is halogen , preferably bromine
- R 1 , s, p, X and L are as defined in general formula (IV).
- Typical intermediate compounds of the present disclosure include, but are not limited to:
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (I) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- Ring A, ring B, L, R 1 to R 4 , p and q are as defined in the general formula (I).
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (II) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- Ring A, ring B, ring C, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (II).
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (Ii) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- Ring A, ring B, ring C, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (Ii).
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (III) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- L, X, R 1 , R 4a , R c , R d , p, n and s are as defined in general formula (III).
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- a compound represented by general formula (IVA) or a salt thereof reacts with a compound represented by general formula (IIIB) or a salt thereof to obtain a compound represented by general formula (IV) or a pharmaceutically acceptable salt thereof,
- X, L, R 1 , R 4a , p, n and s are as defined in general formula (IV).
- Another aspect of the present disclosure relates to a method for preparing a compound represented by general formula (Ii) or a pharmaceutically acceptable salt thereof, the method comprising the following steps:
- Ring D is a 3- to 8-membered heterocyclic group containing at least one double bond in the ring, preferably a 5-membered or 6-membered heterocyclic group containing at least one double bond in the ring, more preferably
- Ring C is a 3- to 8-membered heterocyclic group, preferably a 5-membered or 6-membered heterocyclic group, more preferably
- X is halogen , preferably bromine
- Ring A, ring B, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (Ii).
- compositions which contains the general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) of the present disclosure Or a compound shown in Table A or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable carriers, diluents or excipients.
- the present disclosure further relates to compounds shown in general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or their pharmaceutically acceptable salts or including their Use of the pharmaceutical composition of the invention in the preparation of a medicament for inhibiting KAT; wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure further relates to compounds shown in general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or their pharmaceutically acceptable salts or including their
- the pharmaceutical composition of the invention is used in the preparation of medicines for treating and/or preventing KAT-mediated diseases; wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure further relates to compounds shown in general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or their pharmaceutically acceptable salts or including their The purposes of the pharmaceutical composition in the preparation of the medicament for treating and/or preventing cancer;
- said cancer is preferably selected from lung cancer (such as NCSLC, SCLC), mesothelioma, bone cancer, pancreatic cancer, skin cancer, head and neck cancer Cancer, brain cancer, melanoma, anal cancer, liver cancer, breast cancer, fallopian tube cancer, endometrial cancer, cervical cancer, ovarian cancer, vaginal cancer, vulvar cancer, Hodgkin's lymphoma, esophagus cancer, colorectal cancer, small bowel cancer Cancer, stomach cancer, thyroid cancer, parathyroid cancer, adrenal cancer, soft tissue sarcoma, penile cancer, testicular cancer, prostate cancer, leukemia, B cell lymphoma, bladder cancer, urethral cancer, ure
- the present disclosure further relates to a method for inhibiting KAT, which comprises administering an effective amount of general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) to the required patient Or a compound shown in Table A or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising it, wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure also relates to a method for treating and/or preventing KAT-mediated diseases, which comprises administering a therapeutically effective dose and/or a preventively effective dose of general formula (I), general formula (II), general formula ( Ii), the compound shown in general formula (III), general formula (IV) or table A or its pharmaceutically acceptable salt, or the pharmaceutical composition comprising it, wherein said KAT is preferably KAT6, more preferably KAT6A and / or KAT6B, .
- the present disclosure further relates to a method for treating and/or preventing cancer, which comprises administering a therapeutically effective amount and/or a preventively effective amount of general formula (I), general formula (II), general formula (Ii), general formula
- said cancer is preferably selected from lung cancer (such as NCSLC, SCLC), intermediate Skin tumor, bone cancer, pancreatic cancer, skin cancer, head and neck cancer, brain cancer, melanoma, anal cancer, liver cancer, breast cancer, fallopian tube cancer, endometrial cancer, cervical cancer, ovarian cancer, vaginal cancer, vulvar cancer, hormonal cancer Oddkin's lymphoma, esophageal cancer, colorectal cancer, small bowel cancer, gastric cancer, thyroid cancer, parathyroid cancer, adrenal cancer, soft tissue sarcoma, penile cancer, testicular cancer, prostate cancer, leukemia, B-cell lymph
- the present disclosure further relates to a compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising it, for use as a medicament.
- the present disclosure further relates to a compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or a pharmaceutically acceptable salt thereof, Or a pharmaceutical composition comprising it, which is used as a drug for inhibiting KAT, wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure further relates to a compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or a pharmaceutically acceptable salt thereof, Or a pharmaceutical composition comprising the same, which is used for inhibiting KAT, wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure further relates to a compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or a pharmaceutically acceptable salt thereof, Or a pharmaceutical composition comprising it, which is used as a KAT inhibitor, wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure also relates to the compound shown in general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or its pharmaceutically acceptable salt, or A pharmaceutical composition comprising it, which is used for treating and/or preventing KAT-mediated diseases, wherein the KAT is preferably KAT6, more preferably KAT6A and/or KAT6B.
- the present disclosure further relates to a compound represented by general formula (I), general formula (II), general formula (Ii), general formula (III), general formula (IV) or Table A or a pharmaceutically acceptable salt thereof, Or a pharmaceutical composition comprising it, for the treatment and/or prevention of cancer; wherein said cancer is preferably selected from lung cancer (such as NCSLC, SCLC), mesothelioma, bone cancer, pancreatic cancer, skin cancer, head and neck cancer, brain cancer Cancer, melanoma, anal cancer, liver cancer, breast cancer, fallopian tube cancer, endometrial cancer, cervical cancer, ovarian cancer, vaginal cancer, vulvar cancer, Hodgkin's lymphoma, esophagus cancer, colorectal cancer, small bowel cancer, stomach cancer , thyroid cancer, parathyroid cancer, adrenal cancer, soft tissue sarcoma, penile cancer, testicular cancer, prostate cancer, leukemia, B cell lymphoma, bladder cancer, urethral cancer, ureter
- the KAT6 is KAT6A and/or KAT6B.
- the cancer is breast cancer.
- the breast cancer is ER + breast cancer.
- the breast cancer is ER + /HER2 - breast cancer.
- the breast cancer is locally advanced or metastatic ER + /HER2 - breast cancer.
- the lung cancer eg, NCSLC, SCLC
- the lung cancer is non-small cell lung cancer.
- the lung cancer (such as NCSLC, SCLC) is locally advanced or metastatic non-small cell lung cancer.
- the prostate cancer is castration-resistant prostate cancer.
- the prostate cancer is locally advanced or metastatic castration-resistant prostate cancer.
- ER + estrogen receptor positive
- HER2 ⁇ human epidermal growth factor receptor 2 negative
- NSCLC non-small cell lung cancer
- CRPC castration-resistant Prostate cancer
- the active compounds are prepared in a form suitable for administration by any suitable route, and the compositions of the present disclosure are formulated by conventional methods using one or more pharmaceutically acceptable carriers. Accordingly, the active compounds of the present disclosure may be formulated in various dosage forms for oral administration, injection (eg, intravenous, intramuscular or subcutaneous), administration by inhalation or insufflation.
- the disclosed compounds can also be formulated into dosage forms such as tablets, hard or soft capsules, aqueous or oily suspensions, emulsions, injections, dispersible powders or granules, suppositories, lozenges or syrups.
- the active compound is preferably presented in unit dose form, or in such a form that the patient can self-administer it as a single dose.
- a unit dosage form of a compound or composition of the present disclosure may be presented as a tablet, capsule, cachet, bottle, powder, granule, lozenge, suppository, reconstituted powder or liquid.
- a suitable unit dosage may be from 0.1 to 1000 mg.
- the pharmaceutical composition of the present disclosure may contain one or more auxiliary materials selected from the following components: fillers (diluents), binders, wetting agents, disintegrants or excipients wait.
- auxiliary materials selected from the following components: fillers (diluents), binders, wetting agents, disintegrants or excipients wait.
- the compositions may contain from 0.1 to 99% by weight of active compound.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients suitable for the manufacture of tablets.
- excipients may be inert excipients, granulating agents, disintegrants, binders and lubricants.
- These tablets may be uncoated or may be coated by known techniques to mask the taste of the drug or to delay disintegration and absorption in the gastrointestinal tract, thus providing sustained release over an extended period of time.
- Oral formulations can also be provided in soft gelatin capsules, wherein the active ingredient is mixed with an inert solid diluent, or where the active ingredient is mixed with a water-soluble carrier or an oil vehicle.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients are suspending, dispersing or wetting agents. Aqueous suspensions may also contain one or more preservatives, one or more coloring agents, one or more flavoring agents and one or more sweetening agents.
- Oily suspensions can be formulated by suspending the active ingredient in a vegetable or mineral oil.
- the oily suspensions may contain a thickening agent.
- Sweetening and flavoring agents as mentioned above may be added to provide a palatable preparation. These compositions can be preserved by adding antioxidants.
- compositions of the present disclosure may also be in the form of oil-in-water emulsions.
- the oily phase may be vegetable oil, or mineral oil or mixtures thereof.
- Suitable emulsifiers may be naturally occurring phospholipids, and the emulsions may also contain sweetening agents, flavoring agents, preservatives and antioxidants.
- Such formulations may also contain a demulcent, a preservative, coloring agents and antioxidants.
- compositions of the present disclosure may be in the form of sterile injectable aqueous solutions.
- acceptable vehicles or solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- the sterile injectable preparation may be a sterile injectable oil-in-water microemulsion in which the active ingredient is dissolved in an oily phase.
- the injection or microemulsion may be injected into the patient's bloodstream by local bulk injection.
- solutions and microemulsions are preferably administered in a manner that maintains a constant circulating concentration of the disclosed compounds.
- a continuous intravenous delivery device can be used.
- An example of such a device is the Deltec CADD-PLUS.TM. Model 5400 IV pump.
- compositions of the present disclosure may be in the form of sterile injectable aqueous or oily suspensions for intramuscular and subcutaneous administration.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension prepared in a parenterally acceptable non-toxic diluent or solvent.
- sterile fixed oils are conveniently employed as a solvent or suspending medium. For this purpose, any blended and fixed oil may be used.
