WO2019189573A1 - ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法 - Google Patents
ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法 Download PDFInfo
- Publication number
- WO2019189573A1 WO2019189573A1 PCT/JP2019/013602 JP2019013602W WO2019189573A1 WO 2019189573 A1 WO2019189573 A1 WO 2019189573A1 JP 2019013602 W JP2019013602 W JP 2019013602W WO 2019189573 A1 WO2019189573 A1 WO 2019189573A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- cells
- homologous recombination
- dna
- genome
- Prior art date
Links
Images












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/711—Natural deoxyribonucleic acids, i.e. containing only 2'-deoxyriboses attached to adenine, guanine, cytosine or thymine and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New breeds of animals
- A01K67/027—New breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/715—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons
- C07K14/7155—Receptors; Cell surface antigens; Cell surface determinants for cytokines; for lymphokines; for interferons for interleukins [IL]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0647—Haematopoietic stem cells; Uncommitted or multipotent progenitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/07—Animals genetically altered by homologous recombination
- A01K2217/072—Animals genetically altered by homologous recombination maintaining or altering function, i.e. knock in
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/108—Swine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; CARE OF BIRDS, FISHES, INSECTS; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/03—Animal model, e.g. for test or diseases
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
Definitions
- the present invention relates to a genome editing method, a composition, a cell, a cell preparation, and a method for producing a cell preparation.
- homologous recombination is used when a specific gene is inserted into a specific site in the genome of a eukaryotic cell and the target genomic DNA is completely replaced with a desired base sequence.
- a gene having DNA having a sequence homologous to the site for insertion on the genome hereinafter referred to as a homologous arm
- a vector hereinafter referred to as a targeting vector
- homologous recombination occurs between the genomic DNA and the vector, and the desired base sequence of the target genomic DNA can be replaced. This replacement is characterized by error-free.
- Patent Document 1 a genome of a one-cell stage embryo cell is cleaved with Cas9 protein, and a homologous arm that hybridizes to the 5 ′ end and 3 ′ end of the target sequence, respectively, and a nucleic acid insert (introduction) adjacent to the homologous arm.
- a method for introducing the nucleic acid insert into the cell using a targeting vector comprising a foreign gene) is disclosed.
- Non-Patent Document 1 a normal HBB (haemoglobin bin) gene is obtained by homologous recombination using a CRISPR / Cas9 system in combination with a Cas9 protein and an adenovirus-associated vector for hematopoietic stem cells derived from a ⁇ -thalassemia patient. It is disclosed that it has been introduced.
- HBB haemoglobin bin
- Non-Patent Document 2 a search for a low molecular compound that suppresses non-homologous end joining or promotes homologous recombination in gene transfer by homologous recombination using the CRISPR / Cas9 system has been made. It is disclosed that Scr7, L755507 and resveratrol were used as such low molecular weight compounds to promote homologous recombination in pig fetal fibroblasts.
- Non-Patent Document 3 discloses a method of relatively increasing the frequency of homologous recombination by suppressing the expression of KU70, DNA ligase IV and the like by RNA interference and decreasing the frequency of nonhomologous recombination. .
- Non-Patent Document 4 in genome editing using the CRISPR / Cas9 system, a 3 ′ end of a cleaved DNA that is asymmetrically released before Cas9 dissociates from double-stranded DNA and is not complementary to sgRNA.
- a method for increasing the frequency of homologous recombination by providing complementary single-stranded DNA is disclosed.
- Non-homologous end joining and homologous recombination are known as DNA repair mechanisms for DNA double-strand breaks.
- Non-homologous end joining proceeds in a shorter time than homologous recombination. Therefore, in order to increase the frequency of homologous recombination, it is necessary to devise a method for relatively reducing the frequency of occurrence of non-homologous end joining.
- the mechanism of non-homologous end joining itself is suppressed, the ability of cells to repair DNA double-strand breaks is greatly impaired, and thus the risk to living organisms (non-viability and tumorigenesis) becomes excessive. As a result, practical application and clinical application in this direction are difficult.
- the present invention provides a genome editing method, a composition, a cell, a cell preparation, and a method for producing the cell preparation, which can increase the frequency of homologous recombination without impairing the ability of the cell to inherent non-homologous end joining.
- the purpose is to do.
- the present invention is as follows.
- a method for editing a genome in an isolated cell wherein foreign DNA having a homology arm having a length of less than 500 bp at the 5 ′ end and 3 ′ end is converted into a foreign genome upon double strand breakage of the target genomic DNA.
- a genome editing method comprising introducing at least one of 5 'end and 3' end of DNA into a target genome by homologous recombination.
- the genome editing method according to [1] or [2], wherein the cells are blood cells or undifferentiated cells.
- [7] A method for editing genome in an isolated cell, wherein foreign DNA having a homologous arm with a length of less than 500 bp is subjected to homologous recombination on both sides of the 5 ′ side and 3 ′ side of the foreign DNA.
- a genome editing method comprising introducing into a target genome.
- the genome editing method according to [7], wherein the cells are blood cells or undifferentiated cells.
- the genome editing method according to [7] or [8], wherein the cell is a stem cell.
- a composition comprising foreign DNA having a homology arm with a length of less than 500 bp at both ends.
- the composition according to [11] further comprising a target genomic DNA cleaving enzyme or DNA or mRNA encoding the enzyme.
- a method for producing a cell preparation for treating severe combined immunodeficiency comprising a homologous arm having a length of less than 500 bp in a cell and at least part of the wild-type DNA of the target genomic DNA
- a cell characterized by introducing at least one of the 5 ′ end and 3 ′ end of the foreign DNA into the genome of the cell by homologous recombination when the target genomic DNA is double-strand-breaked Preparation method of the preparation.
- the method for producing a cell preparation according to [15] wherein the foreign DNA is introduced into the target genomic DNA by homologous recombination between the 5 ′ end and the 3 ′ end of the foreign DNA.
- a method for producing a cell preparation for treating severe combined immunodeficiency comprising a homologous arm having a length of less than 500 bp in a cell and at least a part of the wild-type DNA of the target genomic DNA
- a method for producing a cell preparation which comprises introducing foreign DNA having both 5 ′ side and 3 ′ side of the foreign DNA into the genome of the cell by homologous recombination.
- the method for producing a cell preparation according to [21] wherein the cells are blood cells or undifferentiated cells.
- a high homologous recombination frequency is realized in the target genome as compared with nonhomologous recombination without impairing the ability of the cell to have nonhomologous end joining.
- FIG. 2 is a schematic diagram for repairing an IL2RG gene mutation in a SCID pig by causing homologous recombination at only one end of the foreign DNA. The 5 'end of the foreign DNA is repaired by non-homologous recombination and the 3' end is repaired by homologous recombination.
- Homologous recombination expresses a fusion protein of LMNB1 and GFP, which localizes to the nuclear membrane. Since GFP is localized in the nuclear membrane in all GFP positive cells, it can be seen that the GFP gene is inserted into the LMNB1 locus only by homologous recombination. It is the result which confirmed the insertion efficiency of the GFP gene in the LMNB1 locus in HEK293T cell by flow cytometry. It is the result of the electrophoresis which confirmed that the GFP gene was inserted only by the homologous recombination in the HPRT locus of the bone marrow stromal cell derived from a human by genomic PCR.
- foreign DNA having a homologous arm having a length of less than 500 bp is obtained by subjecting at least one of the 5 ′ end and the 3 ′ end of the foreign DNA to a homologous group upon double strand breakage of the target genomic DNA. It is a method of introducing into the target genome by replacement.
- the present invention provides a method for genome editing in a cell, wherein a foreign DNA having a homologous arm having a length of less than 500 bp is converted into a 5 ′ end of the foreign DNA upon double strand breakage of the target genomic DNA.
- the double strand of the target genomic DNA is cleaved at a location on the target genomic DNA where foreign DNA is to be introduced.
- the system used for double-strand breakage of the target genomic DNA is not particularly limited, and examples thereof include a CRISPR-Cas9 system, a transcription-activator-like-effector nuclease (TALEN) system, and a Zn finger nuclease system.
- the method for introducing these systems into cells is not particularly limited, and the target genomic DNA cleaving enzyme itself may be introduced into the cells, or the target genomic DNA cleaving enzyme expression vector may be introduced into the cells.
- the system used for double-strand breakage of the target genomic DNA is introduced into the cell simultaneously with the foreign DNA or before or after the foreign DNA.
- the CRISPR-Cas9 system a method of introducing a Cas9 expression vector and an expression vector encoding a guide RNA that induces Cas9 at a site to be cleaved, a recombinant Cas9 protein that is expressed and purified, and a guide RNA And the like.
- the guide RNA may be divided into tracrRNA and crRNA, or may be sgRNA connected to one.
- the CRISPR-Cas9 system is preferable as the system used for double-strand breakage of the target genomic DNA.
- the means for introducing foreign genes, nucleic acids and proteins into cells is not particularly limited, and any of a method using a viral vector, a non-viral introduction method, or other known methods may be used.
- methods using viral vectors include retrovirus vectors, lentivirus vectors, adenovirus vectors, adeno-associated (AAV) virus vectors, herpes virus vectors, Sendai virus vectors, Sindbis virus vectors, and the like.
- the non-viral introduction method include calcium phosphate method, lipofection method, electroporation method, microinjection method, whisker method, plasma method, laser injection method, particle gun method, and Agrobacterium method.
- the cells to be subjected to the genome editing method of the present invention are not particularly limited. Isolated cells are preferred, and examples include animal cells, plant cells, insect cells, fungi such as yeast and mold, and bacteria such as Escherichia coli. Examples of animal cells include animal-derived stem cells, germ cells, germ line cells, cell lines, primary culture cells, and cells derived from stem cells or cells made from primary culture cells. The stem cells may be established cells or primary cultured cells.
- the genome editing method of the present invention is not necessarily limited to isolated cells. The animal individual itself or somatic cells and stem cells within the individual are also targeted.
- Animal-derived stem cells are characterized by (i) having the ability to self-replicate and (ii) having pluripotency.
- Animal stem cells are classified into pluripotent stem cells, multipotent stem cells, oligopotent stem cells, and unipotent stem cells according to the difference in differentiation ability.
- animal stem cells include embryonic stem cells such as ES cells and EG cells, ES-like stem cells such as induced pluripotent stem cells (iPS cells), embryonic stem cells, muse cells, placental stem cells, hematopoietic stem cells, Leaf stem cells (dental pulp-derived mesenchymal stem cells, adipose-derived mesenchymal stem cells, bone marrow-derived mesenchymal stem cells, synovial-derived mesenchymal stem cells, etc.), hair follicle stem cells, mammary stem cells, neural stem cells, satellite cells, and intestinal epithelium Examples include adult stem cells such as stem cells, and germline stem cells such as GS cells. Animal stem cells may be cells obtained by genetic engineering of stem cells.
- Hematopoietic stem cells include pluripotent stem cells that suppress immune rejection by reorganizing human leukocyte type antigen (HLA). Hematopoietic stem cells are preferred as animal cells.
- germ cells and germ line cells include eggs, oocytes, oocyte cells, sperm, spermatocytes, spermatogonia, sperm stem cells (spermatogonial stem cells), primordial germ cells and the like.
- a fertilized egg fertilized by an egg and sperm may be used. Further, it may be a 2-cell to 8-cell embryo in which a fertilized egg is divided, or a morula to blastocyst until implantation.
- Animal cell lines are not particularly limited. Examples of animal cell lines include Chinese hamster ovary tissue-derived cells (CHO cells), African green monkey kidney-derived cell lines (Vero cells), human liver cancer-derived cells (HepG2 cells), and canine kidney tubular epithelial cell-derived cells. Strain (MDCK cell), human fetal kidney cell line (HEK293 cell), and human hepatoma tissue-derived established cell line (huGK-14).
- Animal primary cultured cells are not particularly limited, and may be derived from normal tissue or diseased tissue.
- Examples of primary cultured cells in animals include dermal papilla cells, endothelial cells, epithelial cells, epidermal keratinocytes, melanocytes, cardiomyocytes, smooth muscle cells, skeletal muscle cells, skeletal myoblasts, osteoblasts, chondrocytes, fibers
- Examples include blast cells, hepatocytes, nerve cells, and immune cells such as regulatory T cells, killer T cells, and gamma delta T cells.
- Cells derived from animal stem cells may be stem cells or differentiated cells.
- Examples of cells derived from animal stem cells include iPS cell-derived retinal pigment epithelial cells, iPS cell-derived neurons, iPS cell-derived immune system cells such as iPS cell-derived killer T cells, iPS cell-derived myocardial progenitor cells and iPS cells Examples include derived hepatocytes.
- Cells produced from primary animal cultured cells are not particularly limited.
- a cell prepared from an animal primary cultured cell is a cell obtained by applying genetic engineering to an animal primary cultured cell.
- Examples of cells produced from primary animal cultured cells include CAR (chimeric antigen receptor) -T cells.
- the present inventors have found that the likelihood of homologous recombination does not depend on the species of animal, the target locus, the transgene, or the type of nuclease that cleaves the target genome.
- blood cells or undifferentiated cells are preferred.
- undifferentiated cells stem cells are more preferable, and hematopoietic stem cells are particularly preferable.
- Plant cells are not particularly limited. Examples of plant cells include cells derived from plant meristems or seeds and callus.
- the callus may be any of those derived from plant tissue pieces, those that are inducible to injury, those that are bacterially induced, those that are formed in interspecific hybridization, and cultured cells.
- cells and callus derived from plant meristem or seed are PLT1, PLT5, LBD16, LBD17, LBD18, LBD29, ARR1, ARR21, ESR1, ESR2, WIND1, WIND2, WIND3, WIND4, LEC1, LEC2 , AGL15, BBM, RKD1, RKD2, and at least one of pluripotency markers such as WUS.
- the genome editing method in plant cells will be described later.
- a targeting vector having a homology arm of less than 500 bp at both ends of the foreign DNA is introduced into the cell.
- the homologous arm refers to DNA that is provided at the 5 'end and 3' end of the foreign DNA to be inserted and has a sequence homologous to the site for insertion on the genome.
- the upper limit of the length of the homologous arm is less than 500 bp, preferably 300 bp or less, more preferably 100 bp or less, particularly preferably 50 bp or less, and may be 10 bp or less.
- the introduced homologous arm contributes to homologous recombination at both the 5 ′ end and the 3 ′ end.
- the lower limit of the length of the homologous arm is preferably 5 bp or more, and more preferably 10 bp or more.
- the length of the homologous arm is preferably 5 to 499 bp, more preferably 5 to 300 bp, further preferably 5 to 100 bp, particularly preferably 5 to 50 bp, and most preferably 10 to 50 bp.
- the length of the foreign DNA is not particularly limited as long as it can be inserted into the genome.
- Examples of the length of the foreign DNA include 50 bp to 10 kbp, 100 to 5 kbp, 100 to 1 kbp, and 100 to 500 bp.
- Examples of foreign DNA include wild-type DNA of target sequence, codon optimized sequence DNA, tagged foreign DNA, promoter sequence, transcription termination sequence, functional gene sequence, fluorescent protein marker gene sequence, drug selection gene sequence, multiple cloning A site arrangement, a combination thereof, and the like can be given.
- the type of targeting vector to be used is not particularly limited, and conventionally known ones such as plasmid vectors and virus vectors can be used.
- virus vectors include retrovirus vectors, lentivirus vectors, adenovirus vectors, adeno-associated (AAV) virus vectors, herpes virus vectors, Sendai virus vectors, Sindbis virus vectors, and the like.
- AAV adeno-associated virus vectors
- the targeting vector include various vectors for genome editing using the CRISR / Cas9 system.
- a targeting vector using a HITI (Homology-independent targeted integration) system can be mentioned.
- the length of the homologous arm in a gene targeting vector for homologous recombination is considered to be more efficiently homologous recombination from the viewpoint of increasing the ratio of homologous regions.
- the genome editing method of the present embodiment can realize homologous recombination with a very high frequency relative to non-homologous recombination on both sides of the 5 'end and 3' end of the foreign DNA.
- a targeting vector using a short homologous arm of less than 500 bp double-strand breakage of DNA can be achieved without suppressing the non-homologous end joining mechanism inherent in cells.
- the frequency of homologous recombination is significantly higher than that of non-homologous recombination while maintaining the ability of cells to repair.
- the present invention can produce blood cells that can be used to treat an individual having a disease caused by a gene mutation.
- the disease caused by the gene mutation is not particularly limited.
