WO2022244841A1 - 脳毛細血管内皮様細胞の製造方法およびその利用 - Google Patents
脳毛細血管内皮様細胞の製造方法およびその利用 Download PDFInfo
- Publication number
- WO2022244841A1 WO2022244841A1 PCT/JP2022/020827 JP2022020827W WO2022244841A1 WO 2022244841 A1 WO2022244841 A1 WO 2022244841A1 JP 2022020827 W JP2022020827 W JP 2022020827W WO 2022244841 A1 WO2022244841 A1 WO 2022244841A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- capillary endothelial
- medium
- brain capillary
- brain
- Prior art date
Links
- 210000004781 brain capillary Anatomy 0.000 title claims abstract description 87
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 55
- 210000004027 cell Anatomy 0.000 claims abstract description 307
- 238000000034 method Methods 0.000 claims abstract description 90
- 210000000130 stem cell Anatomy 0.000 claims abstract description 39
- 238000012258 culturing Methods 0.000 claims abstract description 31
- 230000003511 endothelial effect Effects 0.000 claims abstract description 30
- 230000002792 vascular Effects 0.000 claims abstract description 28
- 102000004266 Collagen Type IV Human genes 0.000 claims abstract description 17
- 108010042086 Collagen Type IV Proteins 0.000 claims abstract description 17
- 108010067306 Fibronectins Proteins 0.000 claims abstract description 17
- 239000012634 fragment Substances 0.000 claims abstract description 16
- 108010038862 laminin 10 Proteins 0.000 claims abstract description 15
- 230000004069 differentiation Effects 0.000 claims description 145
- 238000012360 testing method Methods 0.000 claims description 57
- 230000014509 gene expression Effects 0.000 claims description 55
- 230000004888 barrier function Effects 0.000 claims description 52
- 239000000126 substance Substances 0.000 claims description 51
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 claims description 50
- 230000001939 inductive effect Effects 0.000 claims description 46
- 230000008499 blood brain barrier function Effects 0.000 claims description 39
- 210000001218 blood-brain barrier Anatomy 0.000 claims description 39
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 38
- 210000003668 pericyte Anatomy 0.000 claims description 37
- 210000004556 brain Anatomy 0.000 claims description 32
- 210000002889 endothelial cell Anatomy 0.000 claims description 26
- 230000035699 permeability Effects 0.000 claims description 22
- 230000000694 effects Effects 0.000 claims description 21
- 102100037362 Fibronectin Human genes 0.000 claims description 16
- 101001116302 Homo sapiens Platelet endothelial cell adhesion molecule Proteins 0.000 claims description 13
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 claims description 13
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 claims description 11
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 11
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 11
- 239000013589 supplement Substances 0.000 claims description 9
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 4
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 claims 2
- 230000001976 improved effect Effects 0.000 abstract description 24
- 102000016359 Fibronectins Human genes 0.000 abstract 1
- 239000002609 medium Substances 0.000 description 147
- 239000011248 coating agent Substances 0.000 description 60
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 48
- 230000006698 induction Effects 0.000 description 42
- 239000010410 layer Substances 0.000 description 39
- 108090000623 proteins and genes Proteins 0.000 description 39
- 102000004169 proteins and genes Human genes 0.000 description 36
- 239000000306 component Substances 0.000 description 35
- 238000010586 diagram Methods 0.000 description 30
- 101100328892 Arabidopsis thaliana COL4 gene Proteins 0.000 description 25
- 101100237842 Xenopus laevis mmp18 gene Proteins 0.000 description 25
- 230000002441 reversible effect Effects 0.000 description 25
- 238000001727 in vivo Methods 0.000 description 21
- 238000003501 co-culture Methods 0.000 description 20
- 238000012744 immunostaining Methods 0.000 description 19
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 18
- 238000004458 analytical method Methods 0.000 description 18
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 17
- 108010085895 Laminin Proteins 0.000 description 17
- 238000000576 coating method Methods 0.000 description 16
- 239000003550 marker Substances 0.000 description 16
- 238000004113 cell culture Methods 0.000 description 15
- 210000004379 membrane Anatomy 0.000 description 14
- 239000012528 membrane Substances 0.000 description 14
- 238000010195 expression analysis Methods 0.000 description 13
- 238000011156 evaluation Methods 0.000 description 12
- 230000006870 function Effects 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 101001017818 Homo sapiens ATP-dependent translocase ABCB1 Proteins 0.000 description 11
- 230000003203 everyday effect Effects 0.000 description 10
- 108020004999 messenger RNA Proteins 0.000 description 10
- 238000010899 nucleation Methods 0.000 description 10
- 210000002966 serum Anatomy 0.000 description 10
- 108010081589 Becaplermin Proteins 0.000 description 9
- 108010078791 Carrier Proteins Proteins 0.000 description 9
- 102000058063 Glucose Transporter Type 1 Human genes 0.000 description 9
- 108091006296 SLC2A1 Proteins 0.000 description 9
- 230000002708 enhancing effect Effects 0.000 description 9
- 230000003278 mimic effect Effects 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 102100022595 Broad substrate specificity ATP-binding cassette transporter ABCG2 Human genes 0.000 description 8
- 101000823298 Homo sapiens Broad substrate specificity ATP-binding cassette transporter ABCG2 Proteins 0.000 description 8
- 101000896557 Homo sapiens Eukaryotic translation initiation factor 3 subunit B Proteins 0.000 description 8
- 101000988834 Homo sapiens Hypoxanthine-guanine phosphoribosyltransferase Proteins 0.000 description 8
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 8
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 description 8
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 8
- 230000012292 cell migration Effects 0.000 description 8
- 230000000052 comparative effect Effects 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- DLBFLQKQABVKGT-UHFFFAOYSA-L lucifer yellow dye Chemical compound [Li+].[Li+].[O-]S(=O)(=O)C1=CC(C(N(C(=O)NN)C2=O)=O)=C3C2=CC(S([O-])(=O)=O)=CC3=C1N DLBFLQKQABVKGT-UHFFFAOYSA-L 0.000 description 8
- 102100029761 Cadherin-5 Human genes 0.000 description 7
- 101000794587 Homo sapiens Cadherin-5 Proteins 0.000 description 7
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 7
- 239000007640 basal medium Substances 0.000 description 7
- 239000002771 cell marker Substances 0.000 description 7
- 150000002632 lipids Chemical class 0.000 description 7
- 238000012423 maintenance Methods 0.000 description 7
- 210000001082 somatic cell Anatomy 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 210000003556 vascular endothelial cell Anatomy 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 108020004635 Complementary DNA Proteins 0.000 description 6
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 6
- 108010036949 Cyclosporine Proteins 0.000 description 6
- 239000007760 Iscove's Modified Dulbecco's Medium Substances 0.000 description 6
- 238000010804 cDNA synthesis Methods 0.000 description 6
- 230000005779 cell damage Effects 0.000 description 6
- 208000037887 cell injury Diseases 0.000 description 6
- 229960001265 ciclosporin Drugs 0.000 description 6
- 239000002299 complementary DNA Substances 0.000 description 6
- 229920000159 gelatin Polymers 0.000 description 6
- 238000003384 imaging method Methods 0.000 description 6
- 210000003716 mesoderm Anatomy 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 238000011160 research Methods 0.000 description 6
- 108010010803 Gelatin Proteins 0.000 description 5
- 102000003940 Occludin Human genes 0.000 description 5
- 108090000304 Occludin Proteins 0.000 description 5
- 241000209094 Oryza Species 0.000 description 5
- 235000007164 Oryza sativa Nutrition 0.000 description 5
- 238000011529 RT qPCR Methods 0.000 description 5
- 108010022164 acetyl-LDL Proteins 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 230000002490 cerebral effect Effects 0.000 description 5
- 239000011247 coating layer Substances 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 210000001161 mammalian embryo Anatomy 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 235000009566 rice Nutrition 0.000 description 5
- 210000001578 tight junction Anatomy 0.000 description 5
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 4
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 4
- 102100024032 Linker for activation of T-cells family member 1 Human genes 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- 108091006232 SLC7A5 Proteins 0.000 description 4
- 102000000591 Tight Junction Proteins Human genes 0.000 description 4
- 108010002321 Tight Junction Proteins Proteins 0.000 description 4
- 238000010162 Tukey test Methods 0.000 description 4
- NIJJYAXOARWZEE-UHFFFAOYSA-N Valproic acid Chemical compound CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 4
- 150000001413 amino acids Chemical group 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000012141 concentrate Substances 0.000 description 4
- 238000005138 cryopreservation Methods 0.000 description 4
- 229930182912 cyclosporin Natural products 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 210000001671 embryonic stem cell Anatomy 0.000 description 4
- 238000010230 functional analysis Methods 0.000 description 4
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 4
- 108010082117 matrigel Proteins 0.000 description 4
- 239000012533 medium component Substances 0.000 description 4
- 238000001543 one-way ANOVA Methods 0.000 description 4
- 239000012466 permeate Substances 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 239000011148 porous material Substances 0.000 description 4
- 230000008672 reprogramming Effects 0.000 description 4
- HTJNEBVCZXHBNJ-XCTPRCOBSA-H trimagnesium;(2r)-2-[(1s)-1,2-dihydroxyethyl]-3,4-dihydroxy-2h-furan-5-one;diphosphate Chemical compound [Mg+2].[Mg+2].[Mg+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.OC[C@H](O)[C@H]1OC(=O)C(O)=C1O HTJNEBVCZXHBNJ-XCTPRCOBSA-H 0.000 description 4
- -1 various laminins Proteins 0.000 description 4
- 101150079978 AGRN gene Proteins 0.000 description 3
- 102100040026 Agrin Human genes 0.000 description 3
- 108700019743 Agrin Proteins 0.000 description 3
- 239000012583 B-27 Supplement Substances 0.000 description 3
- AQGNHMOJWBZFQQ-UHFFFAOYSA-N CT 99021 Chemical compound CC1=CNC(C=2C(=NC(NCCNC=3N=CC(=CC=3)C#N)=NC=2)C=2C(=CC(Cl)=CC=2)Cl)=N1 AQGNHMOJWBZFQQ-UHFFFAOYSA-N 0.000 description 3
- 102000004057 Claudin-5 Human genes 0.000 description 3
- 108090000582 Claudin-5 Proteins 0.000 description 3
- 102000012422 Collagen Type I Human genes 0.000 description 3
- 108010022452 Collagen Type I Proteins 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101710103506 Platelet-derived growth factor subunit A Proteins 0.000 description 3
- 239000012980 RPMI-1640 medium Substances 0.000 description 3
- 101150086694 SLC22A3 gene Proteins 0.000 description 3
- 229920004890 Triton X-100 Polymers 0.000 description 3
- 239000013504 Triton X-100 Substances 0.000 description 3
- 210000004102 animal cell Anatomy 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 210000001130 astrocyte Anatomy 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 229940096422 collagen type i Drugs 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 238000007865 diluting Methods 0.000 description 3
- 229940000406 drug candidate Drugs 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- TUFFYSFVSYUHPA-UHFFFAOYSA-M rhodamine 123 Chemical compound [Cl-].COC(=O)C1=CC=CC=C1C1=C(C=CC(N)=C2)C2=[O+]C2=C1C=CC(N)=C2 TUFFYSFVSYUHPA-UHFFFAOYSA-M 0.000 description 3
- 238000009331 sowing Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000010257 thawing Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- GUAHPAJOXVYFON-ZETCQYMHSA-N (8S)-8-amino-7-oxononanoic acid zwitterion Chemical compound C[C@H](N)C(=O)CCCCCC(O)=O GUAHPAJOXVYFON-ZETCQYMHSA-N 0.000 description 2
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 108010052946 Activin Receptors Proteins 0.000 description 2
- 102000018918 Activin Receptors Human genes 0.000 description 2
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000019058 Glycogen Synthase Kinase 3 beta Human genes 0.000 description 2
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 2
- 108700021430 Kruppel-Like Factor 4 Proteins 0.000 description 2
- 102100032352 Leukemia inhibitory factor Human genes 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 241000282579 Pan Species 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 238000002123 RNA extraction Methods 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 238000000692 Student's t-test Methods 0.000 description 2
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 238000010372 cloning stem cell Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 210000002257 embryonic structure Anatomy 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 229960000890 hydrocortisone Drugs 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 238000003125 immunofluorescent labeling Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 2
- 238000012758 nuclear staining Methods 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 238000010827 pathological analysis Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- 230000001012 protector Effects 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 238000012827 research and development Methods 0.000 description 2
- 238000010839 reverse transcription Methods 0.000 description 2
- 229910052711 selenium Inorganic materials 0.000 description 2
- 239000011669 selenium Substances 0.000 description 2
- 239000012128 staining reagent Substances 0.000 description 2
- 230000008093 supporting effect Effects 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 230000002123 temporal effect Effects 0.000 description 2
- 150000003505 terpenes Chemical class 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- VUDQSRFCCHQIIU-UHFFFAOYSA-N 1-(3,5-dichloro-2,6-dihydroxy-4-methoxyphenyl)hexan-1-one Chemical compound CCCCCC(=O)C1=C(O)C(Cl)=C(OC)C(Cl)=C1O VUDQSRFCCHQIIU-UHFFFAOYSA-N 0.000 description 1
- BJHCYTJNPVGSBZ-YXSASFKJSA-N 1-[4-[6-amino-5-[(Z)-methoxyiminomethyl]pyrimidin-4-yl]oxy-2-chlorophenyl]-3-ethylurea Chemical compound CCNC(=O)Nc1ccc(Oc2ncnc(N)c2\C=N/OC)cc1Cl BJHCYTJNPVGSBZ-YXSASFKJSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 102100034135 Activin receptor type-1C Human genes 0.000 description 1
- 108010059616 Activins Proteins 0.000 description 1
- 102000005606 Activins Human genes 0.000 description 1
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 1
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- 102100031734 Fibroblast growth factor 19 Human genes 0.000 description 1
- 101710153349 Fibroblast growth factor 19 Proteins 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 102000011787 Histone Methyltransferases Human genes 0.000 description 1
- 108010036115 Histone Methyltransferases Proteins 0.000 description 1
- 101000799193 Homo sapiens Activin receptor type-1C Proteins 0.000 description 1
- 101100281008 Homo sapiens FGF2 gene Proteins 0.000 description 1
- 101000942967 Homo sapiens Leukemia inhibitory factor Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102100020880 Kit ligand Human genes 0.000 description 1
- 101710177504 Kit ligand Proteins 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100446513 Mus musculus Fgf4 gene Proteins 0.000 description 1
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 1
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 1
- 241000220317 Rosa Species 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000008790 VE-cadherin Human genes 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 108010076089 accutase Proteins 0.000 description 1
- 239000000488 activin Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 101150010487 are gene Proteins 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 210000001109 blastomere Anatomy 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 108010018828 cadherin 5 Proteins 0.000 description 1
- 210000001043 capillary endothelial cell Anatomy 0.000 description 1
- 150000001765 catechin Chemical class 0.000 description 1
- ADRVNXBAWSRFAJ-UHFFFAOYSA-N catechin Natural products OC1Cc2cc(O)cc(O)c2OC1c3ccc(O)c(O)c3 ADRVNXBAWSRFAJ-UHFFFAOYSA-N 0.000 description 1
- 235000005487 catechin Nutrition 0.000 description 1
- 239000012592 cell culture supplement Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000007910 cell fusion Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 229930183167 cerebroside Natural products 0.000 description 1
- 150000001784 cerebrosides Chemical class 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 101150027734 cript gene Proteins 0.000 description 1
- 229920003020 cross-linked polyethylene Polymers 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 235000021186 dishes Nutrition 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 210000003981 ectoderm Anatomy 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000001900 endoderm Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 238000013210 evaluation model Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 210000001654 germ layer Anatomy 0.000 description 1
- 150000002327 glycerophospholipids Chemical class 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000005732 intercellular adhesion Effects 0.000 description 1
- 101150111214 lin-28 gene Proteins 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- ZFLWDHHVRRZMEI-UHFFFAOYSA-N methyl 2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-1,4-dihydropyridine-3-carboxylate Chemical compound COC(=O)C1=C(C)NC(C)=C([N+]([O-])=O)C1C1=CC=CC=C1C(F)(F)F ZFLWDHHVRRZMEI-UHFFFAOYSA-N 0.000 description 1
- 210000004925 microvascular endothelial cell Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- FMURUEPQXKJIPS-UHFFFAOYSA-N n-(1-benzylpiperidin-4-yl)-6,7-dimethoxy-2-(4-methyl-1,4-diazepan-1-yl)quinazolin-4-amine;trihydrochloride Chemical compound Cl.Cl.Cl.C=12C=C(OC)C(OC)=CC2=NC(N2CCN(C)CCC2)=NC=1NC(CC1)CCN1CC1=CC=CC=C1 FMURUEPQXKJIPS-UHFFFAOYSA-N 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 150000008442 polyphenolic compounds Chemical class 0.000 description 1
- 235000013824 polyphenols Nutrition 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 210000001811 primitive streak Anatomy 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000003762 quantitative reverse transcription PCR Methods 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 238000010079 rubber tapping Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 238000010374 somatic cell nuclear transfer Methods 0.000 description 1
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 1
- 150000003408 sphingolipids Chemical class 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images






















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/78—Connective tissue peptides, e.g. collagen, elastin, laminin, fibronectin, vitronectin or cold insoluble globulin [CIG]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
- C07K14/50—Fibroblast growth factor [FGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
Definitions
- the present disclosure relates to a method for producing brain capillary endothelial-like cells.
- This application is based on Japanese Patent Application No. 2021-085121 filed on May 20, 2021, the content of which is incorporated herein.
