WO2022234850A1 - 環状ペプチド化合物を含む製剤及びその製造方法 - Google Patents
環状ペプチド化合物を含む製剤及びその製造方法 Download PDFInfo
- Publication number
- WO2022234850A1 WO2022234850A1 PCT/JP2022/019540 JP2022019540W WO2022234850A1 WO 2022234850 A1 WO2022234850 A1 WO 2022234850A1 JP 2022019540 W JP2022019540 W JP 2022019540W WO 2022234850 A1 WO2022234850 A1 WO 2022234850A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formulation
- added
- formula
- composition
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 336
- 238000004519 manufacturing process Methods 0.000 title claims description 24
- 238000002360 preparation method Methods 0.000 title description 74
- 108010069514 Cyclic Peptides Proteins 0.000 title description 10
- 102000001189 Cyclic Peptides Human genes 0.000 title description 10
- 239000000203 mixture Substances 0.000 claims abstract description 442
- 239000000654 additive Substances 0.000 claims abstract description 110
- 230000000996 additive effect Effects 0.000 claims abstract description 97
- 239000012453 solvate Substances 0.000 claims abstract description 64
- 150000003839 salts Chemical class 0.000 claims abstract description 62
- 239000007788 liquid Substances 0.000 claims abstract description 56
- -1 propylene glycol fatty acid ester Chemical class 0.000 claims description 108
- 239000004094 surface-active agent Substances 0.000 claims description 89
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 claims description 56
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 54
- 239000000194 fatty acid Substances 0.000 claims description 54
- 229930195729 fatty acid Natural products 0.000 claims description 54
- 238000000034 method Methods 0.000 claims description 48
- 239000004359 castor oil Substances 0.000 claims description 46
- 235000019438 castor oil Nutrition 0.000 claims description 45
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims description 45
- 229920003171 Poly (ethylene oxide) Polymers 0.000 claims description 44
- 230000002209 hydrophobic effect Effects 0.000 claims description 42
- 238000002156 mixing Methods 0.000 claims description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims description 13
- 229940113116 polyethylene glycol 1000 Drugs 0.000 claims description 12
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 12
- 239000002202 Polyethylene glycol Substances 0.000 claims description 11
- 125000005456 glyceride group Chemical group 0.000 claims description 11
- 229920001223 polyethylene glycol Polymers 0.000 claims description 11
- 239000003963 antioxidant agent Substances 0.000 claims description 10
- 229920001214 Polysorbate 60 Polymers 0.000 claims description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerol Natural products OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 7
- 230000003078 antioxidant effect Effects 0.000 claims description 7
- 235000011187 glycerol Nutrition 0.000 claims description 6
- 238000009472 formulation Methods 0.000 description 183
- 239000000243 solution Substances 0.000 description 118
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 117
- 239000013078 crystal Substances 0.000 description 100
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 99
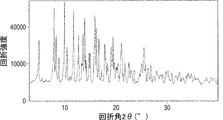
- 238000000634 powder X-ray diffraction Methods 0.000 description 71
- 229940125904 compound 1 Drugs 0.000 description 69
- 238000003756 stirring Methods 0.000 description 62
- 239000002904 solvent Substances 0.000 description 61
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 59
- 238000003746 solid phase reaction Methods 0.000 description 57
- 239000011347 resin Substances 0.000 description 49
- 229920005989 resin Polymers 0.000 description 49
- 239000002245 particle Substances 0.000 description 38
- 239000007790 solid phase Substances 0.000 description 38
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
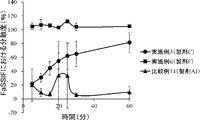
- 230000000052 comparative effect Effects 0.000 description 32
- 238000005406 washing Methods 0.000 description 30
- 230000002829 reductive effect Effects 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 25
- 238000009826 distribution Methods 0.000 description 25
- 238000006243 chemical reaction Methods 0.000 description 23
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 22
- 229920002685 Polyoxyl 35CastorOil Polymers 0.000 description 21
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 239000007963 capsule composition Substances 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 19
- 239000002775 capsule Substances 0.000 description 19
- 239000011259 mixed solution Substances 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 18
- 238000005259 measurement Methods 0.000 description 18
- 108090000765 processed proteins & peptides Proteins 0.000 description 18
- 229960004063 propylene glycol Drugs 0.000 description 18
- 239000006185 dispersion Substances 0.000 description 17
- 239000003814 drug Substances 0.000 description 17
- 230000014759 maintenance of location Effects 0.000 description 17
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 16
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 16
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 16
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- 239000005642 Oleic acid Substances 0.000 description 16
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 16
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 16
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 16
- 238000012360 testing method Methods 0.000 description 16
- GHHURQMJLARIDK-UHFFFAOYSA-N 2-hydroxypropyl octanoate Chemical compound CCCCCCCC(=O)OCC(C)O GHHURQMJLARIDK-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- 230000015572 biosynthetic process Effects 0.000 description 15
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 15
- 238000003786 synthesis reaction Methods 0.000 description 15
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 14
- 102000004196 processed proteins & peptides Human genes 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 150000004665 fatty acids Chemical class 0.000 description 12
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 10
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 235000006708 antioxidants Nutrition 0.000 description 9
- 238000004090 dissolution Methods 0.000 description 9
- 238000002296 dynamic light scattering Methods 0.000 description 9
- 239000007903 gelatin capsule Substances 0.000 description 9
- 239000012299 nitrogen atmosphere Substances 0.000 description 9
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical group OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 8
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 8
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 8
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 8
- 229940068968 polysorbate 80 Drugs 0.000 description 8
- 229920000053 polysorbate 80 Polymers 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 7
- 241000700159 Rattus Species 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 150000001413 amino acids Chemical class 0.000 description 7
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 239000001087 glyceryl triacetate Substances 0.000 description 7
- 235000013773 glyceryl triacetate Nutrition 0.000 description 7
- 239000007902 hard capsule Substances 0.000 description 7
- 235000017557 sodium bicarbonate Nutrition 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 239000007901 soft capsule Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical group OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 7
- 229960000984 tocofersolan Drugs 0.000 description 7
- 229960002622 triacetin Drugs 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 description 6
- 239000011627 DL-alpha-tocopherol Substances 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 238000011049 filling Methods 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
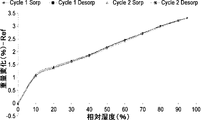
- 238000001179 sorption measurement Methods 0.000 description 6
- 238000003860 storage Methods 0.000 description 6
- 238000010998 test method Methods 0.000 description 6
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 6
- ZPGDWQNBZYOZTI-SFHVURJKSA-N (2s)-1-(9h-fluoren-9-ylmethoxycarbonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 ZPGDWQNBZYOZTI-SFHVURJKSA-N 0.000 description 5
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 101150105104 Kras gene Proteins 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 229940014259 gelatin Drugs 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 5
- 229940011051 isopropyl acetate Drugs 0.000 description 5
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 5
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 239000000825 pharmaceutical preparation Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000008213 purified water Substances 0.000 description 5
- 229910001220 stainless steel Inorganic materials 0.000 description 5
- 239000010935 stainless steel Substances 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- BXRZCDISGRVJCA-KRWDZBQOSA-N (2s)-1-(9h-fluoren-9-ylmethoxycarbonyl)azetidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCN1C(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 BXRZCDISGRVJCA-KRWDZBQOSA-N 0.000 description 4
- QXVFEIPAZSXRGM-DJJJIMSYSA-N (2s,3s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-methylpentanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H]([C@@H](C)CC)C(O)=O)C3=CC=CC=C3C2=C1 QXVFEIPAZSXRGM-DJJJIMSYSA-N 0.000 description 4
- YWWVWXASSLXJHU-AATRIKPKSA-N (9E)-tetradecenoic acid Chemical compound CCCC\C=C\CCCCCCCC(O)=O YWWVWXASSLXJHU-AATRIKPKSA-N 0.000 description 4
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- 101100404726 Arabidopsis thaliana NHX7 gene Proteins 0.000 description 4
- 241000282693 Cercopithecidae Species 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 4
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 4
- 102000057028 SOS1 Human genes 0.000 description 4
- 108700022176 SOS1 Proteins 0.000 description 4
- 101100197320 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RPL35A gene Proteins 0.000 description 4
- 101150100839 Sos1 gene Proteins 0.000 description 4
- UYXTWWCETRIEDR-UHFFFAOYSA-N Tributyrin Chemical compound CCCC(=O)OCC(OC(=O)CCC)COC(=O)CCC UYXTWWCETRIEDR-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 4
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 4
- 238000004455 differential thermal analysis Methods 0.000 description 4
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 4
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 229960004488 linolenic acid Drugs 0.000 description 4
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- 235000021313 oleic acid Nutrition 0.000 description 4
- 238000003305 oral gavage Methods 0.000 description 4
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229920003023 plastic Polymers 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 239000000473 propyl gallate Substances 0.000 description 4
- 235000010388 propyl gallate Nutrition 0.000 description 4
- 229940075579 propyl gallate Drugs 0.000 description 4
- 230000004850 protein–protein interaction Effects 0.000 description 4
- ARIWANIATODDMH-UHFFFAOYSA-N rac-1-monolauroylglycerol Chemical compound CCCCCCCCCCCC(=O)OCC(O)CO ARIWANIATODDMH-UHFFFAOYSA-N 0.000 description 4
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical compound CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000002411 thermogravimetry Methods 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 4
- PVNIQBQSYATKKL-UHFFFAOYSA-N tripalmitin Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCC PVNIQBQSYATKKL-UHFFFAOYSA-N 0.000 description 4
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 4
- 235000015112 vegetable and seed oil Nutrition 0.000 description 4
- 239000008158 vegetable oil Substances 0.000 description 4
- 238000010792 warming Methods 0.000 description 4
- UXLHLZHGQPDMJQ-QHCPKHFHSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-(4-methylphenyl)propanoic acid Chemical compound C1=CC(C)=CC=C1C[C@@H](C(O)=O)NC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 UXLHLZHGQPDMJQ-QHCPKHFHSA-N 0.000 description 3
- FBNFRRNBFASDKS-IBGZPJMESA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-4-oxo-4-prop-2-enoxybutanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CC(=O)OCC=C)C(=O)O)C3=CC=CC=C3C2=C1 FBNFRRNBFASDKS-IBGZPJMESA-N 0.000 description 3
- BUJQSIPFDWLNDC-FQEVSTJZSA-N (2s)-2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]-4-methylpentanoic acid Chemical compound C1=CC=C2C(COC(=O)N(C)[C@@H](CC(C)C)C(O)=O)C3=CC=CC=C3C2=C1 BUJQSIPFDWLNDC-FQEVSTJZSA-N 0.000 description 3
- JOFHWKQIQLPZTC-LBPRGKRZSA-N (2s)-2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]propanoic acid Chemical compound C1=CC=C2C(COC(=O)N(C)[C@@H](C)C(O)=O)C3=CC=CC=C3C2=C1 JOFHWKQIQLPZTC-LBPRGKRZSA-N 0.000 description 3
- CVZUKWBYQQYBTF-ZDUSSCGKSA-N (4s)-4-[(2-methylpropan-2-yl)oxycarbonylamino]-5-oxo-5-phenylmethoxypentanoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CCC(O)=O)C(=O)OCC1=CC=CC=C1 CVZUKWBYQQYBTF-ZDUSSCGKSA-N 0.000 description 3
- ZHKQIADIIYMFOZ-UHFFFAOYSA-N 2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]acetic acid Chemical compound C1=CC=C2C(COC(=O)N(CC(O)=O)C)C3=CC=CC=C3C2=C1 ZHKQIADIIYMFOZ-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical group CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 3
- SHBUUTHKGIVMJT-UHFFFAOYSA-N Hydroxystearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OO SHBUUTHKGIVMJT-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 102000005157 Somatostatin Human genes 0.000 description 3
- 108010056088 Somatostatin Proteins 0.000 description 3
- KPFBUSLHFFWMAI-HYRPPVSQSA-N [(8r,9s,10r,13s,14s,17r)-17-acetyl-6-formyl-3-methoxy-10,13-dimethyl-1,2,7,8,9,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-17-yl] acetate Chemical compound C1C[C@@H]2[C@](CCC(OC)=C3)(C)C3=C(C=O)C[C@H]2[C@@H]2CC[C@](OC(C)=O)(C(C)=O)[C@]21C KPFBUSLHFFWMAI-HYRPPVSQSA-N 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 239000006285 cell suspension Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 150000001923 cyclic compounds Chemical class 0.000 description 3
- 238000003795 desorption Methods 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 229940072106 hydroxystearate Drugs 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 239000012669 liquid formulation Substances 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 210000004379 membrane Anatomy 0.000 description 3
- 229940126701 oral medication Drugs 0.000 description 3
- 230000035699 permeability Effects 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 3
- 229960000553 somatostatin Drugs 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 3
- 238000012982 x-ray structure analysis Methods 0.000 description 3
- HTDWMNDUFNXBIA-FQEVSTJZSA-N (2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]-4-oxo-4-prop-2-enoxybutanoic acid Chemical compound C1=C2C3=CC=CC=C3C(COC(=O)N(C)[C@@H](CC(=O)OCC=C)C(=O)O)C2=CC=C1 HTDWMNDUFNXBIA-FQEVSTJZSA-N 0.000 description 2
- CUNWUEBNSZSNRX-RKGWDQTMSA-N (2r,3r,4r,5s)-hexane-1,2,3,4,5,6-hexol;(z)-octadec-9-enoic acid Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O.CCCCCCCC\C=C/CCCCCCCC(O)=O CUNWUEBNSZSNRX-RKGWDQTMSA-N 0.000 description 2
- AQTUACKQXJNHFQ-LURJTMIESA-L (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]pentanedioate Chemical compound CC(C)(C)OC(=O)N[C@H](C([O-])=O)CCC([O-])=O AQTUACKQXJNHFQ-LURJTMIESA-L 0.000 description 2
- JGWHUIQSEKGLAQ-FQEVSTJZSA-N (2s)-2-cyclopentyl-2-(9h-fluoren-9-ylmethoxycarbonylamino)acetic acid Chemical compound C1([C@H](NC(=O)OCC2C3=CC=CC=C3C3=CC=CC=C32)C(=O)O)CCCC1 JGWHUIQSEKGLAQ-FQEVSTJZSA-N 0.000 description 2
- WOAPUHMPKSQFJV-NRFANRHFSA-N (2s)-2-cyclopentyl-2-[9h-fluoren-9-ylmethoxycarbonyl(methyl)amino]acetic acid Chemical compound C1([C@H](N(C)C(=O)OCC2C3=CC=CC=C3C3=CC=CC=C32)C(O)=O)CCCC1 WOAPUHMPKSQFJV-NRFANRHFSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 2
- UBEIMDKGOYBUKT-FLIQGJDUSA-N 1,2,3-trilinolenoylglycerol Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/C\C=C/C\C=C/CC)COC(=O)CCCCCCC\C=C/C\C=C/C\C=C/CC UBEIMDKGOYBUKT-FLIQGJDUSA-N 0.000 description 2
- HBOQXIRUPVQLKX-BBWANDEASA-N 1,2,3-trilinoleoylglycerol Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/C\C=C/CCCCC)COC(=O)CCCCCCC\C=C/C\C=C/CCCCC HBOQXIRUPVQLKX-BBWANDEASA-N 0.000 description 2
- SKGWNZXOCSYJQL-BUTYCLJRSA-N 1,2,3-tripalmitoleoylglycerol Chemical compound CCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCC)COC(=O)CCCCCCC\C=C/CCCCCC SKGWNZXOCSYJQL-BUTYCLJRSA-N 0.000 description 2
- ODDSXTDNXBAVPQ-UHFFFAOYSA-N 1,2-dihydroxypropyl hexadecanoate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(O)C(C)O ODDSXTDNXBAVPQ-UHFFFAOYSA-N 0.000 description 2
- QHZLMUACJMDIAE-SFHVURJKSA-N 1-hexadecanoyl-sn-glycerol Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](O)CO QHZLMUACJMDIAE-SFHVURJKSA-N 0.000 description 2
- 239000000263 2,3-dihydroxypropyl (Z)-octadec-9-enoate Substances 0.000 description 2
- RRZWKUGIZRDCPB-UHFFFAOYSA-N 2,3-dihydroxypropyl hexanoate Chemical group CCCCCC(=O)OCC(O)CO RRZWKUGIZRDCPB-UHFFFAOYSA-N 0.000 description 2
- FKOKUHFZNIUSLW-UHFFFAOYSA-N 2-Hydroxypropyl stearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(C)O FKOKUHFZNIUSLW-UHFFFAOYSA-N 0.000 description 2
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 2
- ZVTDEEBSWIQAFJ-KHPPLWFESA-N 2-hydroxypropyl (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C)O ZVTDEEBSWIQAFJ-KHPPLWFESA-N 0.000 description 2
- BHIZVZJETFVJMJ-UHFFFAOYSA-N 2-hydroxypropyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCC(C)O BHIZVZJETFVJMJ-UHFFFAOYSA-N 0.000 description 2
- VCNPGCHIKPSUSP-UHFFFAOYSA-N 2-hydroxypropyl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OCC(C)O VCNPGCHIKPSUSP-UHFFFAOYSA-N 0.000 description 2
- RZRNAYUHWVFMIP-GDCKJWNLSA-N 3-oleoyl-sn-glycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-GDCKJWNLSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 2
- YWWVWXASSLXJHU-UHFFFAOYSA-N 9E-tetradecenoic acid Natural products CCCCC=CCCCCCCCC(O)=O YWWVWXASSLXJHU-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 235000019489 Almond oil Nutrition 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- JEBFVOLFMLUKLF-IFPLVEIFSA-N Astaxanthin Natural products CC(=C/C=C/C(=C/C=C/C1=C(C)C(=O)C(O)CC1(C)C)/C)C=CC=C(/C)C=CC=C(/C)C=CC2=C(C)C(=O)C(O)CC2(C)C JEBFVOLFMLUKLF-IFPLVEIFSA-N 0.000 description 2
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 2
- CZMWSWHQFZWAHZ-VWLOTQADSA-N C1=CC=2C(COC(=O)N(CC)[C@@H](CC3=CC=C(C)C=C3)C(=O)O)C3=CC=CC=C3C=2C=C1 Chemical compound C1=CC=2C(COC(=O)N(CC)[C@@H](CC3=CC=C(C)C=C3)C(=O)O)C3=CC=CC=C3C=2C=C1 CZMWSWHQFZWAHZ-VWLOTQADSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 2
- LKUNXBRZDFMZOK-GFCCVEGCSA-N Capric acid monoglyceride Natural products CCCCCCCCCC(=O)OC[C@H](O)CO LKUNXBRZDFMZOK-GFCCVEGCSA-N 0.000 description 2
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 238000003109 Karl Fischer titration Methods 0.000 description 2
- 239000005639 Lauric acid Substances 0.000 description 2
- 235000018330 Macadamia integrifolia Nutrition 0.000 description 2
- 240000000912 Macadamia tetraphylla Species 0.000 description 2
- 235000003800 Macadamia tetraphylla Nutrition 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 235000006679 Mentha X verticillata Nutrition 0.000 description 2
- 235000002899 Mentha suaveolens Nutrition 0.000 description 2
- 235000001636 Mentha x rotundifolia Nutrition 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- NXEIEGXQRYYGCH-QFIPXVFZSA-N OC([C@H](CCC1=CC(F)=C(C(F)(F)F)C(F)=C1)NC(OCC1C2=CC=CC=C2C2=C1C=CC=C2)=O)=O Chemical compound OC([C@H](CCC1=CC(F)=C(C(F)(F)F)C(F)=C1)NC(OCC1C2=CC=CC=C2C2=C1C=CC=C2)=O)=O NXEIEGXQRYYGCH-QFIPXVFZSA-N 0.000 description 2
- 229940123973 Oxygen scavenger Drugs 0.000 description 2
- 235000019482 Palm oil Nutrition 0.000 description 2
- 235000021314 Palmitic acid Nutrition 0.000 description 2
- QHZLMUACJMDIAE-UHFFFAOYSA-N Palmitic acid monoglyceride Natural products CCCCCCCCCCCCCCCC(=O)OCC(O)CO QHZLMUACJMDIAE-UHFFFAOYSA-N 0.000 description 2
- 235000021319 Palmitoleic acid Nutrition 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002675 Polyoxyl Polymers 0.000 description 2
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 2
- 229920002696 Polyoxyl 40 castor oil Polymers 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 229920001219 Polysorbate 40 Polymers 0.000 description 2
- 235000019484 Rapeseed oil Nutrition 0.000 description 2
- 235000019485 Safflower oil Nutrition 0.000 description 2
- IYFATESGLOUGBX-YVNJGZBMSA-N Sorbitan monopalmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O IYFATESGLOUGBX-YVNJGZBMSA-N 0.000 description 2
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- BAECOWNUKCLBPZ-HIUWNOOHSA-N Triolein Natural products O([C@H](OCC(=O)CCCCCCC/C=C\CCCCCCCC)COC(=O)CCCCCCC/C=C\CCCCCCCC)C(=O)CCCCCCC/C=C\CCCCCCCC BAECOWNUKCLBPZ-HIUWNOOHSA-N 0.000 description 2
- PHYFQTYBJUILEZ-UHFFFAOYSA-N Trioleoylglycerol Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCCCCCCCC)COC(=O)CCCCCCCC=CCCCCCCCC PHYFQTYBJUILEZ-UHFFFAOYSA-N 0.000 description 2
- BKZCZANSHGDPSH-KTKRTIGZSA-N [3-(2,3-dihydroxypropoxy)-2-hydroxypropyl] (z)-octadec-9-enoate Chemical group CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)COCC(O)CO BKZCZANSHGDPSH-KTKRTIGZSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 239000008168 almond oil Substances 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 235000013793 astaxanthin Nutrition 0.000 description 2
- MQZIGYBFDRPAKN-ZWAPEEGVSA-N astaxanthin Chemical compound C([C@H](O)C(=O)C=1C)C(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)C(=O)[C@@H](O)CC1(C)C MQZIGYBFDRPAKN-ZWAPEEGVSA-N 0.000 description 2
- 239000001168 astaxanthin Substances 0.000 description 2
- 229940022405 astaxanthin Drugs 0.000 description 2
- 235000021302 avocado oil Nutrition 0.000 description 2
- 239000008163 avocado oil Substances 0.000 description 2
- 229960004217 benzyl alcohol Drugs 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 239000003240 coconut oil Substances 0.000 description 2
- 235000019864 coconut oil Nutrition 0.000 description 2
- 239000000306 component Substances 0.000 description 2
- 235000005687 corn oil Nutrition 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- PHMQYKDOTWAOBI-UHFFFAOYSA-N decanoic acid;propane-1,2-diol Chemical compound CC(O)CO.CCCCCCCCCC(O)=O PHMQYKDOTWAOBI-UHFFFAOYSA-N 0.000 description 2
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 2
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000007922 dissolution test Methods 0.000 description 2
- 229960004756 ethanol Drugs 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 229940085942 formulation r Drugs 0.000 description 2
- 238000003304 gavage Methods 0.000 description 2
- 229940087068 glyceryl caprylate Drugs 0.000 description 2
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 2
- 229940068939 glyceryl monolaurate Drugs 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- LHGVFZTZFXWLCP-UHFFFAOYSA-N guaiacol Chemical compound COC1=CC=CC=C1O LHGVFZTZFXWLCP-UHFFFAOYSA-N 0.000 description 2
- UIMPORZWRQZHLG-UHFFFAOYSA-N hexanoic acid;propane-1,2-diol Chemical group CC(O)CO.CCCCCC(O)=O UIMPORZWRQZHLG-UHFFFAOYSA-N 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- HBOQXIRUPVQLKX-UHFFFAOYSA-N linoleic acid triglyceride Natural products CCCCCC=CCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COC(=O)CCCCCCCC=CCC=CCCCCC HBOQXIRUPVQLKX-UHFFFAOYSA-N 0.000 description 2
- 239000000944 linseed oil Substances 0.000 description 2
- 235000021388 linseed oil Nutrition 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- RZRNAYUHWVFMIP-UHFFFAOYSA-N monoelaidin Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-UHFFFAOYSA-N 0.000 description 2
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 2
- 230000010807 negative regulation of binding Effects 0.000 description 2
- 239000010466 nut oil Substances 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 229960002446 octanoic acid Drugs 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000002540 palm oil Substances 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 description 2
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 description 2
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 2
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 2
- 229940099429 polyoxyl 40 stearate Drugs 0.000 description 2
- 229940068977 polysorbate 20 Drugs 0.000 description 2
- 229940101027 polysorbate 40 Drugs 0.000 description 2
- 229940113124 polysorbate 60 Drugs 0.000 description 2
- 229940026235 propylene glycol monolaurate Drugs 0.000 description 2
- 229940093625 propylene glycol monostearate Drugs 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- 150000004053 quinones Chemical class 0.000 description 2
- LKUNXBRZDFMZOK-UHFFFAOYSA-N rac-1-monodecanoylglycerol Chemical compound CCCCCCCCCC(=O)OCC(O)CO LKUNXBRZDFMZOK-UHFFFAOYSA-N 0.000 description 2
- DCBSHORRWZKAKO-UHFFFAOYSA-N rac-1-monomyristoylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OCC(O)CO DCBSHORRWZKAKO-UHFFFAOYSA-N 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 235000005713 safflower oil Nutrition 0.000 description 2
- 239000003813 safflower oil Substances 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229940035044 sorbitan monolaurate Drugs 0.000 description 2
- 235000011069 sorbitan monooleate Nutrition 0.000 description 2
- 239000001593 sorbitan monooleate Substances 0.000 description 2
- 229940035049 sorbitan monooleate Drugs 0.000 description 2
- 235000011071 sorbitan monopalmitate Nutrition 0.000 description 2
- 239000001570 sorbitan monopalmitate Substances 0.000 description 2
- 229940031953 sorbitan monopalmitate Drugs 0.000 description 2
- 235000011076 sorbitan monostearate Nutrition 0.000 description 2
- 239000001587 sorbitan monostearate Substances 0.000 description 2
- 229940035048 sorbitan monostearate Drugs 0.000 description 2
- 229960005078 sorbitan sesquioleate Drugs 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 238000013112 stability test Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000012916 structural analysis Methods 0.000 description 2
- 150000003900 succinic acid esters Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 2
- 150000003613 toluenes Chemical class 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- LADGBHLMCUINGV-UHFFFAOYSA-N tricaprin Chemical compound CCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCC)COC(=O)CCCCCCCCC LADGBHLMCUINGV-UHFFFAOYSA-N 0.000 description 2
- 229940093633 tricaprin Drugs 0.000 description 2
- MAYCICSNZYXLHB-UHFFFAOYSA-N tricaproin Chemical compound CCCCCC(=O)OCC(OC(=O)CCCCC)COC(=O)CCCCC MAYCICSNZYXLHB-UHFFFAOYSA-N 0.000 description 2
- 229940093609 tricaprylin Drugs 0.000 description 2
- QXTIBZLKQPJVII-UHFFFAOYSA-N triethylsilicon Chemical compound CC[Si](CC)CC QXTIBZLKQPJVII-UHFFFAOYSA-N 0.000 description 2
- 229940081852 trilinolein Drugs 0.000 description 2
- VLPFTAMPNXLGLX-UHFFFAOYSA-N trioctanoin Chemical compound CCCCCCCC(=O)OCC(OC(=O)CCCCCCC)COC(=O)CCCCCCC VLPFTAMPNXLGLX-UHFFFAOYSA-N 0.000 description 2
- PHYFQTYBJUILEZ-IUPFWZBJSA-N triolein Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)COC(=O)CCCCCCC\C=C/CCCCCCCC PHYFQTYBJUILEZ-IUPFWZBJSA-N 0.000 description 2
- 229940117972 triolein Drugs 0.000 description 2
- 229960001947 tripalmitin Drugs 0.000 description 2
- SKGWNZXOCSYJQL-UHFFFAOYSA-N tripalmitoleoyl-sn-glycerol Natural products CCCCCCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCCCCCC)COC(=O)CCCCCCCC=CCCCCCC SKGWNZXOCSYJQL-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical compound C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- REPVLJRCJUVQFA-UHFFFAOYSA-N (-)-isopinocampheol Natural products C1C(O)C(C)C2C(C)(C)C1C2 REPVLJRCJUVQFA-UHFFFAOYSA-N 0.000 description 1
- WMSUFWLPZLCIHP-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 9h-fluoren-9-ylmethyl carbonate Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1COC(=O)ON1C(=O)CCC1=O WMSUFWLPZLCIHP-UHFFFAOYSA-N 0.000 description 1
- XINQFOMFQFGGCQ-UHFFFAOYSA-L (2-dodecoxy-2-oxoethyl)-[6-[(2-dodecoxy-2-oxoethyl)-dimethylazaniumyl]hexyl]-dimethylazanium;dichloride Chemical compound [Cl-].[Cl-].CCCCCCCCCCCCOC(=O)C[N+](C)(C)CCCCCC[N+](C)(C)CC(=O)OCCCCCCCCCCCC XINQFOMFQFGGCQ-UHFFFAOYSA-L 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- FWNZKPKGBYWNJO-KVVVOXFISA-N (z)-octadec-9-enoic acid;propane-1,2-diol Chemical compound CC(O)CO.CCCCCCCC\C=C/CCCCCCCC(O)=O FWNZKPKGBYWNJO-KVVVOXFISA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- SUHKWEINMZXPHI-UHFFFAOYSA-N 1-bromo-2,4-difluoro-3-(trifluoromethyl)benzene Chemical compound FC1=CC=C(Br)C(F)=C1C(F)(F)F SUHKWEINMZXPHI-UHFFFAOYSA-N 0.000 description 1
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- JVKUCNQGESRUCL-UHFFFAOYSA-N 2-Hydroxyethyl 12-hydroxyoctadecanoate Chemical compound CCCCCCC(O)CCCCCCCCCCC(=O)OCCO JVKUCNQGESRUCL-UHFFFAOYSA-N 0.000 description 1
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- HJBLUNHMOKFZQX-UHFFFAOYSA-N 3-hydroxy-1,2,3-benzotriazin-4-one Chemical compound C1=CC=C2C(=O)N(O)N=NC2=C1 HJBLUNHMOKFZQX-UHFFFAOYSA-N 0.000 description 1
- RGLXLXXMVNSEFE-FQEVSTJZSA-N 9H-fluoren-9-ylmethyl (4S)-5-oxo-4-(2-oxo-2-prop-2-enoxyethyl)-1,3-oxazolidine-3-carboxylate Chemical compound C=CCOC(=O)C[C@@H]1N(COC1=O)C(=O)OCC1c2ccccc2-c2ccccc12 RGLXLXXMVNSEFE-FQEVSTJZSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- QOOVAUCGJDRKRX-IBGZPJMESA-N CC(C)(C)OC(=O)N[C@@H](CCC(=O)ON1C(=O)C2=CC=CC=C2C1=O)C(=O)OCC3=CC=CC=C3 Chemical compound CC(C)(C)OC(=O)N[C@@H](CCC(=O)ON1C(=O)C2=CC=CC=C2C1=O)C(=O)OCC3=CC=CC=C3 QOOVAUCGJDRKRX-IBGZPJMESA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101000868152 Homo sapiens Son of sevenless homolog 1 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical class [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 241000920340 Pion Species 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920001304 Solutol HS 15 Polymers 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 235000009470 Theobroma cacao Nutrition 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- RDWDVLFMPFUBDV-PXMDEAMVSA-N [(e)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-tripyrrolidin-1-ylphosphanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.C1CCCN1[P+](N1CCCC1)(O/N=C(C(=O)OCC)\C#N)N1CCCC1 RDWDVLFMPFUBDV-PXMDEAMVSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 238000003016 alphascreen Methods 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- CJPQIRJHIZUAQP-MRXNPFEDSA-N benalaxyl-M Chemical compound CC=1C=CC=C(C)C=1N([C@H](C)C(=O)OC)C(=O)CC1=CC=CC=C1 CJPQIRJHIZUAQP-MRXNPFEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229940116229 borneol Drugs 0.000 description 1
- CKDOCTFBFTVPSN-UHFFFAOYSA-N borneol Natural products C1CC2(C)C(C)CC1C2(C)C CKDOCTFBFTVPSN-UHFFFAOYSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- FNAQSUUGMSOBHW-UHFFFAOYSA-H calcium citrate Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O FNAQSUUGMSOBHW-UHFFFAOYSA-H 0.000 description 1
- 239000001354 calcium citrate Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 125000005131 dialkylammonium group Chemical group 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- DTGKSKDOIYIVQL-UHFFFAOYSA-N dl-isoborneol Natural products C1CC2(C)C(O)CC1C2(C)C DTGKSKDOIYIVQL-UHFFFAOYSA-N 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000002900 effect on cell Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- LCFXLZAXGXOXAP-DAXSKMNVSA-N ethyl (2z)-2-cyano-2-hydroxyiminoacetate Chemical compound CCOC(=O)C(=N/O)\C#N LCFXLZAXGXOXAP-DAXSKMNVSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 102000046752 human SOS1 Human genes 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 210000002850 nasal mucosa Anatomy 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- UQPSGBZICXWIAG-UHFFFAOYSA-L nickel(2+);dibromide;trihydrate Chemical compound O.O.O.Br[Ni]Br UQPSGBZICXWIAG-UHFFFAOYSA-L 0.000 description 1
- 238000011017 operating method Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 125000006353 oxyethylene group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- SQYNKIJPMDEDEG-UHFFFAOYSA-N paraldehyde Chemical compound CC1OC(C)OC(C)O1 SQYNKIJPMDEDEG-UHFFFAOYSA-N 0.000 description 1
- 229960003868 paraldehyde Drugs 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- PARWUHTVGZSQPD-UHFFFAOYSA-N phenylsilane Chemical compound [SiH3]C1=CC=CC=C1 PARWUHTVGZSQPD-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 229920000223 polyglycerol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920001289 polyvinyl ether Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical class CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000004626 scanning electron microscopy Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 238000000371 solid-state nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 238000004441 surface measurement Methods 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000012929 tonicity agent Substances 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 239000006211 transdermal dosage form Substances 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- 235000013337 tricalcium citrate Nutrition 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a formulation containing a cyclic peptide compound having KRAS inhibitory activity and a method for producing the same.
- Non-Patent Document 1 compounds that have been conventionally used as oral drugs should preferably have a molecular weight of 500 g/mol or less.
- Non-Patent Document 2 compounds that have been conventionally used as oral drugs should preferably have a molecular weight of 500 g/mol or less.
- Non-Patent Document 3, 4 Like insulin, which is used to treat hyperglycemia, peptides composed of natural amino acids have poor metabolic stability, and it has been difficult to develop peptides as oral drugs. However, it has been found that the metabolic stability and membrane permeability of peptides are improved by cyclizing peptides or using non-natural amino acids such as N-methylamino acids in peptides (Non-Patent Document 3, 4).
- cyclic peptides containing unnatural amino acids it has become known that cyclic peptides containing N-substituted amino acids in particular can have metabolic stability and membrane permeability, that is, can have drug-likeness ( Patent document 1).
- Non-Patent Document 5 It has been suggested that library compounds of cyclic peptides containing non-natural amino acids are useful for creating inhibitors of protein-protein interactions.
- Patent Document 6 discloses a cyclic peptide that exhibits a pharmacological action. In addition, Patent Document 6 discloses a technology for formulating cyclosporin. Studies on oral formulation of somatostatin have also been conducted (Non-Patent Document 6).
- An object of the present invention is to provide a formulation of a cyclic compound containing an unnatural amino acid.
- An object of the present invention is to provide a method for producing a formulation of a cyclic compound having an efficient RAS inhibitory action.
- Patent Document 4 describes inhibition of binding between RAS and SOS
- Patent Document 5 describes peptides that compete with compounds that bind to RAS. No effect on cells was shown. In addition, these documents do not describe drug-like peptides.
- Patent Documents 6 and 7 describe the formulation of cyclic peptides, they merely describe the formulation of specific compounds.
- Non-Patent Document 2 describes peptides used as medicines, but does not describe drug-like peptides or peptides useful for RAS mutant cancer.
- Non-Patent Documents 3 and 4 describe that peptides containing N-methyl amino acids can be applied as medicines, but do not describe peptides useful for RAS mutant cancer.
- Non-Patent Document 5 describes that cyclic peptides can be applied as medicines, but does not describe peptides useful for RAS mutant cancer.
- Non-Patent Document 6 exemplifies somatostatin and describes the elements necessary for oral formulation, but only somatostatin is described.
- a surfactant is effective as a specific additive, preferably a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the formulation can be made more effective by adding an oily component. It has been found that a mixture of the active ingredient and these additives can be formulated and used in a formulation having a specific dosage form.
- the formulation according to the present invention has excellent stability of the active ingredient in the formulation and excellent dissolution from the formulation.
- the present invention relates to the following.
- Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component.
- the surfactant is a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the hydrophobic surfactant is at least one selected from the group consisting of propylene glycol fatty acid ester, glycerin fatty acid ester, polyglycerin fatty acid ester, sorbitan fatty acid ester, and hydrophobic polyoxyethylene hydrogenated castor oil. , [3] or [4].
- the hydrophobic surfactant comprises a propylene glycol fatty acid ester.
- the propylene glycol fatty acid ester is propylene glycol monocaproate, propylene glycol monocaprylate, propylene glycol monocaprate, propylene glycol monolaurate, propylene glycol monomyristate, propylene glycol monopalmitate, propylene glycol monostearate, and propylene glycol monooleate, which is at least one selected from the group consisting of [5].
- the glycerin fatty acid ester is glyceryl monocaproate, glyceryl monocaprylate, glyceryl monocaprate, glyceryl monolaurate, glyceryl monomyristate, glyceryl monopalmitate, glyceryl monostearate, glyceryl monooleate, and monolinol
- the composition of [5] which is at least one selected from the group consisting of glyceryl acid.
- the composition according to [5] wherein the polyglycerin fatty acid ester is diglyceryl monooleate.
- the sorbitan fatty acid ester is sorbitan monocaprylate, sorbitan monocaprate, sorbitan monolaurate, sorbitan monomyristate, sorbitan monopalmitate, sorbitan monostearate, sorbitan monooleate, sorbitan sesquioleate, and trio
- the composition according to [5] which is at least one selected from the group consisting of sorbitan lactate.
- the hydrophobic polyoxyethylene hydrogenated castor oil is at least one selected from the group consisting of polyoxyethylene hydrogenated castor oil 5 and polyoxyethylene hydrogenated castor oil 10. .
- the hydrophilic surfactant is polyethylene glycol fatty acid ester, polyoxyethylene castor oil, hydrophilic polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate. , caprylocaproyl polyoxyl-8 glyceride, and any combination thereof, the composition according to any one of [3] to [13].
- the hydrophilic surfactant is polyoxyethylene castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, and these The composition according to any one of [3] to [14], which is at least one selected from the group consisting of any combination. [16] The composition according to any one of [3] to [15], wherein the hydrophilic surfactant comprises polyoxyethylene castor oil.
- polyethylene glycol fatty acid ester is at least one selected from the group consisting of polyoxyethylene hydroxystearate, polyethylene glycol monolaurate, polyethylene glycol monostearate, and polyoxyl 40 stearate; The described composition.
- polyoxyethylene castor oil is at least one selected from the group consisting of polyoxyl 30 castor oil, polyoxyl 35 castor oil, and polyoxyl 40 castor oil.
- polyoxyethylene castor oil is Polyoxyl 35 castor oil.
- the hydrophilic polyoxyethylene hydrogenated castor oil is selected from the group consisting of polyoxyethylene hydrogenated castor oil 20, polyoxyethylene hydrogenated castor oil 40, polyoxyethylene hydrogenated castor oil 50, and polyoxyethylene hydrogenated castor oil 60.
- the hydrophilic surfactant is selected from the group consisting of polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, polysorbate 80, and combinations thereof; The composition according to any one of [3] to [15], which is at least one selected. [23] The composition according to any one of [3] to [15], wherein the hydrophilic surfactant comprises polyoxyl 35 castor oil. [24] The composition of [3], wherein the hydrophobic surfactant is a propylene glycol fatty acid ester and the hydrophilic surfactant comprises polyoxyethylene castor oil.
- the hydrophobic surfactant is propylene glycol monocaprylate, and the hydrophilic surfactant is polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl- 8.
- the composition of [3] which is at least one selected from the group consisting of glycerides, polysorbate 80, and combinations thereof.
- oily component is at least one selected from the group consisting of fatty acids, acylglycerols, and combinations thereof.
- oily component contains a fatty acid.
- the fatty acid is selected from the group consisting of caproic acid, caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid;
- the composition of [28], which is at least one of [32] The composition of [28], wherein the fatty acid is at least one selected from the group consisting of oleic acid, linoleic acid, and linolenic acid.
- the acylglycerol is selected from the group consisting of triacetin, tributyrin, tricaproin, tricaprylin, tricaprin, tripalmitin, tripalmitolein, glyceryl tristearate, triolein, trilinolein, trilinolenin, and medium-chain fatty acid triglycerides;
- the composition of [28] which is at least one.
- the vegetable oil is selected from the group consisting of olive oil, almond oil, coconut oil, cocoa butter, macadamia nut oil, avocado oil, safflower oil, soybean oil, linseed oil, rapeseed oil, castor oil, corn oil, and palm oil.
- composition of [3] which contains propylene glycol fatty acid ester as the hydrophobic surfactant, polyoxyethylene castor oil as the hydrophilic surfactant, and fatty acid as the oil component.
- composition of [3] which contains propylene glycol monocaprylate as the hydrophobic surfactant, polyoxyl 35 castor oil as the hydrophilic surfactant, and oleic acid as the oil component.
- the antioxidant is dl- ⁇ -tocopherol, butyrated hydroxytoluene, butyrated hydroxyanisole, propyl gallate, propyl gallate, pharmaceutically acceptable quinones, astaxanthin, and D- ⁇ -tocopheryl polyethylene glycol;
- the composition of [40] which is at least one selected from the group consisting of 1000 succinates.
- the solubilizer is at least one selected from the group consisting of ethanol, propylene glycol, polyethylene glycol 300, polyethylene glycol 400, diethylene glycol monoethyl ether, and combinations thereof; Composition.
- the compound represented by formula (1), or a salt thereof, or a solvate thereof is a compound represented by formula (1) or a hydrate thereof, [1] to [43] A composition according to any one of the preceding claims.
- the composition of [44], wherein the compound represented by formula (1), a salt thereof, or a solvate thereof is a compound represented by formula (1).
- Any one of [1] to [46], wherein the content of the compound represented by formula (1), or a salt thereof, or a solvate thereof is 10% by weight or less in the entire composition.
- a composition according to claim 1. Any one of [1] to [48], wherein the content of the compound represented by formula (1), or a salt thereof, or a solvate thereof is 7% by weight or less in the entire composition.
- a composition according to claim 1. [50] The composition according to any one of [1] to [49], wherein the content of the liquid additive in the entire composition is 50% by weight or more and 97% by weight or less.
- composition according to any one of [1] to [50], wherein the content of the liquid additive in the entire composition is 55% by weight or more and 96% by weight or less.
- the composition according to any one of [1] to [51], wherein the content of the liquid additive in the entire composition is 70% by weight or more and 90% by weight or less.
- the composition according to any one of [3] to [52], wherein the content of the hydrophobic surfactant in the entire composition is 20% by weight or more and 70% by weight or less.
- the composition according to any one of [3] to [53], wherein the content of the hydrophobic surfactant in the entire composition is 25% by weight or more and 65% by weight or less.
- composition according to any one of [1] to [58], wherein the content of the oily component in the entire composition is 0% by weight or more and 50% by weight or less.
- the composition according to any one of [1] to [60], wherein the content of the oily component in the entire composition is 10% by weight or more and 20% by weight or less.
- Any one of [1] to [65], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 5 or more.
- any one of [1] to [66], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 10 or more.
- a composition according to claim 1. Any one of [1] to [66], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 5 to 2000. or the composition according to claim 1.
- the total weight ratio of the liquid additive and the oily component to the compound represented by the formula (1), or a salt thereof, or a solvate thereof is 10 or more, [1] to The composition of any one of [68].
- the weight ratio of the sum of the liquid additive and the oily component to the compound represented by the formula (1) or a salt thereof, or a solvate thereof is 10 to 2000 [1] The composition according to any one of -[69]. [71] The weight ratio of the sum of the liquid additive and the oily component to the compound represented by the formula (1), or a salt thereof, or a solvate thereof is 10 to 1000. [1] The composition according to any one of [70]. [72] The composition according to any one of [1] to [71], wherein the composition is liquid. [73] The composition according to any one of [1] to [72], wherein droplets formed when the composition is dispersed in water have an average particle size of less than 200 nm.
- [78] A method for producing the composition according to any one of [1] to [75], comprising the following steps; (1) Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component; (2) mixing the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component; and (3) A step of obtaining a composition in which the compound represented by the formula (1), a salt thereof, or a solvate thereof is dissolved. [79] the compound represented by formula (1) provided in the step (1), a salt thereof, or a solvate thereof is a hydrate of the compound represented by formula (1); The method of [78].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8.296 in powder X-ray diffraction. , 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.°.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least two peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least three peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 4 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 5 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 6 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 7 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 8 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 9 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern containing at least 10 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern containing at least 11 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 12 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79], having a powder X-ray diffraction pattern containing a peak at 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, and 10.435 in powder X-ray diffraction. ° 11.729 °, 12.704 °, 15.895 °, and 16.643 ° ( ⁇ 0.2 °) having a powder X-ray diffraction pattern comprising at least one peak, in [79] described method.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10 .435° 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least two peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction.
- the hydrate of the compound represented by formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction. .435° 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern containing at least four peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction. .435°, 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern comprising at least five peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10 .435°, 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern comprising at least six peaks [79 ].
- the hydrate of the compound represented by formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, 8 .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least 10 peaks; The method of [79].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, 8 .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having at least 13 peaks.
- the hydrate of the compound represented by the above formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least 15 peaks; The method of [79].
- the hydrate of the compound represented by the above formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895 °, 16.190 °, 16.643 °, 17.813 °, and 19.424 ° ( ⁇ 0.2 °) powder X-ray diffraction pattern containing peaks, the method of [79] .
- a method for producing a pharmaceutical preparation comprising the steps of providing the composition of any one of [1] to [75], and formulating the composition to provide a pharmaceutical preparation.
- Production of a capsule formulation comprising the steps of providing the composition of any one of [1] to [75], and filling the composition into capsules to provide a capsule formulation.
- the composition according to the present invention is excellent in various properties required as a formulation.
- the composition according to the present invention has desirable particle characteristics when a liquid composition is emulsified to form droplets, is extremely stable and dispersible, and is easily absorbed into the body. Are better.
- FIG. 1 is a graph showing the results of powder X-ray diffraction measurement of the hydrate crystals (Form C) of compound 1 obtained in Preparation Example 3.
- FIG. The vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- FIG. 2 shows the results of thermogravimetric and differential thermal analysis of hydrate crystals of compound 1 (Form C).
- the horizontal axis is temperature (°C) and measurement time (minutes), and the right vertical axis is weight change (mg) of the sample in thermogravimetric analysis.
- the left vertical axis represents the heat flow (mW) observed in the differential thermal analysis.
- FIG. 3 shows the crystal structure of the hydrate crystal of Compound 1 (Form C) obtained by single-crystal X-ray structure analysis.
- FIG. 4 shows the results of dynamic water vapor adsorption measurement of hydrate crystals of Compound 1 (Form C). The vertical axis is weight change (%) and the horizontal axis is relative humidity (%).
- “Cycle1 Sorp” black diamonds
- “Cycle1 Desorp” black squares
- Cycle2 Sorp” black triangles
- “Cycle2 Desorp” filled squares) indicates desorption in cycle 2.
- FIG. 5 is a graph showing temporal changes in the dispersibility of formulations according to the present invention (formulations C and F) and a comparative example (formulation AJ) in fasting artificial intestinal fluid (FaSSIF).
- composition of the present invention has the following formula (1): The compound represented by (“(5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-butyl)-18-cyclopentyl-29-(3,5- Difluoro-4-(trifluoromethyl)phenethyl)-36-ethyl-11-isobutyl-N,N,5,6,12,16,19,33-octamethyl-35-(4-methylbenzyl)-4,7 ,10,13,17,20,23,28,31,34,37-undecaoxotetratriacontahydro-2H,4H-spiro[azeto[2,1-u]pyrrolo[2,1-i][ 1,4,7,10,13,16,19,22,25,28,31]undecaazacyclotetratriacontin-21,1′-cyclopentane]-15-carbox
- composition according to the present invention optionally contains an oily component, preferably an oily component.
- the compound that can be used in the present invention is the compound represented by the above formula (1), a salt thereof, or a solvate thereof.
- a salt of Compound 1 can preferably be a chemically or pharmaceutically acceptable salt thereof.
- Compound 1 or a salt thereof that can be used in the present invention can also be a solvate thereof, preferably a chemically or pharmaceutically acceptable solvate thereof.
- the compound used in the present invention may preferably be exclusively in the form of a free form in the composition, but may contain the form of a salt or solvate depending on the form of the composition.
- the salts of compound 1 include, for example, hydrochloride; hydrobromide; hydroiodide; phosphate; sulfonates such as; carboxylates such as acetates, citrates, malate, tartrates, succinates, salicylates; or alkali metal salts such as sodium salts, potassium salts; magnesium salts, calcium salts alkaline earth metal salts such as; ammonium salts such as ammonium salts, alkylammonium salts, dialkylammonium salts, trialkylammonium salts and tetraalkylammonium salts; These salts are produced, for example, by contacting the compound 1 with an acid or base that can be used in the production of pharmaceuticals.
- a solvate refers to a compound formed with a solvent to form a single molecular cluster, and a solvate formed with a solvent that is acceptable for ingestion accompanying administration of a drug. It is not particularly limited. Examples include hydrates, alcoholates (ethanolates, methanolates, 1-propanolates, 2-pronolates, etc.), solvates with a single solvent such as dimethylsulfoxide, as well as , solvates formed with a plurality of solvents for one molecule of the compound, and solvates formed with a plurality of types of solvents for one molecule of the compound. If the solvent is water, it is called a hydrate.
- the solvate of the compound of the present invention is preferably a hydrate, and such a hydrate is specifically a 1-10 hydrate, preferably a 1-5 hydrate, more preferably a 1-3 hydrates.
- the number of water molecules in the hydrate of compound 1 used in the present invention may change due to the desorption of water molecules bound to compound 1 depending on the ambient environment such as temperature and humidity.
- Compound 1 or a salt thereof, or a solvate thereof that can be used in the present invention can be provided in the form of a crystal, an amorphous form, or a mixture thereof, preferably Compound 1 or a salt thereof, or a solvate thereof
- the product may be provided in crystalline form.
- Crystals of Compound 1 or a salt thereof or a solvate thereof that can be used in the present invention preferably include hydrate crystals of Compound 1 (also referred to as Form C).
- Crystals of Compound 1 or a salt thereof, or a solvate thereof can be isolated by techniques known in the art, such as X-ray powder diffraction (XRPD), moisture determination methods (e.g. Karl Fischer method), scanning electron microscopy (SEM). Characterization can be by analytical, solid state NMR, or thermal techniques such as differential scanning calorimetry (DSC), or any other standard quantitative measurement method.
- XRPD X-ray powder diffraction
- moisture determination methods e.g. Karl Fischer method
- SEM scanning electron microscopy
- Characterization can be by analytical, solid state NMR, or thermal techniques such as differential scanning calorimetry (DSC), or any other standard quantitative measurement method.
- the diffraction angle 2 ⁇ in powder X-ray diffraction is preferably a diffraction peak measured using CuK ⁇ radiation.
- a crystalline compound 1 hydrate that can be used in the present invention has a powder X-ray diffraction pattern that includes at least one of the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 4.964°, 7.921°, 8.296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, 17.813° ( ⁇ 0.2°)
- Compound 1 used in the present invention includes all isotopes of Compound 1.
- Isotopes of compound 1 have at least one atom with the same atomic number (number of protons) and a different mass number (sum of proton and neutron numbers), with abundance ratios different from those in nature. It is replaced.
- Examples of isotopes contained in Compound 1 include hydrogen, carbon, nitrogen, oxygen, and fluorine atoms, respectively, 2 H, 3 H, 13 C, 14 C, 15 N, 17 O, 18 O, 18 F and the like are included.
- radioactive isotopes that emit radioactivity and decay such as 3 H and 14 C, are useful in tissue distribution tests of drugs or compounds.
- Stable isotopes are safe to use because they do not decay, their abundance changes little over time, and they are non-radioactive.
- the isotopes of compound 1 can be converted according to conventional methods by replacing the reagents used in the synthesis with reagents containing the corresponding isotopes.
- Acids or bases used to form salts of Compound 1, as well as solvents used to form solvates of Compound 1, can also include all isotopes.
- liquid additive that can be used in the composition according to the present invention is an additive that is pharmaceutically acceptable and capable of dissolving the compound used in the present invention.