- innate immune deficiency in addition to X-SCID in the examples, adenosine deaminase [ADA] deficiency, chronic granulomatosis, X-linked ⁇ -globulin blood) [XLA], ZAP-70 deficiency, high IgM syndrome, IgA deficiency, IgG subclass deficiency, Bloom syndrome, Wiscott-Aldrich syndrome, Ataxia telangiectasia, DiGeorge syndrome), Fanconi anemia, thalassemia, sickle cell anemia, Any of white matter dystrophy, hemophilia, mucopolysaccharidosis and the like may be used.
- the present invention realizes a high homologous recombination frequency compared to non-homologous recombination, various diseases that are difficult to treat by non-homologous recombination but are expected to be treated by homologous recombination, such as giant genes It is extremely useful for the treatment of diseases with mutations (such as muscular dystrophy) and diseases with long gene mutations (such as triplet / repeat diseases such as Huntington's disease).
- the indication of the present invention is not necessarily limited to the treatment of diseases caused by genetic mutations.
- the present invention can be widely used for functional modification of mesenchymal stem cells and T cells. For example, modification of the HLA locus of mesenchymal stem cells and CAR-T cells.
- genome-modified cells prepared by the present invention include various cancers, leukemias, hematopoietic disorders, myelodysplastic syndromes, ischemic diseases such as myocardial infarction / cerebral infarction / occlusive arteriosclerosis, Buerger's disease, peripheral diseases, and severe lower limbs. It can be used for the treatment of ischemia, pulmonary hypertension, autoimmune disease, Rubus nephritis, Crohn's disease, corneal disease, corneal disorder, glaucoma, optic neuropathy, retinitis pigmentosa, macular degeneration and the like.
- the application of the present invention is not limited to medical treatment. For example, it is possible to apply the method for editing genomes of animal cells according to the present invention to the production of beef cattle with a high content of highly unsaturated fatty acids and excellent taste, and the production of tuna with an increased ratio of large trout.
- the present invention relates to a method for genome editing in a cell, wherein a foreign DNA having a homologous arm having a length of less than 500 bp is converted into a 5 ′ end of the foreign DNA upon double strand breakage of the target genomic DNA.
- a method for introducing one of the 3 ′ ends into the genome of the cell by homologous recombination and the other by non-homologous recombination is provided.
- Examples of this embodiment include mouse fertilized eggs and pig bone marrow stromal cells.
- the length of one homologous arm in the foreign DNA is preferably at least twice as long as the other homologous arm, and preferably at least 3 times. More preferably, it is 4 times or more, more preferably 5 times or more.
- the length of the short homologous arm is preferably 50 bp or less, more preferably 30 bp or less, particularly preferably 10 bp or less, and may be 0 bp.
- the length of the long homologous arm is preferably 30 bp or more, more preferably 40 bp or more, and particularly preferably 50 bp or more.
- non-homologous recombination means non-homologous end joining.
- the length of the foreign DNA is not particularly limited as long as it can be inserted into the genome, and examples thereof include 100 bp to 10 kbp.
- the generation mechanism of unilateral homologous recombination in the genome editing method of this embodiment is unknown, but is presumed as follows. Short homologous arms are less likely to undergo homologous recombination than long homologous arms, and double-strand breaks in genomic DNA and short homologous arms are linked by non-homologous recombination mechanisms (non-homologous end joining). . Thus, when one end of the transgene is connected to the genomic DNA, it serves as a fulcrum, and the long homologous arm at the other end is located near the homologous sequence on the genome, and homologous recombination is likely to occur. . That is, it is considered that one end of the foreign DNA is first connected by non-homologous recombination, and then the foreign DNA is integrated into the genome by homologous recombination on the other end side.
- the present invention provides a method for genome editing in an isolated cell, wherein foreign DNA having a homologous arm having a length of less than 500 bp is selected from the 5 ′ side and the 3 ′ side of the foreign DNA, A method for introducing both sides into a target genome by homologous recombination is provided.
- a targeting vector that causes homologous recombination without double-strand breaking the target genomic DNA.
- Such targeting vectors include AAV vectors including single stranded DNA. The same as in the first embodiment, except that the target genomic DNA is not cleaved by double strands.
- composition of the present invention contains foreign DNA having a homology arm with a length of less than 500 bp at both ends.
- composition of the present invention may contain a target genomic DNA cleaving enzyme or DNA or mRNA encoding the enzyme, not a DNA cleaving enzyme, but a nicked or double stranded nick on one side of double-stranded DNA It may be a helicase that separates DNA into single strands.
- examples of the target genomic DNA cleaving enzyme include Cas9, Transcryption activator-like effector nuclease (TALEN), Zn finger nuclease, and the like.
- examples of the DNA or mRNA encoding the enzyme include DNA or mRNA encoding these proteins.
- the composition preferably contains a guide RNA that induces Cas9.
- the composition may also contain an expression vector encoding a guide RNA.
- the length of the homologous arm provided at both ends of the foreign DNA is less than 500 bp, preferably 300 bp or less, as in the above-described [Genome editing method].
- the foreign DNA is the same as that in [Genome editing method] described above, and the composition of the present invention may contain a targeting vector containing the foreign DNA.
- the targeting vector is the same as that in [Genome editing method] described above.
- the foreign DNA described above may be contained in one type of vector or may be contained in a plurality of types of vectors.
- the vector is not particularly limited, and is the same as that in [Genome editing method] described above.
- composition of the present invention is preferably for pharmaceutical use, and more preferably contains a pharmaceutically acceptable carrier.
- the pharmaceutical composition of the present embodiment is, for example, orally in the form of tablets, coated tablets, pills, powders, granules, capsules, solutions, suspensions, emulsions, or injections, suppositories. It is administered parenterally in the form of an external preparation for skin.
- binders such as gelatin, corn starch, gum tragacanth and gum arabic; excipients such as starch and crystalline cellulose; swelling agents such as alginic acid; solvents for injection such as water, ethanol and glycerin; Examples thereof include adhesives such as rubber adhesives and silicone adhesives.
- a pharmaceutically acceptable carrier is used singly or in combination of two or more.
- composition of the present invention may further contain an additive.
- Additives include lubricants such as calcium stearate and magnesium stearate; sweeteners such as sucrose, lactose, saccharin and maltitol; flavoring agents such as peppermint and red oil; stabilizers such as benzyl alcohol and phenol; phosphoric acid Buffers such as salts and sodium acetate; Solubilizing agents such as benzyl benzoate and benzyl alcohol; Antioxidants; Preservatives and the like.
- An additive is used by mixing 1 type individually or 2 types or more.
- composition of the present invention is preferably for the treatment of severe combined immunodeficiency.
- severe combined immunodeficiency disease SCID
- the most common disease type is the X-chain linkage type, which results from mutations in the IL-2 receptor ⁇ gene (IL2RG).
- the foreign DNA contained in the X-chain-linked severe combined immunodeficiency treatment composition contains at least a part of the wild type IL-2 receptor ⁇ gene.
- the target genomic DNA cleaving enzyme it preferably contains a guide RNA that hybridizes to the IL-2 receptor ⁇ gene on the target genome or an expression vector that encodes the guide RNA.
- the gene therapy method of the present invention comprises an enzyme that cleaves a target genomic DNA having a mutation, or a DNA or mRNA that encodes the enzyme, and a wild type of the target genomic DNA that has a homology arm of less than 500 bp at both ends.
- the mutation is caused by exon and intron of the target genomic DNA or partial or complete deletion, substitution, insertion of an arbitrary sequence or the like in the expression regulatory region of the target genomic DNA.
- the administration method is not particularly limited, and may be appropriately determined according to the patient's symptoms, weight, age, sex, and the like.
- tablets, coated tablets, pills, powders, granules, capsules, solutions, suspensions, emulsions and the like are administered orally.
- the injection is administered alone or mixed with a normal fluid such as glucose or amino acid, and is administered intravenously, and further, if necessary, intramedullary, intraarterial, intramuscular, intradermal, subcutaneous or intraperitoneal administration. Is done.
- the dosage of the pharmaceutical composition varies depending on the patient's symptoms, body weight, age, sex, etc., and cannot be determined unconditionally, but in the case of oral administration, for example, 1 ⁇ g to 10 g per day, such as 1 day 0.01 to 2000 mg of active ingredient may be administered per unit. In the case of injections, for example, 0.1 ⁇ g to 1 g per day, for example 0.001 to 200 mg per day, may be administered.
- the cell of the present invention is characterized in that a fragment derived from a foreign DNA remains on the target genome on the 5 ′ side or 3 ′ side of the genomic DNA insertion site.
- one end of the foreign DNA is first connected to one end of the target genomic DNA generated by double-strand break by non-homologous recombination. Therefore, the cell of the present invention has a foreign DNA-derived fragment on the 5 ′ side or 3 ′ side of the genomic DNA insertion site. As shown in the results of FIG.
- the cell preparation of the present invention contains, for example, a cell in which a target genomic DNA having a mutation is edited to a wild type by using the genome editing method of the second embodiment described above. Furthermore, as described in the above [Cell], the cell preparation of the present invention comprises a cell in which a foreign DNA-derived fragment remains on the target genome on the 5 ′ side or 3 ′ side of the genomic DNA insertion site. Including.
- the method for producing a cell preparation of the present invention is a method for producing a cell preparation for treating severe combined immunodeficiency, wherein the cell has a homologous arm having a length of less than 500 bp, and the target genomic DNA is wild-type.
- the target genomic DNA is double-strand-cut, at least one of the 5 ′ end and 3 ′ end of the foreign DNA is introduced into the genome of the cell by homologous recombination. It is a method to do.
- the present invention relates to a method for producing a cell preparation for treating severe combined immunodeficiency, wherein the cell has a homology arm of less than 500 bp in length and said wild type target genomic DNA
- a cell in which foreign DNA having at least a part of DNA is introduced into the genome of the cell by homologous recombination between the 5 ′ end and the 3 ′ end of the foreign DNA at the time of double strand breakage of the target genomic DNA A method for producing a formulation is provided.
- the present invention relates to a method for producing a cell preparation for treating severe combined immunodeficiency, wherein the cell has a homology arm of less than 500 bp in length and said wild type target genomic DNA
- the foreign DNA having at least a part of the DNA is subjected to homologous recombination of one of the 5 ′ end and 3 ′ end of the foreign DNA and non-homologous recombination when the target genomic DNA is double-stranded.
- a method for producing a cell preparation to be introduced into the genome of a cell is provided.
- the present invention relates to a method for producing a cell preparation for treating severe combined immunodeficiency, wherein the cell has a homology arm of less than 500 bp in length and said wild type target genomic DNA
- a method for producing a cell preparation wherein foreign DNA having at least a part of DNA is introduced into the genome of the cell by homologous recombination between 5 ′ side and 3 ′ side of the foreign DNA.
- the cell serving as a host for the cell preparation is not particularly limited, and examples thereof include those described above in [Genome editing method], and may be bone marrow-derived cells taken out from the human body.
- the genome-edited cells are expanded outside the body in this way, they are administered as a cell preparation to a patient by intravenous injection or the like.
- a higher homologous recombination frequency is realized in the target genome than non-homologous recombination.
- a cell preparation can be produced efficiently.
- target genomic DNA having a mutation in bone marrow-derived cells of a patient with severe combined immunodeficiency is restored to a healthy base sequence without error by homologous recombination, and severe combined immunodeficiency is cured.
- Cell preparation can be provided.
- the method for genome editing in plant cells wherein the foreign DNA having a homologous arm with a length of less than 500 bp is converted into a 5 ′ end and a 3 ′ end of the foreign DNA upon double strand breakage of the target genomic DNA.
- the method of introducing into the genome of said cell by homologous recombination in is provided.
- the system used for double-strand breakage of the target genomic DNA is not particularly limited, and is the same as that in the above-mentioned [Genome editing method].
- the method of introducing these systems into cells is not particularly limited, and is the same as that described in the above [Genome editing method].
- the means for introducing a foreign gene into a cell is not particularly limited, and is the same as that in the above-described [Genome editing method].
- Plant cells are not particularly limited, and are the same as those in [Genome editing method] described above.
- plant cells cells derived from meristems or seeds of plants, callus and the like are preferable.
- the upper limit of the length of the homologous arm is less than 500 bp, preferably 300 bp or less, more preferably 100 bp or less, particularly preferably 50 bp or less, and may be 10 bp or less.
- the lower limit of the length of the homologous arm is preferably 5 bp or more, and more preferably 10 bp or more.
- the length of the homologous arm is not particularly limited in the range between the upper limit value and the lower limit value, and is the same as that in [Genome editing method] described above.
- the length of the foreign DNA is not particularly limited as long as it can be inserted into the genome, and is the same as that in the above-mentioned [Genome editing method].
- the type of targeting vector to be used is not particularly limited, and is the same as that in [Genome editing method] described above.
- Genome editing in plant cells may be performed in an in vitro culture system or in planta.
- foreign genes, nucleic acids and proteins are introduced into cells using known methods such as the Agrobacterium method, particle gun method and whisker method for callus or tissue fragments. Is done using.
- the introduction of foreign genes, nucleic acids, and proteins into cells is performed on the exposed immature embryos or the stems of fully mature embryos using a known method.
- a method of introducing into a mature seed embryo using a particle gun method is preferable in terms of efficiency of introduction into a plant body.
- it is applied to barley and potato as cereals, tomato and rape as vegetables, and to carnations, roses, sweet peas, chrysanthemums as florets.
- the genome editing method for plant cells of this embodiment expands the applicability of genome editing technology in the agricultural field. Specific examples include the production of sake rice that is low in carbohydrates and difficult to hangover.
- the donor plasmid has a structure in which the following combinations of homologous arms are added to 155 bp of foreign DNA containing the mutation site.
- the base sequence of the 5'-side 10 bp homology arm is shown below.
- 5′-GGCCCAGGTT-3 ′ (SEQ ID NO: 1)
- a base sequence containing a 5'-side 50 bp homology arm is shown below.
- 5′-CAAAAGGAAATGTGTGGGTGGGGAGGGGTAGTGGGTAAGGGGCCCAGGTT-3 ′ (SEQ ID NO: 2)
- the base sequence of foreign DNA is shown below.
- the lower case is the 5 ′ regulatory region lacking in the SCID pig, and the upper case is the base sequence of Ex1 (exon 1) codon-optimized.
- Ex1 exon 1 codon-optimized.
- the base sequence of the 3 ′ 50 bp homology arm is shown below.
- 5′-GTGGGAAACTGGGACGTTGGGGGTAGGGTTGGTGAGCCGGGGGAGGCTGG-3 ′ (SEQ ID NO: 4)
- the base sequence of the 3 bp 10 bp homologous arm is shown below.
- 5′-GTGGGAAACT-3 ′ (SEQ ID NO: 5)
- the HITI base sequences added to both ends of the homologous arm are shown below.
- 5′-CCTTCGGGTTCAGTCCCACCCCA-3 ′ SEQ ID NO: 6
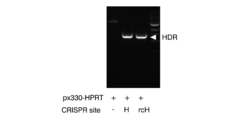
- FIG. 1 shows the result of electrophoresis of the amplified PCR product. As shown in FIG.
- NHEJ Non-homologous end joining
- HDR Homology directed repair
- homologous recombination occurred at a very high frequency (100%) at both the 5 ′ end and 3 ′ end. Further, it was confirmed that the mutant sequence repaired by such homologous recombination is a sequence encoding exon 1 of wild type IL2RG (error-free repair).
- nucleotide sequence of the homologous arm (10 bp) on the 5 'side is the same as SEQ ID NO: 1.
- the base sequence of the 3'-side homology arm (50 bp) is the same as SEQ ID NO: 4.
- the HITI base sequence added to both ends of the homologous arm is the same as SEQ ID NO: 6.
- Cas9 protein, crRNA shown in SEQ ID NO: 7, tracrRNA, and the donor plasmid were introduced by electroporation.
- As the pig-derived bone marrow stromal cells adherent cells obtained by liquid culture of pig bone marrow mononuclear cells were used.
- Genomic DNA was purified from the cells 3 days after electroporation, and DNA insertion was confirmed by PCR.
- a primer used for insertion confirmation a combination of A and B and a combination of C and D shown in FIG. 4 were used.
- 389 bp DNA is amplified when non-homologous recombination occurs.
- PCR using a combination of primers C and D 399 bp DNA is amplified when non-homologous recombination occurs, and 301 bp DNA is amplified when homologous recombination occurs.
- FIG. 5 shows the result of electrophoresis of the PCR product.
