WO2020256045A1 - ヘキサメチル置換/ジメチル置換4,4'-ビス(2-プロペン-1-イルオキシ)-1,1'-ビフェニルの結晶体 - Google Patents
ヘキサメチル置換/ジメチル置換4,4'-ビス(2-プロペン-1-イルオキシ)-1,1'-ビフェニルの結晶体 Download PDFInfo
- Publication number
- WO2020256045A1 WO2020256045A1 PCT/JP2020/023893 JP2020023893W WO2020256045A1 WO 2020256045 A1 WO2020256045 A1 WO 2020256045A1 JP 2020023893 W JP2020023893 W JP 2020023893W WO 2020256045 A1 WO2020256045 A1 WO 2020256045A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- crystal
- temperature
- crystals
- yloxy
- Prior art date
Links
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/215—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring having unsaturation outside the six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F16/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical
- C08F16/12—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical by an ether radical
- C08F16/32—Monomers containing two or more unsaturated aliphatic radicals
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
Definitions
- the present invention relates to hexamethyl-substituted / dimethyl-substituted 4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl crystals.
- a cross-linking agent having an allyl group is widely used.
- Triallyl isocyanurate which is a general-purpose curing agent for modified polyphenylene oxide, has a problem that it is a liquid at room temperature (25 ° C.), easily volatilizes with a solvent at the prepreg formation stage, and has low heat resistance.
- Patent Documents 1 and 2 below describe diallyl ether compounds such as biphenol, 4,4'-dihydroxy-3,3', 5,5'-tetramethylbiphenyl as allyl ether compounds having a biphenol skeleton.
- the present invention has been made against the background of the above-mentioned circumstances, and is a crystal at room temperature (25 ° C.), has better heat resistance and storage stability, and is volatile in the prepreg molding process.
- An object of the present invention is to provide new allyl ethers having a suppressed biphenol skeleton.
- Compound A 2,2', 3,3', 5,5'-hexamethyl-4,4'-bis (2-propene-1-yloxy) -1,1', which is one of allyl ethers having a biphenol skeleton.
- Compound B 3,3'-dimethyl-4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl
- the present invention is as follows. 1.
- a crystal of a compound represented by the following formula (1) (In the formula, R represents a hydrogen atom or a methyl group. However, all four Rs are the same.) 2.
- the compound represented by the formula (1) is 2,2', 3,3', 5,5'-hexamethyl-4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl. Is 1.
- 3. The endothermic peak top temperature by differential scanning calorimetry is in the range of 70 to 75 ° C.
- the compound represented by the formula (1) is 3,3'-dimethyl-4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl.
- the crystal according to. 5. 3.
- the endothermic peak top temperature by differential scanning calorimetry is in the range of 110 to 115 ° C.
- Crystals of "Compound A"(2,2',3,3',5,5'-hexamethyl-4,4'-bis (2-propen-1-yloxy) -1,1'-biphenyl) of the present invention The body or "Compound B"(3,3'-dimethyl-4,4'-bis (2-propen-1-yloxy) -1,1'-biphenyl) is "Compound X" (3) having a similar chemical structure. , 3', 5,5'-Tetramethyl-4,4'-bis (2-propen-1-yloxy) -1,1'-biphenyl) has better crystallinity, so crystallization The operation and filtration operation can be easily performed, and the handling is excellent.
- the endothermic peak top temperature by differential scanning calorimetry is as high as 72 ° C for "Compound X", 73 ° C for the crystal of "Compound A” of the present invention, and 112 ° C for "Compound B".
- the crystal of the present invention can reduce blocking and melt-fixing during transportation and storage in summer. Further, since the crystal of the present invention has a higher 5% weight loss temperature of the compound itself as compared with the conventional general-purpose curing agent triallyl isocyanurate, it has an excellent effect that volatility in the prepreg molding step is suppressed. Demonstrate. Therefore, from the above points, the crystal of the present invention is very useful in industrial use.
- FIG. It is a chart figure which shows the differential scanning calorimetry data of the crystal body of "Compound A” obtained in Example 1.
- FIG. It is a chart figure which shows the powder X-ray diffraction analysis measurement data of the crystal body of "Compound A” obtained in Example 1.
- FIG. It is a photograph which shows the state of "Compound A” (left side) and “Compound X” (right side) before the start of the test of "confirmation test 1 of storage stability at high temperature and handleability of crystal body” in Example. ..
- FIG. It is a chart figure which shows the powder X-ray diffraction analysis measurement data of the crystal body of "Compound B” obtained in Example 2.
- FIG. It is a photograph which shows the state of "Compound B” (left side) and “Compound X” (right side) before the start of the test of "confirmation test 2 of storage stability under high temperature and handleability of crystal body” in Example. ..
- the "Compound A"(2,2',3,3',5,5'-hexamethyl-4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl) of the present invention is used. It is a compound represented by the following chemical formula.
- the "Compound B"(3,3'-dimethyl-4,4'-bis (2-propene-1-yloxy) -1,1'-biphenyl) of the present invention is a compound represented by the following chemical formula.
- the method for producing "Compound A” and “Compound B” of the present invention is not particularly limited, but for example, as shown by the following reaction formula, known 4,4'-dihydroxy-2,2', 3 , 3', 5,5'-hexamethylbiphenyl (hereinafter referred to as "Compound a”) and 4,4'-dihydroxy-3,3'-dimethylbiphenyl (hereinafter referred to as "Compound b").
- Examples thereof include a production method for obtaining a diether compound by reacting with allyl halide such as allyl chloride.
- reaction temperature in the above production method is preferably in the range of 20 to 120 ° C., more preferably in the range of 40 to 80 ° C., and even more preferably in the range of 60 to 70 ° C.
- the reaction pressure is usually carried out under normal pressure, but may be carried out under pressurization or reduced pressure.
- "Compound a" or “Compound b” is phenoxided with a base to enhance nucleophilicity, and the reaction is carried out.
- the base is not particularly limited, but is an alkali metal carbonate such as lithium carbonate, sodium carbonate and potassium carbonate, alkali metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, lithium hydroxide, sodium hydroxide and potassium hydroxide.
- alkali metal hydroxides such as, organic bases such as triethylamine and pyridine can be preferably used. Of these, sodium carbonate and potassium carbonate are preferable.
- the amount of this base used is preferably 1 to 5 equivalents, more preferably 1 to 3 equivalents, still more preferably 1 to 2 equivalents, relative to one hydroxyl group of "Compound a" or "Compound b".
- a catalyst may be used for the purpose of accelerating the reaction.
- the catalyst examples include alkali metal bromide salts such as sodium bromide and potassium bromide, alkali metal iodide salts such as sodium iodide and potassium iodide, and ammonium halide salts such as ammonium bromide and ammonium iodide. ..
- the amount of the catalyst used is preferably 0.05 to 3 equivalents, more preferably 0.1 to 1 equivalents, and 0.2 to 0.5 equivalents, relative to one hydroxyl group of "Compound a" or "Compound b". Is even more preferable.
- reaction solvent In the above production method, the reaction solvent may not be used, but it is preferably used for the reason of improving operability and reaction speed during industrial production.
- the reaction solvent is not particularly limited as long as it is not distilled out from the reaction vessel at the above reaction temperature and is inactive in the reaction.
- ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone and cyclohexanone, tetrahydrofuran, 1,4- Examples thereof include ethers such as dioxane, 1,3-dioxane and diethoxyethane, and aprotic polar solvents such as acetonitrile, dimethyl sulfoxide, dimethylformamide and N-methylpyrrolidone. Each of these organic solvents may be used alone, or two or more of them may be used in combination as appropriate to adjust the polarity. Of these, N-methylpyrrolidone and acetonitrile are preferable.
- the end point of the reaction can be confirmed by liquid chromatography or gas chromatography analysis. It is preferable that the end point of the reaction is the time when the unreacted "Compound a” or “Compound b" disappears and the increase of the target product "Compound A” or “Compound B” is no longer observed.
- the reaction time varies depending on the reaction conditions such as the reaction temperature, but is usually completed in about 1 to 30 hours.
- Compound A or “Compound B” can be obtained by performing post-treatment operations such as separation by column chromatography.
- post-treatment operations such as separation by column chromatography.
- purification by distillation, recrystallization or column chromatography may be carried out according to a conventional method.
- the crystal of the present invention can be obtained by crystallizing the crude product obtained by the above production method.
- the "Compound A” or “Compound B” used in the crystallization step is a crude crystal obtained by treating the reaction solution, a crystal obtained by recrystallizing the crude crystal, or a solution containing "Compound A” or “Compound B". Examples thereof include a residual solution obtained by distilling off the solvent. It may be amorphous.
- Examples of the solvent that can be used for crystallization include aromatic hydrocarbons such as toluene, xylene and benzene, aliphatic hydrocarbons such as hexane, heptane and cyclohexane, ketones such as acetone, methyl ethyl ketone and methyl isobutyl ketone, and methanol. , Alcohols such as ethanol and propanol, esters such as ethyl acetate and butyl acetate, ethers such as tetrahydrofuran and diethyl ether, nitriles such as acetonitrile, amides such as N-methylpyrrolidone, water and the like.
- aromatic hydrocarbons such as toluene, xylene and benzene
- aliphatic hydrocarbons such as hexane, heptane and cyclohexane
- ketones such as acetone, methyl e
- the solvent to be mixed with may be used as an aqueous solution. Further, these crystallization solvents may be used alone or in combination of two or more. Above all, as the solvent used for crystallization, the combined use of acetonitrile and water and the combined use of methyl isobutyl ketone and methanol are preferable.
