WO2017148361A1 - 一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法 - Google Patents
一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法 Download PDFInfo
- Publication number
- WO2017148361A1 WO2017148361A1 PCT/CN2017/075113 CN2017075113W WO2017148361A1 WO 2017148361 A1 WO2017148361 A1 WO 2017148361A1 CN 2017075113 W CN2017075113 W CN 2017075113W WO 2017148361 A1 WO2017148361 A1 WO 2017148361A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutical composition
- composition according
- producing
- weight
- methyl
- Prior art date
Links
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2077—Tablets comprising drug-containing microparticles in a substantial amount of supporting matrix; Multiparticulate tablets
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5089—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
Definitions
- the invention belongs to the field of pharmaceutical preparations, in particular to a method comprising 5-(2-diethylamino-ethyl)-2-(5-fluoro-2-oxo-1,2-dihydro-indole-3- a pharmaceutical composition of a subunit-methyl)-3-methyl-1,5,6,7-tetrahydro-pyrrole[3,2-c]pyridin-4-one or a pharmacologically acceptable salt thereof Preparation.
- EGFR targeted epidermal growth factor receptor
- TKIs clinical tyrosine kinase inhibitors
- TKIs can also inhibit the angiogenic factors of tumor cells and inhibit the signaling of EGFR to tumor vascular endothelial cells, and the "cross-talk" between EGFR and vascular endothelial growth factor receptor (VEGFR) signaling pathways.
- VEGFR vascular endothelial growth factor receptor
- the chemical name of this compound is 5-(2-diethylamino-ethyl)-2-(5-fluoro-2-oxo-1,2-dihydro-indol-3-ylidene-methyl)- 3-methyl-1,5,6,7-tetrahydro-pyrrole[3,2-c]pyridin-4-one, which is known to inhibit tumor growth and angiogenesis and selectively inhibit vascular endothelial growth factor
- the kinase activity of the (VEGF) receptor is clinically useful for the treatment of various tumors such as kidney cancer, gastrointestinal stromal tumors, colorectal cancer, and pancreatic neuroendocrine tumors.
- the compound of the formula (I) or a pharmaceutically acceptable salt thereof is poorly water-soluble and unstable in the presence of moisture, wet granulation cannot be carried out using water as a solvent, and the compound of the formula (I) or a general preparation method is used.
- the pharmaceutically acceptable salt is prepared into a pharmaceutical composition, the composition is difficult to dissolve quickly and maintains a stable quality, and it is necessary to study a suitable method to obtain a stable and rapidly eluting composition.
- the preparation method of the pharmaceutical composition provided by the present invention comprises mixing the active pharmaceutical ingredient and at least one filler to granulate.
- the active pharmaceutical ingredient is 5-(2-diethylamino-ethyl)-2-(5-fluoro-2-oxo-1,2-dihydro-indol-3-ylidene-methyl)-3 Methyl-1,5,6,7-tetrahydro-pyrrole[3,2-c]pyridin-4-one or a pharmaceutically acceptable salt thereof.
- the granulation method described therein may be selected from the group consisting of high-speed shear wet granulation, wet one-step granulation, and dry granulation, preferably dry granulation.
- the filler may be included in an amount ranging from 20% to 95%, preferably from 30% to 90%; more preferably from 40% to 85%; most preferably from 50% to 80%, based on the total weight of the composition.
- the present inventors have surprisingly found that the composition obtained by granulating the active ingredient after mixing with the filler is far superior to the composition obtained by directly mixing or compressing the active ingredient with the filler; without granulation The dissolution of the composition did not meet the requirements.
- the pharmaceutical composition of the present invention has a good dissolution rate by using a granulation process after mixing with a filler.
- purified water preferably 900 ml
- the dissolution rate was measured at a paddle speed of 50 rpm at ° C, and the dissolution rate was 80% or more in 45 minutes.
- the water-soluble filler may be a sugar alcohol, preferably one or more of lactose, glucose, sucrose, mannitol, and sorbitol.
- the water soluble filler is mannitol.
- the above water-soluble filler is in close contact with the active ingredient by the granulation process, and can promote dissolution of the active ingredient and remain stable.
- the content of the water-soluble filler of the present invention is not particularly limited, and in a preferred embodiment of the present invention, the content of the water-soluble filler may range from 20% to 95% by weight based on the total weight of the composition; preferably 30% to 90%. More preferably, it is 40% to 85%; most preferably 50% to 80%.