- fatty acids are also used in the preparation of injectables.
- the disclosed compounds may be administered in the form of suppositories for rectal administration.
- These pharmaceutical compositions can be prepared by mixing the drug with a suitable non-irritating excipient which is solid at ordinary temperatures but liquid in the rectum and will therefore melt in the rectum to release the drug.
- Aqueous suspensions of dispersible powders and granules can be prepared by the addition of water to administer the disclosed compounds.
- These pharmaceutical compositions can be prepared by mixing the active ingredient with a dispersing or wetting agent, suspending agent or one or more preservatives.
- the dosage of the drug to be administered depends on many factors, including but not limited to the following factors: the activity of the specific compound used, the age of the patient, the weight of the patient, the state of health of the patient, the behavior of the patient , patient's diet, administration time, administration method, rate of excretion, combination of drugs, severity of disease, etc.; in addition, the optimal treatment mode such as the mode of treatment, the daily dosage of the compound or the content of the pharmaceutically acceptable saltkinds can be validated against traditional treatment regimens.
- alkyl refers to a saturated aliphatic hydrocarbon group comprising 1 to 20 (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 , 16, 17, 18, 19 or 20) carbon atoms straight or branched (ie C 1-20 alkyl), preferably containing 1 to 12 (eg 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12) carbon atoms (i.e. C 1-20 alkyl), more preferably an alkyl group containing 1 to 6 carbon atoms (i.e. C 1- 6 alkyl).
- Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, n-pentyl, 1,1-dimethylpropyl, 1 ,2-Dimethylpropyl, 2,2-Dimethylpropyl, 1-ethylpropyl, 2-methylbutyl, 3-methylbutyl, n-hexyl, 1-ethyl-2- Methylpropyl, 1,1,2-trimethylpropyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 2,2-dimethylbutyl, 1,3 - Dimethylbutyl, 2-ethylbutyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 2,3-dimethylbutyl, n-heptyl, 2 -Methylhexyl, 3-methylhexyl, 4-methylhex
- Alkyl groups may be substituted or unsubstituted, and when substituted, they may be substituted at any available point of attachment, the substituents being preferably selected from D atoms, halogen, alkoxy, haloalkyl, haloalkoxy, One or more of cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- alkylene refers to a saturated straight or branched chain aliphatic hydrocarbon group, which is a residue derived from the same carbon atom or two different carbon atoms of a parent alkane by removing two hydrogen atoms, which has 1 to 20 (e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20) carbon atoms (i.e. C 1-20 alkylene), preferably 1 to 12 carbon atoms (ie C 1-12 alkylene), more preferably 1 to 6 carbon atoms (ie C 1-6 alkylene).
- C 1-20 alkylene preferably 1 to 12 carbon atoms (ie C 1-12 alkylene), more preferably 1 to 6 carbon atoms (ie C 1-6 alkylene).
- Non-limiting examples include: methylene (-CH 2 -), 1,1-ethylene (-CH(CH 3 )-), 1,2-ethylene (-CH 2 CH 2 )-, 1,1-propylene (-CH(CH 2 CH 3 )-), 1,2-propylene (-CH 2 CH(CH 3 )-), 1,3-propylene (-CH 2 CH 2 CH 2 -), 1,4-butylene (-CH 2 CH 2 CH 2 CH 2 -), etc.
- the alkylene group may be substituted or unsubstituted, and when substituted, it may be substituted at any available point of attachment, the substituents being preferably selected from D atoms, halogen, alkoxy, haloalkyl, haloalkoxy, One or more of cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- heteroalkylene means that one or more -CH 2 - in the alkylene group is replaced by one or more selected from N, O, S, S(O) and S(O) 2 ; wherein
- the alkyl group is as defined above; the heteroalkylene group can be substituted or unsubstituted, and when substituted, the substituent can be substituted at any available attachment point, and the substituent is preferably selected from D atom, halogen, Alkoxy, haloalkyl, haloalkoxy, cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl, and heteroaryl One or more of the bases.
- alkenyl refers to an alkyl compound containing at least one carbon-carbon double bond in the molecule, wherein the definition of the alkyl group is as described above, and it has 2 to 12 (such as 2, 3, 4, 5, 6, 7, 8 , 9, 10, 11 or 12) carbon atoms (ie C 2-12 alkenyl).
- the alkenyl group is preferably an alkenyl group having 2 to 6 carbon atoms (ie, a C 2-6 alkenyl group).
- Alkenyl may be substituted or unsubstituted, and when substituted, the substituent is preferably selected from the group consisting of alkoxy, halo, haloalkyl, haloalkoxy, cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl
- the substituent is preferably selected from the group consisting of alkoxy, halo, haloalkyl, haloalkoxy, cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl
- radical, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl are examples of radical, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- alkynyl refers to an alkyl compound containing at least one carbon-carbon triple bond in the molecule, wherein the definition of the alkyl group is as described above, and it has 2 to 12 (such as 2, 3, 4, 5, 6, 7, 8 , 9, 10, 11 or 12) carbon atoms (ie C 2-12 alkynyl).
- the alkynyl group is preferably an alkynyl group having 2 to 6 carbon atoms (ie, a C 2-6 alkynyl group).
- Alkynyl may be substituted or unsubstituted, and when substituted, the substituent is preferably selected from the group consisting of alkoxy, halo, haloalkyl, haloalkoxy, cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl
- substituent is preferably selected from the group consisting of alkoxy, halo, haloalkyl, haloalkoxy, cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl
- radical, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl are examples of radical, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- cycloalkyl refers to a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent, and the cycloalkyl ring contains 3 to 20 (such as 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20) carbon atoms (ie 3 to 20 membered cycloalkyl), preferably containing 3 to 12 (eg 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12) carbon atoms (ie 3 to 12 membered cycloalkyl), preferably containing 3 to 8 (eg 3, 4, 5, 6, 7 and 8) carbon atoms ( i.e.
- 3 to 8 membered cycloalkyl further preferably comprising 4 to 7 (eg 4, 5, 6 and 7) carbon atoms (ie 4 to 7 membered cycloalkyl), more preferably comprising 3 to 6 (eg 3, 4, 5 and 6) carbon atoms (ie 3 to 6 membered cycloalkyl).
- Non-limiting examples of monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, cycloheptyl, cycloheptatriene Base, cyclooctyl, etc.; polycyclic cycloalkyl includes spirocycloalkyl, fused cycloalkyl and bridged cycloalkyl.
- spirocycloalkyl refers to 5 to 20 membered (e.g. 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 ring atoms, i.e. 5- to 20-membered spirocycloalkyl), a polycyclic group that shares one carbon atom (called a spiro atom) between monocyclic rings, which may contain one or more double bonds. It is preferably 6 to 14 membered (ie 6 to 14 membered spirocycloalkyl), more preferably 7 to 10 membered (eg 7, 8, 9 or 10 membered, ie 7 to 10 membered spirocycloalkyl).
- the spirocycloalkyl group can be divided into single spirocycloalkyl, double spirocycloalkyl or polyspirocycloalkyl, preferably single spirocycloalkyl and double spirocycloalkyl. More preferably, it is a 3-membered/5-membered, 3-membered/6-membered, 4-membered/4-membered, 4-membered/5-membered, 4-membered/6-membered, 5-membered/5-membered or 5-membered/6-membered monospirocycloalkyl group.
- Non-limiting examples of spirocycloalkyl groups include:
- fused cycloalkyl refers to 5 to 20 membered (e.g. 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 ring atoms, i.e. 5 to 20-membered fused cycloalkyl), an all-carbon polycyclic group in which each ring in the system shares an adjacent pair of carbon atoms with other rings in the system, one or more of which may contain one or more bis key. It is preferably 6 to 14 membered (ie, 6 to 14 membered fused cycloalkyl), more preferably 7 to 10 membered (eg 7, 8, 9 or 10 membered, ie 7 to 10 membered fused cycloalkyl).
- bicyclic, tricyclic, tetracyclic or polycyclic fused cycloalkyl preferably bicyclic or tricyclic, more preferably 3-membered/4-membered, 3-membered/5-membered, 3-membered/6-membered , 4 yuan/4 yuan, 4 yuan/5 yuan, 4 yuan/6 yuan, 5 yuan/4 yuan, 5 yuan/5 yuan, 5 yuan/6 yuan, 6 yuan/3 yuan, 6 yuan/4 yuan, 6 yuan Member/5 member and 6 member/6 member bicyclic fused cycloalkyl.
- fused cycloalkyl groups include:
- bridged cycloalkyl refers to 5 to 20 membered (e.g. 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 carbon atoms, i.e. 5- to 20-membered bridged cycloalkyl), any two rings share two carbon atoms not directly connected to the full-carbon polycyclic group, which may contain one or more double bonds.
- it is 6 to 14 membered (ie, 6 to 14 membered bridged cycloalkyl group), more preferably 7 to 10 membered (eg 7, 8, 9 or 10 membered, ie 7 to 10 membered bridged cycloalkyl group).
- bridged cycloalkyl groups preferably bicyclic, tricyclic or tetracyclic, more preferably bicyclic or tricyclic.
- bridged cycloalkyl groups include:
- the cycloalkyl ring includes a cycloalkyl group as described above (including monocyclic cycloalkyl, spirocycloalkyl, fused cycloalkyl and bridged cycloalkyl) fused to an aryl, heteroaryl or heterocycloalkane
- a radical ring where the ring attached to the parent structure is a cycloalkyl, non-limiting examples include etc.; preferably
- Cycloalkyl may be substituted or unsubstituted and when substituted it may be substituted at any available point of attachment, the substituents are preferably selected from halogen, alkyl, alkoxy, haloalkyl, haloalkoxy , cycloalkyloxy, heterocyclyloxy, hydroxyl, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl, and heteroaryl.
- alkoxy refers to -O-(alkyl), wherein alkyl is as defined above.
- alkoxy include: methoxy, ethoxy, propoxy and butoxy.
