WO2015178460A1 - 生体試料中レナリドミドの前処理方法及び分析方法 - Google Patents
生体試料中レナリドミドの前処理方法及び分析方法 Download PDFInfo
- Publication number
- WO2015178460A1 WO2015178460A1 PCT/JP2015/064642 JP2015064642W WO2015178460A1 WO 2015178460 A1 WO2015178460 A1 WO 2015178460A1 JP 2015064642 W JP2015064642 W JP 2015064642W WO 2015178460 A1 WO2015178460 A1 WO 2015178460A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lenalidomide
- enantiomer
- biological sample
- acid
- thalidomide
- Prior art date
Links
Images











Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/94—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving narcotics or drugs or pharmaceuticals, neurotransmitters or associated receptors
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/10—Selective adsorption, e.g. chromatography characterised by constructional or operational features
- B01D15/12—Selective adsorption, e.g. chromatography characterised by constructional or operational features relating to the preparation of the feed
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/10—Selective adsorption, e.g. chromatography characterised by constructional or operational features
- B01D15/16—Selective adsorption, e.g. chromatography characterised by constructional or operational features relating to the conditioning of the fluid carrier
- B01D15/163—Pressure or speed conditioning
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/265—Adsorption chromatography
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/04—Preparation or injection of sample to be analysed
- G01N30/06—Preparation
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/88—Integrated analysis systems specially adapted therefor, not covered by a single one of the groups G01N30/04 - G01N30/86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 - B01D15/36
- B01D15/3833—Chiral chromatography
Definitions
- the present invention is a method for pretreating a biological sample containing an enantiomer of lenalidomide, characterized in that deproteinization is performed under acidic conditions, and the lenalidomide pretreated in this way.
- the present invention relates to a method for quantifying the enantiomer of lenalidomide in a biological sample, characterized in that the biological sample containing the enantiomer is analyzed by high performance liquid chromatography.
- Lenalidomide is a derivative of thalidomide and is widely used as an immunopreparation drug effective for various malignant blood diseases such as multiple myeloma. Lenalidomide has been reported to have an improved toxicity profile and superior immunoprep activity compared to thalidomide (Non-Patent Document 1). The pharmacokinetic properties of lenalidomide have been clarified by HPLC method using reverse phase mode (Non-patent document 2; Non-patent document 3; Non-patent document 4). The final elimination half-life of lenalidomide is about It is estimated to be 3-4 hours.
- Non-patent Document 5 S-pomaridomide (3-amino-phthalimido-glutarimide) is compared with the R form and racemic form.
- corneal neovascularization induced by bFGF or VEGF is significantly inhibited (Non-patent Document 6).
- Enantiomers of thalidomide can be obtained by repeated extraction using an organic solvent, modified amylose (Non-patent Document 8), cellulose (Non-patent Document 7), vancomycin (Non-patent Document 9) or methacrylamide (Non-patent Document 10). They have been separated and quantified by chromatographic methods using selected stationary phases and have been shown to racemize very rapidly in blood. Such racemization was first disclosed by G. Blaschke et al. And reported that the half-life of thalidomide in human plasma is about 10 minutes (Non-patent Document 10). This is extremely short considering that the elimination half-life of thalidomide is 8.7 hours (Non-patent Document 11).
- An object of the present invention is to provide a novel method for pretreating a sample while suppressing racemization and / or degradation of the enantiomer of lenalidomide and maintaining the content of each enantiomer of lenalidomide in the living body.
- the inventor has now obtained the surprising finding that the enantiomer of lenalidomide stays in the living body for a significantly longer time when compared to the enantiomer of thalidomide.
- the thalidomide enantiomer has a very fast racemization rate in vivo, and it was thought that administration of the pure enantiomer had no pharmacological significance, but lenalidomide had an elimination half-life. Considering this, it has been revealed that the pharmacological effect remains in the living body as an enantiomer for a time that cannot be ignored. Therefore, the evaluation of the pharmacokinetic and pharmacological effects of the pure enantiomers of lenalidomide is considered to have a very important meaning.
- thalidomide when analyzing a pure enantiomer of lenalidomide in a biological sample, it is necessary to perform deproteinization in advance in order to prevent a decrease in analysis accuracy due to protein interference.
- the pretreatment of thalidomide has been conventionally performed by extracting a biological sample containing thalidomide one or more times with a strong hydrophobic organic solvent, drying the obtained organic solvent layer, and then re-dissolving in the organic solvent. .
- lenalidomide has a higher polarity than thalidomide and is difficult to dissolve in the organic solvent layer, the conventional method cannot be applied.
- the present inventor conducted extensive studies on the influence of pH on the racemization of the enantiomer of lenalidomide, and as a result, found that the enantiomer of lenalidomide is extremely stable under acidic conditions.
- the present invention has been completed.
- the present invention includes the following inventions.
- [1] A method for pretreating a biological sample containing an enantiomer of lenalidomide, wherein the protein is deproteinized under acidic conditions.
- [2] The method according to [1], wherein the acidic condition is pH 5 or less.
- [3] The acid according to [1] or [2], wherein an acid selected from perchloric acid, trichloroacetic acid, trifluoroacetic acid, metaphosphoric acid, hydrochloric acid, succinic acid, and maleic acid is added.
- Method The biological sample according to any one of [1] to [3], wherein the biological sample is blood, serum, plasma, urine, saliva, breast milk, spinal fluid, semen, tissue, or microsome.
- [8] A method for preserving the enantiomer of lenalidomide, wherein the enantiomer of lenalidomide is maintained in an aqueous buffer under acidic conditions.
- the aqueous buffer is a citrate buffer.
- the method according to any one of [8] to [10], wherein the enantiomer of the lenalidomide is R-form.
- the present invention suppresses racemization and / or degradation of the enantiomer of lenalidomide, pre-treats the sample while maintaining the enantiomer content of lenalidomide in the living body, and enables easy and efficient accurate enantiomer of lenalidomide in the biological sample. Separation and quantification are possible. This will clarify the pharmacokinetic and pharmacological properties of the pure enantiomer of lenalidomide in vivo, and will help to find possible side effects and improved efficacy by administration of the pure enantiomer of lenalidomide. It is done. Therefore, the present invention is convinced that it will greatly contribute to the utilization and development of the enantiomers of lenalidomide in the medical and pharmaceutical industries such as pharmacological efficacy, safety, physical properties, and pharmacokinetic studies.
- the enantiomer eluted in fraction 1 was named LL1, and the enantiomer eluted in fraction 2 was named LL2.
- Chiralpak IA was used for the analysis. It is a chromatogram showing S-thalidomide (a) and R-thalidomide (b).
- S-thalidomide (a) and R-thalidomide (b) The time change of the peak areas of (S * )-LL and (R * )-LL after adding an aqueous buffer at room temperature is shown.
- the time course of enantiomeric excess in aqueous buffer (pH 4-9) at room temperature is shown.
- FIG. 5 is a plot of log 10 (enantiomeric excess) versus time at room temperature.
- the chromatograms of (a) LL enantiomer in water (control) and (b) LL enantiomer in human serum are shown.
- HPLC conditions Column: Chiralpak IA (4.6 mm id ⁇ 250 cm) + Security guard C8 (3.0 mm id ⁇ 4 mm), flow rate: 0.75 mL / min, injection volume: 2 ⁇ L.
- FIG. 5 is a chromatogram after addition of 10 ⁇ g / mL (S * )-LL in human serum at 37 ° C.
- S * The racemization half-life of LL and TD enantiomers in Na-phosphate buffer (pH 7.4, hatched) and human serum (black) is shown.
- the bar graph shows the average value of three tests, and the error bar shows SD. Significant differences are ** p ⁇ 0.01 and *** p ⁇ 0.001.
- Lenalidomide (LL) is a derivative of thalidomide (TD), which is known as a therapeutic agent for multiple myeloma, and is widely used as an immune preparation effective for various malignant blood diseases such as multiple myeloma. It has a chemical structure represented by the following formula, and has an asymmetric center on the dioxopiperidine ring, like thalidomide.
- Lenalidomide combined with dexamethasone, has a high response rate of 60% in relapsed / refractory multiple myeloma and has been approved worldwide, and has been approved in about 50 countries so far, with sales of 400 billion yen The blockbuster medicine exceeds. In Japan, it was approved in 2010, but from the viewpoint of ensuring safety, there is a condition that it must comply with the management procedures of medical personnel, patients, and families and conduct a use-results survey for all cases.
- the present inventor in this optical resolution using a chromatographic method, uses an organic solvent selected from the group consisting of an aprotic solvent, a secondary alcohol, and a mixture thereof as a mobile phase. It was found that the pure enantiomer of lenalidomide can be separated and quantified.
- a method for separating and quantifying enantiomers of lenalidomide which is subjected to sample chromatography comprising an enantiomeric mixture of lenalidomide, and an aprotic solvent, a secondary alcohol and mixtures thereof
- a method is provided that stably optically resolves each enantiomer of lenalidomide from a mixture of lenalidomide enantiomers by using an organic solvent selected from the group consisting of:
- a sample containing a mixture of enantiomers is flowed together with an organic solvent of a mobile phase on a stationary phase carrying a compound having chiral discrimination ability (chiral identifier), and each enantiomer is adsorbed and the difference in retention time is determined.
- It is a method for optically resolving each enantiomer by utilizing. Generally, it is carried out using a high performance liquid chromatography (HPLC) apparatus equipped with an optical resolution column.
- HPLC high performance liquid chromatography
- the aprotic solvent used as the mobile phase is not particularly limited as long as the object of the present invention can be achieved.
- esters such as acetonitrile and ethyl acetate, ketones such as acetone, ethers such as diethyl ether and diisopropyl ether Or the combination of these etc. is mentioned.
- Acetonitrile, ethyl acetate or a combination thereof is preferable.
- the secondary alcohol used as the mobile phase is not particularly limited as long as the object of the present invention can be achieved, and examples thereof include isopropanol, 2-butanol, cyclopentanol, cyclohexanol, and combinations thereof.
- isopropanol is used.
- the optical resolution column used in the method of the present invention is not particularly limited as long as the object of the present invention can be achieved, and it may be a normal phase column, a reverse phase column, a column of other separation mode, or a multi-mode column in which each is combined. Good.
- polysaccharide derivative chiral columns are used.
- the polysaccharide derivative chiral column is a column in which a polysaccharide derivative is immobilized on a carrier as a chiral identifier.
- Examples of the polysaccharide derivative supported on the polysaccharide derivative chiral column include amylose derivatives and cellulose derivatives.
- Chiralpak (registered trademark) IA or Chiralpak (registered trademark) IC is preferably used.
- a pure enantiomer of lenalidomide can be separated and quantified from a mixture of enantiomers of lenalidomide (such as a racemate) without decomposition or racemization.
- thalidomide When separating and quantifying a pure enantiomer of lenalidomide in a biological sample, in general, macromolecules such as proteins that cause a decrease in the separation efficiency of low molecules are removed in advance.
- the pretreatment of thalidomide is conventionally performed by extracting a biological sample containing thalidomide one or more times with a hydrophobic organic solvent (for example, n-hexane / ethyl acetate mixture), and drying the obtained organic solvent layer. This was done by re-dissolving in an organic solvent such as dioxane.
- a hydrophobic organic solvent for example, n-hexane / ethyl acetate mixture
- lenalidomide is more polar than thalidomide and has a low solubility in the organic solvent layer, so that the conventional method has low efficiency and is not suitable.
- the present inventors have conducted extensive studies on the effect of pH on racemization of the enantiomer of lenalidomide, and as a result, the enantiomer of lenalidomide is extremely stable under acidic conditions. I found it. Accordingly, in another aspect of the present invention, there is provided a method for pretreating a biological sample containing an enantiomer of lenalidomide, wherein the biological sample is deproteinized under acidic conditions.
- Such acidic conditions are typically pH 5 or less, preferably in the range of pH 2 to pH 5, and most preferably in the range of pH 4 to pH 5.
- the acid added to the sample to provide acidic conditions is not particularly limited as long as the object of the present invention can be achieved, but is not limited to perchloric acid, trichloroacetic acid, trifluoroacetic acid, metaphosphoric acid, hydrochloric acid, or succinic acid or maleic acid. Dicarboxylic acid is mentioned.
- the biological sample is not particularly limited as long as lenalidomide is contained, and examples thereof include blood, serum, plasma, urine, saliva, breast milk, sweat, spinal fluid, semen, tissue, and microsomes.
- the biological sample containing the pretreated lenalidomide enantiomer is subjected to chromatography, preferably high performance liquid chromatography (HPLC), whereby the lenalidomide enantiomer present in the biological sample can be separated easily and efficiently. It becomes possible to quantify.
- HPLC high performance liquid chromatography
- the enantiomer of lenalidomide can be preserved by maintaining the enantiomer of lenalidomide from which the fraction has been collected in an aqueous buffer under acidic conditions.
- acidic conditions are typically pH 5 or lower, preferably in the range of pH 2 to pH 5.
- the aqueous buffer is not particularly limited as long as acidic conditions can be maintained, and examples thereof include citrate buffer, phosphate buffer, acetate buffer, glycine-hydrochloric acid buffer, MES-HEPES buffer, Tris buffer, and borate buffer. It is done.
- Example 1 Stability of lenalidomide in various solvents
- lenalidomide racemate Selleck Chemicals (US)
- methanol, ethanol, isopropanol, acetonitrile, ethyl acetate was incubated at room temperature or 50 ° C. for 24 hours.
- the following HPLC conditions were used to quantify the enantiomer of lenalidomide and evaluate the stability of lenalidomide in various solvents.
- the peak areas of the enantiomers before and after incubation are shown in FIG. 1 (the first observed peak is enantiomer 1 and the next observed peak is enantiomer 2).
- lenalidomide was extremely unstable, but no decomposition was observed with acetonitrile or ethyl acetate.
- Lenalidomide was also stable in isopropanol. Considering these results, it is considered that the decomposition of lenalidomide is promoted by the weak basicity of the alcohol attacking the ⁇ carbon atom of the carbonyl group. This is consistent with the results of the stability of lenalidomide in acetonitrile and ethyl acetate, which are aprotic solvents.
- Example 2 Separation of Lenalidomide Enantiomers Using Various Solvents
- lenalidomide in alcohol especially methanol and ethanol
- water > pH 7
- solvents is unstable, so a highly stable aprotic solvent should be used as the mobile phase and sample solvent. is there.
- LL lenalidomide
- TD thalidomide
- the mobile phase used an aprotic solvent consisting of acetonitrile (ACN), ethyl acetate (EtOAc), tetrahydrofuran (THF) and t-butyl methyl ether (BME).
- ACN acetonitrile
- EtOAc ethyl acetate
- THF tetrahydrofuran
- BME t-butyl methyl ether
- IPA isopropanol
- the enantiomeric resolution was evaluated using the separation factor (RS) defined in the following equation.
- RS 2 ⁇ (T 2 ⁇ T 1 ) / (W 1 + W 2 )
- numbers 1 and 2 mean that they relate to enantiomer 1 and enantiomer 2, respectively (peak 1 elutes earlier than peak 2)
- T means retention time
- W means peak width.
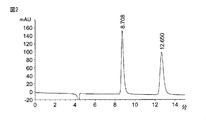
- Table 1 shows the resolution of LL enantiomers when various solvents are used as the mobile phase.
- FIG. 2 shows that the LL enantiomer is well separated when ethyl acetate is used as the mobile phase. Further, when a mobile phase mixed with a certain amount of ethyl acetate was used, the separation degree was superior to those containing no or little ethyl acetate (for example, Nos. 3, 10, 11 and 17). . From these results, it was found that ethyl acetate is a very excellent solvent as a chromatographic mobile phase for LL enantiomer separation.
- Example 3 Preparation of pure enantiomers of lenalidomide Based on the stability of lenalidomide and the results in enantiomeric separation, a method for preparing pure enantiomers of lenalidomide was established. Specifically, the pure enantiomer of lenalidomide was prepared according to the following scheme and HPLC conditions.
- HPLC conditions Apparatus HPD PUMP and LAMBDA1010 (Bischoff) Column: Chiralpak IC (2.0 mm id ⁇ 250 cm, 5 ⁇ m, Daicel Corporation), RT Mobile phase: Ethyl acetate Flow rate: 10 mL / min Injection volume: 2.3 ⁇ L / mL LL racemate in 5000 ⁇ L acetonitrile Detection: UV 254 nm
- the LL enantiomer was completely separated. After fractions were collected and dried, more than 80 mg of pure enantiomer was obtained from 180 mg of racemate. Fractions were dissolved in acetonitrile (1 or 0.1 mg / mL) and stored at ⁇ 25 ° C. Based on the integrated peak area of the chromatogram shown in FIG. 4 (A-2, B-2), fraction 1 and fraction 2 recovered as lenalidomide enantiomer 1 (LL1) and lenalidomide enantiomer 1 (LL2) are: Sufficient enantiomeric purity was 99.02% and 99.96% respectively.
- Example 4 Estimation of absolute configuration for LL1 and LL2 from elution order
- the absolute configuration of LL1 and LL2 compares the order of elution of LL1 and LL2 with commercially available R-thalidomide and S-thalidomide (Sigma Aldrich) Can be reasonably estimated.
- the order of elution was confirmed using the HPLC conditions shown below.
- the enantiomeric excess (EE, [(S ⁇ R) / (S + R) ⁇ 100],%) at each point is plotted in FIG. 7, and the log 10 (EE) versus time plot is shown in FIG.
- FIG. 8 shows an ideal linear approximation.
- the stability of the pure enantiomer showed a very strong dependence on pH, whose half-life was 10 times longer for every pH drop of 1, and at pH 4 it was impossible to estimate the half-life.
- pH 9 rapid racemization and decomposition were observed. This result clearly shows that the enantiomer of lenalidomide is stable under acidic conditions ( ⁇ pH 4).
- Example 6 Establishment of biological sample pretreatment methods So far, there have been many reports on the measurement of thalidomide and lenalidomide in blood (serum and plasma) or urine using HPLC. With respect to TD, measurements of enantiomers in biological samples have been reported and the racemization half-life in vivo or in vitro has been elucidated (Eriksson T et al., Chirality., 1995, 7 (1), p. .44-52 and Knoche B et al., J. Chromatogr. A., 1994, 666, p.235-240). All these measurements are based on the liquid-liquid extraction method. Such a conventional method is shown in scheme (a).
- the aprotic solvent is substantially inactive to racemization and decomposition, it can be measured by this method.
- the solubility of thalidomide and lenalidomide in an organic solvent that is hydrophobic is not high, and thus it has been necessary to repeatedly extract using a large amount of solvent.
- FIG. 9 shows a chromatogram obtained by using the method of scheme (b). As shown in FIG. 9 (b), it was shown that the enantiomer of lenalidomide in serum can be well separated and quantified without being affected by contaminant components such as proteins and can be applied to biological samples.
- Example 7 Racemization of lenalidomide under physiological conditions Using the pure enantiomers obtained, the racemization rate of lenalidomide under biological buffer conditions (pH 7.4, 37 ° C) and human serum (37 ° C) was evaluated according to the following scheme.
- the continuous change of the chromatogram in the incubation of S * -LL is shown in FIG.
- the enantiomer of LL was separated and quantified at a level sufficient to detect the racemized product without being disturbed by other substances in the sample.
- the racemization half-life was calculated in the same manner as described in Example 5. Thereby, the racemization half-life in aqueous buffer was estimated as follows: 272 ⁇ 1 for (S * )-LL, (R * )-LL, (S) -TD and (R) -TD, respectively .3, 267 ⁇ 1.1, 266 ⁇ 10 and 295 ⁇ 24 (min). From the above results, it was revealed that at pH 7.4, the LL enantiomer has a half-life similar to that of TD in an aqueous buffer.
Abstract
本発明の課題は、レナリドミドのエナンチオマーを含有する生体試料を前処理するための新規な方法を提供し、これにより、レナリドミドのエナンチオマーの簡便かつ正確な定量分析方法を確立することにある。 レナリドミドのエナンチオマーを含有する生体試料を酸性条件下で除タンパクすることにより、生体試料中のレナリドミドのエナンチオマーのラセミ化・分解を防ぎ、そして、このようにして前処理した生体試料をHPLCに供することにより、レナリドミドのエナンチオマーを簡便かつ正確に定量することが可能となる。
Description
本発明は、レナリドミドのエナンチオマーを含有する生体試料を前処理するための方法であって、酸性条件下で除タンパクが行われることを特徴とする方法、及び、このようにして前処理したレナリドミドのエナンチオマーを含有する生体試料を高速液体クロマトグラフィーにより分析することを特徴とする、生体試料中のレナリドミドのエナンチオマーを定量するための方法に関する。
レナリドミドは、サリドマイドの誘導体であり、多発性骨髄腫など多様な悪性血液疾患に有効な免疫調製薬として広く用いられている。レナリドミドは、サリドマイドと比較して、改善された毒性プロフィール及び優れた免疫調製活性を有することが報告されている(非特許文献1)。逆相モードを利用したHPLC法によって、レナリドミドの薬物動態学的特性が明らかにされており(非特許文献2; 非特許文献3; 及び非特許文献4)、レナリドミドの最終排泄半減期は、約3~4時間であると推測されている。
また、サリドマイドやその誘導体、類似体に関しては、R体とS体との薬物活性の相違が報告されている。例えば、サリドマイドの鎮静作用はR体のみに見られることが報告されている一方で(非特許文献5)、S-ポマリドミド(3-アミノ-フタリミド-グルタリミド)は、R体やラセミ体と比較して、bFGFやVEGFにより誘発される角膜血管新生を有意に阻害することが報告されている(非特許文献6)。
このようなエナンチオマーにおける活性の違いにもかかわらず、依然として、サリドマイドがR体:S体=50:50のラセミ体混合物として投与されている。この主な理由の1つとして、血中での極めて速いラセミ化特性が挙げられる(非特許文献7)。
サリドマイドのエナンチオマーは、有機溶媒を用いた反復抽出や、修飾アミロース(非特許文献8)、セルロース(非特許文献7)、バンコマイシン(非特許文献9)又はメタクリルアミド(非特許文献10)などのエナンチオ選択型の固定相を用いたクロマトグラフィー法によって分離・定量されており、血中で極めて迅速にラセミ化することが明らかになっている。このようなラセミ化は、G. Blaschke らによって初めて開示され、ヒト血漿中でのサリドマイドのラセミ化半減期は約10分であると報告されている(非特許文献10)。これは、サリドマイドの排出半減期が8.7時間であること(非特許文献11)を考慮すると極めて短いものである。
このような体内動態を示すことから、サリドマイドの純粋なエナンチオマーを投与することに薬理学的な意義がないものと考えられており、サリドマイドに類似した基本骨格を有するレナリドミドについても同様の性質を有するものと考えられてきたため、レナリドミドの純粋なエナンチオマーの薬物動態学的・薬理学的特性については十分に研究されておらず、生体試料中のレナリドミドの純粋なエナンチオマーの分離・定量方法についても、これまでに全く報告されていない。
Hideshima T et al., Ther. Clin. Risk. Manag. 2008, 4(1), p.129-36
Tohnya TM et al., J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci., 2004, 811(2), p.135-41
Chen N et al., Cancer. Chemother. Pharmacol., 2012, 70(5), p.717-25
Chen N et al., J Clin Pharmacol., 2007, 47(12), p.1466-75
Hoeglund P et al., J. Pharmacokinet. Biopharm., 1998 26(4), p.363-83
Lentzsch S et al., Cancer. Res., 2002, 62(8), p.2300-5
Eriksson T et al., Chirality., 1995, 7(1), p.44-52
Meyring M et al., J. Chromatogr. A. 2000, 876(1-2), p.157-67
Murphy-Poulton SF et al., J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci., 2006, 831(1-2), p.48-56
Knoche B et al., J. Chromatogr. A., 1994, 666, p.235-240
Chen TL et al., Drug. Metab. Dispos., 1989, 17(4), p.402-5
本発明の課題は、レナリドミドのエナンチオマーのラセミ化及び/又は分解を抑制し、生体中のレナリドミドのエナンチオマーそれぞれの含量を維持したまま試料を前処理するための新規な方法を提供することにある。
本発明者は、この度、レナリドミドのエナンチオマーが、サリドマイドのエナンチオマーと比較した場合、有意に長い間生体内に滞留する、という驚くべき知見を得た。上述のとおり、サリドマイドのエナンチオマーは生体内におけるラセミ化速度が極めて早いため、純粋なエナンチオマーを投与することに薬理学的な意義がないものと考えられていたが、レナリドミドは、その排泄半減期を考慮すると、薬理効果を無視できない程長い時間エナンチオマーとして生体内に留まっていることが明らかになった。したがって、レナリドミドの純粋なエナンチオマーの薬物動態学的・薬理学的作用の評価は、極めて重要な意味を有するものと考えられる。また、生体試料中のレナリドミドの純粋なエナンチオマーを分析する場合には、タンパク質の妨害による分析精度の低下を防ぐため、あらかじめ、除タンパク処理を行う必要がある。サリドマイドの前処理は、従来、サリドマイドを含む生体試料を強疎水性有機溶媒により、1又は複数回抽出し、得られた有機溶媒層を乾燥させ後、有機溶媒に再溶解させることによって行っていた。しかしながら、レナリドミドはサリドマイドよりも極性が高く、有機溶媒層に溶解しにくいため、従来の方法が適用できなかった。このような課題を解決するために、本発明者は、レナリドミドのエナンチオマーのラセミ化に及ぼすpHの影響について鋭意検討を重ねた結果、レナリドミドのエナンチオマーは、酸性条件下において極めて安定であることを見いだし、本発明を完成した。
具体的には、本発明は以下の発明を包含する。
[1] レナリドミドのエナンチオマーを含有する生体試料を前処理するための方法であって、酸性条件下で生体試料の除タンパクが行われることを特徴とする方法。
[2] 前記酸性条件がpH5以下であることを特徴とする、[1]に記載の方法。
[3] 過塩素酸、トリクロロ酢酸、トリフルオロ酢酸、メタリン酸、塩酸、コハク酸、及びマレイン酸から選択される酸が添加されることを特徴とする、[1]又は[2]に記載の方法。
[4] 前記生体試料が、血液、血清、血漿、尿、唾液、母乳、髄液、精液、組織、又はミクロソームであることを特徴とする、[1]~[3]のいずれかに記載の方法。
[5] 前記レナリドミドのエナンチオマーがS体であることを特徴とする、[1]~[4]のいずれかに記載の方法。
[6] 前記レナリドミドのエナンチオマーがR体であることを特徴とする、[1]~[4]のいずれかに記載の方法。
[7] 生体試料中のレナリドミドのエナンチオマーを定量するための方法であって、[1]~[6]のいずれかに記載の方法によって前処理したレナリドミドのエナンチオマーを含有する生体試料を高速液体クロマトグラフィーにより分離分析することを特徴とする、方法。
[8] レナリドミドのエナンチオマーを保存するための方法であって、レナリドミドのエナンチオマーを水性バッファーにおいて酸性条件下で維持することを特徴とする方法。
[9] 前記酸性条件がpH5以下であることを特徴とする、[8]に記載の方法。
[10] 前記水性バッファーが、クエン酸バッファーであることを特徴とする、[8]又は[9]に記載の方法。
[11] 前記レナリドミドのエナンチオマーがS体であることを特徴とする、[8]~[10]のいずれかに記載の方法。
[12] 前記レナリドミドのエナンチオマーがR体であることを特徴とする、[8]~[10]のいずれかに記載の方法。
[1] レナリドミドのエナンチオマーを含有する生体試料を前処理するための方法であって、酸性条件下で生体試料の除タンパクが行われることを特徴とする方法。
[2] 前記酸性条件がpH5以下であることを特徴とする、[1]に記載の方法。
[3] 過塩素酸、トリクロロ酢酸、トリフルオロ酢酸、メタリン酸、塩酸、コハク酸、及びマレイン酸から選択される酸が添加されることを特徴とする、[1]又は[2]に記載の方法。
[4] 前記生体試料が、血液、血清、血漿、尿、唾液、母乳、髄液、精液、組織、又はミクロソームであることを特徴とする、[1]~[3]のいずれかに記載の方法。
[5] 前記レナリドミドのエナンチオマーがS体であることを特徴とする、[1]~[4]のいずれかに記載の方法。
[6] 前記レナリドミドのエナンチオマーがR体であることを特徴とする、[1]~[4]のいずれかに記載の方法。
[7] 生体試料中のレナリドミドのエナンチオマーを定量するための方法であって、[1]~[6]のいずれかに記載の方法によって前処理したレナリドミドのエナンチオマーを含有する生体試料を高速液体クロマトグラフィーにより分離分析することを特徴とする、方法。
[8] レナリドミドのエナンチオマーを保存するための方法であって、レナリドミドのエナンチオマーを水性バッファーにおいて酸性条件下で維持することを特徴とする方法。
[9] 前記酸性条件がpH5以下であることを特徴とする、[8]に記載の方法。
[10] 前記水性バッファーが、クエン酸バッファーであることを特徴とする、[8]又は[9]に記載の方法。
[11] 前記レナリドミドのエナンチオマーがS体であることを特徴とする、[8]~[10]のいずれかに記載の方法。
[12] 前記レナリドミドのエナンチオマーがR体であることを特徴とする、[8]~[10]のいずれかに記載の方法。
本発明によって、レナリドミドのエナンチオマーのラセミ化及び/又は分解を抑制し、生体中のレナリドミドのエナンチオマー含量を維持したまま試料を前処理し、簡便かつ効率的に正確な生体試料中のレナリドミドのエナンチオマーを分離・定量することが可能となる。これにより、生体内におけるレナリドミドの純粋なエナンチオマーの薬物動態学的特性や薬理学的特性を明らかにし、レナリドミドの純粋なエナンチオマーの投与による副作用低減や薬効向上の可能性を見出すことに役立つものと考えられる。したがって、本発明は、レナリドミドのエナンチオマーの薬理薬効・安全性・物性・薬物動態研究など医療・製薬産業における活用・展開に大きく寄与するものと確信する。
レナリドミド(LL)は、多発性骨髄腫治療薬として知られるサリドマイド(TD)の誘導体であり、多発性骨髄腫など多様な悪性血液疾患に有効な免疫調製薬として広く用いられている。以下の式により表される化学構造を有し、サリドマイドと同様に、ジオキソピペリジン環上に不斉中心を有する。

レナリドミドはデキサメタゾンとの併用により、再発・難治性多発性骨髄腫での奏効率が60%と高く、世界中で認可が進んでおり、これまでに約50ヶ国で認可され、売上は4000億円を超えるブロックバスター薬となっている。日本では2010年に承認されたが、安全性を確保する観点から医療関係者・患者・家族の管理手順遵守や全症例で使用成績調査を実施しなければならないという条件が付いている。レナリドミドはサリドマイドよりも薬効が高く、眠気や痺れ等の副作用は軽微である一方、サリドマイドと同様に催奇性のリスクが依然として存在するため、妊婦への投与は禁忌となっている。レナリドミドはサリドマイドの誘導体であるため、サリドマイドと同様、血中で速やかにラセミ化するものと推測されている。このため、R体:S体=50:50のラセミ体として販売されており、現在、レナリドミドの純粋なエナンチオマーを入手することは困難である。また、ラセミ化を抑制し安定な前処理・分離分析条件も確立されていないことから、レナリドミドの純粋なエナンチオマーの体内動態やラセミ化速度などについては、これまでにほとんど知見がない。
本発明者は、この度、クロマトグラフィー法を用いた光学分割において、非プロトン性溶媒、第二級アルコール及びこれらの混合物から成る群から選択される有機溶媒を移動相として用いることにより、分解やラセミ化を抑制し、レナリドミドの純粋なエナンチオマーを分離・定量できることを見出した。したがって、本発明の態様において、レナリドミドのエナンチオマーを分離・定量するための方法であって、レナリドミドのエナンチオマー混合物を含む試料クロマトグラフィーに供し、そして、非プロトン性溶媒、第二級アルコール及びこれらの混合物から成る群から選択される有機溶媒を移動相として用いることにより、レナリドミドのエナンチオマー混合物からレナリドミドの各エナンチオマーを安定的に光学分割することを特徴とする方法が供される。
クロマトグラフィー法は、不斉識別能を有する化合物(キラル識別子)が担持された固定相にエナンチオマー混合物を含む試料を移動相の有機溶媒と共に流し、各々のエナンチオマーを吸着させ、その保持時間の差を利用して各々のエナンチオマーを光学分割するための方法である。一般には、光学分割用カラムを装着した高速液体クロマトグラフィー(HPLC)装置を用いて行われる。
移動相として用いられる非プロトン性溶媒は、本発明の目的を達成できる限り特に制限されないが、例えば、アセトニトリル、酢酸エチルなどのエステル類、アセトンなどのケトン類、ジエチルエーテル、ジイソプロピルエーテルなどのエーテル類又はこれらの組み合せなどが挙げられる。好ましくは、アセトニトリル、酢酸エチル又はこれらの組み合せである。
移動相として用いられる第二級アルコールは、本発明の目的を達成できる限り特に制限されないが、イソプロパノール、2-ブタノール、シクロペンタノール、又はシクロヘキサノール又はこれらの組み合せが挙げられる。好ましくは、イソプロパノールである。
本発明の方法において用いられる光学分割用カラムは、本発明の目的を達成できる限り特に限定されず、順相カラム、逆相カラム、その他の分離モードのカラムやそれぞれを組合わせたマルチモードカラムでもよい。典型的には、多糖誘導体キラルカラムが用いられる。多糖誘導体キラルカラムとは、キラル識別子として多糖誘導体が担体に固定化されたカラムである。多糖誘導体キラルカラムに担持される多糖誘導体としては、例えば、アミロース誘導体やセルロール誘導体が挙げられる。本発明においては、好ましくはChiralpak(登録商標)IA又はChiralpak(登録商標)ICが用いられる。
本発明の光学分割法により、レナリドミドのエナンチオマー混合体(ラセミ体など)から、分解やラセミ化を伴うことなく、レナリドミドの純粋なエナンチオマーを分離・定量することができる。
生体試料中のレナリドミドの純粋なエナンチオマーを分離・定量する場合には、一般に低分子の分離効率の低下をもたらすタンパク質のような高分子は、あらかじめ取り除く。サリドマイドの前処理は、従来、サリドマイドを含む生体試料を疎水性有機溶媒(例えば、n-ヘキサン/酢酸エチル混液)により、1又は複数回抽出し、得られた有機溶媒層を乾燥させた後、ジオキサンなどの有機溶媒に再溶解させることによって行っていた。しかしながら、レナリドミドはサリドマイドよりも極性が高く、有機溶媒層への溶解度が小さいため、従来の方法では効率が低く不適であった。このような課題を解決するために、本発明者らは、レナリドミドのエナンチオマーのラセミ化に及ぼすpHの影響について鋭意検討を重ねた結果、レナリドミドのエナンチオマーは、酸性条件下において極めて安定であることを見出した。したがって、本発明の別の態様においては、レナリドミドのエナンチオマーを含有する生体試料を前処理するための方法であって、前記生体試料を酸性条件下で除タンパクすることを特徴とする方法が供される。
このような酸性条件は、典型的にはpH5以下であり、好ましくはpH2~pH5の範囲であり、最も好ましくはpH4~pH5の範囲である。
酸性条件をもたらすために試料に加えられる酸は、本発明の目的を達成できる限り特に制限されないが、過塩素酸、トリクロロ酢酸、トリフルオロ酢酸、メタリン酸、塩酸、又はコハク酸やマレイン酸などのジカルボン酸が挙げられる。
生体試料は、レナリドミドが含有されている限り特に制限されないが、例えば、血液、血清、血漿、尿、唾液、母乳、汗、髄液、精液、組織又はミクロソームなどが挙げられる。
このようにして、前処理したレナリドミドのエナンチオマーを含有する生体試料をクロマトグラフィー、好ましくは高速液体クロマトグラフィー(HPLC)に供することにより、生体試料に存在するレナリドミドのエナンチオマーを簡便かつ効率的に分離・定量することが可能となる。
さらに、画分を分取したレナリドミドのエナンチオマーを水性バッファー中で、酸性条件下で維持することにより、レナリドミドのエナンチオマーを保存することが可能となる。このような酸性条件は、典型的にはpH5以下であり、好ましくはpH2~pH5の範囲である。水性バッファーは、酸性条件を維持することができる限り特に制限されないが、例えば、クエン酸バッファー、リン酸バッファー、酢酸バッファー、グリシン-塩酸バッファー、MES-HEPESバッファー、Trisバッファー、又はホウ酸バッファーが挙げられる。
例1.多様な溶媒におけるレナリドミドの安定性
試料として、メタノール、エタノール、イソプロパノール、アセトニトリル、酢酸エチル中の0.5mg/mLのレナリドミドのラセミ体(Selleck Chemicals(米))を室温又は50℃で24時間インキュベートし、以下のHPLC条件を用いて、レナリドミドのエナンチオマーを定量し、多様な溶媒におけるレナリドミドの安定性を評価した。
HPLC条件
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IA(4.6×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH(100%)
流速: 1.0mL/分
注入体積: 5μL
検出: UV 230nm
試料として、メタノール、エタノール、イソプロパノール、アセトニトリル、酢酸エチル中の0.5mg/mLのレナリドミドのラセミ体(Selleck Chemicals(米))を室温又は50℃で24時間インキュベートし、以下のHPLC条件を用いて、レナリドミドのエナンチオマーを定量し、多様な溶媒におけるレナリドミドの安定性を評価した。
HPLC条件
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IA(4.6×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH(100%)
流速: 1.0mL/分
注入体積: 5μL
検出: UV 230nm
インキュベーション前後のエナンチオマーのピーク面積を図1に示す(最初に観察されたピークをエナンチオマー1とし、次に観察されたピークをエナンチオマー2とした)。メタノール及びエタノール中では、レナリドミドは極めて不安定であったが、アセトニトリルや酢酸エチルでは分解が見られなかった。また、レナリドミドは、イソプロパノール中でも安定であった。これらの結果を考慮すると、レナリドミドの分解は、カルボニル基のα炭素原子を攻撃するアルコールの弱塩基性により促進されるものと考えられる。これは、非プロトン性溶媒であるアセトニトリルや酢酸エチルでレナリドミドが安定性を示した結果にも矛盾しない。
例2.多様な溶媒を用いたレナリドミドのエナンチオマーの分離
レナリドミドのエナンチオマーのラセミ化速度を明らかにするために、レナリドミドの純粋なエナンチオマーを分離・定量することは極めて重要である。例1に示されるとおり、アルコール(特に、メタノールとエタノール)や水(>pH7)溶媒中のレナリドミドは不安定であるため、安定性の高い非プロトン性溶媒を移動相や試料溶媒として用いるべきである。
以下のHPLC条件に基づき、表1に示される多様な有機溶媒を移動相として用いた場合のレナリドミド(LL)及びサリドマイド(TD)のエナンチオマーの分離について試験した。試料は、アセトニトリル中に溶解した0.5mg/mLのLL(和光純薬工業)及びTD(シグマアルドリッチ)を用いた。
レナリドミドのエナンチオマーのラセミ化速度を明らかにするために、レナリドミドの純粋なエナンチオマーを分離・定量することは極めて重要である。例1に示されるとおり、アルコール(特に、メタノールとエタノール)や水(>pH7)溶媒中のレナリドミドは不安定であるため、安定性の高い非プロトン性溶媒を移動相や試料溶媒として用いるべきである。
以下のHPLC条件に基づき、表1に示される多様な有機溶媒を移動相として用いた場合のレナリドミド(LL)及びサリドマイド(TD)のエナンチオマーの分離について試験した。試料は、アセトニトリル中に溶解した0.5mg/mLのLL(和光純薬工業)及びTD(シグマアルドリッチ)を用いた。
HPLC条件
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IC(4.6×250cm,5μm,株式会社ダイセル),RT
移動相: 表1に示される溶媒
流速: 1.0mL/分
注入体積: 5μL
検出: UV 230nm又は254nm
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IC(4.6×250cm,5μm,株式会社ダイセル),RT
移動相: 表1に示される溶媒
流速: 1.0mL/分
注入体積: 5μL
検出: UV 230nm又は254nm
移動相は、アセトニトリル(ACN)、酢酸エチル(EtOAc)、テトラヒドロフラン(THF)及びt-ブチルメチルエーテル(BME)から成る非プロトン性溶媒を用いた。また、イソプロパノール(IPA)は第二級アルコールであるが、図1に示されるとおり優れた安定性を有することから同様に試験した。エナンチオマー分離能は、以下の式に規定される分離係数(RS)を用いて評価した。
RS=2×(T2-T1)/(W1+W2)
式中、数字1及び数字2は、それぞれ、エナンチオマー1及びエナンチオマー2に関することを意味し(ピーク1はピーク2よりも早く溶出する)、Tは保持時間を意味し、Wはピーク幅を意味する。移動相として、各種溶媒を用いた場合のLLエナンチオマーの分離度を表1に示す。
RS=2×(T2-T1)/(W1+W2)
式中、数字1及び数字2は、それぞれ、エナンチオマー1及びエナンチオマー2に関することを意味し(ピーク1はピーク2よりも早く溶出する)、Tは保持時間を意味し、Wはピーク幅を意味する。移動相として、各種溶媒を用いた場合のLLエナンチオマーの分離度を表1に示す。
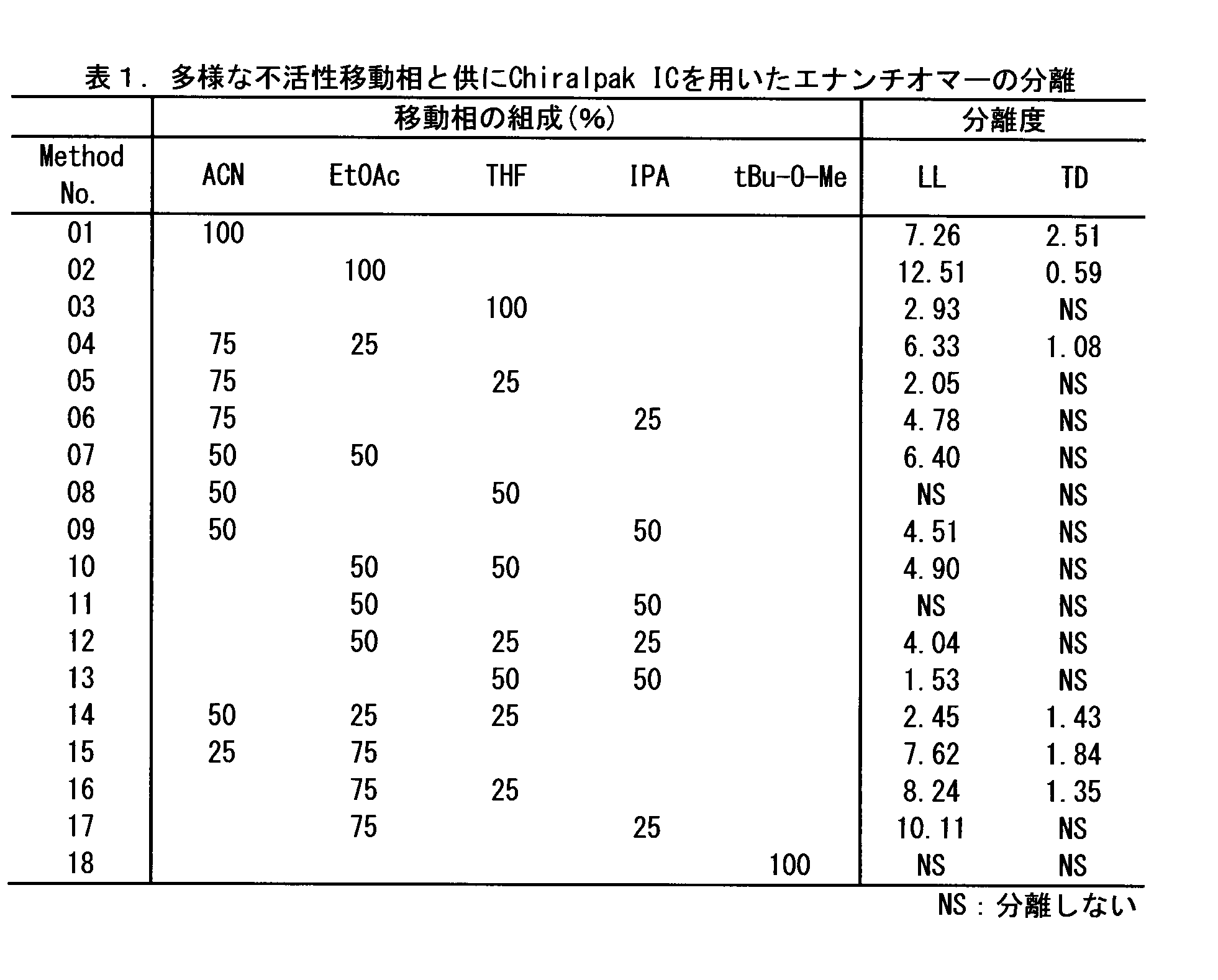
これらの移動相の中で、LLエナンチオマーの分離において最も有効であったものは酢酸エチルであり、酢酸エチルを移動相として用いた場合(表1,No.02)に最も高い分離度が得られた(RS=12.51)。図2には、酢酸エチルを移動相として用いた場合に、LLエナンチオマーが良好に分離することが示されている。さらに、一定量の酢酸エチルが混合された移動相を用いた場合、酢酸エチルを全くあるいは僅かしか含まないものよりも優れた分離度を示した(例えば、No.3,10,11及び17)。これらの結果から、LLエナンチオマー分離のためのクロマトグラフィーの移動相として酢酸エチルは極めて優れた溶媒であることが判明した。
また、アセトニトリルも良好な分離度を示した(表1,No.01,RS=7.26)。さらに、図1に示されるとおり、アセトニトリルも優れた安定性を示すことから、LLエナンチオマー分離のためのクロマトグラフィーの移動相として、良好な溶媒となり得る。
一方で、TDについては、いずれの移動相を用いた場合にも十分な分離が観察されなかった。
例3.レナリドミドの純粋なエナンチオマーの調製
上記レナリドミドの安定性及びエナンチオマー分離における結果に基づき、レナリドミドの純粋なエナンチオマーを調製するための方法を確立した。具体的には、以下のスキーム及びHPLC条件に従い、レナリドミドの純粋なエナンチオマーを調製した。
上記レナリドミドの安定性及びエナンチオマー分離における結果に基づき、レナリドミドの純粋なエナンチオマーを調製するための方法を確立した。具体的には、以下のスキーム及びHPLC条件に従い、レナリドミドの純粋なエナンチオマーを調製した。

HPLC条件
装置: HPD PUMP 及び LAMBDA1010 (Bischoff社) カラム: Chiralpak IC(2.0mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: 酢酸エチル
流速: 10mL/分
注入体積: 5000μLのアセトニトリル中2.3μL/mLのLLラセミ体
検出: UV 254nm
装置: HPD PUMP 及び LAMBDA1010 (Bischoff社) カラム: Chiralpak IC(2.0mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: 酢酸エチル
流速: 10mL/分
注入体積: 5000μLのアセトニトリル中2.3μL/mLのLLラセミ体
検出: UV 254nm
図3に示されるとおり、LLエナンチオマーは完全に分離した。画分を分取し乾燥させた後、180mgのラセミ体から、80mg超の純粋なエナンチオマーが得られた。画分をアセトニトリル(1又は0.1mg/mL)に溶解し、-25℃で保管した。図4(A-2,B-2)に示されたクロマトグラムの積分ピーク面積に基づき、レナリドミドエナンチオマー1(LL1)及びレナリドミドエナンチオマー1(LL2)として回収された画分1及び画分2は、それぞれ99.02%及び99.96%の十分なエナンチオマー純度を示した。
例4.溶出順番からのLL1及びLL2についての絶対配置の推定
LL1及びLL2の絶対配置は、LL1とLL2の溶出の順番を、商業的に入手可能なR-サリドマイド及びS-サリドマイド(シグマアルドリッチ)と比較することにより十分合理的に推定できる。溶出の順番は以下に示すHPLC条件を用いて確認した。
LL1及びLL2の絶対配置は、LL1とLL2の溶出の順番を、商業的に入手可能なR-サリドマイド及びS-サリドマイド(シグマアルドリッチ)と比較することにより十分合理的に推定できる。溶出の順番は以下に示すHPLC条件を用いて確認した。
HPLC条件
装置: ナノスペース SI-2 シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入体積: 0.1mg/mLの(S)-TD,(R)-TD,5μL
検出: UV 230nm
装置: ナノスペース SI-2 シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入体積: 0.1mg/mLの(S)-TD,(R)-TD,5μL
検出: UV 230nm
図5からも明らかなように、S-サリドマイドは、R-サリドマイドよりも早く溶出した。同じHPLC条件下において、LL1がLL2よりも早く溶出することを考慮すれば、LL1の絶対配置はS-エナンチオマーに対応し、LL2の絶対配置はR-エナンチオマーに対応するものと考えられる(以下、LL1及びLL2は、S*-LL及びR*-LLと規定する)。
例5.pHにおけるラセミ化半減期の依存性
レナリドミドのラセミ化半減期及びそのpH依存性を明らかにするために、以下のスキーム及びHPLC条件にしたがい実験を行った。
レナリドミドのラセミ化半減期及びそのpH依存性を明らかにするために、以下のスキーム及びHPLC条件にしたがい実験を行った。


HPLC条件
装置: ナノスペース SI-2 シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入: 5μL
検出: UV 230nm
装置: ナノスペース SI-2 シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm,株式会社ダイセル),RT
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入: 5μL
検出: UV 230nm
多様なpHにおける(S*)-LLの変動を図6に示す。pH6でのインキュベーションでは(R*)-LLが僅かに産生し、7以上のpHではラセミ化が有意に加速した。また、pH9及び8では、(S*)-及び(R*)-LLのピーク面積が低下していることから、これらのpH条件ではLLのラセミ化および分解が生じていることがわかる。
各ポイントの鏡像体過剰率(EE,[(S-R)/(S+R)×100],%)を図7にプロットし、そしてlog10(EE)対時間プロットを図8に示す。サリドマイドに関して報告されるとおり、ラセミ化は擬一次反応であると考えられるため、図8は、理想的な直線近似を示す。
この直線性を用いて、直線近似式(y=ax+b)に基づきEE=50の時間を計算することによりラセミ化半減期を算定した。純粋なエナンチオマーの安定性は、pHにおける極めて強い依存性を示し、その半減期は、pHが1下がるごとに10倍も長くなり、pH4において、半減期の推定は不可能となった。一方、pH9においては、急激なラセミ化と分解が観察された。これの結果は、レナリドミドのエナンチオマーが、酸性条件(<pH4)で安定であることを明確に示すものである。
この直線性を用いて、直線近似式(y=ax+b)に基づきEE=50の時間を計算することによりラセミ化半減期を算定した。純粋なエナンチオマーの安定性は、pHにおける極めて強い依存性を示し、その半減期は、pHが1下がるごとに10倍も長くなり、pH4において、半減期の推定は不可能となった。一方、pH9においては、急激なラセミ化と分解が観察された。これの結果は、レナリドミドのエナンチオマーが、酸性条件(<pH4)で安定であることを明確に示すものである。
例6.生体試料の前処理方法の確立
これまでに、HPLCを用いた血液(血清及び血漿)又は尿中におけるサリドマイドやレナリドミドの測定については多く報告されている。TDに関して、生体試料中におけるエナンチオマーの測定が報告されており、生体内又は試験管内におけるラセミ化半減期は明らかとなっている(Eriksson T et al., Chirality., 1995, 7(1), p.44-52及びKnoche B et al., J. Chromatogr. A., 1994, 666, p.235-240)。これらの測定は全て液-液抽出法に基づくものである。このような、従来の方法をスキーム(a)に示す。非プロトン性溶媒は、ラセミ化や分解に実質的に不活性であるため、この方法により測定が可能である。しかしながら、疎水性である有機溶媒中のサリドマイドやレナリドミドの溶解度は高くないため、多量の溶媒を用いて繰り返し抽出する必要があった。
これまでに、HPLCを用いた血液(血清及び血漿)又は尿中におけるサリドマイドやレナリドミドの測定については多く報告されている。TDに関して、生体試料中におけるエナンチオマーの測定が報告されており、生体内又は試験管内におけるラセミ化半減期は明らかとなっている(Eriksson T et al., Chirality., 1995, 7(1), p.44-52及びKnoche B et al., J. Chromatogr. A., 1994, 666, p.235-240)。これらの測定は全て液-液抽出法に基づくものである。このような、従来の方法をスキーム(a)に示す。非プロトン性溶媒は、ラセミ化や分解に実質的に不活性であるため、この方法により測定が可能である。しかしながら、疎水性である有機溶媒中のサリドマイドやレナリドミドの溶解度は高くないため、多量の溶媒を用いて繰り返し抽出する必要があった。
このため、例5に実証された酸性条件におけるエナンチオマーの安定性に関する知見に基づき、極めて単純かつ効率的なレナリドミドの前処理方法を確立した。本発明の分析法をスキーム(b)に示す。反応を停止させ、かつ同時にタンパク質を沈殿・除去するために酸としてHClO4を用いた。
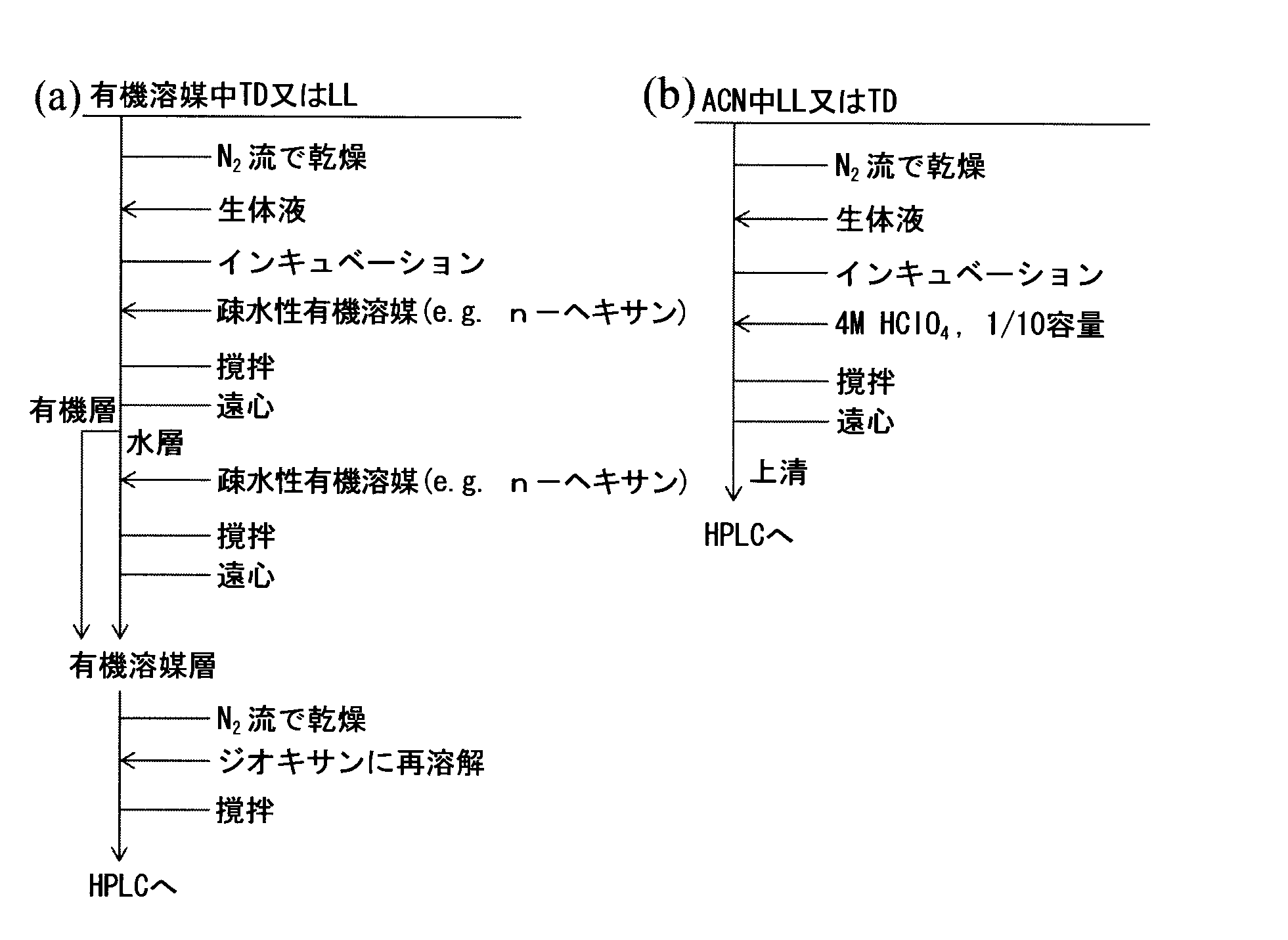
スキーム(b)の方法を用いて得られたクロマトグラムを図9に示す。図9(b)に示されるとおり、血清中のレナリドミドのエナンチオマーはタンパク質などの夾雑成分の影響を受けることなく良好に分離・定量が可能で生体試料にも適用できることが示された。
例7.生理条件におけるレナリドミドのラセミ化
得られた純粋なエナンチオマーを用いて、生体バッファー条件(pH7.4,37℃)及びヒト血清(37℃)におけるレナリドミドのラセミ化速度を、以下のスキームに従い評価した。
得られた純粋なエナンチオマーを用いて、生体バッファー条件(pH7.4,37℃)及びヒト血清(37℃)におけるレナリドミドのラセミ化速度を、以下のスキームに従い評価した。

HPLC条件
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm)+Security guard C8(3.0mm i.d.×4mm)
温度: 40℃
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入: 20μL
検出: UV 230nm
装置: ナノスペース SI-2シリーズ(資生堂)
カラム: Chiralpak IA(4.6mm i.d.×250cm,5μm)+Security guard C8(3.0mm i.d.×4mm)
温度: 40℃
移動相: EtOH/H2O(95/5,v/v)中0.1%ギ酸
流速: 0.75mL/分
注入: 20μL
検出: UV 230nm
図10に示される(S*)-LLのクロマトグラム[(a)バッファー中、(b)血清中0.01mg/mL]からも明らかなように、本発明の方法は、十分な保持・分離と検出感度(シグナル/ノイズ比)を有する。LLを伴わない血清のみに由来するブランク試料(c)は、本発明の方法においてLLの測定を阻害する夾雑物質を含まなかった。図18(b)の(S*)-LLのピーク面積は、図18(a)の91%であった。これは、血清中レナリドミドの前処理において十分な回収率を有することを示す。
S*-LLのインキュベーションにおけるクロマトグラムの連続変化を図11に示す。LLのエナンチオマーは、試料中で他の物質に妨害されることなく、ラセミ化産物を検出するために十分なレベルで分離・定量された。
例5に記載される方法と同様にしてラセミ化半減期を算出した。これにより、水性バッファー中のラセミ化半減期を以下のとおり推定した;(S*)-LL、(R*)-LL、(S)-TD及び(R)-TDについて、それぞれ、272±1.3、267±1.1、266±10及び295±24(分)。以上の結果から、pH7.4において、LLエナンチオマーは、水性バッファー中ではTDと同様の半減期を有することが明らかとなった。
ヒト血清中における半減期もまた、同様に算定した;(S*)-LL、(R*)-LL、(S)-TD及び(R)-TDについて、それぞれ、131±4.9、109±2.0、21.8±1.0及び32.9±25(分)。これらの結果を、図12に示す。ヒト血清中のサリドマイドは、バッファー中と比較して僅か8.2%(S体)及び11.1%(R体)の半減期であり、サリドマイドのラセミ化は、血清中で極度に促進されることが解る。このエナンチオマーに認められたラセミ化の促進は、生体分子、例えば血清中のヒトアルブミンなどのタンパク質によるものと思われる。
血清中のLLに関しては、(S*)-LLが(S)-TDよりも6倍も長いラセミ化半減期を有し、(R*)-LLは(R)-TDよりも3倍長いラセミ化半減期を有するという結果が得られた。これらの結果は、LLエナンチオマー、特に(S*)-LLが、サリドマイドと比較して極めて安定であることを明確に示すものである。なお、TDの場合とは対照的に、(S*)-LLのほうが(R*)-LLよりも安定であった。このようなS-及びR-エナンチオマーの半減期の相違にも注目すべきである。
Claims (12)
- レナリドミドのエナンチオマーを含有する生体試料を前処理するための方法であって、酸性条件下で生体試料の除タンパクが行われることを特徴とする方法。
- 前記酸性条件がpH5以下であることを特徴とする、請求項1に記載の方法。
- 過塩素酸、トリクロロ酢酸、トリフルオロ酢酸、メタリン酸、塩酸、コハク酸、及びマレイン酸から選択される酸が添加されることを特徴とする、請求項1又は2に記載の方法。
- 前記生体試料が、血液、血清、血漿、尿、唾液、母乳、髄液、精液、組織、又はミクロソームであることを特徴とする、請求項1~3のいずれか1項に記載の方法。
- 前記レナリドミドのエナンチオマーがS体であることを特徴とする、請求項1~4のいずれか1項に記載の方法。
- 前記レナリドミドのエナンチオマーがR体であることを特徴とする、請求項1~4のいずれか1項に記載の方法。
- 生体試料中のレナリドミドのエナンチオマーを定量するための方法であって、請求項1~6のいずれか1項に記載の方法によって前処理したレナリドミドのエナンチオマーを含有する生体試料を高速液体クロマトグラフィーにより分離分析することを特徴とする、方法。
- レナリドミドのエナンチオマーを保存するための方法であって、レナリドミドのエナンチオマーを水性バッファーにおいて酸性条件下で維持することを特徴とする方法。
- 前記酸性条件がpH5以下であることを特徴とする、請求項8に記載の方法。
- 前記水性バッファーが、クエン酸バッファーであることを特徴とする、請求項8又は9に記載の方法。
- 前記レナリドミドのエナンチオマーがS体であることを特徴とする、請求項8~10のいずれか1項に記載の方法。
- 前記レナリドミドのエナンチオマーがR体であることを特徴とする、請求項8~10のいずれか1項に記載の方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15796899.1A EP3147661A4 (en) | 2014-05-22 | 2015-05-21 | Method for pretreatment and method for analysis of lenalidomide in biological sample |
| US15/312,777 US10725057B2 (en) | 2014-05-22 | 2015-05-21 | Method for pretreatment and method for analysis of lenalidomide in biological sample |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014106360A JP6566607B2 (ja) | 2014-05-22 | 2014-05-22 | 生体試料中レナリドミドの前処理方法及び分析方法 |
| JP2014-106360 | 2014-05-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015178460A1 true WO2015178460A1 (ja) | 2015-11-26 |
Family
ID=54554120
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/064642 WO2015178460A1 (ja) | 2014-05-22 | 2015-05-21 | 生体試料中レナリドミドの前処理方法及び分析方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10725057B2 (ja) |
| EP (1) | EP3147661A4 (ja) |
| JP (1) | JP6566607B2 (ja) |
| TW (1) | TWI673495B (ja) |
| WO (1) | WO2015178460A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210285975A1 (en) * | 2017-12-25 | 2021-09-16 | Fujirebio Inc. | Method of testing a blood for macrolide immunosuppressant |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3167746A1 (en) | 2020-01-29 | 2021-08-05 | Foghorn Therapeutics Inc. | Compounds and uses thereof |
| US11787800B2 (en) | 2020-07-29 | 2023-10-17 | Foghorn Therapeutics Inc. | BRD9 degraders and uses thereof |
| WO2023283263A1 (en) | 2021-07-06 | 2023-01-12 | Foghorn Therapeutics Inc. | Citrate salt, pharmaceutical compositions, and methods of making and using the same |
| WO2023009719A2 (en) * | 2021-07-28 | 2023-02-02 | Foghorn Therapeutics Inc. | Stability-enhancing compositions and methods of preparing compounds |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6616934B2 (ja) | 2014-05-22 | 2019-12-04 | 株式会社 資生堂 | レナリドミドの光学分割方法 |
-
2014
- 2014-05-22 JP JP2014106360A patent/JP6566607B2/ja not_active Expired - Fee Related
-
2015
- 2015-05-21 WO PCT/JP2015/064642 patent/WO2015178460A1/ja active Application Filing
- 2015-05-21 US US15/312,777 patent/US10725057B2/en active Active
- 2015-05-21 EP EP15796899.1A patent/EP3147661A4/en not_active Withdrawn
- 2015-05-22 TW TW104116549A patent/TWI673495B/zh not_active IP Right Cessation
Non-Patent Citations (3)
| Title |
|---|
| "Zenshori-Enshin Bunri, Japan Patent Office Hyojun Gijutsushu, Japan Patent Office", SHITSURYO BUNSEKI GIJUTSU, 12 May 2006 (2006-05-12), XP008185712 * |
| L. MAHESHWARA REDDY ET AL.: "Development of a Rapid and Sensitive HPLC Assay Method for Lenalidomide Capsules and Its Related Substances", E-JOURNAL OF CHEMISTRY, vol. 9, no. Issue 3, pages 1165 - 1174, XP055238404, ISSN: 0973-4945 * |
| See also references of EP3147661A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210285975A1 (en) * | 2017-12-25 | 2021-09-16 | Fujirebio Inc. | Method of testing a blood for macrolide immunosuppressant |
Also Published As
| Publication number | Publication date |
|---|---|
| US10725057B2 (en) | 2020-07-28 |
| EP3147661A1 (en) | 2017-03-29 |
| US20170192024A1 (en) | 2017-07-06 |
| TW201625942A (zh) | 2016-07-16 |
| TWI673495B (zh) | 2019-10-01 |
| JP2015222201A (ja) | 2015-12-10 |
| EP3147661A4 (en) | 2018-01-17 |
| JP6566607B2 (ja) | 2019-08-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015178461A1 (ja) | レナリドミドの光学分割方法 | |
| WO2015178460A1 (ja) | 生体試料中レナリドミドの前処理方法及び分析方法 | |
| Ribeiro et al. | Enantioseparation of chiral pharmaceuticals in biomedical and environmental analyses by liquid chromatography: An overview | |
| Barclay et al. | Chiral analysis of metoprolol and two of its metabolites, α-hydroxymetoprolol and deaminated metoprolol, in wastewater using liquid chromatography–tandem mass spectrometry | |
| Morante-Zarcero et al. | Comparative HPLC methods for β-blockers separation using different types of chiral stationary phases in normal phase and polar organic phase elution modes. Analysis of propranolol enantiomers in natural waters | |
| Geryk et al. | Enantioselective potential of chiral stationary phases based on immobilized polysaccharides in reversed phase mode | |
| Camacho-Munoz et al. | Enantioselective simultaneous analysis of selected pharmaceuticals in environmental samples by ultrahigh performance supercritical fluid based chromatography tandem mass spectrometry | |
| Barclay et al. | Trace analysis of fluoxetine and its metabolite norfluoxetine. Part II: Enantioselective quantification and studies of matrix effects in raw and treated wastewater by solid phase extraction and liquid chromatography–tandem mass spectrometry | |
| Barclay et al. | Trace analysis of fluoxetine and its metabolite norfluoxetine. Part I: development of a chiral liquid chromatography–tandem mass spectrometry method for wastewater samples | |
| Lee et al. | On the method development of immobilized polysaccharide chiral stationary phases in supercritical fluid chromatography using an extended range of modifiers | |
| Saracino et al. | Analysis of risperidone and its metabolite in plasma and saliva by LC with coulometric detection and a novel MEPS procedure | |
| CN104634887A (zh) | 一种分离和测定替格瑞洛及其光学异构体的方法 | |
| Materazzo et al. | Effect of the water content on the retention and enantioselectivity of albendazole and fenbendazole sulfoxides using amylose-based chiral stationary phases in organic–aqueous conditions | |
| Liu et al. | Studies on the chiral separation of pheniramine and its enantioselective pharmacokinetics in rat plasma by HPLC-MS/MS | |
| CN104133010B (zh) | 高效液相色谱法分离分析阿西马朵林中间体及光学异构体 | |
| Hefnawy et al. | Enantioselective high-performance liquid chromatographic method for the determination of baclofen in human plasma | |
| Sardella et al. | Novel orthogonal liquid chromatography methods to dose neurotransmitters involved in Parkinson's disease | |
| Deconinck et al. | Comparison of three development approaches for Stationary Phase Optimised Selectivity Liquid Chromatography based screening methods Part I: A heterogeneous group of molecules (slimming agents in food supplements) | |
| KWIATKOWSKA-PUCHNIARZ et al. | HPLC METHOD FOR SEPARATING ENANTIOMERS OF IMIDAZOLE DERIVATIVES ñ ANTIFUNGAL COMPOUNDS | |
| Xu et al. | Selective extraction of methenamine from chicken eggs using molecularly imprinted polymers and LC-MS/MS confirmation | |
| CN104133029A (zh) | 一种测定琥珀酸索非那新中间体光学纯度的方法 | |
| JP6842415B2 (ja) | 放射性医薬品からアセトアルデヒドを除去する方法 | |
| CN112415123A (zh) | 一种左乙拉西坦原料或氯化钠注射液中左乙拉西坦对映异构体的检测方法 | |
| Yang et al. | Enantioseparation and impurity determination of the enantiomers of novel phenylethanolamine derivatives by high performance liquid chromatography on amylose stationary phase | |
| CN108226319A (zh) | 一种检测卡巴拉汀贴剂中光学异构体的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15796899 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15312777 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015796899 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015796899 Country of ref document: EP |