WO2015076073A1 - プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 - Google Patents
プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 Download PDFInfo
- Publication number
- WO2015076073A1 WO2015076073A1 PCT/JP2014/078620 JP2014078620W WO2015076073A1 WO 2015076073 A1 WO2015076073 A1 WO 2015076073A1 JP 2014078620 W JP2014078620 W JP 2014078620W WO 2015076073 A1 WO2015076073 A1 WO 2015076073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- component
- fiber
- prepreg
- resin particles
- Prior art date
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/12—Layered products comprising a layer of synthetic resin next to a fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/20—Layered products comprising a layer of synthetic resin characterised by the use of special additives using fillers, pigments, thixotroping agents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/38—Layered products comprising a layer of synthetic resin comprising epoxy resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/22—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed
- B32B5/30—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed one layer being formed of particles, e.g. chips, granules, powder
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/249—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs characterised by the additives used in the prepolymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L61/00—Compositions of condensation polymers of aldehydes or ketones; Compositions of derivatives of such polymers
- C08L61/34—Condensation polymers of aldehydes or ketones with monomers covered by at least two of the groups C08L61/04, C08L61/18 and C08L61/20
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/02—Composition of the impregnated, bonded or embedded layer
- B32B2260/021—Fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/04—Impregnation, embedding, or binder material
- B32B2260/046—Synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2264/00—Composition or properties of particles which form a particulate layer or are present as additives
- B32B2264/02—Synthetic macromolecular particles
- B32B2264/0214—Particles made of materials belonging to B32B27/00
- B32B2264/0264—Polyamide particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/306—Resistant to heat
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/51—Elastic
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/558—Impact strength, toughness
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2605/00—Vehicles
- B32B2605/08—Cars
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2605/00—Vehicles
- B32B2605/12—Ships
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2605/00—Vehicles
- B32B2605/18—Aircraft
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G14/00—Condensation polymers of aldehydes or ketones with two or more other monomers covered by at least two of the groups C08G8/00 - C08G12/00
- C08G14/02—Condensation polymers of aldehydes or ketones with two or more other monomers covered by at least two of the groups C08G8/00 - C08G12/00 of aldehydes
- C08G14/04—Condensation polymers of aldehydes or ketones with two or more other monomers covered by at least two of the groups C08G8/00 - C08G12/00 of aldehydes with phenols
- C08G14/06—Condensation polymers of aldehydes or ketones with two or more other monomers covered by at least two of the groups C08G8/00 - C08G12/00 of aldehydes with phenols and monomers containing hydrogen attached to nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2361/00—Characterised by the use of condensation polymers of aldehydes or ketones; Derivatives of such polymers
- C08J2361/34—Condensation polymers of aldehydes or ketones with monomers covered by at least two of the groups C08J2361/04, C08J2361/18, and C08J2361/20
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2379/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2361/00 - C08J2377/00
- C08J2379/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2463/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2477/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2477/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
Definitions
- the present invention relates to a prepreg, a fiber reinforced composite material, and a particle-containing resin composition used for production thereof.
- the present invention particularly relates to a fiber reinforced composite material for aircraft use, marine use use, automobile use, sports use, and other general industrial use, and a prepreg used for obtaining the composite material.
- Fiber-reinforced composite materials composed of various fibers and matrix resins are widely used for aircraft, ships, automobiles, sporting goods and other general industrial applications because of their excellent mechanical properties. In recent years, the application range of fiber reinforced composite materials has been expanded as the use results have been increased.
- a material using a benzoxazine resin is proposed in, for example, Patent Documents 1 and 2.
- the benzoxazine resin has excellent moisture resistance and heat resistance, but has a problem of poor toughness, and has been devised to compensate for its drawbacks by blending epoxy resin and various resin fine particles.
- Another object of the present invention is to provide a prepreg capable of obtaining a fiber-reinforced composite material, a particle-containing resin composition for obtaining such a prepreg, and a fiber-reinforced composite material.
- the present invention provides (A) a benzoxazine resin, (B) an epoxy resin, and (C) two molecules in a molecule impregnated between the reinforcing fibers and the fibers of the reinforcing fibers.
- Polyamide resin particles comprising a copolymer obtained by copolymerizing caprolactam and laurolactam in a molar ratio of 1: 9 to 3: 7, and caprolactam and laurolactam in a molar ratio of 9: 1 to 7: 3.
- Providing a prepreg comprising a polyamide resin particles made of a copolymer obtained by polymerizing.
- the interlaminar fracture toughness, the CAI and the flexural modulus are high-ordered while using a benzoxazine resin having excellent moisture resistance and heat resistance by laminating and heating under pressure.
- a fiber-reinforced composite material that can be achieved at the same time and can maintain a high glass transition temperature of the resin material can be obtained.
- the present inventors consider the reason why the above-mentioned prepreg can improve the interlaminar fracture toughness, CAI and flexural modulus.
- the presence of the compound having a phenolic hydroxyl group, which is a curing agent for the benzoxazine resin lowers the melting temperature of the polyamide resin particles.
- the melting temperature of the polyamide resin particles becomes too low, the polyamide resin particles are easily melted when the thermosetting resin is cured when the fiber reinforced composite material is produced using the prepreg. It becomes easy to enter the reinforcing fiber layer.
- one polyamide resin particle is difficult to flow under the temperature condition for sufficiently curing the components (A) to (C). It is considered that the particles can be appropriately melted, and as a result, a cured resin layer having excellent adhesion, peeling resistance and bending elasticity is formed between the fiber layers.
- the surface layer is composed of 65 to 78 parts by mass of the component (A) and 22 to 35 parts by mass of the component (B) when the total of the component (A) and the component (B) is 100 parts by mass.
- the component (C) is contained in an amount of 5 to 20 parts by mass
- the component (D) is contained in an amount of 15 to 45 parts by mass.
- a polyamide resin comprising a copolymer in which caprolactam and laurolactam in the surface layer are copolymerized in a molar ratio of 1: 9 to 3: 7 when the content of each component in the surface layer is within the above range.
- the melting temperature of the particles and the polyamide resin particles made of a copolymer obtained by copolymerizing caprolactam and laurolactam in a molar ratio of 9: 1 to 7: 3 can be set to an appropriate range, and caprolactam and laurolactam
- the polyamide resin particles made of a copolymer copolymerized at a molar ratio of 1: 9 to 3: 7 can cause moderate melting while sufficiently preventing the polyamide resin particles from entering the reinforcing fiber layer.
- the flexural modulus can be further improved.
- the present invention also provides a fiber-reinforced composite material obtained by laminating a plurality of prepregs according to the present invention and heating them under pressure.
- the fiber-reinforced composite material of the present invention is obtained from the prepreg according to the present invention, so that it has excellent moisture resistance and heat resistance, and can simultaneously achieve interlaminar fracture toughness, CAI and bending elastic modulus in a high dimension. According to the fiber reinforced composite material of the present invention, the material can be reduced in weight by the excellent physical properties described above.
- the present invention also provides (A) a benzoxazine resin, (B) an epoxy resin, (C) a curing agent having two or more phenolic hydroxyl groups in the molecule, and (D) an average particle size of 5 to 50 ⁇ m.
- a particle-containing resin composition comprising polyamide resin particles comprising a copolymer copolymerized at a molar ratio of 1 to 7: 3.
- the surface layer of the prepreg according to the present invention described above can be produced.
- interlaminar fracture toughness, CAI and flexural modulus can be simultaneously achieved in a high dimension while using a benzoxazine resin having excellent moisture resistance and heat resistance, and the glass transition temperature of the resin material is maintained at a high level.
- a prepreg capable of obtaining a fiber-reinforced composite material that can be obtained, a particle-containing resin composition for obtaining such a prepreg, and a fiber-reinforced composite material can be provided.
- the fiber-reinforced composite material of the present invention can be suitably used for aircraft applications, marine applications, automobile applications, sports applications, and other general industrial applications, and is particularly useful for aircraft applications.
- FIG. 1 is a schematic cross-sectional view for explaining a prepreg according to an embodiment of the present invention.
- a prepreg 10 shown in FIG. 1A includes a reinforcing fiber layer 3 including a reinforcing fiber 1 and a resin composition 2 impregnated between the fibers of the reinforcing fiber 1, and a surface of the reinforcing fiber layer 3.
- the provided polyamide resin particles 4a made of a copolymer obtained by copolymerizing caprolactam and laurolactam at a molar ratio of 1: 9 to 3: 7, and 9: 1 to 7: 3 mol of caprolactam and laurolactam.
- a polyamide resin particle 4b made of a copolymer copolymerized at a ratio and a surface layer 6a containing a resin composition 5 are provided.
- Polyamide resin particles 4 b made of a copolymer copolymerized at a molar ratio of 7: 3 are included in the layer of the resin composition 5.
- 1B is a polyamide resin made of a copolymer obtained by copolymerizing caprolactam and laurolactam in a molar ratio of 1: 9 to 3: 7 instead of the surface layer 6a in the prepreg 10.
- the polyamide resin particles 4b made of a copolymer obtained by copolymerizing particles 4a and caprolactam and laurolactam in a molar ratio of 9: 1 to 7: 3 are on the side opposite to the reinforcing fiber layer 3 of the resin composition 5 layer.
- the structure is the same as that of the prepreg 10 except that the surface layer 6b formed on the surface is provided.
- the resin composition 2 contains (A) a benzoxazine resin, (B) an epoxy resin, and (C) a curing agent having two or more phenolic hydroxyl groups in the molecule.
- the surface layers 6a and 6b are (A) a benzoxazine resin, (B) an epoxy resin, (C) a curing agent having two or more phenolic hydroxyl groups in the molecule, and (D) an average particle size of 5 to Polyamide resin particles containing 50 ⁇ m polyamide resin particles, the polyamide resin particles comprising a copolymer obtained by copolymerizing caprolactam and laurolactam in a molar ratio of 1: 9 to 3: 7, and caprolactam and laurolactam 9 Polyamide resin particles comprising a copolymer copolymerized at a molar ratio of 1 to 7: 3.
- component (A) examples include compounds having a benzoxazine ring represented by the following general formula (A-1).
- R 5 represents a chain alkyl group having 1 to 12 carbon atoms, a cyclic alkyl group having 3 to 8 carbon atoms, an aryl group having 6 to 14 carbon atoms, or an alkyl group having 1 to 12 carbon atoms.
- a chain alkyl group or an aryl group substituted with a halogen is shown.
- a hydrogen atom may be bonded to the bond.
- Examples of the chain alkyl group having 1 to 12 carbon atoms include a methyl group, an ethyl group, a propyl group, an isopropyl group, an n-butyl group, an isobutyl group, and a t-butyl group.
- Examples of the cyclic alkyl group having 3 to 8 carbon atoms include a cyclopentyl group and a cyclohexyl group.
- Examples of the aryl group having 6 to 14 carbon atoms include a phenyl group, a 1-naphthyl group, a 2-naphthyl group, a phenanthryl group, and a biphenyl group.
- Examples of the chain alkyl group having 1 to 12 carbon atoms or the aryl group substituted with halogen include, for example, o-tolyl group, m-tolyl group, p-tolyl group, xylyl group, o-ethylphenyl group, m-ethyl. Examples thereof include a phenyl group, p-ethylphenyl group, ot-butylphenyl group, mt-butylphenyl group, pt-butylphenyl group, o-chlorophenyl group, and o-bromophenyl group.
- R 5 is preferably a methyl group, an ethyl group, a propyl group, a phenyl group, or an o-methylphenyl group among the above examples because it gives good handleability.
- examples of the (A) benzoxazine resin include compounds having a benzoxazine ring represented by the following general formula (A-2).
- L represents an alkylene group or an arylene group.
- benzoxazine resin of the component (A) for example, a monomer represented by the following formula, an oligomer obtained by polymerizing several monomers of the monomer, and at least one monomer represented by the following formula are different from these monomers.
- a reaction product with a compound having a benzoxazine ring having a structure is preferred.
- the component (A) is excellent in flame retardancy because the benzoxazine ring undergoes ring-opening polymerization to form the same skeleton as the phenol resin. Further, excellent mechanical properties such as low water absorption and high elastic modulus can be obtained from the dense structure.
- a component can be used individually by 1 type or in combination of 2 or more types.
- Epoxy resin (hereinafter sometimes referred to as component (B)) used in the present embodiment is blended as a component that controls the viscosity of the composition and increases the curability of the composition.
- component (B) for example, an epoxy resin having a precursor such as an amine, a phenol, a carboxylic acid, or a compound having a carbon-carbon double bond is preferred.
- epoxy resins having amines as precursors include tetraglycidyldiaminodiphenylmethane, glycidyl compounds of xylenediamine, triglycidylaminophenol, and glycidylaniline, and their respective positional isomers and substituted groups with alkyl groups and halogens. Is mentioned.
- the complex viscoelastic modulus ⁇ * at 25 ° C. obtained by a dynamic viscoelasticity measuring device described later is described as the viscosity.
- triglycidylaminophenol Commercially available products of triglycidylaminophenol include, for example, “jER” 630 (viscosity: 750 mPa ⁇ s) (manufactured by Mitsubishi Chemical Corporation), “Araldite” MY0500 (viscosity: 3500 mPa ⁇ s), MY0510 (viscosity: 600 mPa ⁇ s). s) (manufactured by Huntsman Advanced Materials), ELM100 (viscosity: 16000 mPa ⁇ s) (manufactured by Sumitomo Chemical).
- Examples of commercially available glycidyl anilines include GAN (viscosity: 120 mPa ⁇ s) and GOT (viscosity: 60 mPa ⁇ s) (manufactured by Nippon Kayaku Co., Ltd.).
- Examples of the glycidyl ether type epoxy resin having a phenol as a precursor include, for example, bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol S type epoxy resin, epoxy resin having a biphenyl skeleton, phenol novolac type epoxy resin, cresol novolac.
- an alkyl group and a substituent with a halogen are included.
- the epoxy resin which modified the epoxy resin which makes phenols a precursor with urethane or isocyanate is also contained in this type.
- liquid bisphenol A type epoxy resins examples include “jER” 825 (viscosity: 5000 mPa ⁇ s), “jER” 826 (viscosity: 8000 mPa ⁇ s), and “jER” 827 (viscosity: 10,000 mPa ⁇ s).
- JER 828 viscosity: 13000 mPa ⁇ s
- Epiclon registered trademark, the same shall apply hereinafter
- 850 viscosity: 13000 mPa ⁇ s
- YD-128 viscosity: 13000 mPa ⁇ s
- DER-331 viscosity: 13000 mPa ⁇ s
- DER-332 viscosity: 5000 mPa ⁇ s) (Made by Dow Chemical Company).
- Examples of commercially available solid or semi-solid bisphenol A type epoxy resins include “jER” 834, “jER” 1001, “jER” 1002, “jER” 1003, “jER” 1004, “jER” 1004AF, and “jER”. "1007", “jER” 1009 (manufactured by Mitsubishi Chemical Corporation).
- liquid bisphenol F-type epoxy resins examples include “jER” 806 (viscosity: 2000 mPa ⁇ s), “jER” 807 (viscosity: 3500 mPa ⁇ s), and “jER” 1750 (viscosity: 1300 mPa ⁇ s).
- Solid bisphenol F type epoxy resins include, for example, 4004P, “jER” 4007P, “jER” 4009P (manufactured by Mitsubishi Chemical Corporation), “Epototo” YDF2001, “Epototo” YDF2004 (above Nippon Steel) Chemical Co., Ltd.).
- Examples of commercially available bisphenol S-type epoxy resins include EXA-1515 (manufactured by DIC Corporation).
- Examples of commercially available epoxy resins having a biphenyl skeleton include “jER” YX4000H, “jER” YX4000, “jER” YL6616 (manufactured by Mitsubishi Chemical Corporation), NC-3000 (manufactured by Nippon Kayaku Co., Ltd.). ).
- phenol novolac epoxy resins examples include “jER” 152, “jER” 154 (manufactured by Mitsubishi Chemical Corporation), “Epicron” N-740, “Epicron” N-770, “Epicron” N -775 (manufactured by DIC Corporation).
- cresol novolac type epoxy resins examples include “Epiclon” N-660, “Epicron” N-665, “Epicron” N-670, “Epicron” N-673, “Epicron” N-695 (above, DIC Corporation), EOCN-1020, EOCN-102S, EOCN-104S (above, Nippon Kayaku Co., Ltd.).
- Examples of commercially available resorcinol type epoxy resins include “Denacol” (registered trademark, hereinafter the same) EX-201 (viscosity: 250 mPa ⁇ s) (manufactured by Nagase ChemteX Corporation).
- Examples of commercially available epoxy resins having a naphthalene skeleton include “Epiclon” HP4032 (manufactured by DIC Corporation), NC-7000, NC-7300 (manufactured by Nippon Kayaku Co., Ltd.).
- trisphenylmethane type epoxy resins examples include TMH-574 (manufactured by Sumitomo Chemical Co., Ltd.).
- dicyclopentadiene type epoxy resins include, for example, “Epicron” HP7200, “Epicron” HP7200L, “Epicron” HP7200H (above, manufactured by DIC Corporation), “Tactix” (registered trademark) 558 (Huntsman Advanced) -Materials Co., Ltd.), XD-1000-1L, XD-1000-2L (Nippon Kayaku Co., Ltd.).
- Examples of commercially available urethane and isocyanate-modified epoxy resins include AER4152 (produced by Asahi Kasei E-Materials Co., Ltd.) having an oxazolidone ring.
- Examples of the epoxy resin having carboxylic acid as a precursor include glycidyl compound of phthalic acid, glycidyl compound of hexahydrophthalic acid, glycidyl compound of dimer acid, and various isomers thereof.
- diglycidyl phthalate examples include, for example, “Epomic” (registered trademark, the same applies hereinafter) R508 (viscosity: 4000 mPa ⁇ s) (manufactured by Mitsui Chemicals), “Denacol” EX-721 (viscosity: 980 mPas). S) (manufactured by Nagase ChemteX Corporation).
- hexahydrophthalic acid diglycidyl ester Commercially available products of hexahydrophthalic acid diglycidyl ester include, for example, “Epomic” R540 (viscosity: 350 mPa ⁇ s) (manufactured by Mitsui Chemicals), AK-601 (viscosity: 300 mPa ⁇ s) (Nippon Kayaku Co., Ltd.) Co., Ltd.).
- dimer acid diglycidyl esters examples include “jER” 871 (viscosity: 650 mPa ⁇ s) (manufactured by Mitsubishi Chemical Corporation), “Epototo” YD-171 (viscosity: 650 mPa ⁇ s) (Nippon Steel). Chemical Co., Ltd.).
- Examples of the epoxy resin using a compound having a carbon-carbon double bond as a precursor include alicyclic epoxy resins.
- Examples of the alicyclic epoxy resin include (3 ′, 4′-epoxycyclohexane) methyl-3,4-epoxycyclohexanecarboxylate, (3 ′, 4′-epoxycyclohexane) octyl-3,4-epoxycyclohexanecarboxylate, 1-methyl-4- (2-methyloxiranyl) -7-oxabicyclo [4.1.0] heptane.
- an epoxy resin that is liquid at 25 ° C. can be blended from the viewpoint of tack and drape.
- the viscosity at 25 ° C. exceeds 20000 mPa ⁇ s, tack and drape properties may be deteriorated.
- a solid epoxy resin can be blended at 25 ° C.
- an epoxy resin having a high aromatic content is preferable, and examples thereof include an epoxy resin having a biphenyl skeleton, an epoxy resin having a naphthalene skeleton, and a phenol aralkyl type epoxy resin.
- a component can be used individually by 1 type or in combination of 2 or more types.
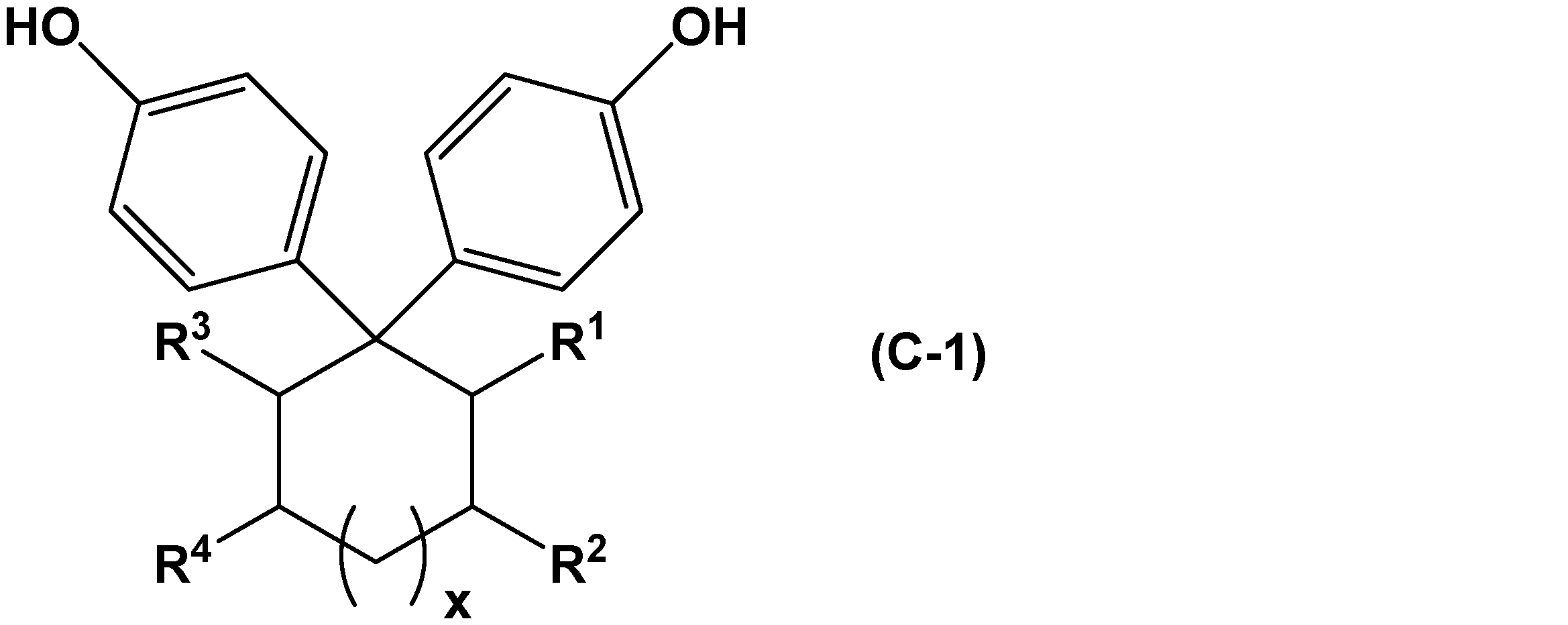
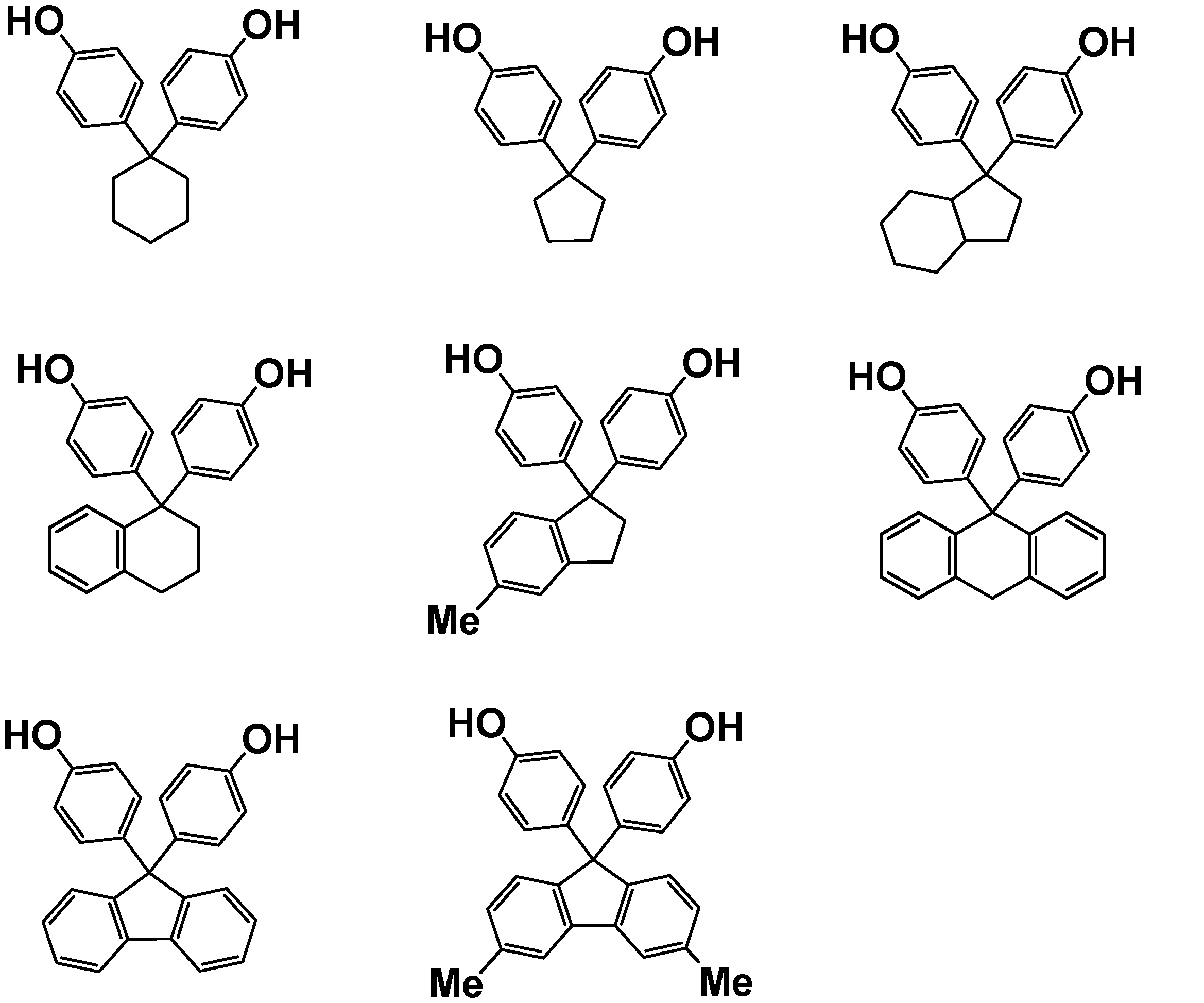
- Examples of the curing agent having two or more phenolic hydroxyl groups in the molecule (C) used in the present embodiment include polyfunctional phenols such as bisphenols. Examples thereof include bisphenol A, bisphenol F, bisphenol S, thiodiphenol, and bisphenols represented by the following general formula (C-1).
- R 1 , R 2 , R 3 and R 4 represent a hydrogen atom or a hydrocarbon group, and when R 1 , R 2 , R 3 or R 4 is a hydrocarbon group, Is a linear or branched alkyl group having 1 to 4 carbon atoms, or adjacent R 1 and R 2 or adjacent R 3 and R 4 are combined to form a substituted or unsubstituted aromatic group having 6 to 10 carbon atoms. A ring or a substituted or unsubstituted alicyclic structure having 6 to 10 carbon atoms is formed, and x represents 0 or 1. ]
- Examples of the curing agent represented by the general formula (C-1) include compounds represented by the following formula.
- bisphenol A bisphenol F
- thiobisphenol hereinafter sometimes referred to as TDP
- 9,9-bis (4-hydroxyphenyl) fluorene hereinafter, it may be referred to as BPF
- 1,1-bis (4-hydroxyphenyl) cyclohexane hereinafter also referred to as BPC
- a component can be used individually by 1 type or in combination of 2 or more types.
- a curing agent other than the component (C) can be used in combination.
- the curing agent that can be used in combination include tertiary aromatic amines such as N, N-dimethylaniline, tertiary aliphatic amines such as triethylamine, imidazole derivatives, and pyridine derivatives. These can be used individually by 1 type or in combination of 2 or more types.
- the polyamide resin particles (D) having an average particle diameter of 5 to 50 ⁇ m used in the present embodiment have (D1) caprolactam and laurolactam of 1: 9 to 3: 7.
- the average particle diameter means an average value of the lengths of the long diameters of each particle measured with 100 particles arbitrarily selected from particles enlarged 200 to 500 times with a scanning electron microscope (SEM). To do.
- the coalescence is called polyamide 6/12 or the like.
- the copolymer may be a random copolymer or a block copolymer.
- the copolymerization ratio (molar ratio) of caprolactam and laurolactam needs to be within the range of 1: 9 to 3: 7, and within the range of 1: 9 to 25:75. More preferably, it is more preferably in the range of 1: 9 to 2: 8.
- the melting point of the polyamide resin particles and the melting temperature of the polyamide resin particles in the particle-containing resin composition can be adjusted to an appropriate range, and the interlaminar fracture toughness, CAI and bending The elastic modulus can be improved.
- the melting point of the polyamide 6/12 resin particles as the component (D1) is preferably 140 ° C. to 170 ° C. from the viewpoint of appropriately melting the polyamide 6/12 resin particles at the time of producing the fiber-reinforced composite. More preferably, it is ⁇ 170 ° C., and further preferably 150 ° C.-170 ° C.
- the melting point of the polyamide resin particles is increased from 25 ° C. to a rate of 10 ° C./min using a differential calorimeter (DSC), and the temperature at the top of the obtained endothermic peak is measured. Is required. When there are a plurality of endothermic peaks, the temperature at the top of the peak with the largest endotherm is taken as the melting point.
- (D1) component polyamide 6/12 resin particles used in the present embodiment commercially available products can be used, for example, Orgasol 4000EXD (registered trademark, manufactured by Arkema Co., Ltd.).
- the copolymerization ratio (molar ratio) of caprolactam and laurolactam must be within the range of 9: 1 to 7: 3, and within the range of 9: 1 to 75:25. More preferably, it is more preferably in the range of 9: 1 to 8: 2.
- the melting point of the polyamide resin particles and the melting temperature of the polyamide resin particles in the particle-containing resin composition can be adjusted to an appropriate range, and the interlaminar fracture toughness, CAI and bending The elastic modulus can be improved.
- the melting point of the polyamide 6/12 resin particles of component (D2) is 170 ° C. or more from the viewpoint of moderately suppressing the polyamide 6/12 resin particles from melting and entering the reinforcing fiber layer during the production of the fiber reinforced composite material. It is preferably 220 ° C, more preferably 175 ° C to 210 ° C, and still more preferably 180 ° C to 205 ° C.
- the melting point of the polyamide resin particles is increased from 25 ° C. to a rate of 10 ° C./min using a differential calorimeter (DSC), and the temperature at the top of the obtained endothermic peak is measured. Is required. When there are a plurality of endothermic peaks, the temperature at the top of the peak with the largest endotherm is taken as the melting point.
- (D2) component polyamide 6/12 resin particles used in the present embodiment commercially available products can be used, for example, Orgasol 3202D (registered trademark, manufactured by Arkema Co., Ltd.).
- the average particle diameter of the polyamide 6/12 resin particles of the component (D1) and the component (D2) is preferably 5 to 50 ⁇ m and more preferably 10 to 30 ⁇ m from the viewpoint of controlling the surface layer thickness.
- the content of the component (A) and the component (B) in the resin composition 2 is such that when the total of the component (A) and the component (B) is 100 parts by mass, the component (A) 65 to 78 parts by mass and the component (B) is preferably 22 to 35 parts by mass.
- the content ratio of the component (A) is less than 65 parts by mass, that is, when the content ratio of the component (B) exceeds 35 parts by mass, the elastic modulus and water resistance of the resulting fiber-reinforced composite tend to decrease.
- the glass transition temperature of the cured resin tends to decrease.
- the content of the component (C) in the resin composition 2 is preferably 5 to 20 parts by mass when the total of the components (A) and (B) is 100 parts by mass, 15 parts by mass is more preferable. If the content of the component (C) is less than 5 parts by mass, it tends to be difficult to sufficiently increase the interlaminar fracture toughness, CAI, and flexural modulus in the fiber-reinforced composite material. There exists a tendency for mechanical properties, such as a glass transition temperature of hardened
- the contents of the component (A) and the component (B) in the surface layers 6a and 6b are the components (A) when the total of the components (A) and (B) is 100 parts by mass. Is 65 to 78 parts by mass, and component (B) is preferably 22 to 35 parts by mass.
- the content ratio of the component (A) is less than 65 parts by mass, that is, when the content ratio of the component (B) exceeds 35 parts by mass, the elastic modulus and water resistance of the resulting fiber-reinforced composite tend to decrease. In addition, the glass transition temperature of the cured resin tends to decrease.
- the content of the component (C) in the surface layers 6a and 6b is preferably 5 to 20 parts by mass when the total of the components (A) and (B) is 100 parts by mass. More preferred is 15 parts by mass.
- the content of the component (C) is less than 5 parts by mass, it tends to be difficult to sufficiently increase the CAI and flexural modulus of the fiber-reinforced composite material.
- the content exceeds 20 parts by mass, the glass transition of the cured product Mechanical properties such as temperature tend to decrease.
- the content of the component (D) in the surface layers 6a and 6b is preferably 15 to 45 parts by mass when the total of the components (A) and (B) is 100 parts by mass, More preferred is 40 parts by mass.
- the content of the component (D) is less than 15 parts by mass, it tends to be difficult to sufficiently increase the interlaminar fracture toughness, CAI, and flexural modulus in the fiber reinforced composite material. The rate tends to decrease.
- the total content of the component (D1) and the component (D2) is preferably in the above range.
- the blending ratio of the component (D1) and the component (D2) is such that the component (D2) is 10 to 1000 parts by mass with respect to 100 parts by mass of the component (D1) in terms of sufficiently increasing the CAI and interlaminar fracture toughness. It is preferably 20 to 500 parts by mass, more preferably 30 to 300 parts by mass.
- the surface layers 6a and 6b in the prepreg of the present embodiment refer to the space between the prepreg surface and the reinforcing fibers of the reinforcing fiber layer, and the content of the component (D) in the surface layer is, for example, from the prepreg surface to the reinforcing fiber layer. It can be calculated based on the contents of the component (A), the component (B), and the component (C) detected up to the reinforcing fiber.
- the surface layer and the reinforcing fiber layer can be blended with other components such as (E) a toughness improver as long as the physical properties are not impaired.
- a toughness improver phenoxy resins “YP-70”, “YP-50”, “FX-316” (registered trademark, manufactured by Nippon Steel & Sumikin Chemical Co., Ltd.), polyethersulfone “Sumika Excel” PES ”(registered trademark, manufactured by Sumitomo Chemical Co., Ltd.).
- nanocarbon examples include carbon nanotubes, fullerenes, and derivatives thereof.
- flame retardant examples include phosphoric acid such as red phosphorus, triphenyl phosphate, tricresyl phosphate, trixylenyl phosphate, cresyl diphenyl phosphate, xylenyl diphenyl phosphate, resorcinol bisphenyl phosphate, bisphenol A bisdiphenyl phosphate, etc.
- examples thereof include esters and boric acid esters.
- the mold release agent examples include silicone oil, stearic acid ester, carnauba wax and the like.
- glass fiber carbon fiber, graphite fiber, aramid fiber, boron fiber, alumina fiber, silicon carbide fiber, or the like can be used. Two or more of these fibers may be mixed and used. In order to obtain a molded product that is lighter and more durable, it is preferable to use carbon fiber or graphite fiber, and it is more preferable to use carbon fiber.
- either a PAN-based carbon fiber or a pitch-based carbon fiber can be used.
- any type of carbon fiber or graphite fiber can be used depending on the application.
- the tensile elastic modulus of the carbon fiber or graphite fiber in the strand tensile test is preferably 150 to 650 GPa, more preferably 200 to 550 GPa, more preferably 230 to 500 GPa.
- the strand tensile test refers to a test performed based on JIS R7601 (1986) after impregnating an epoxy resin into a bundle of carbon fiber or graphite fiber and curing at 130 ° C. for 35 minutes.
- the form of the reinforcing fiber is not particularly limited.
- the long fiber, tow, woven fabric, mat, knit, braid, and a length of less than 10 mm are aligned in one direction. It is possible to use short fibers that are chopped in the same manner.
- the long fiber is a single fiber or a fiber bundle substantially continuous for 10 mm or more.
- a short fiber is a fiber bundle cut into a length of less than 10 mm.
- an array in which reinforcing fiber bundles are aligned in a single direction like the prepreg of this embodiment is most suitable.
- a (woven fabric) arrangement is also applicable.
- the amount of reinforcing fibers per unit area is preferably 25 to 3000 g / m 2 .
- the amount of reinforcing fibers is less than 25 g / m 2, it is necessary to increase the number of laminated layers in order to obtain a predetermined thickness when forming a fiber-reinforced composite material, and the work may be complicated.
- the amount of reinforcing fibers exceeds 3000 g / m 2 , the prepreg drapability tends to deteriorate. If the prepreg is a flat surface or a simple curved surface, the amount of reinforcing fibers may exceed 3000 g / m 2 .
- the fiber content in the prepreg is preferably 30 to 90% by mass, more preferably 35 to 85% by mass, and still more preferably 40 to 80% by mass. If the content is less than 30% by mass, the amount of resin is too large to obtain the advantages of a fiber reinforced composite material excellent in specific strength and specific elastic modulus, or heat generation during curing when molding the fiber reinforced composite material. The amount can be too large. When the content exceeds 90% by mass, poor resin impregnation occurs, and the resulting composite material tends to have many voids.
- FIG.2 and FIG.3 is a schematic cross section for demonstrating the method to manufacture the prepreg which concerns on one Embodiment of this invention.
- the method shown in FIG. 2 is an embodiment of a method for manufacturing the prepreg 10 according to the present embodiment described above.
- a reinforcing fiber bundle 7 in which reinforcing fibers 1 are aligned in one direction is prepared (a), and the reinforcing resin bundle 7 is impregnated with the first resin composition 2 containing the components (A) to (C).
- the reinforcing fiber layer 3 is formed (b), and both surfaces of the reinforcing fiber layer 3 are impregnated with the second resin composition containing the components (A) to (C) and the component (D).
- the prepreg 10 is obtained by forming 6a (c).
- a reinforcing fiber bundle 7 in which the reinforcing fibers 1 are aligned in one direction is prepared (a), and the resin composition containing the components (A) to (D) on both sides of the reinforcing fiber bundle 7. Is impregnated once to form the surface layer 6a made of the resin composition 2 containing the components (D) 4a and 4b and the components (A) to (C) not impregnated in the fiber, and the prepreg 11 is obtained. (B).
- the prepreg 12 shown in FIG. 1B includes, for example, the component (D) on the surface of the reinforcing fiber bundle impregnated with the resin composition after the reinforcing fiber bundle is impregnated with the resin composition containing the components (A) to (C). Can be produced by spraying.
- Each resin composition impregnated into the reinforcing fiber bundle is kneaded with the above components (A) to (C) and other components as necessary, or the above components (A) to (D) and other components as necessary. Can be prepared.
- the kneading method of the resin composition is not particularly limited, and for example, a kneader, a planetary mixer, a twin screw extruder, or the like is used.
- a kneader a planetary mixer, a twin screw extruder, or the like is used.
- the particles are previously diffused into the liquid resin component by a homomixer, three rolls, a ball mill, a bead mill, an ultrasonic wave, or the like.
- heating / cooling, pressurization / depressurization may be performed as necessary at the time of mixing with the matrix resin or pre-diffusion of particles.
- the viscosity of the resin composition is preferably 10 to 20000 Pa ⁇ s at 50 ° C. from the viewpoint of precursor film production. More preferably, it is 10 to 10,000 Pa ⁇ s, and most preferably 50 to 6000 Pa ⁇ s. If it is less than 10 Pa ⁇ s, the tackiness of the resin composition becomes high and it may be difficult to apply. On the other hand, if it exceeds 20000 Pa ⁇ s, it becomes semi-solid and coating becomes difficult.
- Examples of the method of impregnating the resin composition include a wet method in which the resin composition is dissolved in a solvent such as methyl ethyl ketone and methanol to lower the viscosity and impregnated, a hot melt method in which the viscosity is reduced by heating and impregnated (dry method), and the like. Can be mentioned.
- the wet method is a method in which a reinforcing fiber is immersed in a solution of a resin composition, then pulled up, and the solvent is evaporated using an oven or the like.
- the hot melt method is a method in which a reinforcing fiber is impregnated directly with a resin composition whose viscosity is reduced by heating, or a film is prepared by once coating a resin composition on a release paper or the like, and then both sides of the reinforcing fiber.
- it is a method of impregnating a reinforcing fiber with a resin by overlapping the film from one side and heating and pressing.
- the hot melt method is preferable because substantially no solvent remains in the prepreg.
- the prepreg according to the present embodiment can be made into a fiber-reinforced composite material by a method of heat-curing a resin while applying pressure to the laminate after lamination.
- methods for applying heat and pressure include a press molding method, an autoclave molding method, a bagging molding method, a wrapping tape method, and an internal pressure molding method.
- the wrapping tape method is a method of winding a prepreg around a mandrel or other core metal to form a tubular body made of a fiber-reinforced composite material, and is a method suitable for producing a rod-shaped body such as a golf shaft or fishing rod. .
- a prepreg is wound around a mandrel, a wrapping tape made of a thermoplastic film is wound around the outside of the prepreg for fixing and applying pressure, and the resin is heated and cured in an oven, and then a cored bar.
- This is a method for extracting a tube to obtain a tubular body.
- a preform obtained by winding a prepreg on an internal pressure applying body such as a tube made of a thermoplastic resin is set in a mold, and then a high pressure gas is introduced into the internal pressure applying body to apply pressure and at the same time
- the mold is heated and molded.
- This method is preferably used when molding a complicated shape such as a golf shaft, a bad, a racket such as tennis or badminton.
- the particle-containing resin composition containing the components (A) to (D) and other components as necessary can be suitably used for the preparation of the prepreg described above.
- Particle-containing resin composition in which the content of component (D) is 15 to 45 parts by mass, preferably 25 to 40 parts by mass, when the total of component (A) and component (B) is 100 parts by mass can be suitably used as a material for forming the surface layer of the prepreg.
- the glass transition temperature of a cured product obtained by heating the particle-containing resin composition from room temperature to 185 ° C. at a heating rate of 2.0 ° C./min and then curing the same at the same temperature for 2 hours. Is preferably 190 ° C. or higher.
- the temperature for curing the resin by heating the laminate As the temperature for curing the resin by heating the laminate, the polyamide 6/12 resin particles as the component (D1) are appropriately melted, and the polyamide 6/12 resin particles as the component (D2) are melted to form a reinforcing fiber layer. From the viewpoint of moderately suppressing the penetration, the temperature is preferably 160 to 190 ° C, more preferably 170 to 190 ° C, and still more preferably 180 to 190 ° C.
- the temperature at which the resin is cured by heating here refers to the temperature of the prepreg.
- the holding time at the temperature at which the laminate is heated to cure the resin can be 30 minutes to 10 hours, preferably 1 to 6 hours.
- the heating rate up to the heating temperature can be arbitrarily selected, but it is preferably 0.3 to 3 ° C./min from the viewpoint of productivity.
- the pressure at the time of heating is preferably 0.2 to 1.0 MPa, more preferably 0.3 to 0.8 MPa.
- the temperature can be lowered at a rate of -0.3 to -3.0 ° C / min.
- FIG. 4 is a schematic cross-sectional view for explaining a fiber-reinforced composite material according to an embodiment of the present invention.
- a fiber-reinforced composite material 100 shown in FIG. 4 includes reinforcing fibers 1, a cured resin 8, and polyamide resin particles 4a and 4b.
- the fiber-reinforced composite material 100 can be obtained by laminating a plurality of prepregs 10, 11, and 12 and heating them under pressure.
- the polyamide resin particles are shown in the same manner as those in the surface layer of the prepreg, but they are melted by pressurization and heating, and are deformed by flow and bonding between particles.
- the capacity ratio of C 1 in the total amount of the polyamide resin content C 1 contained in the cured resin layer between the reinforcing fiber layers and the polyamide resin content C 2 contained in the reinforcing fiber layer ⁇ C 1 / (C 1 + C 2 ) ⁇ ⁇ 100 is preferably 70% by volume or more, and more preferably 80% by volume or more.
- the content of the polyamide resin is determined by analyzing the cut surface when the fiber reinforced composite material is cut on a surface orthogonal to the direction in which any reinforcing fiber in the fiber reinforced composite material is stretched by microscopic observation, and performing image analysis. This is determined by observing the distribution of the resin.
- the fiber reinforced composite material according to the present embodiment can be obtained by directly impregnating the reinforced fiber base material with the resin composition and curing it.
- a method in which a reinforcing fiber base is placed in a mold, and then a resin composition containing the components (A) to (D) is poured and impregnated and cured, or the reinforcing fiber base and the above (A) to (D) are used.
- the film can be produced by laminating a film comprising a resin composition containing a component) and heating and pressurizing the laminate.
- the film can be obtained by applying a predetermined amount of a resin composition with a uniform thickness on a release paper or a release film in advance.
- the reinforcing fiber base examples include long fibers arranged in one direction, two-way woven fabric, non-woven fabric, mat, knit, braided string, and the like.
- the lamination here includes not only the case where the fiber base materials are simply overlapped but also the case where they are preformed by being attached to various types or core materials.
- a foam core or a honeycomb core is preferably used.
- the foam core urethane or polyimide is preferably used.
- the honeycomb core an aluminum core, a glass core, an aramid core, or the like is preferably used.
- the fiber reinforced composite material according to the present embodiment preferably has a post-impact compressive strength (CAI) measured in accordance with ASTM D7136 and D7137 of 210 MPa or more, and more preferably 220 MPa or more.
- CAI post-impact compressive strength
- Fiber-reinforced composite material according to the present embodiment is preferably measured mode I interlaminar fracture toughness in accordance with ASTM D5528 (G1c) is 210J / m 2 or more, more preferably 250 J / m 2 or more.
- the fiber-reinforced composite material according to the present embodiment preferably has a mode II interlaminar fracture toughness value (G2c) measured according to Composite Materials Handbook 17-1 of 1500 J / m 2 or more, preferably 1800 J / m 2 or more. More preferred.
- G2c mode II interlaminar fracture toughness value
- the glass transition temperature of the cured resin is preferably 180 ° C. or higher, and more preferably 190 ° C. or higher.
- the fiber-reinforced composite material according to the present invention having the above physical properties is suitably used for railway vehicles, aircraft, building members, and other general industrial applications.
- Example 1 to 5 Comparative Examples 1 and 2
- the raw material is heat-mixed in the ratio shown in Table 1, the 1st resin composition (the “1st” composition in a table) which does not contain particles, and the 2nd which contains particles.
- a resin composition (the “second” composition in the table) was obtained.
- the raw materials used here are as shown below.
- the obtained first and second resin compositions were respectively applied to a release paper at 70 to 100 ° C. to obtain an 18 g / m 2 first resin film and a 25 g / m 2 second resin film. .
- the obtained first resin film was supplied from above and below the carbon fibers aligned in one direction and impregnated between the fibers to form a carbon fiber layer.
- the 2nd resin film was laminated from the upper and lower sides of the carbon fiber layer, the surface layer was formed, and the prepreg was produced.
- the amount of carbon fiber per unit area of this prepreg was 150 g / m 2
- the total resin composition amount (matrix resin amount) in the carbon fiber layer and the surface layer was 86 g / m 2 .
- the polyamide 6/12 resin particles as the component (D1) and the polyamide 6/12 resin particles as the component (D2) are heated at a rate of 25 ° C. to 10 ° C./min using a differential calorimeter (DSC).
- the top temperature of the obtained endothermic peak was defined as the melting point of the polyamide resin particles.
- the melting point of the polyamide 6/12 resin particles (20/80) is 162 ° C.
- the melting point of the polyamide 6/12 (80/20) resin particles is 194 ° C.
- the melting point was 187 ° C.
- the melting point of the polyamide 6/12 (90/10) resin particles was 200 ° C.
- the obtained second resin composition was heated in an oven from room temperature to 185 ° C. at a temperature rising rate of 2.0 ° C./min and cured at the same temperature for 2 hours to obtain a cured resin.
- the obtained cured product was measured as the glass transition temperature at the midpoint temperature determined based on JIS K7121 (1987). The results are shown in Table 1.
- the present invention while utilizing a benzoxazine resin having excellent moisture resistance and heat resistance, interlaminar fracture toughness, CAI and flexural modulus can be achieved simultaneously in a high dimension, and glass of resin material It is possible to provide a prepreg capable of obtaining a fiber-reinforced composite material that can maintain a high transition temperature, a particle-containing resin composition for obtaining such a prepreg, and a fiber-reinforced composite material.
- the fiber reinforced composite material can be used for aircraft applications, marine applications, automobile applications, sports applications, and other general industrial applications.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Reinforced Plastic Materials (AREA)
- Laminated Bodies (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Epoxy Resins (AREA)
Abstract
Description
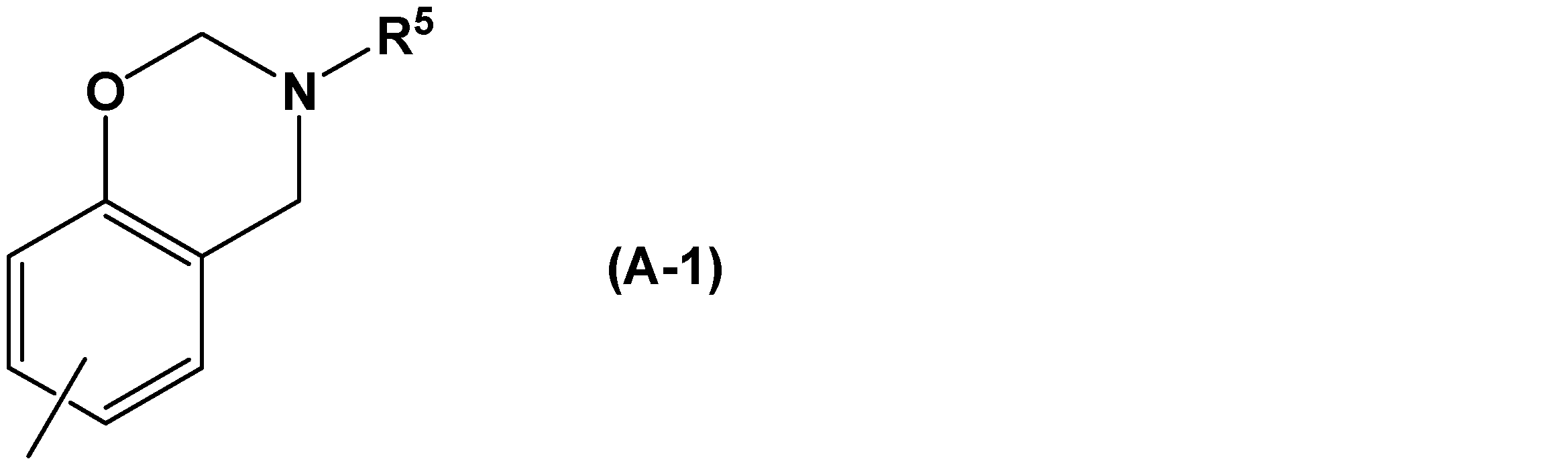



各実施例、比較例について、表1に示す割合で原料を加熱混合し、粒子を含有しない第1の樹脂組成物(表中の「第1」の組成)と、粒子を含有する第2の樹脂組成物(表中の「第2」の組成)を得た。なお、ここで用いた原料は以下に示す通りである。
F-a:ビスフェノールF-アニリン型(F-a型ベンゾオキサジン、四国化成(株)製)
P-a:フェノール-アニリン型(P-a型ベンゾオキサジン、四国化成(株)製)
(B)成分:エポキシ樹脂
2021P:「セロキサイド」(登録商標)2021P(ダイセル化学工業(株)製)
(C)成分:硬化剤
BPF(9,9-ビス(4-ヒドロキシフェニル)フルオレン、大阪ガスケミカル製)
(D)成分:ポリアミド樹脂粒子
(D1)成分
PA6/12(20/80):カプロラクタムとラウロラクタムとを20:80のモル比で共重合させたポリアミド6/12共重合体(ランダム共重合体)からなる粒子(平均粒子径25μm)
(D2)成分
PA6/12(80/20):カプロラクタムとラウロラクタムとを80:20のモル比で共重合させたポリアミド6/12共重合体(ランダム共重合体)からなる粒子(平均粒子径20μm)
PA6/12(75/25):カプロラクタムとラウロラクタムとを75:25のモル比で共重合させたポリアミド6/12共重合体(ランダム共重合体)からなる粒子(平均粒子径20μm)
PA6/12(90/10):カプロラクタムとラウロラクタムとを90:10のモル比で共重合させたポリアミド6/12共重合体(ランダム共重合体)からなる粒子(平均粒子径20μm)
(E)成分:靭性向上剤
YP70:フェノキシ樹脂(YP-70、新日鐵住金化学株式会社製)

得られた第1及び第2の樹脂組成物をそれぞれ離型紙上に70~100℃で塗布し、18g/m2の第1の樹脂フィルム及び25g/m2の第2の樹脂フィルムを得た。得られた第1の樹脂フィルムを、一方向に引き揃えた炭素繊維の上下から供給して繊維間に含浸し、炭素繊維層を形成した。続いて、第2の樹脂フィルムを炭素繊維層の上下からラミネートして表面層を形成し、プリプレグを作製した。このプリプレグの単位面積当たりの炭素繊維量は150g/m2であり、炭素繊維層及び表面層中の合計の樹脂組成物量(マトリックス樹脂量)は86g/m2であった。
上記(D1)成分であるポリアミド6/12樹脂粒子及び(D2)成分であるポリアミド6/12樹脂粒子を、示差熱量計(DSC)を用いて、25℃から10℃/分の速度で昇温し、得られた吸熱ピークのトップの温度をポリアミド樹脂粒子の融点とした。ポリアミド6/12樹脂粒子(20/80)の融点は162℃であり、ポリアミド6/12(80/20)樹脂粒子の融点は194℃であり、ポリアミド6/12(75/25)樹脂粒子の融点は187℃であり、ポリアミド6/12(90/10)樹脂粒子の融点は200℃であった。
得られた第2の樹脂組成物を、示差熱量計(DSC)を用いて、25℃から10℃/分の速度で昇温し、得られた吸熱ピークのトップの温度を第2の樹脂組成物中でのポリアミド樹脂粒子の融解温度とした。結果を表1に示す。表1中の融解温度欄において、二つの融解温度が記載されている例において、上段は(D1)成分の融解温度、下段は(D2)成分の融解温度を示す。また、一例として、実施例3の第2の樹脂組成物のDSCチャートを図5に示す。
得られた第2の樹脂組成物を、オーブン中、室温から2.0℃/分の昇温速度で185℃まで昇温し、同温度で2時間硬化して樹脂硬化物を得た。得られた硬化物を、示差熱量計(DSC)を用いて、JIS K7121(1987)に基づいて求めた中間点温度をガラス転移温度として測定した。結果を表1に示す。
得られた第2の樹脂組成物を、オーブン中、室温から2.0℃/分の昇温速度で185℃まで昇温し、同温度で2時間硬化させ、厚さ2mmの樹脂硬化物を得た。この樹脂硬化物について、JIS J 7171に従い曲げ弾性率測定を行った。結果を表1に示す。
得られたプリプレグを、[+45°/0°/-45°/90°]4s構成で、擬似等方的に32プライ(層)積層し、オートクレーブにて、圧力0.6MPa、室温から2.0℃/分の昇温速度で185℃まで昇温した後、同温度で2時間加熱硬化し、繊維強化複合材料を得た。この繊維強化複合材料について、ASTM D7136及びD7137に従い、縦150mm×横100mmのサンプルを切り出し、サンプルの中心部に6.7J/mmの落錘衝撃を与え、衝撃後圧縮強度(CAI)を求めた。結果を表1に示す。
得られたプリプレグを炭素繊維の方向が同じ方向になるように揃えて26プライ積層し、中央層間(13層目と14層目の間)の一部の領域に、炭素繊維の方向と垂直な積層体側面に予亀裂が導入されるように、カプトンフィルム(1mil)(東レ・デュポン社製)をはさんだ。なお、1milは、1/1000インチで、25.3995μmを示す。これをオートクレーブにて、圧力0.6MPa、室温から2.0℃/分の昇温速度で185℃まで昇温した後、同温度で2時間加熱硬化し、繊維強化複合材料を得た。この繊維強化複合材料について、縦(繊維方向)264.0mm×横25.4mmのサンプルを切り出し、端部にヒンジを接着した試験片を得た。この試験片に対して、ASTM D5528に従い、負荷速度1.0mm/minで、ダブルカンチレバービーム試験を実施し、モードI層間破壊靭性値(G1c)を求めた。結果を表1に示す。
得られたプリプレグを炭素繊維の方向が同じ方向になるように揃えて26プライ積層し、中央層間(13層目と14層目の間)の一部の領域に、炭素繊維の方向と垂直な積層体側面に予亀裂が導入されるように、カプトンフィルム(1mil)(東レ・デュポン社製)をはさんだ。なお、1milは、1/1000インチで、25.3995μmを示す。これをオートクレーブにて、圧力0.6MPa、室温から2.0℃/分の昇温速度で185℃まで昇温した後、同温度で2時間加熱硬化し、繊維強化複合材料を得た。この繊維強化複合材料について、縦(繊維方向)264.0mm×横25.4mmのサンプルを切り出し、試験片を得た。この試験片に対して、Composite Materials Handbook 17-1に従い、負荷速度1.0mm/minで端面切欠き曲げ試験を実施し、モードII層間破壊靭性値(G2c)を求めた。結果を表1に示す。
CAIの測定をする際に得られた繊維強化複合材料中の任意の炭素繊維が伸びる方向に直交する面で繊維強化複合材料を切断したときの切断面を顕微鏡観察(500倍)により分析し、500μm×100μmの範囲について画像解析を行うことでポリアミド粒子の分布を観察することにより、炭素繊維層間の1つの樹脂硬化物に含まれるポリアミド樹脂の含有量C1と、1つの炭素繊維層内に含まれるポリアミド樹脂の含有量C2とを算出した。この測定を、異なる炭素繊維層及び樹脂硬化物の組み合わせとなる任意の5箇所について行い、C1及びC2の5箇所の平均値を用いて、1プリプレグ当たりのC1の容量割合{C1/(C1+C2)}×100を求めた。結果を表1に示す。
更に、CAI、G1c及びG2cについて、昇温速度を0.3℃/分に変更した以外は上記と同様の評価を行った。結果を、2.0℃/分での評価結果と合わせて表2に示す。

Claims (4)
- 強化繊維と、前記強化繊維の繊維間に含浸された、(A)ベンゾオキサジン樹脂、(B)エポキシ樹脂、及び、(C)分子中に2個以上のフェノール性水酸基を有する硬化剤を含有する樹脂組成物と、を含む強化繊維層と、
前記強化繊維層の少なくとも一方の表面上に設けられた、(A)ベンゾオキサジン樹脂、(B)エポキシ樹脂、(C)分子中に2個以上のフェノール性水酸基を有する硬化剤、及び、(D)平均粒子径が5~50μmのポリアミド樹脂粒子を含有する表面層と、
を備え、
前記ポリアミド樹脂粒子は、カプロラクタムとラウロラクタムとを1:9~3:7のモル比で共重合させた共重合体からなるポリアミド樹脂粒子及びカプロラクタムとラウロラクタムとを9:1~7:3のモル比で共重合させた共重合体からなるポリアミド樹脂粒子を含む、プリプレグ。 - 前記表面層は、前記(A)成分と前記(B)成分との合計を100質量部としたときに、前記(A)成分を65~78質量部、前記(B)成分を22~35質量部、前記(C)成分を5~20質量部、及び前記(D)成分を15~45質量部含有する、請求項1に記載のプリプレグ。
- 請求項1又は2に記載のプリプレグを複数積層し、加圧下で加熱して得られる繊維強化複合材料。
- (A)ベンゾオキサジン樹脂と、
(B)エポキシ樹脂と、
(C)分子中に2個以上のフェノール性水酸基を有する硬化剤と、
(D)平均粒子径が5~50μmのポリアミド樹脂粒子と、
を含有し、
前記ポリアミド樹脂粒子は、カプロラクタムとラウロラクタムとを1:9~3:7のモル比で共重合させた共重合体からなるポリアミド樹脂粒子及びカプロラクタムとラウロラクタムとを9:1~7:3のモル比で共重合させた共重合体からなるポリアミド樹脂粒子を含む、粒子含有樹脂組成物。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020167016036A KR20160094984A (ko) | 2013-11-19 | 2014-10-28 | 프리프레그, 섬유 강화 복합 재료 및 입자 함유 수지 조성물 |
| CN201480063133.1A CN105793334A (zh) | 2013-11-19 | 2014-10-28 | 预浸料、纤维强化复合材料及含有颗粒的树脂组合物 |
| EP14864078.2A EP3072919A4 (en) | 2013-11-19 | 2014-10-28 | Prepreg, fibre-reinforced composite material, and particle-containing resin composition |
| US15/037,424 US20160289404A1 (en) | 2013-11-19 | 2014-10-28 | Prepreg, fibre-reinforced composite material, and particle-containing resin composition |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013238896A JP6308756B2 (ja) | 2013-11-19 | 2013-11-19 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
| JP2013-238896 | 2013-11-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015076073A1 true WO2015076073A1 (ja) | 2015-05-28 |
Family
ID=53179342
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/078620 WO2015076073A1 (ja) | 2013-11-19 | 2014-10-28 | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20160289404A1 (ja) |
| EP (1) | EP3072919A4 (ja) |
| JP (1) | JP6308756B2 (ja) |
| KR (1) | KR20160094984A (ja) |
| CN (1) | CN105793334A (ja) |
| WO (1) | WO2015076073A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170021596A1 (en) * | 2015-05-05 | 2017-01-26 | Sunrez Corp. | Fiber Reinforced Core |
| US10442115B2 (en) | 2016-05-25 | 2019-10-15 | Johns Manville | Manufacturing thermoplastic composites and articles |
| EP3604406A4 (en) * | 2017-03-24 | 2020-03-18 | Mitsubishi Chemical Corporation | PREMIX AND FIBER REINFORCED COMPOSITE MATERIAL |
| JP2019099987A (ja) * | 2017-11-29 | 2019-06-24 | 東レ株式会社 | 強化繊維基材、強化繊維積層体および繊維強化樹脂 |
| JP7352850B2 (ja) * | 2018-08-03 | 2023-09-29 | 東レ株式会社 | 繊維強化樹脂 |
| JP7214559B2 (ja) | 2019-04-26 | 2023-01-30 | キヤノン株式会社 | 電子写真感光体、プロセスカートリッジおよび電子写真装置 |
| JP7166995B2 (ja) * | 2019-08-02 | 2022-11-08 | Eneos株式会社 | プリプレグの製造方法及びプリプレグ |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007016121A (ja) | 2005-07-07 | 2007-01-25 | Toray Ind Inc | 複合材料用プリプレグおよび複合材料 |
| JP2010013636A (ja) | 2008-06-03 | 2010-01-21 | Mitsubishi Rayon Co Ltd | 繊維強化複合材料用樹脂組成物およびそれを用いた繊維強化複合材料 |
| JP2010525101A (ja) * | 2007-04-17 | 2010-07-22 | ヘクセル コーポレイション | 熱可塑性粒子のブレンドを含む複合材料 |
| JP2013166855A (ja) * | 2012-02-15 | 2013-08-29 | Jx Nippon Oil & Energy Corp | 繊維強化複合材料 |
| JP2013166854A (ja) * | 2012-02-15 | 2013-08-29 | Jx Nippon Oil & Energy Corp | 繊維強化複合材料 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0694515B2 (ja) * | 1986-12-25 | 1994-11-24 | 東レ株式会社 | プリプレグ |
| GB201008884D0 (en) * | 2010-05-27 | 2010-07-14 | Hexcel Composites Ltd | Improvements in composite materials |
| JP5532549B2 (ja) * | 2008-05-29 | 2014-06-25 | 三菱レイヨン株式会社 | プリプレグおよび繊維強化複合材料の成形方法 |
| CN104254566B (zh) * | 2011-09-30 | 2016-03-23 | 吉坤日矿日石能源株式会社 | 苯并噁嗪树脂组合物和纤维增强复合材料 |
-
2013
- 2013-11-19 JP JP2013238896A patent/JP6308756B2/ja active Active
-
2014
- 2014-10-28 KR KR1020167016036A patent/KR20160094984A/ko not_active Application Discontinuation
- 2014-10-28 US US15/037,424 patent/US20160289404A1/en not_active Abandoned
- 2014-10-28 EP EP14864078.2A patent/EP3072919A4/en not_active Withdrawn
- 2014-10-28 WO PCT/JP2014/078620 patent/WO2015076073A1/ja active Application Filing
- 2014-10-28 CN CN201480063133.1A patent/CN105793334A/zh active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007016121A (ja) | 2005-07-07 | 2007-01-25 | Toray Ind Inc | 複合材料用プリプレグおよび複合材料 |
| JP2010525101A (ja) * | 2007-04-17 | 2010-07-22 | ヘクセル コーポレイション | 熱可塑性粒子のブレンドを含む複合材料 |
| JP2010013636A (ja) | 2008-06-03 | 2010-01-21 | Mitsubishi Rayon Co Ltd | 繊維強化複合材料用樹脂組成物およびそれを用いた繊維強化複合材料 |
| JP2013166855A (ja) * | 2012-02-15 | 2013-08-29 | Jx Nippon Oil & Energy Corp | 繊維強化複合材料 |
| JP2013166854A (ja) * | 2012-02-15 | 2013-08-29 | Jx Nippon Oil & Energy Corp | 繊維強化複合材料 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3072919A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2015098535A (ja) | 2015-05-28 |
| CN105793334A (zh) | 2016-07-20 |
| EP3072919A1 (en) | 2016-09-28 |
| KR20160094984A (ko) | 2016-08-10 |
| EP3072919A4 (en) | 2017-08-02 |
| JP6308756B2 (ja) | 2018-04-11 |
| US20160289404A1 (en) | 2016-10-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6291221B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP6291223B2 (ja) | 繊維強化複合材料の製造方法、プリプレグ、粒子含有樹脂組成物及び繊維強化複合材料 | |
| WO2015076073A1 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP6278951B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP6278950B2 (ja) | 繊維強化複合材料の製造方法 | |
| JP5912920B2 (ja) | 繊維強化複合材料 | |
| JP6308755B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| WO2015076072A1 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP6422857B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP6324373B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 | |
| JP7166995B2 (ja) | プリプレグの製造方法及びプリプレグ | |
| JP6278952B2 (ja) | プリプレグ、繊維強化複合材料及び粒子含有樹脂組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14864078 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15037424 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014864078 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014864078 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167016036 Country of ref document: KR Kind code of ref document: A |