WO2010100993A1 - 位相差フィルム - Google Patents
位相差フィルム Download PDFInfo
- Publication number
- WO2010100993A1 WO2010100993A1 PCT/JP2010/051600 JP2010051600W WO2010100993A1 WO 2010100993 A1 WO2010100993 A1 WO 2010100993A1 JP 2010051600 W JP2010051600 W JP 2010051600W WO 2010100993 A1 WO2010100993 A1 WO 2010100993A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- film
- axis
- present
- degrees
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/08—Cellulose derivatives
- C08L1/10—Esters of organic acids, i.e. acylates
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3083—Birefringent or phase retarding elements
Definitions
- Cellulose ester films, polycarbonate films, polycycloolefin films and the like are used as retardation films for TN type liquid crystal display devices.
- thermoplastic norbornene-based resin film the contrast viewing angle is wide, so that the retardation unevenness is very conspicuous even in the same degree as the normal stretching, and the LCD quality is deteriorated.
- an object of the present invention is to provide a retardation film having a wide contrast viewing angle and small retardation unevenness.
- X axis direction of the in-plane maximum refractive index of the refractive index ellipsoid
- Y axis direction in the plane of the refractive index ellipsoid of the refractive index ellipsoid and perpendicular to the in-plane maximum refractive index
- Z axis refractive index ellipsoid
- B Angle formed by X-axis and Y-axis
- C Angle formed by Y-axis and Z
- any retardation film produced by a single film formation belongs to the retardation film of the present invention.
- a layer provided with a slippery layer by co-casting or co-extrusion belongs to the retardation film of the present invention.
- ⁇ can be measured by, for example, KOBRA31WR manufactured by Oji Scientific Instruments.
- the three axes X, Y, and Z of the refractive index ellipsoid are measured by an ellipsometer (for example, a spectroscopic ellipsometer DVA36VW manufactured by Mizoji Optical Co., Ltd.) and the like, and are angles formed by the respective axes.
- the inclination angle ⁇ of the retardation film of the present invention is in the range of 0 ⁇ ⁇ ⁇ 50 degrees, more preferably 15 ⁇ ⁇ ⁇ 45 degrees.
- the inclined film of the present invention is preferably a cellulose ester film described below composed of a cellulose ester.
- the optical film of the present invention is preferably a cellulose ester film described below, an aromatic terminal polyester compound represented by the following general formula (I), Formula (I) B- (GA) nGB (Wherein B is an arylcarboxylic acid residue, G is an alkylene glycol residue having 2 to 12 carbon atoms, an aryl glycol residue having 6 to 12 carbon atoms, or an oxyalkylene glycol residue having 4 to 12 carbon atoms, A Represents an alkylene dicarboxylic acid residue having 4 to 12 carbon atoms or an aryl dicarboxylic acid residue having 6 to 12 carbon atoms, and n represents an integer of 1 or more.) And at least one compound selected from the group consisting of an ester compound having at least one of the pyranose structure or furanose structure and having all or part of the OH groups
- the acyl group bonded to the hydroxyl group may be linear or branched or may form a ring. Furthermore, another substituent may be substituted. In the case of the same degree of substitution, birefringence decreases when the number of carbon atoms is large. Therefore, the number of carbon atoms is preferably selected from acyl groups having 2 to 6 carbon atoms.
- the cellulose ester preferably has 2 to 4 carbon atoms, more preferably 2 to 3 carbon atoms.
- cellulose ester cellulose acetate, cellulose acetate propionate, cellulose acetate butyrate, or cellulose acetate to which propionate group or butyrate group is bonded in addition to acetyl group such as cellulose acetate propionate butyrate.
- Mixed fatty acid esters can be used.
- butyryl group forming the butyrate may be linear or branched.
- cellulose ester preferably used in the present invention, cellulose acetate, cellulose acetate butyrate, cellulose acetate propionate, and cellulose acetate phthalate are particularly preferably used.
- Preferred cellulose esters other than cellulose acetate phthalate for the present invention preferably satisfy the following formulas (1) and (2).
- Formula (2) 0 ⁇ Y ⁇ 1.5
- X is the degree of substitution of the acetyl group
- Y is the degree of substitution of the propionyl group or butyryl group, or a mixture thereof.
- resins having different degrees of substitution may be mixed and used.
- the mixing ratio is preferably 10:90 to 90:10 (mass ratio).
- cellulose acetate propionate is particularly preferably used.
- the method for measuring the substitution degree of the acyl group can be measured according to ASTM-D817-96.
- the weight average molecular weight Mw and number average molecular weight Mn of the cellulose ester were measured using gel permeation chromatography (GPC).
- the cellulose ester such as cellulose acetate phthalate of the present invention can be produced by a known method. Specifically, it can be synthesized with reference to the method described in JP-A-10-45804.
- Formula (I) B- (GA) nGB (Wherein B is an arylcarboxylic acid residue, G is an alkylene glycol residue having 2 to 12 carbon atoms, an aryl glycol residue having 6 to 12 carbon atoms, or an oxyalkylene glycol residue having 4 to 12 carbon atoms, A Represents an alkylene dicarboxylic acid residue having 4 to 12 carbon atoms or an aryl dicarboxylic acid residue having 6 to 12 carbon atoms, and n represents an integer of 1 or more.)
- an arylcarboxylic acid residue represented by B and an alkylene glycol residue or oxyalkylene glycol residue or arylglycol residue represented by G, an alkylenedicarboxylic acid residue or aryldicarboxylic acid represented by A And is obtained by the same reaction as a normal polyester compound.
- alkylene glycol component having 2 to 12 carbon atoms of the aromatic terminal polyester compound examples include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, 1,2-butanediol, 1,3-butanediol, 1,2-propanediol, 2-methyl 1,3-propanediol, 1,4-butanediol, 1,5-pentanediol, 2,2-dimethyl-1,3-propanediol (Neopentyl glycol), 2,2-diethyl-1,3-propanediol (3,3-dimethylolpentane), 2-n-butyl-2-ethyl-1,3-propanediol (3,3-dimethylol) Heptane), 3-methyl-1,5-pentanediol 1,6-hexanediol, 2,2,4-trimethyl 1 There are 3-pentane
- arylene dicarboxylic acid component having 6 to 12 carbon atoms examples include phthalic acid, terephthalic acid, isophthalic acid, 1,5 naphthalene dicarboxylic acid, and 1,4 naphthalene dicarboxylic acid.
- n is preferably 1 or more and 100 or less, and the number average molecular weight is preferably 300 to 1500, more preferably 400 to 1000.
- aromatic terminal polyester compound that can be used in the present invention are shown below, but the present invention is not limited thereto.
- the proportion of esterification is preferably 70% or more of the OH groups present in the pyranose structure or furanose structure.
- ester compounds are collectively referred to as sugar ester compounds.
- Glucose galactose, mannose, fructose, xylose or arabinose, lactose, sucrose, nystose, 1F-fructosyl nystose, stachyose, maltitol, lactitol, lactulose, cellobiose, maltose, cellotriose, maltotriose, raffinose or kestose .
- ester compound according to the present invention will be given below, but the present invention is not limited thereto.
- the cellulose ester film of the present invention preferably contains the sugar ester compound of the present invention in an amount of 0.5 to 30% by mass of the cellulose ester film in order to suppress the fluctuation of the retardation value and stabilize the display quality.
