WO2022234850A1 - 環状ペプチド化合物を含む製剤及びその製造方法 - Google Patents
環状ペプチド化合物を含む製剤及びその製造方法 Download PDFInfo
- Publication number
- WO2022234850A1 WO2022234850A1 PCT/JP2022/019540 JP2022019540W WO2022234850A1 WO 2022234850 A1 WO2022234850 A1 WO 2022234850A1 JP 2022019540 W JP2022019540 W JP 2022019540W WO 2022234850 A1 WO2022234850 A1 WO 2022234850A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formulation
- added
- formula
- composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
- A61K9/4825—Proteins, e.g. gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a formulation containing a cyclic peptide compound having KRAS inhibitory activity and a method for producing the same.
- Non-Patent Document 1 compounds that have been conventionally used as oral drugs should preferably have a molecular weight of 500 g/mol or less.
- Non-Patent Document 2 compounds that have been conventionally used as oral drugs should preferably have a molecular weight of 500 g/mol or less.
- Non-Patent Document 3, 4 Like insulin, which is used to treat hyperglycemia, peptides composed of natural amino acids have poor metabolic stability, and it has been difficult to develop peptides as oral drugs. However, it has been found that the metabolic stability and membrane permeability of peptides are improved by cyclizing peptides or using non-natural amino acids such as N-methylamino acids in peptides (Non-Patent Document 3, 4).
- cyclic peptides containing unnatural amino acids it has become known that cyclic peptides containing N-substituted amino acids in particular can have metabolic stability and membrane permeability, that is, can have drug-likeness ( Patent document 1).
- Non-Patent Document 5 It has been suggested that library compounds of cyclic peptides containing non-natural amino acids are useful for creating inhibitors of protein-protein interactions.
- Patent Document 6 discloses a cyclic peptide that exhibits a pharmacological action. In addition, Patent Document 6 discloses a technology for formulating cyclosporin. Studies on oral formulation of somatostatin have also been conducted (Non-Patent Document 6).
- An object of the present invention is to provide a formulation of a cyclic compound containing an unnatural amino acid.
- An object of the present invention is to provide a method for producing a formulation of a cyclic compound having an efficient RAS inhibitory action.
- Patent Document 4 describes inhibition of binding between RAS and SOS
- Patent Document 5 describes peptides that compete with compounds that bind to RAS. No effect on cells was shown. In addition, these documents do not describe drug-like peptides.
- Patent Documents 6 and 7 describe the formulation of cyclic peptides, they merely describe the formulation of specific compounds.
- Non-Patent Document 2 describes peptides used as medicines, but does not describe drug-like peptides or peptides useful for RAS mutant cancer.
- Non-Patent Documents 3 and 4 describe that peptides containing N-methyl amino acids can be applied as medicines, but do not describe peptides useful for RAS mutant cancer.
- Non-Patent Document 5 describes that cyclic peptides can be applied as medicines, but does not describe peptides useful for RAS mutant cancer.
- Non-Patent Document 6 exemplifies somatostatin and describes the elements necessary for oral formulation, but only somatostatin is described.
- a surfactant is effective as a specific additive, preferably a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the formulation can be made more effective by adding an oily component. It has been found that a mixture of the active ingredient and these additives can be formulated and used in a formulation having a specific dosage form.
- the formulation according to the present invention has excellent stability of the active ingredient in the formulation and excellent dissolution from the formulation.
- the present invention relates to the following.
- Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component.
- the surfactant is a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the hydrophobic surfactant is at least one selected from the group consisting of propylene glycol fatty acid ester, glycerin fatty acid ester, polyglycerin fatty acid ester, sorbitan fatty acid ester, and hydrophobic polyoxyethylene hydrogenated castor oil. , [3] or [4].
- the hydrophobic surfactant comprises a propylene glycol fatty acid ester.
- the propylene glycol fatty acid ester is propylene glycol monocaproate, propylene glycol monocaprylate, propylene glycol monocaprate, propylene glycol monolaurate, propylene glycol monomyristate, propylene glycol monopalmitate, propylene glycol monostearate, and propylene glycol monooleate, which is at least one selected from the group consisting of [5].
- the glycerin fatty acid ester is glyceryl monocaproate, glyceryl monocaprylate, glyceryl monocaprate, glyceryl monolaurate, glyceryl monomyristate, glyceryl monopalmitate, glyceryl monostearate, glyceryl monooleate, and monolinol
- the composition of [5] which is at least one selected from the group consisting of glyceryl acid.
- the composition according to [5] wherein the polyglycerin fatty acid ester is diglyceryl monooleate.
- the sorbitan fatty acid ester is sorbitan monocaprylate, sorbitan monocaprate, sorbitan monolaurate, sorbitan monomyristate, sorbitan monopalmitate, sorbitan monostearate, sorbitan monooleate, sorbitan sesquioleate, and trio
- the composition according to [5] which is at least one selected from the group consisting of sorbitan lactate.
- the hydrophobic polyoxyethylene hydrogenated castor oil is at least one selected from the group consisting of polyoxyethylene hydrogenated castor oil 5 and polyoxyethylene hydrogenated castor oil 10. .
- the hydrophilic surfactant is polyethylene glycol fatty acid ester, polyoxyethylene castor oil, hydrophilic polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate. , caprylocaproyl polyoxyl-8 glyceride, and any combination thereof, the composition according to any one of [3] to [13].
- the hydrophilic surfactant is polyoxyethylene castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, and these The composition according to any one of [3] to [14], which is at least one selected from the group consisting of any combination. [16] The composition according to any one of [3] to [15], wherein the hydrophilic surfactant comprises polyoxyethylene castor oil.
- polyethylene glycol fatty acid ester is at least one selected from the group consisting of polyoxyethylene hydroxystearate, polyethylene glycol monolaurate, polyethylene glycol monostearate, and polyoxyl 40 stearate; The described composition.
- polyoxyethylene castor oil is at least one selected from the group consisting of polyoxyl 30 castor oil, polyoxyl 35 castor oil, and polyoxyl 40 castor oil.
- polyoxyethylene castor oil is Polyoxyl 35 castor oil.
- the hydrophilic polyoxyethylene hydrogenated castor oil is selected from the group consisting of polyoxyethylene hydrogenated castor oil 20, polyoxyethylene hydrogenated castor oil 40, polyoxyethylene hydrogenated castor oil 50, and polyoxyethylene hydrogenated castor oil 60.
- the hydrophilic surfactant is selected from the group consisting of polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, polysorbate 80, and combinations thereof; The composition according to any one of [3] to [15], which is at least one selected. [23] The composition according to any one of [3] to [15], wherein the hydrophilic surfactant comprises polyoxyl 35 castor oil. [24] The composition of [3], wherein the hydrophobic surfactant is a propylene glycol fatty acid ester and the hydrophilic surfactant comprises polyoxyethylene castor oil.
- the hydrophobic surfactant is propylene glycol monocaprylate, and the hydrophilic surfactant is polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl- 8.
- the composition of [3] which is at least one selected from the group consisting of glycerides, polysorbate 80, and combinations thereof.
- oily component is at least one selected from the group consisting of fatty acids, acylglycerols, and combinations thereof.
- oily component contains a fatty acid.
- the fatty acid is selected from the group consisting of caproic acid, caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid;
- the composition of [28], which is at least one of [32] The composition of [28], wherein the fatty acid is at least one selected from the group consisting of oleic acid, linoleic acid, and linolenic acid.
- the acylglycerol is selected from the group consisting of triacetin, tributyrin, tricaproin, tricaprylin, tricaprin, tripalmitin, tripalmitolein, glyceryl tristearate, triolein, trilinolein, trilinolenin, and medium-chain fatty acid triglycerides;
- the composition of [28] which is at least one.
- the vegetable oil is selected from the group consisting of olive oil, almond oil, coconut oil, cocoa butter, macadamia nut oil, avocado oil, safflower oil, soybean oil, linseed oil, rapeseed oil, castor oil, corn oil, and palm oil.
- composition of [3] which contains propylene glycol fatty acid ester as the hydrophobic surfactant, polyoxyethylene castor oil as the hydrophilic surfactant, and fatty acid as the oil component.
- composition of [3] which contains propylene glycol monocaprylate as the hydrophobic surfactant, polyoxyl 35 castor oil as the hydrophilic surfactant, and oleic acid as the oil component.
- the antioxidant is dl- ⁇ -tocopherol, butyrated hydroxytoluene, butyrated hydroxyanisole, propyl gallate, propyl gallate, pharmaceutically acceptable quinones, astaxanthin, and D- ⁇ -tocopheryl polyethylene glycol;
- the composition of [40] which is at least one selected from the group consisting of 1000 succinates.
- the solubilizer is at least one selected from the group consisting of ethanol, propylene glycol, polyethylene glycol 300, polyethylene glycol 400, diethylene glycol monoethyl ether, and combinations thereof; Composition.
- the compound represented by formula (1), or a salt thereof, or a solvate thereof is a compound represented by formula (1) or a hydrate thereof, [1] to [43] A composition according to any one of the preceding claims.
- the composition of [44], wherein the compound represented by formula (1), a salt thereof, or a solvate thereof is a compound represented by formula (1).
- Any one of [1] to [46], wherein the content of the compound represented by formula (1), or a salt thereof, or a solvate thereof is 10% by weight or less in the entire composition.
- a composition according to claim 1. Any one of [1] to [48], wherein the content of the compound represented by formula (1), or a salt thereof, or a solvate thereof is 7% by weight or less in the entire composition.
- a composition according to claim 1. [50] The composition according to any one of [1] to [49], wherein the content of the liquid additive in the entire composition is 50% by weight or more and 97% by weight or less.
- composition according to any one of [1] to [50], wherein the content of the liquid additive in the entire composition is 55% by weight or more and 96% by weight or less.
- the composition according to any one of [1] to [51], wherein the content of the liquid additive in the entire composition is 70% by weight or more and 90% by weight or less.
- the composition according to any one of [3] to [52], wherein the content of the hydrophobic surfactant in the entire composition is 20% by weight or more and 70% by weight or less.
- the composition according to any one of [3] to [53], wherein the content of the hydrophobic surfactant in the entire composition is 25% by weight or more and 65% by weight or less.
- composition according to any one of [1] to [58], wherein the content of the oily component in the entire composition is 0% by weight or more and 50% by weight or less.
- the composition according to any one of [1] to [60], wherein the content of the oily component in the entire composition is 10% by weight or more and 20% by weight or less.
- Any one of [1] to [65], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 5 or more.
- any one of [1] to [66], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 10 or more.
- a composition according to claim 1. Any one of [1] to [66], wherein the weight ratio of the liquid additive to the compound represented by the formula (1), a salt thereof, or a solvate thereof is 5 to 2000. or the composition according to claim 1.
