WO2018026087A2 - 당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학적 조성물 - Google Patents
당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학적 조성물 Download PDFInfo
- Publication number
- WO2018026087A2 WO2018026087A2 PCT/KR2017/004022 KR2017004022W WO2018026087A2 WO 2018026087 A2 WO2018026087 A2 WO 2018026087A2 KR 2017004022 W KR2017004022 W KR 2017004022W WO 2018026087 A2 WO2018026087 A2 WO 2018026087A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- enclomiphene
- formula
- glucoside
- derivatives
- glucose
- Prior art date
Links
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/18—Acyclic radicals, substituted by carbocyclic rings
Definitions
- the present invention relates to an added enclomiphene, a method for preparing the same, and a pharmaceutical composition comprising the same, and more particularly, clomifene, which is a kind of selective estrogen receptor modulator (SERM) used as an infertility drug.
- SERM selective estrogen receptor modulator
- the present invention relates to a compound in which a sugar is added to an enclomifene (enclomiphene) which is a trans-isomer among structural isomers of the compound, and an enzymatic preparation method of the compound and a pharmaceutical composition comprising the same.
- Clomifene or clomiphene is one of the selective estrogen receptor modulators (SERMs) and is best used to treat infertility in women who induce ovulation in ovulation or oligo-ovulation. It is a frequently used prescription (Practice Committee of the American Society for Reproductive Medicine, a committee opinion.Fertil. Steril. 2013. 100 (2): p341-348).
- SERMs selective estrogen receptor modulators
- Clomiphene was first synthesized by organic synthesis in 1956 and began to be used in the 1960s. It was initially used to treat oligomenorrhea syndrome and was used as an infertility treatment due to ovulation-inducing effects. In vivo, clomiphene acts on the hypothalamic estrogen receptor and interferes with the feedback inhibition of estrogens to the gonadotropin, the chorionic gonadotropin, which in turn leads to hypothalamic-pituitary-hypothalamic- Induces ovulation by promoting activation of the pituitary-gonadal axis (Goldstein, SR) et al., Human Reprod. Update, 2000. 6 (3): p212-224.).
- Clomiphene is also known to be effective in the treatment of secondary hypogonadism in men. This pharmacological function has the advantage of cost savings and convenience of administration compared to conventional testosterone replacement therapy (Hill, S. et al., Drugs 2009. 12 (2): p109-119).
- side effects of clomiphene administration may include reversible ovarian enlargement, blurred vision, or visual disorders such as scotomata, headaches and hot flashes. Hypertrophy, reversible alopecia, and ovarian hyperstimulation syndrome may occur.
- Clomiphene is a trans and cis type geometric isomer of enclomifene (ENCLOM) and zuclomifene, which are structurally similar to the representative SERM tamoxifen. It contributes to the mixed activity of estrogen and anti-estrogen in clomiphene (Adashi EY, Fertil Steril, 1984. 42 (3): p331-344).
- Enclomiphene or Androxal under the brand name
- a simple trans isomer of clomiphene is currently entering Phase III clinical trial. Research is ongoing as a treatment for dependent diabetes (Hill S., et al., Drugs, 2009. 12 (2): p109-119.).
- Enclomiphene is a 4-hydroxyenclomifene (hereinafter referred to as 4-OH) by metabolic processes of cytochrome p450 enzyme system of the microsomes in the liver after human administration similarly to tamoxifen as described above, mainly by CYP2D6 action.
- 4-OH 4-hydroxyenclomifene
- -ENCLOM N-desethyl-4-hydroxyenclomifene
- N-DE-4-OH-ENCLOM N-desethyl-4-hydroxyenclomifene
- the active metabolites show at least a 100-fold higher estrogen receptor antagonistic activity than the original compound ENCLOM, and it can be seen that clomiphene is a kind of prodrug (urdter, TE, et al., Hum. Mol. Genet. , 21 (5): p 1145-1154).
- clomiphene is a kind of prodrug (urdter, TE, et al., Hum. Mol. Genet. , 21 (5): p 1145-1154).
- the development of new derivatives or the development of new prodrugs that can provide higher bioavailability than the bioavailability of existing clomiphene or ENCLOM will be important in the future. In other words, high bioavailability will allow for some reduction in clinical treatment dose, and the low dose may reduce the risk of side effects that may occur in the treatment patient.
- Patents for the preparation and use of enclomiphene and related derivatives have been filed for the most part by Repros Therapeutics, Texas, USA, representative patents of which are disclosed in WO2013020017A1 and WO2014197477A1. These patents disclose the structural novelty and production of metabolites (including 4-OH-ENCLOM and N-DE-4-OH-ENCLOM) that have been biotransformed by various metabolic reactions, including the transclomiphene ENCLOM and human liver metabolism. It includes methods and pharmacological uses. However, in the case of the above patents, the scope of patents for ENCLOM and its metabolites is limited through enzymatic bioconversion, so the preparation of compounds added to sugars to ENCLOM and its active metabolites has not been derived to date. .
- the present invention is a sugar addition tamoxifen derivative which can be applied as a preventive and therapeutic agent for breast cancer as a kind of SERM through Korean Patent Application No. 10-2015-0104325 (name of the invention: a method for preparing a sugar-added estrogen receptor modulator using a sugar transfer enzyme). They have successfully converted their lives.
- GT glycotransferase
- the inventors have referred to as a substrate for ENCLOM human recombinant cytochrome P450 mono-and micro spokes also Jia ditch (Micromonospora rhodorangea )
- Jia ditch Micromonospora rhodorangea
- MrGT2 actinomycetes-derived glycosyltransferase
- An object of the present invention is osteoporosis, vaginal atrophy, dyspareunia, gynecomastia, male secondary hypogonadism and insulin-independent diabetes or lipodystrophy, infertility, male prostatic hyperplasia
- SERM derivatives that can be used for the treatment of diseases such as prostate cancer, ovarian cancer and breast cancer by the one-pot biotransformation method of human-derived recombinant cytochrome enzyme and microorganism-derived recombinant glycotransferase It aims to do it.
- the present invention provides an enclomiphene derivative represented by Formula 1, an isomer thereof or a pharmaceutically acceptable solvate, hydrate or prodrug thereof:
- R 1 is CH 2 CH 3 , CH 2 CH 2 Cl or Cl
- R 2 is N (CH 3 ) 2 , OH or (CH 2 ) 4 N
- R 3 is glucose (glucose), 2 2-deoxy-glucose or galactose.
- the present invention also comprises the steps of (a) reacting an enclomiphene and a sugar donor in the presence of a glycotransferase (MrGT2) represented by the amino acid sequence of SEQ ID NO: 4 with cytochromes to synthesize an enclomiphene derivative represented by Chemical Formula 1 ; And (b) provides a method for producing an enclomiphene derivative represented by the formula (1) comprising the step of recovering the synthesized enclomiphene derivative.
- MrGT2 glycotransferase
- the present invention also provides a pharmaceutical composition for treating estrogen receptor-related diseases comprising an enclomiphene derivative represented by Formula 1, an isomer thereof, or a pharmaceutically acceptable solvate, hydrate or prodrug thereof as an active ingredient.
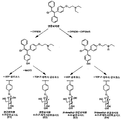
- 1 is a schematic diagram of enclomiphene one-pot biotransformation through human-derived recombinant CYPs and actinomycetes-recombinant glycotransferase MrGT2 enzyme reactions.
- Fig. 2 is a graph showing in vitro antagonistic activity of the enclomiphene derivatives and the estrogen receptor of enclomiphene before bioconversion.
- 3 is a graph comparing the degree of bioavailability improvement of derivatives prepared by analyzing the concentrations of enclomiphene and its active metabolites in experimental animal serum.
- Figure 4 is a graph of pharmacokinetic curves analyzed for 72 hours after administration of the concentration of enclomiphene and its active metabolites in experimental animal serum.
- the present invention isolates the GT coding gene from the genome of the micromonospora Rorandogia strain, constructs a recombinant expression vector and transformant Escherichia coli comprising the same, and then expresses the metabolism of ENCLOM in the expressed sugar transferase.
- Recombinant recombinant CYP2D6 and CYP3A4 / 5 were added, respectively, and uridine diphosphate glucose (UDP-Glc) and thymidine diphosphate 2'-deoxy-glucose, which are enclomiphene and nucleotidyl sugar Sugar addition biosynthesis of ENCLOM derivatives was performed by one-pot reaction in which (thymidine diphosphate 2'-deoxy-glucose, TDP-2'-deoxy-Glc) was reacted with a sugar donor. The structure was then confirmed by mass spectrometry and nuclear magnetic resonance spectrometry instrumental analysis.
- one aspect of the present invention relates to an enclomiphene derivative represented by Formula 1, an isomer thereof, or a pharmaceutically acceptable solvate, hydrate or prodrug thereof.
- R 1 is CH 2 CH 3 , CH 2 CH 2 Cl or Cl
- R 2 is N (CH 3 ) 2 , OH or (CH 2 ) 4 N
- R 3 is glucose (glucose), 2 2-deoxy-glucose or galactose.
- an enclomiphene derivative represented by the following Chemical Formula 1 is reacted with an enclomiphene and a sugar donor in the presence of a glycotransferase (MrGT2) represented by the amino acid sequence of SEQ ID NO: 4 and cytochrome. Synthesizing; And (b) relates to a method for producing an enclomiphene derivative represented by the formula (1) comprising the step of recovering the synthesized enclomiphene derivative.
- MrGT2 glycotransferase
- the enclomiphene derivatives according to the present invention are enclomiphene 4- O -glucoside, N-desethyl-enclomiphene 4- O -glucoside, enclomiphene 4- O- 2'-dioxy glucoside, N-desethyl-enclomiphene 4 -O- 2'-deoxy glucoside.
- the compound of formula 1 according to the present invention may be used in the form of a pharmaceutically acceptable salt, and as the salt, an acid addition salt formed by a pharmaceutically acceptable free acid is useful.
- Inorganic and organic acids can be used as the free acid, hydrochloric acid, bromic acid, sulfuric acid, phosphoric acid, etc. can be used as the inorganic acid, citric acid, acetic acid, lactic acid, maleic acid, fumaric acid, gluconic acid, methanesulfuric acid.