- the content is preferably 5 to 30% by mass.
- the content of the aromatic terminal polyester compound and sugar ester compound represented by the general formula (I) of the present invention can be selected in a mass ratio ranging from 99: 1 to 1:99, and the total amount of both compounds is The content is preferably 1 to 40% by mass relative to the cellulose ester.
- the plasticizer is not particularly limited, but preferably a (meth) acrylic polymer plasticizer, a polycarboxylic acid ester plasticizer, a glycolate plasticizer, a phthalate ester plasticizer, a fatty acid ester plasticizer, It is selected from polyhydric alcohol ester plasticizers, polyester plasticizers and the like.
- At least one is preferably a polyhydric alcohol ester plasticizer.
- the (meth) acrylic polymer used in the present invention preferably exhibits negative birefringence with respect to the stretching direction as a function when contained in an optical compensation film, and the structure is not particularly limited. However, it is preferably a polymer having a weight average molecular weight of 500 or more and 30000 or less obtained by polymerizing an ethylenically unsaturated monomer. In addition, about the birefringence, the presence was confirmed by the following test.
- the refractive index was measured using an Abbe refractometer-4T (manufactured by Atago Co., Ltd.) using a multi-wavelength light source.
- the refractive index ny in the stretching direction and the refractive index in the orthogonal in-plane direction were nx.
- the (meth) acrylic polymer is judged to be negatively birefringent with respect to the stretching direction.
- the (meth) acrylic polymer having a weight average molecular weight of 500 to 30,000 used in the present invention is a (meth) acrylic polymer having an aromatic ring in the side chain or a (meth) acrylic having a cyclohexyl group in the side chain.
- a polymer may be used.
- the polymer has a weight average molecular weight of 500 or more and 30000 or less, and the composition of the polymer is controlled, for example, when the optical compensation film is a cellulose ester film particularly preferable in the present invention, the cellulose ester and the polymer The compatibility with can be improved.
- the polymer Since the polymer has a weight average molecular weight of 500 or more and 30000 or less, it is considered to be between the oligomer and the low molecular weight polymer. In order to synthesize such a polymer, it is difficult to control the molecular weight in normal polymerization, and it is desirable to use a method that can align the molecular weight as much as possible by a method that does not increase the molecular weight too much.
- the (meth) acrylic polymer used in the optical compensation film of the present invention includes an ethylenically unsaturated monomer Xa having no aromatic ring and hydroxyl group in the molecule, and no hydroxyl ring in the molecule.
- the polymer Y is preferably a polymer Y having a weight average molecular weight of 500 or more and 3000 or less obtained by polymerizing the ethylenically unsaturated monomer Ya which is not present and an ethylenically unsaturated monomer copolymerizable with Ya.
- High molecular weight polymer X having a weight average molecular weight of 2,000 to 30,000 obtained by copolymerization of the polymerizable unsaturated monomer Xb and a copolymerizable ethylenically unsaturated monomer excluding Xa and Xb, and more preferably, Contains a low molecular weight polymer Y having a weight average molecular weight of 500 to 3,000 obtained by polymerizing an ethylenically unsaturated monomer Ya having no aromatic ring and an ethylenically unsaturated monomer copolymerizable with Ya It is preferable.
- the polymer X used in the present invention includes an ethylenically unsaturated monomer Xa having no aromatic ring and a hydroxyl group in the molecule and an ethylenically unsaturated monomer Xb having no hydroxyl ring in the molecule and having a hydroxyl group, Xa and Xb.
- Xa is an acrylic or methacrylic monomer that does not have an aromatic ring and a hydroxyl group in the molecule
- Xb is an acrylic or methacrylic monomer that does not have an aromatic ring in the molecule and has a hydroxyl group.
- the polymer X used in the present invention is represented by the following general formula (X).
- Xa represents an ethylenically unsaturated monomer having no aromatic ring and hydroxyl group in the molecule
- Xb represents an ethylenically unsaturated monomer having no aromatic ring and having a hydroxyl group in the molecule
- Xc represents a copolymerizable ethylenically unsaturated monomer excluding Xa and Xb.
- polymer X is preferably a polymer represented by the following general formula (X-1).
- R1 and R3 each represent a hydrogen atom or a methyl group.
- R2 represents an alkyl group having 1 to 12 carbon atoms or a cycloalkyl group.
- R4 represents —CH 2 —, —C 2 H 4 — or —C 3 H 6 —.
- Xc is, [CH 2 -C (-R1) (- CO 2 R2)] representing the a polymerizable monomer unit or [CH 2 -C (-R3) ( - - CO 2 R4-OH)].
- the monomer as a monomer unit constituting the polymer X according to the present invention is listed below, but is not limited thereto.
- a hydroxyl group means not only a hydroxyl group but also a group having an ethylene oxide chain.
- Examples of the ethylenically unsaturated monomer Xa having no aromatic ring and hydroxyl group in the molecule include methyl acrylate, ethyl acrylate, propyl acrylate (i-, n-), and butyl acrylate (n-, i-, s -, T-), pentyl acrylate (n-, i-, s-), hexyl acrylate (n-, i-), heptyl acrylate (n-, i-), octyl acrylate (n-, i -), Nonyl acrylate (n-, i-), myristyl acrylate (n-, i-), acrylic acid (2-ethylhexyl), acrylic acid ( ⁇ -caprolactone), etc.
- the thing changed into acid ester can be mentioned.
- methyl acrylate, ethyl acrylate, methyl methacrylate, ethyl methacrylate, and propyl methacrylate are preferable.
- the ethylenically unsaturated monomer Xb having no hydroxyl ring in the molecule and having a hydroxyl group is preferably acrylic acid or methacrylic acid ester as the monomer unit having a hydroxyl group.
- Xc is not particularly limited as long as it is a monomer other than Xa and Xb and is a copolymerizable ethylenically unsaturated monomer, but preferably has no aromatic ring.
- the molar composition ratio m: n of Xa and Xb is preferably in the range of 99: 1 to 65:35, more preferably in the range of 95: 5 to 75:25.
- P of Xc is 0-10. Xc may be a plurality of monomer units.
- haze tends to occur during film formation, and it is preferable to optimize these and determine the molar composition ratio of Xa and Xb.
- the molecular weight of the high molecular weight polymer X is more preferably 5000 or more and 30000 or less, and still more preferably 8000 or more and 25000 or less.
- the compatibility with the cellulose ester is further improved, and bleeding out under high temperature and high humidity and further haze generation immediately after film formation are suppressed.
- the weight average molecular weight of the polymer X according to the present invention can be adjusted by a known molecular weight adjusting method.
- a molecular weight adjusting method include a method of adding a chain transfer agent such as carbon tetrachloride, lauryl mercaptan, octyl thioglycolate, and the like.
- the weight average molecular weight Mw and the number average molecular weight Mn were measured using gel permeation chromatography (GPC).
- the low molecular weight polymer Y used in the present invention is a polymer having a weight average molecular weight of 500 or more and 3000 or less obtained by polymerizing an ethylenically unsaturated monomer Ya having no aromatic ring.
- a weight average molecular weight of 500 or more is preferred because the residual monomer in the polymer is reduced.
- Ya is preferably an acrylic or methacrylic monomer having no aromatic ring.