- NHEJ indicates a band size corresponding to non-homologous recombination
- HDR indicates a band size corresponding to homologous recombination. That is, in pig-derived bone marrow stromal cells, non-homologous recombination occurred at the 5 ′ end of the foreign DNA, and homologous recombination occurred at the 3 ′ end.
- MCS Multicloning Site
- BSR blasticidin S deaminase
- the base sequence of the 5'-side 50 bp homology arm targeting the Rosa26 region is shown below. 5′-TGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3 ′ (SEQ ID NO: 9)
- the base sequence of the 5 ′ 100 bp homologous arm targeting the Rosa26 region is shown below. 5'-AATACCTTTCTGGGAGTTCTCTGCTGCCTCCTGGCTTCTGAGGACCGCCCTGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3 '(SEQ ID NO: 10)
- the base sequence of the 3 ′ 50 bp homologous arm targeting the Rosa26 region is shown below. 5′-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGT-3 ′ (SEQ ID NO: 12)
- the base sequence of the 3'-side 100 bp homology arm targeting the Rosa26 region is shown below. 5'-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGTCGGGAAGTTTTTTAATAGGGGCAAATAAGGAAAATGGGAGGATAGGTAGT-3 '(SEQ ID NO: 13)
- the crRNA, tracrRNA shown in SEQ ID NO: 16 (5′-ACUCCAGUCUUUCUAGAAGAGUUUUAGAGCUAUGCU-3 ′) targeting the Cas9 protein and Rosa26 region, and the above-mentioned donor plasmid were hematopoietic by electroporation Introduced into stem cells.
- genomic DNA was purified from the cells, and DNA insertion was confirmed by PCR.
- FIG. 6 only bands having a size corresponding to homologous recombination were observed at the 5 ′ end and 3 ′ end. That is, the GFP gene was inserted into the Rosa26 region of the mouse hematopoietic stem cell genome only by homologous recombination.
- the base sequence of the 5 '100 bp homologous arm is shown below. 5'-GGATCGGTGGCTCCATCCTGGCCTCACTGTCCACCTTCCAGCAGATGTGGATCAGCAAGCAGGAGTACGATGAGTCCGGCCCCTCCATCGTGCACCGCAA-3 '(SEQ ID NO: 17)
- the base sequence of the 3 ′ 100 bp homologous arm is shown below. 5'-GGACTGTTACTGAGCTGCGTTTTACACCCTTTCTTTGACAAAACCTAACTTGCGCAGAAAAAAAAAATAAGAGACAACATTGGCATGGCTTTGTTTTT-3 '(SEQ ID NO: 18)
- HITI base sequences added to both ends of the homologous arm are shown below. 5′-AGTCCGCCTAGAAGCACTTGCGG-3 ′ (SEQ ID NO: 19)
- the base sequence of the foreign DNA containing the GFP gene is the same as in Experimental Example 3.
- Cas9 protein, crRNA shown in SEQ ID NO: 20 (5′-AGUCCGCCUAGAAGCACUUGGUUUUAGAGCUAUGCU-3 ′), tracrRNA, and the donor plasmid were introduced into fertilized mouse eggs by microinjection.
- Genomic DNA was extracted from the cells 6 days after microinjection, and DNA insertion was confirmed by PCR.
- FIG. 7A since the cells exhibited green fluorescence when observed with a fluorescence microscope, it was confirmed that the GFP gene was knocked in by homologous recombination at the 5 ′ end. Further, as shown in FIG.
- the base sequence of the 5'-side 60 bp homologous arm is shown below. 5′-GATGAACCAGGTTATGACCTTGATTTATTTTGCATACCTAATCATTATGCTGAGGATTTG-3 ′ (SEQ ID NO: 21)
- the base sequence of the 5'-side 244 bp homology arm is shown below. 5'-CCGGCCTGTTGTTTTCTTACATAATTCATTATCATACCTACAAAGTTAACAGTTACTAATATCATCTTACACCTAAATTTCTCTGATAGACTAAGGTTATTTTTTAACATCTTAATCCAATCAAATGTTTGTATCCTGTAATGCTCTCATTGAAACAGCTATATTTCTTTTTCAGATTAGTAGATGTGTTTTTTTT
- the base sequence of the 3 ′ 61 bp homologous arm is shown below. 5′-GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGG-3 ′ (SEQ ID NO: 23)
- nucleotide sequence of the 3'-side 239 bp homologous arm is shown below. 5'-GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGGTTTTTTACTTTTTCTTGTGTTAATTTCAAACATCAGCAGCTGTTCTGAGTACTTGCTATTTGAACATAAACTAGGCCAACTTATTAAATAACTGATGCTTTTATAGAAAATCTTCTTTTAGGG
- the base sequence of foreign DNA containing the GFP gene is shown below. 5 '-3' (SEQ ID NO: 25)
- HITI base sequences added to both ends of the homologous arm are shown below. 5′-ACCCTTTCCAAATCCTCAGCATAATG-3 ′ (SEQ ID NO: 26)
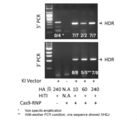
- a plasmid (px330-HPRT) that co-expresses the Cas9 protein, the sgRNA having the recognition sequence shown in SEQ ID NO: 27 (5′-UUAUGCUGAGGAUUUGGAAA-3 ′), and the donor plasmid were introduced into Jurkat cells by electroporation. Three days after electroporation, genomic DNA was purified from the cells, and DNA insertion was confirmed by PCR. As shown in FIG. 8, bands having a size corresponding to homologous recombination were observed at the 5 ′ end and 3 ′ end. That is, knock-in by bilateral homologous recombination was observed in human T cell leukemia cells. It was confirmed by the following sequence that the knock-in was due to error-free homologous recombination.
- TA-cloned PCR product was introduced into E. coli, and each colony formed was sequenced.
- homologous recombination occurred in all clones in 8 clones at the 5 ′ end, and homologous recombination occurred in all clones in 7 clones at the 3 ′ end. confirmed.
- homologous recombination occurs in all clones among 6 clones at the 5 ′ end, and homologous recombination occurs in all clones among 8 clones at the 3 ′ end. confirmed.
- the base sequence of the 5 '101 bp homologous arm is shown below. 5'-CGCCGGTTTGTGCCTTCGGTCCCCGCTTCGCCCCCTGCCGTCCCCTCCTTATCACGGTCCCGCTCGCGGCCTCGCCGCCCCGCTGTCTCCGCCGCCCGCCA-3 '(SEQ ID NO: 28)
- the base sequence of the 3′-side 101 bp homology arm is shown below. 5'-acCCCCGTGCCGCCGCGGATGGGCAGCCGCGCTGGCGGCCCCACCACGCCGCTGAGCCCCACGCGCCTGTCGCGGCTCCAGGAGAAGGAGGAGCTGCGCGA-3 '(SEQ ID NO: 29)
- the base sequence of the foreign DNA containing the GFP gene is shown below. 5 ′-3 ′ (SEQ ID NO: 30)
- HITI base sequences added to both ends of the homologous arm are shown below. 5′-GGGGTCGCAGTCGCCATGGCGGG-3 ′ (SEQ ID NO: 31)
- the rcHITI base sequence added to both ends of the homologous arm (reverse complement of HITI, that is, the guide RNA recognition sequence including the PAM sequence is in the same direction as the genome at both ends of the foreign DNA) is shown below. 5′-CCCGCCATGGCGACTGCGACCCC-3 ′ (SEQ ID NO: 32)
- a plasmid (px330-LMNB1) co-expressing the Cas9 protein, sgRNA having the recognition sequence shown in SEQ ID NO: 33 (5′-GGGGUCGCAGUCGCCAUGGC-3 ′), and the donor plasmid were introduced by lipofection.
- FIG. 9A only the band corresponding to the homologous recombination was recognized in the case where either HITI or rcHITI guide RNA recognition sequence was provided.
- FIG. 9B GFP-positive cells were purified by FACS 4 days after lipofection, and further cultured for 1 week, and then observed with a fluorescence microscope. As a result, the nuclear membrane showed a green color. It was confirmed to be localized in the membrane.
- the donor plasmid is designed so that when both ends homologous recombination occurs, the LMNB1 gene and the GFP gene are fused, and the product is localized in the nuclear membrane. Therefore, homologous recombination occurs at the 5 'end and 3' end. It was confirmed from the fluorescence image that it was happening. As shown in FIG. 9C, GFP-expressing cells were quantified using flow cytometry one week after lipofection. When a donor plasmid added with a nucleotide sequence of HITI or rcHITI was used, 8.9-9 It was confirmed that homologous recombination occurred in 2%.
- a donor plasmid containing a 10-bp homologous arm, a donor plasmid containing a 60-bp homologous arm, and a donor plasmid containing a 240-bp homologous arm was made.
- the base sequence of the 5 '10 bp homologous arm is shown below. 5′-TGAGGATTTG-3 ′ (SEQ ID NO: 34)
- the base sequence of the homologous arm of about 60 bp to about 240 bp on both ends, the base sequence of HITI, and the base sequence of foreign DNA are the same as in Experimental Example 6.
- a plasmid (px330-HPRT) that co-expresses Cas9 protein and sgRNA having the recognition sequence shown in SEQ ID NO: 27, and the donor plasmid were introduced into human-derived bone marrow stromal cells by lipofection. Three days after lipofection, genomic DNA was purified from the cells, and DNA insertion was confirmed by PCR. As shown in FIG. 10, bands having a size corresponding to homologous recombination were observed at the 5 ′ end and 3 ′ end. That is, knock-in in human-derived bone marrow stromal cells was bilateral homologous recombination.
- a donor plasmid containing a homologous arm of 10 bp on both ends In order to knock in the GFP gene at the HPRT locus of human iPS cells, a donor plasmid containing a homologous arm of 10 bp on both ends, a donor plasmid containing a homologous arm of about 60 bp on both ends, and a donor plasmid containing a homologous arm of about 240 bp on both ends were prepared. .
- the base sequence of the homology arm of 10 bp to about 240 bp on both ends, the base sequence of HITI, and the base sequence of foreign DNA are the same as in Experimental Example 7.
- the Cas9 protein, crRNA shown in SEQ ID NO: 36 (5′-UUAUGCUGAGGAUUUGGAAAGUUUUAGAGCUAUGCU-3 ′), tracrRNA, and the donor plasmid were introduced into human iPS cells by electroporation.
- genomic DNA was purified from the cells, and DNA insertion was confirmed by PCR.
- bands having a size corresponding to homologous recombination were observed at the 5 ′ end and 3 ′ end.
- ⁇ Experimental Example 9> The experiment in ⁇ Experimental example 3> was performed using ZFN or TALEN. A donor plasmid containing 100 bp homology arms at both ends was used. The results are shown in FIG. Similarly, when ZFN or TALEN was used, bands having a size corresponding to homologous recombination were observed at the 5 ′ end and 3 ′ end. In the present invention, it was revealed that the target genomic DNA cleaving enzyme is not limited to CRISPR / Cas9, and either ZFN or TALEN may be used.
- high homologous recombination frequency can be realized in the target genome as compared with nonhomologous recombination without impairing the ability of the cell to bind non-homologous ends.
Abstract
単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、外来DNAの5'端と3'端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
Description
本発明は、ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法に関する。
本願は、2018年3月29日に、日本に出願された特願2018-66174号に基づき優先権を主張し、その内容をここに援用する。
本願は、2018年3月29日に、日本に出願された特願2018-66174号に基づき優先権を主張し、その内容をここに援用する。
真核細胞のゲノムの特定部位に、特定遺伝子を挿入して、標的ゲノムDNAを所望の塩基配列にそっくり置換したい場合、一般に相同組換えが利用される。具体的には、挿入したい外来DNAの両端(5’末端及び3’末端)に、ゲノム上の挿入を行う部位と相同な配列を有するDNA(以下、相同アームという。)を備えた遺伝子導入用ベクター(以下、ターゲティングベクターという。)を用いることが行われている。係るベクターを対象細胞に導入すると、ゲノムDNAとベクターとの間で相同組換えが生じ、標的ゲノムDNAの所望の塩基配列を置換することができる。この置換はエラーフリーが特徴である。
しかしながら、哺乳類由来の細胞では、この相同組換えが生じる頻度は、100万分の1と極めて低いため、相同組換えの実用化、特に医療応用面での実用化は非常に困難であった。
近年、ゲノム編集ヌクレアーゼの発見により、ゲノム上の任意の箇所でDNA二重鎖を切断することができるようになった。この結果、ゲノムDNAと導入外来遺伝子との間で相同組換えを誘導しやすくなった(例えば、特許文献1、非特許文献1~2参照。)。
近年、ゲノム編集ヌクレアーゼの発見により、ゲノム上の任意の箇所でDNA二重鎖を切断することができるようになった。この結果、ゲノムDNAと導入外来遺伝子との間で相同組換えを誘導しやすくなった(例えば、特許文献1、非特許文献1~2参照。)。
特許文献1においては、1細胞期胚の細胞のゲノムをCas9タンパク質で切断し、標的配列の5’末端及び3’末端にそれぞれハイブリダイズする相同アームと、当該相同アームに隣接する核酸インサート(導入外来遺伝子)とを備えるターゲティングベクターを用いて、前記核酸インサートを前記細胞中に導入する方法が開示されている。
非特許文献1においては、β-サラセミア患者に由来する造血幹細胞に対して、Cas9タンパク質とアデノウイルス随伴ベクターとを組み合わせたCRISPR/Cas9システムを用い、相同組換えにより正常HBB(haemoglobin beta)遺伝子を導入したことが開示されている。
しかしながら、近年の技術においても、相同組換えの発生頻度よりも非相同組換えの発生頻度が高く、相同組換えの発生頻度は最も高い場合でも10分の1程度である。従って、相同組換えの実用化を目指すにあたっては、更なる相同組換え頻度の向上が必要である。
相同組換えの頻度を上げる試みとして、例えば非特許文献3~4に挙げられる方法が提案されている。
相同組換えの頻度を上げる試みとして、例えば非特許文献3~4に挙げられる方法が提案されている。
非特許文献2においては、CRISPR/Cas9システムを用いた相同組換えによる遺伝子導入において、非相同末端結合を抑制するか、又は相同組換えを促進する低分子化合物の探索がなされている。このような低分子化合物として、Scr7、L755507及びレスベラトールを使用し、ピッグ胎児線維芽細胞において相同組換えを促進したことが開示されている。
非特許文献3においては、KU70やDNAリガーゼIV等の発現をRNA干渉により抑制して、非相同組換えの頻度を下げることにより、相対的に相同組換えの頻度を上げる方法が開示されている。
非特許文献4においては、CRISPR/Cas9システムを用いたゲノム編集において、二重鎖DNAからCas9が解離する前に非対称的に放出される、sgRNAと相補的でない切断DNAの3’末端に対して、相補的な1本鎖DNAを供与することにより、相同組換えの頻度を上げる方法が開示されている。
Nature. 2016 Nov 17;539(7629):384-389. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Dever D. et al.
Sci Rep. 2017; 7: 8943. Small molecules enhance CRISPR/Cas9-mediated homology-directed genome editing in primary cells Guoling Li, et al.
Nat Biotechnol. 2015 May;33(5):543-548. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Chu VT., et al.
Nat Biotechnol. 2016;34:339-344, Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Richerdson C, et al.