- Phonic acid, glyconic acid, succinic acid, tartaric acid, 4-toluenesulfonic acid, galluxuronic acid, embonic acid, glutamic acid, aspartic acid and the like can be used.
- the compound of Formula 1 according to the present invention may include not only pharmaceutically acceptable salts, but also all salts, hydrates, and solvates that may be prepared by conventional methods.
- the compound of Formula 1 according to the present invention may be prepared in crystalline or amorphous form, and when the compound of Formula 1 is prepared in crystalline form, it may be optionally hydrated or solvated.
- the enzyme reaction obtained in the present invention may further comprise the step of separating or purifying by Medium Pressure Liquid Chromatography (MPLC) method.
- MPLC Medium Pressure Liquid Chromatography
- the present invention provides in vitro antagonistic activity and laboratory animals against ER of the original SERMs and derivatives thereof. The bioavailability of the subject was examined, and the antagonist activity and bioavailability were confirmed to be improved.
- the present invention provides a pharmaceutical composition for treating estrogen receptor-related diseases comprising an enclomiphene derivative represented by Formula 1, an isomer thereof, or a pharmaceutically acceptable solvate, hydrate or prodrug thereof as an active ingredient in another aspect. It is about.
- composition according to the invention may further comprise a pharmaceutically acceptable carrier, excipient or diluent.
- the estrogen receptor-related diseases include osteoporosis, vaginal atrophy, dyspareunia, gynecomastia, male hypogonadism, insulin-independent diabetes, indigestion, and infertility.
- Male prostatic hyperplasia prostate cancer, ovarian cancer or breast cancer.
- treatment refers to any action in which the symptoms of the diseases are improved or cured by administration of a pharmaceutical composition comprising a compound represented by Formula 1, or a pharmaceutically acceptable salt thereof.
- the compound of Formula 1 of the present invention or a pharmaceutically acceptable salt thereof exhibits some in vitro antagonistic activity against ER (Experimental Example 1), and the glycoside improves the bioavailability of the enclomiphene derivatives compared to the administration of conventional enclomiphene.
- Example 2 can be used as a formulation that can not only lower the existing dosage, but also improve some of the side effects of long-term administration.
- compositions of the invention may be formulated in oral or parenteral dosage forms according to standard pharmaceutical practice. These formulations may contain, in addition to the active ingredient, additives such as pharmaceutically acceptable carriers, adjuvants or diluents.
- the pharmaceutical composition containing at least one or more of the compound represented by the formula (1), or a pharmaceutically acceptable salt thereof as an active ingredient may be prepared in various oral or parenteral forms as described below. It may be formulated in a dosage form and administered, but is not limited thereto.
- Formulations for oral administration may be, for example, tablets, pills, hard capsules, soft capsules, solutions, suspensions, emulsifiers, syrups, granules, elixirs, and the like.
- Tods dextrose, sucrose, mannitol, sorbitol, cellulose and / or glycine
- lubricants such as silica, talc, magnesium salts of stearic acid, calcium salts of stearic acid and / or polyethylene glycol have.
- Tablets may also contain binders such as magnesium aluminum silicate, starch paste, gelatin, methylcellulose, sodium carboxymethylcellulose and / or polyvinylpyrrolidine, optionally with starch, agar, alginic acid or its sodium salt.
- binders such as magnesium aluminum silicate, starch paste, gelatin, methylcellulose, sodium carboxymethylcellulose and / or polyvinylpyrrolidine, optionally with starch, agar, alginic acid or its sodium salt.
- binders such as magnesium aluminum silicate, starch paste, gelatin, methylcellulose, sodium carboxymethylcellulose and / or polyvinylpyrrolidine, optionally with starch, agar, alginic acid or its sodium salt.
- Formulations for parenteral administration may be by a method of injecting subcutaneous injection, intravenous injection, intramuscular injection or intrathoracic injection.
- at least one compound represented by Formula 1, or a pharmaceutically acceptable salt thereof in order to formulate into a dosage form for parenteral administration, at least one compound represented by Formula 1, or a pharmaceutically acceptable salt thereof, and mixed with water with a stabilizer or buffer to prepare a solution or suspension, It may be prepared in ampule or vial unit dosage forms.
- the composition may contain sterile and / or preservatives, stabilizers, emulsifiers or emulsifiers, auxiliaries such as salts and / or buffers for the control of osmotic pressure, and other therapeutically useful substances, which are conventional methods of mixing, granulating Or according to a coating method.
- the dosage of the compound of the present invention to the human body may vary depending on the age, weight, sex, dosage form, health condition and degree of disease of the patient, and generally based on an adult patient having a weight of 70 kg. It is 0.001-1000 mg / day, Preferably it is 0.01-500 mg / day, It can also divide and administer once a day to several times at regular time intervals according to a decision of a doctor or a pharmacist.
- the present invention also provides a glycotransferase (MrGT2) gene comprising all of the nucleotide sequence of SEQ ID NO.
- GT may be a sugar transfer enzyme MrGT2 derived from a micromonospora rodoranga strain, but is not limited thereto.
- the present invention also provides MrGT2 comprising all of the amino acid sequence of SEQ ID NO.
- the present invention provides a total of four main products obtained by the sequential enzymatic reaction of 1) the actinomycetes-derived recombinant GT enzyme from E. coli and 2) the human-derived recombinant CYP and the recombinant GT (ie, MrGT2) obtained in step 1). It provides a method for preparing a compound represented by the formula (1) comprising the step of obtaining sugar addition enclofen derivatives.
- the preparation method of the present invention may further include the step of separating, purifying, or separating and purifying the enzyme reaction product obtained in step 2) by medium pressure liquid chromatography (MPLC).
- MPLC medium pressure liquid chromatography
- the recombinant sugar transfer enzyme of step 1) may be separated from the micromonospora Rorandogia, but is not limited thereto.
- the recombinant sugar transferase of step 1) is preferably MrGT2, more preferably MrGT2 expressed in E. coli.
- the stationary phase used in the medium pressure liquid chromatography method in step 3) may be reverse phase C18, but is not limited thereto.
- the mobile phase used in the medium pressure liquid chromatography method in step 3) may be a methanol: water: formic acid 65: 35: 0.2 (v / v / v / v) mixed solution, but is not limited thereto.
- the medium pressure liquid chromatography retention time in step 3 may be 14 to 16 minutes, but is not limited thereto.
- the present invention is directed to treating an estrogen receptor-related disease by administering a pharmaceutical composition comprising an enclomiphene derivative represented by Formula 1, an isomer thereof, or a pharmaceutically acceptable solvate, hydrate or prodrug thereof as an active ingredient. It is about how to.
- the present invention provides a pharmaceutical composition for treating estrogen receptor-related diseases comprising an enclomiphene derivative represented by Formula 1, an isomer thereof, or a pharmaceutically acceptable solvate, hydrate or prodrug thereof as an active ingredient. It is about.
- PCR Polymerase chain reaction
- Genomic DNA of the strain and the primers were mixed with Taq DNA polymerase (Taq DNA polymerase) to perform a total of 32 cycles (initial inert at 5 °C, 52.8 °C 69.3 °C gradient reaction at 72 °C, Reaction after 5 minutes).
- Taq DNA polymerase Taq DNA polymerase
- the T-easy vector was isolated and purified from the selected transformed E.
- nucleotide sequence SEQ ID NO: 3
- amino acid sequence inferred therefrom SEQ ID NO: 4
- the base sequence of the total ORF is 1146bp
- the post-translational protein is composed of a total of 381 amino acids (total 41.1kDa).
- the recombinant E. coli was inoculated at 1% by volume in LB medium (Luria Bertani) to which the above-described antibiotic and sorbitol and 2.5 mM betaine were added at a final concentration of 1 M, and then cultured at 30 ° C. (optical density) When the growth is confirmed between 0.6 and 0.8, to induce protein expression, isopropyl-D-thiogalactopyranoside (IPTG; Sigma, St. Louis, MO, USA). Recombinant E. coli strains were further incubated at 22 ° C. for 18 hours.
- the culture medium was centrifuged at 2000 rpm for 10 minutes to recover the cells, and the cells were dissolved in 50 mM sodium phosphate lysis buffer (300 nM NaCl, 10 mM imidazole, 10% glycerol, 1% Triton X-100), Sonication was performed. Thereafter, refrigerated centrifugation at 12000rpm for 20 minutes, the supernatant was collected separately, and some samples were analyzed by 12% SDS-PAGE to confirm the expression level of the recombinant glycotransferase MrGT1.
- 50 mM sodium phosphate lysis buffer 300 nM NaCl, 10 mM imidazole, 10% glycerol, 1% Triton X-100
- the supernatant described above was equilibrated with 50 mM phosphate buffer solution (0.3 M NaCl, 20 mM imidazole), such as Talon-metal affinity resin, Clontech, Mountain View, CA, USA) and incubated for 1 hour at 4 °C. After refrigeration and centrifugation at 2000 rpm for 5 minutes, the resin was introduced into a disposable column and washed with a phosphate buffer solution containing 50 mM imidazole of 10 times the resin volume. Finally, recombinant sugar transfer enzyme MrGT2 bound to the resin was purified with 3 ml of phosphate buffer containing 150 mM imidazole.
- 50 mM phosphate buffer solution 0.3 M NaCl, 20 mM imidazole
- Talon-metal affinity resin such as Talon-metal affinity resin
- Enclomifene (enclomifene or [E] -clomifene, Sigma-Aldrich, St. Louis, MO, USA), used as a substrate, was dissolved in dimethyl sulfoxide (DMSO) at a level of 20 mM, followed by reaction buffer (50 mM).
- DMSO dimethyl sulfoxide
- NMR samples were prepared by dissolving each derivative in 200 ⁇ l of DMSO-d6 and then leaving the solvent in a 5 mm Shigemi Advanced NMR microtube (Sigemi advanced NMR microtube, Sigma, St. Louis, Mo.). 13 C NMR spectra were obtained at 298 K using a Varian INOVA 500 spectrophotometer and chemical shifts were recorded in ppm using TMS as internal reference. All NMR data calculations were performed using Mnova Suite 5.3.2 software and the sugar additions were compared to SERMs using 13 C-NMR spectra of SERM derivatives as substrates.