- the polymer Y used in the present invention is represented by the following general formula (Y).
- the polymer Y according to the present invention is more preferably a polymer represented by the following general formula (Y-1).
- Yb is not particularly limited as long as it is an ethylenically unsaturated monomer copolymerizable with [CH 2 —C (—R 5) (— CO 2 R 6)] which is Ya.
- Yb may be plural.
- k + q 100, q is preferably 1-30.
- An ethylenically unsaturated monomer Ya constituting the polymer Y obtained by polymerizing an ethylenically unsaturated monomer having no aromatic ring is, for example, methyl acrylate, ethyl acrylate, propyl acrylate ( i-, n-), butyl acrylate (n-, i-, s-, t-), pentyl acrylate (n-, i-, s-), hexyl acrylate (n-, i-), acrylic Acid heptyl (n-, i-), octyl acrylate (n-, i-), nonyl acrylate (n-, i-), myristyl acrylate (n-, i-), cyclohexyl acrylate, acrylic acid ( 2-ethylhexyl), acrylic acid ( ⁇ -caprolactone), acrylic acid (2-hydroxyethyl), acrylic acid (2-hydroxypropyl), acrylic acid (3
- Yb is not particularly limited as long as it is an ethylenically unsaturated monomer copolymerizable with Ya.
- vinyl esters include vinyl acetate, vinyl propionate, vinyl butyrate, vinyl valerate, vinyl pivalate, and vinyl caproate.
- Vinyl caprate, vinyl laurate, vinyl myristate, vinyl palmitate, vinyl stearate, vinyl cyclohexanecarboxylate, vinyl octylate, vinyl methacrylate, vinyl crotonate, vinyl sorbate, vinyl cinnamate and the like are preferred.
- Yb may be plural.
- Such polymerization methods include a method using a peroxide polymerization initiator such as cumene peroxide and t-butyl hydroperoxide, a method using a polymerization initiator in a larger amount than normal polymerization, and a mercapto compound in addition to the polymerization initiator. And a method using a chain transfer agent such as carbon tetrachloride, a method using a polymerization terminator such as benzoquinone and dinitrobenzene in addition to the polymerization initiator, and further disclosed in JP-A Nos. 2000-128911 and 2000-344823. Examples thereof include a compound having one thiol group and a secondary hydroxyl group, or a bulk polymerization method using a polymerization catalyst in which the compound and an organometallic compound are used in combination. Used.
- a peroxide polymerization initiator such as cumene peroxide and t-butyl hydroperoxide
- the polymer Y is preferably a polymerization method using a compound having a thiol group and a secondary hydroxyl group in the molecule as a chain transfer agent.
- the terminal of the polymer Y has a hydroxyl group and a thioether resulting from the polymerization catalyst and the chain transfer agent. The compatibility of Y and cellulose ester can be adjusted by this terminal residue.
- Polymers X and Y preferably have a hydroxyl value of 30 to 150 [mgKOH / g].
- sample Xg (about 1 g) is precisely weighed in a flask, and 20 ml of an acetylating reagent (a solution obtained by adding pyridine to 20 ml of acetic anhydride to 400 ml) is accurately added thereto. Attach an air cooling tube to the mouth of the flask and heat in a glycerin bath at 95-100 ° C. After 1 hour and 30 minutes, the mixture is cooled, 1 ml of purified water is added from an air condenser, and acetic anhydride is decomposed into acetic acid.
- an acetylating reagent a solution obtained by adding pyridine to 20 ml of acetic anhydride to 400 ml
- hydroxyl value is calculated by the following formula.
- Hydroxyl value ⁇ (BC) ⁇ f ⁇ 28.05 / X ⁇ + D
- B is the amount (ml) of 0.5 mol / L potassium hydroxide ethanol solution used for the blank test
- C is the amount (ml) of 0.5 mol / L potassium hydroxide ethanol solution used for titration
- f is a factor of a 0.5 mol / L potassium hydroxide ethanol solution
- D is an acid value
- 28.05 is 1/2 of 1 mol amount 56.11 of potassium hydroxide.
- polymer X and polymer Y are both excellent in compatibility with cellulose ester, excellent in productivity without evaporation and volatilization, good retention as a protective film for polarizing plates, low moisture permeability, and dimensional stability. Excellent in properties.
- the polymer X and the polymer Y are 5 mass% or more as a total amount with respect to the total mass of the cellulose ester, the polymer X and the polymer Y have a sufficient effect for adjusting the retardation value Rt. Moreover, if it is 35 mass% or less as a total amount, adhesiveness with polarizer PVA is favorable.
- the polymer X and the polymer Y can be directly added and dissolved as a material constituting the dope solution described later, or can be added to the dope solution after being previously dissolved in an organic solvent for dissolving the cellulose ester.
- R1- (OH) n represents an n-valent organic group
- n represents a positive integer of 2 or more
- the OH group represents an alcoholic and / or phenolic hydroxyl group.
- triethylene glycol triethylene glycol, tetraethylene glycol, dipropylene glycol, tripropylene glycol, sorbitol, trimethylolpropane, and xylitol are preferable.
- aliphatic monocarboxylic acid a fatty acid having a straight chain or a side chain having 1 to 32 carbon atoms can be preferably used.
- the number of carbon atoms is more preferably 1-20, and particularly preferably 1-10.
- acetic acid is contained, the compatibility with the cellulose ester is increased, and it is also preferable to use a mixture of acetic acid and another monocarboxylic acid.
- Preferred aliphatic monocarboxylic acids include acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, enanthic acid, caprylic acid, pelargonic acid, capric acid, 2-ethyl-hexanoic acid, undecylic acid, lauric acid, tridecylic acid, Saturated fatty acids such as myristic acid, pentadecylic acid, palmitic acid, heptadecylic acid, stearic acid, nonadecanoic acid, arachidic acid, behenic acid, lignoceric acid, serotic acid, heptacosanoic acid, montanic acid, melicic acid, laccelic acid, undecylenic acid, olein Examples thereof include unsaturated fatty acids such as acid, sorbic acid, linoleic acid, linolenic acid, and arachidonic acid.
- Examples of preferable alicyclic monocarboxylic acids include cyclopentane carboxylic acid, cyclohexane carboxylic acid, cyclooctane carboxylic acid, and derivatives thereof.
- aromatic monocarboxylic acids examples include those in which 1 to 3 alkoxy groups such as alkyl group, methoxy group or ethoxy group are introduced into the benzene ring of benzoic acid such as benzoic acid and toluic acid, biphenylcarboxylic acid, Examples thereof include aromatic monocarboxylic acids having two or more benzene rings such as naphthalenecarboxylic acid and tetralincarboxylic acid, or derivatives thereof. Benzoic acid is particularly preferable.
- the molecular weight of the polyhydric alcohol ester is not particularly limited, but is preferably 300 to 1500, and more preferably 350 to 750. A higher molecular weight is preferred because it is less likely to volatilize, and a smaller one is preferred in terms of moisture permeability and compatibility with cellulose ester.
- the carboxylic acid used in the polyhydric alcohol ester may be one kind or a mixture of two or more kinds. Moreover, all the OH groups in the polyhydric alcohol may be esterified, or a part of the OH groups may be left as they are.
- the glycolate plasticizer is not particularly limited, but alkylphthalylalkyl glycolates can be preferably used.