- Alkoxy may be optionally substituted or unsubstituted, and when substituted it is preferably one or more of the following groups independently selected from D atom, halogen, alkoxy, haloalkyl, haloalkoxy , cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- heterocyclyl refers to a saturated or partially unsaturated monocyclic or polycyclic ring substituent comprising 3 to 20 ring atoms (e.g. 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 ring atoms, i.e. 3 to 20 membered heterocyclyl), wherein one or more ring atoms are heteroatoms selected from nitrogen, oxygen and sulfur,
- the sulfur can be optionally oxoated (ie to form a sulfoxide or sulfone), excluding the -O-O-, -O-S- or -S-S- ring moieties, the remaining ring atoms being carbon.
- ring atoms ie 3 to 12 membered heterocyclyl
- 1 to 4 eg 1 , 2, 3 and 4
- 3 to 8 ring atoms such as 3, 4, 5, 6, 7 and 8, ie 3 to 8 membered heterocyclyl
- 1-3 for example 1, 2 and 3
- 4 to 7 ring atoms for example 4, 5, 6 and 7, i.e. 4 to 7 membered heterocyclyl
- 1-3 e.g. 1, 2 and 3
- heterocyclyl 3, 4, 5 and 6, i.e. 3 to 6 membered heterocyclyl), wherein 1-3 (e.g. 1 , 2 and 3) are heteroatoms; most preferably contain 5 or 6 ring atoms (ie 5 or 6 membered heterocyclyl), of which 1-2 (eg 1, 2) are heteroatoms.
- monocyclic heterocyclyl groups include pyrrolidinyl, tetrahydropyranyl, 1,2,3,6-tetrahydropyridyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholine base, homopiperazinyl, etc.
- Polycyclic heterocyclyls include spiroheterocyclyls, fused heterocyclyls and bridged heterocyclyls.
- spiroheterocyclyl refers to 5 to 20 membered (e.g. 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 ring atoms, i.e. 5- to 20-membered spiro heterocyclic group), a polycyclic heterocyclic group that shares one atom (called spiro atom) between monocyclic rings, wherein one or more ring atoms are heteroatoms selected from nitrogen, oxygen and sulfur, so
- the sulfur described above can be optionally oxoated (ie to form a sulfoxide or sulfone), with the remaining ring atoms being carbon. It may contain one or more double bonds.
- the spiroheterocyclyl is 6-14 membered (ie 6-14 membered spiroheterocyclyl), more preferably 7-10 membered (eg 7, 8, 9 or 10 membered, ie 7-10 membered spiroheterocyclyl).
- the spiroheterocyclyl can be divided into single spiroheterocyclyl, double spiroheterocyclyl or polyspiroheterocyclyl, preferably single spiroheterocyclyl and double spiroheterocyclyl.
- spiroheterocyclyls include:
- fused heterocyclyl refers to 5 to 20 membered (e.g. 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20 ring atoms, i.e. 5 to 20-membered fused heterocyclic group), each ring in the system shares an adjacent pair of atoms with other rings in the system, and one or more rings may contain one or more double bonds,
- One or more of the ring atoms are heteroatoms selected from nitrogen, oxygen and sulfur, said sulfur being optionally oxoated (ie to form sulfoxides or sulfones), and the remaining ring atoms are carbon.
- it is 6 to 14 membered (ie 6 to 14 membered fused heterocyclic group), more preferably 7 to 10 membered (eg 7, 8, 9 or 10 membered, ie 7 to 10 membered fused heterocyclic group).
- bicyclic, tricyclic, tetracyclic or polycyclic fused heterocyclic groups preferably bicyclic or tricyclic, more preferably 3-membered/4-membered, 3-membered/5-membered, 3-membered/6-membered , 4 yuan/4 yuan, 4 yuan/5 yuan, 4 yuan/6 yuan, 5 yuan/4 yuan, 5 yuan/5 yuan, 5 yuan/6 yuan, 6 yuan/3 yuan, 6 yuan/4 yuan, 6 yuan 5-membered and 6-membered/6-membered bicyclic condensed heterocyclic groups.
- fused heterocyclic groups include:
- bridged heterocyclyl refers to 5 to 14 membered (eg 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 ring atoms, i.e. 5 to 14 membered bridged heterocyclyl), any two A polycyclic heterocyclic group in which two rings share two atoms not directly connected, which may contain one or more double bonds, wherein one or more ring atoms are heteroatoms selected from nitrogen, oxygen and sulfur, said Sulfur can be optionally oxoated (ie to form a sulfoxide or sulfone), with the remaining ring atoms being carbon.
- it can be divided into bicyclic, tricyclic, tetracyclic or polycyclic bridged heterocyclic groups, preferably bicyclic, tricyclic or tetracyclic, more preferably bicyclic or tricyclic.
- bridged heterocyclyl groups include:
- the heterocyclyl ring includes a heterocyclyl as described above (including monocyclic heterocyclyl, spiro heterocyclyl, fused heterocyclyl and bridged heterocyclyl) fused to an aryl, heteroaryl or cycloalkyl On the ring, where the ring attached to the parent structure is a heterocyclyl, non-limiting examples of which include:
- the heterocyclyl group may be substituted or unsubstituted and when substituted it may be substituted at any available point of attachment, the substituents are preferably selected from halogen, alkyl, alkoxy, haloalkyl, haloalkoxy , cycloalkyloxy, heterocyclyloxy, hydroxyl, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl, and heteroaryl.
- aryl refers to a 6 to 14 membered (e.g. 6, 7, 8, 9, 10, 11, 12, 13 or 14 ring atoms, i.e. 6 to 14 membered aryl) all group having a conjugated ⁇ electron system.
- Carbon monocyclic or fused polycyclic (fused polycyclic rings are rings sharing adjacent pairs of carbon atoms) groups, preferably 6 to 10 membered (ie 6 to 10 membered aryl), eg phenyl and naphthyl.
- the aryl ring includes an aryl ring as described above fused to a heteroaryl, heterocyclyl or cycloalkyl ring, wherein the ring bonded to the parent structure is an aryl ring, non-limiting examples of which include :
- Aryl may be substituted or unsubstituted, and when substituted, it may be substituted at any available point of attachment, the substituents being preferably selected from halogen, alkyl, alkoxy, haloalkyl, haloalkoxy, One or more of cycloalkyloxy, heterocyclyloxy, hydroxy, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl and heteroaryl.
- heteroaryl refers to a group containing 1 to 4 (eg 1, 2, 3 and 4) heteroatoms, 5 to 14 ring atoms (eg 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 ring atoms, ie 5 to 14 membered heteroaryl), wherein the heteroatoms are selected from oxygen, sulfur and nitrogen.
- Heteroaryl is preferably 5 to 10 membered (e.g. 5, 6, 7, 8, 9 or 10 membered, i.e. 5 to 10 membered heteroaryl), more preferably 5 or 6 membered (i.e. 5 or 6 membered heteroaryl).
- Aryl such as furyl, thienyl, pyridyl, pyrrolyl, N-alkylpyrrolyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, etc.
- the heteroaryl ring includes a heteroaryl as described above fused to an aryl, heterocyclyl or cycloalkyl ring, wherein the ring attached to the parent structure is a heteroaryl ring, non-limiting examples of which include :
- Heteroaryl may be substituted or unsubstituted and when substituted it may be substituted at any available point of attachment, the substituents are preferably selected from halogen, alkyl, alkoxy, haloalkyl, haloalkoxy , cycloalkyloxy, heterocyclyloxy, hydroxyl, hydroxyalkyl, cyano, amino, nitro, cycloalkyl, heterocyclyl, aryl, and heteroaryl.
- cycloalkyl, heterocyclyl, aryl and heteroaryl groups mentioned above include residues derived by removing one hydrogen atom from a parent ring atom, or residues derived by removing two hydrogen atoms from the same or two different ring atoms of the parent Derivative residues, i.e. "divalent cycloalkyl", “divalent heterocyclyl” (e.g. etc.), "arylene” and "heteroarylene”.
- cycloalkylalkyl refers to an alkyl group substituted by one or more cycloalkyl groups, wherein cycloalkyl and alkyl are as defined above.
- heterocyclylalkyl refers to an alkyl group substituted by one or more heterocyclyl groups, wherein heterocyclyl and alkyl are as defined above.
- heteroarylalkyl refers to an alkyl group substituted by one or more heteroaryl groups, wherein heteroaryl and alkyl are as defined above.
- cycloalkyloxy refers to cycloalkyl-O-, wherein cycloalkyl is as defined above.
- heterocyclyloxy refers to heterocyclyl-O-, wherein heterocyclyl is as defined above.
- alkylthio refers to alkyl-S-, wherein alkyl is as defined above.
- haloalkyl refers to an alkyl group substituted with one or more halogens, wherein alkyl is as defined above.
- haloalkoxy refers to an alkoxy group substituted with one or more halogens, wherein alkoxy group is as defined above.
- alkoxyalkyl refers to an alkyl group substituted by one or more alkoxy groups, wherein alkyl and alkoxy group are as defined above.
- hydroxyalkyl refers to an alkyl group substituted with one or more hydroxy groups, wherein alkyl is as defined above.
- halogen refers to fluorine, chlorine, bromine or iodine.
- hydroxyl refers to -OH.
- mercapto refers to -SH.
- amino refers to -NH2 .
- cyano refers to -CN.
- nitro refers to -NO2 .
- aldehyde refers to -C(O)H
- carboxylate refers to -C(O)O(alkyl), -C(O)O(cycloalkyl)(alkyl)C(O)O- or (cycloalkyl)C(O) O-, wherein alkyl and cycloalkyl are as defined above.
- the disclosed compounds may exist in particular geometric or stereoisomeric forms.
- This disclosure contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers isomers, (D)-isomers, (L)-isomers, and their racemic and other mixtures, such as enantiomerically or diastereomerically enriched mixtures, all of which are within the scope of this disclosure.
- Additional asymmetric carbon atoms may be present in substituents such as alkyl groups. All such isomers, as well as mixtures thereof, are included within the scope of this disclosure.
- Optically active (R)- and (S)-isomers as well as D and L-isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If one enantiomer of a compound of the present disclosure is desired, it can be prepared by asymmetric synthesis or derivatization with chiral auxiliary agents, wherein the resulting diastereomeric mixture is separated and the auxiliary group is cleaved to provide pure desired enantiomer.