- Enclomiphene 4- O -glucoside ( E1 ; 22.3 mg; bioconversion rate 42%; 13 C NMR [125 MHz, DMSO-d6] ⁇ 13.2, 49.7, 54.0, 62.1, 66.8, 71.4, 73.2, 76.8, 81.3, 109.0, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.3, 156.2, 158.3)
- N-desethyl-enclomiphene 4- O -glucoside ( E2 , 20.7 mg; bioconversion 38%; 13 C NMR [125 MHz, DMSO-d6] ⁇ 15.5, 44.2, 49.2, 62.1, 68.9, 71.4, 73.4, 76.8 , 81.5, 109.1, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.3, 156.2, 158.3)
- Enclomiphene 4- O- 2'-dioxy glucoside ( E3 , 13.0 mg; bioconversion rate 24%; 13 C NMR [125 MHz, DMSO-d6] ⁇ 13.2, 37.6, 49.8, 54.1, 62.1, 66.8, 68.9, 71.3 , 81.4, 104.4, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.5, 156.4, 158.6)
- N-desethyl-enclomiphene 4- O- 2'-dioxy glucoside E4 , 11.3 mg; bioconversion rate 20%; 13 C NMR [125 MHz, DMSO-d6] ⁇ 15.5, 37.5, 44.2, 49.3, 62.1, 68.8, 71.4, 81.4, 104.6, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.7, 137.5, 156.6, 158.6)
- the NR peptide ER ⁇ ELISA kit (ActiveMotif, Carlsbad, Calif., USA) was used to test the binding affinity of the biotransformed SERM derivatives to the estrogen receptor ⁇ (ER ⁇ ), and estra as the competition control. Diol (17 ⁇ -estradiol) was used. Detailed experimental methods were performed according to the manufacturer's protocol. Each control dissolved in DMSO was diluted to the buffer contained in the kit and prepared at 25 uM level, and the nuclear extract was prepared according to the kit manufacturer's protocol per well in a 96-well microplate. ) was added to the level of 15 ug. Meanwhile, even in the case of sugar addition derivatives, samples were prepared at a level of 25 uM.
- the bioavailability of the sugar-added enclomiphene derivatives produced by the above example was compared using a clomiphene, which is currently in clinical use, as a control using a mouse model in which the hysterectomy was performed.
- Detailed experimental example was carried out as follows. One week after the hysterectomy was performed on female rats weighing about 150 g, they were used in this experiment. Four dogs were designed into each treatment group, for a total of six groups (ie, one group was a placebo, another group was a clomiphene-treated control group, and the other four groups were each sugar-added derivative treatment group). The body weight before dosing was measured.
- the weight of rats before the administration was constant in all groups, but the average body weight of each group was measured 24 hours after the last administration, and the weight gain was increased in the placebo group (about 28-33 g). It was confirmed.
- the blood concentrations of 4-OH-ECLOM and N-DE-4-OH-ECLOM, the active metabolites of clomiphene were compared with the clomiphene-treated control group.
- the glycosides of the enclomiphene derivatives and the enclomiphene before bioconversion were orally administered to the rats once a week for 3 weeks in the week after hysterectomy, and blood was collected 24 hours after the last administration.
- pharmacokinetic analysis was performed to more accurately confirm the degree of improvement in bioavailability of clomiphene compared to clomiphene.
- the total concentration of enclomiphene and its active metabolites was analyzed by instrumental analysis from 12 sections of plasma samples prepared up to 72 hours in each treatment group, and then the concentration changes of enclomiphene and its active metabolites in plasma with time. Is shown graphically.
- Pharmacokinetic parameters were measured through non-compartmental analysis of WinNonlin Professional software (version 5.1; Pharsight Co., Mountain View, CA, USA). When a pharmacokinetic index of each treatment group the curve maximum blood concentration (C max, peak concentration), maximum blood concentration arrival time (T max, paek time), by measuring the enemy (AUC 72hr, area under curve) parameters in Table 1 Indicated.
- the areas under the curve of the four groups of added clomiphene treatments were higher than the clomiphene treated control groups.
- no noticeable difference was found in the area under the curve between the four sugar additions of the clomiphene derivative treatment groups, but compared to the glucose added clomiphene glucoside derivatives (E1 and E3), clomiphene deoxy was added.
- Treatment with glucoside derivatives (E2 and E4) resulted in slightly higher area under the curve.
- the distribution of the peak blood concentrations of all the treatment groups including the control group did not show a significant difference within the significance level, but the time to reach the peak blood concentration in the remaining four sugar-added derivative treatment groups compared to the clomiphene control group.
- the compound produced in the above Example was formulated as follows.
- the compound After sifting 5.0 mg of the compound, it was mixed with 14.8 mg of lactose, 10.0 mg of polyvinyl pyrrolidone and 0.2 mg of magnesium stearate. The mixture was prepared using a capsule making machine. Filled in 5 gelatin capsules.
- the existing compounds can be improved through the bioconversion of the structurally modified derivatives by the sugar transfer of the existing pharmacologically active compounds, and this improvement can be replaced with the existing prodrugs and new dosage forms of the existing drugs.
- the potential for use as a drug and drug substance, including its applicability, can be identified.
- Formula 1 prepared by one-pot biotransformation of a sequential reaction between a novel recombinant glycotransferase (MrGT2) and a human-derived recombinant CYP enzyme expressed in Escherichia coli derived from a Micromonospora rhodorangea strain according to the present invention.
- the compound of or a pharmaceutically acceptable salt thereof exhibits some in vitro antagonistic activity against the estrogen receptor (ER) and at the same time has an improved bioavailability compared to compounds prior to sugar addition.
- the compound of formula 1 according to the present invention or a pharmaceutically acceptable salt thereof is osteoporosis, vaginal atrophy, dyspareunia, gynecomastia, male hypogonadism and insulin It can be applied as a structural or pharmacologically improved drug for the treatment of diseases such as non-dependent diabetes or lipodystrophy, infertility, male prostate hypertrophy, prostate cancer, ovarian cancer, breast cancer, etc., and according to the administration of existing SERM or enclomiphene It can be used as a new drug formulation that can reduce side effects that may occur.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
본 발명에 따른 화학식 1의 화합물 또는 이의 약학적으로 허용가능한 염은 에스트로겐 수용체α(ERα)에 길항 활성을 유지하면서 생체이용률을 향상시키는 효과가 있는 당부가 엔클로미펜 유도체들로서, 골다공증(osteoporosis), 갱년기성 질 위축증(vaginal atrophy), 성교불쾌증(dyspareunia), 여유증(gynecomastia), 남성 이차 성선기능저하증 및 인슐린 비의존성 당뇨병 혹은 지방이영양증, 불임증, 남성전립선비대증, 전립선암, 난소암, 유방암 등의 에스트로겐 수용체 관련 질환을 치료할 수 있는 프로드럭 혹은 대체 개량 제형으로서 유용하게 사용될 수 있다.
Description
본 발명은 당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학조성물에 관한 것으로서, 더욱 상세하게는 불임치료약으로 사용되고 있는 선택적 에스트로겐 수용체 조절제(selective estrogen receptor modulator, SERM)의 일종인 클로미펜(clomifene)의 구조 이성질체 중 트랜스-이성질체인 엔클로미펜(enclomifene, enclomiphene)에 당이 부가된 화합물 및 상기 화합물의 효소적 제조방법 및 이를 포함하는 약학조성물에 관한 것이다.
클로미펜(clomifene 또는 clomiphene)은 선택적 에스트로겐 수용체 조절제(selective estrogen receptor modulator, 이하 ‘SERM’라 함) 중 하나로서 무배란증(anovulation)이나 희소배란증(oligo-ovulation)에 배란 유도를 일으키는 여성 불임치료에 가장 빈번히 사용되고 있는 처방전이다(Practice Committee of the American Society for Reproductive Medicine, a committee opinion. Fertil. Steril. 2013. 100(2): p341-348).
클로미펜은 1956년 유기 합성법으로 첫 합성된 후 1960년대부터 사용되기 시작하였으며, 초기에는 희발월경(oligomenorrhea) 증후군의 치료로 사용되다가 배란 유도효과에 따른 불임치료제로 사용되고 있다. 생체 내 클로미펜은 시상하부 내 에스트로겐 수용체에 작용하여 융모생식샘자극호르몬인 고나도트로핀(gonadotropin)에 대한 에스트로겐의 피드백 저해(feedback inhibition)를 방해하고, 이는 결국 시상하부-뇌하수체-성선축(hypothalamic-pituitary-gonadal axis)의 활성화를 촉진하여 배란을 유도하게 된다(Goldstein, S.R. et al., Human Reprod. Update, 2000. 6(3): p212-224.). 클로미펜은 또한 남성의 이차 성선기능저하증(hypogonadism)의 치료에 효과적인 것으로 알려져 있다. 이 약리적 기능은 기존 테스토스테론 대체요법(testosterone replacement therapy)과 비교하여 비용 상 절감 및 투여의 편리성이라는 이점이 있다(Hill, S. et al., Drugs 2009. 12(2): p109-119). 반면, 클로미펜 투여에 따른 부작용으로 가역적인 난소 비대증(ovarian enlargement)이나 변시증(blurred vision)이나 암점(scotomata)과 같은 시각 장애 현상, 두통 및 안면홍조(hot flash) 등이 나타날 수 있으며 드문 경우로 간 비대증, 가역적 탈모증 및 난소 고자극 신드롬 등이 나타날 수 있다.
클로미펜은 대표적 SERM인 타목시펜(tamoxifen)과 구조적으로 유사한 트리페닐에틸렌(triphenylethylene) 구조의 트랜스(trans) 및 시스(cis)형의 기하 구조 이성질체인 엔클로미펜(enclomifene, 이하 ENCLOM)과 주클로미펜(zuclomifene)으로 이루어져 있으며, 클로미펜의 에스트로겐성 및 항-에스트로겐성의 혼합된 활성에 기여하고 있다(Adashi E.Y., Fertil Steril, 1984. 42(3): p331-344). 특히, 전술한 바와 같은 클로미펜의 남성 이차 성선기능저하증(secondary hypogonadism) 치료 효과와 관련하여, 최근 클로미펜의 단순 트랜스 이성질체인 엔클로미펜(혹은 브랜드명으로 Androxal)이 임상 3상에 진입 중이며, 이밖에도 인슐린 비의존성 당뇨병에 대한 치료제로서 연구가 진행 중이다(Hill S., et al., Drugs, 2009. 12(2): p109-119.).
엔클로미펜 및 그 구조 대사체에 대한 선행문헌들은 대부분 클로미펜의 체내 투여 후 대사과정 중 나타나는 대사체들에 대한 내용이 대부분이다(Mazzarino M., et al., Eur. J. Mass Spectrom., 2008. 14(3): p171-180; Ganchev B., et al., Anal. Bioanal. Chem., 2011. 400(10): p3429-3441; Murdter, T.E., et al., Hum. Mol. Genet., 2012. 21(5): p1145-1154). 엔클로미펜은 전술한 바 있는 타목시펜과 유사하게 인체 투여 후 간 내 마이크로솜의 사이토크롬 p450 효소계의 대사과정을 통하여, 즉 주로 CYP2D6 작용에 의한 4-하이드록시 엔클로미펜(4-hydroxyenclomifene, 이하 4-OH-ENCLOM)과 CYP2D6와 CYP3A4/5의 연속적 대사 작용에 의한 N-데스에틸-4-하이드록시 엔클로미펜(N-desethyl-4-hydroxyenclomifene, 이하 N-DE-4-OH-ENCLOM) 활성대사체로 대사된다. 상기 활성 대사체들은 원래 화합물인 ENCLOM에 비하여 적어도 100배 이상 높은 에스트로겐 수용체 길항활성을 보이고 있으며 이를 통하여 클로미펜도 프로드럭의 일종임을 알 수 있다(urdter, T.E., et al., Hum. Mol. Genet., 2012. 21(5): p1145-1154). 이와 같이 신규한 유도체의 개발이나 혹은 기존 클로미펜이나 ENCLOM의 생체이용률(bioavailability)보다 높은 생체이용률을 제공할 수 있는 신규한 프로드럭의 개발은 향후 중요하다 할 것이다. 즉, 높은 생체이용률은 임상 처치 용량(dose)의 일부 감소화를 가능케 할 것이며, 그 저용량 투약에 따라서 처치 환자 대상으로 발생 가능한 부작용 위험성을 감소시킬 수 있을 것이다.
SERM의 구조적 변형을 통한 신규 유도체의 개발은 개량신약 및 의약품 전달 시스템(drug delivery system) 개발의 주요 기술로서 인식되고 있으며, 그 중 하나는 그 화학구조 내 수산기(hydroxy function)에 당 분자를 전이시킬 수 있는 당전이 효소(GT)를 이용한 효소적 제조 방법이 있다. 특히, 엔클로미펜과 그 구조적 유사 화합물들의 당전이화는 원래 물질에 비하여 초회 통과(first pass)를 거친 대사(metabolism) 과정에서 혈중 약물 농도를 일정 수준이상으로 유지하여 생체이용률을 향상시킬 수가 있으므로, 의약품의 제형 개량, 신규 제형 개발 및 프로드럭(prodrug)화 등에 유용한 생전환(bio-conversion) 기술이라 할 것이다.
엔클로미펜 및 그 관련 유도체들의 제조 방법 및 용도에 대한 특허는 대부분 미국 텍사스 소재 리프로스 테라퓨틱스(Repros Therapeutics)에 의하여 출원되었으며, 그 대표적인 특허는 국제특허공개 WO2013020017A1 및 WO2014197477A1에 개시되어 있다. 상기 공개특허들은 트랜스형 클로미펜인 ENCLOM과 인체 간 대사를 포함한 다양한 대사반응으로 생전환된 대사체들(4-OH-ENCLOM 및 N-DE-4-OH-ENCLOM을 포함)의 구조적 신규성과 그 제조방법 및 약리적 용도를 포함하여 기재하고 있다. 하지만 상기 특허들의 경우엔 효소적 생전환을 통하여 ENCLOM 및 그 대사체들에 대한 특허 범위가 제한되어 있는 바, ENCLOM 및 그 활성대사체들에 당이 부가한 화합물의 제조는 현재까지 그 유래가 없다.
본 발명자는 대한민국 특허출원 제10-2015-0104325호(발명의 명칭: 당전이 효소를 이용한 당 부가된 에스트로겐 수용체 조절제의 제조방법)를 통해 SERM의 일종으로 유방암 예방 및 치료제로 응용 가능한 당 부가 타목시펜 유도체들의 생전환에 성공한 바 있다. 이를 위하여, SERM 활성을 지닌 타목시펜 구조 유사체들을 기질로 하여 당전이 반응을 가능케 하는 마이크로모노스포라 로도랑지아(Micromonospora
rhodorangea) 방선균 유래 신규 당전이효소(GT)의 발견, 효소적 반응을 통한 당부가 SERM의 생합성, 나아가 당부가 유도체들의 원래 화합물과 비교하여 생체이용률과 같은 제형적 특성의 개선 등을 확인하였다.
이에, 본 발명자들은 ENCLOM을 기질로 하여 인간 재조합 사이토크롬 P450과 마이크로모노스포라 로도랑지아(Micromonospora
rhodorangea) 방선균 유래 재조합 당전이 효소(glycosyltransferase; 이하 MrGT2) 조합을 활용한 원팟(one-pot) 당부가 엔클로미펜 유도체들을 생합성한 결과, 이들 유도체들이 당부가 전의 원래 SERM들과 그 유도체들의 에스트로겐 수용체(ER)에 대한 인 비트로(in vitro) 길항 활성 및 약물동력학적으로 생체이용률이 향상된 것을 확인하고, 본 발명을 완성하였다.
발명의 요약
본 발명의 목적은 골다공증(osteoporosis), 갱년기성 질 위축증(vaginal atrophy), 성교불쾌증(dyspareunia), 여유증(gynecomastia), 남성 이차 성선기능저하증 및 인슐린 비의존성 당뇨병 혹은 지방이영양증, 불임증, 남성전립선비대증, 전립선암, 난소암, 유방암 등의 질환 치료 목적으로 사용될 수 있는 당부가 SERM 유도체를 인간 유래 재조합 사이토크롬효소와 미생물 유래 재조합 당전이효소의 원팟(one-pot) 생전환 방법에 의해 제조하여 제공하는 것을 목적으로 한다.
상기 목적을 달성하기 위하여, 본 발명은 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용 가능한 이들의 용매화물, 수화물 또는 프로드럭을 제공한다:
화학식 1

상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다.
본 발명은 또한, (a) 서열번호 4의 아미노산 서열로 표시되는 당전이효소(MrGT2)와 사이토크롬의 존재하에 엔클로미펜 및 당 공여체를 반응시켜 상기 화학식 1로 표시되는 엔클로미펜 유도체를 합성시키는 단계; 및 (b) 상기 합성된 엔클로미펜 유도체를 회수하는 단계를 포함하는 화학식 1로 표시되는 엔클로미펜 유도체의 제조방법을 제공한다.
본 발명은 또한, 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용 가능한 이들의 용매화물, 수화물 또는 프로드럭을 유효성분으로 포함하는 에스트로겐 수용체 관련 질환 치료용 약학 조성물을 제공한다.
도 1은 인간 유래 재조합 CYP들과 방선균 유래 재조합 당전이효소 MrGT2 효소 반응을 통한 엔클로미펜 원팟 생전환 모식도이다.
도 2는 당부가 엔클로미펜 유도체들과 생전환 전 엔클로미펜의 에스트로겐 수용체에 대한 인비트로 길항 활성을 나타낸 그래프이다.
도 3는 실험동물 혈청 중 엔클로미펜 및 그 활성대사체들의 농도를 분석함으로서 제조된 유도체들의 생체이용률 향상 정도를 비교한 그래프이다.
도 4는 실험동물 혈청 중 엔클로미펜 및 그 활성대사체들의 농도를 투약 후 72시간 동안 분석한 약물동력학 곡선 그래프이다.
발명의 상세한 설명 및 구체적인
구현예
다른 식으로 정의되지 않는 한, 본 명세서에서 사용된 모든 기술적 및 과학적 용어들은 본 발명이 속하는 기술 분야에서 숙련된 전문가에 의해서 통상적으로 이해되는 것과 동일한 의미를 갖는다. 일반적으로, 본 명세서에서 사용된 명명법은 본 기술 분야에서 잘 알려져 있고 통상적으로 사용되는 것이다.
본 발명은 마이크로모노스포라 로도랑지아 균주의 유전체(genome)로부터 GT 암호화 유전자를 분리하고, 이를 포함하는 재조합 발현 벡터 및 형질전환 대장균을 제작한 다음, 상기 발현된 당전이 효소에 ENCLOM의 대사에 관여하는 재조합 CYP2D6와 CYP3A4/5를 각각 첨가하고, 엔클로미펜 및 핵산당(nucleotidyl sugar)인 우리딘 이인산 글루코스(uridine diphosphate glucose; 이하 UDP-Glc)와 티미딘 이인산 2’-디옥시-글루코스(thymidine diphosphate 2’-deoxy-glucose, TDP-2’-deoxy-Glc)를 당 공여체로 반응시킨 원팟 반응에 의하여 당부가 ENCLOM 유도체를 생합성하였다. 그 다음, 질량분석기와 핵자기 공명 스펙트로미트리(nuclear magnetic resonance spectrometry) 기기분석을 통하여 그 구조를 확인하였다.
따라서, 본 발명의 일 관점은 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭에 관한 것이다.
화학식 1

상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다.
또한, 본 발명의 다른 관점에서 (a) 서열번호 4의 아미노산 서열로 표시되는 당전이효소(MrGT2)와 사이토크롬의 존재하에 엔클로미펜 및 당 공여체를 반응시켜 하기 화학식 1로 표시되는 엔클로미펜 유도체를 합성시키는 단계; 및 (b) 상기 합성된 엔클로미펜 유도체를 회수하는 단계를 포함하는 화학식 1로 표시되는 엔클로미펜 유도체의 제조방법에 관한 것이다.
본 발명에 따른 엔클로미펜 유도체는 엔클로미펜 4-O-글루코사이드, N-데스에틸-엔클로미펜 4-O-글루코사이드, 엔클로미펜 4-O-2'-디옥시 글루코사이드, N-데스에틸-엔클로미펜 4-O-2'-디옥시 글루코사이드일 수 있다.
본 발명에 따른 상기 화학식 1의 화합물은 약학적으로 허용가능한 염의 형태로 사용할 수 있으며, 염으로는 약학적으로 허용가능한 유리산(free acid)에 의해 형성된 산 부가염이 유용하다. 유리산으로는 무기산과 유기산을 사용할 수 있으며, 무기산으로는 염산, 브롬산, 황산, 인산 등을 사용할 수 있고, 유기산으로는 구연산, 아세트산, 젖산, 말레산, 푸마린산, 글루콘산, 메탄설폰산, 글리콘산, 숙신산, 타타르산, 4-톨루엔술폰산, 갈룩투론산, 엠본산, 글루탐산, 아스파르트산 등을 사용할 수 있다.
또한, 본 발명에 따른 상기 화학식 1의 화합물은 약학적으로 허용되는 염뿐만 아니라, 통상의 방법에 의해 제조될 수 있는 모든 염, 수화물 및 용매화물을 포함할 수 있다.
또한, 본 발명에 따른 상기 화학식 1의 화합물은 결정 형태 또는 비결정 형태로 제조될 수 있으며, 화학식 1의 화합물이 결정 형태로 제조될 경우, 임의로 수화되거나 용매화될 수 있다.
본 발명에서 수득한 효소반응물을 중압 액체 크로마토그래피(Medium Pressure Liquid Chromatography, MPLC) 방법으로 분리 또는 정제하는 단계를 추가로 포함할 수 있다.
본 발명은 당이 효소적으로 부가된 SERM 유도체들 즉 엔클로펜 유도체의 생화학적 활성을 확인하고자, 당부가 전의 원래 SERM들과 그 유도체들의 ER에 대한 인 비트로(in vitro) 길항 활성 및 실험동물을 대상으로 한 생체이용률을 검사하였으며, 길항 활성 및 생체이용률이 향상된 것을 확인하였다.
따라서, 본 발명은 또 다른 관점에서 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭을 유효성분으로 포함하는 에스트로겐 수용체 관련 질환 치료용 의약 조성물에 관한 것이다.
본 발명에 따른 약학 조성물은 의약적으로 허용가능한 담체, 부형제 또는 희석제를 추가로 포함할 수 있다.
본 발명에서 상기 에스트로겐 수용체 관련 질환은 골다공증(osteoporosis), 갱년기성 질 위축증(vaginal atrophy), 성교불쾌증(dyspareunia), 여유증(gynecomastia), 남성 이차 성선기능저하증, 인슐린 비의존성 당뇨병, 지방이영양증, 불임증, 남성전립선비대증, 전립선암, 난소암 또는 유방암일 수 있다.
본 발명에서 사용되는 용어 "치료"는, 화학식 1로 표시되는 화합물, 또는 이의 약학적으로 허용가능한 염을 포함하는 약학적 조성물의 투여로 상기 질환들의 증세가 호전되거나 완치되는 모든 행위를 의미한다.
본 발명의 상기 화학식 1의 화합물 또는 이의 약학적으로 허용가능한 염은 ER에 대한 일부 인비트로 길항 활성을 나타내고(실험예 1), 기존 엔클로미펜의 투약에 비하여 당부가 엔클로미펜 유도체들의 생체이용률이 향상(실험예 2)됨을 나타내므로 기존 투여량을 낮출 수 있을 뿐 만 아니라 장기 투여에 대한 부작용을 일부 개선할 수 있는 제형으로 유용하게 사용될 수 있다.
본 발명의 약학적 조성물은 표준 약학적 실시에 따라 경구 또는 비경구 투여 형태로 제형화할 수 있다. 이들 제형은 유효성분 이외에 약학적으로 허용가능한 담체, 보조제 또는 희석액 등의 첨가물을 함유할 수 있다.
본 발명의 조성물을 의약품으로 사용하는 경우, 화학식 1로 표시되는 화합물, 또는 이의 약학적으로 허용가능한 염을 적어도 하나 이상 유효성분으로 함유하는 약학적 조성물은 임상 투여시에 하기의 다양한 경구 또는 비경구 투여 형태로 제제화되어 투여될 수 있으나, 이에 한정되지 않는다.
경구 투여용 제형으로는 예를 들면 정제, 환제, 경질 캅셀제, 연질 캅셀제, 액제, 현탁제, 유화제, 시럽제, 과립제, 엘릭시르제 등의 제형일 수 있는데, 이들 제형은 유효성분 외에 희석제(예: 락토즈, 덱스트로즈, 수크로즈, 만니톨, 솔비톨, 셀룰로즈 및/또는 글리신), 활택제(예: 실리카, 탈크, 스테아르산의 마그네슘염, 스테아르산의 칼슘염 및/또는 폴리에틸렌 글리콜)를 함유할 수 있다. 정제는 또한, 마그네슘 알루미늄 실리케이트, 전분 페이스트, 젤라틴, 메틸셀룰로즈, 나트륨 카복시메틸셀룰로즈 및/또는 폴리비닐피롤리딘과 같은 결합제를 함유할 수 있으며, 경우에 따라 전분, 한천, 알긴산 또는 그의 나트륨염과 같은 붕해제 또는 비등 혼합물 및/또는 흡수제, 착색제, 향미제, 및 감미제를 함유할 수 있다.
비경구 투여용 제형으로는 피하 주사, 정맥 주사, 근육 내 주사 또는 흉부 내 주사를 주입하는 방법에 의할 수 있다. 이때, 비경구 투여용 제형으로 제제화하기 위하여, 화학식 1로 표시되는 화합물, 또는 이의 약학적으로 허용가능한 염을 적어도 하나 이상 포함하고, 안정제 또는 완충제와 함께 물에 혼합하여 용액 또는 현탁액으로 제조하며, 이를 앰플 또는 바이알 단위 투여형으로 제조할 수 있다. 상기 조성물은 멸균되고/되거나 방부제, 안정화제, 수화제 또는 유화 촉진제, 삼투압 조절을 위한 염 및/또는 완충제 등의 보조제, 기타 치료적으로 유용한 물질을 함유할 수 있으며, 통상적인 방법인 혼합, 과립화 또는 코팅 방법에 따라 제제화할 수 있다.
또한, 본 발명의 화합물의 인체에 대한 투여량은 환자의 나이, 몸무게, 성별, 투여 형태, 건강 상태 및 질환 정도에 따라 달라질 수 있으며, 몸무게가 70㎏인 성인 환자를 기준으로 할 때, 일반적으로 0.001~1,000㎎/일이고, 바람직하게는 0.01~500㎎/일이며, 의사 또는 약사의 판단에 따라 일정시간 간격으로 1일 1회 내지 수회로 분할 투여할 수도 있다.
또한, 본 발명은 서열번호 3의 염기 서열의 전부를 포함하는 당전이 효소(MrGT2) 유전자를 제공한다.
본 발명의 일 태양에서, 상기 GT는 마이크로모노스포라 로도랑지아 균주 유래의 당전이 효소 MrGT2일 수 있으나, 이에 한정되지 않는다.
또한, 본 발명은 서열번호 4의 아미노산 서열의 전부를 포함하는 MrGT2를 제공한다.
또한, 본 발명은 1) 방선균 유래 재조합 GT 효소를 대장균에서 얻는 단계 및 2) 인간 유래 재조합 CYP와 상기 1) 단계에서 얻어진 재조합 GT(즉, MrGT2)의 순차적 효소반응으로 얻어진 주요 산물 총 4종의 당부가 엔클로펜 유도체들을 얻는 단계를 포함하는 상기 화학식 1로 표시되는 화합물의 제조 방법을 제공한다.
본 발명의 제조방법은 3) 상기 2) 단계에서 얻어진 효소반응 산물을 중압 액체 크로마토그래피(Medium Pressure Liquid Chromatography; 이하 MPLC) 방법으로 분리, 정제, 또는 분리 및 정제하는 단계를 추가적으로 포함할 수 있다.
본 발명에서, 상기 1) 단계의 재조합 당전이 효소는 마이크로모노스포라 로도랑지아로부터 분리할 수 있으나, 이에 한정되지 않는다.
본 발명에서, 상기 1) 단계의 재조합 당전이 효소는 바람직하게는 MrGT2이고, 더욱 바람직하게는 대장균에서 발현된 MrGT2이다.
본 발명에서, 상기 3) 단계에서 중압 액체 크로마토그래피 방법에 사용되는 고정상은 역상 C18일 수 있으나, 이에 한정되지 않는다.
본 발명에서, 상기 3) 단계에서 중압 액체 크로마토그래피 방법에 사용되는 이동상은 메탄올:물:포름산 65:35:0.2(v/v/v/v) 혼합액일 수 있으나, 이에 한정되지 않는다.
본 발명에서, 상기 3) 단계에서 중압 액체 크로마토그래피 유지 시간(Retention time)은 14~16분일 수 있으나, 이에 한정되지 않는다.
본 발명은 또 다른 관점에서 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭을 유효성분으로 포함하는 의약 조성물을 투여하여 에스트로겐 수용체 관련 질환을 치료하는 방법에 관한 것이다.
본 발명은 또 다른 관점에서 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭을 유효성분으로 포함하는 에스트로겐 수용체 관련 질환 치료용 의약 조성물의 용도에 관한 것이다.
이하, 실시예를 통하여 본 발명을 더욱 상세히 설명하고자 한다. 이들 실시예는 오로지 본 발명을 예시하기 위한 것으로, 본 발명의 범위가 이들 실시예에 의해 제한되는 것으로 해석되지 않는 것은 당업계에서 통상의 지식을 가진 자에게 있어서 자명할 것이다.
실시예
실시예 1~2: 당부가 엔클로미펜 유도체들의 제조
(1) 마이크로모노스포라 로도랑지아 유전체로부터 GT 암호화 유전자의 분리
마이크로모노스포라 로도랑지아 균주로부터 유래한 스테롤 당전이 효소를 분리하기 위하여, 우선 마이크로모노스포라 로도랑지아 균주에서 분리한 유전체를 주형으로 사용하고, 기존 마이크로모노스포라 속에서 보고된 유전체의 염기서열을 기초로 하여 제작한 서열번호 1 및 서열번호 2의 프라이머(primer)로 중합효소 연쇄반응(polymerase chain reaction, PCR)을 다음과 같이 실시하였다. N-터미널 프라이머는 하기 서열번호 1의 서열이고, C-터미널 프라이머는 하기 서열번호 2의 서열이다.
서열번호 1
5'-GG CATATG ATCCACGCGCACGACTTCCGGATG-3' (NdeI 제한위치)
서열번호 2
5'-CG CTCGAG ATTCGGCCTGCGCCTCCCACGTCCA-3' (XhoI 제한위치)
상기 균주의 유전체 DNA 및 상기 프라이머들을 Taq DNA 중합효소(Taq DNA polymerase)와 함께 혼합하여 전체 32 사이클을 수행하였다(초기 불활성 98℃에서 5분, 52.8℃에서 69.3℃로 기울기 반응 후, 72℃에서 5분 동안 반응 후 종결). PCR 산물을 정제한 후, pGEMR-T 이지 벡터(pGEMR-T easy vector, Promega, Madison, WI, USA)에 접합한 다음, 대장균 XL1 블루(XL1 blue, Stratagene, La Jolla, CA, USA)에 42℃에서 45초간 열처리한 후, 형질전환을 수행하였다. 선발된 형질전환 대장균으로부터 T-이지 벡터를 분리, 정제한 후 그 염기서열(서열번호 3) 및 이로부터 유추한 아미노산 서열(서열번호 4)을 결정하였다. 전체 ORF의 염기서열은 1146bp이며, 그 번역 후 단백질은 총 381개의 아미노산으로 구성(총 41.1kDa)되어 있다.
서열번호 3

서열번호 4

(2) 대장균에서 재조합 당전이 효소 MrGT2의 발현
상기 T-이지 벡터를 NdeI과 XhoI 제한효소로 처리한 MrGT2 DNA 절편을, 동일한 제한효소들로 처리한 단백질 발현 벡터인 pET-28(a+)(Novagen, Madison, WI, USA)에 삽입한 후, 대장균 BL21(DE3)(Stratagene, La Jolla, CA, USA)에 형질전환시켰다. 이때, 형질전환체의 선발을 위하여 카나마이신 항생제 50ppm을 사용하였다.
상기 재조합 대장균을 전술한 항생제와 최종 농도 1M의 소르비톨(sorbitol) 및 2.5mM 베타인(betaine)이 첨가된 LB 배지(Luria Bertani)에 1% 부피비로 접종한 후, 30℃에서 배양하였고, 광학농도(optical density) 0.6~0.8 사이로 성장이 확인될 때, 단백질 발현을 유도시키고자, 최종 농도 0.5mM의 이소프로필-D-티오갈락토피라노시드(isopropyl-D-thiogalactopyranoside, IPTG; Sigma, St. Louis, MO, USA)를 첨가하였다. 재조합 대장균 균주를 22℃에서 18시간 추가 배양하였다. 배양 후 배양액을 2000rpm에서 10분간 원심분리하여 균체를 회수한 후, 50mM 인산나트륨 용균(lysis) 완충용액(300nM NaCl, 10mM imidazole, 10% glycerol, 1% Triton X-100)에 균체를 용해시키고, 초음파 분해(sonication)를 실시하였다. 이 후, 12000rpm에서 20분간 냉장 원심분리하고, 상등액을 따로 모아 일부 시료를 12% SDS-PAGE로 분석하여 재조합 당전이 효소 MrGT1의 발현 정도를 확인하였다. 발현이 확인된 재조합 단백질을 정제하기 위하여, 전술한 상등액을 50mM 인산 완충용액(0.3M NaCl, 20mM imidazole)으로 평형화된 탈론-금속 친화성 수지(Talon-metal affinity resin, Clontech, Mountain View, CA, USA)와 섞은 후 4℃에서 한시간 동안 배양하였다. 2000rmp에서 5분간 냉장 원심분리한 후, 수지를 일회용 컬럼에 도입한 후, 수지 10배 용량의 50mM 이미다졸을 포함하는 인산 완충용액으로 세척하였다. 최종적으로 150mM 이미다졸을 포함한 인산 완충용액 3 ml로 수지에 결합된 재조합 당전이 효소 MrGT2를 정제하였다.
(3) 인간 유래 재조합 CYP들과 방선균 유래 재조합 MrGT2 연쇄 효소 반응에 의한 엔클로미펜의 당전이 유도체로 생전환, 분리 및 정제
기질로 사용되는 엔클로미펜(enclomifene 혹은 [E]-clomifene, Sigma-Aldrich, St. Louis, MO, 미국)을 20mM 수준으로 디메틸설폭시드(dimethyl sulfoxide; 이하 DMSO)에 용해한 후, 반응 완충용액(50mM 인산 완충용액, 10mM 염화마그네슘)으로 최종 1mM의 농도가 되게 각각을 희석한 후, BD Gentest사 (Woburn, MA, USA)에서 구입한 인간 유래 재조합 CYP(CYP2D6, CYP3A4 그리고 CYP3A5, 각각 20uM 수준), 재조합 당전이 효소 MrGT2 25uM와 핵산당인 UDP-Glc 및 TDP-2’-deoxy-Glc 각각 2mM의 수준으로, 그리고 CYP의 산화 반응을 위한 NADH조효소의 지속적 공급을 위하여 추가적으로 글루코스 6-인산(10mM, Sigma-Aldrich), NADP+(1mM, Sigma-Aldrich) 및 글루코스 6-인산 탈수소효소(8U/ml 수준, Sigma-Aldrich)를 첨가하여 37℃에서 2시간 동안 원팟 반응을 실시하였다(도 1). 반응 후, 동량의 에틸아세테이트를 첨가하여 반응을 정지시킨 후, 6000rpm에서 10분간 원심분리한 후의 상층 유기 용매층만을 모아서 감압건조를 실시하였다. 상기 추출물 내 목적 화합물의 생합성 여부를 확인하기 위하여 HPLC-ESI-MS 분석을 수행 하였다. 이동상으로 메탄올:물:포름산 65:35:0.2(v/v/v/v) 혼합액을 분당 140μl 속도로, 고정상 컬럼으로는 Acquity CSH C18(Waters, 50×1.0mm, 1.7μm; Milford, MA, USA)을 사용하여 분석하였다.
한편, 상기 조추출물로부터 목적하는 당부가 SERM 유도체들의 분리 및 정제를 위하여 하기 콤비플래시 Rf MPLC 시스템을 적용하였다. 역상(reverse-phased) C18 카트리지에 메탄올:물:포름산 65:35:0.2(v/v/v/v) 혼합액을 이동상으로 분당 15ml의 속도로 흘러가는 MPLC 시스템에, 동일한 이동상 6ml에 용해시킨 전술한 조추출물을 주입한 후, 크로마토그램 상 검출된 피크를 자동 분취하였다. 개별 분획을 다시 감압하여 농축한 후, 이온트랩 질량 분석기(LCQ ion-trap mass spectrometer, ThermoFinnigan, San Jose, CA, USA)로 질량 스펙트럼을 분석함으로써 목적하는 당부가 SERM 유도체들을 분리하였다. 기질로 엔클로미펜은 MPLC 시스템 상 약 17~18분의 유지 시간으로 분취되었고, 목적하는 당부가 유도체들은 기질보다 빠른 유지 시간인 14~16분으로 검출되었다. 이들 정제된 당부가 화합물은 도 1에 나타내었다. 핵산당으로 UDP-Glc 당공여체를 이용한 생전환 반응의 수율은 약 40% 정도로 반응이 일어났으나, TDP-2’-deoxy-Glc의 경우엔 그 생전환 수율이 25% 이하 수준으로 상대적으로 낮게 나타난 바, 이는 당전이 효소의 당수용체 및 당공여체 기질들에 대한 특이성의 차이를 제시하였다.
NMR 시료들은 200㎕의 DMSO-d6에 각 유도체들을 용해시킨 후, 5mm 시게미 어드밴스드 NMR 마이크로튜브(Shigemi advanced NMR microtube, Sigma, St. Louis, MO)에 상기 용매를 방치시킴으로써 제조하였다. 13C NMR 스펙트럼은 배리언(Varian) INOVA 500 분광광도계를 이용하여 298 K에서 획득하였고, 화학적 이동은 내부 준거로서 TMS를 이용하여 ppm으로 기록되었다. 모든 NMR 데이타 산출은 Mnova Suite 5.3.2 소프트웨어를 이용하여 수행하였고, 당부가 SERM 유도체들의 13C-NMR 스펙트럼을 기질로 사용한 SERM들과 비교하였다.
엔클로미펜 4-O-글루코사이드(E1; 22.3mg; 생전환율 42%; 13C NMR [125 MHz, DMSO-d6] δ 13.2, 49.7, 54.0, 62.1, 66.8, 71.4, 73.2, 76.8, 81.3, 109.0, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.3, 156.2, 158.3)
N-데스에틸-엔클로미펜 4-O-글루코사이드(E2, 20.7mg; 생전환율 38%; 13C NMR [125 MHz, DMSO-d6] δ 15.5, 44.2, 49.2, 62.1, 68.9, 71.4, 73.4, 76.8, 81.5, 109.1, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.3, 156.2, 158.3)
엔클로미펜 4-O-2'-디옥시 글루코사이드(E3, 13.0mg; 생전환율 24%; 13C NMR [125 MHz, DMSO-d6] δ 13.2, 37.6, 49.8, 54.1, 62.1, 66.8, 68.9, 71.3, 81.4, 104.4, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.8, 137.5, 156.4, 158.6)
N-데스에틸-엔클로미펜 4-O-2'-디옥시 글루코사이드(E4, 11.3mg; 생전환율 20%; 13C NMR [125 MHz, DMSO-d6] δ 15.5, 37.5, 44.2, 49.3, 62.1, 68.8, 71.4, 81.4, 104.6, 114.1, 120.3, 127.6, 128.4, 129.1, 130.1, 131.1, 131.7, 137.5, 156.6, 158.6)
실험예 1: 당부가 엔클로미펜 유도체들의 에스트로겐 수용체α(ERα)에 대한 길항 활성 검정
생전환 된 당전이 SERM 유도체들의 에스트로겐 수용체α(ERα)에 대한 결합 친화도(binding affinity)를 검정하기 위하여 NR peptide ERα ELISA 키트(ActiveMotif, Carlsbad, CA, 미국)를 사용하였으며, 그 경쟁 대조군으로 에스트라디올(17β-estradiol)을 이용하였다. 세부적인 실험 방법은 제조사의 프로토콜에 따라 수행하였다. DMSO에 용해된 각 대조구들을 키트 내 포함되어 있는 완충용액으로 희석하여 25uM 수준으로 준비하였고, 96-웰 마이크로플레이트(96-well microplate) 내 각각의 웰 당 키트 제조사의 프로토콜에 따라서 핵 추출물(nuclear extract)을 15ug 수준으로 첨가하였다. 한편, 당부가 유도체들의 경우에도 25uM 수준으로 시료를 준비하였다. 에스트로겐 수용체α(ERα)에 대한 활성도는 최종 흡광도 450nm의 마이크로플레이트 리더(microplate reader)로 정량화 하였으며, 모든 검정은 최소 삼 반복의 평균 값 및 표준 편차로 표시하였다. 그 결과, 양성 길항 대조구인 엔클로미펜은 25uM 수준에서 ERα에 대한 길항 활성이 뚜렷이 나타났다. 한편, 당부가 엔클로미펜 유도체 총 4종의 경우에도 모두 25uM 수준에서 통계적으로 유의한 길항 활성을 보였다. 위 결과는 상기 당부가 유도체들이 검정 시 첨가되는 핵 추출물 내 ERα 외에 존재하는 대사 효소계(즉, 글루코시다아제(glucosidase) 등)의 반응을 통하여 당부가 이전의 엔클로펜 활성대사체로 전환되어 길항 활성을 제공하고 있음을 제시하고 있다.
따라서, 본 발명의 당부가 엔클로미펜 유도체들은 에스트로겐 수용체α(ERα)에 대하여 모두 엔클로미펜 양성 대조구와 유사한 길항 활성이 유지되고 있음을 확인할 수 있었다.
실험예 2: 당부가 엔클로미펜 유도체들의 생체이용률 비교
상기 실시예에 의해 생성된 당부가 엔클로미펜 유도체들의 생체이용률을 자궁 절개된 실험 쥐 모델을 활용하여 기존 임상 이용 중인 클로미펜을 대조구로 하여 비교하고자 하였다. 세부적인 실험예는 아래와 같이 진행하였다. 약 150g 정도의 암컷 쥐에 자궁 절계 시술을 행한 일주일 후 본 실험에 활용되었다. 4마리를 각각의 하나의 처리 그룹으로 설계하여 총 여섯 그룹(즉, 한 그룹은 플라시보(placebo)이며, 또 다른 한 그룹은 클로미펜 처리 대조 그룹으로, 나머지 네 그룹은 각각의 당부가 유도체 처리 그룹)하여 투약 전 체중을 계측하였다. 하루에 한번 3umole 수준으로 경구 투여로 일주일을 지속한 후, 마지막 투여 후 24시간 이후 각 그룹 내 쥐들의 체중을 측정하는 동시에 채혈을 실시하였다. 채혈 후, 전혈에 동일 부피의 에틸아세테이트로 처리하여 30초간 강하게 혼합한 후, 4000rpm에서 5분간 원심분리를 실시하였다. 이 후, 유기용매 상층을 분리하여 감압건조한 후 분석 전까지 -70℃에 보관하였다. 혈중 활성대사체인 4-OH-ECLOM 및 N-DE-4-OH-ECLOM 그리고 엔클로미펜(ECLOM)의 정량은 본 발명에서 제시한 HPLC-ESI-MS 기기분석으로 실시하였다. 한편, 총 여섯 처리 그룹 중 클로미펜과 당 부가 클로미펜 유도체 처리 그룹 총 5개 그룹의 경우, 생체 이용률의 정밀한 비교를 위하여 마지막 투여 후 0, 1, 2, 4, 6, 12, 18, 24, 36, 48, 60 그리고 72시간까지 채혈을 실시하였다. 시간대 별 채혈 시료들은 상기 시료 추출법에 맞추어 동일하게 준비한 후, 활성대사체인 4-OH-ECLOM 및 N-DE-4-OH-ECLOM 그리고 엔클로미펜(ECLOM)의 정량을 HPLC-ESI-MS 기기분석으로 실시하였다.
그 결과, 투여 전 쥐의 체중은 모든 그룹에서 일정하게 나타났으나, 마지막 투여를 마친 후 24 시간 후에 각 그룹들의 평균 체중을 측정한 바, 플라시보 투여 그룹에서 체중의 증가(약 28~33g 정도)를 확인하였다. 한편, 클로미펜의 활성대사체인 4-OH-ECLOM 및 N-DE-4-OH-ECLOM 혈중 농도는 클로미펜 처리 대조 그룹과 비교하여 모든 처리 그룹에서 그 활성대사체들의 혈중 농도가 다소 높게 유지됨을 확인 할 수 있었다(도 3). 이 때 당부가 엔클로미펜 유도체들과 생전환 전 엔클로미펜을 자궁 절계 시술 후 쥐에 일주일 간 하루에 한번 3umole 수준으로 경구 투여한 다음, 마지막 투여 후 24시간에 채혈하였다. 도 3에 나타낸 바와 같이, 실험예 1의 결과와 마찬가지로 당부가 유도체들이 활성대사체인 4-OH-ECLOM 및 N-DE-4-OH-ECLOM으로 대사되는 프로드럭의 기능이 가능함을 보여주고 있을 뿐만 아니라, 당부가 유도체들의 경우 활성 대사체의 직접적인 경구 투여보다 혈액 순환계에 상대적으로 오랜 기간 잔존할 수 있음을 시사하고 있다.
한편, 보다 정확하게 당부가 클로미펜의 클로미펜 대비 생체이용률의 향상 정도를 확인하기 위하여 약물동력학적 분석을 실시하였다. 즉, 각 처리 그룹에서 준비된 72시간까지의 12구획의 혈장 시료로부터 엔클로미펜 및 그 활성대사체들의 총 농도를 기기분석으로 분석한 후, 시간에 따른 혈장 내 엔클로미펜 및 그 활성대사체들의 농도 변화를 그래프로 나타내었다. 약물동력학 파라미터들은 WinNonlin Professional 소프트웨어(version 5.1; Pharsight Co., Mountain View, CA, USA)의 비구획적 분석(non-compartmental analysis)법을 통하여 계측하였다. 각 처리 그룹들의 약물동력학 지표로 최고 혈중 농도(Cmax, peak concentration), 최고 혈중 농도 도달 시간(Tmax, paek time), 곡선하면적(AUC72hr, area under curve) 파라미터를 계측하여 표 1에 나타내었다.

도 4에 나타낸 바와 같이, 클로미펜 처리 대조 그룹에 비하여, 나머지 당 부가 클로미펜 처리 네 그룹들의 곡선하면적들이 높게 나타났다. 다만, 4종의 당부가 클로미펜 유도체 처리 그룹들 간에 있어서는 곡선하면적에 눈에 띄는 차이를 확인할 수는 없었으나, 글루코스 부가된 클로미펜 글루코사이드 유도체들(E1과 E3)에 비하여 디옥시글루코스 부가된 클로미펜 디옥시글루코사이드 유도체들(E2과 E4)의 처리 결과에서 다소 높은 곡선하면적 값이 나타났다. 한편, 대조 그룹을 포함한 전체 처리 그룹들의 최고 혈중 농도들의 분포는 모두 유의 수준 내로 차이가 나타나지 않았으나, 최고 혈중 농도 도달 시간의 경우엔 클로미펜 대조 처리 그룹에 비하여 나머지 4종의 당 부가 유도체 처리 그룹들에서 다소 높게 나타났다. 이는 각 처리 그룹 내 투여 화합물의 간 대사속도에는 차이가 나타나지 않았으나, 투여 화합물들의 혈 중 머무름 시간에는 차이가 있음을 보여주고 있으며, 곡선하면적 값 비교를 통하여 당 부가된 클로미펜 유도체들의 생체이용률이 당 부가 전 클로미펜의 생체이용률 보다 1.2배에서 1.4배 향상됐음을 제시하는데 성공하였다. 따라서, 당 부가 유도체 내에서도 디옥시글루코사이드 유도체들(E2와 E4)의 상대적으로 향상된 생체이용률은 프로드럭으로의 이용 가능성을 제시하고 있다. 이를 통하여 기존 에스트로겐 수용체 조절제 투약 시 문제점 중 하나인 장기 투약에 따른 문제점을 실시예에 의해 생성된 당부가 엔클로미펜 유도체들을 활용하여 해결할 수 있을 것이다.
상기 실시예에서 생성된 화합물에 대하여 하기와 같이 제제화하였다.
제제예 1: 정제(직접 가압)
화합물 5.0㎎을 체로 친 후, 락토스 14.1㎎, 크로스포비돈(USNF) 0.8㎎ 및 마그네슘 스테아레이트 0.1㎎을 혼합하고 가압하여 정제로 제조하였다.
제제예 2: 정제(습식 조립)
화합물 5.0㎎을 체로 친 후, 락토스 16.0㎎과 녹말 4.0㎎을 섞었다. 폴리솔베이트 800.3㎎을 순수한 물에 녹인 후, 이 용액의 적당량을 첨가하여 미립화하였다. 건조 후에 미립을 체질한 후, 콜로이달 실리콘 디옥사이드 2.7㎎ 및 마그네슘 스테아레이트 2.0㎎과 혼합하였다. 상기 혼합물을 가압하여 정제로 제조하였다.
제제예 3: 분말과 캡슐제
화합물 5.0㎎을 체로 친 후에, 락토스 14.8㎎, 폴리비닐 피롤리돈 10.0㎎ 및 마그네슘 스테아레이트 0.2㎎과 함께 혼합하였다. 상기 혼합물을 캡슐 제조기를 사용하여 단단한 No. 5 젤라틴 캡슐에 채웠다.
이상의 결과에 나타낸 바와 같이, 기존 약리 활성 화합물의 당 전이에 의한 구조 변형 유도체 생전환을 통하여 기존 화합물을 개량할 수 있으며, 이러한 개량화로서 기존 약물의 대체 가능한 프로드럭화 및 신규 제형(dosage form)으로의 응용 가능성을 포함하는 의약품 및 의약품 원료로서의 활용 잠재성을 확인할 수 있다.