- phthalate ester plasticizer examples include diethyl phthalate, dimethoxyethyl phthalate, dimethyl phthalate, dioctyl phthalate, dibutyl phthalate, di-2-ethylhexyl phthalate, dioctyl phthalate, dicyclohexyl phthalate, and dicyclohexyl terephthalate.
- citrate plasticizer examples include acetyl trimethyl citrate, acetyl triethyl citrate, and acetyl tributyl citrate.
- fatty acid ester plasticizers examples include butyl oleate, methylacetyl ricinoleate, and dibutyl sebacate.
- phosphate ester plasticizer examples include triphenyl phosphate, tricresyl phosphate, cresyl diphenyl phosphate, octyl diphenyl phosphate, diphenyl biphenyl phosphate, trioctyl phosphate, tributyl phosphate, and the like.
- the polyvalent carboxylic acid is represented by the following general formula (b).
- Trivalent or higher aromatic polyvalent carboxylic acids such as trimellitic acid, trimesic acid, pyromellitic acid or derivatives thereof, succinic acid, adipic acid, azelaic acid, sebacic acid, oxalic acid, fumaric acid, maleic acid, tetrahydrophthal
- An aliphatic polyvalent carboxylic acid such as an acid, an oxypolyvalent carboxylic acid such as tartaric acid, tartronic acid, malic acid and citric acid can be preferably used.
- the alcohol used in the polyvalent carboxylic acid ester compound that can be used in the present invention is not particularly limited, and known alcohols and phenols can be used.
- an aliphatic saturated alcohol or aliphatic unsaturated alcohol having a straight chain or a side chain having 1 to 32 carbon atoms can be preferably used. More preferably, it has 1 to 20 carbon atoms, and particularly preferably 1 to 10 carbon atoms.
- alicyclic alcohols such as cyclopentanol and cyclohexanol or derivatives thereof
- aromatic alcohols such as benzyl alcohol and cinnamyl alcohol, or derivatives thereof can be preferably used.
- the alcoholic or phenolic hydroxyl group of the oxypolycarboxylic acid may be esterified with a monocarboxylic acid.
- monocarboxylic acids include the following, but the present invention is not limited thereto.
- aliphatic monocarboxylic acid a straight-chain or side-chain fatty acid having 1 to 32 carbon atoms can be preferably used. More preferably, it has 1 to 20 carbon atoms, and particularly preferably 1 to 10 carbon atoms.
- Preferred aliphatic monocarboxylic acids include acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, enanthic acid, caprylic acid, pelargonic acid, capric acid, 2-ethyl-hexanecarboxylic acid, undecylic acid, lauric acid, tridecylic acid, Saturated fatty acids such as myristic acid, pentadecylic acid, palmitic acid, heptadecylic acid, stearic acid, nonadecanoic acid, arachidic acid, behenic acid, lignoceric acid, serotic acid, heptacosanoic acid, montanic acid, melicic acid, and laccelic acid, undecylenic acid, olein Examples thereof include unsaturated fatty acids such as acid, sorbic acid, linoleic acid, linolenic acid, and arachidonic acid.
- Examples of preferable alicyclic monocarboxylic acids include cyclopentane carboxylic acid, cyclohexane carboxylic acid, cyclooctane carboxylic acid, and derivatives thereof.
- aromatic monocarboxylic acids examples include those in which an alkyl group is introduced into the benzene ring of benzoic acid such as benzoic acid and toluic acid, and two or more benzene rings such as biphenyl carboxylic acid, naphthalene carboxylic acid, and tetralin carboxylic acid.
- benzoic acid and toluic acid examples include those in which an alkyl group is introduced into the benzene ring of benzoic acid such as benzoic acid and toluic acid, and two or more benzene rings such as biphenyl carboxylic acid, naphthalene carboxylic acid, and tetralin carboxylic acid.
- the aromatic monocarboxylic acid which has, or those derivatives can be mentioned.
- Particularly preferred are acetic acid, propionic acid, and benzoic acid.
- the alcohol used for the polyvalent carboxylic acid ester that can be used in the present invention may be one kind or a mixture of two or more kinds.
- the acid value of the polyvalent carboxylic acid ester compound that can be used in the present invention is preferably 1 mgKOH / g or less, and more preferably 0.2 mgKOH / g or less. Setting the acid value in the above range is preferable because the environmental fluctuation of retardation is also suppressed.
- the cellulose ester film B according to the present invention can also contain an ultraviolet absorber.
- the ultraviolet absorber is intended to improve durability by absorbing ultraviolet light having a wavelength of 400 nm or less, and the transmittance at a wavelength of 370 nm is particularly preferably 10% or less, more preferably 5% or less. Preferably it is 2% or less.
- the ultraviolet absorber used in the present invention is not particularly limited, for example, oxybenzophenone compounds, benzotriazole compounds, salicylic acid ester compounds, benzophenone compounds, cyanoacrylate compounds, triazine compounds, nickel complex compounds, inorganic powders Examples include the body.
- the UV absorbers preferably used in the present invention are benzotriazole UV absorbers, benzophenone UV absorbers, and triazine UV absorbers, particularly preferably benzotriazole UV absorbers and benzophenone UV absorbers. .
- a discotic compound such as a compound having a 1,3,5 triazine ring is also preferably used as an ultraviolet absorber.
- the polarizing plate protective film according to the present invention preferably contains two or more ultraviolet absorbers.
- a polymeric ultraviolet absorber can be preferably used, and in particular, a polymer type ultraviolet absorber described in JP-A-6-148430 is preferably used.
- the method of adding the UV absorber can be added to the dope after dissolving the UV absorber in an alcohol such as methanol, ethanol or butanol, an organic solvent such as methylene chloride, methyl acetate, acetone or dioxolane or a mixed solvent thereof. Or you may add directly in dope composition.
- an alcohol such as methanol, ethanol or butanol
- an organic solvent such as methylene chloride, methyl acetate, acetone or dioxolane or a mixed solvent thereof.
- inorganic powders that do not dissolve in organic solvents use a dissolver or sand mill in the organic solvent and cellulose ester to disperse them before adding them to the dope.
- the amount of the UV absorber used is not uniform depending on the type of UV absorber, the operating conditions, etc., but when the dry film thickness of the polarizing plate protective film is 30 to 200 ⁇ m, the amount used is 0.5 to the polarizing plate protective film. Is preferably 10 to 10% by mass, and more preferably 0.6 to 4% by mass.
- Antioxidant are also referred to as deterioration inhibitors.
- a liquid crystal image display device or the like is placed in a high humidity and high temperature state, the cellulose ester film may be deteriorated.
- the antioxidant has a role of delaying or preventing the cellulose ester film from being decomposed by, for example, a residual solvent amount of halogen in the cellulose ester film or phosphoric acid of a phosphoric acid plasticizer. It is preferable to make it contain in a film.
- a hindered phenol compound is preferably used.