- the “liquid additive” means that the additive is in a dissolved state in the composition, and those that are not dissolved at the stage of being provided as raw materials for the production of the composition are It can be used as one component of the composition according to the present invention by mixing it with the components of (1) or dissolving it in the manufacturing process by heating or the like.
- an additive that is liquid at room temperature can be preferably used.
- room temperature is used in the ordinary sense in the technical field and is not particularly limited, but unless otherwise specified, for example, preferably 1 to 30° C., more preferably about 15 to 28° C. be.
- Liquid additives that can be used in the present invention preferably include surfactants, and surfactants preferably include hydrophobic surfactants and hydrophilic surfactants. In the present invention, it is desirable to use a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the HLB value (Hydrophilic-Lipophilic Balance value) of the hydrophobic surfactant is preferably less than 10, more preferably 0
- the HLB value of the hydrophilic surfactant is preferably 10 or more, more preferably 10 or more and 30 or less, still more preferably 10 or more and 20 or less.
- the HLB value is a value that represents the degree of affinity of a surfactant for water and oil (an organic compound that is insoluble in water), and is known to those skilled in the art. Values based on known methods can be adopted.
- Hydrophobic surfactants preferably include propylene glycol fatty acid esters, glycerin fatty acid esters, polyglycerin fatty acid esters, sorbitan fatty acid esters, and hydrophobic polyoxyethylene hydrogenated castor oil, etc. These may also be used in combination. can.
- the hydrophobic surfactant propylene glycol fatty acid ester or sorbitan fatty acid ester can be used more preferably, and propylene glycol fatty acid ester can be used more preferably.
- Propylene glycol fatty acid esters preferably include propylene glycol monocaproate, propylene glycol monocaprylate, propylene glycol monocaprate, propylene glycol monolaurate, propylene glycol monomyristate, propylene glycol monopalmitate, propylene glycol monostearate, and propylene glycol monooleate.
- propylene glycol monocaprylate can be used more preferably.
- Glycerin fatty acid esters preferably include glyceryl monocaproate, glyceryl monocaprylate, glyceryl monocaprate, glyceryl monolaurate, glyceryl monomyristate, glyceryl monopalmitate, glyceryl monostearate, glyceryl monooleate, and monolinol. and glyceryl acid.
- Polyglycerol fatty acid esters preferably include diglyceryl monooleate.
- Sorbitan fatty acid esters preferably include sorbitan monocaprylate, sorbitan monocaprate, sorbitan monolaurate, sorbitan monomyristate, sorbitan monopalmitate, sorbitan monostearate, sorbitan monooleate, sorbitan sesquioleate, and trio and sorbitan lactate.
- the hydrophobic polyoxyethylene hydrogenated castor oil is a hydrophobic polyoxyethylene hydrogenated castor oil, for example, a polyoxyethylene hydrogenated castor oil having an HLB value of preferably less than 10, more preferably 0 or more and less than 10. oil.
- Hydrophobic polyoxyethylene hydrogenated castor oil preferably includes polyoxyethylene hydrogenated castor oil 5 and polyoxyethylene hydrogenated castor oil 10 and the like.
- Hydrophilic surfactants preferably include polyethylene glycol fatty acid ester, polyoxyethylene castor oil, hydrophilic polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan fatty acid ester, and D- ⁇ -tocopheryl polyethylene glycol 1000 succinate. , caprylocaproyl polyoxyl-8 glyceride and the like, and these can also be used in combination. Hydrophilic surfactants, more preferably polyoxyethylene castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, or these Combinations can be used, more preferably polyoxyethylene castor oil.
- Polyethylene glycol fatty acid esters preferably include polyoxyethylene hydroxystearate, polyethylene glycol monolaurate, polyethylene glycol monostearate, and polyoxyl 40 stearate.
- the polyoxyethylene castor oil preferably includes polyoxyl 30 castor oil, polyoxyl 35 castor oil, polyoxyl 40 castor oil, and more preferably polyoxyl 35 castor oil.
- the hydrophilic polyoxyethylene hydrogenated castor oil is a hydrophilic polyoxyethylene hydrogenated castor oil, for example, the HLB value is preferably 10 or more, more preferably 10 or more and 30 or less, still more preferably 10 or more. 20 or less polyoxyethylene hydrogenated castor oil.
- Hydrophilic polyoxyethylene hydrogenated castor oils preferably include polyoxyethylene hydrogenated castor oil 20, polyoxyethylene hydrogenated castor oil 40, polyoxyethylene hydrogenated castor oil 50, and polyoxyethylene hydrogenated castor oil 60. .
- Polyoxyethylene sorbitan fatty acid esters preferably include polysorbate 20, polysorbate 40, polysorbate 60, and polysorbate 80.
- hydrophilic surfactant more preferably polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, polysorbate 80, or a combination thereof is preferably used. can be done.
- hydrophobic surfactant preferably propylene glycol monocaprylate
- hydrophilic surfactant preferably polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, capri Rocaproyl polyoxyl-8 glycerides, polysorbate 80, or combinations thereof can be used.
- the surfactant preferably uses a propylene glycol fatty acid ester as a hydrophobic surfactant and contains at least polyoxyethylene castor oil as a hydrophilic surfactant.
- the surfactant is a hydrophobic surfactant. It is desirable to use propylene glycol monocaprylate as the hydrophilic surfactant and to include at least polyoxyl 35 castor oil as the hydrophilic surfactant.
- Oily Component may contain an oily component.
- Oily ingredients preferably include fatty acids, acylglycerols, vegetable oils and combinations thereof. Fatty acids or acylglycerols are more preferably used as the oily component, and fatty acids are more preferably used.
- Fatty acids preferably include caproic acid, caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid.
- caproic acid caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid.
- oleic acid, linoleic acid, and linolenic acid are more preferred, and oleic acid is even more preferred.
- Preferred acylglycerols include triacetin, tributyrin, tricaproin, tricaprylin, tricaprin, tripalmitin, tripalmitolein, glyceryl tristearate, triolein, trilinolein, trilinolenin, and medium-chain fatty acid triglycerides. Among these, triacetin is more preferred.
- Vegetable oils include olive oil, almond oil, coconut oil, cocoa butter, macadamia nut oil, avocado oil, safflower oil, soybean oil, linseed oil, rapeseed oil, castor oil, corn oil, and palm oil.
- oily component oleic acid, triacetin, or a combination thereof can be preferably used, and oleic acid is more preferably used.
- propylene glycol fatty acid ester as a hydrophobic surfactant, contain at least polyoxyethylene castor oil as a hydrophilic surfactant, and fatty acid as an oily component.
- the surfactant is propylene glycol monocaprylate as a hydrophobic surfactant, contains at least polyoxyl 35 castor oil as a hydrophilic surfactant, and oleic acid as an oily component. It is desirable to use
- composition according to the present invention can further contain an antioxidant.
- an antioxidant preferably dl- ⁇ -tocopherol, butyrated hydroxytoluene, butyrated hydroxyanisole, propyl gallate, propyl gallate, pharmaceutically acceptable quinones, astaxanthin, and D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, etc., as well as combinations thereof.
- Antioxidants more preferably include dl- ⁇ -tocopherol.
- composition according to the present invention can further contain a solubilizer.
- Solubilizers preferably include ethanol, propylene glycol, polyethylene glycol 300, polyethylene glycol 400, diethylene glycol monoethyl ether, combinations thereof, and the like.
- the solubilizer is more preferably ethanol or propylene glycol, and still more preferably propylene glycol.
- the content of Compound 1, or a salt thereof, or a solvate thereof is such that it dissolves in the liquid additive and optionally used oily component and exhibits a certain level of effectiveness.
- the content is not particularly limited as long as it is, the content of Compound 1, or a salt thereof, or a solvate thereof with respect to the entire composition, the upper limit is preferably 10% by weight or less, 9% by weight % or less, 8 wt% or less, 7 wt% or less, 6 wt% or less, 5 wt% or less, 4 wt% or less, 3 wt% or less, or 2 wt% or less, more preferably 10 wt% or less, 8 wt% Below, it can be 7% by weight or less.
- the lower limit of the content is not particularly limited, but preferably exceeds 0%, 0.1% by weight or more, 0.2% by weight or more, 0.3% by weight or more, 0.4% by weight or more, 0 0.5 wt% or more, 0.6 wt% or more, 0.7 wt% or more, 0.8 wt% or more, 0.9 wt% or more, or 1 wt% or more, more preferably 0 .1 wt% or more, 0.2 wt% or more, 0.3 wt% or more, 0.4 wt% or more, or 0.5 wt% or more. Further, the content can be within any combination of the above lower limit and upper limit.
- the content range is preferably 0.1 wt% to 10 wt%, 0.2 wt% to 10 wt%, 0.3 wt% to 10 wt%, 0.4 wt% or more. It can be 10 wt % or less, 0.5 wt % or more and 10 wt % or less, and the like.
- a liquid additive preferably a surfactant
- a surfactant may be contained so that Compound 1 or a salt thereof, or a solvate thereof can be dissolved, and the content is particularly limited.
- the content of the liquid additive in the entire composition is preferably 50% to 97% by weight, more preferably 55% to 96% by weight, and still more preferably 70% to 90% by weight. can do.
- the content of the hydrophobic surfactant in the entire composition is preferably 20% by weight or more and 70% by weight or less, more preferably 25% by weight or more and 65% by weight or less, and still more preferably 30% by weight. % or more and 55% or less by weight.
- the content of the hydrophilic surfactant in the entire composition is preferably 20% by weight or more and 40% by weight or less, more preferably 25% by weight or more and 35% by weight or less, still more preferably 28% by weight. % by weight or more and 33% by weight or less.
- the weight ratio of the hydrophobic surfactant to the hydrophilic surfactant is preferably 0.5 to 3.0, more preferably is 1.0 to 2.5, more preferably 1.5 to 2.0.
- the content of the oily component in the entire composition is preferably 0% by weight or more and 50% by weight or less, more preferably 1% by weight or more and 40% by weight or less, and still more preferably 10% by weight or more. 20% by weight or less.
- the content of the antioxidant with respect to the entire composition is not particularly limited as long as it does not adversely affect the pharmaceutical properties of the formulation, but is preferably 0.01% by weight. As described above, the content is more preferably 0.01% to 5% by weight, more preferably 0.1% to 5% by weight, and still more preferably 0.1% to 2% by weight.
- the content of the solubilizer in the composition as a whole is not particularly limited as long as it does not adversely affect the pharmaceutical properties of the formulation, but is preferably 1% by weight or more and 20% by weight. % by weight or less, more preferably about 2% to 15% by weight.
- the weight ratio of the liquid additive to the compound represented by formula (1), or a salt thereof, or a solvate thereof is not particularly limited as long as compound 1 or a salt thereof, or a solvate thereof can be dissolved, but is preferably 5 or more, more preferably 10 or more.
- the upper limit of the weight ratio is not particularly limited, it can be, for example, preferably 2000 or less, more preferably 1000 or less.
- the weight ratio can be in the range of any combination of the lower limit and the upper limit. can.
- Liquid additives are preferably surfactants.
- the total weight ratio of the liquid additive and the oily component to the compound represented by formula (1), a salt thereof, or a solvate thereof is not particularly limited as long as the compound 1 or a salt thereof, or a solvate thereof can be dissolved, but is preferably 10 or more.
- the upper limit of the weight ratio is not particularly limited, it can be, for example, preferably 2000 or less, more preferably 1000 or less.
- the weight ratio can range in any combination of lower and upper limits, for example the weight ratio can be preferably 10-2000, more preferably 10-2000.
- Liquid additives are preferably surfactants.
- composition of the present invention may be liquid, gel or semi-solid, preferably liquid.
- composition according to the present invention is characterized in that droplets formed when the composition is dispersed in a liquid such as water have a small average particle size and a narrow particle size distribution.
- a certain dilution ratio for example, the composition is 0.01 to 1% by volume with respect to the entire mixed solution
- the mixed solution is stirred at a temperature suitable for handling and/or at a temperature that does not affect the stability of the substances contained in the composition, for example, near room temperature (about 25 ° C.), and the composition of the present invention is dispersed in the solution.
- the average particle size of the droplets formed when the droplets are formed is preferably less than 200 nm, more preferably 10 nm or more and less than 200 nm, still more preferably 50 nm or more and less than 200 nm.
- the composition of the present invention preferably has a polydispersity index (PDI), which is an index representing particle size distribution, of less than 0.5, more preferably 0.4 or less, and still more preferably 0.3. It is below.
- PDI polydispersity index
- the average particle size and polydispersity index of the compound in the composition can be determined using known methods such as dynamic light scattering (DLS).
- a solution simulating the internal environment e.g., dissolution test fluid 1 of the Japanese Pharmacopoeia, pH 1.2, body simulating temperature (e.g., 36-37°C) used for disintegration test of enteric preparations
- a solution simulating the internal environment e.g., dissolution test fluid 1 of the Japanese Pharmacopoeia, pH 1.2, body simulating temperature (e.g., 36-37°C) used for disintegration test of enteric preparations
- the mixed solution e.g., when the composition of the present invention is dispersed in the solution by stirring under a certain dilution ratio (for example, the composition is 0.01 to 1% by volume with respect to the entire mixed solution)
- the particle size distribution of the droplets to be formed can similarly exhibit characteristics such that the average particle size of the droplets is small and the width of the particle size distribution is narrow. That is, droplets having a small average particle size and a narrow particle size distribution can be formed even in the gastrointestinal tract.
- Such a composition of the present invention can have excellent dispersibility in liquid and can have excellent absorption into the body.
- a pharmaceutical formulation according to the present invention is a pharmaceutical formulation comprising a composition containing compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component.
- the pharmaceutical formulation according to the present invention comprises the compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component, and optionally an antioxidant, a solubilizer, etc., in addition to It can be produced by introducing a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier commonly used excipients, binders, lubricants, coloring agents, flavoring agents, and if necessary, stabilizers, emulsifiers, absorption enhancers, pH adjusters, preservatives, etc. can be used.
- Ingredients that are generally used as raw materials for pharmaceutical formulations can be blended and formulated by a conventional method.
- the administration of the formulation according to the present invention may be either oral administration or parenteral administration. Oral administration is preferred, but the administration method is not limited to oral administration.
- compound 1 of the present invention or a salt thereof, or a solvate thereof a liquid additive, an optional oily component, etc., and optionally a binder, a disintegrant, a lubricant, etc.
- a liquid additive for example, compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, an optional oily component, etc., and optionally a binder, a disintegrant, a lubricant, etc.
- an agent, a coloring agent, a flavoring agent, etc. it is made into a liquid formulation, a capsule formulation, etc. by a conventional method.
- the composition of the present invention is preferably a liquid, it can be an injection or a capsule in liquid formulations.
- composition of the present invention when used as a capsule, the capsules commonly used in capsule formulations can be used.
- the type of capsule is not particularly limited, and those commonly used in this technical field can be used.
- examples of capsules include hard capsules (hard capsules) and soft capsules (soft capsules).
- a hard capsule usually consists of a cap and a body, and can be produced by putting a cap on the body filled with a formulation.
- raw materials for hard capsules for example, those containing gelatin, hydroxypropylmethylcellulose, pullulan, or a mixture thereof can be preferably used. , No. 1, No. 0, No. 00, and No. 000.
- Soft capsules can be produced, for example, by enveloping a formulation with a base such as gelatin.
- Raw materials for soft capsules that can be preferably used include, for example, gelatin, starch, carrageenan, agar, glycerin, sorbitol, or mixtures thereof.
- Excipients include, for example, lactose, cornstarch, sucrose, glucose, mannitol, sorbitol, crystalline cellulose, and silicon dioxide.
- binders include polyvinyl alcohol, polyvinyl ether, methylcellulose, ethylcellulose, gum arabic, tragacanth, gelatin, shellac, hydroxypropylmethylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, polypropyleneglycol-polyoxyethylene-block polymer, and meglumine. be done.
- disintegrants examples include starch, agar, gelatin powder, crystalline cellulose, calcium carbonate, sodium hydrogencarbonate, calcium citrate, dextrin, pectin, carboxymethylcellulose/calcium, and the like.
- lubricants examples include magnesium stearate, talc, polyethylene glycol, silica, hydrogenated vegetable oils, and the like.
- coloring agents those that are permitted to be added to pharmaceuticals are used, and as flavoring agents, cocoa powder, mint brain, aromatic powder, mint oil, borneol, cinnamon powder, etc. are used.
- compound 1 used in the present invention or a salt thereof, or a solvate thereof may be added with a pH adjuster, a solubilizer, a tonicity agent, etc., if necessary. Add solubilizers, stabilizers, etc., and formulate in a conventional manner.
- parenteral administration specifically includes an injection dosage form, a nasal dosage form, a pulmonary dosage form, a transdermal dosage form, and the like.
- injection dosage forms include systemic or local administration by intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection, and the like.
- nasal dosage forms include systemic or local administration, such as by absorption of active ingredients in the formulation through the nasal mucosa or administration of the formulation through the nasal cavity.
- transpulmonary dosage forms can be administered systemically or locally, such as by administering the formulation to the lungs through the trachea.
- transdermal administration forms include systemic or local administration, such as by applying the formulation to the skin.
- the administration method can be appropriately selected according to the patient's age and symptoms.
- the dosage of the pharmaceutical formulation containing Compound 1 or its salt or solvate thereof used in the present invention can be selected, for example, in the range of 0.001 mg to 100 mg per kg of body weight per dose.
- doses can be selected in the range of 0.1 to 1000 mg/body per patient, but are not necessarily limited to these figures.
- the dosage and administration method vary depending on the patient's body weight, age, symptoms, etc., but can be appropriately selected by those skilled in the art.
- the method for producing the composition according to the present invention comprises: (1) Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component (Step 1); (2) mixing the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component (step 2); and (3) A step of obtaining a composition in which the compound represented by formula (1), or a salt thereof, or a solvate thereof is dissolved (step 3).
- compound 1 or a salt thereof, or a solvate thereof supplied in step (1) is preferably a hydrate of the compound represented by formula (1). , more preferably one or more water molecules of the compound represented by formula (1).
- the number of water molecules contained in the hydrate of the compound represented by formula (1) can be desorbed depending on the surrounding environment such as temperature and humidity.
- Compound 1 or a salt thereof, or a solvate thereof supplied in step (1) is preferably Compound 1, a salt thereof, or a solvate thereof 1 It is a crystal, more preferably a hydrate crystal of the compound represented by formula (1).
- the water molecules in the hydrate crystals of the compound represented by formula (1) preferably contain one or more water molecules, but the number of water molecules desorbs depending on the surrounding environment such as temperature and humidity. What can be done is described above.
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15.895°, 16.190°, 16. It can have an X-ray powder diffraction pattern comprising at least one peak at 643°, 17.813°, and 19.424° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.813° ( ⁇ 0.2°) can have an X-ray powder diffraction pattern comprising at least one peak of
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.813° ( ⁇ 0.2°) It is more preferable to have a powder X-ray diffraction pattern containing a peak of
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15.895°, 16.190°, 16. It is even more preferred to have an X-ray powder diffraction pattern comprising peaks at 643°, 17.813° and 19.424° ( ⁇ 0.2°).
- the analysis by powder X-ray diffraction of the present invention can be performed, for example, according to a conventional method such as the "powder X-ray diffraction measurement method" described in the Japanese Pharmacopoeia (15th revision). Further, according to the Japanese Pharmacopoeia, it is explained that the diffraction angles 2 ⁇ of the same crystal form usually match within the range of ⁇ 0.2°. Therefore, the present invention includes not only crystals in which the diffraction angles of peaks in X-ray powder diffraction match perfectly, but also crystals in which the diffraction angles of peaks match with an error of about ⁇ 0.2°.
- each component each component of the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component, and, if necessary, a pharmaceutical Ingredients that are usually used as excipients and the like are put into a known stirring and mixing device or the like and mixed.
- the mixing temperature and mixing time of each component are not particularly limited as long as they do not adversely affect the components.
- the mixing temperature is preferably 0 to 50° C., more preferably 10 to 30° C., and the mixing time is preferably about 5 minutes to 60 minutes.
- a method for producing a pharmaceutical formulation according to the present invention can include a step of providing the composition of the present invention, and a step of formulating the composition to provide a pharmaceutical formulation.
- the method for producing a pharmaceutical formulation according to the present invention can include the steps of providing the composition of the present invention and filling the composition into capsules to produce capsule formulations.
- the method for producing a pharmaceutical preparation according to the present invention can include the step of filling capsules with the composition obtained by the method for producing the composition to provide a capsule formulation. That is, it can include a step of filling capsules with the composition obtained by steps (1) to (3) in the method for producing the composition to provide a capsule formulation.
- the meaning of the term “and/or” includes any combination in which “and” and “or” are appropriately combined.
- “A, B, and/or C” includes the following seven variations: (i) A, (ii) B, (iii) C, (iv) A and B, (v) A and C, (vi) B and C, (vii) A, B, and C.
- the term "about” when used in combination with a numerical value means a range of +10% and -10% of the numerical value.
- to indicating a range includes values at both ends thereof, for example, "A to B” means a range of A or more and B or less.
- the unit of molecular weight in this specification is "g/mol". Molecular weight units may be omitted in this specification.
- Reagents such as liquid additives, surfactants, oleaginous components, solubilizers, antioxidants, or solvents used in the practice of this invention were obtained without purification from commercial suppliers, except as specifically noted. Using.
- the solubility of the compound represented by formula (1), or a salt thereof, or a solvate thereof is measured by adding an additive or formulation to the compound, followed by shaking or stirring. Dissolution was confirmed and calculated from the amount of liquid added. It is also possible to measure with an apparatus such as high performance liquid chromatography.
- Test Method 1 Particle Size Distribution Measurement by Dynamic Scattering (DLS) Particle size distribution is determined by dynamic scattering (DLS) using ZETASIZER Nano-ZS (Malvern). A sample is placed in a disposable cell (small volume cuvette), set in ZETASIZER Nano-ZS (Malvern), and particle size distribution is determined by dynamic scattering (DLS). The average particle size (Z-average size) and polydispersity index (PDI) are used as indicators of the particle size distribution. Z-average is the average particle diameter based on the scattering intensity using the cumulant method. PDI is an index representing the width of the particle size distribution, and is shown in the range of 0 to 1.
- dispersions with a PDI of 0.1 or less are monodisperse
- dispersions with a PDI between 0.1 and 0.5 are It is believed to have a narrow distribution.
- dispersions with a PDI greater than 0.5 are considered polydisperse.
- Test method 3 Calculation of pharmacokinetic parameters Plasma is separated from the collected blood by centrifugation, deproteinized with acetonitrile, and the concentration in plasma is measured using an LC-MS/MS apparatus. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were determined by non-compartmental analysis using pharmacokinetic analysis software Phoenix WinNonlin 8.2 (Certara L.P.). calculate.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Preparation example 1 Preparation of Compound 1 Compound 1 ("(5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-butyl)-18-cyclopentyl-29 -(3,5-difluoro-4-(trifluoromethyl)phenethyl)-36-ethyl-11-isobutyl-N,N,5,6,12,16,19,33-octamethyl-35-(4-methyl) benzyl)-4,7,10,13,17,20,23,28,31,34,37-undecaoxotetratriacontahydro-2H,4H-spiro[azeto[2,1-u]pyrrolo[2 ,1-i][1,4,7,10,13,16,19,22,25,28,31]undecaazacyclotetratriacontin-21,1′-cyclopentane
- Fmoc-Asp(OAl)-OH ((2S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-4-oxo-4-prop-2-enoxybutanoic acid, CAS No. 146982-24-3) ( 200 g, 506 mmol), p-toluenesulfonic acid (5.7 g, 0.05 equivalent) and paraformaldehyde (45.6 g, 3 equivalent) were mixed with toluene and stirred at 110° C. for 16 hours. The reaction mixture was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate and washed twice with an aqueous sodium hydrogencarbonate solution.
- the reaction mixture was diluted with ethyl acetate (422 mL), washed twice with hydrochloric acid (1 mol/L, 422 mL), and the resulting aqueous phase was extracted twice with ethyl acetate (422 mL). All the organic phases were mixed and washed in order with water (422 mL), saturated aqueous sodium bicarbonate/water mixture (1:1, 422 mL), and saturated brine/water mixture (1:1, 422 mL). The resulting organic phase was dried over sodium sulfate and evaporated under reduced pressure. DCM (512 mL) was added to the resulting residue and stirred for 0.5 hours.
- the solution was drained from the frit after 10 minutes of shaking.
- DMF (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- a solution of triethylamine hydrochloride (7.96 g, 57.8 mmol) in DCM (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- DCM (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- DMF (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit. This DMF washing step of the resin was repeated one more time.
- Nickel bromide trihydrate (NiBr 2.3H 2 O) (13.5 g, 49.7 mmol, 0.3 eq) and 4,4′-di-tert-butyl-2,2′-bipyridyl (dtbbpy, CAS No. 72914-19-3) (13.3 g, 49.7 mmol, 0.3 equivalent) was added to DMA (400 mL) and stirred at 50° C. for 3 hours under a nitrogen atmosphere to prepare a Ni solution.
- TfOH trifluoromethanesulfonic acid
- Acetonitrile/water 400/400 mL was added to the residue and the pH was adjusted to 7 with aqueous sodium hydroxide (48%).
- Fmoc-OSu (36.6 g, 108.6 mmol, 0.9 equivalents) was added to this solution, the pH was adjusted to 8.0 with an aqueous sodium hydroxide solution (48%), and the mixture was stirred at room temperature for 16 hours.
- the reaction solution was filtered while washing with acetonitrile/water (1/1) to remove solid components, the filtrate was diluted with acetonitrile, and adjusted to be acidic with 6 mol/L aqueous hydrochloric acid.
- Multipep RSi peptide synthesizer
- the compound 1217-c-resin obtained above (200 mg per solid-phase reaction vessel) was added to 30 solid-phase reaction vessels and set in a peptide synthesizer.
- Dichloromethane (DCM) was added to all 30 solid-phase reactors and allowed to stand for 1 hour to swell the resin. The solvent was then drained from the frit.
- NMP solution of Fmoc-Hph(4-CF3-35-F2)-OH (0.45 mol/L) and HOAt (0.375 mol/L) (for one solid-phase reaction vessel 0.6 mL) and a DMF solution of N,N'-diisopropylcarbodiimide (DIC) (10 v/v%, 0.72 mL per solid phase reaction vessel) were mixed in a mixing vial of the synthesizer, and then 30 vials. It was added to all solid phase reactors and allowed to stand at 40° C. for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- DIC N,N'-diisopropylcarbodiimide
- NMP solution of Fmoc-MeGly-OH (0.6 mol/L) and HOAt (0.375 mol/L) (0.6 mL per solid-phase reaction vessel) and N,N'-diisopropylcarbodiimide (DIC) in DMF (10 v/v%, 0.72 mL per solid-phase reaction vessel) was mixed in the mixing vial of the synthesizer and then added to all 30 solid-phase reaction vessels. °C for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- NMP solution of Fmoc-MeAla-OH (0.6 mol/L) and HOAt (0.375 mol/L) (0.6 mL per solid-phase reaction vessel) and N,N'-diisopropylcarbodiimide (DIC) in DMF (10 v/v%, 0.72 mL per solid-phase reaction vessel) was mixed in the mixing vial of the synthesizer and then added to all 30 solid-phase reaction vessels. °C for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- a mixed solution of saturated aqueous ammonium chloride solution (40 mL) and water (40 mL) was added to the resulting solution, and the mixture was extracted with isopropyl acetate (350 mL).
- the obtained organic phase was washed with a mixed solution of saturated aqueous sodium hydrogencarbonate solution (40 mL) and water (40 mL) and a mixed solution of saturated brine (40 mL) and water (40 mL) in that order, dried over sodium sulfate, and dried under reduced pressure. Evaporation of solvent gave 3.36 g of residue.
- Preparation example 2 Preparation of seed crystals used in Preparation Example 3
- the amorphous compound 1 (122.3 mg) obtained in Preparation Example 1 was dissolved in DMSO (0.612 mL), and this solution (0.015 mL) was heated to -20°C. was lyophilized for 2 days at A water-acetonitrile mixture (3:1, 0.015 mL) was added to the obtained lyophilized product, and the mixture was shaken and stirred at room temperature for 7 days to obtain hydrate crystals of Compound 1 (Form C).
- the reaction mixture was filtered under pressure using a filter cloth (PF-020), and the inside of the reaction vessel and the filter were filtered using a filter (CCF-G100-E1N).
- Acetone (7.5 kg) and purified water ( 7.5 kg) of the mixed solution was used to wash the obtained crystals.
- the obtained crystals were washed with purified water (17.0 kg x 2) filtered using a filter (CCF-G100-E1N), the pressure of the filtration device that collected the crystals was reduced, and the external temperature of the filtration device was set to 70°C. and dried the crystals for 17 hours. Further, the crystals were dried at an external temperature of room temperature to 30° C. for 27 hours.
- the dry powder was recovered from the filter to obtain a white powder (2.6 kg).
- the resulting white powder was confirmed to have the same structure as the compound obtained in Preparation Example 1, "Synthesis of compound 1 (cyclization and purification of peptide)".
- HPLC analysis conditions Equipment: Waters ACQUITY UPLC H-Class Column: ACQUITY UPLC CSH C18 (Waters), 2.1 mm ID x 150 mm, 1.7 ⁇ m
- Mobile phase 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
- Elution method B) 20% (0 min) ⁇ 100% (24 min) ⁇ 100% (29 min) ⁇ 20% (29.1 min) ⁇ 20% (34 min)
- Flow rate 0.3 mL/min
- Powder X-ray analysis was performed using the XRPD equipment shown below.
- XRPD measurement condition measurement device X'pert-pro MPD (manufactured by PANalytical) Radiation source: CuK ⁇ Tube voltage: 45 kV Tube current: 40mA Scanning range: 3-40° Scanning speed: 4.2°/min Sampling width: 0.017°
- the water content of the hydrate crystals of Compound 1 (Form C) obtained in the same manner as in Preparation Example 3 was measured by Karl Fischer titration. Measurement was performed using CA-310 (manufactured by Nitto Seiko Analytic Tech) after acclimating the sample in a laboratory environment. As a result of the measurement, the water content of the hydrate crystals of Compound 1 (Form C) was 6.50 wt%.
- the hydrate crystal of compound 1 (Form C) is a hydrate crystal that has water molecules in its crystal structure. was confirmed.
- a hydrate crystal of compound 1 (Form C) obtained in the same manner as in Preparation Example 3 was subjected to dynamic water vapor adsorption measurement.
- the results are shown in FIG. Measurement device: DVS Intrinsic (manufactured by Surface Measurement Systems) Temperature: 25°C Relative humidity (%) measurement points: Cycle 1: 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%); Cycle 2: 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%); Cycle 2: 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%) Threshold: 0.001 dm/dt (%/min) Minimum sorption time: 10 minutes Maximum sorption time: 1440 minutes
- the hydrate crystal of Compound 1 is a hydrate crystal that changes in weight by 3.3% as the hydration number changes in the relative humidity range of 0 to 95%. rice field.
- Compound I The hydrate crystals of compound 1 (Form C) obtained in Preparation Example 3 (hereinafter sometimes referred to as “compound I”) were used for the preparation and evaluation of the following compositions or formulations.
- a compound used as an active ingredient in Examples is described as “compound I”
- compound I dissolved in a liquid additive or the like means compound 1 in the free form.
- Test Example 1 Evaluation of solubility of compound I in various additives The solubility of compound I in various additives was determined. Various liquid additives were added to Compound I and stirred, and when Compound I did not dissolve, further additives were added and stirred. This operation was repeated until compound I dissolved, and the approximate solubility was obtained from the amount of additive required to dissolve compound I. Since Polyoxyethylene (15) hydroxystearate (Solutol HS15) is solid at room temperature (RT), it was liquefied by heating to 37° C. to determine its solubility. Solubility of other additives was determined at room temperature. Table 2 shows the results. As a result, PG monocaprylate was found to exhibit high solubility in compound I.
- Test Example 2 Evaluation of solubility of compound I in each formulation
- the solubility of compound I in each of additive mixtures 1 to 4 was measured. Assuming the migration of water from the capsule shell when formulated as a capsule, the solubility of an additive mixture containing 3% water was also measured. As a result, as shown in Table 4, it was found that additive mixtures 1 to 4 were capable of dissolving compound I at a high concentration of 100 mg/mL or more.
- the solubility of compound I decreased to 60 to 70 mg / mL. decided to evaluate.
- additive mixtures 5 to 45 were prepared based on the formulations in Tables 5 to 10.
- Formulations A to V which are compositions of the present invention, were prepared by the following method.
- Example 1 (Preparation of Formulation A) Compound I was weighed into a vial and 1 mL of Additive Mixture 1 was added per 50 mg of Compound I. Formulation A was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 2 (Preparation of Formulation B) Compound I was weighed into a vial and 1 mL of Additive Mixture 2 was added per 50 mg of Compound I. Formulation B was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 3 (Preparation of Formulation C) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 50 mg of Compound I. Formulation C was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 4 (Preparation of Formulation D) Compound I was weighed into a vial and 1 mL of Additive Mixture 4 was added per 50 mg of Compound I. Formulation D was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 5 (Preparation of Formulation E) Compound I was weighed into a vial and 1 mL of Additive Mixture 7 was added per 50 mg of Compound I. Formulation E was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 6 (Preparation of Formulation F) Compound I was weighed into a vial and 1 mL of Additive Mixture 19 was added per 50 mg of Compound I. Formulation F was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 7 (Preparation of Formulation G) Compound I was weighed into a vial and 1 mL of Additive Mixture 20 was added per 50 mg of Compound I. Formulation G was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 8 (Preparation of Formulation H) Compound I was weighed into a vial and 1 mL of Additive Mixture 21 was added per 50 mg of Compound I. Formulation H was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 9 (Preparation of Formulation I) Compound I was weighed into a vial and 1 mL of Additive Mixture 25 was added per 50 mg of Compound I. Formulation I was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 10 (Preparation of Formulation J) Compound I was weighed into a vial and 1 mL of Additive Mixture 26 was added per 50 mg of Compound I. Formulation J was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 11 (Preparation of Formulation K) Compound I was weighed into a vial and 1 mL of Additive Mixture 28 was added per 50 mg of Compound I. Formulation K was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 12 (Preparation of Formulation L) Compound I was weighed into a vial and 1 mL of Additive Mixture 29 was added per 50 mg of Compound I. Formulation L was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 13 (Preparation of Formulation M) Compound I was weighed into a vial and 1 mL of Additive Mixture 30 per 50 mg of Compound I was added. Formulation M was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 14 (Preparation of Formulation N) Compound I was weighed into a vial and 1 mL of Additive Mixture 32 was added per 50 mg of Compound I. Formulation N was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 15 (Preparation of Formulation O) Compound I was weighed into a vial and 1 mL of Additive Mixture 33 was added per 50 mg of Compound I. Formulation O was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 16 (Preparation of Formulation P) Compound I was weighed into a vial and 1 mL of Additive Mixture 34 was added per 50 mg of Compound I. Formulation P was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 17 (Preparation of Formulation Q) Compound I was weighed into a vial and 1 mL of Additive Mixture 35 was added per 50 mg of Compound I. Formulation Q was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 18 (Preparation of Formulation R) Compound I was weighed into a vial and 1 mL of Additive Mixture 40 was added per 50 mg of Compound I. Formulation R was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 19 (Preparation of Formulation S) Compound I was weighed into a vial and 1 mL of Additive Mixture 41 was added per 50 mg of Compound I. Preparation S was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 20 (Preparation of Formulation T) Compound I was weighed into a vial and 1 mL of Additive Mixture 42 was added per 50 mg of Compound I. Preparation T was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 21 (Preparation of Formulation U) Compound I was weighed into a vial and 1 mL of Additive Mixture 44 was added per 50 mg of Compound I. Formulation U was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 22 (Preparation of Formulation V) Compound I was weighed into a vial and 1 mL of Additive Mixture 45 was added per 50 mg of Compound I. Formulation V was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 1 (Preparation of Formulation W) Compound I was weighed into a vial and 1 mL of Additive Mixture 8 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation W was obtained.
- Comparative Example 2 (Preparation of Formulation X) Compound I was weighed into a vial and 1 mL of Additive Mixture 9 was added per 50 mg of Compound I. Preparation X was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 3 (Preparation of Formulation Y) Compound I was weighed into a vial and 1 mL of Additive Mixture 10 was added per 50 mg of Compound I. Formulation Y was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 4 (Preparation of Formulation Z) Compound I was weighed into a vial and 1 mL of Additive Mixture 13 was added per 50 mg of Compound I. Formulation Z was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 5 (Preparation of Formulation AA) Compound I was weighed into a vial and 1 mL of Additive Mixture 14 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AA was obtained.
- Comparative Example 6 (Preparation of Formulation AB) Compound I was weighed into a vial and 1 mL of Additive Mixture 15 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AB was obtained.
- Comparative Example 7 (Preparation of Formulation AC) Compound I was weighed into a vial and 1 mL of Additive Mixture 16 was added per 50 mg of Compound I. Formulation AC was obtained after confirming complete dissolution of compound I by stirring using a stirrer.
- Comparative Example 8 (Preparation of Formulation AD) Compound I was weighed into a vial and 1 mL of Additive Mixture 17 was added per 50 mg of Compound I. Formulation AD was obtained after confirming complete dissolution of Compound I by stirring using a stirrer.
- Comparative Example 9 (Preparation of Formulation AE) Compound I was weighed into a vial and 1 mL of Additive Mixture 18 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AE was obtained.
- Comparative Example 10 (Preparation of Formulation AF) Compound I was weighed into a vial and 1 mL of Additive Mixture 22 was added per 50 mg of Compound I. Formulation AF was obtained after confirming complete dissolution of Compound I by stirring using a stirrer.
- Comparative Example 11 (Preparation of Formulation AG) Compound I was weighed into a vial and 1 mL of Additive Mixture 23 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring with a stirrer, Formulation AG was obtained.
- Comparative Example 12 (Preparation of Formulation AH) Compound I was weighed into a vial and 1 mL of Additive Mixture 24 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AH was obtained.
- Comparative Example 13 (Preparation of Formulation AI) Compound I was weighed into a vial and 1 mL of Additive Mixture 27 was added per 50 mg of Compound I. Formulation AI was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 14 (Preparation of Formulation AJ) Compound I was weighed into a vial and 1 mL of Additive Mixture 31 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AJ was obtained.
- Comparative Example 15 (Preparation of Formulation AK) Compound I was weighed into a vial and 1 mL of Additive Mixture 36 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AK was obtained.
- Comparative Example 16 (Preparation of Formulation AL) Compound I was weighed into a vial and 1 mL of Additive Mixture 37 was added per 50 mg of Compound I. After confirming that compound I was completely dissolved by stirring using a stirrer, preparation AL was obtained.
- Comparative Example 17 (Preparation of Formulation AM) Compound I was weighed into a vial and 1 mL of Additive Mixture 38 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AM was obtained.
- Comparative Example 18 (Preparation of Formulation AN) Compound I was weighed into a vial and 1 mL of Additive Mixture 39 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AN was obtained.
- Comparative Example 19 (Preparation of Formulation AO) Compound I was weighed into a vial and 1 mL of Additive Mixture 43 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AO was obtained.
- Test Example 3 (particle size distribution measurement by DLS) Water was then added to Formulations A through V prepared in Examples 1-22 and Formulations W through Formulations AO prepared in Comparative Examples 1-19 to form dispersions.
- the particle size of droplets generated in the dispersion was measured to evaluate the particle size distribution at room temperature (approximately 25°C). 4 ⁇ L of each formulation was placed in a vial, and 396 ⁇ L of water was added.
- the average particle size (Z-average size) and polydispersity index (PDI) were used as indices representing the particle size distribution.
- PDI is an index representing the width of the particle size distribution and is shown in the range of 0 to 1.
- dispersions with a PDI of 0.1 or less are monodisperse
- dispersions with a PDI between 0.1 and 0.5 are It is believed to have a narrow distribution.
- dispersions with a PDI greater than 0.5 are considered polydisperse.
- Table 16 all of Formulations A to V were good dispersions with a small average particle size (less than 200 nm) and a narrow distribution of PDI of less than 0.5.
- formulations W to AO had a large average particle size (200 nm or more) and were polydisperse with a PDI exceeding 0.5.
- Test Example 4 80°C stability test
- 10 mg each of formulations A, C, F, H, and J prepared in Examples 1, 3, 6, 8, and 10 were placed in a glass vial, placed in a brown bottle containing silica gel and an oxygen scavenger, and sealed. Each brown bottle was placed in a constant temperature bath at 80° C. and stored for 2 weeks.
- compound I showed a high survival rate of 95% or more in all formulations, indicating that it is a formulation that can be developed as a drug.
- compound I showed a high survival rate of 95% or more in any formulation, and it was found to be a formulation that can be developed as a drug.
- Formulation C dispersed immediately after the start of the test.
- Formulation F also showed a degree of dispersion of more than 80% after 60 minutes, and was found to be a good formulation compared to formulation AJ.
- Example 23 (Preparation of Capsule Formulation AP for Rat Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. The solution in which the compound was dissolved was filled into No. 9 gelatin capsules to produce Formulation AP.
- Example 24 (Preparation of Capsule Formulation AQ for Rat Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 19 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. The solution in which the compound was dissolved was filled into No. 9 gelatin capsules to produce Formulation AQ.
- Comparative Example 20 (Preparation of capsule formulation AR for rat administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 31 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. Formulation AR was prepared by filling a No. 9 gelatin capsule with the solution in which the compound was dissolved.
- Comparative Example 21 (Preparation of solution preparation AS for rat administration) Compound I was weighed into a vial and dissolved by adding 1 mL of dimethylsulfoxide (DMSO) per 10 mg of compound I. Furthermore, 0.5 mL of polyoxyl 35 castor oil and 8.5 mL of water were added and well stirred and mixed to prepare a solution formulation AS.
- DMSO dimethylsulfoxide
- Test Example 7 (rat absorption evaluation) Capsule formulations AP and AQ of Examples 23 and 24 and capsule formulations AR and solution formulations AS of Comparative Examples 20 and 21 were orally administered to rats (Wister Hannover, male) at a dose of 10 mg/kg, four of each formulation.
- Formulations AP, AQ and AR were administered by oral gavage as capsules as they were, and by oral gavage with water at a volume of 5 mL/kg.
- Solution formulation AS was administered by oral gavage as is. About 0.5, 1, 2, 4, 7, 24 and 48 hours after administration, blood is collected, and the concentration of compound I contained in the plasma is quantified by LC-MS/MS by the method shown in test method 3. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were calculated.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Table 20 shows the AUC and Cmax after administration of each formulation.
- Capsule formulations AP and AQ of Examples 23, 24 were found to exhibit higher AUC and Cmax than Capsule formulation AR of Comparative Example 20.
- the capsule formulation AP of Example 23 exhibited comparable AUC and Cmax compared to the solution formulation AS of Comparative Example 21.
- capsule formulation AT and solution formulation AU were prepared to evaluate absorption in monkeys.
- Example 25 (Preparation of Capsule Formulation AT for Monkey Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 50 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer.
- Formulation AT was prepared by filling a No. 1 gelatin capsule with the solution in which the compound was dissolved.
- Comparative Example 22 (Preparation of solution formulation AU for monkey administration) Compound I was weighed into a vial and dissolved by adding 1 mL of dimethylsulfoxide (DMSO) per 15 mg of compound I. Further, 0.25 mL of polyoxyl 35 castor oil and 8.75 mL of water were added and well stirred and mixed to prepare a solution formulation AU.
- DMSO dimethylsulfoxide
- Test Example 8 (monkey absorbability evaluation) The capsule formulation AT of Example 25 and the solution formulation AU of Comparative Example 22 were orally administered to cynomolgus monkeys at a dose of 3 mg/kg, four of each formulation. Capsule formulation AT was administered by gavage as is, and water was additionally administered by gavage at a volume of 4 mL/kg. The solution formulation AU was administered by oral gavage as is. About 0.5, 1, 2, 4, 7, 24 and 48 hours after administration, blood is collected, and the concentration of compound I contained in the plasma is quantified by LC-MS/MS by the method shown in test method 3. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were calculated.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Table 21 shows the AUC and Cmax after administration of each formulation. Capsule formulation AT of Example 25 was found to exhibit similar AUC and Cmax as compared to solution formulation AU of Comparative Example 22.
- Example 26 (Production of Capsules (Hard Capsules) 1) Weigh compound I in a stainless steel container, add 51.5 mg of Propylene glycol monocaprylate, 13.3 mg of Oleic acid, 31.1 mg of Polyoxyl 35 castor oil and 0.2 mg of dl- ⁇ -tocopherol per 0.5 mg of compound I, and homogenizer was used to stir and mix. The solution in which the compound was dissolved was filled into No. 3 gelatin capsules and band-sealed to produce Formulation AV.
- Example 27 (Production of Capsules (Hard Capsules) 2) Weigh compound I in a stainless steel container, add 123.6 mg of Propylene glycol monocaprylate, 31.9 mg of Oleic acid, 74.6 mg of Polyoxyl 35 castor oil and 0.5 mg of dl- ⁇ -tocopherol per 12 mg of compound I, Stirred and mixed using a homogenizer. The solution in which the compound was dissolved was filled into No. 3 gelatin capsules and band-sealed to prepare Formulation AW.
- Example 28 (Production of Capsules (Soft Capsules) 3) Compound I was weighed into a stainless steel container, and 206.4 mg of Propylene glycol monocaprylate, 53.3 mg of Oleic acid and 124.6 mg of Polyoxyl 35 castor oil and 0.8 mg of dl- ⁇ -tocopherol were added per 19.1 mg of Compound I. In addition, they were stirred and mixed using a stirring blade. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AX.
- Example 29 (Production of Capsules (Soft Capsules) 4) Weigh Compound I in a stainless steel container, add 46.4 mg of Propylene glycol monocaprylate, 12.0 mg of Oleic acid and 28.0 mg of Polyoxyl 35 castor oil per 4.5 mg of Compound I, and stir and mix using a stirring blade. did. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AY.
- Example 30 (Production of Capsules (Soft Capsules) 5) Weigh compound I in a stainless steel container, add 61.8 mg of Propylene glycol monocaprylate, 16.0 mg of Oleic acid and 37.3 mg of Polyoxyl 35 castor oil per 6.0 mg of compound I, stir and mix using a stirring blade did. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AZ.
- Test Example 9 particle size distribution measurement by DLS
- the particle size distribution of Formulation AV, Formulation AW, Formulation AX, Formulation AY, and Formulation AZ prepared in Examples 26-30 was evaluated when dispersed in water.
- the particle size distribution was determined by dynamic scattering (DLS) using the method shown in Test Method 1. Similar to Test Example 3, the average particle size (Z-average size) and the polydispersity index (PDI) were used as indices representing the particle size distribution.
- DLS dynamic scattering
- Formulation AV, Formulation AW, Formulation AX, Formulation AY, and Formulation AZ all had small average particle sizes (less than 200 nm) and good dispersion showing narrow distributions with a PDI of less than 0.5. was the body.
- Test Example 10 Protein-protein interaction inhibitory activity of Kras and SOS1 using AlphaScreen (KrasG12D-SOS PPI) Protein-protein interaction inhibition (PPI) of Kras and SOS1 can be performed by methods known to those skilled in the art using equipment, reagents, etc. available from commercial suppliers. . Specifically, biotin-tagged human Kras and His-tagged human SOS1 enzymes expressed in E. coli and loaded with GDP after purification were used to transfer nickel-bound donor beads to streptavidin-bound acceptor beads. Measured by energy transfer.
- This measurement utilizes the fact that when the donor beads are irradiated with light of 680 nm, energy is transferred to the acceptor beads via singlet oxygen, and light of 520 to 620 nm is detected from the acceptor beads. .
- a 50% inhibitory concentration (IC50 value) was calculated from the inhibition rate against the control group containing no Compound 1.
- the IC50 value for Compound 1 was 0.0010 ⁇ M.
- Compound 1 was shown to inhibit the protein-protein interaction between Kras and SOS1. Inhibition of binding between K-ras and SOS1 is known to inhibit signal transduction downstream of Kras. Therefore, it was suggested that Compound 1 also has cell proliferation inhibitory activity.
- AsPC-1 cell growth inhibitory activity can be performed by methods known to those skilled in the art using equipment, reagents, cell lines, etc. available from commercial suppliers. Specifically, after serially diluting Compound 1 with dimethylsulfoxide, 200 nL was dispensed into a U-bottom 96-well plate using Labcyte Echo. For the human pancreatic cancer strain AsPC-1, a cell suspension was prepared in RPMI-1640 medium supplemented with 15% fetal bovine serum to give a cell suspension of 1000 cells/100 ⁇ L.
- CGI cell growth inhibition activity
- composition of the present invention can be made excellent in various properties required as a formulation.
- compositions containing such specific ingredients have desirable particle characteristics, good stability and dispersibility of the active ingredient, and good absorption by the body. Therefore, the composition according to the present invention is suitable for use as a pharmaceutical preparation.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
〔1〕下記式(1):

