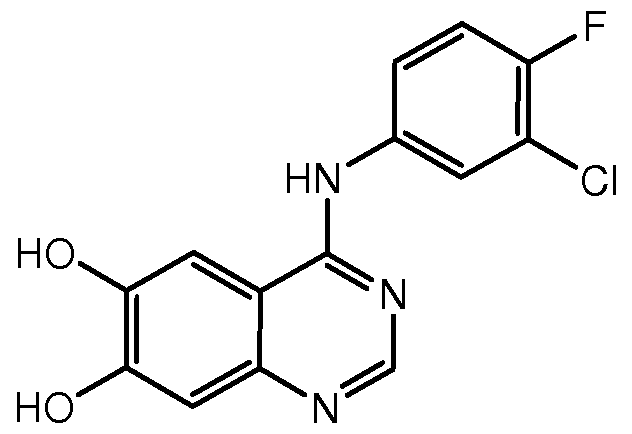
一种抑制肿瘤细胞增殖的喹唑啉衍生物、 喹唑啉配合物蛋白激酶抑制剂 及它们的制备方法
技术领域
本发明涉及一种抑制肿瘤细胞增殖的喹唑啉衍生物、 喹唑啉配合物蛋白激酶抑制 剂及它们的制备方法。 背景技术
临床使用的抗肿瘤化学药物包括细胞毒性药物和分子靶向药物两大类。细胞毒性抗 肿瘤药物(如顺铂等)都是非特异性的,它们在抑制和杀伤异常增殖的肿瘤细胞的同时, 对其它增殖较快的正常细胞也同样产生抑制和杀伤作用, 由此产生的毒副作用以及肿瘤 细胞对药物先天或获得性的抗药性等问题一直是制约细胞毒性化疗药物临床应用的瓶 颈。
近十年来, 针对肿瘤细胞特异的增殖、 分化和凋亡机制, 高选择性的分子靶向治 疗药物得到迅速发展。但是, 许多对肿瘤细胞高表达的蛋白激酶有很好抑制活性的小分 子化合物, 由于水溶性差或毒副作用大, 以及易产生抗药性而不能被临床应用。 发明内容
本发明的目的在于提供一种具有分子靶向特性的可以作为酪氨酸蛋白激酶抑制剂 的喹唑啉衍生物、 喹唑啉配合物及它们的制备方法, 所述喹唑啉衍生物的结构上含有潜 在金属配位位点, 能与细胞毒性的金属类抗肿瘤化合物配位, 以改善酪氨酸蛋白激酶抑 制剂的水溶性, 同时降低细胞毒性药物的毒副作用。 该类蛋白激酶抑制剂喹唑啉衍生物 不仅有很好的激酶抑制效果, 而且在与钌、 铂等金属配位得到喹唑啉配合物后仍然表现 出良好的抑制肿瘤细胞增殖性能。 而且, 在外加表皮生长因子 EGF 的条件下, 大多数 化合物对表皮生长因子受体 EGFR过表达的肿瘤细胞 (如人乳腺癌细胞株 (敏感型) MCF-7/S) 的增殖表现出更好的抑制活性, 说明表皮生长因子受体 EGFR (—种酪氨酸 蛋白激酶)是本发明提供的喹唑啉衍生物、 喹唑啉配合物蛋白激酶抑制剂对肿瘤细胞增 殖起抑制作用的靶标之一。
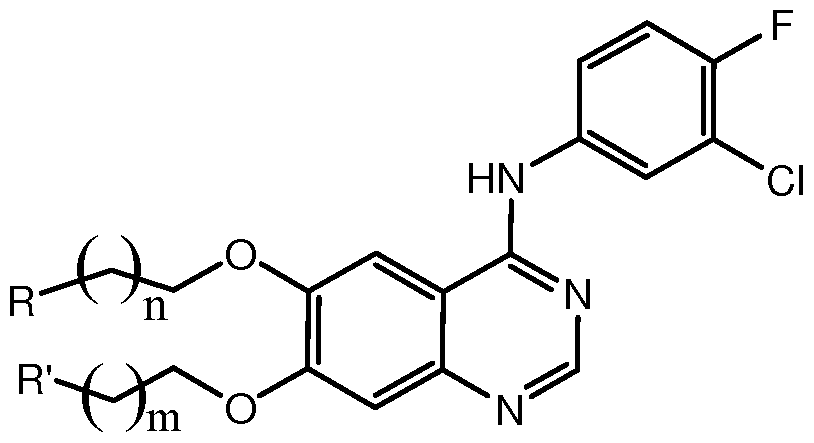
本发明提供了一种 式 (1)表示的分子结构:
Π)
在通式 (1)中, R和 R'中的至少一个为含有能够与贵金属配位的原子的基团, m和 n相同或不同, 为 0-5的整数。
本发明还提供了该喹唑啉衍生物的制备方法,其中,该方法包括提供第一反应物 A, 所述第一反应物 A为式(a)所示的化合物, 式(a) 中 。。和 Run相同或不同, 独立地 选自氢或甲基, 并且其中至少一个为氢; 该方法包括用式(Q1 )所示的基团取代式(a) 中的 。。位置的氢或甲基和 /或用式(Q2)所示的基团取代式(a) 中的 R1()1位置的氢或 甲基; R和 R'中的至少一个为含有能够与贵金属配位的原子的基团, m和 n相同或不同, -5的整数;
R
本发明还提供了一种喹唑啉配合物蛋白激酶抑制剂, 所述喹唑啉配合物由含贵金 属的可配位化合物和能够与该可配位化合物中的贵金属配位的配体形成, 其中, 所述配 体为本发明提供的所述的喹唑啉衍生物。
本发明还提供了一种喹唑啉配合物蛋白激酶抑制剂的制备方法, 该方法包括将含 贵金属的可配位化合物与配体进行配位,所述配体为由本发明提供的方法制备得到的喹 唑啉衍生物。
本发明提供的喹唑啉衍生物为一类酪氨酸蛋白激酶抑制剂 Iressa衍生物,其结构中 含有潜在金属配位位点,能够取代细胞毒性金属配合物(如钌有机金属配合物、顺铂等) 中的一个或多个配体, 而且所述喹唑啉衍生物与细胞毒性的金属钌 (Π、 III) 和 /或铂 形成作为酪氨酸蛋白激酶抑制剂的喹唑啉配合物,使一批由于水溶性差而不能作为分子 靶向药物的高活性蛋白激酶抑制剂重新成为候选化合物;通过一个或多个分子靶向药物 单元的引入降低细胞毒性金属抗肿瘤药物的使用剂量和频率,达到降低单一靶向细胞毒 性药物毒副作用的目的。 而且, "单分子多靶点"作用机制的引入将有助于降低肿瘤细 胞对药物产生抗药性。
并且, 采用两种体外分析方法考察本发明提供的喹唑啉衍生物以及喹唑啉配合物 对酪氨酸蛋白激酶活性抑制情况后得知:
酶联免疫吸附分析(ELISA)结果表明: 本发明提供的喹唑啉衍生物以及喹唑啉配 合物对蛋白激酶表皮生长因子 EGFR的磷酸化作用有很好的抑制活性。
同时, 对肿瘤细胞增殖影响实验的结果表明: 本发明提供的喹唑啉衍生物以及喹 唑啉配合物酪氨酸蛋白激酶抑制剂, 分别对人乳腺癌细胞株 (抗药型) MCF-7/A、 人乳 腺癌细胞株 (敏感型) MCF-7/S、 前列腺癌细胞 PC-3、 角朊细胞 Colo-16、 人非小细胞
肺癌细胞株 A549等多种肿瘤细胞增殖均表现出良好的抑制效果。 且在外加表皮生长因 子 EGF条件下, 本发明提供的喹唑啉衍生物以及喹唑啉配合物酪氨酸蛋白激酶抑制剂 对表皮生长因子受体 EGFR过表达的肿瘤细胞(如人乳腺癌细胞株(敏感型) MCF-7/S ) 的细胞增殖也表现出更高的抑制效果。 附图说明
图 1-a为编号为 JLY2009的化合物的 X射线衍射晶体结构图, 图 1-b为与其对应 的化合物的结构式;
图 2为在 ELISA测试条件下测得编号为 LQ 1001的参比化合物 IC5Q=4nM的曲线图; 图 3为在 ELISA测试条件下测得编号为 JLY1002的化合物 IC5Q=60.2nM的曲线图; 图 4为在 ELISA测试条件下测得编号为 JLY2007的化合物 IC5Q=7.5nM曲线图; 图 5为在 ELISA测试条件下编号为 ZW1001的化合物 IC5Q=4.6nM的曲线图; 图 6为在 ELISA测试条件下编号为 ZW2001的化合物 IC5Q』lnM的曲线图; 图 7为编号为 JLY2007的化合物在 MCF-7/S + EGF 抑制肿瘤细胞增殖的测试条件 下, 测得的 IC5Q=24.48 M的曲线图;
图 8为编号为 ZW2004的化合物在 MCF-7/S + EGF 抑制肿瘤细胞增殖的测试条件 下, 测得的 IC5。=33.87 M的曲线图。 具体实施方式
根据本发明提供的 式 (1)表示的分子结构:
(1)
在通式 (1)中, R和 R'中的至少一个为含有能够与贵金属配位的原子的基团, m和 n相同或不同, 为 0-5的整数。
其中, R和 /或 R'中的至少一个为含有能够与贵金属配位的原子的基团, 例如, R 和 /或 R'中能够与贵金属配位的原子可以为氧原子、 氮原子和硫原子中的一种或多种。 优选情况下, R和 /或 R'中能够与贵金属配位的原子为氧原子和 /或氮原子; 所述氧原子 可以为羟基中的氧原子, 所述氮原子可以为胺基中的氮原子或芳香杂环上的氮原子。 具 体来说, 所述胺基可以为脂肪族胺基, 所述芳香杂环可以选自咪唑环、 吡啶环、 2,2'-联 吡啶环、 菲洛啉环和 8-羟基喹啉环中的一种。 R和 /或 R'中能够与贵金属配位的原子个 数可以根据配位的需要适当选择, 通常可以为 1-3个。
按照本发明, 所述贵金属通常可以为钌和 /或铂等。
按照本发明, 在通式 (1)中, 所述具有配位功能的基团 R和 R'可以是各种能与金属 特别是细胞毒性的金属钌形成配位体的基团。
优选情况下, 按照本发明的一种具体实施方式, m为 0, R'为氢, 且所述具有配位 功能的基团 R选自稠杂环亚胺基或者取代的稠杂环亚胺基、胺基烷基亚胺基、环上具有 叔胺基的具有咪唑类五元杂环结构的基团、六元芳杂环亚胺基或者取代的六元芳杂环亚 胺基中的任意一种, 且所述亚胺基或叔胺基上的氮与 6位氧所在的烷基链连接。 δΡ, 稠 杂环亚胺基或者取代的稠杂环亚胺基、六元芳杂环亚胺基或者取代的六元芳杂环亚胺基 中的亚胺基上的氮用于与 6位氧所在的烷基链连接, 环上的氮原子、 氧原子以及硫原子 中的至少一个能够与贵金属配位;胺基烷基亚胺基的亚胺基上的氮用于与 6位氧所在的 烷基链连接, 且胺基和亚胺基上的氮原子均能够与贵金属配位; 环上具有叔胺基的具有 咪唑类五元杂环结构的基团的叔胺基上的氮与 6位氧所在的烷基链连接, 咪唑类五元杂 环结构上的其它氮原子、 氧原子以及硫原子中的至少一个能够与贵金属配位。
具体来说,所述稠杂环亚胺基或者取代的稠杂环亚胺基具有通式 (2)-(7)中任意一个 所示的结构:
(5) (6) (7)
其中, 在上述通式 (5)-(7)中, n可以各自为 0-3的整数。优选情况下, 所述稠杂环亚 胺基或者取代的稠杂环亚胺基具有通式 (2)和 (16)中任意一个所示的结构:
(2) (16)。
(8)
其中, 在通式 (8)中, d为 2-5的整数, 、 R2和 R3各自独立地选自氢原子和碳原 子数为 1-3 的烷基中的任意一种。 更优选情况下, 所述胺基烷基亚胺基具有通式 (17)所 示的结构:
Η,Ν^^ ^肌
(17)。
所述环上具有叔胺基的具有咪唑类五元杂环结构的基团具有通式 (9)所示的结构:
(9)
其中, 在通式 (9)中, 、 R5和 各自独立地选自氢原子和碳原子数为 1-3的烷基 中的一种; 优选情况下, 所述环上具有叔胺基的具有咪唑类五元杂环结构的基团具有通 式 (18)
所述六元芳杂环亚胺基或者取代的六元芳杂环亚胺基具有通式 (10)-(14)所示的结 构:
(11) (12)
(13) (14)
其中, 在通式 CIO)中, R- Rs、 R 、 R
1Q和 R
u各自独立地为氢原子、 亚胺基、 碳原 子数为 1-3的烷基中的一种, 且 R
7、 R
8、 R
9、 。和 R
u中的至少一个基团为亚胺基; 在通式 (11)-(14)中, t各自为 0-3的整数。 优选情况下, 所述六元芳杂环亚胺基或者取代 的六元 亚胺基可以由下述通式 (19)-(20)中的任意一种表示:
(19) (20)。
其中, 在通式 (19)中, 氨基的位置可以位于吡啶氮的对位、 间位或邻位, 如下述结 构式所示:
按照本发明, 所述喹唑啉衍生物的制备方法包括提供第一反应物 A, 所述第一反 应物 A为式(a)所示的化合物, 式(a) 中 。。和 Run相同或不同, 独立地选自氢或甲 基, 并且其中至少一个为氢; 该方法包括用式 (Q1 ) 所示的基团取代式 (a) 中的 R
1QQ 位置的氢或甲基和 /或用式 (Q2) 所示的基团取代式 (a) 中的 R
1()1位置的氢或甲基; R 和 R'中的至少一个为含有能够与贵金属配位的原子的基团, m和 n相同或不同, 为 0-5 的整数;
(a) (Ql ) (Q2)。
其中, R和 /或 R'中能够与贵金属配位的原子与前述所定义的内容相同。
当 m为 0, R'为氢, R为具有配位功能的基团时, 按照本发明提供的酪氨酸蛋白 激酶的制备方法, 在方式 (1 ) 中, 所述第一有机胺选自烷基二胺或取代的烷基二胺以 及环上具有亚胺基的具有咪唑类五元杂环结构的化合物中的任意一种; 在方式 (2) 中, 所述第二有机胺选自稠杂环基取代的胺或者取代的稠杂环取代的胺以及六元芳杂环基 取代的胺或者取代的六元芳杂环取代的胺中的任意一种。
优选情况下, 所述稠杂环基取代的胺或者取代的稠杂环取代的胺为通式 (21)-(26) 所示, 所述环上具有亚胺基的具有咪唑类五元杂环结构的化合物为通式 (27)所示, 六元 芳杂环基取代的胺或者取代的六元芳杂环取代的胺为通式 (28)-(32)所示, 所述烷基二胺 或取
(32) (33)
其中, 在通式 (;24)-(;26)中, r可以各自为 0-3 的整数; 在通式 (27)中, 、 R5和 可以各自独立地选自氢原子和碳原子数为 1-3的烷基中的任意一种; 在通式 (28)中, R7、 R8、 R9、 。禾卩 Ru可以各自独立地为氢原子、亚胺基、碳原子数为 1-3的烷基中的一种, 且 R7、 R8、 R9、 。和 Ru中的至少一个基团为胺基; 在通式 (29)-(32)中, t可以各自为 0-3的整数; 在通式 (33)中, d可以为 2-5的整数, 、 R2和 R3可以各自独立地选自氢 原子和碳原子数为 1-3的烷基中的任意一种。
优选情况下, 所述第一有机胺为如通式 (34)至 (35)所示化合物中的任意一种; 所述 第二有机 一种:
(38) (39)。
即, 所述第一有机胺中通式 (33)所述的化合物优选为如通式 (34)所示化合物, 所述
第一有机胺中通式 (27)所述的化合物优选为如通式 (35)所示化合物, 所述第二有机胺中 通式 (24)所述的化合物优选为如通式 (37)所示的化合物,所述第二有机胺中通式 (28)所述 的化合物优选为如通式 (38)所示化合物, 所述第二有机胺中通式 (29)所述的化合物优选 为如通式 (39)所示化合物。
按照本发明提供的方法, 所述第一有机胺和第二有机胺均可以商购得到, 也可以 按照本各种常规的制备方法制备得到。
按照本发明提供的方法, R1QQ为氢、 R1Q1为甲基,用式(Q1 )所示的基团取代式(a) 中的氢的方法包括方式 (一) 和方式 (二):
按照本发明, 方式 (一) 包括:
( 1 )在第一有机溶剂的存在下, 将第一反应物 A与二卤代烷接触反应, 生成中间 体产物 B, 所述二卤代烷由下式(k)所示, 所述中间体产物 B由下式(b )所示, 其中, X、 X
(2 ) 在第二有机溶剂的存在下和縮合反应条件下, 将步骤 (1 ) 得到的中间体产 物 B与含有能够与贵金属配位的原子的第一有机胺加热回流,縮合反应的条件使得中间 体产物 B中 6位的卤代烷氧基中的卤原子与所述第一有机胺发生縮合反应。
在方式 (一) 中:
在步骤(1 ) 中, 将第一反应物 A与二卤代烷接触反应的条件可以包括反应的温度 和反应的时间, 所述反应温度可以在较宽的温度范围内进行, 优选情况下, 反应的温度 可以为 50-100°C, 更优选 70-90°C ; 反应时间的延长有利于反应物的转化率或反应产物 的收率的提高,但是反应时间过长对反应物的转化率或反应产物的收率的提高幅度并不 明显, 因此, 一般情况下, 所述反应的时间可以为 1-10小时, 更优选 2-6小时。
所述第一反应物 A与二卤代烷的摩尔比为可以 1 : ( 3-8), 优选为 1 : ( 3-4.5 )。 为了进一步促进反应向正方向进行, 在步骤(1 ) 中, 将第一反应物 A与二卤代烷 接触反应优选在缚酸剂的存在下进行, 所述缚酸剂的用量的可选择范围较宽, 只要能够 起到促使第一反应物 A与二卤代烷的反应进一步向正方向进行的目的即可, 优选情况 下, 所述缚酸剂与第一反应物 A的摩尔比为 (3-8 ) : 1。
所述卤代烷可以选自二卤乙烷、 二卤丙浣、 二卤丁烷和二卤戊烷中的一种或多种, 具体来说, 可以选自二溴乙烷、 二溴丙烷、 二溴丁烷和二溴戊烷中的一种或多种。
在步骤 (2) 中, 将步骤 (1 ) 得到的中间体产物 B与第一有机胺加热回流的条件
可以包括加热回流的温度和时间, 所述温度一般为 50-95 °C, 时间通常为 1-10小时, 优 选为 2-6小时。
所述中间体产物 B与第一有机胺的摩尔比可以为 1 : ( 1-10), 优选为 1 : ( 1-8)。 为了进一步促进反应向正方向进行, 在步骤(2) 中, 将中间体产物 B与第一有机 胺的加热回流优选在缚酸剂的存在下进行, 所述缚酸剂的用量的可选择范围较宽, 只要 能够起到促使中间体产物 B与第一有机胺的加热回流进一步向正方向进行的目的即可, 优选情况下, 所述缚酸剂与第一有机胺的摩尔比为 (3-8 ) : 1。
所述縮合反应的条件只要使得中间体产物 B中 6位的卤代烷氧基中的卤素与具有 配位功能的基团的化合物中的胺基或亚胺基发生縮合反应即可。
当所述第一有机胺为由通式 (33 ) 特别是由通式 (34 ) 表示的化合物时, 只要在 加热回流的条件下即可以保证使其与所述中间体产物发生縮合反应。当所述第一有机胺 为由通式 (27)特别是由通式 (35 )表示的化合物时, 所述縮合反应的条件还包括在催 化剂的存在下进行, 以进一步促进反应的进行。 所述催化剂可以为各种碱类物质及相转 移催化剂, 例如, KI和 (C4H9)4NBr等催化剂中的一种或多种。
所述第一有机溶剂与第二有机溶剂可以各自独立地选自 N,N-二甲基甲酰胺 (DMF) 和乙腈中的一种或多种; 以第一反应物与二卤代烷的总量为 1000毫克为基准, 所述第 一有机溶剂的用量可以为 4-20毫升; 以中间体产物与第二反应物的总量为 1000毫克为 基准, 第二有机溶剂的用量可以为 10-60毫升。
所述缚酸剂的种类可以为本领域技术人员所公知的各种常规的缚酸剂, 例如, 所 述缚酸剂可以选自 K2C03、 CsC03、 NaOH和三乙胺的一种或多种。
按照本发明, 方式 (二) 包括:
( 1 )在第一有机溶剂的存在下, 将第一反应物 A与卤代羧酸酯接触反应, 生成中 间体产物 C, 所述卤代羧酸酯由下式 (1) 所示, 所述中间体产物 C如下式 (c) 所示, 其中
( 1) ( c)
(2) 在碱催化下, 将步骤 (1 ) 得到的中间体产物 C水解, 得到中间产物 D, 所 述中间产物 D如下式 (d) 所示, 并将中间产物 D进行卤化反应得到中间产物 E, 所述 中间产物 E如下式 (e) 所示; 将所述中间产物 E与第二有机胺接触反应, 所述第二有 机胺为由具有配位功能的基团的化合物,接触反应的条件使得中间产物 E中 6位的烷氧 基酰卤中的卤原子与第二有机胺发生縮合反应, 得到縮合产物 F, 所述縮合产物 F由下
式 (
(3 ) 将步骤 (2) 得到的縮合产物 F中的 6位的烷氧基酰胺中的羰基还原成亚烷 基;
按照本发明提供的方法, 在方式 (二) 中:
在步骤(1 ) 中, 将第一反应物 A与卤代羧酸酯接触反应的条件包括反应的温度和 反应的时间, 所述反应温度可以在较宽的温度范围内进行, 优选情况下, 反应的温度可 以温度为 10-60°C,优选为 20-50°C ; 反应时间的延长有利于反应物的转化率或反应产物 的收率的提高,但是反应时间过长对反应物的转化率或反应产物的收率的提高幅度并不 明显, 因此, 一般情况下, 所述反应的时间可以为 0.3-5小时, 更优选 0.5-4小时。
所述第一反应物与卤代羧酸酯的摩尔比可以为 1 : ( 1-1.5 ), 优选为 1 : ( 1-1.1 )。 为了进一步促进反应向正方向进行, 在步骤 (1 ) 中, 将第一反应物与卤代羧酸酯 的接触反应优选在缚酸剂的存在下进行, 所述缚酸剂的用量的可选择范围较宽, 只要能 够起到促使第一反应物与卤代羧酸酯的反应进一步向正方向进行的目的即可,优选情况 下, 所述缚酸剂与第一反应物 A的用量摩尔比为 (2-5 ) :1。
按照本发明, 所述卤代羧酸酯可以选自卤代乙酸乙酯、 卤代乙酸甲酯和卤代丙酮 酸乙酯中的一种或多种。具体来说, 可以选自溴代乙酸乙酯、溴代乙酸甲酯和溴代丙酮 酸乙酯中的一种或多种。
以第一反应物与卤代羧酸酯的总量为 1000毫克为基准, 所述第一有机溶剂的用量 一般为 10-20毫升。
在步骤 (2) 中, 在碱催化下, 将步骤 (1 ) 得到的中间产物 C水解的条件可以为 常规的将酯水解为酸的条件。 例如, 水解的温度可以为 20-60°C, 优选为 25-40°C, 水解 的时间可以为 1-15小时, 优选为 2-6小时, 所述碱一般可以选自 NaOH、 LiOH和 KOH 中的一种或多种, 所述碱的用量一般可以为中间产物 C的物质的量的 3-5倍。 通常该水
解反应在混合溶剂的存在下进行,如水-甲醇-四氢呋喃的混合溶液,以反应物总量为 1000 毫克为基准所述混合溶剂的总用量为 60-150毫升; 混合溶剂的体积比可以为 1 : ( 1-2) : (2-4)。
在步骤 (2) 中, 将中间产物 D进行卤化反应的方法包括将中间产物 D与氯化亚 砜接触反应, 所述接触反应的条件一般包括反应的温度为 25-75 °C, 反应的时间为 1-5 小时, 氯化亚砜的用量为中间产物 D (4-(3'-氯 -4'氟-苯氨基) -6-烷氧基羧酸 -7-甲氧基喹 唑啉) 的物质的量的 5-15 倍; 为了便于水解产物的溶解, 所述反应优选在第一有机溶 剂的存在下进行, 其用量可以根据水解产物的溶解情况而定。 更优选情况下, 在将中间 产物 D进行卤化的步骤中, 为了进一步促进该反应向正方向进行,所述反应还包括在缚 酸剂, 优选为在吡啶的存在下进行, 吡啶的用量可以为 1-3滴 (约 0.5-2毫摩尔)。
将中间产物 D与氯化亚砜接触反应得到的中间产物 E与第二有机胺的接触反应优 选在第三有机溶剂的存在下进行, 接触反应的条件包括反应的温度可以为 3-30°C, 反应 的时间可以为 2-8小时, 所述中间产物 E与第二有机胺的摩尔比为 1 : ( 1-2), 优选为 1 : ( 1.1-1.5 )。 所述第三有机溶剂可以选自二氯甲烷 (CH2C12) 和 /或三氯甲烷, 以中间产 物 E与第二有机胺的总量为 1000毫克为基准, 所述第三有机溶剂的用量为 30-60毫升。
在步骤 (3 ) 中, 将步骤 (2) 得到的縮合产物 F在第四有机溶剂的存在下使 6位 的烷氧基酰胺中的羰基还原的方法为将硼氢化钠与縮合产物 F加热回流,加热回流的条 件通常包括温度为 40-60°C, 时间为 6-20小时, 硼氢化钠的用量可以为縮合产物 F的物 质的量的 2-4倍。 所述第四有机溶剂可以选自 THF和 /或二氧六环, 以硼氢化钠与縮合 产物 F的总量为 1000毫克为基准, 第四有机溶剂的用量为 50-80毫升。 此外, 所述还 原反应优选在酸性环境下进行, 例如, 在惰性气氛中, 向反应体系中加入三氟乙酸 (TFA), 以促进反应向正反向进行 (三氟乙酸的用量可以为 1-3滴 (约 0.5-2毫摩尔)。 所述惰性气氛可以为不与反应物与产物反应的任何惰性气氛, 例如, 氮气以及元素周期 表中的零族气体中的至少一种, 所述惰性气氛可以为流动气氛也可以为静态气氛。
按照本发明提供的方法,为了进一步促进反应向正方向进行,所述将第一反应物 A 与卤代羧酸酯的接触反应以及将中间产物 E 与第二有机胺的接触反应均优选缚酸剂的 存在下进行, 所述缚酸剂与第一反应物 A 的用量摩尔比可以为 (2-5 ) :1 , 所述缚酸剂 与中间产物 E的用量摩尔比可以为 (2-5 ) :1。 所述缚酸剂的种类如前述内容所述。
按照本发明提供的方法, R1QQ为氢、 R1Q1为甲基,用式(Q1 )所示的基团取代式(a) 中的 R1(K)位置的氢并用式 (Q2) 所示的基团取代式 (a) 中的 R1Q1位置的甲基的方法包 括:
( 1 )在惰性气体保护下, 将第一反应物 A与熔融的吡啶盐酸盐接触反应, 生成中 间体产物 H, 所述中间体产物 H如下式 (h) 表示的化合物;
(h)
(2) 在第一有机溶剂的存在下, 将步骤 (1 ) 得到的中间体产物 H与卤代脂肪醇 接触反应得到中间体产物 I, 所述中间体产物 I由下式 (i) 表示的化合物, 并将中间产 物 I进行卤代反应, 得到中间产物 J, 所述中间产物 J由下式 (j ) 所示的化合物, 将中 间产
ω (ρ。
其中, 在步骤 a) 中, 将第一反应物 A与熔融的吡啶盐酸盐接触反应的条件包括 反应温度和反应时间, 所述反应温度可以为 150-185°C, 所述反应时间可以为 2-5小时。
所述惰性气氛可以为不与反应物与产物反应的任何惰性气氛, 例如, 氮气以及元 素周期表中的零族气体中的至少一种, 所述惰性气氛可以为流动气氛也可以为静态气 在步骤( 1 )中,所述第一反应物 A与熔融的吡啶盐酸盐的摩尔比可以为 1 : ( 15-25 )。 在步骤 (2) 中, 将步骤 (1 ) 得到的中间体产物 H与卤代脂肪醇接触反应的条件 可以包括反应的温度为 40-60°C, 反应的时间为 5-15小时。所述中间体产物 H与卤代脂 肪醇的摩尔比可以为 1 : (3-8 )。 所述卤代脂肪醇的碳原子数可以为 1-5, 例如, 所述卤 代脂肪醇可以选自 2-卤代乙醇、 3-卤代丙醇和 4-卤代丁醇中的一种或几种, 具体来说, 可以选自 2-溴乙醇、 3-溴丙醇和 4-溴丁醇中的一种或几种。
在步骤(2)中, 将中间产物 I进行卤代反应的方法包括在第五有机溶剂的存在下, 将中间产物 D与三卤化磷接触反应,接触反应的条件包括反应温度和反应时间,所述反 应温度可以为 90-110°C, 反应时间可以为 1-10小时; 中间产物 D与三卤化磷的摩尔比 可以为 1 : ( 1.2-2.5 )。
所述第五有机溶剂可以选自氯苯、 吡啶和 N,N-二甲基甲酰胺中的一种或多种。 以 中间产物 D 与三卤化磷的总量为 1000 毫克为基准, 所述第五有机溶剂的用量可以为 20-80毫升。
为了进一步促进反应向正方向进行, 将中间产物 I进行的卤代反应优选在缚酸剂, 优选为吡啶的存在下进行, 所述缚酸剂的用量为中间产物 I物质的量的 3-8倍。
在步骤 (2) 中, 将中间产物 J与氨进行氨解反应的方法包括在第六有机溶剂的存 在下, 将中间产物 J与氨接触反应, 中间产物 J与氨的摩尔比可以为 1 : ( 10-30)。
将中间产物 J与氨接触反应,进行氨解的条件一般包括反应温度和反应时间,所述 反应温度可以为 25-50°C, 反应时间可以为 5-15小时。
所述第六有机溶剂可以选自甲醇、 乙醇和异丙醇中的一种或多种, 以中间产物 J 的量为 1000毫克为基准, 所述第六有机溶剂的用量可以为 20-50毫升。
为了提高最终产物的纯度, 该方法还包括分离和纯化所述中间产物的步骤, 所述 分离和纯化的方法可以采用本领域常规的分离方法以及纯化方法, 例如, 所述分离的方 法包括过滤、 离心、 萃取等, 所述纯化的方法包括柱层析分离、 重结晶等方法, 具体操 作条件和方法为本领域技术人员所公知, 在这里不再赘述。
在有机合成过程中的一些常规的操作例如除去溶剂、 洗涤和干燥的方法均可以采 取常规的操作方法进行, 例如, 除去溶剂的方法可以为减压蒸馏的方法。 洗涤的方法可 以为用水、 异丙醇、 乙醚等进行洗涤, 以除去部分未反应的原料。 干燥的方法和条件为 本领域技术人员所公知, 例如, 所述干燥的温度可以为 40-80°C, 优选为 50-60°C ; 干燥 的时间可以为 2-12小时, 优选为 5-8小时。
按照本发明, 所述喹唑啉配合物蛋白激酶抑制剂由含贵金属的可配位化合物和能 够与该可配位化合物中的贵金属配位的配体形成, 其中, 所述配体为本发明提供的所述 喹唑啉衍生物。
按照本发明, 所述喹唑啉配合物蛋白激酶抑制剂可以分别由下述四种通式表示, 即:
按照本发明的一种具体实施方式, 所述喹唑啉配合物蛋白激酶抑制剂由 AG(X'Y')Z表示:
ΧΎ'为权利要求 1中通式 (1 ) 所示的喹唑啉衍生物形成的基团, 其中, m为 0,
R'为氢, R为权利要求 8中由结构式(5 ) - ( 7) 中任意一种所示的稠杂环亚胺基或者取 代的稠杂环亚胺基; 且所述亚胺基上的氮与通式 (1 ) 中 6位氧所在的烷基链连接; 所 述稠杂环上的氮以及羟基上的氧与 G配位; 优选情况下, R为权利要求 9中式(16)所 示的结构;
其中, Z可以选自卤素、 -SCN、 -N3和-SCN中的一种基团; A可以选自苯、 联苯、 异丙基甲苯和环烷并苯中的一种; B为 Cl—, PF6-或 BF4-; G优选为钌。
具体来说, ΧΎ', 即由通式 (1 )所示的喹唑啉衍生物形成的基团可以分别为如下 述结构式所示的基团:
按照本发明的另一种具体实施方式, 所述喹唑啉配合物蛋白激酶抑制剂还可以由 通式 [AG(XY)Z]+B-表示:
XY为权利要求 1中通式(1 )所示的喹唑啉衍生物形成的基团, 其中, m为 0, R' 为氢, R为权利要求 8中由结构式(2) - (4) 中所示的稠杂环亚胺基或者取代的稠杂环 亚胺基和由结构式 (8) 所示的胺基烷基亚胺基以及由通式 (11 ) - ( 14) 所示的六元芳 杂环亚胺基或者取代的六元芳杂环亚胺基中任意一种, 且所述亚胺基上的氮与通式(1 ) 中 6位氧所在的烷基链连接; 其中, 所述稠杂环上的两个氮原子可与 G配位, 或者, 所 述氨基烷基亚胺基上的两个氮原子可与 G配位,或者,所述六元芳杂环上的两个氮原子 可与 G配位。 优选情况下, R为权利要求 9中由式 (2) 所示的结构、 式 (17) 所示的 结构或者式 (20) 所示的结构;
或者, R和 R'均为 -NH2 ; n为 1-3的整数, m为 1-3的整数, 其中, R与 R'上的氮 可与 G配位;
其中, Z可以选自卤素、 -SCN、 -N3和 -SCN中的一种基团; A可以选自苯、 联苯、 异丙基甲苯和环烷并苯中的一种; B为 Cl—, PF6-或 BF4-; G优选为钌。
具体来说, XY, 即由通式 (1 ) 所示的喹唑啉衍生物形成的基团可以分别为如下 述结构式所示的基团:
按照本发明的另一种具体实施方式, 所述喹唑啉配合物蛋白激酶抑制剂还可以由 通式 [AG(X1Y1)Z1]+B-表示:
X1Y1为碳原子数为 1-5的烷基二胺基, Z1为权利要求 1中通式 (1 ) 所示的喹唑 啉衍生物形成的基团, 其中, m为 0, R'为氢, R为权利要求 8中由通式 (9)所示的环 上具有叔胺基的具有咪唑类五元杂环结构的基团以及由通式(10)所示的六元芳杂环亚 胺基或者取代的六元芳杂环亚胺基中的任意一种,且所述亚胺基或叔胺基上的氮与通式 ( 1 ) 中 6位氧所在的烷基链连接; 其中, 所述咪唑类五元杂环结构上的除叔胺基之外 的氮原子可与 G配位, 或者, 所述六元芳杂环上的氮原子可与 G配位;
优选情况下, R为权利要求 9中由式 (18) 或式 (19)所示的结构; X1Y1为碳原 子数为 1-2的烷基二胺基;
其中, A可以选自苯、 联苯、 异丙基甲苯和环烷并苯中的一种; B为 Cl—, PF6-或 BF4"; G优选为钌。
具体来说, Zl, 即由通式 (1)所示的喹唑啉衍生物形成的基团可以分别为如下述 结构式所示的基团:
按照本发明的另一种具体实施方式, 所述喹唑啉配合物蛋白激酶抑制剂为由
G(M)W表示:
M为权利要求 1中通式 (1) 所示的喹唑啉衍生物形成的基团, 其中, m为 0, R' 为氢, R为权利要求 8中由通式(2) - (7)所示的稠杂环亚胺基或者取代的稠杂环亚胺 基、 由通式 (8)所示的胺基烷基亚胺基、 由通式 (9)所示结构的环上具有叔胺基的具 有咪唑类五元杂环结构的基团、 由通式 (10) - (14) 中任意一个所示结构的六元芳杂 环亚胺基或者取代的六元芳杂环亚胺基中的任意一种, W选自卤素和 DMSO中的至少 一种; G为钌;
优选情况下, R为权利要求 9中式(17)或(18)所示的结构; W为卤素和 DMSO;
所述稠杂环上的氮以及羟基上的氧与 G配位, 或者所述稠杂环上的两个氮原子与 G配位; 或者, 所述氨基烷基亚胺基上的两个氮原子与 G配位; 或者, 所述六元芳杂环 上的两个氮原子可与 G配位; 或者, 所述 R与 R'上的氮与 G配位; 或者, 所述咪唑类 五元杂环结构上的除叔胺基之外的氮原子与 G配位; 或者,所述六元芳杂环上的氮原子 与 G配位。
具体来说, M, 即由通式 (1 ) 所示的喹唑啉衍生物形成的基团已经在上述内容中 详细列举, 因此, 在此不再赘述。
按照本发明, 所述喹唑啉配合物蛋白激酶抑制剂的合成路线主要分为两类: 一、 制备有机金属钌基配合物系列: 由通式 ARu(X'Y')Z、 [ARu(XY)Z]+B-或 [ARU(X1Y1)Z1]+B-表示的机金属钌基配合物, 其中, A可以选自苯、 对甲基异丙基苯、 联苯、 环烷并苯等芳烃。
情况 (一):
(A)由含有两个 A基团的卤化钌芳烃二聚体 [(T!6-Arene)RuCl2】2与含两个配位原子 的 ΧΎ'或 XY或 X1Y1基团的螯合配体(即优选为含有乙二胺、联吡啶、 8-羟基喹啉类、 菲洛琳类等结构的 Iressa衍生物) 进行反应制备。 其中, Z为 Cl, B-为 PF6-。
[(T|6-Arene)RuCl2]2 + Iressa衍生物双齿配体 ►
(B)由含有两个 A基团的卤化钌芳烃二聚体 [(T!6-Arene)RuCl2】2先与含两个配位原 子的 X1Y1 的螯合配体分子 (X1Y1 优选为乙二胺) 反应, 再与含单个配位原子的 Z1 基团的单齿配体(即含吡啶、咪唑等结构的 Iressa衍生物)进行反应制备。其中, B- =PF6-。
Iressa衍生物单齿配体
[(r|6-Arene)RuCl2]2 + en ► ► [ARU(X1Y1)Z1]+B- 二、 制备 NAMIA类 Ru ( II、 III) 配合物, 由通式 Ru(M)W表示;
M为权利要求 1中通式 (1 )所示的喹唑啉衍生物形成的基团 (即, 包括上述含两 个配位原子的 ΧΎ'或 ΧΥ或 X1Y1基团的螯合配体 (即优选为含乙二胺、 联吡啶、 8- 羟基喹啉类、 菲洛琳类等结构的 Iressa衍生物) 以及含单个配位原子的 Z1基团的单齿 配体 (即含吡啶、 咪唑等结构的 Iressa衍生物)), W选自卤素和 DMSO中的至少一种。
RuC1 .3H O NAMIA类配合物 Ru(M)W
按照本发明的一种具体实施方式, 由通式 AG(X'Y')Z或通式 [AG(XY)Z]+B-表示的 喹唑啉配合物蛋白激酶抑制剂的制备方法包括:
将含有两个 A基团的卤化钌芳烃二聚体与含有两个配位原子的螯合配体在醇或醇 的水溶液中接触,接触反应的条件使得所述卤化钌芳烃二聚体中的钌与螯合配体中的两 个配位原子螯合配位。
其中, 所述含有两个 A基团的卤化钌芳烃二聚体与含有两个配位原子的螯合配体 的摩尔比可以为 1 :1-3, 所述接触条件包括接触的温度可以为 20-50°C, 接触的时间可以 为 0.5-2小时; 以所述含有两个 A基团的卤化钌芳烃二聚体和含有两个配位原子的螯合 配体的总量为 100毫克为基准, 所述醇或醇的水溶液的用量为 30-50毫升, 所述醇优选 为甲醇, 所述醇的水溶液优选为甲醇与水的混合溶液。 优选情况下, 将反应产物从反应 产物混合物中分离的方法可以采用各种常规的方法, 例如, 在反应产物混合物中加入 NH4PF6, 充分溶解, 浓縮反应液使反应产物析出沉淀, 过滤, 所述 NH4PF6的用量可以 为该步骤 (3 ) 中的氯化钌芳烃二聚体物质的量的 6-20倍。
优选情况下, 由通式 AG(X'Y')Z或通式 [AG(XY)Z]+B-表示的蛋白酶抑制剂的制备 方法还包括用 -SCN、 -N3、 -SCH3、 -SH、 吡啶基、 由碳原子数为 1-3的烷基中的一种或 几种基团取代的吡啶基、 咪唑基和由碳原子数为 1-3的烷基中的一种或几种基团取代的 咪唑基中的一种基团将由含有两个 A基团的卤化钌芳烃二聚体中的钌与螯合配体中的 两个配位原子螯合配位后得到的反应产物中的卤素离子取代的步骤。
其中, 所述将由含有两个 A基团的卤化钌芳烃二聚体中的钌与螯合配体中的两个 配位原子螯合配位后得到的反应产物中的卤素离子取代的方法可以采用各种常规的方 法, 优选情况下, 该方法包括: 在第九有机溶剂中, 将所述反应产物与 AgPF6或 AgBF4 在室温, 如 20-50°C下混合 0.5-2小时 (上述反应产物与 AgPF6或 AgBF4的摩尔比一般 为 1 :0.95-1.05 ), 过滤, 将滤液与碱金属的硫氰酸盐、 碱金属的叠氮化物、 碱金属的硫 甲基盐、 碱金属的硫氢盐、 碳原子数为 1-3的饱和羧酸、 吡啶、 由碳原子数为 1-3的烷 基中的一种或几种基团取代的吡啶、 咪唑和由碳原子数为 1-3烷基中的一种或几种基团 取代的咪唑中的一种混合;
所述碱金属的硫氰酸盐、 碱金属的叠氮化物、 碱金属的硫甲基盐、 碱金属的硫氢
盐、 碳原子数为 1-3的饱和羧酸、 吡啶、 由碳原子数为 1-3的烷基中的一种或几种基团 取代的吡啶、 咪唑和由碳原子数为 1-3烷基中的一种或几种基团取代的咪唑中的一种的 用量没有特别限定, 只要保证能够将卤素离子沉淀并用上述碱金属的硫氰酸盐、 碱金属 的叠氮化物、碱金属的硫甲基盐、碱金属的硫氢盐、碳原子数为 1-3的饱和羧酸、吡啶、 由碳原子数为 1-3的烷基中的一种或几种基团取代的吡啶、 咪唑和由碳原子数为 1-3烷 基中的一种或几种基团取代的咪唑阴离子取代即可, 一般情况下, 所述碱金属的硫氰酸 盐、 碱金属的叠氮化物、 碱金属的硫甲基盐、 碱金属的硫氢盐、 碳原子数为 1-3的饱和 羧酸、 吡啶、 由碳原子数为 1-3的烷基中的一种或几种基团取代的吡啶、 咪唑和由碳原 子数为 1-3 烷基中的一种或几种基团取代的咪唑中的一种与所述反应产物的摩尔比为 1-5 : 1; 优选为 1-3 : 1。
以所述反应产物和所述碱金属的硫氰酸盐、 碱金属的叠氮化物、 碱金属的硫甲基 盐、 碱金属的硫氢盐、 碳原子数为 1-3的饱和羧酸、 吡啶、 由碳原子数为 1-3的烷基中 的一种或几种基团取代的吡啶、 咪唑和由碳原子数为 1-3烷基中的一种或几种基团取代 的咪唑中的一种的总量为 100毫克为基准, 所述第九有机溶剂的用量为 30-50毫升; 所 述第九有机溶剂为甲醇和 /或乙醇。
按照本发明, 由通式 AG(X'Y')Z以及由 [AG(XY)Z]+B-表示的喹唑啉配合物蛋白激 酶抑制剂中,用于螯合配位的含有两个配位原子的配体已经在上述内容中进行过详细描 述, 在此不再赘述。
按照本发明的一种具体实施方式, 由通式 [AG(X1Y1)Z1]+B-表示的蛋白酶抑制剂的 制备方法包括:
( 1 )将含有两个 A基团的卤化钌芳烃二聚体与碳原子数为 1-5的烷基二胺在醇或 醇的水溶液中接触,接触反应的条件使得所述含有两个 A基团的卤化钌芳烃二聚体中的 钌与烷基二胺中的两个配位原子氮螯合配位;
( 2 ) 用含有单个配位原子的单齿配体将步骤 (1 ) 中的由含有两个 A基团的卤化 钌芳烃二聚体中的钌与碳原子数为 1-5的烷基二胺中的两个配位原子氮螯合配位后所得 到的反应产物中的卤素离子取代的步骤。
步骤 (1 ) 中, 所述含有两个 A基团的卤化钌芳烃二聚体与碳原子数为 1-5的烷基 二胺的摩尔比为 1 : 1-3, 接触的温度可以为 20-50°C, 接触的时间为 0.5-2小时; 以所述 含有两个 A基团的卤化钌芳烃二聚体和碳原子数为 1-5的烷基二胺的总量为 100毫克为 基准, 所述醇或醇的水溶液的用量可以为 30-50毫升, 所述醇优选为甲醇, 所述醇的水 溶液优选为甲醇的水溶液。
步骤(2 ) 中, 所述将由含有两个 A基团的卤化钌芳烃二聚体中的钌与碳原子数为 1-5 的烷基二胺中的两个配位原子氮螯合配位后所得到的反应产物中的卤素离子取代的 方法可以采用各种常规的方法, 例如, 所述取代的方法包括: 在第九有机溶剂中, 将所 述反应产物与 AgPF6或 AgBF4在室温, 如 20-50°C下混合 0.5-2小时 (上述反应产物与
AgPF6或 §8?4的摩尔比一般为 1 :0.95-1.05 ) 混合, 过滤, 将滤液与具有单个配位原子 的单齿配体混合; 所述具有单个配位原子的单齿配体的用量没有特别限定, 只要保证能 够将卤素离子沉淀并用上述具有单个配位原子的单齿配体中的一种取代即可,一般情况 下,所述含有单个配位原子的单齿配体与所述反应产物的摩尔比为 1-5: 1,优选为 1-3: 1。 以所述反应产物和含有单个配位原子的单齿配体的总量为 100毫克为基准,所述第九溶 剂的用量可以为 30-50毫升; 所述第九有机溶剂为甲醇和 /或乙醇。
按照本发明, 由通式 [AG(X1Y1)Z1]+B-表示的喹唑啉配合物蛋白激酶抑制剂中, 用 于配位的含有单个配位原子的单齿配体已经在上述内容中进行过详细描述,在此不再赘 述。
按照本发明, 所述含有两个 A基团的卤化钌芳烃二聚体可以商购得到, 也可以按 照本领域技术人员公知的方法制备得到。例如,所述含有两个 A基团的卤化钌芳烃二聚 体的制备方法包括:
( 1 ) 在液氨和低级醇的混合液中, 将芳烃与碱金属混合, 得到二氢化芳烃;
(2)将二氢化芳烃与卤化钌在第七有机溶剂中接触, 接触反应的条件使反应得到 卤化钌芳烃二聚体。
其中, 所述含有两个 A基团的卤化钌芳烃二聚体的制备方法的步骤(1 ) 中, 所述 将芳烃还原为二氢化芳烃的反应为本领域技术人员公知的伯奇反应, 反应的条件和方法 为本领域技术人员所公知, 例如, 碱金属, 如钠、 钾或锂与反应物芳烃的摩尔比可以为 (4-8): 1 , 液氨、 低级醇与反应物芳烃的摩尔比可以为 (200-300):(10-15): 1, 所述低级醇可 以选自甲醇、乙醇、异丙醇和丁醇中的一种或几种;所述反应的温度可以为 -78至 -50°C, 所述反应的时间可以为 1-3小时。
一般, 在进行伯奇反应后所得到的反应产物为二氢化芳烃和部分未反应芳烃原料 的混合物, 即使通过减压蒸馏的方法除去部分溶剂和未反应的原料, 也不能从该反应产 物混合物中将二氢化芳烃完全分离出来, 因此, 其实是采用该反应产物混合物作为后续 反应的原料进行反应的, 通过核磁共振分析能够得到, 在该反应产物混合物中, 二氢化 芳烃的纯度一般在 60-90重量%左右。
其中, 所述含有两个 A基团的卤化钌芳烃二聚体的制备方法的步骤(2) 中, 所述 步骤 (1 ) 得到的含有二氢化芳烃的反应产物混合物的用量使得其中所述二氢化芳烃与 卤化钌 (III) 的摩尔比可以为 3-5: 1, 所述接触条件包括接触的温度可以为 60-90°C, 接 触的时间可以为 1-12小时; 所述第七有机溶剂可以选自乙醇和 /或甲醇; 以二氢化芳烃 与卤化钌(III)的总量为 100毫克为基准, 所述第七有机溶剂的用量可以为 30-50毫升; 然后, 可以通过过滤, 洗涤的方法分离出卤化钌芳烃二聚体。
优选情况下,所述含有两个 A基团的卤化钌芳烃二聚体中的 A基团选自苯、联苯、 异丙基甲苯和环烷并苯中的一种。
按照本发明的一种具体实施方式, 由通式 G(M)W表示的喹唑啉配合物蛋白酶抑制
剂的制备方法包括:
( 1 )在第十有机溶剂的存在下, 将卤化钌与盐酸水溶液和 DMSO的混合物, 或者 将卤化钌与 DMSO加热回流, 使反应得到 DMSO配位的钌化合物;
(2) 将步骤 (1 ) 得到的 DMSO配位的钌化合物与含有单个或两个配位原子的喹 唑啉衍生物配体在醇或醇的水溶液或醇的盐酸溶液中接触, 接触反应的条件使 DMSO 配位的钌化合物中的钌与所述配体中的单个或两个配位原子配位。
按照本发明, 步骤 (1 ) 中, 所述加热回流的温度可以为 70°C至 200°C, 加热回流 的时间可以为 3-6小时; 所述卤化钌与盐酸水溶液中氯化氢的摩尔比可以为 1 :(40-80), 所述卤化钌与 DMSO的摩尔比可以为 1 :(40-80); 所述第十有机溶剂可以为常规使用的 有机溶剂, 例如, 可以选自甲醇、 乙醇、 异丙醇中的一种或多种; 以卤化钌与盐酸水溶 液和 DMSO的总量为 2000毫克为基准, 所述第十有机溶剂的用量可以为 30-50毫升。
按照本发明, 步骤(2) 中, 所述 DMSO配位的钌化合物与含有两个配位原子的喹 唑啉衍生物螯合配体的摩尔比可以为 1 :1-3, 接触的温度可以为 20-50°C, 接触的时间可 以为 0.5-6小时;以 DMSO配位的钌化合物和含有两个配位原子的螯合配体的总量为 100 毫克为基准, 所述醇或醇的水溶液的用量可以为 3-10毫升; 所述醇优选为乙醇, 所述 醇的水溶液优选为乙醇的水溶液。
或者, 按照本发明, 步骤(2) 中, 所述 DMSO配位的钌化合物与含有单个配位原 子的单齿配体的摩尔比可以为 1 :1-3, 所述接触的温可以为 20-50°C, 接触的时间可以为
0.5-6小时; 以 DMSO配位的钌化合物和含有单个配位原子的喹唑啉衍生物配体的总量 为 100毫克为基准, 所述醇或醇的盐酸溶液的用量可以为 8-20毫升; 所述醇优选为乙 醇。
按照本发明, 由通式 G(M)W表示的喹唑啉配合物蛋白激酶抑制剂中, 用于配位的 所述含有两个配位原子的螯合配体以及含有单个配位原子的单齿配体已经在上述内容 中进行过详细描述, 在此不再赘述。
以上详细描述了本发明的优选实施方式, 但是, 本发明并不限于上述实施方式中 的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型, 这些简单变型均属于本发明的保护范围。
另外需要说明的是, 在上述具体实施方式中所描述的各个具体技术特征, 在不矛 盾的情况下, 可以通过任何合适的方式进行组合, 为了避免不必要的重复, 本发明对各 种可能的组合方式不再另行说明。
此外, 本发明的各种不同的实施方式之间也可以进行任意组合, 只要其不违背本 发明的思想, 其同样应当视为本发明所公开的内容。
下面将通过具体实施例对本发明进行进一步的详细描述。
实施例 1
本实施例用于说明本发明提供的喹唑啉衍生物配体及喹唑啉配合物蛋白激酶抑制 剂
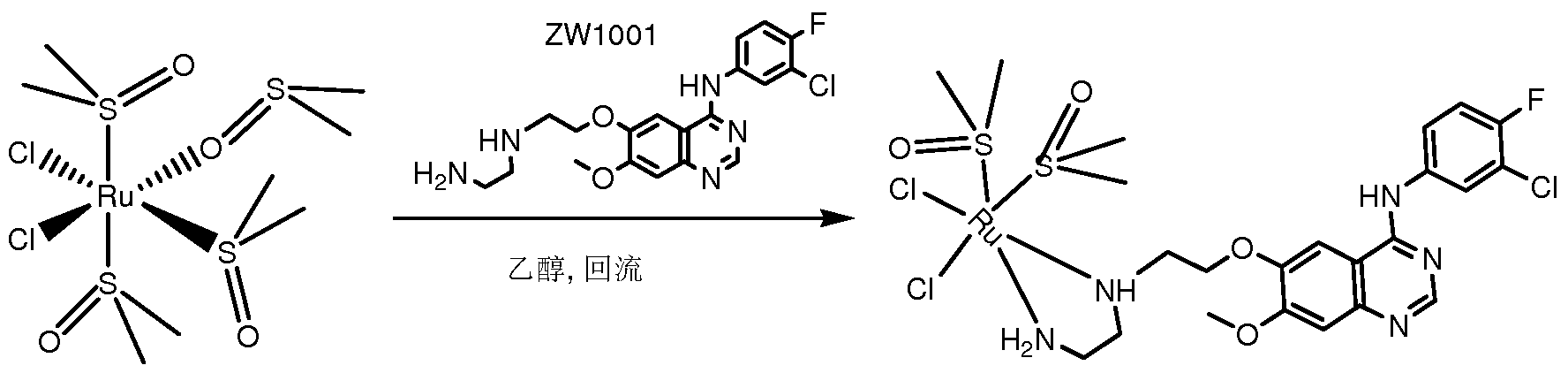
( 1 )将 2.0g上述反应式中的第一反应物 A (4-(3'-氯 -4'-氟-苯氨基) -6-羟基 -7-甲氧 基喹唑啉 (购自南京安格医药化工有限公司)、 5.0g缚酸剂无水碳酸钾置于 30ml DMF 中, 控制油浴温度为 87°C, 搅拌 15分钟, 然后滴加 2mL 1,2-二溴乙烷, 保持温度继续 反应 4.5小时。反应毕, 冷却至室温, 抽滤, 收集滤液, 搅拌下缓慢倾入 120mL冷水中, 有黏稠状物质析出, 乙酸乙酯 50mIX3萃取, 合并萃取液, 用 30mL水洗一次, 无水硫 酸钠干燥。 硅胶柱层析分离产品 (乙酸乙酯 /石油醚 4:1 ), 得到中间体产物 ZW1001-M 淡黄色粉末 0.5g, 收率为 17%。
1H-NMR (DMSO-de, 400MHz), 5(ppm): 9.55(1H, s), 8.52(1H, s),8.12(lH, dd, Ji=6.7Hz, J2=2.4Hz), 7.85(1H, s),7.78(lH, dd, Ji=8.6Hz, J2=4.6Hz), 7.48(1H, t, Ji= J2=9.0Hz): 7.24(1H, s), 4.50(2H, t, Ji=J2=5.5Hz), 3.93(5H, m); ESI-MS: m/z 428.61, 430.61 ( [M]+)。
采用 ELISA测试条件, 测定了由本实施例得到的中间产物编号为 ZW1001-M的化
ZW1001-M ZW1001
(2)将 0.80g由步骤( 1 )得到的中间体产物 ZW1001-M(4-(3'-氯 -4'-氟-苯氨基) -6-0 溴乙氧基) -7-甲氧基喹唑啉)与 ImL蒸馏过的乙二胺(购自北京化学试剂公司),在 40mL 乙腈中加热回流 3小时, 加热回流的温度为 80°C ; 停止反应后, 自然冷却析出结晶, 抽 滤, 滤饼用乙腈洗涤一次, 干燥得到编号为 ZW1001的白色固体 0.6g, 收率为 67%。
ESI-MS: m/z 406.9 ([M+H]+)。
图 5为在 ELISA测试条件下测得的编号为 ZW1001的化合物 IC
5Q为 4.6nM的 IC
5Q 曲线图, 说明该抑制剂对 EGFR蛋白激酶抑制剂具有良好的抑制活性。
(3 ) 将 0.2g由步骤 (2) 制备得到的产物 ZW1001置于 50mL圆底烧瓶中, 加入 无水甲醇 12mL溶解, 加入无水碳酸钾 0.2g, 在室温 (25°C ) 下搅拌 0.5h, 常压滤去不 溶物, 收集滤液, 加入钌芳烃二聚体 (钌对甲基异丙基苯二聚体, 购自东京化成公司) 0.12g, 然后在室温 (25°C ) 下搅拌反应 7h。 反应完成后加入 0.4g六氟磷酸铵, 室温搅 拌 0.5h。 硅胶柱层析分离产物 (甲醇 /二氯甲烷体积比为 1 :20), 得到红色油状物, 柱层 析产物进一步用薄层层析分离纯化(甲醇 /二氯甲烷体积比为 1 :10),得到编号为 ZW2001 的淡黄色粉末 0.10g, 收率为 35%。
1H-NMR (DMSO-de, 400MHz), 5(ppm): 1H-NMR δ (ppm): 9.62(1H, s), 8.54(1H, s), 8.15(1H, d), 7.94(1H, s), 7.80(1H, m), 7.45(1H, m), 7.31(1H, s), 6.60(2H, m), 5.78(1H, m), 5.64(4H, m), 4.46(2H, d), 4.00(3H, s), 3.81(1H, m), 3.51(1H, m), 2.85(2H, m), 2.72(3H, m),2.32(lH, s), 2.23(3H, s), 2.02(1H, s), 1.98(3H, m).;
ESI-MS: 676.1(M+), 640.1(M-C1)+
图 6为在 ELISA测试条件下测得的编号为 ZW2001的化合物的 IC5Q为 81nM的 IC5o 曲线图, 说明该抑制剂对 EGFR蛋白激酶抑制剂具有良好的抑制活性。 实施例 2-6
本实施例用于说明本发明提供的喹唑啉衍生物配体及喹唑啉配合物蛋白激酶抑制 剂的制备。
在实施例 2和实施例 3中, 分别按照实施例 1的方法制备配体和配合物, 不同的 是: 在步骤 (3 ) 中, 分别用 O.lg钌苯二聚体 (购自东京化成公司)、 0.15g钌联苯二聚 体代替实施例 1 中步骤 (3 ) 中所用的对甲基异丙基苯二聚体, 得到具有下述结构的编 号分别为 ZW2002、 ZW2003的喹唑啉配合物蛋白激酶抑制剂。
其中, 所述钌联苯二聚体的制备方法为: 在液氨和乙醇的混合液中, 在 -78°C下, 将联苯与金属钠混合 1小时(液氨、 乙醇、 联苯与钠的摩尔比为 250:10:1 :5 ), 得到二氢 化联苯; 然后将反应产物在 150°C下进行减压蒸馏, 除去溶剂和部分未反应的原料, 通 过核磁共振分析得到, 反应产物混合物中, 二氢化联苯的纯度在 70重量%左右;
(2) 将含有二氢化联苯的反应产物混合物与氯化钌在乙醇中接触, 其中, 含有二 氢化联苯的反应产物混合物的用量使得二氢化联苯与氯化钌的摩尔比为 5:1, 接触的温 度为 80°C, 接触的时间为 8小时, 以 100毫克二氢化联苯与氯化钌的总重量计, 乙醇的
用量为 60毫升 过滤, 用甲醇洗涤得到三氯化钌联苯二聚体。
ZW2002
1H-NMR δ (ppm): 8.61(1H, s), 8.12(2H,d), 8.10(1H, s), 7.77(1H, s), 7.40(4H, m): 6.85(1H, m), 6.25(1H, m), 5.69(1H, m), 4.45(3H, s), 4.02(4H, m), 2.50(2H, m), 2.33(1H, d): 2.02(1H, d)
MALDI-TOF: 620.1 M+), 584.31(M-C1)+
ZW2003
1H-NMR δ (ppm): 8.63(1H, s), 8.11(1H, s), 8.10(1H, s), 7.78(3H, m), 7.46(5H, m), 7.31(1H, s), 6.78(1H, d), 6.28(2H, m), 6.04(1H, m), 5.89(1H, m), 4.38(2H, s), 4.06(3H, s), 3.74(2H, s), 3.69(2H, s), 2.02(2H, s).
MALDI-TOF: 696.1 (M+),660.3(M-C1)+ 在实施例 4中, 分别按照实施例 1的方法制备配体和配合物, 不同的是: 在步骤 (1 ) 中, 用 4.8mL 1.3 二溴丙烷代替 1,2-二溴乙烷, 得到中间体产物 ZW1002-M淡黄色粉末 1.3g, 收率为 37.8%; 溶剂由丙酮代替 DMF;
在步骤 (2) 中, 将 0.70g由步骤 (1 ) 得到的中间体产物 ZW1002-M (4-(3'-氯 -4'- 氟-苯氨基) -6-(2-溴丙氧基) -7-甲氧基喹唑啉) 与 1.3mL蒸馏过的乙二胺 (购自北京化学 试剂公司), 在 40mL乙腈中室温 (25°C ) 反应 9小时; 停止反应后, 自然冷却析出结 晶,抽滤,滤饼用乙腈洗涤一次,干燥得到编号为 ZW1002的白色固体 5g,收率为 65.62%; 在步骤 (3 ) 中, 将 0.3g由步骤 (2) 制备得到的产物 ZW1002置于 50mL圆底烧
瓶中, 加入无水甲醇 12mL溶解, 加入无水碳酸钾 0.3g, 在室温 (25°C ) 下搅拌 0.5h, 常压滤去不溶物, 收集滤液, 加入钌芳烃二聚体 (钌对甲基异丙基苯二聚体, 购自东京 化成公司) 0.22g, 然后在室温 (25°C ) 下搅拌反应 7h。 反应完成后加入 0.4g六氟磷酸 铵, 室温搅拌 0.5h。 硅胶柱层析分离产物 (甲醇 /二氯甲烷体积比为 1 :20), 得到红色油 状物, 柱层析产物进一步用薄层层析分离纯化 (甲醇 /二氯甲烷体积比为 1 :10), 得到编 号为 ZW2004的淡黄色粉末 0.15g, 收率为 23%。
ZW2004
MALDI-TOF: 690.6(M+), 655.3(M-C1)+
图 8为编号为 ZW2004的化合物在 MCF-7/S + EGF 抑制肿瘤细胞增殖的测试条件 下, 测得的 IC5Q=33.87 uM的曲线图, 说明该抑制剂对 EGFR蛋白激酶抑制剂具有良好 的抑制活性。 在实施例 5-6中, 分别按照实施例 4的方法制备配体和配合物, 不同的是: 在步骤 (3 ) 中, 分别用 0.3g钌苯二聚体 (购自东京化成公司)、 0.25g钌联苯二聚体 (按照实 施例 3中所述的方法制备得到) 代替实施例 1中步骤 (3 ) 中所用的对甲基异丙基苯二 聚体, 得到具有下述结构的编号分别为 ZW2005、 ZW2006的喹唑啉配合物蛋白激酶抑 制剂。
+ PF6
ZW2005
1H-NMR δ (ppm): 8.81(1H, s), 8.04(2H, m), 7.72(1H, m), 7.55(1H, m), 7.26(2H, m) 6.85(1H, m), 6.65(2H, m), 5.85(1H, s), 4.00(3H, s),
MALDI-TOF:634.3 (M+), 598.1(M-C1)+
ZW2006
MALDI-TOF:709.1 (M+), 674.3(M-C1)+ 实施例 7
本实施例用于说明本发明提供的喹唑啉衍生物配体及喹唑啉配合物蛋白激酶抑制 剂的制备。
A C
( 1 ) 将 0.652g的溴代乙酸乙酯, 1.2g 的第一反应物 A (4-(3-氯 -4-氟-苯氨基) -6- 羟基 -7-甲氧基喹唑啉), 1.3g的缚酸剂 K2C03与 20ml的 DMF混合, 在 40°C下, 搅拌 约 2h后,用 TLC薄层析跟踪观察反应进程,至第一反应物 A色斑接近消失则反应完全,
过滤反应液, 将 DMF滤液, 滴加到 100ml的蒸馏水中, 立刻析出大量的黄色沉淀, 过 滤出析出物后, 真空干燥, 用混合溶剂二氯甲浣 /甲醇 (体积比为 20: 1 ) 重结晶, 再重 结晶, 可得到中间产物 C浅黄色的粉末, 真空干燥后, 产物称重 l . lg, 产率为 70%。
-MS: m/z 406.2 ([M+H]+); 444.2 ([M+K]+) 。
C D
(2)将 NaOH约 88mg, 2.2mmol以及步骤( 1 )得到的 446.6mg中间体产物 C (分 散在同样重量比的水 -甲醇 -四氢呋喃混合溶剂 20ml中) 与 30ml水-甲醇-四氢呋喃 (体 积比为 1 : 1 :3 ) 的混合溶剂混合, 在室温下, 搅拌反应 12小时。 反应停止后, 真空旋蒸 浓縮反应液至 10ml, 用质量百分比浓度为 25%的 HC1溶液调节至酸性, 析出大量的白 色絮状物,减压抽滤,将滤饼真空干燥,得到中间产物 D。产物称重 415mg,产率为 82%。
MS (EI, 80 eV) m/z 377 (M++)
( 3 )在装有回流冷凝管(上端附有无水氯化钙干燥管,并连接一导气管通入 NaOH 饱和液吸收尾气)的 100ml的三口烧瓶中加入 5mmol步骤(2)得到的中间产物 D, 3.5ml (约 40mmol) 氯化亚砜, 滴入吡啶一滴 (约 0.6mmol), 另外补加 1-2 滴 DMF (约 0.5-2.0mmol), 以帮助羧酸溶解, 置于油浴中加热, 充分搅拌, 加热约 50分钟后升温至 75 °C, 维持在 70-75 °C (2-3小时), 至无气体逸出, 反应完毕, 减压蒸馏出去过量的氯 化亚砜后, 冷却, 得到中间产物 E, 并将中间产物 E溶于约 10ml的无水二氯甲烷中, 并置于 50ml的恒压漏斗中, 称取 5-甲基胺 -2,2'-联吡啶 0.77g和缚酸剂三乙胺 0.64g (三 者物质的量比约为 1.2:1.0: 1.5 ), 一起溶于 30ml的二氯甲烷中, 装入 100ml三口烧瓶中, 冰浴下搅拌, 缓慢滴入恒压漏斗中的中间产物 E的溶液, 10分钟滴完后, 反应温度保
持在 5°C左右, 继续搅拌 3小时后, 反应完毕, 过滤, 除去沉淀, 合并滤液, 减压浓縮, 可得目标产物粗品, 用乙醇重结晶, 或再次重结晶, 得纯品。 产物称重 1.32g, 产率为 48.5%。
ESI-MS: m/z 544([M+H]+)
(4)将硼氢化钠 (380mg, lOmmol)悬浮于 100 mL干燥 THF中, 氩气保护下滴 入三氟乙酸 TFA (2mL) , 室温搅拌至无气泡, 滴加步骤(3 )得到的目标产物 (544mg 约 lmmol) 在 50 mLTHF的溶液, 加热回流 2小时, 加水 100 mL淬灭反应, 乙酸乙酯 萃取, 有机层用无水硫酸钠干燥, 减压浓縮干, 经硅胶柱层析分离(二氯甲浣 /甲醇体积 比为 20/1 ) , 得白色 160mg目标产物, 收率 32%。
ESI-MS: m/z 531.2 ([M+H]+)。 实施例 8
本实施例用于说明本发明提供的喹唑啉衍生物配体及喹唑啉配合物蛋白激酶抑制 剂的
A H
取 0.5g约 1.56mmol的第一反应物 A (4-( 氯 -4-氟-苯氨基) -6-羟基 -7-甲氧基喹唑 啉), 置于 50mL圆底烧瓶中, 加入吡啶盐酸盐固体 3.0g, 氩气保护下油浴升温至 170 。C, 磁力搅拌, 反应物渐渐熔化, 保持温度反应 4h。 反应毕, 冷却至室温, 加入 30mL 水, 加热回流 lOmin, 冷却抽滤, 滤饼干燥后用无水甲醇重结晶, 得到中间产物 H黄绿 色粉末 0.36g, 收率 75%。
ESI-MS: m/z 306.8( [M+H]+)
H I
( 1 ) 将 1.33g 上述步骤得到的中间产物 H (4-(3-氯 -4-氟-苯氨基) -6,7-二羟基喹唑 啉), 7.0g无水碳酸钾与 70ml丙酮混合, 控制油浴温度 50°C, 加热回流, 搅拌 15min, 然后滴加 3mL 2-溴乙醇, 保持温度继续反应 10h。 反应毕, 冷却至室温, 抽滤, 收集滤 液, 滤液浓縮。 硅胶柱层析分离产品 (甲醇 /二氯甲烷体积比为 1 :15 ), 得到中间产物 I 白色粉末 0.8g, 收率为 46.8%。
ESI-MS检测: m/z 394.8( [M+H]+)
I J
(2) 将 0.4g步骤 (1 ) 得到的中间产物 I与 15mL干燥的氯苯和 0.5mL吡啶, 在 室温 (25°C ) 下搅拌, 得到悬浊液。 另取 0.15mL三溴化磷, 加入 3mL氯苯稀释, 在室 温(25°C )下缓慢滴加到上述悬浊液中, 滴加完毕后, 加热至回流反应 3小时, TLC显 示反应物 I色斑接近消失则反应完全。 冷却至室温, 反应液依次用饱和碳酸氢钠溶液、 饱和氯化钠溶液洗涤, 无水硫酸镁干燥, 浓縮得到中间产物 J淡黄色黏稠物, 硅胶柱层 析分离 (甲醇 /二氯甲烷体积比为 1 :15 ), 得到产物 (9) 白色粉末 0.25g, 收率为 47%。
ESI-MS检测: m/z 520.40( [M+H]+) 542.4 ( [M+Na]+)
J
(3 ) 在室温 (25°C ) 下, 将 0.5g步骤 (2) 得到的中间产物 J, 与饱和的氨 -甲醇 溶液 15mL混合 (中间产物 J与氨的用量摩尔比为 1 :20), 然后在室温 (25°C ) 下搅拌 反应 10h, TLC显示反应物 J的色斑接近消失则反应完全。 旋蒸浓縮后得到白色粉末, 冷水洗涤一次, 甲醇水重结晶得到产物白色粉末 0.28g, 收率为 76%。
ESI-MS检测: m/z 392.83( [M+H]+) 414.74 ( [M+Na]+) 实施例 9
本实施例用于说明本发明提供的喹唑啉衍生物配体及喹唑啉配合物蛋白激酶抑制 剂的制备。
合成:
ZW1 001 -M JLY1002
将 414 mg (6 mmol) 咪唑 (购自北京化学试剂公司), TBAB (四丁基溴化铵) 32 mg (购自北京化学试剂公司), NaOH 480 mg加入到 30ml乙腈中, 加热回流 1小时(加 热回流的温度为 80°C ) 后, 滴加入 2587 mg (6 mmol) 由实施例 1制得的中间体产物 ZW1001-M, 继续搅拌回流 3小时, 加热回流的温度为 80°C。 停止反应, 旋蒸出溶剂, 向残留物中加入 25 ml水和 25 ml乙酸乙酯, 有白色固体在乙酸乙酯和水层之间析出, 滤出固体, 用水和乙酸乙酯洗涤后, 将产物在室温下真空干燥 20 小时, 得到编号为 JLY1002的白色固体 1.16g, 收率为 70%。
ESI-MS: m/z 414.7 ([M+H]+); 436.6 ([M+Na]+)。
图 3为在 ELISA测试条件下测得的编号为 JLY1002的化合物的 IC5Q为 60.2nM的 IC50曲线图, 说明该抑制剂对 EGFR蛋白激酶具有很好的激酶抑制活性。
喹唑啉配合物蛋白激酶抑制剂编号为 JLY2008及 JLY2007的化合物的合成: 合成的路线如下:
+
0)2H]
JLY2008
[trans-RuCl4(Me2SO)2][(Me2SO)2H]的合成:
乙醇 HC1(37%) i
RuCl,»3H90 [(DMSO)2H] ++
回流, 3h DMSO
I
将 lOOmg RuCl3-3H20加入到 30 ml乙醇中, 形成悬浮液, 并加热回流 3 h (加热回 流的温度为 80°C ), 形成深绿色溶液。 将得到的溶液通过滤纸除去可能存在的未溶解的 固体, 并用旋转蒸发仪浓縮至 2mL。 将 0.75 mL盐酸水溶液 (质量百分比浓度为 37%) 和 1.5 mL DMSO加入到该混合液中, 在 80 °C下放置 30 min, 形成亮橙色溶液。
将上述得到的溶液冷却至室温 (25°C ), 加入 10 ml丙酮, 并从溶液中分离析出的 橙红色晶体。 加入少量乙醚可加速晶体的析出。 通过过滤收集上述晶体, 并用 20 ml温 度为零下 4°C的冷丙酮溶剂洗涤, 再用 10 ml乙醚洗涤, 最后在室温 (25°C ) 下真空干 编号为 JLY2008的化合物的合成:
JLY2008
将 20mg (0.036 mmol)上述制备的 [tra/w-RuCl4(Me2SO)2][(Me2SO)2H]在室温 (25 °C )下加入到 4 mL 乙醇 /盐酸 (0.1 M) 中, 搅拌 5 min, 加入 29.8 mg (0.072 mmol)上述 制备得到的编号为 JLY1002的喹唑啉衍生物配体, 约 10分钟后有固体析出, 继续搅拌 4ho停止反应, 过滤,滤饼依次用水, 乙醇和乙醚洗涤,真空干燥。得黄色产物 10.6 mg。 产率为 40%。
ESI-MS (negative): m/z 735.2 [RuinCl4(DMSO)(L2)]", 241.8 [RumCl4]". Anal. Calcd for C22H24Cl5FN503RuS (735.86): C, 35.91; H, 3.29; N, 9.52. Found: C, 35.88; H, 3.70; N, 8.93. 编号为 JLY2007的化合物的合成:
将 55.6 mg (0.1 mmol)上述制备的 [tra/w-RuCl4(Me2SO)2][(Me2SO)2H]在室温 (25 °C ) 下加入到 4 mL乙醇中, 搅拌 5 min, 加入 40.58 mg (0.1 mmol) 实施例 1制备得到 的编号为 ZW1001的喹唑啉衍生物配体, 约 10分钟后, 有固体析出, 继续搅拌 30 min。 然后加入 4 mL水, 继续搅拌 30 min。 停止反应, 滤出固体, 用乙醇和乙醚洗涤, 真空 干燥, 得到黄色产物 43 mg。 产率为 62%。
ESI-MS (positive): m/z 693.1 [RuinCl3(DMSO)(L5)]+, 615.12 [RuinCl3(L5)]+, 505.14 [Rum(L5)]+. Anal. Calcd for C21H27CwFN503RuS'3H20 (745.46): C, 33.83; H, 4.46; N, 9.39. Found: C, 33.8; H, 4.13; N, 9.18. 图 4为在 ELISA测试条件下测得的编号为 JLY2007的化 合物的 IC5Q为 7.5nM的 IC5Q曲线图, 说明该抑制剂对 EGFR蛋白激酶具有很好的激酶 抑制活性。
图 7为编号为 JLY2007的化合物在 MCF-7/S + EGF 抑制肿瘤细胞增殖的测试条件 下, 测得的 IC5Q=24.48 uM的曲线图。 无 EGF条件下, 该化合物的 IC5O100, 几乎没 有抑制活性。 在外加 EGF后, 肿瘤细胞的增殖受到极大的抑制 IC5Q=24.48 uM。 说明该
化合物的抑制活性与 EGF有关联, EGFR可能是化合物的作用靶标之 喹唑啉配合物蛋白激酶抑制剂编号为 JLY2009的化合物的合成:
合成路线如下:
JLY2009
Cis-RuCl2(Me2SO)4的合
将 100 mg RuC13-3H20加入到 1 ml二甲基亚砜中, 加热回流 5 min (加热回流的 温度为 189°C ), 形成亮黄色透明溶液。 冷却后加入 15 mL丙酮, 并用旋转蒸发仪浓縮 到原体积的一半, 析出黄色晶体。 通过过滤收集上述晶体, 并用丙酮和乙醚洗涤, 最后 在室温 (25°C ) 下真空干燥。 编号为 JLY2009的化合物的合成:
JLY2009
将 48.5 mg (0.1 mmol) £^-Ru Cl2(DMSO)4加入到 10 mL乙醇中, 在 80°C下加热回 流。 加入 40.58 mg (0.1 mmol) 实施例 1制备得到的编号为 ZW1001的喹唑啉衍生物配 体, 继续在上述温度下回流搅拌 6h。 有固体析出, 滤出固体, 用乙醇和乙醚洗涤, 真空 干燥。 获得产物 JLY2009 40 mg, 产率为 55%。
ESI-MS (positive): m/z 736.21 [RunCl2(DMSO)2(L)]+, 658.17 [RunCl2(DMSO)(L)]+, 580.14 [RunCl2(L5)]+, 542.15 [RunCl(L)]+, 506.17 [Run(L)]+. 1H NMR: (DMSO- ) ^(ppm): 9.49 (s, 1H), 8.48 (s, 1H), 8.11-8.09 (m, 1H), 7.85 (s, 1H), 7.81-7.77 (m, 1H), 7.45-7.40 (t, 1H), 7.20 (s, 1H), 4.42-4.38 (m, 2H), 4.33-4.30 (m, 1H), 4.24-4.21 (m, 1H), 3.92 (s, 3H), 3.88-3.83 (m, 1H), 3.44-3.37 (m, 1H), 3.29 (s, 6H), 3.23 (s, 3H), 3.12-3.11 (m, 8H).
将 JLY2009溶于 DMSO/acetone (体积比为 1 :5 ) 的混合溶液中, 用乙醚慢慢挥发 进入, 析出 JLY2009的单晶。
图 1-a为编号为 JLY2009的化合物的 X射线衍射晶体结构图, 图 1-b为与其对应 的化合物的结构式。 实施例 10
该实施例用于说明对由实施例 1-9 制备得到的喹唑啉衍生物以及喹唑啉配合物蛋 白激酶抑制剂的体外活性测试。
一、 酶联免疫吸附分析 ( Enzyme Immunosorbent Assay, ELISA)
应用酶联免疫吸附分析(Enzyme Immunosorbent Assay, ELISA)分别测定由实施例
1 -9合成的化合物的激酶抑制活性(所合成化合物的浓度分别为 40μΜ(微摩尔 /升)、 4μΜ、 400ηΜ (纳摩尔 /升)、 40ηΜ、 4ηΜ、 400ρΜ (皮摩尔 /升)、 40ρΜ、 4ρΜ)。 使用 CST公 司的蛋白激酶检测试剂盒: PTPlB(Tyr66) Biotinylated Peptide作为激酶 EGFR( Epidermal Growth Factor Receptor) 的底物。 分别加入由实施例 1-9合成的化合物的激酶抑制剂, 使用 Spectramax M5 (USA, Molecular Devices)型酶标仪, 采用分光光度法, 测定特定波 长 450nm处的光吸收 OD值,根据下式计算化合物对细胞的抑制生长率,通过 OriginPro 7.0 数据处理软件由上述抑制生长率的数值得到 δ曲线, 考察本发明所合成的化合物的 激酶抑制剂对激酶底物的磷酸化反应的抑制程度, 从而得到 IC50值 (即对激酶底物的 磷酸化抑制程度达到 50%时的激酶抑制剂的浓度值)。 实验结果是两次独立的平行实验 的平均值, 变动一般为 ±15%。 各化合物的物化性质和酶活性抑制测试结果 IC5Q值如下
表 1所^。
0D激酶 - 0D测
抑制生长率 X 100 %
0D激酶 _0D空白
ELISA实验所测试的化合物及化合物编号如下:
1、 :C16H13ClFN3O2,M.W.= 333.7
如图 2所示, 在 ELISA测试条件下测得的编号为 LQ 1001的参比化合物的 IC50值 为 4 nM, 表明对蛋白激酶 EGFR的磷酸化作用具有良好的抑制效果。
2、 JLY1002 : C
2oHi
7ClFN
50
2, M.W=413.8 ί=\
.W=691.4
6>W00:〇ν6·7 Z11M Μ42-=
9' S8=A W 'n¾d¾
s ^13
£¾
I£3
:S003AZ l 01
CSM80/llOZN3/X3d 69CSS0/ZT0Z OAV
上表中, 易溶 DMSO是指在常温常压下, 在 DMSO中的溶解度不低于 1.0克, 微 溶于水是指在常温常压下, 在水中的溶解度为 0.09-0.10 克, 溶于甲醇、 乙醇是指在常
温常压下, 在甲醇中的溶解度为 1.0-9.9克, 在乙醇中的溶解度为 1.0-9.9克。
从上表 1 中的结果可以看出, 本发明提供的喹唑啉衍生物及其与钌形成的喹唑啉 配合物, 对蛋白激酶表皮生长因子受体 EGFR均表现出良好的抑制活性。 二、 对多种肿瘤细胞增殖影响实验:
1、 细胞毒实验
人乳腺癌细胞株 (抗药型) MCF-7/A、 人乳腺癌细胞株 (敏感型) MCF-7/S、 前 列腺癌细胞 PC-3、 角朊细胞 Colo-16、 人非小细胞肺癌细胞株 A549等细胞培养于含 10 重量% fetal bovine serum (FBS, Hyclone, USA) 的 RPMI1640培养基 (Invitrogen, USA), 并加入浓度为 lOOng/ml的 epidermal growth factor (EGF)刺激其生长。 2-3天后收集处于 对数生长期的细胞,接种于 96孔板(6500细胞 /孔 /100 ul ,含 lOOng/mlEGF的 RPMI1640) 培养 24 h, 然后分别加入梯度浓度的化合物(200、 100、 50、 25、 12.5、 6.25、 1 μΜ/0。 相同体积的含有 1重量%二甲基亚砜 (Dimethyl sulfoxide, DMSO)的 RPMI1640设为实验 对照组; 空白组为只有培养液但不加细胞。 各浓度组分别设 3个平行孔。 继续培养 48 h 后, SRB法检测细胞的存活率。 结果分别如表 2和表 3所示。
2、 外加 EGF条件下的细胞毒实验
人乳腺癌细胞株 (敏感型) MCF-7/S、 人非小细胞肺癌细胞株 A549 培养于含 10 重量0 /0 fetal bovine serum (FBS, Hyclone, USA) 的 HAM'S/F-12培养基(HyClone, USA), 并在此培养基中加入 lOOng/ml epidermal growth factor (EGF, Sigma, USA),上述细胞购自 中国科学院上海生命科学研究院细胞资源中心, 置于 C02恒温培养箱, 培养 3-5天后用 于实验。 结果分别如表 2所示。
其中, 磺基罗丹明 B (SRB)法检测化合物细胞毒实验:
收集处于对数生长期的细胞, 分别接种于 96 孔板中 (6500 细胞 /孔 /100 ul, 含 lOOng/ml EGF的 RPMI1640), 培养 24 h后, 分别加入梯度浓度的化合物 (200、 100、 50、 25、 12.5、 6.25、 1 ( μΜ/L) ); 将相同体积 100ng/ml EGF的 RPMI1640添加质量百 分比浓度为 1%的二甲基亚砜 (Dimethyl sulfoxide, DMSO)设为实验对照组; 只有培养液 但不加细胞为空白组。 各浓度组分别设 3个平行孔。 继续培养 48 h后, SRB法检测细 胞的存活率。每孔加入 50 ul 10%的预冷 (4°C )的三氯乙酸(Trichloroacetic acid,TCA), 经 4°C固定 1小时, 用水清洗 5次后, 充分干燥后加入 ΙΟΟμΙ的质量百分比浓度为 0.4% 的 SRB溶液 (Sigma, USA), 置于 37°C避光染色 30 min。 质量百分比浓度为 1%的%醋 酸清洗 4-5数次, 晾干,加入 200 μΐ的 Tris溶液 (10 mM, pH 10.5)充分溶解后置酶标仪测 定吸光度 (吸收波长为 570 nm)。
按照以下公式计算生长抑制率 (IR): IR(%) = [1- (实验组 A值-空白组 A值) I (对照 组 A值-空白组 A值)] X 100%, 并采用 Origin 6.0软件计算 IC50值。
说明:
01 , 蛋白激酶抑制剂, 分子靶向药物
-Ru, 金属类抗肿瘤药物, 细胞毒性抗肿瘤药物 <
阳性对照物 3: 紫杉醇, 细胞毒性抗肿瘤药物。
表 2: 人乳腺癌细胞株(抗药型) MCF-7/A、 人乳腺癌细胞株(敏感型) MCF-7/S、 MCF-7/S+EGF。
表 3: 前列腺癌细胞 PC-3、 角朊细胞 Colo-16、 非小细胞肺癌细胞 A549。
从上表 2和表 3中的结果可以看出, 本发明提供的喹唑啉衍生物以及喹唑啉配合 物蛋白激酶抑制剂, 对人乳腺癌细胞株(抗药型) MCF-7/A、 人乳腺癌细胞株(敏感型) MCF-7/S、 前列腺癌细胞 PC-3、 角朊细胞 Colo-16、 人非小细胞肺癌细胞株 A549等多 种肿瘤细胞增殖均表现出良好的抑制活性。 并且在外加表皮生长因子 EGF条件下, 所 述化合物对表皮生长因子受体 EGFR过表达的肿瘤细胞 (如人乳腺癌细胞株 (敏感型) MCF-7/S) 的细胞增殖表现出更高的抑制活性, 说明表皮生长因子受体 EGFR (—种酪 氨酸蛋白激酶)是本发明提供的喹唑啉衍生物、 喹唑啉配合物对肿瘤细胞增殖起抑制作 用的靶标之一。