しかしながら、非特許文献3~4における方法によっても、相同組換えの頻度は依然として低く、未だ改善の余地がある。
DNA二重鎖切断に対するDNA修復機序には、非相同末端結合と相同組換えとが知られている。非相同末端結合は相同組換えより短時間で進行する。従って、相同組換えの頻度を上げるためには、非相同末端結合の発生頻度を相対的に低減させる工夫が必要となる。しかし、非相同末端結合の機構そのものを抑制すると、DNAの二重鎖切断に対する細胞の修復能を大きく損なうこととなるため、生命体に対するリスク(生存不能や腫瘍化)が過大となる。この結果、この方向性での実用化・臨床応用は困難である。
本発明は、細胞が本来有する非相同末端結合の能力を損なわず、相同組換えの頻度を上昇させることができる、ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法を提供することを目的とする。
発明者らは、外来DNAの5’末端と3’末端において、一般には相同組換えに用いないような短い相同アームをターゲッティングベクターに採用すると、標的ゲノムDNAの二重鎖切断時、非相同組換えに対する相同組換えの頻度が著しく高いことを見出し、本発明を完成させた。
即ち、本発明は以下のとおりである。
[1]単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを5’端と3’端に有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
[2]前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを前記標的ゲノム導入する、[1]に記載のゲノム編集方法。
[3]前記細胞が、血球系細胞又は未分化細胞である、[1]又は[2]に記載のゲノム編集方法。
[4]前記細胞が、幹細胞である、[1]~[3]のいずれかに記載のゲノム編集方法。
[5]前記細胞が、造血幹細胞である、[1]~[4]のいずれかに記載のゲノム編集方法。
[6]前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを前記標的ゲノムに導入する、[1]に記載のゲノム編集方法。
[1]単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを5’端と3’端に有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
[2]前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを前記標的ゲノム導入する、[1]に記載のゲノム編集方法。
[3]前記細胞が、血球系細胞又は未分化細胞である、[1]又は[2]に記載のゲノム編集方法。
[4]前記細胞が、幹細胞である、[1]~[3]のいずれかに記載のゲノム編集方法。
[5]前記細胞が、造血幹細胞である、[1]~[4]のいずれかに記載のゲノム編集方法。
[6]前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを前記標的ゲノムに導入する、[1]に記載のゲノム編集方法。
[7]単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、外来DNAの5’側と3’側のうち、両側を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
[8]前記細胞が、血球系細胞又は未分化細胞である、[7]に記載のゲノム編集方法。
[9]前記細胞が、幹細胞である、[7]又は[8]に記載のゲノム編集方法。
[10]前記細胞が、造血幹細胞である、[7]~[9]のいずれかに記載のゲノム編集方法。
[8]前記細胞が、血球系細胞又は未分化細胞である、[7]に記載のゲノム編集方法。
[9]前記細胞が、幹細胞である、[7]又は[8]に記載のゲノム編集方法。
[10]前記細胞が、造血幹細胞である、[7]~[9]のいずれかに記載のゲノム編集方法。
[11]500bp未満の長さの相同アームを両端に有する外来DNAを含有することを特徴とする組成物。
[12]更に標的ゲノムDNA切断酵素又は前記酵素をコードするDNA若しくはmRNAを含有する、[11]に記載の組成物。
[13]医薬用である、[11]又は[12]に記載の組成物。
[14]重症複合免疫不全症治療用である、[11]~[13]のいずれかに記載の組成物。
[12]更に標的ゲノムDNA切断酵素又は前記酵素をコードするDNA若しくはmRNAを含有する、[11]に記載の組成物。
[13]医薬用である、[11]又は[12]に記載の組成物。
[14]重症複合免疫不全症治療用である、[11]~[13]のいずれかに記載の組成物。
[15]重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、前記細胞のゲノム中に導入することを特徴とする細胞製剤の製造方法。
[16]前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを標的ゲノムDNAに導入する、[15]に記載の細胞製剤の製造方法。
[17]前記細胞が、血球系細胞又は未分化細胞である、[15]又は[16]に記載の細胞製剤の製造方法。
[18]前記細胞が、幹細胞である、[15]~[17]のいずれかに記載の細胞製剤の製造方法。
[19]前記細胞が、造血幹細胞である、[15]~[18]のいずれかに記載の細胞製剤の製造方法。
[20]前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを標的ゲノムDNAに導入する、[15]に記載の細胞製剤の製造方法。
[16]前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを標的ゲノムDNAに導入する、[15]に記載の細胞製剤の製造方法。
[17]前記細胞が、血球系細胞又は未分化細胞である、[15]又は[16]に記載の細胞製剤の製造方法。
[18]前記細胞が、幹細胞である、[15]~[17]のいずれかに記載の細胞製剤の製造方法。
[19]前記細胞が、造血幹細胞である、[15]~[18]のいずれかに記載の細胞製剤の製造方法。
[20]前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを標的ゲノムDNAに導入する、[15]に記載の細胞製剤の製造方法。
[21]重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、前記外来DNAの5’側と3’側のうち、両方を相同組換えにより、前記細胞のゲノム中に導入することを特徴とする細胞製剤の製造方法。
[22]前記細胞が、血球系細胞又は未分化細胞である、[21]に記載の細胞製剤の製造方法。
[23]前記細胞が、幹細胞である、[21]又は[22]に記載の細胞製剤の製造方法。
[24]前記細胞が、造血幹細胞である、[21]~[23]のいずれかに記載の細胞製剤の製造方法。
[25]細胞の標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片を有することを特徴とする細胞。
[26][25]に記載の細胞を含有することを特徴とする細胞製剤。
[22]前記細胞が、血球系細胞又は未分化細胞である、[21]に記載の細胞製剤の製造方法。
[23]前記細胞が、幹細胞である、[21]又は[22]に記載の細胞製剤の製造方法。
[24]前記細胞が、造血幹細胞である、[21]~[23]のいずれかに記載の細胞製剤の製造方法。
[25]細胞の標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片を有することを特徴とする細胞。
[26][25]に記載の細胞を含有することを特徴とする細胞製剤。
本発明によれば、細胞が本来有する非相同末端結合の能力を損なわず、標的ゲノムにおいて、非相同組換えに比して高い相同組換え頻度を実現する。
[ゲノム編集方法]
本発明のゲノム編集方法は、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入する方法である。
本発明のゲノム編集方法は、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入する方法である。
<第一実施形態>
一実施形態において、本発明は、細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’末端と3’末端における相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
一実施形態において、本発明は、細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’末端と3’末端における相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
本実施形態において、先ず、標的ゲノムDNA上の、外来DNAを導入したい箇所で、係る標的ゲノムDNAの二重鎖を切断する。標的ゲノムDNAの二重鎖切断に用いるシステムとしては、特に限定されず、CRISPR-Cas9システム、Transcription activator-like effector nuclease(TALEN)システム、及びZnフィンガーヌクレアーゼシステム等が挙げられる。これらのシステムの細胞への導入方法としては、特に限定されず、標的ゲノムDNA切断酵素自体を細胞に導入してもよく、標的ゲノムDNA切断酵素発現ベクターを細胞に導入してもよい。標的ゲノムDNAの二重鎖切断に用いるシステムは、外来DNAと同時又は外来DNAの前若しくは後に細胞に導入される。
例えば、CRISPR-Cas9システムにおいては、Cas9発現ベクターと、切断したい箇所にCas9を誘導するガイドRNAをコードする発現ベクターと、を細胞に導入する方法や、発現精製した組み換えCas9タンパク質と、ガイドRNAと、を細胞に導入する方法等が挙げられる。ガイドRNAはtracrRNAとcrRNAの2つに分かれていてもよく、1本につながっているsgRNAであっても良い。
本実施形態において、標的ゲノムDNAの二重鎖切断に用いるシステムは、CRISPR-Cas9システムが好ましい。
本実施形態において、標的ゲノムDNAの二重鎖切断に用いるシステムは、CRISPR-Cas9システムが好ましい。
本実施形態において、外来遺伝子、核酸及びタンパク質の細胞への導入手段としては、特に限定されず、ウイルスベクターを用いる方法、又は非ウイルス導入法、又はその他公知の方法のいずれであってもよい。ウイルスベクターを用いる方法としては、例えば、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴(AAV)ウイルスベクター、ヘルペスウイルスベクター、センダイウイルスベクター、シンドビスウイルスベクター等が挙げられる。非ウイルス導入法としては、例えば、リン酸カルシウム法、リポフェクション法、エレクトロポレーション法、マイクロインジェクション法、ウィスカー法、プラズマ法、レーザーインジェクション法、パーティクルガン法及びアグロバクテリウム法等が挙げられる。
本実施形態において、本発明のゲノム編集方法の対象となる細胞は、特に限定されない。単離された細胞が好ましく、動物細胞、植物細胞、昆虫細胞、酵母及びカビ等の真菌、並びに大腸菌等の細菌等が挙げられる。
動物細胞の例として、動物由来の幹細胞、生殖細胞、生殖系列細胞、株化細胞、初代培養細胞、及び、幹細胞から誘導された細胞又は初代培養細胞から作製された細胞が挙げられる。前記幹細胞はまた、株化された細胞であっても、初代培養細胞であってもよい。
本発明のゲノム編集方法は、必ずしも単離された細胞に限らない。動物個体そのものあるいは個体内の体細胞や幹細胞も対象となる。
動物細胞の例として、動物由来の幹細胞、生殖細胞、生殖系列細胞、株化細胞、初代培養細胞、及び、幹細胞から誘導された細胞又は初代培養細胞から作製された細胞が挙げられる。前記幹細胞はまた、株化された細胞であっても、初代培養細胞であってもよい。
本発明のゲノム編集方法は、必ずしも単離された細胞に限らない。動物個体そのものあるいは個体内の体細胞や幹細胞も対象となる。
動物由来の細胞としては、幹細胞が好ましい。動物由来の幹細胞は、(i)自己複製能を有し、(ii)多分化能を有するという特徴を有する。
動物の幹細胞は、その分化能の違いによって、多能性(pluripotent)幹細胞、多能性(multipotent)幹細胞、少能性(oligopotent)幹細胞、及び単能性(unipotent)幹細胞に分けられる。
動物の幹細胞の例として、ES細胞及びEG細胞等の胚性幹細胞、誘導多能性(induced pluripotent)幹細胞(iPS細胞)等のES様幹細胞、胎生幹細胞、ミューズ細胞、胎盤幹細胞、造血幹細胞、間葉系幹細胞(歯髄由来間葉系幹細胞、脂肪由来間葉系幹細胞、骨髄由来間葉系幹細胞及び滑膜由来間葉系幹細胞等)、毛包幹細胞、乳腺幹細胞、神経幹細胞、サテライト細胞及び腸管上皮幹細胞等の成体幹細胞、並びにGS細胞等の生殖系幹細胞が挙げられる。
動物の幹細胞は、幹細胞に対して遺伝子操作を加えた細胞であってもよい。このような細胞として、例えば、ヒト白血球型抗原(HLA)を改編することにより免疫拒絶反応を抑えた多能性幹細胞等が挙げられる。
動物細胞としては、造血幹細胞が好ましい。
生殖細胞、生殖系列細胞の例として、卵子、卵母細胞、卵原細胞、精子、精母細胞、精原細胞、精子幹細胞(精原幹細胞)、始原生殖細胞等が挙げられる。卵子と精子が受精した受精卵でもよい。また、受精卵が分裂した、2細胞~8細胞胚でもよく、着床するまでの桑実胚~胚盤胞でもよい。
動物の幹細胞は、幹細胞に対して遺伝子操作を加えた細胞であってもよい。このような細胞として、例えば、ヒト白血球型抗原(HLA)を改編することにより免疫拒絶反応を抑えた多能性幹細胞等が挙げられる。
動物細胞としては、造血幹細胞が好ましい。
生殖細胞、生殖系列細胞の例として、卵子、卵母細胞、卵原細胞、精子、精母細胞、精原細胞、精子幹細胞(精原幹細胞)、始原生殖細胞等が挙げられる。卵子と精子が受精した受精卵でもよい。また、受精卵が分裂した、2細胞~8細胞胚でもよく、着床するまでの桑実胚~胚盤胞でもよい。
動物の株化細胞は、特に限定されない。動物の株化細胞の例として、チャイニーズハムスター卵巣組織由来細胞(CHO細胞)、アフリカミドリザル腎臓由来株化細胞(Vero細胞)、ヒト肝癌由来細胞(HepG2細胞)、イヌ腎臓尿細管上皮細胞由来の細胞株(MDCK細胞)、ヒト胎児腎細胞株(HEK293細胞)及びヒト肝癌組織由来樹立細胞株(huGK-14)等が挙げられる。
動物の初代培養細胞は、特に限定されず、正常組織由来又は疾患組織由来のいずれであってもよい。動物の初代培養細胞の例として、毛乳頭細胞、内皮細胞、上皮細胞、表皮角化細胞、メラノサイト、心筋細胞、平滑筋細胞、骨格筋細胞、骨格筋芽細胞、骨芽細胞、軟骨細胞、線維芽細胞、肝細胞、神経細胞、並びに、制御性T細胞、キラーT細胞及びガンマ・デルタT細胞等の免疫細胞等が挙げられる。
動物の幹細胞から誘導された細胞は、幹細胞であっても、分化した細胞であってもよい。動物の幹細胞から誘導された細胞の例として、iPS細胞由来網膜色素上皮細胞、iPS細胞由来神経細胞、iPS細胞由来キラーT細胞等のiPS細胞由来免疫系細胞、iPS細胞由来心筋前駆細胞及びiPS細胞由来肝細胞等が挙げられる。
動物の初代培養細胞から作製された細胞は、特に限定されない。典型的には、動物の初代培養細胞から作製された細胞は、動物の初代培養細胞に遺伝子操作を加えた細胞である。動物の初代培養細胞から作製された細胞の例として、CAR(chimeric antigen receptor)-T細胞等が挙げられる。
実施例において後述するように、本発明者は、相同組換えの起きやすさは、動物種や標的遺伝子座や導入遺伝子や、標的ゲノムを切断するヌクレアーゼの種類によらないことを見出した。
本実施形態においては、血球系細胞又は未分化細胞が好ましい。未分化細胞としては、幹細胞がより好ましく、造血幹細胞が特に好ましい。
本実施形態においては、血球系細胞又は未分化細胞が好ましい。未分化細胞としては、幹細胞がより好ましく、造血幹細胞が特に好ましい。
植物細胞は、特に限定されない。植物細胞の例として、植物の分裂組織又は種子に由来する細胞及びカルス等が挙げられる。カルスは、植物組織片から誘導されたもの、傷害誘導性のもの、細菌誘導性のもの、種間交雑において形成されたもの、及び培養細胞のいずれであってもよい。典型的には、植物の分裂組織又は種子に由来する細胞及びカルスは、PLT1、PLT5、LBD16、LBD17、LBD18、LBD29、ARR1、ARR21、ESR1、ESR2、WIND1、WIND2、WIND3、WIND4、LEC1、LEC2、AGL15、BBM、RKD1、RKD2、及びWUS等の多能性マーカーの少なくとも1つを発現するという特徴を有する。植物細胞におけるゲノム編集方法については、後述する。
次いで、本実施形態において、外来DNAの両端に、500bp未満の相同アームを備えるターゲティングベクターを細胞に導入する。相同アームとは、挿入しようとする外来DNAの5’端及び3’端に設けられ、ゲノム上の挿入を行う部位と相同な配列を有するDNAをいう。
相同アームの長さの上限値は、500bp未満であり、300bp以下が好ましく、100bp以下がより好ましく、50bp以下が特に好ましく、10bp以下であってもよい。導入された相同アームは、5’端及び3’端の両側において相同組換えに寄与する。
相同アームの長さの下限値は、5bp以上が好ましく、10bp以上がより好ましい。
相同アームの長さの下限値は、5bp以上が好ましく、10bp以上がより好ましい。
相同アームの長さは、5~499bpが好ましく、5~300bpがより好ましく、5~100bpがさらに好ましく、5~50bpが特に好ましく、10~50bpが最も好ましい。
外来DNAの長さは、ゲノム上に挿入できる長さであれば特に限定されない。前記外来DNAの長さとしては、例えば、50bp~10kbp、100~5kbp、100~1kbp及び100~500bp等が挙げられる。外来DNAの例としては、標的配列の野生型DNA、コドン最適化配列DNA、タグ付加外来DNA、プロモーター配列、転写終結配列、機能的遺伝子配列、蛍光タンパク質マーカー遺伝子配列、薬剤選択遺伝子配列、マルチクローニングサイト配列、及びこれらの組合せ等が挙げられる。
本実施形態において、使用するターゲティングベクターの種類は、特に限定されず、プラスミドベクター、ウイルスベクター等、従来公知のものを用いることができる。
ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴(AAV)ウイルスベクター、ヘルペスウイルスベクター、センダイウイルスベクター、シンドビスウイルスベクター、等が挙げられる。
ターゲティングベクターの例として、CRISR/Cas9システムを利用したゲノム編集用の各種ベクターが挙げられ、例えば、HITI(Homology-independent targeted integration)システムを利用したターゲティングベクターが挙げられる。
ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴(AAV)ウイルスベクター、ヘルペスウイルスベクター、センダイウイルスベクター、シンドビスウイルスベクター、等が挙げられる。
ターゲティングベクターの例として、CRISR/Cas9システムを利用したゲノム編集用の各種ベクターが挙げられ、例えば、HITI(Homology-independent targeted integration)システムを利用したターゲティングベクターが挙げられる。
通常、相同組換えのための遺伝子ターゲティングベクターにおける相同アームの長さは、相同領域の比率を上げるという観点から、長い方が効率よく相同組換えが行われると考えられる。しかしながら、本発明者らは、5’末端及び3’末端の両方に短い相同アームを備える外来DNAが、標的ゲノムDNAの二重鎖切断時、非相同組み換えよりも著しく高い頻度で相同組み換えを生じるという、全く予想外の結果が得られることを見出した。
本実施形態のゲノム編集方法により、外来DNAの5’末端及び3’末端の両側において、非相同組換えに対して極めて高い頻度の相同組換えを実現できる。具体的には、本発明の方法において、500bp未満の短い相同アームを使用したターゲティングベクターを使用することにより、細胞が本来有する非相同末端結合の機構を抑制することなく、DNAの二重鎖切断に対する細胞の修復能を維持したまま、非相同組み換えよりも著しく高い相同組換えの頻度を実現する。
従って、本実施形態のゲノム編集方法は、医療及び産業へのゲノム編集技術の応用可能性を広げるものである。本発明は、遺伝子変異に起因する疾患を有する個体を治療するために用いることのできる血液系細胞を作製することができる。遺伝子変異に起因する疾患は、特に限定されないが、例えば、先天性免疫不全症(実施例のX-SCIDのほか、 アデノシンデアミナーゼ[ADA]欠損症、慢性肉芽腫症、X連鎖無γ-グロブリン血症[XLA]、ZAP-70欠損症、高IgM症候群、IgA欠損症、IgGサブクラス欠損症、Bloom症候群、Wiscott-Aldrich症候群、Ataxia telangiectasia、DiGeorge症候群)、Fanconi貧血、サラセミア、鎌状赤血球貧血症、白質ジストロフィー、血友病、ムコ多糖症等のいずれであってもよい。本発明が非相同組換えに比して高い相同組換え頻度を実現するものであることから、非相同組換えでは治療が難しいが相同組換えによって治療の期待される各種疾患、たとえば、巨大遺伝子に変異のある疾患(筋ジストロフィーなど)や、遺伝子変異が長大な疾患(ハンチントン病等のトリプレット・リピート病など)の治療に極めて有用である。本発明の適応は、必ずしも遺伝子変異に起因する疾患の治療に限らない。たとえば、本発明は、間葉系幹細胞やT細胞の機能改変のために広く利用できる。たとえば、間葉系幹細胞やCAR-T細胞のHLA遺伝子座の改変など。これら本発明によって作成したゲノム改変細胞は、各種がん、白血病、造血障害、骨髄異形成症候群、心筋梗塞・脳梗塞・閉塞性動脈硬化症等の虚血性疾患、バージャー病、末梢疾患、重症下肢虚血、肺高血圧症、自己免疫疾患、ルーブス腎炎、クローン病、角膜疾患、角膜障害、緑内障、視神経障害、網膜色素変性症、黄斑変性等の治療に供することができる。本発明の応用は医療に限らない。たとえば、高不飽和脂肪酸を多く含み食味に優れた肉牛及び大トロの比率を増したマグロの作出等への、本発明による動物細胞のゲノム編集方法の応用可能性等が挙げられる。
<第二実施形態>
一実施形態において、本発明は、細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
一実施形態において、本発明は、細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
本実施形態の例としては、マウス受精卵やピッグ骨髄間質細胞等が挙げられる。
本実施形態のゲノム編集の効率を上げる一手段として、外来DNAにおける一方の相同アームの長さは、他方の相同アームの長さの2倍以上であることが好ましく、3倍以上であることがより好ましく、4倍以上であることが更に好ましく、5倍以上であることが特に好ましい。
短い相同アームの長さとしては、50bp以下が好ましく、30bp以下がより好ましく、10bp以下が特に好ましく、0bpであってもよい。長い相同アームの長さとしては、30bp以上が好ましく、40bp以上がより好ましく、50bp以上が特に好ましい。
導入された短い相同アームは、非相同組換えに寄与し、長い相同アームは、相同組換えに寄与する。本実施形態において、非相同組換えとは、非相同末端結合を意味する。
外来DNAの長さとしては、ゲノム上に挿入できる長さであれば特に限定されず、例えば100bp~10kbpが挙げられる。
短い相同アームの長さとしては、50bp以下が好ましく、30bp以下がより好ましく、10bp以下が特に好ましく、0bpであってもよい。長い相同アームの長さとしては、30bp以上が好ましく、40bp以上がより好ましく、50bp以上が特に好ましい。
導入された短い相同アームは、非相同組換えに寄与し、長い相同アームは、相同組換えに寄与する。本実施形態において、非相同組換えとは、非相同末端結合を意味する。
外来DNAの長さとしては、ゲノム上に挿入できる長さであれば特に限定されず、例えば100bp~10kbpが挙げられる。
本実施形態のゲノム編集方法における、片側相同組換えの発生機序は不明だが、次のように推測している。短い相同アームは、長い相同アームに比べれば相同組換えが起こりづらく、ゲノムDNAの二重鎖切断された箇所と、短い相同アームとが非相同組換えの機序(非相同末端結合)によってつながる。こうして、導入遺伝子の一端がゲノムDNAにつながったことで、そこが支点となって、他端の長い相同アームは、ゲノム上の相同配列近傍に位置するようになり、相同組換えが生じやすくなる。即ち、外来DNAの一端がまず非相同組換えによってつながり、その後他端側の相同組換えによって、外来DNAがゲノムに組み込まれるものと考えられる。
<第三実施形態>
一実施形態において、本発明は、単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、外来DNAの5’側と3’側のうち、両側を相同組換えにより、標的ゲノムに導入する方法を提供する。
一実施形態において、本発明は、単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、外来DNAの5’側と3’側のうち、両側を相同組換えにより、標的ゲノムに導入する方法を提供する。
本実施形態においては、ターゲティングベクターとして、標的ゲノムDNAを二重鎖切断せずとも、相同組換えを起すものを用いる。係るターゲティングベクターとしては、一本鎖DNAを包含するAAVベクターが挙げられる。
標的ゲノムDNAを二重鎖切断しないこと以外は、第一実施形態と同様である。
標的ゲノムDNAを二重鎖切断しないこと以外は、第一実施形態と同様である。
[組成物]
本発明の組成物は、500bp未満の長さの相同アームを両端に有する外来DNAを含有する。
本発明の組成物は、500bp未満の長さの相同アームを両端に有する外来DNAを含有する。
本発明の組成物は、標的ゲノムDNA切断酵素又は前記酵素をコードするDNA若しくはmRNAを含有してもよく、DNA切断酵素ではなく、二本鎖DNAの片側にニックを入れるニッケースや、二本鎖DNAを一本鎖に分離するヘリケースでもよい。
本発明において、標的ゲノムDNA切断酵素としては、Cas9、Transcription activator-like effector nuclease(TALEN)、Znフィンガーヌクレアーゼ等が挙げられる。また、前記酵素をコードするDNA又はmRNAとしては、これらのタンパク質をコードするDNA又はmRNAが挙げられる。
標的ゲノムDNA切断酵素として、Cas9を用いる場合には、組成物は、Cas9を誘導するガイドRNAを含有することが好ましい。また、組成物は、ガイドRNAをコードする発現ベクターを含有してもよい。
本発明において、外来DNAの両端に設けられる相同アームの長さは500bp未満であり、300bp以下であることが好ましく、上述した[ゲノム編集方法]におけるものと同様である。
本発明において、外来DNAは、上述した[ゲノム編集方法]におけるものと同様であり、本発明の組成物は、外来DNAを含むターゲティングベクターを含んでいてもよい。ターゲティングベクターについては、上述した[ゲノム編集方法]におけるものと同様である。
本発明において、上述した外来DNAは、一種のベクターに含まれるものでもよく、複数種のベクターに含まれるものでもよい。ベクターは特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
本発明の組成物は、医薬用であることが好ましく、薬学的に許容される担体を含むことがより好ましい。本実施形態の医薬用組成物は、例えば、錠剤、被覆錠剤、丸剤、散剤、顆粒剤、カプセル剤、液剤、懸濁剤、乳剤等の形態で経口的に、あるいは、注射剤、坐剤、皮膚外用剤等の形態で、非経口的に投与される。
薬学的に許容される担体としては、医薬組成物の製剤に通常用いられるものを特に制限なく用いることができる。より具体的には、例えば、ゼラチン、コーンスターチ、トラガントガム及びアラビアゴム等の結合剤;デンプン及び結晶性セルロース等の賦形剤;アルギン酸等の膨化剤;水、エタノール及びグリセリン等の注射剤用溶剤;ゴム系粘着剤及びシリコーン系粘着剤等の粘着剤等が挙げられる。薬学的に許容される担体は、1種を単独で又は2種以上を混合することにより用いられる。
本発明の組成物は、更に添加剤を含んでいてもよい。添加剤としては、ステアリン酸カルシウム及びステアリン酸マグネシウム等の潤滑剤;ショ糖、乳糖、サッカリン及びマルチトール等の甘味剤;ペパーミント及びアカモノ油等の香味剤;ベンジルアルコール及びフェノール等の安定剤;リン酸塩及び酢酸ナトリウム等の緩衝剤;安息香酸ベンジル及びベンジルアルコール等の溶解補助剤;酸化防止剤;防腐剤等が挙げられる。
添加剤は、1種を単独で又は2種以上を混合することにより用いられる。
添加剤は、1種を単独で又は2種以上を混合することにより用いられる。
本発明の組成物は、重症複合免疫不全症治療用であることが好ましい。重症複合免疫不全症(SCID)において、最も頻度の高い病型は、X鎖連鎖型であり、IL-2受容体γ遺伝子(IL2RG)の変異に起因する。
本発明において、X鎖連鎖型重症複合免疫不全症治療用組成物に含まれる外来DNAは、野生型IL-2受容体γ遺伝子の少なくとも一部を含むことが好ましい。また、標的ゲノムDNA切断酵素として、Cas9を用いる場合には、標的ゲノム上のIL-2受容体γ遺伝子にハイブリダイズするガイドRNA又はガイドRNAをコードする発現ベクターを含有することが好ましい。
本発明の組成物を使用することにより、本発明のゲノム編集方法を提供することが可能となる。
[遺伝子治療方法]
本発明の遺伝子治療方法は、変異を有する標的ゲノムDNAを切断する酵素又は前記酵素をコードするDNA若しくはmRNAと、500bp未満の長さの相同アームを両端に備えた、前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAと、を含有する医薬組成物を対象に投与する方法である。
本発明の遺伝子治療方法は、変異を有する標的ゲノムDNAを切断する酵素又は前記酵素をコードするDNA若しくはmRNAと、500bp未満の長さの相同アームを両端に備えた、前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAと、を含有する医薬組成物を対象に投与する方法である。
本発明において、変異は、標的ゲノムDNAのエクソン及びイントロン、又は標的ゲノムDNAの発現調節領域における、一部又は全部の欠失、置換、任意の配列の挿入等により生じる。
本発明において、投与方法は特に限定されず、患者の症状、体重、年齢、性別等に応じて適宜決定すればよい。例えば、錠剤、被覆錠剤、丸剤、散剤、顆粒剤、カプセル剤、液剤、懸濁剤、乳剤等は経口投与される。また、注射剤は、単独で、又はブドウ糖、アミノ酸等の通常の補液と混合して静脈内投与され、更に必要に応じて、骨髄内、動脈内、筋肉内、皮内、皮下又は腹腔内投与される。
本発明において、医薬組成物の投与量は、患者の症状、体重、年齢、性別等によって異なり、一概には決定できないが、経口投与の場合には、例えば1日あたり1μg~10g、例えば1日あたり0.01~2000mgの有効成分を投与すればよい。また、注射剤の場合には、例えば1日あたり0.1μg~1g、例えば1日あたり0.001~200mgの有効成分を投与すればよい。
[細胞]
第二実施形態において、本発明の細胞は、その標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片が残存することを特徴とする。上述した第二実施形態のゲノム編集方法によれば、外来DNAの一端がまず非相同組換えによって、二重鎖切断によって生じた標的ゲノムDNAの一端とつながる。そのため、本発明の細胞は、外来DNAのゲノム挿入部位の5’側または3’側に、外来DNA由来の断片をもつ。図4の結果に示されるように、標的ゲノムDNAの5’端で非相同組換えが起きた場合には、外来DNAのゲノム挿入部位の5’側に外来DNA由来の断片を有する。標的ゲノムDNAの3’側で非相同組換えが起きた場合には、外来DNAのゲノム挿入部位の3’側に外来DNA由来の断片を有する。
第二実施形態において、本発明の細胞は、その標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片が残存することを特徴とする。上述した第二実施形態のゲノム編集方法によれば、外来DNAの一端がまず非相同組換えによって、二重鎖切断によって生じた標的ゲノムDNAの一端とつながる。そのため、本発明の細胞は、外来DNAのゲノム挿入部位の5’側または3’側に、外来DNA由来の断片をもつ。図4の結果に示されるように、標的ゲノムDNAの5’端で非相同組換えが起きた場合には、外来DNAのゲノム挿入部位の5’側に外来DNA由来の断片を有する。標的ゲノムDNAの3’側で非相同組換えが起きた場合には、外来DNAのゲノム挿入部位の3’側に外来DNA由来の断片を有する。
[細胞製剤]
本発明の細胞製剤は、上述した第二実施形態のゲノム編集方法を用いることにより、例えば、変異を有する標的ゲノムDNAが野生型に編集された細胞を含有する。更に、上記[細胞]で述べたように、本発明の細胞製剤は、その標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片が残存する細胞を含む。
本発明の細胞製剤は、上述した第二実施形態のゲノム編集方法を用いることにより、例えば、変異を有する標的ゲノムDNAが野生型に編集された細胞を含有する。更に、上記[細胞]で述べたように、本発明の細胞製剤は、その標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片が残存する細胞を含む。
[細胞製剤の製造方法]
本発明の細胞製剤の製造方法は、重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、前記細胞のゲノム中に導入する方法である。
本発明の細胞製剤の製造方法は、重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、前記細胞のゲノム中に導入する方法である。
一実施形態において、本発明は、重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、両方を相同組換えにより、前記細胞のゲノム中に導入する細胞製剤の製造方法を提供する。
一実施形態において、本発明は、重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、前記細胞のゲノム中に導入する細胞製剤の製造方法を提供する。
一実施形態において、本発明は、重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、前記外来DNAの5’側と3’側のうち、両方を相同組換えにより、前記細胞のゲノム中に導入する細胞製剤の製造方法を提供する。
細胞製剤の宿主となる細胞は、特に限定されず、上述した[ゲノム編集方法]におけるものと同様のものが挙げられ、人体から取り出した骨髄由来細胞でもよい。
このようにして、ゲノム編集した細胞を体外で増殖させた後、細胞製剤として、静脈注射等により、患者に投与する。
本発明の細胞製剤の製造方法を用いることにより、標的ゲノムにおいて、非相同組換えに比して高い相同組換え頻度を実現する。この結果、効率よく細胞製剤を製造することができる。
本発明の細胞製剤の製造方法により、重症複合免疫不全症の患者の骨髄由来細胞における変異を有する標的ゲノムDNAを相同組換えによってエラーなく健常の塩基配列に修復し、重症複合免疫不全症を根治する細胞製剤を提供することが可能となる。
[植物細胞のゲノム編集方法]
一実施形態において、植物細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’末端と3’末端における相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
一実施形態において、植物細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’末端と3’末端における相同組換えにより、前記細胞のゲノム中に導入する方法を提供する。
本実施形態において、標的ゲノムDNAの二重鎖切断に用いるシステムは、特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。これらのシステムの細胞への導入方法についても、特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
本実施形態において、外来遺伝子の細胞への導入手段については、特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
植物細胞は、特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。植物細胞としては、植物の分裂組織又は種子に由来する細胞及びカルス等が好ましい。
相同アームの長さの上限値は、500bp未満であり、300bp以下が好ましく、100bp以下がより好ましく、50bp以下が特に好ましく、10bp以下であってもよい。相同アームの長さの下限値は、5bp以上が好ましく、10bp以上がより好ましい。相同アームの長さは、この上限値と下限値との間の範囲で特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
外来DNAの長さとしては、ゲノム上に挿入できる長さであれば特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
本実施形態において、使用するターゲティングベクターの種類は、特に限定されず、上述した[ゲノム編集方法]におけるものと同様である。
植物細胞におけるゲノム編集は、in vitro培養系で行われても、in plantaで行われてもよい。in vitro培養系でのゲノム編集方法においては、外来遺伝子、核酸及びタンパク質の細胞への導入は、カルス又は組織片に対して、アグロバクテリウム法、パーティクルガン法及びウィスカー法等の公知の方法を用いて行われる。in plantaにおけるゲノム編集方法においては、外来遺伝子、核酸及びタンパク質の細胞への導入は、露出させた未熟胚や完熟胚の茎頂に対して、公知の方法を用いて行われる。コムギ、イネ、トウモロコシ又はダイズ等の穀物類では、植物体への導入効率から、パーティクルガン法を用いて完熟種子胚に導入する方法が好ましい。その他、穀物類として大麦、ジャガイモ、野菜類としてトマト、アブラナ、また花卉類としてカーネーション、バラ、スイートピー、キク等に適用される。
本実施形態の植物細胞におけるゲノム編集方法は、農業分野でのゲノム編集技術の応用可能性を広げるものである。具体的には、炭水化物が少なく二日酔いしにくい酒米の作出等が挙げられる。
以下、実験例により本発明をさらに詳細に説明するが、本発明はこれらの例によって限定されるものではない。
<実験例1>
[ドナープラスミドの構築]
Watanabe M et al., PLoS One., 2013 Oct 9;8(10):e76478.に記載されるSCIDモデルピッグ由来造血幹細胞のゲノム上のInterleukin-2 receptor gamma遺伝子(以下、IL2RGともいう。)における5’制御領域からエクソン1途中までの変異(85bp及び1bpの欠損、2bp及び1bpの塩基置換)を修復すべく、ドナープラスミド(HITIターゲティグベクター)を用いてドナープラスミドを作製した。ドナープラスミドの構造を図1に示す。ドナープラスミドは、変異部位を含む155bpの外来DNAに、以下の組合せの相同アームを付加した構造を有する。
(a)5’末端が10bp、3’末端が50bpの相同アーム
(b)5’末端、3’末端ともに50bpの相同アーム
(c)5’末端が50bp、3’末端が10bpの相同アーム
(d)5’末端、3’末端ともに10bpの相同アーム
[ドナープラスミドの構築]
Watanabe M et al., PLoS One., 2013 Oct 9;8(10):e76478.に記載されるSCIDモデルピッグ由来造血幹細胞のゲノム上のInterleukin-2 receptor gamma遺伝子(以下、IL2RGともいう。)における5’制御領域からエクソン1途中までの変異(85bp及び1bpの欠損、2bp及び1bpの塩基置換)を修復すべく、ドナープラスミド(HITIターゲティグベクター)を用いてドナープラスミドを作製した。ドナープラスミドの構造を図1に示す。ドナープラスミドは、変異部位を含む155bpの外来DNAに、以下の組合せの相同アームを付加した構造を有する。