- BMECs Brain microvascular endothelial cells (BMECs), which are one of the constituent cells of the blood-brain barrier (BBB), release substances through strong intercellular adhesion and the expression of efflux transporters. Inhibits non-specific entry into the brain parenchyma. In drug discovery, this strong barrier function inhibits the migration of drug candidates to the parenchymal (nerve) side of the brain, and drug development may be discontinued. Therefore, a screening model that can evaluate pharmacokinetics in the human BBB is desired.
- iPS cells human induced pluripotent stem cells
- Brain capillary endothelial-like cells obtained by inducing differentiation of human iPS cells using the method described in Patent Document 1 have extremely weak properties as vascular endothelial cells (ECs), and furthermore, as epithelial cells. It is suggested that the properties are different from those of BMECs in the human body because they also have properties.
- the inventors of the present application have found that, in conventional methods, factors such as vascular endothelial growth factor (hereinafter also referred to as "VEGF”) that are essential for differentiation into vascular cells are added. Therefore, it was considered that differentiation into BMECs was induced without going through vascular endothelial progenitor cells (EPCs), which are precursors of BMECs.
- EPCs vascular endothelial progenitor cells
- the inventors of the present application have found that induction of differentiation into BMECs via EPCs is effective in producing cells with improved imitation (similarity) to in vivo BMECs. rice field. Furthermore, the inventors of the present invention have invented a technique that enables the production of brain capillary endothelial-like cells with improved imitation to in vivo BMECs through intensive research.
- the present invention can be realized as the following forms.
- a method for producing brain capillary endothelial-like cells includes a culture step of culturing vascular endothelial progenitor cells using a laminin511 fragment, fibronectin, and collagen type IV. According to this method for producing brain capillary endothelial-like cells, it is possible to produce brain capillary endothelial-like cells that mimic the in vivo brain capillary endothelial cells.
- a medium containing B27 (registered trademark) supplement, A 83-01, and Fibroblast Growth Factor-2 (FGF2) is used. It may be cultured using According to this method for producing brain capillary endothelial-like cells, the barrier function of brain capillary endothelial-like cells can be further improved.
- the vascular endothelial progenitor cells may be cells induced to differentiate from pluripotent stem cells.
- the vascular endothelial progenitor cells may be cells induced to differentiate from human induced pluripotent stem cells. According to this method for producing brain capillary endothelial-like cells, it is possible to easily produce brain capillary endothelial-like cells that mimic brain capillary endothelial cells in the human body.
- the vascular endothelial progenitor cells may be cells induced to differentiate using a vascular endothelial growth factor.
- vascular endothelial progenitor cells are cells that have been induced to differentiate from pluripotent stem cells using vascular endothelial growth factor. It is possible to produce brain capillary endothelial-like cells with improved cell mimicry.
- the vascular endothelial progenitor cells in the culturing step, may be co-cultured with brain pericytes. According to this method for producing brain capillary endothelial-like cells, vascular endothelial progenitor cells are co-cultured with brain pericytes. like cells can be produced.
- the brain pericytes may be cells differentiated from pluripotent stem cells using a medium containing A83-01. According to this method for producing cerebral capillary endothelial-like cells, the use of a medium containing A83-01 can improve mimicking of in vivo cerebral pericytes.
- the cell layer of brain capillary endothelial-like cells obtained by the method for producing brain capillary endothelial-like cells of the above aspect is used to allow a test substance to permeate the blood-brain barrier.
- a method is provided for assessing sexuality. According to this form of method for evaluating blood-brain barrier permeability, since a cell layer of brain capillary endothelial-like cells that mimic brain capillary endothelial cells in vivo is used, the permeability of the test substance through the blood-brain barrier is Evaluation accuracy can be improved.
- the method for evaluating the blood-brain barrier permeability of the test substance in the form described above may include the following steps (i) to (iii): (i) providing the cell layer; (ii) contacting the cell layer with the test substance; (iii) evaluating the blood-brain barrier permeability of the test substance by quantifying the test substance that has permeated the cell layer; According to this method of evaluating the blood-brain barrier permeability of a test substance, the accuracy of evaluation of the blood-brain barrier permeability of the test substance can be further improved.
- the cell layer of brain capillary endothelial-like cells obtained by the method for producing brain capillary endothelial-like cells of the above aspect is used to prepare the blood-brain barrier barrier of the test substance.
- Methods are provided for assessing impact on function.
- a cell layer of brain capillary endothelial-like cells that mimic brain capillary endothelial cells in vivo is used. It is possible to improve the evaluation accuracy of the effect on the blood-brain barrier barrier function.
- a brain capillary endothelial-like cell obtained by the above method for producing a brain capillary endothelial-like cell.
- the PECAM1 expression level is higher than the primary cultured brain capillary endothelial cells and the immortalized brain capillary endothelial cells, and the TEER value is 50 ⁇ cm 2 or more.
- a method for inducing differentiation of brain capillary endothelial-like cells includes a culture step of culturing vascular endothelial progenitor cells using a laminin511 fragment, fibronectin, and collagen type IV. According to this method of inducing differentiation of brain capillary endothelial-like cells, it is possible to obtain brain capillary endothelial-like cells that mimic brain capillary endothelial cells in vivo.
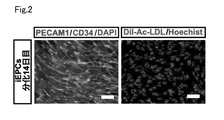
- FIG. 10 is an explanatory diagram showing the results of confirming the properties of iEPCs on day 14 of differentiation.
- Explanatory drawing showing a comparison of electrical resistance values of non-frozen iEPCs and frozen iEPCs.
- Explanatory diagram showing the results of examination of iBMELCs differentiation induction conditions (types of medium components).
- Explanatory diagram showing the results of examination of conditions for inducing differentiation into iBMELCs (concentrations of medium components).
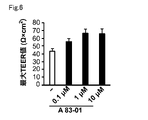
- Explanatory diagram showing the results of examination of conditions for inducing differentiation into iBMELCs concentration of A 83-01).
- Explanatory diagram showing the results of examination of conditions for inducing differentiation into iBMELCs (coating component species).
- Explanatory diagram showing the results of examining conditions for inducing differentiation into iBMELCs (LN511F concentration).
- Explanatory drawing which shows the result of gene expression analysis.
- Explanatory drawing showing a comparison of electrical resistance values between iEPCs and immortalized cells.
- Explanatory diagram showing the results of functional analysis of P-glycoprotein (P-gp).
- Explanatory diagram for explaining the outline of co-culture of iEPCs and iBPCs.
- Explanatory diagram showing changes in TEER values over time due to co-culture of iEPCs and iBPCs.
- Explanatory drawing which shows the result of the immunostaining of the brain pericyte marker protein.
- Explanatory drawing which shows the analysis result of the brain pericyte marker gene expression level.
- Explanatory drawing which shows the result of the immunostaining of the brain pericyte marker protein.
- Explanatory drawing which shows the expression-analysis result of a brain pericyte marker protein.
- Microscopic images showing the cell migration and proliferation abilities of iBPCs.
- Explanatory drawing showing the cell migration ability and
- a method for producing brain microvascular endothelial-like cells (hereinafter also referred to as "BMELCs”) is provided.
- This production method includes a culturing step of culturing vascular endothelial progenitor cells (hereinafter also referred to as "EPCs”) using Laminin511 fragment, Fibronectin, and collagen type IV.
- the EPCs used in the method of the present disclosure may be cells induced to differentiate from pluripotent stem cells, cells derived from the body, or immortalized cells.
- inducing differentiation refers to working to differentiate along a specific cell lineage.
- pluripotency refers to the ability to differentiate into all cells that make up the body (pluripotency), and to produce daughter cells that have the same differentiation potential as self through cell division. It refers to a cell that also has the ability (self-renewal ability).
- Pluripotency can be evaluated by transplanting cells to be evaluated into nude mice and examining the presence or absence of teratoma formation containing cells of each of the three germ layers (ectoderm, mesoderm, and endoderm).
- pluripotent stem cells examples include embryonic stem cells (ES cells), embryonic germ cells (EG cells), induced pluripotent stem cells (iPS cells), and the like. Pluripotent stem cells are not limited to these as long as they are cells having both pluripotency and self-renewal ability. From the standpoint of availability and ethics, it is preferable to use iPS cells as pluripotent stem cells. Mammalian cells are preferably used as pluripotent stem cells. Examples of mammals include, but are not limited to, primates such as humans and chimpanzees, and rodents such as mice and rats. Human cells are more preferably used as pluripotent stem cells. Therefore, it is particularly preferable to use human iPS cells as pluripotent stem cells. By using EPCs induced to differentiate from human iPS cells, it is possible to easily produce BMELCs that mimic human BMECs in vivo.
- EPCs induced to differentiate from human iPS cells it is possible to easily produce BMELCs that mimic human BM
- ES cells can be established, for example, by culturing an early embryo before implantation, an inner cell mass constituting the early embryo, a single blastomere, etc. (Manipulating the Mouse Embryo A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1994); Thomson, J. A. et al., Science, 282, 1145-1147 (1998)).
- an early embryo an early embryo produced by nuclear transfer of the nucleus of a somatic cell may be used (Wilmut et al. (Nature, 385, 810 (1997)), Cibelli et al. (Science, 280, 1256 (1998)), Akira Iriya et al.
- ES cells are available from archives or are commercially available.
- human ES cells are available from Kyoto University Institute for Frontier Medical Sciences (eg, KhES-1, KhES-2 and KhES-3), WiCell Research Institute, ESI BIO, and the like.
- ES cells can be established by culturing primordial germ cells in the presence of LIF, bFGF, SCF, etc. (Matsui et al., Cell, 70, 841-847 (1992), Shamblott et al. USA, 95 (23), 13726-13731 (1998), Turnpenny et al., Stem Cells, 21 (5), 598-609, (2003)).
- iPS cells are cells with pluripotency (multipotency) and proliferative potential, which are produced by reprogramming somatic cells through the introduction of reprogramming factors. iPS cells exhibit properties similar to ES cells. Somatic cells used to generate iPS cells are not particularly limited, and may be differentiated somatic cells or undifferentiated stem cells. In addition, the origin is not particularly limited, but somatic cells of mammals (for example, primates such as humans and chimpanzees, and rodents such as mice and rats), particularly preferably somatic cells of humans, are used. iPS cells can be produced by various methods reported so far. In addition, it is naturally envisioned that an iPS cell production method that will be developed in the future will be applied.
- the most basic method of producing iPS cells is to introduce four transcription factors, Oct3/4, Sox2, Klf4 and c-Myc, into cells using viruses (Takahashi K, Yamanaka S : Cell 126 (4), 663-676, 2006; Takahashi, K, et al.: Cell 131 (5), 861-72, 2007).
- human iPS cells were established by introducing four factors, Oct4, Sox2, Lin28 and Nonog (Yu J, et al: Science 318(5858), 1917-1920, 2007). 3 factors excluding c-Myc (Nakagawa M, et al: Nat. Biotechnol.
- Cells that have undergone transformation to iPS cells, that is, reprogramming (reprogramming), are pluripotent stem cell markers (undifferentiated markers) such as Fbxo15, Nanog, Oct/4, Fgf-4, Esg-1 and Cript. Expression or the like can be selected as an index. Selected cells are collected as iPS cells.
- iPS cells can also be provided, for example, from Kyoto University or the RIKEN BioResource Center, a national research and development agency.
- iPS cells can be provided by, for example, Reprocell.
- Differentiation induction of pluripotent stem cells such as iPS cells into EPCs may be performed using, for example, the method described in International Publication No. 2020/179380 or the method described in JP-A-2018-110548. .
- it includes a step of differentiating pluripotent stem cells into EPCs, and a step of purifying EPCs by utilizing the difference in adhesion ability between EPCs and other cells that make up the cell population obtained in that step. good too.
- pluripotent stem cells are cultured under conditions that induce differentiation into EPCs.
- pluripotent stem cells differentiate into EPCs via mesderm two stages of differentiation induction described below, that is, a step of differentiating pluripotent stem cells into mesoderm, and differentiating the obtained cells into EPCs.
- BMP4 bone morphogenetic factor 4
- VEGF vascular endothelial growth factor
- bFGF vascular endothelial growth factor
- Other culture conditions (culture temperature, etc.) in the step of differentiating pluripotent stem cells into EPCs may be conditions generally employed in animal cell culture. For example, it may be cultured in an environment such as 37°C and 5% CO 2 .
- a basal medium for example, serum-free vascular endothelial cell medium (Human Endothelial-SFM), Dulbecco's modified Eagle medium (D-MEM), Iscove's modified Dulbecco's medium (IMDM), MEM ⁇ medium, Ham F12 medium (HamF12) , Glasgow basal medium, RPMI1640 medium, MCDB107 medium and the like may be used, and two or more basal media may be mixed and used such as a mixed medium such as D-MEM and Ham's F12 medium.
- other arbitrary components such as serum, serum substitutes, antibiotics, and supplements may be added to the medium.
- the differentiation into EPCs can be determined or evaluated, for example, using the expression of vascular endothelial progenitor cell markers as an index.
- Vascular endothelial progenitor cell markers include, for example, PECAM1 (CD31), CD34, CDH5 (VE-Cadherin), FLK1 (VEGFR-2) and the like.
- CD34 is specific to EPCs and is a particularly useful vascular endothelial progenitor cell marker.
- EPCs In the process of purifying EPCs, the ratio of EPCs that make up the cell population is increased. EPCs can be purified by preferentially or selectively detaching and removing other cells with low adhesion ability by utilizing the fact that EPCs exhibit higher adhesion ability than other cells.
- Purified cells may be cultured for maintenance and proliferation, or may be stored until use.
- the storage method may follow a conventional method, for example, using TC protector (DS Pharma Biomedical), Cell Banker (Xenoac), Stem Cell Banker (Xenoac), Cell Reserver One (Nacalai), etc. It may be stored frozen at -80°C or the like. EPCs stocks can be made by cryopreservation.
- a culturing step of culturing EPCs using a Laminin511 fragment, Fibronectin, and collagen type IV (hereinafter also referred to as “step A”) including.
- step A the three components of Laminin511 fragment (hereinafter also referred to as "LN511F"), Fibronectin (hereinafter also referred to as "FBN”), and collagen type IV (hereinafter also referred to as “COL4") are used as a coating agent.
- the coating agent includes a basement membrane component and is used to form a coating layer on a culture vessel for culturing cells.
- a coat layer containing these three components can be formed.
- forming a coat layer is also referred to as “coating”.
- cells are sometimes cultured on a coat layer formed with a coating agent for the purpose of improving cell viability and growth rate, promoting differentiation induction, cell selection, and the like.
- the barrier function of BMELCs can be significantly improved by using LN511F in addition to FBN and COL4.
- Laminin is a heterotrimeric molecule consisting of three subunit chains: ⁇ -chain, ⁇ -chain and ⁇ -chain. Five types of ⁇ -chain, ⁇ 1 to ⁇ 5, three types of ⁇ -chain, ⁇ 1- ⁇ 3, and three types of ⁇ -chain, ⁇ 1- ⁇ 3, are known.
- RNA ⁇ NM_000426, NM_001079823 (Subunit alpha2), NM_002291 (Subunit beta1), NM_002292 (Subunit beta2), NM_000228 (Subunit beta3), NM_001318046, NM_001318047, NM_001318048, NM_007356 (Subunit beta4), NM_002293 ( Subunit gamma 1), NM_005562, NM_018891 (Subunit gamma 2) are known.
- Laminin511 is a laminin molecule consisting of ⁇ 5, ⁇ 1, and ⁇ 1 subunit chains.
- Laminin511 fragment (LN511F) means a fragment (E8 fragment) of Laminin511 corresponding to the integrin binding site.
- LN511F in the present specification also includes proteins that are highly identical to LN511F.
- a protein highly identical to LN511F refers to an amino acid sequence that is 80% or more identical to the amino acid sequence of LN511F in the amino acid sequence that constitutes such a protein.
- proteins with high identity to LN511F also include proteins with longer amino acid sequences than LN511F. From the viewpoint of improving the barrier function of BMELCs, the homology of proteins with high identity to LN511F is preferably 85% or more, more preferably 90% or more, and even more preferably 95% or more. .
- the content of LN511F is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, the coating agent used in step A preferably contains 1 ⁇ g/mL or more, more preferably 5 ⁇ g/mL or more. Preferably, the content is more preferably 10 ⁇ g/mL or more. Moreover, the content of LN511F in the coating agent used in step A is preferably 200 ⁇ g/mL or less, more preferably 100 ⁇ g/mL or less, from the viewpoint of economy.
- the content of FBN is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, the coating agent used in step A preferably contains 10 ⁇ g/mL or more, more preferably 100 ⁇ g/mL or more. preferable.
- the FBN content in the coating agent used in step A is preferably 500 ⁇ g/mL or less, more preferably 200 ⁇ g/mL or less, from the viewpoint of economy.
- the content of COL4 is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, the coating agent used in step A preferably contains 100 ⁇ g/mL or more, more preferably 400 ⁇ g/mL or more. preferable. Moreover, the content of COL4 in the coating agent used in step A is preferably 2000 ⁇ g/mL or less, more preferably 1000 ⁇ g/mL or less, from the viewpoint of economy.
- the total content of the three components LN511F, FBN and COL4 is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, the coating agent used in step A preferably contains a total of 100 ⁇ g / mL or more. A total content of 300 ⁇ g/mL or more is more preferable, and a total content of 500 ⁇ g/mL or more is more preferable.
- the total content of the three components LN511F, FBN and COL4 is preferably 2000 ⁇ g/mL or less in total in the coating agent used in step A from the viewpoint of economy, and is 1000 ⁇ g/mL or less in total. is more preferred.
- the content ratio of LN511F and FBN is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, it is preferably 1:1 to 1:100, and 1:2 to More preferably 1:20.
- the content ratio of LN511F and COL4 is not particularly limited, but from the viewpoint of enhancing the barrier function of BMELCs, it is preferably 1:4 to 1:400, and 1:8 to More preferably 1:80.
- the total content of the three components LN511F, FBN, and COL4 is preferably 20 ng or more, more preferably 100 ng or more, per 1 cm 2 of the area of the culture dish, ie, the coat area of the coat layer.
- the total content of the three components LN511F, FBN, and COL4 is preferably 200 ⁇ g or less, more preferably 40 ⁇ g or less per 1 cm 2 of coating area of the coating layer.
- the culture dish any form of culture dish can be used, such as a petri dish or a cell culture plate having a plurality of wells.
- the coating agent and the coat layer may contain other optional components within the range that does not impair the effects of the invention. Examples of optional components include, but are not limited to, vitronectin, various laminins, agrin, gelatin, poly-L-lysine, and the like.
- the above step A can be performed using various media.
- the medium include serum-free vascular endothelial cell medium (Human Endothelial-SFM), Dulbecco's Modified Eagle Medium (D-MEM), Iscove's Modified Dulbecco's Medium (IMDM), MEM ⁇ medium, Ham F12 medium (HamF12), Glasgow Basic media such as basal medium, RPMI1640 medium and MCDB107 medium can be mentioned.
- As a medium two or more basal mediums may be mixed and used.
- a medium containing B27 (registered trademark) supplement hereinafter also referred to as "B27s"
- B27s registered trademark
- FGF2 Fibroblast Growth Factor-2
- B27s is a cell culture supplement available from Thermo Fisher Scientific. From the viewpoint of enhancing the barrier function of BMELCs, the B27s content in the medium used in step A is preferably 0.5% or more, more preferably 2.5% or more, and 7% or more. is more preferred. In addition, the content of B27s in the medium used in step A is preferably 15% or less, more preferably 10% or less, from the viewpoint of economy.
- a 83-01 is a type of TGF- ⁇ inhibitor. More specifically, A83-01 is a TGF- ⁇ type I/activin receptor-like kinase (ALK5), type I activin/nodal receptor-like kinase (ALK4), type I nodal receptor-like kinase (ALK7) It is a selective inhibitor. From the viewpoint of enhancing the barrier function of BMELCs, A 83-01 is preferably contained in the medium used in step A at 10 nM or more, more preferably at 0.1 ⁇ M or more. Moreover, the content of A 83-01 in the medium used in step A is preferably 100 ⁇ M or less, more preferably 10 ⁇ M or less, from the viewpoint of economy.
- FGF2 means fibroblast growth factor 2 and is also called basic fibroblast growth factor (bFGF).
- the FGF2 may be human FGF2, such as human recombinant FGF2.
- FGF2 is preferably contained in the medium used in step A at 1 ng/mL or more, more preferably at 10 ng/mL or more.
- the content of FGF2 in the medium used in step A is preferably 10 ⁇ g/mL or less, more preferably 1 ⁇ g/mL or less, from the viewpoint of economy.
- the medium used in step A may contain other optional components as long as they do not interfere with the effects of the invention.
- Optional ingredients include, but are not limited to, serum (fetal bovine serum, human serum, sheep serum, etc.), serum replacements (Knockout serum replacement (KSR), etc.), antibiotics (penicillin, streptomycin, etc.), supplements ( ITS-G supplement), L-glutamine, L-ascorbic acid phosphate magnesium salt n-hydrate, non-essential amino acids (NEAA), 2-mercaptoethanol, Chemically Defined Lipid Concentrate (Gibco), etc. can.
- the culture period in step A is not particularly limited, but may be, for example, 1 to 10 days, preferably 2 to 5 days.
- This culture period can suppress the deterioration of the efficiency of inducing differentiation into BMELCs, and can enhance the barrier function of BMELCs.
- subculture may be performed during differentiation induction. For example, when the cells become confluent or subconfluent, a part of the cells may be collected and transferred to another culture container to continue the culture.
- pre-treat the cells with a ROCK inhibitor Ra-associated coiled-coil forming kinase/Rho-binding kinase
- Y-27632 ROCK inhibitor
- culture temperature may be those generally employed in the culture of animal cells.
- it may be cultured in an environment of 37°C and 5% CO 2 .
- three-dimensional culture may be performed using a three-dimensional culture plate or the like including a coating layer coated with the coating agent, without being limited to the method of two-dimensionally culturing cells using a culture dish or the like.
- the method for producing BMELCs in the present disclosure may include a step of inducing differentiation of pluripotent stem cells into vascular endothelial progenitor cells (hereinafter also referred to as “step B”) before the culture step (step A). . Further, step B may be a step of inducing differentiation of pluripotent stem cells into vascular endothelial progenitor cells using vascular endothelial growth factor.
- EPCs may be co-cultured with brain pericytes (hereinafter also referred to as "BPCs").
- brain pericytes also called pericytes, constitute the blood-brain barrier together with brain capillary endothelial cells.
- co-culture of EPCs and BPCs means a state in which EPCs and BPCs are cultured together.
- EPCs and BPCs may be present in the same medium (culture solution) or in different media (culture solutions).
- EPCs and BPCs may be in contact with each other or may be in non-contact, preferably in non-contact.
- the non-contact state is, for example, a state in which EPCs and BPCs are separated from each other on the surface side and the back side via a microporous support membrane or the like, or a state in which the EPCs and BPCs are separated from each other in the medium via a support This corresponds to a state in which they exist separately separated from each other.
- the support is not particularly limited, but may be composed of, for example, a support film and a support.
- the support is a member for supporting EPCs and BPCs and fixing the support membrane to the culture vessel.
- the support may in particular be a top hat-like shaped cell culture insert.
- the support membrane is preferably microporous, and the pore size is preferably such that cells cannot pass through, but the protrusions of cells, medium, etc. can pass through.
- the supporting membrane may be made of any material that does not interfere with the maintenance, survival, or differentiation induction of cells.
- BPCs used for co-culture with EPCs may be cells induced to differentiate from pluripotent stem cells, cells derived from the body, or immortalized cells. From the viewpoint of availability, it is preferable to use cells induced to differentiate from pluripotent stem cells as BPCs, and from the viewpoint of ethics, it is more preferable to use iPS cells.
- the method for producing BMELCs in the present disclosure may include a step of inducing differentiation of pluripotent stem cells into brain pericytes (hereinafter also referred to as "step C") before the culture step (step A).
- pluripotent stem cells are cultured under conditions that induce differentiation into BPCs.
- two-step differentiation induction described below may be performed so that pluripotent stem cells differentiate into BPCs via mesderm. That is, a step of differentiating pluripotent stem cells into mesoderm and a step of differentiating the obtained cells into BPCs may be performed.
- the step of differentiating pluripotent stem cells into mesoderm may be performed, for example, by the method described above.
- a platelet-derived growth factor (PDGF-BB) or the like may be used as a differentiation-inducing factor to induce differentiation into BPCs.
- culture conditions may be those generally employed in the culture of animal cells. Culture conditions may be set to, for example, 37° C., 5% CO 2 and the like.
- a basal medium for example, serum-free vascular endothelial cell medium (Human Endothelial-SFM), Dulbecco's modified Eagle medium (D-MEM), Iscove's modified Dulbecco's medium (IMDM), MEM ⁇ medium, Ham F12 medium (HamF12) , Glasgow basal medium, RPMI1640 medium, MCDB107 medium, EGM-2 basal medium, etc.
- a coating agent such as Vitronectin-N (VTN-N) may be used.
- the differentiation into BPCs can be judged and evaluated, for example, using the expression of the pericyte marker as an indicator.
- Pericyte markers include, for example, PDGFR- ⁇ , NG2, ⁇ -SMA and the like. From the viewpoint of mimicking in vivo cells, BPCs are preferably negative for ⁇ -SMA expression. Therefore, the negative expression of ⁇ -SMA can be used as a useful index to distinguish between brain pericytes and peripheral pericytes.
- step A may be cells induced to differentiate from pluripotent stem cells using a medium containing A83-01. Therefore, step C may include a step of inducing differentiation of pluripotent stem cells into brain pericytes using a medium containing A83-01. BPC mimicry can be further enhanced by using media containing A 83-01.
- a 83-01 is preferably contained at 10 nM or more, more preferably 0.1 ⁇ M or more, in the medium used in step C from the viewpoint of improving the cell migration ability and cell proliferation ability of BPCs. .
- the content of A 83-01 in the medium used in step C is preferably 100 ⁇ M or less, more preferably 10 ⁇ M or less, from the viewpoint of economy.
- step C and step A may be performed continuously. That is, the culture vessel such as the cell culture insert used in step C may be used as it is, and step A may be performed continuously while replacing the medium as appropriate. More specifically, first, in step C, pluripotent stem cells may be seeded on the outside (back surface of the bottom) of the cell culture insert to induce differentiation of the pluripotent stem cells into BPCs. It is preferable to add A83-01 to the medium during the differentiation induction. EPCs may then be seeded on the inside (bottom top) of the cell culture insert and co-cultured with BPC using LN511F, FBN and COL4.
- EPCs are induced to differentiate into BMELCs.
- EPCs can be expanded and cryopreserved, and thus have excellent supplyability. Therefore, according to the method of the present disclosure for inducing differentiation of EPCs into BMELCs, BMELCs can be easily produced because frozen cells can be used as differentiation sources.
- the method of the present disclosure for inducing differentiation of EPCs into BMELCs it is possible to suppress variations in performance due to differences in cell lots produced.
- a monolayer (cell layer) of BMELCs is formed by continuously culturing the BMELCs produced by differentiation induction including step A above under culture conditions suitable for the maintenance and proliferation of BMELCs.
- a cell layer with excellent barrier function can be obtained.
- the barrier function of the cell layer obtained by the methods of the present disclosure can be characterized by strong tight junctions and the maintenance of tight junctions over an extended period of time.
- the barrier function of the cell layer obtained by the method of the present disclosure can be further characterized by having the function of important or characteristic drug transporters (eg, BCRP, P-gp, GLUT1) in BMECs.
- important or characteristic drug transporters eg, BCRP, P-gp, GLUT1
- tight junction markers for example, Claudin5, Occludin, ZO-1
- Claudin5, Occludin, ZO-1 for example, Claudin5, Occludin, ZO-1
- a cell layer of BMELCs can be formed on the semipermeable membrane (porous membrane) by culturing on the semipermeable membrane.
- This aspect is particularly effective when the cell layer of BMELCs obtained by the production method of the present disclosure is used for various assays.
- a culture vessel for example, Transwell (registered trademark) provided by Corning
- a culture insert having a culture surface made of a semi-permeable membrane
- Another aspect of the present disclosure relates to the use of BMELCs produced by inducing differentiation by the above method.
- BMELCs produced by inducing differentiation by the above method.
- it is possible to obtain a cell layer composed of BMELCs, which can be used in a BBB model.
- BBB pharmacokinetic research, pathological analysis, differentiation research, etc.
- pharmacokinetic study in the BBB it can be used for a new BBB pharmacokinetic evaluation model, etc.
- pathological analysis for example, it can be used for a pathological reproduction model using disease iPS cells for diseases related to BBB disruption.
- an assay using a cell layer composed of BMELCs prepared by the method of the present disclosure can be used to evaluate the intracerebral penetration (blood-brain barrier permeability) of a test substance such as a drug or drug candidate.
- the evaluation accuracy can be improved.
- the permeability of the test substance through the BBB in the human body can be easily predicted in vitro.
- a method for evaluating the BBB permeability of a test substance using the cell layer of BMELCs obtained by the above production method (hereinafter referred to as "BBB permeability evaluation method of the present disclosure” ) is provided.
- BBB permeability evaluation method of the present disclosure the following steps (i) to (iii) are performed.
- Step of preparing a cell layer of BMELCs obtained by the production method of the present disclosure (ii) Step of contacting the cell layer with a test substance (iii) Quantifying the test substance that permeates the cell layer Evaluating the blood-brain barrier permeability of the test substance by
- a cell layer of BMELCs obtained by the production method of the present disclosure is prepared.
- other cells pericytes, astrocytes, etc.
- a culture vessel with a culture insert is used to form a cell layer of BMELCs in the culture insert (a cell layer is formed on the bottom top surface of the culture insert), and adhered to the bottom back surface of the culture insert.
- pericyte-adherent co-culture system culture pericytes in the compartment between the culture insert and the well (pericyte-non-adherent co-culture system), or adhere to the bottom surface of the culture insert.
- Astrocytes are cultured in the compartment between the culture insert and the well (pericyte-adhesive/astrocyte-nonadherent co-culture system), or astrocytes are cultured attached to the bottom underside of the culture insert. (Astrocyte-adherent co-culture system).
- Contact in step (ii) is typically performed by adding the test substance to the medium.
- the timing of addition of the test substance is not particularly limited. Therefore, after starting culture in a medium containing no test substance, the test substance may be added at a certain point, or culture may be started in advance in a medium containing the test substance.
- test substances typically are drugs or drug candidates are used as test substances.
- the test substance is not particularly limited, and organic compounds or inorganic compounds with various molecular sizes can be used as the test substance.
- organic compounds include nucleic acids, peptides, proteins, lipids (simple lipids, complex lipids (phosphoglycerides, sphingolipids, glycosylglycerides, cerebrosides, etc.), prostaglandins, isoprenoids, terpenes, steroids, polyphenols, catechins, vitamins (B1, B2, B3, B5, B6, B7, B9, B12, C, A, D, E, etc.) may be used as the test substance.
- test substance By simultaneously adding more than one type of test substance, interaction between test substances, synergistic action, etc. may be investigated.
- the test substance may be derived from a natural product or synthesized.
- an efficient assay system can be constructed using, for example, a combinatorial synthesis technique.
- the period of contact with the test substance can be set arbitrarily.
- the duration of contact is, for example, 10 minutes to 3 days, preferably 1 hour to 1 day.
- step (iii) the test substance that permeates the cell layer is quantified.
- a culture vessel equipped with a culture insert such as Transwell (registered trademark)
- the test substance permeating the culture insert that is, the upper vessel (culture insert) or the lower vessel through the cell layer
- the test substance that has migrated into the (well) is quantified by a measurement method suitable for the test substance.
- measurement methods include mass spectrometry, liquid chromatography, and immunological techniques (e.g., Fluorescent Immunoassay (FIA), Enzyme Immunoassay (EIA)). can be done.
- FIA Fluorescent Immunoassay
- EIA Enzyme Immunoassay
- the membrane permeability of the test substance is evaluated based on the quantitative results (the amount of the test substance that permeates the cell layer) and the amount of the test substance used (typically, the amount added to the medium).
- absorption by the cell layer absorption
- effects on the cell layer e.g., effects on barrier function
- effects on the expression or function of transporters e.g., BCRP and P-gp
- Effects on barrier function can be evaluated by measuring TEER values, permeation tests using non-absorbable markers, and the like.
- the effect on transporter expression can be evaluated by immunological techniques, Western blotting, flow cytometry, etc., and the effect on transporter function can be evaluated, for example, by an activity test using a substrate. can.
- the present disclosure provides another use of the cell layer of BMELCs produced by the production method of the present disclosure, an evaluation method targeting or targeting the BBB function, that is, evaluating the effect of the test substance on the BBB barrier function.
- the evaluation method is useful, for example, as a search means for substances that reinforce (improve) the barrier function, substances that protect the barrier function, substances that regulate the barrier function, and the like. It can also be used for evaluation of toxicity to the BBB.
- a step of preparing a cell layer and a step of contacting the cell layer with a test substance are performed, and then the effect of the cell layer on the barrier function is evaluated.
- the method for evaluating the effect on barrier function is as described above.
- brain capillary endothelial-like cells obtained by the production method described above are provided.
- the expression level of PECAM1 is higher than primary cultured brain capillary endothelial cells and immortalized brain capillary endothelial cells, and the transendothelial electrical resistance (TEER value) is 50 ⁇ cm.
- TEER value transendothelial electrical resistance
- Brain capillary endothelial-like cells that are 2 or more are provided. Such brain capillary endothelial-like cells can be obtained, for example, by the production method described above.
- the mRNA expression level or the protein expression level may be used. Quantification of the expression level of mRNA can be performed using, for example, RT-qPCR.
- the protein expression level can be quantified by, for example, an immunostaining method.
- the TEER value can be measured using Millicell ERS-2 (chopstick type) according to the manual attached to the device. The TEER value should be measured before changing the medium. More specifically, first, the culture dish is allowed to stand at room temperature for about 15 minutes to return the internal temperature of the medium to room temperature, and then the TEER value of the cell culture insert without seeding the cells is measured and used as a blank. . Then, after measuring the TEER value of the cell culture insert seeded with cells, the value obtained by subtracting the TEER value of the blank can be obtained as the actual measurement value.
- FIG. 1 is an explanatory diagram showing a differentiation protocol from human iPS cells (hereinafter also referred to as "hiPSCs”) to human iPS cell-derived vascular endothelial progenitor cells (hereinafter also referred to as "iEPCs").
- hiPSCs are differentiated into iEPCs via primitive streak, mesoderm, and immature iEPCs, and then purified. manufactured iEPCs.
- the iEPCs thus produced were induced to differentiate into human iPS cell-derived brain capillary endothelial-like cells (hereinafter also referred to as "iBMELCs").
- iBMELCs human iPS cell-derived brain capillary endothelial-like cells
- VTN-N Vitronectin-N
- D-PBS D-PBS
- a gelatin coating was made by leaving a 0.1% gelatin solution at 37°C for 1 hour or at 4°C overnight.
- Fibronectin manufactured by Wako Pure Chemical Industries
- collagen type IV manufactured by Nitta Gelatin Inc., was diluted with D-PBS (-) to 400 ⁇ g/mL and allowed to stand at 37° C. for 2 to 4 hours.
- Coating with three components was performed by adding one component to the mixture of fibronectin and collagen type IV.
- the third component includes VTN-N (manufactured by Thermo Fisher Scientific), laminin 221 fragment (LN221F: iMatrix221 manufactured by Nippi Incorporated), laminin 411 fragment (LN411F: iMatrix411 manufactured by Nippi Incorporated), laminin 511 fragment (LN511F: manufactured by Nippi Incorporated).
- laminin fragment mix (mixture of laminin 221F, laminin 411F and laminin 511F), laminin 211 (manufactured by Veritas), laminin 411 (manufactured by Veritas), laminin 511 (manufactured by Veritas), agrin (manufactured by R&D), laminin mix (laminin 211, a mixture of laminin 411 and laminin 511).
- concentrations of VTN-N, LN221F, and LN411F were 10 ⁇ g/mL.
- the concentration of LN511F was 10-200 ⁇ g/mL.
- Human immortalized cell line hCMEC/D3 was obtained from Merck Millipore, hCMEC/D3 medium (5% fetal bovine serum (Sigma-Aldrich), 5 ⁇ g/mL L-ascorbic acid phosphate magnesium salt n-hydrate , 1% chemically defined lipid concentrate (Thermo Fisher Scientific), 10 ⁇ M HEPES solution (Sigma-Aldrich), 1% penicillin-streptomycin solution (Biological Industries), 1.4 ⁇ M hydrocortisone (Wako) and 1 ng/mL FGF2 (PeproTech, Inc.) containing Endothelial Cell Basal Medium-2 (Lonza)).
- TrypLE Select was used and seeded on a collagen type I coated culture dish at 5.0 ⁇ 10 3 cells/cm 2 . Medium exchange was performed once every two days.
- modified DMEM/F12 (0.1% chemically defined lipid concentrate, 0.1 ⁇ insulin-transferrin-selenium (manufactured by Thermo Fisher Scientific), 2 mM GlutaMAX (manufactured by Sigma-Aldrich), 450 mM 1-thioglycerol (manufactured by Sigma-Aldrich), DMEM/F12 containing 50 ⁇ g/mL of L-ascorbic acid phosphate magnesium salt n-hydrate and 1 ⁇ penicillin-streptomycin solution) and 5 ⁇ M CHIR-99021 (Focus Biomolecules) were added to culture for 1 day.
- the cells were cultured for one day in a medium containing modified DMEM/F12 supplemented with 50 ng/mL of FGF2. Thereafter, the cells were cultured for 3 days in a medium containing modified DMEM/F12 supplemented with 50 ng/mL VEGF (Biolegend) and 25 ng/mL BMP4 (Prospec). During this period, the medium was changed every day. On day 5, cells treated with 10 ⁇ M Y-27632 for 1 hour were dissociated with TrypLE Select, passaged to a new gelatin-coated culture dish at 3.5 ⁇ 10 4 cells/cm 2 , and modified Human endothelial (HE)-SFM.
- HE Human endothelial
- the cells were washed with D-PBS (-) three times or more, treated with TrypLE Select again, iEPCs were exfoliated, collected in a tube, centrifuged at 100 xg for 5 minutes, and re-inoculated on a new culture dish.
- the cells were collected in a medium in a 15 mL tube or a 50 mL tube and centrifuged at 100 xg for 5 minutes. They were then resuspended in medium and replated on VTN-N-coated culture dishes at 1 ⁇ 10 4 cells/cm 2 or 1.5 ⁇ 10 4 cells/cm 2 .
- D-PBS D-PBS
- TrypLE Select was added and observed under a microscope at room temperature. When the extra cells began to detach, the cells were strongly tapped several times, the detachment solution was aspirated, and the cells were washed once with D-PBS (-). Cultured until day 14 of differentiation.
- the coating of the culture dish includes two components, FBN and COL4, or three components obtained by adding one of VTN-N, LN221, LN411F, and LN511F to these two components, or LN411F and LN511F. and four components were used.
- the third component laminin 211, laminin 411, laminin 511, agrin, laminin mix, laminin fragment mix, and hepex were also used.
- Non-frozen or frozen iEPCs (14 days of differentiation) were seeded, HE-SFM (+ 1 ⁇ penicillin - streptomycin, hereinafter omitted) medium supplemented with 2-10% KSR and 20 ng/mL FGF2, 5% HE-SFM Medium supplemented with FBS (manufactured by Sigma-Aldrich Corporation) and 20 ng/mL FGF2, medium supplemented with 5% Platelet-poor plasma-derived bovine serum (PDS) (manufactured by Alfa Aesar) and 20 ng/mL FGF2 in HE-SFM , HE-SFM supplemented with 2-10% B27 (registered trademark) supplement (Thermo Fisher Scientific) and 20 ng/mL FGF2, or HE-SFM with 7.5% B27 supplement, 0.1-10 ⁇ M A 83-01 and The cells were cultured in a medium supplemented with 20 ng/mL FGF2 to induce differentiation into brain capillary-like
- the medium was changed on the first day after seeding, and thereafter, the medium was changed every other day.
- 1 ⁇ 10 5 cells were added to a culture dish coated with 100 ⁇ g/mL Fibronectin, 400 ⁇ g/mL collagen type IV, and 10 ⁇ g/mL LN511F.
- HE-SFM at 7.5 Culture was performed in a medium containing % B27 supplement, 1 ⁇ M A 83-01 and 20 ng/mL FGF2.
- Human immortalized brain capillary endothelial cells hCMEC/D3 were similarly induced to differentiate into brain capillary-like cells using differentiation induction condition A described later.
- HE-SFM based medium 1% PDS and 1 ⁇ penicillin-streptomycin solution (Biological Industries USA, Inc.) containing HE-SFM
- the medium was changed every day.
- the differentiated cells were washed with D-PBS (-), an Accutase cell detachment solution was added, and the cells were allowed to stand at 37°C for 20 minutes. After that, the cells collected using the medium were centrifuged at 100 ⁇ g for 5 minutes to prepare a cell suspension, and then placed on a Fibronectin + collagen type IV coated well plate or Transwell (registered trademark) (12-well, pore size: 1 ⁇ m).
- HE-SFM based medium supplemented with 10 ⁇ M RA and 20 ng/mL FGF2.
- the medium was changed to HE-SFM based medium to which RA and FGF2 were not added, and cultured.
- iEPCs differentiated from hiPSCs by the above method were analyzed by an immunostaining method in order to confirm their properties as EPCs.
- PECAM1 and CD34 were used as antibodies in this immunostaining.
- iBMELCs differentiated from frozen iEPCs by the above method using the following differentiation induction condition A, and as a comparative example, a coating agent containing Fibronectin 100 ⁇ g / mL and collagen type IV 400 ⁇ g / mL, 7.5% KSR and Cells cultured in HE-SFM medium supplemented with 20 ng/mL FGF2 were subjected to protein expression analysis by immunostaining.
- PECAM1, CDH5, ZO-1, CLAUDIN5, P-gp, BCRP and GLUT1 were used as antibodies in this immunostaining.
- Cells on a 96-well plate were washed three times with 0.1% BSA-containing D-PBS (-), fixed in 4% paraformaldehyde for 15 minutes, and washed again with 0.1% BSA-containing D-PBS (-). and D-PBS (-) containing 0.1 w/v% Triton X-100 for 5 minutes. Subsequently, after washing with D-PBS (-) containing 0.1% BSA, the primary antibody was allowed to react overnight at 4°C.
- the plate was washed three times with D-PBS (-) containing 0.1% BSA, and reacted with a secondary antibody (Anti-Rabbit, Anti-Mouse) diluted 200-fold at room temperature for 60 minutes. After that, the plate was washed three times with D-PBS (-) containing 0.1% BSA, and reacted with 1 ⁇ g/mL DAPI (Dojindo), which is a nuclear staining reagent, for 5 minutes. After that, it was washed three times with D-PBS (-) and analyzed by Operetta High-Content Imaging System (manufactured by PerkinElmer). Details of the antibodies used in immunostaining are shown in Table 1.
- Coating agent 100 ⁇ g/mL Fibronectin, 400 ⁇ g/mL collagen type IV, 10 ⁇ g/mL LN511F Medium: HE-SFM supplemented with 7.5% B27® supplement, 1 ⁇ M A 83-01 and 20 ng/mL FGF2
- Acetylated LDL uptake test was performed on iEPCs differentiated from hiPSCs in order to confirm their properties as EPCs. iEPCs induced to differentiate from hiPSCs (on day 14 of differentiation) were reacted with Dil-labeled acetylated LDL at a final concentration of 10 ⁇ g/mL for 5 hours, and then reacted with fluorescently labeled Hoechist33342 for 30 minutes. and analyzed with the Operetta High-Content Imaging System.
- the relative maximum TEER value of the frozen group was determined.
- maximum TEER value means the maximum TEER value measured on each day from 2 days after sowing to 10 days after sowing.
- the TEER value was measured using Millicell ERS-2 (chopstick type) according to the attached manual.
- the medium volume was 600 ⁇ L apical and 1800 ⁇ L basolateral.
- sample “B27” the medium contained an additional 7.5% B27s
- in sample “B27+A 83-01” the medium contained an additional 7.5% B27s and 1 ⁇ M A 83-01
- in sample “B27+A 83-01+LN511F ” further contained 7.5% B27s and 1 ⁇ M A 83-01 in the medium and 10 ⁇ g/mL LN511F in the coating agent.
- the number of samples was 3 for each.
- Cells on Transwell (registered trademark) (12-well, pore size: 1 ⁇ m) were used for the permeation test.
- Four days after seeding the medium was replaced with a transport buffer (HBSS containing 10 mM HEPE solution) and cultured at 37°C for 20 minutes.
- HBSS containing 10 mM HEPE solution
- Transport buffer containing 1 mg/mL FD4 or 300 ⁇ M LY was added apically. After incubation for 60 minutes at 37°C, 100 ⁇ L of solution was collected from the basolateral side. The volume of transport buffer was 500 ⁇ L apical and 1500 ⁇ L basolateral. The fluorescence intensity of FD4 or LY was measured with a Synergy HTX multimode plate reader (BioTek) and analyzed with Gen 5 data analysis software (BioTek).
- the protein expression level was analyzed using the Operetta High-Content Imaging System, the cells immunostained as described above were observed with an Operetta microscope, and the fluorescence intensity was calculated using Harmony (registered trademark) high-content analysis software. did. Based on the protein expression level in the comparative example, the relative expression level of the protein in the iBMELCs was determined.
- RNA extraction was performed according to the attached manual of Agencourt (registered trademark) RNAdvance Tissue Kit (manufactured by Beckman Coulter, Inc.).
- RNAdvance Tissue Kit manufactured by Beckman Coulter, Inc.
- cDNA complementary DNA
- ReverTra Ace registered trademark
- qPCR RT Master Mix manufactured by Toyobo Co., Ltd.
- RT-q PCR KAPA SYBR Fast qPCR Kit (manufactured by Nippon Genetics Co., Ltd.) was used, cDNA was used as a template, and the reaction was carried out according to the attached manual. The results obtained were corrected using HPRT1 as an endogenous control. Taking the expression level of mRNA in hBMECs as a reference and setting it to 100, relative expression levels of mRNA in iBMELCs, iBMELCs 2 and hCMEC/D3 were determined.
- the CDH5 forward primer sequence is shown in SEQ ID NO: 1
- the CDH5 reverse primer sequence is shown in SEQ ID NO: 2
- the MDR1 forward primer sequence is shown in SEQ ID NO: 3
- MDR1 The reverse primer sequence for GLUT1 is shown in SEQ ID NO: 4
- the BCRP forward primer sequence is shown in SEQ ID NO: 5
- the BCRP reverse primer sequence is shown in SEQ ID NO: 6
- the GLUT1 forward primer sequence is shown in SEQ ID NO: 7.
- the sequence of the reverse primer for GLUT1 is shown in SEQ ID NO: 8
- the sequence of the forward primer for Occludin is shown in SEQ ID NO: 9
- the sequence of the reverse primer for Occludin is shown in SEQ ID NO: 10
- the sequence of the forward primer for ZO-1 is shown in SEQ ID NO: 9.
- the sequence of the reverse primer for ZO-1 is shown in SEQ ID NO: 12
- the sequence of the forward primer for LAT1 is shown in SEQ ID NO: 13
- the sequence of the reverse primer for LAT1 is shown in SEQ ID NO: 14, and the forward primer for HPRT1.
- SEQ ID NO: 15 the sequence of the reverse primer for HPRT1 is shown in SEQ ID NO: 16
- sequence of the forward primer for PECAM1 is shown in SEQ ID NO: 17
- sequence of the reverse primer for PECAM1 is shown in SEQ ID NO: 18.
- P glycoprotein (P -gp) As an efflux transporter, P glycoprotein (P -gp), a substrate uptake assay was performed.
- Cells on Transwell (registered trademark) (12-well, pore size: 1 ⁇ m) were used for substrate uptake studies.
- Rhodamine 123 was used as a substrate.
- Media was removed and pre-incubated for 15 minutes at 37°C in transport buffer. The number of samples was 3. Incubation was carried out at 37° C. for 60 min in transport buffer containing 10 ⁇ M rhodamine 123 in the presence or absence of 10 ⁇ M cyclosporin A (CsA), an inhibitor of P-gp.
- CsA cyclosporin A
- Fig. 2 is an explanatory diagram showing the results of confirming the properties of iEPCs on the 14th day of differentiation.
- the images on the left side of the page show the results of protein expression analysis by immunostaining
- the images on the right side of the page show the results of the acetylated LDL uptake test.
- a scale bar of 100 ⁇ m is shown in all images. From the results shown in Fig. 2, it was confirmed that the vascular endothelial progenitor cell markers PECAM1 and CD34 were expressed, and that the acetylated LDL uptake function, which is one of the functions of vascular endothelial progenitor cells, was confirmed. found to have. That is, it was found that the cells differentiated and purified by the above method have the properties of vascular endothelial progenitor cells.
- FIG. 3 is an explanatory diagram showing a comparison of the electrical resistance values of non-frozen iEPCs and frozen iEPCs.
- the vertical axis indicates the relative maximum TEER value ( ⁇ cm 2 ) when the maximum TEER value of the non-frozen group is set to 1.0. From the results shown in FIG. 3, there was no significant difference in the maximum TEER values between the non-frozen group and the frozen group, indicating that the frozen iEPCs had a barrier function equivalent to that of the non-frozen iEPCs. Therefore, iEPCs induced to differentiate from hiPSCs were cryopreserved to prepare iEPCs stocks, and it was found that the iEPCs stocks can be used for the preparation of iBMELCs.
- FIGS. 4 to 10 are explanatory diagrams showing the results of examination of conditions for inducing differentiation from iEPCs to iBMELCs.
- Differentiation induction conditions are the type of medium components in FIG. 4, the concentration of medium components in FIG. 5, the concentration of A83-01 in FIG. shows the results of examining the number of cells.
- the vertical axis in FIGS. 4 to 6 and 8 to 10 indicates the maximum TEER value ( ⁇ cm 2 ), and the vertical axis in FIG. 7 indicates the maximum TEER value ( ⁇ cm 2 ) is set to 1.0, and the relative maximum TEER value of each sample is shown.
- Figure 4 shows the results of a study in which 5% each of KSR, FBS, PDS or B27s was added to HE-SFM medium containing FBN and COL4 as coating agents and 20 ng/mL of FGF2. From the results shown in FIG. 4, it was found that the barrier function of iBMELCs can be particularly improved by inducing differentiation by adding B27s to the medium.
- Figure 5 shows the results of a study in which 2 to 10% of KSR or B27s was added to HE-SFM medium containing FBN and COL4 as coating agents and 20 ng/mL of FGF2. From the results shown in Fig. 5, it was found that the barrier function of iBMELCs improved as the content of B27s in the medium increased, and that the barrier function of iBMELCs with a content of 7.5% was as high as that with a content of 10%. rice field. It was also found that the addition of B27s improved the barrier function of iBMELCs more than the addition of KSR at any content.
- Fig. 6 shows the result of adding 0.1 ⁇ M, 1 ⁇ M or 10 ⁇ M of A 83-01 to HE-SFM medium containing 20 ng/mL of FGF2 and 7.5% of B27s as coating agents, FBN and COL4. is shown. From the results shown in FIG. 6, it was found that the barrier function of iBMELCs was improved when A 83-01 was added compared to when A 83-01 was not added. It was also found that A 83-01 with a content of 1 ⁇ M exhibits a high barrier function equivalent to that with a content of 10 ⁇ M.
- Fig. 7 shows the results of investigations (number of samples: 1) in which 10 ⁇ g/mL of various components were added to the two components of FBN and COL4 as coating agents.
- HE-SFM supplemented with 7.5% B27s, 1 ⁇ M A83-01 and 20 ng/mL FGF2 was used as the medium. From the results shown in FIG. 7, it was found that the barrier function of iBMELCs can be particularly improved by adding LN511F to the coating agent.
- Fig. 8 shows the results of a study in which 10 ⁇ g/mL of various components were added to the two components of FBN and COL4 as coating agents.
- HE-SFM supplemented with 7.5% B27s, 1 ⁇ M A83-01 and 20 ng/mL FGF2 was used as the medium. From the results shown in FIG. 8, it was found that the barrier function of iBMELCs can be particularly improved by adding LN511F to the coating agent.
- Fig. 9 shows the results of investigations in which 10 ⁇ g/mL, 50 ⁇ g/mL, 100 ⁇ g/mL or 200 ⁇ g/mL of LN511F was added to the two components of FBN and COL4 as coating agents. Also shown are the results of a study in which 100 ⁇ g/mL each of LN411F and LN511F were added to the two components of FBN and COL4. HE-SFM supplemented with 7.5% B27s, 1 ⁇ M A83-01 and 20 ng/mL FGF2 was used as the medium. From the results shown in FIG.
- the coating agent containing 10 ⁇ g/mL of LN511F exhibited a high barrier function equivalent to that of the coating agent containing 200 ⁇ g/mL.
- iBMELCs using a coating layer with only LN511F added to the two components of FBN and COL4 were better than those with a coating layer using both LN411F and LN511F in the two components of FBN and COL4. It was found that the barrier function of
- FIG. 10 shows the results of a study in which different numbers of iEPCs were seeded under the differentiation-inducing condition A described above. According to the results shown in FIG. 10, the maximum TEER value was highest when the number of cells was 1 ⁇ 10 5 cells/well, indicating that the barrier function of iBMELCs can be further improved by setting a low cell density. rice field.
- FIG. 11 is an explanatory diagram showing temporal changes in the TEER value under each differentiation-inducing condition.
- the vertical axis indicates the TEER value ( ⁇ cm 2 ), and the horizontal axis indicates the number of days elapsed after sowing.
- a coating agent containing two components, FBN and COL4, and a HE-SFM medium containing 20 ng/mL FGF2 were common conditions.
- iBMELCs differentiated using a coating agent containing LN511F showed a higher TEER value over a long period of time than iBMELCs differentiated using a coating agent not containing LN511F. rice field. That is, iBMELCs differentiated with a coating agent containing LN511F were found to have a higher long-term barrier function than iBMELCs differentiated with a coating agent not containing LN511F.
- FIG. 12 is an explanatory diagram showing the results of permeation tests for FD4 and LY under different differentiation induction conditions.
- the vertical axis indicates the transmission coefficient P app (10 ⁇ 6 cm / sec).
- the four differentiation-inducing conditions used in FIG. 12 are the same as the differentiation-inducing conditions used in the analysis of changes in TEER values over time. From the results shown in FIG. 12, iBMELCs differentiated using a coating agent containing LN511F do not contain LN511F, regardless of whether FD4 or LY, which are indicators of permeation by the paracellular pathway, are targeted for permeation.
- iBMELCs differentiated using a coating agent It was found to show a lower permeability coefficient than iBMELCs differentiated using a coating agent. That is, it was found that iBMELCs differentiated using a coating agent containing LN511F have a higher barrier function than iBMELCs differentiated using a coating agent not containing LN511F.
- iBMELCs differentiated with a coating agent containing LN511F in addition to FBN and COL4 were compared with iBMELCs differentiated with a coating agent that did not contain LN511F. It was suggested that it has a high barrier function. Therefore, iBMELCs produced by a production method including a culture step of culturing iEPCs using LN511F, FBN and COL4 were found to be useful as a model of human BBB.
- differentiation induction using a medium containing B27s can improve the barrier function of iBMELCs more than differentiation induction using a medium not containing B27s.
- the addition of A 83-01 to the medium could further improve the barrier function of iBMELCs compared to the case of not adding A 83-01 to the medium.
- FIG. 13 is an explanatory diagram showing protein expression analysis results by immunostaining and protein expression level analysis results by Operetta High-Content Imaging System.
- the upper side of FIG. 13 shows images obtained by immunofluorescence staining for each BMECs marker with a scale bar of 50 ⁇ m, and the lower side of FIG. 13 shows relative protein expression levels.
- the relative protein expression level is shown as a relative value when the protein expression level in the cells of the comparative example (described as "iEPCs" in FIG. 13) is set to 1.0 as a reference.
- the results of protein expression analysis by immunostaining revealed the following.
- iBMELCs have the properties of vascular endothelial cells.
- iBMELCs showed higher expression levels of BMECs markers such as tight junction markers ZO-1 and CLAUDIN5, drug efflux transporter markers P-gp and BCRP, and glucose transporter marker GLUT1 compared to the cells of the comparative example. Rose. Therefore, it was suggested that the iBMELCs produced by the differentiation-inducing method described above strongly possess characteristics of cerebral capillary endothelial cells.
- FIG. 14 is an explanatory diagram showing the results of gene expression analysis by the RT-q PCR method.
- the vertical axis in FIG. 14 indicates the relative mRNA expression level when the mRNA expression level in primary human BMECs (hBMECs) is set to 100.
- iBMELCs are differentiated cells (iBMELCs 2), hBMECs, and human immortal cells induced by a method modified from the report of Lippmann et al. BMECs (hCMEC/D3) showed significantly higher expression levels.
- iBMELCs showed a significantly higher expression level of GLUT1 than hBMECs and hCMEC/D3, and an expression level equivalent to that of iBMELCs2. It was also confirmed that iBMELCs expressed P-gp at the same level as hBMECs.
- iBMELCs produced by the above differentiation induction method have the properties of brain capillary endothelial cells and have a high degree of mimicry with in vivo BMECs. Therefore, it was found that iBMELCs produced by the above differentiation induction method are useful as a model of human BBB by exhibiting properties similar to those of in vivo BMECs.
- FIG. 15 is an explanatory diagram showing a comparison of electrical resistance values between iEPCs and immortalized cells.
- the vertical axis in FIG. 15 indicates the maximum TEER value ( ⁇ cm 2 ), "+” indicates the samples induced to differentiate using the above differentiation induction condition A, and "-" indicates the comparison.
- the sample is cultured under the above-described conditions different from the differentiation-inducing condition A. From the results shown in FIG. 15, it was found that, regardless of whether iPS cells or immortalized cells were used, the cells induced to differentiate by the above method using the differentiation-inducing condition A exhibited a high TEER value.
- brain capillary endothelial-like cells with a barrier function that mimics brain capillary endothelial cells are produced by inducing differentiation by the above culture method. shown that it can be done.
- the results shown in FIG. 15 show that iBMELCs having a higher barrier function can be produced by inducing differentiation using iPS cells compared to the case of inducing differentiation using immortalized cells. was done.
- FIG. 16 is an explanatory diagram showing the results of functional analysis of P-glycoprotein (P-gp).
- the vertical axis indicates the relative amount of substrate accumulation when the amount of substrate accumulation in the absence of cyclosporine is set to 1.0, and "-" indicates samples in the absence of cyclosporine. "+” indicates samples in the presence of cyclosporine.
- iBMELCs induced to differentiate by the above method accumulate more substrate in the presence of cyclosporine, which is an inhibitor of efflux transporters, compared to the absence of such an inhibitor. was confirmed. Therefore, iBMELCs induced to differentiate by the above method were found to have the function of P-gp.
- FIG. 17 is an explanatory diagram for explaining the outline of co-culture of iEPCs and iBPCs.
- FIG. 17 shows a schematic outline of a method of inducing differentiation of human iPS cells (hiPSCs) into human iPS cell-derived brain pericytes (hereinafter also referred to as “iBPCs”) and coculturing the iBPCs with the above-described iEPCs.
- hiPSCs human iPS cell-derived brain pericytes
- iBPCs human iPS cell-derived brain pericytes
- iEPCs were seeded on the surface of the cell culture insert and co-cultured in a BMECs differentiation medium. From day 13 to day 19 of differentiation induction, that is, from day 1 to day 7 of co-culture, the barrier function was evaluated by measuring and analyzing the TEER value.
- Materials 802-3G strain obtained from Reprocell Co., Ltd. was used as hiPSCs for inducing differentiation into iBPCs.
- the Matrigel GFR coating for maintenance culture of hiPSCs was prepared by diluting Matrigel GFR (manufactured by Corning Incorporated) 30-fold with human iPS cell maintenance medium on ice and then standing at 37° C. for 30 minutes or more.
- the hiPSCs were cultured on a VTN-N-coated culture dish using mTeSR plus medium (manufactured by StemCell Technologies) as a medium.
- VTN-N coating on the above culture dish VTN-N (manufactured by Thermo Fisher Scientific) was diluted with D-PBS (-) to 1 ⁇ g/cm 2 and then heated at 37°C for 1-2 hours. It was prepared by standing still.
- iBPCs differentiated day 4
- EGM-2 EGM-2 at 10 ⁇ M.
- Y-27632, 50 ng/mL of PDGF-BB and 1 ⁇ M of A83-01 were added for 2 hours.
- the iBPC-inoculated cell culture insert was placed in a 12-well companion plate, and 10 ⁇ M Y-27632 and 50 ng/mL PDGF-BB were added to 600 ⁇ L of EGM-2 on the apical side and 1800 ⁇ L on the basal side. Cultivation was carried out in the culture medium.
- EGM-2 was cultured in a medium containing 50 ng/mL PDGF-BB, and from day 8 of differentiation, EGM-2 was added with 50 ng/mL PDGF-BB and 1 ⁇ M Culture was performed in a medium supplemented with A83-01. During this period, the medium was replaced every day.
- frozen iEPCs day 14 of differentiation
- the cells were cultured in a medium supplemented with FGF2 and 19 days of differentiation, and induced to differentiate into cerebral capillary-like cells. During this period, the medium was replaced every day.
- FIG. 18 is an explanatory diagram showing temporal changes in TEER values in iBMELCs with and without coculture.
- the vertical axis indicates the TEER value ( ⁇ cm 2 ), and the horizontal axis indicates the number of days elapsed after co-culture (iEPCs alone culture without co-culture corresponds to co-culture). number of days).
- the sample co-cultured with iBPC is indicated as "EPC+PC”
- EPC sample cultured with iEPCs alone without co-culture with iBPC
- Fig. 18 The results shown in Fig. 18 showed the following.
- the TEER value of iBMELCs co-cultured with iBPCs was significantly higher than the TEER value of iBMELCs obtained by culturing iEPCs alone at the 1% level by two-way repeated measures analysis of variance. A difference was observed, indicating an increase in TEER values over time. Therefore, it was found that the tight junction function of iBMELCs can be further improved by co-culture with iBPCs.
- modified DMEM/F12 (0.1% chemically defined lipid concentrate, 0.1 ⁇ insulin-transferrin-selenium (manufactured by Thermo Fisher Scientific), 2 mM GlutaMAX (manufactured by Sigma-Aldrich), 450 mM 1-thioglycerol (manufactured by Sigma-Aldrich), DMEM/F12 containing 50 ⁇ g/mL of L-ascorbic acid phosphate magnesium salt n-hydrate and 1 ⁇ penicillin-streptomycin solution) and 5 ⁇ M CHIR-99021 (manufactured by Focus Biomolecules) were added to culture for 1 day.
- EGM-2 EGM containing 2% FBS, 0.2 ⁇ g/mL Hydrocortisone, 10 ng/mL FGF2, 20 ng/mL R3-IGF-1, 1 ⁇ g/mL Ascorbic acid, 5 ng/mL hEGF, gentamicin, amphotericinB -2 basal medium
- 50 ng/mL PDGF-BB and 0.5 ng/mL VEGF were added for 2 days.
- the medium was changed every day.
- Table 2 Details of the antibodies used in immunostaining are shown in Table 2.
- “Target” indicates the name of the antibody
- “Source” indicates the name of the manufacturer of the antibody
- “Catalog number” indicates the catalog number of the antibody
- “Species” indicates the origin of the antibody
- “ Dilution” indicates the dilution ratio in terms of volume.
- RNA extraction was performed according to the attached manual of Agencourt (registered trademark) RNAdvance Tissue Kit (manufactured by Beckman Coulter, Inc.).
- RNA complementary DNA
- ReverTra Ace registered trademark
- qPCR RT Master Mix manufactured by Toyobo Co., Ltd.
- KAPA SYBR Fast qPCR Kit manufactured by Nippon Genetics Co., Ltd.
- cDNA was used as a template
- HPRT1 HPRT1
- the sequence of the PDGFR- ⁇ forward primer is shown in SEQ ID NO: 19
- the sequence of the PDGFR- ⁇ reverse primer is shown in SEQ ID NO: 20
- the NG2 forward primer sequence is shown in SEQ ID NO: 21.
- the sequence of the reverse primer for NG2 is shown in SEQ ID NO: 22
- the sequence of the forward primer for ⁇ -SMA is shown in SEQ ID NO: 23
- the sequence of the reverse primer for ⁇ -SMA is shown in SEQ ID NO: 24
- the forward primer for HPRT1 is shown in SEQ ID NO: 15
- the sequence of the reverse primer for HPRT1 is shown in SEQ ID NO: 16.
- a 83-01 was further added to the medium of iBPCs (X (+)) that had been induced to differentiate by adding A 83-01, and A 83-01 was added.
- A83-01 was not added to the culture medium of iBPCs (X(-)) that had been induced to differentiate without the use of A83-01.
- FIG. 19 is an explanatory diagram showing the results of immunostaining of brain pericyte marker protein.
- FIG. 19 shows the results of immunostaining on differentiation day 8 and differentiation day 12 for iBPCs (X( ⁇ )) induced to differentiate without addition of A83-01.
- a scale bar of 100 ⁇ m is shown in all images.
- PDGFR- ⁇ , NG2, and ⁇ -SMA protein expression increased over time on day 8 and day 12 of differentiation. It is known that the expression of ⁇ -SMA is weak in cerebral pericytes in vivo.
- FIG. 20 is an explanatory diagram showing analysis results of brain pericyte marker gene expression levels.
- the vertical axis in FIG. 20 indicates the relative mRNA expression level when the mRNA expression level in human primary pericytes is set to 1.
- "*" indicates that a significant difference was observed at the 5% level by multiple comparison (Tukey's test) in one-way ANOVA
- "**" indicates multiple comparison in one-way ANOVA.
- (Tukey's test) indicates that a significant difference was observed at the 1% level
- "***” indicates a significant difference at the 0.1% level by multiple comparison (Tukey's test) in one-way analysis of variance. indicates that From the results shown in FIG. 20, the following was found. In other words, we found that the addition of A 83-01 to the culture medium that induces differentiation into iBPCs can suppress the gene expression of ⁇ -SMA to the same level as that of primary human pericytes.
- FIG. 21 is an explanatory diagram showing the results of immunostaining of brain pericyte marker protein.
- images obtained by immunofluorescent staining for each marker are shown with a scale bar of 100 ⁇ m.
- FIG. 22 is an explanatory diagram showing the results of expression analysis of brain pericyte marker proteins.
- FIG. 22 shows the analysis result of the expression level of each marker protein by Operetta High-Content Imaging System.
- the vertical axis in FIG. 22 shows the relative value of the average fluorescence intensity when the average fluorescence intensity of the marker protein in iBPCs (X(-)) induced to differentiate without the addition of A 83-01 is set to 1. ing.
- "*" indicates that a significant difference was observed at the 5% level in the Student's t-test
- "**" indicates that a significant difference was observed at the 1% level in the Student's t-test. indicates
- FIG. 23 is a microscopic image showing the cell migration/proliferation ability of iBPCs.
- the double-headed arrow in FIG. 23 indicates the distance of the cell-damaged portion, in other words, the range where no cells exist.
- a scale bar of 100 ⁇ m is also shown in each image.
- the border of the cell-damaged portion at the time of cell damage (0 hours after cell damage) is indicated by a thick line, and the border of the cell that has changed due to the subsequent culture is indicated by a thin line.
- FIG. 24 is an explanatory diagram showing the cell migration ability/proliferation ability of iBPCs.
- the vertical axis in FIG. 24 indicates the relative value when the distance of the cell-damaged portion is set to 1 with the time of cell damage (0 hours after culture after cell damage) as the reference.
- brain capillary endothelial-like cells with improved imitation of in vivo brain capillary endothelial cells.
- vascular endothelial progenitor cells of frozen cells can be used as differentiation sources, so brain capillary endothelial-like cells can be easily produced. Therefore, it is possible to stably supply brain capillary endothelial-like cells, thereby contributing to the establishment of a supply system of brain capillary endothelial-like cells for pharmaceutical companies and research institutes.
- the BBB model constructed using the present disclosure can be used, for example, in evaluation systems such as efficacy/safety of pharmaceuticals.
- SEQ ID NO: 1 Description of Artificial Sequence: CDH5 Forward Primer SEQ ID NO: 2: Description of Artificial Sequence: CDH5 Reverse Primer SEQ ID NO: 3: Description of Artificial Sequence: MDR1 Forward Primer SEQ ID NO: 4: Description of Artificial Sequence: MDR1 Reverse Primer SEQ ID NO: 5: Description of Artificial Sequence: BCRP Forward Primer SEQ ID NO: 6: Description of Artificial Sequence: BCRP Reverse Primer SEQ ID NO: 7: Description of Artificial Sequence: GLUT1 Forward Primer SEQ ID NO: 8: Description of Artificial Sequence: GLUT1 Reverse Primer SEQ ID NO: 9: Artificial Sequence Description: Occludin Forward Primer SEQ ID NO: 10: Artificial Sequence Description: Occludin Reverse Primer SEQ ID NO: 11: Artificial Sequence Description: ZO-1 Forward Primer SEQ ID NO: 12: Artificial Sequence Description: ZO-1 Reverse Primer SEQ ID NO 13: Artificial Sequence Description: LAT1 Forward Primer SEQ ID NO: 14
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Biomedical Technology (AREA)
- Toxicology (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Gastroenterology & Hepatology (AREA)
- General Engineering & Computer Science (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
(i)前記細胞層を用意する工程;
(ii)前記細胞層に前記被検物質を接触させる工程;
(iii)前記細胞層を透過した前記被検物質を定量することにより、前記被検物質の血液脳関門透過性を評価する工程。
この形態の被検物質の血液脳関門透過性を評価する方法によれば、被験物質の血液脳関門における透過性の評価精度をさらに向上できる。
(i)本開示の製造方法により得られたBMELCsの細胞層を用意する工程
(ii)前記細胞層に被検物質を接触させる工程
(iii)前記細胞層を透過した被検物質を定量することにより、被検物質の血液脳関門透過性を評価する工程
(1)培養dishのコーティング
hiPSCsをiEPCsに分化誘導する際の培養dishのコーティングとして、Vitronectin-N (VTN-N)コーティングは、VTN-N(Thermo Fisher Scientific製)を1μg/cm2となるようD-PBS (-) で希釈し、37℃で1~2時間静置した。Gelatinコーティングは、0.1% gelatin溶液を37℃で1時間または4℃で一晩静置した。iEPCsをiBMELCsに分化誘導する際の培養dishのコーティングとして、Fibronectinとcollagen type IVとの2成分のコーティングは、Fibronectin(Wako Pure Chemical Industries製)が100μg/mL、collagen type IV(Nitta Gelatin Inc.製)が400μg/mLとなるようにD-PBS (-)で希釈し、37℃で2~4時間静置した。また、3成分によるコーティングは、Fibronectinとcollagen type IVとの混合液に1成分を追加してコーティングを行なった。この3成分目には、VTN-N(Thermo Fisher Scientific製)、ラミニン221断片(LN221F:Nippi Incorporated製 iMatrix221)、ラミニン411断片(LN411F:Nippi Incorporated製 iMatrix411)、ラミニン511断片(LN511F:Nippi Incorporated製 iMatrix511)、ラミニン断片mix(ラミニン221F、ラミニン411Fおよびラミニン511Fの混合物)、ラミニン211(Veritas製)、ラミニン411(Veritas製)、ラミニン511(Veritas製)、アグリン(R&D製)、ラミニンmix(ラミニン211、ラミニン411およびラミニン511の混合物)が該当する。VTN-N、LN221F、LN411Fの濃度は、10μg/mLとした。LN511Fの濃度は、10~200μg/mLとした。また、LN411FおよびLN511Fをいずれも100μg/mLとなるように加えた4成分によるコーティングも行った。また、ヒト不死化細胞株の培養に用いるCollagen type Iコーティングは、0.01% collagen type I溶液(新田ゼラチン製)で浸し、吸引後、静置して乾燥させた。Lippmannらの報告に準じた方法において用いるMatrigel GFRコーティングは、氷上でMatrigel GFR(Corning Incorporated製)をヒトiPS細胞の維持培地で30倍希釈し、37℃で30分以上静置した。
ヒトiPS細胞として610B1株(Riken Cell Bankより入手)を用い、VTN-Nをコートした培養dish上で、Essential 8 flex medium (Thermo Fisher Scientific製)を培地として用いて培養を行った。610B1株の継代時の剥離液には、0.5M EDTA (pH 8.0) またはTrypLE Select (Thermo Fisher Scientific製) を用いた。剥離にTrypLE Selectを用いた場合、継代直後にY-27632を10μM添加して翌日培地交換をした。
Essential 8 Flex medium中にて培養されたヒトiPS細胞を、TrypLE Selectで剥離し、2 × 104 cells/cm2の密度で、VTN-Nコート培養dish上に播種し、Y-27632(Focus Biomolecules製)を10μM 含むEssential 8 Flex mediumで24時間培養した。その後、modified DMEM/F12 (0.1% chemically defined lipid concentrate、0.1 × insulin - transferrin - selenium (Thermo Fisher Scientific製)、2 mM GlutaMAX (Sigma-Aldrich製)、450 mM 1-thioglycerol (Sigma-Aldrich製)、50μg/mL of L-ascorbic acid phosphate magnesium salt n-hydrateおよび1 × penicillin - streptomycin solutionを含むDMEM/F12) に5 μMのCHIR-99021(Focus Biomolecules製)を添加した培地で1日培養した。その後、modified DMEM/F12に、50 ng/mLのFGF2を添加した培地で1日培養した。その後、modified DMEM/F12に50 ng/mL VEGF(Biolegend製)および25 ng/mL BMP4(Prospec製)を添加した培地で3日間培養した。この間毎日培地交換を行った。5日目に、10μM Y-27632にて1時間処理した細胞をTrypLE Selectで解離させ、3.5 × 104 cells/cm2で新しい gelatinコート培養dishに継代し、modified Human endothelial (HE)-SFM {HE-SFM (Thermo Fisher Scientific製) + 5% knockout serum replacement (Thermo Fisher Scientific製) + 1× penicillin - streptomycin}に50 ng/mLのVEGFと10 ng/mLのFGF2を添加した培地にて分化誘導を行った。この間毎日培地交換した。
10μMのY-27632にて1時間処理した分化8日目の細胞を、TrypLE Selectで45~60秒間処理し、iEPCs以外の細胞 (extra cells) をある程度剥がし、培養dishを数回弾くように叩く (タッピングする) ことで、完全にextra cellsを剥がした。この時点で、培養dishの一番端の細胞が剥がれにくい場合、培養dishの端の細胞を、一周円を描くように吸引した。その後D-PBS (-) で3回以上洗い、再びTrypLE Selectで処理し、iEPCsを剥離した後、tubeに回収し、100 × gで5分間遠心して新しい培養dishに再播種した。
純化後の細胞は、modified Human endothelial SFMに10 ng/mL EGF (GenScript製)、20 ng/mL FGF2、10 μM Y-27632、0.5 μM A 83-01(Adooq Bioscoence製)、3 μM CHIR-99021を加えた培地で培養を行った。培地交換は、播種後1日目 (分化9日目および12日目) において行った。また、継代は、分化11日目で行った。継代時は、D-PBS (-) で一回洗浄した後、37℃においてTrypLE Selectで5分以上処理し、細胞を剥離させた。その後、培地で細胞を15 mL tubeもしくは50 mL tubeに回収し、100 × gで5分間遠心した。その後、培地を用いて再懸濁させ、VTN-Nをコートした培養dishに、1 × 104 cells/cm2または1.5 × 104 cells/cm2で再播種した。分化11日目の時点でextra cellsの残存率が多く、純化しきれていなかったと判断した場合、D-PBS (-) にて洗浄した後、TrypLE Selectを加え、常温にて顕微鏡で観察し、extra cellsが剥がれ始めた段階で数回強くタッピングし、剥離液を吸引し、D-PBS (-) にて一回洗浄した後、再度TrypLE Selectを加え、通常の継代法を適用した。分化14日目まで培養した。
継代時と同様の操作で、分化14日目のiEPCsの細胞ペレットを作製し、TC Protector(DSファーマバイオメディカル社製)にて再懸濁後、-80℃のディープフリーザーにて凍結し、保存した。解凍時は、37℃の温浴にて半融解させ、あらかじめ温めておいた15 mL tubeもしくは50 mL tubeに入った10 mLの培地の中から1 mLを半融解の細胞懸濁液に注ぎ、完全に融解させた後、tubeに戻し、100 × gで5分間遠心した。その後、培地で再懸濁し、wellプレートまたはTranswell(登録商標)に播種した。また、hCMEC/D3から調製された血管内皮前駆細胞についても、同様の方法により凍結保存・解凍した。
コーティング剤および培地への添加物を互いに異ならせた条件において、iEPCsをiBMELCsへと分化誘導した。培養dishのコーティングには、上述のように、FBNとCOL4との2成分、またはこれら2成分にVTN-N、LN221、LN411F、LN511Fのうちのいずれか1成分を追加した3成分もしくはLN411FとLN511Fとを加えた4成分を用いた。また、3成分目として、ラミニン211、ラミニン411、ラミニン511、アグリン、ラミニンmix、ラミニン断片mix、hep exも用いた。非凍結・凍結iEPCs (分化14日目) を播種し、HE-SFM (+1 × penicillin - streptomycin 以降省略) に2~10% KSRおよび20 ng/mL FGF2を添加した培地、HE-SFMに5% FBS(Sigma-Aldrich Corporation製)および20 ng/mL FGF2を添加した培地、HE-SFMに5% Platelet-poor plasma-derived bovine serum (PDS) (Alfa Aesar製) および20ng/mL FGF2を添加した培地、HE-SFMに2~10% B27(登録商標) supplement (Thermo Fisher Scientific製) および20 ng/mL FGF2を添加した培地、もしくはHE-SFMに7.5% B27 supplement、0.1~10μM A 83-01および20 ng/mL FGF2を添加した培地で培養し、脳毛細血管様細胞へと分化誘導した。播種後1日目に培地交換し、その後は1日おきに培地交換した。また、培養dishにiEPCsを播種する際の細胞密度を検討するために、100μg/mLのFibronectin、400μg/mLのcollagen type IV、10μg/mLのLN511Fでコーティングした培養dishに、1 × 105 cells/cm2、3 × 105 cells/cm2、5 × 105 cells/cm2、7 × 105 cells/cm2、9 × 105 cells/cm2にて播種を行い、HE-SFMに7.5% B27 supplement、1μM A 83-01および20 ng/mL FGF2を添加した培地にて培養を行った。また、ヒト不死化脳毛細血管内皮細胞(hCMEC/D3)についても、同様に、後述する分化誘導条件Aを用いて脳毛細血管様細胞へと分化誘導した。
Lippmannらの報告を改変したプロトコールを基に行なった。ヒトiPS細胞をMatrigel-GFRにてコーティングした6-wellプレートに播種し、35 ng/mLのFGF2を添加したStemSure hPSC medium (wako製) にて3日間または4日間培養した。この間培地交換は毎日行なった。分化開始日 (60-70%コンフルエント) に培地をDMEM/F12 based medium (iPS cell medium) に変更し、6日間培養した。この間培地交換は毎日行なった。分化6日目に培地を10 μM のall-trans-retinoic acid (RA)(Tocris Bioscience製) および20 ng/mLのFGF2を含有するHE-SFM based medium (1% PDSおよび1 × Penicillin-streptomycin solution(Biological Industries USA, Inc.製)を含有するHE-SFM) でさらに2日間培養した。この間毎日培地交換を行なった。分化8日目に分化細胞をD-PBS (-) で洗浄し、Accutase cell detachment solutionを加え、37℃で20分間静置した。その後培地を用いて回収した細胞を100 × gで5分間遠心し、細胞懸濁液を作製後、Fibronectin + collagen type IVコートwellプレートまたはTranswell(登録商標)(12-well, pore size: 1μm) に播種し、10μMのRAおよび20 ng/mLのFGF2を添加したHE-SFM based mediumにて培養した。分化9日目に培地をRAとFGF2非添加のHE-SFM based mediumに変更し、培養した。
上記の方法によりhiPSCsから分化誘導されたiEPCsに対し、EPCsとしての性状を確認するために、免疫染色法による解析を行なった。この免疫染色おける抗体には、PECAM1およびCD34を用いた。また、以下の分化誘導条件Aを用いた上記方法により凍結iEPCsから分化誘導されたiBMELCsと、比較例として、Fibronectin 100μg/mLとcollagen type IV 400μg/mLとを含むコーティング剤と、7.5% KSRと20 ng/mL FGF2とを添加したHE-SFM培地を用いて培養した細胞とに対し、免疫染色によるタンパク質発現解析を行なった。この免疫染色おける抗体には、PECAM1、CDH5、ZO-1、CLAUDIN5、P-gp、BCRPおよびGLUT1を用いた。96-wellプレート上の細胞を、0.1% BSA含有D-PBS (-) で3回洗浄後、4% paraformaldehyde中で15分間固定し、再度0.1% BSA含有D-PBS (-) で洗浄した後、0.1 w/v% Triton X-100含有D-PBS (-) にて5分間透過処理を行った。続いて、0.1% BSA含有D-PBS (-) で洗浄した後、一次抗体を4℃で1晩反応させた。その後、0.1% BSA含有D-PBS (-) で3回洗浄し、室温で二次抗体(Anti-Rabbit、Anti-Mouse)を200倍希釈で60分間反応させた。その後、0.1% BSA含有D-PBS (-) で3回洗浄し、核染色試薬である1μg/mLのDAPI(Dojindo製)を5分間反応させた。その後、D-PBS (-) で3回洗浄し、Operetta High-Content Imaging System (PerkinElmer製) にて解析した。免疫染色において用いた抗体の詳細を、表1に示す。
コーティング剤:100μg/mLのFibronectin、400μg/mLのcollagen type IV、10μg/mLのLN511F
培地:7.5% B27(登録商標)supplement、1μM A 83-01および20 ng/mL FGF2を添加したHE-SFM

hiPSCsから分化誘導されたiEPCsに対し、EPCsとしての性状を確認するために、アセチル化LDLの取り込み試験を行った。hiPSCsから分化誘導されたiEPCs(分化14日目)に、Dilで標識されたアセチル化LDLを終濃度10 μg/mLで5時間反応させ、その後、蛍光標識であるHoechist33342を30分間反応させ、培地で4回洗浄し、Operetta High-Content Imaging Systemにて解析した。
hiPSCsから分化誘導されたiEPCsにおける凍結保存の影響を確認するために、凍結の有無による経内皮電気抵抗(TEER値)の違いを比較した。分化14日目のiEPCsを上記の方法により凍結し、解凍させた凍結群と、分化14日目のiEPCsを凍結しなかった非凍結群とを、上記の分化誘導条件Aを用いた上記方法によりiBMELCsへと分化誘導させ、TEER値を測定した。凍結群と非凍結群のサンプル数は、それぞれ3とした。非凍結群の最大TEER値を基準として、凍結群の相対最大TEER値を求めた。本明細書において、「最大TEER値」とは、播種後2日目から播種後10日目までの各日に測定したTEER値のうち、最大の値を意味している。TEER値は、Millicell ERS-2 (チョップスティック型) を使用し、添付マニュアルに従い測定した。培地の液量は、頂端側で600 μL、基底側で1800 μLとした。
コーティング剤および培地を互いに異ならせた各誘導条件を用いて上記方法により凍結iEPCsから分化誘導されたiBMELCsに対し、TEER値を解析することによりバリア機能を評価した。サンプル数は、特に断らない限り、それぞれ3とした。TEER値の測定は、上記のとおり行った。
互いに異なる4つの誘導条件を用いて上記方法により凍結iEPCsから分化誘導されたiBMELCsに対し、傍細胞経路による透過の指標となるFD4およびLYを透過対象として、透過試験を行った。4つの分化誘導条件では、FBNとCOL4との2成分を含むコーティング剤と20 ng/mLのFGF2を含有するHE-SFM培地とが共通条件であり、サンプル「KSR」では培地にさらに7.5% KSRを含有させ、サンプル「B27」では培地にさらに7.5% B27sを含有させ、サンプル「B27+A 83-01」では培地にさらに7.5% B27sと1μM A 83-01とを含有させ、サンプル「B27+A 83-01+LN511F」では培地にさらに7.5% B27sと1μM A 83-01とを、コーティング剤にはさらに10μg/mL LN511Fを含有させた。サンプル数は、それぞれ3とした。透過試験にはTranswell(登録商標) (12-well, pore size: 1μm) 上の細胞を用いた。播種後4日目に、培地をtransport buffer (10 mM HEPE solutionを含むHBSS) に置き換え、37℃で20分間培養した。1 mg/mL FD4または300 μM LYを含有するtransport bufferを頂端側に加えた。37℃で60分間インキュベートした後、100 μLの溶液を側底側から回収した。トランスポートバッファーの液量は頂端側で500 μL、基底側で1500 μLとした。FD4またはLYの蛍光強度をSynergy HTX multimode plate reader (BioTek) で測定し、Gen 5 data analysis software (BioTek) にて解析した。
脳毛細血管内皮細胞マーカータンパク質発現量の解析を行なった。サンプルとしては、上記の分化誘導条件Aを用いて上記方法により凍結iEPCsから分化誘導されたiBMELCs(播種後4日目)と、比較例として、Fibronectin 100μg/mLとcollagen type IV 400μg/mLとを含むコーティング剤と、7.5% KSRと20 ng/mL FGF2とを添加したHE-SFM培地を用いて培養した細胞(播種後4日目)とを用いた。サンプル数は、それぞれ3とした。タンパク質発現量の解析には、Operetta High-Content Imaging Systemを用い、上記のとおり免疫染色を施した細胞をオペレッタ顕微鏡にて観察し、Harmony(登録商標) high-content analysis softwareにて蛍光強度を算出した。比較例におけるタンパク質の発現量を基準として、iBMELCsにおけるタンパク質の相対的発現量を求めた。
脳毛細血管内皮細胞マーカー遺伝子発現量の解析を、RT-q PCRにて行なった。サンプルとして、上記の分化誘導条件Aを用いて上記方法により凍結iEPCsから分化誘導されたiBMELCs(播種後4日目、サンプル数3)、上記Lippmannらの報告を改変した方法により分化誘導された細胞(以下、「iBMELCs 2」とも記載、サンプル数3)、ヒト不死化
BMECs(以下、「hCMEC/D3」とも記載、サンプル数3)、ヒト初代BMECs(以下、「hBMECs」とも記載、サンプル数1)を用いた。RNA抽出は、Agencourt(登録商標) RNAdvance Tissue Kit(Beckman Coulter, Inc.製)の添付マニュアルに従い行った。逆転写反応として、相補的 DNA (cDNA)の合成を、ReverTra Ace(登録商標) qPCR RT Master Mix(Toyobo Co., Ltd.製)を使用し、添付マニュアルに従い行なった。RT-q PCRには、KAPA SYBR Fast qPCR Kit(Nippon Genetics Co., Ltd.製)を用い、cDNAを鋳型にして、反応は添付マニュアルに従い行なった。得られた結果を、内在性コントロールとしてHPRT1を用いて補正した。hBMECsにおけるmRNAの発現量を基準として100とし、iBMELCs、iBMELCs 2およびhCMEC/D3におけるmRNAの相対的発現量を求めた。
iEPCsと不死化細胞とのTEER値を比較検討した。上記の分化誘導条件Aを用いて上記方法により凍結iEPCsから分化誘導されたiBMELCsおよびhCMEC/D3-分化誘導有り (播種後4日目)と、比較例として、Fibronectin 100μg/mLとcollagen type IV 400μg/mLとを含むコーティング剤と、7.5% KSRと20 ng/mL FGF2とを添加したHE-SFM培地を用いて培養したiEPCsおよびhCMEC/D3-分化誘導無し(播種後4日目)とを用いた。サンプル数は、それぞれ3とした。TEER値の測定は上記のとおり行い、最大TEER値を比較した。
上記の分化誘導条件Aを用いて上記方法により凍結iEPCsから分化誘導されたiBMELCs(播種後4日目)に対して、排出トランスポーターとしてのP糖タンパク質(P-gp)の機能解析のために、基質取り込み試験を行なった。基質取り込み試験には、Transwell(登録商標) (12-well, pore size: 1μm) 上の細胞を用いた。基質としては、rhodamine 123を用いた。培地を除去し、トランスポートバッファー中にて37℃で15分間プレインキュベートした。なお、サンプル数は3とした。P-gpの阻害剤である10 μMシクロスポリンA (CsA)の存在下または非存在下で、10 μM rhodamine 123を含有するトランスポートバッファー中にて37℃で60分間インキュベートした。その後、細胞をPBSで三回洗浄し、5 w/v% Triton X-100含有 PBSで溶解し、rhodamine 123の蛍光強度を、Synergy HTXマルチモードプレートリーダーで測定し、Gen5データ分析ソフトウェアで解析した。
(1)iEPCs の性状確認結果
図2は、分化14日目のiEPCsの性状確認の結果を示す説明図である。図2において、紙面左側の画像は、免疫染色法によるタンパク質発現解析の結果を示しており、紙面右側の画像は、アセチル化LDL取り込み試験の結果を示している。いずれの画像においても、100μmのスケールバーが示されている。図2に示す結果から、血管内皮前駆細胞マーカーであるPECAM1およびCD34が発現していることを確認でき、また、血管内皮前駆細胞の機能の一つであるアセチル化LDLの取込機能を細胞が有していることがわかった。すなわち、上記方法にて分化および純化された細胞が、血管内皮前駆細胞の性状を有していることがわかった。
図3は、非凍結iEPCsおよび凍結iEPCsの電気抵抗値を比較して示す説明図である。図3において、縦軸は、非凍結群の最大TEER値を基準として1.0とした場合の相対最大TEER値(Ω×cm2)を示している。図3に示す結果から、非凍結群と凍結群との最大TEER値に有意差はなく、凍結iEPCsが非凍結iEPCsと同等のバリア機能を有することがわかった。したがって、hiPSCsから分化誘導されたiEPCsを凍結保存してiEPCsストックを作成し、そのiEPCsストックをiBMELCsの調製に利用できることがわかった。
図4~10は、iEPCsからiBMELCsへの分化誘導条件の検討結果を示す説明図である。分化誘導条件として、図4では培地成分の種類、図5では培地成分の濃度、図6ではA 83-01の濃度、図7および図8ではコーティング成分種、図9ではLN511Fの濃度、図10では細胞数の検討結果を示している。図4~図6、図8~図10における縦軸は、最大TEER値(Ω×cm2)を示し、図7における縦軸はcontrol(FBNとCOL4との2成分のみ)の最大TEER値(Ω×cm2)を基準として1.0とした場合の各サンプルの相対最大TEER値を示している。
図12は、各分化誘導条件におけるFD4およびLYの透過試験の結果を示す説明図である。図12において、縦軸は、透過係数 Papp(10-6 cm / sec)を示している。図12において用いた4つの分化誘導条件は、TEER値の経時的変化の解析において用いた分化誘導条件と同じである。図12に示す結果から、傍細胞経路による透過の指標となるFD4とLYとのいずれを透過対象とした場合においても、LN511Fを含有するコーティング剤を用いて分化誘導したiBMELCsは、LN511Fを含有しないコーティング剤を用いて分化誘導したiBMELCsよりも、低い透過係数を示すことがわかった。すなわち、LN511Fを含有するコーティング剤を用いて分化誘導したiBMELCsは、LN511Fを含有しないコーティング剤を用いて分化誘導したiBMELCsよりも、高いバリア機能を有することがわかった。
図13は、免疫染色によるタンパク質発現解析結果と、Operetta High-Content Imaging Systemによるタンパク質発現量の解析結果とを示す説明図である。図13の紙面上側は、50μmのスケールバーとともに、各BMECsマーカーに対する免疫蛍光染色法によって得られた画像を示しており、図13の紙面下側は、相対タンパク質発現量を示している。相対タンパク質発現量は、比較例の細胞(図13において「iEPCs」と記載)におけるタンパク質発現量を基準として1.0とした場合の相対値として示している。免疫染色によるタンパク質発現解析結果から、以下のことがわかった。すなわち、血管内皮細胞マーカーとして代表的なPECAM1およびCDH5が、iBMELCsの細胞膜上に局在しているため、iBMELCsが血管内皮細胞としての性質を有していることが示唆された。また、iBMELCsは、比較例の細胞と比較して、タイトジャンクションマーカーのZO-1およびCLAUDIN5、薬物排出トランスポーターマーカーのP-gpおよびBCRP、グルコーストランスポーターマーカーのGLUT1等のBMECsマーカーの発現量が上昇した。したがって、上記の分化誘導方法により製造されたiBMELCsは、脳毛細血管内皮細胞としての特徴を強く有することが示唆された。
図14は、RT-q PCR法による遺伝子発現解析の結果を示す説明図である。図14の縦軸は、ヒト初代BMECs(hBMECs)におけるmRNA発現量を基準として100とした場合の相対mRNA発現量を示している。図14に示す結果から、iBMELCsは、PECAM1、CDH5、ZO-1、OCCLUDIN、BCRPの各BMECsマーカーについて、Lippmannらの報告を改変した方法により分化誘導された細胞(iBMELCs 2)やhBMECs、ヒト不死化BMECs(hCMEC/D3)よりも優位に高い発現量を示すことがわかった。また、iBMELCsは、GLUT1について、hBMECsおよびhCMEC/D3よりも優位に高い発現量を示すとともに、iBMELCs 2と同等の発現量を示した。また、iBMELCsは、P-gpについても、hBMECsと同等に発現することが確認できた。
図15は、iEPCsと不死化細胞とにおける電気抵抗値を比較して示す説明図である。図15における縦軸は、最大TEER値(Ω×cm2)を示しており、「+」は、上記の分化誘導条件Aを用いて分化誘導したサンプルを示しており、「-」は、比較例として、分化誘導条件Aとは異なる上述の条件により培養したサンプルであることを示している。図15に示す結果から、iPS細胞と不死化細胞とのいずれを用いた場合においても、分化誘導条件Aを用いて上記方法により分化誘導した細胞では高いTEER値を示すことがわかった。すなわち、iPS細胞由来と不死化細胞由来とのいずれの場合においても、上記の培養方法により分化誘導を行うことにより、脳毛細血管内皮細胞を模倣したバリア機能を有する脳毛細血管内皮様細胞を製造できることが示された。また、図15に示す結果から、iPS細胞を用いて分化誘導を行うことにより、不死化細胞を用いて分化誘導を行った場合と比較して、より高いバリア機能を有するiBMELCsを製造できることが示された。
図16は、P糖タンパク質(P-gp)の機能解析の結果を示す説明図である。図9および図16において、縦軸は、シクロスポリンの非存在下における基質蓄積量を基準として1.0とした場合の相対的基質蓄積量を示しており、「-」は、シクロスポリン非存在下のサンプルを示し、「+」は、シクロスポリン存在下のサンプルを示している。図16に示す結果から、上記方法により分化誘導されたiBMELCsは、排出トランスポーターに対する阻害剤であるシクロスポリンの存在下において、かかる阻害剤の非存在下と比較して基質の蓄積量が多くなることが確認できた。したがって、上記方法により分化誘導されたiBMELCsは、P-gpの機能を有することがわかった。
以上の結果より、上記方法を用いて分化誘導を行うことにより、血管内皮細胞としての性質を有し、生体内の脳毛細血管内皮細胞に対する模倣性(類似性)を向上させた脳毛細血管内皮様細胞を製造できることがわかった。
図17は、iEPCsとiBPCとの共培養の概要を説明するための説明図である。図17では、ヒトiPS細胞(hiPSCs)をヒトiPS細胞由来脳ペリサイト(以下、「iBPC」とも呼ぶ)へと分化誘導し、そのiBPCと上述のiEPCsとを共培養する方法の概要が模式的に示されている。後述するように、セルカルチャーインサートの裏面の膜上にiBPCを播種した後、分化誘導8日目に培地にA 83-01を加え、ペリサイト分化培地にて培養を行った。その後、分化誘導12日目にセルカルチャーインサートの表側にiEPCsを播種して、BMECs分化培地にて共培養した。分化誘導13日目から19日目まで、すなわち共培養1日目から7日目まで、TEER値を測定して解析することにより、バリア機能を評価した。
iBPCに分化誘導させるためのhiPSCとしては、802-3G株(株式会社リプロセルより入手)を用いた。このhiPSCを維持培養する際のMatrigel GFRコーティングは、氷上でMatrigel GFR(Corning Incorporated製)をヒトiPS細胞の維持培地で30倍希釈し、その後、37℃で30分以上静置して作製した。このhiPSCを、VTN-Nをコートした培養dish上で、mTeSR plus medium(StemCell Technologies製)を培地として用いて培養を行った。802-3G株の継代時の剥離液には、0.5M EDTA(pH 8.0)またはTrypLE Select(Thermo Fisher Scientific製)を用いた。剥離にTrypLE Selectを用いた場合、継代直後にY-27632を10μM添加して翌日培地交換した。なお、上記培養dishへのVTN-Nコーティングは、VTN-N(Thermo Fisher Scientific製)を1μg/cm2となるようにD-PBS(-)で希釈し、その後、37℃で1~2時間静置して作製した。
後述する方法によってhiPSCから分化4日目まで分化誘導させたiBPCを用いた。培養dishのコーティングとして、セルカルチャーインサート内部(apical)側の膜上には、100μg/mLのFibronectin、400μg/mLのcollagen type IV、10μg/mLのLN511F の3成分を用いた。培養dishのコーティングとして、セルカルチャーインサート外部(basal)側の膜上には、1μg/cm2のVTN-Nを用いた。10 cm 培養皿の上にセルカルチャーインサートを裏返した状態で静置し、その後、iBPC(分化4日目)を1.2×104 cells/cm2にて膜上に播種し、EGM-2に10μMのY-27632と50 ng/mLのPDGF-BBと1μMのA 83-01とを添加した培地にて2時間培養を行った。その後、iBPCを播種したセルカルチャーインサートを12 wellコンパニオンプレートに設置し、apical側に600μL、basal側に1800μLのEGM-2に、10μMのY-27632と50 ng/mLのPDGF-BBとを添加した培地にて培養を行った。分化5日目からは、EGM-2に50 ng/mLのPDGF-BBを添加した培地にて培養を行い、分化8日目からは、EGM-2に50 ng/mLのPDGF-BBと1μM A 83-01とを添加した培地にて培養を行った。この間毎日培地交換した。分化12日目に凍結iEPCs (分化14日目) を1×105 cells/cm2にてapical側に播種し、HE-SFMに7.5% B27 supplementと1μMのA 83-01と20 ng/mLのFGF2とを添加した培地で分化19日目まで培養し、脳毛細血管様細胞へと分化誘導した。この間毎日培地交換した。
上記方法により凍結iEPCsおよびiBPCを共培養した条件下で分化誘導されたiBMELCsに対して、TEER値を解析することによりバリア機能を評価した。解析には、Two-way Repeated-Measures ANOVAを用いた。また、上記方法により凍結iEPCsを単独で培養した場合におけるiBMELCsに対しても、同様にTEER値を測定した。サンプル数は、それぞれ3とした。TEER値は、Millicell ERS-2 (チョップスティック型) を使用し、添付マニュアルに従い測定した。培地の液量は、頂端側で600μL、基底側で1800μLとした。
hiPSCからiBPCへの分化誘導に及ぼすA 83-01の影響を調べた。
上述のhiPSCを用いてiBPCへの分化誘導を行った。mTeSR plus medium中にて培養されたヒトiPS細胞を、TrypLE Selectで剥離した後、2×104 cells/cm2の密度で、VTN-Nコート培養dish上に播種し、Y-27632(Focus Biomolecules製)を10μM含むmTeSR plus mediumで24時間培養した。その後、modified DMEM/F12(0.1% chemically defined lipid concentrate、0.1×insulin-transferrin-selenium (Thermo Fisher Scientific製)、2 mM GlutaMAX(Sigma-Aldrich製)、450 mM 1-thioglycerol(Sigma-Aldrich製)、50μg/mL of L-ascorbic acid phosphate magnesium salt n-hydrateおよび1×penicillin-streptomycin solutionを含むDMEM/F12)に5μMのCHIR-99021(Focus Biomolecules製)を添加した培地で1日培養した。その後、modified DMEM/F12に、50 ng/mLのFGF2を添加した培地で1日培養した。その後、EGM-2(2% FBS、0.2μg/mL Hydrocortisone、10 ng/mL FGF2、20 ng/mL R3-IGF-1、1μg/mL Ascorbic acid、5 ng/mL hEGF、Gentamicin、amphotericinBを含むEGM-2 basal medium)に50 ng/mL PDGF-BBおよび0.5 ng/mL VEGFを添加した培地で2日間培養した。この間毎日培地交換を行った。4日目に、10μM Y-27632にて1時間処理した細胞をTrypLE Selectで解離させた後、1.25×104 cells/cm2で新しい VTN-Nコート培養dishに継代し、EGM-2培地に50 ng/mLのPDGF-BBを添加した培地にて分化誘導を行った。この間毎日培地交換した。8日目に、EGM-2に50 ng/mLのPDGF-BB、1μMのA 83-01を添加した培地にて分化誘導を行った。この間毎日培地交換した。以下の説明では、培地にA 83-01を添加して分化誘導されたiBPCsを、「X(+)」とも記載し、培地にA 83-01を添加せずに分化誘導されたiBPCsを、「X(-)」とも記載する。
上記の方法によりhiPSCsから分化誘導されたiBPC(X(+)およびX(-))に対し、BPCとしての性状を確認するために、免疫染色法による解析を行った。この免疫染色における抗体には、PDGFR-β、NG2およびα-SMAを用いた。96-wellプレート上の細胞を、0.1% BSA含有D-PBS(-)で3回洗浄後、4% paraformaldehyde中で15分間固定し、その後、再度0.1% BSA含有D-PBS(-)で3回洗浄した後、0.1w/v% Triton X-100含有D-PBS(-)にて5分間透過処理を行った。続いて、0.1% BSA含有D-PBS(-)で洗浄した後、一次抗体を4℃で1晩反応させた。その後、0.1% BSA含有D-PBS(-)で3回洗浄した後に、室温で二次抗体(Anti-Rabbit)を200倍希釈で60分間反応させた。その後、0.1% BSA含有D-PBS (-) で3回洗浄し、核染色試薬である1μg/mLのDAPI(Dojindo製)を5分間反応させた。その後、D-PBS (-) で3回洗浄した後に、Operetta High-Content Imaging System (PerkinElmer製) にて解析した。免疫染色において用いた抗体の詳細を、表2に示す。なお、表2において、「Target」は抗体名を示し、「Source」は抗体の製造会社名を示し、「Catalog number」は抗体のカタログナンバーを示し、「Species」は抗体の由来を示し、「Dilution」は体積換算における希釈率を示す。

脳ペリサイトマーカー遺伝子発現量の解析をRT-q PCRにて行った。サンプルとして、上記の方法においてA 83-01を添加して分化誘導された分化12日目のiBPCs(X(+))、上記の方法においてA 83-01を添加せずに分化誘導された分化12日目のiBPCs(X(-))、ヒト初代ペリサイト(以下、「hPC」とも記載する)を用いた。サンプル数は、いずれも3とした。RNA抽出は、Agencourt(登録商標) RNAdvance Tissue Kit(Beckman Coulter, Inc.製)の添付マニュアルに従い行った。逆転写反応として、相補的 DNA (cDNA)の合成を、ReverTra Ace(登録商標) qPCR RT Master Mix(Toyobo Co., Ltd.製)を使用し、装置の添付マニュアルに従い行なった。RT-q PCRには、KAPA SYBR Fast qPCR Kit(Nippon Genetics Co., Ltd.製)を用い、cDNAを鋳型にして、反応は装置の添付マニュアルに従い行なった。得られた結果を、内在性コントロールとしてHPRT1を用いて補正した。hPCにおけるmRNAの発現量を基準として1とし、X(+)、X(-)におけるmRNAの相対的発現量を求めた。解析には、One-way ANOVAおよびTukey’s testを用いた。
サンプルとして、上記の方法においてA 83-01を添加して分化誘導されたiBPCs(X(+))と、上記の方法においてA 83-01を添加せずに分化誘導されたiBPCs(X(-))とを用いた。サンプル数は、いずれも1とした。上記の方法により6-well プレート上で分化誘導されたiBPC(分化12日目)に、10μLピペットチップを用いて縦に傷をつけた。その後、EGM-2に50 ng/mLのPDGF-BBを添加した培地を用いて培養を行った。その培養の際に、A 83-01を添加して分化誘導されたiBPCs(X(+))の培地には、0.1~10μMのA 83-01をさらに添加し、A 83-01を添加せずに分化誘導されたiBPCs(X(-))の培地には、A 83-01を添加しなかった。細胞損傷後における培養0、12、24時間後の細胞遊走の様子を明視野観察にて確認し、Image Jにて、損傷面積の算出を行った。
図19は、脳ペリサイトマーカータンパク質の免疫染色の結果を示す説明図である。図19では、A 83-01を添加せずに分化誘導されたiBPCs(X(-))について、分化8日目と分化12日目とにおける免疫染色の結果が示されている。いずれの画像においても、100μmのスケールバーが示されている。図19に示す結果によれば、分化8日目と分化12日目において、経時的にPDGFR-β、NG2、α-SMAのタンパク質発現が強くなっていることが分かった。なお、生体内の脳ペリサイトでは、α-SMAの発現が弱いことが知られている。
また、いずれの画像においても、100μmのスケールバーが示されている。図23に示される画像では、図示の便宜上、細胞損傷時(細胞損傷後における培養0時間後)における細胞損傷部分の境界を太線で示し、その後の培養によって推移した細胞の境界を細線で示している。図24は、iBPCの細胞遊走能・増殖能を示す説明図である。図24の縦軸は、細胞損傷部分の距離を、細胞損傷時(細胞損傷後における培養0時間後)を基準として1とした場合の相対値を示している。
配列番号2:人工配列の説明:CDH5 リバースプライマー
配列番号3:人工配列の説明:MDR1 フォワードプライマー
配列番号4:人工配列の説明:MDR1 リバースプライマー
配列番号5:人工配列の説明:BCRP フォワードプライマー
配列番号6:人工配列の説明:BCRP リバースプライマー
配列番号7:人工配列の説明:GLUT1 フォワードプライマー
配列番号8:人工配列の説明:GLUT1 リバースプライマー
配列番号9:人工配列の説明:Occludin フォワードプライマー
配列番号10:人工配列の説明:Occludin リバースプライマー
配列番号11:人工配列の説明:ZO-1 フォワードプライマー
配列番号12:人工配列の説明:ZO-1 リバースプライマー
配列番号13:人工配列の説明:LAT1 フォワードプライマー
配列番号14:人工配列の説明:LAT1 リバースプライマー
配列番号15:人工配列の説明:HPRT1 フォワードプライマー
配列番号16:人工配列の説明:HPRT1 リバースプライマー
配列番号17:人工配列の説明:PECAM1 フォワードプライマー
配列番号18:人工配列の説明:PECAM1 リバースプライマー
配列番号19:人工配列の説明:PDGFR-β フォワードプライマー
配列番号20:人工配列の説明:PDGFR-β リバースプライマー
配列番号21:人工配列の説明:NG2 フォワードプライマー
配列番号22:人工配列の説明:NG2 リバースプライマー
配列番号23:人工配列の説明:α-SMA フォワードプライマー
配列番号24:人工配列の説明:α-SMA リバースプライマー
Claims (13)
- 脳毛細血管内皮様細胞の製造方法であって、
Laminin511断片と、Fibronectinと、collagen type IVと、を用いて血管内皮前駆細胞を培養する培養工程を含む、
脳毛細血管内皮様細胞の製造方法。 - 請求項1に記載の製造方法において、
前記培養工程では、B27(登録商標) supplementと、A 83-01と、Fibroblast Growth Factor-2(FGF2)と、を含む培地を用いて培養する、
脳毛細血管内皮様細胞の製造方法。 - 請求項1または請求項2に記載の製造方法において、
前記血管内皮前駆細胞は、多能性幹細胞から分化誘導された細胞である、
脳毛細血管内皮様細胞の製造方法。 - 請求項3に記載の製造方法において、
前記血管内皮前駆細胞は、ヒト人工多能性幹細胞から分化誘導された細胞である、
脳毛細血管内皮様細胞の製造方法。 - 請求項3または請求項4に記載の製造方法において、
前記血管内皮前駆細胞は、血管内皮増殖因子を用いて分化誘導された細胞である、
脳毛細血管内皮様細胞の製造方法。 - 請求項1から請求項5までのいずれか一項に記載の製造方法において、
前記培養工程では、前記血管内皮前駆細胞を脳ペリサイトと共培養する、
脳毛細血管内皮様細胞の製造方法。 - 請求項6に記載の製造方法において、
前記脳ペリサイトは、A 83-01を含む培地を用いて多能性幹細胞から分化誘導された細胞である、
脳毛細血管内皮様細胞の製造方法。 - 請求項1から請求項7までのいずれか一項に記載の製造方法により得られた脳毛細血管内皮様細胞の細胞層を用いて、被検物質の血液脳関門透過性を評価する方法。
- 請求項8に記載の方法において、以下の工程(i)~(iii)を含む、方法:
(i)前記細胞層を用意する工程;
(ii)前記細胞層に前記被検物質を接触させる工程;
(iii)前記細胞層を透過した前記被検物質を定量することにより、前記被検物質の血液脳関門透過性を評価する工程。 - 請求項1から請求項7までのいずれか一項に記載の製造方法により得られた脳毛細血管内皮様細胞の細胞層を用いて、被検物質の血液脳関門バリア機能への影響を評価する方法。
- 請求項1から請求項7までのいずれか一項に記載の製造方法で得られた、脳毛細血管内皮様細胞。
- 請求項11に記載の脳毛細血管内皮様細胞において、
PECAM1の発現量が、初代培養脳毛細血管内皮細胞および不死化脳毛細血管内皮細胞よりも大きく、
TEER値が、50Ω×cm2以上である、
脳毛細血管内皮様細胞。 - 脳毛細血管内皮様細胞の分化誘導方法であって、
Laminin511断片と、Fibronectinと、collagen type IVと、を用いて血管内皮前駆細胞を培養する培養工程を含む、
脳毛細血管内皮様細胞の分化誘導方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023522717A JPWO2022244841A1 (ja) | 2021-05-20 | 2022-05-19 | |
| EP22804750.2A EP4342980A1 (en) | 2021-05-20 | 2022-05-19 | Method for producing brain capillary endothelial-like cells, and use therefor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-085121 | 2021-05-20 | ||
| JP2021085121 | 2021-05-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022244841A1 true WO2022244841A1 (ja) | 2022-11-24 |
Family
ID=84141602
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/020827 WO2022244841A1 (ja) | 2021-05-20 | 2022-05-19 | 脳毛細血管内皮様細胞の製造方法およびその利用 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP4342980A1 (ja) |
| JP (1) | JPWO2022244841A1 (ja) |
| WO (1) | WO2022244841A1 (ja) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007166915A (ja) * | 2005-12-19 | 2007-07-05 | Masami Niwa | 血液脳関門インヴィトロ・モデル、病態血液脳関門インヴィトロ・モデル、及びこれを用いた薬物スクリーニング方法、病態血液脳関門機能解析方法、病因解析方法 |
| US20170283772A1 (en) * | 2016-04-05 | 2017-10-05 | Wisconsin Alumni Research Foundation | Methods for differentiation of human pluripotent stem cells to brain microvascular endothelial cells |
| JP2018110548A (ja) | 2017-01-11 | 2018-07-19 | 公立大学法人名古屋市立大学 | 血管内皮前駆細胞の調製方法 |
| WO2019208736A1 (ja) * | 2018-04-27 | 2019-10-31 | 公立大学法人名古屋市立大学 | 多能性幹細胞から脳毛細血管内皮細胞への分化誘導方法 |
| WO2020179380A1 (ja) | 2019-03-06 | 2020-09-10 | 公立大学法人名古屋市立大学 | 血管内皮前駆細胞の調製及び拡大培養 |
| JP2021085121A (ja) | 2019-11-28 | 2021-06-03 | 東レ株式会社 | 嵩高糸 |
-
2022
- 2022-05-19 EP EP22804750.2A patent/EP4342980A1/en active Pending
- 2022-05-19 WO PCT/JP2022/020827 patent/WO2022244841A1/ja active Application Filing
- 2022-05-19 JP JP2023522717A patent/JPWO2022244841A1/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007166915A (ja) * | 2005-12-19 | 2007-07-05 | Masami Niwa | 血液脳関門インヴィトロ・モデル、病態血液脳関門インヴィトロ・モデル、及びこれを用いた薬物スクリーニング方法、病態血液脳関門機能解析方法、病因解析方法 |
| US20170283772A1 (en) * | 2016-04-05 | 2017-10-05 | Wisconsin Alumni Research Foundation | Methods for differentiation of human pluripotent stem cells to brain microvascular endothelial cells |
| JP2018110548A (ja) | 2017-01-11 | 2018-07-19 | 公立大学法人名古屋市立大学 | 血管内皮前駆細胞の調製方法 |
| WO2019208736A1 (ja) * | 2018-04-27 | 2019-10-31 | 公立大学法人名古屋市立大学 | 多能性幹細胞から脳毛細血管内皮細胞への分化誘導方法 |
| WO2020179380A1 (ja) | 2019-03-06 | 2020-09-10 | 公立大学法人名古屋市立大学 | 血管内皮前駆細胞の調製及び拡大培養 |
| JP2021085121A (ja) | 2019-11-28 | 2021-06-03 | 東レ株式会社 | 嵩高糸 |
Non-Patent Citations (39)
| Title |
|---|
| "Manipulating the Mouse Embryo A Laboratory Manual", 1994, COLD SPRING HARBOR LABORATORY PRESS |
| AKIRA IRIYA ET AL., PROTEIN, NUCLEIC ACID, ENZYME, vol. 44, 1999, pages 892 |
| AOKI HIROMASA, YAMASHITA MISAKI, HASHITA TADAHIRO, IWAO TAKAHIRO, MATSUNAGA TAMIHIDE: "Laminin 221 fragment is suitable for the differentiation of human induced pluripotent stem cells into brain microvascular endothelial-like cells with robust barrier integrity", FLUIDS AND BARRIERS OF THE CNS, vol. 17, no. Article no 25, 1 December 2020 (2020-12-01), pages 1 - 11, XP093008662, DOI: 10.1186/s12987-020-00186-4 * |
| BAGUISI ET AL., NATURE BIOTECHNOLOGY, vol. 17, 1999, pages 456 |
| CHUNG Y. ET AL., CELL STEM CELL, vol. 2, 2008, pages 113 - 117 |
| IWAO TAKAHIRO, HASHITA TADAHIRO, MATSUNAGA TAMIHIDE: "Generation of cells supporting drug discovery research using human iPS cells", DRUG DELIVERY SYSTEM, vol. 35, no. 4, 1 January 2020 (2020-01-01), pages 319 - 330, XP093008661 * |
| KIM DKIM CHMOON JI ET AL., CELL STEM CELL, vol. 4, 2009, pages 472 - 476 |
| KIM ET AL., CELL STEM CELL, vol. 1, 2007, pages 346 - 352 |
| KIM J B ET AL., CELL, vol. 136, no. 3, 2009, pages 411 - 419 |
| KIM J B ET AL., NATURE, vol. 454, no. 7204, 2008, pages 646 - 650 |
| KLIMANSKAYA I. ET AL., NATURE, vol. 444, 2006, pages 481 - 485 |
| LIPPMANN: "Matrigel GFR (manufactured by Corning Incorporated) was diluted 30 times with a human iPS cell maintenance medium on ice, and allowed to stand at 37°C for 30 minutes or longer.", CELL CULTURE |
| MATSUI ET AL., CELL, vol. 70, 1992, pages 841 - 847 |
| MOTALLEBNEJAD PEDRAM, AZARIN SAMIRA M.: "Chemically defined human vascular laminins for biologically relevant culture of hiPSC-derived brain microvascular endothelial cells", FLUIDS AND BARRIERS OF THE CNS, vol. 17, no. Article no. 54, 1 December 2020 (2020-12-01), pages 1 - 16, XP093008666, DOI: 10.1186/s12987-020-00215-2 * |
| NAKAGAWA M ET AL., NAT. BIOTECHNOL., vol. 26, no. 11, 2008, pages 1269 - 1275 |
| NAKAJIMA ET AL., STEM CELLS, vol. 25, 2007, pages 983 - 985 |
| NATURE GENETICS, vol. 22, 1999, pages 127 |
| PROC. NATL. ACAD. SCI. USA, vol. 96, 1999, pages 14984 |
| REVAZOVA ET AL., CLONING STEM CELLS, vol. 10, 2008, pages 11 - 24 |
| REVAZOVA ET AL., CLONING STEM CELLS, vol. 9, 2007, pages 432 - 449 |
| RIDEOUT III ET AL., NATURE GENETICS, vol. 24, no. 109, 2000 |
| SHAMBLOTT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 95, no. 23, 1998, pages 13726 - 13731 |
| SILVA J ET AL., PLOS. BIOL., vol. 6, no. 10, 2008, pages 253 |
| SRIRAM G. ET AL.: "Efficient differentiation of human embryonic stem cells to arterial and venous endothelial cells under feeder- and serum-free conditions.", STEM CELL RESEARCH & THERAPY, vol. 6, 2015, pages 261, XP055630854, DOI: 10.1186/s13287-015-0260-5 |
| STADTFELD M ET AL., SCIENCE, vol. 322, no. 5903, 2008, pages 949 - 953 |
| STRELCHENKO N. ET AL., REPROD BIOMED ONLINE., vol. 9, 2004, pages 623 - 629 |
| TACHIBANA ET AL.: "Human Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer", CELL, 2013 |
| TAKAHASHI KYAMANAKA S, CELL, vol. 126, no. 4, 2006, pages 663 - 676 |
| TAKAHASHI, K ET AL., CELL, vol. 131, no. 5, 2007, pages 861 - 72 |
| THOMSON, J. A. ET AL., SCIENCE, vol. 280, 1998, pages 1256 - 1147 |
| TURNPENNY ET AL., STEM CELLS, vol. 21, no. 5, 2003, pages 598 - 609 |
| WAKAYAMA ET AL., NATURE, vol. 394, no. 369, 1998 |
| WASSARMAN, P. M. ET AL., METHODS IN ENZYMOLOGY, vol. 365, 2003 |
| WILMUT ET AL., NATURE, vol. 385, no. 810, 1997 |
| WOLTJEN KMICHAEL IPMOHSENI P ET AL., NATURE, vol. 458, 2009, pages 771 - 775 |
| YU J ET AL., SCIENCE, vol. 318, no. 5858, 2007, pages 1917 - 1920 |
| YU JHU KSMUGA-OTTO KTIAN S ET AL., SCIENCE, vol. 324, 2009, pages 797 - 801 |
| YUSA KRAD RTAKEDA J ET AL., NAT METHODS, vol. 6, 2009, pages 363 - 369 |
| ZHANG X. ET AL., STEM CELLS, vol. 24, 2006, pages 2669 - 2676 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4342980A1 (en) | 2024-03-27 |
| JPWO2022244841A1 (ja) | 2022-11-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6678107B2 (ja) | 膵前駆細胞の増殖方法 | |
| JP7161775B2 (ja) | 中間中胚葉細胞から腎前駆細胞への分化誘導方法、および多能性幹細胞から腎前駆細胞への分化誘導方法 | |
| JP7166631B2 (ja) | 新規軟骨細胞誘導方法 | |
| JP5896421B2 (ja) | 多能性幹細胞から骨格筋または骨格筋前駆細胞への分化誘導法 | |
| US11746332B2 (en) | Method for producing renal progenitor cells | |
| AU2016320991A1 (en) | Method for producing retinal pigment epithelial cells | |
| JP6694215B2 (ja) | 新規軟骨細胞誘導方法 | |
| US20130052729A1 (en) | Method for preparing induced paraxial mesoderm progenitor (ipam) cells and their use | |
| US20210332329A1 (en) | Novel renal progenitor cell marker and method for concentrating renal progenitor cells using same | |
| JP7251812B2 (ja) | 多能性幹細胞から脳毛細血管内皮細胞への分化誘導方法 | |
| JP6646311B2 (ja) | 多能性幹細胞から中胚葉前駆細胞および血液血管前駆細胞への分化誘導法 | |
| WO2011060342A2 (en) | Cardiac differentiation of human pluripotent stem cells under defined conditions using matrix overlay methods | |
| JP2023100987A (ja) | 多能性幹細胞由来の板状軟骨およびその製造方法 | |
| WO2022244841A1 (ja) | 脳毛細血管内皮様細胞の製造方法およびその利用 | |
| KR102049211B1 (ko) | 내피세포의 제조 방법 | |
| WO2022168908A1 (ja) | 多能性幹細胞由来の陰窩-絨毛様構造を有する腸管細胞の製造方法及びその用途 | |
| JP7437766B2 (ja) | 中内胚葉系への分化抵抗性が解除された多能性幹細胞の作製方法 | |
| WO2021161991A1 (ja) | 多能性幹細胞を脳毛細血管内皮様細胞へと分化誘導するためのコーティング剤およびその利用 | |
| JP2015159785A (ja) | 多能性幹細胞から脳血管内皮細胞を製造する方法 | |
| JP7072756B2 (ja) | 多能性幹細胞から中胚葉前駆細胞および血液血管前駆細胞への分化誘導法 | |
| US20220135940A1 (en) | Method for producing kidney structure having dendritically branched collecting duct from pluripotent stem cells | |
| JP2024074875A (ja) | 血管内皮前駆細胞の調製及び拡大培養 | |
| JP2020115771A (ja) | 多能性幹細胞から軟骨組織を製造する方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22804750 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2023522717 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022804750 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022804750 Country of ref document: EP Effective date: 20231220 |