- the amount of the crystallization solvent used is largely related to the solubility of "Compound A” or "Compound B" in each solvent, but is preferably about 0.5 to 10 times by weight with respect to the target product.
- two or more kinds of solvents are mixed and used, they can be appropriately adjusted in consideration of the solubility. For example, two kinds of solvents are mixed in a ratio of 1:10 to 10: 1, preferably 1: 5.
- the temperature at which the crystals are precipitated is preferably 20 to 50 ° C, more preferably 20 to 25 ° C.
- the final cooling temperature is preferably 0 to 20 ° C, more preferably 10 to 20 ° C.
- the operation in the crystallization step uses the above crystallization solvent, for example, cooling crystallization or poor solvent addition crystallization (solubility in a solution in which "Compound A” or “Compound B” is dissolved in a good solvent. It can be carried out by crystallization) in which a poor solvent is added to lower the temperature. Above all, it is preferable to carry out by crystallization with poor solvent addition. Poor solvent addition The method of adding the poor solvent in the case of crystallization can be appropriately adjusted according to the precipitation of crystals, but the required amount of the poor solvent may be added continuously or collectively. You may. By drying the crystals obtained by crystallization, the solvent used in crystallization can be removed.
- This drying can be carried out on the crystals obtained by crystallization at preferably 40 to 70 ° C. under reduced pressure, more preferably 40 to 50 ° C. under reduced pressure. Drying may be carried out under normal pressure or reduced pressure, but when carried out industrially, it is more efficient to carry out under reduced pressure because the solvent used in crystallization can be removed.
- the crystal of "Compound A” of the present invention preferably has an endothermic peak top temperature in the range of 70 to 75 ° C. by differential scanning calorimetry. Among them, a crystal having an endothermic peak top temperature in the range of 71 to 74.5 ° C. is preferable, and a crystal having a heat absorption peak top temperature in the range of 71 to 74 ° C. is more preferable.
- the crystal of "Compound B” of the present invention preferably has an endothermic peak top temperature in the range of 110 to 115 ° C. by differential scanning calorimetry. Among them, a crystal having an endothermic peak top temperature in the range of 111 to 114 ° C.
- Each crystal of the present invention usually has a purity in the range of 95-100%. The purity is preferably in the range of 97 to 100%, more preferably in the range of 98 to 100%.
- the present invention can easily carry out the crystallization operation and the filtration operation, and exhibits the effect of being excellent in handleability.
- the endothermic peak top temperature of the differential scanning calorific value analysis is 52 ° C for "Compound X", 73 ° C for the crystal of "Compound A” of the present invention, and 112 ° C for the crystal of "Compound B".
- the crystal of the present invention can reduce blocking and melt sticking during transportation and storage in summer.
- the crystal of the present invention has a higher 5% weight loss temperature of the compound itself than the conventional general-purpose curing agent triallyl isocyanurate, it has an excellent effect of suppressing volatility in the prepreg molding process. Demonstrate. Therefore, from the above points, it is very useful in industrial use.
- DSC Differential scanning calorimetry
- the crystals were precisely weighed in an aluminum pan, and measured using a differential scanning calorimetry device (manufactured by Shimadzu Corporation: DSC-60) under the following operating conditions using aluminum oxide as a control. (Operating conditions) Heating rate: 10 ° C / min Measurement temperature range: 30-300 ° C Measurement atmosphere: open, nitrogen 50 mL / min Sample amount: 2-3 mg 2. 2.
- Purity analyzer Prominence UFLC (Liquid Chromatography) manufactured by Shimadzu Corporation Pump: LC-20AD Column oven: CTO-20A Detector: SPD-20A Column: HALO C18 (inner diameter 3 mm, length 75 mm) Oven temperature: 50 ° C Flow rate: 0.7 mL / min Mobile phase: (A) 0.2% by volume acetic acid aqueous solution, (B) methanol gradient condition: (A)% by volume (time from the start of analysis) 50% (0min) ⁇ 100% (7.5min) ⁇ 100% (15min) Sample injection volume: 10 ⁇ L Detection wavelength: 280 nm 3. 3.
- Powder X-ray diffraction (XRD) 0.1 g of the crystal was filled in the sample filling portion of the glass test plate, and the measurement was performed using the following device and the following conditions.
- the 5% weight loss temperature of the obtained crystal of "Compound A” was measured and found to be 242 ° C.
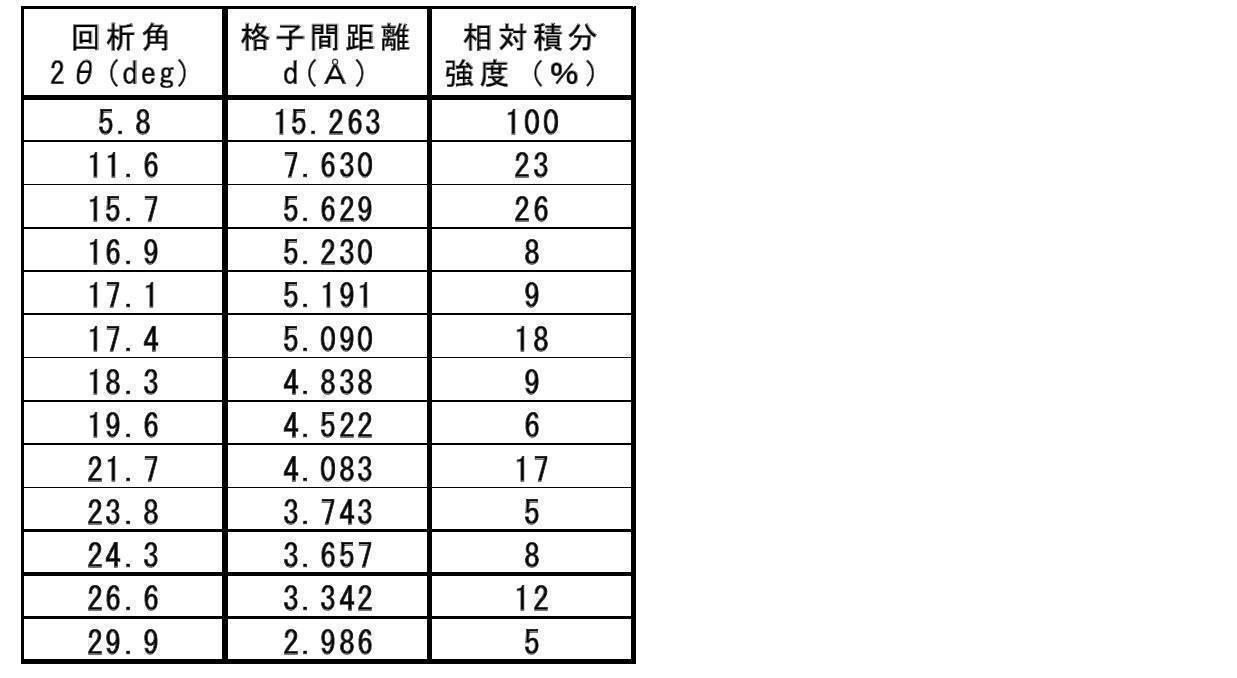
- Table 1 shows the main peaks of XRD (those having a relative intensity of more than 5%).
- the characteristic 2 ⁇ (deg) of “Compound A” obtained in Example 1 is 5.8, 11.6, 15.7, 17.4, 18.3, 19.6. , 21.7, 24.3, 26.6.
- FIG. 2 shows a chart (partially enlarged view) showing the powder X-ray diffraction analysis measurement data.
- the 5% weight loss temperature of the obtained crystal of "Compound B” was measured and found to be 234 ° C.
- Table 2 shows the main peaks of XRD (those having a relative intensity of more than 4%).
- the characteristic 2 ⁇ (deg) of “Compound B” obtained in Example 1 was 7.2, 14.5, and 21.7.
- FIG. 8 shows a chart (partially enlarged view) showing the powder X-ray diffraction analysis measurement data.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Liquid Crystal Substances (AREA)
Abstract
常温(25℃)では結晶体であり、より優れた耐熱性や保存安定性を有し、さらに、プリプレグ成形工程における揮発性が抑制された、ビフェノール骨格を有する新たなアリルエーテル類を提供することを課題とする。解決手段として、下記式(1)で表される化合物の結晶体を提供する。 (式中、Rは水素原子またはメチル基を表す。ただし、4つのRは全て同一である。)
Description
本発明は、ヘキサメチル置換/ジメチル置換4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’ -ビフェニルの結晶体に関する。
熱硬化性樹脂の硬化剤として、アリル基を有する架橋剤が汎用されている。変性ポリフェニレンオキシドの汎用硬化剤であるトリアリルイソシアヌレートは、常温(25℃)で液体であり、プリプレグ形成段階で溶媒と共に揮発しやすく、耐熱性が低いという問題点がある。
下記特許文献1、2には、ビフェノール骨格を有するアリルエーテル化合物類として、ビフェノール、4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル等のジアリルエーテル化合物について記載されており、そのうち、3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルの製造例が記載されている。当該化合物について、本発明者らが検討したところ、融点が約50℃と、トリアリルイソシアヌレートと比較して融点が高く、耐熱性が改善されていることが確認された。しかしながら、プリプレグ成形工程では、通常130~180度程度の高温にさらされることから、それでもなお、当該化合物の揮発によるプリプレグ製造設備の汚染が懸念された。また、運搬途中や保管中における高温条件下では、融解が起こり結晶同士くっつき、取り扱い性が低下するおそれもあった。
これらの状況から、耐熱性、保存安定性に優れ、プリプレグ成形工程における揮発性が抑制された化学物質が求められていた。
下記特許文献1、2には、ビフェノール骨格を有するアリルエーテル化合物類として、ビフェノール、4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル等のジアリルエーテル化合物について記載されており、そのうち、3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルの製造例が記載されている。当該化合物について、本発明者らが検討したところ、融点が約50℃と、トリアリルイソシアヌレートと比較して融点が高く、耐熱性が改善されていることが確認された。しかしながら、プリプレグ成形工程では、通常130~180度程度の高温にさらされることから、それでもなお、当該化合物の揮発によるプリプレグ製造設備の汚染が懸念された。また、運搬途中や保管中における高温条件下では、融解が起こり結晶同士くっつき、取り扱い性が低下するおそれもあった。
これらの状況から、耐熱性、保存安定性に優れ、プリプレグ成形工程における揮発性が抑制された化学物質が求められていた。
本発明は、上述した事情を背景としてなされたものであって、常温(25℃)では結晶体であり、より優れた耐熱性や保存安定性を有し、さらに、プリプレグ成形工程における揮発性が抑制された、ビフェノール骨格を有する新たなアリルエーテル類の提供を課題とする。
ビフェノール骨格を有するアリルエーテル類の1つである、2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(以下、「化合物A」という。)または、3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(以下、「化合物B」という。)は、製造されたことを報告する文献等は未だなく、その具体的な物性や化学的な性状等については未だ知られていない。
本発明者は、上述の課題解決のために「化合物A」と「化合物B」について鋭意検討した結果、「化合物A」と「化合物B」それぞれの製造及び結晶の取得に初めて成功し、さらに、その化学的性状は従来公知の化合物である3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(以下、「化合物X」という。)よりも優れた耐熱性、高温下での保存安定性を示し、さらに結晶体は良好な取り扱い性を有することを見出し、本発明を完成させた。
すなわち、本発明にかかる結晶体を提供することにより、上記課題を解決することができるものである。
本発明者は、上述の課題解決のために「化合物A」と「化合物B」について鋭意検討した結果、「化合物A」と「化合物B」それぞれの製造及び結晶の取得に初めて成功し、さらに、その化学的性状は従来公知の化合物である3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(以下、「化合物X」という。)よりも優れた耐熱性、高温下での保存安定性を示し、さらに結晶体は良好な取り扱い性を有することを見出し、本発明を完成させた。
すなわち、本発明にかかる結晶体を提供することにより、上記課題を解決することができるものである。
本発明は以下の通りである。
1.下記式(1)で表される化合物の結晶体。
 (式中、Rは水素原子またはメチル基を表す。ただし、4つのRは全て同一である。)
(式中、Rは水素原子またはメチル基を表す。ただし、4つのRは全て同一である。)
2.式(1)で表される化合物が、2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、1.に記載の結晶体。
3.示差走査熱量分析による吸熱ピークトップ温度が70~75℃の範囲である2.に記載の結晶体。
4.式(1)で表される化合物が、3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、1.に記載の結晶体。
5.示差走査熱量分析による吸熱ピークトップ温度が110~115℃の範囲である4.に記載の結晶体。
1.下記式(1)で表される化合物の結晶体。
2.式(1)で表される化合物が、2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、1.に記載の結晶体。
3.示差走査熱量分析による吸熱ピークトップ温度が70~75℃の範囲である2.に記載の結晶体。
4.式(1)で表される化合物が、3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、1.に記載の結晶体。
5.示差走査熱量分析による吸熱ピークトップ温度が110~115℃の範囲である4.に記載の結晶体。
本発明の「化合物A」(2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)の結晶体または「化合物B」(3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)は、類似化学構造を有する「化合物X」(3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)に比べて、結晶性が良好であることから、晶析操作、ろ過操作を簡便に実施することができ、取り扱い性に優れている。
また、示差走査熱量分析による吸熱ピークトップ温度は、「化合物X」が52℃であるのに対して、本発明の「化合物A」の結晶体は73℃、「化合物B」は112℃と高く、本発明の結晶体は、夏場の運搬や保管時におけるブロッキングや溶融固着などを低減することができる。
さらに、本発明の結晶体は、従来の汎用硬化剤であるトリアリルイソシアヌレートに比べて、化合物自体の5%重量減少温度が高いため、プリプレグ成形工程における揮発性が抑制されるという優れた効果を発揮する。
よって、上記の点から、本発明の結晶体は、工業的な使用において非常に有用である。
また、示差走査熱量分析による吸熱ピークトップ温度は、「化合物X」が52℃であるのに対して、本発明の「化合物A」の結晶体は73℃、「化合物B」は112℃と高く、本発明の結晶体は、夏場の運搬や保管時におけるブロッキングや溶融固着などを低減することができる。
さらに、本発明の結晶体は、従来の汎用硬化剤であるトリアリルイソシアヌレートに比べて、化合物自体の5%重量減少温度が高いため、プリプレグ成形工程における揮発性が抑制されるという優れた効果を発揮する。
よって、上記の点から、本発明の結晶体は、工業的な使用において非常に有用である。
以下、本発明を詳細に説明する。
本発明の「化合物A」(2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)は、下記化学式で表される化合物である。

本発明の「化合物B」(3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)は、下記化学式で表される化合物である。

本発明の「化合物A」(2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)は、下記化学式で表される化合物である。
<製造方法>
本発明の「化合物A」、「化合物B」の製造方法については、特に制限はないが、例えば、下記反応式で示されるように、公知の4,4’-ジヒドロキシ-2,2’,3,3’,5,5’-ヘキサメチルビフェニル(以下、「化合物a」という。)や、4,4’-ジヒドロキシ-3,3’-ジメチルビフェニル(以下、「化合物b」という。)と、塩化アリル等のハロゲン化アリルとを反応させてジエーテル体を得る製造方法が挙げられる。
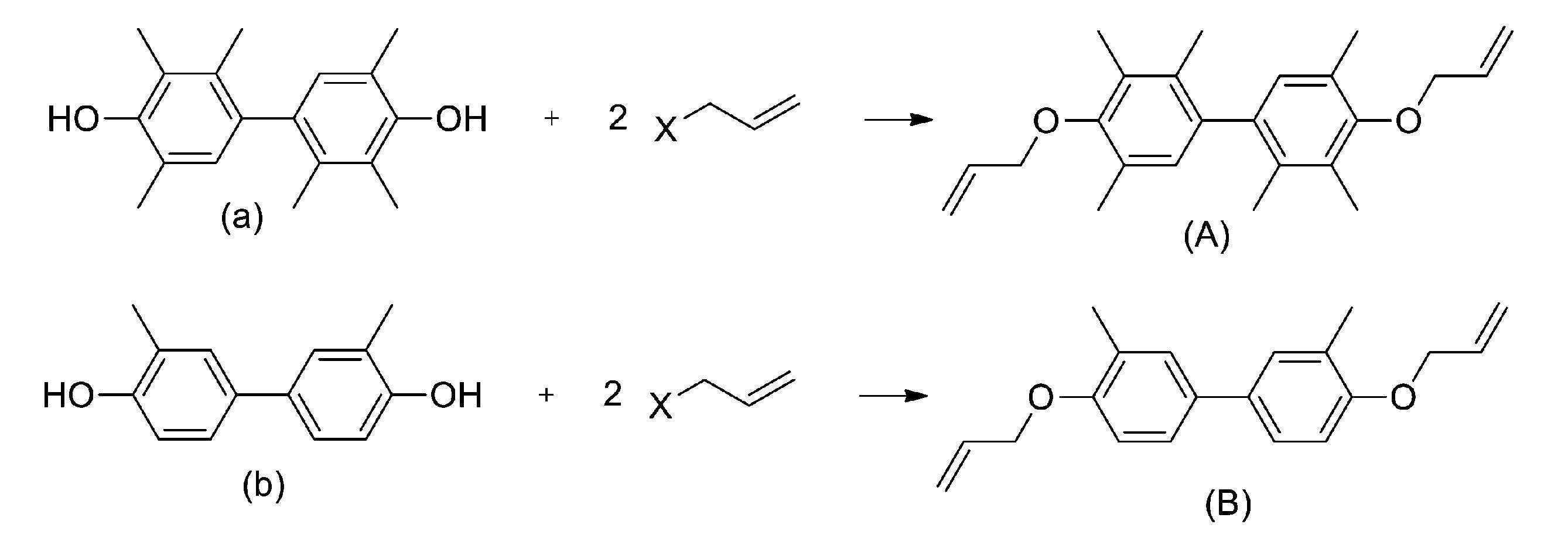