- a diastereoisomeric salt is formed with an appropriate optically active acid or base, and then a diastereomeric salt is formed by a conventional method known in the art. Diastereomeric resolution is performed and the pure enantiomers are recovered. Furthermore, the separation of enantiomers and diastereomers is usually accomplished by the use of chromatography using chiral stationary phases, optionally in combination with chemical derivatization methods (e.g. amines to amino groups formate).
- the bond Indicates unassigned configuration, i.e. if chiral isomers exist in the chemical structure, the bond can be or both Two configurations.
- the bond If the configuration is not specified, it can be the Z configuration or the E configuration, or both configurations.
- tautomer or tautomeric form refers to structural isomers of different energies that can interconvert via a low energy barrier.
- proton tautomers also known as prototropic tautomers
- lactam-lactim equilibrium is between A and B as shown below.
- the present disclosure also includes certain isotopically labeled compounds of the disclosure that are identical to those described herein, but wherein one or more atoms are replaced by an atom of an atomic mass or mass number different from that normally found in nature.
- isotopes that can be incorporated into compounds of the present disclosure include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, iodine, and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 13 N, 15 N, 15 O, 17 O, 18 O, 31 P, 32 P, 35 S, 18 F, 123 I, 125 I and 36 Cl, etc.
- the compounds of the present disclosure may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute the compounds.
- compounds can be labeled with radioactive isotopes, such as tritium ( 3 H), and heavy hydrogen can be used to replace hydrogen to form deuterated drugs.
- radioactive isotopes such as tritium ( 3 H)
- heavy hydrogen can be used to replace hydrogen to form deuterated drugs.
- the bond formed by deuterium and carbon is stronger than the bond formed by ordinary hydrogen and carbon.
- Deuterated drugs have the advantages of reducing toxic and side effects, increasing drug stability, enhancing curative effect, and prolonging the biological half-life of drugs. All permutations of isotopic composition of the disclosed compounds, whether radioactive or not, are included within the scope of the present disclosure.
- substitution with heavier isotopes such as deuterium may confer certain therapeutic advantages resulting from greater metabolic stability (e.g. increased in vivo half-life or reduced dosage requirements), and thus in some cases
- deuterium substitution may be partial or complete, partial deuterium substitution meaning at least one hydrogen is replaced by at least one deuterium.
- deuterium when a position is specifically designated as deuterium (D), the position is understood to have an abundance of deuterium (i.e., at least 10 % deuterium incorporation).
- exemplary compounds having a natural abundance greater than deuterium can be at least 1000 times more abundant deuterium, at least 2000 times more abundant deuterium, at least 3000 times more abundant deuterium, at least 4000 times more abundant deuterium, at least 5000 times more abundant deuterium, at least 6000 times more abundant deuterium, or more abundant deuterium.
- the present disclosure also includes various deuterated forms of compounds of formula (I). Each available hydrogen atom attached to a carbon atom can be independently replaced by a deuterium atom.
- deuterated starting materials can be used in the preparation of deuterated forms of compounds of formula (I), or they can be synthesized using conventional techniques using deuterated reagents, including but not limited to deuterated borane, trideuterated Borane tetrahydrofuran solution, deuterated lithium aluminum hydride, deuterated ethyl iodide and deuterated methyl iodide, etc.
- Optional or “optionally” means that the subsequently described event or circumstance can but need not occur, and that the description includes instances where the event or circumstance occurs or does not occur.
- a heterocyclic group optionally substituted with an alkyl group means that an alkyl group may but need not be present, and the description includes cases where the heterocycle group is substituted with an alkyl group and cases where the heterocycle group is not substituted with an alkyl group .
- Substituted means that one or more hydrogen atoms in a group, preferably 1 to 5, more preferably 1 to 3 hydrogen atoms are independently substituted by a corresponding number of substituents. Possible or impossible substitutions can be determined (by experiment or theory) by those skilled in the art without undue effort. For example, an amino or hydroxyl group with free hydrogen may be unstable when bonded to a carbon atom with an unsaturated (eg, ethylenic) bond.
- “Pharmaceutical composition” means a mixture containing one or more compounds described herein, or a physiologically/pharmaceutically acceptable salt or prodrug thereof, and other chemical components, and other components such as a physiologically/pharmaceutically acceptable carrier and excipients.
- the purpose of the pharmaceutical composition is to promote the administration to the organism, facilitate the absorption of the active ingredient and thus exert biological activity.
- “Pharmaceutically acceptable salt” refers to the salt of the disclosed compound, which is safe and effective when used in mammals, and has proper biological activity. Salts can be prepared separately during the final isolation and purification of the compounds, or by reacting the appropriate group with a suitable base or acid.
- Bases commonly used to form pharmaceutically acceptable salts include inorganic bases, such as sodium hydroxide and potassium hydroxide, and organic bases, such as ammonia. Acids commonly used to form pharmaceutically acceptable salts include inorganic acids as well as organic acids.
- the terms “therapeutically effective amount”, “inhibitory effective amount” or “prophylactically effective amount” refer to the amount of drug or agent that is sufficient to achieve or partially achieve the desired effect.
- the determination of the effective amount varies from person to person, depending on the age and general condition of the recipient, and also depends on the specific active substance. The appropriate effective amount in each case can be determined by those skilled in the art according to routine experiments.
- the term "pharmaceutically acceptable” means those compounds, materials, compositions and/or dosage forms, which are suitable for use in contact with patient tissues without undue toxicity, irritation, allergic reaction or Other problems or complications that have a reasonable benefit/risk ratio and are valid for the intended use.
- the compound represented by general formula (I) of the present disclosure, or the preparation method of pharmaceutically acceptable salt thereof, comprises the following steps:
- L, ring A, ring B, R 1 to R 4 , p and q are as defined in the general formula (I).
- the preparation method of the compound represented by the general formula (II) of the present disclosure, or a pharmaceutically acceptable salt thereof, comprises the following steps:
- Ring A, ring B, ring C, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (II).
- the preparation method of the compound represented by the general formula (Ii) of the present disclosure, or a pharmaceutically acceptable salt thereof, comprises the following steps:
- Ring A, ring B, ring C, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (Ii).
- the preparation method of the compound represented by the general formula (III) of the present disclosure or a pharmaceutically acceptable salt thereof comprises the following steps:
- L, X, R 1 , R 4a , R c , R d , p, n and s are as defined in general formula (III).
- the preparation method of the compound represented by the general formula (IV) of the present disclosure or a pharmaceutically acceptable salt thereof comprises the following steps:
- X, L, R 1 , R 4a , p, n and s are as defined in general formula (IV).
- the preparation method of the compound represented by the general formula (II) of the present disclosure or a pharmaceutically acceptable salt thereof comprises the following steps:
- the first step the compound represented by the general formula (IIA') or its salt and the compound represented by the general formula (IIB') or its salt, under the action of a base (such as n-butyllithium), nucleophilic addition occurs Reaction, obtains the compound or its pharmaceutically acceptable salt shown in general formula (IIa);
- a base such as n-butyllithium
- Second step the compound represented by general formula (IIa) or its salt undergoes chlorination reaction (for example under the effect of PCl or SOCl 2 ), to obtain the compound represented by general formula (IIb) or its pharmaceutically acceptable salt ;
- the third step the compound represented by the general formula (IIb) or its pharmaceutically acceptable salt undergoes a reduction reaction under the action of a metal (such as zinc powder or iron powder) to obtain the compound represented by the general formula (II) or its pharmaceutically acceptable salt the salt used;
- a metal such as zinc powder or iron powder
- X is halogen , preferably bromine
- Ring A, ring B, ring C, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (II).
- the preparation method of the compound represented by the general formula (Ii) of the present disclosure or a pharmaceutically acceptable salt thereof comprises the following steps:
- Ring D is a 3- to 8-membered heterocyclic group containing at least one double bond in the ring, preferably a 5-membered or 6-membered heterocyclic group containing at least one double bond in the ring, more preferably
- Ring C is a 3- to 8-membered heterocyclic group, preferably a 5-membered or 6-membered heterocyclic group, more preferably
- X is halogen , preferably bromine
- Ring A, ring B, L, R 1 , R 2 , R 4a , p, q and n are as defined in the general formula (Ii).
- the bases include organic bases and inorganic bases
- the organic bases include but not limited to triethylamine, pyridine, 3,5-lutidine, N,N- Diisopropylethylamine, n-butyllithium, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, sodium acetate, potassium acetate, sodium tert-butoxide, potassium tert-butoxide or 1,8 -diazabicycloundec-7-ene
- the inorganic bases include but not limited to sodium hydride, potassium phosphate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydroxide, lithium hydroxide and potassium hydroxide ; the base described in scheme one to four, preferably selected from pyridine, bis(trimethylsilyl)amide lithium and 3,5-lutidine; the base described in scheme five, preferably n-butyl Lithium; the base described in scheme six, preferably potassium carbonate.
- the metal catalysts include but are not limited to palladium acetate, tetrakis(triphenylphosphine)palladium, tris(dibenzylideneacetone)dipalladium, 1,1'-bis(diphenylphosphino)bis Ferrocenepalladium(II)chloride, bis(acetonitrile)palladium(II)chloride and palladium on carbon, preferably palladium acetate.
- the ligands include but not limited to triphenylphosphine, tri(o-tolyl)phosphine and 1,1'-binaphthyl-2,2'-bisdiphenylphosphine (BINAP), preferably Triphenylphosphine.
- the reaction temperature is 100-150°C, preferably 120°C.
- the reaction time is 0.5-6 hours, preferably 2-3 hours, more preferably 3 hours.
- the reactions of the above schemes 1 to 6 are preferably carried out in a solvent, and the solvents used include but are not limited to: ethylene glycol dimethyl ether, acetic acid, methanol, ethanol, acetonitrile, n-butanol, toluene, tetrahydrofuran, dichloromethane, petroleum ether , ethyl acetate, n-hexane, dimethyl sulfoxide, 1,4-dioxane, water, N,N-dimethylformamide, N,N-dimethylacetamide, 1,2-di Bromoethane, pyridine and mixtures thereof.