(a)5’末端が10bp、3’末端が50bpの相同アーム
(b)5’末端、3’末端ともに50bpの相同アーム
(c)5’末端が50bp、3’末端が10bpの相同アーム
(d)5’末端、3’末端ともに10bpの相同アーム
5’側の10bpの相同アームの塩基配列を以下に示す。
5’-GGCCCAGGTT-3’(配列番号1)
5’側の50bpの相同アームを含む塩基配列を以下に示す。
5’-CAAAAGGAAATGTGTGGGTGGGGAGGGGTAGTGGGTAAGGGGCCCAGGTT-3’(配列番号2)
5’-GGCCCAGGTT-3’(配列番号1)
5’側の50bpの相同アームを含む塩基配列を以下に示す。
5’-CAAAAGGAAATGTGTGGGTGGGGAGGGGTAGTGGGTAAGGGGCCCAGGTT-3’(配列番号2)
外来DNAの塩基配列を以下に示す。なお、小文字がSCIDピッグで欠損している5’制御領域、大文字がコドンオプティマイズされたEx1(エクソン1)の塩基配列である。
5’-cctgacacagtctacacccaggaaacaaggagtaagcgccATGCTCAAACCCCCCCTCCCCGTCAAGTCTCTCCTCTTCCTCCAGCTCCCTCTGCTCGGCGTCGGCCTCAATCCTAAGGTCCTCACCCACAGCGGCAACGAGGACATCACCGCTG-3’(配列番号3)
5’-cctgacacagtctacacccaggaaacaaggagtaagcgccATGCTCAAACCCCCCCTCCCCGTCAAGTCTCTCCTCTTCCTCCAGCTCCCTCTGCTCGGCGTCGGCCTCAATCCTAAGGTCCTCACCCACAGCGGCAACGAGGACATCACCGCTG-3’(配列番号3)
3’側の50bpの相同アームの塩基配列を以下に示す。
5’-GTGGGAAACTGGGACGTTGGGGGTAGGGTTGGTGAGCCGGGGGAGGCTGG-3’(配列番号4) 3’側の10bpの相同アームの塩基配列を以下に示す。
5’-GTGGGAAACT-3’(配列番号5)
相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-CCTTCGGGTTCAGTCCCACCCCA-3’(配列番号6)
5’-GTGGGAAACTGGGACGTTGGGGGTAGGGTTGGTGAGCCGGGGGAGGCTGG-3’(配列番号4) 3’側の10bpの相同アームの塩基配列を以下に示す。
5’-GTGGGAAACT-3’(配列番号5)
相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-CCTTCGGGTTCAGTCCCACCCCA-3’(配列番号6)
[CRISPR-Cas9を用いた、野生型IL2RG-Ex1遺伝子のIL2RG欠損ピッグ造血幹細胞への導入]
Cas9タンパク質、配列番号7(5’-UGGGGUGGGACUGAACCCGAGUUUUAGAGCUAUGCU-3’)に示されるcrRNA、tracrRNA(Alt-R(登録商標) CRISPR-Cas9 tracrRNA、カタログ番号1072534、Integrated DNA Technologies社製)、及び上記ドナープラスミドを、エレクトロポレーションによりIL2RG欠損ピッグ造血幹細胞へ導入した。ピッグ造血幹細胞は、ピッグ骨髄中の各種(CD3、CD16、及びCD45RA)分化マーカー陰性細胞(Lin-)を用いた。
Cas9タンパク質、配列番号7(5’-UGGGGUGGGACUGAACCCGAGUUUUAGAGCUAUGCU-3’)に示されるcrRNA、tracrRNA(Alt-R(登録商標) CRISPR-Cas9 tracrRNA、カタログ番号1072534、Integrated DNA Technologies社製)、及び上記ドナープラスミドを、エレクトロポレーションによりIL2RG欠損ピッグ造血幹細胞へ導入した。ピッグ造血幹細胞は、ピッグ骨髄中の各種(CD3、CD16、及びCD45RA)分化マーカー陰性細胞(Lin-)を用いた。
[ゲノムPCRによる挿入の確認]
エレクトロポレーションの7日後に細胞からゲノムDNAを精製し、PCRによる外来DNA挿入の確認を行った。挿入確認に用いたプライマーは図1に示すAとBの組み合わせ、及びCとDの組み合わせを用いた。5’端の相同組換えが起きると、プライマーAとBの組み合わせによるPCRにより335bpのDNAが増幅される。3’端の相同組換えが起きると、プライマーCとDの組み合わせによるPCRにより、197bpのDNAが増幅される。
図2に、増幅されたPCR産物を電気泳動した結果を示す。図2に示されるように、5’末端及び3’末端において、相同組換えが生じたこと示すサイズのバンドのみが認められた。図2中、NHEJ(Non-homologous end joining)は非相同組換えに相当するバンドサイズを示し、HDR (Homology directed repair)は相同組換えに相当するバンドサイズを示す。
エレクトロポレーションの7日後に細胞からゲノムDNAを精製し、PCRによる外来DNA挿入の確認を行った。挿入確認に用いたプライマーは図1に示すAとBの組み合わせ、及びCとDの組み合わせを用いた。5’端の相同組換えが起きると、プライマーAとBの組み合わせによるPCRにより335bpのDNAが増幅される。3’端の相同組換えが起きると、プライマーCとDの組み合わせによるPCRにより、197bpのDNAが増幅される。
図2に、増幅されたPCR産物を電気泳動した結果を示す。図2に示されるように、5’末端及び3’末端において、相同組換えが生じたこと示すサイズのバンドのみが認められた。図2中、NHEJ(Non-homologous end joining)は非相同組換えに相当するバンドサイズを示し、HDR (Homology directed repair)は相同組換えに相当するバンドサイズを示す。
[シーケンスによる組み換え方法の確認]
TAクローニングしたPCR産物を大腸菌に導入し、形成された各コロニーのシーケンスを行った。
使用した相同アームの組合せは以下の通りである。
(a)5’末端が10bp、3’末端が50bpの相同アーム
(b)5’末端、3’末端ともに50bpの相同アーム
(c)5’末端が50bp、3’末端が10bpの相同アーム
(d)5’末端、3’末端ともに10bpの相同アーム
5’末端では、組み換えが生じた(a)4クローン、(b)8クローン、(c)7クローン、(d)6クローン中、全てのクローンにおいて相同組換えが起きており、3’末端では、組み換えが生じた(a)8クローン、(b)7クローン、(c)8クローン、(d)10クローン中、全てのクローンにおいて相同組換えが起きており、5’末端、3’末端共に極めて高い頻度(100%)で、相同組換えが起きていることが確認された。また、かかる相同組換えにより修復された変異配列は、野生型IL2RGのエクソン1をコードする配列であること(エラーフリーの修復)が確認された。
TAクローニングしたPCR産物を大腸菌に導入し、形成された各コロニーのシーケンスを行った。
使用した相同アームの組合せは以下の通りである。
(a)5’末端が10bp、3’末端が50bpの相同アーム
(b)5’末端、3’末端ともに50bpの相同アーム
(c)5’末端が50bp、3’末端が10bpの相同アーム
(d)5’末端、3’末端ともに10bpの相同アーム
5’末端では、組み換えが生じた(a)4クローン、(b)8クローン、(c)7クローン、(d)6クローン中、全てのクローンにおいて相同組換えが起きており、3’末端では、組み換えが生じた(a)8クローン、(b)7クローン、(c)8クローン、(d)10クローン中、全てのクローンにおいて相同組換えが起きており、5’末端、3’末端共に極めて高い頻度(100%)で、相同組換えが起きていることが確認された。また、かかる相同組換えにより修復された変異配列は、野生型IL2RGのエクソン1をコードする配列であること(エラーフリーの修復)が確認された。
[IL2RG-Ex1遺伝子を導入したIL2RG欠損ピッグ造血幹細胞のSCIDピッグへの移植及び検出]
上記のゲノム修復を行ったIL2RG欠損ピッグ造血幹細胞を、同一個体のIL2RG欠損ピッグへ自家移植した。移植の4週間後、当該個体の末梢血由来の細胞を採取し、相同組換えによってゲノム修復された細胞の有無をPCR解析で確認した。その結果、相同組換えに相当するバンドのみが検出され、非相同組換えに相当するバンドは検出されなかった(図3)。さらに、得られたPCR産物のシーケンス解析を行ったところ、SCIDピッグの遺伝子変異が、相同組換えによりエラーフリーで健常塩基配列に修復されていることが確認された。これは、当該ピッグの遺伝子異常がもっぱらターゲティングベクターの相同組換えによって修復されたことを示す。
上記のゲノム修復を行ったIL2RG欠損ピッグ造血幹細胞を、同一個体のIL2RG欠損ピッグへ自家移植した。移植の4週間後、当該個体の末梢血由来の細胞を採取し、相同組換えによってゲノム修復された細胞の有無をPCR解析で確認した。その結果、相同組換えに相当するバンドのみが検出され、非相同組換えに相当するバンドは検出されなかった(図3)。さらに、得られたPCR産物のシーケンス解析を行ったところ、SCIDピッグの遺伝子変異が、相同組換えによりエラーフリーで健常塩基配列に修復されていることが確認された。これは、当該ピッグの遺伝子異常がもっぱらターゲティングベクターの相同組換えによって修復されたことを示す。
<実験例2>
[ドナープラスミドの構築]
Watanabe M et al., PLoS One., 2013 Oct 9;8(10):e76478.に記載されるSCIDモデルピッグ由来骨髄間質細胞のゲノム上のIL2RGを修復すべく、図4に示すドナープラスミド(ターゲティグベクター)を作製した。
[ドナープラスミドの構築]
Watanabe M et al., PLoS One., 2013 Oct 9;8(10):e76478.に記載されるSCIDモデルピッグ由来骨髄間質細胞のゲノム上のIL2RGを修復すべく、図4に示すドナープラスミド(ターゲティグベクター)を作製した。
5’側の相同アーム(10bp)の塩基配列は、配列番号1と同様である。
Ex1(エクソン1)の塩基配列は、配列番号3と同様である。
3’側の相同アーム(50bp)の塩基配列は、配列番号4と同様である。
相同アームの両端に付加するHITI塩基配列は、配列番号6と同様である。
[CRISPR-Cas9を用いた、野生型IL2RG-Ex1遺伝子のIL2RG欠損ピッグ細胞への挿入]
Cas9タンパク質、配列番号7に示されるcrRNA、tracrRNA、及び上記ドナープラスミドをエレクトロポレーションにより導入した。ピッグ由来骨髄間質細胞は、ピッグ骨髄単核球を液体培養して得られる付着細胞を用いた。
Cas9タンパク質、配列番号7に示されるcrRNA、tracrRNA、及び上記ドナープラスミドをエレクトロポレーションにより導入した。ピッグ由来骨髄間質細胞は、ピッグ骨髄単核球を液体培養して得られる付着細胞を用いた。
[ゲノムPCRによる挿入の確認]
エレクトロポレーション3日後の細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。挿入確認に用いたプライマーは図4に示すAとBの組み合わせ、及びCとDの組み合わせを用いた。プライマーAとBの組み合わせによるPCRでは、非相同組換えが起きると389bpのDNAが増幅される。プライマーCとDの組み合わせによるPCRでは、非相同組換えが起きると399bpのDNAが増幅され、相同組換えが起きると301bpのDNAが増幅される。
図5に、PCR産物を電気泳動した結果を示す。図5に示されるように、5’末端では、非相同組換えに相当するサイズのバンドが、3’末端では、相同組換え及び非相同組換えに相当するサイズのバンドが認められた。図5中、NHEJは非相同組換えに相当するバンドサイズを示し、HDRは相同組換えに相当するバンドサイズを示す。すなわち、ピッグ由来骨髄間質細胞では、外来DNAの5’端では非相同組換え、3’端では相同組換えという片側相同組み換えを生じた。これは、相同アームの短い側の5’端(10bp)では非相同組換えが、相同アームの長い側の3’端(50bp)では相同組換えが生じたことを示す。
エレクトロポレーション3日後の細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。挿入確認に用いたプライマーは図4に示すAとBの組み合わせ、及びCとDの組み合わせを用いた。プライマーAとBの組み合わせによるPCRでは、非相同組換えが起きると389bpのDNAが増幅される。プライマーCとDの組み合わせによるPCRでは、非相同組換えが起きると399bpのDNAが増幅され、相同組換えが起きると301bpのDNAが増幅される。
図5に、PCR産物を電気泳動した結果を示す。図5に示されるように、5’末端では、非相同組換えに相当するサイズのバンドが、3’末端では、相同組換え及び非相同組換えに相当するサイズのバンドが認められた。図5中、NHEJは非相同組換えに相当するバンドサイズを示し、HDRは相同組換えに相当するバンドサイズを示す。すなわち、ピッグ由来骨髄間質細胞では、外来DNAの5’端では非相同組換え、3’端では相同組換えという片側相同組み換えを生じた。これは、相同アームの短い側の5’端(10bp)では非相同組換えが、相同アームの長い側の3’端(50bp)では相同組換えが生じたことを示す。
[シーケンスによる組み換え方法の確認]
TAクローニングしたPCR産物を大腸菌に導入し、形成された各コロニーのシーケンスを行った。5’末端では、5クローン中、全てのクローンにおいて、非相同組換えが起きており、3’末端では、7クローン中、6クローンという高い頻度で、相同組換えが起きていることが確認された。
TAクローニングしたPCR産物を大腸菌に導入し、形成された各コロニーのシーケンスを行った。5’末端では、5クローン中、全てのクローンにおいて、非相同組換えが起きており、3’末端では、7クローン中、6クローンという高い頻度で、相同組換えが起きていることが確認された。
<実験例3>
マウス造血幹細胞ゲノムのRosa26領域、β-Actin (Actb)遺伝子座に、MCS (Multicloning Site)、GFP、blasticidin S deaminase (BSR)遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端50bpの相同アームを含むドナープラスミド、及び両端100bpの相同アームを含むドナープラスミドを作製した。マウス造血幹細胞は、マウス骨髄中の各種(CD5、CD45R、CD11b、Gr-1、7-4、及びTer-119)分化マーカー陰性細胞(Lin-)を用いた。
マウス造血幹細胞ゲノムのRosa26領域、β-Actin (Actb)遺伝子座に、MCS (Multicloning Site)、GFP、blasticidin S deaminase (BSR)遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端50bpの相同アームを含むドナープラスミド、及び両端100bpの相同アームを含むドナープラスミドを作製した。マウス造血幹細胞は、マウス骨髄中の各種(CD5、CD45R、CD11b、Gr-1、7-4、及びTer-119)分化マーカー陰性細胞(Lin-)を用いた。
Rosa26領域をターゲットとする、5’側の10bpの相同アームの塩基配列を以下に示す。
5’- TGCAACTCCA-3’(配列番号8)
5’- TGCAACTCCA-3’(配列番号8)
Rosa26領域をターゲットとする、5’側の50bpの相同アームの塩基配列を以下に示す。
5’-TGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3’(配列番号9)
5’-TGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3’(配列番号9)
Rosa26領域をターゲットとする、5’側の100bpの相同アームの塩基配列を以下に示す。
5’-AATACCTTTCTGGGAGTTCTCTGCTGCCTCCTGGCTTCTGAGGACCGCCCTGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3’(配列番号10)
5’-AATACCTTTCTGGGAGTTCTCTGCTGCCTCCTGGCTTCTGAGGACCGCCCTGGGCCTGGGAGAATCCCTTCCCCCTCTTCCCTCGTGATCTGCAACTCCA-3’(配列番号10)
Rosa26領域をターゲットとする、3’側の10bpの相同アームの塩基配列を以下に示す。
5’- ACAGGTGTAA-3’(配列番号11)
5’- ACAGGTGTAA-3’(配列番号11)
Rosa26領域をターゲットとする、3’側の50bpの相同アームの塩基配列を以下に示す。
5’-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGT-3’(配列番号12)
5’-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGT-3’(配列番号12)
Rosa26領域をターゲットとする、3’側の100bpの相同アームの塩基配列を以下に示す。
5’-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGTCGGGAAGTTTTTTAATAGGGGCAAATAAGGAAAATGGGAGGATAGGTAGT-3’(配列番号13)
5’-ACAGGTGTAAAATTGGAGGGACAAGACTTCCCACAGATTTTCGGTTTTGTCGGGAAGTTTTTTAATAGGGGCAAATAAGGAAAATGGGAGGATAGGTAGT-3’(配列番号13)
GFP遺伝子を含む外来DNAの塩基配列を以下に示す。
5’-actagttctagcatctgtagggcgcagtagtccagggtttccttgatgatgtcatacttatcctgtcccttttttttccacagctcgcggttgaggacaaactcttcgcgcatgcggatccggtaccATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAgaattc-3’(配列番号14)
5’-actagttctagcatctgtagggcgcagtagtccagggtttccttgatgatgtcatacttatcctgtcccttttttttccacagctcgcggttgaggacaaactcttcgcgcatgcggatccggtaccATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTACAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAGCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAgaattc-3’(配列番号14)
Rosa26領域をターゲットとする場合の、相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-CCATCTTCTAGAAAGACTGGAGT-3’(配列番号15)
5’-CCATCTTCTAGAAAGACTGGAGT-3’(配列番号15)
実験例1及び2と同様の方法で、Cas9タンパク質、Rosa26領域をターゲットとする配列番号16(5’- ACUCCAGUCUUUCUAGAAGAGUUUUAGAGCUAUGCU-3’)に示されるcrRNA、tracrRNA、及び上記ドナープラスミドをエレクトロポレーションによりマウス造血幹細胞に導入した。
エレクトロポレーション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図6に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドだけが認められた。すなわち、マウス造血幹細胞ゲノムのRosa26領域に、GFP遺伝子が相同組換えのみにより挿入された。
エレクトロポレーション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図6に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドだけが認められた。すなわち、マウス造血幹細胞ゲノムのRosa26領域に、GFP遺伝子が相同組換えのみにより挿入された。