本発明の「化合物A」、「化合物B」の製造方法については、特に制限はないが、例えば、下記反応式で示されるように、公知の4,4’-ジヒドロキシ-2,2’,3,3’,5,5’-ヘキサメチルビフェニル(以下、「化合物a」という。)や、4,4’-ジヒドロキシ-3,3’-ジメチルビフェニル(以下、「化合物b」という。)と、塩化アリル等のハロゲン化アリルとを反応させてジエーテル体を得る製造方法が挙げられる。
(原料)
原料の1つである「化合物a」または「化合物b」は、市販されているので、本発明の「化合物A」または「化合物B」を製造するために、その市販品を使用してもよいし、公知の製造方法に従い製造したものを使用しても良い。その純度は、特に限定されないが、通常、95%以上が好ましく、99%以上がより好ましい。
もう1つの原料であるハロゲン化アリルとしては、塩化アリル、臭化アリル、ヨウ化アリル等が挙げられる。中でも、反応効率の点から臭化アリルが好ましい。ハロゲン部分が異なる2種以上のハロゲン化アリルを併用してもよい。
このハロゲン化アリルの使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して1~10当量、好ましくは1~5当量、より好ましくは1~3当量使用する。
原料の1つである「化合物a」または「化合物b」は、市販されているので、本発明の「化合物A」または「化合物B」を製造するために、その市販品を使用してもよいし、公知の製造方法に従い製造したものを使用しても良い。その純度は、特に限定されないが、通常、95%以上が好ましく、99%以上がより好ましい。
もう1つの原料であるハロゲン化アリルとしては、塩化アリル、臭化アリル、ヨウ化アリル等が挙げられる。中でも、反応効率の点から臭化アリルが好ましい。ハロゲン部分が異なる2種以上のハロゲン化アリルを併用してもよい。
このハロゲン化アリルの使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して1~10当量、好ましくは1~5当量、より好ましくは1~3当量使用する。
(反応条件)
上記製造方法における反応温度は、20~120℃の範囲が好ましく、40~80℃の範囲がより好ましく、60~70℃の範囲がさらに好ましい。反応温度が高いと、副生成物が増加し「化合物A」または「化合物B」の収率が低下する傾向がある。また、反応温度が低すぎると、反応速度が低下し効率的ではない。
反応圧力は、通常、常圧下で行われるが、加圧または減圧下で行ってもよい。
上記製造方法では、「化合物a」または「化合物b」を塩基によりフェノキシド化して、求核性を高めて、反応を行う。塩基としては、特に限定されないが、炭酸リチウム、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩、炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩、水酸化リチウム、水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物のほか、トリエチルアミン、ピリジン等の有機塩基などが好適に使用することができる。中でも、炭酸ナトリウムや炭酸カリウムが好ましい。この塩基の使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して1~5当量が好ましく、1~3当量がより好ましく、1~2当量がさらに好ましい。
さらに、反応を促進させる目的で触媒を使用してもよい。触媒としては、臭化ナトリウム、臭化カリウム等の臭化アルカリ金属塩、ヨウ化ナトリウム、ヨウ化カリウム等のヨウ化アルカリ金属塩、臭化アンモニウム、ヨウ化アンモニウム等のハロゲン化アンモニウム塩が挙げられる。触媒の使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して0.05~3当量が好ましく、0.1~1当量がより好ましく、0.2~0.5当量がさらに好ましい。
上記製造方法における反応温度は、20~120℃の範囲が好ましく、40~80℃の範囲がより好ましく、60~70℃の範囲がさらに好ましい。反応温度が高いと、副生成物が増加し「化合物A」または「化合物B」の収率が低下する傾向がある。また、反応温度が低すぎると、反応速度が低下し効率的ではない。
反応圧力は、通常、常圧下で行われるが、加圧または減圧下で行ってもよい。
上記製造方法では、「化合物a」または「化合物b」を塩基によりフェノキシド化して、求核性を高めて、反応を行う。塩基としては、特に限定されないが、炭酸リチウム、炭酸ナトリウム、炭酸カリウム等のアルカリ金属炭酸塩、炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属炭酸水素塩、水酸化リチウム、水酸化ナトリウム、水酸化カリウム等のアルカリ金属水酸化物のほか、トリエチルアミン、ピリジン等の有機塩基などが好適に使用することができる。中でも、炭酸ナトリウムや炭酸カリウムが好ましい。この塩基の使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して1~5当量が好ましく、1~3当量がより好ましく、1~2当量がさらに好ましい。
さらに、反応を促進させる目的で触媒を使用してもよい。触媒としては、臭化ナトリウム、臭化カリウム等の臭化アルカリ金属塩、ヨウ化ナトリウム、ヨウ化カリウム等のヨウ化アルカリ金属塩、臭化アンモニウム、ヨウ化アンモニウム等のハロゲン化アンモニウム塩が挙げられる。触媒の使用量は、「化合物a」または「化合物b」の1つのヒドロキシル基に対して0.05~3当量が好ましく、0.1~1当量がより好ましく、0.2~0.5当量がさらに好ましい。
(反応溶媒)
上記製造方法において、反応溶媒は使用しなくてもよいが、工業的生産時の操作性や反応速度の向上などの理由で使用することが好ましい。反応溶媒としては、上記反応温度において反応容器から留出せず、反応に不活性であれば特に制限はなく、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン類、テトラヒドロフラン、1,4-ジオキサン、1,3-ジオキサン、ジエトキシエタン等のエーテル類、アセトニトリル、ジメチルスルホキシド、ジメチルホルムアミド、N-メチルピロリドン等の非プロトン性極性溶媒等が挙げられる。これらの有機溶媒は、それぞれ単独で用いてもよいし、また、極性を調整するために適宜2種以上を併用してもよい。中でも、N-メチルピロリドン、アセトニトリルが好ましい。
上記製造方法において、反応溶媒は使用しなくてもよいが、工業的生産時の操作性や反応速度の向上などの理由で使用することが好ましい。反応溶媒としては、上記反応温度において反応容器から留出せず、反応に不活性であれば特に制限はなく、例えば、アセトン、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン類、テトラヒドロフラン、1,4-ジオキサン、1,3-ジオキサン、ジエトキシエタン等のエーテル類、アセトニトリル、ジメチルスルホキシド、ジメチルホルムアミド、N-メチルピロリドン等の非プロトン性極性溶媒等が挙げられる。これらの有機溶媒は、それぞれ単独で用いてもよいし、また、極性を調整するために適宜2種以上を併用してもよい。中でも、N-メチルピロリドン、アセトニトリルが好ましい。
(反応後処理)
反応の終点は、液体クロマトグラフィーまたはガスクロマトグラフィー分析にて確認することができる。未反応の「化合物a」または「化合物b」が消失し、目的物「化合物A」または「化合物B」の増加が認められなくなった時点を反応の終点とするのが好ましい。反応時間は、反応温度等の反応条件により異なるが、通常1~30時間程度で終了する。
反応終了後、反応液から目的とする「化合物A」または「化合物B」を精製、単離するのが好ましく、例えば、常法に従い、反応終了後、中和、水洗、晶析、ろ過、蒸留、カラムクロマトグラフィーによる分離などの後処理操作を行い、「化合物A」または「化合物B」を得ることができる。さらに純度を高めるため、常法に従い蒸留や再結晶、カラムクロマトグラフィーによる精製を行ってもよい。
反応の終点は、液体クロマトグラフィーまたはガスクロマトグラフィー分析にて確認することができる。未反応の「化合物a」または「化合物b」が消失し、目的物「化合物A」または「化合物B」の増加が認められなくなった時点を反応の終点とするのが好ましい。反応時間は、反応温度等の反応条件により異なるが、通常1~30時間程度で終了する。
反応終了後、反応液から目的とする「化合物A」または「化合物B」を精製、単離するのが好ましく、例えば、常法に従い、反応終了後、中和、水洗、晶析、ろ過、蒸留、カラムクロマトグラフィーによる分離などの後処理操作を行い、「化合物A」または「化合物B」を得ることができる。さらに純度を高めるため、常法に従い蒸留や再結晶、カラムクロマトグラフィーによる精製を行ってもよい。
(晶析)
本発明の結晶体は、上記製造方法により得られた粗生成物を、晶析することにより得ることができる。
晶析工程に用いる「化合物A」または「化合物B」としては、反応液を処理して得られる粗結晶、該粗結晶を再結晶した結晶、「化合物A」または「化合物B」を含む溶液から溶媒を留出除去された残液等が挙げられる。非晶質のものでもよい。
晶析に使用できる溶媒としては、例えば、トルエン、キシレン、ベンゼン等の芳香族炭化水素類、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素類、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン類、メタノール、エタノール、プロパノール等のアルコール類、酢酸エチル、酢酸ブチル等のエステル類、テトラヒドロフラン、ジエチルエーテル等のエーテル類、アセトニトリル等のニトリル類、N-メチルピロリドン等のアミド類、水などが挙げられ、水と混和する溶媒に関しては水溶液として用いても良い。また、これらの晶析溶媒は、単独で用いても良いし、2種類以上混合して使用しても良い。中でも、晶析に使用する溶媒としては、アセトニトリルと水の併用並びにメチルイソブチルケトンとメタノールの併用が好適である。