- the pharmaceutical composition of the invention has good stability, and the degradation product is placed at a temperature of 25 ° C and a relative humidity of 75% for 10 days, and the degradation product is as small as 0.5%, or placed at a temperature of 25 ° C and a relative humidity of 90%.
- the day degradant is as small as 1%.
- the pharmaceutical composition is excellent in stability, and when other fillers are used, stability may be poor.
- the pharmacologically acceptable salt of the active ingredient may be selected from the group consisting of hydrochloride, malate, hydrobromide, p-toluenesulfonate, methanesulfonate, sulfate or
- the ethanesulfonate salt is preferably a malate salt in the present invention.
- the active ingredient may be included in an amount ranging from 3% to 40%, preferably from 5% to 30%, most preferably from 10% to 20%, based on the total weight of the composition.
- the pharmaceutical composition provided by the present invention may contain other fillers such as starch, pregelatinized starch, dextrin, microcrystalline cellulose, and the like.
- the amount of other fillers is from about 5 to 50% by weight based on the total weight of the composition.
- the pharmaceutical composition provided by the present invention may contain a disintegrating agent selected from the group consisting of croscarmellose sodium, sodium carboxymethyl starch, low-substituted hydroxypropylcellulose, and crospovidone. One or several.
- the disintegrant content is preferably from about 1% to 20% based on the total weight of the composition.
- the pharmaceutical composition provided by the present invention may further comprise one or more lubricants to aid in filling the capsule or tableting.
- the lubricant may be selected from the group consisting of talc, magnesium stearate, sodium stearyl fumarate, zinc stearate, glyceryl behenate, sodium lauryl sulfate, hydrogenated vegetable oil, colloidal silica, and the like.
- the content of the lubricant is preferably from about 0.5% to 5%, based on the total weight of the composition.
- the method of preparing the pharmaceutical composition comprises the steps of:
- a disintegrant selected from one of croscarmellose sodium, sodium carboxymethyl starch, low-substituted hydroxypropylcellulose, and crospovidone or Multiple
- a lubricant selected from one or more of magnesium stearate, sodium stearyl fumarate, colloidal silica and talc.
- the method of preparing the pharmaceutical composition of the present invention further comprises the step of tableting or filling the dry granules into capsules.
- the invention is preferably prepared as a hard capsule.
- the composition can be caused to dissolve more rapidly.
- the particle size of the active ingredient is determined by a laser particle size analyzer, and d0.9 is required to be less than 100 ⁇ m, preferably less than 80 ⁇ m, more preferably less than 60 ⁇ m, and most preferably less than 40 ⁇ m.
- Figure 1 shows the dissolution profiles of the capsules of Examples 1 to 4 and Comparative Examples 3 to 4 in purified water.
- Figure 2 shows the dissolution profiles of the capsules of Examples 5 to 8 and Comparative Examples 6 to 8 in purified water.
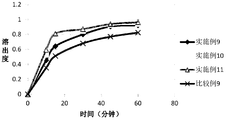
- Figure 3 shows the dissolution profiles of the capsules of Examples 9 to 11 and Comparative Example 9 in purified water.
- compound A 5-(2-Diethylamino-ethyl)-2-(5-fluoro-2-oxo-1,2-dihydro-indole-3-ylidene-methyl)-3-methyl -1,5,6,7-tetrahydro-pyrrole[3,2-c]pyridin-4-one malate (hereinafter referred to as compound A), pregelatinized starch, lactose, crospovidone, Silica, according to the ratio of the prescriptions of Examples 1 to 4 in Table 1, uniformly mixed, dry granulation by dry granulator, adding the prescribed amount of magnesium stearate, mixing evenly, and the obtained total mixed granules were poured. Capsules are prepared and capsules are prepared.
- Compound A microcrystalline cellulose, crospovidone, silica and calcium hydrogen phosphate or pregelatinized starch were uniformly mixed according to the prescription ratios of Comparative Examples 1 and 2 in Table 1, and dried by a dry granulator. After the granulation, a prescribed amount of magnesium stearate is added, uniformly mixed, and the obtained total mixed granules are filled and capsuled to prepare a capsule.
- Compound A pregelatinized starch, lactose, crospovidone, silica and magnesium stearate were uniformly mixed according to the prescription ratio of Comparative Example 3 in Table 1, and the obtained mixed powder was filled and filled, and prepared. Capsules.
- Compound A, lactose, crospovidone, silica, and magnesium stearate were uniformly mixed according to the prescription ratio of Comparative Example 4 in Table 1, and the obtained mixed powder was filled and filled to prepare a capsule.
