WO2014010681A1 - Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component - Google Patents
Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component Download PDFInfo
- Publication number
- WO2014010681A1 WO2014010681A1 PCT/JP2013/069008 JP2013069008W WO2014010681A1 WO 2014010681 A1 WO2014010681 A1 WO 2014010681A1 JP 2013069008 W JP2013069008 W JP 2013069008W WO 2014010681 A1 WO2014010681 A1 WO 2014010681A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- layer
- current collector
- resin layer
- electrode structure
- Prior art date
Links
- 238000003860 storage Methods 0.000 title claims description 21
- 239000011255 nonaqueous electrolyte Substances 0.000 title claims description 19
- 230000005611 electricity Effects 0.000 title 1
- 229920005989 resin Polymers 0.000 claims abstract description 145
- 239000011347 resin Substances 0.000 claims abstract description 145
- 239000002245 particle Substances 0.000 claims abstract description 41
- 239000000758 substrate Substances 0.000 claims abstract description 28
- 238000000034 method Methods 0.000 claims abstract description 22
- 239000011149 active material Substances 0.000 claims description 38
- -1 acrylic ester Chemical class 0.000 claims description 24
- 229920005672 polyolefin resin Polymers 0.000 claims description 21
- 239000004743 Polypropylene Substances 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 14
- 229920001155 polypropylene Polymers 0.000 claims description 14
- 239000011737 fluorine Substances 0.000 claims description 12
- 229910052731 fluorine Inorganic materials 0.000 claims description 12
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 claims description 11
- 239000007772 electrode material Substances 0.000 claims description 9
- 229920013716 polyethylene resin Polymers 0.000 claims description 7
- 150000001733 carboxylic acid esters Chemical class 0.000 claims description 4
- 229920001225 polyester resin Polymers 0.000 claims description 4
- 239000004645 polyester resin Substances 0.000 claims description 4
- 229920002803 thermoplastic polyurethane Polymers 0.000 claims description 4
- 239000004925 Acrylic resin Substances 0.000 claims description 3
- 229920000178 Acrylic resin Polymers 0.000 claims description 3
- 239000003822 epoxy resin Substances 0.000 claims description 3
- 229920000647 polyepoxide Polymers 0.000 claims description 3
- JHPBZFOKBAGZBL-UHFFFAOYSA-N (3-hydroxy-2,2,4-trimethylpentyl) 2-methylprop-2-enoate Chemical compound CC(C)C(O)C(C)(C)COC(=O)C(C)=C JHPBZFOKBAGZBL-UHFFFAOYSA-N 0.000 claims description 2
- 239000006185 dispersion Substances 0.000 description 24
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 17
- 239000011230 binding agent Substances 0.000 description 13
- 239000011888 foil Substances 0.000 description 12
- 239000000463 material Substances 0.000 description 12
- 239000004020 conductor Substances 0.000 description 11
- 125000004185 ester group Chemical group 0.000 description 11
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 10
- 239000002033 PVDF binder Substances 0.000 description 10
- 229910052782 aluminium Inorganic materials 0.000 description 10
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 10
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 229910052799 carbon Inorganic materials 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 8
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- 239000011248 coating agent Substances 0.000 description 8
- 238000000576 coating method Methods 0.000 description 8
- 229910052744 lithium Inorganic materials 0.000 description 8
- 229910001416 lithium ion Inorganic materials 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- 238000002156 mixing Methods 0.000 description 8
- 239000004698 Polyethylene Substances 0.000 description 7
- 238000000113 differential scanning calorimetry Methods 0.000 description 7
- 208000028659 discharge Diseases 0.000 description 7
- 229920001577 copolymer Polymers 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 229920000573 polyethylene Polymers 0.000 description 6
- 229910052802 copper Inorganic materials 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 239000011889 copper foil Substances 0.000 description 5
- 230000004927 fusion Effects 0.000 description 5
- 230000020169 heat generation Effects 0.000 description 5
- 238000012423 maintenance Methods 0.000 description 5
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000006230 acetylene black Substances 0.000 description 3
- 239000006229 carbon black Substances 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000010439 graphite Substances 0.000 description 3
- 229910002804 graphite Inorganic materials 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 239000002931 mesocarbon microbead Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 238000009783 overcharge test Methods 0.000 description 3
- 229920002493 poly(chlorotrifluoroethylene) Polymers 0.000 description 3
- 239000005023 polychlorotrifluoroethylene (PCTFE) polymer Substances 0.000 description 3
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 3
- 239000004810 polytetrafluoroethylene Substances 0.000 description 3
- 229920001780 ECTFE Polymers 0.000 description 2
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 125000005396 acrylic acid ester group Chemical group 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 2
- 238000007600 charging Methods 0.000 description 2
- 238000005238 degreasing Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 description 2
- 229920000840 ethylene tetrafluoroethylene copolymer Polymers 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000000155 melt Substances 0.000 description 2
- 125000005397 methacrylic acid ester group Chemical group 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229920002620 polyvinyl fluoride Polymers 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 230000000630 rising effect Effects 0.000 description 2
- 239000010731 rolling oil Substances 0.000 description 2
- 238000007788 roughening Methods 0.000 description 2
- 239000000779 smoke Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 230000003746 surface roughness Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- SKYXLDSRLNRAPS-UHFFFAOYSA-N 1,2,4-trifluoro-5-methoxybenzene Chemical compound COC1=CC(F)=C(F)C=C1F SKYXLDSRLNRAPS-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 229910000838 Al alloy Inorganic materials 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229910000881 Cu alloy Inorganic materials 0.000 description 1
- 239000005058 Isophorone diisocyanate Substances 0.000 description 1
- 229910012851 LiCoO 2 Inorganic materials 0.000 description 1
- 229910015643 LiMn 2 O 4 Inorganic materials 0.000 description 1
- 229910014689 LiMnO Inorganic materials 0.000 description 1
- 229910013290 LiNiO 2 Inorganic materials 0.000 description 1
- 229910013870 LiPF 6 Inorganic materials 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229920001807 Urea-formaldehyde Polymers 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 230000004323 axial length Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 1
- 239000003990 capacitor Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004581 coalescence Methods 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000010280 constant potential charging Methods 0.000 description 1
- 238000010277 constant-current charging Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000003851 corona treatment Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- QHGJSLXSVXVKHZ-UHFFFAOYSA-N dilithium;dioxido(dioxo)manganese Chemical compound [Li+].[Li+].[O-][Mn]([O-])(=O)=O QHGJSLXSVXVKHZ-UHFFFAOYSA-N 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 239000008151 electrolyte solution Substances 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000006232 furnace black Substances 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- NIMLQBUJDJZYEJ-UHFFFAOYSA-N isophorone diisocyanate Chemical compound CC1(C)CC(N=C=O)CC(C)(CN=C=O)C1 NIMLQBUJDJZYEJ-UHFFFAOYSA-N 0.000 description 1
- GELKBWJHTRAYNV-UHFFFAOYSA-K lithium iron phosphate Chemical compound [Li+].[Fe+2].[O-]P([O-])([O-])=O GELKBWJHTRAYNV-UHFFFAOYSA-K 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 150000007974 melamines Chemical class 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 239000007773 negative electrode material Substances 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 229920001083 polybutene Polymers 0.000 description 1
- 229920005678 polyethylene based resin Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005673 polypropylene based resin Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000007774 positive electrode material Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000011164 primary particle Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 238000004439 roughness measurement Methods 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- 150000003628 tricarboxylic acids Chemical class 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- 229920001567 vinyl ester resin Polymers 0.000 description 1
Images






Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/66—Current collectors
- H01G11/68—Current collectors characterised by their material
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/64—Carriers or collectors
- H01M4/66—Selection of materials
- H01M4/665—Composites
- H01M4/667—Composites in the form of layers, e.g. coatings
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/26—Electrodes characterised by their structure, e.g. multi-layered, porosity or surface features
- H01G11/28—Electrodes characterised by their structure, e.g. multi-layered, porosity or surface features arranged or disposed on a current collector; Layers or phases between electrodes and current collectors, e.g. adhesives
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/54—Electrolytes
- H01G11/56—Solid electrolytes, e.g. gels; Additives therein
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/64—Carriers or collectors
- H01M4/66—Selection of materials
- H01M4/668—Composites of electroconductive material and synthetic resins
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/021—Physical characteristics, e.g. porosity, surface area
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2200/00—Safety devices for primary or secondary batteries
- H01M2200/10—Temperature sensitive devices
- H01M2200/106—PTC
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/64—Carriers or collectors
- H01M4/66—Selection of materials
- H01M4/661—Metal or alloys, e.g. alloy coatings
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Definitions
- the present invention relates to a current collector, an electrode structure, a nonaqueous electrolyte battery, or a power storage component.
- lithium battery Due to the high energy density, the use of lithium batteries in electronic devices such as mobile phones and laptop computers is expanding.
- lithium cobaltate, lithium manganate, lithium iron phosphate and the like are used as the positive electrode active material, and graphite is used as the negative electrode active material.
- a lithium battery is generally composed of an electrode made of these active materials, a separator that is a porous sheet, and an electrolyte solution in which a lithium salt is dissolved.
- Such a lithium secondary battery has a high battery capacity and output, good charge / discharge characteristics, and a relatively long service life.
- Lithium batteries have the advantage of high energy density, but have a problem with safety because they use non-aqueous electrolyte.
- the non-aqueous electrolyte since the non-aqueous electrolyte is included, the components of the non-aqueous electrolyte are decomposed as the heat is generated, so that the internal pressure increases and the battery may swell.
- problems such as heat generation may occur.
- problems such as heat generation may occur due to the occurrence of an internal short circuit.
- the method in which the PTC element is attached to the positive electrode cap portion has a problem that an increase in the short circuit current cannot be suppressed when an internal short circuit occurs and the temperature rises.
- the separator incorporated in the lithium battery has a function of suppressing an increase in the short-circuit current by closing the pores of the separator by melting the resin at the time of abnormal heat generation and decreasing the ionic conductivity. .
- the separator at a location away from the heat generating portion does not always melt, and there is a possibility that a short circuit may occur due to the separator contracting due to heat.
- the means for preventing heat generation due to internal short circuit still leaves room for improvement.
- Patent Document 1 a CC foil (carbon coat) having a PTC function in which a resin layer to which conductive particles are added using a fluororesin or an olefin resin as a binder is formed on a conductive substrate. Foil) has been proposed.
- Patent Document 2 discloses a PTC layer in which carbon black is added to a polymer (for example, polyethylene).
- Patent Document 3 discloses a resin layer composed of a CC layer (carbon coat layer) having a surface roughness Ra (arithmetic mean roughness) of 0.5 to 1.0 ⁇ m.
- the expression of the PTC function is due to the thermal expansion of the binder resin.
- the capacity of the battery or the power storage component is increased, there is no room for thermal expansion of the PTC layer or the resin layer because the amount of the active material is increased and the density of the active material layer is also increased.
- the present invention has been made in view of the above circumstances, and it is an object of the present invention to provide a current collector having a PTC layer having room for thermal expansion at the time of temperature rise while ensuring sufficient conductivity at a normal temperature.
- surface of the electroconductive base material is provided.
- the resin layer includes an organic resin and conductive particles.
- the amount of the resin layer deposited on the conductive substrate is 0.5 to 20 g / m 2 .
- Rz (ten-point average roughness) of the surface of the resin layer is 0.4 to 10 ⁇ m.
- the Sm (average interval of irregularities) on the surface of the resin layer is 5 to 200 ⁇ m.
- the average resistance of the resin layer measured by the two-terminal method is 0.5 to 50 ⁇ .
- an electrode structure comprising the above-described current collector and an active material layer or an electrode material layer provided on the resin layer of the current collector.
- this electrode structure includes the above-described current collector, sufficient conductivity can be ensured during normal operation, and a sufficient safety function can be achieved during an internal short circuit.
- a nonaqueous electrolyte battery or a power storage component comprising the above current collector and an active material layer or an electrode material layer provided on the resin layer of the current collector. Is done.
- this non-aqueous electrolyte battery or power storage component includes the above-described current collector, sufficient conductivity can be ensured during normal operation, and a sufficient safety function can be achieved during an internal short circuit.
- FIG. 1 is a cross-sectional view showing the structure of the electrode structure according to the embodiment.
- the electrode structure 110 of this embodiment includes a current collector 100.
- the current collector 100 includes a conductive substrate 102 and a resin layer 103 provided on at least one surface of the conductive substrate 102.
- the electrode structure 110 of the present embodiment further includes an active material layer (or electrode material layer) 105 provided on the resin layer 103 of the current collector 100.
- the resin layer 103 has a roughened surface 109 as will be described later.
- FIG. 2 is a cross-sectional view for explaining problems of an electrode structure having a conventional PTC layer.
- the electrode structure 210 having the conventional PTC layer 203 if the conductive paths 211 are too small, there is a problem that the resistance at the normal temperature is too high and the output characteristics of the battery or the power storage component are deteriorated.
- the conductive path 211 is easily cut when the binder resin 207 of the PTC layer 203 is thermally expanded (when the film thickness of the PTC layer 203 is increased). In addition, it provides a sufficient safety function when an internal short circuit occurs. However, there is a problem that the resistance of the PTC layer 203 at room temperature is too high and sufficient conduction cannot be obtained between the conductive base material 202 and the active material layer 205.
- FIG. 3 is a cross-sectional view for explaining problems of an electrode structure having a conventional PTC layer.
- the electrode structure 310 including the conventional PTC layer 303 when the conductive paths 311 are formed densely and the conductivity is increased more than necessary, there are few conductive paths 311 that are cut even when the binder resin 307 expands and melts. There is.
- the PTC function is manifested by thermal expansion of the binder resin.
- the capacity of the battery or the power storage component is increased, there is a problem that there is no room for thermal expansion of the PTC layer because the amount of the active material is increased and the density of the active material layer is increased.
- FIG. 4 is a cross-sectional view for explaining a difference between the electrode structure according to the embodiment and a conventional electrode structure including a PTC layer.
- the electrode structure 110 according to the present embodiment includes the conductive substrate 102 and the resin layer 103 provided on at least one surface of the conductive substrate 102.
- the resin layer 103 has a roughened surface 109 as will be described later.
- the electrode structure 110 of the present embodiment further includes an active material layer (or electrode material layer) 105 provided on the resin layer 103.
- the active material layer 105 includes an active material 121, a conductive material 123, and a binder 125.
- the resin layer 103 includes an organic resin 107 and conductive particles 111.
- the amount of the resin layer 103 attached to the conductive substrate 102 is 0.5 to 20 g / m 2 .
- the Rz (ten-point average roughness) of the roughened surface 109 of the resin layer 103 is 0.4 to 10 ⁇ m.
- the Sm (average interval of unevenness) of the roughened surface 109 of the resin layer 103 is 5 to 200 ⁇ m.
- the average resistance of the resin layer 103 measured by the two-terminal method is 0.5 to 50 ⁇ .
- FIG. 5 is a cross-sectional view illustrating a mechanism in which the resistance of the PTC layer of the electrode structure according to the embodiment rapidly increases.
- this electrode structure 110 when used, when the temperature in the nonaqueous electrolyte battery or the power storage component reaches the vicinity of the melting point of the organic resin 107, the organic resin 107 expands in volume and is dispersed in the resin layer 103. The conductivity is lowered because the contact between the particles 111 is peeled off.
- the electrode structure 110 of this embodiment has the resin layer 103 which has the special roughening surface 109, it is in securing moderate space on the resin layer 103 surface as a room of thermal expansion of the resin layer 103. Has succeeded. Therefore, even if the volume change at the time of melting of the organic resin 107 is large, it can be thermally expanded without any problem, and good PTC characteristics can be obtained. That is, when the internal temperature of the nonaqueous electrolyte battery or the power storage component reaches the vicinity of the melting point of the organic resin 107 due to heat generated when the nonaqueous electrolyte battery or the power storage component is overcharged, the resistance of the resin layer 103 rapidly rises and becomes conductive.
- the electric current between the conductive substrate 102 and the active material layer 105 is interrupted. Therefore, if this electrode structure 110 is used, a sufficient safety function can be exhibited at the time of an internal short circuit of the nonaqueous electrolyte battery or the power storage component.
- the electrode structure 110 that simultaneously exhibits a good balance between the two effects of ensuring sufficient electrical conductivity at normal temperatures and having room for thermal expansion when the temperature rises. These two effects are difficult to achieve at the same time in the conventional electrode structures 210 and 310 described with reference to FIGS.
- the electrode structure 110 of the present embodiment since the electrode structure 110 of the present embodiment has the resin layer 103 having the special roughened surface 109, it has succeeded in satisfying these two conflicting effects at the same time.
- various metal foils for nonaqueous electrolyte batteries or power storage components can be used.
- various metal foils for the positive electrode and the negative electrode can be used.
- aluminum, copper, stainless steel, nickel and the like can be used.
- aluminum and copper are preferable from the balance between high conductivity and cost.
- aluminum means aluminum and an aluminum alloy
- copper means pure copper and a copper alloy.
- the aluminum foil can be used on the secondary battery positive electrode side, the secondary battery negative electrode side or the electric double layer capacitor electrode, and the copper foil can be used on the secondary battery negative electrode side.
- A1085 material which is a pure aluminum type, and A3003 material can be used.
- A1085 material which is a pure aluminum type can be used.
- copper foil is the same also as copper foil, although it does not specifically limit, Rolled copper foil and electrolytic copper foil are used preferably.
- the thickness of the conductive substrate 102 is not particularly limited, but is preferably 5 ⁇ m or more and 50 ⁇ m or less. If the thickness is less than 5 ⁇ m, the strength of the foil may be insufficient and it may be difficult to form the conductive substrate 102 or the like. On the other hand, if it exceeds 50 ⁇ m, the other constituent elements, particularly the active material layer 105 or the electrode material layer, must be thinned. In particular, when the power storage component such as a nonaqueous electrolyte battery or a power storage component is used, the active material layer In some cases, the thickness of 105 must be reduced, and a necessary and sufficient capacity cannot be obtained.
- the resin layer 103 of the present embodiment is a PTC (Positive temperature coefficient) layer including an organic resin 107 and conductive particles 111 laminated on the surface of the conductive substrate 102.
- PTC Positive temperature coefficient
- the adhesion amount of the resin layer 103 of the present embodiment on the conductive substrate 102 is 0.5 to 20 g / m 2 . If it is less than 0.5 g / m ⁇ 2 >, an uncoated part will generate
- the adhesion amount of the resin layer 103 may be, for example, 0.5, 1, 2, 5 , 10, 15, 20 g / m 2 , and is within a range between any two of the numerical values exemplified here. May be.
- the Rz (ten-point average roughness) of the roughened surface 109 of the resin layer 103 of the present embodiment is 0.4 to 10 ⁇ m. If Rz is less than 0.4 ⁇ m, there is insufficient room for thermal expansion. When Rz exceeds 10 ⁇ m, the contact with the active material layer 105 is poor, and the contact battery performance or the contact storage performance is insufficient.
- Rz of the resin layer 103 may be 0.4, 0.7, 1, 2, 5, 10 ⁇ m, for example, or may be within a range between any two of the numerical values exemplified here.
- Sm (average interval of irregularities) of the roughened surface 109 of the resin layer 103 of the present embodiment is preferably 5 to 200 ⁇ m. If Sm is less than 5 ⁇ m, there is insufficient room for thermal expansion. If Sm exceeds 200 ⁇ m, the surface shape is too flat and there is insufficient room for thermal expansion.
- the Sm of the resin layer 103 may be, for example, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180, 200 ⁇ m, and is exemplified here. It may be within a range between any two of the numerical values.
- the average resistance measured by the two-terminal method of the resin layer 103 of this embodiment is 0.5 to 50 ⁇ . If the average resistance is within this range, it is possible to avoid the problem that the conductive path is cut off due to the expansion of the organic resin 107 due to the temperature rise in the temperature range lower than the temperature to be shut down, and the conductive path can be connected at the last minute. .
- the initial resistance of the resin layer 103 is a resistance value measured by Mitsubishi Analitech's resistivity meter Loresta EP (two-terminal method) (measured from above the resin layer 103 formed on the conductive base material 102) of 0.5 to 50 ⁇ . If it is. If it is less than 0.5 ⁇ , the resistance rise at the time of temperature rise is insufficient. When it exceeds 50 ⁇ , the performance as a battery or a storage battery is insufficient.
- the resin used as the organic resin 107 of the resin layer 103 is not particularly limited, but is selected from the group consisting of a fluorine resin, an olefin resin, an epoxy resin, an acrylic resin, a polyester resin, and a urethane resin. More than seeds can be used. Among these, it is particularly preferable to use a fluorine resin or an olefin resin.
- the fluorine-based resin used as the organic resin 107 of the resin layer 103 is a resin containing a fluorine resin as a resin component, and may be composed of only a fluorine resin. It may contain a resin.
- the fluororesin is a resin containing fluorine, such as polyvinylidene fluoride (PVDF), polytetrafluoroethylene (PTFE), tetrafluoroethylene-perfluoroalkyl vinyl ether copolymer (PFA), tetrafluoroethylene-hexafluoropropylene copolymer.
- PVDF polyvinylidene fluoride
- PTFE polytetrafluoroethylene
- PFA tetrafluoroethylene-perfluoroalkyl vinyl ether copolymer
- Fluororesins such as coalescence (FEP), polychlorotrifluoroethylene (PCTFE), tetrafluoroethylene-ethylene copolymer (ETFE), chlorotrifluoroethylene-ethylene copolymer (ECTFE), and polyvinyl fluoride (PVF);
- FEP coalescence
- PCTFE polychlorotrifluoroethylene
- ETFE tetrafluoroethylene-ethylene copolymer
- ECTFE chlorotrifluoroethylene-ethylene copolymer
- PVF polyvinyl fluoride
- PVDF polyvinylidene fluoride
- the fluororesin can be used in an amount of 100% by mass when the entire resin component is 100% by mass, but can also be used in combination with other resin components. It is preferable to contain 40 mass% or more normally with respect to all the resin components, Preferably it contains 50 mass% or more. This is because if the blending amount of the fluororesin is too small, control of the conductive particles described later will not be successful, and it will be difficult to reliably combine a shutdown function and excellent high rate characteristics.
- the ratio of the fluororesin is, for example, 40, 50, 60, 70, 80, 90, 100% by mass, and may be within the range of any two numerical values exemplified here.
- the weight average molecular weight of the fluororesin is, for example, 30,000 to 1,000,000, specifically, for example, 30,000, 40,000, 50,000, 60,000, 80,000, 90,000, 100,000, 150,000. , 200,000, 300,000, 400,000, 500,000, 700,000, 800,000, 900,000, 1 million, and may be in the range between any two of the numerical values exemplified here.
- a weight average molecular weight means what was measured by GPC (gel permeation chromatograph).
- the fluororesin preferably has a carboxyl group or a carboxylic ester group (hereinafter simply referred to as “ester group”). This is because the adhesion between the conductive substrate 102 and the resin layer 103 can be improved. Further, when the fluororesin has an ester group, the adhesion between the fluororesin and conductive particles (eg, carbon particles) is improved.
- the fluororesin has a carboxyl group (—COOH) or an ester group (—COOR, R is, for example, a hydrocarbon having 1 to 5 carbon atoms) is not particularly limited.
- the fluororesin is a carboxyl group or an ester. It may be a copolymer of a monomer having a group and a monomer containing fluorine, and the fluororesin may be a mixture of a fluororesin and a resin having a carboxyl group or an ester group. The resin may be modified with a compound having a carboxyl group or an ester group.
- the method for modifying the fluororesin is not particularly limited, but in one example, as disclosed in JP-A-2002-304997, radiation is emitted to the fluororesin to desorb fluorine atoms to generate radicals. And the method of graft-polymerizing the compound which has a carboxyl group or an ester group on a fluororesin by mixing a fluororesin and the compound which has a carboxyl group or an ester group in that state is mentioned.
- the value of the ratio of the number of carboxyl groups or ester groups to the number of fluorine atoms in the fluorine-based resin is not particularly limited, but is, for example, 0.1 to 5, and preferably 0.5 to 2.
- this ratio is specifically, for example, 0.1, 0.2, 0.5, 1, 1.5, 2, 3, 4, 5, and between any two of the numerical values exemplified here. It may be within the range.
- the monomer (or compound) having a carboxyl group or an ester group include acrylic acid, methacrylic acid, and esters thereof (eg, methyl methacrylate).
- the olefin resin used as the organic resin 107 of the resin layer 103 is a resin containing an olefin resin as a resin component, and may be composed of only an olefin resin. It may contain a resin.
- the olefin resin include polyethylene, polypropylene, polybutene, polystyrene and the like. These can be used singly or in combination of two or more, but polyethylene and polypropylene are particularly preferable in that they can surely have both a shutdown function and excellent high rate characteristics.
- this olefinic resin can be used in an amount of 100% by mass, but can also be used in combination with other resin components. It is preferable to contain 40 mass% or more normally with respect to all the resin components, Preferably it contains 50 mass% or more. This is because if the blending amount of the olefin resin is too small, control of conductive particles described later will not be successful, and it will be difficult to reliably combine a shutdown function and excellent high rate characteristics.
- the ratio of the olefin resin is, for example, 40, 50, 60, 70, 80, 90, 100% by mass, and may be within the range of any two numerical values exemplified here.
- the weight average molecular weight of the olefin resin is, for example, 10,000 to 1,000,000, specifically, for example, 10,000, 30,000, 40,000, 50,000, 60,000, 80,000, 90,000, 100,000. 150,000, 200,000, 300,000, 400,000, 500,000, 600,000, 700,000, 800,000, 900,000, 1 million, and within the range between any two of the numerical values exemplified here Also good.
- a weight average molecular weight means what was measured by GPC (gel permeation chromatograph).
- This olefin resin is preferably modified with carboxylic acid or the like to have a carboxyl group or a carboxylic ester group (hereinafter simply referred to as “ester group”). This is because the adhesion between the conductive substrate 102 and the resin layer 103 can be improved. Moreover, when an olefin resin has an ester group, the adhesiveness of an olefin resin and electroconductive particle (example: carbon particle) improves.
- the carboxylic acid for modifying the olefinic resin may be any of monocarboxylic acid, dicarboxylic acid, and tricarboxylic acid.
- acetic acid, acetic anhydride, maleic acid, maleic anhydride, trimellitic acid, trimellitic anhydride, Fumaric acid, adipic acid, glutaric acid, succinic acid, malonic acid, oxalic acid and the like can be used. This is because the more carboxyl group, the better the adhesion to the conductive substrate, but the dispersibility of the conductive particles may be lowered and the resistance may be increased.
- this olefin resin into a copolymer with acrylic acid ester and methacrylic acid ester.
- the acrylic ester and methacrylic ester preferably further have a glycidyl group.
- the olefin resin is preferably a mixture of a polypropylene resin and a polyethylene resin.
- the mixing ratio of the polypropylene resin and the polyethylene resin is defined by a peak appearing in DSC (differential scanning calorimetry). This is because simply adding a predetermined amount of polypropylene-based resin and polyethylene-based resin may change the crystal state and the like, and a desired result may not be obtained.
- DSC differential scanning calorimetry
- a coating film made of a mixed system of a polypropylene resin and a polyethylene resin and a conductive material is thermally analyzed by DSC and melted from the melting peak area of each resin (the area surrounded by the thermal melting peak and straight line of each resin).
- the amount of heat is determined, and the ratio of addition is defined by the ratio of the heat of fusion of each resin to the heat of fusion of both.
- the addition ratio of the polypropylene resin is (the heat of fusion of the polypropylene resin) / (the heat of fusion of the polypropylene resin + the heat of fusion of the polyethylene resin) ⁇ 100 (%), and this ratio is 95% to 30%. % Is preferred.
- Conductive Particles As the conductive particles 111 used in the resin layer 103 of the present embodiment, known conductive fine particles such as carbon powder and metal powder can be used, among which furnace black, acetylene black, and ketjen. Carbon black such as black is preferred. In particular, it is preferable that the electrical resistance of the powder is 100% green compact and 1 ⁇ 10 ⁇ 1 ⁇ or less, and the above can be used in combination as necessary.
- the particle size is not particularly limited, but the primary particle size is preferably about 10 to 100 nm.
- the blending amount of the conductive particles 111 is not particularly limited, but is preferably blended so that the volume% value occupied by the conductive particles 111 is 5 to 50% when the entire resin layer 103 is 100%. . If the blending amount of the conductive particles 111 is too small, the number of contact points between the conductive particles 111 is small, and the electrical resistance at normal temperature is increased. When there are too many compounding quantities of the electroconductive particle 111, the contact between the electroconductive particles 111 will be maintained also at the time of temperature rising, and it will become difficult to exhibit a shutdown function. This value is, for example, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50%, and may be within a range between any two of the numerical values exemplified here.
- the surface shape (surface roughness) of the roughened surface 109 of the resin layer 103 of this embodiment is obtained by dispersing the conductive particles 111 by a predetermined dispersion method, and further by predetermined baking conditions. It is obtained by coating with.
- the dispersion state of the conductive particles 111 of the present embodiment is realized by the following dispersion method, for example.
- the dispersion of the conductive material has been carried out mainly with the aim of finely and evenly dispersing, but in this embodiment, a suitably agglomerated dispersion state is preferable, and the method will be described below.
- Dispersers, planetary mixers, ball mills, and the like can be used as the dispersers.
- a disperser is used will be described.
- the dispersion state of the conductive particles 111 of the present embodiment can be achieved, for example, by pre-dispersing the conductive particles 111 in a solution of a fluorine-based resin or an olefin-based resin, and further performing this dispersion.
- a conductive material is added to the resin liquid so that the concentration of the conductive material in the solid content (resin solid content + conductive material) is 10 to 70% by volume, and 2 to 60 at a rotational speed of 300 to 5000 rpm.
- the amount of conductive particles in the pre-dispersion is less than 10% by volume, the particle size after the main dispersion may become too small, and if it exceeds 70% by volume, the particle size after the main dispersion may become too large. If the rotation speed of the preliminary dispersion is lower than 300 rpm, the particle diameter after the main dispersion may become too large, and if it exceeds 5000 rpm, the particle diameter after the main dispersion may become too small. If the pre-dispersion stirring time is shorter than 2 minutes, the particle size after the main dispersion may become too large, and if longer than 60 minutes, the particle size after the main dispersion may become too small.
- the resin solution is added to the pre-dispersion paste so that the conductive material addition amount in the resin layer is 5 to 50% by volume (the solid content in the resin liquid (resin solid content + conductive material)). So that the concentration of the conductive material is 5 to 50% by volume).
- stirring is performed at a rotational speed of 500 to 8000 rpm for 10 to 120 minutes. If the rotational speed of this dispersion is lower than 500 rpm, the particle size may be too large, and if it is higher than 8000 rpm, the particle size may be too small. If the stirring time of this dispersion is shorter than 10 minutes, the particle size may be too large, and if longer than 120 minutes, the particle size may be too small.
- the method for manufacturing the current collector 100 of the present embodiment is not particularly limited as long as the resin layer 103 is formed on the conductive base material 102 by a known method. It is also effective to perform a pretreatment so that the adhesiveness of the conductive substrate 102 is improved.
- rolling oil or wear powder may remain, and adhesion with the resin layer may deteriorate.
- rolling oil or wear powder By removing degreasing by degreasing or the like, adhesion with the resin layer 103 can be improved.
- the adhesion with the resin layer 103 can be improved by a dry activation treatment such as a corona discharge treatment.
- the electrode structure 110 of the present embodiment includes an active material layer 105 including an active material, which is laminated on the resin layer 103. Since the electrode structure 110 includes the active material layer 105 containing the active material particles 121 on the current collector 100, good discharge rate characteristics can be obtained.
- the electrode structure 110 of this embodiment can be obtained by forming an active material layer or an electrode material layer on at least one surface of the current collector 100 of this embodiment.
- the electrode structure for an electrical storage component in which the electrode material layer is formed will be described later.
- an electrode structure for a nonaqueous electrolyte battery for example, a lithium ion secondary battery (for example, a lithium ion secondary battery) using the electrode structure 110 and a separator, a nonaqueous electrolyte solution, or the like ( Battery components).
- a member other than the current collector 100 can be a known non-aqueous battery member.
- the active material layer 105 formed as the electrode structure 110 in the present embodiment may be conventionally proposed for a non-aqueous electrolyte battery.
- the current collector 100 of the present embodiment using aluminum as the positive electrode, LiCoO 2 , LiMnO 2 , LiNiO 2 or the like as the active material 121, carbon black such as acetylene black as the conductive particles 123, etc.
- the positive electrode structure of this embodiment can be obtained by applying and drying a paste dispersed in PVDF or water-dispersed PTFE as the binder 125.
- the negative electrode structure 110 for example, graphite, graphite, mesocarbon microbeads or the like are used as the active material 121 for the current collector 110 of the present embodiment using copper as the conductive substrate 102, and these are increased.
- the negative electrode structure of the present embodiment can be obtained by coating and drying a paste mixed with SBR as a binder after being dispersed in CMC as a sticking agent as a material for forming the active material layer 105.
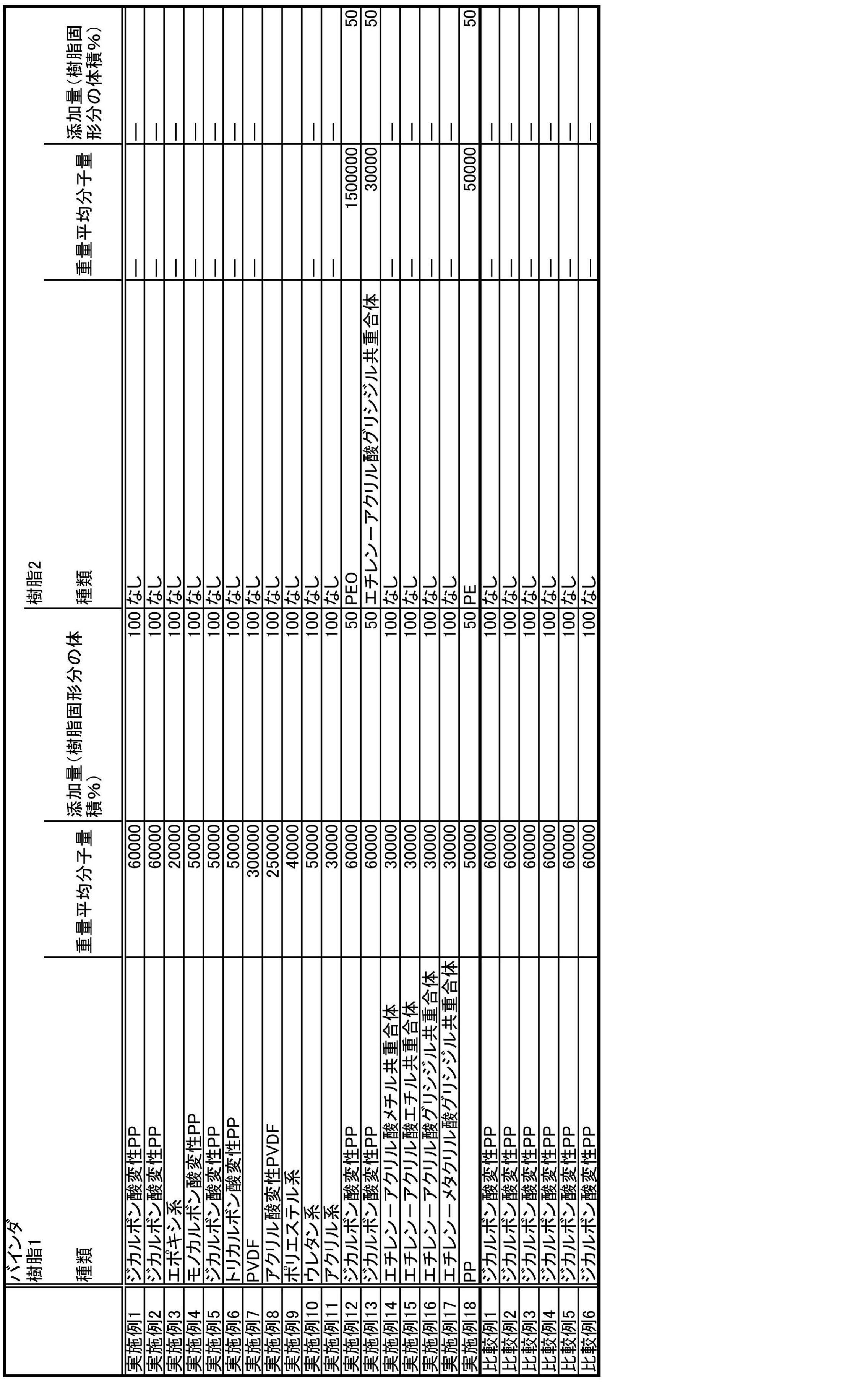
- ⁇ Formation of resin layer on conductive substrate> Based on Table 1, coating amounts using various resins were prepared.
- As the olefin resin an aqueous emulsion having a solid content of 5 to 40% by weight was used.
- the mixture of polypropylene and polyethylene in Example 18 was mixed so that the addition ratio of polypropylene was 50% based on the DSC chart of the coating film.
- the PVDF resin used was an N-methyl-2-pyrrolidone solution having a solid content of 10 to 15% by weight.
- As the epoxy resin a bisphenol A type resin and a urea resin dissolved in methyl ethyl ketone at a solid content of 20% by weight were used.
- As the polyester resin a polyester resin produced by an esterification reaction of isophthalic acid and ethylene glycol and dissolved in xylene at a solid content of 20% by weight was used.
- the urethane resin a urethane resin obtained by reacting isophorone diisocyanate and polyethylene glycol and butylated melamine dissolved in xylene at a solid content of 20% by weight was used.
- an aqueous emulsion having a solid content of 20% by weight of a copolymer of butyl acrylate, acrylonitrile, and acrylamide was used as the acrylic resin. All molecular weights were measured by GPC (gel permeation chromatography) in the state of a resin solution.
- the paste obtained by pre-dispersion and main dispersion based on Table 2 was applied to an aluminum foil and baked at a baking temperature (base material arrival temperature) of 100 ° C. and a baking time (in-furnace time) of 120 seconds.
- a CC layer carbon coat layer
- ⁇ Evaluation method> (1) Amount of coating The coated foil was cut into 100 mm squares, and the mass was measured. After removing the coating film, the mass was measured again, and the adhesion amount was calculated from the difference. Table 3 shows the measurement results.
- (2) Roughness measurement Measured with a rough clock (SE-30D manufactured by Kosaka Laboratory) (reference length 2.5 mm, cut-off ⁇ c 0.8 mm, drive speed 0.1 mm / s). Based on JIS B0601, Rz (ten-point average roughness) and Sm (average interval of unevenness) were measured. Table 3 shows the measurement results (average of n 5).
- the capacity maintenance ratio measurement (high rate characteristic) was 0.80 or more, and the result of the overcharge test was good with no change in the battery.
- the capacity maintenance rate measurement (high rate characteristic) is less than 0.80 or the overcharge test result is smoke (a state where the shutdown function is insufficient and smoke is emitted from the battery), which is not suitable for practical use. Therefore, in Examples 1 to 18, since the CC layer (carbon coat layer) having a special roughened surface is provided, the CC layer (carbon coat layer) is used as a room for thermal expansion of the CC layer (carbon coat layer). It has succeeded in securing an appropriate space on the surface. Therefore, even if the volume change at the time of melting
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Composite Materials (AREA)
- Power Engineering (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Manufacturing & Machinery (AREA)
- Cell Electrode Carriers And Collectors (AREA)
- Battery Electrode And Active Subsutance (AREA)
Abstract
Description
図1は、実施形態に係る電極構造体の構造を示した断面図である。本実施形態の電極構造体110は、集電体100を備えている。この集電体100は、導電性基材102と、その導電性基材102の少なくとも片面に設けられている樹脂層103と、を備える。また、本実施形態の電極構造体110は、この集電体100の樹脂層103上に設けられている活物質層(または電極材層)105をさらに備える。なお、この樹脂層103は、後述するように粗面化表面109を有している。 <Overall configuration of electrode>
FIG. 1 is a cross-sectional view showing the structure of the electrode structure according to the embodiment. The
図2は、従来のPTC層を備える電極構造体の問題点を説明するための断面図である。従来のPTC層203を備える電極構造体210では、導電パス211を少なくしすぎると常温での抵抗が高すぎて電池または蓄電部品の出力特性が低下する問題がある。 <Problems of conventional PTC layer>
FIG. 2 is a cross-sectional view for explaining problems of an electrode structure having a conventional PTC layer. In the
図4は、実施形態に係る電極構造体が従来のPTC層を備える電極構造体と異なる点を説明するための断面図である。すでに説明したように、本実施形態の電極構造体110は、導電性基材102と、その導電性基材102の少なくとも片面に設けられている樹脂層103と、を備える。なお、この樹脂層103は、後述するように粗面化表面109を有している。また、本実施形態の電極構造体110は、この樹脂層103上に設けられている活物質層(または電極材層)105をさらに備える。また、この活物質層105は、活物質121と、導電材123と、バインダ125を含む。 <Roughened surface of resin layer>
FIG. 4 is a cross-sectional view for explaining a difference between the electrode structure according to the embodiment and a conventional electrode structure including a PTC layer. As already described, the
本実施形態の導電性基材102としては、非水電解質電池または蓄電部品用の各種金属箔が使用可能である。具体的には、正極用、負極用の種々の金属箔を使用することができ、例えば、アルミニウム、銅、ステンレス、ニッケルなどが使用可能である。その中でも導電性の高さとコストのバランスからアルミニウム、銅が好ましい。なお、本明細書において、アルミニウムは、アルミニウム及びアルミニウム合金を意味し、銅は純銅および銅合金を意味する。本実施形態において、アルミニウム箔は二次電池正極側、二次電池負極側または電気二重層キャパシタ電極、銅箔は二次電池負極側に用いることができる。アルミニウム箔としては、特に限定されないが、純アルミ系であるA1085材や、A3003材など種々のものが使用できる。また、銅箔としても同様であり、特に限定されないが、圧延銅箔や電解銅箔が好んで用いられる。 <Conductive substrate>
As the
本実施形態の樹脂層103は、導電性基材102の表面上に積層されてなる有機樹脂107と導電性粒子111とを含むPTC(Positive temperature coefficient)層である。 <Resin layer>
The
本実施形態の樹脂層103の導電性基材102上の付着量は、0.5~20g/m2である。0.5g/m2未満では未塗工部が発生し、電池性能または蓄電性能が不足する。20g/m2を超えると、抵抗が大きすぎて電池性能または蓄電性能が不足する。樹脂層103の付着量は、例えば、0.5、1、2、5、10、15、20g/m2であってもよく、ここで例示した数値の何れか2つの間の範囲内であってもよい。 (1) Structure of roughened surface The adhesion amount of the
この樹脂層103の有機樹脂107として用いられるフッ素系樹脂は、樹脂成分としてフッ素樹脂を含む樹脂であり、フッ素樹脂のみからなるものであってもよく、フッ素樹脂と別の樹脂とを含有するものであってもよい。フッ素樹脂は、フッ素を含む樹脂であり、ポリフッ化ビニリデン(PVDF)、ポリテトラフルオロエチレン(PTFE)、テトラフルオロエチレン-パーフルオロアルキルビニルエーテル共重合体(PFA)、テトラフルオロエチレン-ヘキサフルオロプロピレン共重合体(FEP)、ポリクロロトリフルオロエチレン(PCTFE)、テトラフルオロエチレン-エチレン共重合体(ETFE)、クロロトリフルオロエチレン-エチレン共重合体(ECTFE)、ポリフッ化ビニル(PVF)等のフッ素樹脂及びその誘導体、PCTFE、テトラフルオロエチレンなどのフルオロオレフインにシクロヘキシルビニルエーテルやカルボン酸ビニルエステルを共重合したフッ素共重合体等が例示される。また、これらは1種単独でも2種以上を組み合わせても用いることができるが、特にポリフッ化ビニリデン(PVDF)がシャットダウン機能と優れたハイレート特性を確実に兼ね備えることができる点で好ましい。 (2) Fluorine-based resin The fluorine-based resin used as the
この樹脂層103の有機樹脂107として用いられるオレフィン系樹脂は、樹脂成分としてオレフィン樹脂を含む樹脂であり、オレフィン樹脂のみからなるものであってもよく、オレフィン樹脂と別の樹脂とを含有するものであってもよい。オレフィン系樹脂としては、ポリエチレン、ポリプロピレン、ポリブテン、ポリスチレン等が例示される。また、これらは1種単独でも2種以上を組み合わせても用いることができるが、特にポリエチレン、ポリプロピレンがシャットダウン機能と優れたハイレート特性を確実に兼ね備えることができる点で好ましい。 (3) Olefin resin The olefin resin used as the
本実施形態の樹脂層103に用いる導電性粒子111は、公知の炭素粉末、金属粉末などの導電性微粒子が使用可能であるが、その中でもファーネスブラック,アセチレンブラック,ケッチェンブラック等のカーボンブラックが好ましい。特に粉体での電気抵抗が、100%の圧粉体で1×10-1Ω以下のものが好ましく、必要に応じて上記のものを組み合わせて使用できる。その粒子サイズに特に制限はないが、一次粒子径としては概ね10~100nmが好ましい。 (4) Conductive Particles As the
本実施形態の樹脂層103の粗面化表面109の表面形状(表面粗度)は所定の分散方法によって導電性粒子111を分散し、さらに所定の焼付条件にて塗工することにより得られる。 (5) Method for Producing Roughened Surface The surface shape (surface roughness) of the roughened
本実施形態の電極構造体110は、樹脂層103の上に積層されている、活物質を含む活物質層105を備える。この電極構造体110は、上記の集電体100上に活物質粒子121を含有する活物質層105を備えているため、良好な放電レート特性が得られる。 <Active material layer>
The
表1に基づいて各種樹脂を用いた塗量を調合した。オレフィン系樹脂はそれぞれの固形分5~40重量%の水系エマルションを用いた。実施例18のポリプロピレンとポリエチレンの混合物は塗膜のDSCチャートを元にした、ポリプロピレンの添加割合が50%になるように混合した。 <Formation of resin layer on conductive substrate>
Based on Table 1, coating amounts using various resins were prepared. As the olefin resin, an aqueous emulsion having a solid content of 5 to 40% by weight was used. The mixture of polypropylene and polyethylene in Example 18 was mixed so that the addition ratio of polypropylene was 50% based on the DSC chart of the coating film.
AB:アセチレンブラック
PP:ポリプロピレン
PE:ポリエチレン
PEO:ポリエチレンオキサイド
PVDF:ポリフッ化ビニリデン In Tables 1 to 3 below, the meaning of each abbreviation is as follows.
AB: Acetylene black PP: Polypropylene PE: Polyethylene PEO: Polyethylene oxide PVDF: Polyvinylidene fluoride

(1)付着量
塗工箔を100mm角に切断し、質量を測定した。塗膜を除去後、再度質量を測定し、その差から付着量を算出した。測定結果を表3に示す。
(2)粗度測定
粗時計(小坂研究所製SE-30D)にて測定した(基準長さ2.5mm、カットオフλc0.8mm、ドライブスピード0.1mm/s)。JIS B0601に準拠して、Rz(十点平均粗さ)、Sm(凹凸の平均間隔)を測定した。(n=5の平均)測定結果を表3に示す。 <Evaluation method>
(1) Amount of coating The coated foil was cut into 100 mm squares, and the mass was measured. After removing the coating film, the mass was measured again, and the adhesion amount was calculated from the difference. Table 3 shows the measurement results.
(2) Roughness measurement Measured with a rough clock (SE-30D manufactured by Kosaka Laboratory) (reference length 2.5 mm, cut-off λc 0.8 mm, drive speed 0.1 mm / s). Based on JIS B0601, Rz (ten-point average roughness) and Sm (average interval of unevenness) were measured. Table 3 shows the measurement results (average of n = 5).
初期抵抗を三菱化学アナリテック製ロレスタEP(二端子法)による抵抗値として、アルミ箔に形成したCC層(カーボンコート層)の上から測定した。(n=10の平均)測定結果を表3に示す。 (3) Resistance The initial resistance was measured from the top of the CC layer (carbon coat layer) formed on the aluminum foil as a resistance value by Loresta EP (two-terminal method) manufactured by Mitsubishi Chemical Analytech. Table 3 shows the measurement results (average of n = 10).
(4-1)電池の作製
(4-1-1)正極の作製
上記の方法にて作製した樹脂層を有する集電体に活物質ペースト(LiMn2O4/AB/PVDF=89.5/5/5.5、溶媒NMP(N-メチル-2-ピロリドン))を塗布し、乾燥した。さらにプレスをかけて、厚さ60μmの活物質層を形成した。 (4) Capacity maintenance rate (4-1) Battery production (4-1-1) Production of positive electrode An active material paste (LiMn 2 O 4 / AB / PVDF = 89.5 / 5 / 5.5, solvent NMP (N-methyl-2-pyrrolidone)) was applied and dried. Further, pressing was performed to form an active material layer having a thickness of 60 μm.
厚さ10μmの銅箔に活物質ペースト(MCMB(メソカーボンマイクロビーズ)/AB/PVDF=93/2/5、溶剤NMP)を塗布し、乾燥した。さらにプレスをかけて、厚さ40μmの活物質層を形成した。 (4-1-2) Production of Negative Electrode An active material paste (MCMB (mesocarbon microbeads) / AB / PVDF = 93/2/5, solvent NMP) was applied to a 10 μm thick copper foil and dried. Further, pressing was performed to form an active material layer having a thickness of 40 μm.
この正極、負極、電解液(1M LiPF6、EC(エチレンカーボネート)/MEC(メチルエチルカーボネート)=3/7)、セパレータ(厚さ25μm、微孔ポリエチレンフィルム)を捲回して、各極にリードを溶接して各極端子に接続し、ケースに挿入して円筒型リチウムイオン電池(φ18mm×軸方向長さ65mm)を得た。 (4-1-3) Production of Cylindrical Lithium Ion Battery This positive electrode, negative electrode, electrolyte (1M LiPF 6 , EC (ethylene carbonate) / MEC (methyl ethyl carbonate) = 3/7), separator (thickness 25 μm, A microporous polyethylene film) was wound, a lead was welded to each electrode, connected to each electrode terminal, and inserted into a case to obtain a cylindrical lithium ion battery (φ18 mm × axial length 65 mm).
この円筒型リチウムイオン電池を用い、0.25mA/cm2にて4.2Vまで定電流定電圧充電後、0.25mA/cm2と5mA/cm2にて定電流放電を行い、それぞれの放電容量から放電維持率=(5mA/cm2の放電容量)/(0.25mA/cm2の放電容量)を算出した。容量維持率が0.8以上あれば、ハイレートでの使用も可能である。測定結果を表3に示す。 (4-2) Capacity maintenance rate measurement (high rate characteristics)
Using this cylindrical lithium ion battery, after constant current and constant voltage charging to 4.2V at 0.25 mA / cm 2, a constant current discharge at 0.25 mA / cm 2 and 5 mA / cm 2, each of the discharge From the capacity, discharge retention ratio = (discharge capacity of 5 mA / cm 2 ) / (discharge capacity of 0.25 mA / cm 2 ) was calculated. If the capacity maintenance rate is 0.8 or more, it can be used at a high rate. Table 3 shows the measurement results.
上記の円筒型リチウムイオン電池を用い、4.2Vまで充電電圧1.5mA/cm2で定電流定電圧充電後、満充電状態の円筒型リチウムイオン電池にさらに250%充電になるまで5Aで充電し、円筒型リチウムイオン電池の挙動を調査した。測定結果を表3に示す。 (4-3) Overcharge test Using the above-described cylindrical lithium ion battery, after charging with constant current and constant voltage at a charging voltage of 1.5 mA / cm 2 up to 4.2 V, the battery was further charged with a fully charged cylindrical lithium ion battery. The battery was charged with 5A until it reached% charge, and the behavior of the cylindrical lithium ion battery was investigated. Table 3 shows the measurement results.

実施例1~18では、容量維持率測定(ハイレート特性)は0.80以上および過充電試験の結果は電池に変化なく良好であり、いずれも実用に耐えるのに対し、比較例1~6では、容量維持率測定(ハイレート特性)が0.80未満または過充電試験結果が発煙(シャットダウン機能が不十分で電池から発煙した状態)となっており、いずれも実用に適さない。このことから、実施例1~18では、特殊な粗面化表面を有するCC層(カーボンコート層)を有するため、CC層(カーボンコート層)の熱膨張の余地としてCC層(カーボンコート層)表面に適度な空間を確保することに成功している。そのため、各種有機樹脂の融解時の体積変化が大きくても問題なく熱膨張でき、良好なPTC特性を得ることができることがわかる。 <Consideration of results>
In Examples 1 to 18, the capacity maintenance ratio measurement (high rate characteristic) was 0.80 or more, and the result of the overcharge test was good with no change in the battery. The capacity maintenance rate measurement (high rate characteristic) is less than 0.80 or the overcharge test result is smoke (a state where the shutdown function is insufficient and smoke is emitted from the battery), which is not suitable for practical use. Therefore, in Examples 1 to 18, since the CC layer (carbon coat layer) having a special roughened surface is provided, the CC layer (carbon coat layer) is used as a room for thermal expansion of the CC layer (carbon coat layer). It has succeeded in securing an appropriate space on the surface. Therefore, even if the volume change at the time of melting | dissolving of various organic resin is large, it can be understood that it can expand | swell without a problem and can obtain a favorable PTC characteristic.
102 導電性基材
103 樹脂層
105 活物質層
107 有機樹脂
109 粗面化表面
110 電極構造体
111 導電性粒子
121 活物質
123 導電材
125 バインダ
202 導電性基材
203 PTC層
205 活物質層
207 バインダ樹脂
210 電極構造体
211 導電パス
302 導電性基材
303 PTC層
305 活物質層
307 バインダ樹脂
310 電極構造体
311 導電パス DESCRIPTION OF
Claims (8)
- 導電性基材と、
前記導電性基材の少なくとも片面に設けられている樹脂層と、
を備える集電体であって、
前記樹脂層が、有機樹脂と導電性粒子とを含み、
前記樹脂層の前記導電性基材上の付着量が0.5~20g/m2であり、
前記樹脂層の表面のRz(十点平均粗さ)が0.4~10μmであり、
前記樹脂層の表面のSm(凹凸の平均間隔)が5~200μmであり、
前記樹脂層の二端子法で測定した抵抗の平均が0.5~50Ωである、
集電体。 A conductive substrate;
A resin layer provided on at least one side of the conductive substrate;
A current collector comprising:
The resin layer includes an organic resin and conductive particles,
The amount of adhesion of the resin layer on the conductive substrate is 0.5 to 20 g / m 2 ;
Rz (ten-point average roughness) of the surface of the resin layer is 0.4 to 10 μm,
Sm (average interval of irregularities) on the surface of the resin layer is 5 to 200 μm,
The average resistance measured by the two-terminal method of the resin layer is 0.5 to 50Ω,
Current collector. - 前記有機樹脂が、フッ素系樹脂、オレフィン系樹脂、エポキシ系樹脂、アクリル系樹脂、ポリエステル系樹脂、ウレタン系樹脂からなる群から選ばれる1種以上である、
請求項1に記載の集電体。 The organic resin is at least one selected from the group consisting of a fluorine resin, an olefin resin, an epoxy resin, an acrylic resin, a polyester resin, and a urethane resin.
The current collector according to claim 1. - 前記フッ素系樹脂が、カルボキシル基またはカルボン酸エステルを有する、
請求項2に記載の集電体。 The fluororesin has a carboxyl group or a carboxylic acid ester;
The current collector according to claim 2. - 前記オレフィン系樹脂が、カルボキシル基またはカルボン酸エステルを有する、
請求項2に記載の集電体。 The olefin resin has a carboxyl group or a carboxylic ester.
The current collector according to claim 2. - 前記オレフィン系樹脂が、アクリル酸エステルまたはメタクリル酸エステルを少なくとも1種含む、
請求項2に記載の集電体。 The olefin resin contains at least one acrylic ester or methacrylic ester,
The current collector according to claim 2. - 前記オレフィン系樹脂が、ポリプロピレン系樹脂およびポリエチレン系樹脂を含む、
請求項2に記載の集電体。 The olefin resin includes a polypropylene resin and a polyethylene resin,
The current collector according to claim 2. - 請求項1~6のいずれかに記載の集電体と、
前記集電体の前記樹脂層上に設けられている活物質層または電極材層と、
を備える、電極構造体。 A current collector according to any one of claims 1 to 6;
An active material layer or an electrode material layer provided on the resin layer of the current collector;
An electrode structure comprising: - 請求項1~7のいずれかに記載の集電体と、
前記集電体の前記樹脂層上に設けられている活物質層または電極材層と、
を備える、非水電解質電池または蓄電部品。 A current collector according to any one of claims 1 to 7;
An active material layer or an electrode material layer provided on the resin layer of the current collector;
A non-aqueous electrolyte battery or a power storage component.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380036476.4A CN104428929B (en) | 2012-07-13 | 2013-07-11 | Collector, electrode assembly, nonaqueous electrolyte battery or electric power storage parts |
| KR1020157004063A KR20150032335A (en) | 2012-07-13 | 2013-07-11 | Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component |
| EP13816526.1A EP2874213B1 (en) | 2012-07-13 | 2013-07-11 | Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component |
| JP2014524872A JPWO2014010681A1 (en) | 2012-07-13 | 2013-07-11 | Current collector, electrode structure, non-aqueous electrolyte battery or power storage component |
| US14/414,422 US20150294802A1 (en) | 2012-07-13 | 2013-07-11 | Current collector, electrode structure and non-aqueous electrolyte battery or electrical storage device |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-157672 | 2012-07-13 | ||
| JP2012157672 | 2012-07-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014010681A1 true WO2014010681A1 (en) | 2014-01-16 |
Family
ID=49916122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/069008 WO2014010681A1 (en) | 2012-07-13 | 2013-07-11 | Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20150294802A1 (en) |
| EP (1) | EP2874213B1 (en) |
| JP (1) | JPWO2014010681A1 (en) |
| KR (1) | KR20150032335A (en) |
| CN (1) | CN104428929B (en) |
| TW (1) | TW201414069A (en) |
| WO (1) | WO2014010681A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014077384A1 (en) * | 2012-11-19 | 2014-05-22 | 古河電気工業株式会社 | Collector, electrode, secondary cell, and capacitor |
| CN104409681A (en) * | 2014-11-19 | 2015-03-11 | 上海航天电源技术有限责任公司 | Preparation method of lithium ion battery pole piece containing PTC coating |
| KR101730219B1 (en) * | 2014-04-04 | 2017-05-11 | 주식회사 엘지화학 | Current collector for lithium secondary battery and electorde comprising the same |
| JP2017084507A (en) * | 2015-10-23 | 2017-05-18 | 日産自動車株式会社 | Electrode and method of manufacturing the same |
| JP2017224562A (en) * | 2016-06-17 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition, backing layer-attached current collector for power storage device, electrode for power storage device and power storage device |
| JP2017224469A (en) * | 2016-06-15 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition for forming backing layer of electrode for nonaqueous electrolyte secondary battery, and use thereof |
| JP2017224407A (en) * | 2016-06-13 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition, backing layer-attached current collector for nonaqueous electrolyte secondary battery, electrode for nonaqueous electrolyte secondary battery, and nonaqueous electrolyte secondary battery |
| CN110783521A (en) * | 2018-07-27 | 2020-02-11 | 丰田自动车株式会社 | Electrode for solid-state battery and solid-state battery |
| JP2021086782A (en) * | 2019-11-29 | 2021-06-03 | グンゼ株式会社 | Resin current collector |
| JPWO2022208682A1 (en) * | 2021-03-30 | 2022-10-06 | ||
| JP2022168728A (en) * | 2021-04-26 | 2022-11-08 | プライムプラネットエナジー&ソリューションズ株式会社 | Electrode collector and secondary battery |
| US11532823B2 (en) * | 2016-06-14 | 2022-12-20 | Solvay Sa | Flexible battery |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102050250B1 (en) * | 2015-09-09 | 2019-12-17 | 주식회사 엘지화학 | Electrode for Secondary Battery with Improved Production Processability |
| JP6666223B2 (en) | 2016-09-21 | 2020-03-13 | 株式会社東芝 | Negative electrode, non-aqueous electrolyte battery, battery pack, and vehicle |
| WO2018083917A1 (en) * | 2016-11-04 | 2018-05-11 | 日産自動車株式会社 | Electrode for cell, and cell |
| CN113053562B (en) * | 2017-01-27 | 2023-03-31 | 昭和电工材料株式会社 | Insulating coated conductive particle, anisotropic conductive film and method for producing same, connection structure and method for producing same |
| JP7074284B2 (en) * | 2017-02-10 | 2022-05-24 | 三井化学株式会社 | Positive electrode and non-aqueous electrolyte secondary battery |
| JP6747577B2 (en) * | 2017-03-14 | 2020-08-26 | 株式会社村田製作所 | Lithium ion secondary battery |
| JP6724861B2 (en) * | 2017-05-26 | 2020-07-15 | トヨタ自動車株式会社 | Electrode current collector and all-solid-state battery |
| JP7149520B2 (en) * | 2017-10-31 | 2022-10-07 | パナソニックIpマネジメント株式会社 | Batteries and battery stacks |
| CN109755463B (en) * | 2017-11-08 | 2020-12-29 | 宁德时代新能源科技股份有限公司 | Electrode pole piece, electrochemical device and safety coating |
| US11469412B2 (en) * | 2018-05-02 | 2022-10-11 | Lg Energy Solution, Ltd. | Anode for lithium metal battery, manufacturing method of the same, lithium metal battery including the same |
| KR102080653B1 (en) | 2018-05-23 | 2020-02-24 | 삼성전기주식회사 | Coil component |
| GB202106834D0 (en) * | 2021-05-13 | 2021-06-30 | Dupont Teijin Films Us Lp | Metallised films |
| US20230112382A1 (en) * | 2021-09-30 | 2023-04-13 | Medtronic, Inc. | Resistive current collector coating |
| EP4379839A1 (en) * | 2022-06-09 | 2024-06-05 | Contemporary Amperex Technology Co., Limited | Positive electrode plate, secondary battery, battery module, battery pack and electric apparatus |
| CN115832188B (en) * | 2022-07-22 | 2024-09-06 | 宁德时代新能源科技股份有限公司 | Pole piece, battery monomer, battery, electricity utilization device and pole piece manufacturing method |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62209803A (en) * | 1986-03-10 | 1987-09-16 | 日本メクトロン株式会社 | Circuit device |
| JPH10199574A (en) * | 1996-12-28 | 1998-07-31 | Japan Storage Battery Co Ltd | Nonaqueous electrolyte battery |
| JPH10241665A (en) | 1996-12-26 | 1998-09-11 | Mitsubishi Electric Corp | Electrode and battery using the same |
| JP2001357854A (en) | 2000-06-13 | 2001-12-26 | Matsushita Electric Ind Co Ltd | Nonaqueous secondary battery |
| JP2002304997A (en) | 2001-02-01 | 2002-10-18 | Mitsubishi Materials Corp | Lithium ion polymer secondary battery and synthesizing method of binder used for adhesion layer thereof |
| JP2005174653A (en) * | 2003-12-09 | 2005-06-30 | Sanyo Electric Co Ltd | Lithium secondary battery and manufacturing method thereof |
| JP2005317468A (en) * | 2004-04-30 | 2005-11-10 | Nissan Motor Co Ltd | Bipolar electrode, method of manufacturing bipolar electrode, bipolar battery, battery pack and vehicle with these mounted thereon |
| JP2006032395A (en) * | 2004-07-12 | 2006-02-02 | Shin Etsu Polymer Co Ltd | Ptc element and its manufacturing method |
| JP2009176599A (en) * | 2008-01-25 | 2009-08-06 | Panasonic Corp | Nonaqueous electrolyte secondary battery |
| JP2010212167A (en) | 2009-03-12 | 2010-09-24 | Toyota Motor Corp | Current collecting foil, battery, vehicle, battery using equipment, and method of manufacturing current collecting foil |
| JP2011048932A (en) * | 2009-08-25 | 2011-03-10 | Nissan Motor Co Ltd | Bipolar secondary battery |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3973003B2 (en) * | 1998-04-13 | 2007-09-05 | Tdk株式会社 | Sheet-type electrochemical element |
| US6159635A (en) * | 1998-09-29 | 2000-12-12 | Electrofuel Inc. | Composite electrode including current collector |
| WO2002084764A1 (en) * | 2001-04-10 | 2002-10-24 | Mitsubishi Materials Corporation | Lithium ion polymer secondary battery, its electrode and method for synthesizing polymer compound in binder used in adhesion layer thereof |
| JP5219339B2 (en) * | 2006-02-28 | 2013-06-26 | 三洋電機株式会社 | Lithium secondary battery |
| CN102648546A (en) * | 2009-09-25 | 2012-08-22 | 大金工业株式会社 | Positive electrode current collector laminate for lithium secondary battery |
| EP2835851A4 (en) * | 2012-04-04 | 2016-03-09 | Uacj Corp | Collector, electrode structure, nonaqueous electrolyte battery, and electricity storage component |
-
2013
- 2013-07-11 US US14/414,422 patent/US20150294802A1/en not_active Abandoned
- 2013-07-11 KR KR1020157004063A patent/KR20150032335A/en active IP Right Grant
- 2013-07-11 EP EP13816526.1A patent/EP2874213B1/en not_active Not-in-force
- 2013-07-11 CN CN201380036476.4A patent/CN104428929B/en not_active Expired - Fee Related
- 2013-07-11 WO PCT/JP2013/069008 patent/WO2014010681A1/en active Application Filing
- 2013-07-11 JP JP2014524872A patent/JPWO2014010681A1/en active Pending
- 2013-07-12 TW TW102125127A patent/TW201414069A/en unknown
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62209803A (en) * | 1986-03-10 | 1987-09-16 | 日本メクトロン株式会社 | Circuit device |
| JPH10241665A (en) | 1996-12-26 | 1998-09-11 | Mitsubishi Electric Corp | Electrode and battery using the same |
| JPH10199574A (en) * | 1996-12-28 | 1998-07-31 | Japan Storage Battery Co Ltd | Nonaqueous electrolyte battery |
| JP2001357854A (en) | 2000-06-13 | 2001-12-26 | Matsushita Electric Ind Co Ltd | Nonaqueous secondary battery |
| JP2002304997A (en) | 2001-02-01 | 2002-10-18 | Mitsubishi Materials Corp | Lithium ion polymer secondary battery and synthesizing method of binder used for adhesion layer thereof |
| JP2005174653A (en) * | 2003-12-09 | 2005-06-30 | Sanyo Electric Co Ltd | Lithium secondary battery and manufacturing method thereof |
| JP2005317468A (en) * | 2004-04-30 | 2005-11-10 | Nissan Motor Co Ltd | Bipolar electrode, method of manufacturing bipolar electrode, bipolar battery, battery pack and vehicle with these mounted thereon |
| JP2006032395A (en) * | 2004-07-12 | 2006-02-02 | Shin Etsu Polymer Co Ltd | Ptc element and its manufacturing method |
| JP2009176599A (en) * | 2008-01-25 | 2009-08-06 | Panasonic Corp | Nonaqueous electrolyte secondary battery |
| JP2010212167A (en) | 2009-03-12 | 2010-09-24 | Toyota Motor Corp | Current collecting foil, battery, vehicle, battery using equipment, and method of manufacturing current collecting foil |
| JP2011048932A (en) * | 2009-08-25 | 2011-03-10 | Nissan Motor Co Ltd | Bipolar secondary battery |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2874213A4 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014077384A1 (en) * | 2012-11-19 | 2014-05-22 | 古河電気工業株式会社 | Collector, electrode, secondary cell, and capacitor |
| KR101730219B1 (en) * | 2014-04-04 | 2017-05-11 | 주식회사 엘지화학 | Current collector for lithium secondary battery and electorde comprising the same |
| CN104409681A (en) * | 2014-11-19 | 2015-03-11 | 上海航天电源技术有限责任公司 | Preparation method of lithium ion battery pole piece containing PTC coating |
| JP2017084507A (en) * | 2015-10-23 | 2017-05-18 | 日産自動車株式会社 | Electrode and method of manufacturing the same |
| JP2017224407A (en) * | 2016-06-13 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition, backing layer-attached current collector for nonaqueous electrolyte secondary battery, electrode for nonaqueous electrolyte secondary battery, and nonaqueous electrolyte secondary battery |
| US11532823B2 (en) * | 2016-06-14 | 2022-12-20 | Solvay Sa | Flexible battery |
| JP2017224469A (en) * | 2016-06-15 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition for forming backing layer of electrode for nonaqueous electrolyte secondary battery, and use thereof |
| JP2017224562A (en) * | 2016-06-17 | 2017-12-21 | 東洋インキScホールディングス株式会社 | Conductive composition, backing layer-attached current collector for power storage device, electrode for power storage device and power storage device |
| CN110783521A (en) * | 2018-07-27 | 2020-02-11 | 丰田自动车株式会社 | Electrode for solid-state battery and solid-state battery |
| JP2021086782A (en) * | 2019-11-29 | 2021-06-03 | グンゼ株式会社 | Resin current collector |
| JP2021165393A (en) * | 2019-11-29 | 2021-10-14 | グンゼ株式会社 | Resin current collector |
| WO2021106300A1 (en) * | 2019-11-29 | 2021-06-03 | グンゼ株式会社 | Resin collector |
| JP7405796B2 (en) | 2019-11-29 | 2023-12-26 | グンゼ株式会社 | resin current collector |
| JPWO2022208682A1 (en) * | 2021-03-30 | 2022-10-06 | ||
| WO2022208682A1 (en) * | 2021-03-30 | 2022-10-06 | Tdk株式会社 | Power storage device electrode and lithium ion secondary battery |
| JP7392828B2 (en) | 2021-03-30 | 2023-12-06 | Tdk株式会社 | Electrodes for power storage devices and lithium ion secondary batteries |
| JP2022168728A (en) * | 2021-04-26 | 2022-11-08 | プライムプラネットエナジー&ソリューションズ株式会社 | Electrode collector and secondary battery |
| JP7304380B2 (en) | 2021-04-26 | 2023-07-06 | プライムプラネットエナジー&ソリューションズ株式会社 | Electrode current collector and secondary battery |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2874213A1 (en) | 2015-05-20 |
| JPWO2014010681A1 (en) | 2016-06-23 |
| CN104428929B (en) | 2017-10-24 |
| TW201414069A (en) | 2014-04-01 |
| EP2874213B1 (en) | 2017-02-22 |
| CN104428929A (en) | 2015-03-18 |
| EP2874213A4 (en) | 2015-08-05 |
| KR20150032335A (en) | 2015-03-25 |
| US20150294802A1 (en) | 2015-10-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014010681A1 (en) | Collector, electrode structure, nonaqueous electrolyte cell, and electricity storage component | |
| JP6220784B2 (en) | Current collector, electrode, secondary battery and capacitor | |
| US20150280241A1 (en) | Collector, electrode structure, nonaqueous electrolyte battery, conductive filler, and electrical storage device | |
| WO2014077384A1 (en) | Collector, electrode, secondary cell, and capacitor | |
| WO2013151046A1 (en) | Collector, electrode structure, nonaqueous electrolyte battery, and electricity storage component | |
| TWI578603B (en) | A current collector, an electrode structure, and a power storage unit | |
| WO2014157405A1 (en) | Collector, electrode structure, battery and capacitor | |
| JP2015204221A (en) | Current collector, electrode structure, and electricity storage component | |
| JP2006182925A (en) | Core/shell type fine particle and lithium secondary battery including the fine particle | |
| WO2018164094A1 (en) | Collector for electricity storage devices, method for producing same, and coating liquid used in production of same | |
| JP5985161B2 (en) | Current collector, electrode structure, non-aqueous electrolyte battery, and power storage component | |
| CN114447341A (en) | Collector for electricity storage device, method for producing same, and coating liquid used for production thereof | |
| JP5780871B2 (en) | Current collector, electrode structure, non-aqueous electrolyte battery, and power storage component | |
| JP6130018B2 (en) | Current collector, electrode structure, non-aqueous electrolyte battery, and power storage component | |
| JP2020198275A (en) | Current collector for all-solid battery, and all-solid battery |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13816526 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014524872 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14414422 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013816526 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013816526 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20157004063 Country of ref document: KR Kind code of ref document: A |