- 2,6-di-t-butyl-p-cresol, pentaerythrityl-tetrakis [3- (3,5-di- -T-butyl-4-hydroxyphenyl) propionate] triethylene glycol-bis [3- (3-t-butyl-5-methyl-4-hydroxyphenyl) propionate], 1,6-hexanediol-bis [3 -(3,5-di-t-butyl-4-hydroxyphenyl) propionate], 2,4-bis- (n-octylthio) -6- (4-hydroxy-3,5-di-t-butylanilino)- 1,3,5-triazine, 2,2-thio-diethylenebis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate], oct Decyl-3- (3,5-di-t-butyl-4-hydroxyphenyl
- 2,6-di-t-butyl-p-cresol, pentaerythrityl-tetrakis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate], triethylene glycol-bis [3 -(3-tert-butyl-5-methyl-4-hydroxyphenyl) propionate] is preferred.
- hydrazine-based metal deactivators such as N, N′-bis [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionyl] hydrazine and tris (2,4-di- A phosphorus processing stabilizer such as t-butylphenyl) phosphite may be used in combination.
- the amount of these compounds added is preferably 1 ppm to 1.0%, more preferably 10 to 1000 ppm in terms of mass ratio with respect to the cellulose derivative.
- the cellulose ester film according to the present invention preferably contains fine particles.
- examples of inorganic compounds include silicon dioxide, titanium dioxide, aluminum oxide, zirconium oxide, calcium carbonate, calcium carbonate, talc, clay, calcined kaolin, calcined calcium silicate, and hydrated silicic acid. Mention may be made of calcium, aluminum silicate, magnesium silicate and calcium phosphate. Further, fine particles of an organic compound can also be preferably used.
- the average primary particle size of the fine particles is preferably 5 to 400 nm, and more preferably 10 to 300 nm.
- the content of these fine particles in the polarizing plate protective film is preferably 0.01 to 1% by mass, particularly preferably 0.05 to 0.5% by mass. In the case of a polarizing plate protective film having a multilayer structure by the co-casting method, it is preferable to contain fine particles of this addition amount on the surface.
- Silicon dioxide fine particles are commercially available, for example, under the trade names Aerosil R972, R972V, R974, R812, 200, 200V, 300, R202, OX50, TT600 (manufactured by Nippon Aerosil Co., Ltd.). it can.
- Zirconium oxide fine particles are commercially available, for example, under the trade names Aerosil R976 and R811 (manufactured by Nippon Aerosil Co., Ltd.).
- Examples of the polymer include silicone resin, fluororesin and acrylic resin. Silicone resins are preferable, and those having a three-dimensional network structure are particularly preferable. For example, Tospearl 103, 105, 108, 120, 145, 3120 and 240 (manufactured by Toshiba Silicone Co., Ltd.) It is marketed by name and can be used.
- additives may be batch-added to a dope that is a cellulose ester-containing solution before film formation, or an additive solution may be separately prepared and added in-line.
- an additive solution may be separately prepared and added in-line.
- a preferable amount of the cellulose ester is 1 to 10 parts by mass, and more preferably 3 to 5 parts by mass with respect to 100 parts by mass of the solvent.
- a static mixer manufactured by Toray Engineering
- SWJ Toray static type in-tube mixer Hi-Mixer
- the cellulose ester film according to the present invention can be preferably used regardless of whether it is a film produced by a solution casting method or a film produced by a melt casting method.
- the cellulose ester film of the present invention is prepared by dissolving a cellulose ester and an additive in a solvent to prepare a dope, casting a dope onto an endless metal support that moves infinitely, and casting the dope. It is carried out by a step of drying as a web, a step of peeling from a metal support, a step of stretching or maintaining the width, a step of further drying, and a step of winding up the finished film.
- the concentration of cellulose ester in the dope is preferably higher because the drying load after casting on the metal support can be reduced. However, if the concentration of cellulose ester is too high, the load during filtration increases and the filtration accuracy is poor. Become.
- the concentration that achieves both of these is preferably 10 to 35% by mass, and more preferably 15 to 25% by mass.
- a preferable range of the mixing ratio of the good solvent and the poor solvent is 70 to 98% by mass for the good solvent and 2 to 30% by mass for the poor solvent.
- the good solvent and the poor solvent change depending on the average acetylation degree (acetyl group substitution degree) of the cellulose ester.
- the good solvent and the poor solvent change depending on the average acetylation degree (acetyl group substitution degree) of the cellulose ester.
- the good solvent and the poor solvent change depending on the average acetylation degree (acetyl group substitution degree) of the cellulose ester.
- the cellulose ester acetate ester acetyl group substitution degree 2.4
- cellulose Acetate propionate is a good solvent
- cellulose acetate (acetyl group substitution degree 2.8) is a poor solvent.
- the good solvent used in the present invention is not particularly limited, and examples thereof include organic halogen compounds such as methylene chloride, dioxolanes, acetone, methyl acetate, and methyl acetoacetate. Particularly preferred is methylene chloride or methyl acetate.
- the poor solvent used in the present invention is not particularly limited, but for example, methanol, ethanol, n-butanol, cyclohexane, cyclohexanone and the like are preferably used.
- the dope preferably contains 0.01 to 2% by mass of water.
- the recovery solvent may contain trace amounts of additives added to the cellulose ester, such as plasticizers, UV absorbers, polymers, monomer components, etc., but these are preferably reused even if they are included. Can be purified and reused if necessary.
- a general method can be used. When heating and pressurization are combined, it is possible to heat above the boiling point at normal pressure.
- a method in which a cellulose ester is mixed with a poor solvent and wetted or swollen, and then a good solvent is added and dissolved is also preferably used.
- the heating temperature with the addition of the solvent is preferably higher from the viewpoint of the solubility of the cellulose ester, but if the heating temperature is too high, the required pressure increases and the productivity deteriorates.
- the preferred heating temperature is 45 to 120 ° C, more preferably 60 to 110 ° C, and still more preferably 70 ° C to 105 ° C.
- the pressure is adjusted so that the solvent does not boil at the set temperature.
- a cooling dissolution method is also preferably used, whereby the cellulose ester can be dissolved in a solvent such as methyl acetate.
- the cellulose ester solution is filtered using an appropriate filter medium such as filter paper.
- an appropriate filter medium such as filter paper.
- the filter medium it is preferable that the absolute filtration accuracy is small in order to remove insoluble matters and the like, but there is a problem that the filter medium is likely to be clogged if the absolute filtration accuracy is too small.
- a filter medium with an absolute filtration accuracy of 0.008 mm or less is preferable, a filter medium with 0.001 to 0.008 mm is more preferable, and a filter medium with 0.003 to 0.006 mm is still more preferable.
- the material of the filter medium there are no particular restrictions on the material of the filter medium, and ordinary filter media can be used. However, plastic filter media such as polypropylene and Teflon (registered trademark), and metal filter media such as stainless steel do not drop off fibers. preferable.
- Bright spot foreign matter means that when two polarizing plates are placed in a crossed Nicol state, an optical film or the like is placed between them, light is applied from one polarizing plate side, and observation is performed from the other polarizing plate side. It is a point (foreign matter) where light from the opposite side appears to leak, and the number of bright spots having a diameter of 0.01 mm or more is preferably 200 / cm 2 or less.
- it is 100 pieces / cm 2 or less, still more preferably 50 pieces / m 2 or less, still more preferably 0 to 10 pieces / cm 2 . Further, it is preferable that the number of bright spots of 0.01 mm or less is small.
- the dope can be filtered by a normal method, but the method of filtering while heating at a temperature not lower than the boiling point of the solvent at normal pressure and in a range where the solvent does not boil under pressure is the filtration pressure before and after filtration.