<実験例4>
マウス受精卵のActb遺伝子座に、GFP遺伝子をノックインすべく、両端100bpの相同アームを含むドナープラスミドを作製した。5’側の相同アームの外側(5’端側)に、ストップコドンを備えているため、5’端で相同組換えが生じた場合にGFPが発現する。
マウス受精卵のActb遺伝子座に、GFP遺伝子をノックインすべく、両端100bpの相同アームを含むドナープラスミドを作製した。5’側の相同アームの外側(5’端側)に、ストップコドンを備えているため、5’端で相同組換えが生じた場合にGFPが発現する。
5’側の100bpの相同アームの塩基配列を以下に示す。
5’-GGATCGGTGGCTCCATCCTGGCCTCACTGTCCACCTTCCAGCAGATGTGGATCAGCAAGCAGGAGTACGATGAGTCCGGCCCCTCCATCGTGCACCGCAA-3’(配列番号17)
5’-GGATCGGTGGCTCCATCCTGGCCTCACTGTCCACCTTCCAGCAGATGTGGATCAGCAAGCAGGAGTACGATGAGTCCGGCCCCTCCATCGTGCACCGCAA-3’(配列番号17)
3’側の100bpの相同アームの塩基配列を以下に示す。
5’-GGACTGTTACTGAGCTGCGTTTTACACCCTTTCTTTGACAAAACCTAACTTGCGCAGAAAAAAAAAAAATAAGAGACAACATTGGCATGGCTTTGTTTTT-3’(配列番号18)
5’-GGACTGTTACTGAGCTGCGTTTTACACCCTTTCTTTGACAAAACCTAACTTGCGCAGAAAAAAAAAAAATAAGAGACAACATTGGCATGGCTTTGTTTTT-3’(配列番号18)
相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-AGTCCGCCTAGAAGCACTTGCGG-3’(配列番号19)
5’-AGTCCGCCTAGAAGCACTTGCGG-3’(配列番号19)
GFP遺伝子を含む外来DNAの塩基配列は、実験例3と同様である。
Cas9タンパク質、配列番号20(5’-AGUCCGCCUAGAAGCACUUGGUUUUAGAGCUAUGCU-3’)に示されるcrRNA、tracrRNA、及び上記ドナープラスミドを、マイクロインジェクションによりマウス受精卵に導入した。
マイクロインジェクション6日後に細胞からゲノムDNAを抽出し、PCRによるDNA挿入の確認を行った。図7Aに示されるように、蛍光顕微鏡で観察すると細胞が緑色蛍光を呈していることから、5’端ではGFP遺伝子が相同組換えによりノックインされていることが確認された。また、図7Bに示されるように、5’末端では相同組換えに相当するサイズのPCRバンドが認められ、3’末端では、非相同組換えに相当するサイズのPCRバンドが認められた。すなわち、マウス受精卵では片側相同組換えによりノックインされた。この結果は、片側相同組換えには、相同アームの長さが両端で必ずしも異なる必要はないことを示す。
Cas9タンパク質、配列番号20(5’-AGUCCGCCUAGAAGCACUUGGUUUUAGAGCUAUGCU-3’)に示されるcrRNA、tracrRNA、及び上記ドナープラスミドを、マイクロインジェクションによりマウス受精卵に導入した。
マイクロインジェクション6日後に細胞からゲノムDNAを抽出し、PCRによるDNA挿入の確認を行った。図7Aに示されるように、蛍光顕微鏡で観察すると細胞が緑色蛍光を呈していることから、5’端ではGFP遺伝子が相同組換えによりノックインされていることが確認された。また、図7Bに示されるように、5’末端では相同組換えに相当するサイズのPCRバンドが認められ、3’末端では、非相同組換えに相当するサイズのPCRバンドが認められた。すなわち、マウス受精卵では片側相同組換えによりノックインされた。この結果は、片側相同組換えには、相同アームの長さが両端で必ずしも異なる必要はないことを示す。
<実験例5>
ヒトT細胞性白血病細胞(Jurkat細胞)ゲノムのHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
ヒトT細胞性白血病細胞(Jurkat細胞)ゲノムのHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
5’側の60bpの相同アームの塩基配列を以下に示す。
5’-GATGAACCAGGTTATGACCTTGATTTATTTTGCATACCTAATCATTATGCTGAGGATTTG-3’(配列番号21)
5’-GATGAACCAGGTTATGACCTTGATTTATTTTGCATACCTAATCATTATGCTGAGGATTTG-3’(配列番号21)
5’側の244bpの相同アームの塩基配列を以下に示す。
5’-CCGGCCTGTTGTTTTCTTACATAATTCATTATCATACCTACAAAGTTAACAGTTACTAATATCATCTTACACCTAAATTTCTCTGATAGACTAAGGTTATTTTTTAACATCTTAATCCAATCAAATGTTTGTATCCTGTAATGCTCTCATTGAAACAGCTATATTTCTTTTTCAGATTAGTGATGATGAACCAGGTTATGACCTTGATTTATTTTGCATACCTAATCATTATGCTGAGGATTTG-3’(配列番号22)
5’-CCGGCCTGTTGTTTTCTTACATAATTCATTATCATACCTACAAAGTTAACAGTTACTAATATCATCTTACACCTAAATTTCTCTGATAGACTAAGGTTATTTTTTAACATCTTAATCCAATCAAATGTTTGTATCCTGTAATGCTCTCATTGAAACAGCTATATTTCTTTTTCAGATTAGTGATGATGAACCAGGTTATGACCTTGATTTATTTTGCATACCTAATCATTATGCTGAGGATTTG-3’(配列番号22)
3’側の61bpの相同アームの塩基配列を以下に示す。
5’- GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGG-3’(配列番号23)
5’- GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGG-3’(配列番号23)
3’側の239bpの相同アームの塩基配列を以下に示す。
5’-GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGGTTTTTTACTTTTTCTTGTGTTAATTTCAAACATCAGCAGCTGTTCTGAGTACTTGCTATTTGAACATAAACTAGGCCAACTTATTAAATAACTGATGCTTTCTAAAATCTTCTTTATTAAAAATAAAAGAGGAGGGCCTTACTAATTACTTAGTATCAGTTGTGGTATAGTGGGACTC-3’(配列番号24)
5’-GAAAGGGTGTTTATTCCTCATGGACTAATTATGGACAGGTAAGTAAGATCTTAAAATGAGGTTTTTTACTTTTTCTTGTGTTAATTTCAAACATCAGCAGCTGTTCTGAGTACTTGCTATTTGAACATAAACTAGGCCAACTTATTAAATAACTGATGCTTTCTAAAATCTTCTTTATTAAAAATAAAAGAGGAGGGCCTTACTAATTACTTAGTATCAGTTGTGGTATAGTGGGACTC-3’(配列番号24)
GFP遺伝子を含む外来DNAの塩基配列を以下に示す。
5’-gaattcATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTtCAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGcGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAcCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGgTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAggatcc-3’(配列番号25)
5’-gaattcATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTtCAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGcGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAcCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGgTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAggatcc-3’(配列番号25)
相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-ACCCTTTCCAAATCCTCAGCATAATG-3’(配列番号26)
5’-ACCCTTTCCAAATCCTCAGCATAATG-3’(配列番号26)
Cas9タンパク質と配列番号27(5’-UUAUGCUGAGGAUUUGGAAA-3’)に示される認識配列を持つsgRNAを共発現するプラスミド(px330-HPRT)、及び上記ドナープラスミドをエレクトロポレーションによりJurkat細胞に導入した。
エレクトロポレーション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図8に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。すなわち、ヒトT細胞性白血病細胞では、両側相同組換えによるノックインが認められた。当該ノックインがエラーフリーの相同組換えによるものであることは、下記の通りシーケンスによって確認した。
エレクトロポレーション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図8に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。すなわち、ヒトT細胞性白血病細胞では、両側相同組換えによるノックインが認められた。当該ノックインがエラーフリーの相同組換えによるものであることは、下記の通りシーケンスによって確認した。
TAクローニングしたPCR産物を大腸菌に導入し、形成された各コロニーのシーケンスを行った。60bpアームの場合、5’末端では、8クローン中、全てのクローンにおいて、相同組換えが起きており、3’末端では、7クローン中、全てのクローンにおいて、相同組換えが起きていることが確認された。240bpアームの場合、5’末端では、6クローン中、全てのクローンにおいて、相同組換えが起きており、3’末端では、8クローン中、全てのクローンにおいて、相同組換えが起きていることが確認された。
<実験例6>
ヒト胎児腎臓細胞株(HEK293T)ゲノムのLMNB1遺伝子座に、GFP遺伝子をノックインすべく、両端101bpの相同アームを含むドナープラスミドを作製した。
ヒト胎児腎臓細胞株(HEK293T)ゲノムのLMNB1遺伝子座に、GFP遺伝子をノックインすべく、両端101bpの相同アームを含むドナープラスミドを作製した。
5’側の101bpの相同アームの塩基配列を以下に示す。
5’-CGCCGGTTTGTGCCTTCGGTCCCCGCTTCGCCCCCTGCCGTCCCCTCCTTATCACGGTCCCGCTCGCGGCCTCGCCGCCCCGCTGTCTCCGCCGCCCGCCA-3’(配列番号28)
5’-CGCCGGTTTGTGCCTTCGGTCCCCGCTTCGCCCCCTGCCGTCCCCTCCTTATCACGGTCCCGCTCGCGGCCTCGCCGCCCCGCTGTCTCCGCCGCCCGCCA-3’(配列番号28)
3’側の101bpの相同アームの塩基配列を以下に示す。
5’-acCCCCGTGCCGCCGCGGATGGGCAGCCGCGCTGGCGGCCCCACCACGCCGCTGAGCCCCACGCGCCTGTCGCGGCTCCAGGAGAAGGAGGAGCTGCGCGA-3’(配列番号29)
5’-acCCCCGTGCCGCCGCGGATGGGCAGCCGCGCTGGCGGCCCCACCACGCCGCTGAGCCCCACGCGCCTGTCGCGGCTCCAGGAGAAGGAGGAGCTGCGCGA-3’(配列番号29)
GFP遺伝子を含む外来DNAの塩基配列を以下に示す。
5’-gatcTGACAATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTtCAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGcGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAcCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGgTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGGCCGGCTCCGgtac-3’(配列番号30)
5’-gatcTGACAATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGTCCGGCGAGGGCGAGGGCGATGCCACCTACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGACCTACGGCGTGCAGTGCTTCAGCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTGGTGAACCGCATCGAGCTGAAGGGCATCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTtCAACAGCCACAACGTCTATATCATGGCCGACAAGCAGAAGAACGGCATCAAGGcGAACTTCAAGATCCGCCACAACATCGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAACCACTACCTGAcCACCCAGTCCGCCCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGgTGGAGTTCGTGACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGGCCGGCTCCGgtac-3’(配列番号30)
相同アームの両端に付加するHITI塩基配列を以下に示す。
5’-GGGGTCGCAGTCGCCATGGCGGG-3’(配列番号31)
5’-GGGGTCGCAGTCGCCATGGCGGG-3’(配列番号31)
相同アームの両端に付加するrcHITI塩基配列(HITIのreverse complement、つまりPAM配列を含むガイドRNA認識配列が外来DNA両端にゲノムと同方向となっている)を以下に示す。
5’-CCCGCCATGGCGACTGCGACCCC-3’(配列番号32)
5’-CCCGCCATGGCGACTGCGACCCC-3’(配列番号32)
Cas9タンパク質と配列番号33(5’-GGGGUCGCAGUCGCCAUGGC-3’)に示される認識配列を持つsgRNAを共発現するプラスミド(px330-LMNB1)、及び上記ドナープラスミドをリポフェクションにより導入した。
図9Aに示されるように、HITIまたはrcHITIのどちらのガイドRNA認識配列を備える場合においても、相同組換えに相当するバンドのみが認められた。
図9Bに示されるように、リポフェクション4日後にGFP陽性細胞をFACSによりGFP陽性細胞を純化し、更に1週間培養した後、蛍光顕微鏡で観察すると核膜が緑色を呈していることからGFPが核膜に局在していることが確認された。上記ドナープラスミドは、両端相同組換えが起きるとLMNB1遺伝子とGFP遺伝子が融合し、その産物は核膜に局在するように設計されているため、5’末端及び3’末端で相同組換えが起きていることが蛍光画像からも確認された。
また、図9Cに示されるように、リポフェクション後1週間でフローサイトメトリーを用いてGFP発現細胞を定量したところ、HITIまたはrcHITIの塩基配列を付加したドナープラスミドを用いた場合、8.9~9.2%で相同組換えが起きていることが確認された。
図9Aに示されるように、HITIまたはrcHITIのどちらのガイドRNA認識配列を備える場合においても、相同組換えに相当するバンドのみが認められた。
図9Bに示されるように、リポフェクション4日後にGFP陽性細胞をFACSによりGFP陽性細胞を純化し、更に1週間培養した後、蛍光顕微鏡で観察すると核膜が緑色を呈していることからGFPが核膜に局在していることが確認された。上記ドナープラスミドは、両端相同組換えが起きるとLMNB1遺伝子とGFP遺伝子が融合し、その産物は核膜に局在するように設計されているため、5’末端及び3’末端で相同組換えが起きていることが蛍光画像からも確認された。
また、図9Cに示されるように、リポフェクション後1週間でフローサイトメトリーを用いてGFP発現細胞を定量したところ、HITIまたはrcHITIの塩基配列を付加したドナープラスミドを用いた場合、8.9~9.2%で相同組換えが起きていることが確認された。
<実験例7>
ヒト由来骨髄間質細胞のHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
ヒト由来骨髄間質細胞のHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
5’側の10bpの相同アームの塩基配列を以下に示す。
5’- TGAGGATTTG-3’(配列番号34)
5’- TGAGGATTTG-3’(配列番号34)
3’側の10bpの相同アームの塩基配列を以下に示す。
5’- GAAAGGGTGT-3’(配列番号35)
5’- GAAAGGGTGT-3’(配列番号35)
両端約60bp~約240bpの相同アームの塩基配列、HITIの塩基配列、及び外来DNAの塩基配列は、実験例6と同様である。
Cas9タンパク質と配列番号27に示される認識配列を持つsgRNAを共発現するプラスミド(px330-HPRT)、及び上記ドナープラスミドを、リポフェクションによりヒト由来骨髄間質細胞に導入した。
リポフェクション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図10に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。すなわち、ヒト由来骨髄間質細胞におけるノックインは両側相同組換えであった。
Cas9タンパク質と配列番号27に示される認識配列を持つsgRNAを共発現するプラスミド(px330-HPRT)、及び上記ドナープラスミドを、リポフェクションによりヒト由来骨髄間質細胞に導入した。
リポフェクション3日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図10に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。すなわち、ヒト由来骨髄間質細胞におけるノックインは両側相同組換えであった。
<実験例8>
ヒトiPS細胞のHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
ヒトiPS細胞のHPRT遺伝子座に、GFP遺伝子をノックインすべく、両端10bpの相同アームを含むドナープラスミド、両端約60bpの相同アームを含むドナープラスミド、両端約240bpの相同アームを含むドナープラスミドを作製した。
両端10bp~約240bpの相同アームの塩基配列、HITIの塩基配列、及び外来DNAの塩基配列は、実験例7と同様である。
実験例1同様、Cas9タンパク質、配列番号36 (5’- UUAUGCUGAGGAUUUGGAAAGUUUUAGAGCUAUGCU -3’)に示されるcrRNA、tracrRNA、及び上記ドナープラスミドをエレクトロポレーションによりヒトiPS細胞に導入した。
エレクトロポレーション4日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図11に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。また、シーケンスにより相同組換えであることを確認した。すなわち、ヒトiPS細胞におけるノックインは両側相同組換えであった。
実験例1同様、Cas9タンパク質、配列番号36 (5’- UUAUGCUGAGGAUUUGGAAAGUUUUAGAGCUAUGCU -3’)に示されるcrRNA、tracrRNA、及び上記ドナープラスミドをエレクトロポレーションによりヒトiPS細胞に導入した。
エレクトロポレーション4日後に細胞からゲノムDNAを精製し、PCRによるDNA挿入の確認を行った。図11に示されるように、5’末端及び3’末端で、相同組換えに相当するサイズのバンドが認められた。また、シーケンスにより相同組換えであることを確認した。すなわち、ヒトiPS細胞におけるノックインは両側相同組換えであった。
<実験例9>
<実験例3>における実験を、ZFNやTALENを用いて行った。両端100bpの相同アームを含むドナープラスミドを用いた。結果を図12に示す。ZFNやTALENを用いた場合にも同様に、5’末端及び3’末端で、相同組換えに相当するサイズ
のバンドが認められた。本発明において、標的ゲノムDNA切断酵素としては、CRISPR/Cas9に限らず、ZFNまたはTALENのいずれを使用してもかまわないことが明らかとなった。
<実験例3>における実験を、ZFNやTALENを用いて行った。両端100bpの相同アームを含むドナープラスミドを用いた。結果を図12に示す。ZFNやTALENを用いた場合にも同様に、5’末端及び3’末端で、相同組換えに相当するサイズ
のバンドが認められた。本発明において、標的ゲノムDNA切断酵素としては、CRISPR/Cas9に限らず、ZFNまたはTALENのいずれを使用してもかまわないことが明らかとなった。
上記実験例において、<実験例1>と同様に、シーケンスにより組み換え方法を確認した結果を表1に示す。カッコ内は相同組換えの効率を示す。
表1に示すように、動物種や標的遺伝子座によらず、相同組換えによるノックインが行われることが確認された。
表1に示すように、動物種や標的遺伝子座によらず、相同組換えによるノックインが行われることが確認された。

本発明によれば、細胞が本来有する非相同末端結合の能力を損なわず、標的ゲノムにおいて、非相同組換えに比して高い相同組換え頻度を実現できる。
Claims (26)
- 単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを5’端と3’端に有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
- 前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを前記標的ゲノム導入する、請求項1に記載のゲノム編集方法。
- 前記細胞が、血球系細胞又は未分化細胞である、請求項1又は2に記載のゲノム編集方法。
- 前記細胞が、幹細胞である、請求項1~3のいずれか一項に記載のゲノム編集方法。
- 前記細胞が、造血幹細胞である、請求項1~4のいずれか一項に記載のゲノム編集方法。
- 前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを前記標的ゲノムに導入する、請求項1に記載のゲノム編集方法。
- 単離された細胞中のゲノム編集方法であって、500bp未満の長さの相同アームを有する外来二本鎖DNAを、外来DNAの5’側と3’側のうち、両側を相同組換えにより、標的ゲノムに導入することを特徴とするゲノム編集方法。
- 前記細胞が、血球系細胞又は未分化細胞である、請求項7に記載のゲノム編集方法。
- 前記細胞が、幹細胞である、請求項7又は8に記載のゲノム編集方法。
- 前記細胞が、造血幹細胞である、請求項7~9のいずれか一項に記載のゲノム編集方法。
- 500bp未満の長さの相同アームを両端に有する外来二本鎖DNAを含有することを特徴とする組成物。
- 更に標的ゲノムDNA切断酵素又は前記酵素をコードするDNA若しくはmRNAを含有する、請求項11に記載の組成物。
- 医薬用である、請求項11又は12に記載の組成物。
- 重症複合免疫不全症治療用である、請求項11~13のいずれか一項に記載の組成物。
- 重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、標的ゲノムDNAの二重鎖切断時、外来DNAの5’端と3’端のうち、少なくとも一方を相同組換えにより、前記細胞のゲノム中に導入することを特徴とする細胞製剤の製造方法。
- 前記外来DNAの5’端と3’端のうち、両方を相同組換えにより、外来DNAを標的ゲノムDNAに導入する、請求項15に記載の細胞製剤の製造方法。
- 前記細胞が、血球系細胞又は未分化細胞である、請求項15又は16に記載の細胞製剤の製造方法。
- 前記細胞が、幹細胞である、請求項15~17のいずれか一項に記載の細胞製剤の製造方法。
- 前記細胞が、造血幹細胞である、請求項15~18のいずれか一項に記載の細胞製剤の製造方法。
- 前記外来DNAの5’端と3’端のうち、一方を相同組換えにより、他方を非相同組換えにより、外来DNAを標的ゲノムDNAに導入する、請求項15に記載の細胞製剤の製造方法。
- 重症複合免疫不全症を治療するための細胞製剤の製造方法であって、細胞において、500bp未満の長さの相同アームを有し且つ前記標的ゲノムDNAの野生型DNAの少なくとも一部を有する外来DNAを、前記外来DNAの5’側と3’側のうち、両方を相同組換えにより、前記細胞のゲノム中に導入することを特徴とする細胞製剤の製造方法。
- 前記細胞が、血球系細胞又は未分化細胞である、請求項21に記載の細胞製剤の製造方法。
- 前記細胞が、幹細胞である、請求項21又は22に記載の細胞製剤の製造方法。
- 前記細胞が、造血幹細胞である、請求項21~23のいずれか一項に記載の細胞製剤の製造方法。
- 細胞の標的ゲノム上、外来DNAのゲノム挿入部位の5’側又は3’側に、外来DNA由来の断片を有することを特徴とする細胞。
- 請求項25に記載の細胞を含有することを特徴とする細胞製剤。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/042,819 US20210071202A1 (en) | 2018-03-29 | 2019-03-28 | Genome editing method, composition, cell, cell preparation, and method for producing cell preparation |
| CN201980034209.0A CN112534051A (zh) | 2018-03-29 | 2019-03-28 | 基因组编辑方法、组合物、细胞、细胞制剂、以及细胞制剂的制造方法 |
| JP2020509319A JP7469806B2 (ja) | 2018-03-29 | 2019-03-28 | ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法 |
| EP19774261.2A EP3778888A4 (en) | 2018-03-29 | 2019-03-28 | GENOME EDITING METHODS, COMPOSITION, CELL, CELL PREPARATIONS AND METHODS OF MAKING CELL PREPARATIONS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-066174 | 2018-03-29 | ||
| JP2018066174 | 2018-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019189573A1 true WO2019189573A1 (ja) | 2019-10-03 |
Family
ID=68060174
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/013602 WO2019189573A1 (ja) | 2018-03-29 | 2019-03-28 | ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20210071202A1 (ja) |
| EP (1) | EP3778888A4 (ja) |
| JP (1) | JP7469806B2 (ja) |
| CN (1) | CN112534051A (ja) |
| WO (1) | WO2019189573A1 (ja) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016081923A2 (en) | 2014-11-21 | 2016-05-26 | Regeneron Pharmaceuticals, Inc. | METHODS AND COMPOSITIONS FOR TARGETED GENETIC MODIFICATION USING PAIRED GUIDE RNAs |
| JP2018066174A (ja) | 2016-10-19 | 2018-04-26 | アルプス電気株式会社 | 位置判定装置、位置判定方法及びプログラム並びにキーレスエントリーシステム |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102648489B1 (ko) * | 2015-04-06 | 2024-03-15 | 더 보드 어브 트러스티스 어브 더 리랜드 스탠포드 주니어 유니버시티 | Crispr/cas-매개 유전자 조절을 위한 화학적으로 변형된 가이드 rna |
| US10563226B2 (en) | 2015-05-13 | 2020-02-18 | Seattle Children's Hospital | Enhancing endonuclease based gene editing in primary cells |
| EP3329001B1 (en) * | 2015-07-28 | 2021-09-22 | Danisco US Inc. | Genome editing systems and methods of use |
| EP3383409A4 (en) * | 2015-12-02 | 2019-10-02 | The Regents of The University of California | COMPOSITIONS AND METHODS FOR MODIFYING TARGET NUCLEIC ACID |
| EP3652312A1 (en) | 2017-07-14 | 2020-05-20 | Editas Medicine, Inc. | Systems and methods for targeted integration and genome editing and detection thereof using integrated priming sites |
-
2019
- 2019-03-28 WO PCT/JP2019/013602 patent/WO2019189573A1/ja active Search and Examination
- 2019-03-28 US US17/042,819 patent/US20210071202A1/en active Pending
- 2019-03-28 EP EP19774261.2A patent/EP3778888A4/en active Pending
- 2019-03-28 JP JP2020509319A patent/JP7469806B2/ja active Active
- 2019-03-28 CN CN201980034209.0A patent/CN112534051A/zh active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016081923A2 (en) | 2014-11-21 | 2016-05-26 | Regeneron Pharmaceuticals, Inc. | METHODS AND COMPOSITIONS FOR TARGETED GENETIC MODIFICATION USING PAIRED GUIDE RNAs |
| JP2018066174A (ja) | 2016-10-19 | 2018-04-26 | アルプス電気株式会社 | 位置判定装置、位置判定方法及びプログラム並びにキーレスエントリーシステム |
Non-Patent Citations (8)
| Title |
|---|
| MORGAN, A. RICHARD ET AL.: "Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned", CELL STEM CELL, vol. 21, 2017, pages 574 - 590, XP085274783, doi:10.1016/j.stem.2017.10.010 * |
| NAT BIOTECHNOL., vol. 33, no. 5, May 2015 (2015-05-01), pages 543 - 548 |
| NAT BIOTECHNOL., vol. 34, 2016, pages 339 - 344 |
| NATURE, vol. 539, no. 7629, 17 November 2016 (2016-11-17), pages 384 - 389 |
| SCI REP., vol. 7, 2017, pages 8943 |
| See also references of EP3778888A4 |
| WATANABE M ET AL., PLOS ONE., vol. 8, no. 10, 9 October 2013 (2013-10-09), pages e76478 |
| ZHANG, JIANPING ET AL.: "Efficient precise knock in with a double cut HDR donor after CRISPR/Cas9- mediated double-stranded DNA cleavage", GENOME BIOLOGY, vol. 18, no. 35, 2017, pages 1 - 18, XP055399694 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2019189573A1 (ja) | 2021-04-15 |
| CN112534051A (zh) | 2021-03-19 |
| EP3778888A1 (en) | 2021-02-17 |
| JP7469806B2 (ja) | 2024-04-17 |
| US20210071202A1 (en) | 2021-03-11 |
| EP3778888A4 (en) | 2022-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7197363B2 (ja) | ヌクレアーゼを使用するヒト神経幹細胞のゲノム編集 | |
| JP5175814B2 (ja) | ヒト人工染色体(hac)ベクター | |
| JP2021166542A (ja) | 疾患を処置するための遺伝子改変された細胞、組織および臓器 | |
| JP2021505187A (ja) | 遺伝子編集のためのcpf1関連方法及び組成物 | |
| JP2019528691A (ja) | ゲノム編集エンハンサー | |
| JP2019525759A (ja) | Bcl11aホーミングエンドヌクレアーゼバリアント、組成物、および使用方法 | |
| WO2008013067A1 (fr) | Vecteur de chromosome artificiel humain (cah), et substance pharmaceutique à base de cellules humaines contenant un vecteur de chromosome artificiel humain (cah) | |
| JP7198823B2 (ja) | 全身においてタンパク質を発現させるためのプラットフォームとしての遺伝子工学操作された造血幹細胞 | |
| WO2021084533A1 (en) | Pam-reduced and pam-abolished cas derivatives compositions and uses thereof in genetic modulation | |
| US20230346836A1 (en) | Genetically engineered t cells with disrupted casitas b-lineage lymphoma proto-oncogene-b (cblb) and uses thereof | |
| JP2021529184A (ja) | 遺伝子治療 | |
| Watanabe et al. | Adeno-associated virus-mediated delivery of genes to mouse spermatogonial stem cells | |
| US20210037797A1 (en) | Inducible disease models methods of making them and use in tissue complementation | |
| WO2019189573A1 (ja) | ゲノム編集方法、組成物、細胞、細胞製剤、及び細胞製剤の製造方法 | |
| JP2022512674A (ja) | 人工トランス活性化因子による選択 | |
| US20220364072A1 (en) | Fusion protein that improves gene editing efficiency and application thereof | |
| WO2021212686A1 (zh) | RHO-adRP基于基因编辑的方法和组合物 | |
| US20220184137A1 (en) | Correction of Beta-Thalassemia Phenotype by Genetically Engineered Hematopoietic Stem Cell | |
| US20220213488A1 (en) | Correction of the two most prevalent ush2a mutations by genome editing | |
| CN111518785B (zh) | 一种高活性piggyBac转座酶及其应用 | |
| KR102472344B1 (ko) | 프라임 편집을 이용한 개의 고관절 이형성증 치료용 조성물 | |
| WO2023021506A1 (en) | Homology dna repair enhancement of cas derivatives compositions and uses thereof in genetic modulation | |
| Zainalbden | In vitro correction of hbb v6g mutation in lymphocyte cells of patients with sickle cell anemia using genome-editing crispr/cas9 technique | |
| JP2022531930A (ja) | 栄養要求性調節可能な細胞を使用する方法および組成物 | |
| CN117363649A (zh) | 基因整合方法及应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19774261 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2020509319 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019774261 Country of ref document: EP Effective date: 20201029 |