〔2〕前記液体添加剤が、界面活性剤である、〔1〕に記載の組成物。
〔3〕前記界面活性剤が、疎水性界面活性剤、および親水性界面活性剤の組み合わせである、〔2〕に記載の組成物。
〔4〕前記疎水性界面活性剤のHLB値が0以上10未満であり、前記親水性界面活性剤のHLB値が10以上30以下である、〔3〕に記載の組成物。
〔5〕前記疎水性界面活性剤が、プロピレングリコール脂肪酸エステル、グリセリン脂肪酸エステル、ポリグリセリン脂肪酸エステル、ソルビタン脂肪酸エステル、および疎水性ポリオキシエチレン硬化ヒマシ油からなる群より選択される少なくとも1種である、〔3〕または〔4〕に記載の組成物。
〔6〕前記疎水性界面活性剤が、プロピレングリコール脂肪酸エステルおよびソルビタン脂肪酸エステルからなる群より選択される少なくとも1種である、〔3〕~〔5〕のいずれか一項に記載の組成物。
〔7〕前記疎水性界面活性剤が、プロピレングリコール脂肪酸エステルを含む、〔3〕~〔6〕のいずれか一項に記載の組成物。
〔8〕前記プロピレングリコール脂肪酸エステルが、モノカプロン酸プロピレングリコール、モノカプリル酸プロピレングリコール、モノカプリン酸プロピレングリコール、モノラウリン酸プロピレングリコール、モノミリスチン酸プロピレングリコール、モノパルミチン酸プロピレングリコール、モノステアリン酸プロピレングリコール、およびモノオレイン酸プロピレングリコールからなる群より選択される少なくとも1種である、〔5〕に記載の組成物。
〔9〕前記グリセリン脂肪酸エステルが、モノカプロン酸グリセリル、モノカプリル酸グリセリル、モノカプリン酸グリセリル、モノラウリン酸グリセリル、モノミリスチン酸グリセリル、モノパルミチン酸グリセリル、モノステアリン酸グリセリル、モノオレイン酸グリセリル、およびモノリノール酸グリセリルからなる群より選択される少なくとも1種である、〔5〕に記載の組成物。
〔10〕前記ポリグリセリン脂肪酸エステルが、モノオレイン酸ジグリセリルである、〔5〕に記載の組成物。
〔11〕前記ソルビタン脂肪酸エステルが、モノカプリル酸ソルビタン、モノカプリン酸ソルビタン、モノラウリン酸ソルビタン、モノミリスチン酸ソルビタン、モノパルミチン酸ソルビタン、モノステアリン酸ソルビタン、モノオレイン酸ソルビタン、セスキオレイン酸ソルビタン、およびトリオレイン酸ソルビタンからなる群より選択される少なくとも1種である、〔5〕に記載の組成物。
〔12〕前記疎水性ポリオキシエチレン硬化ヒマシ油が、ポリオキシエチレン硬化ヒマシ油 5およびポリオキシエチレン硬化ヒマシ油 10からなる群より選択される少なくとも1種である、〔5〕に記載の組成物。
〔13〕前記疎水性界面活性剤が、モノカプリル酸プロピレングリコールである、〔3〕~〔7〕のいずれか一項に記載の組成物。
〔14〕前記親水性界面活性剤が、ポリエチレングリコール脂肪酸エステル、ポリオキシエチレンヒマシ油、親水性ポリオキシエチレン硬化ヒマシ油、ポリオキシエチレンソルビタン脂肪酸エステル、D-α-トコフェリルポリエチレングリコール1000コハク酸塩、カプリロカプロイルポリオキシル-8 グリセリド、およびこれらのいずれかの組合せからなる群より選択される少なくとも1種である、〔3〕~〔13〕のいずれか一項に記載の組成物。
〔15〕前記親水性界面活性剤が、ポリオキシエチレンヒマシ油、ポリオキシエチレンソルビタン脂肪酸エステル、D-α-トコフェリルポリエチレングリコール1000コハク酸塩、カプリロカプロイルポリオキシル-8 グリセリド、およびこれらのいずれかの組合せからなる群より選択される少なくとも1種である、〔3〕~〔14〕のいずれか一項に記載の組成物。
〔16〕前記親水性界面活性剤が、ポリオキシエチレンヒマシ油を含む、〔3〕~〔15〕のいずれか一項に記載の組成物。
〔17〕前記ポリエチレングリコール脂肪酸エステルが、ポリオキシエチレンヒドロキシステアレート、モノラウリン酸ポリエチレングリコール、モノステアリン酸ポリエチレングリコール、およびステアリン酸ポリオキシル40からなる群より選択される少なくとも1種である、〔14〕に記載の組成物。
〔18〕前記ポリオキシエチレンヒマシ油が、ポリオキシル30ヒマシ油、ポリオキシル35ヒマシ油、およびポリオキシル40ヒマシ油からなる群から選択される少なくとも1種である、〔14〕に記載の組成物。
〔19〕前記ポリオキシエチレンヒマシ油が、ポリオキシル35ヒマシ油である、〔14〕に記載の組成物。
〔20〕前記親水性ポリオキシエチレン硬化ヒマシ油が、ポリオキシエチレン硬化ヒマシ油20、ポリオキシエチレン硬化ヒマシ油40、ポリオキシエチレン硬化ヒマシ油50、およびポリオキシエチレン硬化ヒマシ油60からなる群より選択される少なくとも1種である、〔14〕に記載の組成物。
〔21〕前記ポリオキシエチレンソルビタン脂肪酸エステルが、ポリソルベート20、ポリソルベート40、ポリソルベート60、およびポリソルベート80からなる群より選択される少なくとも1種である、〔14〕に記載の組成物。
〔22〕前記親水性界面活性剤が、ポリオキシル35ヒマシ油、D-α-トコフェリルポリエチレングリコール1000コハク酸塩、カプリロカプロイルポリオキシル-8 グリセリド、ポリソルベート80、およびこれらの組合せからなる群より選択される少なくとも1種である、〔3〕~〔15〕のいずれか一項に記載の組成物。
〔23〕前記親水性界面活性剤が、ポリオキシル35ヒマシ油を含む、〔3〕~〔15〕のいずれか一項に記載の組成物。
〔24〕前記疎水性界面活性剤がプロピレングリコール脂肪酸エステルであり、前記親水性界面活性剤がポリオキシエチレンヒマシ油を含む、〔3〕に記載の組成物。
〔25〕前記疎水性界面活性剤がモノカプリル酸プロピレングリコールであり、前記親水性界面活性剤がポリオキシル35ヒマシ油、D-α-トコフェリルポリエチレングリコール1000コハク酸塩、カプリロカプロイルポリオキシル-8 グリセリド、ポリソルベート80、およびこれらの組合せからなる群より選択される少なくとも1種である、〔3〕に記載の組成物。
〔26〕前記疎水性界面活性剤がモノカプリル酸プロピレングリコールであり、前記親水性界面活性剤がポリオキシル35ヒマシ油を含む、〔3〕に記載の組成物。
〔27〕組成物が油性成分を含む、〔1〕~〔26〕のいずれか一項に記載の組成物。
〔28〕前記油性成分が、脂肪酸、アシルグリセロール、植物油およびこれらの組合せからなる群より選択される少なくとも1種である、〔1〕~〔27〕のいずれか一項に記載の組成物。
〔29〕前記油性成分が、脂肪酸、アシルグリセロール、およびこれらの組合せからなる群より選択される少なくとも1種である、〔1〕~〔28〕のいずれか一項に記載の組成物。
〔30〕前記油性成分が脂肪酸を含む、〔1〕~〔29〕のいずれか一項に記載の組成物。
〔31〕前記脂肪酸が、カプロン酸、カプリル酸、カプリン酸、ラウリン酸、ミリスチン酸、ミリストレイン酸、パルミチン酸、パルミトレイン酸、ステアリン酸、オレイン酸、リノール酸、およびリノレン酸からなる群より選択される少なくとも1種である、〔28〕に記載の組成物。
〔32〕前記脂肪酸が、オレイン酸、リノール酸、およびリノレン酸からなる群より選択される少なくとも1種である、〔28〕に記載の組成物。
〔33〕前記アシルグリセロールが、トリアセチン、トリブチリン、トリカプロイン、トリカプリリン、トリカプリン、トリパルミチン、トリパルミトレイン、トリステアリン酸グリセリン、トリオレイン、トリリノレイン、トリリノレニン、および中鎖脂肪酸トリグリセリドからなる群より選択される少なくとも1種である、〔28〕に記載の組成物。
〔34〕前記アシルグリセロールがトリアセチンである、〔28〕に記載の組成物。
〔35〕前記植物油が、オリーブ油、アーモンド油、ヤシ油、カカオ脂、マカデミアナッツ油、アボカド油、サフラワー油、ダイズ油、アマニ油、ナタネ油、ヒマシ油、コーン油、およびパーム油からなる群より選択される少なくとも1種である、〔28〕に記載の組成物。
〔36〕前記油性成分が、オレイン酸またはトリアセチンである、〔1〕~〔35〕のいずれか一項に記載の組成物。
〔37〕前記油性成分が、オレイン酸である、〔1〕~〔36〕のいずれか一項に記載の組成物。
〔38〕疎水性界面活性剤としてプロピレングリコール脂肪酸エステルを含み、親水性界面活性剤としてポリオキシエチレンヒマシ油を含み、油性成分として脂肪酸を含む、〔3〕に記載の組成物。
〔39〕疎水性界面活性剤としてモノカプリル酸プロピレングリコールを含み、親水性界面活性剤としてポリオキシル35ヒマシ油を含み、油性成分としてオレイン酸を含む、〔3〕に記載の組成物。
〔40〕さらに抗酸化剤を含む、〔1〕~〔39〕のいずれか一項に記載の組成物。
〔41〕前記抗酸化剤が、dl-α-トコフェロール、ブチレート化ヒドロキシトルエン、ブチレート化ヒドロキシアニソール、プロピルガレート、没食子酸プロピル、医薬として許容されうるキノン、アスタキサンチン、およびD-α-トコフェリルポリエチレングリコール1000コハク酸塩からなる群より選択される少なくとも1種である、〔40〕に記載の組成物。
〔42〕さらに可溶化剤を含む、〔1〕~〔41〕のいずれか一項に記載の組成物。
〔43〕前記可溶化剤が、エタノール、プロピレングリコール、ポリエチレングリコール300、ポリエチレングリコール400、ジエチレングリコールモノエチルエーテル、およびこれらの組合せからなる群より選択される少なくとも1種である、〔42〕に記載の組成物。
〔44〕前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が、式(1)で表される化合物またはその水和物である、〔1〕~〔43〕のいずれか一項に記載の組成物。
〔45〕前記水和物が、式(1)で表される化合物の水和物である、〔44〕に記載の組成物。
〔46〕前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が、式(1)で表される化合物である、〔44〕に記載の組成物。
〔47〕前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物の組成物全体に対する含有率が、10重量%以下である、〔1〕~〔46〕のいずれか一項に記載の組成物。
〔48〕前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物の組成物全体に対する含有率が、8重量%以下である、〔1〕~〔47〕のいずれか一項に記載の組成物。
〔49〕前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物の組成物全体に対する含有率が、7重量%以下である、〔1〕~〔48〕のいずれか一項に記載の組成物。
〔50〕前記液体添加剤の組成物全体に対する含有率が、50重量%以上97重量%以下である、〔1〕~〔49〕のいずれか一項に記載の組成物。
〔51〕前記液体添加剤の組成物全体に対する含有率が、55重量%以上96重量%以下である、〔1〕~〔50〕のいずれか一項に記載の組成物。
〔52〕前記液体添加剤の組成物全体に対する含有率が、70重量%以上90重量%以下である、〔1〕~〔51〕のいずれか一項に記載の組成物。
〔53〕前記疎水性界面活性剤の組成物全体に対する含有率が、20重量%以上70重量%以下である、〔3〕~〔52〕のいずれか一項に記載の組成物。
〔54〕前記疎水性界面活性剤の組成物全体に対する含有率が、25重量%以上65重量%以下である、〔3〕~〔53〕のいずれか一項に記載の組成物。
〔55〕前記疎水性界面活性剤の組成物全体に対する含有率が、30重量%以上55重量%以下である、〔3〕~〔54〕のいずれか一項に記載の組成物。
〔56〕前記親水性界面活性剤の組成物全体に対する含有率が、20重量%以上40重量%以下である、〔3〕~〔55〕のいずれか一項に記載の組成物。
〔57〕前記親水性界面活性剤の組成物全体に対する含有率が、25重量%以上35重量%以下である、〔3〕~〔56〕のいずれか一項に記載の組成物。
〔58〕前記親水性界面活性剤の組成物全体に対する含有率が、28重量%以上33重量%以下である、〔3〕~〔57〕のいずれか一項に記載の組成物。
〔59〕前記油性成分の組成物全体に対する含有率が、0重量%以上50重量%以下である、〔1〕~〔58〕のいずれか一項に記載の組成物。
〔60〕前記油性成分の組成物全体に対する含有率が、1重量%以上40重量%以下である、〔1〕~〔59〕のいずれか一項に記載の組成物。
〔61〕前記油性成分の組成物全体に対する含有率が、10重量%以上20重量%以下である、〔1〕~〔60〕のいずれか一項に記載の組成物。
〔62〕前記油性成分が、脂肪酸を含む、〔1〕~〔61〕のいずれか一項に記載の組成物。
〔63〕前記疎水性界面活性剤の前記親水性界面活性剤に対する重量比が、0.5~3.0である、〔3〕~〔62〕のいずれか一項に記載の組成物。
〔64〕前記疎水性界面活性剤の前記親水性界面活性剤に対する重量比が、1.0~2.5である、〔3〕~〔63〕のいずれか一項に記載の組成物。
〔65〕前記疎水性界面活性剤の前記親水性界面活性剤に対する重量比が、1.5~2.0である、〔3〕~〔64〕のいずれか一項に記載の組成物。
〔66〕前記液体添加剤の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、5以上である、〔1〕~〔65〕のいずれか一項に記載の組成物。
〔67〕前記液体添加剤の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、10以上である、〔1〕~〔66〕のいずれか一項に記載の組成物。
〔68〕前記液体添加剤の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、5~2000である、〔1〕~〔66〕のいずれか一項に記載の組成物。
〔69〕前記液体添加剤および前記油性成分の合計の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、10以上である、〔1〕~〔68〕のいずれか一項に記載の組成物。
〔70〕前記液体添加剤および前記油性成分の合計の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、10~2000である、〔1〕~〔69〕のいずれか一項に記載の組成物。
〔71〕前記液体添加剤および前記油性成分の合計の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、10~1000である、〔1〕~〔70〕のいずれか一項に記載の組成物。
〔72〕前記組成物が液体である、〔1〕~〔71〕のいずれか一項に記載の組成物。
〔73〕前記組成物を水に分散させたときに形成される液滴の平均粒子径が、200nm未満である、〔1〕~〔72〕のいずれか一項に記載の組成物。
〔74〕前記液滴の平均粒子径が、10nm以上200nm未満である、〔73〕に記載の組成物。
〔75〕前記液滴の平均粒子径が、50nm以上200nm以下である、〔74〕に記載の組成物。
〔76〕〔1〕~〔75〕のいずれか一項に記載の組成物を含有する、医薬製剤。
〔77〕前記製剤の剤型がカプセル剤である、〔76〕に記載の医薬製剤。
〔78〕下記の工程を含む、〔1〕~〔75〕のいずれか1項に記載の組成物の製造方法;
(1)下記式(1):