- the increase in the difference (referred to as differential pressure) is small and preferable.
- the preferred temperature is 45 to 120 ° C, more preferably 45 to 70 ° C, and still more preferably 45 to 55 ° C.
- the metal support in the casting process is preferably a mirror-finished surface, and a stainless steel belt or a drum whose surface is plated with a casting is preferably used as the metal support.
- the preferred support temperature is 0 to 55 ° C, more preferably 25 to 50 ° C.
- the amount of residual solvent when peeling the web from the metal support is preferably 10 to 150% by mass, more preferably 20 to 40% by mass or 60 to 130% by mass. Particularly preferred is 20 to 30% by mass or 70 to 120% by mass.
- the amount of residual solvent is defined by the following formula.
- Residual solvent amount (% by mass) ⁇ (MN) / N ⁇ ⁇ 100 Note that M is the mass of a sample collected during or after the production of the web or film, and N is the mass after heating M at 114 ° C. for 1 hour.
- the web is peeled off from the metal support, and further dried, and the residual solvent amount is preferably 1% by mass or less, more preferably 0.1% by mass or less, Particularly preferred is 0 to 0.01% by mass or less.
- a roll drying method (a method in which webs are alternately passed through a plurality of rolls arranged above and below) and a method in which the web is dried while being conveyed by a tenter method are employed.
- the cellulose ester film of the present invention it is particularly preferable to perform stretching in the width direction (lateral direction) by a tenter method in which both ends of the web are held with clips or the like. Peeling is preferably performed at a peeling tension of 300 N / m or less.
- the means for drying the web is not particularly limited, and can be generally performed with hot air, infrared rays, a heating roll, microwave, or the like, but is preferably performed with hot air in terms of simplicity.
- drying temperature in the web drying process is increased stepwise from 40 to 200 ° C.
- the film thickness of the cellulose ester film is not particularly limited, but 10 to 200 ⁇ m is used.
- the film thickness is particularly preferably 10 to 100 ⁇ m. More preferably, it is 20 to 60 ⁇ m.
- the retardation film of the present invention is a biaxial retardation film having an optically distorted axis, and has a refractive index inclined to the minimum or maximum direction, and generates shear stress on the film surface. It can also be produced by performing a rolling process (also called a shear rolling process or a shearing process).
- the method, the method of controlling the peeling angle and peeling tension when peeling the film from the substrate (drum, belt, etc.) at the time of film formation, or the axis tilting treatment by wind described in Reference 4 and the film transporting by roller It is also possible to produce by shear rolling using a roller that does not rotate / drive the opposite side of the roller.
- These treatments are generally preferably carried out at a film treatment temperature in the range of Tg (glass transition temperature of the film) ⁇ 50 ° C. to Tg + 40 ° C.
- Tg glass transition temperature of the film
- variations occur, and it is necessary to further devise the process in order to produce it stably.
- a process for generating a shearing stress in an oblique direction is performed with another side of the width.
- One side of the film is subjected to the rolling process with a roller that generates shear stress in the opposite direction and a roller that rotates in the conveying direction after the processing between the rollers of different peripheral speeds is performed once.
- the other surface can be produced by a method such as shearing in an oblique direction with a residual solvent difference.
- the retardation film of the present invention expresses a necessary function by generating a shear stress and deforming it by giving a difference in speed or path length between one side of the film and the other side.
- this process is performed with a roller, drum, or belt sandwiched, if the film edge is subjected to a knurling process such as embossing in advance, the processing width should be carried out only inside the knurling part. Can do. You may slit an edge part after processing and knurling again.
- the retardation film of the present invention can be processed by a plurality of rollers or drums, but these must be different in surface hardness or elastic modulus, such as resin, elastic metal, rubber-coated metal, or metal-coated rubber. Are preferably combined.
- the surface material of the hard roller is hard chrome plated, nickel plated, or ceramic super hard material tungsten-carbide.
- the material has good releasability and can be polished to a mirror surface.
- the surface roughness is 0.2 s or less, particularly preferably 0.1 s or less.
- the retardation film of the present invention may be processed by changing the temperature of upper and lower rollers (or drums) to be processed at different temperatures, and may have a difference of about 150 ° C., for example.
- rollers (or drums) having different surface hardness are used and a temperature difference is given to the rollers, it is preferable to increase the high hardness side at a high temperature because it is easy to stabilize from the viewpoint of control.
- rollers when the elasticity of the rollers is different and it is preferable to make a difference in peripheral speed, it is preferable to drive the roller on the lower elastic side at a low speed or rotate around.
- the treatment may be performed with the solvent remaining on the web.
- the roller peripheral speed is important for the ratio of the peripheral speeds of the two rollers.
- the value of (high speed side speed / low speed side speed) is preferably 1.05 to 1.80, and preferably 1.60 or less. More preferred.
- the temperature at which the treatment of the present invention is carried out is preferably carried out at or above the glass transition point of the material used. If the temperature is at or below the glass transition point, variations are likely to occur during processing, making it difficult to ensure stability.
- a metal belt may be used.
- the average value is usually within ⁇ 10%, preferably within ⁇ 5%, more preferably within ⁇ 1%, and particularly preferably within ⁇ 0.5%.
- a horizontal uniaxial stretching method using a tenter method a compression stretching method between rollers, a longitudinal uniaxial stretching method using two sets of rollers having different circumferences, or a stretching method using a biaxial stretching method combining a horizontal uniaxial and a longitudinal uniaxial. Etc. can be used.
- the stretching speed is usually 0.1 to 200% / second, preferably 0.1 to 100% / second.
- stretching may be performed in two directions at the same time, or may be performed in a direction different from the first stretching direction after uniaxial stretching.
- the stretched film may be cooled as it is, but it is preferably at least 1 second in a temperature atmosphere of Tg-20 ° C. to Tg.
- the knurling method can process a metal ring having an uneven pattern on its side surface by heating or pressing.
- the grip part of the clip of the both ends of a film is deform
- the yellow index Ye at the end in the width direction of the film immediately after melt extrusion and the yellow index Yc at the center of the film preferably satisfy the following formula, and more preferably, Ye / Yc is 3.0 or less. If Ye / Yc is greater than 5.0, the color of the produced film increases when the film edge is cut off and reused as a raw material.
- the yellow index at the end is defined as the maximum value within 30 mm from both ends in the width direction of the film.
- the film thickness variation of the cellulose ester film of the present invention is preferably in the range of ⁇ 3%, more preferably ⁇ 1%.
- the glass transition temperature Tg mentioned here is a temperature increase / decrease process once again after temperature increase / decrease at 10 ° C./min using a differential scanning calorimeter (DSC-7 type manufactured by Perkin Elmer). The Tg at that time was measured and measured, and the intermediate glass transition temperature (Tmg) determined according to JIS K7121 (1987) was used.
- the polarizing plate can be produced by a general method.
- the cellulose ester film of the present invention is preferably subjected to alkali saponification treatment, and the treated film is bonded to at least one surface of a polarizer prepared by immersing and stretching in an iodine solution using a completely saponified polyvinyl alcohol aqueous solution. .
- the cellulose ester film of the present invention may be used on the other surface, or another optical film or a polarizing plate protective film may be used.
- a commercially available cellulose ester film can be used as the optical film and the polarizing plate protective film used on the other surface.
- the optically anisotropic layer can be formed by the method described in JP-A-2003-98348. By using it in combination with the cellulose ester film of the present invention, a polarizing plate having excellent flatness and a stable viewing angle expansion effect can be obtained.
- a polarizer which is a main component of a polarizing plate, is an element that allows only light of a plane of polarization in a certain direction to pass.
- a typical polarizer currently known is a polyvinyl alcohol-based polarizing film, which is polyvinyl alcohol.
- iodine is dyed on a system film and one in which dichroic dye is dyed.
- the polarizer is formed by forming a polyvinyl alcohol aqueous solution into a film and dyeing the film by uniaxial stretching or dyeing or uniaxially stretching, and then performing a durability treatment with a boron compound.
- a polarizing plate On the surface of the polarizer, one side of the cellulose ester film of the present invention is bonded to form a polarizing plate. It is preferably bonded with an aqueous adhesive mainly composed of completely saponified polyvinyl alcohol or the like.
- the long cellulose ester film produced by the melt casting film-forming method according to the present invention can be bonded with a long polarizer (polarizing film) by subjecting it to alkali saponification treatment.
- the productive effect is obtained with a long length of 1, and the longer the length is 1500 m, 2500 m, and 5000 m, the higher the productive effect of manufacturing the polarizing plate.
- the polarizing plate using the cellulose ester film of this invention is excellent in rework property, the effect that a polarizing plate yield improves can also be acquired.
- the polarizing plate containing the cellulose ester film of the present invention can exhibit high display quality as compared with a normal polarizing plate.
- the polarizing plate of the present invention can be used for TN mode, OCB (Optical Compensated Bend) mode, IPS (In-Plane Switching) mode, and the like.
- the liquid crystal display device is applied as a device for colorization and moving image display, and the display quality is improved by the present invention, and the contrast is improved and the resistance of the polarizing plate is improved. .
- Example 1 ⁇ Production of retardation film>
- the conditions for carrying out the shearing treatment were in the range of Tg-30 ° C. to Tg + 100 ° C. in the production of all retardation films.
- the cellulose esters used are listed in Table 1.
- ⁇ Production of Cellulose Ester Film 101> ⁇ Fine particle dispersion 1> Fine particles (Aerosil R812 manufactured by Nippon Aerosil Co., Ltd.) 11 parts by weight Ethanol 89 parts by weight The above was stirred and mixed with a dissolver for 50 minutes, and then dispersed with Manton Gorin.
- Fine particle addition liquid 1 The fine particle dispersion 1 was slowly added to the dissolution tank containing methylene chloride with sufficient stirring. Further, the particles were dispersed by an attritor so that the secondary particles had a predetermined particle size. This was filtered through Finemet NF manufactured by Nippon Seisen Co., Ltd. to prepare a fine particle additive solution 1.
- a main dope solution having the following composition was prepared. First, methylene chloride and ethanol were added to the pressure dissolution tank. Cellulose esters A and B were added to a pressure dissolution tank containing a solvent while stirring. This is completely dissolved with heating and stirring. This was designated as Azumi Filter Paper No. The main dope solution was prepared by filtration using 244.
- the dope solution was uniformly cast on a stainless steel belt support at a temperature of 33 ° C. and a width of 1500 mm. The temperature of the stainless steel belt was controlled at 30 ° C.
- the solvent was evaporated until the residual solvent amount reached 105%, and peeling was performed from the stainless steel band support with a peeling tension of 162 N / m.
- the web is conveyed at a speed of 85 m / min, and the web is set to a pressure of 2.1 MPa (contact width is 6 mm) with a metal roller having a diameter of 35 cm and an elastic metal roller.
- the speed of the elastic metal roller was made the same as the conveying speed, the speed of the metal roller was set to 98 m / min, and the first shearing process was performed.
- the residual solvent when passing through this roller was 1.2% by mass.
- the second shearing process by the metal roller (driving roller) / elastic metal roller (following roller) whose rotation direction is 50 degrees and the conveyance direction, and further, the conveyance direction is -50 degrees A third shearing treatment was performed with a metal roller / elastic metal roller. During the second shearing process, the speed of the metal roller was set to 99 m / min, and during the third shearing process, the rotation speed of the metal roller was set to 98.5 m / min. The set temperatures of the metal rollers were all 185 ° C.
- stretching was performed in a direction perpendicular to the transport direction by a tenter.
- the stretching temperature was set at 155 ° C.
- the stretching ratio was 1.08 times
- the stretching speed was 15% / second.
- drying is terminated while conveying the drying zone at 120 ° C. and 130 ° C. with a number of rolls, and slitting to a width of 1.75 m, A knurling process with a width of 10 mm and a height of 7 ⁇ m was applied to both ends of the film, and the film was wound around a core having an inner diameter of 6 inches with an initial tension of 220 N / m and a final tension of 110 N / m to obtain a retardation film 101 with a film thickness of 42 ⁇ m.
- phase difference film 102> In the production of the phase difference film 101, the speed of the metal roller in the second and third shearing treatments was 104 m / min, and the stretching by the tenter was 1.2 times (stretching speed was 40% / second). A retardation film 102 having a thickness of 34 ⁇ m was produced in the same manner as the retardation film 101.
- ⁇ Phase difference films 103, 104, 105, 106> In the production of the retardation film 101, a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- the phase difference film 103 having a film thickness of 50 ⁇ m at a shearing process of 1st, 2nd and 3rd times, a metal roller speed of 85.6 m / min, a stretching ratio of 1.20 times with a tenter, a stretching temperature of 165 ° C.
- a retardation film 104 having a film thickness of 60 ⁇ m was prepared by setting the temperature of the metal roller to 195 ° C., the stretching ratio of the tenter to 1.15 times, and the stretching temperature to 145 ° C. Both residual solvents during the metal roller treatment were 2.2% by mass.
- the stretching condition of the retardation film 103 in the tenter is 1.55 times at 175 ° C., the retardation film 105 having a film thickness of 25 ⁇ m, the second metal roller temperature of the retardation film 103 is 195 ° C., and the third temperature is 175
- a retardation film 106 having a thickness of 70 ⁇ m was produced at a temperature of 0 ° C.
- Preparation of retardation film 107> In the production of the retardation film 101, a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- the film thickness is the same as that of the phase difference film 102 except that the second treatment angle of the metal roller is 5 degrees with respect to the conveying direction and the speed of the metal roller is 121 m / min.
- a 45 ⁇ retardation film 107 was produced.
- ⁇ Preparation of retardation films 108 and 109> In the production of the retardation film 101, a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- Retardation film 108 speed of metal belt Min 86.5M, except that the draw ratio of the tenter to 1.9 times to produce a retardation film 109 having a thickness of 34 ⁇ m in the same manner as the phase difference film 101.
- ⁇ Preparation of retardation film 110> In the production of the retardation film 101, a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- the first treatment of the shearing process was performed using a 2.8 mm metal belt made of SUS instead of the metal roller, the pressure was set to 2.8 MPa, and the contact width was set to 15 cm. The process was performed at a speed of 88 m / min, an atmospheric temperature of 200 ° C., a tenter stretching temperature of 175 ° C., and a stretching ratio of 1.70 times.
- a retardation film 110 was produced.