晶析溶媒の使用量は、各溶媒における「化合物A」または「化合物B」の溶解度が大きく関係するが、目的物に対して概略0.5~10重量倍が好ましい。2種類以上混合して使用する場合には、溶解度を考慮して適宜調整することができるが、例えば、2種類の溶媒を1:10~10:1の割合の範囲で、好ましくは1:5~5:1の割合の範囲で、より好ましくは1:3~3:1の割合の範囲で使用することができる。
結晶を析出させる温度としては20~50℃が好ましく、20~25℃がより好ましい。結晶を析出させる際は、種晶を用いなくてもよいが、種晶を用いた方が好ましく、種晶なしで析出させた結晶を種晶として用いればよい。最終の冷却温度としては、0~20℃が好ましく、10~20℃がより好ましい。上記温度まで冷却後、析出した結晶をろ過操作により分離する。
晶析工程における操作は、上記晶析溶媒を使用して、例えば、冷却晶析や貧溶媒添加晶析(良溶媒に「化合物A」または「化合物B」が溶けている溶液に対して、溶解度を低くするような貧溶媒を添加する晶析)により行うことができる。中でも、貧溶媒添加晶析により行うことが好適である。貧溶媒添加晶析により行う場合の貧溶媒の添加方法は、結晶の析出に応じて適宜調整することができるが、必要な貧溶媒の量を連続的に添加してもよいし、一括に添加してもよい。
晶析により得られた結晶を乾燥することにより、晶析において使用した溶媒を除去することができる。この乾燥は、晶析により得られた結晶を、減圧下好ましくは40~70℃、より好ましくは減圧下40~50℃において実施することができる。乾燥する際は常圧でも減圧下でも良いが、工業的に実施する場合には、減圧下において実施する方がより効率的に、晶析において使用した溶媒を除去できることからも好適である。
本発明の結晶体は、上記製造方法により得られた粗生成物を、晶析することにより得ることができる。
晶析工程に用いる「化合物A」または「化合物B」としては、反応液を処理して得られる粗結晶、該粗結晶を再結晶した結晶、「化合物A」または「化合物B」を含む溶液から溶媒を留出除去された残液等が挙げられる。非晶質のものでもよい。
晶析に使用できる溶媒としては、例えば、トルエン、キシレン、ベンゼン等の芳香族炭化水素類、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族炭化水素類、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン類、メタノール、エタノール、プロパノール等のアルコール類、酢酸エチル、酢酸ブチル等のエステル類、テトラヒドロフラン、ジエチルエーテル等のエーテル類、アセトニトリル等のニトリル類、N-メチルピロリドン等のアミド類、水などが挙げられ、水と混和する溶媒に関しては水溶液として用いても良い。また、これらの晶析溶媒は、単独で用いても良いし、2種類以上混合して使用しても良い。中でも、晶析に使用する溶媒としては、アセトニトリルと水の併用並びにメチルイソブチルケトンとメタノールの併用が好適である。
晶析溶媒の使用量は、各溶媒における「化合物A」または「化合物B」の溶解度が大きく関係するが、目的物に対して概略0.5~10重量倍が好ましい。2種類以上混合して使用する場合には、溶解度を考慮して適宜調整することができるが、例えば、2種類の溶媒を1:10~10:1の割合の範囲で、好ましくは1:5~5:1の割合の範囲で、より好ましくは1:3~3:1の割合の範囲で使用することができる。
結晶を析出させる温度としては20~50℃が好ましく、20~25℃がより好ましい。結晶を析出させる際は、種晶を用いなくてもよいが、種晶を用いた方が好ましく、種晶なしで析出させた結晶を種晶として用いればよい。最終の冷却温度としては、0~20℃が好ましく、10~20℃がより好ましい。上記温度まで冷却後、析出した結晶をろ過操作により分離する。
晶析工程における操作は、上記晶析溶媒を使用して、例えば、冷却晶析や貧溶媒添加晶析(良溶媒に「化合物A」または「化合物B」が溶けている溶液に対して、溶解度を低くするような貧溶媒を添加する晶析)により行うことができる。中でも、貧溶媒添加晶析により行うことが好適である。貧溶媒添加晶析により行う場合の貧溶媒の添加方法は、結晶の析出に応じて適宜調整することができるが、必要な貧溶媒の量を連続的に添加してもよいし、一括に添加してもよい。
晶析により得られた結晶を乾燥することにより、晶析において使用した溶媒を除去することができる。この乾燥は、晶析により得られた結晶を、減圧下好ましくは40~70℃、より好ましくは減圧下40~50℃において実施することができる。乾燥する際は常圧でも減圧下でも良いが、工業的に実施する場合には、減圧下において実施する方がより効率的に、晶析において使用した溶媒を除去できることからも好適である。
<本発明の結晶体>
本発明の「化合物A」の結晶体は、示差走査熱量分析による吸熱ピークトップ温度が70~75℃の範囲にあることが好ましい。中でも、当該吸熱ピークトップ温度が71~74.5℃の範囲にある結晶体が好ましく、71~74℃の範囲にある結晶体がより好ましい。
本発明の「化合物B」の結晶体は、示差走査熱量分析による吸熱ピークトップ温度が110~115℃の範囲にあることが好ましい。中でも、当該吸熱ピークトップ温度が111~114℃の範囲にある結晶体が好ましく、111~113℃の範囲にある結晶体がより好ましい。
本発明のそれぞれの結晶体は、通常、その純度が95~100%の範囲にある。その純度は97~100%の範囲が好ましく、98~100%の範囲がより好ましい。
「化合物A」(2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)の結晶体または「化合物B」(3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)の結晶体は、類似化学構造を有する「化合物X」(3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)に比べて、結晶性が良好であることを見出し、本発明を完成させたものである。そして、本発明は、晶析操作、ろ過操作を簡便に実施することができ、取り扱い性に優れるという効果を発揮する。
また、示差走査熱量分析による吸熱ピークトップ温度は、「化合物X」は52℃であるのに対して、本発明の「化合物A」の結晶体は73℃、「化合物B」の結晶体は112℃と高く、本発明の結晶体は、夏場の運搬や保管時におけるブロッキングや溶融固着などを低減することができる。
さらに、本発明の結晶体は、従来の汎用硬化剤であるトリアリルイソシアヌレートに比べて化合物自体の5%重量減少温度が高いため、プリプレグ成形工程における揮発性が抑制されるという優れた効果を発揮する。
よって、上記の点から、工業的な使用において非常に有用である。
本発明の「化合物A」の結晶体は、示差走査熱量分析による吸熱ピークトップ温度が70~75℃の範囲にあることが好ましい。中でも、当該吸熱ピークトップ温度が71~74.5℃の範囲にある結晶体が好ましく、71~74℃の範囲にある結晶体がより好ましい。
本発明の「化合物B」の結晶体は、示差走査熱量分析による吸熱ピークトップ温度が110~115℃の範囲にあることが好ましい。中でも、当該吸熱ピークトップ温度が111~114℃の範囲にある結晶体が好ましく、111~113℃の範囲にある結晶体がより好ましい。
本発明のそれぞれの結晶体は、通常、その純度が95~100%の範囲にある。その純度は97~100%の範囲が好ましく、98~100%の範囲がより好ましい。
「化合物A」(2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)の結晶体または「化合物B」(3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)の結晶体は、類似化学構造を有する「化合物X」(3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル)に比べて、結晶性が良好であることを見出し、本発明を完成させたものである。そして、本発明は、晶析操作、ろ過操作を簡便に実施することができ、取り扱い性に優れるという効果を発揮する。
また、示差走査熱量分析による吸熱ピークトップ温度は、「化合物X」は52℃であるのに対して、本発明の「化合物A」の結晶体は73℃、「化合物B」の結晶体は112℃と高く、本発明の結晶体は、夏場の運搬や保管時におけるブロッキングや溶融固着などを低減することができる。
さらに、本発明の結晶体は、従来の汎用硬化剤であるトリアリルイソシアヌレートに比べて化合物自体の5%重量減少温度が高いため、プリプレグ成形工程における揮発性が抑制されるという優れた効果を発揮する。
よって、上記の点から、工業的な使用において非常に有用である。
以下、本発明を実施例により具体的に説明するが、本発明はこれら実施例に限定されるものではない。
分析方法は以下の通りである。
<分析方法>
1.示差走査熱量分析(DSC)
結晶体をアルミパンに精密に秤量し、示差走査熱量測定装置((株)島津製作所製:DSC-60)を用いて、酸化アルミニウムを対照として下記操作条件により測定した。
(操作条件)
昇温速度 :10℃/min
測定温度範囲:30~300℃
測定雰囲気 :開放、窒素50mL/min
サンプル量 :2~3mg
2.純度分析
装置:(株)島津製作所製 ProminenceUFLC(液体クロマトグラフィー)
ポンプ :LC-20AD
カラムオーブン :CTO-20A
検出器 :SPD-20A
カラム :HALO C18(内径3mm、長さ75mm)
オーブン温度 :50℃
流量 :0.7mL/min
移動相 :(A)0.2体積%酢酸水溶液、(B)メタノール
グラジエント条件:(A)体積%(分析開始からの時間)
50%(0min)→100%(7.5min)→100%(15min)