(2)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物、液体添加剤、および任意で油性成分を混合する工程;および、
(3)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が溶解した組成物を得る工程。
〔79〕前記工程(1)において提供される式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が、式(1)で表される化合物の水和物である、〔78〕に記載の方法。
〔80〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも1つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-1〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも2つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-2〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも3つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-3〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも4つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-4〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも5つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-5〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも6つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-6〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも7つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-7〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも8つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-8〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも9つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-9〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも10個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-10〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも11個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-11〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のうちの、少なくとも12個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔80-12〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折 において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、および17.813°(±0.2°)のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも1つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-1〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも2つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-2〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも3つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-3〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも4つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-4〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも5つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-5〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のうちの、少なくとも6つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-6〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、7.921°、9.956°、10.435°11.729°、12.704°、15.895°、および16.643°(±0.2°)のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-7〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、14.752°、14.968°、15.895°、16.190°、16.643°、17.813°、および19.424°(±0.2°)のうちの、少なくとも7つのピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-8〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、14.752°、14.968°、15.895°、16.190°、16.643°、17.813°、および19.424°(±0.2°)のうちの、少なくとも10個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-9〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、14.752°、14.968°、15.895°、16.190°、16.643°、17.813°、および19.424°(±0.2°)のうちの、少なくとも13個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-10〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、14.752°、14.968°、15.895°、16.190°、16.643°、17.813°、および19.424°(±0.2°)のうちの、少なくとも15個のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔81-10〕前記式(1)で表される化合物の水和物が結晶であり、該結晶は、粉末X線回折において、回折角2θとして、4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、14.752°、14.968°、15.895°、16.190°、16.643°、17.813°、および19.424°(±0.2°)のピークを含む粉末X線回折パターンを有する、〔79〕に記載の方法。
〔82〕〔1〕~〔75〕のいずれか1項に記載の組成物を提供する工程、および該組成物を製剤化して、医薬製剤を提供する工程、を含む、医薬製剤の製造方法。
〔83〕〔1〕~〔75〕のいずれか1項に記載の組成物を提供する工程、および該組成物をカプセルに充填して、カプセル製剤を提供する工程、を含む、カプセル製剤の製造方法。

本発明で用い得る化合物は、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物である。化合物1の塩は、好ましくはその化学的もしくは薬学的に許容される塩であることができる。また本発明で用い得る化合物1またはその塩は、それらの溶媒和物、好ましくはその化学的もしくは薬学的に許容される溶媒和物であることができる。本発明で用いられる化合物は、組成物中においては、好ましくは専らフリー体の態様であり得るが、組成物の態様によっては、塩または溶媒和物の態様を含む場合がある。
4.964°、7.921°、8.296°、8.855°、9.956°、10.435°、11.729°、12.704°、13.552°、13.901°、15.895°、16.643°、17.813°(±0.2°)
本発明に係る組成物に用い得る「液体添加剤」は、医薬的に許容されるものであり、本発明で用いる化合物を溶解させることのできる添加剤である。本発明においては、「液体添加剤」とは、添加剤が組成物中において溶解した状態にあることを意味し、組成物の製造の原料として提供される段階において溶解していないものは、他の成分との混合や加熱等により製造過程で溶解させて、本発明に係る組成物の一成分とし得る。液体添加剤としては、室温において液体である添加剤を好ましく使用することができる。
本明細書において、「室温」は、本技術分野における通常の意味で用いられ特に限定されないが、特段の記載がない限り、例えば、好ましくは1~30℃、より好ましくは15~28℃程度である。
本発明に係る組成物においては、油性成分を含有させることができる。油性成分としては、好ましくは、脂肪酸、アシルグリセロール、植物油およびこれらの組合せが挙げられる。油性成分としては、さらに好ましくは脂肪酸またはアシルグリセロールを用いることができ、より好ましくは脂肪酸を用いることができる。
本発明に係る医薬製剤は、本発明の化合物1もしくはその塩、またはそれらの溶媒和物と、液体添加剤、および任意で油性成分を含有する組成物を含む、医薬製剤である。
本発明に係る組成物の製造方法は、
(1)下記式(1):

(2)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物、液体添加剤、および任意で油性成分を混合する工程(工程2);および、
(3)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が溶解した組成物を得る工程(工程3)を含む。

Triacetin:トリアセチン
Oleic acid:オレイン酸
Propylene glycol(PG) monocaprylate:モノカプリル酸プロピレングリコール
Glycerol monooleate:モノオレイン酸グリセロール
Sorbitan trioleate:トリオレイン酸ソルビタン
Polyoxyl 35 castor oil:ポリオキシル35ヒマシ油
Polyoxyethylene (15) hydroxystearate:ポリオキシエチレンヒドロキシステアレート
PEG-8 Caprylic/Capric Glycerides:カプリロカプロイルポリオキシル-8 グリセリド
Polysorbate 80:ポリソルベート80
Propylene glycol(PG):プロピレングリコール
EtOH:エタノール
PEG400:ポリエチレングリコール400
D-α-Tocopherol polyethylene glycol 1000 succinate:D-α-トコフェリルポリエチレングリコール1000コハク酸塩
dl-α-tocopherol:dl-α-トコフェロール
t-Bu:tert-ブチル
CSA:(+)-10-カンファースルホン酸
DBU:1,8-ジアザビシクロ[5.4.0]-7-ウンデセン
DCM:ジクロロメタン
DCE:1,2-ジクロロエタン
DMA:ジメチルアセトアミド
DMF:N,N-ジメチルホルムアミド
DIC:N,N’-ジイソプロピルカルボジイミド
DIPEA:N,N-ジイソプロピルエチルアミン
DMAP:N,N-ジメチル-4-アミノピリジン
DMSO:ジメチルスルホキシド
EDTA:エチレンジアミン四酢酸
Fmoc:9-フルオレニルメチルオキシカルボニル
Fmoc-OSu:炭酸N-スクシンイミジル9-フルオレニルメチル
HOAt:1-ヒドロキシ-7-アザベンゾトリアゾール
HOBt:1-ヒドロキシベンゾトリアゾール
HOOBt:3,4-ジヒドロ-3-ヒドロキシ-4-オキソ-1,2,3-ベンゾトリアジン
MTBE:メチルtert-ブチルエーテル NMP:N-メチル-2-ピロリドン
oxyma:シアノ(ヒドロキシイミノ)酢酸エチル
TES:トリエチルシラン
TFA:トリフルオロ酢酸
TFE:2,2,2-トリフルオロエタノール
THF:テトラヒドロフラン
TfOH:トリフルオロメタンスルホン酸
TsOH:p-トルエンスルホン酸
WSCI・HCl:1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩
ZETASIZER Nano-ZS (Malvern)を用いて動的散乱(DLS)による粒子径分布を求める。サンプルをディスポーザブルセル(少量量キュベット)に入れ、ZETASIZER Nano-ZS(Malvern)にセットして動的散乱(DLS)による粒子径分布求める。粒子径分布を表す指標として、平均粒子径(Z-averageサイズ)と多分散性指数(polydispersity index, PDI)を用いる。Z-averageはキュムラント法を用いた散乱強度基準の平均粒子径である。また、PDIは粒子径分布の幅を表す指標であり、0から1の範囲で示される。PDI=0は粒子径の分布が無い懸濁液を表し、0.1以下のPDIを示す分散体は単分散であり、PDIが0.1から0.5の間の値を示す分散体は狭い分布を有すると考えられる。一方、PDIが0.5を超える分散体は多分散であると考えられる。
製剤の空腹時人工腸液(FaSSIF)への分散性能を推定するためにμDISS Profiler(Pion Inc.)を用いて分散プロファイルを解析する。各製剤100μLを37°Cに保温された10mLのFaSSIF表面に加え、200rpmの速度で攪拌した。5、10、15、20、25、30および60分の時点でガイドプラスチックチューブを通して溶液50μLを容器の中央から分取し、UPLCによって対象化合物の濃度を決定する。次の式により、分散度(%)を求める。
分散度(%)=(分取した溶液中の化合物の濃度/製剤全量が均一に分散したときの化合物の濃度)X100
採取した血液は遠心分離により血漿を分離し、アセトニトリルによる除タンパク処理後、LC-MS/MS装置を用いて血漿中濃度を測定する。得られた血漿中濃度推移より、薬物動態解析ソフトPhoenix WinNonlin 8.2(Certara L. P.)を用いてノンコンパートメント解析により血漿中薬物濃度対時間曲線下面積(AUC)および最高血漿中濃度(Cmax)を算出する。
化合物1の製造
下記の構造を有する化合物1(「(5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-ブチル)-18-シクロペンチル-29-(3,5-ジフルオロ-4-(トリフルオロメチル)フェネチル)-36-エチル-11-イソブチル-N,N,5,6,12,16,19,33-オクタメチル-35-(4-メチルベンジル)-4,7,10,13,17,20,23,28,31,34,37-ウンデカオキソテトラトリアコンタヒドロ-2H,4H-スピロ[アゼト[2,1-u]ピロロ[2,1-i][1,4,7,10,13,16,19,22,25,28,31]ウンデカアザシクロテトラトリアコンチン-21,1'-シクロペンタン]-15-カルボキサミド」)は下記のスキーム1に従って合成した。