- Compound A pregelatinized starch, mannitol, crospovidone, and silica were uniformly mixed according to the prescription ratios of Examples 5 to 8 in Table 2, and dry granulation was carried out by a dry granulator. Prescription amount Magnesium stearate was mixed well, and the obtained total mixed granules were filled into capsules to prepare capsules, and capsules of Examples 5 to 8 were prepared.
- Compound A pregelatinized starch, calcium hydrogen phosphate, crospovidone, and silica were uniformly mixed according to the prescription ratio of Comparative Example 5 in Table 2, and dry granulation was carried out by a dry granulator, and then a prescription was added. The amount of magnesium stearate was uniformly mixed, and the obtained total mixed particles were filled into capsules to prepare capsules, and the capsule of Comparative Example 5 was prepared.
- Compound A pregelatinized starch, mannitol, crospovidone, silica and magnesium stearate were uniformly mixed according to the prescription ratio of Comparative Example 6 in Table 2, and the obtained mixed powder was filled and filled.
- the capsule of Comparative Example 6 was prepared.
- Comparative Example 7 Compound A, mannitol, crospovidone, silica and magnesium stearate were uniformly mixed according to the prescription ratio of Comparative Example 7 in Table 2, and the obtained mixed powder was filled and filled to prepare Comparative Example 7. Capsules. According to the prescription ratio of Comparative Example 8 in Table 2, compound A, pregelatinized starch, mannitol and crospovidone were uniformly mixed, and then dried ethanol was added for wet granulation, dried, and then granulated, and silica was added thereto. The capsule of Comparative Example 8 was prepared by uniformly mixing with magnesium stearate and filling the obtained mixture.
- the dissolution rate of the capsules of Examples 1 to 8 and Comparative Examples 3, 4, 6, 7, and 8 was measured according to the dissolution degree and the release degree measurement method (Chinese Pharmacopoeia 2015 Edition, Fourth Edition, General Law No. 0931, second method). 900 ml of purified water was used as the dissolution medium, and the dissolution test was carried out at 37 ⁇ 0.5 ° C at a paddle speed of 50 rpm. The results showed that in the capsules of Examples 1 to 8 which were prepared by the dry granulation process, Compound A was quickly dissolved, and the capsules of Comparative Examples 3 to 4 and Comparative Examples 6 to 8 which were not prepared by the dry granulation process were used. Compound A dissolves slowly and is incomplete.
- the dissolution profile is shown in Figures 1 and 2.
- Example 9 to 11 and Comparative Example 9 in Table 3 were respectively pregelatinized starch, mannitol, crospovidone, and silica at the prescription ratio of Example 6 in Example 2.
- the mixture was uniformly mixed, and after dry granulation by a dry granulator, a prescribed amount of magnesium stearate was added and uniformly mixed, and the obtained total mixed granules were filled and filled to prepare capsules, and Examples 9 to 11 and Comparative Example 9 were prepared. Capsules.
- Example 9 Example 10
- Example 11 Compound A particle size distribution d0.9 128 ⁇ m 67 ⁇ m 55 ⁇ m 37 ⁇ m
- the particle size distribution of Compound A in Table 3 was measured by Malvern laser particle size analyzer Mastersizer 2000.
- the particle refractive index was 1.520
- the sampler was Scirocco 2000 (A)
- the analysis mode was general (fine powder).
- the sensitivity was normal.
- the dissolution rates of the capsules of Examples 9 to 11 and Comparative Example 9 were measured according to the dissolution degree and the release degree measurement method (Chinese Pharmacopoeia 2015 Edition, Fourth Edition, General Law No. 0931, second method). 900 ml of purified water was used as the dissolution medium, and the dissolution test was carried out at 37 ⁇ 0.5 ° C at a paddle speed of 50 rpm. The results show that in the examples 9 to 11, the particle size distribution d0.9 of the compound A with the particle size distribution d0.9 less than 100 ⁇ m becomes smaller, and the dissolution rate of the capsule is gradually increased, indicating the particle size distribution d0 of the compound A. The smaller the 9 is, the faster the capsule dissolves. On the other hand, the compound A of Comparative Example 9 had a particle diameter of more than 100 ⁇ m, and dissolution was relatively slow.
- the dissolution profile is shown in Figure 3.
- Example 4 and Example 6 and the capsules of Comparative Example 1, Comparative Example 2, and Comparative Example 5 were respectively placed in an environment of a temperature of 25 ° C, a relative humidity of 75%, a temperature of 25 ° C, and a relative humidity of 90%.
- the cells were allowed to stand for 5 days and 10 days in an open condition, and then the formation of the degradation product was measured by an HPLC method.
- the results showed that Example 4, Example 6 containing lactose or mannitol, compared with Comparative Example 1, Comparative Example 2 and Comparative Example 5 containing no lactose and mannitol, the rate of degradation product formation was significantly lower in a high-humidity environment. It shows that when the capsule contains lactose or mannitol, it is more stable in a high-humidity environment.
- Table 4 The test results are shown in Table 4.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Inorganic Chemistry (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明提供了一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法。具体而言,本发明提供了一种药物组合物的制备方法,包括将5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药理学上可接受的盐与至少一种填充剂混合后进行制粒的步骤。本发明的药物组合物具有溶出迅速、稳定性良好的特点。
Description
本发明属于药物制剂领域,具体涉及一种含有5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药理学上可接受的盐的药物组合物的制备方法。
随着分子生物学技术的进展和从细胞受体与增殖调控的分子水平对肿瘤发病机制认识的进一步深入,针对细胞受体、关键基因和调控分子为靶点的治疗开始进入临床,人们称之为“分子靶向治疗”。这些领域包括具有靶向性的表皮生长因子受体(EGFR)阻滞剂、针对某些特定细胞标志物的单克隆抗体、针对某些癌基因和癌的细胞遗传学标志的药物、抗肿瘤血管生成的药物、抗肿瘤疫苗和基因治疗等。
首先进入临床的酪氨酸激酶抑制剂(TKIs)的抗肿瘤作用机制可能通过以下途径实现:抑制肿瘤细胞的损伤修复、使细胞分裂阻滞在G1期、诱导和维持细胞凋亡、抗新生血管形成等。EGFR过度表达常预示病人预后差、转移快、对化疗药物抗拒、激素耐药、生存期较短等。TKIs还可通过下调肿瘤细胞的血管生成因子以及抑制EGFR对肿瘤血管内皮细胞的信号传导,EGFR和血管内皮生长因子受体(VEGFR)两种信号传导通路的“交叉对话”,为临床同时抑制这两种传导通路提供了合理的依据。临床试验结果显示,多靶点抑制剂在治疗方面优于单靶点抑制剂,多靶点联合阻断信号传导是肿瘤治疗和药物开发新的发展方向。
到目前为止,美国FDA批准多种多靶点TKIs上市,如:索拉非尼(sorafenib)、凡德他尼(vandetanib)和Sunitinib(Sutent,SU-11248),其中Sunitinib于2006年1月份批准上市,治疗GIST和晚期肾癌。由于目前临床上除了伊马替尼外,没有治疗晚期GIST的药物,治疗肾癌的药物也很少,所以Sunitinib的结果令人鼓舞。WO2007085188公开了一种与Sunitinib类似的化合物,如下式(I)所示,其可能更好地应用于上述肿瘤的治疗。该化合物化学名为5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮,已知其具有抑制参与肿瘤增殖和血管生成,能够选择性抑制血管内皮生长因子(VEGF)受体的激酶活性,临床上可用于肾癌、胃肠间质瘤、结直肠癌和胰腺神经内分泌瘤等多种肿瘤的治疗。

由于式(I)化合物或其可药用盐水溶性差,且在水分存在的情况下不稳定,不能使用水作为溶剂进行湿法制粒,而在使用一般的制备方法将式(I)化合物或其可药用盐制备成药物组合物时,组合物又难以迅速溶出并且保持质量稳定,需要研究合适的方法获得稳定且溶出迅速的组合物。
发明内容
本发明的目的在于提供一种制备稳定性良好同时溶出迅速的药物组合物的方法,并且该制备方法工艺简单,适合工艺化大生产。
本发明提供的药物组合物的制备方法包括将活性药物成分和至少一种填充剂混合后制粒。活性药物成分为5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药学上可接受的盐。其中所述的制粒方法可以选自高速剪切湿法制粒、湿法一步制粒、干法制粒,优选干法制粒。所述填充剂的含量范围可以是基于组合物总重量计20%-95%;优选30%-90%;更优选40%-85%;最优选50%-80%
本发明惊奇地发现,当将活性成分与填充剂混合后制粒得到的组合物溶出远远优于直接将活性成分与填充剂直接混合灌装或压片得到的组合物;不经过制粒的组合物溶出无法满足要求。
本发明的药物组合物由于采用与填充剂混合后制粒的工艺,药物溶出度良好,按照中国药典2015年版四部通则0931第二法,使用纯化水(优选900ml)作为溶出介质,在37±0.5℃下以50rpm的桨速进行溶出度测定,45分钟内溶出度大于等于80%。
所述水溶性填充剂可以为糖醇类,优选乳糖、葡萄糖、蔗糖、甘露醇、山梨醇中的一种或几种。
在本发明优选的实施方案中,所述水溶性填充剂为甘露醇。
上述水溶性填充剂通过制粒过程与活性成分紧密接触,能够促进活性成分的溶出,并保持稳定。本发明的水溶性填充剂含量没有特别限制,在本发明优选的实施方案中,所述水溶性填充剂的含量范围可以是基于组合物总重量计20%-95%;优选30%-90%;更优选40%-85%;最优选50%-80%。
本发明的药物组合物具有良好的稳定性,温度25℃、相对湿度75%的条件下放置10天降解产物小等于0.5%,或在温度25℃、相对湿度90%的条件下放置10
天降解物小等于1%。当采用上述水溶性填充剂时,所述药物组合物稳定性良好,而使用其它填充剂时,则可能造成稳定性不佳。
本发明的药物组合物中,所述活性成分的药理学上可接受的盐可以选自盐酸盐、苹果酸盐、氢溴酸盐、对甲苯磺酸盐、甲磺酸盐、硫酸盐或乙磺酸盐,本发明优选苹果酸盐。基于组合物的总重量,所述活性成分的含量范围可以是基于组合物总重量计3%-40%;优选5%-30%;最优选10-20%。
本发明提供的药物组合物中可以含有其他填充剂,例如淀粉、预胶化淀粉、糊精、微晶纤维素等一种或多种。基于组合物的总重量,其他填充剂的含量为约5-50wt%。
本发明提供的药物组合物中可以含有崩解剂,所述崩解剂选自交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素和交联聚维酮中的一种或几种。基于组合物的总重量,所述崩解剂含量优选为约1%~20%。
本发明提供的药物组合物中还可包含一种或多种润滑剂,有助于灌装胶囊或压片。润滑剂可选自滑石粉、硬脂酸镁、硬脂富马酸钠、硬脂酸锌、山嵛酸甘油酯、月桂基硫酸钠、氢化植物油、胶体二氧化硅等。基于组合物的总重量计,润滑剂的含量优选为约0.5%~5%。
在本发明特别优选的实施方案中,所述药物组合物的制备方法,包含如下步骤:
将10-20wt%的5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药理学上可接受的盐与30-80wt%的乳糖或甘露醇混合后进行干法制粒,压片制成片剂或灌装制成胶囊;其中活性物质的粒径分布范围d(0.9)优选小于60μm,最优选小于40μm;所述组合物还包括:
1)任选地5-50wt%的预胶化淀粉;
2)1-30wt%的崩解剂,所述崩解剂选自交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素和交联聚维酮中的一种或多种;
3)0.5-5wt%的润滑剂,所述润滑剂选自硬脂酸镁、硬脂富马酸钠、胶体二氧化硅和滑石粉的一种或多种。
本发明的药物组合物的制备方法还包括将干颗粒压片制成片剂或灌装制成胶囊的步骤。本发明优选制备成硬胶囊剂。
本发明的药物组合物中所含活性成分的粒径分布符合一定要求时,可促使组合物更加快速地溶出。采用激光粒度仪测定活性成分的粒径,d0.9需小于100μm,优选小于80μm,更优选小于60μm,最优选小于40μm。
图1显示实施例1至4以及比较例3至4的胶囊在纯化水中的溶出曲线。
图2显示实施例5至8以及比较例6至8的胶囊在纯化水中的溶出曲线。
图3显示实施例9至11以及比较例9的胶囊在纯化水中的溶出曲线。
通过以下实施例和实验例进一步详细说明本发明。这些实施例和实验例仅用于说明性目的,而并不用于限制本发明的范围。
实施例1~4、比较例1~4
将5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮的苹果酸盐(以下简称为化合物A)、预胶化淀粉、乳糖、交联聚维酮、二氧化硅,按表1中实施例1~4的处方比例,混合均匀,采用干法制粒机进行干法制粒后,加入处方量的硬脂酸镁,混合均匀,将得到的总混颗粒灌装胶囊,制备胶囊剂。
将化合物A、微晶纤维素、交联聚维酮、二氧化硅与磷酸氢钙或预胶化淀粉,按表1中比较例1和2的处方比例混合均匀,采用干法制粒机进行干法制粒后,加入处方量的硬脂酸镁,混合均匀,将得到的总混颗粒灌装胶囊,制备胶囊剂。
将化合物A、预胶化淀粉、乳糖、交联聚维酮、二氧化硅和硬脂酸镁,按表1中比较例3的处方比例,混合均匀,将得到的混合粉末灌装胶囊,制备胶囊剂。
将化合物A、乳糖、交联聚维酮、二氧化硅和硬脂酸镁,按表1中比较例4的处方比例,混合均匀,将得到的混合粉末灌装胶囊,制备胶囊剂。
表1

单位:质量%
实施例5~8、比较例5~8
将化合物A、预胶化淀粉、甘露醇、交联聚维酮、二氧化硅,按表2中实施例5~8的处方比例,混合均匀,采用干法制粒机进行干法制粒后,加入处方量的
硬脂酸镁,混合均匀,将得到的总混颗粒灌装胶囊,制备胶囊剂,制备实施例5~8的胶囊剂。
将化合物A、预胶化淀粉、磷酸氢钙、交联聚维酮、二氧化硅,按表2中比较例5的处方比例,混合均匀,采用干法制粒机进行干法制粒后,加入处方量的硬脂酸镁,混合均匀,将得到的总混颗粒灌装胶囊,制备胶囊剂,制备比较例5的胶囊剂。
将化合物A、预胶化淀粉、甘露醇、交联聚维酮、二氧化硅和硬脂酸镁,按表2中比较例6的处方比例,混合均匀,将得到的混合粉末灌装胶囊,制备比较例6的胶囊剂。
将化合物A、甘露醇、交联聚维酮、二氧化硅和硬脂酸镁,按表2中比较例7的处方比例,混合均匀,将得到的混合粉末灌装胶囊,制备比较例7的胶囊剂。按表2中比较例8的处方比例,将化合物A、预胶化淀粉、甘露醇、交联聚维酮混合均匀后,加入无水乙醇进行湿法制粒,干燥后整粒,加入二氧化硅和硬脂酸镁,混合均匀,将得到的混合物灌装胶囊,制备比较例8的胶囊剂。
表2

单位:质量%
实验例1:溶出实验
根据溶出度与释放度测定法(中国药典2015年版四部通则0931第二法),对实施例1~8和比较例3、4、6、7、8的胶囊剂进行溶出度测定。使用900ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,采用干法制粒工艺制备的处方中实施例1~8的胶囊剂中,化合物A溶出迅速,未采用干法制粒工艺制备的比较例3~4和比较例6~8的胶囊剂中,化合物A溶出缓慢且不完全。
溶出曲线图见图1和图2。
实施例9~11、比较例9
以2中实施例6的处方比例,分别将表3中实施例9~11及比较例9的不同粒径水平的化合物A与预胶化淀粉、甘露醇、交联聚维酮、二氧化硅混合均匀,采用干法制粒机进行干法制粒后,加入处方量的硬脂酸镁,混合均匀,将得到的总混颗粒灌装胶囊,制备胶囊剂,制备实施例9~11及比较例9的胶囊剂。
表3
| 样号 | 比较例9 | 实施例9 | 实施例10 | 实施例11 |
| 化合物A粒径分布d0.9 | 128μm | 67μm | 55μm | 37μm |
备注:表3中化合物A的粒径分布是采用马尔文激光粒度测定仪Mastersizer2000测定,颗粒折射率为1.520,进样器为Scirocco2000(A),分析模式为通用(细粉),灵敏度为正常。
实验例2:溶出实验
根据溶出度与释放度测定法(中国药典2015年版四部通则0931第二法),对实施例9~11和比较例9的胶囊剂进行溶出度测定。使用900ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,实施例9~11中,随着粒径分布d0.9小于100μm的化合物A的粒径分布d0.9变小,胶囊剂的溶出速度逐步加快,说明化合物A的粒径分布d0.9越小,胶囊剂溶出越迅速。而比较例9的化合物A粒径大于100μm,溶出比较迟缓。
溶出曲线图见图3。
实验例3:稳定性研究
将实施例4、实施例6的胶囊剂和比较例1、比较例2、比较例5的胶囊剂,分别置于温度25℃、相对湿度75%和温度25℃、相对湿度90%的环境下,在开放条件中放置5天、10天,然后采用HPLC法测定降解物的生成。结果表明含有乳糖或甘露醇的实施4、实施例6,相比不含乳糖和甘露醇的比较例1、比较例2和比较例5,在高湿环境中,降解物生成的速度明显更低,说明当胶囊剂中含有乳糖或甘露醇时,在高湿环境中更加的稳定。试验结果见表4。
表4

Claims (14)
- 一种药物组合物的制备方法,包括将作为活性物质的5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药理学上可接受的盐与至少一种填充剂混合后进行制粒的步骤。
- 根据权利要求1所述的药物组合物的制备方法,其中所述的制粒方法选自高速剪切湿法制粒、湿法一步制粒、干法制粒,优选干法制粒。
- 根据权利要求1所述的药物组合物的制备方法,其特征在于,还包括将干颗粒压片制成片剂或灌装制成胶囊的步骤。
- 根据权利要求1所述的药物组合物的制备方法,其中所述填充剂为水溶性填充剂。
- 根据权利要求4所述的药物组合物的制备方法,其中所述水溶性填充剂为糖醇类,优选乳糖、葡萄糖、蔗糖、甘露醇、山梨醇中的一种或几种。
- 根据权利要求1所述的药物组合物的制备方法,其中所述填充剂的含量为基于组合物总重量计20%-95%;优选30%-90%;更优选40%-85%;最优选50%-80%。
- 根据权利要求1所述的药物组合物的制备方法,所述组合物按照中国药典2015年版四部通则0931第二法,使用纯化水作为溶出介质,在37±0.5℃下以50rpm的桨速进行溶出度测定,45分钟内溶出度大于等于80%。
- 根据权利要求1至6任一项所述的药物组合物的制备方法,其中所述活性物质的粒径,采用激光粒度仪测定,d0.9小于100μm,优选小于80μm,更优选小于60μm,最优选小于40μm。
- 根据权利要求1所述的药物组合物的制备方法,其中所述组合物还含有崩解剂,所述崩解剂选自交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素和交联聚维酮中的一种或几种,优选其含量为基于组合物总重量计的1-20%。
- 根据权利要求1所述的药物组合物的制备方法,其中所述组合物还含有润滑剂,选自滑石粉、硬脂酸镁、硬脂酸锌、硬脂富马酸钠、山嵛酸甘油酯、月桂醇硫酸钠、氢化植物油和胶体二氧化硅中的一种或几种,优选其含量为基于组合物总重量计的0.5-5%。
- 根据权利要求1至10任一项所述的药物组合物的制备方法,其中所述药理学上可接受的盐选自盐酸盐、苹果酸盐、氢溴酸盐、对甲苯磺酸盐、甲磺酸盐、硫酸盐或乙磺酸盐;优选苹果酸盐。
- 根据权利要求1所述的药物组合物的制备方法,所述活性物质含量为基于组合物总量计3%-40%;优选5%-30%;最优选10-20%。
- 一种药物组合物的制备方法,包含如下步骤:将10-20wt%的5-(2-二乙胺基-乙基)-2-(5-氟-2-氧代-1,2-二氢-吲哚-3-亚基-甲基)-3-甲基-1,5,6,7-四氢-吡咯[3,2-c]吡啶-4-酮或其药理学上可接受的盐与30-80wt%的乳糖或甘露醇混合后进行干法制粒,压片制成片剂或灌装制成胶囊;其中活性物质的粒径分布范围d(0.9)优选小于60μm,最优选小于40μm;所述组合物还包括:1)任选地5-50wt%的预胶化淀粉;2)1-30wt%的崩解剂,所述崩解剂选自交联羧甲基纤维素钠、羧甲淀粉钠、低取代羟丙基纤维素和交联聚维酮中的一种或多种;3)0.5-5wt%的润滑剂,所述润滑剂选自硬脂酸镁、硬脂富马酸钠、胶体二氧化硅和滑石粉的一种或多种。
- 根据权利要求1至13任一项所述的药物组合物的制备方法,所述药物组合物为片剂或胶囊。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780001554.5A CN107530332B (zh) | 2016-03-01 | 2017-02-28 | 一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法 |
| US16/080,086 US10966964B2 (en) | 2016-03-01 | 2017-02-28 | Method for preparing pharmaceutical composition comprising pyrrolo-fused six-membered heterocyclic compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610115984 | 2016-03-01 | ||
| CN201610115984.7 | 2016-03-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017148361A1 true WO2017148361A1 (zh) | 2017-09-08 |
Family
ID=59743491
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2017/075113 WO2017148361A1 (zh) | 2016-03-01 | 2017-02-28 | 一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10966964B2 (zh) |
| CN (1) | CN107530332B (zh) |
| TW (1) | TWI738729B (zh) |
| WO (1) | WO2017148361A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115381791A (zh) * | 2022-09-21 | 2022-11-25 | 迪沙药业集团有限公司 | 一种盐酸氟桂利嗪药物组合物 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111184693B (zh) * | 2019-05-29 | 2023-07-21 | 百济神州(苏州)生物科技有限公司 | 一种raf激酶抑制剂制剂及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101007814A (zh) * | 2006-01-27 | 2007-08-01 | 上海恒瑞医药有限公司 | 吡咯并六元杂环化合物及其在医药上的用途 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090136570A1 (en) * | 2006-01-20 | 2009-05-28 | Bhagwant Rege | Taste-Masked Tablets and Granules |
| JP5256047B2 (ja) | 2006-01-27 | 2013-08-07 | シャンハイ ヘンルイ ファーマシューティカル カンパニー リミテッド | ピロロ[3,2−c]ピリジン−4−オン2−インドリノン(indolinone)プロテインキナーゼ阻害剤 |
| EP2468258A1 (en) * | 2010-12-22 | 2012-06-27 | LEK Pharmaceuticals d.d. | Process for the preparation of a pharmaceutical composition comprising a low soluble pharmaceutically active ingredient |
-
2017
- 2017-02-24 TW TW106106405A patent/TWI738729B/zh active
- 2017-02-28 US US16/080,086 patent/US10966964B2/en active Active
- 2017-02-28 CN CN201780001554.5A patent/CN107530332B/zh active Active
- 2017-02-28 WO PCT/CN2017/075113 patent/WO2017148361A1/zh active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101007814A (zh) * | 2006-01-27 | 2007-08-01 | 上海恒瑞医药有限公司 | 吡咯并六元杂环化合物及其在医药上的用途 |
Non-Patent Citations (1)
| Title |
|---|
| LIU, BING ET AL.: "Clinical Efficacy of Famitinib Malate for Treatment of Metastatic Renal Cell Carcinoma-a Report of 9 Case", ACADEMIC JOURNAL OF SECOND MILITARY MEDICAL UNIVERSITY, vol. 36, no. 12, 31 December 2015 (2015-12-31), pages 1348 - 1351, XP055414110 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115381791A (zh) * | 2022-09-21 | 2022-11-25 | 迪沙药业集团有限公司 | 一种盐酸氟桂利嗪药物组合物 |
| CN115381791B (zh) * | 2022-09-21 | 2023-06-16 | 迪沙药业集团有限公司 | 一种盐酸氟桂利嗪药物组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107530332A (zh) | 2018-01-02 |
| CN107530332B (zh) | 2018-09-28 |
| TWI738729B (zh) | 2021-09-11 |
| TW201731507A (zh) | 2017-09-16 |
| US20190060290A1 (en) | 2019-02-28 |
| US10966964B2 (en) | 2021-04-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6876758B2 (ja) | ブルトンチロシンキナーゼの阻害剤を含む剤形組成物 | |
| US20200108008A1 (en) | Olaparib oral sustained and controlled release pharmaceutical composition and uses thereof | |
| JP6571105B2 (ja) | 汎rafキナーゼ阻害剤の薬学的製剤、その調製プロセス、及び使用方法 | |
| EP2646000B1 (en) | Chemoembolization composition comprising anti-angiogenic agents | |
| US9901583B2 (en) | Method of treating non-small cell lung cancer and/or small cell lung cancer using thienotriazolodiazepine compounds | |
| TW201113050A (en) | 3-cyanoquinoline tablet formulations and uses thereof | |
| CN105377299A (zh) | 用于治疗前列腺癌的包含二氢吡嗪并-吡嗪化合物和雄激素受体拮抗剂的组合疗法 | |
| WO2020098774A1 (zh) | 一种包含parp抑制剂的药物组合物 | |
| WO2017148361A1 (zh) | 一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物的制备方法 | |
| WO2017148359A1 (zh) | 一种含有吡咯并六元杂环化合物或其可药用盐的药物组合物 | |
| CN106176752B (zh) | 色瑞替尼药物组合物 | |
| CN114832112A (zh) | 含有alk激酶抑制剂的药物制剂组合物及其制备方法 | |
| JP2020519581A (ja) | 肥満細胞疾患の処置のための方法及び医薬組成物 | |
| US20240226103A1 (en) | Solid dispersion of a her2 inhibitor | |
| TW202206074A (zh) | 藥物配製物 | |
| TW202110453A (zh) | 嘧啶基胺基-吡唑化合物之修飾釋放調配物及治療方法 | |
| WO2022138717A1 (ja) | 経口固形製剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17759212 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17759212 Country of ref document: EP Kind code of ref document: A1 |