試料注入量 :10μL
検出波長 :280nm
3.NMR分析
フーリエ変換核磁気共鳴AVANCE III HD 400(BRUKER製)を用い、重水素化クロロホルム(CDCl3)中で合成物の1H-NMRスペクトルを測定した。
4.5%重量減少温度の分析(示差熱・熱重量分析装置:DTG)
結晶体8~12mgをアルミパンに秤量し、示差熱・熱重量分析装置((株)島津製作所製:DTG-60A)を用いて、下記操作条件により測定した。
(操作条件)
昇温速度 :10℃/min
測定温度範囲:30~400℃
測定雰囲気 :開放、窒素50mL/min
5.粉末X線回析(XRD)
結晶体0.1gをガラス試験板の試料充填部に充填し、下記装置と下記条件により測定した。
装置 :(株)リガク製:SmartLab(粉末X線回折装置)
X線源 :CuKα
スキャン軸 :2θ/θ
モード :連続
測定範囲 :2θ=5°~90°
ステップ :0.01°
スピード計測時間 :2θ=33.9°/min
IS :2/3
RS :20.00mm
出力 :40kV、30mA
分析方法は以下の通りである。
<分析方法>
1.示差走査熱量分析(DSC)
結晶体をアルミパンに精密に秤量し、示差走査熱量測定装置((株)島津製作所製:DSC-60)を用いて、酸化アルミニウムを対照として下記操作条件により測定した。
(操作条件)
昇温速度 :10℃/min
測定温度範囲:30~300℃
測定雰囲気 :開放、窒素50mL/min
サンプル量 :2~3mg
2.純度分析
装置:(株)島津製作所製 ProminenceUFLC(液体クロマトグラフィー)
ポンプ :LC-20AD
カラムオーブン :CTO-20A
検出器 :SPD-20A
カラム :HALO C18(内径3mm、長さ75mm)
オーブン温度 :50℃
流量 :0.7mL/min
移動相 :(A)0.2体積%酢酸水溶液、(B)メタノール
グラジエント条件:(A)体積%(分析開始からの時間)
50%(0min)→100%(7.5min)→100%(15min)
試料注入量 :10μL
検出波長 :280nm
3.NMR分析
フーリエ変換核磁気共鳴AVANCE III HD 400(BRUKER製)を用い、重水素化クロロホルム(CDCl3)中で合成物の1H-NMRスペクトルを測定した。
4.5%重量減少温度の分析(示差熱・熱重量分析装置:DTG)
結晶体8~12mgをアルミパンに秤量し、示差熱・熱重量分析装置((株)島津製作所製:DTG-60A)を用いて、下記操作条件により測定した。
(操作条件)
昇温速度 :10℃/min
測定温度範囲:30~400℃
測定雰囲気 :開放、窒素50mL/min
5.粉末X線回析(XRD)
結晶体0.1gをガラス試験板の試料充填部に充填し、下記装置と下記条件により測定した。
装置 :(株)リガク製:SmartLab(粉末X線回折装置)
X線源 :CuKα
スキャン軸 :2θ/θ
モード :連続
測定範囲 :2θ=5°~90°
ステップ :0.01°
スピード計測時間 :2θ=33.9°/min
IS :2/3
RS :20.00mm
出力 :40kV、30mA
<実施例1>
2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物A」)の合成

「化合物a」10.0g、アセトニトリル25.0g、炭酸カリウム15.0gを4つ口フラスコに仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル12.1gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、メタノール25.0g、水50.0gを加え、析出した結晶をろ過し、粉末24.1gを取得した。
得られた粉末にアセトニトリル20.0gを室温(20℃)で添加し完全に溶解させ、そこに水40.0gを一括で添加したところ結晶が析出した。この結晶をろ過し、乾燥を行い「化合物A」の結晶体を9.2g取得した。NMR分析により、得られた結晶体は目的物であることを確認した。
収率は「化合物a」に対して、71.0mol%であった。
高速液体クロマトグラフィー測定による純度は98.0%、示差走査熱量分析による吸熱ピークトップ温度は73℃であった。
示差走査熱量分析データを示すチャート図を図1に示す(試料量:2.311mg)。
1H-NMR:1.91(s、6H、Me×2)、2.24(s、6H、Me×2)、2.27(s、6H、Me×2)、4.32-4.34(td、4H、CH2)、5.27-5.31(td、2H、allyl)、5.45-5.51(td、2H、allyl)、6.11-6.21(td、2H、allyl)、6.78(s、2H、Ar).
得られた「化合物A」の結晶体について、5%重量減少温度を測定したところ、242℃であった。
また、XRDの主なピーク(5%を超える相対強度を有するもの)を表1に示す。表1に示すとおり、実施例1で得られた「化合物A」の特徴的な2θ(deg)は、5.8、11.6、15.7、17.4、18.3、19.6、21.7、24.3、26.6であった。この粉末X線回折分析測定データを示すチャート図(部分拡大図)を図2に示す。
2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物A」)の合成
得られた粉末にアセトニトリル20.0gを室温(20℃)で添加し完全に溶解させ、そこに水40.0gを一括で添加したところ結晶が析出した。この結晶をろ過し、乾燥を行い「化合物A」の結晶体を9.2g取得した。NMR分析により、得られた結晶体は目的物であることを確認した。
収率は「化合物a」に対して、71.0mol%であった。
高速液体クロマトグラフィー測定による純度は98.0%、示差走査熱量分析による吸熱ピークトップ温度は73℃であった。
示差走査熱量分析データを示すチャート図を図1に示す(試料量:2.311mg)。
1H-NMR:1.91(s、6H、Me×2)、2.24(s、6H、Me×2)、2.27(s、6H、Me×2)、4.32-4.34(td、4H、CH2)、5.27-5.31(td、2H、allyl)、5.45-5.51(td、2H、allyl)、6.11-6.21(td、2H、allyl)、6.78(s、2H、Ar).
得られた「化合物A」の結晶体について、5%重量減少温度を測定したところ、242℃であった。
また、XRDの主なピーク(5%を超える相対強度を有するもの)を表1に示す。表1に示すとおり、実施例1で得られた「化合物A」の特徴的な2θ(deg)は、5.8、11.6、15.7、17.4、18.3、19.6、21.7、24.3、26.6であった。この粉末X線回折分析測定データを示すチャート図(部分拡大図)を図2に示す。
<実施例2>
3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物B」)の合成

「化合物b」10.0g、アセトニトリル25g、炭酸カリウム15gを4つ口フラスコに仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル12.1gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、メタノール25g、水50gを加え、析出した結晶をろ過し、粉末21.3gを取得した。
得られた粉末をアセトニトリル100gでリスラリーを行い、それをろ過して粉末16.9gを得た。次いで、得られた粉末にメチルイソブチルケトン50gを室温(20℃)で添加し完全に溶解させ、そこにメタノール50gを一括で添加して、結晶を析出させた。得られた結晶をろ過し、乾燥を行い「化合物B」の結晶体を5.0g取得した。NMR分析により、得られた結晶体は目的物であることを確認した。収率は「化合物b」に対して、36mol%であった。
高速液体クロマトグラフィー測定による純度は99.4%、示差走査熱量分析による吸熱ピークトップ温度は112℃であった。
示差走査熱量分析データを示すチャート図を図7に示す。
1H-NMR:2.33(s、6H、Me×2)、4.58-4.60(td、4H、CH2)、5.29-5.32(td、2H、allyl)、5.44-5.49(td、2H、allyl)、6.06-6.16(m、2H、=CH-)、6.86-6.88(d、2H、Ar)、7.31-7.34(dd、2H、Ar)、7.35-7.36(d、2H、Ar).
得られた「化合物B」の結晶体について、5%重量減少温度を測定したところ、234℃であった。
また、XRDの主なピーク(4%を超える相対強度を有するもの)を表2に示す。表2に示すとおり、実施例1で得られた「化合物B」の特徴的な2θ(deg)は、7.2、14.5、21.7であった。この粉末X線回折分析測定データを示すチャート図(部分拡大図)を図8に示す。
3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物B」)の合成
得られた粉末をアセトニトリル100gでリスラリーを行い、それをろ過して粉末16.9gを得た。次いで、得られた粉末にメチルイソブチルケトン50gを室温(20℃)で添加し完全に溶解させ、そこにメタノール50gを一括で添加して、結晶を析出させた。得られた結晶をろ過し、乾燥を行い「化合物B」の結晶体を5.0g取得した。NMR分析により、得られた結晶体は目的物であることを確認した。収率は「化合物b」に対して、36mol%であった。
高速液体クロマトグラフィー測定による純度は99.4%、示差走査熱量分析による吸熱ピークトップ温度は112℃であった。
示差走査熱量分析データを示すチャート図を図7に示す。
1H-NMR:2.33(s、6H、Me×2)、4.58-4.60(td、4H、CH2)、5.29-5.32(td、2H、allyl)、5.44-5.49(td、2H、allyl)、6.06-6.16(m、2H、=CH-)、6.86-6.88(d、2H、Ar)、7.31-7.34(dd、2H、Ar)、7.35-7.36(d、2H、Ar).
得られた「化合物B」の結晶体について、5%重量減少温度を測定したところ、234℃であった。
また、XRDの主なピーク(4%を超える相対強度を有するもの)を表2に示す。表2に示すとおり、実施例1で得られた「化合物B」の特徴的な2θ(deg)は、7.2、14.5、21.7であった。この粉末X線回折分析測定データを示すチャート図(部分拡大図)を図8に示す。

<比較例1>
実施例1と同じ製造方法による3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物X」)の結晶体の合成トライ1
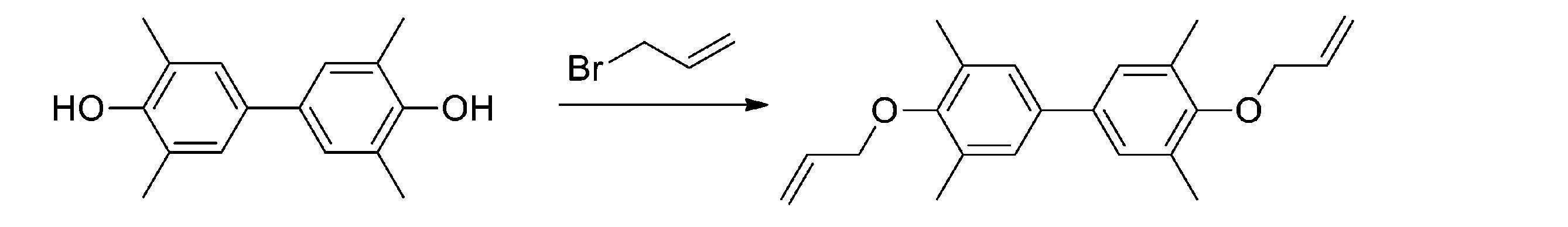
4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル1.1g、アセトニトリル2.5g、炭酸カリウム1.7gを試験管に仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル2.4gを滴下した。24時間撹拌後、温度を室温まで下げ、メタノール2.5g、水5gを加えたが、溶液が2層に分離し「化合物X」の結晶を得ることはできなかった。
実施例1と同じ製造方法による3,3’,5,5’-テトラメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニル(「化合物X」)の結晶体の合成トライ1
<比較例2>
「化合物X」の結晶体の合成トライ2
4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル1.1g、アセトン2.5g、炭酸カリウム1.7gを試験管に仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル2.4gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、酢酸ブチル5.0g、水5.0g加えて撹拌し、静置後水層を除去した。残った油層に水5.0gを加えて撹拌し、静置後水層を除去する操作を2回繰り返して、無機塩を除去した。そして、残った油層の溶媒を減圧蒸留により除去した。この蒸留残渣を室温で10時間程度放置したが、結晶が析出しなかった。
そこで、-20℃の冷凍庫で24時間程度冷却したところ、オイルの全体が結晶状に固化しており、「化合物X」の結晶0.9gを取得することができた。
「化合物X」の結晶体の合成トライ2
4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル1.1g、アセトン2.5g、炭酸カリウム1.7gを試験管に仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル2.4gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、酢酸ブチル5.0g、水5.0g加えて撹拌し、静置後水層を除去した。残った油層に水5.0gを加えて撹拌し、静置後水層を除去する操作を2回繰り返して、無機塩を除去した。そして、残った油層の溶媒を減圧蒸留により除去した。この蒸留残渣を室温で10時間程度放置したが、結晶が析出しなかった。
そこで、-20℃の冷凍庫で24時間程度冷却したところ、オイルの全体が結晶状に固化しており、「化合物X」の結晶0.9gを取得することができた。
<比較例3>
「化合物X」の結晶体の合成トライ3
4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル27.6g、N-メチルピロリドン70.0g、炭酸カリウム47.2gを4つ口フラスコに仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル44.6gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、シクロヘキサン61.2g、水134.0g加えて撹拌し、静置後水層を除去した。残った油層に水60.0gを加えて撹拌し、静置後水層を除去する操作を3回繰り返して、無機塩を除去した。そして、残った油層の溶媒を減圧蒸留により除去した。
この蒸留残渣を25℃まで冷却後、上記比較例2で取得した種結晶を添加し、メタノール39.8g加え、結晶を析出させた。この結晶をろ過し、乾燥を行い「化合物X」の結晶体を15.6g取得した。
比較例1~3の結果より、「化合物X」は種結晶を使用しないと、本発明の結晶体である「化合物A」または「化合物B」と同様の方法では、結晶体が得られないことが明らかとなった。
収率は4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニルに対して、52.8mol%であった。
高速液体クロマトグラフィー測定による純度は97.2%、示差走査熱量分析による吸熱ピークトップ温度は52℃であった。
得られた「化合物X」の結晶体について、5%重量減少温度を測定したところ、245℃であった。
「化合物X」の結晶体の合成トライ3
4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニル27.6g、N-メチルピロリドン70.0g、炭酸カリウム47.2gを4つ口フラスコに仕込み、60℃まで昇温し、同温で3時間撹拌した。次いで、反応液の温度を60~70℃に保ちながら、臭化アリル44.6gを滴下した。24時間撹拌後、反応液の温度を室温(20℃)まで下げ、シクロヘキサン61.2g、水134.0g加えて撹拌し、静置後水層を除去した。残った油層に水60.0gを加えて撹拌し、静置後水層を除去する操作を3回繰り返して、無機塩を除去した。そして、残った油層の溶媒を減圧蒸留により除去した。
この蒸留残渣を25℃まで冷却後、上記比較例2で取得した種結晶を添加し、メタノール39.8g加え、結晶を析出させた。この結晶をろ過し、乾燥を行い「化合物X」の結晶体を15.6g取得した。
比較例1~3の結果より、「化合物X」は種結晶を使用しないと、本発明の結晶体である「化合物A」または「化合物B」と同様の方法では、結晶体が得られないことが明らかとなった。
収率は4,4’-ジヒドロキシ-3,3’,5,5’-テトラメチルビフェニルに対して、52.8mol%であった。
高速液体クロマトグラフィー測定による純度は97.2%、示差走査熱量分析による吸熱ピークトップ温度は52℃であった。
得られた「化合物X」の結晶体について、5%重量減少温度を測定したところ、245℃であった。
<高温下での保存安定性並びに結晶体の取り扱い性の確認試験1>
「化合物A(本発明の結晶体)」
実施例1で得られた「化合物A」の結晶体0.1gを、直径約3cmの円筒形の容量20mLのガラス製サンプル瓶に入れ、ふたをしたものを3つ用意した。図3の左側にその1つの状態を示す。
それぞれ40℃、50℃、60℃に保温された水バスに入れ、20時間保持した。その後、サンプル瓶を逆さにして結晶体の流動性を確認した。図4、5、6の左側にその状態を示す。
その結果、40℃、50℃、60℃何れの温度で保管した結晶体もサンプル瓶の底にほとんど張り付くことなく、結晶体の0.098gが逆さにしたサンプル瓶のふた部分に落下した。
すなわち、サンプル瓶からの「化合物A」の結晶体の排出率は98%であった。
「化合物X」
次に、上記比較例3で得られた「化合物X」の結晶体を使用して同様に試験を行い、「化合物X」の保存安定性並びに結晶体の取り扱い性を確認した。図3の右側に試験開始前の状態を示す。
その結果、50℃並びに60℃で保管した結晶体は、溶融が起こり液状に変化した。図5、6の右側にその状態を示す。
40℃で保管した結晶体は、サンプル瓶の底に固まりになって張り付いており、結晶体の0.051gが落下した。図4の右側にその状態を示す。
すなわち、サンプル瓶からの「化合物X」の結晶体の排出率は51%であった。
(結果)
以上のことから、本発明の「化合物A」の結晶体は、従来公知の「化合物X」の結晶体と比べて、高温下における保存安定性並びに結晶体の取り扱い性に優れることが明らかになった。
「化合物A(本発明の結晶体)」
実施例1で得られた「化合物A」の結晶体0.1gを、直径約3cmの円筒形の容量20mLのガラス製サンプル瓶に入れ、ふたをしたものを3つ用意した。図3の左側にその1つの状態を示す。
それぞれ40℃、50℃、60℃に保温された水バスに入れ、20時間保持した。その後、サンプル瓶を逆さにして結晶体の流動性を確認した。図4、5、6の左側にその状態を示す。
その結果、40℃、50℃、60℃何れの温度で保管した結晶体もサンプル瓶の底にほとんど張り付くことなく、結晶体の0.098gが逆さにしたサンプル瓶のふた部分に落下した。
すなわち、サンプル瓶からの「化合物A」の結晶体の排出率は98%であった。
「化合物X」
次に、上記比較例3で得られた「化合物X」の結晶体を使用して同様に試験を行い、「化合物X」の保存安定性並びに結晶体の取り扱い性を確認した。図3の右側に試験開始前の状態を示す。
その結果、50℃並びに60℃で保管した結晶体は、溶融が起こり液状に変化した。図5、6の右側にその状態を示す。
40℃で保管した結晶体は、サンプル瓶の底に固まりになって張り付いており、結晶体の0.051gが落下した。図4の右側にその状態を示す。
すなわち、サンプル瓶からの「化合物X」の結晶体の排出率は51%であった。
(結果)
以上のことから、本発明の「化合物A」の結晶体は、従来公知の「化合物X」の結晶体と比べて、高温下における保存安定性並びに結晶体の取り扱い性に優れることが明らかになった。
<高温下での保存安定性並びに結晶体の取り扱い性の確認試験2>
「化合物B(本発明の結晶体)」
実施例2で得られた「化合物B」の結晶体を使用して、「高温下での保存安定性並びに結晶体の取り扱い性の確認試験1」と同様の試験を行った。図9の左側に試験開始前の状態を示す。
それぞれ40℃、70℃に保温された水バスに入れ、20時間保持した。その後、サンプル瓶を逆さにして結晶体の流動性を確認した。図10、11の左側にその状態を示す。
その結果、40℃、70℃何れの温度で保管した結晶体もサンプル瓶の底にほとんど張り付くことなく、結晶体が逆さにしたサンプル瓶のふた部分に落下した。
サンプル瓶からの「化合物B」の結晶体の排出率は、40℃で保管した場合97%であり、70℃で保管した場合は99%であった。
「化合物X」
次に、上記比較例3で得られた「化合物X」の結晶体を使用して同様に試験を行い、「化合物X」の保存安定性並びに結晶体の取り扱い性を確認した。図9の右側に試験開始前の状態を示す。
その結果、70℃で保管した結晶体は、溶融が起こり液状に変化した。図11の右側にその状態を示す。
40℃で保管した結晶体は、サンプル瓶の底に固まりになって張り付いており、結晶体の一部が落下した。図10の右側にその状態を示す。
サンプル瓶からの「化合物X」の結晶体の排出率は38%であった。
(結果)
以上のことから、本発明の「化合物B」の結晶体は、従来公知の「化合物X」の結晶体と比べて、高温下における保存安定性並びに結晶体の取り扱い性に優れることが明らかになった。
「化合物B(本発明の結晶体)」
実施例2で得られた「化合物B」の結晶体を使用して、「高温下での保存安定性並びに結晶体の取り扱い性の確認試験1」と同様の試験を行った。図9の左側に試験開始前の状態を示す。
それぞれ40℃、70℃に保温された水バスに入れ、20時間保持した。その後、サンプル瓶を逆さにして結晶体の流動性を確認した。図10、11の左側にその状態を示す。
その結果、40℃、70℃何れの温度で保管した結晶体もサンプル瓶の底にほとんど張り付くことなく、結晶体が逆さにしたサンプル瓶のふた部分に落下した。
サンプル瓶からの「化合物B」の結晶体の排出率は、40℃で保管した場合97%であり、70℃で保管した場合は99%であった。
「化合物X」
次に、上記比較例3で得られた「化合物X」の結晶体を使用して同様に試験を行い、「化合物X」の保存安定性並びに結晶体の取り扱い性を確認した。図9の右側に試験開始前の状態を示す。
その結果、70℃で保管した結晶体は、溶融が起こり液状に変化した。図11の右側にその状態を示す。
40℃で保管した結晶体は、サンプル瓶の底に固まりになって張り付いており、結晶体の一部が落下した。図10の右側にその状態を示す。
サンプル瓶からの「化合物X」の結晶体の排出率は38%であった。
(結果)
以上のことから、本発明の「化合物B」の結晶体は、従来公知の「化合物X」の結晶体と比べて、高温下における保存安定性並びに結晶体の取り扱い性に優れることが明らかになった。
<比較例4>
市販品のトリアリルイソシアヌレート(東京化成工業(株)製)の5%重量減少温度を測定したところ、176℃であった。
このことから、本発明の「化合物A」または「化合物B」の結晶体は、従来汎用されているトリアリルイソシアヌレートと比べて著しく重量減少温度が高く、熱安定性に優れ、揮発分による汚染を抑制することができることが明らかになった。
市販品のトリアリルイソシアヌレート(東京化成工業(株)製)の5%重量減少温度を測定したところ、176℃であった。
このことから、本発明の「化合物A」または「化合物B」の結晶体は、従来汎用されているトリアリルイソシアヌレートと比べて著しく重量減少温度が高く、熱安定性に優れ、揮発分による汚染を抑制することができることが明らかになった。
Claims (5)
- 下記式(1)で表される化合物の結晶体。
(式中、Rは水素原子またはメチル基を表す。ただし、4つのRは全て同一である。)
- 式(1)で表される化合物が、2,2’,3,3’,5,5’-ヘキサメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、請求項1に記載の結晶体。
- 示差走査熱量分析による吸熱ピークトップ温度が70~75℃の範囲である請求項2に記載の結晶体。
- 式(1)で表される化合物が、3,3’-ジメチル-4,4’-ビス(2-プロペン-1-イルオキシ)-1,1’-ビフェニルである、請求項1に記載の結晶体。
- 示差走査熱量分析による吸熱ピークトップ温度が110~115℃の範囲である請求項4に記載の結晶体。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-114197 | 2019-06-20 | ||
| JP2019114197 | 2019-06-20 | ||
| JP2019237031 | 2019-12-26 | ||
| JP2019-237031 | 2019-12-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020256045A1 true WO2020256045A1 (ja) | 2020-12-24 |
Family
ID=74040834
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/023893 WO2020256045A1 (ja) | 2019-06-20 | 2020-06-18 | ヘキサメチル置換/ジメチル置換4,4'-ビス(2-プロペン-1-イルオキシ)-1,1'-ビフェニルの結晶体 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202110779A (ja) |
| WO (1) | WO2020256045A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023026845A1 (ja) * | 2021-08-23 | 2023-03-02 | 本州化学工業株式会社 | 硬化性樹脂組成物、ワニス、プリプレグ、硬化物 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010073960A1 (ja) * | 2008-12-26 | 2010-07-01 | 昭和電工株式会社 | エポキシ化合物の製造方法 |
| WO2010084823A1 (ja) * | 2009-01-22 | 2010-07-29 | チッソ株式会社 | 液晶組成物および液晶表示素子 |
| JP2014034580A (ja) * | 2012-08-07 | 2014-02-24 | Ajinomoto Co Inc | 樹脂組成物 |
| JP2014169428A (ja) * | 2013-02-05 | 2014-09-18 | Nippon Kayaku Co Ltd | アリルエーテル樹脂およびその製造方法 |
| US20190194408A1 (en) * | 2017-12-25 | 2019-06-27 | Industrial Technology Research Institute | Thermally conductive resin, resin composition, prepreg, and copper clad laminate |
| US20190194383A1 (en) * | 2017-12-25 | 2019-06-27 | Iteq Corporation | Resin composition, prepreg, and copper clad laminate |
| CN110938357A (zh) * | 2019-10-29 | 2020-03-31 | 联茂(无锡)电子科技有限公司 | 多层结构以及基板的制造方法 |
-
2020
- 2020-06-18 WO PCT/JP2020/023893 patent/WO2020256045A1/ja active Application Filing
- 2020-06-19 TW TW109120810A patent/TW202110779A/zh unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010073960A1 (ja) * | 2008-12-26 | 2010-07-01 | 昭和電工株式会社 | エポキシ化合物の製造方法 |
| WO2010084823A1 (ja) * | 2009-01-22 | 2010-07-29 | チッソ株式会社 | 液晶組成物および液晶表示素子 |
| JP2014034580A (ja) * | 2012-08-07 | 2014-02-24 | Ajinomoto Co Inc | 樹脂組成物 |
| JP2014169428A (ja) * | 2013-02-05 | 2014-09-18 | Nippon Kayaku Co Ltd | アリルエーテル樹脂およびその製造方法 |
| US20190194408A1 (en) * | 2017-12-25 | 2019-06-27 | Industrial Technology Research Institute | Thermally conductive resin, resin composition, prepreg, and copper clad laminate |
| US20190194383A1 (en) * | 2017-12-25 | 2019-06-27 | Iteq Corporation | Resin composition, prepreg, and copper clad laminate |
| CN110938357A (zh) * | 2019-10-29 | 2020-03-31 | 联茂(无锡)电子科技有限公司 | 多层结构以及基板的制造方法 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023026845A1 (ja) * | 2021-08-23 | 2023-03-02 | 本州化学工業株式会社 | 硬化性樹脂組成物、ワニス、プリプレグ、硬化物 |
| KR20240044418A (ko) | 2021-08-23 | 2024-04-04 | 혼슈우 카가쿠고교 가부시키가이샤 | 경화성 수지 조성물, 바니시, 프리프레그, 경화물 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202110779A (zh) | 2021-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6426226B2 (ja) | 9,9−ビス(4−(2−ヒドロキシエトキシ)フェニル)フルオレンの結晶体 | |
| WO2021015083A1 (ja) | 2,2'-ビス(エトキシカルボニルメトキシ)-1,1'-ビナフチルの結晶体 | |
| KR20240018699A (ko) | 2,2'-비스(카르복시메톡시)-1,1'-비나프틸의 결정체 | |
| WO2020256045A1 (ja) | ヘキサメチル置換/ジメチル置換4,4'-ビス(2-プロペン-1-イルオキシ)-1,1'-ビフェニルの結晶体 | |
| JP7295692B2 (ja) | 2,2’-ビス(カルボキシメトキシ)-1,1’-ビナフチルの結晶体 | |
| JP7516701B2 (ja) | ビナフチルカルボン酸類の製造方法 | |
| US11091461B2 (en) | Method for producing crystal of uracil compound | |
| JP2006188449A (ja) | 環式ジスルホン酸エステルの製造方法 | |
| WO2022138178A1 (ja) | 1,1,1-トリス(4-ヒドロキシ-3,5-ジメチルフェニル)エタンの結晶体及びその製造方法 | |
| JP6890871B1 (ja) | エステル基含有酸二無水物誘導体の製造方法 | |
| JP4799892B2 (ja) | シクロヘキサン誘導体およびその製造方法 | |
| JP7027541B2 (ja) | ジシクロヘキサンジカルボン酸ジエステルの製造方法およびジシクロヘキサンジカルボン酸の製造方法 | |
| JP2020164523A (ja) | 9,9−ビス(6−ヒドロキシ−2−ナフチル)フルオレンの結晶多形およびその製造方法 | |
| JP7535367B1 (ja) | ビナフチル骨格を有するジエステル化合物の結晶及びその製造方法 | |
| KR102702691B1 (ko) | 2,2'-비스(카르복시메톡시)-1,1'-비나프틸의 결정체 | |
| KR102236738B1 (ko) | 1,1-비스(4-(2-히드록시에톡시)페닐)-3,3,5-트리메틸시클로헥산의 결정체 및 그의 제조방법 | |
| WO2023112604A1 (ja) | イソソルビド-ビス(トリメリテートアンハイドライド)の結晶及びその製造方法 | |
| JP4390193B2 (ja) | エポキシ樹脂の製造法 | |
| WO2023112605A1 (ja) | イソマンニド-ビス(トリメリテートアンハイドライド)の結晶及びその製造方法 | |
| JP7315386B2 (ja) | ビナフタレン骨格を有する化合物およびその結晶多形体、ならびにその製造方法 | |
| WO2023176687A1 (ja) | ビフェナントレン化合物又はそのアルカリ金属塩 | |
| WO2023176736A1 (ja) | 新規な1,3-ビス(1-メチル-1-フェニルエチル)ベンゼン化合物 | |
| WO2020004207A1 (ja) | 9,9-ビス(4-ヒドロキシフェニル)-2,3-ベンゾフルオレンの結晶体 | |
| TWI631104B (zh) | 2-胺基菸鹼酸苄酯衍生物之製造方法 | |
| JP4507398B2 (ja) | 3−ハロメチルオキセタン化合物の合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20825881 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20825881 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |