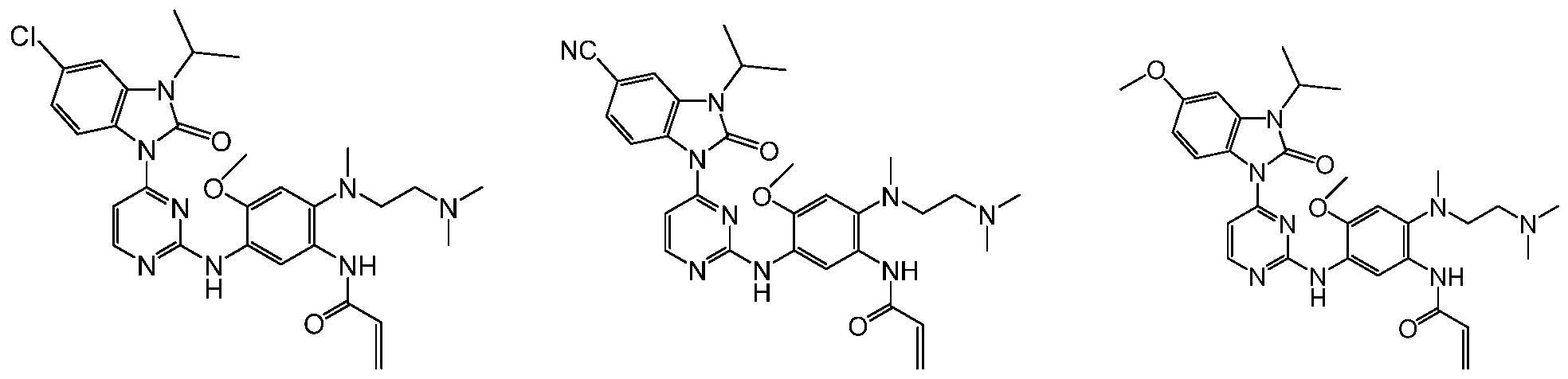
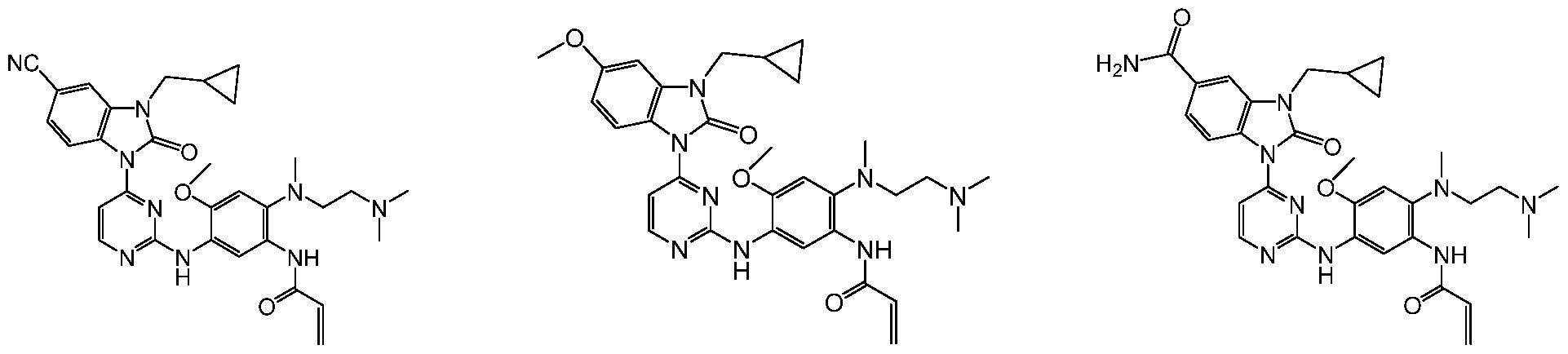
2,4-二含氮基团取代嘧啶类化合物及其制备方法和应用
技术领域
本发明涉及药物化学技术领域, 特别是涉及一种 2,4-二含氮基团取代嘧啶类化合物及其 制备方法和应用。 背景技术
表皮生长因子受体 (Epidermal Growth Factor Receptor, EGFR) 家族的受体酪氨酸激酶 (Tyrosine Kinase,TK), 包括 EGFR (HERD , HER2 (ErbB2) , HER3 (ErbB3 ) 和 HER4 (ErbB4), 参与调节多种发育, 代谢和生理过程。 EGFR受体激活使 EGFR的胞内结构域磷 酸化, 导致 Ras/有丝分裂原活化蛋白激酶信号通路、 PI3K/AKT通路和信号转导与转录激活 因子的信号转导通路的激活 (J ClinOncol 2008;26: 1742 - 1751 )。
在肿瘤细胞中, EGFR基因突变和 EGFR蛋白的表达增加, 使 EGFR TK激活, 促进肿瘤 细胞生存、 增殖、 侵袭和转移。 表皮生长因子受体酪氨酸激酶抑制剂 (TKI), 如厄洛替尼, 吉非替尼和埃克替尼, 是可逆的竞争性抑制剂, 能够竞争性的结合酪氨酸激酶结构域中受体 腺苷三磷酸的结合位点。 这些药物在 EGFR基因激活突变的非小细胞肺癌 (NSCLC) 患者中 有显著的治疗效果。 表皮生长因子受体基因的序列分析表明, 90%EGFR激活突变为外显子 19的缺失突变和外显子 21的 L858R突变 (J ClinOncol 2008;26: 1742 - 1751 )。 在这些 EGFR 激活突变的 NSCLC 病人中, 厄洛替尼和吉非替尼的反应率大约为 70% (Clin Cancer Res 2006;12:3908 - 3914)。 携带 EGFR激活突变的 NSCLC 占西方人种的 10-15%和亚洲人种的 30-40% (Nat Rev Cancer 2010; 10: 760-774; Oncogene 2009; 28 (Suppl 1): S24 - S31.)。 且此 类第一代可逆性、 竞争性的 EGFR抑制剂由于同时抑制皮肤和胃肠道野生型 EGFR, 会引起 皮疹和腹泻等副作用 (Nat Clin Pract Oncol 2008; 5: 268-278; CurrOncol2011; 18: 126 - 138)。
这些携带 EGFR激活突变的 NSCLC患者在服用吉非替尼、 厄洛替尼或埃克替尼 6-14个 月后,病人病情恶化(N Engl J Med 2010; 362: 2380-2388; Lancet Oncol 2012; 13:239-246)。 50-60%的获得性耐药性病人发生了第二点 T790M耐药突变。 野生型 EGFR基因 20外显子第 790个氨基酸位点是苏氨酸 (T), 而 T790M突变是 EGFR基因 20外显子第 790位点由一个 庞大体积的蛋氨酸(M)替代了苏氨酸(T)。 Τ790Μ突变改变了 ATP的亲和性, 导致第一代 的 EGFR-TKI (酪氨酸激酶抑制剂)不能有效阻止信号,从而产生了耐药性(N Eng J Med 2005; 352:786-792; PLoS Med 2005; 2:e73; Oncogene 2009; 28(Suppl 1): S24 - S31.)。
第二代 EGFR抑制剂如 Dacomitinib、 阿法替尼、 Neratinib、 XL647 等, 为不可逆性 EGFR-TKI。 能够非特异性抑制野生型 EGFR, 激活型 L858R突变以及耐药型 T790M突变。 虽然这类化合物对 EGFR T790M突变型有活性, 但临床中未能克服 T790M突变产生的耐药 性 (Lancet Oncol 2012; 13:528-538; J Clin Oncol 2013; 31 :3335-3341)。 又由于其非选择性抑 制野生型 EGFR, 导致产生剂量限制毒性 (DLT), 因此, 这些药物在体内不能达到有效抑制 T790M的浓度。
第三代 EGFR抑制剂能够选择性抑制 T790M耐药突变, 并对野生型 EGFR抑制作用小。
如 Rociletinib (CO- 1686), AZD9291和 HM61713等均是选择性不可逆 T790M EGFR抑制剂(J Med Chem2014 ; 57:8249-8267; Cancer Discov 2014; 4: 1046-1061; Cancer Discov 2013; 3: 1404-1405; Mol Cancer Ther 2014; 13: 1468-1479; )。 Osimertinib (AZD9291), 和 Rociletinib (CO- 1686) 1临床用于 T790M导致的 EGFR-TKI耐药病人展现出了良好的安全性和抗肿瘤活 性 (N Engl J Med. 2015;372: 1689-1699)。
由于第三代 EGFR抑制剂选择性抑制突变型 EGFR, 而对野生型 EGFR抑制作用小, 在 临床前研究中显示出很强的抗肿瘤活性同时, 在诸如皮疹和腹泻等毒性方面剂量限制毒性 (DLT) 比第一、 二代 EGFR抑制剂明显降低。 发明内容
基于此, 有必要针对上述问题, 提供一类化合物, 该类化合物能够选择性抑制 T790M EGFR , 包括单点突变 T790M和双点突变如 L858R/T790M, exl9del/T790M等。同时对 EGFR 单点激活突变如 L858R和 exl9del也有抑制活性, 并且, 这类化合物对野生型 EGFR抑制作 用弱, 即具有很好的选择性, 不会产生剂量限制毒性的问题。
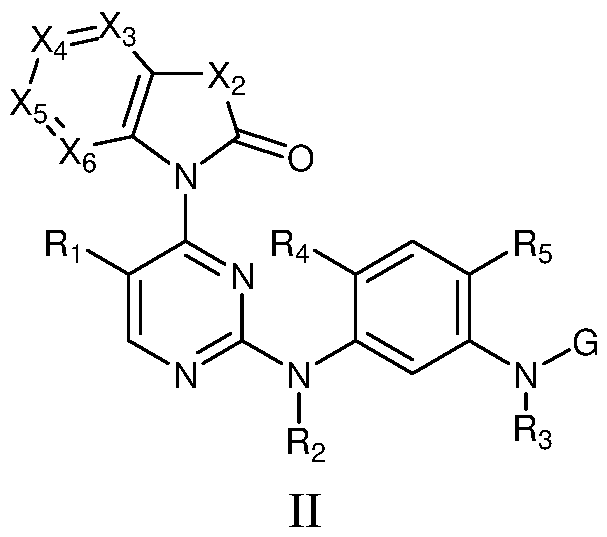
一种式 I所示的 2,4-二含氮基团取代嘧啶类化合物或者其药学上可接受的盐或立体异构 体-
其中: 和 X
2之间的虚线表示 和 X
2之间可选单键或双键;
选自: H, C1-C6焼基, C3-C6环垸基, C3-C6环垸基取代的甲基, 卤素取代的 C1-C4 垸基, 羟基取代的 C1-C4垸基, C1-C3垸氧基取代的 C1-C4垸基, 氨基取代的 C1-C4垸基, C1-C3垸基胺基取代 C1-C4垸基, 卤素, 硝基, 羟基, C1-C6垸氧基, C1-C6垸硫基, C1-C6 亚砜基, C1-C6砜基, 氰基, 氨基, C1-C3垸基取代的胺基, 酯基, 酰基, 酰胺基, 羧基;
R2, R3分别独立选自: H, C1-C6垸基;
R4选自: H, OH, C1-C6焼基, C1-C6垸氧基, 羟基取代的 C1-C4垸基, C1-C3垸氧基 取代的 C1-C4垸氧基;
Ri6, R17分别独立选自: H, C1-C6垸基, C1-C6垸氧基;
Z选自: C, N或 0, 且当 Z为 0时, 1 8不存在;
m选自: 0, 1或 2;
n选自: 1, 2或 3 ;
p选自: 1, 2, 3, 4, 5或 6;
R6, R7, 1 8分别独立选自: H, C1-C6焼基, C3-C6环垸基, C3-C6环垸基取代的甲基, 苯甲基, 苯基, 酰基, 甲磺基, 卤素取代的 C1-C4垸基, 羟基取代的 C1-C4垸基, C1-C3垸 氧基的取代 C1-C4垸基, 氨基取代的 C1-C4垸基, C1-C3垸基胺基取代 C1-C4垸基, 氨基, C1-C3垸基取代的胺基, 羟基, C1-C6垸氧基;
Xi选自: N, C=0或 C-R9 ;
R9选自: H, C1-C6垸基, 卤素取代的 C1-C6垸基, 含 0,N,S杂原子的 C1-C6垸基, 卤 素, 氰基, 氨基;
X2选自: N, N-R10, C-Rio;
Rio选自: H, 卤素, C1-C6垸基, C2-C6烯基, C2-C6炔基, C3-C6环垸基, C3-C6环 垸基甲基, 卤素取代的 C1-C6垸基, 含 0,N,S杂原子的 C1-C6垸基;
X3, X4, X5, X6分别独立选自: N, C-Rn;
Ru选自: H, C1-C6焼基, 羟基取代的 C1-C4垸氧基, C1-C3垸氧基取代的 C1-C4垸氧 基, C3-C6环垸基, C3-C6环垸基甲基, 卤素取代的 C1-C4垸基, 羟基取代的 C1-C4垸基, C1-C3垸氧基取代的 C1-C4垸基, 氨基取代的 C1-C4垸基, C1-C3垸基胺基取代的 C1-C4垸 基,卤素, 硝基, 羟基, C1-C6垸氧基, C1-C6垸硫基, C1-C6亚砜基, C1-C6砜基, 氰基, 氨基, C1-C3垸基取代的胺基, 酯基, 酰基, 酰胺基, C1-C3垸基取代的酰胺基, 羧基;
Ri2, R13分别独立选自: H, C1-C6垸基;
R
14选自: H, C1-C6垸基, C1-C3垸氧基取代的 C1-C4垸基, 氨基取代的 C1-C4垸基 C1-C3垸基胺基取代的 C1-C4垸基, 杂环取代的 C1-C4垸基; 并且, 当1^选自: H, 卤素或氰基; R
4选自: C1-C6垸氧基; G选自:
Rio选自: H, C1-C6焼基, C3-C6环垸基, 卤素取代的 C1-C6垸基时; Rii不为: H, C1-C6 垸基, C3-C6环垸基, 卤素取代的 C1-C4垸基, 或 C1-C6砜基;
或者 当1^选自: H, 卤素或氰基; R4选自: C1-C6垸氧基; G选自:
Ru选自: H, C1-C6焼基, C3-C6环垸基, 卤素取代的 C1-C4垸基, C1-C6砜基时, R1()不为 H, C1-C6垸基, C3-C6环垸基, 或卤素取代的 C1-C6垸基。
在 一些实施例中, 选自如下式 II或式 III所示
其中: Xi选自: N, C-R9;
X2, X3, X4, X5, X6如上所述;
Ri, R2, R3, R4, R5, R9, G如上所述。
在其中一些实施例中, X3, X5, X6分别独立选自: N, CH; X4选自: C-R11 ; Ru如上所 述'
在其中一些实施例中, 选自如
其中: X2选自: N-R1();
R10选自: C1-C6垸基, C2-C6烯基, C3-C6环垸基, C3-C6环垸基甲基;
X3, X5, X6分别独立选自: CH;
X4选自: C-R11 ;
Ru选自: C1-C6垸氧基, 卤素, 氰基。
Ri, R2, R3, R4, R5, G如上所述。
在其中一些实施例中, 选
X3选自: N;
X4如权利要求 1所述;
X5, X6选自: CH;
Ri, R2, R3, R4, R5, R9, G如上所述。
在其中一些实施例中, X2分别独立选自: C或 N-R10 ;
Rio选自: C1-C6垸基, C2-C6烯基, C3-C6环垸基, C3-C6环垸基甲基;
X3选自 CH或 N;
X5, X6分别独立选自: CH;
X4选自: C-R11 ;
Ru选自: C1-C6垸氧基, 卤素, 氰基;
Ri, R2, R3选自: H;
R4选自: 甲氧基;
在其中一些实施例中, 选自: H, F, Cl, Br, 甲基, 三氟甲基, 甲氧基, 氰基, 羟基, 二 甲胺基, 酰胺基;
R2, R3选自: H;
R4选自: H, 甲氧基, 乙氧基, 甲基, 乙基, OH, 甲氧基乙氧基;
Rl2, Ri3选自: H;
R14选自: H, C1-C6垸基, C1-C3垸氧基取代的 C1-C4垸基, 氨基取代的 C1-C4垸基, C1-C3垸基胺基取代的 C1-C4垸基, 杂环取代的 C1-C4垸基。
在其中一 , R5选自如下
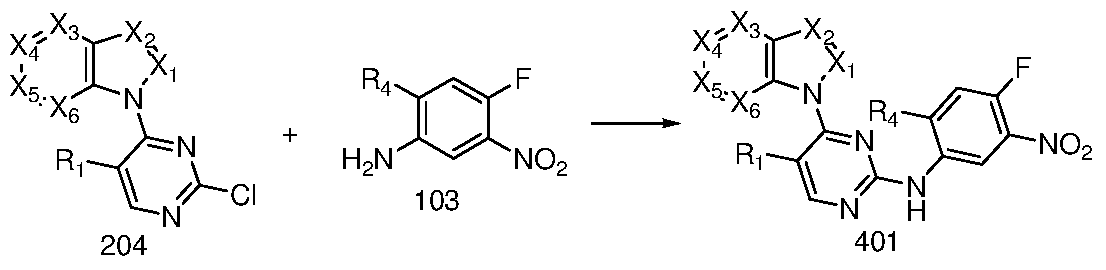
本发明还公开了上述的 2,4-二含氮基团取代嘧啶类化合物的制备方法, 包括以下步骤: 将中间体 101 中的硝基还原得到中间体 102, 并在酸性条件下与硝酸盐反应得到中间体 103, 中间体 103 中的氨基被溴替换得到中间体 104, 采用 R5氮上连有氢的中间体与中间体 104反应, 得到中间体 105, 备用; 或将中间体 103与1 5氮上连有氢的中间体反应, 生成中 间体 106, 备用;
反应线路如下:
将中间体 201与中间体 202或 203或 301反应, 分别得到中间体 204 205, 或 302, 用 氨水取代中间体 204上的氯, 得到中间体 205, 将中间体 302中的硫氧化得到中间体 303, 中 间体 303与氨水反应得到中间体 205, 中间体 205备用;
反应线路如下:
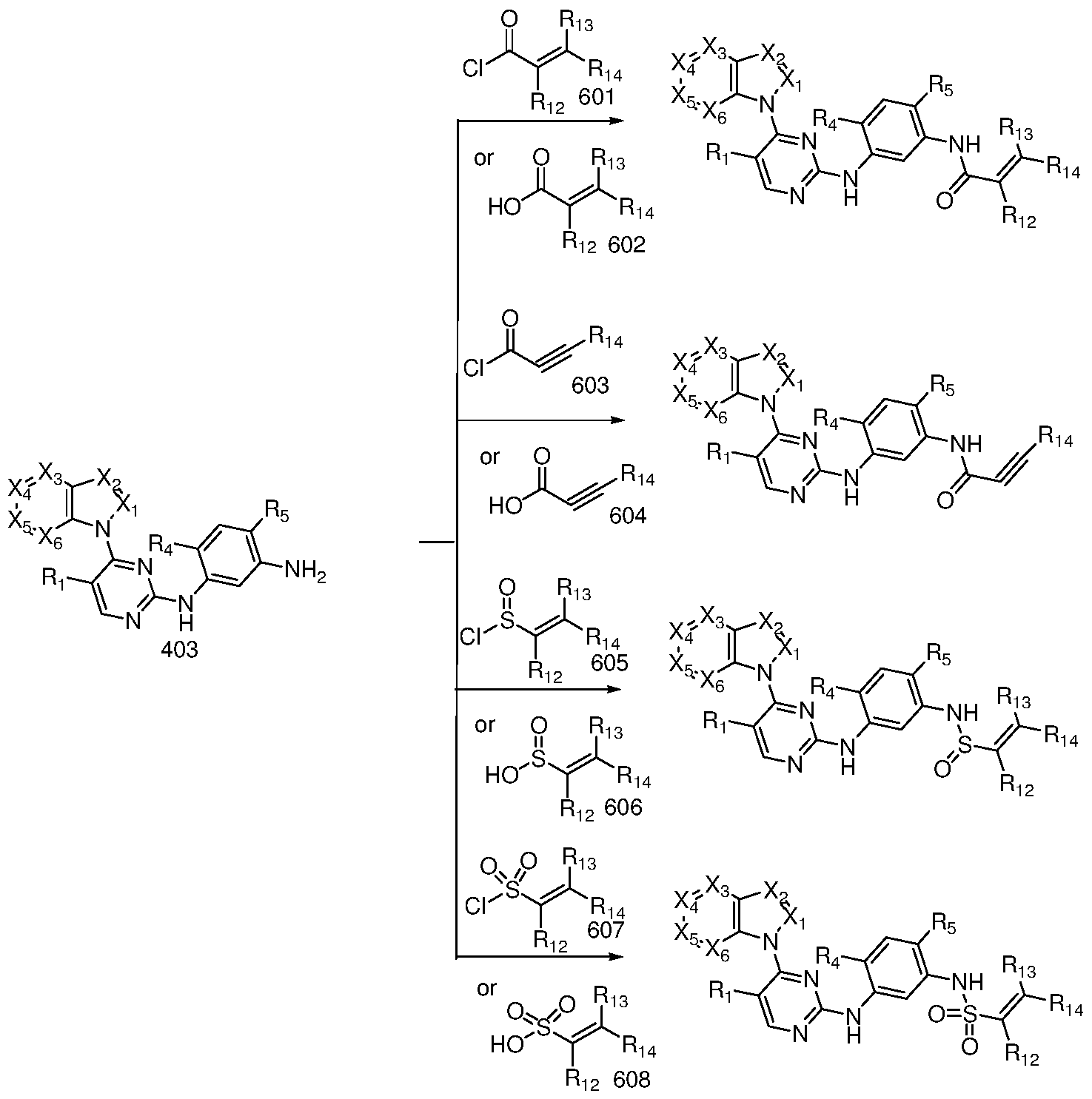
将中间体 204与中间体 103反应, 得到中间体 401, 采用 R5氮上连有氢的化合物与中间 体 401反应, 得到中间体 402; 或将中间体 204与中间体 106反应, 得到中间体 402; 或将中 间体 205与中间体 105反应,得到中间体 402,再将中间体 402的硝基还原,得到中间体 403 备用;
反应路线如下:
1〇2
R4.
I02
205
将中间体 403与含有 G基团的中间体反应, 反应路线如下:
即得目标化合物。
本发明还公开了上述的 2,4-二含氮基团取代嘧啶类化合物的制备方法, 包括以下步骤: 将中间体 101 中的硝基还原得到中间体 102, 并在酸性条件下与硝酸盐反应得到中间体 103, 中间体 103 中的氨基被溴替换得到中间体 104, 采用 R5氮上连有氢的中间体与中间体 104反应, 得到中间体 105, 备用; 中间体 103也可与 R5氮上连有氢的化合物反应, 生成中 间体 106, 备用;
反应线路如下:
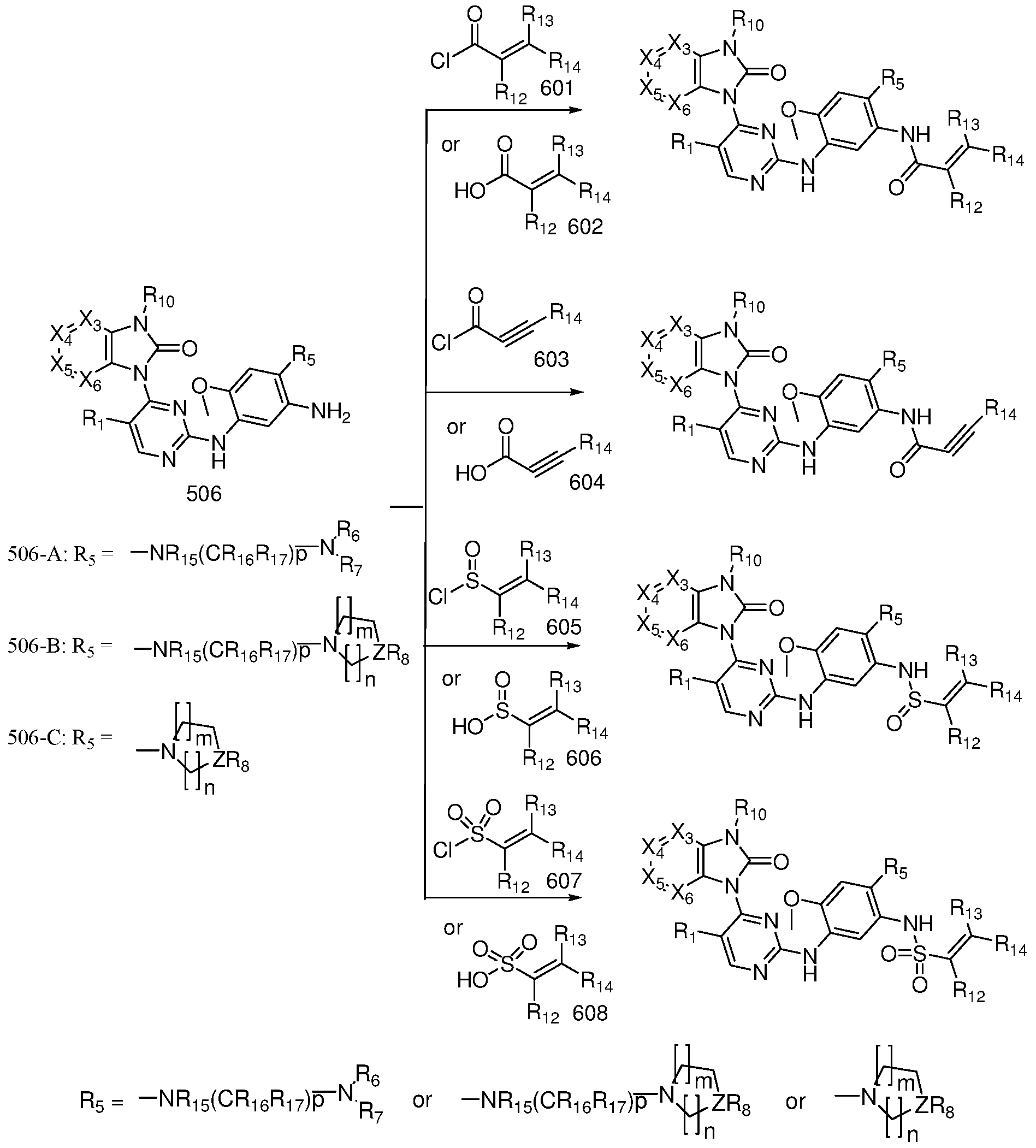
将中间体 501在 Ν,Ν'-羰基二咪唑 (CDI) 存在的条件下关环, 得到中间体 502, 中间体 502与中间体 202反应, 得到中间体 503, 以氨水中的氨基将中间体 503中的氯取代, 得到中 间体 504, 将该中间体 504与中间体 105反应, 得到中间体 505 ; 或将中间体 503与中间体 106反应, 得到中间体 505 ; 或将中间体 503与中间体 103反应, 所获得的产物中的氟用带有 氨基的 1 5反应得到中间体 505 ; 将中间体 505的硝基还原, 得到中间体 506, 备用;
反应路线如下:
得中间体 506与含有 G基团的中
即得目标化合物。
本发明还公开了上述的 2,4-二含氮基团取代嘧啶类化合物或者其药学上可接受的盐或立 体异构体在制备预防和治疗肿瘤药物中的应用。
在其中一些实施例中, 所述肿瘤为实体肿瘤。
在其中一些实施例中, 所述肿瘤为 EGFR基因突变的恶性肿瘤。
在其中一些实施例中, 所述肿瘤为携带 T790M EGFR突变的非小细胞肺癌 (NSCLC)。 本发明还公开了一种防治肿瘤的药物组合物, 包括作为活性成份的上述 2,4-二含氮基团 取代嘧啶类化合物或者其药学上可接受的盐或立体异构体, 以及药学上可接受的载体。
在其中一些实施例中, 所述肿瘤为携带 T790M EGFR突变的非小细胞肺癌 (NSCLC)。 与现有技术相比, 本发明具有以下有益效果:
本发明的一种 2,4-二含氮基团取代嘧啶类化合物, 是一系列新的化合物, 这类化合物选 择性抑制 T790M EGFR, 包括单点突变 T790M和双点突变如 L858R/T790M, exl9del/T790M。 同时对 EGFR单点激活突变如 L858R和 exl9del也有抑制活性。
并且, 这类化合物不但对 L858R/T790M EGFR突变的细胞如 H1975有很强的抗细胞增 殖的活性, 而且对野生型 EGFR细胞如 LOVO及 H358的抗细胞增殖的活性弱, 具有高度的 选择性。
该类 2,4-二含氮基团取代嘧啶类化合物具有潜力成为用作 EGFR 突变的恶性肿瘤, 尤其 是携带 T790M EGFR 突变的 NSCLC治疗的药物。 附图说明
图 1为实验例中化合物 91在 H1975移植肿瘤小鼠口服给药 (30 mg/kg) 后血浆和肿瘤组 织药物浓度药时曲线图;
图 2为实验例中化合物 91口服给药抑制携带 T790突变的 H1975 移植肿瘤 EGFR磷酸化 示意图;
图 3为实验例中化合物 20、 50、 91、 118 抑制携带 T790突变的 H1975 移植肿瘤生长量 效关系 (肿瘤体积变化);
图 4为实验例中化合物 50剂量依赖性抑制携带 T790突变的 H1975 移植肿瘤生长量效关 系;
图 5为实验例中化合物 91剂量依赖性抑制携带 T790突变的 H1975 移植肿瘤生长量效关 系。 具体实施方式
以下结合具体实施例和附图对本发明做进一步的解释说明, 但并不对本发明造成任何限 制。
本文所用术语 "垸基"意指包括具有特定碳原子数目的支链的和直链的饱和脂肪烃基。 例如, "C1-C6垸基" 中 "C1-C6" 的定义包括以直链或支链排列的具有 1、 2、 3、 4、 5或 6 个碳原子的基团。 例如, "C1-C6垸基"具体包括甲基、 乙基、 正丙基、 异丙基、 正丁基、 叔 丁基、 异丁基、 戊基、 己基。
术语 "环垸基"指具有特定碳原子数目的单环饱和脂肪烃基。 例如 "环垸基"包括环丙 基、 甲基-环丙基、 2 , 2 -二甲基-环丁基、 2 -乙基-环戊基、 环己基等。
术语 "垸氧基"指垸基与氧直接连接的基团, 如甲氧基, 乙氧基等。
术语 "垸硫基"指垸基与硫直接连接的基团。
术语 "C1-C3垸基胺基取代 C1-C4垸基"指的是具有 1、 2、 3个碳原子的垸基与氮原子 相连后其氮原子再与具有 1、 2、 3、 4个碳原子的垸基与氮原子相连的基团,例如甲胺基甲基, 甲胺基乙基, 二甲胺基甲基等。
术语"杂环"包括饱和的含有杂原子的环垸基和杂芳基, 其中杂原子可以选自氮、 硫和氧 及氮、 硫、 磷的任何氧化态形式, 优选含 N的饱和杂环垸基, 如哌啶等。
术语 "取代的"是指用指定取代基的基团置换特定结构中的氢基。 本发明包括式 I -III化合物的游离形式, 也包括其药学上可接受的盐及立体异构体。 可通 过常规化学方法自含有碱性部分或酸性部分的本发明化合物合成本发明的药学上可接受的 盐。 通常, 通过离子交换色谱或通过游离碱和化学计算量或过量的所需盐形式的无机或有机 酸在适当溶剂或多种溶剂的组合中反应制备碱性化合物的盐。 类似的, 通过和适当的无机或 有机碱反应形成酸性化合物的盐。
因此, 本发明化合物的药学上可接受的盐包括通过碱性本发明化合物和无机或有机酸反 应形成的本发明化合物的常规无毒盐。例如, 常规的无毒盐包括自无机酸例如盐酸、 氢溴酸、 硫酸、 氨基磺酸、 磷酸、 硝酸等制备的盐, 也包括自有机酸例如乙酸、 丙酸、 琥珀酸、 乙醇 酸、 硬脂酸、 乳酸、 苹果酸、 酒石酸、 柠檬酸、 抗坏血酸、 扑酸、 马来酸、 羟基马来酸、 苯 乙酸、 谷氨酸、 苯甲酸、 水杨酸、 对氨基苯磺酸、 2 —乙酰氧基一苯甲酸、 富马酸、 甲苯磺 酸、 甲磺酸、 乙垸二磺酸、 草酸、 羟乙基磺酸、 三氟乙酸等制备的盐。
如果本发明化合物为酸性的, 则适当的 "药学上可接受的盐"指通过药学上可接受的无 毒碱包括无机碱及有机碱制备的盐。 得自无机碱的盐包括铝盐、 铵盐、 钙盐、 铜盐、 铁盐、 亚铁盐、 锂盐、 镁盐、 锰盐、 亚锰盐、 钾盐、 钠盐、 锌盐等。 特别优选铵盐、 钙盐、 镁盐、 钾盐和钠盐。 得自药学上可接受的有机无毒碱的盐, 所述碱包括伯胺、 仲胺和叔胺的盐, 取 代的胺包括天然存在的取代胺、 环状胺及碱性离子交换树脂例如精氨酸、 甜菜碱、 咖啡因、 胆碱、 Ν , Ν '—二苄基乙二胺、 二乙胺、 2 —二乙基氨基乙醇、 2—二甲基氨基乙醇、 氨基乙 醇、 乙醇胺、 乙二胺、 Ν—乙基吗啉、 Ν—乙基哌啶、 葡萄糖胺、 氨基葡萄糖、 组氨酸、 羟 钴胺、 异丙基胺、 赖氨酸、 甲基葡萄糖胺、 吗啉、 哌嗪,哌啶、 呱咤、 多胺树脂、 普鲁卡因、 嘌呤、 可可碱、 三乙胺、 三甲胺、 三丙胺、 氨基丁三醇等。
除在文献中已知的或在实验程序中例证的标准方法外, 可采用如下合成方案 (方案 1-6) 中的方法制备本发明化合物。 结合下述的合成方案, 能够对本发明中所述的化合物以及合成 方法进行更好的理解。 所述的合成方案描述了可以用于制备本发明中所述的化合物的方法, 所述的方法仅仅是为说明目的的说明性方案描述, 并不构成对本发明所具有的范围的限制。
方案一
106-B: R5 :
-B:R
5 = —
NR
15(CR
16R
17)p- -NR
15(CR
16R
17)p-N††ZR,
万案二
91755
实施例 1 : 中间体 103, 104, 105和 106的制备 (按照方案一线路制备)
步骤 la:4-氟 -2-甲氧基苯胺 (4-fluoro-2-methoxyaniline) (中间体 102)的制备: 将 2-甲氧基 -4-氟硝基苯 (中间体 101)(20克, 0.12摩尔, 1.0当量),氯化铵 (13克, 0.24摩尔, 2.0当量) 和 水 (50毫升) 溶于乙醇 (200毫升) , 升温到 55 °C后, 往里分批加入铁粉 (13克, 0.24摩尔, 2.0当量) 。 升温到 85 °C反应两个小时后, 冷却到室温, 过滤, 旋干溶剂。 将残留物溶于乙酸 乙酯, 用水洗, 饱和食盐水洗, 无水硫酸钠干燥, 过滤, 旋干溶剂得到浅绿色产物 4-氟 -2-甲
氧基苯胺 (15克, 收率: 91%)。 LCMS (ESI): m々142[M + H ]+。
步骤 lb: 4-氟 -2-甲氧基 -5-硝基苯胺 (4-fluoro-2-methoxy-5-nitroaniline) (中间体 103)的制备: 冰浴条件下,往浓硫酸 (150毫升)中滴加 4-氟 -2-甲氧基苯胺 (中间体 102)(15克, 106.4毫摩尔, 1.0当量) , 滴加过程要控制温度在 0°C左右。 当形成的固体完全溶解后, 往里分批加入硝酸 钾 (11克, 106.4毫摩尔, 1.0当量), 在此条件下反应一小时。 将反应液倒入冰水中, 氢氧化 钠调节 pH到碱性, 析出大量固体, 过滤, 水洗, 石油醚洗, 晾干得到棕黄色固体 4-氟 -2-甲 氧基 -5-硝基苯胺 (18 克, 收率: 77%)。 LCMS (ESI): m々187 [M + H ]+。
步骤 lc: 1-溴 -4-氟 -2-甲氧基 -5-硝基苯 (l-bromo-4-fluoro-2-methoxy-5-nitrobenzene)(中间 体 104)的制备: 在氮气氛围下,将溴化铜 (3.568克, 16毫摩尔, 1.5当量)和亚硝酸叔丁酯 (3.399 克, 33毫摩尔, 3 当量)混合于乙腈 (250毫升;), 升温至 50°C, 然后将化合物 4-氟 -2-甲氧基 -5- 硝基苯胺 (103)(2克, 11毫摩尔, 1当量)的乙腈 (20毫升)溶液滴加至体系中, 再搅拌反应 2.5 小时。 反应冷却至室温, 用乙酸乙酯和水萃取, 有机相用水洗, 盐水洗, 无水硫酸钠干燥, 减压浓缩, 得到棕色油状物。 油状物用硅胶柱层析纯化 (石油醚 /乙酸乙酯 =10/1), 得到黄色固 体 1-溴 -4-氟 -2-甲氧基 -5-硝基苯 (1.87克, 收率: 69.57%)。
步骤 Id: N1-(4-溴 -5-甲氧基 -2-硝基苯基;) -Ν^ί^,Ν2-三甲基乙垸 -1,2-二胺 (N1-(4-bromo-5-methoxy-2-nitrophenyl)- N^N^N -trimethylethane-l'S-diamine) (中间体 105-A-5) 的制备: 将化合物 1-溴 -4-氟 -2-甲氧基 -5-硝基苯 (中间体 104X1.87 克, 7.48 毫摩尔, 1 当 量) ,Ν,Ν-二异丙基乙基胺 (2.41克, 17.8毫摩尔, 2.5当量) 和 Ν,Ν,Ν'-三甲基乙二胺 (1.146 克, 11.22毫摩尔, 1.5当量)的乙醇 (50毫升)溶液加热至回流, 搅拌反应 16小时。 反应结束 后, 冷却至室温, 二氯甲垸萃取, 有机相用水洗和盐水洗, 无水硫酸钠干燥, 减压浓缩, 得 到红色油状物 ^-(4-溴 -5-甲氧基 -2-硝基苯基) -Ν^ί^,Ν2-三甲基乙垸 -1,2-二胺 (2.3 克, 粗品) , 直接用于下一步反应。
步骤 le : N1^- (二甲胺基) 乙基) -5-甲氧基 - N1-甲基 -2-硝基苯基) -1, 4-二胺 ( N1-(2-(dimethylamino)ethyl)-5-methoxy-N1-methyl-2-nitrobenzene-l,4-diamine ) ( 中 间体 106-A-11 ) 的制备: 将化合物 4-氟 -2-甲氧基 -5-硝基苯胺 (中间体 103)(2克, 10.75毫摩尔, 1 当量) , DIPEA (2.77克, 21.5毫摩尔, 2当量)和 Ν,Ν,Ν-三甲基乙二胺 (1.644克, 16.125 毫 摩尔, 1.5当量)混合于 50毫升四氢呋喃中。 混合物在回流的条件下搅拌反应 3天。 反应结束 后, 冷却至室温, 用二氯甲垸和水萃取, 有机相用水洗, 盐水洗, 无水硫酸钠干燥, 减压浓 缩, 硅胶柱层析 (二氯甲焼 /甲醇 /氨水 =100/1/0.01)纯化, 得到红色油状物 Ν1-^- (二甲胺基) 乙基) -5-甲氧基 - Ν1-甲基 -2-硝基苯基) -1, 4-二胺 (1.8 克, 粗品)。 实施例 2: 中间体 204的制备 (按照方案二线路制备)
1-(2-氯 -5-甲氧基嘧啶 -4-基) -1Η-吲哚( l-(2-chloro-5-methoxypyrimidin-4-yl)-lH-indole) (中 间体 204-6) 的制备:将吲哚 (201-6) (I克, 8.55毫摩尔, 1.0当量)溶于四氢呋喃 (30毫升), 氮气保护, 零度条件下, 往里分批加入叔丁醇钾 (1.44克, 12.82毫摩尔, 1.5 当量)。 室温条 件下, 反应一小时。再次冷却到室温, 往里滴加 2,4-二氯 -5-甲氧基嘧啶 (202-6) (2.3克, 12.82 毫摩尔, 1.5 当量)溶于四氢呋喃 (20毫升)溶液, 滴加完后, 升到室温反应过夜。 将其倒入水 中, 乙酸乙酯萃取, 饱和食盐水洗, 无水硫酸钠干燥, 旋干, 石油醚洗涤得到米色固体 1-(2-
氯 -5-甲氧基嘧啶 -4-基) -1H-吲哚 (2.2克, 收率: 100%)。 LCMS (ESI): m/z 260 [M + H ]+。 下列中间体(204-8, 204-10, 204-11)可按上述合成中间体 204-6的方法制备, 仅是将其 中的中间体 201-6及 202-6替换为相应的中间体 201和 202。
中间体 204-8的制备: 吲哚 (201-6) 和 2,4-二氯 -5-甲基嘧啶 (202-8)反应, 制备得到 1 -(2- 氯 -5-甲基嘧啶 -4-基) -1H-吲哚( l-(2-chloro-5-methyl-pyrimidin-4-yl)-lH-indole) (中间体 204-8)。 LCMS (ESI): m/z244[M + H ]+。
中间体 204-10的制备: 吲哚 (201-6)禾 P 2,4-二氯 -Ν,Ν-二甲基嘧啶 -5-胺 (202-10)反应, 制 备得到 2-氯 -4-(1Η-吲哚 -1-基 )-Ν,Ν-二甲基嘧啶 -5-胺 ( 2-chloro-4-( lH-indol- 1 -yl)-N,N- dimethylpyrimidin-5-amine) (中间体 204-10)。 LCMS (ESI): m/z273[M + H ]+。
中间体 204-11的制备: 吲哚 (201-6) 和 2,4-二氯 -N-甲基嘧啶 -5-胺 (202-11)反应, 制备得 至 IJ 2-氯 -4-(1Η-吲哚 -1-基) -N-甲基嘧啶 -5-胺 ( 2-chloro-4-(lH-indol-l-yl)-N-methylpyrimidin- 5-amine) (中间体 204-11)。 LCMS (ESI): m/z259[M + H ]+。 实施例 3: 中间体 205的制备 (按照方案二线路或方案三线路制备)
中间体 205可按三种方法制备, 方法 3-1, 方法 3-2和方法 3-3, 描述如下:
方法 3-1 (方案二线路) :
4-(1Η-吲哚 -1-基) -5-甲氧基嘧啶 -2-胺(4-(lH-indol-l-yl)-5-methoxypyrimidin-2-amine) (中 间体 205-6)的制备: 把化合物 1-(2-氯 -5-甲氧基嘧啶 -4-基) -1H-吲哚(中间体 204-6) (1克, 3.9 毫摩尔, 1.0当量), 氨水 (2毫升)和 Ν,Ν-二异丙基乙胺 (1毫升)溶于 N-甲基吡咯垸酮 (10 毫 升;), 在闷罐中, 升温到 110°C反应过夜。 反应液冷却到室温, 倒入水中, 乙酸乙酯萃取, 饱 和食盐水洗,无水硫酸钠干燥,旋干,残留物用硅胶柱层析纯化 (洗脱剂:石油醚 /乙酸乙酯 20/1 至 1/1)得到白色固体 4-(1Η-吲哚 -1-基) -5-甲氧基嘧啶 -2-胺 (240毫克,收率 :26%)。LCMS (ESI): m々241[M + H]+。
方法 3-2 (方案二线路) :
4-(1Η-吲哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (4-(lH-indol-l-yl)-5-(trifluoromethyl)pyrimidin- 2-amine) (中间体 205-5) 的制备: 在室温下, 将氢化钠 (210毫克, 7.0毫摩尔, 5.0当量) 加入到 15毫升 N-甲基吡咯垸酮, 于冰水浴中冷却, 加入吲哚 (201-6) (410毫克, 3.5毫摩 尔, 2.5当量),于室温下搅拌 1小时后,于冰浴中冷却,将 2-氨基 -4-氯 -5-三氟甲基嘧啶(203-5)
(279毫克, 1.4毫摩尔, 1.0当量) 溶解于 1毫升 N-甲基吡咯垸酮, 加入到上述混合物中, 室温下搅拌 1小时。 加入水和乙酸乙酯, 分液, 干燥有机相, 减压浓缩, 经硅胶柱层析纯化
(洗脱剂: 石油醚 /乙酸乙酯 4/1至 1/1), 得到黄色固体 4-(1Η-吲哚 -1-基;) - 5-三氟甲基嘧啶 -2- 胺 (220毫克, 收率: 56%)。 LCMS (ESI): m/z 279[M + H ]+。
4-(5-甲氧基 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-胺 ( 4-(5-methoxy-lH-pyrrolo[3,2-b] pyridin- 1 -yl)pyrimidin-2-amine ) (中间体 205-20) 的制备: 将化合物 5-甲氧基 -1H-吡咯 [3,2-b] 吡啶(201-20) (200毫克, 1.35毫摩尔, 1当量), 碳酸铯 (877.5毫克, 2.7毫摩尔, 2当量)和 2-氨基 -4-氯嘧啶 (203-20) (261 毫克, 2.025 毫摩尔, 1.5 当量) 混合于 N-甲基吡咯垸酮 (5 毫升;)。 混合物加热至 150°C搅拌反应 2小时。 反应结束后, 冷却至室温, 用水淬灭, 过滤,
滤渣用水洗,减压干燥得到灰色固体 (300毫克,收率: 92.13%)。LCMS (ESI): m/z 242[M + H ]+ 0 4-(5-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (4-(5-fluoro-lH-indol-l-yl)pyrimidin-2-amine ) (中间体 205-28)的制备: 将 5-氟吲哚(201-28 )(500毫克, 3.7毫摩尔,1.0当量),2-氨基 -4-氯嘧啶(203-28 ) (480毫克, 5.1毫摩尔, 1.0当量)和碳酸铯 (2.4克, 7.4毫摩尔, 2.0当量) 溶于 NMP (50毫 升;), 150°C下反应 1 小时。 冷却到室温, 将其倒入水中, 过滤, 水洗, 干燥得到粉红色固体 4-(5-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (500毫克, 收率: 59%)。 LCMS (ESI): m/z229 [M + H ]+。
方法 3-3 (方案三线路) :
4-(5-硝基 -吲哚 -1-基) -嘧啶 -2-氨基(4-(5-Nitro-indol-l-yl)-pyrimidin-2-ylamine)(中间体 205-34)的制备: 室温下将 5-硝基 -1H-吲哚 (201-34 ) (3克, 18.5毫摩尔) 和 4-氯 -2-甲硫基- 嘧啶 (301-34 ) (2.98克, 18.5毫摩尔)溶于 Ν,Ν-二甲基甲酰胺( 100毫升), 然后加入碳酸钾 ( 5.1克, 37毫摩尔)。 加热到 80°C反应过夜, 然后加入水 500毫升, 直接抽滤, 真空干燥, 然后得到灰色固体 1-(2-甲硫基 -嘧啶 -4-基) -5-硝基 -1H-吲哚 (302-34 ) ( 5.2克, 收率 98%)。
LCMS (ESI): m/z 287[M + H ]+。 将化合物 302-34 ( 0.5克, 1.7毫摩尔) 溶于二氯甲垸 (15毫 升), 然后加入间氯过氧苯甲酸 (0.3克, 1.7毫摩尔)。 常温下搅拌 4小时, 然后加入饱和碳 酸钠水溶液 20毫升, 用二氯甲垸 (20毫升) 萃取 3次, 干燥浓缩之后得到黄色固体 1-(2-甲 硫亚砜基 -嘧啶 -4-基) -5-硝基 -1H-吲哚 (303-34 ( 0.51克, 收率 98%)。 LCMS (ESI): m/z 303 [M + H ]+。 将化合物 303-34 (0.51克, 1.69毫摩尔)溶于四氢呋喃 (10毫升), 然后加入 20%的氨 水 (2毫升)。 封管下 80°C反应过夜, 加入 20毫升水, 用二氯甲垸 (20毫升) 萃取 3次, 浓 缩干燥之后得到黄色固体 4-(5-硝基 -吲哚 -1-基) -嘧啶 -2-氨基(205-34 ) ( 0.37克, 收率 99.5%)。
LCMS (ESI): mlz 256[M + H ]+。
4-(5- ((叔丁基二苯基硅)氧基) -1H-吲哚 -1 -基)嘧啶 -2-胺 (4-(5-((tert-butyldiphenylsilyl)oxy)- lH-indol- 1 -yl)pyrimidin-2-amine ) (中间体 205-36)的制备:在氮气的氛围下, 将化合物 5- ((叔丁 基二苯基硅)氧基) -1H-吲哚(201-36 ) (800毫克, 2.15毫摩尔, 1.2当量), 2-甲硫基 -4-氯嘧啶 ( 301-34 )(286毫克, 1.79毫摩尔, 1当量),碳酸铯 (1160毫克, 3.57毫摩尔,2当量), Pd2(dba)3(77 毫克, 0.086毫摩尔, 0.05当量)和 Xantphos(91毫克, 0.16毫摩尔, 0.09当量)混合于 20毫升 甲苯中, 在 110°C下搅拌反应 1 小时。 反应结束后, 冷却至室温, 减压浓缩, 硅胶柱层析纯 化 (石油醚 /乙酸乙酯 =20/1), 得到灰色固体 5- ((叔丁基二苯基硅;)氧基; )-1-(2-甲硫基嘧啶 -4- 基) -1H-吲哚(302-36 ) (980毫克, 粗品)。 LCMS (ESI): m々 496 [M + H ]+。 在氮气的氛围下和 冰水浴中, 将化合物 302-36(240毫克, 0.485毫摩尔, 1当量)溶解于 5毫升二氯甲垸中。 然 后将间氯过氧苯甲酸 (118毫克, 0.528毫摩尔, 1.2当量)的二氯甲垸 (2毫升)溶液滴加入其中。 混合物在室温下反应 30分钟。 反应结束后, 用饱和碳酸钠淬灭, 二氯甲垸和水萃取, 有机相 用无水硫酸钠干燥, 减压浓缩, 得到灰色固体 5- ((叔丁基二苯基硅;)氧基; )-1-(2-甲基亚磺酰基 嘧啶 -4-基) -1H-吲哚(303-36 ) (300毫克, 粗品) 。 LCMS (ESI): m々 512 [M + H ]+。在闷罐中, 将化合物 303-36(300毫克, 0.587毫摩尔, 1当量)和氨水 (;5毫升) 溶解于 10毫升四氢呋喃 中。 混合物在 80°C下搅拌反应 2小时。 反应结束后, 冷却至室温, 减压浓缩, 硅胶柱层析纯 化 (石油醚 /乙酸乙酯 =1/1), 得到白色固体 4-(5- ((叔丁基二苯基硅;)氧基) -1H-吲哚 -1-基;)嘧啶 -2- 胺 (205-36 ) (200毫克, 收率: 73.53%)。 LCMS (ESI): m/z 464 [M + H ]+。
4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-胺 (4-(5-methoxy-lH-indol-l-yl)pyrimidin-2-amine) (中
间体 205-27)的制备:在冰浴的条件下, 将化合物 5-甲氧基吲哚 (201-27) (500 毫克, 3.4 毫 摩尔, 1当量)溶解于 N-甲基吡咯垸酮 (5 毫升)。 然后加入氢化钠 (272 毫克, 6.8 毫摩尔, 2 当量), 混合物在室温下搅拌 30分钟。 将化合物 2-甲硫基 -4-氯嘧啶 (301-15) (544 毫克, 3.4 毫摩尔, 1当量)滴加入体系中, 再搅拌反应 1小时。 反应结束后, 加水淬灭, 过滤, 滤渣用 水洗, 干燥, 得到黄色固体甲氧基 -1-(2-甲硫基嘧啶 -4-基) -1H-吲哚(302-27) (1.02 克, 粗品)。 LCMS (ESI): m/z 272 [M + H]+。 将化合物 5-甲氧基 -1-(2-甲硫基嘧啶 -4-基) -1H-吲哚 (302-27) (500 毫克, 1.845 毫摩尔, 1当量)溶于二氯甲垸 (20毫升) , 氮气保护条件下, 降温到 0°C左 右往里滴加间氯过氧苯甲酸 (374.4 毫克, 1.845 毫摩尔, 1.2当量)溶于二氯甲垸 (2毫升)。 滴 加完后, 室温条件下反应 1小时。 反应液用碳酸钠水溶液洗, 饱和食盐水洗, 无水硫酸钠干 燥, 旋干得到黄色固体 5-甲氧基 -1-(2-甲基亚磺酰基嘧啶 -4-基;) -1H-吲哚 (303-27) (580毫克, 粗品)。 LCMS (ESI): m/z 288 [M + H ]+。 把化合物 5-甲氧基 -l-(2-甲基亚磺酰基嘧啶 -4-基) -1H- 吲哚 (580 毫克, 2.02 毫摩尔, 1当量) 和氨水 (5毫升) 溶于四氢呋喃 (10 毫升),在闷罐中, 升温到 80°C反应 2小时。 反应液冷却到室温, 旋干, 硅胶柱层析 (二氯甲焼 /甲醇 =200/1)得到 淡黄色固体 4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-胺(205-27) (407毫克, 收率: 89.92%)。 LCMS (ESI): m/z241[M + H 下列中间体 (205-8, 205-10) 可按上述合成方法 3-1 中的中间体 205-6的方法制备, 仅 是将其中的中间体 204-6替换为相应的中间体 204。
中间体 205-8的制备: 合成方法如实施例 3中方法 3-1中的中间体 205-6的制备, 仅是将 其中 1-(2-氯 -5-甲氧基嘧啶 -4-基) -1H-吲哚 (204-6) 替换为 1-(2-氯 -5-甲基嘧啶 -4-基) -1H-吲哚 (204-8),制备得到 4-(1Η-吲哚 -1-基) -5-甲基嘧啶 -2-胺 ( 4-(lH-indol-l-yl)-5-methylpyrimidin- 2-amine) (中间体 205-8)。 LCMS (ESI): m/z 225 [M + H ]+。
中间体 205-10的制备: 合成方法如实施例 3中方法 3-1中的中间体 205-6的制备, 仅是 将其中 1—(2-氯 -5-甲氧基嘧啶 -4-基) -1H-吲哚(204-10)替换为 2-氯 -4-(1Η-吲哚 -1-基) -Ν,Ν-二甲 基嘧啶 -5-胺 (204-10),制备得到 4-(1Η-吲哚 -1-基 )-Ν5,Ν5-二甲基嘧啶 -2,5-二胺 (4-(lH-indol-l-yl)-N5,N5-dimethylpyrimidine-2,5-diamine) (中间体 205-10)。 LCMS (ESI): m/z 254 [M + H]+。
下列中间体(205-29, 205-30, 205-31, 205-124)可按上述合成方法 3-2中的中间体 205-28 的方法制备, 仅是将其中中间体 201-28和 203-28替换为相应的中间体 201和 203。
中间体 205-29的制备: 合成方法如实施例 3中方法 3-2中的中间体 205-28的制备, 仅是 将其中加入 5-氟吲哚 (201-28) 替换为 6-氟-1 吲哚(201-29),将 2-氨基 -4-氯 -5-三氟甲基嘧啶 (203-5) 替换为 2-氨基 -4-氯嘧啶 (203-28) 制备得到 4-(6-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (4-(6-fluoro-lH-indol-l-yl)pyrimidin-2-amine) (中间体 205-29)。 LCMS (ESI): m/z229[M + H]+。
中间体 205-30的制备: 合成方法如实施例 3中方法 3-2中的中间体 205-28的制备, 仅是 将其中加入 5-氟吲哚 (201-28) 替换为 4-氟 -1H-吲哚 (201-30),将 2-氨基 -4-氯 -5-三氟甲基嘧啶 (203-5) 替换为 2-氨基 -4-氯嘧啶 (203-28) 制备得到 4-(4-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (4-(4-fluoro-lH-indol-l-yl)pyrimidin-2-amine) (中间体 205-30)。 LCMS (ESI): m/z229[M +
H]+。
中间体 205-31的制备: 合成方法如实施例 3中方法 3-2中的中间体 205-28的制备, 仅是 将其中加入 5-氟吲哚 (201-28)替换为 5-氯吲哚 (201-31),将 2-氨基 -4-氯 -5-三氟甲基嘧啶(203-5) 替换为 2-氨基 -4-氯嘧啶 ( 203-28 ) 制备得到. 4-(5-氯 -1H-吲哚 -1-基)嘧啶 -2-胺 (4-(5-chloro-lH-indol-l-yl)pyrimidin-2-amine) (中间体 205-31)。 LCMS (ESI): m/z245[M + H]+。
中间体 205-124的制备: 合成方法如实施例 3中方法 3-2中的中间体 205-20的制备, 仅 是将其中加入 5-甲氧基 -1H-吡咯 [3,2-b]吡啶 (201-20) 替换为 5-氯 -1H-吡咯 [3,2-b]吡啶 (201-124), 制备得到.(4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-胺 (中间体 205-124)。 LCMS (ESI): m/z246 [M + H]+。 下列中间体 (205-39) 可按上述合成方法 3-3中的中间体 205-34的方法制备, 仅是将其 中的中间体 201-34替换为相应的中间体 201。
中间体 205-39的制备: 合成方法如实施例 3中方法 3-3中的中间体 205-34的制备, 仅是 将其中 5-硝基 -1H-吲哚 (201-34)替换为 (1H-吲哚 -5-基)甲醇 (201-39),制备得到 (1-(2-氨基嘧啶 -4- 基) -1H-吲哚 -5-基)甲醇(l-(2-aminopyrimidin-4-yl)-lH-indol-5-yl)methanol) (中间体 205-39)。 LCMS (ESI): m/z241 [M + H ]+。 下列中间体 (205-37, 205-38) 可按上述合成方法 3-3中的中间体 205-36的方法制备, 仅是将其中中间体 201-36替换为相应的中间体 201。
中间体 205-37的制备: 合成方法如实施例 3中方法 3-3中中间体 205-36的制备, 仅是将 其中 5- ((叔丁基二苯基硅)氧基) -1H-吲哚 (201-36) 替换为 5-(2- ((叔丁基二苯基硅基)氧基)乙 氧基) -1H-吲哚 (201-37),制备得到 4-(5-(2- ((叔丁基二苯基硅基)氧基)乙氧基) -1H-吲哚 -1-基)嘧 啶 -2-胺 4-(5-(2-((tert-butyldiphenylsilyl)oxy)ethoxy)-lH-indol- 1 -yl)pyrimidin-2-amine (中间体 205-37)。 LCMS (ESI): m/z 509 [M + H]+。
中间体 205-38的制备: 合成方法如实施例 3中方法 3-3中中间体 205-36的方法制备, 仅 是将其中 5- ((叔丁基二苯基硅)氧基) -1H-吲哚 (201-36) 替换为 5-(2-甲氧乙氧基) -1H-吲哚 (201-38), 制 备 得 至 IJ 4-(5-(2- 甲 氧 乙 氧 基 )-1Η- 吲 哚 -1- 基 ) 嘧 啶 -2- 胺 (4-(5-(2-methoxyethoxy)-lH-indol-l-yl)pyrimidin-2-amine) (;中间体 205-38)。 LCMS (ESI): m/z 285 [M + H]+。 下列中间体 (205-33, 205-46) 可按上述合成方法 3-3中的中间体 205-27的方法制备, 仅是将其中的中间体 201-27替换为相应的中间体 201。
中间体 205-33的制备: 合成方法如实施例 3中方法 3-3中中间体 205-27的制备, 仅是将 其中 5-甲氧基吲哚 (201-27) 替换为 5-氰基吲哚 (201-33),制备得到 1-(2-胺基嘧啶 -4-基) -1H- 吲哚 -5-氰基( l-(2-aminopyrimidin-4-yl)-lH-indole-5-carbonitrile) (中间体 205-33)。LCMS (ESI): m/z236 [M + H]+。
中间体 205-46的制备: 合成方法如实施例 3中方法 3-3中中间体 205-27的制备, 仅是将
其中 5-甲氧基吲哚(201-27 )替换为 3-氯吲哚 C201-46),制备得到 N-4-O氯 -1H-吲哚 -1-基)嘧啶 -2-胺 4-(3-chloro-lH-indol-l-yl)pyrimidin-2-amine (中间体 205-46)。 LCMS (ESI): m/z245 [M + H ]+。 实施例 4: 中间体 403的制备 (按照方案四线路制备)
中间体 403可按三种方法制备, 方法 4-1, 方法 4-2和方法 4-3, 描述如下:
方法 4-1 (通过中间体 103和 204制备):
步骤 4-la : N-(4-氟 -2-甲氧基 -5-硝基苯基) -4-(1Η-吲哚 -1-基)嘧啶 -2-胺 ( N-(4-fluoro-2- methoxy-5-nitrophenyl)-4-(lH-indol-l-yl)pyrimidm-2-amine) (中间体 401-77)的制备: 在闷罐中, 化合物 1-(2-氯嘧啶 -4-基) -1H-吲哚(204-6 ) (500毫克, 2.18毫摩尔, 1当量),对甲苯磺酸 (450.47 毫克, 2.616毫摩尔, 1.2当量)和化合物 4-氟 -2-甲氧基 -5-硝基 -苯胺 (103 ) (405.5毫克, 2.18 毫摩尔, 1当量)的叔丁醇 (15毫升)溶液混合体系在 110°C下搅拌反应 16小时。 反应冷却至室 温,减压浓缩,得到黄色固体。黄色固体用 2N氢氧化钠水溶液和石油醚 /乙酸乙酯 10/1洗涤, 减压浓缩,得到黄色固体 N-(4-氟 -2-甲氧基 -5-硝基苯基; HK1H-吲哚 -1-基)嘧啶 -2-胺 (570毫克, 收率: 68.88%)。 LCMS (ESI): m/z 380 [M + H ]+。
步骤 4-lb : N-(4-(3-二甲胺基吖啶 -1-基) -2-甲氧基 -5-硝基苯基) -(4-(1Η-吲哚 -1-基)嘧啶 -2- 胺 (N-(4-(3-(dimethylamino)azetidin-l-yl)-2-methoxy-5-nitrophenyl)-4-(lH-indol-l-yl)pyrimidin-2 -amine) (中间体 402-C-77)的制备: 将化合物 N-(4-氟 -2-甲氧基 -5-硝基苯基) -4-(1Η-吲哚 -1-基) 嘧啶 -2-胺 (401-6 ) (150毫克, 0.4毫摩尔, 1.0当量)溶解于 30毫升四氢呋喃中。 然后分别加 入 Ν,Ν-二异丙基乙胺 (153毫克, 1.2毫摩尔, 3.0当量)和 3-二甲胺基 -氮杂环丁垸盐酸盐 (81 毫克, 0.6毫摩尔, 1.5当量), 加热至 80°C下搅拌反应 5小时。 反应结束后, 冷却至室温, 用 乙酸乙酯和水萃取, 有机相用水洗, 盐水洗, 无水硫酸钠干燥, 减压浓缩, 最后硅胶柱层析 纯化 (二氯甲垸 /甲醇 =150/ 100/1), 得到红色固体 ,N-(4-(3-二甲胺基吖啶 -1 -基) -2-甲氧基 -5- 硝基苯基) -(4-(1Η-吲哚 -1-基)嘧啶 -2-胺 (402-C-77 ) (170 毫克, 收率: 93%)。 LCMS (ESI): m々460[M + H ]+。
步骤 4-l c: -(4-(111-吲哚 -1-基)嘧啶 -2-基) -4-(3-二甲胺基吖啶 -1-基) -6-甲氧基苯 -1,3-二胺 Ni- -GH-indol-l-y pyrimidin-S-yl -p^dimethylamino^zetidin-l-y -S-methoxybenzene-l -di amine (中间体 403-C-77)的制备: 将 N-(4-(3-二甲胺基吖啶 -1-基) -2-甲氧基 -5-硝基苯 基) -(4-(1Η-吲哚 -1-基)嘧啶 -2-胺(402-C-77 ) (170毫克, 0.37毫摩尔, 1.0当量) 和氯化铵 (65 毫克, 1.2 毫摩尔, 8.0当量) 混合于 30毫升乙醇和 7毫升水中, 加热至 50°C, 然后加入还 原铁粉 (165毫克, 2.96毫摩尔, 8.0当量)。 加热至 80°C, 反应 2小时。 反应完成后, 过滤, 将滤液浓缩, 再用二氯甲垸萃取, 有机相用无水硫酸钠干燥, 减压旋干, 得到产品 -(4-(1 吲哚 -1-基)嘧啶 -2-基) -4-(3-二甲胺基吖啶 -1-基) -6-甲氧基苯 -1,3-二胺 (179毫克,粗品)。 LCMS (ESI): m々430[M + H ]+。
方法 4-2 (通过中间体 105和 205制备):
步骤 4-2a: -(4-(111-吲哚 -1-基) - 5-三氟甲基嘧啶 -2-基) -N4-(2-二甲胺乙基) -2-甲氧基 -N4- 甲基 -5-硝基苯 -1,4-二胺 ( ^-(^(lH-indol-l-yl S- rifluoromethyDpyrimidm -yi N4-^- (dimethylamino)ethyl)-2-methoxy-N4-methyl-5-nitrobenzene-l,4-diamine ) (中 I司体 402-A-5 ) 的
制备: 氮气保护下, 将 4-(1Η-吲哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) (265毫克, 0.95毫 摩尔, 1.2当量), ^-(4-溴 -5-甲氧基 -2-硝基苯基) -N^N^N2-三甲基乙焼 - 1 ,2-二胺( 105-A- 1 ) ( 266 毫克, 0.80毫摩尔, 1.0当量), 碳酸铯 (782毫克, 2.4毫摩尔, 3.0当量), 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (46毫克, 0.08毫摩尔, 0.1当量)和三 (二亚苄基丙酮)二钯 (46毫克, 0.04 毫摩尔, 0.05当量)加入到 35毫升甲苯中, 回流反应 6个小时。 冷却至室温, 过滤, 所得滤 液减压浓缩, 经硅胶柱层析纯化 (洗脱剂: 二氯甲垸 /甲醇 50/1 至 10/1 ), 得到红色固体 -(4-(111-吲哚 -1-基) - 5-三氟甲基嘧啶 -2-基) -N4-(2-二甲胺乙基 )-2-甲氧基 -N4-甲基 -5-硝基苯 -1,4-二胺(152毫克, 收率: 36%)。 LCMS (ESI): m々530[M + H ]+。
步骤 4-2b: ^^ΙΗ-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1-^-二甲胺乙基) -5-甲氧基 -N1- 甲 基 苯 -1,2,4- 三 胺 ( ^^-(^(lH-indol-l-yO-S- rifluoromethyDpyrimidm -yi N1-^- (dimethylamino)ethyl)-5-methoxy-N1-methylbenzene-l,2,4-tri amine) (中间体 403-A-5 ) 的制备: 将化合物 -(4-(111-吲哚 -1-基) - 5-三氟甲基嘧啶 -2-基) -N4-(2-二甲胺乙基) -2-甲氧基 -N4-甲基 -5-硝基苯 -1,4-二胺 (402-5 ) ( 140毫克, 0.26毫摩尔, 1.0当量) 和氯化铵 ( 114毫克, 2.12 毫摩尔, 8.0当量)加入到 30毫升乙醇和 8毫升水的混合液中, 加热至 50 °C, 分批加入还原 性铁粉 (127毫克, 2.26毫摩尔, 8.5当量), 加热至回流, 反应 1个小时。 反应冷却至室温, 减压浓缩,用二氯甲垸萃取,有机相用无水硫酸钠干燥,减压浓缩,得到棕色油状物 Ν4-(4-(1Η- 口引噪—μ基)— 5-三氟甲基嘧啶 -2-基) -Ν1-^-二甲胺乙基) -5-甲氧基 -Ν1-甲基苯 -1,2,4-三胺 (130毫 克, 收率: 98%)。 LCMS (ESI): m々500[M + H ]+。
方法 4-3 (通过中间体 204和 106制备):
步骤 4-3a: N2-{4-{[2- (二甲基氨基)乙基] (甲基)氨基 }-2-甲氧基 -5-硝基苯 }-4-(1Η-吲哚 -1- 基)-N5-甲基嘧啶 -2,5-二胺 ( N2-(4-((2-(dimethylammo)ethyl)(methyl)ammo)-2-methoxy- 5-nitrophenyl)-4-(lH-indol-l-yl)-N5-methylpyrimidine-2,5-diamine) (中间体 402-A-11 ) 的制备: 将 2-氯 -4-(1Η-吲哚 -1-基) -N-甲基嘧啶 -5-胺(204-11 ) (268毫克, 1.036毫摩尔)溶解于甲苯(40 毫升)中,依次加入 (二甲基氨基;)乙基] -5-甲氧基 -N1-甲基 -2-硝基苯 -1,4-二胺(106-A-11 ) (278毫克, 1.036毫摩尔, 1.0当量)、 三 (二亚苄基丙酮)二钯 (94.8毫克, 0.1036毫摩尔, 0.1 当量)、碳酸铯(675毫克, 2.072毫摩尔, 2.0当量)和 4,5-双 (二苯基膦) -9,9-二甲基氧杂蒽(60 毫克, 0.1036毫摩尔, 0.1当量), 然后在氮气保护下搅拌, 并在 100°C下搅拌 4个小时。 混合 液冷却至室温, 浓缩, 然后通过柱层析分离提纯 (二氯甲垸 /甲醇 =40: 1 ), 得到黄色固体 N2-{4-{[2- (二甲基氨基)乙基] (甲基)氨基 }-2-甲氧基 -5-硝基苯 }-4-(1Η-吲哚 -1-基) -N5-甲基嘧啶 -2,5-二胺 (263毫克, 收率: 51.8%)。 LCMS (ESI): m/z491 [M + H ]+。
步骤 4-3b: Ν4-[4-(1Η-吲哚 -1-基) -5- (甲基氨基)嘧啶 -2-基] -N1-^- (二甲基氨基) 乙基] -5-甲 氧基 -N1- 甲基苯 -1,2,4-三胺 ( Nt -GH-indol-l-yO-S- methylammo^yrimidmJ-yl N1- (2-(dimethylamino)ethyl)-5-methoxy-N1-methylbenzene-l,2,4-tri amine ) (中间体 403- A- 11) 的制 备: 将 N2-{4-{[2- (二甲基氨基)乙基] (甲基)氨基 }-2-甲氧基 -5-硝基苯 }-4-(1Η-吲哚 -1-基) -N5-甲 基嘧啶 -2,5-二胺(402-A-11 ) (260毫克, 0.537 毫摩尔, 1.0当量)溶解在甲醇(40 毫升) 中, 加入锌粉 (279毫克, 4.297 毫摩尔, 8.0当量)和氯化铵 (230毫克, 4.297毫摩尔, 8.0当量), 然后混合物在 60°C搅拌反应四个小时, 冷却到室温, 加入二氯甲垸稀释, 过滤, 滤液用水和 饱和食盐水洗涤。 有机相用无水硫酸钠干燥, 浓缩得到黄色固体 Ν4-[4-(1Η-吲哚 -1-基;) -5- (甲
基氨基;)嘧啶 -2-基] -N1-^- (二甲基氨基) 乙基] -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (226 毫克, 粗 品)。 LCMS (ESI): m/z461 [M + H ]+。
下列中间体 ( 403-C-78 , 403-C-79, 403-B-80, 403-B-81 , 403-B-82, 403-C-84 ) 可按上 述合成方法 4-1制备。
中间体 403-C-78的制备: 合成方法如实施例 4方法 4-1中 403-C-77的合成, 仅是将其中 化合物 3-二甲胺基 -氮杂环丁垸盐酸盐替换为 (S)-N,N-二甲胺基四氢吡咯 -3-胺,制备得到 (S N^ - IH-吲哚 -1-基)嘧啶 -2-基) -4-(3- (二甲胺基)四氢吡咯 -1-基)- 6-甲氧基苯 -1, 3-二胺 ((S iS^- -GH-indol-l-y pyrimidin-S-yl -p-Wimethylamino^yrrolidin-l-yi S-methoxybenzen e-l,3-diamine)(403-C-78)。 LCMS (ESI): m/z444[M + H ]+。
中间体 403-C-79的制备: 合成方法如实施例 4方法 4-1中 403-C-77的合成, 仅是将其中 化合物 3-二甲胺基 -氮杂环丁垸盐酸盐替换为 (R)-N,N-二甲胺基四氢吡咯 -3-胺,制备得到 ( -^-(4-(111-吲哚 -1-基)嘧啶 -2-基) -4-(3- (二甲胺基)四氢吡咯 -1-基)- 6-甲氧基苯 -1, 3-二胺 ( (R)-Nl -(4-( lH-indol- 1 -yl)pyrimidin-2-yl)-4-(3 -(dimethylamino)pyrrolidin- 1 -yl)-6-methoxybenz ene-l,3-diamine) (化合物 403-C-79)。 LCMS (ESI): m/z444[M + H ]+。
中间体 403-B-80的制备: 合成方法如实施例 4中方法 4-1。 将 N-(4-氟 -2-甲氧基 -5-硝基 苯) -4-(1Η-吲哚 -1-基)嘧啶 -2-胺 (401-6) (250毫克, 0.659毫摩尔, 1.0当量) 溶解在乙醇 (30 毫升) 中, 加入 N, N-二异丙基乙胺 (255毫克, 1.978毫摩尔, 1.0当量) 和 N-甲基 -2- (吡咯 垸 -1-基;)乙胺 (380毫克, 粗品), 然后混合液在封管中加热到 100°C反应过夜。 反应结束后, 冷却到室温, 加入乙酸乙酯稀释。 有机相用水和饱和食盐水洗涤, 用无水硫酸钠干燥, 浓缩 并通过柱层析(洗脱剂: 二氯甲焼 /甲醇 60/1至 50/1 )纯化, 得到黄色固体 -[4-(111-吲哚 -1- 基)嘧啶 -2-基] -2-甲氧基 -N4-甲基 -5-硝基 -N4-[2- (吡咯垸 -1-基)乙基]苯基 -1,4-二胺 (402-B-80 ) (257毫克, 收率: 80.0%)。 LCMS (ESI): m/z 488 [M + H ]+。再将 -[4-(111-吲哚 -1-基)嘧啶 -2- 基] -2-甲氧基 -N4-甲基 -5-硝基 -N4-[2- (吡咯垸 -1-基)乙基]苯基 -1,4-二胺 (402-B-80) (257 毫克, 0.527毫摩尔, 1.0当量) 溶解在甲醇 (40毫升) 中, 加入锌粉 (274毫克, 4.216毫摩尔, 8.0 当量)和氯化铵(225毫克, 4.216毫摩尔, 8.0当量), 然后混合物在 60°C搅拌反应四个小时, 冷却到室温, 加入二氯甲垸稀释, 过滤, 滤液用水和饱和食盐水洗涤。 有机相用无水硫酸钠 干燥,浓缩得到黄色固体 Ν4-[4-(1Η-吲哚 -1-基) 嘧啶 -2-基] -5-甲氧基 -N1-甲基 -N1-^- (吡咯垸 -1- 基 ) 乙 基 ] 苯 基 -1,2,4-三 胺 (N4-(4-(lH-indol-l-yl)pyrimidin-2-yl)-5-methoxy-N1-methyl- N^i -Cpyrrolidin- 1 -yl)ethyl)benzene- 1 ,2,4-triamine) (403-B-80) ( 169 毫克,粗品)。 LCMS (ESI): m/z 458 [M + H ]+。
中间体 403-B-81的制备: 合成方法如实施例 4方法 4-1中 403-B-80的合成, 仅是将其中 化合物 N-甲基 -2- (吡咯垸 -1-基)乙胺替换为 N-甲基 -2- (哌啶 -1-基)乙垸 -1-胺,制备得到 Ν 4-[4-(1Η-吲哚 -1-基)嘧啶 -2-基] -5-甲氧基 -N1-甲基 -N1-^- (哌啶 -1-基)乙基]苯 -1,2,4-三胺 ( ^-(^(lH-indol-l-yDpyrimidin-S-y -S-methoxy-Ni-methyl-Ni-p piperidin-l-y ethy benzene -1,2,4-triamine) (中间体 403-B-81)。 LCMS (ESI): m/z472 [M + H ]+。
中间体 403-B-82的制备: 合成方法如实施例 4方法 4-1中 403-B-80的合成, 仅是将其中 化合物 N-甲基 -2- (吡咯垸小基)乙胺替换为甲基 -(2-吗啉 -4-基-乙基) -胺,制备得到 N4-(4-吲哚 -1- 基 -嘧啶 -2-基)-5-甲氧基 -N1- 甲基 -^-(2-吗啉 -4-基 -乙基) -苯基 -1,2,4-三胺
(N -(4-Indol-l-yl-pyrimidin-2-yl)-5-methoxy-N -methyl-N -(2-morpholin-4-yl-ethyl)-benzene- 1 ,2,
4- triamine) (中间体 403-B-82)。 LCMS (ESI): m々474[M + H ]+。
中间体 403-C-84的制备: 合成方法如实施例 4方法 4-1中 403-C-77的合成, 仅是将其中 化合物 3-二甲胺基 -氮杂环丁垸盐酸盐替换为 N-甲基哌嗪,制备得到 N^WIH-吲哚 -1-基)嘧啶 -2-基)-6-甲氧基 -4-(4-甲基哌嗪 -1-基)苯 -1,3-二胺(^-(^(lH-indol-l-yDpyrimidm -yl)^- methoxy-4-(4-methylpiperazin-l-yl)benzene-l,3-diamine ) (;中间体 403-C-79)。 LCMS (ESI): m/z444 [M + H ]+。 下列中间体 ( 403-A-6, 403-A-8 , 403-A-10 , 402-A-20, 403-A-27, 403-A-28 , 403-A-29, 403-A-30, 403-A-31 , 403-Α-33 , 403-Α-34, 403-Α-36, 403-Α-37, 403-Α-38 , 403-Α-39, 403-A-41 , 403-Α-46, 403-A-124 )可按上述合成方法 4-2制备, 仅是将其中的中间体 205-5替换为相应的 中间体 205。
中间体 403-Α-6的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲哚 小基)—5—三氟甲基嘧啶—2—胺 (205-5 ) 替换为 4-(1Η-吲哚 -1-基) -5-甲氧基嘧啶 -2-胺 (205-6 ) , 制备得到 Ν4-(4-(1Η-吲哚 -1-基) -5-甲氧基嘧啶 -2-基) -Ν1^-二甲胺乙基) -5-甲氧基 -Ν1-甲基苯 -1,2,4- 三 胺 ( N4-(4-(lH-indol-l-yl)-5-methoxypyrimidin-2-yl)-N1-(2-(dimethylamino)ethyl)-
5- methoxy-N methylbenzene-lA^triamine) (中间体 403-A-6)。 LCMS (ESI): m/z 462 [M + H ]+。
中间体 403-A-8的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲哚 -1-S) - 5-三氟甲基嘧啶 -2-胺 ( 205-5 ) 替换为 4-(1Η-吲哚 -1-基) -5-甲基嘧啶 -2-胺 ( 205-8 ) , 制备得到 Ν4-(4-(1Η-吲哚 -1-基) -5-甲基嘧啶 -2-基) -Nl-(2-二甲胺乙基 )-5-甲氧基 -N1-甲基苯 -1,2,4- 三 胺 N4-(4-(lH- indol-l-yl)-5-methylpyrimidin-2-yl)-Nl-(2-(dimethylamino)ethyl)-5- methoxy-Nl -methylbenzene- 1 ,2,4-tri amine (中间体 403-A-8)。 LCMS (ESI): m/z 446 [M + H ]+。
中间体 403-A-10的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1—基)—5-三氟甲基嘧啶 -2-胺(205-5 )4-(1Η-吲哚 -1-基) -N5,N5-二甲基嘧啶 -2,5-二胺(205-10 ), 制备得到 N4-[5- (二甲基氨基 )-4-(1Η-吲哚 -1-基) 嘧啶 -2-基] -N1-^- (二甲基氨基) 乙基] -5-甲氧 基 -N1-甲基苯 -1,2,4-三胺 ( N s^dimethylamino ^GH-indol-l-yDpyrimidin -yD-N p- (dimethylamino)ethyl)-5-methoxy-N1-methylbenzene-l,2,4-tri amine (中间体 403-A-10))。 LCMS (ESI): m々475[M + H ]+。
中间体 403-A-20的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5-甲氧基 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2- 胺 ( 205-20 ) ,^-(2-二甲胺乙基) -5-甲氧基 -N4-(4-(5-甲氧基 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2- 基)- N1-甲基 -2-硝基苯 -1,4-二胺 (N^p-Wimethylamim^ethyl S-methoxy-Nt - -methoxy- lH-pyrrolo[3,2-b]pyridin-l-yl)pyrimidin-2-yl)-Nl-methyl-2-nitrobenzene-l,4-diamine ) (中 I司体 403-A-20)。 LCMS (ESI): m/z 493 [M + H ]+。 中间体 403-A-27的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺(205-5 )替换为 4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-胺(205-27 ) , 制备得到 N1-^-二甲胺乙基) -5-甲氧基 -N4-(4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-基)— N1-甲基苯
- 1 ,2,4-三 I¾ (N -(2-(dimethylamino)ethyl)-5-methoxy-N -(4-(5-methoxy-lH-indol-l - yl)pyrimidin- - y -N^methylbenzene- 1 ,2,4-triamine) (中间体 403-A-27)。 LCMS (ESI): m/z 462[M + H ]+。
中间体 403-A-28的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (205-28 ) , 制备得到 ^-(2-二甲胺乙基) -N4-(4-(5-氟 -1H-吲唑小基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4- 三 胺 ( N1-(2-(dimethylamino)ethyl)-N4-(4 5-fluoro-lH-indol-l-yl)pyrimidin-2-yl)-5-methoxy- N^methylbenzene- 1 ,2,4-triamine ) (中间体 403-A-28)。 LCMS (ESI): m/z 450[M + H ]+。
中间体 403-A-29的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(6-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (205-29) , 制备得到 ^-(2-二甲胺乙基) -N4-(4-(6-氟 -1H-吲唑小基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4- 三胺 ( N1-(2-(dimethylamino)ethyl)-N4-(4-(6-fluoro-lH-indol-l-yl)pyrimidin-2-yl)-5-methoxy-N1 -methylbenzene- 1 ,2,4-triamine ) (中间体 403-A-29)。 LCMS (ESI): m/z 450[M + H ]+。
中间体 403-A-30的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(4-氟 -1H-吲哚 -1-基)嘧啶 -2-胺 (205-30) , 制备得到 N1^-二甲胺乙基) -N4-(4-(4-氟 -1H-吲唑小基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4- 三胺 ( N1-(2 dimethylamino)ethyl)-N4-(4-(4-fluoro-lH-indol-l-yl)pyrimidin-2-yl)-5-methoxy-N1- methylbenzene- 1 ,2,4-triamine ) (中间体 403-A-30)。 LCMS (ESI): m/z 450 [M + H ]+。
中间体 403-A-31的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5-氯 -1H-吲哚 -1-基)嘧啶 -2-胺 (205-31 ) , 制备得到 N4-(4-(5-氯 -1H-吲唑 -1-基)嘧啶 -2-基) -^-(2-二甲胺乙基)- 5-甲氧基 -N1-甲基苯 -1, 2,4-三胺 (N4-(4-(5-chloro-lH-indol-l-yl)pyrimidin-2-yl)-N1-(2-(dimethylamino)ethyl)-5-methoxy- N^methylbenzene- 1 ,2,4-triamine ) (中间体 403-A-31)。 LCMS (ESI): 466[M + H ]+。
中间体 403-A-33的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚小基)- 5—三氟甲基嘧啶—2—胺(205—5 )替换为 1-(2-胺基嘧啶 -4-基) -1H-吲哚 -5-氰基(205-33 ) , 制备得到 1-(2-((5-胺基 -4-((2- (二甲胺基)乙基) (甲基)胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基) -1H- 口引噪 -5-氰基 (l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino) pyrimidin-4-yl)-lH-indole-5-carbonitrile) (中间体 403-A-33)。 LCMS (ESI): m/z 457[M + H ]+。
中间体 403-A-34的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚小基)- 5—三氟甲基嘧啶—2—胺 (205-5 ) 替换为 4-(5-硝基 -吲哚 -1-基) -嘧啶 -2-氨基 (205-34) , 制备得到 N1-^-二甲氨基-乙基) -5-甲氧基 -N1-甲基 -N4-[4-(5-硝基 -吲哚 -1-基) -嘧啶 -2-基] -苯基 -1,2,4- 三 胺 ( N1-(2-dimethylamino-ethyl)-5-methoxy-N1-methyl-N4-[4-(5-nitro-indol-l-yl)- pyrimidin-2-yl]-benzene- 1 ,2,4-triamine ) (中间体 403-A-34)。 LCMS (ESI): m/z 477 [M + H ]+。
中间体 403-A-36的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚小基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5- ((叔丁基二苯基硅)氧基) -1H-吲哚小基) 嘧啶 -2-胺 (205-36) ,制备得到 1-(2-((5-胺基 -4-((2-二甲胺乙基) (甲基) 胺基) -2-甲氧基苯基) 胺基)嘧啶 -4-基)-1Η-吲哚 -5-醇 ( l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-
2- methoxyphenyl)amino)pyrimidin-4-yl)-lH-indol-5-ol ) (;中间体 403-A-36)。 LCMS (ESI): m/z 448[M + H ]+。
中间体 403-A-37的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚- i -基) - 5-三氟甲基嘧啶 -2-胺(205-5 )替换为 4-(5-(2- ((叔丁基二苯基硅基)氧基)乙氧基 )-1Η- 吲哚 -1-基)嘧啶 -2-胺 (205-37) ,制备得到 N4-4-(5-(2- ((叔丁基二苯基硅基)氧基)乙氧基) -1H-吲 哚 -1-基)嘧啶 -2-基 (2-((2-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (Nt - -p-i tert-butyldiphenylsilyDoxy thoxy lH-indol-l-yDpyrimidin-S-yl N p^dimethyla mino)ethyl)-5-methoxy-N1-methylbenzene-l,2,4-triamine)(中间体 403-A-37)。 LCMS (ESI): m々730[M + H ]+。
中间体 403-A-38的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚- i -基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5-(2-甲氧乙氧基) -1H-吲哚 -1-基)嘧啶 -2-胺
(205-38) ,制备得到 N1- (2-((2-二甲胺乙基) -5-甲氧基 -N4 -4-(5-(2-甲氧乙氧基) -1H-吲哚 -1-基) 嘧啶 -2-基) -N1-甲基苯 -1, 2,4-三胺 ( N p-Wimethylammo^hyO-S-methoxy-N4^^- (2-methoxyethoxy)-lH-indol-l-yl)pyrimidin-2-yl)-N1-methylbenzene-l,2,4-tri amine ) (中 间体 403-A-38 LCMS (ESI): m/z730[M + H ]+。
中间体 403-A-39的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚- i -基) - 5-三氟甲基嘧啶 -2-胺 ( 205-5 ) 替换为 (1-(2-氨基嘧啶 -4-基) -1H-吲哚 -5-基)甲醇
(205-39) ,制备得到 (1-(2-((5-胺基 -4-((2-二甲胺乙基)(甲基) 胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基 )-1Η- 吲 哚 -5-基) 甲 醇 (l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxyphenyl)amino)pyrimidin-4-yl)-lH-indol-5-yl)methanol (中间体 403-A-39)。LCMS (ESI): m/z462[M + H
中间体 403-A-41的制备:将化合物 1-(2-胺基嘧啶-4-基)-111-吲哚-5-氰基(205-33 )(120 毫 克, 0.51 毫摩尔, 1.2当量),化合物 N1-^溴 -5-甲氧基 -2-硝基苯基) -Ν^ί^,Ν2-三甲基乙垸 -1,2- 二胺 (105-Α-1)(139.4 毫克, 0.42 毫摩尔, 1当量),碳酸铯 (273 毫克, 0.84 毫摩尔, 2当量), 三 (二亚苄基丙酮)二钯 (19 毫克, 0.021 毫摩尔, 0.05当量)和 4,5-双二苯基膦 -9,9-二甲基氧 杂蒽 (21.85 毫克, 0.038 毫摩尔, 0.09当量)溶于 20毫升甲苯,氮气保护条件下,升温到 110°C 反应 6小时。 冷却到室温, 旋干溶剂, 残留物硅胶柱层析 (二氯甲焼 /甲醇 =100/1)纯化得到红 色固体 1-(2-((4-((2- (二甲胺基)乙基; 甲基)胺基) -2-甲氧基 -5-硝基苯基)胺基)嘧啶 -4-基) -1H-吲 哚 -5-氰基 (402-A-33 ) (200 毫克, 收率: 98.03%)。 LCMS (ESI): m/z 487 [M + H ]+。 将上述 得到的化合物(500毫克, 1.03毫摩尔, 1当量)溶解到乙醇(10毫升)二甲基亚砜(1毫升) 的混合溶剂中, 加入氢氧化钠 (1毫升, 1M水溶液), 冰浴下搅拌均匀, 缓慢滴加双氧水 (1 毫升, 30%水溶液), 冰浴下继续搅拌 5分钟, 用饱和亚硫酸钠水溶液将反应淬灭, 用乙酸乙 酯萃取后, 通过硅胶柱层析分离提纯得到 1-(2-((4-((2- (二甲胺基)乙基; 甲基)胺基; )-2-甲氧基 -5-硝基苯基)胺基)嘧啶 -4-基) -1H-吲哚 -5-甲酰胺(402-A-41 ) (417毫克, 0.827毫摩尔, 收率: 80.3%) LCMS (ESI): m々 505[M + H ]+。 将上述得到的化合物 (417毫克, 0.827毫摩尔, 1当 量) 溶解到乙醇 (30毫升) 氯化铵水溶液 (3毫升) 的混合溶剂中, 加热到 60°C, 加入还原 铁粉 (185毫克, 3.0毫摩尔, 4当量) 继续搅拌 2小时后过滤, 用二氯甲垸萃取旋干得到的 粗品通过硅胶柱层析分离提纯得到 1-(2-((5-胺基 -4-((2- (二甲胺基)乙基; 甲基)胺基; )-2-甲氧基 苯基)胺基)嘧啶 -4-基;) -1H-吲哚 -5-甲酰胺
( l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-
4-yl)-lH-indole-5-carboxamide) ( 403-A-41 ) ( 377毫克, 0.794毫摩尔,收率: 96.0% )LCMS (ESI): m/z [M + H ]+。
中间体 403-A-46的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η-吲 哚 -1-基) - 5-三氟甲基嘧啶 -2-胺(205-5 )替换为 N-4-(3-氯 -1H-吲哚 -1-基)嘧啶 -2-胺(205-46 ) , 制备得到 N4-(4-(3-氯 -1H-吲哚 -1-基)嘧啶 -2-基) -N1-^-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4- 三 胺 (IS^KS-chloro-lH-indol-l-y pyrimidin-S-y -NiKdimethylamino^thy -S-methoxy-Ni-methy lbenzene-l,2,4-triamine) (中间体 403-A-46)。 LCMS (ESI): m/z 466[M + H ]+。
中间体 403-A-124的制备: 合成方法如实施例 4中方法 4-2, 仅是将其中化合物 4-(1Η- 吲哚 -1-基) - 5-三氟甲基嘧啶 -2-胺 (205-5 ) 替换为 4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-胺 ( 205-124 ) ,制备得到 N4-(4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-基) -N1-^-二甲胺乙基) -5- 甲氧基 -N1-甲基苯 -1,2,4-三胺 (Nt - -chloro-lH-pyrrolol^^-Wpyridm-l-yDpyrimidm -yl N1- (2-(dimethylamino)ethyl)-5-methoxy-N1-methylbenzene-l,2,4-tri amine) (中间体 403-A-124)。 LCMS (ESI): m/z 467 [M + H ]+。 实施例 5: 中间体 506的制备 (按照方案五线路制备)
中间体 506可按三种方法制备, 方法 5-1, 方法 5-2和方法 5-3, 描述如下:
方法 5-1: 通过中间体 503和 504制备。
方法 5-2 (通过中间体 503和 106制备):
步骤 5-2a: 6-氯 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同(6-chloro-l -methyl- lH-benzo[d]imidazol- 2(3H)-one) (中间体 502-51)的制备:将 5-氯 -N1-甲基苯 -1,2-二胺 ( 501-51 ) (3.3克, 21毫摩尔,
1.0当量)溶解于 150毫升二氯甲垸中,加入 4.4毫升三乙胺 (31.5毫摩尔, 1.5当量),冷却至 0°C。 在氮气保护下, 缓慢滴加溶于 30毫升二氯甲垸的三光气溶液(2.6克, 8.4毫摩尔, 0.4当量), 反应半小时。 将反应液用碳酸钠溶液调节 pH为 7~8, 有固体析出, 过滤, 干燥, 即得到产品 得到产品 6-氯 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (2.65克,收率: 69%)。 LCMS (ESI): m/z 183[M + H ]+。
步骤 5-2b : 5-氯 -l-(2-氯嘧啶 -4-基)- 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 ( S-chloro-l- p-chloropyrimidin^-yl S-methyl-lH-benzol^imidazol pH one) (;中间体 503-51的制备): 首 先将 6-氯 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 ( 502-51 ) ( 500 毫克, 2.7毫摩尔, 1.0当量)和碳 酸铯 (670毫克, 4.05毫摩尔, 1.5当量)混合于 70毫升 Ν,Ν-二甲基甲酰胺中。在氮气保护下, 加入 2,4-二氯嘧啶 (410毫克, 2.7 毫摩尔, 1.0当量) , 室温搅拌反应 2小时。 反应完成后, 加入大量的水, 有固体析出, 过滤, 干燥, 即得到产品 5-氯 -1-(2-氯嘧啶 -4-基) - 3-甲基 -1Η-苯 并 [d]咪唑 -2(3Η)-酮 (410 毫克, 收率: 50%)。 LCMS (ESI): m/z 295 [Μ + Η ]+。
步骤 5-2c: 5-氯 -1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) - 3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 5-chloro-l-(2-(4-((2-(dimethylamino)ethyl)(methyl) amino)- 2-methoxy-5-nitrophenylamino)pyrimidin-4-yl)-3-methyl-lH-benzo[d]imidazol-2(3H)-one ) (中间 体 505-A-51)的制备:将 5-氯 -1-(2-氯嘧啶 -4-基)- 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同(503-51 ) (240 毫克, 0.81 毫摩尔, 1.0 当量), N1-^-二甲胺乙基) -5-甲氧基 -N1-甲基 -2-硝基苯 -1,4-二胺
( 106-A-l l ) (216毫克, 0.81毫摩尔, 1.0当量), 碳酸铯 (528毫克, 1.62毫摩尔, 2.0当量), 三 (二亚苄基丙酮)二钯 (40毫克, 0.04 毫摩尔, 0.05当量), 4,5-双二苯基膦 -9,9-二甲基氧杂蒽 (48毫克, 0.08毫摩尔, 0.1当量) 混合于 50毫升甲苯中。 在氮气保护下, 将反应置于预先加 热好的 110°C油浴中, 加热, 搅拌反应 3小时。 减压浓缩, 最后通过硅胶柱层析纯化 (二氯 甲焼 /甲醇 =300/1 150/1 ) 得到产品 5-氯 -1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基 苯胺基)嘧啶 -4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (150毫克, 收率: 35%)。 LCMS (ESI): m/z 527[M + H ]+。
步骤 5-2d: l-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氯 -3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( l-(2-(5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2- methoxyphenylamino)pyrimidin-4-yl)-5-chloro-3-methyl-lH-benzo[d]imidazol-2(3H)-one ) (中间 体 506-A-51)的制备:将 5-氯 -1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 505-A-51 ) (150毫克, 0.28毫摩尔, 1.0当量)和氯 化铵 (126毫克, 2.38毫摩尔, 8.5当量) 混合于 30毫升乙醇和 7毫升水中, 加热至 50°C。 然后加入还原铁粉 (125毫克, 2.24毫摩尔, 8.0当量), 加热至 80°C, 反应 2小时。 反应完成 后, 过滤, 将滤液浓缩, 再用二氯甲垸萃取, 有机相用无水硫酸钠干燥, 减压旋干, 最后通 过硅胶柱层析纯化 (二氯甲焼 /甲醇 =150/1 50/1 ) 得到产品 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲 基;)胺基; )-2-甲氧基苯胺基)嘧啶 -4-基;) -5-氯 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (140毫克, 收率: 99%) MS (ESI): m/z497[M + H ]+。 下列中间体 ( 506-A-52, 506-A-53 , 506-A-57, 506-A-99, 506-A-133, 506-A-135 ) 可按方 法 5-2制备。 仅是将其中中间体 503-51替换为相应的中间体 503。
中间体 506-A-52的制备: 合成方法如实施例 5中方法 5-2, 仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基)— 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (503-51 ) 替换为 1-(2-氯嘧啶 -4-基) -3-甲基 -2- 氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -5-甲腈(503-52 ),制备得到 1-(2-((5-胺基 -4-((2-二甲胺乙基 )(甲 基)胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基) -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -5-甲腈 ( l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin- 4-yl)-3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazole-5-carbonitrile) (中间体 506-A-52)。LCMS (ESI): m々488[M + H ]+。
中间体 506-A-53的制备: 合成方法如实施例 5中方法 5-2, 仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮(503-51 )替换为 1-(2-氯嘧啶 -4-基) -5-甲氧基 -3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (503-53 ) ,制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺 基)-2-甲氧基苯胺基)嘧啶 -4-基)-5-甲氧基 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4- yl)-5-methoxy-3-methyl-lH-benzo[d]imidazol-2(3H)-one) (;中间体 506-A-53)。 LCMS (ESI): m/z 493 [M + H ]+。
中间体 506-A-57的制备: 合成方法如实施例 5中方法 5-2, 仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮(503-51 )替换为 1-(2-氯 -5-甲氧基嘧啶 -4-基) -3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (503-57 ) ,制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺
基) -2-甲氧基苯胺基 )-5-甲氧基嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (l-(2-((5-amino-4- ((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)-5-methoxypyrimidin-4-yl)-3- methyl-lH-benzo[d]imidazol-2(3H)-one) (中间体 506-A-57)。 LCMS (ESI): m/z 493 [M + H ]+。
中间体 506-A-99的制备: 合成方法如实施例 5中方法 5-2, 仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基)— 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (503-51 ) 替换为 5-氯 -1-(2-氯嘧啶 -4-基) -3-环 丙基 -1,3-二氢 -2H-苯唑 [d]咪唑 -2-酮 (503-99), 制备得到 1-(2-((5-氨基 -4-((2- (二甲氨基)乙基) 甲基)氨基) -2-甲氧基苯基)氨基)嘧啶 -4-基) -5-氯 -3-环丙基 -1,3-二氢 -2H-苯并 [d]咪唑 -2-酮 (l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4- yl)-5-chloro-3-cyclopropyl-l,3-dihydro-2H-benzo[d]imidazol-2-one ) (中间体 506-A-99)。 LCMS (ESI): m々 523 [M + H ]+。
中间体 506-A-133的制备:合成方法如实施例 5中方法 5-2,仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮(503-51 )替换为 1-(2-氯 -5-甲氧基嘧啶 -4-基) -3- 异丙基 -1H -苯并 [d]咪唑 -2(3H)-酉同 l-(2-chloro-5-methoxypyrimidin-4-yl)-3-isopropyl-lH-benzo [d]imidazol-2(3H)-one ( 503-133 ), 制备得到 1-(2-(4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基 -5- 胺基苯胺基)-5- 甲 氧基嘧 啶 -4-基)-3-异丙基 -1H -苯并 [d]咪唑 -2(3H)-酮 l-(2-((4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-aminophenyl)amino)-5-methoxyp yrimidin-4-yl)-3-isopropyl-lH-benzo[d]imidazol-2(3H)-one (中间体 506-A-133)。 LCMS (ESI): m/z 521 [M + H ]+。
中间体 506-A-135的制备:合成方法如实施例 5中方法 5-2,仅是将其中化合物 5-氯 -1-(2- 氯嘧啶—4-基) - 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (503-51 ) 替换为 1-(2-氯 -5- (二甲氨基)嘧啶 -4- 基)-3-异丙基 -1,3-二氢 -2H-苯并咪唑 -2-酮(l -(2-chloro-5-(dimethylamino)pyrimidin-4-yl)-3- isopropyl-l,3-dihydro-2H-benzo[d]imidazol-2-one) ( 503-135 ) , 制备得到 1-(2-((5-氨基 -4-((2- (二 甲基氨基;)乙基 X甲基)氨基; )-2-甲氧基苯基)氨基; )-5- (二甲基氨基;)嘧啶 -4-基;) -3-异丙基 -1,3-二氢 -2H-苯并咪唑 -2-酮 (l-(2-((5-ammo-4-((2-(dimethylamino)ethyl)(methyl)ammo)-2-methoxyphenyl) amino)-5-(dimethylamino)pyrimidin-4-yl)-3-isopropyl-l,3-dihydro-2H-benzo[d]imidazol-2-one) (中间体 506-A-135)。 LCMS (ESI): m/z 534 [M + H ]+。 方法 5-3 (通过中间体 503和 103制备):
步骤 5-3a : 1-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酉同 ( l-p-methylbut -enyl lH- benzo imidazol pH one) (中间体 502-62)的制备: 将邻硝基苯胺 (5 克, 36毫摩尔, 1.0 当量) 溶解于 100毫升 N-甲基吡咯垸酮中, 加入碳酸铯 (23克, 72毫摩尔, 2.0当量)。 然 后往混合液中滴加 1-溴 -3-甲基 -2-丁烯 (7克, 46.8毫摩尔, 1.3当量), 加热至 100°C, 反应 过夜。 加水淬灭, 乙酸乙酯萃取, 饱和食盐水洗涤多次, 无水硫酸钠干燥, 减压浓缩, 得到 黄色油状物, 最后用硅胶柱层析纯化 (乙酸乙酯 /石油醚 =1/5~1/3 ), 得到产品 N1^-甲基 -2- 丁烯)苯 -1,2-二胺 (501-62 ) ( 3克, 收率: 40%)。 LCMS (ESI ):m々207[M + H ]+。
将化合物 501-62 (1.28克, 7.2毫摩尔, 1.0当量;)溶解于 100毫升四氢呋喃中, 加入 1.5 毫升三乙胺 (10.8毫摩尔, 1.5当量)。 在氮气保护下, 加入 Ν,Ν'-羰基二咪唑 (3.5克, 21.6毫 摩尔, 3.0当量;), 加热至 50°C, 反应过夜。 反应完成, 用二氯甲垸萃取, 减压浓缩。 最后用
硅胶柱层析纯化(二氯甲焼 /甲醇 =200/l~100/l )得到 1-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)- 酮 (1.3克, 收率: 89%)。 LCMS (ESI ):m々203[M + H ]+。
步骤 5-3b : 1 - (2-氯嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 ( l-(2-chloropyrimidin-4-yl)-3-(3-methylbut-2-enyl)-lH-benzo[d]imidazol-2(3H)-one ) (中间体 503-62)的制备:首先将 1-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酉同 (502-63 ) (600毫克, 2.97 毫摩尔, 1.0当量)和碳酸铯 (1.93克, 5.94毫摩尔, 2.0当量)混合于 10毫升 Ν,Ν-二甲基甲酰 胺中。 在氮气保护下, 加入 2,4-二氯嘧啶 (659毫克, 4.45毫摩尔, 1.5当量) , 室温搅拌反应 2小时。 反应完成后, 加入大量的水, 有固体析出, 过滤, 干燥, 即得到产品 1 - (2-氯嘧啶 -4- 基) -3-(3-甲基 -2-丁烯) -1Η-苯并 [d]咪唑 -2(3Η)-酮 (755毫克,收率: 81%)。LCMS (ESI):m/z315[M + H ]+。
步骤 5-3c: 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -3-(3- 甲基 -2-丁烯)-1Η-苯并 [d]咪唑 -2(3H)-酉同 l-(2-(4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxy-5-nitrophenylamino)pyrimidin-4-yl)-3-(3-methylbut-2-enyl)-lH-benzo[d]imidazol-2(3H )-one (中间体 505-A-62)的制备:将化合物 1 - (2-氯嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪 唑 -2(3H)-酉同 (503-62) (350毫克, 1.27毫摩尔, 1.0当量), 4-氟 -2-甲氧基 -5-硝基苯胺 (103 ) (261毫克, 1.4毫摩尔, 1.3当量)混合于 60毫升的异丙醇中, 加入浓盐酸 (1.5 毫升)。 混合物 在 100°C下搅拌反应过夜。 反应结束后, 冷却至室温, 减压浓缩, 得到黄色固体。 所得固体 用乙酸乙酯溶解, 加入饱和碳酸钠溶液, 中和剩余的酸。 有机相减压浓缩, 用石油醚 /乙酸乙 酯 (10/1)重结晶, 得到黄色固体 (213毫克, 收率: 41%)。 LCMS (ESI): m々465[M + H ]+。 将化 合物上述得到的化合物 (213毫克, 0.458毫摩尔, 1.0当量)溶解于 30毫升四氢呋喃中。 然后 将 Ν,Ν,Ν-三甲基乙二胺 (140毫克, 1.37毫摩尔, 3.0当量)和 Ν, Ν-二异丙基乙胺 (177毫克, 1.37毫摩尔, 3.0当量)加入到反应体系中, 在 80°C下搅拌反应过夜。 反应结束后, 冷却至室 温, 用乙酸乙酯和水萃取, 有机相用水洗, 饱和食盐水洗涤多次, 无水硫酸钠干燥, 减压浓 缩, 最后硅胶柱层析纯化 (二氯甲焼 /甲醇 =200/1 100/1), 得到红色固体, 即产品 1-(2-(4-((2- 二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪 唑 -2(3H)-酮 (132毫克, 收率: 52%) 。 LCMS (ESI): m々547[M + H ]+。 步骤 5-3d: 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -3-(3- 甲 基 -2- 丁 烯 )-1Η- 苯 并 [d] 咪 唑 -2(3H)- 酮 l-(2-(5-amino-4-((2-(dimethylamino) ethyl)(methyl)amino)-2-methoxyphenylamino)pyrimidin-4-yl)-3-(3-methylbut-2-enyl)-lH-benzo[d] imidazol-2(3H)-one(中间体 506- A-62)的制备:将 1 -(2-(4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮(505-A-62) (132毫克, 0.24毫摩尔, 1.0当量) 和氯化铵 (108毫克, 2.04毫摩尔, 8.5当量) 混合于 40毫升乙醇和 10毫升水中, 加热至 50°C, 然后加入还原铁粉 (108毫克, 1.92毫摩尔, 8.0当量)。 加热至 80°C, 反应 2小时。 反应完成后, 过滤, 将滤液浓缩, 再用二氯甲垸萃取, 有机相用无水硫 酸钠干燥,减压旋干,最后硅胶柱层析纯化(二氯甲焼 /甲醇 =200/1 50/1 )得到产品 1-(2-(4-((2- 二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪 唑 -2(3H)-酮 (110毫克, 收率: 88%) LCMS (ESI):m々517[M + H ]+。
下列中间体 ( 506-A-50, 506-A-63, 506-A-91, 506-A-98, 506-A-101, 506-A-102, 506-A-105, 506-A-118, 506-A-120, 506-A-125, 506-A-127,506-A-128, 506-A-132, 506-A-138 , 506-A-139 ) 可按方法 5-3制备。 仅是将其中中间体 502-62替换为相应的中间体 502。
中间体 506-A-50的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 6-氟 -1-甲基 -1,3-二氢 -2H-苯并 [d]咪唑 -2- 酮 (502-50 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4- 基 )-5-氟 -3-甲基 -1,3-二氢 -2H-苯并 [d]咪唑 -2-酮 ( l-(2-((5-amino-4-((2-(dimethylamino) ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4-yl)-5-fluoro-3-methyl-l,3-dihydro-2H- benzo[d]imidazol-2-one) (;中间体 506-A-50)。 LCMS (ESI): m/z 481 [M + H ]+。
中间体 506-A-62的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酉同(502-62 )替换为 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)- 酮 (502-62 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4- 基)-3-(3-甲基 -2-丁烯)-1Η-苯并 [d]咪唑 -2(3H)-酉同 ( l-(2-(5-amino-4-((2-(dimethylamino) ethyl)(methyl)amino)-2-methoxyphenylamino)pyrimidin-4-yl)-3-(3-methylbut-2-enyl)-lH-benzo[d] imidazol-2(3H)-one) (中间体 506-A-62)。 LCMS (ESI): m/z5 \ 7 [M + H ]+。
中间体 506-A-63的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 —2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酉同 (502-62 ) 替换为 1-环丙甲基 -1,3-二氢 -2H-苯并 [d]咪唑 -2- 酮 (502-63 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4- 基)— 3-环丙甲基 -1,3-二氢 -2H-苯并 [d]咪唑 -2-酉同(l-(2-((5-amino-4-((2-(dimethylammo)ethyl) (methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4-yl)-3-(cyclopropylmethyl)-l,3-dihydro-2H-b enzo[d]imidazol-2-one) (中间体 506-A-63)。 LCMS (ESI): m/z503 [M + H ]+。
中间体 506-A-91的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 6-氟 -1-异丙基 -1,3-二氢 -2H -苯并 [d]咪唑 -2(3H)-酮 ( 502-91 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基苯胺基) 嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮(l-(2-(5-amino-4-((2-(dimethylamino)ethyl) (methyl)amino)-2-methoxyphenylamino)pyrimidin-4-yl)-5-fluoro-3-isopropyl-lH-benzo[d]imidazo l-2(3H)-one) (中间体 506-A-91)。 LCMS (ESI): m/z509 [M + H ]+。
中间体 506-A-98的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 1-环丙基 -6-氟 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 502-98 ) , 制备得到 1-{2-{5-胺基 -4-{ [2- (二甲胺基) 乙基: |( 甲基) 胺基 }-2-甲氧基苯胺基 } 嘧啶 -4-基}-3-环丙基 -5-氟 -1H-苯并 [d]咪唑 -2(3H)- 酉同 (l-(2-(5-amino-4-((2-(dimethylamino)ethyl) (methyl)amino)-2-methoxyphenylamino)pyrimidin-4-yl)-3-cyclopropyl-5-fluoro-lH-benzo[d]imida zol-2(3H)-one) (中间体 506-A-98)。 LCMS (ESI): m/z 507 [M + H ]+。
中间体 506-A-101的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1-(3-甲 基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 3-环丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 -5-氰基 (502-101 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺 基)嘧啶—4-基)-3-环丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -5-氰基(l-(2-((5-amino-4-((2- (dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4-yl)-3-cyclopropyl-2-o
xo-2,3-dihydro-lH-benzo[d]imidazole-5-carbonitrile) (中间体 506-A-101)。 LCMS (ESI): m/z 514 [M + H ]+。
中间体 506-A-102的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1-(3-甲 基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 1-环丙基 -6-甲氧基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 502-102 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基苯胺基) 嘧啶 -4-基)-3-环丙基 -5-甲氧基 1,3-二氢 -2H-苯并 [d]咪唑 -2-酮(l-(2-((5-amino-4-((2- (dimethylamino)ethyl)(methyl)amino)-2-methoxyphenyl)amino)pyrimidin-4-yl)-3-cyclopropyl-5-m ethoxy-l,3-dihydro-2H-benzo[d]imidazol-2-one) (中间体 506-A-102)。 LCMS (ESI): m/z 519 [M + H ]+。
中间体 506-A-105的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1-(3-甲 基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 1-环丙甲基 -6-氟 -1,3-二氢 -2H-苯并 [d] 咪唑 -2(3H)-酮 (502-105 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基苯 胺基)嘧啶 -4-基)- 3-环丙甲基 -5-氟 -1H-苯并 [d]咪唑 -2(3H)-酮 ( l-(2-(5-amino-4-((2- (dimethylamino)ethyl)(methyl)amino)-2-methoxyphenylamino)pyrimidin-4-yl)-3-(cyclopropylmeth yl)-5-fluoro-lH-benzo[d]imidazol-2(3H)-one) (;中间体 506-A-105)。 LCMS (ESI): m/z 521 [M + H ]+。
中间体 506-A-118 的制备 (合成方法如实施例 5 中方法 5-3):将 4-氟 -2-甲基 -5-硝基苯胺 ( 103-118 ) 040毫克, 2.0毫摩尔, 1.0当量), 1-(2-氯嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)- 酮 (600毫克, 2.3毫摩尔, 1.15当量) 和浓盐酸 (5毫升, 59.0毫摩尔, 29.5当量) 加入到 100毫升异丙醇中, 于 100°C下反应过夜。 冷却至室温, 饱和碳酸钠水溶液中和, 用乙酸乙 酯萃取水相, 用水和饱和食盐水洗, 无水硫酸钠干燥, 减压浓缩, 经过硅胶柱层析纯化 (洗 脱剂: 石油醚: 乙酸乙酯 =1 : 1 ), 得到白色固体 1-(2-(4-氟 -2-甲基 -5-硝基苯胺基)嘧啶 -4-基) -3- 甲基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酮 (120毫克, 收率: 15%)。 LCMS (ESI): m/z 395[M + H ]+。 将上述固体 ( 120毫克, 0.30毫摩尔, 1.0当量), Ν,Ν,Ν'-三甲基 -1,2-乙二胺 ( 0.08毫升, 0.60 毫摩尔, 2.0当量)和二异丙基乙胺 (0.10毫升, 0.60毫摩尔, 2.0当量)加入到 30毫升四氢 呋喃中, 加热回流, 搅拌过夜。 冷却至室温, 减压浓缩, 残留物溶解于乙酸乙酯, 用水和饱 和食盐水洗有机相,无水硫酸钠干燥,减压浓缩,得到红色固体 1-(2-(4-((2-二甲胺乙基 X甲基) 胺基) -2-甲基 -5-硝基苯胺基)嘧啶 -4-基) -3-甲基 -1,3-二氢 -2Η -苯并 [d]咪唑 -2-酮 ( 505-A-118 ) ( 140毫克, 收率: 98.6%)。 LCMS (ESI): m々476[M + H ]+。 将上述得到的固体 (140毫克, 0.30毫摩尔, 1.0当量) 和氯化铵 (128毫克, 2.40毫摩尔, 8.0当量) 加入到 30毫升乙醇和 6毫升水的混合液中,加热至 65°C,分批加入还原性铁粉(143毫克, 2.55毫摩尔, 8.5当量), 加热至回流, 反应 1个小时。 反应冷却至室温, 加入二氯甲垸, 过滤, 滤液用水和饱和食盐 水洗, 有机相用无水硫酸钠干燥, 减压浓缩, 经过硅胶柱层析纯化 (洗脱剂: 二氯甲垸: 甲 醇 =40: 1 15 : 1 ), 得到棕色固体 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲基苯胺基)嘧啶 -4-基) -3-甲基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酉同(506-A-118 72毫克,收率: 54%)。 LCMS (ESI): m/z446 [M + H ]+。
中间体 506-A-120的制备:合成方法如实施例 5中方法 5-3中的中间体 506-A-118的制备, 仅是将其中化合物 4-氟 -2-甲基 -5-硝基苯胺 (103-118)替换为 4-氟 -2- ( 2-甲氧基 - 乙氧基) -5-
硝基 - 苯胺 (103-120 ) , 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-(2-甲氧基- 乙 氧 基 ) - 苯 胺 基 ) 嘧 啶 -4- 基 )-3- 甲 基 -1,3- 二 氢 -2H - 苯 并 咪 唑 -2- 酮 ( l-{2-[5-Amino-4-[(2-dimethylamino-ethyl)-methyl-amino]-2-(2-methoxy-ethoxy)-phenylamino] -pyrimidin-4-yl}-3-methyl-l,3-dihydro-benzoimidazol-2-one) (中间体 506-A-120)。 LCMS (ESI): m/z507 [M + H ]+。
中间体 506-A-125的制备:合成方法如实施例 5中方法 5-3中的中间体 506-A-118的制备, 仅是将其中化合物 4-氟 -2-甲基 -5-硝基苯胺 (103-118)替换为 4-氟 -3-硝基 -苯胺 (103-125 ) , 制 备得到 1-(2-(3-胺基 -4-((2-二甲胺乙基 X甲基)胺基)苯胺基)嘧啶 -4-基) - 3-甲基 -1,3-二氢 -2H-苯 并 [d] 咪 唑 -2- 酉同 l-(2-((3-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)phenyl)amino) pyrimidin-4-yl)-3 -methyl- 1 ,3 -dihydro-2H-benzo[d]imidazol-2-one(中间体 506-A-125)。 LCMS (ESI): m/z433 [M + H ]+。
中间体 506-A-127的制备:合成方法如实施例 5中方法 5-3中的中间体 506-A-118的制备, 仅是将其中化合物 1-(2-氯嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮替换为 1-(2-氯嘧啶 -4- 基) -3-异丙基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酮, 制备得到 1-(2-(5-胺基 -4-((2-二甲胺乙基) (甲基) 胺基)-2-甲基苯胺基)嘧啶 -4-基)-3-异丙基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酮 ( l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methylphenyl)amino)pyrimidin-4- yl)-3-isopropyl-l,3-dihydro-2H-benzo[d]imidazol-2-one ) (;中间体 506-A-127)。 LCMS (ESI): m/z475 [M + H ]+。
中间体 506-A-128的制备:合成方法如实施例 5中方法 5-3中的中间体 506-A-125的制备, 仅是将其中化合物 1-(2-氯嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮替换为 1-(2-氯嘧啶 -4- 基) -3-异丙基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酮, 制备得到 1-(2-(4-((2-二甲胺乙基) (甲基)胺基) -3- 胺 基 苯 胺 基 ) 嘧 啶 -4- 基 )-3- 异 丙 基 -1H - 苯 并 [d] 咪 唑 -2(3H)- 酮 (l-(2-((4-((2-(dimethylamino)ethyl)(methyl)amino)-3-aminophenyl)amino)pyrimidin-4-yl)-3-isopro pyl-lH-benzo[d]imidazol-2(3H)-one) (中间体 506-A-128)。 LCMS (ESI): m々461 [M + H ]+。
中间体 506-A-132的制备:合成方法如实施例 5中方法 5-3中的中间体 506-A-125的制备, 仅是将其中化合物 4-氟 -3-硝基苯胺 (103-125)替换为 4-氟 -2-异丙基 -5-硝基苯胺 (103-132), 制 备得到得到 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-异丙基苯胺基)嘧啶 -4-基) -3-甲基 -1,3-二氢 -2H -苯并 [d]咪唑 -2-酮 (l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl) amino)- 2-isopropylphenyl)amino)pyrimidin-4-yl)-3-methyl-l,3-dihydro-2H-benzo[d]imidazol-2-one ) (中 间体 506-A-127)。 LCMS (ESI): m々475 [M + H ]+。
中间体 506-A-138的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮(502-62 )替换为 1-异丙基 -6-甲氧基 -1H -苯并 [d]咪唑 -2(3H)- 酮 (502-138 ) , 制备得到 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-胺基苯胺基)嘧啶 -4- 基) -3 -异丙基 -5 -甲氧基 - 1 H -苯并 [d]咪唑 -2(3H)-酉同( 1 -(2-((4-((2-(dimethylamino)ethyl)(methyl) amino)-2-methoxy-5-aminophenyl)amino)pyrimidin-4-yl)-3-isopropyl-5-methoxy-lH-benzo[d]imi dazol-2(3H)-one) (中间体 506-A-138)。 LCMS (ESI): m/z52 \ [M + H ]+。
中间体 506-A-139的制备: 合成方法如实施例 5中方法 5-3, 仅是将其中化合物 1 -(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酮 (502-62 ) 替换为 1 -(2-氯嘧啶 -4-基)- 5-羟基 -3-异丙基 -1H -
苯并 [d]咪唑 -2(3H)-酮 (502-139 ) , 制备得到 1-(2-(4-((2-二甲胺乙基 )(甲基)胺基) -2-甲氧基 -5- 胺基苯胺基)嘧啶 -4-基) -5-羟基 -3-异丙基 -1H -苯并 [d]咪唑 -2(3H)-酮 ( 1-(2-((4-((2- (dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-aminophenyl)amino)pyrimidin-4-yl)-5-hydrox y-3-isopropyl-lH-benzo[d]imidazol-2(3H)-one) (中间体 506-A-139)。 LCMS (ESI): m/zS01 [M + H ]+。 实施例 6: Ν-(5-((4-(1Η-吲哚 -1-基) -5-三氟甲基嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲 基)胺基) -4-甲氧基苯基)丙烯酰胺 (N-(5-((4-(lH-indol-l-yl)-5-(trifluoromethyl) pyrimidin-2-yl) amino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 5)的制 备 (按照方案六线路制备)
将化合物 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^-二甲胺乙基) -5-甲氧基 -N1-甲 基苯 -1,2,4-三胺 (403-A-5) ( 90毫克, 0.26毫摩尔, 1.0当量), 三乙胺 ( 0.09毫升, 0.65毫摩 尔, 2.5当量), HATU ( 118毫克, 0.31毫摩尔, 1.2当量) 和丙烯酸 (602-3) ( 23毫克, 0.31 毫摩尔, 1.2当量) 加入到 15毫升二氯甲垸中, 于室温下搅拌 0.5小时。 反应停止搅拌, 加 入饱和碳酸钠水溶液, 分液, 用二氯甲垸萃取水相两次, 合并有机相, 依次用稀盐酸、 饱和 食盐水洗, 干燥, 减压浓缩, 经薄层制备纯化 (洗脱剂: 二氯甲焼 /甲醇 25/1 ), 得到微黄色 固体 Ν-(5-((4-(1Η-吲哚 -1-基) -5-三氟甲基嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基)胺 基) -4-甲氧基苯基)丙烯酰胺 (25毫克, 收率: 17%)。
该化合物 5的表征数据为: LCMS (ESI): m/z 554[M + H ]+。熔点: 161-162摄氏度; 1H MR (500 MHz, DMSO-de) δ 9.98 (s, 1H), 9.61 (s, 1H), 8.86 (s, 1H), 8.32 (s, 1H), 7.71 (s, 1H), 7.58 (s, 1H), 7.48 (s, 1H), 7.11 (s, 2H), 7.00 (s, 1H), 6.73 (s, 1H), 6.42 (s, 1H), 6.23 (d, J = 16.9 Hz, 1H), 5.75 (d, = 9.9 Hz, 1H), 3.79 (s, 3H), 2.87 (s, 2H), 2.69 (s, 3H), 2.21 (s, 8H)。 实施例 7: N-(5-((4-(lH-吲哚 -1-基) -5-甲氧基嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲基) 胺 基 )-4- 甲 氧 基 苯 基 ) 丙 烯 酰 胺 (N-(5-((4-(lH-indol-l-yl)-5-methoxypyrimidin- 2-yl)amino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 6) 的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 Ν4-(4-(1Η-吲哚 -1-基) -5-甲氧基 嘧啶 -2-基) -^-(2-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-6 ) (270毫克, 0.58毫 摩尔, 1.0当量),得到米黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基) -5-甲氧基嘧啶 -2-基)胺基) -2-((2- (二甲 胺基)乙基; 甲基)胺基; )-4-甲氧基苯基)丙烯酰胺 (35毫克, 收率: 12%)。
该化合物 6的表征数据为: LCMS (ESI): m/z516 [M + H ]+。 熔点: 143.3-147.8摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.64 (s, 1H), 8.45 (s, 1H), 8.22 (s, 1H), 8.00 (d, = 19.9 Hz, 2H), 7.57 (s, 1H), 7.11 (s, 2H), 7.01 (s, 1H), 6.70 (s, 1H), 6.38 (m, 1H), 6.21 (d, J = 16.6 Hz, 1H), 5.74 (d, = 9.2 Hz, 1H), 3.83 (d, = 34.2 Hz, 6H), 2.86 (s, 2H), 2.71 (s, 3H), 2.29 (s, 2H), 2.19 (s, 6H)。 实施例 8: Ν-(5-((4-(1Η-吲哚 -1-基) -5-甲基嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基)
胺基 )-4- 甲 氧基苯基)丙烯酰胺 ( N-(5-((4-(lH-indol-l-yl)-5-methylpyrimidin-2-yl) amino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 8)的制 备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 Ν4-(4-(1Η-吲哚 -1-基) -5-甲基嘧 啶 -2-基) -N1^-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (99毫克, 0.22毫摩尔, 1.0当量), 得到米黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基) -5-甲基嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲基) 胺基) -4-甲氧基苯基)丙烯酰胺 (60毫克, 收率: 54.5%)。
该化合物 8的表征数据为: LCMS (ESI): m/z500 [M + H ]+ D 1H NMR (500 MHz, DMSO-d6) δ 9.99 (s, 1H), 8.61 (s, 1H), 8.46 (s, 1H), 8.32 (s, 1H), 7.74 (d, = 3.4 Hz, 1H), 7.68 (d, J = 7.3 Hz, 1H), 7.60 (dd, J = 6.3, 2.4 Hz, 1H), 7.11 (m, 2H), 6.98 (s, 1H), 6.69 (d, = 3.3 Hz, 1H), 6.39 (dd, = 16.8, 10.1 Hz, 1H), 6.23 (dd, = 16.9, 1.8 Hz, 1H), 5.74 (m, 1H), 3.80 (s, 3H), 2.86 (s, 2H), 2.67 (d, = 9.8 Hz, 3H), 2.30 (s, 2H), 2.20 (s, 6H), 2.17 (s, 3H)。 实施例 9: N-{5-[5- (二甲基氨基 )-4-(1Η-吲哚 -1-基)嘧啶 -2-基氨基 ]-2-{[2- (二甲基氨基)乙 基 ](甲基)氨基 }-4-甲氧基苯}丙烯酰胺 (N-(5-(5-(dimethylamino)-4-(lH-indol-l-yl)pyrimidin- 2-ylamino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide ) (化合物 10) 的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N4-[5- (二甲基氨基 )-4-(1Η-吲哚 -1-基) 嘧啶 -2-基] -N1-^- (二甲基氨基) 乙基] -5-甲氧基 -N1-甲基苯 -1,2,4-三胺(403-A-10) (277 毫克, 0.591毫摩尔, 1.0当量) ,得到黄色固体 N-{5-[5- (二甲基氨基 )-4-(1Η-吲哚 -1-基)嘧啶 -2- 基氨基 ]-2-{[2- (二甲基氨基;)乙基] (甲基;)氨基 }-4-甲氧基苯)丙烯酰胺 (60毫克, 收率: 19.2%) 该化合物 10的表征数据为: LCMS (ESI): m/z529[M + H ]+。熔点: 154-155摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.69 (s, 1H), 8.38 (s, 1H), 8.13 (m, 2H), 7.87 (dd, = 6.0, 3.3 Hz, 1H), 7.57 (m, 1H), 7.10 (m, 2H), 6.98 (s, 1H), 6.69 (d, J = 3.5 Hz, 1H), 6.39 (dd, J = 16.9, 10.1 Hz, 1H), 6.23 (dd, = 16.9, 1.8 Hz, 1H), 5.74 (dd, = 8.7, 3.0 Hz, 1H), 3.81 (s, 3H), 2.86 (s, 2H), 2.69 (s, 3H), 2.47 (s, 6H), 2.29 (s, 2H), 2.20 (s, 6H)。 实施例 10: Ν-{5-[4-(1Η-吲哚 -1-基) -5- (甲基氨基)嘧啶 -2-基氨基 ]-2-{[2- (二甲基氨基) 乙 基] (甲基) 氨基 }-4-甲氧基苯}丙烯酰胺 (N-(5-(4-(lH-indol-l-yl)-5-(methylamino)pyrimidin- 2-ylamino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 11) 的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 Ν4-[4-(1Η-吲哚 -1-基) -5- (甲基氨 基)嘧啶 -2-基] -N1-^- (二甲基氨基) 乙基] -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-11 ) (216 毫 克, 0.4695 毫摩尔, 1.0当量) ,得到黄色固体 Ν-{5-[4-(1Η-吲哚 -1-基) -5- (甲基氨基)嘧啶 -2-基 氨基] -2-{[2- (二甲基氨基) 乙基] (甲基) 氨基 }-4-甲氧基苯}丙烯酰胺(30毫克, 收率: 12.4%)。)
该化合物 11的表征数据为: LCMS (ESI): m/z515 [M + H ]+。 熔点 154-156摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.85 (s, 1H), 8.17 (s, 1H), 7.85 (d, J = 3.4 Hz, 1H), 7.61 (m, 3H), 7.14 (dt, J = 19.9, 7.0 Hz, 2H), 6.95 (s, 1H), 6.71 (d, J = 3.3 Hz, 1H), 6.38 (dd, J = 16.9, 10.1 Hz, 1H), 6.24 (d, = 15.6 Hz, 1H), 5.74 (m, 1H), 4.70 (m, 1H), 3.81 (d, = 8.3 Hz, 3H), 2.85 (s, 2H), 2.68 (m, 6H), 2.26 (s, 2H), 2.19 (s, 6H)。 实施例 11 : N-(2-((2- (二甲胺基)乙基)(甲基)胺基) -4-甲氧基 5-((4-(5-甲氧基 -1H-吲哚 -1-基) 嘧啶 -2-基)胺基)苯基)丙烯酰胺(N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy- 5-((4-(5-methoxy-lH-indol-l-yl)pyrimidin-2-yl)amino)phenyl)acrylamide)(化合物 27)的制备 (按 照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 -(2-二甲胺乙基) -5-甲氧基 -N4-(4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-基)— N1-甲基苯 -1,2,4-三胺 (403-A-27)(100 毫克, 0.206 毫摩尔, 1当量),得到黄色固体 N-(2-((2- (二甲胺基)乙基)(甲基)胺基) -4-甲氧基 5-((4-(5-甲氧基 -1H-吲哚 -1-基)嘧啶 -2-基)胺基)苯基)丙烯酰胺。 (67毫克, 收率: 59.98%)。
该化合物 27的表征数据为: LCMS (ESI): m/z5 \6 [M + H ]+。熔点: 117-118摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.55 (d, J = 12.9 Hz, 2H), 8.33 (m, 2H), 8.09 (s, 1H), 7.07 (m, 3H), 6.67 (d, = 11.2 Hz, 2H), 6.40 (dd, = 16.2, 11.0 Hz, 1H), 6.19 (d, = 16.9 Hz, 1H), 5.72 (d, = 10.0 Hz, 1H), 3.76 (s, 6H), 2.92 (s, 2H), 2.75 (d, = 1.8 Hz, 3H), 2.34 (s, 2H), 2.21 (s, 6H)。 实施例 12: ^2-((2-二甲胺乙基 )(甲基)胺基) -(5—((4-(5—氟 -1H-吲唑 -1-基)嘧啶 -2-基)胺基) -4-甲氧基苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (5-fluoro- ΙΗ-indol- 1 -yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide ) (化合物 28)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N (2-二甲胺乙基) -N4-(4-(5-氟 -1H-吲唑 -1-基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4-三胺(403-A-28 ) (400毫克, 0.9毫摩尔, 1.0 当量),得到黄色固体 N-2-((2-二甲胺乙基) (甲基)胺基) -(5-((4-(5-氟 -1H-吲唑 -1-基)嘧啶 -2- 基)胺基) -4-甲氧基苯基)丙烯酰胺 (230毫克, 收率: 51%)。
该化合物 28的表征数据为: LCMS (ESI): m/z504 [M + H ]+。 熔点: 168-170摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.65 (s, 1H), 8.51 (s, 1H), 8.41 (m, 2H), 8.20 (s, 1H), 7.37 (d, = 7.7 Hz, 1H), 7.12 (d, = 4.9 Hz, 1H), 7.06 (s, 1H), 6.87 (s, 1H), 6.76 (s, 1H), 6.41 (m, 1H), 6.19 (d, = 16.8 Hz, 1H), 5.73 (d, = 9.2 Hz, 1H), 3.78 (s, 3H), 2.92 (s, 2H), 2.75 (s, 3H), 2.35 (s, 2H), 2.21 (s, 6H)。 实施例 13: N-2-((2-二甲胺乙基) (甲基)胺基) -(5—((4-(6—氟 -1H-吲唑 -1-基)嘧啶 -2-基)胺基) -4- 甲 氧 基 苯 基 ) 丙 烯 酰 胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (6-fluoro- lH-indol- 1 -yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide ) (化合物 29)的制备
(按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N (2-二甲胺乙基) -N4-(4-(6-氟 -1H-吲唑 -1-基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4-三胺 (403-A-29) (162毫克, 0.36毫摩 尔, 1当量),得到黄色固体 N-2-((2-二甲胺乙基) (甲基) 胺基) -(5-((4-(6-氟 -1H-吲唑 -1-基)嘧啶 -2- 基)胺基) -4-甲氧基苯基)丙烯酰胺 (60毫克, 收率: 33.15%)。
该化合物 29的表征数据为: LCMS (ESI): m々504 [M + H ]+。 熔点: 164-165摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.78 (s, 1H), 8.48 (s, 1H), 8.38 (d, = 5.7 Hz, 1H), 8.22 (d, = 9.7 Hz, 1H), 8.12 (d, = 3.7 Hz, 1H), 7.57 (dd, = 8.6, 5.7 Hz, 1H), 7.11 (d, = 5.7 Hz: 1H), 7.01 (m, 2H), 6.78 (d, = 3.6 Hz, 1H), 6.37 (dd, = 16.9, 10.2 Hz, 1H), 6.17 (dd, = 16.9, 1.8 Hz, 1H), 5.71 (m, 1H), 3.76 (s, 3H), 2.90 (s, 2H), 2.74 (s, 3H), 2.36 (s, 2H), 2.22 (s, 6H)。 实施例 14: N-2-((2-二甲胺乙基 )(甲基)胺基) -(5—((4-(4—氟 -1H-吲唑 -1-基)嘧啶 -2-基)胺基) -4- 甲 氧 基 苯 基 ) 丙 烯 酰 胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (4-fluoro- lH-indol- 1 -yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide ) (化合物 30)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N (2-二甲胺乙基) -N4-(4-(4-氟 -1H-吲唑 -1-基)嘧啶 -2-基) -5-甲氧基 -N1-甲基苯 -1, 2,4-三胺 (403-A-30) (195毫克, 0.43毫摩 尔, 1当量),得到黄色固体 N-2-((2-二甲胺乙基) (甲基) 胺基) -(5-((4-(4-氟 -1H-吲唑 -1-基)嘧啶 -2- 基)胺基) -4-甲氧基苯基)丙烯酰胺 (60毫克, 收率: 33.15%)。
该化合物 30的表征数据为: LCMS (ESI): m/z504 [M + H ]+。熔点: 165.5-166.5摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.69 (s, 1H), 8.52 (s, 1H), 8.42 (d, = 5.6 Hz, 1H), 8.21 (dd, J = 27.4, 5.8 Hz, 2H), 7.15 (d, J = 5.7 Hz, 1H), 7.07 (m, 2H), 6.96 (dd, J = 9.8, 8.2 Hz, 1H), 6.84 (d, = 3.6 Hz, 1H), 6.40 (dd, = 16.9, 10.2 Hz, 1H), 6.20 (dd, = 16.9, 1.8 Hz, 1H), 5.72 (dd, = 10.2, 1.6 Hz, 1H), 3.77 (s, 3H), 2.91 (t, = 5.7 Hz, 2H), 2.74 (d, = 10.0 Hz, 3H), 2.35 (d, = 5.3 Hz, 2H), 2.21 (s, 6H)。 实施例 l5: N-(5-((4-(5-氯 -1H-吲唑 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲基)胺基) -4-甲氧基苯基)丙烯酰胺 N-(5-((4-(5-chloro-lH-indol-l-yl)pyrimidin-2-yl)amino)-2-((2- (dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide (化合物 31)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N4-(4-(5-氯 -1H-吲唑 -1-基)嘧啶 —2—基) -^-(2—二甲胺乙基)— 5-甲氧基 -N1-甲基苯 1 2,4-三胺 (300毫克, 0.65毫摩尔, 1.0当量), 得到米黄色固体 N-(5-((4-(5-氯 -1H-吲唑 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲基) 胺基) -4-甲氧基苯基)丙烯酰胺 (85毫克, 收率: 25%)。
该化合物 31的表征数据为: LCMS (ESI): m々520 [M + H ]+。 熔点: 150-153摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.67 (s, 1H), 8.51 (s, 1H), 8.41 (d, = 5.6 Hz, 2H),
8.20 (d, J = 3.6 Hz, 1H), 7.65 (d, J = 2. l Hz, 1H), 7.12 (d, = 5.7 Hz, 1H), 7.05 (m, 2H), 6.76 (d, J = 3.5 Hz, 1H), 6.41 (dd, = 16.9, 10.2 Hz, 1H), 6.18 (dd, = 17.0, 1.8 Hz, 1H), 5.73 (dd, = 10.2,
1.6 Hz, 1H), 3.77 (d, = 7.7 Hz, 3H), 2.93 (t, = 5.7 Hz, 2H), 2.75 (d, = 8.8 Hz, 3H), 2.35 (t, = 5.5 Hz, 2H), 2.22 (s, 6H)。 实施例 16: N-5-((4-(5-氰基 -1H-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基) 胺基) -4-甲氧基—苯基)丙烯酰胺 (N-(5-((4-(5-cyano-lH-indol-l-yl)pyrimidin-2-yl) amino)-2-((2- (dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide)(化合物 33)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 1-(2-((5-胺基 -4-((2- (二甲胺基)乙 基)(甲基)胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基) -1H-吲哚 -5-氰基 (403-A-33)(100 毫克, 0.394 毫 摩尔, 1 当量),得到黄色固体 N-5-((4-(5-氰基 -1H-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基) 乙基; 甲基)胺基) -4-甲氧基-苯基)丙烯酰胺。 (35 毫克, 收率: 31.53%)。
该化合物 33的表征数据为: LCMS (ESI): m/z511 [M + H ]+。熔点: 127-128摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.83 (s, 1H), 8.59 (s, 1H), 8.46 (m, 2H), 8.32 (d, J =
3.7 Hz, 1H), 8.14 (d, = 1.1 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.18 (d, J = 5.7 Hz, 1H), 7.07 (s, 1H), 6.90 (d, = 3.6 Hz, 1H), 6.41 (dd, = 17.0, 10.2 Hz, 1H), 6.17 (dd, = 16.9, 1.8 Hz, 1H), 5.73 (dd, J = 10.2, 1.7 Hz, 1H), 3.78 (s, 3H), 2.92 (t, J = 5.8 Hz, 2H), 2.76 (s, 3H), 2.34 (t, J = 5.7 Hz, 2H), 2.21 (s, 6H)。 实施例 17: N-{2-[(2-二甲氨基-乙基) -甲基 -氨基 ]-4-甲氧基 -5-[4-(5-硝基 -吲哚 -1-基) -嘧啶 -2-基氨基] -苯基 } -丙烯酰胺 ( N-{2-[(2-Dimethylamino-ethyl)-methyl-amino]-4-methoxy- 5-[4-(5-nitro-indol-l-yl)-pyrimidin-2-ylamino]-phenyl}-acrylamide) (化合物 34)的制备 (按照方 案六线路制备)
将 N1^-二甲氨基-乙基) -5-甲氧基 -N1-甲基 -N4-[4-(5-硝基-吲哚- 1 -基) -嘧啶 -2-基] -苯基 -1,2,4-三胺 ( 403-A-34 ) ( 0.13克, 0.27毫摩尔) 和三乙胺 ( 55毫克, 0.55毫摩尔) 溶于四氢 呋喃 (10毫升), 降温至零下 70°C, 搅拌慢慢滴加丙烯酰氯 (25毫克, 0.27毫摩尔)。 在零 下 70 °C反应 2小时, 然后加入甲醇 (1毫升) 和水 (15毫升)。 用二氯甲垸 (15毫升) 萃取 三次, 有机相干燥浓缩之后, 用二氯甲垸: 甲醇 (10: 1 ) 柱层析得到黄色固体 N-{2-[(2-二甲 氨基-乙基) -甲基 -氨基 ]-4-甲氧基 -5-[4-(5-硝基 -吲哚 -1-基;) -嘧啶 -2-基氨基] -苯基 丙烯酰胺 (90 毫克, 收率 62.2%)。
该化合物 34 的表征数据为: LCMS (ESI): mlz 531 [M + H ]+。 1H NMR (500 MHz, DMSO-de) δ 10.11 (s, 1H), 8.80 (s, 1H), 8.53 (m, 4H), 8.36 (d, = 3.6 Hz, 1H), 7.88 (d, = 8.0 Hz, 1H), 7.19 (d, J = 5.6 Hz, 1H), 7.09 (s, 1H), 7.04 (d, J = 3.6 Hz, 1H), 6.39 (dd, J = 16.9, 10.2 Hz, 1H), 6.17 (dd, J = 17.0, 1.7 Hz, 1H), 5.71 (m, 1H), 3.78 (s, 3H), 2.94 (t, J = 5.7 Hz, 2H), 2.77 (s, 3H), 2.35 (t, = 5.7 Hz, 2H), 2.21 (s, 6H)。 实施例 IS: (2-((2-二甲胺乙基 )(甲基) 胺基) -5—((4-(5—羟基 -1H-吲哚 -1-基)嘧啶 -2-基)胺
基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (5-hydroxy- ΙΗ-indol- 1 -yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide ) (化合物 36)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 1-(2-((5-胺基 -4-((2-二甲胺乙基) (甲基) 胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基) -1H-吲哚 -5-醇 (403-A-36) (111毫克, 0.248毫摩 尔, 1 当量), 得到黄色固体 N-(2-((2-二甲胺乙基 )(甲基) 胺基) -5—((4-(5—羟基 -1H-吲哚 -1-基) 嘧啶 -2-基)胺基) -4-甲氧基苯基)丙烯酰胺 (55毫克, 收率: 44.12%)。
该化合物 36的表征数据为: LCMS (ESI): m々 502 [M + H ]+。 熔点: 218-219摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.00 (s, 1H), 8.53 (d, = 10.0 Hz, 2H), 8.33 (d, =
5.7 Hz, 1H), 8.17 (d, = 8.8 Hz, 1H), 8.03 (d, = 3.6 Hz, 1H), 7.05 (d, = 4.7 Hz, 2H), 6.88 (d, = 2.3 Hz, 1H), 6.58 (m, 2H), 6.39 (dd, = 16.9, 10.2 Hz, 1H), 6.20 (dd, = 16.9, 1.8 Hz, 1H), 5.73 (d, = 11.7 Hz, 1H), 3.77 (s, 3H), 2.91 (t, = 5.6 Hz, 2H), 2.76 (s, 3H), 2.34 (t, = 5.4 Hz, 2H), 2.22 (s: 6H)。 实施例 l9: N-(2-((2-二甲胺乙基) (甲基) 胺基) -5-((4-(5-(2-羟乙氧基) -1H-吲哚 -1-基)嘧啶 -2-基)胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (5-(2-hydroxyethoxy)-lH-indol-l-yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide) (化合物
37)的制备 (按照方案六线路制备)
将 N 4-4-(5-(2- ((叔丁基二苯基硅基)氧基)乙氧基) -1H-吲哚 -1-基)嘧啶 -2-基) -N1- (2-((2-二 甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-37 ) ( 320毫克, 0.41毫摩尔, 1.0当量), 三乙胺 (0.1毫升, 0.8毫摩尔, 2.0当量), HATU ( 182毫克, 0.48毫摩尔, 1.2当量) 和丙 烯酸 (35毫克, 0.48毫摩尔, 1.2当量)加入到 25毫升二氯甲垸中, 于室温下搅拌 0.5小时。 反应停止搅拌, 加入饱和碳酸钠水溶液, 分液, 用二氯甲垸萃取水相两次, 合并有机相, 依 次用稀盐酸、 饱和食盐水洗, 干燥, 减压浓缩, 经硅胶柱层析纯化 (洗脱剂: 二氯甲焼 /甲醇 40/1 至 15/1 ), 得到黄色固体 N-5-((4-(5-(2- ((叔丁基二苯基硅基)氧基)乙氧基) -1H-吲哚 -1-基) 嘧啶 -2-基;)胺基; )- (2-((2-二甲胺乙基 X甲基)胺基; ) -4-甲氧基苯基)丙烯酰胺 (200毫克, 收率: 62%)。 MS (ESI): m/z 784(M + H)+。
将上述得到的化合物 (200毫克, 0.25毫摩尔, 1.0当量) 溶解于 20毫升四氢呋喃中, 加入四丁基氟化铵 (300毫克, 1.15毫摩尔, 4.6当量)。 室温下, 搅拌反应 1小时, 减压浓 缩, 二氯甲垸萃取, 水洗六次, 无水硫酸钠干燥, 减压浓缩, 经硅胶柱层析纯化 (洗脱剂: 二氯甲焼 /甲醇 40/1至 15/1 ), 得到黄色固体 N-(2-((2-二甲胺乙基) (甲基) 胺基) -5-((4-(5-(2-羟 乙氧基;) -1H-吲哚 -1-基;)嘧啶 -2-基;)胺基; ) -4-甲氧基苯基)丙烯酰胺 (30毫克, 22%)。
该化合物 37的表征数据为: LCMS (ESI): /?t/Z 546 [M + H ]+。 熔点: 65-67摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.55 (d, J = 14.9 Hz, 2H), 8.32 (dd, J = 28.8, 7.0 Hz, 2H), 8.09 (d, = 3.1 Hz, 1H), 7.07 (dd, = 13.9, 8.1 Hz, 3H), 6.68 (t, = 7.7 Hz, 2H), 6.40 (dd, = 16.9, 10.2 Hz, 1H), 6.20 (d, = 16.8 Hz, 1H), 5.72 (d, = 10.1 Hz, 1H), 4.80 (s, 1H), 3.98 (t, =
4.8 Hz, 2H), 3.77 (s, 3H), 3.72 (d, = 3.6 Hz, 2H), 2.92 (s, 2H), 2.75 (s, 3H), 2.35 (s, 2H), 2.21 (s, 6H)。
实施例 20: N-(2-((2-二甲胺乙基 X甲基) 胺基) -5-((4-(5-(2-甲氧乙氧基) -1H-吲哚 -1-基)嘧 啶 -2-基)胺基) -4-甲氧基苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)- 4-methoxy-5-((4-(5-(2-methoxyethoxy)-lH-indol-l-yl)pyrimidin-2-yl)amino)phenyl)aciylamide ) (化合物 38)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5) N1- (2-((2-二甲胺乙基) -5-甲氧基 -N4 —4-(5-(2-甲氧乙氧基) -1H-吲哚 -1-基)嘧啶 -2-基) -N1-甲基苯 -1, 2,4-三胺 (403-A-38 ) ( 140毫克, 0.28毫摩尔, 1.0当量), 得到黄色固体 N-(2-((2-二甲胺乙基 X甲基)胺基) -5-((4-(5-(2-甲氧乙 氧基) -1H-吲哚 -1-基)嘧啶 -2-基;)胺基) -4-甲氧基苯基)丙烯酰胺 (30毫克, 收率: 19%)。
该化合物 38的表征数据为: LCMS (ESI): m/z 560[M + H ]+。熔点: 59-61摄氏度; 1H MR (500 MHz, DMSO-de) δ 10.03 (s, 1H), 8.56 (s, 2H), 8.35 (s, 2H), 8.10 (s, 1H), 7.09 (s, 3H), 6.69 (s, 2H), 6.43 (s, 1H), 6.21 (s, 1H), 5.73 (s, 1H), 4.09 (s, 3H), 3.73 (d, J = 54.8 Hz, 7H), 2.95 (s, 2H), 2.75 (s, 3H), 2.39 (m, 2H), 2.25 (s, 6H)。 实施例 21 : N-(2-((2-二甲胺乙基) (甲基) 胺基) -5-((4-(5-(2-羟甲基) -1H-吲哚 -1-基)嘧啶 -2- 基)胺基) -4-甲氧基苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5- ((4-(5-(hydroxymethyl)-lH-indol-l-yl)pyrimidin-2-yl)amino)-4-methoxyphenyl)aciylamide) (化合 物 39)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5) 替换为 (1-(2-((5-胺基 -4-((2-二甲胺乙基) (甲基) 胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基) -1H-吲哚 -5-基)甲醇 (403-A-39 ) ( 120毫克, 0.26 毫摩尔, 1.0当量), 得到黄色固体 N-(2-((2-二甲胺乙基 X甲基) 胺基) -5-((4-(5-(2-羟甲基 )-1Η- 吲哚 -1-基)嘧啶 -2-基)胺基) -4-甲氧基苯基)丙烯酰胺 (25毫克, 收率: 19%)。
该化合物 39的表征数据为: LCMS (ESI): m/z 516[M + H ]+。熔点: 67-68摄氏度; 1H MR (500 MHz, DMSO-dg) δ 10.08 (s, 1H), 8.57 (s, 2H), 8.35 (d, = 26.2 Hz, 2H), 8.11 (s, 1H), 7.51 (s, 1H), 7.08 (d, J = 26.0 Hz, 3H), 6.74 (s, 1H), 6.39 (s, 1H), 6.19 (d, J = 16.3 Hz, 1H), 5.73 (s, 1H), 5.07 (s, 1H), 4.53 (s, 2H), 3.78 (s, 3H), 2.92 (s, 2H), 2.76 (s, 3H), 2.35 (s, 2H), 2.22 (s, 6H)。 实施例 22: l-(2-((5-丙烯酰胺 -4-((2- (二甲胺基)乙基)(甲基;)胺基; )-2-甲氧基苯基)胺基)嘧啶 -4-基)-1Η-吲哚 -5-甲酰胺 ( -(2-((5-acrylamido-4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxyphenyl)amino)pyrimidin-4-yl)-lH-indole-5-carboxamide) (化合物 41)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 1-(2-((5-胺基 -4-((2- (二甲胺基)乙 基; 甲基)胺基) -2-甲氧基苯基)胺基)嘧啶 -4-基;) -1H-吲哚 -5-甲酰胺 (100毫克, 0.21毫摩尔, 1 当量), 得到米黄色固体 1-(2-((5-丙烯酰胺 -4-((2- (二甲胺基)乙基) (甲基)胺基) -2-甲氧基苯基) 胺基)嘧啶 -4-基) -1H-吲哚 -5-甲酰胺 (60毫克, 0.114毫摩尔, 收率: 54.3%)。
该化合物 41的表征数据为: LCMS (ESI): m/z 529[M + H ]+。熔点: 121.3-125.1摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.68 (s, 1H), 8.52 (s, 1H), 8.42 (d, = 5.6 Hz, 2H), 8.21 (d, = 3.6 Hz, 1H), 8.13 (s, 1H), 7.88 (s, 1H), 7.65 (d, = 8.8 Hz, 1H), 7.16 (t, = 8.9 Hz, 2H), 7.06 (s, 1H), 6.84 (d, J = 3.5 Hz, 1H), 6.40 (dd, J = 16.8, 10.2 Hz, 1H), 6.19 (d, J = 17.0 Hz, 1H), 5.71 (d, = 10.5 Hz, 1H), 3.78 (s, 3H), 2.93 (s, 2H), 2.77 (s, 3H), 2.36 (s, 2H), 2.21 (s, 6H)。 实施例 23 : N-(5-((4-(3-氯 -1H-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲基) 胺 基)-4-甲氧基苯基)丙烯酰胺 N-(5-((4-(3-chloro-lH-indol-l-yl)pyrimidin-2-yl)amino)-2- ((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide (化合物 46)的制备 (按照 方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N4-(4-(3-氯 -1H-吲哚 -1-基)嘧啶 -2-基) -^-(2-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-46) ( 216毫克, 0.46毫摩尔, 1.0当量),得到浅黄色固体 N-(5-((4-(3-氯 -1H-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲 基) 胺基) -4-甲氧基苯基)丙烯酰胺 (50毫克, 收率: 29%)。
该化合物 46的表征数据为: LCMS (ESI): m/z 520[M + H ]+。 熔点: 142-144摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.67 (s, 1H), 8.56 (s, 1H), 8.43 (s, 3H), 7.55 (d, = 7.2 Hz, 1H), 7.28 (s, 1H), 7.21 (s, 1H), 7.15 (d, = 5.1 Hz, 1H), 7.06 (s, 1H), 6.41 (s, 1H), 6.22 (d, J = 16.5 Hz, 1H), 5.73 (d, J = 9.5 Hz, 1H), 3.78 (s, 3H), 2.94 (s, 2H), 2.72 (d, J = 29.5 Hz, 3H), 2.37 (s, 2H), 2.24 (s, 6H)。 实施例 24 : N-(5-(4-(5-氯 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -2-((2-二甲胺乙基 X甲基)胺基) -4-甲氧基苯基)丙烯酰胺 (N-(5-(4-(5-chloro-3-methyl-2-oxo-2,3- dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-ylamino)-2-((2-(dimethylamino)ethyl)(methyl)ami no)-4-methoxyphenyl)acrylamide ) (化合物 51)的制备 (按照方案七线路制备)
将 1-(2-(5-胺基 -4-《2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氯 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 506-A-51 ) (140毫克, 0.28毫摩尔, 1.0当量), 三乙胺 (57毫克, 0.56毫摩尔,2.0当量) ,2-(7-偶氮苯并三氮唑) -Ν,Ν,Ν',Ν'-四甲基脲六氟磷酸酯 (117毫克, 0.308 毫摩尔, 1.1当量)溶于二氯甲垸 C20 毫升)中, 然后加入丙烯酸 (22毫克, 0.308毫摩尔, 1.1 当量), 室温反应。 反应半小时后, 往里加入 30 毫升水, 萃取, 有机相用碳酸钠水溶液洗涤, 无水硫酸钠干燥, 旋干, 残留物用硅胶柱层析纯化 (二氯甲垸 /甲醇 =500/1 150/1 ) 得到米黄 色固体 Ν-(5-(4-(5-氯 -3-甲基 -2-氧代 -2,3-二氢 -1Η-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -2-((2-二甲胺 乙基 X甲基)胺基; )-4-甲氧基苯基)丙烯酰胺 (23毫克, 收率: 14.8%)。
该化合物 51的表征数据为: LCMS (ESI): m/z 551 [M + H ]+。熔点: 70-71摄氏度; 1H MR (500 MHz, DMSO-de) δ 10.02 (s, 1H), 8.72 (s, 1H), 8.42 (m, 2H), 8.08 (s, 1H), 7.65 (d, J = 5.6 Hz, 1H), 7.36 (s, 1H), 7.05 (s, 1H), 6.87 (d, = 8.5 Hz, 1H), 6.40 (dd, = 17.0, 10.2 Hz, 1H), 6.17 (d, J = 16.9 Hz, 1H), 5.72 (d, = 10.1 Hz, 1H), 3.75 (s, 3H), 3.35 (s, 3H), 2.92 (s, 2H), 2.74 (s, 3H), 2.34 (s, 2H), 2.21 (s, 6H)。
实施例 25: N-(5-(( 4-(5-氰基 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-基)胺 基) -2-((2-二甲胺乙基 )(甲基)胺基) -4-甲氧基苯基)丙烯酰胺(N-(5-((4-(5-cyano-3-methyl-2-oxo- 2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-2-((2-(dimethylamino)ethylXmethyl) amino)-4-methoxyphenyl)acrylamide) (化合物 52)的制备 (按照方案七线路制备)
合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧 基苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-((5-胺 基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧基苯基)胺基)嘧啶 -4-基;) -3-甲基 -2-氧代 -2,3-二氢 -1H- 苯并 [d]咪唑 -5-甲腈(506-A-52) (65.4毫克, 0.134毫摩尔, 1当量),得到黄色固体 N-(5-(( 4-(5- 氰基 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲基) 胺基) -4-甲氧基苯基)丙烯酰胺 (40毫克, 收率: 55.06%)。
该化合物 52的表征数据为: LCMS (ESI): m々 542 [M + H ]+。 熔点: 183-185摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.79 (s, 1H), 8.48 (d, = 5.3 Hz, 1H), 8.39 (s, 1H), 8.23 (s, 1H), 7.77 (s, 1H), 7.62 (d, = 5.2 Hz, 1H), 7.31 (d, = 7.3 Hz, 1H), 7.05 (s, 1H), 6.41 (dd, J = 16.7, 10.2 Hz, 1H), 6.17 (d, = 17.0 Hz, 1H), 5.72 (d, = 10.1 Hz, 1H), 3.77 (s, 3H), 3.39 (s, 3H), 2.92 (s, 2H), 2.75 (s, 3H), 2.35 (s, 2H), 2.21 (s, 6H)。 实施例 26: N -2-((2-二甲胺乙基 X甲基)胺基)- 4-甲氧基 -5-((4-(5-甲氧基 -3-甲基 -2-氧代 -2,3- 二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-基)胺基) 苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl) (methyl)amino)-4-methoxy-5-((4-(5-methoxy-3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l- yl)pyrimidin-2-yl)amino)phenyl)acrylamide) (化合物 53)的制备 (按照方案七线路制备)
合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧 基苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-(5-胺 基 -4-((2-二甲胺乙基) (甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-甲氧基 -3-甲基 -1H-苯并 [d]咪 唑 -2(3H)-酉同 (506-A-53 ) (170毫克, 0.35毫摩尔, 1.0当量), 得到米黄色固体 N -2-((2-二甲 胺乙基) (甲基)胺基)- 4-甲氧基 -5-((4-(5-甲氧基 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基) 嘧啶 -2-基;)胺基) 苯基)丙烯酰胺 (80毫克, 收率: 42%)。
该化合物 53的表征数据为: LCMS (ESI): m々547 [M + H ]+。熔点: 75-87摄氏度; 1H MR (500 MHz, DMSO-de) δ 10.04 (s, 1H), 8.65 (s, 1H), 8.40 (m, 2H), 7.98 (d, = 7.7 Hz, 1H), 7.69 (d, = 5.7 Hz, 1H), 7.05 (s, 1H), 6.84 (d, = 2.3 Hz, 1H), 6.39 (dd, = 16.9, 10.1 Hz, 2H), 6.18 (dd, J = 17.0, 1.6 Hz, 1H), 5.73 (m, 1H), 3.76 (t, J = 8.5 Hz, 6H), 3.34 (s, 3H), 2.92 (t, J = 5.5 Hz, 2H), 2.75 (s, 3H), 2.35 (d, = 5.2 Hz, 2H), 2.21 (s, 6H)。 实施例 27: N-(2-((2-二甲胺乙基 X甲基)胺基) -4-甲氧基 -5-((5-甲氧基 -4-(3-甲基 -2-氧代 -2,3- 二氢 - 1 H-苯并 [d]咪唑 - 1 -基)嘧啶 -2-基)胺基)苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethy 1) (methyl)amino)-4-methoxy-5-((5-methoxy-4-(3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l- yl)pyrimidin-2-yl)amino)phenyl)acrylamide) (化合物 57)的制备 (按照方案七线路制备)
合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧 基苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-(5-胺 基 -4-((2-二甲胺乙基) (甲基)胺基) -2-甲氧基苯胺基 )-5-甲氧基嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪
唑 -2(3H)-酉同 (506-A-57 ) (400毫克, 0.81毫摩尔, 1.0当量), 得到黄色固体 N-(5-((5-氯 -4-(3- 甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-基)胺基) -2-((2-二甲胺乙基) (甲基)胺基) -4- 甲氧基苯基)丙烯酰胺 (15毫克, 收率: 45.08%)。
该化合物 57的表征数据为: LCMS (ESI): m々547 [M + H ]+。 熔点: 168-172摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.99 (s, 1H), 8.63 (s, 1H), 8.58 (s, 1H), 8.20 (s, 1H), 7.21 (d, = 7.8 Hz, 1H), 7.13 (t, = 7.6 Hz, 1H), 7.08 (d, = 7.7 Hz, 1H), 7.01 (t, = 7.6 Hz, 1H), 6.96 (s, 1H), 6.37 (dd, = 16.9, 10.1 Hz, 1H), 6.22 (d, = 17.0 Hz, 1H), 5.73 (d, = 10.5 Hz, 1H), 3.81 (d, = 9.0 Hz, 6H), 3.37 (s, 3H), 2.83 (t, J = 5.6 Hz, 2H), 2.68 (s, 3H), 2.27 (t, J = 5.4 Hz, 2H), 2.18 (s, 6H)。 实施例 28: (Ε)-Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基)胺 基)-4-甲氧基苯基) -2-丁烯酰胺((E)-N-(5-((4-(lH-indol-l -yl)pyrimidin-2-yl)amino)-2- ((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)but-2-enamide) (化合物 66)的制备(按 照方案六线路制备)
常温下, 将巴豆酸 (602-66 ) ( 24毫克, 0.277毫摩尔, 1.2当量) 三乙胺 (58.3毫克, 0.58 毫摩尔, 2.5 当量) 和 HATU ( 2-(7-偶氮苯并三氮唑) -Ν,Ν,Ν',Ν'-四甲基脲六氟磷酸酯) ( 114毫克, 0.3毫摩尔, 1.3当量) 溶于二氯甲垸 (10毫升), 搅拌半小时后, 将 Ν4-(4-(1Η- 吲哚 -1-基)嘧啶 -2-基) -Ν1^-二甲胺乙基) -5-甲氧基 -Ν1-甲基苯 -1,2,4-三胺(403-A-1 ) ( 100毫克, 0.232毫摩尔 , 1当量) 溶于二氯甲垸 (2毫升) 滴加到反应中, 搅拌反应 1小时。 反应液在水 相和乙酸乙酯中萃取分离, 有机相用盐水洗涤后用无水硫酸钠干燥, 真空下浓缩, 再用硅胶 柱层析 (洗脱剂: 二氯甲焼 /甲醇 80/1 ) 分离提纯得到 (Ε)-Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基) 胺基; )-2-((2- (二甲胺基)乙基)(甲基)胺基; )-4-甲氧基苯基; ) -2-丁烯酰胺 (50毫克, 0.1毫摩尔, 收 率: 43.1%)。
该化合物 66的表征数据为: LCMS (ESI): m/z 500[M + H ]+;熔点: 150.2-152.8摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.57 (d, J = 20.3 Hz, 2H), 8.38 (d, J = 5.6 Hz, 2H), 8.13 (d, = 3.5 Hz, 1H), 7.58 (d, = 7.7 Hz, 1H), 7.10 (m, 4H), 6.74 (m, 2H), 6.07 (d, = 15.2 Hz, 1H), 3.76 (s, 3H), 2.90 (t, J = 5.3 Hz, 2H), 2.74 (s, 3H), 2.35 (t, J = 5.4 Hz, 2H), 2.22 (s, 6H), 1.87 (d, = 6.7 Hz, 3H)。 实施例 29: (E)-N-(5-((4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基)胺 基)-4-甲氧基苯基) -2-戊烯酰胺 ((E)-N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)-2- ((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)pent-2-enamide)(化合物 67)的制备 (按照方案六线路制备)
合成方法如实施例 28, 仅是将巴豆酸 (602-66 ) 替换为反式 -2-戊烯酸 (602-67) ( 26毫 克, 0.25 毫摩尔, 1.2 当量) ,得到微黄色固体 (Ε)-Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺 基) -2-((2- (二甲胺基)乙基; 甲基)胺基) -4-甲氧基苯基) -2-戊烯酰胺 (19毫克, 收率: 18%)。
该化合物 67的表征数据为: LCMS (ESI): m/z 514[M + H ]+。熔点: 124.3-126.2摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.89 (s, 1H), 8.65 (s, 1H), 8.50 (s, 1H), 8.38 (d, J = 5.7 Hz, 2H), 8.13 (d, = 3.6 Hz, 1H), 7.58 (d, = 7.7 Hz, 1H), 7.10 (ddd, = 35.6, 16.6, 9.3 Hz, 4H), 6.80 (m,
2H), 6.08 (dd, J = 17.2, 10.2 Hz, 1H), 3.77 (s, 3H), 2.94 (s, 2H), 2.74 (s, 3H), 2.24 (dd, = 22.1 15.0 Hz, 8H), 1.23 (s, 2H), 1.05 (t, = 7.4 Hz, 3H)。 实施例 30: N-(5-((4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲基)胺基) -4- 甲 氧 基 苯 基 ) 甲 基 丙 烯 酰 胺 (N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)-2- ((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)methacrylamide)(化合物 68)的制备 (按照方案六线路制备)
合成方法如实施例 28, 仅是将巴豆酸 (602-66 ) 替换为甲基丙烯酸 (602-68X24毫克, 0.28毫摩尔, 1.2当量),得到米黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺 基;)乙基; 甲基)胺基; )-4-甲氧基苯基) 甲基丙烯酰胺 (70毫克, 收率: 60%)。
该化合物 68的表征数据为: LCMS (ESI): m々500 [M + H ]+;熔点: 145.7-147.1摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.81 (s, 1H), 8.59 (s, 1H), 8.53 (s, 1H), 8.40 (d, = 5.7 Hz, 2H), 8.14 (d, J = 3.7 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.12 (m, 4H), 6.77 (d, J = 3.6 Hz, 1H), 5.79 (s, 1H), 5.50 (s, 1H), 3.79 (s, 3H), 3.01 (s, 2H), 2.71 (s, 3H), 2.17 (s, 8H), 1.97 (s, 3H)。 实施例 31 : (E)-N-(5-((4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基) (甲基)胺 基) -4-甲氧基苯基) -4-二甲胺基丁 -2-烯酰胺 ((E)-N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)- 2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)-4-(dimethylamino)but-2-enamideX 化合物 69)的制备 (按照方案六线路制备)
合成方法如实施例 28, 仅是将巴豆酸 (602-66 ) 替换为反式 -4-二甲氨基巴豆酸盐酸盐 (602-69) ( 37毫克, 0.22毫摩尔, 1.2当量),得到棕色固体 (Ε)-Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2- 基;)胺基; )-2-((2- (二甲胺基)乙基)(甲基;)胺基; )-4-甲氧基苯基; )-4-二甲胺基丁 -2-烯酰胺 (40毫克, 收率: 40%)。
该化合物 69的表征数据为: LCMS (ESI): m/z 543 [M + H ]+。熔点: 116.2-118.1摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.50 (m, 4H), 8.13 (s, 1H), 7.59 (s, 1H), 7.08 (d, = 44.8 Hz, 4H), 6.72 (d, = 41.2 Hz, 2H), 6.30 (s, 1H), 3.79 (s, 3H), 3.29 (s, 2H), 3.12 (s, 2H), 2.99 (s: 2H), 2.73 (s, 3H), 2.35 (s, 6H), 2.22 (s, 6H)。 实施例 32: (Ε)-Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基氨基 ]-2-{ [2- (二甲基氨基)乙基] (甲基)氨 基) -4-甲氧基苯基 } -4-哌啶 - 1 -基 }丁 -2-烯酰胺( (E)-N-(5-(4-( IH-indol- 1 -yl)pyrimidin-2-ylamino)- 2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)-4-(piperidin- 1 -yl)but-2-enamide )
(化合物 70)的制备 (按照方案六线路制备)
将 Ν 4-[4-(1Η-吲哚 -1-基)嘧啶 -2-基] -^-[2- (二甲基氨基)乙基] -5-甲氧基 -N1-甲基苯 -1,2,4- 三胺(403-1 ) ( 145毫克, 0.336毫摩尔, 1.0当量)溶解在乙腈((30毫升)中, 依次加入 Ν,Ν- 二异丙基乙胺 (87毫克, 0.672毫摩尔, 2.0当量), HATU ( 2-(7-偶氮苯并三氮唑) -四甲基脲 六氟磷酸酯)(128毫克, 0.336毫摩尔, 1.0当量)和 (Ε)-4- (哌啶 -1-基)丁 -2-烯酸(602-70 ) ( 100 毫克, 粗品) 并在室温下搅拌反应两个小时。 反应结束后加入饱和碳酸钠水溶液终止反应, 用二氯甲垸 (3 x80毫升) 萃取, 合并有机相并用无水硫酸钠干燥, 浓缩并通过柱层析 (洗脱 剂: 二氯甲焼 /甲醇 60/1至 15/1 ) 纯化, 得到灰色固体 (Ε)-Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基氨
基] -2-{ [2- (二甲基氨基;)乙基] (甲基;)氨基; )-4-甲氧基苯基 }-4-哌啶 -1-基)丁 -2-烯酰胺 (60毫克, 收 率: 30.6%)。
该化合物 70的表征数据为: MS (ESI): m/z583 [M + H ]+。 1H MR (500 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.57 (s, 1H), 8.42 (m, 3H), 8.13 (d, = 3.6 Hz, 1H), 7.58 (d, = 7.4 Hz, 1H), 7.13 (m, 3H), 7.03 (s, 1H), 6.77 (d, = 3.5 Hz, 1H), 6.69 (m, 1H), 6.36 (s, 1H), 3.79 (s, 3H), 3.16 (s, 2H): 3.04 (s, 2H), 2.71 (s, 3H), 2.42 (s, 12H), 1.54 (s, 4H), 1.40 (s, 2H)。 实施例 33: N-(5-((4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲基)胺基) -4- 甲氧基苯基) -2-丁炔酰胺 (N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)- 2-((2-(dimethylamino) ethylXmethyl)amino)-4-methoxyphenyl)but-2-ynamideX化合物 73)的制备 (按照方案六线路制 备)。
合成方法如实施例 28, 仅是将巴豆酸 (602-66 ) 替换为 2-丁炔酸 (604-73) ( 21毫克, 0.25毫摩尔, 1.2当量) , 得到微黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲 胺基)乙基; 甲基)胺基; )-4-甲氧基苯基; ) -2-丁炔酰胺 (48毫克, 收率: 46%)。
该化合物 73的表征数据为: LCMS (ESI): m/z 498 [M + H ]+。熔点: 189.7-191.8摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.60 (s, 1H), 8.39 (m, 3H), 8.11 (d, = 3.6 Hz, 1H): 7.59 (d, = 7.6 Hz, 1H), 7.13 (m, 4H), 6.77 (d, = 3.5 Hz, 1H), 3.77 (s, 3H), 2.90 (s, 2H), 2.75 (s, 3H), 2.31 (d, = 24.4 Hz, 8H), 2.03 (s, 3H)。 实施例 34: N-(5-((4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙基)(甲基)胺基) -4- 甲氧基苯基) -2-戊炔酰胺 (N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)-2- ((2-(dimethylamino) ethyl)(methyl)amino)-4-methoxyphenyl)pent-2-ynamide) (化合物 74)的制备 (按照方案六线路制 备)
合成方法如实施例 28, 仅是将巴豆酸 (602-66 ) 替换为 2-戊炔酸 (25毫克, 0.25毫摩 尔, 1.2 当量) , 得到微黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((2- (二甲胺基)乙 基; 甲基)胺基) -4-甲氧基苯基) -2-戊炔酰胺 (27毫克, 收率: 25%)。
该化合物 74的表征数据为: LCMS (ESI): m/z 512[M + H ]+。熔点: 149.4-151.7摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.92 (s, 1H), 8.60 (s, 1H), 8.39 (d, J = 5.6 Hz, 3H), 8.11 (d, = 3.6 Hz, 1H), 7.59 (d, = 7.6 Hz, 1H), 7.13 (m, 4H), 6.77 (d, = 3.5 Hz, 1H), 3.78 (s, 3H), 2.96 (s, 2H), 2.74 (s, 3H), 2.41 (dt, = 27.1, 13.5 Hz, 8H), 1.24 (s, 2H), 1.16 (t, J = 1.5 Hz, 3H)。 实施例 35: N-(5-《4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-《3-二甲胺基吖啶 -1-基) -4-甲氧基苯 基)丙烯酰胺 N-(5-(4-(lH-indol-l-yl)pyrimidin-2-ylamino)-2-(3-(dimethylamino)azetidin- 1-yl)- 4-methoxyphenyl)acrylamide (化合物 77)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 -(4-(111-吲哚 -1-基)嘧啶 -2- 基)—4-(3-二甲胺基吖啶 -1-基) -6-甲氧基苯 -1,3-二胺 (403-C-77 ) (179毫克, 0.416毫摩尔, 1.0 当量), 得到米黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基) -2-((3-二甲胺基吖啶 -1-基) -4- 甲氧基苯基)丙烯酰胺 (45毫克, 收率: 22%)。
该化合物 77的表征数据为: LCMS (ESI): m々484 [M + H ]+;熔点: 81-83摄氏度; 1H MR (500 MHz, DMSO-de) δ 9.26 (s, 1H), 8.41 (s, 2H), 8.34 (d, J = 5.6 Hz, 1H), 8.07 (d, J = 3.6 Hz, 1H), 7.58 (d, = 7.5 Hz, 1H), 7.46 (s, 1H), 7.17 (dt, = 19.2, 7.2 Hz, 2H), 7.06 (d, = 5.7 Hz, 1H), 6.76 (d, = 3.5 Hz, 1H), 6.47 (dd, = 17.1, 10.2 Hz, 1H), 6.20 (m, 2H), 5.66 (d, = 11.8 Hz, 1H), 3.97 (t, = 7.0 Hz, 2H), 3.78 (s, 3H), 3.59 (t, = 6.6 Hz, 2H), 3.08 (m, 1H), 2.09 (s, 6H)。 实施例 36: (S)-N-(5-((4-(4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-(3- (二甲胺基)四氢吡咯 -1- 基) - 4- 甲 氧基苯基)丙烯酰胺 ( (S)-N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)- 2-(3 -(dimethylamino)pyrrolidin- 1 -yl)-4-methoxyphenyl)acrylamide ) (化合物 78)的制备 (按照方 案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 (SH^- - IH-吲哚 -1-基)嘧啶 -2- 基) -4-(3- (二甲胺基)四氢吡咯 -1-基) - 6-甲氧基苯 -1, 3-二胺 (403-C-78 ) (130毫克, 0.293毫摩 尔, 1当量), 得到黄色固体 (S)-N-(5-((4-(4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-(3- (二甲胺基)四 氢吡咯 -1-基;) - 4-甲氧基苯基)丙烯酰胺 (100毫克, 收率: 68.49%)。
该化合物 78的表征数据为: LCMS (ESI): m々 498 [M + H ]+; 熔点: 161-162摄氏度; 1H雇 R (500 MHz, DMSO-de) δ 9.32 (s, 1H), 8.43 (s, 2H), 8.35 (d, J = 5.6 Hz, 1H), 8.08 (d, J =
3.5 Hz, 1H), 7.58 (d, = 8.5 Hz, 2H), 7.16 (d, = 3.6 Hz, 2H), 7.07 (d, = 5.7 Hz, 1H), 6.76 (d, = 3.4 Hz, 1H), 6.51 (m, 2H), 6.17 (d, = 17.0 Hz, 1H), 5.66 (d, = 10.3 Hz, 1H), 3.79 (s, 3H), 3.39 (dd, J = 15.9, 9.0 Hz, 1H), 3.22 (dd, J = 16.5, 7.4 Hz, 3H), 2.71 (s, 1H), 2.18 (s, 6H), 2.09 (s, 1H), 1.74 (dd, = 20.0, 10.1 Hz, 1H)。 实施例 37: (R)-N-(5-((4-(4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-(3- (二甲胺基)四氢吡咯 -1- 基)- 4-甲氧基苯基)丙烯酰胺 ((R)-N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)-2- (3-(dimethylamino)pyrrolidin-l-yl)-4-methoxyphenyl)acrylamide) (化合物 79)的制备 (按照方案 六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 (RH^- - IH-吲哚 -1-基)嘧啶 -2- 基) -4-(3- (二甲胺基)四氢吡咯 -1-基) - 6-甲氧基苯 -1, 3-二胺 (403-C-79) (135毫克, 0.305毫摩 尔, 1当量), 得到黄色固体 (R)-N-(5-((4-(4-(lH-吲哚 -1-基)嘧啶 -2-基)胺基) -2-(3- (二甲胺基)四 氢吡咯 -1-基) - 4-甲氧基苯基)丙烯酰胺 (69毫克, 收率: 56.67%)。
该化合物 79的表征数据为: LCMS (ESI): m々 498 [M + H ]+; 熔点: 161-162摄氏度; 1H雇 R (500 MHz, DMSO-de) δ 9.32 (s, 1H), 8.43 (s, 2H), 8.35 (d, J = 5.6 Hz, 1H), 8.08 (d, J =
3.6 Hz, 1H), 7.58 (d, J = 9.0 Hz, 2H), 7.16 (m, 2H), 7.07 (d, J = 5.7 Hz, 1H), 6.76 (d, J = 3.5 Hz, 1H), 6.51 (m, 2H), 6.17 (d, = 17.1 Hz, 1H), 5.67 (d, = 10.3 Hz, 1H), 3.79 (s, 3H), 3.39 (dd, = 15.9, 9.2 Hz, 1H), 3.22 (dd, = 16.3, 7.3 Hz, 3H), 2.71 (m, 1H), 2.18 (s, 6H), 2.09 (s, 1H), 1.74 (dd, = 20.4, 9.6 Hz, 1H)。 实施例 38: Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基氨基 ]-4-甲氧基 -2-{甲基 [2- (吡咯垸 -1-基)乙基:
氨 基 } 苯 基 } 丙 烯 酰 胺 ( N-(5-(4-(lH-indol-l-yl)pyrimidin-2-ylamino)-4-methoxy- 2-(methyl(2-(pyrrolidin-l-yl)ethyl)amino)phenyl)acrylamide) (化合物 80)的制备(按照方案六线 路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 Ν4-[4-(1Η-吲哚 -1-基) 嘧啶 -2- 基] -5-甲氧基 -N1-甲基 -N^2- (吡咯垸 -1-基) 乙基] 苯基 -I,2,4-三胺(403-B-80 ) ( 169毫克, 0.369 毫摩尔, 1.0 当量), 得到黄色固体 Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基氨基 ]-4-甲氧基 -2-{甲基 [2- (吡咯垸 -1-基)乙基]氨基 }苯基 }丙烯酰胺 (25毫克, 收率: 13.3%)。
该化合物 80的表征数据为: LCMS (ESi;>: m/z 512 [M + H ]+ ; 熔点: 151-153摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.59 (s, 1H), 8.48 (s, 1H), 8.39 (d, J = 5.6 Hz, 2H), 8.13 (d, = 3.7 Hz, 1H), 7.58 (d, = 7.6 Hz, 1H), 7.11 (ddd, = 30.8, 17.7, 11.4 Hz, 4H), 6.76 (d, = 3.6 Hz, 1H), 6.45 (m, 1H), 6.19 (d, J = 16.9 Hz, 1H), 5.73 (m, 1H), 3.78 (s, 3H), 3.00 (s, 2H), 2.74 (s, 3H), 2.50 (d, = 1.7 Hz, 6H), 1.73 (s, 4H)。 实施例 39: N-{5-(4-吲哚 -1-基 -嘧啶 -2-基氨基 )-4-甲氧基 -2- [甲基 -(2-吗啉 -4-基-乙基) -氨 基 ] - 苯 基 } - 丙 烯 酰 胺 (N- { 5-(4-Indol- 1 -yl-pyrimidin-2-ylamino)-4-methoxy-2-[methyl- (2-morpholin-4-yl-ethyl)-amino]-phenyl}-acrylamideX化合物 82)的制备 (按照方案六线路制备) 合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N4-(4-吲哚 -1-基 -嘧啶 -2-基) -5-甲 氧基 -N1-甲基 -^—(2-吗啉 -4-基-乙基) -苯基 -1,2,4-三胺 (403-B-82 ) ( 0.12克, 0.25毫摩尔), 得 到黄色固体 Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基氨基 ]-4-甲氧基 -2-{甲基 [2- (吡咯垸 -1-基)乙基]氨 基 }苯基 }丙烯酰胺 (25毫克, 收率: 13.3%)。
该化合物 82 的表征数据为: LCMS (ESI): mlz 528[M + H ]+; 1H MR (500 MHz, DMSO-de) δ 9.29 (s, 1H), 8.58 (s, 1H), 8.39 (m, 3H), 8.12 (d, = 3.7 Hz, 1H), 7.58 (d, = 7.2 Hz, 1H), 7.12 (m, 3H), 7.01 (s, 1H), 6.76 (d, = 3.6 Hz, 1H), 6.61 (dd, = 16.9, 10.2 Hz, 1H), 6.19 (dd, J = 17.0, 1.7 Hz, 1H), 5.73 (d, = 10.6 Hz, 1H), 3.79 (s, 3H), 3.55 (m, 4H), 3.03 (t, = 6.6 Hz, 2H), 2.74 (s, 3H), 2.41 (t, = 6.6 Hz, 2H), 2.34 (s, 4H)。 实施例 40: Ν-(5-((4-(1Η-吲哚 -1-基)嘧啶 -2-基)胺基)- 4-甲氧基 -2-(4-甲基哌嗪 -1-基)苯基) 丙炼酰胺 N-(5-((4-(lH-indol-l-yl)pyrimidin-2-yl)amino)-4-methoxy-2-(4-methylpiperazin-l-yl) phenyl)acrylamide (化合物 84)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^- 二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 ^-(4-(1 吲哚 -1-基)嘧啶 -2-基) -6-甲氧基 -4-(4-甲基哌嗪 -1-基)苯 -1,3-二胺 (403-C-84 ) ( 180毫克, 0.41毫摩尔, 1.0当量), 得到浅黄色固体 Ν-(5-((4-(1Η-吲哚 -1-基;)嘧啶 -2-基)胺基) - 4-甲氧基 -2-(4-甲基哌嗪小基)苯基) 丙烯酰胺 (40毫克, 收率: 20%)。
该化合物 84的表征数据为: LCMS (ESI): m/z 484[M + H ]+ ; 熔点: 204-205摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.63 (s, 1H), 8.39 (t, J = 8.8 Hz, 2H), 8.21 (s, 1H), 8.12 (d, = 3.5 Hz, 1H), 7.58 (d, = 7.3 Hz, 1H), 7.14 (m, 3H), 6.90 (s, 1H), 6.77 (d, = 3.4 Hz,
1H), 6.62 (dd, = 16.9, 10.2 Hz, 1H), 6.18 (d, = 17.1 Hz, 1H), 5.71 (d, = 10.3 Hz, 1H), 3.78 (s: 3H), 2.91 (s, 4H), 2.58 (s, 4H), 2.28 (s, 3H)。 实施例 41 : N-(2- ((2-二甲胺乙基 X甲基)胺基) -4-甲氧基 -5-(4-(5-甲氧基 -1H-吡咯 [3,2-b]吡 啶 -1-基)嘧啶 -2-胺基)-苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)- 4-methoxy-5-((4-(5-methoxy-lH-pyrrolo[3,2-b]pyridin-l-yl)pyrimidin-2-yl)amino)phenyl)acrylami de ) (化合物 20)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^-二 甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N1-^-二甲胺乙基) - 5-甲氧基- N4-(4-(5-甲氧基 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-基) -N1-甲基苯 -1,2,4-三胺 (403-A-20 ) (132 毫克, 0.286毫摩尔, 1当量),得到白色固体 N-(2- ((2-二甲胺乙基 X甲基)胺基) -4-甲氧基 -5-(4-(5- 甲氧基 -1H-吡咯 [3,2-b]吡啶 -1-基;)嘧啶 -2-胺基) -苯基;)丙烯酰胺 (90毫克, 收率: 61.05%)。
该化合物 20的表征数据为: LCMS (ESI): ιη/ζ5 Π [Μ + Η ]+。熔点 177-178摄氏度; 1H MR (500 MHz, DMSO-de) δ 10.05 (s, 1H), 8.71 (s, 1H), 8.61 (s, 1H), 8.49 (s, 1H), 8.40 (d, = 5.0 Hz, 1H), 8.32 (s, 1H), 7.14 (d, J = 5.1 Hz, 1H), 7.05 (s, 1H), 6.74 (s, 1H), 6.43 (m, 2H), 6.18 (d, J = 16.8 Hz, 1H), 5.72 (d, J = 9.8 Hz, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 2.91 (s, 2H), 2.75 (s, 3H), 2.34 (s, 2H), 2.21 (s, 6H)。 实施例 42: N-(2-((2-二甲胺乙基 X甲基)胺基)- 5-(4-(5-氟 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl)(methyl) amino)-5-((4-(5-fluoro-3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)ami no)-4-methoxyphenyl)acrylamide ) (化合物 50)的制备 (按照方案七线路制备)
步骤 42a: 5-氟N1-甲基苯-l,2-二胺(5-fluoro-N1-methylbenzene-l,2-diamme)(中间体501-50 ) 的制备: 将 2,4-二氟硝基苯 (10克, 62.9毫摩尔, 1.0当量)溶于四氢呋喃 (150毫升), 冰浴条 件下往里快速滴加 40%甲胺水溶液 (9.8克, 125.8毫摩尔, 2.0当量)。 室温反应 4小时, 液质 检测反应完全, 往里加入石油醚 (50毫升)和乙酸乙酯 (50毫升;), 用水洗、 2摩尔每升盐酸洗、 碳酸氢钠水溶液洗、 饱和食盐水洗, 无水硫酸钠干燥, 旋干溶剂得到黄色固体产物 5-氟 N- 甲基 -2-硝基苯胺 (10.7克, 收率: 100%)。 该化合物表征数据为: LCMS (ESI): m/z 171 [M + H ]+。 上述得到的化合物 (10.5克, 61.8毫摩尔, 1.0当量), 铁粉 (17.3克, 309毫摩尔, 5.0当 量), 氯化铵 (16.5克, 309毫摩尔, 5.0当量)和水 (50毫升)溶于乙醇 (200 毫升), 放到 85 °C油 浴中反应 3小时。液质检测反应完全, 将反应液冷却到室温, 倒入 200毫升水中, 再加入 200 毫升二氯甲垸, 搅拌 5分钟, 垫硅藻土过滤, 分液。 有机相用无水硫酸钠干燥, 旋干溶剂, 得到红色液体产物 5-氟 N1-甲基苯 -1,2-二胺 (8.5克,收率: 98.2%)。该化合物表征数据为: LCMS (ESI): m/z 141 [M + H ]+。
步骤 42b : 6-氟 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同(6-fluoro-l -methyl- ΙΗ-benzo [d]imidazol- 2(3H)-one) (中间体 502-50)的制备:将 5-氟 N1-甲基苯 -1,2-二胺 (501-50) (4.2克, 30.0毫摩尔, 1.0当量)溶于二氯甲垸 (100毫升), 加入三乙胺 (6.1克, 60.0毫摩尔, 2.0当量), 氮气保护, 冰浴冷却到 0°C左右,往里滴加三光气 (3.1克, 10.5毫摩尔, 0.35当量)溶于二氯甲垸 (50毫升)
溶液, 温度控制小于 5度。 滴加完后, 冰浴控制温度 0°C左右反应半小时, 液质检测反应完 全,加入 30毫升甲醇,搅拌 10分钟后,倒入 100毫升水中,分液,水相用二氯甲焼 /甲醇 =3/1 (100 毫升)萃取, 有机相合并, 无水硫酸钠干燥, 旋干, 加入 100毫升二氯甲垸析晶, 过滤, 滤饼 二氯甲垸洗涤, 真空干燥得到粉色固体产物 6-氟 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (4.2 克, 收率: 84.0%)。 该化合物表征数据为: LCMS (ESI): m/z l67 [M + H ]+。
步骤 42c: 1 - (2-氯嘧啶 -4-基) -5-氟 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (l-(2-chloropyrimidin-4-yl)-5-fluoro-3-methyl-lH-benzo[d]imidazol-2(3H)-one) (;中间体 503-50) 的制备: 将 6-氟 -1-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (502-50) (7.0克, 42.2毫摩尔, 1.0 当量) 和 2,4-二氯嘧啶 (9.4克, 63.3毫摩尔, 1.5当量)溶于 DMF (200毫升), 往里加入碳酸铯 (20.7 克, 63.3毫摩尔, 1.5当量), 水浴冰水浴控制温度小于 20 °C反应 4小时。 倒入 600毫升水中, 搅拌 30分钟 (冷却到室温), 过滤, 残留物用水洗, 200毫升石油醚洗涤, 200毫升甲醇洗, 收集固体, 用石油醚 /乙酸乙酯 =2/1(150毫升)加热打浆, 冷却到室温完全析晶, 过滤, 滤饼用 石油醚 /乙酸乙酯 =2/1溶液洗涤, 干燥得到白色固体 1 - (2-氯嘧啶 -4-基) -5-氟 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (10.1克, 收率: 86.3%)。 该化合物表征数据为: LCMS (ESI): m/z 279 [M + H ]+。
步骤 42d: 1-(2-(4-《2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -5-氟 -3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 ( l-(2-((4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxy- 5-nitrophenyl)amino)pyrimidin-4-yl)-5-fluoro-3-methyl-lH-benzo[d]imidazol- 2(3H)-one) (中 I司体 505-A-50)的制备: 将 1 - (2-氯嘧啶 -4-基) -5-氟 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (503-50) (10 克, 35.9毫摩尔, 1.0当量)和 4-氟 -2-甲氧基 -5-硝基苯胺 (103) (7.3克, 39.5毫摩尔, 1.1当量) 溶于异丙醇 (400毫升)溶液, 往里滴加浓盐酸 (8毫升;), 升温到 85 °C反应 48小时。液质检测反 应完全, 冷却到室温, 加入 100毫升水, 再加入 2摩尔每升的氢氧化钠水溶液调节 PH>8, 搅 拌 10分钟, 过滤, 滤饼用甲醇洗、 乙酸乙酯洗, 真空干燥得到淡黄色固体产物 1-(2-(4-氟 -2- 甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -5-氟 3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (12.0克,收率: 77.9%)。 该化合物表征数据为: LCMS (ESI): m/z 429 [M + H ]+。 上述得到的化合物 (6.5克, 15.2毫摩 尔, 1.0当量), Ν,Ν,Ν-三甲基乙二胺 (3.1克, 30.4毫摩尔, 2.0当量)和 Ν,Ν-二异丙基乙胺 (5.9 克, 45.6毫摩尔, 3.0当量)溶于 100 毫升四氢呋喃, 再加入 Ν-甲基吡咯垸酮 (20毫升)。 将 反应体系加热至回流, 搅拌反应 18小时。 冷却至室温, 加入 100毫升乙酸乙酯后用水洗, 饱和食盐水洗, 无水硫酸镁干燥, 旋干溶剂得到黄色粗品, 粗品用甲醇打浆, 过滤, 滤饼用 甲醇洗涤得到黄色固体 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4- 基) -5-氟 -3-甲基 -1Η-苯并 [d]咪唑 -2(3Η)-酮 (7.0克,收率: 90.9%)。该化合物表征数据为: LCMS (ESI): m/z 511 [M + H ]+。
步骤 42e: 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3- 甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 ( l-(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxyphenyl)amino)pyrimidin-4-yl)-5-fluoro-3-methyl-lH-benzo[d]imidazol-2(3H)-one ) (中 间体 506-A-50)的制备: 将 1-(2-(4-《2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4- 基)—5-氟 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (505-A-50) (7.0克, 13.7毫摩尔, 1.0当量),铁粉 (3.8 克, 68.5毫摩尔, 5.0当量),氯化铵 (3.7克, 68.5毫摩尔, 5.0当量)和水 (20毫升)溶于乙醇 (100
毫升), 升温到 85 °C反应 3小时。液质检测反应完全, 得 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基;)嘧啶 -4-基;) -5-氟 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮。 该化合物表征数据 为: LCMS (ESI): m/z481 [M + H ]+。
步骤 42f: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 - 1 -基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl) amino)-5-((4-(5-fluoro-3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)ami no)-4-methoxyphenyl)acrylamide ) (化合物 50)的制备: 方法 1 : 上述得到的 1-(2-(5-胺基 -4-((2- 二甲胺乙基 X甲基;)胺基; )-2-甲氧基苯胺基)嘧啶 -4-基;) -5-氟 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 506-A-50)反应液冰盐浴降温冷却到 0°C左右后, 滴加 3-氯丙酰氯 (3.5克, 27.4毫摩尔, 2.0 当量)溶于四氢呋喃 (20毫升)溶液。 滴加完后, 温度控制在 0°C左右搅拌 30分钟。 液质检测 未完全反应, 加入 Ν,Ν-二异丙基乙胺 (5.3克, 41.1毫摩尔, 3.0当量), 搅拌 10分钟后, 补加 3-氯丙酰氯 (3.5克, 27.4毫摩尔, 2.0当量)溶于四氢呋喃 (20毫升)溶液。 滴加完后, 温度控 制在 0°C左右搅拌 30分钟。 液质检测反应完全, 加入 200毫升水和 200毫升二氯甲垸, 搅拌 10分钟, 布氏漏斗中垫硅藻土过滤, 分液, 水相再用 100毫升二氯甲垸萃取, 合并有机相, 旋干溶剂得到灰白色残留物。 残留物用 100毫升乙酸乙酯打浆, 过滤, 乙酸乙酯洗涤, 得到 白色中间体。 将中间体溶于 100毫升乙腈, 再往里加入 10毫升三乙胺, 回流反应 18小时。 液质检测反应完全, 冷却到室温, 加入 100毫升水, 搅拌 1小时使固体充分析出, 过滤, 滤 饼用水洗、 石油醚洗, 把滤饼溶于 50毫升二氯甲垸, 无水硫酸钠干燥, 过滤, 旋干溶剂得到 白色固体产物 N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (4.7克, 收率: 75.1%)。 方法 2: 将 1-(2-(5- 胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 ( 506-A-50) ( 100 毫克, 0.21 毫摩尔, 1 当量), 2-(7-偶氮苯并三氮唑) -Ν,Ν,Ν',Ν'- 四甲基脲六氟磷酸酯 (103毫克, 0.27毫摩, 1.3当量) 和三乙胺 (64毫克, 0.63毫摩尔, 3 当量) 溶于二氯甲垸 (20毫升) 中, 将丙烯酸 (18毫克, 0.252毫摩尔, 1.2当量) 溶于二氯 甲垸 (1 毫升) 缓慢滴入反应, 搅拌 2小时后, 将反应液浓缩, 通过硅胶柱层析 (洗脱剂: 二氯甲焼 /甲醇 =100/1 )分离提纯得到白色固体 Ν-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3- 甲基 -2-氧代 -2,3-二氢 -1Η-苯并 [d]咪唑 -1-基;)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (80毫克, 收率: 71.4%)
该化合物 50的表征数据为: LCMS (ESI): /ii/z 535 [M + H ]+。 熔点: 208-210摄氏度; 1H 雇 R (500 MHz, DMSO-de) δ 10.01 (s, 1H), 8.72 (s, 1H), 8.42 (m, 2H), 8.11 (s, 1H), 7.66 (d, = 5.6 Hz, 1H), 7.20 (dd, J = 8.9, 2.6 Hz, 1H), 7.04 (s, 1H), 6.65 (t, J = 8.2 Hz, 1H), 6.40 (dd, J = 16.9, 10.2 Hz, 1H), 6.18 (dd, J = 17.0, 1.9 Hz, 1H), 5.72 (dd, J = 10.2, 1.7 Hz, 1H), 3.75 (d, J = 4.4 Hz, 3H), 3.34 (d, = 8.6 Hz, 3H), 2.91 (t, = 5.9 Hz, 2H), 2.75 (s, 3H), 2.34 (dd, = 11.2, 5.4 Hz, 2H), 2.20 (s, 6H)。 实施例 43: N-(2-((2-二甲胺乙基 X甲基)胺基) -4-甲氧基 -5-(4-(3-(3-甲基 -2-丁烯) -2-氧代 -2,3- 二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino) ethyl)(methyl)amino)-4-methoxy-5-(4-(3-(3-methylbut-2-enyl)-2-oxo-2,3-dihydro-lH-benzo[d]imi
dazol-l-yl)pyrimidin-2-ylamino)phenyl)acrylamide) (化合物 62)的制备 (按照方案七线路制备) 合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧基 苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -3-(3-甲基 -2-丁烯) -1H-苯并 [d]咪唑 -2(3H)-酉同 (506-A-62) (110毫克, 0.2毫摩尔, 1.0当量), 得到米黄色固体 N-(2-((2-二甲胺乙 基 X甲基)胺基) -4-甲氧基 -5-(4-(3-(3-甲基 -2-丁烯) -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 (60毫克, 收率: 49%)。
该化合物 62的表征数据为: LCMS (ESI): m/z 571 [M + H ]+。 熔点: 187-188摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.70 (s, 1H), 8.42 (d, J = 5.5 Hz, 2H), 8.10 (s, 1H), 7.67 (d, J = 5.4 Hz, 1H), 7.13 (d, J = 10.0 Hz, 2H), 7.04 (s, 1H), 6.88 (s, 1H), 6.39 (dd, J = 16.8, 10.2 Hz, 1H), 6.18 (d, = 16.8 Hz, 1H), 5.72 (d, = 10.1 Hz, 1H), 5.24 (s, 1H), 4.47 (d, = 6.2 Hz, 2H), 3.75 (s, 3H), 2.90 (s, 2H), 2.74 (s, 3H), 2.37 (d, J = 28.5 Hz, 2H), 2.21 (s, 6H), 1.80 (d, J = 20.1 Hz, 3H), 1.68 (s, 3H)。 实施例 44: N-(5-(4-(3-环丙甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1 -基)嘧啶 -2-胺基) -2-((2- 二甲胺乙基)(甲基)胺基 )-4-甲氧基苯基)丙烯酰胺 ( N-(5-((4-(3-(cyclopropylmethyl)- 2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-2-((2-(dimethylamino)ethyl)( methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 63)的制备 (按照方案七线路制备)
合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧基 苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) - 3-环丙甲基 -1,3-二氢 -2H-苯并 [d] 咪唑 -2-酮 (506-A-63 ) ( 529毫克, 1.05毫摩尔, 1当量), 得到米黄色固体 N-(5-(4-(3-环丙甲 基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -2-((2-二甲胺乙基 X甲基)胺基) -4-甲氧 基苯基)丙烯酰胺 (300毫克, 收率: 51.4%)。
该化合物 63的表征数据为: LCMS (ESI): m/z 557 [M + H ]+。熔点: 68-72摄氏度; 1H MR (500 MHz, DMSO-de) δ 10.04 (s, 1H), 8.70 (s, 1H), 8.43 (d, J = 5.6 Hz, 2H), 8.10 (d, = 7.1 Hz, 1H), 7.67 (d, = 5.6 Hz, 1H), 7.30 (d, = 7.9 Hz, 1H), 7.15 (t, = 7.7 Hz, 1H), 7.05 (s, 1H), 6.89 (t, = 7.8 Hz, 1H), 6.39 (dd, = 16.9, 10.2 Hz, 1H), 6.18 (d, = 16.9 Hz, 1H), 5.72 (d, = 10.3 Hz, 1H), 3.77 (m, 5H), 2.91 (t, J = 5.5 Hz, 2H), 2.75 (s, 3H), 2.34 (s, 2H), 2.21 (s, 6H), 0.47 (m, 2H), 0.40 (m, 2H)。 实施例 45 : (Ε)-Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-胺基] -2-{ [2- (二甲基胺基)乙基] (甲基)胺 基 }-4-甲基苯 }-4-甲氧基丁 -2-烯酰胺 ((E)-N-(5-(4-(lH-indol-l-yl)pyrimidin-2-ylamino)- 2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)-4-methoxybut-2-enamide ) (化合 物 71)的制备 (按照方案六线路制备)
将化合物 Ν4-(4-(1Η-吲哚 -1-基)嘧啶 -2-基) -Nl-(2-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4- 三胺 (403-1 ) ( 200毫克, 0.464毫摩尔, 1.0当量) 溶解于乙腈 (50毫升)中, 然后加入 Ν,Ν- 二异丙基乙基胺 (120毫克, 0.928毫摩尔, 2当量)、 (Ε)-4-溴 -2-丁烯酸 (602-71 ) (92毫克, 0.5568毫摩尔, 1.2当量)和 2-(7-偶氮苯并三氮唑) -Ν,Ν,Ν',Ν'-四甲基脲六氟磷酸酯(211毫克,
0.5568毫摩尔, 1.2当量), 然后在室温下搅拌 4个小时。 当反应结束时, 加入碳酸钠水溶液, 再用二氯甲垸(100毫升 X 3 )萃取, 将有机层合并, 真空浓缩。将残余物通过柱层析 (洗脱剂: 二氯甲垸 /甲醇 50/1 至 30/1)纯化, 得到黄色油状物 (Ε)-Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-胺 基] -2-{ [2- (二甲基胺基)乙基] (甲基)胺基 }-4-甲基苯基}-4-溴丁 -2-烯酰胺 (200 毫克,收率: 74.5%)。 MS (ESI): m々578 (M+l)+.将上述所得的化合物 (200毫克, 0.346毫摩尔, 1.0当量) 溶解在甲醇 (25毫升) 中, 加入甲醇钠 (38毫克, 0.692毫摩尔, 2.0当量), 然后在室温下 搅拌过夜。 当反应结束时, 加入水, 再用二氯甲垸 (100毫升 x 3 ) 萃取, 将有机层合并, 真 空浓缩。 将残余物通过柱层析(洗脱剂: 二氯甲垸 /甲醇 50/1)纯化, 得到黄色固体 (Ε)-Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-胺基] -2-{ [2- (二甲基胺基)乙基] (甲基)胺基 }-4-甲基苯 }-4-甲 氧基丁 -2-烯酰胺 (40毫克, 收率: 21%)。
该化合物 71的表征数据为: LCMS (ESI): m/z530 [M + H ]+。熔点: 193-200摄氏度; 1H MR (500 MHz, CDC13) δ 9.51 (s, 1H), 8.48 (d, J = 5.6 Hz, 1H), 8.24 (d, J = 8.3 Hz, 1H), 8.12 (s, 1H), 7.62 (d, = 7.4 Hz, 2H), 7.28 (m, 3H), 7.21 (t, = 7.2 Hz, 1H), 6.97 (m, 2H), 6.74 (m, 2H), 4.16 (d, = 2.8 Hz, 2H), 3.90 (s, 3H), 3.43 (s, 3H), 3.08 (s, 2H), 2.64 (m, 11H)。 实施例 46: Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基胺基 ]-4-甲氧基 -2-{甲基 [2- (哌啶 -1-基)乙基] 胺 基 } 苯 基 } 丙 烯 酰 胺 (N-(5-(4-(lH-indol-l-yl)pyrimidin-2-ylamino)-4-methoxy-2- (methyl(2-(piperidin -l-yl)ethyl)amino)phenyl)acrylamide)(化合物 81)的制备 (按照方案六线路 制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^-二 甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 Ν4-[4-(1Η-吲哚 -1-基)嘧啶 -2-基] -5- 甲氧基 -N1-甲基 -^-[2- (哌啶 -1-基)乙基]苯 -1,2,4-三胺 ( 403-B-81 ) ( 150毫克, 0.318毫摩尔, 1.0 当量), 得到黄色固体 Ν-{5-[4-(1Η-吲哚 -1-基)嘧啶 -2-基胺基 ]-4-甲氧基 -2-{甲基 [2- (哌啶 -1-基) 乙基]胺基 }苯基 }丙烯酰胺 (25毫克, 收率: 10.3%)。
该化合物 81的表征数据为: LCMS (ESI): m/z 526 [M + H ]+; 熔点: 131-136摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.57 (s, 1H), 8.39 (m, 3H), 8.12 (d, = 3.7 Hz, 1H), 7.58 (d, J = 7.2 Hz, 1H), 7.12 (m, 3H), 7.00 (s, 1H), 6.76 (d, J = 3.6 Hz, 1H), 6.59 (dd, J = 16.8, 10.2 Hz, 1H), 6.19 (m, 1H), 5.73 (m, 1H), 3.79 (s, 3H), 3.03 (d, = 5.1 Hz, 2H), 2.72 (s, 3H), 2.35 (m, 6H), 1.49 (s, 4H), 1.36 (d, = 19.9 Hz, 2H)。 实施例 47: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3-异丙基 -2-氧代 -2,3-二氢 -1H-苯 并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl)(methyl) amino)-5-(4-(5-fluoro-3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-ylami no)-4-methoxyphenyl)acrylamide) (化合物 91)的制备 (按照方案七线路制备)
步骤 47a: 5-氟 N1-异丙基苯 -1,2-二胺 (S-fluoro-N^isopropylbenzene-l^diamine) (中间体 501-91 ) 的制备: 将 2, 4-二氟硝基苯(10克, 62. 9毫摩尔, 1. 0当量), 异丙胺(4. 8克, 81. 8 毫摩尔, 1. 5当量)和碳酸钾(17. 4克, 125. 8毫摩尔, 2. 0当量)溶于 DMF (150毫升), 室温反 应 18小时。 液质检测反应完全, 倒入 400毫升水中, 搅拌 30分钟充分析晶, 过滤, 滤饼用
水洗, 收集滤饼真空干燥得到黄色固体产物 5-氟 N-异丙基 -2-硝基苯胺胺 (11. 5克, 收率: 92. 4%)。 该化合物表征数据为: LCMS CESi;>: m/z199[M + H ]+。 将上述得到的化合物 (6.1克, 30.8毫摩尔, 1.0当量;), 铁 (8.6克, 154毫摩尔, 5.0当量;), 氯化铵 (8.2克, 154毫摩尔, 3.0 当量)和水 (20毫升)溶于乙醇 (100 毫升), 氮气保护条件下, 升温到 85 °C反应 3小时。 液质检 测反应完全, 将反应液冷却到室温, 倒入 200毫升水中, 再加入 200毫升二氯甲垸, 搅拌 5 分钟, 垫硅藻土过滤, 分液。 有机相用无水硫酸钠干燥, 旋干溶剂, 得到红色液体产物 5-氟 N1-异丙基苯 -1,2-二胺 (4.5克, 收率: 86.5%)。该化合物表征数据为: LCMS (ESI): m/z169[M + H ]+。
步骤 47b: 6-氟 -1-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (6-fluoro-l-isopropyl-lH-benzo
[d]imidazol- 2(3H)-one) (中间体 502-91)的制备: 将 5-氟 N1-异丙基苯 -1, 2-二胺 (501-91 ) (4. 5克, 26. 8毫摩尔, 1. 0当量)溶于二氯甲垸(100毫升), 加入三乙胺(5. 5克, 53. 6毫摩 尔, 2. 0当量),氮气保护,冰浴冷却到 0°C左右,往里滴加三光气(2. 8克, 9. 38毫摩尔, 0. 35 当量)溶于二氯甲垸 (50毫升)溶液, 温度控制小于 5°C。 滴加完后, 冰浴控制温度 0°C左右反 应半小时, 液质检测反应完全, 往里加入 10毫升甲醇搅拌 10分钟淬灭反应。 反应液用水洗, 饱和食盐水洗, 无水硫酸镁干燥, 旋干得到棕色固体产物 6-氟 -1-异丙基 -1H-苯并 [d]咪唑 -2 (3H) _酮(4. 6克, 收率: 88. 5%)。 该化合物表征数据为: LCMS (ESI): m/z195[M + H ]+。
步骤 47c: 1 - (2-氯嘧啶 -4-基) -5-氟 3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮
(l-(2-chloropyrimidin-4-yl)-5-fluoro-3-isopropyl-lH-benzo[d]imidazol-2(3H)-one) (中间体 503-91) 的制备: 将 6-氟 -1-异丙基 -1H-苯并 [d]咪唑 -2 (3H) _酮(502-91 ) (4. 6克, 23. 7毫摩尔, 1. 0 当量)和 2, 4-二氯嘧啶(5. 3克, 35. 6毫摩尔, 1. 5当量)溶于 DMF (80毫升), 往里加入碳酸 铯 (11. 6克, 35. 6毫摩尔, 1. 5当量), 室温反应过夜。倒入 500毫升水中, 搅拌 30分钟(冷 却到室温), 过滤, 残留物用水洗, 200毫升石油醚洗涤, 收集固体, 加入 100毫升甲醇打浆, 过滤, 滤饼用甲醇洗, 收集固体真空干燥得到粉红色产物 1 - (2-氯嘧啶 -4-基) -5-氟 3-异 丙基 -1H-苯并 [d]咪唑 -2 (3H) _酮(6. 1克, 收率: 83. 6%)。 该化合物表征数据为: LCMS (ESI): m/z307[M + H ]+。
步骤 47d: 1-(2-(4-《2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -5-氟 -3- 异丙基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (l-(2-((4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxy- 5-nitrophenyl)amino)pyrimidin-4-yl)-5-fluoro-3-isopropyl-lH-benzo[d]imidazol- 2(3H)-one) (中间体 505-A-91)的制备: 将 1 - (2-氯嘧啶 -4-基) -5-氟 3-异丙基 -1H-苯并 [d] 咪唑 -2 (3H) _酮 (503-91) (2. 8克, 9. 13毫摩尔, 1. 0当量)和 4-氟 -2-甲氧基 -5-硝基苯胺(103) (1. 87克, 10毫摩尔, 1. 1当量)溶于异丙醇(100毫升)溶液, 往里滴加浓盐酸(2毫升), 升 温到 85°C反应 18小时。 液质检测反应完全, 冷却到室温, 过滤, 滤饼用石油醚(100毫升) 洗涤, 收集固体, 真空干燥得到土色固体产物 1- (2- (4-氟 -2-甲氧基 -5-硝基苯胺基)嘧啶 -4- 基) -5-氟 3-异丙基 -1H-苯并 [d]咪唑 -2 (3H) _酮(3. 4克,收率: 81. 0%)。该化合物表征数据为: LCMS (ESI): m/z457[M + H ]+。 上述得到的化合物(3. 4克, 7. 46毫摩尔, 1. 0当量), N, N, N- 三甲基乙二胺 (1. 5克, 14. 92毫摩尔, 2. 0当量)和 N,N-二异丙基乙胺(2. 9克, 22. 38毫摩 尔, 3. 0当量)溶于 80 毫升四氢呋喃。 将反应体系加热至回流, 搅拌反应 18小时。 冷却至室 温, 加入 80毫升乙酸乙酯后用水洗, 饱和食盐水洗, 无水硫酸镁干燥, 旋干溶剂, 得到黄色
固体 l- (2- (4- ( (2-二甲胺乙基)(甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -5-氟 -3- 异丙基 -1H-苯并 [d]咪唑 -2 (3H) _酮(3. 7克,收率: 92. 5%)。该化合物表征数据为: LCMS (ESI): m/z539[M + H ]+。
步骤 47e: 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3- 异丙基 - 1H-苯并 [d]咪唑 -2(3H)-酮 ( 1 -(2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)amino)- 2-methoxyphenyl)amino)pyrimidin-4-yl)-5-fluoro-3-isopropyl-lH-benzo[d]imidazol-2(3H)-one ) (中间体 506-A-91)的制备:将 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-硝基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (505-A-91 ) (3.7克, 6.88毫摩尔, 1.0当量), 铁 (1.9克, 34.4毫摩尔, 5.0当量;), 氯化铵 (1.8克, 34.4毫摩尔, 3.0当量;)和水 (12毫升;)溶于 乙醇 (60毫升), 升温到 85 °C反应 3小时。 液质检测反应完全, 得 1-(2-(5-胺基 -4-((2-二甲胺乙 基 X甲基)胺基; )-2-甲氧基苯胺基;)嘧啶 -4-基;) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮。 该化合 物表征数据为: LCMS (ESI): m/z509 [M + H ]+。
步骤 47f: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl) (methyl)amino)-5-(4-(5-fluoro-3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin -2-ylammo)-4-methoxyphenyl)acrylamide)(化合物 91)的制备:
方法 1 : 上述得到的 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧基苯胺基;)嘧啶 -4- 基)—5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮(506-A-91 )反应液冰盐浴降温冷却到 0°C左右后, 滴加 3-氯丙酰氯 (1.8克, 13.76毫摩尔, 2.0当量)溶于四氢呋喃 (10毫升)。 滴加完后, 温度 控制在 0°C左右搅拌 30分钟。 液质检测完全反应, 加入 100毫升水和 100毫升二氯甲垸, 搅 拌 10分钟, 布氏漏斗中垫硅藻土过滤, 分液, 水相再用 50毫升二氯甲垸萃取, 合并有机相, 旋干溶剂得到乳白色残留物。 残留物用 50毫升乙酸乙酯打浆, 过滤, 乙酸乙酯洗涤, 得到白 色中间体。 将中间体溶于 50毫升乙腈, 再往里加入 5毫升三乙胺, 回流反应 18小时。 液质 检测反应完全, 冷却到室温, 倒入 300毫升水中, 搅拌 1小时使固体充分析出, 过滤, 滤饼 用水洗, 把滤饼溶于 100毫升二氯甲垸, 用饱和食盐水洗涤, 无水硫酸钠干燥, 旋干溶剂得 到白色固体产物 N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(5-氟 -3-异丙基 -2-氧代 -2,3-二氢 -1H-苯 并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (2.9克,收率: 75.1%)。方法 2:将 1 -(2-(5- 胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪 唑 -2(3H)-酮 (506-A-91) (400 毫克, 0.787毫摩尔, 1.0当量) 溶解于 50毫升二氯甲垸中, 加入 三乙胺 (0.328毫升, 2.36 毫摩尔, 3.0当量)。 冷却至 -70°C, 滴加溶于 10毫升二氯甲垸的 丙烯酰氯 (601-1) (109毫克, 1.18 毫摩尔, 1.5当量) 溶液, 反应 15分钟。 加入甲醇淬灭反 应, 然后用二氯甲垸萃取。 无水硫酸钠干燥, 减压浓缩, 残留物硅胶柱层析纯化 (洗脱剂: 二氯甲焼 /甲醇 400/1至 150/1 ) 得到白色固体 N-(2-((2-二甲胺乙基 X甲基)胺基)- 5-(4-(5-氟 -3- 异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基;)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (224 毫 克, 收率: 50%)。
该化合物 91的表征数据为: LCMS (ESI): m/z 563 [M + H ]+。 熔点: 225-227摄氏度; 1H 雇 R (500 MHz, DMSO-de) δ 10.01 (s, 1H), 8.70 (s, 1H), 8.41 (m, 2H), 8.11 (s, 1H), 7.62 (d, = 5.6 Hz, 1H), 7.32 (d, = 9.3 Hz, 1H), 7.04 (s, 1H), 6.65 (t, = 8.9 Hz, 1H), 6.40 (dd, = 16.9, 10.2
Hz, 1H), 6.18 (d, = 16.8 Hz, 1H), 5.72 (d, = 10.3 Hz, 1H), 4.64 (m, 1H), 3.76 (s, 3H), 2.90 (d, J = 5.2 Hz, 2H), 2.74 (s, 3H), 2.34 (s, 2H), 2.20 (s, 6H), 1.47 (d, J = 6.9 Hz, 6H)。 实施例 48 : N-{5-[4-(3-环丙基 -5-氟 -2-氧 -2,3-二氢苯并 [d]咪唑 -1-基)嘧啶 -2-基胺 基 ]-2-{ [2-(二 甲 胺 基) 乙 基 ] ( 甲 基) 胺 基 }-4- 甲 氧 基 苯基 } 丙 烯 酰胺 N-(5-(4-(3-cyclopropyl-5-fluoro-2-oxo-2,3-dihydrobenzo[d]imidazol-l-yl)pyrimidin-2-ylamino)-2- ((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide (化合物 98)的制备 (按照 方案七线路制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-{2-{5-胺基 -4-{ [2- (二甲胺基) 乙基: |( 甲基) 胺基 }-2-甲氧基苯胺基 } 嘧啶 -4-基}-3-环丙基 -5- 氟 -1H-苯并 [d]咪唑 -2(3H)- 酮 ( 506-A-98 ) (868毫克, 1.72毫摩尔, 1.0当量), 得到黄色固体 N-{5-[4-(3-环丙基 -5-氟 -2-氧 -2,3-二氢苯并 [d]咪唑 -1-基)嘧啶 -2-基胺基 ]-2-{ [2- (二甲胺基)乙 基: K甲基)胺基 }-4-甲氧基苯基)丙烯酰胺为黄色固体 (300毫克, 31.1%)。
该化合物 98的表征数据为: LCMS (ESI): m/z 561 [M + H ]+。 熔点: 181-184摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.69 (s, 1H), 8.41 (m, 2H), 8.08 (s, 1H), 7.62 (d, J = 5.6 Hz, 1H), 7.11 (dd, = 8.8, 2.6 Hz, 1H), 7.04 (s, 1H), 6.66 (t, = 8.4 Hz, 1H), 6.40 (dd, = 16.9, 10.2 Hz, 1H), 6.18 (dd, = 16.9, 1.8 Hz, 1H), 5.73 (dd, = 14.6, 4.4 Hz, 1H), 3.75 (s, 3H), 2.91 (t, J = 52 Hz, 3H), 2.74 (s, 3H), 2.34 (s, 2H), 2.20 (s, 6H), 1.07 (m, 2H), 0.93 (m, 2H)。 实施例 49 : N-(5-((4-(5-氯 -3-环丙基 -2-氧代 -2,3-二氢 -IH-苯并咪唑 -1-基)嘧啶 -2-基)氨 基)-2-((2-(二甲氨基)乙基 X甲基)氨基)-4-甲氧基苯基)丙烯酰胺 ( N-(5-((4-(5-chlor0- 3-cyclopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-2-((2-(dimethyl amino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 99)的制备(按照方案七线路 制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-((5-氨基 -4-((2- (二甲氨基)乙基)甲基)氨基) -2-甲氧基苯基)氨基)嘧啶 -4-基) -5-氯 -3-环丙基 -1,3-二氢 -2H-苯并 [d]咪唑 -2-酮(506-A-99 ) ( 160毫克, 0.31毫摩尔),得到黄色固体 N-(5-((4-(5- 氯 -3—环丙基 -2—氧代—2,3—二氢- 1H-苯并咪唑 -1-基)嘧啶 -2-基)氨基) -2-((2- (二甲氨基)乙基 X甲基) 氨基)—4-甲氧基苯基)丙烯酰胺 (40毫克, 23%)。
该化合物 99的表征数据为: LCMS (ESI): /ii/z 577 [M + H ]+。 熔点: 124-126摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.03 (s, IH), 8.70 (s, IH), 8.42 (m, 2H), 8.04 (s, IH), 7.60 (d, J = 5.2 Hz, IH), 7.26 (s, IH), 7.05 (s, IH), 6.87 (d, = 8.1 Hz, IH), 6.39 (dd, J = 16.6, 10.3 Hz, IH), 6.18 (d, = 16.8 Hz, IH), 5.72 (d, = 9.9 Hz, IH), 3.75 (s, 3H), 2.91 (s, 3H), 2.74 (s, 3H), 2.34 (s, 2H), 2.21 (s, 6H), 1.07 (d, = 5.5 Hz, 2H), 0.93 (s, 2H)。 实施例 50 : N-(5-(4-(5-氰基 -3-环丙基 -2-氧代 -2,3-二氢 -IH-苯并 [d]咪唑 -1-基)嘧啶 -2-胺 基)-2-((2-二甲胺乙基)(甲基)胺基)-4-甲氧基苯基)丙烯酰胺 ( N-(5-((4-(5-Cyan0-
3-cyclopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-2-((2-(dimethyl amino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide ) (化合物 101)的制备(按照方案七线 路制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -3-环丙基 -2-氧代 -2,3- 二氢 -1H-苯并 [d]咪唑 -5-氰基 (506-A-101 ) ( 200毫克, 0.39毫摩尔, 1.0当量), 得到淡黄色 固体 N-(5-(4-(5-氰基 -3-环丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -2-((2-二甲 胺乙基 X甲基)胺基; )-4-甲氧基苯基)丙烯酰胺 (130毫克, 收率: 56%)。
该化合物 101的表征数据为: LCMS (ESI): m/z568 [M + H ]+。 熔点: 177-179摄氏度; 1H MR (500 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.77 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.30 (s, 1H), 8.21 (s, 1H), 7.68 (s, 1H), 7.57 (d, = 5.5 Hz, 1H), 7.35 (d, = 7.2 Hz, 1H), 7.03 (s, 1H), 6.56 (s, 1H), 6.19 (d, = 16.8 Hz, 1H), 5.72 (d, = 10.3 Hz, 1H), 3.78 (s, 3H), 2.97 (m, 3H), 2.65 (m, 5H), 2.36 (s, 6H), 1.09 (d, = 5.8 Hz, 2H), 0.95 (s, 2H)。 实施例 51 : N-(5-(4-(3-环丙基 -5-甲氧基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺 基)-2-((2-二甲胺乙基 X甲基)胺基 )-4-甲氧基苯基)丙烯酰胺 ( N-(5-((4-(3-Cycl0piOpyl- 5-methoxy-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-2-((2-(dimethylam ino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide) (化合物 102)的制备 (按照方案七线路 制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基;)胺基; )-2-甲氧基苯胺基)嘧啶 -4-基; )-3-环丙基 -5-甲氧基 1,3-二氢 -2H-苯并 [d]咪唑 -2-酮 (506-A-102 ) (420毫克, 0.81毫摩尔, 1.0当量), 得到米黄色 固体 N-(5-(4-(3-环丙基 -5-甲氧基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -2-((2-二 甲胺乙基 X甲基)胺基; )-4-甲氧基苯基)丙烯酰胺 (250毫克, 收率: 54%)。
该化合物 102的表征数据为: LCMS (ESI): m/z 573 [M + H ]+。 熔点: 144-146摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.63 (s, 1H), 8.39 (m, 2H), 7.96 (d, J = 8.5 Hz, 1H> 7.64 (d, = 5.6 Hz, 1H), 7.05 (s, 1H), 6.80 (d, = 2.3 Hz, 1H), 6.39 (m, 2H), 6.18 (dd, = 16.9, 1.4 Hz, 1H), 5.72 (d, = 11.1 Hz, 1H), 3.75 (d, = 7.1 Hz, 6H), 2.89 (m, 3H), 2.75 (s, 3H), 2.34 (t, = 5.7 Hz, 2H), 2.20 (s, 6H), 1.07 (q, = 6.9 Hz, 2H), 0.92 (m, 2H)。 实施例 52 : N-(5-(4-(3-环丙甲基 -5-氟 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1 -基)嘧啶 -2-胺 基) -2-((2-二甲胺乙基 )(甲基)胺基) -4-甲氧基苯基)丙烯酰胺 (N-(5-(4-(3-(cyclopropylmethyl)- 5-fluoro-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-ylamino)-2-((2-(dimethylamino )ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide ) (化合物 105)的制备(按照方案七线路制备) 合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基苯胺基)嘧啶 -4-基) - 3-环丙甲基 -5-氟 -1H-
苯并 [d]咪唑 -2(3H)-酮(506-A-105 )(520 毫克, 1毫摩尔, 1.0当量),得到米黄色固体 N-(5-(4-(3- 环丙甲基 -5-氟 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑小基)嘧啶 -2-胺基) -2-((2-二甲胺乙基 X甲基)胺 基) -4-甲氧基苯基)丙烯酰胺 (180毫克, 收率: 31%)。
该化合物 105的表征数据为: LCMS (ESI): m/z 563 [M + H ]+。 熔点: 168-169摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.72 (s, 1H), 8.42 (m, 2H), 8.11 (s, 1H), 7.65 (d, J = 4.9 Hz, 1H), 7.31 (d, = 7.8 Hz, 1H), 7.05 (s, 1H), 6.66 (s, 1H), 6.40 (dd, = 16.3, 10.2 Hz, 1H), 6.18 (d, = 16.9 Hz, 1H), 5.72 (d, = 9.6 Hz, 1H), 3.76 (s, 5H), 2.91 (s, 2H), 2.75 (s, 3H), 2.34 (s, 2H), 2.20 (s, 6H), 1.23 (s, 1H), 0.45 (m, 4H)。
实施例 53: N-(2-((2-二甲胺乙基 X甲基)胺基) -4-甲基 -5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H-苯 并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl) (methyl)amino)-4-methyl-5-((4-(3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin- 2-yl)amino)phenyl)acrylamide ) (化合物 118)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲基苯胺基)嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酉同 (506-A-118 ) ( 72毫克, 0.16毫摩尔, 1.0当量), 得到白色固体 N-(2-((2-二甲胺乙 基 X甲基)胺基) -4-甲基 -5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基) 丙烯酰胺 (15毫克, 收率: 18.7%)。
该化合物 118的表征数据为: LCMS (ESI): m/z 501 [M + H ]+。 熔点: 208-210摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.11 (s, 1H), 9.06 (s, 1H), 8.42 (d, = 5.6 Hz, 1H), 8.33 (s, 1H), 7.96 (s, 1H), 7.69 (d, J = 5.6 Hz, 1H), 7.16 (m, 3H), 6.80 (d, J = 7.0 Hz, 1H), 6.40 (dd, J = 16.8, 10.3 Hz, 1H), 6.20 (d, = 16.9 Hz, 1H), 5.74 (d, = 11.2 Hz, 1H), 3.35 (s, 3H), 2.87 (s, 2H), 2.72 (s, 3H), 2.36 (s, 2H), 2.18 (m, 9H)。 实施例 54 : N-(2-((2-二甲胺乙基 X甲基)胺基) -4-(2-甲氧基-乙氧基) -5-(4-(3-甲基 -2-氧代 -2,3-二氢-苯并咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 ( N-{2-[(2-Dimethylamino-ethyl) -methyl-amino]-4-(2-methoxy-ethoxy)-5-[4-(3-methyl-2-oxo-2,3-dihydro-benzoimidazol-l-yl)-pyri midin-2-ylamino]-phenyl } -acrylamide ) (化合物 120)的制备 (按照方案七线路制备)
合成方法如实施例 24, 仅是将其中 1-(2-((5-胺基 -4-((2-二甲胺乙基 X甲基)胺基; )-2-甲氧基 苯基)胺基) -5-氟嘧啶 -4-基) -3-甲基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-55 ) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-(2-甲氧基-乙氧基) -苯胺基)嘧啶 -4-基) -3-甲基 -1 ,3-二氢 -2H - 苯并咪唑 -2-酮 (506-A-120 ) (250毫克, 0.49毫摩尔, 1.0当量), 得到白色蜡状固体 N-(2-((2- 二甲胺乙基 X甲基)胺基; )-4-(2-甲氧基-乙氧基) -5-(4-(3-甲基 -2-氧代 -2,3-二氢-苯并咪唑 -1-基;)嘧 啶 -2-胺基)苯基)丙烯酰胺 (28毫克, 收率: 10.2%)。
该化合物 120的表征数据为: LCMS (ESI): m/z56\ [M + H ]+。熔点: 71-78摄氏度; 1H MR (500 MHz, DMSO-dg) δ 10.04 (s, 1H), 8.60 (s, 1H), 8.52 (s, 1H), 8.45 (d, = 5.6 Hz, 1H), 8.10 (d, J = 7.4 Hz, 1H), 7.71 (d, J = 5.6 Hz, 1H), 7.17 (m, 2H), 7.07 (s, 1H), 6.90 (t, J = 7.6 Hz, 1H), 6.39
(dd, J = 16.9, 10.2 Hz, 1H), 6.18 (dd, J = 16.9, 1.8 Hz, 1H), 5.72 (m, 1H), 4.08 (m, 2H), 3.52 (m, 2H), 3.38 (d, = 9.6 Hz, 3H), 3.19 (s, 3H), 2.89 (t, = 5.7 Hz, 2H), 2.72 (s, 3H), 2.33 (s, 2H), 2.21 (s, 6H)。 实施例 55: N-(5-(4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-胺基) -2- ((2-二甲胺乙基 )(甲基) 胺 基 )-4- 甲 氧 基 苯基)丙 烯 酰胺 N-(5-((4-(5-chloro-lH-pyrrolo[3,2-b]pyridin-l-yl) pyrimidin-2-yl)amino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide (化合物 124)的制备 (按照方案六线路制备)
合成方法如实施例 6, 仅是将其中 Ν4-(4-(1Η-吲哚 -1-基)- 5-三氟甲基嘧啶 -2-基) -N1^-二 甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (403-A-5)替换为 N4-(4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1- 基)嘧啶 -2-基) -Nl-(2-二甲胺乙基) -5-甲氧基 -N1-甲基苯 -1,2,4-三胺 (200毫克, 0.43毫摩尔, 1.0 当量),米黄色固体 N-(5-(4-(5-氯 -1H-吡咯 [3,2-b]吡啶 -1-基)嘧啶 -2-胺基) -2- ((2-二甲胺乙基 )(甲 基)胺基) -4-甲氧基苯基)丙烯酰胺 (140毫克, 收率: 63%)。
该化合物 124的表征数据为: LCMS (ESI )m々521 [M + H ]+。 熔点: 127 129摄氏度; 1H MR (500 MHz, DMSO) δ 10.12 (s, 1H), 9.03 (s, 1H), 8.37 (d, J = 5.6 Hz, 2H), 8.29 (s, 1H), 8.08 (d, J = 3.6 Hz, 1H), 7.56 (d, J = 7.7 Hz, 1H), 7.24 (s, 1H), 7.10 (m, 2H), 6.98 (t, J = 7.5 Hz, 1H), 6.75 (d, = 3.6 Hz, 1H), 6.40 (dd, = 16.9, 10.2 Hz, 1H), 6.19 (dd, = 16.9, 1.7 Hz, 1H), 5.74 (dd, J = 10.2, 1.6 Hz, 1H), 2.89 (t, = 5.3 Hz, 2H), 2.74 (s, 3H), 2.36 (s, 2H), 2.20 (d, = 23.4 Hz, 9H). 实施例 56: N-(2-((2-二甲胺乙基 X甲基)胺基) -5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪 唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺(N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5-((4- (3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)phenyl)acrylamide)( 化合物 125)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 2, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(3-胺基 -4-((2-二甲胺乙基 X甲基)胺基)苯胺基)嘧啶 -4-基) - 3-甲基 -1,3-二氢 -2H-苯并 [d]咪 唑 -2-酮(506-A-125 ) ( 110毫克, 0.26毫摩尔, 1当量),得到白色固体 N-(2-((2-二甲胺乙基 )(甲 基)胺基) -5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 (50 毫克, 0.10毫摩尔, 收率: 38.5%)。
该化合物 125的表征数据为: LCMS (ESI): m/z 487[M + H ]+。 熔点: 190~193摄氏度; 1H 雇 R (500 MHz, DMSO-de) δ 10.18 (s, 1H), 9.63 (s, 1H), 8.51 (dd, = 17.1, 9.4 Hz, 3H), 7.76 (d, J = 5.6 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.25 (m, 3H), 7.07 (t, J = 7.0 Hz, 1H), 6.40 (dd, J = 16.9, 10.1 Hz, 1H), 6.24 (d, = 16.8 Hz, 1H), 5.77 (d, = 10.2 Hz, 1H), 3.39 (s, 3H), 2.84 (t, = 5.5 Hz, 2H), 2.66 (d, = 23.7 Hz, 3H), 2.29 (t, = 5.1 Hz, 2H), 2.22 (d, = 12.1 Hz, 6H). 实施例 57: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 -1-基)嘧啶 -2-胺基) -4-甲基苯基)丙烯酰胺 N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5- ((4-(3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)-4-methylphe nyl)acrylamide (化合物 127)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基)胺基) -2-甲基苯胺基)嘧啶 -4-基) -3-异丙基 -1,3-二氢 -2H - 苯并 [d]咪唑 -2-酮 (506-A-127) ( 360毫克, 0.77毫摩尔, 1.0当量), 得到白色固体 N-(2-((2-二 甲胺乙基) (甲基)胺基)- 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4- 甲基苯基)丙烯酰胺 (15毫克, 收率: 8%)。
该化合物 127的表征数据为: LCMS (ESI) : m/z 529 [M + 1]+。 熔点: 179 181摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.04 (s, 1H), 8.38 (d, J = 43.5 Hz, 2H), 7.97 (s, 1H), 7.65 (s, 1H), 7.33 (d, J = 6.6 Hz, 1H), 7.23 (s, 1H), 7.09 (s, 1H), 6.77 (s, 1H), 6.40 (m, 1H), 6.20 (d, J = 16.5 Hz, 1H), 5.74 (d, J = 8.9 Hz, 1H), 4.66 (s, 1H), 2.86 (s, 2H), 2.72 (s, 3H), 2.34 (s, 2H), 2.18 (d, = 32.8 Hz, 9H), 1.47 (d, = 5.0 Hz, 6H). 实施例 58: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 1 基)嘧啶—2-胺基)苯基)丙烯酰胺 ( N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-5- ((4-(3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)phenyl)acryla mide ) (化合物 128)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(4-((2-二甲胺乙基) (甲基)胺基) -3-胺基苯胺基)嘧啶 -4-基) -3-异丙基 -1H -苯并 [d]咪唑 -2(3H)-酮 (506-A-128)(650毫克, 1.33毫摩尔, 1.0当量),得到白色固体 N-(2-((2-二甲胺乙基) (甲 基)胺基) - 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 (390毫克, 收率: 54%)。
该化合物 128的表征数据为: LCMS (ESI) : m/z 515 [M + 1]+; 熔点: 85~90摄氏度; 1H 雇 R (500 MHz, DMSO-d6) δ 10.19 (s, 1H), 9.62 (s, 1H), 8.51 (dd, = 16.1, 5.7 Hz, 3H), 7.73 (d, J = 5.6 Hz, 1H), 7.53 (dd, J = 8.6, 1.7 Hz, 1H), 7.40 (d, J = 7.9 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.19 (t, = 7.7 Hz, 1H), 7.05 (t, = 7.8 Hz, 1H), 6.40 (dd, = 16.9, 10.1 Hz, 1H), 6.25 (dd, = 16.9. 1.8 Hz, 1H), 5.77 (m, 1H), 4.71 (dd, J = 13.9, 7.0 Hz, 1H), 2.83 (t, J = 5.6 Hz, 2H), 2.68 (s, 3H), 2.29 (t, = 5.6 Hz, 2H), 2.20 (s, 6H), 1.51 (d, = 6.9 Hz, 6H). 实施例 59: N-(2-((2-二甲胺乙基 )(甲基)胺基) -4-异丙基- 5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H- 苯并 [d]咪唑 -1 -基)嘧啶 -2-胺基)苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl)(methyl)amino)- 4-isopropyl-5-((4-(3-methyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amino)p henyl)acrylamide)(化合物 132)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91) 替换为 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-异丙基 -5-胺基苯胺基)嘧啶 -4-基) -3-甲基 -1,3-二氢 -2H - 苯并 [d]咪唑 -2-酉同 (506-A-132) ( 500毫克, 0.99毫摩尔, 1.0当量), 得到黄色固体 N-(2-((2- 二甲胺乙基 X甲基)胺基) -4-异丙基- 5-(4-(3-甲基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基)苯基)丙烯酰胺 (73毫克, 收率: 16%)。
该化合物 132的表征数据为: LCMS (ESI) : m/z 529 [M + 1]+; 1H MR (500 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.02 (s, 1H), 8.40 (d, = 5.6 Hz, 1H), 8.24 (s, 1H), 7.83 (s, 1H), 7.66 (d = 5.6 Hz, 1H), 7.28 (s, 1H), 7.13 (m, 2H), 6.73 (s, 1H), 6.40 (dd, = 16.9, 10.2 Hz, 1H), 6.19 (dd, J = 16.9, 1.7 Hz, 1H), 5.73 (d, = 11.6 Hz, 1H), 3.34 (s, 3H), 3.13 (dt, = 13.7, 6.8 Hz, 1H), 2.92 (dd, = 23.6, 18.3 Hz, 2H), 2.76 (s, 3H), 2.37 (s, 2H), 2.21 (s, 6H), 1.09 (t, = 9.4 Hz, 6H). 实施例 60: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d] 咪唑 - 1 -基) -5-甲氧基嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino)ethyl) (methyl)amino)-5-((4-(3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)-5-methoxypyrimi din-2-yl)amino)-4-methoxyphenyl)acrylamide) (化合物 133)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-胺基苯胺基) -5-甲氧基嘧啶 -4-基) -3-异丙基 -1H -苯并 [d]咪唑 -2(3H)-酉同(506-A-133 ) (660毫克, 1.2毫摩尔, 1.0当量),得到白色固体 N-(2-((2- 二甲胺乙基 X甲基)胺基) - 5-(4-(3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基) -5-甲氧基嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (310毫克, 收率: 42.7%)。
该化合物 133的表征数据为: LCMS(ESI) : m/z 575 [M + 1]+.熔点; 92~95摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.65 (s, 1H), 8.58 (s, 1H), 8.20 (s, 1H), 7.36 (d, = 7.9 Hz, 1H), 7.03 (m, 4H), 6.30 (ddd, J = 18.4, 17.0, 5.8 Hz, 2H), 5.74 (d, = 11.4 Hz, 1H), 4.64 (m, 1H), 3.81 (d, = 6.0 Hz, 6H), 2.83 (t, = 5.7 Hz, 2H), 2.68 (s, 3H), 2.27 (t, = 5.7 Hz, 2H), 2.18 (s, 6H), 1.49 (d, = 6.9 Hz, 6H). 实施例 61: N-(5-((5- (二甲基氨基 )-4-(3-异丙基 -2-酮 -2,3-二氢 -1H-苯并咪唑 -1-基)嘧啶 -2- 基;)氨基) -2-((2- (二甲基氨基)乙基 X甲基)氨基; )-4-甲氧基苯基)丙烯酰胺 (N-(5-((5- (dimethylamino)-4-(3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)pyrimidin-2-yl)amin o)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide)(化合物 135)的制备 (按照方案七线路制备)
合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-(2-((5-氨基 -4-((2- (二甲基氨基)乙基) (甲基)氨基) -2-甲氧基苯基)氨基) -5- (二甲基氨基)嘧啶 -4- 基) -3-异丙基 -1,3-二氢 -2H-苯并咪唑 -2-酮,得到白色固体 N-(5-((5- (二甲基氨基 )-4-(3-异丙基 -2- 酮 -2,3-二氢 - 1H-苯并咪唑 -1 -基;)嘧啶 -2-基;)氨基) -2-((2- (二甲基氨基)乙基 )(甲基;)氨基) -4-甲氧基 苯基)丙烯酰胺 (90毫克, 收率: 13.9%)。
该化合物 135的表征数据为: LCMS (ESI): m/z 588 [M + 1]+.熔点: 114.6~116.2摄氏度; 1H MR (500 MHz, DMSO) δ 10.15 (s, 1H), 9.69 (s, 1H), 8.48 (s, 1H), 8.41 (s, 1H), 8.10 (s, 1H), 7.36 (d, J = 7.9 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H), 6.98 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 6.3 Hz, 2H), 6.22 (d, J = 16.9 Hz, 1H), 5.69 (d, J = 10.6 Hz, 1H), 4.63 (dt, J = 13.8, 6.9 Hz, 1H), 3.82 (s, 3H), 3.15 (d, = 63.2 Hz, 4H), 2.71 (d, = 42.9 Hz, 6H), 2.55 (s, 3H), 2.50 (s, 6H), 1.47 (d, = 6.9 Hz, 6H).
实施例 62: N-(2-((2-二甲胺乙基 X甲基)胺基) - 5-(4-(3-异丙基 -5-甲氧基 -2-氧代 -2,3-二氢 - 1H-苯并 [d]咪唑 - 1 -基)嘧啶 -2-胺基) -4-甲氧基苯基)丙烯酰胺 (N-(2-((2-(dimethylamino) ethyl)(methyl)amino)-5-((4-(3 sopropyl-5-methoxy-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl) pyrimidin-2-yl)amino)-4-methoxyphenyl)acrylamide )(化合物 138)的制备(按照方案七线路制备) 合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-(2-(4-((2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-胺基苯胺基)嘧啶 -4-基) -3-异丙基 -5-甲氧基 -1H -苯并 [d]咪唑 -2(3H)-酮 (1.3克, 2.36毫摩尔, 1.0当量),得到白色固体 N-(2-((2-二甲胺乙基 )(甲 基;)胺基) - 5-(4-(3-异丙基 -5-甲氧基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基;)嘧啶 -2-胺基) -4-甲氧 基苯基)丙烯酰胺 (940毫克, 收率: 65.7%)。
该化合物 138的表征数据为: LCMS (ESI) : m/z 575 [M + 1]+; 熔点: 95~98摄氏度; 1H MR (500 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.63 (s, 1H), 8.40 (m, 2H), 8.00 (d, J = 8.5 Hz, 1H), 7.66 (d, = 5.7 Hz, 1H), 7.05 (s, 1H), 6.90 (d, = 2.4 Hz, 1H), 6.40 (m, 2H), 6.19 (dd, = 17.0, 1.8 Hz, 1H), 5.73 (dd, = 14.4, 4.3 Hz, 1H), 4.64 (m, 1H), 3.75 (s, 6H), 2.91 (t, = 5.7 Hz, 2H), 2.75 (s: 3H), 2.34 (t, = 5.6 Hz, 2H), 2.20 (s, 6H), 1.47 (d, = 6.9 Hz, 6H). 实施例 63: N-(2-((2-二甲胺乙基 )(甲基)胺基) - 5-(4-(5-羟基 -3-异丙基 -2-氧代 -2,3-二氢 -1H- 苯并 [d]咪唑 -1-基;)嘧啶 -2-胺基;) -4-甲氧基苯基)丙烯酰胺(N-OCOCdimethylamino) ethyl)(methyl)amino)-5-((4-(5-hydroxy-3-isopropyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-l-yl)p yrimidin-2-yl)amino)-4-methoxyphenyl)acrylamideX化合物 139)的制备 (按照方案七线路制备) 合成方法如实施例 47步骤 47f方法 1, 仅是将其中 1-(2-(5-胺基 -4-((2-二甲胺乙基 X甲基) 胺基) -2-甲氧基苯胺基)嘧啶 -4-基) -5-氟 -3-异丙基 -1H-苯并 [d]咪唑 -2(3H)-酮 (506-A-91)替换为 1-(2-(4-《2-二甲胺乙基 X甲基)胺基) -2-甲氧基 -5-胺基苯胺基)嘧啶 -4-基) -5-羟基 -3-异丙基 -1H - 苯并 [d]咪唑 -2(3H)-酮 (35毫克, 0.065毫摩尔, 1.0当量),得到白色固体 N-(2-((2-二甲胺乙基) (甲 基)胺基) - 5-(4-(5-羟基 -3-异丙基 -2-氧代 -2,3-二氢 -1H-苯并 [d]咪唑 -1-基)嘧啶 -2-胺基) -4-甲氧基 苯基)丙烯酰胺 (20毫克, 收率: 54.8%)。
该化合物 139的表征数据为: LCMS (ESI) : m/z 561 [M + l]+ ; 熔点: 115 117摄氏度; 1H MR (500 MHz, DMSO) δ 10.06 (s, 1H), 9.35 (s, 1H), 8.62 (s, 1H), 8.38 (d, = 15.4 Hz, 2H), 7.89 (s, 1H), 7.66 (s, 1H), 7.03 (s, 1H), 6.69 (s, 1H), 6.26 (dd, = 67.7, 30.1 Hz, 3H), 5.73 (s, 1H), 4.58 (s, 1H), 3.74 (s, 3H), 2.90 (s, 2H), 2.75 (s, 3H), 2.34 (s, 2H), 2.18 (d, = 26.7 Hz, 6H), 1.45 (s, 6H). 实验例
一、 酶活性抑制实验
1、 实验方法
( 1 ) EGFR T790M/L858R活性抑制实验。
采用 Caliper 迁移率变动检测技术 (Caliper mobility shift assay) 测定 EGFR T790M/L858R蛋白激酶活性 (参见 J Biomol Screen 14:31, 2009)。
实验方法为: 将待测化合物用 DMSO 溶解后用激酶缓冲液 (50 mM HEPES-pH7.5,
0.0015% Brij-35 , 10 mM MgCl2, 2 mM DTT)稀释, 在 384孔板中加入 5 μΐ的 10%DMSO溶 解的 5倍反应终浓度的化合物,无化合物对照孔是 5μ1的 10% DMSO, 无酶活性对照孔是 5μ1 激酶缓冲液。加入 10 μΐ 的稀释 2.5倍后的 EGFR Τ790Μ L858R (Invitrogen , Cat.No PR8911A, Lot. 1498821A) 酶溶液后在室温下孵育 10分钟, 再加入 10 μΐ 的稀释 2.5倍后的底物溶液 Peptide FAM-P22 (GL Biochem, Cat.No. 112393, Lot. No. P130408-ZB112393)。 28°C下孵育 60 分钟后加 25 μΐ 终止液 (lOO mM HEPES, ρΗ 7.5, 0.015% Brij-35 , 0.2% Coating Reagent #3 ) 终止反应。 在 Caliper EZ Reader II ( Caliper Life Sciences) 上读取转化率数据。 把转化率转化 成抑制率数据。
其中 max是指 DMSO无化合物对照孔的转化率, min是指无酶活对照的转化率。 以化合 物浓度和抑制率为横纵坐标, 绘制曲线,使用 XLFit excel add-in version 4.3.1软件拟合曲线并 计算 IC50。 抑制率%= (max-转化率 )/(max-min) X 100。
(2) EGFR T790M活性抑制实验
采用 Caliper迁移率变动检测技术 (Caliper mobility shift assay)测定 EGFR T790M 蛋白 激酶活性 (参见 J Biomol Screen 14:31, 2009) 。
实验方法为: 将待测化合物用 DMSO 溶解后用激酶缓冲液 (50 mM HEPES-pH 7.5, 0.0015% Brij-35 , 10 mM MgCl2, 2 mM DTT)稀释, 在 384孔板中加入 5 μΐ的 10%DMSO溶 解的 5倍反应终浓度的化合物,无化合物对照孔是 5μ1的 10% DMSO, 无酶活性对照孔是 5μ1 激酶缓冲液。 加入 10 μΐ 的稀释 2.5倍后的 EGFR_T790M (Invitrogen , Cat.No PR8052A, Lot. 1229180E)酶溶液后在室温下孵育 10分钟, 再加入 10 μΐ 的稀释 2.5倍后的底物溶液 Peptide FAM-P22 (GL Biochem, Cat. No. 112393, Lot. No. P130408-ZB 112393)。 28°C下孵育 60 分钟后 力口 25 μ1 终止液 (lOO mM HEPES, ρΗ 7.5, 0.015% Brij-35 , 0.2% Coating Reagent #3 ) 终止反 应。 Caliper EZ Reader II ( Caliper Life Sciences) 上读取转化率数据。 把转化率转化成抑制率 数据。
其中 max是指 DMSO无化合物对照孔的转化率, min是指无酶活对照的转化率。 以化合 物浓度和抑制率为横纵坐标, 绘制曲线,使用 XLFit excel add-in version 4.3.1软件拟合曲线并 计算 IC50。 抑制率%= (max-转化率 )/(max-min) X 100。
2、 实验结果
计算出上述实验中各化合物的半数抑制浓度 (IC5Q), 结果如下表所示。
表 1. 酶活性抑制结果 (IC5())
37 V 38 V
39 V 41 V
46 IV 50 V
51 V 52 V
53 V 57 IV
62 V 63 V
66 III 67 II
68 III 69 V
70 IV 71 IV
73 IV 74 IV
77 IV 78 IV
79 V 80 IV
81 IV 82 IV
84 III 91 V
98 V 99 IV
101 V 102 V
105 V 118 V
120 IV 124 V
125 V 127 V
128 V 132 IV
133 IV 135 IV
138 IV 139 V
AZD9291 V V
CO-1686 IV IV
其中: I> 100 nM, ΙΟΟηΜ >II> 50 nM, 50 nM>III> 10 nM, 10nM>IV> InM, V< lnM。
从上表中可以看出,这类化合物能选择性抑制 T790M EGFR , 包括单点突变 T790M和双 点突变如 T790M/L858R。 二、 肿瘤细胞增殖抑制实验
1、 实验方法
采用 CellTiter-Glo发光细胞活力检测试剂盒法(Promega, #G7572)测定三磷酸腺苷( ATP) 的含量来评估细胞活力。
人非小细胞肺癌肿瘤细胞株 H1975携带 L858R和 T790M表皮生长因子受体 (EGFR) 双重突变。 人结肠癌细胞株 LOVO和人非小细胞肺癌 H358表达野生型 EGFR。 人非小细胞 肺癌细胞株 HCC827携带外显子 19的缺失突变(exl9del)。 这些细胞株购买自上海复旦 IBS 细胞资源中心和美国菌种保藏中心 (ATCC)。
实验方法为:用胰酶将细胞从细胞培养盘上消化和 DPBS培养基重悬后用 Scepter自动细 胞计数仪 (Millipore #PHCC00000) 计数测定细胞密度。 将细胞稀释成每毫升含 44,000个细 胞的溶液。调整密度后的细胞溶液以每孔 90微升加入细胞实验板中。孔板置于 37°C、 5% C02
培养箱培养 24小时后加入不同浓度的待试化合物。细胞在 10%胎牛血清存在下与化合物一起 培养 72小时。使用 CellTiter-Glo® Luminescent Cell Viability Assay kit (详见厂家说明书) 测定 ATP 的含量来评估细胞生长抑制。
简要来讲, 每个孔中加入 30 w lCellTiter-Glo®试剂, 摇板 10 分钟, 诱导细胞裂解, 用 Fluoroskan Ascent FL (Thermo) 检测记录萤光信号。 从二甲基亚砜处理 72小时的细胞得到最 大的信号值。 从单独的培养基(细胞数为零)得到最小信号值。 抑制率% = (最大信号值 - 化 合物信号值) I (最大信号值 - 最小信号值) X 100。 使用 GraphPad Prism V5.0 (GraphPad Software, San Diego, CA) 软件处理数据。 通过 S形剂量 -反应曲线拟合计算 IC5Q值。
2、 实验结果
计算出上述实验中各化合物的 IC5Q, 结果如下表所示。
表 2. 肿瘤细胞增殖抑制结果
77 IV I I
78 IV I I
79 III I I
80 V I I
82 III I
91 V II
98 V II
99 V II
101 V II
102 V II
105 V II
118 V II
124 V II
125 V II
127 V I
128 V II
132 V I
133 V I
135 IV I
138 V II
139 V II
AZD9291 V V I II
CO-1686 IV II I II 其中: I> 1 μΜ, 1μΜ>ΙΙ>0.1 μΜ, 0.1 μΜ>ΙΙΙ> 0.05 μΜ, 0.05μΜ>ΐν> Ο.ΟΙμΜ, ν≤0.01μΜ。
AZD9291和 CO-1686为阳性对照药。
从上表中可以看出, 这类化合物对 L858R/T790M EGFR突变的细胞如 H1975有很强的 抗细胞增殖的活性,对携带外显子 19的缺失突变的 HCC827也有很强的抑制活性。但对野生 型 EGFR细胞如 LOVO及 H358的抗细胞增殖的活性弱, 具有高度的选择性。 三、 药代动力学 (PK) 实验
1、 实验方法
雄性 SD大鼠, 体重 250-300克; 或雄性比格犬, 9-10 kg, 试验前过夜禁食。 待试化合 物溶解在 30%磺丁基 -β-环糊精 (SBE-p-CD) 中, 大鼠以 20 mg/kg和比格犬以 5 mg/kg 的剂 量灌胃给药。 给药后 15 分钟、 30 分钟和 1、 2、 3、 4、 6、 8 及 24 小时取血, 每时间点约 取血 0.3 ml, 置于含 K2-EDTA (乙二胺四乙酸二钾)的离心管中, 离心处理(2,000 g, 10 分 钟, 4 °C ) 取血浆, 储存在 _80°C的超低温冰箱中。 50μί的血浆样品与 5微升内标 (IS) 混 合, 用乙酸乙酯萃取。 真空干燥后残留物重新溶于乙腈中。 对样品进行过滤, 并注入到 LC-MS/MS分析。
2、 实验结果
本发明提供的化合物 20、 50、 62、 91、 124在大鼠经口服给药后, 吸收良好, 血液暴露 量效高。这类化合物 Tmax为 1.0-3.7小时; T1/2半衰期为 3.0-6.8小时。 Cmax 为 84. -349.7 ng/ml (表 3 )。化合物 50和化合物 90在比格犬口服 Cmax分别为 428.0和 109.2 ng/ml, AUC0-24 分 别为 1423.4 和 785.6 hrX ng/ml) (表 4)。表中 Tmax是指达峰时间, Cmax是指最大血药浓度, T1/2为半衰期, AUCo.24是指 0-24 小时时间 -浓度曲线下面积, AUClrf是指 0-Inf时间-浓度曲 线下面积。
表 3. 大鼠口服给药 (20 mg/kg) 药代动力学
四、 人 H1975非小细胞肺癌移植肿瘤小鼠模型实验
Balb/C裸鼠购自北京维通利华实验动物技术有限公司, 饲养于 SPF动物房。培养皿中 人非小细胞肺癌肿瘤细胞株 H1975用胰蛋白酶 -EDTA (0.25%胰蛋白酶, 1 mM EDTA) 收 集后, 用不含血清的 PBS溶液清洗。 最后细胞由不含血清的培养基中稀释后植入。 只有 大于 90 %的存活率(台盼蓝排斥)的单细胞悬液才可用于注射。采用 1ml的注射器和 25G 注射器针头,将悬浮在 0.1毫升的不含血清的培养基中 500万个细胞注入每只小鼠右前侧 翼皮下区域并小心避开血管。 在植入一周左右即可测量出肿瘤大小。 使用卡尺测量肿瘤 的大小。 并用以下公式计算肿瘤体积: 肿瘤体积= (长 X宽 2)/2 。 五、 肿瘤模型药代动力学实验
1. 实验方法
当 H1975移植肿瘤体积达到 400 mm3左右, 将动物分为 6个口服给药组, 即赋形剂对照 组 (0 小时) 、 给药后 30分钟、 2小时、 4小时、 8小时和 24小时组 (每时间点 3个动物)。 化合物 91以 3 mg/ml的浓度溶解在 30%磺丁基 -β-环糊精 (SBE- β -CD ) 以及 1.0摩尔等值的 盐酸 (pH 3-4 ) 中, 以 30 mg/kg的剂量灌胃给药。 小鼠在上述时间点经 C02安乐死处置后, 经心脏采血置于含 K2-EDTA 的离心管中, 离心后收集血浆。 并采集肿瘤组织, 立即在液氮 中冷却。 随后放置在 -80 ° C保存, 直至药代动力学分析。
药代动力学分析: 血浆和肿瘤组织匀浆样品与 5微升内标 (IS ) 混合, 用乙酸乙酯萃取。 真空干燥后残留物重新溶于乙腈中。 对样品进行过滤, 并注入到 LC-MS/MS分析。
2. 实验结果
如图 1和下表所示,化合物 91 口服给药后迅速吸收,血浆和肿瘤组织峰值时间分别为 0.5 小时和 4小时、 Cmax分别为 1723.3 ng/ml 禾 P 868.0 ng/ml 。 肿瘤组织中 AUC0-24h (6434.6 ng/ml*hr)和血浆 AUC0-24h (5095.6 ng/ml*hr)相当。
表 5. 化合物 91在 H1975移植肿瘤小鼠口服给药 (30 mg/kg) 后药代动力学参数
1. 实验方法
当 H1975移植肿瘤体积达到 2000 mm3左右, 将动物分为 4个口服给药组, 即赋形剂对照 组 (0 小时) 、 给药后 2小时、 4小时、 和 8小时组 (每时间点 3-4个动物)。 化合物 91以 3 mg/ml的浓度溶解在 30%磺丁基 -β-环糊精 (SBE- P -CD ) 以及 1.0摩尔等值的盐酸 (ρΗ 3-4 ) 中, 以 30 mg/kg的剂量灌胃给药。 小鼠在上述时间点经 C02安乐死处置后, 采集肿瘤组织, 立即在液氮中冷却。 随后放置在 -80 ° C保存。
取 20mg肿瘤组织在 300μ1蛋白质提取液体 (RIPA, CST, #9806) 中匀浆。 该提取液还加入 磷酸酶抑制剂(1 : 100 v/v, Protease Inhibitor Cocktail,sigma, #P8340)和 lOOmM/L PMSF(1 : 100 v/v)。 组织裂解液在 4°C以 12,000 rpm离心 15分钟。 收集 200μ1上清液保存在 -80 ° C。 采用 Bradford法测定蛋白浓度 (Bey otime, #P0006),加入上样缓冲液(Beyotime, #P0015L )于 100 °C 煮 5 min, 以 8%-10% SDS-PAGE 电泳分离蛋白后转移至 PVDF 膜, 膜用含 5%的 BSA(Beyotime, #ST023)封闭 60 min, 力口 β -actin Antibody ( CST, #4970)、 Mouse Anti-EGFR Antibody ( BD, #610017 )、 或 Human Phospho-EGFR/ErbB 1 (Y1068) Mouse Antibody ( R&D, #MAB3570 ) 于 4 °C下孵育过夜, 然后用 1 x TBST 液洗膜 3 X 5 min。 分别以荧光二抗 IRDye@800CW Goat ( polyclonal ) Anti-Mouse lgG (H+L), Highly Cross Adsorbed (LI-COR,
926-32210)和 IRDye@680CW Goat (polyclonal) Anti-Rabbit lgG(H+L), Highly Cross Adsorbed (LI-COR, #926-68071 )室温下避光孵育 2 h后再洗膜,洗涤条件同上。最后将膜置于 LI-COR Odyssey红外荧光扫描成像系统上检测成像。
2. 实验结果
如图 2所示, 化合物 91以 30 mg/kg 口服给药后, 抑制携带 L858R和 T790MEGFR双重 突变的人非小细胞肺癌肿瘤细胞株 H1975 EGFR的磷酸化, 2小时和 4小时抑制作用强力, 8 小时仍有明显抑制。 六、 肿瘤模型药效学实验
1. 实验方法
实验研究化合物 20、 50、 91、 118在 H1975移植肿瘤模型抑制肿瘤生长。 当 H1975肿瘤 体积达到平均 260mm3左右,将动物分为 5个口服给药组, 即赋形剂对照组和化合物 20、 50、 91、 118口服给药组 (n=8/组)。 将化合物分别溶解在 30%磺丁基 -β-环糊精 (SBE- P -CD) 以 及 1.0摩尔等值的盐酸 (ρΗ 3-4) 中, 以 10 ml/kg的剂量灌胃给药, 每日一次, 连续给药 16 天。
进一步实验评价化合物 50和 91抑制 H1975移植肿瘤生长的量效关系, 将动物分为 8个 口服给药组, 即赋形剂对照组, AZD9291 阳性对照组 (最大耐受量 25 mg/kg) , 化合物 50 化合物和化合物 91 10mg/kg、 20 mg/kg和 40 mg/kg 口服给药组(n=7/组)。将化合物分别溶 解在 30%磺丁基 -β-环糊精(SBE- p -CD) 以及 1.0摩尔等值的盐酸(ρΗ 3-4) 中, 以 10 ml/kg 的剂量灌胃给药, 每日一次, 连续给药 22天。
2. 实验结果
赋形剂组给药后 16天因肿瘤体积超过 2000 mm3, 终止实验。 如图 3所示, 化合物 20、 50、 91、 118 30 mg/kg qd口服给药抑制 H1975移植肿瘤生长, 使肿瘤消退。 化合物 50和化 合物 91给药后 16天, 导致肿瘤基本消失 (T/C值分别为 -97%和- 95 %; P <0.001 )。 与给 药前比较, 各给药组动物体重无明显下降。
化合物 50和化合物 91口服给药剂量依赖性抑制 H1975移植肿瘤生长。连续给药 22天后, 化合物 50 和化合物 91高剂量(40mg/kg)使肿瘤完全消失,在 1/4 最大耐受量(10 mg/kg) 时, 亦可导致肿瘤缩小 (T/C值分别为 -65%和 -73%)。 两个化合物在各个剂量组对体重影 响较小, 最大体重下降小于 5%, 如图 4-5和下表所示。
表 6. 在 H1975移植肿瘤模型化合物 91和 50抗肿瘤活性和对体重的影响
化合物 50, 10 mg/kg, po, qd 7 -65 <0. 001 0. 0 化合物 50, 20 mg/kg, po, qd 7 -95 <0. 001 -2. 0 化合物 50, 40 mg/kg, po, qd 7 -100 <0. 001 -4. 4 化合物 91, 10 mg/kg, po, qd 7 -73 <0. 001 1. 6 化合物 91, 20 mg/kg, po, qd 7 -100 <0. 001 1. 2 化合物 91, 40 mg/kg, po, qd 7 -100 <0. 001 -3. 5 以上所述实施例的各技术特征可以进行任意的组合, 为使描述简洁, 未对上述实施例中 的各个技术特征所有可能的组合都进行描述, 然而, 只要这些技术特征的组合不存在矛盾, 都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式, 其描述较为具体和详细, 但并不能因 此而理解为对发明专利范围的限制。 应当指出的是, 对于本领域的普通技术人员来说, 在不 脱离本发明构思的前提下, 还可以做出若干变形和改进, 这些都属于本发明的保护范围。 因 此, 本发明专利的保护范围应以所附权利要求为准。