LCMS(ESI)m/z=408(M+H)+
保持時間:1.407分(分析条件SMDmethod_20)
LCMS(ESI)m/z=410(M+H)+
保持時間:1.956分(分析条件SMDmethod_05)
窒素雰囲気下、氷冷下にてWSCI・HCl(27.4g,143mmol)のDMF(217mL)溶液にHOBt(17.72g,131mmol)を加え、更に化合物aa033b(48.8g,119mmol)をDCM(90mL)とDMF(90mL)の混合溶液として加え、0℃で30分攪拌した。そこにジメチルアミンのTHF溶液(2mol/l,65.6mL,131mmol)を滴下にて加え、0℃で30分攪拌した。反応液を酢酸エチル(488mL)で希釈し、有機相を塩酸(1mol/L,390mL)で2回洗浄し、続いて水で洗浄し、更に飽和炭酸水素ナトリウム水溶液と水の混合溶液(1:1,488mL))で2回洗浄し、さらに、飽和食塩水と水の混合溶液(1:1、488mL)で1回洗浄後、得られた有機相を無水硫酸ナトリウムで乾燥し、減圧下溶媒留去して化合物aa011-aを得た。(51.16g,収率98%)。
LCMS(ESI)m/z=437.0(M+H)+
保持時間:1.262分(分析条件SMDFA05)

LCMS(ESI)m/z=378(M+H)+
保持時間:1.01分(分析条件SQDFA05)
LCMS(ESI)m/z=380(M+H)+
保持時間:0.92分(分析条件SQDFA05)
化合物aa079(42.2g,111mmol)とOxyma(19.99g,141mmol)のDMF(391mL)溶液に、WSCI・HCl(31.5g,164mmol)を室温にて加え、30分攪拌して溶液Aを得た。
LCMS(ESI)m/z=598.2(M+Na)+
保持時間:1.320分(分析条件SMDAM05)
窒素雰囲気下、室温にて化合物1217-a(55.55g,96mmol)のDCM(193mL)溶液に、テトラキス(トリフェニルホスフィン)パラジウム(0)(1.115g,0.965mmol)を加え、更にフェニルシラン(8.31mL、67.5mmol)を滴下にて加え、30分攪拌した。反応液をMTBE(556ml)で希釈し、飽和炭酸水素ナトリウム水溶液と水の混合溶液(1:1,556ml)で抽出した。得られた有機相を水(278ml)で抽出した。水相を混合し、DCM(556ml)を加えた。そこにリン酸(56.7g,579mmol)を滴下にて加えてpHを2~3に調整し、有機相を分離後、水相をDCM(556ml)にて抽出した。得られた有機相を混合し、飽和食塩水と水の混合溶液(1:1、556ml)で洗浄後、硫酸ナトリウムで乾燥し、減圧下溶媒留去して化合物1217-bを得た。(48.87g,収率95%)
LCMS(ESI)m/z=536(M+H)+
保持時間:1.138分(分析条件SMDAM05)
フィルター付きの反応容器に2-クロロトリチルクロライドレジン(SUNRESIN社から購入、1.36mmol/g,114g、155mmol)をセットし、DCM(1140mL)を加え、25℃で45分攪拌後、フィルターから溶媒を排出した。反応容器に化合物1217-b(48.87g,91mmol)とメタノール(29.6mL,730mmol)とDIPEA(76mL,438mmol)のDCM(798mL)溶液を加え、25℃で60分攪拌し、フィルターから溶液を排出した。続けて、反応容器にメタノール(111mL,2737mmol)とDIPEA(76mL,438mmol)のDCM(684mL)溶液を加え、25℃で90分攪拌し、フィルターから溶液を排出した。反応容器にDCM(570mL)を加えて5分攪拌し、フィルターから溶液を排出した。このレジンの洗浄操作を更に4回繰り返し、得られたレジンを減圧下乾燥して化合物1217-b-resinを得た(140.5g)。本実施例に記載のレジンの定量法により、担持量を0.482mmol/gと算出した。
上記で得られたレジン(0.482mmol/g、60g、28.92mmol)をプラスチック製固相反応容器にセットした。室温にて、この固相反応容器にDCM(600mL)を加え、5分振盪した後、溶媒をフリットから排出した。この固相反応容器にDMF(420mL)を加え、5分振盪した後、溶媒をフリットから排出した。このレジンの洗浄工程を更に1回繰り返した。この固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、420mL)を添加しFmoc基の脱保護を行った。10分振盪後に溶液をフリットから排出した。この固相反応容器にDMF(420mL)を加え、5分振盪した後、溶液をフリットから排出した。この固相反応容器にトリエチルアミン塩酸塩(7.96g、57.8mmol)のDCM(420mL)溶液を加え、5分振盪した後、溶液をフリットから排出した。この固相反応容器にDCM(420mL)を加え、5分振盪した後、溶液をフリットから排出した。この固相反応容器にDMF(420mL)を加え、5分振盪した後、溶液をフリットから排出した。このDMFによるレジンの洗浄工程を更に1回繰り返した。


LCMS(ESI)m/z=505.2(M+Na)+
保持時間:0.992分(分析条件SMDmethod_16)
LCMS(ESI)m/z=496(M+Na)+
保持時間:1.544分(分析条件SMDmethod_15)
保持時間:3.538分(分析条件SMDmethod_14)
1H-NMR(300MHz,DMSO-d6)δ12.69(s,1H),7.90(d,J=7.5Hz,2H),7.78-7.54(m,3H),7.48-7.20(m,6H),4.33(d,J=6.3Hz,2H),4.24(t,J=6.9Hz,1H),3.97-3.84(m,1H),2.79-2.65(m,2H),2.15-2.00(m,1H),2.00-1.83(m,1H)

LCMS(ESI)m/z=428(M+H)+
保持時間:1.03分(分析条件SQDFA05)
LCMS(ESI)m/z=430(M+H)+
保持時間:0.95分(分析条件SQDFA05)
30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、30℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-Pro-OH(CAS番号71989-31-6)(0.6mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて4時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-Hph(4-CF3-35-F2)-OH(化合物aa134)(0.45mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて2.5時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-MeGly-OH(0.6mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて2.5時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、前述のとおり製造したFmoc-EtPhe(4-Me)-OH(0.6mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて2.5時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-Aze(2)-OH(0.6mol/L)とHOOBt(0.375mol/L)のNMPとDMSOの混合溶液(7:3)(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、60℃にて5時間静置した。その後、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を30本すべての固相反応容器に対して添加し、60℃にて5時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-MeAla-OH(0.6mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて2.5時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。
上記で得られたレジンを含む30本すべての固相反応容器にジアザビシクロウンデセン(DBU)のDMF溶液(2v/v%、固相反応容器1本に対して1.4mL)を添加し、35℃に加温して10分後に溶液をフリットから排出した。この30本すべての固相反応容器にDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このDMFによるレジンの洗浄工程を更に3回繰り返した。続いて、Fmoc-Ile-OH(0.6mol/L)とHOAt(0.375mol/L)のNMP溶液(固相反応容器1本に対して0.6mL)と、N,N’-ジイソプロピルカルボジイミド(DIC)のDMF溶液(10v/v%、固相反応容器1本に対して0.72mL)を合成機のmixing vialで混合した後に30本すべての固相反応容器に対して添加し、40℃にて10時間静置した。その後、溶液をフリットから排出した。30本すべての固相反応容器に対してDMF(固相反応容器1本に対して1.4mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に2回繰り返した。続いて、30本すべての固相反応容器に対してDCM(固相反応容器1本に対して1.6mL)を加え、溶媒をフリットから排出した。このレジンの洗浄工程を更に5回繰り返した。30本すべての固相反応容器からレジンを回収し、混合して続く操作を行った。
上記で得られたレジンを200mLのプラスチック製固相反応容器に加え、ここにDCM(60mL)を加え、30℃で5分振盪した後、溶媒をフリットから排出した。この固相反応容器にトルエン(50mL)を加え、30℃で5分振盪した後、溶媒をフリットから排出した。このトルエンによるレジンの洗浄工程を更に1回繰り返した。この固相反応容器にジアザビシクロウンデセン(DBU)のトルエン溶液(2v/v%、45mL)を添加し、30℃で5分振盪後に溶液をフリットから排出した。
上記により得られたレジンを含む固相反応容器に、2,2,2-トリフルオロエタノール(TFE)(60mL)とDCM(60mL)とDIPEA(0.909mL)の混合溶液を加え、室温にて2時間振盪した。その後、溶液をフリットから回収した。この固相反応容器に2,2,2-トリフルオロエタノール(TFE)(30mL)とDCM(30mL)の混合溶液を加え、室温にて5分間振盪後、溶液をフリットから回収した。更にこの固相反応容器に、2,2,2-トリフルオロエタノール(TFE)(30mL)とDCM(30mL)の混合溶液を加え、室温にて5分間振盪後、溶液をフリットから回収した。回収したすべての溶液を混合し、減圧下にて溶媒留去して化合物1217-dを粗生成物として得た。(3.85g)
LCMS(ESI)m/z=1453.9(M-H)-
保持時間:0.67分(分析条件SQDAA50)
上記により得られた化合物1217-d(3.85g)を酢酸イソプロピル(529mL)とDIPEA(0.915mL、5.24mmol)の混合液に溶解し、HCTU(O-(1H-6-クロロベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウム ヘキサフルオロリン酸、CAS番号330645-87-9)(1.805g、4.36mmol)を加え、室温にて21時間攪拌した。その後、溶液量が約半分になるまで減圧下溶媒留去した。得られた溶液に、飽和塩化アンモニウム水溶液(40mL)と水(40mL)の混合溶液を加え、酢酸イソプロピル(350mL)で抽出した。得られた有機相を飽和炭酸水素ナトリウム水溶液(40mL)と水(40mL)の混合溶液、飽和食塩水(40mL)と水(40mL)の混合溶液で順に洗浄後、硫酸ナトリウムで乾燥し、減圧下溶媒留去して3.36gの残渣を得た。得られた残渣を逆相シリカゲルカラムクロマトグラフィー(Daisogel SP-120-40/60-ODS-RPS、溶出液としてアセトニトリル(0.1%のギ酸を含む)/水(0.1%のギ酸を含む)を使用)で精製し、目的物を含む溶出液を凍結乾燥することでアモルファス状態の化合物1(1.36g、収率34%)を得た。得られた化合物1のマススペクトルの値と液体クロマトグラフィーの保持時間は下記のとおりであった。
LCMS(ESI)m/z=1437.7(M+H)+
保持時間:7.496分(分析条件SSC-A-AF-01)
調製例3で使用した種結晶の調製
調製例1において得られたアモルファス状態の化合物1(122.3mg)をDMSO(0.612mL)に溶解させ、この溶解液(0.015mL)を-20℃で2日間凍結乾燥した。得られた凍結乾燥物に水-アセトニトリル混合液(3:1,0.015mL)を加え、室温にて7日間振とう攪拌することで化合物1の水和物結晶(Form C)を得た。
化合物1の結晶化:((5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-ブチル)-18-シクロペンチル-29-(3,5-ジフルオロ-4-(トリフルオロメチル)フェネチル)-36-エチル-11-イソブチル-N,N,5,6,12,16,19,33-オクタメチル-35-(4-メチルベンジル)-4,7,10,13,17,20,23,28,31,34,37-ウンデカオキソテトラトリアコンタヒドロ-2H,4H-スピロ[アゼト[2,1-u]ピロロ[2,1-i][1,4,7,10,13,16,19,22,25,28,31]ウンデカアザシクロテトラトリアコンチン-21,1'-シクロペンタン]-15-カルボキサミド)の水和物結晶 Form Cの合成
窒素で置換した化合物1を含む溶液が入った反応釜に入れ、反応釜の外温を40℃に設定し、フィルター(CCF-G100-D1N)を用いて濾過した精製水(10.9kg)を加えた。アセトン(59.2g)/水(61.2g)の混合液に、調製例2と同様の操作により得られた、化合物1の粉砕結晶(10.2g)を加えて得た懸濁液を反応釜に加えた。懸濁液の入った容器をアセトン(59.2g)/水(61.2g)の混合液で洗いこみながら反応釜に加えた後、2時間1分間撹拌した。フィルター(CCF-G100-D1N)を用いて濾過した精製水(2.7kg)を加え、7時間10分間撹拌した。さらに、アセトン(59.2g)/水(61.2g)の混合液に、調製例2と同様の操作により、得られた化合物1の粉砕結晶(10.2g)を加えて得た懸濁液を反応釜に加えた。懸濁液の入った容器をアセトン(59.2g)/水(61.2g)の混合液で洗いこみながら、反応釜に加え、12時間40分間撹拌した。フィルター(CCF-G100-D1N)を用いて濾過した精製水(2.7kg)を加え、2時間撹拌した。反応釜の外温を40℃から25℃へ1時間かけて降温後、反応混合物を18時間44分間攪拌した。反応混合物を、濾布(PF-020)を用いて加圧濾過し、反応釜内と濾過機を、フィルター(CCF-G100-E1N)を用いて濾過したアセトン(7.5kg)と精製水(7.5kg)の混合液で洗浄しながら得られた結晶を洗浄した。得られた結晶を、フィルター(CCF-G100-E1N)を用いて濾過した精製水(17.0kgx2)で洗浄し、結晶を回収した濾過装置を減圧し、濾過装置の外温を70℃に設定して結晶を17時間乾燥した。さらに、外温を室温~30℃にて結晶を27時間乾燥した。乾燥末を濾過機から回収し、白色の粉末(2.6kg)を得た。
HPLC分析条件
装置:Waters ACQUITY UPLC H-Class
カラム:ACQUITY UPLC CSH C18 (Waters), 2.1 mm ID×150 mm, 1.7 μm
移動相:0.05% TFA/water (A)、0.05% TFA/MeCN (B)
溶出法:B) 20%(0 min)→100%(24 min)→100%(29 min)→20%(29.1 min)→20%(34 min)
流速:0.3 mL/min
カラム温度:50 ℃
検出波長:220 nm(PDA)
HPLC分析による保持時間:18.199分
XRPD測定条件
測定装置:X’pert-pro MPD(PANalytical社製)
線源:CuKα
管電圧:45 kV
管電流:40 mA
走査範囲:3~40°
走査速度:4.2°/分
サンプリング幅:0.017°
測定装置:TGA/DSC 3+(Mettler Toledo製)
測定範囲:25~350℃
昇温速度:10℃/分
雰囲気:乾燥窒素
測定装置:Rigaku R-AXIS RAPID-II with a VariMax Cu diffractometer(リガク社製)
対陰極:Cu
管電圧:40 kV
管電流:30 mA
温度:-180℃
測定:構造解析に十分な回折斑点が得られると考えらえるストラテジー、露光時間で測定を行った。
構造解析:初期構造決定は直接法(SIR2004、CrystalStructure、Rigaku)で行い、構造精密化はfull-matrix least-squares法(SHELXL-2017/1、APEX3、Bruker)で行った。全ての非水素原子は異方性温度因子で精密化した。水分子の水素原子はリストレインを用いて適切な位置に置き、結合している酸素原子の1.5倍の大きさの等方性温度因子で精密化した。そのほかの水素原子はライディングモデルを用いて適切な位置に置き、結合している非水素原子の1.2倍の大きさの等方性温度因子とした。
結果を図3に示す。
測定装置:DVS Intrinsic(Surface Measurement Systems製)
温度:25℃
相対湿度(%)測定点:
サイクル1:0、10、20、30、40、50、60、70、80、90、95、90、80、70、60、50、40、30、20、10、0(%);
サイクル2:10、20、30、40、50、60、70、80、90、95、90、80、70、60、50、40、30、20、10、0(%)
閾値:0.001 dm/dt(%/分)
最小収着時間:10分
最大収着時間:1440分
試験例1(各種添加剤への化合物Iの溶解度評価)
各種添加剤への化合物Iの溶解度を求めた。化合物Iに液体状の各種添加剤を添加して攪拌し、化合物Iが溶けない場合はさらに添加剤を添加して攪拌した。この操作を化合物Iが溶けるまで繰り返し、化合物Iを溶かすために必要な添加剤量からおおよその溶解度を求めた。なお、Polyoxyethylene (15) hydroxystearate (Solutol HS15)は室温(R.T.)で固体であるため37℃に加熱して液体にして溶解度を求めた。その他の添加剤は室温で溶解度を求めた。結果を表2に示す。その結果、PG monocaprylateは化合物Iに対して高い溶解性を示すことが判明した。

高い溶解性を示したPG monocaprylateを用いた添加剤混合液を表3の処方に基づいて調製した。次に、各添加剤混合液1~4への化合物Iの溶解度を測定した。カプセル剤として製剤化した際のカプセル剤皮からの水分移行を想定し、水を3%添加した添加剤混合液についても溶解度を測定した。その結果、表4に示すとおり、添加剤混合液1~4は化合物Iを100mg/mL以上の高い濃度で溶かすことができることが判明した。また、水を各添加剤混合液に添加すると、化合物Iの溶解度が60~70mg/mLに低下したことから、以降の化合物Iの各添加剤混合液への溶解度評価には水を添加して評価することとした。









表12の処方に基づき、以下の方法により本発明組成物である製剤A~Vを調製した。

化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液1を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Aを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液2を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Bを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液3を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Cを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液4を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Dを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液7を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Eを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液19を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Fを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液20を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Gを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液21を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Hを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液25を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Iを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液26を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Jを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液28を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Kを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液29を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Lを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液30を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Mを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液32を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Nを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液33を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Oを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液34を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Pを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液35を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Qを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液40を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Rを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液41を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Sを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液42を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Tを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液44を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Uを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液45を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Vを得た。

表14の処方に基づき、以下の方法により本発明組成物である製剤W~AOを調製した。

化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液8を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Wを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液9を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Xを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液10を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Yを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液13を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤Zを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液14を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AAを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液15を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤ABを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液16を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤ACを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液17を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤ADを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液18を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AEを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液22を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AFを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液23を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AGを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液24を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AHを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液27を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AIを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液31を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AJを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液36を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AKを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液37を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤ALを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液38を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AMを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液39を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤ANを得た。
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液43を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認して製剤AOを得た。

次に、実施例1から22で調製した製剤Aから製剤Vおよび比較例1から19で調製した製剤Wから製剤AOに水を加え、分散体を生成させた。分散体中に生じた液滴の粒子径を測定し、室温(約25℃)における粒子径分布を評価した。バイアルに各製剤4μLを入れ、さらに水を396μL加えてたのち、攪拌したサンプルについて試験法1に示した方法で動的散乱(DLS)による粒子径分布求めた。粒子径分布を表す指標として、平均粒子径(Z-averageサイズ)と多分散性指数(polydispersity index, PDI)を用いた。DLSにおいて、PDIは粒子径分布の幅を表す指標でであり、0から1の範囲で示される。PDI=0は粒子径の分布が無い懸濁液を表し、0.1以下のPDIを示す分散体は単分散であり、PDIが0.1から0.5の間の値を示す分散体は狭い分布を有すると考えられる。一方、PDIが0.5を超える分散体は多分散であると考えられる。表16に示したとおり、製剤AからVはいずれも平均粒子径も小さく(200nm未満)かつPDIが0.5未満の狭い分布を示す良好な分散体であった。一方、製剤Wから製剤AOの平均粒子径は大きく(200nm以上)、PDIが0.5を超えて多分散であった。

医薬品の長期安定性を見積もるため安定性試験を行った。まず実施例1、3、6、8、10で調製した製剤A、C、F、H、Jそれぞれ10mgをガラスバイアルにとり、シリカゲルおよび脱酸素剤を入れた褐色瓶に入れて密閉した。各褐色瓶を80℃の恒温槽にいれ、2週間保存した。保存前後の化合物Iの濃度をUPLCで定量し、次の式により残存率を求めた。
残存率(%)=(保存後の化合物Iの濃度/保存前の化合物I濃度)X100

自己乳化製剤は液体であるため、医薬品として開発するためには主にカプセル剤に充填される。そこで実施例1、3、6で調製した製剤A、C、Fそれぞれ10mgを硬カプセルにとり、シリカゲルおよび脱酸素剤を入れた褐色瓶に入れて密閉した。各褐色瓶を40℃の恒温槽にいれ、6か月間保存した。保存前後の化合物Iの濃度をUPLCで定量し、次の式により残存率を求めた。
残存率(%)=(保存後の化合物Iの濃度/保存前の化合物I濃度)X100

試験法2で示した分散性評価方法にしたがって、製剤C、Fおよび比較として製剤AJの空腹時人工腸液(FaSSIF)への分散性能を推定するために、制御された温度と攪拌速度(37℃、200 rpm)を提供するμDISS装置を用いて分散プロファイルを解析した。各製剤100μLを37℃に保温された10mLのFaSSIF表面に加え、200 rpmの速度で攪拌した。5、10、15、20、25、30および60分の時点でガイドプラスチックチューブを通して溶液50μLを容器の中央から分取し、UPLCによって化合物Iの濃度を決定した。次の式により、分散度(%)を求めた。
分散度(%)=(分取した溶液中の化合物Iの濃度/製剤全量が均一に分散したときの化合物Iの濃度)X100
実施例23(ラット投与用カプセル製剤APの作製)
化合物Iをバイアルに秤取し、化合物I 60mgあたり1mLの添加剤混合液3を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認した。化合物が溶解した溶液を9号ゼラチンカプセルに充填して製剤APを作製した。
化合物Iをバイアルに秤取し、化合物I 60mgあたり1mLの添加剤混合液19を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認した。化合物が溶解した溶液を9号ゼラチンカプセルに充填して製剤AQを作製した。
化合物Iをバイアルに秤取し、化合物I 60mgあたり1mLの添加剤混合液31を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認した。化合物が溶解した溶液を9号ゼラチンカプセルに充填して製剤ARを作製した。
化合物Iをバイアルに秤取し、化合物I 10mgあたり1mLのジメチルスルホキシド(DMSO)を加えて溶解させた。さらにポリオキシル35ヒマシ油を0.5mL、水を8.5mL加えてよく攪拌、混合して溶液製剤ASを作製した。

実施例23、24のカプセル製剤AP、AQおよび比較例20、21のカプセル製剤ARおよび溶液製剤ASをラット(Wister Hannover, male)に10 mg/kgの容量で各製剤4例ずつ経口投与した。製剤AP、AQおよびARはカプセル剤をそのまま強制経口投与し、さらに水を5mL/kgの容量で強制経口投与した。溶液製剤ASはそのまま強制経口投与した。投与後、約0.5、1、2、4、7、24および48時間後に採血を行い、試験法3に示した方法で血漿中に含まれる化合物Iの濃度をLC-MS/MSによって定量し、血漿中薬物濃度対時間曲線下面積(AUC)および最高血漿中濃度(Cmax)を算出した。

実施例25(サル投与用カプセル製剤ATの作製)
化合物Iをバイアルに秤取し、化合物I 50mgあたり1mLの添加剤混合液3を加えた。撹拌子を用いて攪拌して化合物Iが完全に溶解することを確認した。化合物が溶解した溶液を1号ゼラチンカプセルに充填して製剤ATを作製した。
化合物Iをバイアルに秤取し、化合物I 15mgあたり1mLのジメチルスルホキシド(DMSO)を加えて溶解させた。さらにポリオキシル35ヒマシ油を0.25mL、水を8.75mL加えてよく攪拌、混合して溶液製剤AUを作製した。
実施例25のカプセル製剤ATおよび比較例22の溶液製剤AUをカニクイザル(cynomolgus monkey)に3mg/kgの容量で各製剤4例ずつ経口投与した。カプセル製剤ATはそのまま強制経口投与し、さらに水を4mL/kgの容量で強制経口投与した。溶液製剤AUはそのまま強制経口投与した。投与後、約0.5、1、2、4、7、24および48時間後に採血を行い、試験法3に示した方法で血漿中に含まれる化合物Iの濃度をLC-MS/MSによって定量し、血漿中薬物濃度対時間曲線下面積(AUC)および最高血漿中濃度(Cmax)を算出した。

化合物Iをステンレス容器に秤取し、化合物I 0.5mgあたり51.5mgのPropylene glycol monocaprylate、13.3 mgのOleic acid、31.1 mgのPolyoxyl 35 castor oil および0.2 mgのdl-α-tocopherol を加え、ホモジナイザーを用いて攪拌、混合した。化合物が溶解した溶液を3号ゼラチンカプセルに充填し、バンドシールして製剤AVを作製した。
化合物Iをステンレス容器に秤取し、化合物I 12mgあたり123.6mgのPropylene glycol monocaprylate、31.9mgのOleic acid、74.6mgのPolyoxyl 35 castor oil および0.5mgのdl-α-tocopherol を加え、ホモジナイザーを用いて攪拌、混合した。化合物が溶解した溶液を3号ゼラチンカプセルに充填し、バンドシールして製剤AWを作製した。
化合物Iをステンレス容器に秤取し、化合物I 19.1mgあたり206.4mgのPropylene glycol monocaprylate、53.3mgのOleic acidおよび124.6mgのPolyoxyl 35 castor oilおよび0.8mgのdl-α-tocopherolを加え、攪拌羽を用いて攪拌、混合した。化合物が溶解した溶液をソフトゼラチンカプセルに充填し、製剤AXを作製した。
化合物Iをステンレス容器に秤取し、化合物I 4.5mgあたり46.4mgのPropylene glycol monocaprylate、12.0mgのOleic acidおよび28.0mgのPolyoxyl 35 castor oilを加え、攪拌羽を用いて攪拌、混合した。化合物が溶解した溶液をソフトゼラチンカプセルに充填し、製剤AYを作製した。
化合物Iをステンレス容器に秤取し、化合物I 6.0mgあたり61.8mgのPropylene glycol monocaprylate、16.0mgのOleic acidおよび37.3mgのPolyoxyl 35 castor oilを加え、攪拌羽を用いて攪拌、混合した。化合物が溶解した溶液をソフトゼラチンカプセルに充填し、製剤AZを作製した。

次に、実施例26~30で調製した製剤AV、製剤AW、製剤AX、製剤AY、および製剤AZを水に分散したときの粒子径分布を評価した。バイアルに日本薬局方溶出試験第1液, pH1.2を9.9mL入れ、さらに各製剤100μLを加えたのち攪拌したサンプルを100μL分取して、日本薬局方溶出試験第1液, pH1.2を0.9mL入れたバイアルに加えたのち攪拌した。これらのサンプルについて、試験法1に示した方法で動的散乱(DLS)による粒子径分布を求めた。粒子径分布を表す指標として、試験例3と同様に、平均粒子径(Z-averageサイズ)と多分散性指数(polydispersity index, PDI)を用いた。表23に示したとおり、製剤AV、製剤AW、製剤AX、製剤AY、および製剤AZはいずれも平均粒子径も小さく(200 nm未満)かつPDIが0.5未満の狭い分布を示す良好な分散体であった。

AlphaScreenを用いたKrasとSOS1のタンパクタンパク相互作用阻害活性(KrasG12D-SOS PPI)
KrasとSOS1のタンパク-タンパク相互作用阻害活性(protein-protein interaction inhibition; PPI)は、商業的供給業者より入手可能な装置、試薬などを用い、当業者であれば公知の方法により実施可能である。具体的には、大腸菌で発現し、精製後にGDPをローディングしたビオチンタグ付きヒトKrasとHisタグ付きヒトSOS1エンザイムを用い、ニッケルを結合させたドナービーズからストレプトアビジンを結合させたアクセプタービーズへのエネルギー伝達により測定した。ドナービーズに680nmの光を照射することで、一重項酸素を介したアクセプタービーズへのエネルギー伝達が起こり、アクセプタービーズから520~620nmの光が検出されることを本測定は利用している。化合物1を含まない対照群に対する阻害率より50%阻害濃度(IC50値)を算出した。
AsPC-1細胞生育阻害活性は、商業的供給業者より入手可能な装置、試薬、細胞株などを用い、当業者であれば公知の方法により実施可能である。具体的には、化合物1をジメチルスルホキシドにて系列希釈後、Labcyte Echoを用いて200nLをU底96穴プレートに分注した。ヒトすい臓がん株AsPC-1は、RPMI-1640培地に15%牛胎児血清を添加した培地で、1000細胞/100μLとなるよう細胞懸濁液を調製した。この細胞懸濁液を、化合物1を添加済みプレートに1ウェルあたり100μL分注し、37℃、5%炭酸ガスインキュベーターにて培養した。96時間後、80μLのCellTiter-Glo(プロメガ)を各ウェルに添加し、蛍光を測定した。化合物1無添加の対照に対する化合物1添加時の増殖阻害率より、被験化合物の細胞生育阻害活性(cell growth inhibition, CGI)を50%増殖阻害濃度(IC50値)として算出した。化合物1のIC50値(μM)は、0.02μM以下であり、抗腫瘍効果を持つことが示された。
Claims (15)
- 下記式(1):
で表される化合物、もしくはその塩、またはそれらの溶媒和物と、液体添加剤、および任意で油性成分を含有する、組成物。
- 前記液体添加剤が、界面活性剤である、請求項1に記載の組成物。
- 前記界面活性剤が、疎水性界面活性剤、および親水性界面活性剤の組み合わせである、請求項2に記載の組成物。
- 前記疎水性界面活性剤のHLB値が0以上10未満であり、前記親水性界面活性剤のHLB値が10以上30以下である、請求項3に記載の組成物。
- 前記疎水性界面活性剤が、プロピレングリコール脂肪酸エステル、グリセリン脂肪酸エステル、ポリグリセリン脂肪酸エステル、ソルビタン脂肪酸エステル、および疎水性ポリオキシエチレン硬化ヒマシ油からなる群より選択される少なくとも1種である、請求項3または4に記載の組成物。
- 前記親水性界面活性剤が、ポリエチレングリコール脂肪酸エステル、ポリオキシエチレンヒマシ油、親水性ポリオキシエチレン硬化ヒマシ油、ポリオキシエチレンソルビタン脂肪酸エステル、D-α-トコフェリルポリエチレングリコール1000コハク酸塩、カプリロカプロイルポリオキシル-8 グリセリド、およびこれらのいずれかの組合せからなる群より選択される少なくとも1種である、請求項3~5のいずれか一項に記載の組成物。
- 組成物が油性成分を含む、請求項1~6のいずれか一項に記載の組成物。
- さらに抗酸化剤を含む、請求項1~7のいずれか一項に記載の組成物。
- 前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が、式(1)で表される化合物またはその水和物である、請求項1~8のいずれか一項に記載の組成物。
- 前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物の組成物全体に対する含有率が、10重量%以下である、請求項1~9のいずれか一項に記載の組成物。
- 前記液体添加剤の組成物全体に対する含有率が、50重量%以上97重量%以下である、請求項1~10のいずれか一項に記載の組成物。
- 前記液体添加剤の、前記式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物に対する重量比が、5以上である、請求項1~11のいずれか一項に記載の組成物。
- 請求項1~12のいずれか一項に記載の組成物を含有する、医薬製剤。
- 下記の工程を含む、請求項1~12のいずれか1項に記載の組成物の製造方法;
(1)下記式(1):
で表される化合物、もしくはその塩、またはそれらの溶媒和物、液体添加剤、および任意で油性成分を提供する工程;
(2)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物、液体添加剤、および任意で油性成分を混合する工程;および、
(3)該式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が溶解した組成物を得る工程。 - 前記工程(1)において提供される式(1)で表される化合物、もしくはその塩、またはそれらの溶媒和物が、式(1)で表される化合物の水和物である、請求項14に記載の方法。
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112023019622A BR112023019622A2 (pt) | 2021-05-07 | 2022-05-06 | Preparação contendo composto de peptídeo cíclico e método para a produção do mesmo |
| EP22798954.8A EP4299069A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| IL306018A IL306018A (en) | 2021-05-07 | 2022-05-06 | A preparation containing a cyclic peptide compound and a method for its production |
| CN202280029838.6A CN117241817A (zh) | 2021-05-07 | 2022-05-06 | 包含环状肽化合物的制剂及其制备方法 |
| KR1020237032063A KR20230169093A (ko) | 2021-05-07 | 2022-05-06 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| KR1020237001877A KR102582225B1 (ko) | 2021-05-07 | 2022-05-06 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| JP2022548967A JP7181442B1 (ja) | 2021-05-07 | 2022-05-06 | 環状ペプチド化合物を含む製剤及びその製造方法 |
| CA3219090A CA3219090A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| AU2022270498A AU2022270498A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| JP2022184220A JP2023015334A (ja) | 2021-05-07 | 2022-11-17 | 環状ペプチド化合物を含む製剤及びその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-079015 | 2021-05-07 | ||
| JP2021079015 | 2021-05-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022234850A1 true WO2022234850A1 (ja) | 2022-11-10 |
Family
ID=83932759
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/019540 WO2022234850A1 (ja) | 2021-05-07 | 2022-05-06 | 環状ペプチド化合物を含む製剤及びその製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP4299069A1 (ja) |
| JP (2) | JP7181442B1 (ja) |
| KR (2) | KR102582225B1 (ja) |
| CN (1) | CN117241817A (ja) |
| AU (1) | AU2022270498A1 (ja) |
| BR (1) | BR112023019622A2 (ja) |
| CA (1) | CA3219090A1 (ja) |
| IL (1) | IL306018A (ja) |
| TW (1) | TW202308679A (ja) |
| WO (1) | WO2022234850A1 (ja) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122059A1 (en) | 2011-03-04 | 2012-09-13 | New York University | Hydrogen bond surrogate macrocycles as modulators of ras |
| WO2013100132A1 (ja) | 2011-12-28 | 2013-07-04 | 中外製薬株式会社 | ペプチド化合物の環化方法 |
| WO2016071515A1 (en) | 2014-11-07 | 2016-05-12 | Sigmoid Pharma Limited | Compositions comprising cyclosporin |
| WO2017181061A1 (en) | 2016-04-15 | 2017-10-19 | Ra Pharmaceuticals, Inc. | Ras binding peptides and methods of use |
| WO2018031730A2 (en) | 2016-08-11 | 2018-02-15 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Peptide inhibitors of phosphoglycerate mutase and methods of use |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
-
2022
- 2022-05-06 WO PCT/JP2022/019540 patent/WO2022234850A1/ja active Application Filing
- 2022-05-06 IL IL306018A patent/IL306018A/en unknown
- 2022-05-06 AU AU2022270498A patent/AU2022270498A1/en active Pending
- 2022-05-06 EP EP22798954.8A patent/EP4299069A1/en active Pending
- 2022-05-06 BR BR112023019622A patent/BR112023019622A2/pt unknown
- 2022-05-06 CA CA3219090A patent/CA3219090A1/en active Pending
- 2022-05-06 TW TW111117094A patent/TW202308679A/zh unknown
- 2022-05-06 JP JP2022548967A patent/JP7181442B1/ja active Active
- 2022-05-06 CN CN202280029838.6A patent/CN117241817A/zh active Pending
- 2022-05-06 KR KR1020237001877A patent/KR102582225B1/ko active IP Right Grant
- 2022-05-06 KR KR1020237032063A patent/KR20230169093A/ko unknown
- 2022-11-17 JP JP2022184220A patent/JP2023015334A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122059A1 (en) | 2011-03-04 | 2012-09-13 | New York University | Hydrogen bond surrogate macrocycles as modulators of ras |
| WO2013100132A1 (ja) | 2011-12-28 | 2013-07-04 | 中外製薬株式会社 | ペプチド化合物の環化方法 |
| WO2016071515A1 (en) | 2014-11-07 | 2016-05-12 | Sigmoid Pharma Limited | Compositions comprising cyclosporin |
| WO2017181061A1 (en) | 2016-04-15 | 2017-10-19 | Ra Pharmaceuticals, Inc. | Ras binding peptides and methods of use |
| WO2018031730A2 (en) | 2016-08-11 | 2018-02-15 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Peptide inhibitors of phosphoglycerate mutase and methods of use |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
Non-Patent Citations (11)
| Title |
|---|
| "Japanese Pharmacopeia", article "Powder X-ray Diffractometry" |
| ACC. CHEM. RES, vol. 41, 2008, pages 1331 - 1342 |
| ADV. DRUG DEL. REV, vol. 23, 1997, pages 3 - 25 |
| ANGEW. CHEM. INT. ED., vol. 52, 2013, pages 254 - 269 |
| CAS , no. 136552-06-2 |
| CAS, no. 330645-87-9 |
| CAS, no. 72914-19-3 |
| CHEM. REV., vol. 119, 2019, pages 10360 - 10391 |
| FUTURE MED. CHEM, vol. 1, 2009, pages 1289 - 1310 |
| KENTARO FURUMOTO, RYUSUKE TAKANO, SATOSHI TANIDA, TAKAMITSU UEDO, KOJI SHIRAKI, NORIYUKI TAKADA, SHOICHI HIGO: "P-5 Establishment of the high-throughput additive screening method for the purpose of improving solubility of the insoluble compound", JOURNAL OF PHARMACEUTICAL SCIENCE AND TECHNOLOGY, JAPAN, vol. 70, no. Suppl., 1 January 2010 (2010-01-01), JP , pages 192, XP009540883, ISSN: 0372-7629 * |
| THER. DELIV, vol. 8, no. 10, 2017, pages 867 - 878 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4299069A1 (en) | 2024-01-03 |
| KR20230020548A (ko) | 2023-02-10 |
| JP2023015334A (ja) | 2023-01-31 |
| CA3219090A1 (en) | 2022-11-10 |
| KR102582225B1 (ko) | 2023-09-22 |
| KR20230169093A (ko) | 2023-12-15 |
| AU2022270498A1 (en) | 2023-10-05 |
| CN117241817A (zh) | 2023-12-15 |
| IL306018A (en) | 2023-11-01 |
| TW202308679A (zh) | 2023-03-01 |
| JPWO2022234850A1 (ja) | 2022-11-10 |
| BR112023019622A2 (pt) | 2023-11-14 |
| JP7181442B1 (ja) | 2022-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2310363B1 (en) | Anticancer oral formulation | |
| ES2929526T3 (es) | Sal cristalina de L-arginina del ácido (R)-2-(7-(4-ciclopentil-3-(trifluorometil)benciloxi)-1,2,3,4-tetrahidrociclo-penta[b]indol-3-il)acético para su uso en trastornos asociados al receptor S1P1 | |
| WO2022121670A1 (zh) | Tolebrutinib的晶型及其制备方法和用途 | |
| US10370406B2 (en) | Salts and polymorphs of SCY-078 | |
| TWI696621B (zh) | 原肌球蛋白相關激酶(trk)抑制劑之醫藥調配物 | |
| EA027869B1 (ru) | Стабилизированная композиция такролимуса | |
| US20060286166A1 (en) | Tadalafil having a large particle size and a process for preparation thereof | |
| AU2014336929A1 (en) | Salt and crystal forms of PLK-4 inhibitor | |
| WO2015054793A1 (en) | Salt and crystal forms of plk-4 inhibitor | |
| WO2021007172A1 (en) | Formulations of rbp4 inhibitors and methods of use | |
| RU2627702C2 (ru) | Кристаллические формы 1-(3-трет-бутил-1-п-толил-1н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1н-индазол-5-илокси)бензил) мочевины гидрохлорида | |
| JP7181442B1 (ja) | 環状ペプチド化合物を含む製剤及びその製造方法 | |
| KR20210009153A (ko) | 자가나노유화 약물전달시스템을 이용한 타다라필의 경구용 고형제 조성물 및 이의 제조방법 | |
| WO2019137027A1 (zh) | Galunisertib的晶型及其制备方法和用途 | |
| CN113372332B (zh) | 一种奥希替尼新晶型 | |
| WO2022161507A1 (zh) | Brepocitinib甲苯磺酸盐的晶型及其制备方法和用途 | |
| WO2019105217A1 (zh) | Galunisertib的晶型及其制备方法和用途 | |
| WO2023069458A1 (en) | (s)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclo hepta[b]indole-4-carboxamide, and related crystalline forms, compositions, and methods thereof | |
| CN114615977A (zh) | 丙酮酸激酶r(pkr)活化组合物 | |
| EP1385521A2 (en) | Pharmaceutical composition containing pde v inhibitors and surfactants | |
| CN117343073A (zh) | 含噻吩并环类衍生物、其制备方法及其在医药上的应用 | |
| AU2002234622A1 (en) | Pharmaceutical composition containing PDE V inhibitors and surfactants |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022548967 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22798954 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20237001877 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 306018 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022270498 Country of ref document: AU Ref document number: AU2022270498 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022798954 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112023019622 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2022270498 Country of ref document: AU Date of ref document: 20220506 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2022798954 Country of ref document: EP Effective date: 20230929 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202392566 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3219090 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2023/013099 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 112023019622 Country of ref document: BR Kind code of ref document: A2 Effective date: 20230925 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |