WO2012057139A1 - トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 - Google Patents
トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 Download PDFInfo
- Publication number
- WO2012057139A1 WO2012057139A1 PCT/JP2011/074551 JP2011074551W WO2012057139A1 WO 2012057139 A1 WO2012057139 A1 WO 2012057139A1 JP 2011074551 W JP2011074551 W JP 2011074551W WO 2012057139 A1 WO2012057139 A1 WO 2012057139A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- iridium complex
- general formula
- formula
- producing
- chemical formula
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System compounds of the platinum group
- C07F15/0033—Iridium compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/342—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising iridium
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1007—Non-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1011—Condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1044—Heterocyclic compounds characterised by ligands containing two nitrogen atoms as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1092—Heterocyclic compounds characterised by ligands containing sulfur as the only heteroatom
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2101/00—Properties of the organic materials covered by group H10K85/00
- H10K2101/10—Triplet emission
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
Definitions
- the present invention is a method for producing a trisorthometalated iridium complex that is suitably used as a material for an organic electroluminescent element, and in particular, a facial-trisortometalated iridium complex having high luminous efficiency and excellent thermal stability.
- the present invention relates to a method for producing a trisorthometalated iridium complex capable of increasing the production ratio.
- Patent Document 1 discloses a method of synthesizing a chlorine-bridged iridium dimer from iridium trichloride n-hydrate as shown in the reaction formula (Chemical Formula 1), and producing a trisorthometalated iridium complex using this as a raw material. ing.
- Patent Document 2 as shown in the reaction formula (Chemical Formula 2), a chlorine-bridged iridium dimer is synthesized from iridium trichloride n-hydrate, and an iridium complex coordinated with acetylacetone is synthesized from the chlorine-bridged iridium dimer.
- a method for producing a trisorthometalated iridium complex from a starting material is disclosed.
- Patent Document 3 As shown in the reaction formula (Chemical Formula 3), a chlorine-bridged iridium dimer was synthesized from iridium trichloride n-hydrate, and an iridium complex coordinated with acetonitrile was synthesized from the chlorine-bridged iridium dimer. Discloses a method for producing a trisorthometalated iridium complex from a starting material.
- Patent Documents 1 to 3 are methods in which an orthometalated iridium complex such as a chlorine-bridged iridium dimer and a bidentate organic ligand such as 2-phenylpyridine are mixed and then reacted by heating.
- an orthometalated iridium complex such as a chlorine-bridged iridium dimer and a bidentate organic ligand such as 2-phenylpyridine are mixed and then reacted by heating.
- Patent Document 4 glycerin as a reaction solvent is heated at 130 ° C. to 140 ° C. for 2 hours and then allowed to cool to 100 ° C. to cool 1-phenylisoquinoline and Ir (acac) 3 (acac: acetylacetone).
- a method for producing a trisorthometalated iridium complex by putting it into glycerin and allowing it to react by heating at around 210 ° C. for 7 hours is disclosed.
- Patent Document 5 a mixture of a bidentate organic ligand and glycerin is heated at 150 ° C. for about 60 minutes and allowed to cool, then Ir (acac) 3 is added, and the temperature is raised to 200 ° C. to tris ortho.
- a method for producing a metalated iridium complex is disclosed.
- Patent Documents 1 to 3 are methods in which an orthometalated iridium complex such as a chlorine-bridged iridium dimer and a bidentate organic ligand such as 2-phenylpyridine are mixed and then heated.
- a meridional isomer which is a geometric isomer, is generated as a by-product, so that it is not easy to obtain a facial isomer with high purity.
- the manufacturing method described in Patent Document 4 is a method in which glycerin as a reaction solvent is heated and allowed to cool in advance, and then the iridium raw material and the bidentate organic ligand are added to the cooled glycerin and heated. It is.
- Patent Document 5 is a method in which glycerin containing a bidentate organic ligand is heated and allowed to cool in advance, and then an iridium raw material is added to the cooled glycerin to cause a heating reaction.
- the production methods described in Patent Documents 4 and 5 are similar to the present invention, but the heating of glycerin, which is a reaction solvent performed in advance, is intended to remove water contained in glycerin, and is not a preliminary method of the present invention. The purpose of heating itself is different.
- the iridium raw material which can be used is limited to Ir (acac) 3 , and it cannot be said that it is a highly general manufacturing method, The reaction is completely different from the manufacturing method which concerns on this invention.
- the tris orthometalated iridium complex has two types of geometric isomers, a facial body and a meridional body, but it has been clarified that the facial body is superior in terms of luminous efficiency and stability ( For example, see Non-Patent Document 1.)
- it is difficult to easily separate facial isomers and meridional isomers and in order to obtain a facial isomer, it is necessary to combine and repeat laborious operations such as recrystallization, column chromatography, or sublimation purification. It was necessary to perform processing.
- the production methods described in Patent Documents 1 to 3 described above cannot completely solve the problem that a meridional isomer that is a geometric isomer is generated as a by-product. Therefore, in a method for producing a trisorthometalated iridium complex, a method for selectively producing a facial body while suppressing the formation of a meridional body that is not very preferable as an organic electroluminescent element material is desired.
- An object of the present invention is to provide a conventional tri-ortho-metalated iridium complex, particularly a facial-tris-ortho-metalated iridium complex, which is preferably used as an organic electroluminescent device material, by reacting by heating after mixing a reaction substrate. Compared with a manufacturing method, it is providing the new manufacturing method for obtaining highly selectively.
- a second object of the present invention is to use a trisorthometalated iridium complex containing a high ratio of facial-trisorthometalated iridium complex, and to use the luminescent material having excellent luminous efficiency and durability, and the luminescent material. Providing a light emitting device.
- the present inventors have intensively studied a method for producing a trisorthometalated iridium complex.
- the production ratio of the facial isomer and the meridional isomer in the above-mentioned known production method surprisingly depends greatly on the addition method of the orthometalated iridium complex, which is an iridium raw material, and the bidentate organic ligand. I found it.
- a conventional production method Patent Document is obtained by preheating a reaction solvent, and post-adding two reaction substrates of an iridium raw material and a bidentate organic ligand to the preheated reaction solvent.
- the tris-orthometalated iridium complex in the facial form has been shown to have better luminous efficiency and stability than the geometric isomer, the meridional form, the tris-ortho-metalated iridium complex produced by the present invention It has been found that a highly efficient and highly durable light-emitting element can be produced by using the complex as a light-emitting material, and the present invention has been completed.
- the method for producing a trisorthometalated iridium complex comprises an orthometalated iridium complex represented by the general formula (Chemical Formula 4) and a bidentate organic ligand represented by the general formula (Chemical Formula 5) as a reaction substrate.
- a trisorthometalated iridium complex represented by the general formula (Chemical Formula 6) is produced, wherein the reaction solvent is preheated (1), and the preheated reaction solvent is ortho-reacted. It has a step (2) of adding a metalated iridium complex and a bidentate organic ligand, and a step (3) of reacting the orthometalated iridium complex and a bidentate organic ligand in order. To do.
- L a is .Z 1 and Z 2 .A 1 .m 2 .m 1 is representative of the one or two which represents a counter anion represents 0 or 1 representing the ligand each independently represents a non-metallic atomic group necessary for forming a 5- or 6-membered ring.
- formed by ring further optionally .L 1 also form a condensed ring with another ring is single Y 1 represents a nitrogen atom or a carbon atom, and when Y 1 is a nitrogen atom, Q 1 represents that the carbon atom and Y 1 are bonded by a single bond.
- the method for producing a trisorthometalated iridium complex according to the present invention includes a form in which the orthometalated iridium complex is a compound represented by the general formula (Formula 7).
- the orthometalated iridium complex is a compound represented by the general formula (Formula 7).
- X represents a halogen atom.
- Z 1 , Z 2 , Y 1 , Q 1 and L 1 are the same as those in the general formula (Chemical Formula 4).
- the L a is encompasses form a mono-anionic ligand.
- the L a is encompasses form a neutral ligand.
- the method for producing a trisorthometalated iridium complex according to the present invention includes a form in which the orthometalated iridium complex is a compound represented by the general formula (Chemical Formula 8).
- R 1 to R 3 represent a hydrogen atom, a deuterium atom or a substituent.
- Z 1 , Z 2 , Y 1 , Q 1 and L 1 are each represented by the general formula (Chemical Formula 4). It is synonymous with the case.)
- the method for producing a trisorthometalated iridium complex according to the present invention includes a form in which the orthometalated iridium complex is a compound represented by the general formula (Chemical Formula 9).
- the orthometalated iridium complex is a compound represented by the general formula (Chemical Formula 9).
- a 1 , Z 1 , Z 2 , Y 1 , Q 1 and L 1 are each synonymous with the case of the general formula (Chemical Formula 4).
- the bidentate organic ligand is at least one selected from (7) to (17) represented by the general formula (Formula 10). Include. (In the formulas (7) to (17) shown in the general formula (Chemical Formula 10), R 4 to R 102 represent a hydrogen atom, a deuterium atom or a substituent.)
- the reaction temperature in the step (3) is preferably in the range of 100 to 300 ° C.
- the generation ratio of the facial body can be further increased.
- the preheating temperature in the step (1) is not higher than the reaction temperature in the step (3) and is in the range of 100 to 300 ° C. preferable.
- the generation ratio of the facial body can be further increased.
- the step (3) is preferably performed under microwave irradiation. Since it can heat up in a short time, the production
- the luminescent material according to the present invention comprises a trisorthometalated iridium complex manufactured by the manufacturing method according to the present invention.
- the light emitting device according to the present invention uses the light emitting material according to the present invention.
- the present invention relates to a conventional production method in which a facial-tris orthometalated iridium complex, which is preferably used as an organic electroluminescent element material, among tris orthometalated iridium complexes, is heated and reacted after mixing a reaction substrate.
- a new manufacturing method for obtaining with high selectivity can be provided.
- the present invention provides a light emitting material excellent in luminous efficiency and durability using a trisorthometalated iridium complex containing a high ratio of facial-trisorthometalated iridium complex, and a light emitting device using the light emitting material. Can be provided.
- Z 1 and Z 2 each independently represents a nonmetallic atom group necessary for forming a 5-membered ring or a 6-membered ring.
- the ring formed may have a substituent, and may form a condensed ring with another ring.
- substituent include a halogen atom, an alkyl group, a substituted alkyl group, a phenoxy group, a substituted phenoxy group, an aryl group, a substituted aryl group, an alkoxy group, a substituted alkoxy group, a dialkylamino group, and a substituted dialkylamino group.
- a halogen atom an alkyl group having 1 to 30 carbon atoms, a substituted alkyl group having 1 to 30 carbon atoms, a phenoxy group having 6 to 30 carbon atoms, a substituted phenoxy group having 6 to 30 carbon atoms, and 6 to 30 carbon atoms.
- a halogen atom an alkyl group having 1 to 10 carbon atoms, a substituted alkyl group having 1 to 10 carbon atoms, an aryl group having 6 to 10 carbon atoms, a substituted aryl group having 6 to 10 carbon atoms, or 1 to 10 carbon atoms.
- a substituted alkoxy group having 1 to 10 carbon atoms is particularly preferred.
- the 5-membered or 6-membered ring formed by Z 1 is preferably an aromatic ring or a heteroaromatic ring, and more preferably an aromatic ring.
- the 5-membered ring or 6-membered ring formed by Z 1 is, for example, quinoline ring, benzoquinoline ring, quinoxaline ring, isoquinoline ring, phenanthridine ring, phenazine ring, acridine ring, triazole ring, imidazophenanthridine ring, phthalazine Ring, quinazoline ring, naphthyridine ring, cinnoline ring, perimidine ring, phenanthroline ring, benzimidazole ring, benzoxazole ring, benzthiazole ring, imidazole ring, thiazole ring, oxazole ring, pyrrole ring, oxadiazole ring, thiadiazole ring,
- a pyrrole ring, a pyridine ring, a naphthalene ring, a fluorene ring, a dibenzothiophene ring, a dibenzofuran ring, a carbazole ring, or a benzene ring is preferable.
- a naphthalene ring, a fluorene ring, a dibenzothiophene ring, a dibenzofuran ring, a carbazole ring or a benzene ring is more preferable, and a benzene ring is particularly preferable.
- the 5-membered ring or 6-membered ring formed by Z 2 is preferably a heteroaromatic ring.
- the 5-membered or 6-membered ring formed by Z 2 is, for example, an imidazole ring, thiazole ring, oxazole ring, pyrrole ring, oxadiazole ring, thiadiazole ring, pyrazole ring, 1,2,3-triazole ring, 1, Examples include 2,4-triazole ring, selenazole ring, pyridine ring, pyrimidine ring, pyrazine ring or pyridazine ring.
- imidazole ring, thiazole ring, 1,2,3-triazole ring, 1,2,4-triazole ring, oxazole ring, pyrrole ring, pyrazole ring, pyridine ring or pyrimidine ring are preferable, and pyrazole ring or pyridine ring Is more preferable.
- L 1 represents a single bond or a divalent group.
- the divalent group include —C (R) (R ′) —, —N (R) —, —O—, —P (R) —, and —S—.
- R and R ′ represent a hydrogen atom or a substituent.
- the substituent include a halogen atom, an aliphatic group, an aromatic group, a heterocyclic group, a cyano group, and a nitro group.
- L 1 is preferably a single bond or —C (R) (R ′) —, and R and R ′ are a hydrogen atom, an aliphatic group or an aromatic group.
- L 1 is particularly preferably a single bond.
- Y 1 represents a nitrogen atom or a carbon atom.
- Q 1 indicates that the bond between the carbon atom and Y 1 is a single bond.
- Q 1 indicates a double bond between the carbon atom and Y 1 .
- Q 1 represents a bond between atoms.
- Y 1 is a nitrogen atom
- Q 1 represents that the carbon atom and Y 1 are bonded by a single bond.
- Y 1 is a carbon atom
- Q 1 represents that the carbon atom and Y 1 are bonded by a double bond.
- L a is as long as the ligand does not matter that the monodentate ligand or bidentate ligand.
- La may contain a metal (for example, iridium), and may form a so-called binuclear complex.
- L a is an anionic ligand or a neutral ligand is preferable.
- a monoanionic ligand or a neutral ligand is particularly preferred.
- Examples of the monodentate anionic ligand include a halogen ligand, a hydroxy ligand, an alkoxide ligand, a phenoxide ligand, a thiocyanate ligand, a cyanate ligand, and an isocyanate ligand.
- Examples of the bidentate anionic ligand include a ⁇ -diketonate ligand, an acetylacetonate ligand, an acetic acid ligand, and a picolinic acid ligand.
- Monodentate neutral ligands include nitrile ligands (eg acetonitrile ligands, propionitrile ligands), sulfoxide ligands (eg dimethyl sulfoxide ligands), amide ligands (eg , Dimethylformamide ligand), ether ligand (eg, tetrahydrofuran ligand), water ligand, ammonia ligand, amine ligand, piperidine ligand, pyridine ligand, pyrazine ligand It is.
- the bidentate neutral ligand include a bipyridine ligand, a phenanthroline ligand, a dipyridylamine ligand, and an ethylenediamine ligand.
- m 1 represents 1 or 2.
- L a is a monodentate ligand
- L a is a bidentate ligand
- m 2 represents 0 or 1.
- a 1 represents a counter anion.
- the counter anion is not particularly limited as long as it is anionic, but a counter anion is preferable.
- Counter anions include, for example, F ⁇ , Cl ⁇ , Br ⁇ , I ⁇ , BF 4 ⁇ , PF 6 ⁇ , ClO 4 ⁇ , CF 3 CF 2 CF 2 COO ⁇ , CF 3 SO 3 ⁇ , CF 3 CO 2 ⁇ . , CH 3 CO 2 ⁇ , SCN ⁇ , CH 3 SO 3 ⁇ , ClO 4 ⁇ , SbF 6 — .
- Cl ⁇ , PF 6 ⁇ , BF 4 ⁇ , PF 6 ⁇ , and CF 3 SO 3 — are preferable.
- X represents a halogen atom.
- they are a chlorine atom or a bromine atom, More preferably, it is a chlorine atom.
- R 1 to R 102 represent a hydrogen atom, a deuterium atom or a substituent.
- substituents include an alkyl group (preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, and particularly preferably 1 to 10 carbon atoms.
- phenyloxy, 1-naphthyloxy, 2-naphthyloxy), a heterocyclic oxy group preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, and particularly preferably 1 carbon atom.
- pyridyloxy, pyrazyloxy, pyrimidyloxy, quinolyloxy an acyl group (preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, and particularly preferably 1 to 12 carbon atoms).
- An acyloxy group (preferably having 2 to 30 carbon atoms, more preferably 2 to 20 carbon atoms, particularly preferably 2 to 10 carbon atoms such as acetoxy and benzoyloxy), acylamino group (preferably 2-30 carbon atoms, more preferably 2-20 carbon atoms, particularly preferably 2-10 carbon atoms, and examples thereof include acetylamino and benzoylamino), alkoxycarbonylamino groups (preferably having 2-2 carbon atoms) 30, more preferably 2 to 20 carbon atoms, particularly preferably 2 to 12 carbon atoms, such as methoxycarbonylamino), aryloxycarbonylamino group (preferably 7 to 30 carbon atoms, more preferably 7 to 20 carbon atoms, particularly preferably 7 to 12 carbon atoms, such as phenyloxycarbonylamino ), A sulfonylamino group (preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, particularly preferably 1 to 12 carbon
- Sulfamoyl groups (preferably having 0 to 30 carbon atoms, more preferably 0 to 20 carbon atoms, particularly preferably 0 to 12 carbon atoms, such as sulfamoyl, methylsulfamoyl, dimethylsulfamoyl,
- a carbamoyl group (preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, particularly preferably 1 to 12 carbon atoms, such as carbamoyl, methylcarbamoyl, diethylcarbamoyl, phenyl).
- Carbamoyl an alkylthio group (preferably carbon) 1-30, more preferably 1-20 carbon atoms, particularly preferably 1-12 carbon atoms such as methylthio and ethylthio), arylthio groups (preferably 6-30 carbon atoms, more preferably carbon atoms) 6 to 20, particularly preferably 6 to 12 carbon atoms, including, for example, phenylthio), a heterocyclic thio group (preferably 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, particularly preferably carbon atoms).
- the number of carbon atoms is 1 to 12, and examples thereof include pyridylthio, 2-benzimidazolylthio, 2-benzoxazolylthio, and 2-benzthiazolylthio), sulfonyl groups (preferably having 1 to 30 carbon atoms, Preferably it has 1 to 20 carbon atoms, particularly preferably 1 to 12 carbon atoms, and examples thereof include mesyl and tosyl), sulfinyl group (preferably Has 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, particularly preferably 1 to 12 carbon atoms, and examples thereof include methanesulfinyl and benzenesulfinyl.
- Ureido groups preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, particularly preferably 1 to 12 carbon atoms, and examples thereof include ureido, methylureido and phenylureido
- phosphoric acid An amide group preferably having 1 to 30 carbon atoms, more preferably 1 to 20 carbon atoms, and particularly preferably 1 to 12 carbon atoms, such as diethyl phosphoric acid amide and phenyl phosphoric acid amide
- a hydroxy group preferably Mercapto group, halogen atom (for example, fluorine atom, chlorine atom, bromine atom, iodine atom), cyano group, sulfo group, carboxyl group, nitro group, trifluoromethyl group, hydroxamic acid group, sulfino group, A hydrazino group, an imino group, a heterocyclic group (preferably having 1 to 30 carbon atoms, more preferably
- a silyl group (preferably having 3 to 40 carbon atoms, more preferably 3 to 30 carbon atoms, and particularly preferably 3 to 24 carbon atoms), and examples thereof include trimethylsilyl and triphenylsilyl. )
- a silyloxy group preferably having 3 to 40 carbon atoms, more preferably 3 to 30 carbon atoms, particularly preferably 3 to 24 carbon atoms, and examples thereof include trimethylsilyloxy and triphenylsilyloxy).
- Preferred substituents are a cyano group, a trifluoromethyl group, a halogen atom, an alkyl group, an alkoxy group, an aryl group, an amino group, or a heterocyclic group.
- R 1 and R 3 are more preferably an alkyl group having 1 to 10 carbon atoms, particularly preferably an alkyl group having 1 to 5 carbon atoms, among those exemplified above as R 1 to R 102 .
- Alkyl groups are, for example, methyl, ethyl, iso-propyl, tert-butyl. Among these, methyl and tert-butyl are more preferable, and methyl is most preferable.
- R 2 is more preferably a hydrogen atom, a deuterium atom or an alkyl group having 1 to 10 carbon atoms, particularly preferably a hydrogen atom or an alkyl group having 1 to 5 carbon atoms, as exemplified above as R 1 to R 102 .
- a hydrogen atom is most preferred.
- R 4 to R 102 are hydrogen atoms, deuterium atoms, cyano groups, trifluoromethyl groups, fluorine atoms, alkyl groups having 1 to 30 carbon atoms, 1 to 30 carbon atoms, among those exemplified above as R 1 to R 102. More preferably an alkoxy group having 30 carbon atoms, an aryl group having 6 to 30 carbon atoms, and an amino group having 0 to 30 carbon atoms, a hydrogen atom, a fluorine atom, an alkyl group having 1 to 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, An amino group having 0 to 20 carbon atoms is particularly preferred.
- the method for producing a tris orthometalated iridium complex includes an orthometalated iridium complex represented by the general formula (Chemical Formula 4) as a reaction substrate and a bidentate organic coordination represented by the general formula (Chemical Formula 5).
- a step (2) of adding the orthometalated iridium complex and the bidentate organic ligand and a step (3) of reacting the orthometalated iridium complex and the bidentate organic ligand are sequentially provided.
- the orthometalated iridium complex represented by the general formula (Formula 4) is represented by the compound represented by the general formula (Formula 7), the compound represented by the general formula (Formula 8), or the general formula (Formula 9). A compound is preferred.
- Compound represented by the general formula (Formula 7) is the iridium dinuclear complex halogen ligand is coordinated as L a.
- the halogen ligand include chlorine, bromine, iodine, and fluorine.
- a form in which chlorine is coordinated hereinafter sometimes referred to as a chlorine-bridged iridium dimer
- Compound represented by the general formula (Formula 8) is ortho-metalated iridium complex ⁇ - diketonate ligand as L a is coordinated.
- Compound represented by the general formula (Formula 9) acetonitrile ligand is coordinated ortho-metalated iridium complex as L a.
- the bidentate organic ligand represented by the general formula (Chemical Formula 5) is a bidentate organic ligand capable of forming an iridium-nitrogen bond or an iridium-carbon bond.
- the bidentate organic ligand is preferably at least one selected from (7) to (17) shown in the general formula (Formula 10).
- a bidentate organic ligand represented by (7), (8), (9) or (15) represented by the general formula (Chemical Formula 10) is more preferable, and (7), (9 ) Or (15) is particularly preferred, and the bidentate organic ligand represented by (7) or (15) represented by the general formula (Formula 10) is particularly preferred.
- Other examples of the bidentate organic ligand include bidentate organic ligands described in International Publication No. 01/041512, International Publication No. 02/15645, and Japanese Patent Application Laid-Open No. 2001-247859.
- the heating means for raising the temperature to the predetermined reaction temperature in the step (3) is not particularly limited, and a conventional external heating method such as an oil bath, a mantle heater, a block heater, or a heating medium circulation jacket or the like. Any of the microwave irradiation methods can be applied. However, it is preferable to select the microwave irradiation method in order to obtain a higher facial body selectivity in a shorter time.
- the microwave frequency is not particularly limited, but is preferably 300 MHz to 300 GHz, more preferably 500 MHz to 10000 MHz, particularly preferably 2000 MHz to 3000 MHz, and particularly preferably 2400 MHz to 2500 MHz.
- the reaction time in the case of heating by the microwave irradiation method depends on the output of the microwave reactor, the organic ligand, the type of solvent used and the amount of liquid, but is preferably 1 minute to 180 minutes, and 3 minutes to 120 minutes. Is more preferable, 5 minutes to 90 minutes is particularly preferable, and 10 minutes to 60 minutes is particularly preferable.
- the output of the microwave is preferably 1 W to 15 kW. More preferably, it is 100 W to 10 kW, particularly preferably 500 W to 8 kW, and particularly preferably 1 kW to 6 kW.
- the reaction time depends on the organic ligand, the type of solvent used and the amount of liquid, but is preferably 10 minutes to 96 hours, and 1 hour to 72 hours. Is more preferable, and 1 to 48 hours is particularly preferable, and 1 to 24 hours is particularly preferable.
- the reaction solvent used in this embodiment is not particularly limited, but alcohol solvents, protic solvents, aprotic solvents, hydrocarbon solvents, nitrile solvents, ionic solvents, and the like are preferably used.
- the boiling point of the reaction solvent used is preferably 100 ° C to 300 ° C, more preferably 150 ° C to 285 ° C, particularly preferably 160 ° C to 250 ° C, and particularly preferably 180 ° C to 230 ° C.
- reaction solvent examples include 2-ethoxyethanol, DMF (N, N-dimethylformamide), diglyme, dodecane, ethylene glycol, 1,2-propanediol, 1,3-propanediol, 1,3- Examples include butanediol and glycerin.
- 2-ethoxyethanol, DMF, diglyme, dodecane, ethylene glycol, 1,2-propanediol, 1,3-propanediol, and 1,3-butanediol are preferable, and ethylene glycol, 1,2-propanediol, 1 Diols such as 1,3-propanediol and 1,3-butanediol are particularly preferred.
- These reaction solvents can be used alone or as a mixed solvent containing two or more.
- the reaction solvent is preheated, and includes two kinds of an orthometalated iridium complex represented by the general formula (Formula 4) and a bidentate organic ligand represented by the general formula (Formula 5).
- the reaction substrate is added later to cause the reaction, and at this time, the two reaction substrates may be uniformly dissolved or uniformly dispersed in a solvent in advance.
- any one of the two reaction substrates may be added to the reaction solvent preheated first.
- the bidentate organic ligand may be added, or after the bidentate organic ligand is added, the orthometalated iridium complex may be added. Further, the orthometalated iridium complex and the bidentate organic ligand may be added simultaneously.
- the two reaction substrates may be mixed in advance and then added to the reaction solvent.
- the steps (1) to (3) are preferably performed in an inert gas atmosphere, and particularly preferably performed in a nitrogen atmosphere or an argon atmosphere.
- the reaction temperature in the step (3) is preferably in the range of 100 ° C to 300 ° C.
- the reaction temperature is more preferably in the range of 145 ° C. to 300 ° C., particularly preferably in the range of 145 ° C. to 285 ° C., particularly preferably in the range of 160 ° C. to 250 ° C., and 180 ° C. to A range of 230 ° C. is particularly preferred.
- the reaction temperature is less than 100 ° C., the production ratio of meridional forms tends to increase.
- reaction temperature exceeds 300 degreeC there exists a possibility that a decomposition reaction may advance easily and a yield may fall.
- the preheating temperature in the step (1) is equal to or lower than the reaction temperature in the step (3), and a temperature between the preheating temperature and the reaction temperature. It is preferable that the difference is small.
- the temperature difference between the preheating temperature and the reaction temperature is preferably 100 ° C. or less, more preferably 50 ° C. or less, particularly preferably 20 ° C. or less, and 10 ° C. or less. Is particularly preferred. When the temperature difference exceeds 100 ° C., it takes a long time to reach the reaction temperature, the production ratio of the meridional body is increased, and the production ratio of the facial body may be lowered.
- the preheating temperature in the step (1) is not higher than the reaction temperature in the step (3) and is in the range of 100 to 300 ° C. Is preferred.
- the preheating temperature is more preferably in the range of 145 ° C to 300 ° C, particularly preferably in the range of 145 ° C to 285 ° C, more preferably in the range of 160 ° C to 250 ° C, and 180 ° C. A range of ⁇ 230 ° C. is particularly preferred.
- the preheating temperature is less than 100 ° C., the production ratio of meridional bodies tends to increase.
- preheating temperature exceeds 300 degreeC a decomposition reaction will advance easily and there exists a possibility that a yield may fall.
- the preheating time is preferably less than 60 minutes, more preferably less than 30 minutes, particularly preferably less than 15 minutes, and particularly preferably 5 minutes after reaching the desired temperature. Less than is more particularly preferable.
- the purpose of the preheating is not, for example, removing water in the reaction solvent, but a preparation step for the step (2). Therefore, when the desired temperature is reached, the next step ( It is preferable to proceed to 2).
- step (1) after the preliminary heating, it is preferable to perform the following step (2) and step (3) without passing through the cooling step.
- the cooling does not include a temporary temperature decrease such as a temperature decrease due to the interruption of microwave irradiation between the steps (1) and (2) and a temperature decrease due to the addition of a chemical.
- the cooling process in the present specification refers to a process of lowering the temperature with the intention of leaving or cooling until reaching a predetermined temperature. Therefore, it is preferable that the temperature decrease is, for example, less than 30 ° C., more preferably less than 20 ° C., and particularly preferably less than 10 ° C.
- the production method increases the production ratio of the facial body by utilizing the fact that the temperature generated by the facial body is higher than the reaction temperature generated by the meridional body. Therefore, it is preferable to set the temperature of the mixture to a higher temperature in the initial state of the reaction starting from when the two kinds of reaction substrates are added afterwards to the preheated reaction solvent.
- the temperature of the reaction solution (liquid containing the reaction solvent and the reaction substrate) when the reaction substrate is mixed is preferably 105 ° C. or higher. More preferably, it is 155 degreeC or more, Most preferably, it is 180 degreeC or more.
- the manufacturing method according to this embodiment is usually performed at normal pressure, but may be performed under pressure or under reduced pressure as necessary.
- an inorganic base containing an alkali metal for example, potassium carbonate, sodium carbonate, sodium hydroxide, potassium hydrogen carbonate
- an organic amine is used to promote the reaction.
- a base such as diethylamine, triethylamine, triisobutylamine, or triethanolamine may be added to the reaction.
- the other reaction substrate in order to accelerate the reaction, added AgCF 3 SO 3, AgCF 3 COO , AgClO 4, AgBF 4, AgBPh 4 or AgPF 6 silver compound such as You may go.
- the amount of the bidentate organic ligand represented by the general formula (Formula 5) is stoichiometric with respect to the orthometalated iridium complex represented by the general formula (Formula 4).
- the ratio is not particularly limited as long as it is higher than the ratio, but 1 to 100 equivalents are preferable.
- the orthometalated iridium complex represented by the general formula (Formula 4) is the general formula (Formula 7), it is more preferably 2 to 100 equivalents, particularly preferably 5 to 80 equivalents, and particularly preferably 10 to 70 equivalents.
- orthometalated iridium complex represented by the general formula (Formula 4) is the general formula (Formula 8)
- 1 to 30 equivalents are more preferable
- 1 to 10 equivalents are particularly preferable
- 1 to 5 equivalents are particularly preferable
- 1 to 30 equivalents are more preferable
- 1 to 10 equivalents are particularly preferable
- 1 to 5 equivalents are particularly preferable.
- the method of preheating in step (1) is not particularly limited, and is, for example, a conventional external heating method or microwave irradiation method such as an oil bath, a mantle heater, a block heater, or a heating medium circulation jacket.
- the microwave irradiation method is preferable in that the temperature can be increased in a shorter time.
- the microwave output is preferably in the range of 0.2 kW to 100 kW per liter of reaction solution, more preferably in the range of 0.5 kW to 50 kW per liter of reaction solution, and particularly preferably in the range of 2 kW to 20 kW per liter of reaction solution. .
- the stirring of the reaction solution is not particularly limited, but for example, a method of stirring by bubbling an inert gas, a method of using a magnetic stirrer, a stirring blade, or the like is preferably used.
- the trisorthometalated iridium complex obtained by the production method according to the present embodiment has a geometric isomer of a facial isomer and a meridional isomer, and the generation ratio thereof is determined by proton nuclear magnetic resonance (proton NMR: Nuclear Magnetic Resonance). ), High performance liquid chromatography (HPLC: High performance liquid chromatography) and the like.
- microwave irradiation methods there are single mode and multi mode as microwave irradiation methods. In the manufacturing method according to the present embodiment, both of them can be used, but the multimode is more preferable.
- the iridium valence of the trisorthometalated iridium complex that is the iridium raw material and product used in the production method according to the present embodiment is preferably trivalent.
- microwave irradiation apparatus used in the manufacturing method according to this embodiment, all commercially available products or conventionally known devices can be used. Moreover, it is preferable to perform reaction by attaching a cooling pipe to the upper part of a microwave irradiation apparatus.
- the material of the reaction vessel used in the production method according to this embodiment is not particularly limited, and examples thereof include borosilicate glass, quartz glass, and polytetrafluoroethylene (for example, Teflon (registered trademark)).
- the orthometalated iridium complexes represented by the general formulas (Chemical Formula 4) and (Chemical Formula 7) to (Chemical Formula 9) can be produced by known methods.
- Known methods include, for example, the methods described in JP-A No. 2002-105055, JP-T 2008-505076, or WO 2009/073246.
- the manufacturing method according to the present embodiment has a high practical value when the microwave irradiation method is adopted as the heating means.
- the microwave irradiation method As a heating means, with the microwave output as it is, the temperature rise rate of the reaction solution becomes slow, and it is longer than before the scale-up until the desired reaction temperature is reached. It takes time.
- the meridional form is likely to be produced at a lower reaction temperature than the facial form, and the scale-up may facilitate the formation of the meridional form.
- the light emitting material according to the present embodiment is composed of a tris ortho-metalated iridium complex manufactured by the manufacturing method according to the present embodiment.
- the light emitting material is suitable as a material for the light emitting layer of the organic electroluminescent element, for example.
- the light emitting element according to the present embodiment uses the light emitting material according to the present embodiment.
- the organic electroluminescent element usually has a structure in which an anode, a hole transport layer, a light emitting layer, an electron transport layer, and a cathode are sequentially laminated on a substrate made of a glass plate, a plastic plate, or the like.
- the formation method of a light emitting layer is not specifically limited, It can form by well-known methods, such as a vapor deposition film-forming method and a wet film-forming method.
- the light emitting device is a surface light emitter such as a flat panel display such as an OA computer or a wall-mounted television, an in-vehicle display device, a mobile phone display, a copying machine, a liquid crystal display, and a backlight light source of instruments It can be applied to a light source, a display board, and a marker lamp.
- a surface light emitter such as a flat panel display such as an OA computer or a wall-mounted television, an in-vehicle display device, a mobile phone display, a copying machine, a liquid crystal display, and a backlight light source of instruments It can be applied to a light source, a display board, and a marker lamp.
- Example 1 Production of Trisorthometalated Iridium Complex (T-9) 240 mL of special grade ethylene glycol was placed in a 500 mL two-necked flask and set in a microwave reactor (Microsynth, manufactured by Milestone General). After argon gas was blown into this for 25 minutes, 1 kW microwave (2450 MHz) was irradiated while magnetically stirring the reaction solvent, and the temperature was raised to boiling (around 198 ° C. to 200 ° C.) in about 3 minutes, and preheating was performed. did.
- a microwave reactor Merosynth, manufactured by Milestone General
- the product was analyzed by proton NMR (manufactured by JEOL, JNM-ECX 400: 400 MHz, in DMSO-d 6 ). As a result, the facial product and the meridional product were a mixture of 90:10 (molar ratio).
- the reaction scheme is shown in the reaction formula (Formula 12).
- T-1 Trisorthometalated Iridium Complex
- 150 mL of special grade ethylene glycol was placed in a 500 mL two-necked flask and set in a microwave reactor (Microsynth, manufactured by Milestone General). After argon gas was blown into the reaction solution for 20 minutes, 300 W microwave (2450 MHz) was irradiated while the reaction solution was magnetically stirred, and the temperature was raised from room temperature to boiling (around 198 ° C to 200 ° C) in about 7 minutes. And preheated.
- the production methods according to the present invention (Examples 1 and 2), compared with the conventional production methods (Comparative Examples 1 and 2), suppress the formation of meridional bodies, and the purity of the facial bodies is high. It became clear that it improved. Since the tris-orthometalated iridium complex in the facial form has been shown to be superior in luminous efficiency and stability to the meridional form, which is the geometric isomer, the tris produced by the production method according to the present invention. By using an orthometalated iridium complex as a light-emitting element material, a light-emitting element with high efficiency and high durability can be manufactured.
- the manufacturing method can greatly contribute to a reduction in manufacturing cost, and has a great practical advantage.
Abstract
本発明の目的は、トリスオルトメタル化イリジウム錯体の中でも、とりわけ有機電界発光素子材料として好適に用いられるフェイシャル‐トリスオルトメタル化イリジウム錯体を、反応基質を混合した後で加熱して反応させる従来の製造方法と比較して、高選択的に得るための新たな製造方法を提供することである。本発明に係るトリスオルトメタル化イリジウム錯体の製造方法は、反応基質として一般式(化4)で表されるオルトメタル化イリジウム錯体と一般式(化5)で表される2座有機配位子とを反応させて、一般式(化6)で表されるトリスオルトメタル化イリジウム錯体を製造する方法であって、反応溶媒を予備加熱する工程(1)と、該予備加熱した反応溶媒にオルトメタル化イリジウム錯体と2座有機配位子とを添加する工程(2)と、オルトメタル化イリジウム錯体と2座有機配位子とを反応させる工程(3)と、を順次有する。
Description
本発明は、有機電界発光素子用材料として好適に用いられるトリスオルトメタル化イリジウム錯体を製造する方法であり、特には、発光効率が高く熱安定性に優れたフェイシャル‐トリスオルトメタル化イリジウム錯体の生成比率を高めることができるトリスオルトメタル化イリジウム錯体の製造方法に関する。
現在、有機電界発光素子は、次世代ディスプレイ技術又は照明技術として注目されており、有機電界発光素子用発光材料の開発が活発に進められている。発光材料は、蛍光材料と燐光材料との2つのタイプに分類できるが、より高い発光効率を示す燐光材料に注目が集まっている。その中でも、トリス(2‐フェニルピリジン)イリジウムに代表されるトリスオルトメタル化イリジウム錯体は、高い発光効率を示し、また、良好な熱安定性を示すことから有望な材料群である。これまで、このようなトリスオルトメタル化イリジウム錯体を得るために、数多くの製造方法が開示されている(例えば、特許文献1~5を参照。)。
特許文献1には、反応式(化1)に示すように、3塩化イリジウムn水和物から塩素架橋イリジウムダイマーを合成し、これを原料にトリスオルトメタル化イリジウム錯体を製造する方法が開示されている。

特許文献2には、反応式(化2)に示すように、3塩化イリジウムn水和物から塩素架橋イリジウムダイマーを合成し、塩素架橋イリジウムダイマーからアセチルアセトンが配位したイリジウム錯体を合成し、これを原料にトリスオルトメタル化イリジウム錯体を製造する方法が開示されている。

特許文献3には、反応式(化3)に示すように、3塩化イリジウムn水和物から塩素架橋イリジウムダイマーを合成し、塩素架橋イリジウムダイマーからアセトニトリルが配位したイリジウム錯体を合成し、これを原料にトリスオルトメタル化イリジウム錯体を製造する方法が開示されている。

特許文献1~3に記載の製造方法は、塩素架橋イリジウムダイマーなどのオルトメタル化イリジウム錯体と2‐フェニルピリジンなどの2座有機配位子とを混合した後、加熱反応させる方法である。
特許文献4には、反応溶媒であるグリセリンを130℃~140℃で2時間加熱した後、100℃まで放冷し、1‐フェニルイソキノリンとIr(acac)3(acac:アセチルアセトン)とを放冷したグリセリンに入れ、210℃付近で7時間加熱反応させてトリスオルトメタル化イリジウム錯体を製造する方法が開示されている。特許文献5には、2座有機配位子とグリセリンとの混合物を150℃で約60分間加熱し、放冷した後、Ir(acac)3を添加し、200℃に昇温してトリスオルトメタル化イリジウム錯体を製造する方法が開示されている。
J.Am.Chem.Soc.,2003年、125巻、7377頁
特許文献1~3に記載の製造方法は、塩素架橋イリジウムダイマーなどのオルトメタル化イリジウム錯体と2‐フェニルピリジンなどの2座有機配位子とを混合した後、加熱反応させる方法である。しかし、これらの方法では、幾何異性体であるメリジオナル体が副生成物として生成するためフェイシャル体を高純度で得ることは容易ではない。一方、特許文献4に記載の製造方法は、予め反応溶媒であるグリセリンを加熱して放冷した後、イリジウム原料と2座有機配位子とを放冷したグリセリンに添加して加熱反応させる方法である。特許文献5に記載の製造方法は、予め2座有機配位子を含むグリセリンを加熱して放冷した後、イリジウム原料を放冷したグリセリンに添加して加熱反応させる方法である。特許文献4及び5に記載の製造方法は、本発明と類似しているが、予め行う反応溶媒であるグリセリンの加熱は、グリセリンに含まれる水を除くことを目的としており、本発明とは予備加熱の目的そのものが異なっている。また、使用可能なイリジウム原料がIr(acac)3に限定され、一般性の高い製造方法とはいえず、その反応は、本発明に係る製造方法とは全く相違している。
トリスオルトメタル化イリジウム錯体には、フェイシャル体及びメリジオナル体の2種類の幾何異性体が存在するが、フェイシャル体の方が発光効率及び安定性の点で優れていることが明らかになっている(例えば、非特許文献1を参照。)。しかし、フェイシャル体及びメリジオナル体の幾何異性体を簡便に分離するのは困難であり、フェイシャル体を得るためには、再結晶、カラムクロマトグラフィー、又は昇華精製など手間のかかる作業を組み合わせて繰り返す後処理を行う必要があった。前述した特許文献1~3に記載された製造方法は、幾何異性体であるメリジオナル体が副生成物として生成する問題を完全に解決したとはいえない。したがって、トリスオルトメタル化イリジウム錯体の製造方法にあっては、有機電界発光素子材料としてあまり好ましくないメリジオナル体の生成を抑制し、フェイシャル体を選択的に製造する方法が渇望されている。
本発明は、前記従来技術の事情に鑑みなされたものである。本発明の目的は、トリスオルトメタル化イリジウム錯体の中でも、とりわけ有機電界発光素子材料として好適に用いられるフェイシャル‐トリスオルトメタル化イリジウム錯体を、反応基質を混合した後で加熱して反応させる従来の製造方法と比較して、高選択的に得るための新たな製造方法を提供することである。
本発明の第二の目的は、フェイシャル‐トリスオルトメタル化イリジウム錯体を高比率で含むトリスオルトメタル化イリジウム錯体を用いて、発光効率及び耐久性に優れた発光材料と、その発光材料を用いた発光素子とを提供することである。
本発明者らは、前記課題を解決すべく、トリスオルトメタル化イリジウム錯体の製造方法を鋭意検討してきた。その結果、前述した公知の製造方法におけるフェイシャル体とメリジオナル体との生成比率は、意外にもイリジウム原料であるオルトメタル化イリジウム錯体と2座有機配位子との添加方法に大きく依存することを見出した。すなわち、反応溶媒を予備加熱し、その予備加熱した反応溶媒に、イリジウム原料及び2座有機配位子の2種の反応基質を後添加して加熱反応させることで、従来の製造方法(特許文献1~3に記載した製造方法のように2種の反応基質を混合した後で、加熱反応させる方法)よりも、メリジオナル体の生成が抑制され、フェイシャル体の純度が大きく向上することを明らかにした。これまで、トリスオルトメタル化イリジウム錯体の幾何異性体であるフェイシャル体とメリジオナル体との生成比率について、イリジウム原料であるオルトメタル化イリジウム錯体と2座有機配位子との添加方法に依存することは開示されておらず、本実験事実は、発明者らの緻密な実験の積み重ねによって得られた重要、かつ、新たな知見である。このように、本発明者らは、有機電界発光素子材料として好適に用いられるフェイシャル‐トリスオルトメタル化イリジウム錯体を従来の製造方法よりも高純度に製造する方法を開発することに成功し、本発明を完成するに至った。
フェイシャル体のトリスオルトメタル化イリジウム錯体は、その幾何異性体であるメリジオナル体よりも発光効率及び安定性で優れていることが明らかにされているので、本発明によって製造されたトリスオルトメタル化イリジウム錯体を発光材料として用いることで、高効率、かつ、高耐久性の発光素子を作製することができることを見出し、本発明を完成するに至った。
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法は、反応基質として一般式(化4)で表されるオルトメタル化イリジウム錯体と一般式(化5)で表される2座有機配位子とを反応させて、一般式(化6)で表されるトリスオルトメタル化イリジウム錯体を製造する方法であって、反応溶媒を予備加熱する工程(1)と、該予備加熱した反応溶媒にオルトメタル化イリジウム錯体と2座有機配位子とを添加する工程(2)と、オルトメタル化イリジウム錯体と2座有機配位子とを反応させる工程(3)と、を順次有することを特徴とする。

(一般式(化4)中、Laは配位子を表す。A1は対アニオンを表す。m1は1又は2を表す。m2は0又は1を表す。Z1及びZ2は、それぞれ独立に、5員環又は6員環を形成するに必要な非金属原子群を表す。さらに、形成される環は更に別の環と縮合環を形成してもよい。L1は単結合又は2価の基を表す。Y1は窒素原子又は炭素原子を表す。Y1が窒素原子のときは、Q1は炭素原子とY1とが単結合で結合していることを表す。Y1が炭素原子のときは、Q1は炭素原子とY1とが2重結合で結合していることを表す。)

(一般式(化5)中、Z1、Z2、Y1、Q1及びL1は各々一般式(化4)の場合と同義である。)

(一般式(化6)中、Z1、Z2、Y1、Q1及びL1は各々一般式(化4)の場合と同義である。)
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、オルトメタル化イリジウム錯体が、一般式(化7)で表される化合物である形態を包含する。

(一般式(化7)中、Xはハロゲン原子を表す。Z1、Z2、Y1、Q1及びL1は各々一般式(化4)の場合と同義である。)
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記Laが、モノアニオン性配位子である形態を包含する。
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記Laが、中性配位子である形態を包含する。
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、オルトメタル化イリジウム錯体が、一般式(化8)で表わされる化合物である形態を包含する。

(一般式(化8)中、R1~R3は水素原子、重水素原子又は置換基を表す。Z1、Z2、Y1、Q1及びL1は各々一般式(化4)の場合と同義である。)
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、オルトメタル化イリジウム錯体が、一般式(化9)で表わされる化合物である形態を包含する。

(一般式(化9)中、A1、Z1、Z2、Y1、Q1及びL1は各々一般式(化4)の場合と同義である。)
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、2座有機配位子が、一般式(化10)に示す(7)~(17)の中から選ばれる少なくとも1つである形態を包含する。

(一般式(化10)に示す(7)~(17)中、R4~R102は水素原子、重水素原子又は置換基を表す。)
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(3)の反応温度が、100~300℃の範囲であることが好ましい。フェイシャル体の生成比率をより高めることができる。
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(1)の予備加熱温度が、前記工程(3)の反応温度以下であり、かつ、100~300℃の範囲であることが好ましい。フェイシャル体の生成比率をより高めることができる。
本発明に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(3)をマイクロ波照射下で行うことが好ましい。短時間で昇温することができるため、フェイシャル体の生成比率をより高めることができる。
本発明に係る発光材料は、本発明に係る製造方法で製造したトリスオルトメタル化イリジウム錯体からなる。
本発明に係る発光素子は、本発明に係る発光材料を用いてなる。
本発明は、トリスオルトメタル化イリジウム錯体の中でも、とりわけ有機電界発光素子材料として好適に用いられるフェイシャル‐トリスオルトメタル化イリジウム錯体を、反応基質を混合した後で加熱して反応させる従来の製造方法と比較して、高選択的に得るための新たな製造方法を提供することができる。また、本発明は、フェイシャル‐トリスオルトメタル化イリジウム錯体を高比率で含むトリスオルトメタル化イリジウム錯体を用いて、発光効率及び耐久性に優れた発光材料と、その発光材料を用いた発光素子とを提供することができる。
次に、本発明について実施形態を示して詳細に説明するが、本発明はこれらの記載に限定して解釈されない。本発明の効果を奏する限り、実施形態は種々の変形をしてもよい。
本明細書において、一般式(化4)~(化9)及び(化10)の(7)~(17)に記載した記号(Z1,Z2,Y1,Q1,L1、La、m1、m2、A1、X、R1~R102)について、次に詳しく説明する。
Z1及びZ2はそれぞれ独立に、5員環又は6員環を形成するのに必要な非金属原子群を表す。形成される環は、置換基を有していてもよく、また更に別の環と縮合環を形成してもよい。置換基としては、例えば、ハロゲン原子、アルキル基、置換アルキル基、フェノキシ基、置換フェノキシ基、アリール基、置換アリール基、アルコキシ基、置換アルコキシ基、ジアルキルアミノ基又は置換ジアルキルアミノ基である。好ましくは、ハロゲン原子、炭素数1~30のアルキル基、炭素数1~30の置換アルキル基、炭素数6~30のフェノキシ基、炭素数6~30の置換フェノキシ基、炭素数6~30のアリール基、炭素数6~30の置換アリール基、炭素数1~30のアルコキシ基、炭素数1~30の置換アルコキシ基、炭素数2~30のジアルキルアミノ基又は炭素数2~30の置換ジアルキルアミノ基である。より好ましくは、ハロゲン原子、炭素数1~10のアルキル基、炭素数1~10の置換アルキル基、炭素数6~10のアリール基、炭素数6~10の置換アリール基、炭素数1~10のアルコキシ基、炭素数1~10の置換アルコキシ基である。特に好ましくは、ハロゲン原子、炭素数1~4のアルキル基又は炭素数1~4の置換アルキル基である。
Z1が形成する5員環又は6員環としては、芳香族環又は複素芳香族環が好ましく、芳香族環がより好ましい。Z1が形成する5員環又は6員環は、例えば、キノリン環、ベンゾキノリン環、キノキサリン環、イソキノリン環、フェナントリジン環、フェナジン環、アクリジン環、トリアゾール環、イミダゾフェナントリジン環、フタラジン環、キナゾリン環、ナフチリジン環、シンノリン環、ペリミジン環、フェナントロリン環、ベンズイミダゾール環、ベンズオキサゾール環、ベンズチアゾール環、イミダゾール環、チアゾール環、オキサゾール環、ピロール環、オキサジアゾール環、チアジアゾール環、ピラゾール環、1,2,3‐トリアゾール環、1,2,4‐トリアゾール環、ピリジン環、ピリミジン環、ピラジン環、ピリダジン環、フラン環、チオフェン環、ナフタレン環、フルオレン環、ジベンゾチオフェン環、ジベンゾフラン環、カルバゾール環又はベンゼン環が挙げられる。これらのうち、ピロール環、ピリジン環、ナフタレン環、フルオレン環、ジベンゾチオフェン環、ジベンゾフラン環、カルバゾール環又はベンゼン環が好ましい。ナフタレン環、フルオレン環、ジベンゾチオフェン環、ジベンゾフラン環、カルバゾール環又はベンゼン環がより好ましく、ベンゼン環が特に好ましい。
Z2が形成する5員環又は6員環としては、複素芳香族環が好ましい。Z2が形成する5員環又は6員環は、例えば、イミダゾール環、チアゾール環、オキサゾール環、ピロール環、オキサジアゾール環、チアジアゾール環、ピラゾール環、1,2,3‐トリアゾール環、1,2,4‐トリアゾール環、セレナゾール環、ピリジン環、ピリミジン環、ピラジン環又はピリダジン環が挙げられる。これらのうち、イミダゾール環、チアゾール環、1,2,3‐トリアゾール環、1,2,4‐トリアゾール環、オキサゾール環、ピロール環、ピラゾール環、ピリジン環又はピリミジン環が好ましく、ピラゾール環又はピリジン環がさらに好ましい。
L1は、単結合又は2価の基を表す。2価の基としては、例えば、-C(R)(R’)-,-N(R)-,-O-,-P(R)-又は-S-が挙げられる。ここで、R及びR’は水素原子又は置換基を表す。置換基としては、例えば、ハロゲン原子、脂肪族基、芳香族基、複素環基、シアノ基、ニトロ基である。L1は、好ましくは単結合、又は-C(R)(R’)-であって、R及びR’が水素原子、脂肪族基又は芳香族基の場合である。L1として特に好ましくは単結合である。
Y1は、窒素原子又は炭素原子を表す。Y1が窒素原子の時は、Q1は炭素原子とY1との間の結合が単結合であることを示す。Y1が炭素原子の時は、Q1は炭素原子とY1との間が2重結合であることを示す。
Q1は、原子間の結合を表す。Y1が窒素原子のときは、Q1は炭素原子とY1とが単結合で結合していることを表す。Y1が炭素原子のときは、Q1は炭素原子とY1とが2重結合で結合していることを表す。
Laは配位子であれば、単座配位子又は2座配位子であることを問わない。Laに金属(例えば、イリジウム)が含まれても構わなく、いわゆる複核錯体を形成してもよい。
また、Laは、アニオン性配位子又は中性配位子が好ましい。モノアニオン性配位子又は中性配位子が特に好ましい。
次に、具体的に好ましいLaの構造を示す。単座のアニオン性配位子としては、例えば、ハロゲン配位子、ヒドロキシ配位子、アルコキシド配位子、フェノキシド配位子、チオシアネート配位子、シアネート配位子又はイソシアネート配位子である。2座のアニオン性配位子としては、例えば、β‐ジケトナート配位子、アセチルアセトナート配位子、酢酸配位子又はピコリン酸配位子である。
単座の中性配位子としては、ニトリル配位子(例えば、アセトニトリル配位子、プロピオニトリル配位子)、スルホキシド配位子(例えば、ジメチルスルホキシド配位子)、アミド配位子(例えば、ジメチルホルムアミド配位子)、エーテル配位子(例えば、テトラヒドロフラン配位子)、水配位子、アンモニア配位子、アミン配位子、ピペリジン配位子、ピリジン配位子、ピラジン配位子である。2座の中性配位子としては、例えば、ビピリジン配位子、フェナントロリン配位子、ジピリジルアミン配位子、エチレンジアミン配位子である。
m1は、1又は2を表す。Laが単座配位子のときはm1=2が好ましく、Laが2座配位子のときはm1=1が好ましい。
m2は、0又は1を表す。Laが単座のモノアニオン性配位子のときはm2=1が好ましく、Laが2座のモノアニオン性配位子のときはm2=0が好ましく、Laが中性配位子のときはm2=1が好ましい。
A1は、対アニオンを表す。対アニオンとしては、アニオン性であれば特に制限はないが、対モノアニオンが好ましい。対アニオンは、例えば、F-,Cl-,Br-,I-,BF4
-,PF6
-,ClO4
-、CF3CF2CF2COO-、CF3SO3
-、CF3CO2
-,CH3CO2
-,SCN-、CH3SO3
-,ClO4
-,SbF6
-が挙げられる。この中でも好ましくは、Cl-,PF6
-,BF4
-,PF6
-,CF3SO3
-である。
Xは、ハロゲン原子を表す。好ましくは塩素原子又は臭素原子であり、さらに好ましくは塩素原子である。
R1~R102は、水素原子、重水素原子又は置換基を表す。置換基としては、例えば、アルキル基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~10であり、例えば、メチル、エチル、iso‐プロピル、tert‐ブチル、n‐オクチル、n‐デシル、n‐ヘキサデシル、シクロプロピル、シクロペンチル、シクロヘキシルが挙げられる。)、アルケニル基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~10であり、例えば、ビニル、アリル、2‐ブテニル、3‐ペンテニルが挙げられる。)、アルキニル基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~10であり、例えば、プロパルギル、3‐ペンチニルが挙げられる。)、アリール基(好ましくは炭素数6~30、より好ましくは炭素数6~20、特に好ましくは炭素数6~12であり、例えば、フェニル、p‐メチルフェニル、ナフチル、アントラニルが挙げられる。)、アミノ基(好ましくは炭素数0~30、より好ましくは炭素数0~20、特に好ましくは炭素数0~10であり、例えば、アミノ、メチルアミノ、ジメチルアミノ、ジエチルアミノ、ジベンジルアミノ、ジフェニルアミノ、ジトリルアミノが挙げられる。)、アルコキシ基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~10であり、例えば、メトキシ、エトキシ、ブトキシ、2‐エチルヘキシロキシが挙げられる。)、アリールオキシ基(好ましくは炭素数6~30、より好ましくは炭素数6~20、特に好ましくは炭素数6~12であり、例えば、フェニルオキシ、1‐ナフチルオキシ、2‐ナフチルオキシが挙げられる。)、ヘテロ環オキシ基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、ピリジルオキシ、ピラジルオキシ、ピリミジルオキシ、キノリルオキシが挙げられる。)、アシル基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、アセチル、ベンゾイル、ホルミル、ピバロイルが挙げられる。)、アルコキシカルボニル基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~12であり、例えば、メトキシカルボニル、エトキシカルボニルが挙げられる。)、アリールオキシカルボニル基(好ましくは炭素数7~30、より好ましくは炭素数7~20、特に好ましくは炭素数7~12であり、例えば、フェニルオキシカルボニルが挙げられる。)、アシルオキシ基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~10であり、例えば、アセトキシ、ベンゾイルオキシが挙げられる。)、アシルアミノ基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~10であり、例えば、アセチルアミノ、ベンゾイルアミノが挙げられる。)、アルコキシカルボニルアミノ基(好ましくは炭素数2~30、より好ましくは炭素数2~20、特に好ましくは炭素数2~12であり、例えば、メトキシカルボニルアミノが挙げられる。)、アリールオキシカルボニルアミノ基(好ましくは炭素数7~30、より好ましくは炭素数7~20、特に好ましくは炭素数7~12であり、例えば、フェニルオキシカルボニルアミノが挙げられる。)、スルホニルアミノ基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、メタンスルホニルアミノ、ベンゼンスルホニルアミノが挙げられる。)、スルファモイル基(好ましくは炭素数0~30、より好ましくは炭素数0~20、特に好ましくは炭素数0~12であり、例えば、スルファモイル、メチルスルファモイル、ジメチルスルファモイル、フェニルスルファモイルが挙げられる。)、カルバモイル基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、カルバモイル、メチルカルバモイル、ジエチルカルバモイル、フェニルカルバモイルが挙げられる。)、アルキルチオ基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、メチルチオ、エチルチオが挙げられる。)、アリールチオ基(好ましくは炭素数6~30、より好ましくは炭素数6~20、特に好ましくは炭素数6~12であり、例えば、フェニルチオが挙げられる。)、ヘテロ環チオ基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、ピリジルチオ、2‐ベンズイミゾリルチオ、2‐ベンズオキサゾリルチオ、2‐ベンズチアゾリルチオが挙げられる。)、スルホニル基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、メシル、トシルが挙げられる。)、スルフィニル基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、メタンスルフィニル、ベンゼンスルフィニルが挙げられる。)、ウレイド基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、ウレイド、メチルウレイド、フェニルウレイドが挙げられる。)、リン酸アミド基(好ましくは炭素数1~30、より好ましくは炭素数1~20、特に好ましくは炭素数1~12であり、例えば、ジエチルリン酸アミド、フェニルリン酸アミドが挙げられる。)、ヒドロキシ基、メルカプト基、ハロゲン原子(例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。)、シアノ基、スルホ基、カルボキシル基、ニトロ基、トリフルオロメチル基、ヒドロキサム酸基、スルフィノ基、ヒドラジノ基、イミノ基、ヘテロ環基(好ましくは炭素数1~30、より好ましくは炭素数1~12であり、ヘテロ原子としては、例えば、窒素原子、酸素原子、硫黄原子であり、具体的には、イミダゾリル、ピリジル、キノリル、フリル、チエニル、ピペリジル、モルホリノ、ベンズオキサゾリル、ベンズイミダゾリル、ベンズチアゾリル、カルバゾリル基、アゼピニル基などが挙げられる。)、シリル基(好ましくは炭素数3~40、より好ましくは炭素数3~30、特に好ましくは炭素数3~24であり、例えば、トリメチルシリル、トリフェニルシリルが挙げられる。)、シリルオキシ基(好ましくは炭素数3~40、より好ましくは炭素数3~30、特に好ましくは炭素数3~24であり、例えば、トリメチルシリルオキシ、トリフェニルシリルオキシが挙げられる。)が挙げられる。好ましい置換基は、シアノ基、トリフルオロメチル基、ハロゲン原子、アルキル基、アルコキシ基、アリール基、アミノ基、又はヘテロ環基である。
R1及びR3としては、R1~R102として前記に例示した中でも炭素数1~10のアルキル基がより好ましく、炭素数1~5のアルキル基が特に好ましい。アルキル基は、例えば、メチル、エチル、iso‐プロピル、tert‐ブチルである。この中で、メチル、tert‐ブチルがより好ましく、メチルが最も好ましい。
R2としては、R1~R102として前記に例示した中でも水素原子、重水素原子又は炭素数1~10のアルキル基がより好ましく、水素原子又は炭素数1~5のアルキル基が特に好ましく、水素原子が最も好ましい。
R4~R102としては、R1~R102として前記に例示した中でも水素原子、重水素原子、シアノ基、トリフルオロメチル基、フッ素原子、炭素数1~30のアルキル基、炭素数1~30のアルコキシ基、炭素数6~30のアリール基、炭素数0~30のアミノ基がより好ましく、水素原子、フッ素原子、炭素数1~20のアルキル基、炭素数6~20のアリール基、炭素数0~20のアミノ基が特に好ましい。
本実施形態に係るトリスオルトメタル化イリジウム錯体の製造方法は、反応基質として一般式(化4)で表されるオルトメタル化イリジウム錯体と一般式(化5)で表される2座有機配位子とを反応させて、一般式(化6)で表されるトリスオルトメタル化イリジウム錯体を製造する方法であって、反応溶媒を予備加熱する工程(1)と、該予備加熱した反応溶媒にオルトメタル化イリジウム錯体と2座有機配位子とを添加する工程(2)と、オルトメタル化イリジウム錯体と2座有機配位子とを反応させる工程(3)と、を順次有する。
一般式(化4)で表されるオルトメタル化イリジウム錯体は、一般式(化7)で表される化合物、一般式(化8)で表される化合物又は一般式(化9)で表わされる化合物であることが好ましい。
一般式(化7)で表される化合物は、Laとしてハロゲン配位子が配位したイリジウム二核錯体である。ハロゲン配位子としては、例えば、塩素、臭素、ヨウ素、フッ素である。この中で、塩素が配位した形態(以降、塩素架橋イリジウムダイマーということもある。)が特に好ましい。一般式(化8)で表される化合物は、Laとしてβ‐ジケトナート配位子が配位したオルトメタル化イリジウム錯体である。一般式(化9)で表される化合物は、Laとしてアセトニトリル配位子が配位したオルトメタル化イリジウム錯体である。
一般式(化5)で表わされる2座有機配位子は、イリジウム‐窒素結合又はイリジウム‐炭素結合を形成し得る2座有機配位子であることを特徴としている。2座有機配位子は、一般式(化10)に示す(7)~(17)の中から選ばれる少なくとも1つであることが好ましい。一般式(化10)に示す(7)、(8)、(9)又は(15)で表わされる2座有機配位子がより好ましく、一般式(化10)に示す(7)、(9)又は(15)で表わされる2座有機配位子が特に好ましく、一般式(化10)に示す(7)又は(15)で表わされる2座有機配位子がとりわけ好ましい。その他、2座有機配位子として、国際公開第01/041512号、国際公開第02/15645号及び特開2001-247859号に記載の2座有機配位子を挙げることができる。
本発明において、工程(3)において所定の反応温度に昇温するための加熱手段は、特に限定されず、オイルバス、マントルヒーター、ブロックヒーター又は熱媒循環式ジャケットなどの従来の外部加熱方式又はマイクロ波照射方式のいずれも適用することができる。ただし、より短時間で、より高いフェイシャル体選択率を得るにはマイクロ波照射方式を選択することが好ましい。
マイクロ波の周波数については、特に制限はないが、300MHz~300GHzが好ましく、500MHz~10000MHzがより好ましく、2000MHz~3000MHzが特に好ましく、2400MHz~2500MHzがとりわけ好ましい。
マイクロ波照射方式による加熱の場合の反応時間は、マイクロ波反応装置の出力、有機配位子、用いる溶媒の種類及び液量に依存するが、1分~180分が好ましく、3分~120分がより好ましく、5分~90分が特に好ましく、10分~60分がとりわけ好ましい。
マイクロ波は、所望の反応温度に達しても、出力を変動(低下)させることなく、所定の一定出力で照射し続けることが好ましい。マイクロ波の出力は、1W~15kWであることが好ましい。より好ましくは、100W~10kWであり、特に好ましくは、500W~8kWであり、とりわけ好ましくは、1kW~6kWである。
一方、オイルバス、マントルヒーターなどによる外部加熱の場合には、反応時間は、有機配位子、用いる溶媒の種類及び液量に依存するが、10分~96時間が好ましく、1時間~72時間がより好ましく、1時間~48時間が特に好ましく、1時間~24時間がとりわけ好ましい。
本実施形態で用いられる反応溶媒としては、特に制限はないが、アルコール系溶媒、プロトン性溶媒、非プロトン性溶媒、炭化水素系溶媒、ニトリル系溶媒、イオン性溶媒などが好ましく用いられる。用いられる反応溶媒の沸点としては、100℃~300℃が好ましく、150℃~285℃がより好ましく、160℃~250℃が特に好ましく、180℃~230℃がとりわけ好ましい。このような反応溶媒としては、例えば、2‐エトキシエタノール、DMF(N,N‐ジメチルホルムアミド)、ジグライム、ドデカン、エチレングリコール、1,2‐プロパンジオール、1,3‐プロパンジオール、1,3‐ブタンジオール、グリセリンが挙げられる。その中でも2‐エトキシエタノール、DMF、ジグライム、ドデカン、エチレングリコール、1,2‐プロパンジオール、1,3‐プロパンジオール、1,3‐ブタンジオールが好ましく、エチレングリコール、1,2‐プロパンジオール、1,3‐プロパンジオール、1,3‐ブタンジオールなどのジオールが特に好ましい。これらの反応溶媒は、単独で又は2種以上を含む混合溶媒として用いることができる。
本実施形態では、反応溶媒を予備加熱し、その中に、一般式(化4)で表わされるオルトメタル化イリジウム錯体と一般式(化5)で表わされる2座有機配位子の2種の反応基質とを後添加して反応させるが、この際、2種の反応基質については、予め溶媒に均一に溶解又は均一に分散してもよい。ただし、予備加熱した反応溶媒の温度の低下を最小限とするために、溶解又は分散させる溶媒の量は最小限とすることが好ましい。最も好ましくは、2種の反応基質の両方を、溶媒に溶解又は分散させずに添加することが好ましい。
本実施形態では、前記2種の反応基質のうち、どちらを先に予備加熱した反応溶媒に添加しても構わない。オルトメタル化イリジウム錯体を添加した後、2座有機配位子を添加してもよいし、2座有機配位子を添加した後、オルトメタル化イリジウム錯体を添加してもよい。さらには、オルトメタル化イリジウム錯体と2座有機配位子とを同時に添加しても構わない。また、前記2種の反応基質を事前に混ぜた後、反応溶媒に添加してもよい。
本実施形態に係る製造方法では、工程(1)~工程(3)を不活性ガス雰囲気下で行うことが好ましく、窒素雰囲気下又はアルゴン雰囲気下で行うことが特に好ましい。
本実施形態に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(3)の反応温度が、100℃~300℃の範囲であることが好ましい。反応温度は、145℃~300℃の範囲であることがより好ましく、145℃~285℃の範囲であることが特に好ましく、160℃~250℃の範囲であることがより特に好ましく、180℃~230℃の範囲であることがとりわけ好ましい。反応温度が100℃未満では、メリジオナル体の生成比率が高くなる傾向にある。反応温度が300℃を超えると、分解反応が進行しやすくなり収率が低下するおそれがある。
本実施形態に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(1)の予備加熱温度が、前記工程(3)の反応温度以下であり、予備加熱温度と反応温度との間の温度差が小さいことが好ましい。予備加熱温度と前記反応温度との間の温度差が、100℃以下であることが好ましく、50℃以下であることがより好ましく、20℃以下であることが特に好ましく、10℃以下であることがとりわけ好ましい。温度差が100℃を超えると、反応温度に達するまでの時間が長くなり、メリジオナル体の生成比率が高まり、フェイシャル体の生成比率が低くなるおそれがある。
本実施形態に係るトリスオルトメタル化イリジウム錯体の製造方法では、前記工程(1)の予備加熱温度が、前記工程(3)の反応温度以下であり、かつ、100~300℃の範囲であることが好ましい。予備加熱温度は、145℃~300℃の範囲であることがより好ましく、145℃~285℃の範囲であることが特に好ましく、160℃~250℃の範囲であることがより特に好ましく、180℃~230℃の範囲であることがとりわけ好ましい。予備加熱温度が、100℃未満では、メリジオナル体の生成比率が高くなる傾向にある。予備加熱温度が300℃を超えると、分解反応が進行しやすくなり収率が低下するおそれがある。
本実施形態に係る製造方法では、予備加熱時間は、所望の温度に到達した後、60分未満であることが好ましく、30分未満であることがより好ましく、15分未満が特に好ましく、5分未満がより特に好ましい。本実施形態では、予備加熱の目的が、例えば反応溶媒中の水を除去することではなく、工程(2)への準備工程であるため、所望の温度に到達すれば、すぐに次の工程(2)へ進むことが好ましい。
工程(1)において、予備加熱した後は、放冷工程を経ずに、続く工程(2)及び工程(3)を行うことが好ましい。ここで、放冷には、工程(1)と工程(2)との間にマイクロ波照射の中断したことによる温度低下、薬品を添加したことによる温度低下などの一時的な温度低下を含まない。すなわち、本明細書における放冷工程とは、所定の温度になるまで放置する又は冷却するなど意図して温度を低下させる工程をいう。したがって、温度低下は、例えば、30℃未満にとどめることが好ましく、より好ましくは、20℃未満、特に好ましくは、10℃未満である。本実施形態に係る製造方法は、フェイシャル体が生成する温度が、メリジオナル体が生成する反応温度よりも高いことを利用して、フェイシャル体の生成比率を高めるものである。したがって、予備加熱した反応溶媒に2種の反応基質を後添加した時を開始時とする反応の初期状態において、混合物の温度をより高い温度とすることが好ましい。具体的には、工程(2)において、反応基質を混合した時の反応溶液(反応溶媒及び反応基質を含む液)の温度が105℃以上であることが好ましい。より好ましくは、155℃以上であり、特に好ましくは、180℃以上である。
本実施形態に係る製造方法は、通常、常圧で行われるが、必要に応じ加圧下又は減圧下で行っても構わない。
本実施形態に係る製造方法では、前記反応基質の他に、反応を促進させるために、アルカリ金属を含む無機塩基(例えば、炭酸カリウム、炭酸ナトリウム、水酸化ナトリウム、炭酸水素カリウム)、又は有機アミン(例えば、ジエチルアミン、トリエチルアミン、トリイソブチルアミン、トリエタノールアミン)などの塩基を添加して行ってもよい。
本実施形態に係る製造方法では、前記反応基質の他に、反応を促進させるために、AgCF3SO3、AgCF3COO、AgClO4、AgBF4、AgBPh4又はAgPF6などの銀化合物を添加して行ってもよい。
本実施形態に係る製造方法において、一般式(化5)で表わされる2座有機配位子の使用量は、一般式(化4)で表わされるオルトメタル化イリジウム錯体に対して、化学量論比以上であれば特に制限ないが、1~100当量が好ましい。一般式(化4)で表わされるオルトメタル化イリジウム錯体が一般式(化7)である場合は、2~100当量がより好ましく、5~80当量が特に好ましく、10~70当量がとりわけ好ましい。一般式(化4)で表わされるオルトメタル化イリジウム錯体が一般式(化8)である場合は、1~30当量がより好ましく、1~10当量が特に好ましく、1~5当量がとりわけ好ましい。一般式(化4)で表わされるオルトメタル化イリジウム錯体が一般式(化9)である場合は、1~30当量がより好ましく、1~10当量が特に好ましく、1~5当量がとりわけ好ましい。
工程(1)の予備加熱の方法は、特に限定されず、例えば、オイルバス、マントルヒーター、ブロックヒーター又は熱媒循環式ジャケットなど従来の外部加熱方式又はマイクロ波照射方式である。この中で、より短時間で昇温できる点で、マイクロ波照射方式が好ましい。
マイクロ波の出力は、反応溶液1リットル当たり0.2kW~100kWの範囲が好ましく、反応溶液1リットル当たり0.5kW~50kWの範囲がより好ましく、反応溶液1リットル当たり2kW~20kWの範囲が特に好ましい。
反応溶液の攪拌については、特に限定されないが、例えば、不活性ガスを通気することによって攪拌する方法、マグネチックスターラー、攪拌羽根などを用いる方法が好ましく用いられる。
本実施形態に係る製造方法によって得られるトリスオルトメタル化イリジウム錯体については、フェイシャル体とメリジオナル体との幾何異性体が存在するが、その生成比率についてはプロトン核磁気共鳴(プロトンNMR:Nuclear Magnetic Resonance)、高速液体クロマトグラフィー(HPLC:High performance liquid chromatography)などを用いて分析することができる。
マイクロ波照射の方法として、シングルモード又はマルチモードが存在する。本実施形態に係る製造方法では、そのどちらとも利用できるが、マルチモードの方がより望ましい。
本実施形態に係る製造方法で用いられるイリジウム原料及び生成物であるトリスオルトメタル化イリジウム錯体のイリジウムの価数については、3価であることが好ましい。
本実施形態に係る製造方法で用いられるマイクロ波照射装置については、市販品又は従来公知のものが全て使用できる。また、マイクロ波照射装置の上部に冷却管を取り付けて反応を行うことが好ましい。
本実施形態に係る製造方法で用いられる反応容器の材質については、特に限定されず、ホウケイ酸ガラス、石英ガラス、ポリテトラフルオロエチレン(例えば、テフロン(登録商標))などが挙げられる。
一般式(化4)及び(化7)~(化9)で表わされるオルトメタル化イリジウム錯体については、公知の方法で製造することができる。公知の方法は、例えば、特開2002-105055号、特表2008-505076号又はWO2009/073246号に記載の方法が挙げられる。
本実施形態に係る製造方法によって、トリスオルトメタル化イリジウム錯体の中でも特に発光効率の高いフェイシャル体が、従来製造方法を用いた場合と比較して、純度良く得られる理由は明確ではないが、一般式(化4)で表わされるオルトメタル化イリジウム錯体及び一般式(化5)で表わされる2座有機配位子の反応を、予備加熱した反応溶媒に、前記2種の反応基質を後添加して行うことによって、熱平衡的に有利なフェイシャル体の生成確率が向上したためではないかと本発明者らは推測している。一方、従来の製造方法(イリジウム原料と2座有機配位子と反応溶媒とを室温で混合し、加熱反応させる方法)を用いると、速度論的に有利なメリジオナル体が生成し易いものと考えられる。
本実施形態に係る製造方法は、加熱手段としてマイクロ波照射法を採用したときに実用的な価値が高い。加熱手段としてマイクロ波照射法を用いてスケールアップする場合、そのままのマイクロ波出力では、反応溶液の昇温速度が遅くなり、所望の反応温度に到達するまでに、スケールアップ前と比較して長い時間がかかってしまう。トリスオルトメタル化イリジウム錯体の製造の場合、メリジオナル体はフェイシャル体より低い反応温度で生成しやすく、スケールアップを行うとメリジオナル体がより生成しやすくなる可能性がある。そこで、スケールアップ前と同じ昇温速度を保つためには、マイクロ波の照射エネルギーを反応液量に対応して大きくすることが必要であるが、マイクロ波発振器の出力を無制限に大きくすることはできないし、また製造コストの点でも不利となる。本発明の製造方法では、予備加熱した反応溶媒中へ2種の反応基質を後添加して反応させるので、所望の反応温度まで迅速に昇温させることができ、フェイシャル体選択性を高めることが可能となる。
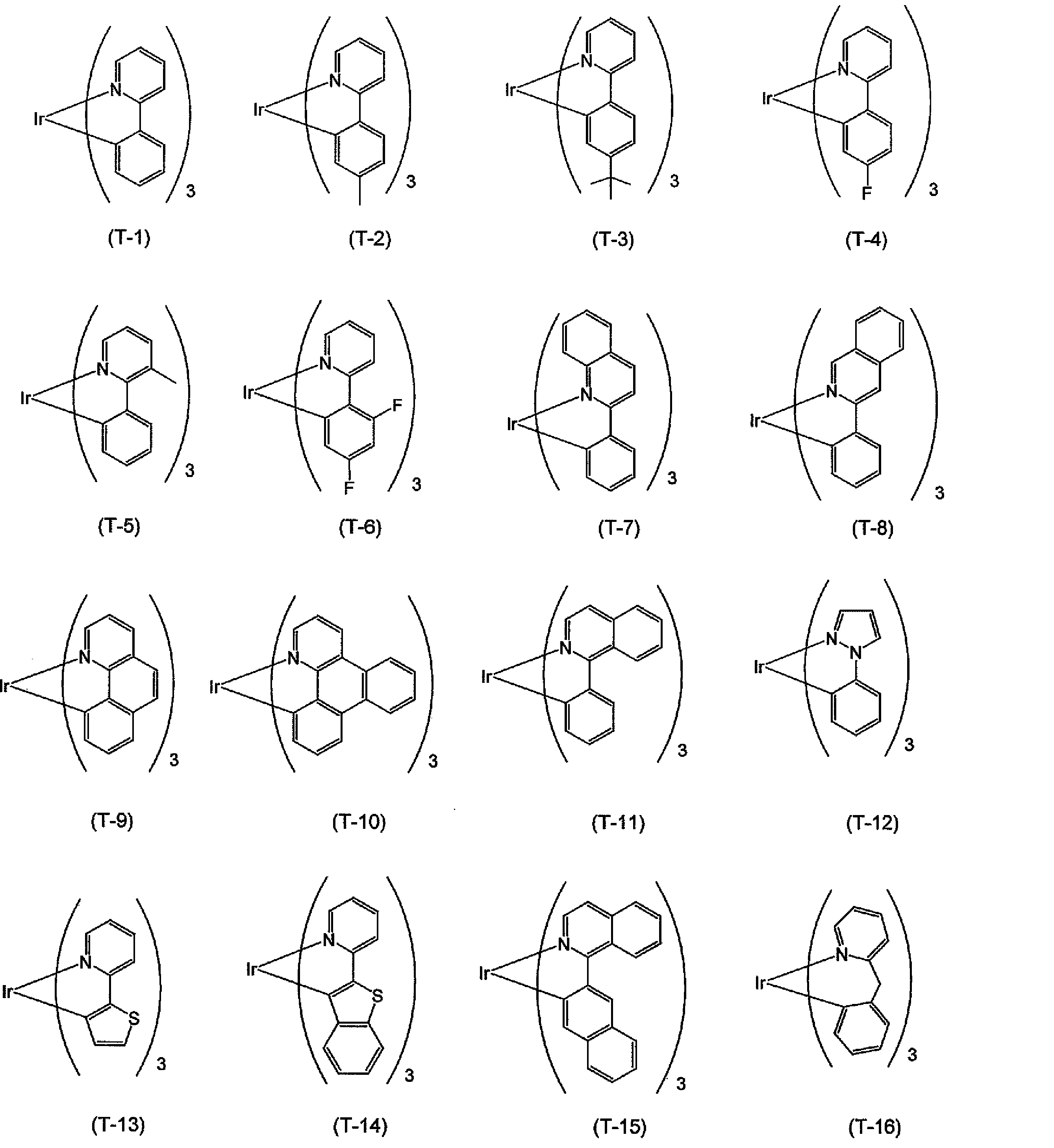
本実施形態に係る製造方法によって製造される一般式(化6)で表わされるトリスオルトメタル化イリジウム錯体の例を化学式(化11)の(T‐1)~(T‐16)に示すが、本発明はこれに限定されるものではない。
本実施形態に係る発光材料は、本実施形態に係る製造方法で製造したトリスオルトメタル化イリジウム錯体からなる。発光材料は、例えば、有機電界発光素子の発光層の材料として好適である。
本実施形態に係る発光素子は、本実施形態に係る発光材料を用いてなる。有機電界発光素子は、通常、ガラス板、プラスチック板などからなる基板上に陽極と、正孔輸送層と、発光層と、電子輸送層と、陰極とを、順次積層した構造を有する。発光層の形成方法は、特に限定されず、蒸着成膜法、湿式成膜法など公知の方法で形成することができる。本実施形態に係る製造方法で製造したトリスオルトメタル化イリジウム錯体を発光素子の発光層に含有することで、従来よりも発光効率及び耐久性に優れた発光素子とすることができる。したがって、本実施形態に係る発光素子は、例えば、OAコンピュータ、壁掛けテレビなどのフラットパネル・ディスプレイ、車載表示素子、携帯電話表示、複写機、液晶ディスプレイ、計器類のバックライト光源などの面発光体としての特徴を生かした光源、表示板、標識灯へ応用することができる。
次に、実施例を示しながら本発明についてさらに詳細に説明するが、本発明は実施例に限定して解釈されない。
<実施例1>トリスオルトメタル化イリジウム錯体(T-9)の製造
特級エチレングリコール240mLを500mLの二口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。これにアルゴンガスを25分間吹き込んだ後、反応溶媒を磁気攪拌しながら1kWのマイクロ波(2450MHz)を照射し、沸騰状態(198℃~200℃付近)まで約3分間で昇温させて予備加熱した。ここで、一旦マイクロ波照射を中断し、反応溶液にベンゾ[h]キノリン13.7gと塩素架橋イリジウムダイマー(D-9)2.97gとを粉末状態で順に添加し、更にアルゴン雰囲気下で1kWのマイクロ波を20分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥し、化学式(化11)に記載の(T-9)を得た(収量3.51g、収率95%)。生成物をプロトンNMR(JEOL社製、JNM-ECX400:400MHz、DMSO‐d6中)で分析した結果、フェイシャル体とメリジオナル体とが90:10(モル比)の混合物であった。反応スキームを反応式(化12)に示す。

特級エチレングリコール240mLを500mLの二口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。これにアルゴンガスを25分間吹き込んだ後、反応溶媒を磁気攪拌しながら1kWのマイクロ波(2450MHz)を照射し、沸騰状態(198℃~200℃付近)まで約3分間で昇温させて予備加熱した。ここで、一旦マイクロ波照射を中断し、反応溶液にベンゾ[h]キノリン13.7gと塩素架橋イリジウムダイマー(D-9)2.97gとを粉末状態で順に添加し、更にアルゴン雰囲気下で1kWのマイクロ波を20分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥し、化学式(化11)に記載の(T-9)を得た(収量3.51g、収率95%)。生成物をプロトンNMR(JEOL社製、JNM-ECX400:400MHz、DMSO‐d6中)で分析した結果、フェイシャル体とメリジオナル体とが90:10(モル比)の混合物であった。反応スキームを反応式(化12)に示す。
<比較例1>トリスオルトメタル化イリジウム錯体(T-9)の製造
ベンゾ[h]キノリン14.3gと塩素架橋イリジウムダイマー(D-9)2.99gと特級エチレングリコール240mLとを500mLの二口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを25分間吹き込んだ後、反応溶液を磁気攪拌しながら1kWのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約3分間で昇温させ、更にアルゴン雰囲気下で1kWのマイクロ波を20分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥し、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-9)を得た(収量3.48g、収率94%)。生成物をプロトンNMR(JEOL社製、JNM-ECX400:400MHz、DMSO‐d6中)で分析した結果、フェイシャル体とメリジオナル体とが66:34(モル比)の混合物であった。
ベンゾ[h]キノリン14.3gと塩素架橋イリジウムダイマー(D-9)2.99gと特級エチレングリコール240mLとを500mLの二口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを25分間吹き込んだ後、反応溶液を磁気攪拌しながら1kWのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約3分間で昇温させ、更にアルゴン雰囲気下で1kWのマイクロ波を20分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥し、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-9)を得た(収量3.48g、収率94%)。生成物をプロトンNMR(JEOL社製、JNM-ECX400:400MHz、DMSO‐d6中)で分析した結果、フェイシャル体とメリジオナル体とが66:34(モル比)の混合物であった。
<実施例2>トリスオルトメタル化イリジウム錯体(T-1)の製造
特級エチレングリコール150mLを500mLの2口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを20分間吹き込んだ後、反応溶液を磁気攪拌しながら300Wのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約7分間で昇温させ予備加熱した。ここで、一旦マイクロ波照射を中断して、反応溶液に2‐フェニルピリジン15.0gと塩素架橋イリジウムダイマー(D-1)1.50gとを粉末状態で添加し、更にアルゴン雰囲気下で300Wのマイクロ波を45分照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥することで、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-1)を得た(収量1.783g、収率97%)。生成物をHPLC(島津製作所社製、Prominence、検出波長:300nm)で分析した結果、フェイシャル体とメリジオナル体とが99.6:0.4(モル比)の混合物であった。反応スキームを反応式(化13)に示す。

特級エチレングリコール150mLを500mLの2口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを20分間吹き込んだ後、反応溶液を磁気攪拌しながら300Wのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約7分間で昇温させ予備加熱した。ここで、一旦マイクロ波照射を中断して、反応溶液に2‐フェニルピリジン15.0gと塩素架橋イリジウムダイマー(D-1)1.50gとを粉末状態で添加し、更にアルゴン雰囲気下で300Wのマイクロ波を45分照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥することで、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-1)を得た(収量1.783g、収率97%)。生成物をHPLC(島津製作所社製、Prominence、検出波長:300nm)で分析した結果、フェイシャル体とメリジオナル体とが99.6:0.4(モル比)の混合物であった。反応スキームを反応式(化13)に示す。
<比較例2>トリスオルトメタル化イリジウム錯体(T-1)の製造
2‐フェニルピリジン15.0gと塩素架橋イリジウムダイマー(D-1)1.50gと特級エチレングリコール150mLとを500mLの2口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを20分間吹き込んだ後、反応溶液を磁気攪拌しながら300Wのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約7分間で昇温させ、更にアルゴン雰囲気下で300Wのマイクロ波を45分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥することで、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-1)を得た(収量1.780g、収率97%)。生成物をHPLC(島津製作所社製Prominence、検出波長:300nm)で分析した結果、フェイシャル体とメリジオナル体とが99.1:0.9(モル比)の混合物であった。
2‐フェニルピリジン15.0gと塩素架橋イリジウムダイマー(D-1)1.50gと特級エチレングリコール150mLとを500mLの2口フラスコに入れ、マイクロ波反応装置(マイルストーンゼネラル社製、Microsynth)にセットした。この反応溶液にアルゴンガスを20分間吹き込んだ後、反応溶液を磁気攪拌しながら300Wのマイクロ波(2450MHz)を照射し、室温から沸騰状態(198℃~200℃付近)まで約7分間で昇温させ、更にアルゴン雰囲気下で300Wのマイクロ波を45分間照射して、198℃~200℃付近で反応させた。反応溶液を室温まで冷却後、反応溶液を濾過し黄色固体を得た。この黄色固体をメタノール、純水、更にメタノールで洗浄後、真空乾燥することで、化学式(化11)に記載のトリスオルトメタル化イリジウム錯体(T-1)を得た(収量1.780g、収率97%)。生成物をHPLC(島津製作所社製Prominence、検出波長:300nm)で分析した結果、フェイシャル体とメリジオナル体とが99.1:0.9(モル比)の混合物であった。
以上の実施例より、本発明に係る製造方法(実施例1~2)は、従来の製造方法(比較例1~2)と比較して、メリジオナル体の生成は抑制され、フェイシャル体の純度が向上することが明らかになった。フェイシャル体のトリスオルトメタル化イリジウム錯体は、その幾何異性体であるメリジオナル体よりも発光効率及び安定性で優れていることが明らかにされているので、本発明に係る製造方法で製造されたトリスオルトメタル化イリジウム錯体を発光素子材料として用いることで、高効率、かつ、高耐久性の発光素子を作製することができる。また、発光素子材料として好ましくないメリジオナル体を、種々の精製手法(再結晶、カラムクロマトグラフィー、昇華精製など)で低減することは、非常に手間と時間を要することであることから、本発明の製造方法は、製造コスト低減にも大きく寄与でき、実用的利点は大である。
Claims (12)
- 反応基質として一般式(化4)で表されるオルトメタル化イリジウム錯体と一般式(化5)で表される2座有機配位子とを反応させて、一般式(化6)で表されるトリスオルトメタル化イリジウム錯体を製造する方法であって、
反応溶媒を予備加熱する工程(1)と、
該予備加熱した反応溶媒にオルトメタル化イリジウム錯体と2座有機配位子とを添加する工程(2)と、
オルトメタル化イリジウム錯体と2座有機配位子とを反応させる工程(3)と、
を順次有することを特徴とするトリスオルトメタル化イリジウム錯体の製造方法。



- オルトメタル化イリジウム錯体が、一般式(化7)で表される化合物であることを特徴とする請求項1に記載のトリスオルトメタル化イリジウム錯体の製造方法。

- 前記Laが、モノアニオン性配位子であることを特徴とする請求項1に記載のトリスオルトメタル化イリジウム錯体の製造方法。
- 前記Laが、中性配位子であることを特徴とする請求項1に記載のトリスオルトメタル化イリジウム錯体の製造方法。
- オルトメタル化イリジウム錯体が、一般式(化8)で表わされる化合物であることを特徴とする請求項1又は3に記載のトリスオルトメタル化イリジウム錯体の製造方法。

- オルトメタル化イリジウム錯体が、一般式(化9)で表わされる化合物であることを特徴とする請求項1又は4に記載のトリスオルトメタル化イリジウム錯体の製造方法。

- 2座有機配位子が、一般式(化10)に示す(7)~(17)の中から選ばれる少なくとも1つであることを特徴とする請求項1~6のいずれか一つに記載のトリスオルトメタル化イリジウム錯体の製造方法。

- 前記工程(3)の反応温度が、100~300℃の範囲であることを特徴とする請求項1~7のいずれか一つに記載のトリスオルトメタル化イリジウム錯体の製造方法。
- 前記工程(1)の予備加熱温度が、前記工程(3)の反応温度以下であり、かつ、100~300℃の範囲であることを特徴とする請求項1~8のいずれか一つに記載のトリスオルトメタル化イリジウム錯体の製造方法。
- 前記工程(3)を、マイクロ波照射下で行うことを特徴とする請求項1~9のいずれか一つに記載のトリスオルトメタル化イリジウム錯体の製造方法。
- 請求項1~10のいずれか一つに記載の製造方法で製造したトリスオルトメタル化イリジウム錯体からなる発光材料。
- 請求項11に記載の発光材料を用いた発光素子。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012540878A JP5881216B2 (ja) | 2010-10-28 | 2011-10-25 | トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-242752 | 2010-10-28 | ||
| JP2010242752 | 2010-10-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012057139A1 true WO2012057139A1 (ja) | 2012-05-03 |
Family
ID=45993846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/074551 WO2012057139A1 (ja) | 2010-10-28 | 2011-10-25 | トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5881216B2 (ja) |
| WO (1) | WO2012057139A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017221848A1 (ja) * | 2016-06-24 | 2017-12-28 | 国立研究開発法人産業技術総合研究所 | ハロゲン架橋イリジウムダイマーの製造方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105055A (ja) * | 2000-09-29 | 2002-04-10 | Fuji Photo Film Co Ltd | イリジウム錯体またはその互変異性体の製造方法 |
| JP2007070290A (ja) * | 2005-09-07 | 2007-03-22 | Tama Tlo Kk | オルトメタル化イリジウム錯体の製造方法及びヨウ素架橋イリジウムダイマー錯体とその製造方法 |
| JP2008505076A (ja) * | 2004-06-29 | 2008-02-21 | イーストマン コダック カンパニー | 有機金属シクロメタル化遷移金属錯体の合成 |
-
2011
- 2011-10-25 WO PCT/JP2011/074551 patent/WO2012057139A1/ja active Application Filing
- 2011-10-25 JP JP2012540878A patent/JP5881216B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002105055A (ja) * | 2000-09-29 | 2002-04-10 | Fuji Photo Film Co Ltd | イリジウム錯体またはその互変異性体の製造方法 |
| JP2008505076A (ja) * | 2004-06-29 | 2008-02-21 | イーストマン コダック カンパニー | 有機金属シクロメタル化遷移金属錯体の合成 |
| JP2007070290A (ja) * | 2005-09-07 | 2007-03-22 | Tama Tlo Kk | オルトメタル化イリジウム錯体の製造方法及びヨウ素架橋イリジウムダイマー錯体とその製造方法 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017221848A1 (ja) * | 2016-06-24 | 2017-12-28 | 国立研究開発法人産業技術総合研究所 | ハロゲン架橋イリジウムダイマーの製造方法 |
| JP2017226633A (ja) * | 2016-06-24 | 2017-12-28 | 国立研究開発法人産業技術総合研究所 | ハロゲン架橋イリジウムダイマーの製造方法 |
| US10844086B2 (en) | 2016-06-24 | 2020-11-24 | National Institute Of Advanced Industrial Science And Technology | Method for producing halogen-crosslinked iridium dimer |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5881216B2 (ja) | 2016-03-09 |
| JPWO2012057139A1 (ja) | 2014-05-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5881215B2 (ja) | トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 | |
| JP4756273B2 (ja) | オルトメタル化イリジウム錯体の製造方法ならびに製造されたイリジウム錯体からなる発光材料 | |
| KR100783711B1 (ko) | 금속 화합물 및 이를 포함하는 유기 전계 발광 소자 | |
| JP5692805B2 (ja) | イリジウム錯体の製造方法ならびに製造されたイリジウム錯体からなる発光材料 | |
| US10053479B2 (en) | Raw material and production method for cyclometalated iridium complex | |
| US8034934B2 (en) | Process for producing ortho-metalated complex of iridium with homoligand | |
| US8436172B2 (en) | Material selecting method upon purifying iridium complex by sublimation | |
| JP5881216B2 (ja) | トリスオルトメタル化イリジウム錯体の製造方法及びそれを用いた発光材料並びに発光素子 | |
| JP6703222B2 (ja) | シクロメタル化イリジウム錯体の製造方法、及び、有機イリジウム材料からなるシクロメタル化イリジウム錯体の前駆体 | |
| JP2017226633A (ja) | ハロゲン架橋イリジウムダイマーの製造方法 | |
| JP6978784B2 (ja) | シクロメタル化イリジウム錯体の製造方法 | |
| JP6651168B2 (ja) | シクロメタル化イリジウム錯体の製造方法 | |
| WO2017221849A1 (ja) | シクロメタル化イリジウム錯体の製造方法、及び、当該方法に好適に用いられる新規なイリジウム化合物 | |
| WO2016111256A1 (ja) | シクロメタル化イリジウム錯体の原料及び製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11836272 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012540878 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11836272 Country of ref document: EP Kind code of ref document: A1 |