- the total weight ratio of the liquid additive and the oily component to the compound represented by the formula (1), or a salt thereof, or a solvate thereof is 10 or more, [1] to The composition of any one of [68].
- the weight ratio of the sum of the liquid additive and the oily component to the compound represented by the formula (1) or a salt thereof, or a solvate thereof is 10 to 2000 [1] The composition according to any one of -[69]. [71] The weight ratio of the sum of the liquid additive and the oily component to the compound represented by the formula (1), or a salt thereof, or a solvate thereof is 10 to 1000. [1] The composition according to any one of [70]. [72] The composition according to any one of [1] to [71], wherein the composition is liquid. [73] The composition according to any one of [1] to [72], wherein droplets formed when the composition is dispersed in water have an average particle size of less than 200 nm.
- [78] A method for producing the composition according to any one of [1] to [75], comprising the following steps; (1) Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component; (2) mixing the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component; and (3) A step of obtaining a composition in which the compound represented by the formula (1), a salt thereof, or a solvate thereof is dissolved. [79] the compound represented by formula (1) provided in the step (1), a salt thereof, or a solvate thereof is a hydrate of the compound represented by formula (1); The method of [78].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8.296 in powder X-ray diffraction. , 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.°.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least two peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least three peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 4 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 5 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 6 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 7 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 8 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 9 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern containing at least 10 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern containing at least 11 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79] having an X-ray powder diffraction pattern comprising at least 12 peaks within 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8 in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and The method of [79], having a powder X-ray diffraction pattern containing a peak at 17.813° ( ⁇ 0.2°).
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, and 10.435 in powder X-ray diffraction. ° 11.729 °, 12.704 °, 15.895 °, and 16.643 ° ( ⁇ 0.2 °) having a powder X-ray diffraction pattern comprising at least one peak, in [79] described method.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10 .435° 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least two peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction.
- the hydrate of the compound represented by formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction. .435° 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern containing at least four peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction. .435°, 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern comprising at least five peaks [79 ].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10 .435°, 11.729°, 12.704°, 15.895°, and 16.643° ( ⁇ 0.2°) with a powder X-ray diffraction pattern comprising at least six peaks [79 ].
- the hydrate of the compound represented by formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 7.921°, 9.956°, 10°, and 10° in powder X-ray diffraction.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction.
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, 8 .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least 10 peaks; The method of [79].
- the hydrate of the compound represented by the formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, 8 .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having at least 13 peaks.
- the hydrate of the compound represented by the above formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895°, 16.190°, 16.643°, 17.813°, and 19.424° ( ⁇ 0.2°) having a powder X-ray diffraction pattern comprising at least 15 peaks; The method of [79].
- the hydrate of the compound represented by the above formula (1) is a crystal, and the crystal has diffraction angles 2 ⁇ of 4.964°, 7.921°, and 8° in powder X-ray diffraction. .296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15 .895 °, 16.190 °, 16.643 °, 17.813 °, and 19.424 ° ( ⁇ 0.2 °) powder X-ray diffraction pattern containing peaks, the method of [79] .
- a method for producing a pharmaceutical preparation comprising the steps of providing the composition of any one of [1] to [75], and formulating the composition to provide a pharmaceutical preparation.
- Production of a capsule formulation comprising the steps of providing the composition of any one of [1] to [75], and filling the composition into capsules to provide a capsule formulation.
- the composition according to the present invention is excellent in various properties required as a formulation.
- the composition according to the present invention has desirable particle characteristics when a liquid composition is emulsified to form droplets, is extremely stable and dispersible, and is easily absorbed into the body. Are better.
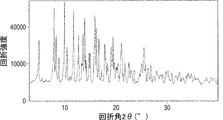
- FIG. 1 is a graph showing the results of powder X-ray diffraction measurement of the hydrate crystals (Form C) of compound 1 obtained in Preparation Example 3.
- FIG. The vertical axis is the diffraction intensity, and the horizontal axis is the diffraction angle 2 ⁇ (°).
- FIG. 2 shows the results of thermogravimetric and differential thermal analysis of hydrate crystals of compound 1 (Form C).
- the horizontal axis is temperature (°C) and measurement time (minutes), and the right vertical axis is weight change (mg) of the sample in thermogravimetric analysis.
- the left vertical axis represents the heat flow (mW) observed in the differential thermal analysis.
- FIG. 3 shows the crystal structure of the hydrate crystal of Compound 1 (Form C) obtained by single-crystal X-ray structure analysis.
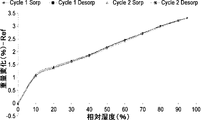
- FIG. 4 shows the results of dynamic water vapor adsorption measurement of hydrate crystals of Compound 1 (Form C). The vertical axis is weight change (%) and the horizontal axis is relative humidity (%).
- “Cycle1 Sorp” black diamonds
- “Cycle1 Desorp” black squares
- Cycle2 Sorp” black triangles
- “Cycle2 Desorp” filled squares) indicates desorption in cycle 2.
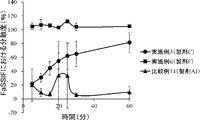
- FIG. 5 is a graph showing temporal changes in the dispersibility of formulations according to the present invention (formulations C and F) and a comparative example (formulation AJ) in fasting artificial intestinal fluid (FaSSIF).
- composition of the present invention has the following formula (1): The compound represented by (“(5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-butyl)-18-cyclopentyl-29-(3,5- Difluoro-4-(trifluoromethyl)phenethyl)-36-ethyl-11-isobutyl-N,N,5,6,12,16,19,33-octamethyl-35-(4-methylbenzyl)-4,7 ,10,13,17,20,23,28,31,34,37-undecaoxotetratriacontahydro-2H,4H-spiro[azeto[2,1-u]pyrrolo[2,1-i][ 1,4,7,10,13,16,19,22,25,28,31]undecaazacyclotetratriacontin-21,1′-cyclopentane]-15-carbox
- composition according to the present invention optionally contains an oily component, preferably an oily component.
- the compound that can be used in the present invention is the compound represented by the above formula (1), a salt thereof, or a solvate thereof.
- a salt of Compound 1 can preferably be a chemically or pharmaceutically acceptable salt thereof.
- Compound 1 or a salt thereof that can be used in the present invention can also be a solvate thereof, preferably a chemically or pharmaceutically acceptable solvate thereof.
- the compound used in the present invention may preferably be exclusively in the form of a free form in the composition, but may contain the form of a salt or solvate depending on the form of the composition.
- the salts of compound 1 include, for example, hydrochloride; hydrobromide; hydroiodide; phosphate; sulfonates such as; carboxylates such as acetates, citrates, malate, tartrates, succinates, salicylates; or alkali metal salts such as sodium salts, potassium salts; magnesium salts, calcium salts alkaline earth metal salts such as; ammonium salts such as ammonium salts, alkylammonium salts, dialkylammonium salts, trialkylammonium salts and tetraalkylammonium salts; These salts are produced, for example, by contacting the compound 1 with an acid or base that can be used in the production of pharmaceuticals.
- a solvate refers to a compound formed with a solvent to form a single molecular cluster, and a solvate formed with a solvent that is acceptable for ingestion accompanying administration of a drug. It is not particularly limited. Examples include hydrates, alcoholates (ethanolates, methanolates, 1-propanolates, 2-pronolates, etc.), solvates with a single solvent such as dimethylsulfoxide, as well as , solvates formed with a plurality of solvents for one molecule of the compound, and solvates formed with a plurality of types of solvents for one molecule of the compound. If the solvent is water, it is called a hydrate.
- the solvate of the compound of the present invention is preferably a hydrate, and such a hydrate is specifically a 1-10 hydrate, preferably a 1-5 hydrate, more preferably a 1-3 hydrates.
- the number of water molecules in the hydrate of compound 1 used in the present invention may change due to the desorption of water molecules bound to compound 1 depending on the ambient environment such as temperature and humidity.
- Compound 1 or a salt thereof, or a solvate thereof that can be used in the present invention can be provided in the form of a crystal, an amorphous form, or a mixture thereof, preferably Compound 1 or a salt thereof, or a solvate thereof
- the product may be provided in crystalline form.
- Crystals of Compound 1 or a salt thereof or a solvate thereof that can be used in the present invention preferably include hydrate crystals of Compound 1 (also referred to as Form C).
- Crystals of Compound 1 or a salt thereof, or a solvate thereof can be isolated by techniques known in the art, such as X-ray powder diffraction (XRPD), moisture determination methods (e.g. Karl Fischer method), scanning electron microscopy (SEM). Characterization can be by analytical, solid state NMR, or thermal techniques such as differential scanning calorimetry (DSC), or any other standard quantitative measurement method.
- XRPD X-ray powder diffraction
- moisture determination methods e.g. Karl Fischer method
- SEM scanning electron microscopy
- Characterization can be by analytical, solid state NMR, or thermal techniques such as differential scanning calorimetry (DSC), or any other standard quantitative measurement method.
- the diffraction angle 2 ⁇ in powder X-ray diffraction is preferably a diffraction peak measured using CuK ⁇ radiation.
- a crystalline compound 1 hydrate that can be used in the present invention has a powder X-ray diffraction pattern that includes at least one of the following peaks as a diffraction angle 2 ⁇ in powder X-ray diffraction. 4.964°, 7.921°, 8.296°, 8.855°, 9.956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, 17.813° ( ⁇ 0.2°)
- Compound 1 used in the present invention includes all isotopes of Compound 1.
- Isotopes of compound 1 have at least one atom with the same atomic number (number of protons) and a different mass number (sum of proton and neutron numbers), with abundance ratios different from those in nature. It is replaced.
- Examples of isotopes contained in Compound 1 include hydrogen, carbon, nitrogen, oxygen, and fluorine atoms, respectively, 2 H, 3 H, 13 C, 14 C, 15 N, 17 O, 18 O, 18 F and the like are included.
- radioactive isotopes that emit radioactivity and decay such as 3 H and 14 C, are useful in tissue distribution tests of drugs or compounds.
- Stable isotopes are safe to use because they do not decay, their abundance changes little over time, and they are non-radioactive.
- the isotopes of compound 1 can be converted according to conventional methods by replacing the reagents used in the synthesis with reagents containing the corresponding isotopes.
- Acids or bases used to form salts of Compound 1, as well as solvents used to form solvates of Compound 1, can also include all isotopes.
- liquid additive that can be used in the composition according to the present invention is an additive that is pharmaceutically acceptable and capable of dissolving the compound used in the present invention.
- the “liquid additive” means that the additive is in a dissolved state in the composition, and those that are not dissolved at the stage of being provided as raw materials for the production of the composition are It can be used as one component of the composition according to the present invention by mixing it with the components of (1) or dissolving it in the manufacturing process by heating or the like.
- an additive that is liquid at room temperature can be preferably used.
- room temperature is used in the ordinary sense in the technical field and is not particularly limited, but unless otherwise specified, for example, preferably 1 to 30° C., more preferably about 15 to 28° C. be.
- Liquid additives that can be used in the present invention preferably include surfactants, and surfactants preferably include hydrophobic surfactants and hydrophilic surfactants. In the present invention, it is desirable to use a combination of a hydrophobic surfactant and a hydrophilic surfactant.
- the HLB value (Hydrophilic-Lipophilic Balance value) of the hydrophobic surfactant is preferably less than 10, more preferably 0
- the HLB value of the hydrophilic surfactant is preferably 10 or more, more preferably 10 or more and 30 or less, still more preferably 10 or more and 20 or less.
- the HLB value is a value that represents the degree of affinity of a surfactant for water and oil (an organic compound that is insoluble in water), and is known to those skilled in the art. Values based on known methods can be adopted.
- Hydrophobic surfactants preferably include propylene glycol fatty acid esters, glycerin fatty acid esters, polyglycerin fatty acid esters, sorbitan fatty acid esters, and hydrophobic polyoxyethylene hydrogenated castor oil, etc. These may also be used in combination. can.
- the hydrophobic surfactant propylene glycol fatty acid ester or sorbitan fatty acid ester can be used more preferably, and propylene glycol fatty acid ester can be used more preferably.
- Propylene glycol fatty acid esters preferably include propylene glycol monocaproate, propylene glycol monocaprylate, propylene glycol monocaprate, propylene glycol monolaurate, propylene glycol monomyristate, propylene glycol monopalmitate, propylene glycol monostearate, and propylene glycol monooleate.
- propylene glycol monocaprylate can be used more preferably.
- Glycerin fatty acid esters preferably include glyceryl monocaproate, glyceryl monocaprylate, glyceryl monocaprate, glyceryl monolaurate, glyceryl monomyristate, glyceryl monopalmitate, glyceryl monostearate, glyceryl monooleate, and monolinol. and glyceryl acid.
- Polyglycerol fatty acid esters preferably include diglyceryl monooleate.
- Sorbitan fatty acid esters preferably include sorbitan monocaprylate, sorbitan monocaprate, sorbitan monolaurate, sorbitan monomyristate, sorbitan monopalmitate, sorbitan monostearate, sorbitan monooleate, sorbitan sesquioleate, and trio and sorbitan lactate.
- the hydrophobic polyoxyethylene hydrogenated castor oil is a hydrophobic polyoxyethylene hydrogenated castor oil, for example, a polyoxyethylene hydrogenated castor oil having an HLB value of preferably less than 10, more preferably 0 or more and less than 10. oil.
- Hydrophobic polyoxyethylene hydrogenated castor oil preferably includes polyoxyethylene hydrogenated castor oil 5 and polyoxyethylene hydrogenated castor oil 10 and the like.
- Hydrophilic surfactants preferably include polyethylene glycol fatty acid ester, polyoxyethylene castor oil, hydrophilic polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan fatty acid ester, and D- ⁇ -tocopheryl polyethylene glycol 1000 succinate. , caprylocaproyl polyoxyl-8 glyceride and the like, and these can also be used in combination. Hydrophilic surfactants, more preferably polyoxyethylene castor oil, polyoxyethylene sorbitan fatty acid ester, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, or these Combinations can be used, more preferably polyoxyethylene castor oil.
- Polyethylene glycol fatty acid esters preferably include polyoxyethylene hydroxystearate, polyethylene glycol monolaurate, polyethylene glycol monostearate, and polyoxyl 40 stearate.
- the polyoxyethylene castor oil preferably includes polyoxyl 30 castor oil, polyoxyl 35 castor oil, polyoxyl 40 castor oil, and more preferably polyoxyl 35 castor oil.
- the hydrophilic polyoxyethylene hydrogenated castor oil is a hydrophilic polyoxyethylene hydrogenated castor oil, for example, the HLB value is preferably 10 or more, more preferably 10 or more and 30 or less, still more preferably 10 or more. 20 or less polyoxyethylene hydrogenated castor oil.
- Hydrophilic polyoxyethylene hydrogenated castor oils preferably include polyoxyethylene hydrogenated castor oil 20, polyoxyethylene hydrogenated castor oil 40, polyoxyethylene hydrogenated castor oil 50, and polyoxyethylene hydrogenated castor oil 60. .
- Polyoxyethylene sorbitan fatty acid esters preferably include polysorbate 20, polysorbate 40, polysorbate 60, and polysorbate 80.
- hydrophilic surfactant more preferably polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, caprylocaproyl polyoxyl-8 glyceride, polysorbate 80, or a combination thereof is preferably used. can be done.
- hydrophobic surfactant preferably propylene glycol monocaprylate
- hydrophilic surfactant preferably polyoxyl 35 castor oil, D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, capri Rocaproyl polyoxyl-8 glycerides, polysorbate 80, or combinations thereof can be used.
- the surfactant preferably uses a propylene glycol fatty acid ester as a hydrophobic surfactant and contains at least polyoxyethylene castor oil as a hydrophilic surfactant.
- the surfactant is a hydrophobic surfactant. It is desirable to use propylene glycol monocaprylate as the hydrophilic surfactant and to include at least polyoxyl 35 castor oil as the hydrophilic surfactant.
- Oily Component may contain an oily component.
- Oily ingredients preferably include fatty acids, acylglycerols, vegetable oils and combinations thereof. Fatty acids or acylglycerols are more preferably used as the oily component, and fatty acids are more preferably used.
- Fatty acids preferably include caproic acid, caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid.
- caproic acid caprylic acid, capric acid, lauric acid, myristic acid, myristoleic acid, palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid, and linolenic acid.
- oleic acid, linoleic acid, and linolenic acid are more preferred, and oleic acid is even more preferred.
- Preferred acylglycerols include triacetin, tributyrin, tricaproin, tricaprylin, tricaprin, tripalmitin, tripalmitolein, glyceryl tristearate, triolein, trilinolein, trilinolenin, and medium-chain fatty acid triglycerides. Among these, triacetin is more preferred.
- Vegetable oils include olive oil, almond oil, coconut oil, cocoa butter, macadamia nut oil, avocado oil, safflower oil, soybean oil, linseed oil, rapeseed oil, castor oil, corn oil, and palm oil.
- oily component oleic acid, triacetin, or a combination thereof can be preferably used, and oleic acid is more preferably used.
- propylene glycol fatty acid ester as a hydrophobic surfactant, contain at least polyoxyethylene castor oil as a hydrophilic surfactant, and fatty acid as an oily component.
- the surfactant is propylene glycol monocaprylate as a hydrophobic surfactant, contains at least polyoxyl 35 castor oil as a hydrophilic surfactant, and oleic acid as an oily component. It is desirable to use
- composition according to the present invention can further contain an antioxidant.
- an antioxidant preferably dl- ⁇ -tocopherol, butyrated hydroxytoluene, butyrated hydroxyanisole, propyl gallate, propyl gallate, pharmaceutically acceptable quinones, astaxanthin, and D- ⁇ -tocopheryl polyethylene glycol 1000 succinate, etc., as well as combinations thereof.
- Antioxidants more preferably include dl- ⁇ -tocopherol.
- composition according to the present invention can further contain a solubilizer.
- Solubilizers preferably include ethanol, propylene glycol, polyethylene glycol 300, polyethylene glycol 400, diethylene glycol monoethyl ether, combinations thereof, and the like.
- the solubilizer is more preferably ethanol or propylene glycol, and still more preferably propylene glycol.
- the content of Compound 1, or a salt thereof, or a solvate thereof is such that it dissolves in the liquid additive and optionally used oily component and exhibits a certain level of effectiveness.
- the content is not particularly limited as long as it is, the content of Compound 1, or a salt thereof, or a solvate thereof with respect to the entire composition, the upper limit is preferably 10% by weight or less, 9% by weight % or less, 8 wt% or less, 7 wt% or less, 6 wt% or less, 5 wt% or less, 4 wt% or less, 3 wt% or less, or 2 wt% or less, more preferably 10 wt% or less, 8 wt% Below, it can be 7% by weight or less.
- the lower limit of the content is not particularly limited, but preferably exceeds 0%, 0.1% by weight or more, 0.2% by weight or more, 0.3% by weight or more, 0.4% by weight or more, 0 0.5 wt% or more, 0.6 wt% or more, 0.7 wt% or more, 0.8 wt% or more, 0.9 wt% or more, or 1 wt% or more, more preferably 0 .1 wt% or more, 0.2 wt% or more, 0.3 wt% or more, 0.4 wt% or more, or 0.5 wt% or more. Further, the content can be within any combination of the above lower limit and upper limit.
- the content range is preferably 0.1 wt% to 10 wt%, 0.2 wt% to 10 wt%, 0.3 wt% to 10 wt%, 0.4 wt% or more. It can be 10 wt % or less, 0.5 wt % or more and 10 wt % or less, and the like.
- a liquid additive preferably a surfactant
- a surfactant may be contained so that Compound 1 or a salt thereof, or a solvate thereof can be dissolved, and the content is particularly limited.
- the content of the liquid additive in the entire composition is preferably 50% to 97% by weight, more preferably 55% to 96% by weight, and still more preferably 70% to 90% by weight. can do.
- the content of the hydrophobic surfactant in the entire composition is preferably 20% by weight or more and 70% by weight or less, more preferably 25% by weight or more and 65% by weight or less, and still more preferably 30% by weight. % or more and 55% or less by weight.
- the content of the hydrophilic surfactant in the entire composition is preferably 20% by weight or more and 40% by weight or less, more preferably 25% by weight or more and 35% by weight or less, still more preferably 28% by weight. % by weight or more and 33% by weight or less.
- the weight ratio of the hydrophobic surfactant to the hydrophilic surfactant is preferably 0.5 to 3.0, more preferably is 1.0 to 2.5, more preferably 1.5 to 2.0.
- the content of the oily component in the entire composition is preferably 0% by weight or more and 50% by weight or less, more preferably 1% by weight or more and 40% by weight or less, and still more preferably 10% by weight or more. 20% by weight or less.
- the content of the antioxidant with respect to the entire composition is not particularly limited as long as it does not adversely affect the pharmaceutical properties of the formulation, but is preferably 0.01% by weight. As described above, the content is more preferably 0.01% to 5% by weight, more preferably 0.1% to 5% by weight, and still more preferably 0.1% to 2% by weight.
- the content of the solubilizer in the composition as a whole is not particularly limited as long as it does not adversely affect the pharmaceutical properties of the formulation, but is preferably 1% by weight or more and 20% by weight. % by weight or less, more preferably about 2% to 15% by weight.
- the weight ratio of the liquid additive to the compound represented by formula (1), or a salt thereof, or a solvate thereof is not particularly limited as long as compound 1 or a salt thereof, or a solvate thereof can be dissolved, but is preferably 5 or more, more preferably 10 or more.
- the upper limit of the weight ratio is not particularly limited, it can be, for example, preferably 2000 or less, more preferably 1000 or less.
- the weight ratio can be in the range of any combination of the lower limit and the upper limit. can.
- Liquid additives are preferably surfactants.
- the total weight ratio of the liquid additive and the oily component to the compound represented by formula (1), a salt thereof, or a solvate thereof is not particularly limited as long as the compound 1 or a salt thereof, or a solvate thereof can be dissolved, but is preferably 10 or more.
- the upper limit of the weight ratio is not particularly limited, it can be, for example, preferably 2000 or less, more preferably 1000 or less.
- the weight ratio can range in any combination of lower and upper limits, for example the weight ratio can be preferably 10-2000, more preferably 10-2000.
- Liquid additives are preferably surfactants.
- composition of the present invention may be liquid, gel or semi-solid, preferably liquid.
- composition according to the present invention is characterized in that droplets formed when the composition is dispersed in a liquid such as water have a small average particle size and a narrow particle size distribution.
- a certain dilution ratio for example, the composition is 0.01 to 1% by volume with respect to the entire mixed solution
- the mixed solution is stirred at a temperature suitable for handling and/or at a temperature that does not affect the stability of the substances contained in the composition, for example, near room temperature (about 25 ° C.), and the composition of the present invention is dispersed in the solution.
- the average particle size of the droplets formed when the droplets are formed is preferably less than 200 nm, more preferably 10 nm or more and less than 200 nm, still more preferably 50 nm or more and less than 200 nm.
- the composition of the present invention preferably has a polydispersity index (PDI), which is an index representing particle size distribution, of less than 0.5, more preferably 0.4 or less, and still more preferably 0.3. It is below.
- PDI polydispersity index
- the average particle size and polydispersity index of the compound in the composition can be determined using known methods such as dynamic light scattering (DLS).
- a solution simulating the internal environment e.g., dissolution test fluid 1 of the Japanese Pharmacopoeia, pH 1.2, body simulating temperature (e.g., 36-37°C) used for disintegration test of enteric preparations
- a solution simulating the internal environment e.g., dissolution test fluid 1 of the Japanese Pharmacopoeia, pH 1.2, body simulating temperature (e.g., 36-37°C) used for disintegration test of enteric preparations
- the mixed solution e.g., when the composition of the present invention is dispersed in the solution by stirring under a certain dilution ratio (for example, the composition is 0.01 to 1% by volume with respect to the entire mixed solution)
- the particle size distribution of the droplets to be formed can similarly exhibit characteristics such that the average particle size of the droplets is small and the width of the particle size distribution is narrow. That is, droplets having a small average particle size and a narrow particle size distribution can be formed even in the gastrointestinal tract.
- Such a composition of the present invention can have excellent dispersibility in liquid and can have excellent absorption into the body.
- a pharmaceutical formulation according to the present invention is a pharmaceutical formulation comprising a composition containing compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component.
- the pharmaceutical formulation according to the present invention comprises the compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component, and optionally an antioxidant, a solubilizer, etc., in addition to It can be produced by introducing a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier commonly used excipients, binders, lubricants, coloring agents, flavoring agents, and if necessary, stabilizers, emulsifiers, absorption enhancers, pH adjusters, preservatives, etc. can be used.
- Ingredients that are generally used as raw materials for pharmaceutical formulations can be blended and formulated by a conventional method.
- the administration of the formulation according to the present invention may be either oral administration or parenteral administration. Oral administration is preferred, but the administration method is not limited to oral administration.
- compound 1 of the present invention or a salt thereof, or a solvate thereof a liquid additive, an optional oily component, etc., and optionally a binder, a disintegrant, a lubricant, etc.
- a liquid additive for example, compound 1 of the present invention or a salt thereof, or a solvate thereof, a liquid additive, an optional oily component, etc., and optionally a binder, a disintegrant, a lubricant, etc.
- an agent, a coloring agent, a flavoring agent, etc. it is made into a liquid formulation, a capsule formulation, etc. by a conventional method.
- the composition of the present invention is preferably a liquid, it can be an injection or a capsule in liquid formulations.
- composition of the present invention when used as a capsule, the capsules commonly used in capsule formulations can be used.
- the type of capsule is not particularly limited, and those commonly used in this technical field can be used.
- examples of capsules include hard capsules (hard capsules) and soft capsules (soft capsules).
- a hard capsule usually consists of a cap and a body, and can be produced by putting a cap on the body filled with a formulation.
- raw materials for hard capsules for example, those containing gelatin, hydroxypropylmethylcellulose, pullulan, or a mixture thereof can be preferably used. , No. 1, No. 0, No. 00, and No. 000.
- Soft capsules can be produced, for example, by enveloping a formulation with a base such as gelatin.
- Raw materials for soft capsules that can be preferably used include, for example, gelatin, starch, carrageenan, agar, glycerin, sorbitol, or mixtures thereof.
- Excipients include, for example, lactose, cornstarch, sucrose, glucose, mannitol, sorbitol, crystalline cellulose, and silicon dioxide.
- binders include polyvinyl alcohol, polyvinyl ether, methylcellulose, ethylcellulose, gum arabic, tragacanth, gelatin, shellac, hydroxypropylmethylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, polypropyleneglycol-polyoxyethylene-block polymer, and meglumine. be done.
- disintegrants examples include starch, agar, gelatin powder, crystalline cellulose, calcium carbonate, sodium hydrogencarbonate, calcium citrate, dextrin, pectin, carboxymethylcellulose/calcium, and the like.
- lubricants examples include magnesium stearate, talc, polyethylene glycol, silica, hydrogenated vegetable oils, and the like.
- coloring agents those that are permitted to be added to pharmaceuticals are used, and as flavoring agents, cocoa powder, mint brain, aromatic powder, mint oil, borneol, cinnamon powder, etc. are used.
- compound 1 used in the present invention or a salt thereof, or a solvate thereof may be added with a pH adjuster, a solubilizer, a tonicity agent, etc., if necessary. Add solubilizers, stabilizers, etc., and formulate in a conventional manner.
- parenteral administration specifically includes an injection dosage form, a nasal dosage form, a pulmonary dosage form, a transdermal dosage form, and the like.
- injection dosage forms include systemic or local administration by intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection, and the like.
- nasal dosage forms include systemic or local administration, such as by absorption of active ingredients in the formulation through the nasal mucosa or administration of the formulation through the nasal cavity.
- transpulmonary dosage forms can be administered systemically or locally, such as by administering the formulation to the lungs through the trachea.
- transdermal administration forms include systemic or local administration, such as by applying the formulation to the skin.
- the administration method can be appropriately selected according to the patient's age and symptoms.
- the dosage of the pharmaceutical formulation containing Compound 1 or its salt or solvate thereof used in the present invention can be selected, for example, in the range of 0.001 mg to 100 mg per kg of body weight per dose.
- doses can be selected in the range of 0.1 to 1000 mg/body per patient, but are not necessarily limited to these figures.
- the dosage and administration method vary depending on the patient's body weight, age, symptoms, etc., but can be appropriately selected by those skilled in the art.
- the method for producing the composition according to the present invention comprises: (1) Formula (1) below: or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component (Step 1); (2) mixing the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component (step 2); and (3) A step of obtaining a composition in which the compound represented by formula (1), or a salt thereof, or a solvate thereof is dissolved (step 3).
- compound 1 or a salt thereof, or a solvate thereof supplied in step (1) is preferably a hydrate of the compound represented by formula (1). , more preferably one or more water molecules of the compound represented by formula (1).
- the number of water molecules contained in the hydrate of the compound represented by formula (1) can be desorbed depending on the surrounding environment such as temperature and humidity.
- Compound 1 or a salt thereof, or a solvate thereof supplied in step (1) is preferably Compound 1, a salt thereof, or a solvate thereof 1 It is a crystal, more preferably a hydrate crystal of the compound represented by formula (1).
- the water molecules in the hydrate crystals of the compound represented by formula (1) preferably contain one or more water molecules, but the number of water molecules desorbs depending on the surrounding environment such as temperature and humidity. What can be done is described above.
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15.895°, 16.190°, 16. It can have an X-ray powder diffraction pattern comprising at least one peak at 643°, 17.813°, and 19.424° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.813° ( ⁇ 0.2°) can have an X-ray powder diffraction pattern comprising at least one peak of
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 7.921°, 9.956°, 10.435°, 11.729°, and 12.704 in powder X-ray diffraction. °, 15.895°, and 16.643° ( ⁇ 0.2°).
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 15.895°, 16.643°, and 17.813° ( ⁇ 0.2°) It is more preferable to have a powder X-ray diffraction pattern containing a peak of
- Crystals of the hydrate of the compound represented by formula (1) have diffraction angles 2 ⁇ of 4.964°, 7.921°, 8.296°, 8.855°, and 9.964° in powder X-ray diffraction. 956°, 10.435°, 11.729°, 12.704°, 13.552°, 13.901°, 14.752°, 14.968°, 15.895°, 16.190°, 16. It is even more preferred to have an X-ray powder diffraction pattern comprising peaks at 643°, 17.813° and 19.424° ( ⁇ 0.2°).
- the analysis by powder X-ray diffraction of the present invention can be performed, for example, according to a conventional method such as the "powder X-ray diffraction measurement method" described in the Japanese Pharmacopoeia (15th revision). Further, according to the Japanese Pharmacopoeia, it is explained that the diffraction angles 2 ⁇ of the same crystal form usually match within the range of ⁇ 0.2°. Therefore, the present invention includes not only crystals in which the diffraction angles of peaks in X-ray powder diffraction match perfectly, but also crystals in which the diffraction angles of peaks match with an error of about ⁇ 0.2°.
- each component each component of the compound represented by formula (1), or a salt thereof, or a solvate thereof, a liquid additive, and optionally an oily component, and, if necessary, a pharmaceutical Ingredients that are usually used as excipients and the like are put into a known stirring and mixing device or the like and mixed.
- the mixing temperature and mixing time of each component are not particularly limited as long as they do not adversely affect the components.
- the mixing temperature is preferably 0 to 50° C., more preferably 10 to 30° C., and the mixing time is preferably about 5 minutes to 60 minutes.
- a method for producing a pharmaceutical formulation according to the present invention can include a step of providing the composition of the present invention, and a step of formulating the composition to provide a pharmaceutical formulation.
- the method for producing a pharmaceutical formulation according to the present invention can include the steps of providing the composition of the present invention and filling the composition into capsules to produce capsule formulations.
- the method for producing a pharmaceutical preparation according to the present invention can include the step of filling capsules with the composition obtained by the method for producing the composition to provide a capsule formulation. That is, it can include a step of filling capsules with the composition obtained by steps (1) to (3) in the method for producing the composition to provide a capsule formulation.
- the meaning of the term “and/or” includes any combination in which “and” and “or” are appropriately combined.
- “A, B, and/or C” includes the following seven variations: (i) A, (ii) B, (iii) C, (iv) A and B, (v) A and C, (vi) B and C, (vii) A, B, and C.
- the term "about” when used in combination with a numerical value means a range of +10% and -10% of the numerical value.
- to indicating a range includes values at both ends thereof, for example, "A to B” means a range of A or more and B or less.
- the unit of molecular weight in this specification is "g/mol". Molecular weight units may be omitted in this specification.
- Reagents such as liquid additives, surfactants, oleaginous components, solubilizers, antioxidants, or solvents used in the practice of this invention were obtained without purification from commercial suppliers, except as specifically noted. Using.
- the solubility of the compound represented by formula (1), or a salt thereof, or a solvate thereof is measured by adding an additive or formulation to the compound, followed by shaking or stirring. Dissolution was confirmed and calculated from the amount of liquid added. It is also possible to measure with an apparatus such as high performance liquid chromatography.
- Test Method 1 Particle Size Distribution Measurement by Dynamic Scattering (DLS) Particle size distribution is determined by dynamic scattering (DLS) using ZETASIZER Nano-ZS (Malvern). A sample is placed in a disposable cell (small volume cuvette), set in ZETASIZER Nano-ZS (Malvern), and particle size distribution is determined by dynamic scattering (DLS). The average particle size (Z-average size) and polydispersity index (PDI) are used as indicators of the particle size distribution. Z-average is the average particle diameter based on the scattering intensity using the cumulant method. PDI is an index representing the width of the particle size distribution, and is shown in the range of 0 to 1.
- dispersions with a PDI of 0.1 or less are monodisperse
- dispersions with a PDI between 0.1 and 0.5 are It is believed to have a narrow distribution.
- dispersions with a PDI greater than 0.5 are considered polydisperse.
- Test method 3 Calculation of pharmacokinetic parameters Plasma is separated from the collected blood by centrifugation, deproteinized with acetonitrile, and the concentration in plasma is measured using an LC-MS/MS apparatus. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were determined by non-compartmental analysis using pharmacokinetic analysis software Phoenix WinNonlin 8.2 (Certara L.P.). calculate.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Preparation example 1 Preparation of Compound 1 Compound 1 ("(5S,8S,11S,15S,18S,23aS,29S,35S,37aS)-8-((S)-sec-butyl)-18-cyclopentyl-29 -(3,5-difluoro-4-(trifluoromethyl)phenethyl)-36-ethyl-11-isobutyl-N,N,5,6,12,16,19,33-octamethyl-35-(4-methyl) benzyl)-4,7,10,13,17,20,23,28,31,34,37-undecaoxotetratriacontahydro-2H,4H-spiro[azeto[2,1-u]pyrrolo[2 ,1-i][1,4,7,10,13,16,19,22,25,28,31]undecaazacyclotetratriacontin-21,1′-cyclopentane
- Fmoc-Asp(OAl)-OH ((2S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-4-oxo-4-prop-2-enoxybutanoic acid, CAS No. 146982-24-3) ( 200 g, 506 mmol), p-toluenesulfonic acid (5.7 g, 0.05 equivalent) and paraformaldehyde (45.6 g, 3 equivalent) were mixed with toluene and stirred at 110° C. for 16 hours. The reaction mixture was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate and washed twice with an aqueous sodium hydrogencarbonate solution.
- the reaction mixture was diluted with ethyl acetate (422 mL), washed twice with hydrochloric acid (1 mol/L, 422 mL), and the resulting aqueous phase was extracted twice with ethyl acetate (422 mL). All the organic phases were mixed and washed in order with water (422 mL), saturated aqueous sodium bicarbonate/water mixture (1:1, 422 mL), and saturated brine/water mixture (1:1, 422 mL). The resulting organic phase was dried over sodium sulfate and evaporated under reduced pressure. DCM (512 mL) was added to the resulting residue and stirred for 0.5 hours.
- the solution was drained from the frit after 10 minutes of shaking.
- DMF (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- a solution of triethylamine hydrochloride (7.96 g, 57.8 mmol) in DCM (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- DCM (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit.
- DMF (420 mL) was added to the solid phase reaction vessel and shaken for 5 minutes before draining the solution through the frit. This DMF washing step of the resin was repeated one more time.
- Nickel bromide trihydrate (NiBr 2.3H 2 O) (13.5 g, 49.7 mmol, 0.3 eq) and 4,4′-di-tert-butyl-2,2′-bipyridyl (dtbbpy, CAS No. 72914-19-3) (13.3 g, 49.7 mmol, 0.3 equivalent) was added to DMA (400 mL) and stirred at 50° C. for 3 hours under a nitrogen atmosphere to prepare a Ni solution.
- TfOH trifluoromethanesulfonic acid
- Acetonitrile/water 400/400 mL was added to the residue and the pH was adjusted to 7 with aqueous sodium hydroxide (48%).
- Fmoc-OSu (36.6 g, 108.6 mmol, 0.9 equivalents) was added to this solution, the pH was adjusted to 8.0 with an aqueous sodium hydroxide solution (48%), and the mixture was stirred at room temperature for 16 hours.
- the reaction solution was filtered while washing with acetonitrile/water (1/1) to remove solid components, the filtrate was diluted with acetonitrile, and adjusted to be acidic with 6 mol/L aqueous hydrochloric acid.
- Multipep RSi peptide synthesizer
- the compound 1217-c-resin obtained above (200 mg per solid-phase reaction vessel) was added to 30 solid-phase reaction vessels and set in a peptide synthesizer.
- Dichloromethane (DCM) was added to all 30 solid-phase reactors and allowed to stand for 1 hour to swell the resin. The solvent was then drained from the frit.
- NMP solution of Fmoc-Hph(4-CF3-35-F2)-OH (0.45 mol/L) and HOAt (0.375 mol/L) (for one solid-phase reaction vessel 0.6 mL) and a DMF solution of N,N'-diisopropylcarbodiimide (DIC) (10 v/v%, 0.72 mL per solid phase reaction vessel) were mixed in a mixing vial of the synthesizer, and then 30 vials. It was added to all solid phase reactors and allowed to stand at 40° C. for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- DIC N,N'-diisopropylcarbodiimide
- NMP solution of Fmoc-MeGly-OH (0.6 mol/L) and HOAt (0.375 mol/L) (0.6 mL per solid-phase reaction vessel) and N,N'-diisopropylcarbodiimide (DIC) in DMF (10 v/v%, 0.72 mL per solid-phase reaction vessel) was mixed in the mixing vial of the synthesizer and then added to all 30 solid-phase reaction vessels. °C for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- NMP solution of Fmoc-MeAla-OH (0.6 mol/L) and HOAt (0.375 mol/L) (0.6 mL per solid-phase reaction vessel) and N,N'-diisopropylcarbodiimide (DIC) in DMF (10 v/v%, 0.72 mL per solid-phase reaction vessel) was mixed in the mixing vial of the synthesizer and then added to all 30 solid-phase reaction vessels. °C for 2.5 hours. The solution was then drained from the frit. DMF (1.4 mL per solid phase reactor) was added to all 30 solid phase reactors and the solvent was drained through the frit. This resin washing step was repeated two more times.
- a mixed solution of saturated aqueous ammonium chloride solution (40 mL) and water (40 mL) was added to the resulting solution, and the mixture was extracted with isopropyl acetate (350 mL).
- the obtained organic phase was washed with a mixed solution of saturated aqueous sodium hydrogencarbonate solution (40 mL) and water (40 mL) and a mixed solution of saturated brine (40 mL) and water (40 mL) in that order, dried over sodium sulfate, and dried under reduced pressure. Evaporation of solvent gave 3.36 g of residue.
- Preparation example 2 Preparation of seed crystals used in Preparation Example 3
- the amorphous compound 1 (122.3 mg) obtained in Preparation Example 1 was dissolved in DMSO (0.612 mL), and this solution (0.015 mL) was heated to -20°C. was lyophilized for 2 days at A water-acetonitrile mixture (3:1, 0.015 mL) was added to the obtained lyophilized product, and the mixture was shaken and stirred at room temperature for 7 days to obtain hydrate crystals of Compound 1 (Form C).
- the reaction mixture was filtered under pressure using a filter cloth (PF-020), and the inside of the reaction vessel and the filter were filtered using a filter (CCF-G100-E1N).
- Acetone (7.5 kg) and purified water ( 7.5 kg) of the mixed solution was used to wash the obtained crystals.
- the obtained crystals were washed with purified water (17.0 kg x 2) filtered using a filter (CCF-G100-E1N), the pressure of the filtration device that collected the crystals was reduced, and the external temperature of the filtration device was set to 70°C. and dried the crystals for 17 hours. Further, the crystals were dried at an external temperature of room temperature to 30° C. for 27 hours.
- the dry powder was recovered from the filter to obtain a white powder (2.6 kg).
- the resulting white powder was confirmed to have the same structure as the compound obtained in Preparation Example 1, "Synthesis of compound 1 (cyclization and purification of peptide)".
- HPLC analysis conditions Equipment: Waters ACQUITY UPLC H-Class Column: ACQUITY UPLC CSH C18 (Waters), 2.1 mm ID x 150 mm, 1.7 ⁇ m
- Mobile phase 0.05% TFA/water (A), 0.05% TFA/MeCN (B)
- Elution method B) 20% (0 min) ⁇ 100% (24 min) ⁇ 100% (29 min) ⁇ 20% (29.1 min) ⁇ 20% (34 min)
- Flow rate 0.3 mL/min
- Powder X-ray analysis was performed using the XRPD equipment shown below.
- XRPD measurement condition measurement device X'pert-pro MPD (manufactured by PANalytical) Radiation source: CuK ⁇ Tube voltage: 45 kV Tube current: 40mA Scanning range: 3-40° Scanning speed: 4.2°/min Sampling width: 0.017°
- the water content of the hydrate crystals of Compound 1 (Form C) obtained in the same manner as in Preparation Example 3 was measured by Karl Fischer titration. Measurement was performed using CA-310 (manufactured by Nitto Seiko Analytic Tech) after acclimating the sample in a laboratory environment. As a result of the measurement, the water content of the hydrate crystals of Compound 1 (Form C) was 6.50 wt%.
- the hydrate crystal of compound 1 (Form C) is a hydrate crystal that has water molecules in its crystal structure. was confirmed.
- a hydrate crystal of compound 1 (Form C) obtained in the same manner as in Preparation Example 3 was subjected to dynamic water vapor adsorption measurement.
- the results are shown in FIG. Measurement device: DVS Intrinsic (manufactured by Surface Measurement Systems) Temperature: 25°C Relative humidity (%) measurement points: Cycle 1: 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%); Cycle 2: 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%); Cycle 2: 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 0 (%) Threshold: 0.001 dm/dt (%/min) Minimum sorption time: 10 minutes Maximum sorption time: 1440 minutes
- the hydrate crystal of Compound 1 is a hydrate crystal that changes in weight by 3.3% as the hydration number changes in the relative humidity range of 0 to 95%. rice field.
- Compound I The hydrate crystals of compound 1 (Form C) obtained in Preparation Example 3 (hereinafter sometimes referred to as “compound I”) were used for the preparation and evaluation of the following compositions or formulations.
- a compound used as an active ingredient in Examples is described as “compound I”
- compound I dissolved in a liquid additive or the like means compound 1 in the free form.
- Test Example 1 Evaluation of solubility of compound I in various additives The solubility of compound I in various additives was determined. Various liquid additives were added to Compound I and stirred, and when Compound I did not dissolve, further additives were added and stirred. This operation was repeated until compound I dissolved, and the approximate solubility was obtained from the amount of additive required to dissolve compound I. Since Polyoxyethylene (15) hydroxystearate (Solutol HS15) is solid at room temperature (RT), it was liquefied by heating to 37° C. to determine its solubility. Solubility of other additives was determined at room temperature. Table 2 shows the results. As a result, PG monocaprylate was found to exhibit high solubility in compound I.
- Test Example 2 Evaluation of solubility of compound I in each formulation
- the solubility of compound I in each of additive mixtures 1 to 4 was measured. Assuming the migration of water from the capsule shell when formulated as a capsule, the solubility of an additive mixture containing 3% water was also measured. As a result, as shown in Table 4, it was found that additive mixtures 1 to 4 were capable of dissolving compound I at a high concentration of 100 mg/mL or more.
- the solubility of compound I decreased to 60 to 70 mg / mL. decided to evaluate.
- additive mixtures 5 to 45 were prepared based on the formulations in Tables 5 to 10.
- Formulations A to V which are compositions of the present invention, were prepared by the following method.
- Example 1 (Preparation of Formulation A) Compound I was weighed into a vial and 1 mL of Additive Mixture 1 was added per 50 mg of Compound I. Formulation A was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 2 (Preparation of Formulation B) Compound I was weighed into a vial and 1 mL of Additive Mixture 2 was added per 50 mg of Compound I. Formulation B was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 3 (Preparation of Formulation C) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 50 mg of Compound I. Formulation C was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 4 (Preparation of Formulation D) Compound I was weighed into a vial and 1 mL of Additive Mixture 4 was added per 50 mg of Compound I. Formulation D was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 5 (Preparation of Formulation E) Compound I was weighed into a vial and 1 mL of Additive Mixture 7 was added per 50 mg of Compound I. Formulation E was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 6 (Preparation of Formulation F) Compound I was weighed into a vial and 1 mL of Additive Mixture 19 was added per 50 mg of Compound I. Formulation F was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 7 (Preparation of Formulation G) Compound I was weighed into a vial and 1 mL of Additive Mixture 20 was added per 50 mg of Compound I. Formulation G was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 8 (Preparation of Formulation H) Compound I was weighed into a vial and 1 mL of Additive Mixture 21 was added per 50 mg of Compound I. Formulation H was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 9 (Preparation of Formulation I) Compound I was weighed into a vial and 1 mL of Additive Mixture 25 was added per 50 mg of Compound I. Formulation I was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 10 (Preparation of Formulation J) Compound I was weighed into a vial and 1 mL of Additive Mixture 26 was added per 50 mg of Compound I. Formulation J was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 11 (Preparation of Formulation K) Compound I was weighed into a vial and 1 mL of Additive Mixture 28 was added per 50 mg of Compound I. Formulation K was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 12 (Preparation of Formulation L) Compound I was weighed into a vial and 1 mL of Additive Mixture 29 was added per 50 mg of Compound I. Formulation L was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 13 (Preparation of Formulation M) Compound I was weighed into a vial and 1 mL of Additive Mixture 30 per 50 mg of Compound I was added. Formulation M was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 14 (Preparation of Formulation N) Compound I was weighed into a vial and 1 mL of Additive Mixture 32 was added per 50 mg of Compound I. Formulation N was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 15 (Preparation of Formulation O) Compound I was weighed into a vial and 1 mL of Additive Mixture 33 was added per 50 mg of Compound I. Formulation O was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 16 (Preparation of Formulation P) Compound I was weighed into a vial and 1 mL of Additive Mixture 34 was added per 50 mg of Compound I. Formulation P was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 17 (Preparation of Formulation Q) Compound I was weighed into a vial and 1 mL of Additive Mixture 35 was added per 50 mg of Compound I. Formulation Q was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 18 (Preparation of Formulation R) Compound I was weighed into a vial and 1 mL of Additive Mixture 40 was added per 50 mg of Compound I. Formulation R was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 19 (Preparation of Formulation S) Compound I was weighed into a vial and 1 mL of Additive Mixture 41 was added per 50 mg of Compound I. Preparation S was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 20 (Preparation of Formulation T) Compound I was weighed into a vial and 1 mL of Additive Mixture 42 was added per 50 mg of Compound I. Preparation T was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Example 21 (Preparation of Formulation U) Compound I was weighed into a vial and 1 mL of Additive Mixture 44 was added per 50 mg of Compound I. Formulation U was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Example 22 (Preparation of Formulation V) Compound I was weighed into a vial and 1 mL of Additive Mixture 45 was added per 50 mg of Compound I. Formulation V was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 1 (Preparation of Formulation W) Compound I was weighed into a vial and 1 mL of Additive Mixture 8 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation W was obtained.
- Comparative Example 2 (Preparation of Formulation X) Compound I was weighed into a vial and 1 mL of Additive Mixture 9 was added per 50 mg of Compound I. Preparation X was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 3 (Preparation of Formulation Y) Compound I was weighed into a vial and 1 mL of Additive Mixture 10 was added per 50 mg of Compound I. Formulation Y was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 4 (Preparation of Formulation Z) Compound I was weighed into a vial and 1 mL of Additive Mixture 13 was added per 50 mg of Compound I. Formulation Z was obtained after confirming that compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 5 (Preparation of Formulation AA) Compound I was weighed into a vial and 1 mL of Additive Mixture 14 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AA was obtained.
- Comparative Example 6 (Preparation of Formulation AB) Compound I was weighed into a vial and 1 mL of Additive Mixture 15 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AB was obtained.
- Comparative Example 7 (Preparation of Formulation AC) Compound I was weighed into a vial and 1 mL of Additive Mixture 16 was added per 50 mg of Compound I. Formulation AC was obtained after confirming complete dissolution of compound I by stirring using a stirrer.
- Comparative Example 8 (Preparation of Formulation AD) Compound I was weighed into a vial and 1 mL of Additive Mixture 17 was added per 50 mg of Compound I. Formulation AD was obtained after confirming complete dissolution of Compound I by stirring using a stirrer.
- Comparative Example 9 (Preparation of Formulation AE) Compound I was weighed into a vial and 1 mL of Additive Mixture 18 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AE was obtained.
- Comparative Example 10 (Preparation of Formulation AF) Compound I was weighed into a vial and 1 mL of Additive Mixture 22 was added per 50 mg of Compound I. Formulation AF was obtained after confirming complete dissolution of Compound I by stirring using a stirrer.
- Comparative Example 11 (Preparation of Formulation AG) Compound I was weighed into a vial and 1 mL of Additive Mixture 23 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring with a stirrer, Formulation AG was obtained.
- Comparative Example 12 (Preparation of Formulation AH) Compound I was weighed into a vial and 1 mL of Additive Mixture 24 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AH was obtained.
- Comparative Example 13 (Preparation of Formulation AI) Compound I was weighed into a vial and 1 mL of Additive Mixture 27 was added per 50 mg of Compound I. Formulation AI was obtained after confirming that Compound I was completely dissolved by stirring using a stirrer.
- Comparative Example 14 (Preparation of Formulation AJ) Compound I was weighed into a vial and 1 mL of Additive Mixture 31 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AJ was obtained.
- Comparative Example 15 (Preparation of Formulation AK) Compound I was weighed into a vial and 1 mL of Additive Mixture 36 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AK was obtained.
- Comparative Example 16 (Preparation of Formulation AL) Compound I was weighed into a vial and 1 mL of Additive Mixture 37 was added per 50 mg of Compound I. After confirming that compound I was completely dissolved by stirring using a stirrer, preparation AL was obtained.
- Comparative Example 17 (Preparation of Formulation AM) Compound I was weighed into a vial and 1 mL of Additive Mixture 38 was added per 50 mg of Compound I. After confirming complete dissolution of Compound I by stirring with a stirrer, Formulation AM was obtained.
- Comparative Example 18 (Preparation of Formulation AN) Compound I was weighed into a vial and 1 mL of Additive Mixture 39 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AN was obtained.
- Comparative Example 19 (Preparation of Formulation AO) Compound I was weighed into a vial and 1 mL of Additive Mixture 43 was added per 50 mg of Compound I. After confirming that Compound I was completely dissolved by stirring using a stirrer, Formulation AO was obtained.
- Test Example 3 (particle size distribution measurement by DLS) Water was then added to Formulations A through V prepared in Examples 1-22 and Formulations W through Formulations AO prepared in Comparative Examples 1-19 to form dispersions.
- the particle size of droplets generated in the dispersion was measured to evaluate the particle size distribution at room temperature (approximately 25°C). 4 ⁇ L of each formulation was placed in a vial, and 396 ⁇ L of water was added.
- the average particle size (Z-average size) and polydispersity index (PDI) were used as indices representing the particle size distribution.
- PDI is an index representing the width of the particle size distribution and is shown in the range of 0 to 1.
- dispersions with a PDI of 0.1 or less are monodisperse
- dispersions with a PDI between 0.1 and 0.5 are It is believed to have a narrow distribution.
- dispersions with a PDI greater than 0.5 are considered polydisperse.
- Table 16 all of Formulations A to V were good dispersions with a small average particle size (less than 200 nm) and a narrow distribution of PDI of less than 0.5.
- formulations W to AO had a large average particle size (200 nm or more) and were polydisperse with a PDI exceeding 0.5.
- Test Example 4 80°C stability test
- 10 mg each of formulations A, C, F, H, and J prepared in Examples 1, 3, 6, 8, and 10 were placed in a glass vial, placed in a brown bottle containing silica gel and an oxygen scavenger, and sealed. Each brown bottle was placed in a constant temperature bath at 80° C. and stored for 2 weeks.
- compound I showed a high survival rate of 95% or more in all formulations, indicating that it is a formulation that can be developed as a drug.
- compound I showed a high survival rate of 95% or more in any formulation, and it was found to be a formulation that can be developed as a drug.
- Formulation C dispersed immediately after the start of the test.
- Formulation F also showed a degree of dispersion of more than 80% after 60 minutes, and was found to be a good formulation compared to formulation AJ.
- Example 23 (Preparation of Capsule Formulation AP for Rat Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. The solution in which the compound was dissolved was filled into No. 9 gelatin capsules to produce Formulation AP.
- Example 24 (Preparation of Capsule Formulation AQ for Rat Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 19 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. The solution in which the compound was dissolved was filled into No. 9 gelatin capsules to produce Formulation AQ.
- Comparative Example 20 (Preparation of capsule formulation AR for rat administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 31 was added per 60 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer. Formulation AR was prepared by filling a No. 9 gelatin capsule with the solution in which the compound was dissolved.
- Comparative Example 21 (Preparation of solution preparation AS for rat administration) Compound I was weighed into a vial and dissolved by adding 1 mL of dimethylsulfoxide (DMSO) per 10 mg of compound I. Furthermore, 0.5 mL of polyoxyl 35 castor oil and 8.5 mL of water were added and well stirred and mixed to prepare a solution formulation AS.
- DMSO dimethylsulfoxide
- Test Example 7 (rat absorption evaluation) Capsule formulations AP and AQ of Examples 23 and 24 and capsule formulations AR and solution formulations AS of Comparative Examples 20 and 21 were orally administered to rats (Wister Hannover, male) at a dose of 10 mg/kg, four of each formulation.
- Formulations AP, AQ and AR were administered by oral gavage as capsules as they were, and by oral gavage with water at a volume of 5 mL/kg.
- Solution formulation AS was administered by oral gavage as is. About 0.5, 1, 2, 4, 7, 24 and 48 hours after administration, blood is collected, and the concentration of compound I contained in the plasma is quantified by LC-MS/MS by the method shown in test method 3. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were calculated.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Table 20 shows the AUC and Cmax after administration of each formulation.
- Capsule formulations AP and AQ of Examples 23, 24 were found to exhibit higher AUC and Cmax than Capsule formulation AR of Comparative Example 20.
- the capsule formulation AP of Example 23 exhibited comparable AUC and Cmax compared to the solution formulation AS of Comparative Example 21.
- capsule formulation AT and solution formulation AU were prepared to evaluate absorption in monkeys.
- Example 25 (Preparation of Capsule Formulation AT for Monkey Administration) Compound I was weighed into a vial and 1 mL of Additive Mixture 3 was added per 50 mg of Compound I. It was confirmed that Compound I was completely dissolved by stirring using a stirrer.
- Formulation AT was prepared by filling a No. 1 gelatin capsule with the solution in which the compound was dissolved.
- Comparative Example 22 (Preparation of solution formulation AU for monkey administration) Compound I was weighed into a vial and dissolved by adding 1 mL of dimethylsulfoxide (DMSO) per 15 mg of compound I. Further, 0.25 mL of polyoxyl 35 castor oil and 8.75 mL of water were added and well stirred and mixed to prepare a solution formulation AU.
- DMSO dimethylsulfoxide
- Test Example 8 (monkey absorbability evaluation) The capsule formulation AT of Example 25 and the solution formulation AU of Comparative Example 22 were orally administered to cynomolgus monkeys at a dose of 3 mg/kg, four of each formulation. Capsule formulation AT was administered by gavage as is, and water was additionally administered by gavage at a volume of 4 mL/kg. The solution formulation AU was administered by oral gavage as is. About 0.5, 1, 2, 4, 7, 24 and 48 hours after administration, blood is collected, and the concentration of compound I contained in the plasma is quantified by LC-MS/MS by the method shown in test method 3. The area under the plasma drug concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) were calculated.
- AUC area under the plasma drug concentration vs. time curve
- Cmax maximum plasma concentration
- Table 21 shows the AUC and Cmax after administration of each formulation. Capsule formulation AT of Example 25 was found to exhibit similar AUC and Cmax as compared to solution formulation AU of Comparative Example 22.
- Example 26 (Production of Capsules (Hard Capsules) 1) Weigh compound I in a stainless steel container, add 51.5 mg of Propylene glycol monocaprylate, 13.3 mg of Oleic acid, 31.1 mg of Polyoxyl 35 castor oil and 0.2 mg of dl- ⁇ -tocopherol per 0.5 mg of compound I, and homogenizer was used to stir and mix. The solution in which the compound was dissolved was filled into No. 3 gelatin capsules and band-sealed to produce Formulation AV.
- Example 27 (Production of Capsules (Hard Capsules) 2) Weigh compound I in a stainless steel container, add 123.6 mg of Propylene glycol monocaprylate, 31.9 mg of Oleic acid, 74.6 mg of Polyoxyl 35 castor oil and 0.5 mg of dl- ⁇ -tocopherol per 12 mg of compound I, Stirred and mixed using a homogenizer. The solution in which the compound was dissolved was filled into No. 3 gelatin capsules and band-sealed to prepare Formulation AW.
- Example 28 (Production of Capsules (Soft Capsules) 3) Compound I was weighed into a stainless steel container, and 206.4 mg of Propylene glycol monocaprylate, 53.3 mg of Oleic acid and 124.6 mg of Polyoxyl 35 castor oil and 0.8 mg of dl- ⁇ -tocopherol were added per 19.1 mg of Compound I. In addition, they were stirred and mixed using a stirring blade. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AX.
- Example 29 (Production of Capsules (Soft Capsules) 4) Weigh Compound I in a stainless steel container, add 46.4 mg of Propylene glycol monocaprylate, 12.0 mg of Oleic acid and 28.0 mg of Polyoxyl 35 castor oil per 4.5 mg of Compound I, and stir and mix using a stirring blade. did. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AY.
- Example 30 (Production of Capsules (Soft Capsules) 5) Weigh compound I in a stainless steel container, add 61.8 mg of Propylene glycol monocaprylate, 16.0 mg of Oleic acid and 37.3 mg of Polyoxyl 35 castor oil per 6.0 mg of compound I, stir and mix using a stirring blade did. The solution in which the compound was dissolved was filled into soft gelatin capsules to produce Formulation AZ.
- Test Example 9 particle size distribution measurement by DLS
- the particle size distribution of Formulation AV, Formulation AW, Formulation AX, Formulation AY, and Formulation AZ prepared in Examples 26-30 was evaluated when dispersed in water.
- the particle size distribution was determined by dynamic scattering (DLS) using the method shown in Test Method 1. Similar to Test Example 3, the average particle size (Z-average size) and the polydispersity index (PDI) were used as indices representing the particle size distribution.
- DLS dynamic scattering
- Formulation AV, Formulation AW, Formulation AX, Formulation AY, and Formulation AZ all had small average particle sizes (less than 200 nm) and good dispersion showing narrow distributions with a PDI of less than 0.5. was the body.
- Test Example 10 Protein-protein interaction inhibitory activity of Kras and SOS1 using AlphaScreen (KrasG12D-SOS PPI) Protein-protein interaction inhibition (PPI) of Kras and SOS1 can be performed by methods known to those skilled in the art using equipment, reagents, etc. available from commercial suppliers. . Specifically, biotin-tagged human Kras and His-tagged human SOS1 enzymes expressed in E. coli and loaded with GDP after purification were used to transfer nickel-bound donor beads to streptavidin-bound acceptor beads. Measured by energy transfer.
- This measurement utilizes the fact that when the donor beads are irradiated with light of 680 nm, energy is transferred to the acceptor beads via singlet oxygen, and light of 520 to 620 nm is detected from the acceptor beads. .
- a 50% inhibitory concentration (IC50 value) was calculated from the inhibition rate against the control group containing no Compound 1.
- the IC50 value for Compound 1 was 0.0010 ⁇ M.
- Compound 1 was shown to inhibit the protein-protein interaction between Kras and SOS1. Inhibition of binding between K-ras and SOS1 is known to inhibit signal transduction downstream of Kras. Therefore, it was suggested that Compound 1 also has cell proliferation inhibitory activity.
- AsPC-1 cell growth inhibitory activity can be performed by methods known to those skilled in the art using equipment, reagents, cell lines, etc. available from commercial suppliers. Specifically, after serially diluting Compound 1 with dimethylsulfoxide, 200 nL was dispensed into a U-bottom 96-well plate using Labcyte Echo. For the human pancreatic cancer strain AsPC-1, a cell suspension was prepared in RPMI-1640 medium supplemented with 15% fetal bovine serum to give a cell suspension of 1000 cells/100 ⁇ L.
- CGI cell growth inhibition activity
- composition of the present invention can be made excellent in various properties required as a formulation.
- compositions containing such specific ingredients have desirable particle characteristics, good stability and dispersibility of the active ingredient, and good absorption by the body. Therefore, the composition according to the present invention is suitable for use as a pharmaceutical preparation.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22798954.8A EP4299069A4 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| US18/289,392 US20240366711A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| KR1020237032063A KR20230169093A (ko) | 2021-05-07 | 2022-05-06 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| JP2022548967A JP7181442B1 (ja) | 2021-05-07 | 2022-05-06 | 環状ペプチド化合物を含む製剤及びその製造方法 |
| KR1020237001877A KR102582225B1 (ko) | 2021-05-07 | 2022-05-06 | 환상 펩타이드 화합물을 포함하는 제제 및 그의 제조 방법 |
| BR112023019622A BR112023019622A2 (pt) | 2021-05-07 | 2022-05-06 | Preparação contendo composto de peptídeo cíclico e método para a produção do mesmo |
| AU2022270498A AU2022270498A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| IL306018A IL306018A (en) | 2021-05-07 | 2022-05-06 | A preparation containing a cyclic peptide compound and a method for its production |
| CA3219090A CA3219090A1 (en) | 2021-05-07 | 2022-05-06 | Preparation containing cyclic peptide compound and method for producing same |
| MX2023013099A MX2023013099A (es) | 2021-05-07 | 2022-05-06 | Preparacion que contiene un compuesto de peptido ciclico y metodo para su produccion. |
| CN202280029838.6A CN117241817A (zh) | 2021-05-07 | 2022-05-06 | 包含环状肽化合物的制剂及其制备方法 |
| JP2022184220A JP2023015334A (ja) | 2021-05-07 | 2022-11-17 | 環状ペプチド化合物を含む製剤及びその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-079015 | 2021-05-07 | ||
| JP2021079015 | 2021-05-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022234850A1 true WO2022234850A1 (ja) | 2022-11-10 |
Family
ID=83932759
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/019540 Ceased WO2022234850A1 (ja) | 2021-05-07 | 2022-05-06 | 環状ペプチド化合物を含む製剤及びその製造方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20240366711A1 (enExample) |
| EP (1) | EP4299069A4 (enExample) |
| JP (2) | JP7181442B1 (enExample) |
| KR (2) | KR20230169093A (enExample) |
| CN (1) | CN117241817A (enExample) |
| AU (1) | AU2022270498A1 (enExample) |
| BR (1) | BR112023019622A2 (enExample) |
| CA (1) | CA3219090A1 (enExample) |
| IL (1) | IL306018A (enExample) |
| MX (1) | MX2023013099A (enExample) |
| TW (1) | TW202308679A (enExample) |
| WO (1) | WO2022234850A1 (enExample) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024085235A1 (ja) * | 2022-10-20 | 2024-04-25 | 中外製薬株式会社 | 環状ペプチドの結晶の製造方法 |
| WO2024219480A1 (ja) * | 2023-04-20 | 2024-10-24 | 中外製薬株式会社 | 環状ペプチド化合物の製造方法 |
| US12312379B2 (en) | 2021-05-07 | 2025-05-27 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing cyclic compounds comprising N-substituted amino acid residues |
| US12371454B2 (en) | 2019-11-07 | 2025-07-29 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide compound having Kras inhibitory action |
| US12410212B2 (en) | 2022-05-06 | 2025-09-09 | Chugai Seiyaku Kabushiki Kaisha | Cyclic compound having selective KRAS inhibitory effect on HRAS and NRAS |
| US12415835B2 (en) | 2011-12-28 | 2025-09-16 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122059A1 (en) | 2011-03-04 | 2012-09-13 | New York University | Hydrogen bond surrogate macrocycles as modulators of ras |
| WO2013100132A1 (ja) | 2011-12-28 | 2013-07-04 | 中外製薬株式会社 | ペプチド化合物の環化方法 |
| WO2016071515A1 (en) | 2014-11-07 | 2016-05-12 | Sigmoid Pharma Limited | Compositions comprising cyclosporin |
| WO2017181061A1 (en) | 2016-04-15 | 2017-10-19 | Ra Pharmaceuticals, Inc. | Ras binding peptides and methods of use |
| WO2018031730A2 (en) | 2016-08-11 | 2018-02-15 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Peptide inhibitors of phosphoglycerate mutase and methods of use |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130034597A1 (en) * | 2011-02-04 | 2013-02-07 | Aegis Therapeutics Llc | Orally bioavailable peptide drug compositions and methods thereof |
| AR125800A1 (es) * | 2021-05-07 | 2023-08-16 | Chugai Pharmaceutical Co Ltd | Métodos para producir compuestos cíclicos que comprendan residuos de aminoácidos sustituidos con n |
-
2022
- 2022-05-06 US US18/289,392 patent/US20240366711A1/en active Pending
- 2022-05-06 IL IL306018A patent/IL306018A/en unknown
- 2022-05-06 CA CA3219090A patent/CA3219090A1/en active Pending
- 2022-05-06 WO PCT/JP2022/019540 patent/WO2022234850A1/ja not_active Ceased
- 2022-05-06 KR KR1020237032063A patent/KR20230169093A/ko active Pending
- 2022-05-06 TW TW111117094A patent/TW202308679A/zh unknown
- 2022-05-06 EP EP22798954.8A patent/EP4299069A4/en active Pending
- 2022-05-06 MX MX2023013099A patent/MX2023013099A/es unknown
- 2022-05-06 JP JP2022548967A patent/JP7181442B1/ja active Active
- 2022-05-06 CN CN202280029838.6A patent/CN117241817A/zh active Pending
- 2022-05-06 KR KR1020237001877A patent/KR102582225B1/ko active Active
- 2022-05-06 AU AU2022270498A patent/AU2022270498A1/en active Pending
- 2022-05-06 BR BR112023019622A patent/BR112023019622A2/pt unknown
- 2022-11-17 JP JP2022184220A patent/JP2023015334A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012122059A1 (en) | 2011-03-04 | 2012-09-13 | New York University | Hydrogen bond surrogate macrocycles as modulators of ras |
| WO2013100132A1 (ja) | 2011-12-28 | 2013-07-04 | 中外製薬株式会社 | ペプチド化合物の環化方法 |
| WO2016071515A1 (en) | 2014-11-07 | 2016-05-12 | Sigmoid Pharma Limited | Compositions comprising cyclosporin |
| WO2017181061A1 (en) | 2016-04-15 | 2017-10-19 | Ra Pharmaceuticals, Inc. | Ras binding peptides and methods of use |
| WO2018031730A2 (en) | 2016-08-11 | 2018-02-15 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Peptide inhibitors of phosphoglycerate mutase and methods of use |
| WO2018225864A1 (ja) | 2017-06-09 | 2018-12-13 | 中外製薬株式会社 | 膜透過性の高い環状ペプチド化合物、及びこれを含むライブラリ |
| WO2020122182A1 (ja) | 2018-12-12 | 2020-06-18 | 中外製薬株式会社 | 分子内水素結合可能な官能基を有するアミノ酸とそれらのアミノ酸を含むペプチド化合物、およびそれらの製造方法 |
| WO2021090855A1 (ja) * | 2019-11-07 | 2021-05-14 | 中外製薬株式会社 | Kras阻害作用を有する環状ペプチド化合物 |
Non-Patent Citations (11)
| Title |
|---|
| "Japanese Pharmacopeia", article "Powder X-ray Diffractometry" |
| ACC. CHEM. RES, vol. 41, 2008, pages 1331 - 1342 |
| ADV. DRUG DEL. REV, vol. 23, 1997, pages 3 - 25 |
| ANGEW. CHEM. INT. ED., vol. 52, 2013, pages 254 - 269 |
| CAS , no. 136552-06-2 |
| CAS, no. 330645-87-9 |
| CAS, no. 72914-19-3 |
| CHEM. REV., vol. 119, 2019, pages 10360 - 10391 |
| FUTURE MED. CHEM, vol. 1, 2009, pages 1289 - 1310 |
| KENTARO FURUMOTO, RYUSUKE TAKANO, SATOSHI TANIDA, TAKAMITSU UEDO, KOJI SHIRAKI, NORIYUKI TAKADA, SHOICHI HIGO: "P-5 Establishment of the high-throughput additive screening method for the purpose of improving solubility of the insoluble compound", JOURNAL OF PHARMACEUTICAL SCIENCE AND TECHNOLOGY, JAPAN, vol. 70, no. Suppl., 1 January 2010 (2010-01-01), JP , pages 192, XP009540883, ISSN: 0372-7629 * |
| THER. DELIV, vol. 8, no. 10, 2017, pages 867 - 878 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12415835B2 (en) | 2011-12-28 | 2025-09-16 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| US12371454B2 (en) | 2019-11-07 | 2025-07-29 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide compound having Kras inhibitory action |
| US12312379B2 (en) | 2021-05-07 | 2025-05-27 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing cyclic compounds comprising N-substituted amino acid residues |
| US12410212B2 (en) | 2022-05-06 | 2025-09-09 | Chugai Seiyaku Kabushiki Kaisha | Cyclic compound having selective KRAS inhibitory effect on HRAS and NRAS |
| WO2024085235A1 (ja) * | 2022-10-20 | 2024-04-25 | 中外製薬株式会社 | 環状ペプチドの結晶の製造方法 |
| WO2024219480A1 (ja) * | 2023-04-20 | 2024-10-24 | 中外製薬株式会社 | 環状ペプチド化合物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7181442B1 (ja) | 2022-11-30 |
| CN117241817A (zh) | 2023-12-15 |
| TW202308679A (zh) | 2023-03-01 |
| MX2023013099A (es) | 2023-11-17 |
| EP4299069A4 (en) | 2025-03-05 |
| EP4299069A1 (en) | 2024-01-03 |
| US20240366711A1 (en) | 2024-11-07 |
| KR20230020548A (ko) | 2023-02-10 |
| KR20230169093A (ko) | 2023-12-15 |
| JP2023015334A (ja) | 2023-01-31 |
| AU2022270498A1 (en) | 2023-10-05 |
| JPWO2022234850A1 (enExample) | 2022-11-10 |
| IL306018A (en) | 2023-11-01 |
| CA3219090A1 (en) | 2022-11-10 |
| BR112023019622A2 (pt) | 2023-11-14 |
| KR102582225B1 (ko) | 2023-09-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7181442B1 (ja) | 環状ペプチド化合物を含む製剤及びその製造方法 | |
| EP2310363B1 (en) | Anticancer oral formulation | |
| TWI696621B (zh) | 原肌球蛋白相關激酶(trk)抑制劑之醫藥調配物 | |
| RS67259B1 (sr) | Kompozicije koje aktiviraju piruvat kinazu r (pkr) | |
| US10370406B2 (en) | Salts and polymorphs of SCY-078 | |
| EA027869B1 (ru) | Стабилизированная композиция такролимуса | |
| WO2022121670A1 (zh) | Tolebrutinib的晶型及其制备方法和用途 | |
| EP3996714A1 (en) | Formulations of rbp4 inhibitors and methods of use | |
| CN110325536A (zh) | Janus激酶抑制剂的晶体形式 | |
| WO2015054793A1 (en) | Salt and crystal forms of plk-4 inhibitor | |
| RU2627702C2 (ru) | Кристаллические формы 1-(3-трет-бутил-1-п-толил-1н-пиразол-5-ил)-3-(5-фтор-2-(1-(2-гидроксиэтил)-1н-индазол-5-илокси)бензил) мочевины гидрохлорида | |
| EA049912B1 (ru) | Препарат, содержащий циклическое пептидное соединение, и способы его приготовления | |
| WO2025040810A1 (en) | Therapeutic compounds, a method of manufacturing thereof as well as uses thereof | |
| KR20210009153A (ko) | 자가나노유화 약물전달시스템을 이용한 타다라필의 경구용 고형제 조성물 및 이의 제조방법 | |
| CN113372332B (zh) | 一种奥希替尼新晶型 | |
| US20250243176A1 (en) | (s)-1-(1-acryloylpiperidin-3-yl)-2-fluoro-5,6,7,8,9,10-hexahydrocyclo hepta[b]indole-4-carboxamide, and related crystalline forms, compositions, and methods thereof | |
| CN103169657A (zh) | 一种含灯盏花素磷脂复合物的自乳化剂及其制备方法和用途 | |
| WO2025235577A1 (en) | Forms and formulations of substituted 3-((3-amino)piperidine-2,6-dione compounds | |
| WO2019137027A1 (zh) | Galunisertib的晶型及其制备方法和用途 | |
| HK40079381A (en) | Novel salts and polymorphs of scy-078 | |
| WO2019105217A1 (zh) | Galunisertib的晶型及其制备方法和用途 | |
| Hao et al. | Sustained Dissolution and Pharmacokinetics of Risperidone Microsuspensions Via Cocrystallization with Kaempferol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022548967 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22798954 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20237001877 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 306018 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022270498 Country of ref document: AU Ref document number: AU2022270498 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022798954 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112023019622 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2022270498 Country of ref document: AU Date of ref document: 20220506 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2022798954 Country of ref document: EP Effective date: 20230929 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202392566 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280029838.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3219090 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2023/013099 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 112023019622 Country of ref document: BR Kind code of ref document: A2 Effective date: 20230925 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |