WO2015137408A1 - ヘテロアリールカルボン酸エステル誘導体の製造方法及びその製造中間体 - Google Patents
ヘテロアリールカルボン酸エステル誘導体の製造方法及びその製造中間体 Download PDFInfo
- Publication number
- WO2015137408A1 WO2015137408A1 PCT/JP2015/057178 JP2015057178W WO2015137408A1 WO 2015137408 A1 WO2015137408 A1 WO 2015137408A1 JP 2015057178 W JP2015057178 W JP 2015057178W WO 2015137408 A1 WO2015137408 A1 WO 2015137408A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound represented
- acceptable salt
- group
- chemically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
Definitions
- the present invention relates to a novel process for producing heteroarylcarboxylic acid ester derivatives and production intermediates thereof. More specifically, the present invention relates to an efficient method for producing a heteroarylcarboxylic ester derivative as a therapeutic agent for diabetes or a production intermediate thereof, and a production intermediate useful for such a production method.
- Patent Document 1 and Patent Document 2 disclose heteroarylcarboxylic acid ester derivatives included in the following formula (I), and representative compounds thereof are It has been reported to show an excellent effect of suppressing blood sugar elevation in a diabetic animal model.
- Patent Document 1 discloses a method shown in the following scheme as a general method for synthesizing a heteroarylcarboxylic acid ester derivative included in the general formula (I). (See Patent Document 1 for symbols in the formula.)
- heteroarylcarboxylic acid ester derivative (F) in which X in the general formula (I) is a lower alkylene group or a lower alkenylene group, A is —OR 5, and R 5 is a lower alkyl group should be prepared as follows: Can do.
- the target heteroarylcarboxylic acid ester derivative (F) in which X represents a lower alkenylene group can be produced.
- X is performed by treating with a catalyst such as 10% palladium / carbon in a hydrogen atmosphere in a solvent that does not adversely affect the reaction, such as methanol, ethanol, or ethyl acetate.
- a catalyst such as 10% palladium / carbon in a hydrogen atmosphere in a solvent that does not adversely affect the reaction, such as methanol, ethanol, or ethyl acetate.
- a heteroarylcarboxylic acid ester derivative (F) in which can represent a lower alkylene group can be produced.
- a known method can be applied to the esterification reaction, and examples thereof include (1) a method using an acid halide and (2) a method using a condensing agent.
- the method using an acid halide is, for example, in a solvent such as dichloromethane that does not adversely affect the reaction or in the absence of a solvent, for example, in the presence or absence of a catalyst such as N, N-dimethylformamide.
- a solvent such as dichloromethane that does not adversely affect the reaction or in the absence of a solvent, for example, in the presence or absence of a catalyst such as N, N-dimethylformamide.
- the acid chloride obtained by reacting with thionyl chloride or oxalyl chloride is reacted with an alcohol in the presence of a base such as pyridine or triethylamine in a solvent that does not adversely affect the reaction such as dichloromethane or tetrahydrofuran. Is done.
- the method using a condensing agent is carried out in the presence of a base such as pyridine or triethylamine in a solvent that does not adversely affect the reaction such as tetrahydrofuran, N, N-dimethylformamide, or dichloromethane.
- a base such as pyridine or triethylamine
- a solvent that does not adversely affect the reaction such as tetrahydrofuran, N, N-dimethylformamide, or dichloromethane.
- the reaction is carried out using a condensing agent such as 1-ethyl-3- (3′-dimethylaminopropyl) carbodiimide (WSC) or 1,3-dicyclohexylcarbodiimide.
- WSC 1-ethyl-3- (3′-dimethylaminopropyl) carbodiimide
- 1,3-dicyclohexylcarbodiimide 1,3-dicyclohexylcarbodiimide
- the heteroarylcarboxylic acid ester derivative (i) in which A is —OR 5 and R 5 is a hydrogen atom the Wittig reagent (G) (wherein E 2 is methyl) instead of the Wittig reagent (B)
- G the Wittig reagent
- Patent Document 2 there is a similar description as a general synthesis method of the heteroarylcarboxylic acid ester derivative contained in the general formula (I).
- the present invention provides a production method suitable for industrialization of a heteroarylcarboxylic acid ester derivative represented by the following formula (9) and an intermediate useful for the production thereof.
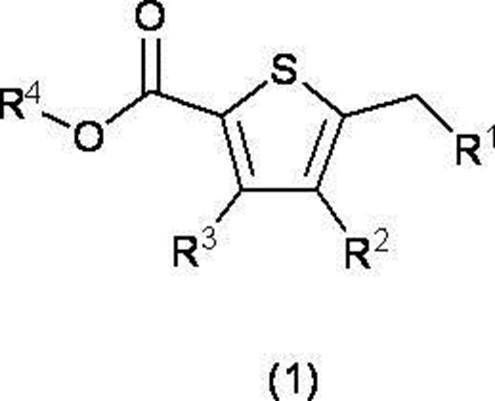
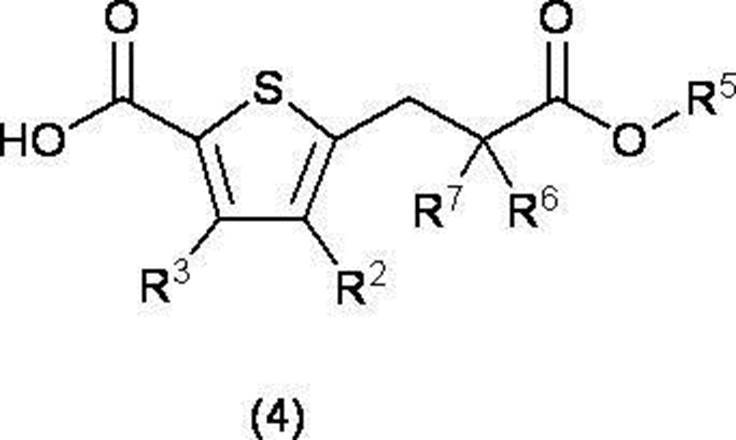
- R 2 and R 3 are the same or different and each independently represents a hydrogen atom or a lower alkyl group; R 5 represents a tert-butyl group, R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded.
- C 3-8 forms a cycloalkane ring
- the compound represented by the formula (1) and the compound represented by the formula (2) are reacted in the presence of (A) an alkyl lithium or (B) a lithium complex prepared from an organic amine and an alkyl lithium. And obtaining a compound represented by formula (3)
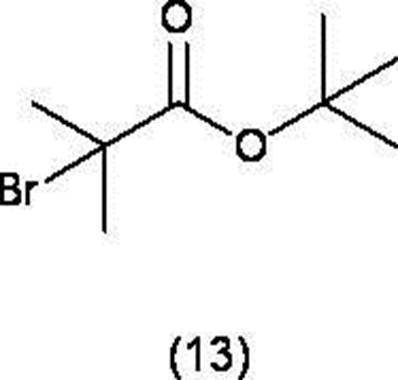
- a compound represented by formula (12) and a compound represented by formula (13) are converted into (A) n-butyllithium or (B) an organic amine selected from diisopropylamine and diisopropylethylamine and n-butyl. Reacting in the presence of a lithium complex prepared from lithium to obtain a compound represented by formula (10),
- R 2 and R 3 are the same or different and each independently represents a hydrogen atom or a lower alkyl group which may have a substituent;
- R 5 represents a lower alkyl group,
- R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded.
- C 3-8 forms a cycloalkane ring
- a compound represented by formula (5) and a compound represented by formula (2) are reacted using a base, and an ester derivative represented by formula (4) or a chemically acceptable product thereof Obtaining salt
- R 2 and R 3 are the same or different and each independently represents a hydrogen atom or a lower alkyl group which may have a substituent;
- R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded.
- R 8 , R 9 , R 10 and R 11 are the same or different and each independently represents a hydrogen atom or a halogen atom
- a compound represented by formula (8) or a chemically acceptable salt thereof is deprotected under acidic conditions, and a heteroarylcarboxylic acid ester derivative represented by formula (9) or a chemical compound thereof Converting to an acceptable salt.
- a process for producing a heteroarylcarboxylic ester derivative represented by formula (9) or a chemically acceptable salt thereof, comprising the following steps (r) to (t):
- R 2 and R 3 are the same or different and each independently represents a hydrogen atom or a lower alkyl group which may have a substituent;
- R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded.
- R 8 , R 9 , R 10 and R 11 are the same or different and each independently represents a hydrogen atom or a halogen atom
- a compound represented by formula (8), or a chemically acceptable salt thereof is deprotected with HCl, HBr, sulfuric acid, methanesulfonic acid or trifluoroacetic acid, A step of converting to an aryl carboxylic acid ester derivative or a chemically acceptable salt thereof.
- a process for producing a heteroarylcarboxylic ester derivative represented by formula (17) or a chemically acceptable salt thereof, comprising the following steps (u) to (w):
- a compound represented by formula (15) or a chemically acceptable salt thereof is deprotected in the presence of HCl, and a heteroarylcarboxylic acid ester derivative represented by formula (17), or a chemical thereof Converting to an acceptable salt.
- a compound represented by the following formula (20) or a chemically acceptable salt thereof is deprotected in the presence of HCl, and a heteroarylcarboxylic acid ester derivative represented by formula (17), or a chemical thereof Converting to an acceptable salt.
- R 4a represents a methyl group, an ethyl group, an isopropyl group or an n-butyl group; R 6a and R 7a are the same and represent a methyl group or an ethyl group, or R 6a and R 7a together with the carbon atom to which they are attached form a cyclopropane ring, a cyclobutane ring or a cyclopentane ring
- R 6a and R 7a represent a methyl group, or a chemically acceptable salt thereof.
- R 4a represents a methyl group, or a chemically acceptable salt thereof.
- R 5a represents a methyl group, an ethyl group, an isopropyl group, a tert-butyl group or an n-butyl group; R 6a and R 7a are the same and represent a methyl group or an ethyl group, or R 6a and R 7a together with the carbon atom to which they are attached form a cyclopropane ring, a cyclobutane ring or a cyclopentane ring)
- R 6a and R 7a represent a methyl group, or a chemically acceptable salt thereof.
- R 5a represents a tert-butyl group, or a chemically acceptable salt thereof.
- R 2 and R 3 are the same or different and each independently represents a hydrogen atom or a lower alkyl group which may have a substituent;
- R 5 represents a lower alkyl group,
- R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded.
- R 8 , R 9 , R 10 and R 11 are the same or different and each independently represents a hydrogen atom or a halogen atom)
- R 2 and R 3 represent a hydrogen atom
- R 6 and R 7 are the same or different and each independently represents a methyl group, an ethyl group or a propyl group, or R 6 and R 7 together with the carbon atom to which they are bonded, a cyclopropane ring, a cyclobutane ring Or form a cyclopentane ring,
- the present invention provides a production method suitable for mass synthesis of heteroaryl carboxylic acid ester derivatives and a novel intermediate.
- the product can be crystallized by using an ester derivative protected with a lower alkyl group as an intermediate, and the product can be easily isolated and purified by filtration separation. be able to.
- the production method of the present invention it is possible to produce a heteroarylcarboxylic acid ester derivative that is a target compound with high yield and high purity.
- substituent means “substituted or unsubstituted”.
- position and number of substituents are arbitrary and are not particularly limited.
- the number of substituents is preferably 1 to 5, more preferably 1 to 3.
- substituents may be the same or different.
- substituents include a halogen atom, a cyano group, a phenyl group, a lower alkyl group, a lower acyl group, a lower alkoxyl group, and a lower alkylthio group.
- the “lower alkyl group” refers to a linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
- C 3-8 cycloalkane ring refers to a cycloalkane ring having 3 to 8 carbon atoms. Examples thereof include a cyclopropane ring, a cyclobutane ring, a cyclopentane ring, a cyclohexane ring, a cycloheptane ring, and a cyclooctane ring.
- the “lower acyl group” refers to an acyl group having a linear, branched or cyclic alkyl group or alkenyl group having 1 to 6 carbon atoms.
- the “lower alkoxyl group” refers to an alkoxyl group having a linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- methoxy group, ethoxy group, n-propoxy group, n-butoxy group, n-pentyloxy group, n-hexyloxy group isopropoxy group, isobutoxy group, sec-butoxy group, tert-butoxy group, cyclopropyloxy Group, cyclobutyloxy group, cyclopentyloxy group and cyclohexyloxy group.
- the “lower alkylthio group” refers to an alkylthio group having a linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- R 2 and R 3 are the same or different and each independently represent a hydrogen atom or a lower alkyl group which may have a substituent.
- R 2 and R 3 represent a hydrogen atom.
- R 6 and R 7 are the same or different and each independently represents a hydrogen atom or an optionally substituted lower alkyl group, or R 6 and R 7 together with the carbon atom to which they are bonded. C 3-8 cycloalkane ring is formed.
- R 6 and R 7 are the same and represent a methyl group, an ethyl group or a propyl group, or R 6 and R 7 together with the carbon atom to which they are attached, a cyclopropane ring, a cyclobutane ring or a cyclo Forms a pentane ring. More preferably, R 6 and R 7 represent a methyl group.
- R 4 represents a lower alkyl group.
- R 4 represents a methyl group, an ethyl group, an isopropyl group or an n-butyl group. More preferably, R 4 represents a methyl group.
- R 5 represents a lower alkyl group.
- R 5 represents a tert-butyl group.
- R 8 , R 9 , R 10 and R 11 are the same or different and each represents a hydrogen atom or a halogen atom.
- R 8 , R 9 and R 11 represent a hydrogen atom and R 10 represents a fluorine atom.
- X represents a halogen atom.
- X represents a chlorine atom.
- a pharmaceutically acceptable salt is preferred.
- pharmaceutically acceptable salts include ammonium salts, salts with alkali metals such as sodium and potassium, alkaline earth such as calcium and barium, for compounds having an acidic group such as a carboxyl group. Salts with other metals, magnesium salts, aluminum salts, zinc salts, salts with organic amines such as triethylamine, ethanolamine, morpholine, pyrrolidine, piperidine, piperazine, dicyclohexylamine, salts with basic amino acids such as arginine and lysine Can be mentioned.
- salts with inorganic acids such as hydrochloric acid, sulfuric acid, phosphoric acid, nitric acid, hydrobromic acid, acetic acid, citric acid, benzoic acid, maleic acid, fumaric acid, tartaric acid, succinic acid , Salts with organic carboxylic acids such as trifluoroacetic acid, tannic acid, butyric acid, hibenzic acid, pamoic acid, enanthic acid, decanoic acid, teocric acid, salicylic acid, lactic acid, oxalic acid, mandelic acid, malic acid, methanesulfonic acid, Mention may be made of salts with organic sulfonic acids such as benzenesulfonic acid and p-toluenesulfonic acid.
- organic carboxylic acids such as trifluoroacetic acid, tannic acid, butyric acid, hibenzic acid, pamoic acid, enanthic acid, decanoic
- Examples of the salt used in the present invention include chemically acceptable salts in addition to those listed above as pharmaceutically acceptable salts, and chemically acceptable salts with chemically acceptable acids. Salts with acceptable bases are included.
- Salts with chemically acceptable acids used in the present invention include inorganic acids (eg, hydrochloric acid, sulfuric acid, phosphoric acid, nitric acid, hydrobromic acid, etc.), organic carboxylic acids (eg, carbonic acid, acetic acid, citric acid).
- inorganic acids eg, hydrochloric acid, sulfuric acid, phosphoric acid, nitric acid, hydrobromic acid, etc.
- organic carboxylic acids eg, carbonic acid, acetic acid, citric acid.
- Acid benzoic acid, maleic acid, fumaric acid, tartaric acid, succinic acid, trifluoroacetic acid, tannic acid, butyric acid, decanoic acid, salicylic acid, lactic acid, oxalic acid, mandelic acid, malic acid, etc.), organic sulfonic acid (eg, methane) And salts with sulfonic acid, p-toluenesulfonic acid, benzenesulfonic acid and the like.
- organic sulfonic acid eg, methane
- salts with sulfonic acid p-toluenesulfonic acid, benzenesulfonic acid and the like.
- Salts with chemically acceptable bases include alkali metal salts (eg, sodium salts, potassium salts, lithium salts, etc.), alkaline earth metal salts (eg, calcium salts, barium salts, etc.), metal salts (eg, , Magnesium salts, aluminum salts, etc.).
- alkali metal salts eg, sodium salts, potassium salts, lithium salts, etc.
- alkaline earth metal salts eg, calcium salts, barium salts, etc.
- metal salts eg, , Magnesium salts, aluminum salts, etc.
- the compounds of the present invention also include solvates thereof such as hydrates and alcohol adducts.
- solvates thereof such as hydrates and alcohol adducts.
- examples of the alcohol adduct include methanol solvate, ethanol solvate, and isopropyl alcohol solvate.
- a method for producing the heteroarylcarboxylic acid ester derivative represented by the general formula (9) is shown below.
- the present invention is a method for producing a compound represented by the formula (9) or a chemically acceptable salt thereof, wherein the following steps (a) to (f) are used. .
- Step (a) This reaction is preferably performed in the presence of a base.
- the base used in the reaction include (A) alkyl lithium, or (B) a lithium complex prepared from an organic amine such as diisopropylamine and diisopropylethylamine and alkyllithium. It is preferable to use a lithium complex prepared from (A) n-butyllithium or (B) an organic amine such as diisopropylamine or diisopropylethylamine and n-butyllithium.
- the amount of the base used is preferably about 1.0 mol to about 2.0 mol with respect to 1 mol of the compound of the formula (1).
- the reaction solvent ethers and hydrocarbons can be used, and among them, tetrahydrofuran is preferable.
- the reaction temperature is preferably -78 ° C to 0 ° C.
- the reaction time is usually about 30 minutes.
- the reagent used for deprotection is preferably a metal hydroxide such as sodium hydroxide.
- the metal hydroxide include alkali metal hydroxides such as sodium hydroxide, lithium hydroxide, and potassium hydroxide.
- the amount of metal hydroxide used is preferably about 1.0 mol to about 2.0 mol with respect to 1 mol of the diester of formula (3).
- the reaction solvent a mixed solvent of alcohol and water is preferable.
- the alcohol include methanol and ethanol.
- the reaction temperature is preferably 10 to 40 ° C.
- the reaction time is usually about 12 hours to about 24 hours.
- Step (c) This reaction is preferably performed in the presence of a base.
- the base used is preferably a lithium complex prepared from an organic amine such as diisopropylamine or 2,2,6,6-tetramethylpiperidine and n-butyllithium and an alkali metal hydride.
- the lithium complex lithium diisopropylamine (LDA) is preferable.
- LDA lithium diisopropylamine
- NaH sodium hydride
- the amount of the lithium complex to be used is preferably about 1.0 mol to about 1.2 mol with respect to 1 mol of the compound of the formula (5), and the amount of alkali metal hydride (eg, sodium hydride) is also About 1 mol to about 1.2 mol is preferable with respect to 1 mol of the compound of 5).
- the reaction solvent ethers and hydrocarbons can be used, and among them, tetrahydrofuran is preferable.
- the reaction temperature is preferably -78 ° C to 0 ° C.
- the reaction time is usually about 30 minutes.
- Step (d) As the reaction solvent for acid halogenation, acetate is preferable, and as the acid halogenating agent, thionyl chloride or oxalyl chloride is preferable. As the additive, N, N-dimethylformamide (DMF) or N-methylpyrrolidone (NMP) is preferable.
- the amount of the acid halogenating agent to be used is preferably about 1.0 mol to about 1.5 mol per 1 mol of the compound of the formula (4).
- the reaction temperature is between 0 ° C. and 45 ° C., preferably 10 ° C. to 30 ° C.
- the reaction time is usually about 30 minutes to about 2 hours.
- the diester derivative represented by the formula (8) is obtained by reacting the acid halide (6) obtained in the step (d) with the amidinophenol derivative (7), preferably in the presence of a base.
- a reaction solvent acetonitrile is preferable.
- the base include organic bases such as pyridine, triethylamine, diisopropylethylamine, and lutidine.
- pyridine is preferred.
- the amount of the base to be used is generally about 2.0 mol to about 3.0 mol with respect to 1 mol of acid halide (6).
- the amount of the amidinophenol derivative (7) to be used is usually preferably about 1.0 mol to about 1.2 mol with respect to 1 mol of the acid halide (6).
- the reaction temperature is between ⁇ 60 ° C. and 30 ° C., preferably ⁇ 25 ° C. to 10 ° C.
- the reaction time is usually about 30 minutes.
- TFA trifluoroacetic acid
- the amount of TFA to be used is generally about 2.0 mol to about 3.0 mol with respect to 1 mol of acid halide (6).
- the temperature for dropping and crystallization is preferably from 0 ° C to 20 ° C.
- the time for crystallization is usually from about 2 to about 24 hours.
- the precipitated diester derivative (8) or a salt thereof can be easily isolated and purified by filtration separation.
- Step (f) The heteroester carboxylate derivative represented by the formula (9) can be obtained by deprotecting the diester derivative (8) obtained in the step (e) under acidic conditions.
- the acid used for the reaction include HCl, HBr, sulfuric acid, methanesulfonic acid, trifluoroacetic acid and the like.
- it is HCl.
- the reaction solvent 1,4-dioxane and acetone are preferable, and it is preferable to add water as an additive.
- the reaction temperature is preferably 10 ° C to 60 ° C.
- the reaction time is usually about 6 hours to about 24 hours.
- the heteroarylcarboxylic acid ester derivative can be crystallized (crystallized) and can be isolated and purified by filtration separation.
- reaction system means a reaction mixture in which a reaction has been performed, or a reaction liquid obtained by filtering insoluble matters (for example, a catalyst) from the reaction mixture.
- the compounds of formula (1) and formula (5) can be produced according to a method known per se.
- the present invention also provides the following intermediates used in the method of the present invention.
- R 4a represents a methyl group, an ethyl group, an isopropyl group or an n-butyl group
- R 6a and R 7a are the same and represent a methyl group or an ethyl group, or R 6a and R 7a together with the carbon atom to which they are attached form a cyclopropane ring, a cyclobutane ring or a cyclopentane ring
- Preferable embodiments of the compound represented by the formula (20) include the following compounds.
- a compound having the following structure or a chemically acceptable salt thereof is provided.
- a compound having the following structure or a chemically acceptable salt thereof is provided.
- a compound represented by the following formula (10) or a chemically acceptable salt thereof is a compound represented by the following formula (10) or a chemically acceptable salt thereof.
- a compound represented by the following formula (21) or a chemically acceptable salt thereof is a compound represented by the following formula (21) or a chemically acceptable salt thereof.
- R 5a represents a methyl group, an ethyl group, an isopropyl group, a tert-butyl group or an n-butyl group
- R 6a and R 7a are the same and represent a methyl group or an ethyl group, or R 6a and R 7a together with the carbon atom to which they are attached form a cyclopropane ring, a cyclobutane ring or a cyclopentane ring
- Preferable embodiments of the compound represented by the formula (21) include the following compounds.
- a compound having the following structure or a chemically acceptable salt thereof is provided.
- a compound having the following structure or a chemically acceptable salt thereof is provided.
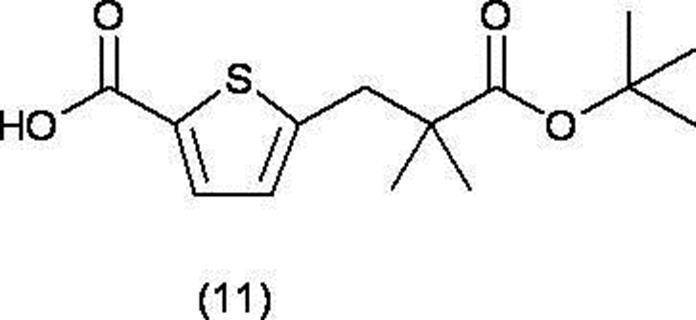
- a compound represented by the following formula (11) or a chemically acceptable salt thereof is a compound represented by the following formula (11) or a chemically acceptable salt thereof.
- the compound of formula (20), the compound of formula (10), the compound of formula (21), the compound of formula (11), the compound of formula (8) and the compound of formula (15) are heteroarylcarboxylic acid ester derivatives ( It is useful as an intermediate for producing 9).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
Abstract
Description



[1] 以下の工程(g)と(h)を含む、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:

R5は、tert-ブチル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成する)
(g) 式(1)で表される化合物と式(2)で表される化合物を、(A)アルキルリチウム、又は(B)有機アミンとアルキルリチウムから調製されるリチウム錯体の存在下で反応させ、式(3)で表される化合物を得る工程



R4は、メチル基、エチル基、イソプロピル基又はn-ブチル基を表し、
R12は、水素原子又はハロゲン原子を表し、
その他の各記号は前記で定義したとおりである)、
(h) 水及びアルコールを含む溶媒中で、式(3)で表される化合物を金属ヒドロキシドを用いて加水分解し、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程。
[2] アルキルリチウムがn-ブチルリチウムである、前記[1]記載の製造方法。
[3] 以下の工程(k)と(l)を含む、式(11)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:




[4] 工程(m)を含む、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:

R5は、低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成する)
(m) 式(5)で表される化合物と式(2)で表される化合物を、塩基を用いて反応させ、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程


その他の記号は前記で定義したとおりである)。
[5] 工程(n)を含む、式(11)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:



R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す)
(o) 式(4)で表される化合物を酸ハロゲン化剤と反応させ、式(6)で表される酸ハライドに変換する工程


Xは、ハロゲン原子を表し、
その他の記号は前記で定義したとおりである)、
(p) 式(6)で表される酸ハライドを、式(7)で表される化合物と反応させ、式(8)で表される化合物、又はその化学的に許容しうる塩へ変換する工程


(q) 式(8)で表される化合物、又はその化学的に許容しうる塩を酸性条件にて脱保護し、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩へと変換する工程。
[7] 以下の工程(r)から(t)を含む、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩の製造方法:

R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す)
(r) 式(4)で表される化合物を塩化チオニル又は塩化オキサリルと反応させ、式(16)で表される酸クロライドに変換する工程


その他の記号は前記で定義したとおりである)、
(s) 式(16)で表される酸クロライドを有機塩基存在下、式(7)で表される化合物と反応させ、式(8)で表される化合物、又はその化学的に許容しうる塩へ変換する工程


(t) 式(8)で表される化合物、又はその化学的に許容しうる塩をHCl、HBr、硫酸、メタンスルホン酸又はトリフルオロ酢酸で脱保護し、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩へと変換する工程。
[8] 以下の工程(u)から(w)を含む、式(17)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩の製造方法:




[9] 下記式(20)で表される化合物、又はその化学的に許容しうる塩。

R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する)
[10] R6a及びR7aが、メチル基を表す、前記[9]記載の化合物、又はその化学的に許容しうる塩。
[11] R4aが、メチル基を表す、前記[9]記載の化合物、又はその化学的に許容しうる塩。
[12] 下記式(10)で表される化合物、又はその化学的に許容しうる塩。


R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する)
[14] R6a及びR7aが、メチル基を表す、前記[13]記載の化合物、又はその化学的に許容しうる塩。
[15] R5aが、tert-ブチル基を表す、前記[13]記載の化合物、又はその化学的に許容しうる塩。
[16] 下記式(11)で表される化合物、又はその化学的に許容しうる塩。


R5は、低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す)
[18] 式(8)中、R2及びR3が、水素原子を表し、
R6及びR7が、同一又は異なって、それぞれ独立して、メチル基、エチル基又はプロピル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにシクロプロパン環、シクロブタン環又はシクロペンタン環を形成する、
前記[17]記載の化合物、又はその化学的に許容しうる塩。
[19] 下記式(15)で表される化合物、又はその化学的に許容しうる塩。

R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表す。好ましくは、R2及びR3は、水素原子を表す。
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成する。好ましくは、R6及びR7は、同一であり、メチル基、エチル基又はプロピル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する。更に好ましくは、R6及びR7は、メチル基を表す。
R4は、低級アルキル基を表す。好ましくは、R4は、メチル基、エチル基、イソプロピル基又はn-ブチル基を表す。更に好ましくは、R4は、メチル基を表す。
R5は、低級アルキル基を表す。好ましくは、R5は、tert-ブチル基を表す。
R8、R9、R10及びR11は、同一又は異なって、それぞれ、水素原子又はハロゲン原子を表す。好ましくは、R8、R9及びR11は水素原子を表し、R10はフッ素原子を表す。
Xは、ハロゲン原子を表す。好ましくは、Xは塩素原子を表す。
本発明は、式(9)で示される化合物又はその化学的に許容しうる塩を製造する方法であって、以下の工程(a)から(f)を用いることを特徴とする製造方法である。









工程(a)
本反応は、塩基存在下で行うことが好ましい。
反応に用いる塩基としては、(A)アルキルリチウム、又は(B)ジイソプロピルアミン、ジイソプロピルエチルアミンなどの有機アミンとアルキルリチウムから調製されるリチウム錯体が挙げられる。(A)n-ブチルリチウム、又は(B)ジイソプロピルアミン、ジイソプロピルエチルアミンなどの有機アミンとn-ブチルリチウムから調製されるリチウム錯体を用いることが好ましい。塩基の使用量は、式(1)の化合物1モルに対して、約1.0モルから約2.0モルが好ましい。
反応溶媒としては、エーテル類、炭化水素類を用いることができ、その中でもテトラヒドロフランが好ましい。
反応温度は-78℃から0℃が好ましい。反応時間は、通常、約30分間である。
脱保護に用いる試薬としては、水酸化ナトリウムなどの金属ヒドロキシドが好ましい。金属ヒドロキシドとしては、水酸化ナトリウム、水酸化リチウム、水酸化カリウムなどのアルカリ金属ヒドロキシドが挙げられる。金属ヒドロキシドの使用量は、式(3)のジエステル1モルに対して、約1.0モルから約2.0モルが好ましい。
反応溶媒としては、アルコールと水の混合溶媒が好ましい。アルコールとしては、メタノール、エタノールなどが挙げられる。
反応温度は10℃から40℃が好ましい。反応時間は、通常、約12時間から約24時間である。
本反応は、塩基存在下で行うことが好ましい。
用いる塩基としては、ジイソプロピルアミン、2,2,6,6-テトラメチルピペリジンなどの有機アミンとn-ブチルリチウムから調製されるリチウム錯体とアルカリ金属水素化物が好ましい。リチウム錯体としては、リチウムジイソプロピルアミン(LDA)が好ましい。アルカリ金属水素化物としては、水素化ナトリウム(NaH)が好ましい。リチウム錯体の使用量は、式(5)の化合物1モルに対して、約1.0モルから約1.2モルが好ましく、アルカリ金属水素化物(例、水素化ナトリウム)の量も、式(5)の化合物1モルに対して、約1.0モルから約1.2モルが好ましい。
反応溶媒としては、エーテル類、炭化水素類を用いることができ、その中でもテトラヒドロフランが好ましい。
反応温度は、-78℃から0℃が好ましい。反応時間は、通常、約30分間である。
酸ハロゲン化の反応溶媒としては、酢酸エステルが好ましく、酸ハロゲン化剤としては、塩化チオニル又は塩化オキサリルが好ましい。また、添加剤としては、N,N-ジメチルホルムアミド(DMF)やN-メチルピロリドン(NMP)が好ましい。
酸ハロゲン化剤の使用量は、式(4)の化合物1モルに対して、好ましくは約1.0モルから約1.5モルが好ましい。
反応温度は0℃から45℃までの間であり、10℃から30℃が好ましい。反応時間は、通常、約30分間から約2時間である。
工程(d)で得られた酸ハライド(6)を、好ましくは塩基存在下、アミジノフェノール誘導体(7)と反応させることで、式(8)で表されるジエステル誘導体が得られる。反応溶媒としては、アセトニトリルが好ましい。
塩基としては、ピリジン、トリエチルアミン、ジイソプロピルエチルアミン、ルチジンなどの有機塩基が挙げられる。塩基としては、ピリジンが好ましい。塩基の使用量は、酸ハライド(6)1モルに対して、通常、約2.0モルから約3.0モルである。
アミジノフェノール誘導体(7)の使用量は、酸ハライド(6)1モルに対して、通常、約1.0モルから約1.2モルが好ましい。
反応温度は-60℃から30℃までの間であり、-25℃から10℃が好ましい。反応時間は、通常、約30分間である。
反応混合液に、トリフルオロ酢酸(TFA)及び水を滴下することで、反応系中からジエステル誘導体(8)をTFA塩として結晶化(晶析)させることが可能であり、ろ過分離により単離精製することができる。TFAの使用量は、酸ハライド(6)1モルに対して、通常、約2.0モルから約3.0モルである。
滴下及び晶析の温度は0℃から20℃が好ましい。晶析の時間は通常、約2から約24時間である。
析出したジエステル誘導体(8)又はその塩は、ろ過分離により簡便に単離精製することができる。
工程(e)で得られたジエステル誘導体(8)を、酸性条件にて脱保護することで、式(9)で表されるヘテロアリールカルボン酸エステル誘導体が得られる。
反応に用いる酸としては、HCl、HBr、硫酸、メタンスルホン酸、トリフルオロ酢酸などが挙げられる。好ましくは、HClである。
反応溶媒としては、1,4-ジオキサン、アセトンが好ましく、添加剤として水を加えることが好ましい。
反応温度は、10℃から60℃が好ましい。反応時間は、通常、約6時間から約24時間である。
反応により、ヘテロアリールカルボン酸エステル誘導体の結晶化(晶析)が可能であり、ろ過分離により単離精製することができる。
下記式(20)で表される化合物、又はその化学的に許容しうる塩。

R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する)
以下の構造を有する化合物、又はその化学的に許容しうる塩。



R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する)
以下の構造を有する化合物、又はその化学的に許容しうる塩。

(分析条件)
以下の実施例における分析は、下記の測定装置を用いて、常法に従って行った。
(1)1H-NMR
装置:AvanceIII 400, Burker社製
(2)高速液体クロマトグラフィー(HPLC)
HPLC:LC-2010AHT(島津製作所製)
使用カラム:InertSustain C18 φ4.6 mm×150 mm,粒径3 μm (GL-Sciences製)
検出波長:UV 254 nm
カラム温度:40℃
注入量:10 μL
分析時間:18分
流速:1.5 mL/min
溶離液:A液: 水/トリフルオロ酢酸混液(1000:1)
B液: アセトニトリル/トリフルオロ酢酸混液(1000:1)
グラジエント:0min: 20%B, 0→13min: 20→90%B, 13→15min: 90%B,
15→15.1min: 90→20%B, 15.1→18min: 20%B
なお、合成の各工程においての収率は、標準品のHPLCエリア面積値をもとに算出した値である。
5-(2-tert-ブトキシカルボニル-2-メチルプロピル)チオフェン-2-カルボン酸 メチルエステルの合成

1H NMR (400 MHz, Chloroform-d) δ 7.62 (d, J = 3.8 Hz, 1H), 6.79 (d, J = 3.8 Hz, 1H), 3.85 (s, 3H), 3.04 (s, 2H), 1.45 (s, 9H), 1.18 (s, 6H).
5-(2-tert-ブトキシカルボニル-2-メチルプロピル)チオフェン-2-カルボン酸の合成

1H NMR (400 MHz, Chloroform-d) δ 7.70 (d, J = 3.8 Hz, 1H), 6.83 (d, J = 3.8 Hz, 1H), 3.06 (s, 2H), 1.46 (s, 9H), 1.19 (s, 6H).
5-(2-tert-ブトキシカルボニル-2-メチルプロピル)チオフェン-2-カルボン酸の合成

1H NMR (400 MHz, Chloroform-d) δ 7.71 (d, J = 3.8 Hz, 1H), 6.83 (d, J = 3.8 Hz, 1H), 3.06 (s, 2H), 1.46 (s, 9H), 1.19 (s, 6H).
5-(2-tert-ブトキシカルボニル-2-メチルプロピル)チオフェン-2-カルボン酸 4-カルバムイミドイル-2-フルオロフェニルエステルの合成

1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 2H), 9.45 (s, 2H), 8.02 - 7.91 (m, 2H), 7.81 - 7.73 (m, 2H), 7.09 (d, J = 3.8 Hz, 1H), 3.13 (s, 2H), 1.42 (s, 9H), 1.15 (s, 6H).
3-[5-(4-カルバムイミドイル-2-フルオロフェノキシ)カルボニル-2-チエニル]-2,2-ジメチルプロパン酸 塩酸塩の合成

1H NMR (400 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.62 (s, 2H), 9.47 (s, 2H), 8.08 - 7.90 (m, 2H), 7.87 - 7.71 (m, 2H), 7.09 (d, J = 3.8 Hz, 1H), 3.14 (s, 2H), 1.17 (s, 6H).
3-[5-(4-カルバムイミドイル-2-フルオロフェノキシ) カルボニル-2-チエニル]-2,2-ジメチルプロパン酸 塩酸塩の精製

1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 9.54 (s, 2H), 9.36 (s, 2H), 8.10 - 7.87 (m, 2H), 7.84 - 7.68 (m, 2H), 7.09 (d, J = 3.8 Hz, 1H), 3.14 (s, 2H), 1.16 (s, 6H).
Claims (19)
- 以下の工程(g)と(h)を含む、式(4)で表されるエステル誘導体、又はその化学的に許容
しうる塩の製造方法:

(式中、R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は低級アルキル基を表し、
R5は、tert-ブチル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成する)
(g) 式(1)で表される化合物と式(2)で表される化合物を、(A)アルキルリチウム、又は(B)有機アミンとアルキルリチウムから調製されるリチウム錯体の存在下で反応させ、式(3)で表される化合物を得る工程



(式中、R1は、ハロゲン原子を表し、
R4は、メチル基、エチル基、イソプロピル基又はn-ブチル基を表し、
R12は、水素原子又はハロゲン原子を表し、
その他の各記号は前記で定義したとおりである)、
(h) 水及びアルコールを含む溶媒中で、式(3)で表される化合物を金属ヒドロキシドを用いて加水分解し、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程。 - アルキルリチウムがn-ブチルリチウムである、請求項1記載の製造方法。
- 以下の工程(k)と(l)を含む、式(11)で表されるエステル誘導体、又はその化学的に許容
しうる塩の製造方法:

(k) 式(12)で表される化合物と式(13)で表される化合物を、(A)n-ブチルリチウム、又は(B)ジイソプロピルアミン及びジイソプロピルエチルアミンから選ばれる有機アミンとn-ブチルリチウムから調製されるリチウム錯体の存在下で反応させ、式(10)で表される化合物を得る工程、



(l) 水及びアルコールを含む溶媒中で、式(10)で表される化合物を、水酸化ナトリウム、水酸化リチウム及び水酸化カリウムから選ばれる金属ヒドロキシドを用いて加水分解し、式(11)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程。 - 工程(m)を含む、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:

(式中、R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表し、
R5は、低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成する)
(m) 式(5)で表される化合物と式(2)で表される化合物を、塩基を用いて反応させ、式(4)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程


(式中、R12はハロゲン原子を表し、
その他の記号は前記で定義したとおりである)。 - 工程(n)を含む、式(11)で表されるエステル誘導体、又はその化学的に許容しうる塩の製造方法:

(n) 式(14)で表される化合物と式(13)で表される化合物を、水素化ナトリウム及びリチウムジイソプロピルアミドを用いて反応させ、式(11)で表されるエステル誘導体、又はその化学的に許容しうる塩を得る工程。


- 以下の工程(o)から(q)を含む、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩の製造方法:

(式中、R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す)
(o) 式(4)で表される化合物を酸ハロゲン化剤と反応させ、式(6)で表される酸ハライドに変換する工程


(式中、R5は、低級アルキル基を表し、
Xは、ハロゲン原子を表し、
その他の記号は前記で定義したとおりである)、
(p) 式(6)で表される酸ハライドを、式(7)で表される化合物と反応させ、式(8)で表される化合物、又はその化学的に許容しうる塩へ変換する工程


(式中、記号は前記で定義したとおりである)、
(q) 式(8)で表される化合物、又はその化学的に許容しうる塩を酸性条件にて脱保護し、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩へと変換する工程。 - 以下の工程(r)から(t)を含む、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩の製造方法:

(式中、R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す)
(r) 式(4)で表される化合物を塩化チオニル又は塩化オキサリルと反応させ、式(16)で表される酸クロライドに変換する工程

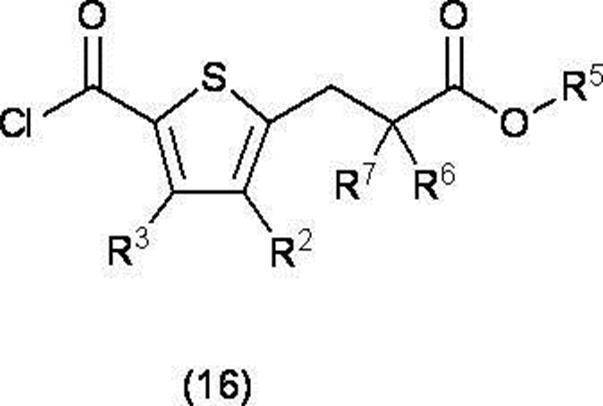
(式中、R5は、低級アルキル基を表し、
その他の記号は前記で定義したとおりである)、
(s) 式(16)で表される酸クロライドを有機塩基存在下、式(7)で表される化合物と反応させ、式(8)で表される化合物、又はその化学的に許容しうる塩へ変換する工程


(式中、各記号は前記で定義したとおりである)、
(t) 式(8)で表される化合物、又はその化学的に許容しうる塩をHCl、HBr、硫酸、メタンスルホン酸又はトリフルオロ酢酸で脱保護し、式(9)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩へと変換する工程。 - 以下の工程(u)から(w)を含む、式(17)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩の製造方法:

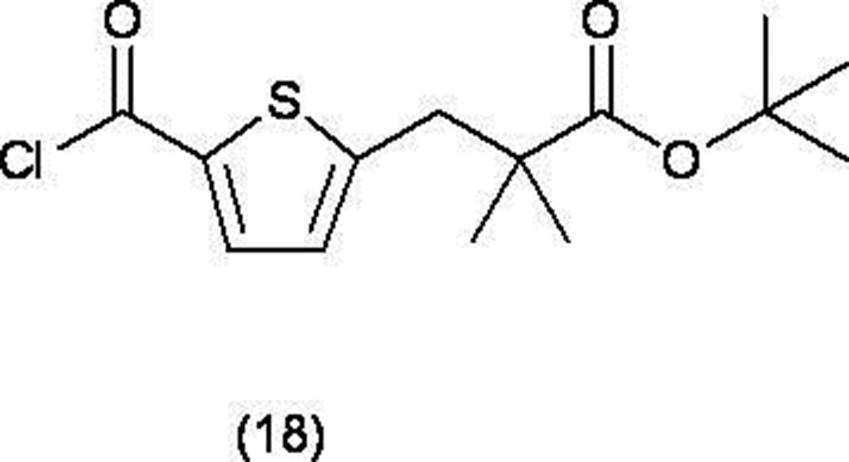
(u) 式(11)で表される化合物を塩化チオニルと反応させ、式(18)で表される酸クロライドに変換する工程、


(v) 式(18)で表される酸クロライドをピリジン存在下、式(19)で表される化合物と反応させ、式(15)で表される化合物、又はその化学的に許容しうる塩へ変換する工程、


(w) 式(15)で表される化合物、又はその化学的に許容しうる塩をHCl存在下で脱保護し、式(17)で表されるヘテロアリールカルボン酸エステル誘導体、又はその化学的に許容しうる塩へと変換する工程。 - 下記式(20)で表される化合物、又はその化学的に許容しうる塩。

(式中、R4aは、メチル基、エチル基、イソプロピル基又はn-ブチル基を表し、
R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する) - R6a及びR7aが、メチル基を表す、請求項9記載の化合物、又はその化学的に許容しうる塩。
- R4aが、メチル基を表す、請求項9記載の化合物、又はその化学的に許容しうる塩。
- 下記式(10)で表される化合物、又はその化学的に許容しうる塩。

- 下記式(21)で表される化合物、又はその化学的に許容しうる塩。

(式中、R5aは、メチル基、エチル基、イソプロピル基、tert-ブチル基又はn-ブチル基を表し、
R6a及びR7aは、同一であり、メチル基又はエチル基を表すか、又はR6a及びR7aは、それらが結合する炭素原子とともに、シクロプロパン環、シクロブタン環又はシクロペンタン環を形成する) - R6a及びR7aが、メチル基を表す、請求項13記載の化合物、又はその化学的に許容しうる塩。
- R5aが、tert-ブチル基を表す、請求項13記載の化合物、又はその化学的に許容
しうる塩。 - 下記式(11)で表される化合物、又はその化学的に許容しうる塩。

- 下記式(8)で表される化合物、又はその化学的に許容しうる塩。

(式中、R2及びR3は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表し、
R5は、低級アルキル基を表し、
R6及びR7は、同一又は異なって、それぞれ独立して、水素原子又は置換基を有してもよい低級アルキル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにC3-8シクロアルカン環を形成し、
R8、R9、R10及びR11は、同一又は異なって、それぞれ独立して、水素原子又はハロゲン原子を表す) - 式(8)中、R2及びR3が、水素原子を表し、
R6及びR7が、同一又は異なって、それぞれ独立して、メチル基、エチル基又はプロピル基を表すか、又はR6及びR7は、それらが結合する炭素原子とともにシクロプロパン環、シクロブタン環又はシクロペンタン環を形成する、
請求項17記載の化合物、又はその化学的に許容しうる塩。 - 下記式(15)で表される化合物、又はその化学的に許容しうる塩。

Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11201607558YA SG11201607558YA (en) | 2014-03-11 | 2015-03-11 | Method for producing heteroarylcarboxylic acid ester derivative, and production intermediate of same |
| EP15760831.6A EP3118196A4 (en) | 2014-03-11 | 2015-03-11 | Method for producing heteroarylcarboxylic acid ester derivative, and production intermediate of same |
| JP2016507799A JPWO2015137408A1 (ja) | 2014-03-11 | 2015-03-11 | ヘテロアリールカルボン酸エステル誘導体の製造方法及びその製造中間体 |
| CN201580013071.8A CN106458965A (zh) | 2014-03-11 | 2015-03-11 | 杂芳基羧酸酯衍生物的制造方法及其制造中间体 |
| IL247709A IL247709A0 (en) | 2014-03-11 | 2016-09-08 | A method for the preparation of a derivative of a heteroaryl carboxylic acid ester, and the preparation of building materials thereof |
| US15/260,800 US20160376248A1 (en) | 2014-03-11 | 2016-09-09 | Method for producing heteroarylcarboxylic acid ester derivative, and production intermediate of same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-048091 | 2014-03-11 | ||
| JP2014048091 | 2014-03-11 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/260,800 Continuation US20160376248A1 (en) | 2014-03-11 | 2016-09-09 | Method for producing heteroarylcarboxylic acid ester derivative, and production intermediate of same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015137408A1 true WO2015137408A1 (ja) | 2015-09-17 |
Family
ID=54071849
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/057178 Ceased WO2015137408A1 (ja) | 2014-03-11 | 2015-03-11 | ヘテロアリールカルボン酸エステル誘導体の製造方法及びその製造中間体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20160376248A1 (ja) |
| EP (1) | EP3118196A4 (ja) |
| JP (1) | JPWO2015137408A1 (ja) |
| CN (1) | CN106458965A (ja) |
| IL (1) | IL247709A0 (ja) |
| SG (1) | SG11201607558YA (ja) |
| WO (1) | WO2015137408A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022039178A1 (ja) | 2020-08-19 | 2022-02-24 | Eaファーマ株式会社 | 脂肪性肝疾患の治療薬 |
| WO2025075132A1 (ja) * | 2023-10-05 | 2025-04-10 | Eaファーマ株式会社 | 化合物の製造方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999059992A1 (en) * | 1998-05-19 | 1999-11-25 | Aventis Pharma Deutschland Gmbh | Thienyl substituted acylguanidines as inhibitors of bone resorption and vitronectin receptor antagonists |
| WO2011071048A1 (ja) * | 2009-12-07 | 2011-06-16 | 味の素株式会社 | ヘテロアリールカルボン酸エステル誘導体 |
| WO2012169579A1 (ja) * | 2011-06-07 | 2012-12-13 | 味の素株式会社 | ヘテロ環カルボン酸エステル誘導体 |
| WO2013187533A1 (en) * | 2012-06-14 | 2013-12-19 | Ajinomoto Co., Inc. | Heteroarylcarboxylic acid ester derivative |
-
2015
- 2015-03-11 EP EP15760831.6A patent/EP3118196A4/en not_active Withdrawn
- 2015-03-11 SG SG11201607558YA patent/SG11201607558YA/en unknown
- 2015-03-11 CN CN201580013071.8A patent/CN106458965A/zh active Pending
- 2015-03-11 WO PCT/JP2015/057178 patent/WO2015137408A1/ja not_active Ceased
- 2015-03-11 JP JP2016507799A patent/JPWO2015137408A1/ja not_active Withdrawn
-
2016
- 2016-09-08 IL IL247709A patent/IL247709A0/en unknown
- 2016-09-09 US US15/260,800 patent/US20160376248A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999059992A1 (en) * | 1998-05-19 | 1999-11-25 | Aventis Pharma Deutschland Gmbh | Thienyl substituted acylguanidines as inhibitors of bone resorption and vitronectin receptor antagonists |
| WO2011071048A1 (ja) * | 2009-12-07 | 2011-06-16 | 味の素株式会社 | ヘテロアリールカルボン酸エステル誘導体 |
| WO2012169579A1 (ja) * | 2011-06-07 | 2012-12-13 | 味の素株式会社 | ヘテロ環カルボン酸エステル誘導体 |
| WO2013187533A1 (en) * | 2012-06-14 | 2013-12-19 | Ajinomoto Co., Inc. | Heteroarylcarboxylic acid ester derivative |
Non-Patent Citations (2)
| Title |
|---|
| GREENE T.W. ET AL., PROTECTING GROUPS IN ORGANIC SYNTHESIS, 1999, pages 404 - 8 * |
| See also references of EP3118196A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022039178A1 (ja) | 2020-08-19 | 2022-02-24 | Eaファーマ株式会社 | 脂肪性肝疾患の治療薬 |
| US12023320B2 (en) | 2020-08-19 | 2024-07-02 | Ea Pharma Co., Ltd. | Therapeutic agent for fatty liver disease |
| WO2025075132A1 (ja) * | 2023-10-05 | 2025-04-10 | Eaファーマ株式会社 | 化合物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| SG11201607558YA (en) | 2016-10-28 |
| US20160376248A1 (en) | 2016-12-29 |
| EP3118196A1 (en) | 2017-01-18 |
| JPWO2015137408A1 (ja) | 2017-04-06 |
| EP3118196A4 (en) | 2018-02-21 |
| CN106458965A (zh) | 2017-02-22 |
| IL247709A0 (en) | 2016-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113227061A (zh) | 贝派地酸的新盐和多晶型物 | |
| TW201311629A (zh) | 用於製備前列腺素醯胺之新穎方法 | |
| CN100528821C (zh) | 反式-4-氨基-1-环己烷羧酸衍生物的制备方法 | |
| JP2019147763A (ja) | プロリンアミド化合物の製造方法 | |
| WO2015137408A1 (ja) | ヘテロアリールカルボン酸エステル誘導体の製造方法及びその製造中間体 | |
| EP2958893B1 (en) | Asymmetric synthesis of a substituted pyrrolidine-2-carboxamide | |
| CN101171251A (zh) | 氨曲南的制备方法 | |
| KR100990046B1 (ko) | 신규한 몬테루카스트 4-할로 벤질아민염 및 이를 이용한 몬테루카스트 나트륨염의 제조방법 | |
| JPWO2015137407A1 (ja) | ヘテロアリールカルボン酸エステル誘導体の製造方法、その製造中間体及び結晶 | |
| JP4769802B2 (ja) | ジアステレオ選択的な還元的アミノ化の方法 | |
| KR20150066777A (ko) | 광학활성 인돌린 유도체 및 이의 제조방법 | |
| EP1918275A1 (en) | Production method of diphenylalanine - NI(II) complex | |
| CN107629039B (zh) | 氘代丙烯酰胺的制备方法和中间体 | |
| TWI920144B (zh) | 八氫噻吩并喹啉化合物之製造方法及其製造中間體 | |
| JP5205971B2 (ja) | テトラヒドロピラン化合物の製造方法 | |
| JP2010530397A (ja) | アミド形成の改善方法 | |
| KR100413172B1 (ko) | 퀴놀리논 유도체의 제조방법 | |
| JP2011520876A (ja) | HMG−CoA還元阻害剤の製造のためのキラル中間体の製造方法 | |
| JP2025101851A (ja) | ハロカルボン酸誘導体及びその製造方法、カルボン酸塩化物及びその製造方法、並びに、チオラクトン誘導体の製造方法 | |
| JP4752121B2 (ja) | ニトリル誘導体の製造方法、その中間体および中間体の製造方法 | |
| JP2013129642A (ja) | 光学活性3,4−ビス(アルキルオキシカルボニル)−1,6−ヘキサン二酸誘導体の製造法 | |
| JP2022025121A (ja) | プロリンアミド化合物の製造方法 | |
| JP4058960B2 (ja) | インダゾール誘導体及びその製法 | |
| JP2023100917A (ja) | プロリンアミド化合物の製造方法 | |
| CN118420530A (zh) | 一种立他司特中间体的合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15760831 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016507799 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 247709 Country of ref document: IL |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015760831 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015760831 Country of ref document: EP |