본 발명에 따라 마이크로모노스포라 로도랑지아(Micromonospora rhodorangea) 균주 유래의 대장균 내 발현된 신규한 재조합 당전이효소(MrGT2)와 인간 유래 재조합 CYP 효소들의 순차적인 반응을 원팟 생전환함으로써 제조한 화학식 1의 화합물 또는 이의 약학적으로 허용가능한 염은 에스트로겐 수용체(estrogen receptor, ER)에 대한 일부 인비트로(in vitro) 길항 활성을 보이는 동시에 당 부가 전 화합물들과 비교시 생체이용률이 향상된 효과가 있다.
본 발명에 따른 화학식 1의 화합물 또는 이의 약학적으로 허용가능한 염은 골다공증(osteoporosis), 갱년기성 질 위축증(vaginal atrophy), 성교불쾌증(dyspareunia), 여유증(gynecomastia), 남성 이차 성선기능저하증 및 인슐린 비의존성 당뇨병 혹은 지방이영양증, 불임증, 남성전립선비대증, 전립선암, 난소암, 유방암 등의 질환의 치료를 목적으로 한 구조적 또는 약리적으로 개량된 의약품으로 응용화가 가능한 동시에 기존 SERM 혹은 엔클로미펜의 투여에 따라 발생할 수 있는 부작용을 감소시킬 수 있는 신규 의약품 제형으로 활용이 가능하다.
이상으로 본 발명 내용의 특정한 부분을 상세히 기술하였는 바, 당업계의 통상의 지식을 가진 자에게 있어서 이러한 구체적 기술은 단지 바람직한 실시 양태일 뿐이며, 이에 의해 본 발명의 범위가 제한되는 것이 아닌 점은 명백할 것이다. 따라서, 본 발명의 실질적인 범위는 첨부된 청구항들과 그것들의 등가물에 의하여 정의된다고 할 것이다.
전자파일 첨부하였음.
Claims (9)
- 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭:화학식 1
 상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다.
상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다. - 제1항에 있어서, 상기 엔클로미펜 유도체는 엔클로미펜 4-O-글루코사이드, N-데스에틸-엔클로미펜 4-O-글루코사이드, 엔클로미펜 4-O-2'-디옥시 글루코사이드 또는 N-데스에틸-엔클로미펜 4-O-2'-디옥시 글루코사이드인 것을 특징으로 하는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭.
- 다음 단계를 포함하는 하기 화학식 1로 표시되는 엔클로미펜 유도체의 제조방법:(a) 서열번호 4의 아미노산 서열로 표시되는 당전이효소(MrGT2)와 사이토크롬의 존재하에 엔클로미펜 및 당 공여체를 반응시켜 하기 화학식 1로 표시되는 엔클로미펜 유도체를 합성시키는 단계; 및(b) 상기 합성된 엔클로미펜 유도체를 회수하는 단계:화학식 1
 상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다.
상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다. - 제3항에 있어서, 상기 엔클로미펜 유도체는 엔클로미펜 4-O-글루코사이드, N-데스에틸-엔클로미펜 4-O-글루코사이드, 엔클로미펜 4-O-2'-디옥시 글루코사이드, N-데스에틸-엔클로미펜 4-O-2'-디옥시 글루코사이드인 것을 특징으로 하는 제조방법.
- 제3항에 있어서, 수득한 효소반응물을 중압 액체 크로마토그래피(Medium Pressure Liquid Chromatography, MPLC) 방법으로 분리 또는 정제하는 단계를 추가로 포함하는 제조방법.
- 제1항의 화학식 1로 표시되는 엔클로미펜 유도체, 이의 이성질체 또는 약학적으로 허용가능한 이들의 용매화물, 수화물 또는 프로드럭을 유효성분으로 포함하는 에스트로겐 수용체 관련 질환 치료용 약학 조성물:화학식 1
 상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다.
상기 화학식 1에서 R1은 CH2CH3, CH2CH2Cl 또는 Cl이고, R2는 N(CH3)2, OH 또는 (CH2)4N이며, R3는 글루코스(glucose), 2-데옥시글루코스(2-deoxy-glucose) 또는 갈락토스(galactose)이다. - 제3항에 있어서, 상기 엔클로미펜 유도체는 엔클로미펜 4-O-글루코사이드, N-데스에틸-엔클로미펜 4-O-글루코사이드, 엔클로미펜 4-O-2’-디옥시 글루코사이드, N-데스에틸-엔클로미펜 4-O-2’-디옥시 글루코사이드인 것을 특징으로 하는 약학 조성물.
- 제6항에 있어서, 의약적으로 허용가능한 담체, 부형제 또는 희석제를 추가로 포함하는 것을 특징으로 하는 약학 조성물.
- 제6항에 있어서, 상기 에스트로겐 수용체 관련 질환은 골다공증(osteoporosis), 갱년기성 질 위축증(vaginal atrophy), 성교불쾌증(dyspareunia), 여유증(gynecomastia), 남성 이차 성선기능저하증, 인슐린 비의존성 당뇨병, 지방이영양증, 불임증, 남성전립선비대증, 전립선암, 난소암 또는 유방암인 것을 특징으로 하는 약학 조성물.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2016-0100098 | 2016-08-05 | ||
| KR1020160100098A KR101970326B1 (ko) | 2016-08-05 | 2016-08-05 | 당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학적 조성물 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2018026087A2 true WO2018026087A2 (ko) | 2018-02-08 |
| WO2018026087A3 WO2018026087A3 (ko) | 2018-08-09 |
Family
ID=61074169
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2017/004022 WO2018026087A2 (ko) | 2016-08-05 | 2017-04-13 | 당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학적 조성물 |
Country Status (2)
| Country | Link |
|---|---|
| KR (1) | KR101970326B1 (ko) |
| WO (1) | WO2018026087A2 (ko) |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027127A1 (en) * | 1999-10-08 | 2001-04-19 | Strakan Group Plc | Tamoxifen and tamoxifen analogue glycosides and uses thereof |
| KR20010108719A (ko) * | 2000-05-31 | 2001-12-08 | 추후보정 | 유방암의 예방 또는 치료용 조성물 및 그 예방 또는 치료방법 |
| CA2698808A1 (en) * | 2007-09-13 | 2009-03-19 | Concert Pharmaceuticals, Inc. | Synthesis of deuterated catechols and benzo[d][1,3] dioxoles and derivatives thereof |
| KR101717212B1 (ko) * | 2015-07-23 | 2017-03-16 | 고려대학교 산학협력단 | 당전이 효소를 이용한 당 부가된 에스트로겐 수용체 조절제의 제조방법 |
-
2016
- 2016-08-05 KR KR1020160100098A patent/KR101970326B1/ko active IP Right Grant
-
2017
- 2017-04-13 WO PCT/KR2017/004022 patent/WO2018026087A2/ko active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2018026087A3 (ko) | 2018-08-09 |
| KR101970326B1 (ko) | 2019-04-18 |
| KR20180016108A (ko) | 2018-02-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011132830A1 (en) | A new kanamycin compound, kanamycin-producing streptomyces species bacterium, and method of producing kanamycin | |
| CN111094288B (zh) | 具有mgat-2抑制活性的稠合环衍生物 | |
| EP3421467A1 (en) | 5-phenylazaindole derivative having ampk activating effect | |
| KR20200120928A (ko) | 소베티롬의 유도체 | |
| WO2018026087A2 (ko) | 당 부가된 엔클로미펜, 이의 제조방법 및 이를 포함하는 약학적 조성물 | |
| KR20030024913A (ko) | 아릴피페라진 유도체 및 정신작용 약제로서의 이의 용도 | |
| WO2014185717A1 (ko) | 신규한 에포싸일론 유도체 및 그의 용도 | |
| CN113929698B (zh) | 二芳基庚烷二聚体及其药物组合物与其制备方法和应用 | |
| EP4289856A1 (en) | Crystal form of 3-hydroxy-5-pregnane-20-one derivative, and preparation method therefor and use thereof | |
| AU2004213097A1 (en) | Benzofuran oxyethylamines serving as antidepressant drugs and anxiolytic drugs | |
| WO2019031754A1 (ko) | 당 부가된 오스페미펜, 이의 제조방법 및 이를 유효성분으로 함유하는 약학적 조성물 | |
| KR101705390B1 (ko) | 이중환 구조를 갖는 신규한 gpcr 효능제 | |
| WO2022055176A1 (ko) | 수두 또는 대상포진 백신 조성물 및 이를 이용하는 방법 | |
| WO2018066947A1 (ko) | 지방산이 결합된 엔테카비어 유도체 화합물 및 이의 약학적 용도 | |
| KR101717212B1 (ko) | 당전이 효소를 이용한 당 부가된 에스트로겐 수용체 조절제의 제조방법 | |
| EP3445741A1 (en) | Antimicrobial agents | |
| WO2012093859A2 (ko) | 폴리사이클릭 펩타이드 화합물을 포함하는 항균용 조성물 및 이의 생산방법 | |
| WO2015069086A1 (ko) | 인삼 분획물 또는 이로부터 분리된 진세노사이드를 함유하는 시르투인 활성화로 치료되는 질환의 예방 또는 치료용 조성물 | |
| KR20210125982A (ko) | 신경변성 질환의 치료를 위한 2-플루오린화된 담즙산 | |
| EP4004013A1 (en) | Dinucleotide compounds for treating cancers and medical uses thereof | |
| AU2011276265A1 (en) | Novel antibacterial compounds, methods of making them, and uses thereof | |
| WO2020054946A1 (ko) | 스트렙토미세스로부터 유래된 새로운 화합물 및 용도 | |
| WO2018135699A1 (ko) | 신규한 에르고스텐올 글리코시드 유도체 | |
| WO2024080509A1 (ko) | Igf-1 수용체 결합 압타머 및 이의 용도 | |
| WO2017073854A1 (ko) | 단순 페놀릭 배당 유도체, 이의 제조방법 및 이를 함유하는 피부미백 또는 주름개선용 조성물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17837139 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17837139 Country of ref document: EP Kind code of ref document: A2 |