- the solvents used include but are not limited to: ethylene glycol dimethyl ether, acetic acid, methanol, ethanol, acetonitrile, n-butanol, toluene, tetrahydrofuran, dichloromethane, petroleum ether , ethyl acetate,
- NMR nuclear magnetic resonance
- MS mass spectroscopy
- MS was determined with Agilent 1200/1290 DAD-6110/6120 Quadrupole MS liquid mass spectrometer (manufacturer: Agilent, MS model: 6110/6120 Quadrupole MS), waters ACQuity UPLC-QD/SQD (manufacturer: waters, MS Model: waters ACQuity Qda Detector/waters SQ Detector), THERMO Ultimate 3000-Q Exactive (manufacturer: THERMO, MS model: THERMO Q Exactive).
- HPLC High performance liquid chromatography
- Chiral HPLC analysis was performed using an Agilent 1260 DAD high performance liquid chromatograph.
- Chiral preparative chromatography uses a Shimadzu LC-20AP preparative chromatograph.
- the CombiFlash rapid preparation instrument uses Combiflash Rf200 (TELEDYNE ISCO).
- the thin-layer chromatography silica gel plate uses Yantai Huanghai HSGF254 or Qingdao GF254 silica gel plate.
- the specification of the silica gel plate used in thin-layer chromatography (TLC) is 0.15mm-0.2mm, and the specification of thin-layer chromatography separation and purification products is 0.4mm. ⁇ 0.5mm.
- Silica gel column chromatography generally uses Yantai Huanghai silica gel 200-300 mesh silica gel as the carrier.
- the known starting materials of the present disclosure can be adopted or synthesized according to methods known in the art, or can be purchased from ABCR GmbH&Co.KG, Acros Organics, Aldrich Chemical Company, Shaoyuan Technology (Shanghai) Co., Ltd., Darui Chemical Products, Shanghai Titan Technology, Aladdin, Anaiji Chemicals, China National Pharmaceutical Group Co., Ltd., Adamas Reagent Co., Ltd., Sigma-Aldrich (Shanghai) Trading Co., Ltd., Shanghai Pide Pharmaceutical Technology Co., Ltd., Shanghai Haohong Bio Pharmaceutical Technology Co., Ltd., Thermo Fisher Scientific (China) Technology Co., Ltd. and other companies.
- the reactions can all be carried out under an argon atmosphere or a nitrogen atmosphere.
- the argon atmosphere or nitrogen atmosphere means that the reaction bottle is connected to an argon or nitrogen balloon with a volume of about 1 L.
- the hydrogen atmosphere means that the reaction bottle is connected to a hydrogen balloon with a capacity of about 1L.
- the pressurized hydrogenation reaction uses Parr 3916EKX hydrogenation instrument and Qinglan QL-500 hydrogen generator or HC2-SS hydrogenation instrument.
- the hydrogenation reaction is usually vacuumized and filled with hydrogen, and the operation is repeated 3 times.
- the solution refers to an aqueous solution.
- reaction temperature is room temperature, which is 20°C to 30°C.
- the monitoring of the reaction process in the embodiment adopts thin-layer chromatography (TLC), the developer used for reaction, the eluent system of the column chromatography that purifies compound adopts and the developer system of thin-layer chromatography comprise: A: n-hexane/ethyl acetate system, B: dichloromethane/methanol system, the volume ratio of the solvent is adjusted according to the polarity of the compound, and can also be adjusted by adding a small amount of basic or acidic reagents such as triethylamine and acetic acid.
- TLC thin-layer chromatography
- reaction solution was cooled to room temperature, filtered under reduced pressure, the filtrate was diluted with ethyl acetate (500 mL), washed with saturated sodium chloride solution (30 mL ⁇ 5), the obtained organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure , to obtain the title product 1b (36.9 g, yield: 100%).
- the product was directly used in the next reaction without further purification.
- reaction solution was quenched with water (10 mL), extracted with ethyl acetate (50 mL ⁇ 3), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the obtained product was purified by silica gel column chromatography with eluent system A As a residue, the title product If (1.11 g, yield: 94.7%) was obtained.
- reaction solution was quenched with water (5 mL), concentrated under reduced pressure, diluted with ethyl acetate (150 mL), washed with saturated sodium chloride solution (10 mL ⁇ 2).
- the obtained organic phase was dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography with eluent system A to obtain the title product 1 g (800 mg, yield: 71.8%).
- reaction solution was cooled to room temperature, concentrated under reduced pressure, diluted with ethyl acetate (150 mL), washed with saturated sodium chloride solution (20 mL ⁇ 3), and the obtained organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure and used The resulting residue was purified by silica gel column chromatography with eluent system A to give the title product 1j (289 mg, yield: 51.4%).
- reaction solution was cooled to room temperature, quenched with water (1 mL), diluted with ethyl acetate (100 mL), washed with saturated sodium chloride solution (20 mL ⁇ 2), and the obtained organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed Concentrated, and the resulting residue was purified by silica gel column chromatography with eluent system A to obtain the title product 1k (350 mg, yield: 91.9%).
- reaction solution was cooled to room temperature, quenched with water (1 mL), diluted with ethyl acetate (100 mL), washed with saturated sodium chloride solution (50 mL ⁇ 2), and the obtained organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed After concentration, the resulting residue was purified by silica gel column chromatography with eluent system A to give the title product 2d (1.84 g, yield: 99.5%).
- reaction solution was cooled to room temperature, the reaction solution was filtered, the filtrate was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography with eluent system A to obtain the title product 4d (503 mg, yield: 87.2%).
- reaction solution was cooled to room temperature, quenched with water (1 mL), diluted with ethyl acetate (30 mL), washed with saturated sodium chloride solution (30 mL ⁇ 2), combined organic phases, dried over anhydrous sodium sulfate, filtered, and the filtrate was Concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography with eluent system A to obtain the title product 4e (323 mg, yield: 73.2%).
- reaction solution was quenched with saturated sodium bisulfite solution (20mL), washed with saturated sodium chloride solution (60mL ⁇ 2), combined organic phases, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure, and purified by silica gel column chromatography The resulting residue was purified with eluent system A to give the title product 5b (4.8 g, yield: 51.7%).
- reaction solution was cooled to room temperature, quenched with water (5 mL), diluted with ethyl acetate (60 mL), washed with saturated sodium chloride solution (30 mL ⁇ 2), combined organic phases, dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed After concentration, the resulting residue was purified by silica gel column chromatography with eluent system A to give the title product 5c (3.2 g, yield: 73.2%).
- 4-bromo-3-methoxyaniline 11a (2.0 g, 9.90 mmol), 37% aqueous formaldehyde (9.0 g, 97.8 mmol), acetic acid (9.0 g, 150 mmol), acetonitrile (30 mL), room temperature Stir for 30 minutes, add sodium cyanoborohydride (800 mg, 12.7 mmol) under ice-cooling, and stir at room temperature for 16 hours.
- reaction solution was diluted with ethyl acetate (100 mL), then washed with water (100 mL) and saturated aqueous sodium chloride solution (100 mL), respectively, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography with eluent system A to obtain the title Product 14c (2.4 g, 1 NMR showed ⁇ 10% Br in the isopropyl para isomer, overall yield: 77.7%, sent directly to the next step).
- reaction solution was washed with saturated sodium bisulfite solution (20 mL) and saturated sodium chloride solution (20 mL), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography with eluent system A to obtain the title product 17b (390.0 mg, yield: 78.0%).
- 2-Methoxy-4-methylbenzenesulfonyl chloride 22a (78mg, 35mmol) and compound 1n (30mg, 0.12mmol) were dissolved in pyridine (5.0mL), and 4-dimethylaminopyridine (3mg, 0.02mmol ), replacing nitrogen 3 times.
- the reaction solution was microwaved at 120° C. for 3 hours.
- test examples are not meant to limit the scope of the present disclosure.
- Test example 1 KAT6 enzyme activity detection (AlphaScreen method)
- EDTA 0.5M
- pH 8.0 pH 8.0
- RNase-free Thermofisher, AM9260G
- Acetyl-CoA (Ac-CoA, CAYMAN, Cat.No.16160)
- AlphaScreen Streptavidin Donor beads (AlphaScreen Streptavidin Donor beads) 5mg (PerkinElmer, 6760002)
- AlphaScreen Protein A Acceptor beads (AlphaScreen Protein A Acceptor beads), 5mg (PerkinElmer, 6760137M)
- Acetylated-Lysine Antibody (Acetylated-Lysine Antibody#9441) (CST 9441S)
- 1 ⁇ detection buffer 100mM Tris-HCL, pH7.8; 15mM NaCl; 1mM EDTA; 0.01% Tween-20; 1mM DTT; 0.01% m/v ovalbumin.
- KAT enzyme solution 1 ⁇ detection buffer to prepare a final concentration of 1.25nM.
- Ac-CoA and H3 mixed substrate Prepare a mixed substrate of Ac-CoA with a final concentration of 1000 nM and H3 with a final concentration of 55 nM in 1 ⁇ detection buffer.
- Detection reagent AlphaScreen protein A acceptor beads with a final concentration of 8 ng/ ⁇ L prepared in 1 ⁇ detection buffer, AlphaScreen streptavidin donor beads at 8 ng/ ⁇ L, acetylated lysine antibody diluted 1:1500, 100 ⁇ M Anacardic acid.
- the disclosed compound has a good inhibitory effect on KAT6A.
- Test example 2 KAT6B enzyme activity detection (AlphaScreen method)
- Bovine serum albumin (Sanko, A500023-0100)
- Acetyl-CoA (Ac-CoA, CAYMAN, Cat.No.16160)
- AlphaScreen protein A receptor beads 5 mg (PerkinElmer, 6760137M)
- a. 1 ⁇ buffer 2 50mM Tris-HCl, pH7.8; 0.1mM EDTA; 0.01% v/v Tween-20; 1mM DTT; 0.01% m/v bovine serum albumin.
- c.Ac-CoA and H3 mixed substrate buffer 2 to prepare a final concentration of 30nM Ac-CoA and 30nM H3 mixed substrate.
- Detection reagent Prepare AlphaScreen protein A acceptor beads with a final concentration of 8ng/ ⁇ L in buffer 2, AlphaScreen streptavidin donor beads with a final concentration of 8ng/ ⁇ L, acetylated lysine antibody diluted 1:1000, and 100 ⁇ M anacardic acid .
- the disclosed compound has a good inhibitory effect on KAT6B.
- Test example 3 U2OS cell H3K23 acetylation IF detection (Immunofluorescence)
- Bovine serum albumin (BSA) (Sangon Biotech, A500023-0100)
- Triton X-100 (Solarbio, T8200)
- Blocking buffer PBS (Shanghai Yuanpei) + BSA (final concentration 1%) + Triton X-100 (final concentration 0.5%).
- Washing buffer PBS (20 ⁇ PBS diluted to 1 ⁇ PBS)+Tween-20 (final concentration 0.1%).
- Secondary antibody solution goat anti-rabbit IgG (H+L), Superclonal TM recombinant secondary antibody, Alexa Fluor 488 at a dilution ratio of 1:1000, Hoechst 33342 at a dilution ratio of 1:5000, diluted with blocking buffer.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
















































































































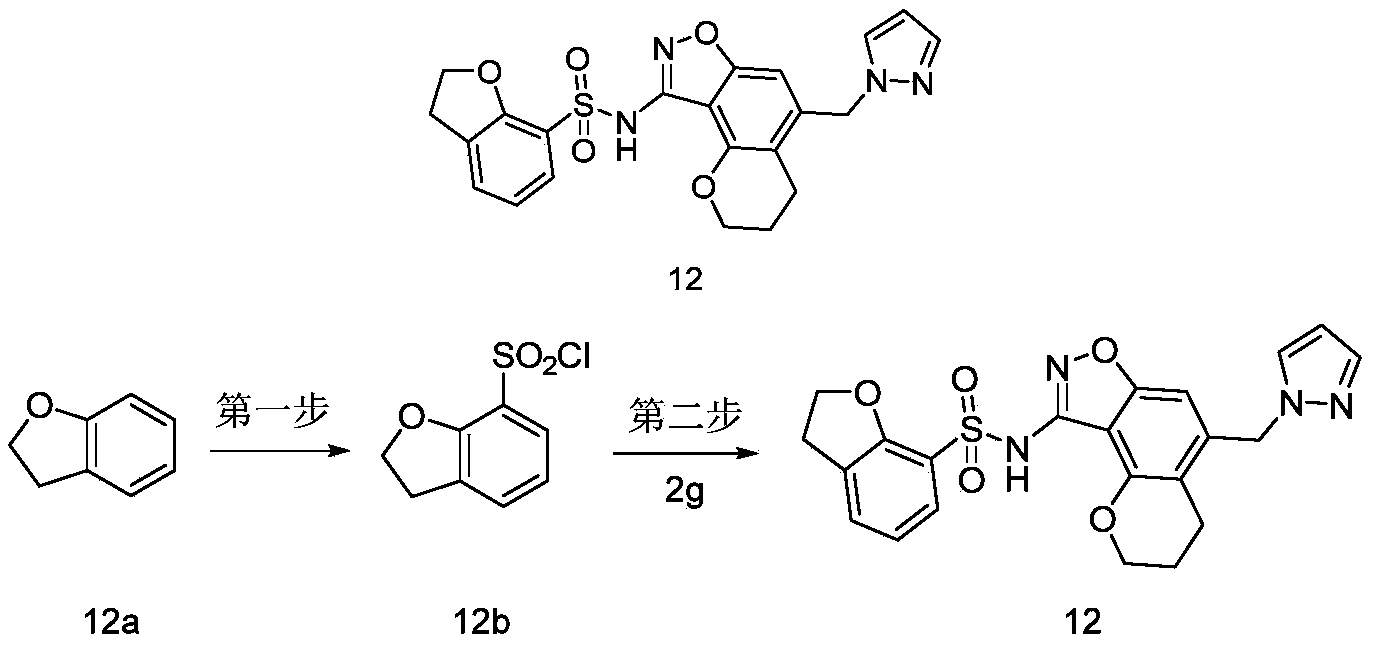

















| 化合物编号 | KAT6A/IC 50(nM) |
| 1 | 0.6 |
| 2 | 0.3 |
| 3 | 0.5 |
| 4 | 4.2 |
| 5 | 1.0 |
| 6 | 0.4 |
| 7 | 0.5 |
| 8 | 0.7 |
| 9 | 0.7 |
| 10 | 0.9 |
| 11 | 1.6 |
| 12 | 12.5 |
| 13 | 0.3 |
| 15 | 1.2 |
| 16 | 1.9 |
| 17 | 10.5 |
| 18 | 12.5 |
| 19 | 2.7 |
| 20 | 2.9 |
| 21 | 4.6 |
| 22 | 4.7 |
| 23 | 4.8 |
| 26 | 12.2 |
| 26-1 | 5.2 |
| 化合物编号 | KAT6B/IC 50(nM) |
| 1 | 1.1 |
| 2 | 0.7 |
| 3 | 1.4 |
| 5 | 1.5 |
| 化合物编号 | H3K23 ac/IC 50(nM) | 最大抑制率/Imax% |
| 1 | 1.1 | 101.8 |
| 2 | 0.4 | 98.5 |
| 3 | 0.9 | 106.3 |
| 4 | 1.5 | 105.5 |
| 5 | 0.6 | 104.2 |
| 7 | 1.4 | 97.4 |
| 8 | 2.8 | 99.4 |
| 9 | 0.75 | 99.5 |
| 10 | 1.2 | 106.3 |
| 11 | 2.1 | 103.1 |
| 20 | 4.6 | 99.4 |
| 22 | 2.9 | 104.8 |
| 化合物编号 | ZR-75-1/IC 50(nM) | 最大抑制率/Imax% |
| 1 | 1.4 | 96 |
| 2 | 0.4 | 96 |
| 3 | 1.9 | 95 |
| 5 | 0.9 | 98 |
| 6 | 0.6 | 95.4 |
| 7 | 1.6 | 95.6 |
| 8 | 2.6 | 96.3 |
| 9 | 1.6 | 96.2 |
| 10 | 2.95 | 94.1 |
| 20 | 8.6 | 94.9 |
| 23 | 10.9 | 90.8 |


Claims (20)
- 一种通式(I)所示的化合物或其可药用的盐:
 其中:环A选自环烷基、杂环基、芳基和杂芳基;环B为环烷基或杂环基;L为化学键、亚烷基或杂亚烷基;其中所述的亚烷基或杂亚烷基各自独立地任选被选自羟基、卤素、烷基、卤代烷基、羟烷基、烷氧基和卤代烷氧基中的一个或多个取代基所取代;各个R 1、各个R 2、R 3相同或不同,且各自独立地选自氢原子、卤素、氰基、硝基、氧代基、烯基、炔基、烷基、烷氧基、环烷基、杂环基、芳基、杂芳基、-OR 5、-C(O)R 6、-C(O)OR 6、-OC(O)R 6、-NHC(O)OR 6、-NR 7R 8、-C(O)NR 7R 8、-S(O) rR 6和-S(O) rNR 7R 8;其中所述的烯基、炔基、烷基、烷氧基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;R 4为氢原子或
其中:环A选自环烷基、杂环基、芳基和杂芳基;环B为环烷基或杂环基;L为化学键、亚烷基或杂亚烷基;其中所述的亚烷基或杂亚烷基各自独立地任选被选自羟基、卤素、烷基、卤代烷基、羟烷基、烷氧基和卤代烷氧基中的一个或多个取代基所取代;各个R 1、各个R 2、R 3相同或不同,且各自独立地选自氢原子、卤素、氰基、硝基、氧代基、烯基、炔基、烷基、烷氧基、环烷基、杂环基、芳基、杂芳基、-OR 5、-C(O)R 6、-C(O)OR 6、-OC(O)R 6、-NHC(O)OR 6、-NR 7R 8、-C(O)NR 7R 8、-S(O) rR 6和-S(O) rNR 7R 8;其中所述的烯基、炔基、烷基、烷氧基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;R 4为氢原子或 环C选自环烷基、杂环基、芳基和杂芳基;R 0选自氢原子、羟基、卤素、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基和环烷基;各个R 4a相同或不同,且各自独立地选自氢原子、羟基、卤素、氰基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基、杂环基氧基和-NR 9R 10;R 5、R 6相同或不同,且各自独立地选自氢原子、烯基、炔基、烷基、环烷基、杂环基、芳基和杂芳基;其中所述的烯基、炔基、烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取 代基所取代;R 7、R 8、R 9、R 10相同或不同,且各自独立地选自氢原子、烷基、环烷基、杂环基、芳基和杂芳基;其中所述的烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;或者,R 7和R 8与相连接的N原子一起形成一个杂环基,或者R 9和R 10与相连接的N原子一起形成一个杂环基,所述的杂环基任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;p为0、1、2、3或4;q为0、1、2、3或4;m为0、1、2、3或4;n为0、1、2、3或4;且r为0、1或2。
环C选自环烷基、杂环基、芳基和杂芳基;R 0选自氢原子、羟基、卤素、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基和环烷基;各个R 4a相同或不同,且各自独立地选自氢原子、羟基、卤素、氰基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基、杂环基氧基和-NR 9R 10;R 5、R 6相同或不同,且各自独立地选自氢原子、烯基、炔基、烷基、环烷基、杂环基、芳基和杂芳基;其中所述的烯基、炔基、烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取 代基所取代;R 7、R 8、R 9、R 10相同或不同,且各自独立地选自氢原子、烷基、环烷基、杂环基、芳基和杂芳基;其中所述的烷基、环烷基、杂环基、芳基和杂芳基各自独立地任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;或者,R 7和R 8与相连接的N原子一起形成一个杂环基,或者R 9和R 10与相连接的N原子一起形成一个杂环基,所述的杂环基任选被选自羟基、卤素、氰基、氨基、硝基、氧代基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环基烷基、环烷基氧基和杂环基氧基中的一个或多个取代基所取代;p为0、1、2、3或4;q为0、1、2、3或4;m为0、1、2、3或4;n为0、1、2、3或4;且r为0、1或2。 - 根据权利要求1所述的通式(I)所示的化合物或其可药用的盐,其中环C为芳基或杂芳基。
- 根据权利要求1或2所述的通式(I)所示的化合物或其可药用的盐,其中R 3为氢原子。
- 根据权利利要求1至3中任一项所述的通式(I)所示的化合物或其可药用的盐,其中R 4为环C为5至10元杂芳基,优选为5元或6元杂芳基;R 4a和n如权利要求1中所定义。

- 根据权利要求1至4中任一项所述的通式(I)所示的化合物或其可药用的盐,其为通式(II)所示的化合物或其可药用的盐:
 其中:环C为5至10元杂芳基,优选为5元或6元杂芳基;环A、环B、L、R 1、R 2、R 4a、p、q和n如权利要求1中所定义。
其中:环C为5至10元杂芳基,优选为5元或6元杂芳基;环A、环B、L、R 1、R 2、R 4a、p、q和n如权利要求1中所定义。 - 根据权利要求1至5中任一项所述的通式(I)所示的化合物或其可药用的盐,其中环A为6至10元芳基,优选选自苯基、

- 根据权利要求1至6中任一项所述的通式(I)所示的化合物或其可药用的盐,其中环B为4至7元杂环基。
- 根据权利要求1至7中任一项所述的通式(I)所示的化合物或其可药用的盐,其为通式(III)所示的化合物或其可药用的盐:
 其中:X选自O、CR aR b和C=O;各个R a、R b、R c和R d相同或不同,且各自独立地选自氢原子、羟基、卤素、氰基、氨基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基氧基和杂环基氧基;或者,R c、R d与所连碳原子一起形成一个C=O;s为0、1、2或3;L、R 1、R 4a、p和n如权利要求1中所定义。
其中:X选自O、CR aR b和C=O;各个R a、R b、R c和R d相同或不同,且各自独立地选自氢原子、羟基、卤素、氰基、氨基、烯基、炔基、烷基、卤代烷基、羟烷基、烷氧基、卤代烷氧基、环烷基、杂环基、芳基、杂芳基、环烷基氧基和杂环基氧基;或者,R c、R d与所连碳原子一起形成一个C=O;s为0、1、2或3;L、R 1、R 4a、p和n如权利要求1中所定义。 - 根据权利要求8所述的通式(I)所示的化合物或其可药用的盐,其中R c和R d相同或不同,且各自独立地为氢原子或卤素;和/或X为O或CH 2;和/或s为1或2。
- 根据权利要求1至9中任一项所述的通式(I)所示的化合物或其可药用的盐,其中各个R 1相同或不同,且各自独立地选自氢原子、羟基、卤素、C 1-6烷基、C 1-6卤代烷基、C 1-6羟烷基、C 1-6烷氧基、C 1-6卤代烷氧基、C 1-6烷氧基C 1-6烷基和-C(O)OCH 3和-NR 7R 8;R 7和R 8相同或不同,且各自独立地为氢原子或C 1-6烷基;优选地,各个R 1相同或不同,且各自独立地选自氢原子、卤素、C 1-6烷基、C 1-6 卤代烷基、C 1-6烷氧基、C 1-6卤代烷氧基和-NR 7R 8;R 7和R 8相同或不同,且各自独立地为氢原子或C 1-6烷基;更优选地,各个R 1相同或不同,且各自独立地为C 1-6烷氧基。
- 根据权利要求1至7和10中任一项所述的通式(I)所示的化合物或其可药用的盐,其中各个R 2相同或不同,且各自独立地选自氢原子、卤素、C 1-6烷基和C 1-6烷氧基,优选为氢原子或卤素。
- 根据权利要求1至11中任一项所述的通式(I)所示的化合物或其可药用的盐,其中各个R 4a相同或不同,且各自独立地选自氢原子、羟基、卤素、C 1-6烷基、C 1-6羟烷基和C 1-6烷氧基,优选为氢原子。
- 根据权利要求1至12中任一项所述的通式(I)所示的化合物或其可药用的盐,其中L为化学键或-CH 2-,优选为化学键。
- 根据权利要求1至13中任一项所述的通式(I)所示的化合物或其可药用的盐,其选自如下化合物:


- 一种通式(IA)所示的化合物或其盐:
 其中:环B、R 2、R 3、R 4和q如权利要求1中所定义。
其中:环B、R 2、R 3、R 4和q如权利要求1中所定义。 - 化合物或其盐,选自以下化合物或其盐:

- 一种制备根据权利要求1所述的通式(I)所示的化合物或其可药用的盐的方法,所述方法包括:
 通式(IA)所示的化合物或其盐与通式(IB)所示的化合物或其盐反应,得到通式(I)所示的化合物或其可药用的盐,其中:环A、环B、L、R 1至R 4、p和q如权利要求1中所定义。
通式(IA)所示的化合物或其盐与通式(IB)所示的化合物或其盐反应,得到通式(I)所示的化合物或其可药用的盐,其中:环A、环B、L、R 1至R 4、p和q如权利要求1中所定义。 - 一种药物组合物,所述药物组合物含有根据权利要求1至14中任一项所述的通式(I)所示的化合物或其可药用的盐,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
- 根据权利要求1至14中任一项所述的通式(I)所示的化合物或其可药用的盐或根据权利要求18所述的药物组合物在制备用于抑制KAT的药物中的用途;其中所述KAT优选为KAT6,更优选为KAT6A和/或KAT6B。
- 根据权利要求1至14中任一项所述的通式(I)所示的化合物或其可药用的盐或根据权利要求18所述的药物组合物在制备用于治疗和/或预防癌症的药物中的用途,其中所述的癌症优选选自肺癌、间皮瘤、骨癌、胰腺癌、皮肤癌、头颈癌、脑癌、黑色素瘤、肛门癌、肝癌、乳腺癌、输卵管癌、子宫内膜癌、宫颈癌、卵巢癌、阴道癌、外阴癌、霍奇金淋巴瘤、食道癌、结直肠癌、小肠癌、胃癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、阴茎癌、睾丸癌、前列腺癌、白血病、B细胞淋巴瘤、膀胱癌、尿道癌、输尿管癌、肾细胞癌、肾盂癌、中枢神经系统肿瘤(CNS)、原发性CNS淋巴瘤、脊髓肿瘤、胶质细胞瘤、脑胶质瘤、垂体腺瘤和鳞状细胞癌;更优选选自乳腺癌、前列腺癌、肺癌、胰腺癌、卵巢癌、宫颈癌、子宫内膜癌、膀胱癌、脑胶质瘤、B细胞淋巴瘤、肝癌和白血病;其中所述乳腺癌优选为ER +乳腺癌或ER +/HER2 -乳腺癌;其中所述肺癌优选为非小细胞肺癌;其中所述前列腺癌优选为去势抵抗性前列腺癌。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22855464.8A EP4385989A1 (en) | 2021-08-10 | 2022-08-10 | Sulfonamide derivative, preparation method therefor and medical use thereof |
| JP2024507166A JP2024529068A (ja) | 2021-08-10 | 2022-08-10 | スルファミド誘導体、その調製方法及びその医薬的使用 |
| CN202280046780.6A CN117597341A (zh) | 2021-08-10 | 2022-08-10 | 磺酰胺衍生物、其制备方法及其在医药上的应用 |
| CA3228411A CA3228411A1 (en) | 2021-08-10 | 2022-08-10 | Sulfonamide derivative, preparation method therefor and medical use thereof |
| KR1020247007144A KR20240046530A (ko) | 2021-08-10 | 2022-08-10 | 설폰아미드 유도체, 이의 제조 방법 및 이의 의학적 용도 |
| AU2022325367A AU2022325367A1 (en) | 2021-08-10 | 2022-08-10 | Sulfonamide derivative, preparation method therefor and medical use thereof |
| MX2024001777A MX2024001777A (es) | 2021-08-10 | 2022-08-10 | Derivado de sulfonamida, metodo de preparacion y uso medico del mismo. |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110913249.1 | 2021-08-10 | ||
| CN202110913249 | 2021-08-10 | ||
| CN202111142090 | 2021-09-28 | ||
| CN202111142090.4 | 2021-09-28 | ||
| CN202111500231.5 | 2021-12-09 | ||
| CN202111500231 | 2021-12-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023016484A1 true WO2023016484A1 (zh) | 2023-02-16 |
Family
ID=85200573
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/111395 WO2023016484A1 (zh) | 2021-08-10 | 2022-08-10 | 磺酰胺衍生物、其制备方法及其在医药上的应用 |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP4385989A1 (zh) |
| JP (1) | JP2024529068A (zh) |
| KR (1) | KR20240046530A (zh) |
| CN (1) | CN117597341A (zh) |
| AU (1) | AU2022325367A1 (zh) |
| CA (1) | CA3228411A1 (zh) |
| MX (1) | MX2024001777A (zh) |
| TW (1) | TW202321263A (zh) |
| WO (1) | WO2023016484A1 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023114710A1 (en) * | 2021-12-13 | 2023-06-22 | Aurigene Oncology Limited | Fused benzoisoxazolyl compounds as kat6a inhibitors |
| US11976075B2 (en) | 2022-03-28 | 2024-05-07 | Isosterix, Inc. | Inhibitors of the MYST family of lysine acetyl transferases |
| WO2024165035A1 (zh) * | 2023-02-10 | 2024-08-15 | 江苏恒瑞医药股份有限公司 | 一种磺酰胺衍生物结晶形式及其制备方法 |
| US12091406B2 (en) | 2021-11-16 | 2024-09-17 | Insilico Medicine Ip Limited | Lysine acetyltransferase 6A (KAT6A) inhibitors and uses thereof |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1065267A (zh) * | 1991-03-28 | 1992-10-14 | 美国辉瑞有限公司 | 杂环环胺衍生物 |
| US20020028834A1 (en) * | 1991-03-28 | 2002-03-07 | Anabella Villalobos | Heterocyclic-cyclic amine derivatives |
| CN1639146A (zh) * | 2002-02-22 | 2005-07-13 | 拜尔药品公司 | 用于治疗过度增生疾病的稠合三环杂环 |
| CN101287493A (zh) * | 2005-08-18 | 2008-10-15 | 根马布股份公司 | 采用cd4结合肽和辐射的疗法 |
| WO2016198507A1 (en) | 2015-06-09 | 2016-12-15 | Monash University | Aryl sulfonohydrazides |
| WO2017218960A1 (en) | 2016-06-17 | 2017-12-21 | Fronthera U.S. Pharmaceuticals Llc | Hemoglobin modifier compounds and uses thereof |
| WO2019043139A1 (en) | 2017-08-31 | 2019-03-07 | Ctxt Pty Limited | FUSED [1,2,4] THIADIAZINE DERIVATIVES AS KAT INHIBITORS OF THE MYST FAMILY |
| WO2019108824A1 (en) | 2017-11-29 | 2019-06-06 | Epizyme, Inc. | Myst family histone acetyltransferase inhibitors |
| WO2019243491A1 (en) | 2018-06-20 | 2019-12-26 | Ctxt Pty Limited | Compounds |
| WO2020002587A1 (en) | 2018-06-28 | 2020-01-02 | Ctxt Pty Limited | Compounds |
| WO2020048548A1 (zh) * | 2018-09-07 | 2020-03-12 | 正大天晴药业集团股份有限公司 | 一种作用于crbn蛋白的三并环类化合物 |
| WO2020069322A1 (en) | 2018-09-28 | 2020-04-02 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| WO2020216701A1 (en) | 2019-04-25 | 2020-10-29 | Bayer Aktiengesellschaft | Acyl sulfonamides for treating cancer |
| WO2020254946A1 (en) | 2019-06-18 | 2020-12-24 | Pfizer Inc. | Benzisoxazole sulfonamide derivatives |
| WO2020254989A1 (en) | 2019-06-19 | 2020-12-24 | Pfizer Inc. | Cycloalkyl and heterocycloalkyl benzisoxazole sulfonamide derivatives |
-
2022
- 2022-08-10 JP JP2024507166A patent/JP2024529068A/ja active Pending
- 2022-08-10 EP EP22855464.8A patent/EP4385989A1/en active Pending
- 2022-08-10 CN CN202280046780.6A patent/CN117597341A/zh active Pending
- 2022-08-10 MX MX2024001777A patent/MX2024001777A/es unknown
- 2022-08-10 AU AU2022325367A patent/AU2022325367A1/en active Pending
- 2022-08-10 WO PCT/CN2022/111395 patent/WO2023016484A1/zh active Application Filing
- 2022-08-10 CA CA3228411A patent/CA3228411A1/en active Pending
- 2022-08-10 KR KR1020247007144A patent/KR20240046530A/ko unknown
- 2022-08-10 TW TW111130097A patent/TW202321263A/zh unknown
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1065267A (zh) * | 1991-03-28 | 1992-10-14 | 美国辉瑞有限公司 | 杂环环胺衍生物 |
| US20020028834A1 (en) * | 1991-03-28 | 2002-03-07 | Anabella Villalobos | Heterocyclic-cyclic amine derivatives |
| CN1639146A (zh) * | 2002-02-22 | 2005-07-13 | 拜尔药品公司 | 用于治疗过度增生疾病的稠合三环杂环 |
| CN101287493A (zh) * | 2005-08-18 | 2008-10-15 | 根马布股份公司 | 采用cd4结合肽和辐射的疗法 |
| WO2016198507A1 (en) | 2015-06-09 | 2016-12-15 | Monash University | Aryl sulfonohydrazides |
| WO2017218960A1 (en) | 2016-06-17 | 2017-12-21 | Fronthera U.S. Pharmaceuticals Llc | Hemoglobin modifier compounds and uses thereof |
| WO2019043139A1 (en) | 2017-08-31 | 2019-03-07 | Ctxt Pty Limited | FUSED [1,2,4] THIADIAZINE DERIVATIVES AS KAT INHIBITORS OF THE MYST FAMILY |
| WO2019108824A1 (en) | 2017-11-29 | 2019-06-06 | Epizyme, Inc. | Myst family histone acetyltransferase inhibitors |
| WO2019243491A1 (en) | 2018-06-20 | 2019-12-26 | Ctxt Pty Limited | Compounds |
| WO2020002587A1 (en) | 2018-06-28 | 2020-01-02 | Ctxt Pty Limited | Compounds |
| WO2020048548A1 (zh) * | 2018-09-07 | 2020-03-12 | 正大天晴药业集团股份有限公司 | 一种作用于crbn蛋白的三并环类化合物 |
| WO2020069322A1 (en) | 2018-09-28 | 2020-04-02 | Praxis Precision Medicines, Inc. | Ion channel modulators |
| WO2020216701A1 (en) | 2019-04-25 | 2020-10-29 | Bayer Aktiengesellschaft | Acyl sulfonamides for treating cancer |
| WO2020254946A1 (en) | 2019-06-18 | 2020-12-24 | Pfizer Inc. | Benzisoxazole sulfonamide derivatives |
| WO2020254989A1 (en) | 2019-06-19 | 2020-12-24 | Pfizer Inc. | Cycloalkyl and heterocycloalkyl benzisoxazole sulfonamide derivatives |
Non-Patent Citations (1)
| Title |
|---|
| J. MED. CHEM., vol. 53, 2010, pages 7035 - 7047 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12091406B2 (en) | 2021-11-16 | 2024-09-17 | Insilico Medicine Ip Limited | Lysine acetyltransferase 6A (KAT6A) inhibitors and uses thereof |
| WO2023114710A1 (en) * | 2021-12-13 | 2023-06-22 | Aurigene Oncology Limited | Fused benzoisoxazolyl compounds as kat6a inhibitors |
| US11976075B2 (en) | 2022-03-28 | 2024-05-07 | Isosterix, Inc. | Inhibitors of the MYST family of lysine acetyl transferases |
| WO2024165035A1 (zh) * | 2023-02-10 | 2024-08-15 | 江苏恒瑞医药股份有限公司 | 一种磺酰胺衍生物结晶形式及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN117597341A (zh) | 2024-02-23 |
| EP4385989A1 (en) | 2024-06-19 |
| CA3228411A1 (en) | 2023-02-16 |
| JP2024529068A (ja) | 2024-08-01 |
| MX2024001777A (es) | 2024-02-29 |
| TW202321263A (zh) | 2023-06-01 |
| AU2022325367A1 (en) | 2024-02-08 |
| KR20240046530A (ko) | 2024-04-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023016484A1 (zh) | 磺酰胺衍生物、其制备方法及其在医药上的应用 | |
| CN111902417B (zh) | 一种二芳基巨环化合物、药物组合物以及其用途 | |
| KR20180096823A (ko) | 신규한 소염제 | |
| WO2020200069A1 (zh) | 吡咯并杂环类衍生物、其制备方法及其在医药上的应用 | |
| WO2022105852A1 (zh) | 三嗪二酮类衍生物、其制备方法及其在医药上的应用 | |
| TW201934554A (zh) | 吡唑并[1,5-a][1,3,5]三<img align="absmiddle" height="18px" width="27px" file="d10999.TIF" alt="其他非圖式 d10999.TIF" img-content="tif" orientation="portrait" inline="yes"></img>-2-胺類衍生物、其製備方法及其在醫藥上的應用 | |
| WO2022247920A1 (zh) | 喹啉胺类化合物、其制备方法及其在医药上的应用 | |
| CN115594695A (zh) | 大环类化合物、其制备方法及其在医药上的应用 | |
| CN111094254B (zh) | 杂芳基类衍生物、其制备方法及其在医药上的应用 | |
| WO2020221209A1 (zh) | 一种cd73抑制剂,其制备方法和应用 | |
| WO2023066371A1 (zh) | 含氮的四环化合物、其制备方法及其在医药上的应用 | |
| WO2023072297A1 (zh) | 含氮的四环化合物、其制备方法及其在医药上的应用 | |
| CN115611877A (zh) | 磺酰胺类化合物、其制备方法及其在医药上的应用 | |
| WO2023030335A1 (zh) | 作为tyk2/jak1假激酶结构域抑制剂的化合物及合成和使用方法 | |
| TW201823231A (zh) | 氮雜雙環基取代的三唑類衍生物、其製備方法及其在醫藥上的應用 | |
| CN112996783A (zh) | 2-氨基嘧啶类衍生物、其制备方法及其在医药上的应用 | |
| CN112341434B (zh) | PI3K/mTOR蛋白降解靶向嵌合体类化合物及其制备方法和医药用途 | |
| WO2024140754A1 (zh) | 一种萘酰胺类化合物、其制备方法及其应用 | |
| WO2023011581A1 (zh) | 含氮杂环化合物、其制备方法及其在医药上的应用 | |
| WO2022161447A1 (zh) | 二甲酰胺类化合物、其制备方法及其在医药上的应用 | |
| WO2023186058A1 (zh) | 吲唑类化合物、其制备方法及其在医药上的应用 | |
| WO2024104398A1 (zh) | 吡啶氮氧化物类衍生物及其药物组合物、制备方法和用途 | |
| WO2022135360A1 (zh) | 嘌呤酮衍生物、其制备方法及其在医药上的应用 | |
| TW202333698A (zh) | 喹啉胺類化合物、其製備方法及其在醫藥上的應用 | |
| CN118344364A (zh) | 氨基取代的稠合杂芳基类化合物、其制备方法及其在医药上的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22855464 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280046780.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022325367 Country of ref document: AU Ref document number: AU2022325367 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2401000743 Country of ref document: TH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024507166 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3228411 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2022325367 Country of ref document: AU Date of ref document: 20220810 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112024002510 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202490333 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20247007144 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202400353T Country of ref document: SG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202427016473 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022855464 Country of ref document: EP Effective date: 20240311 |
|
| ENP | Entry into the national phase |
Ref document number: 112024002510 Country of ref document: BR Kind code of ref document: A2 Effective date: 20240207 |