- ⁇ Preparation of retardation film 111> In the production of the retardation film 101, a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- the first treatment of the shearing process was performed using a 2.8 mm metal belt made of SUS instead of the metal roller, the pressure was set to 2.6 MPa, and the contact width was set to 15 cm.
- the film was processed at a speed of 88 m / min, an atmospheric temperature of 186 ° C., a tenter stretching temperature of 158 ° C., and a stretching ratio of 1.50 times.
- a retardation film 111 was produced.
- the KC8UX manufactured by Konica Minolta Opto Co., Ltd. was similarly saponified on one side of the polarizer, and the cellulose ester film of the present invention that had been subjected to the alkali saponification treatment was bonded to the opposite side with a completely saponified polyvinyl alcohol 5% aqueous solution.
- the polarizer was bonded and dried so that the transmission axis of the polarizer and the in-plane slow axis of the film were parallel to each other.
- the polarizing plate 107 was produced by rotating the retardation film 107 by 90 degrees so that the in-plane slow axis of the retardation film and the transmission axis of the polarizer were orthogonal to each other.
- the elastic metal roller was set at the same speed as the conveying speed, and the metal roller was sheared so that the speed of the metal roller was 2.5 m / min.
- the second treatment by the metal roller / elastic metal roller whose rotation direction is the conveyance direction and the 50 degree direction, and the third treatment by the metal roller / elastic metal roller whose rotation direction is the ⁇ 50 degree direction. was processed.
- the speed of the metal roller was set to 2.7 m / min
- the rotation speed of the metal roller was set to 2.75 m / min.
- the set temperatures of the metal rollers were all 165 ° C.
- the retardation film 112 was produced by setting the stretching temperature to 148 ° C., the stretching ratio to 1.15 times, and the stretching speed to 15% / second.
- the speed of the elastic metal roller was set to be the same as the conveying speed, and the speed of the metal roller was set to 3.2 m / min.
- the second treatment by the metal roller / elastic metal roller whose rotation direction is the conveyance direction and the 50 degree direction, and the third treatment by the metal roller / elastic metal roller whose rotation direction is the ⁇ 50 degree direction. was processed.
- the speed of the metal roller was set to 2.9 m / min
- the rotation speed of the metal roller was set to 2.85 m / min.
- the set temperatures of the metal rollers were all 155 ° C.
- a retardation film 201 was produced in the same manner as the retardation film 112 except that film formation was performed in the same manner as the retardation film 112 and the second and third roller treatments were not performed.
- the saponified KC8UX manufactured by Konica Minolta Opto Co., Ltd. is used as a fully saponified polyvinyl alcohol 5% aqueous solution as an adhesive, and the transmission axis of the polarizer and the film Each of them was bonded and dried so that the in-plane slow axes were parallel to each other.
- a dope was produced and formed in the same manner except that the composition of the main dope solution was changed to the following.
- the retardation film 203 is manufactured by setting the speed of the metal roller to 15 m / min, setting the atmosphere temperature to 210 ° C., setting the stretching temperature in the tenter to 178 ° C., and the stretching ratio to 2.1 times. did.
- ⁇ Preparation of retardation films 204 and 205> The film was formed in the same manner as the phase difference film 101, and the second roller treatment and the third roller treatment were not performed, and the stretching conditions in the tenter were the stretching temperature of 130 ° C., the stretching ratio of 1.21 times, and 114 ° C. Retardation films 204 and 205 were produced at a double magnification.
- polarizing plate 207 a polarizing plate was prepared such that the slow axis and the transmission axis of the polarizer were parallel to each other through an adhesive layer on a polarizing plate using Konica Minolta Op KC8UX.
- ⁇ Gradation inversion> Using a wedge that displays white to black in 8 steps, observe the angle at which the brightness begins to change up, down, sideways, and diagonally down, ⁇ over 30 degrees in all directions, and down over 30 degrees in all other directions The direction is 20 to 30 degrees, and the direction is 20 degrees in any one direction.
- ⁇ Frame unevenness> The display device was treated at 50 ° C. and 90% RH for 200 hours. Immediately after the display device was taken out, it was displayed in black and allowed to stand at room temperature for 2 hours, and was judged by the magnitude and strength of unevenness generated on the entire surface and the frame shape. ⁇ If there is no noticeable unevenness or a non-problematic non-uniformity occurs in one place, ⁇ If there is a non-practical unevenness in two places ⁇ If there is a noticeable unevenness in one place What was there was marked with x.
Landscapes
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- General Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polarising Elements (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
Abstract
本発明の目的は、コントラスト視野角が広く、位相差ムラの小さい位相差フィルムを提供することにある。 本発明の目的は、光学的に2軸の位相差フィルムであってX軸、Y軸、Z軸、A、B、C、βを下記のように定義するとき、次の(1)~(6)の関係を全て有することを特徴とする位相差フィルム。 (1)0≦β≦50度 (2)B=90度 (3)A≠90度 (4)C≠90度 (5)80度<A<90度 (6)80度<C<90度 X軸:屈折率楕円体の面内最大屈折率の方向 Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向 Z軸:屈折率楕円体の屈折率最小方向 A:X軸とZ軸のなす角度 B:X軸とY軸のなす角度 C:Y軸とZ軸のなす角度 β:フィルムの法線方向とZ軸のなす角度 によって達成された。
Description
本発明は、位相差フィルムに関し、より詳しくは、コントラスト視野角および位相差ムラに優れた位相差フィルムフィルムに関する。
セルロースエステルフィルム、ポリカーボネートフィルム、ポリシクロオレフィンフィルム等は、TN型液晶表示装置用の位相差フィルムとして使用されている。
TN型液晶表示装置は、中間調が反転するといった問題がある。そのため特許文献1では、円盤状化合物のチルト角を制御することでこの問題の解決を図っているが、この方法も中間調反転を無くすためには十分ではなく、実用的な視野角はまだ非常に狭いままであった。
一方、ポリカーボネートフィルムでは製膜時に強制的に剪断をかけ、フィルム厚み方向の光軸を傾斜されることにより位相差を発現させた、TN型液晶表示装置に適した位相差フィルム(以下、傾斜フィルムともいう)が提案されている(特許文献2~3)。
特許文献4では、光弾性係数が大きいことにより位相差の発現性に不安定なポリカーボネート、ポリエステル、トリアセチルセルロースの代わりに、熱可塑性ノルボルネン系樹脂(ポリシクロオレフィンフィルムの一種)を使用した技術が開示されている。
しかしながら、熱可塑性ノルボルネン系樹脂フィルムでは、コントラスト視野角が広くなるため、位相差ムラが通常延伸と同程度でも非常に目立ち易くなり、LCD品位の低下となる。
さらに、一軸を傾斜させただけではコントラスト視野角が十分に広がらず、この問題を解決するには傾斜した位相差層が、光学的に二軸の必要あることがわかった。
従って本発明の目的は、コントラスト視野角が広く、位相差ムラの小さい位相差フィルムを提供することにある。
本発明の上記課題は以下の構成により達成される。
1.光学的に2軸の位相差フィルムであってX軸、Y軸、Z軸、A、B、C、βを下記のように定義するとき、次の(1)~(6)の関係を全て有することを特徴とする位相差フィルム。
(1)0≦β≦50度
(2)B=90度
(3)A≠90度
(4)C≠90度
(5)80度<A<90度
(6)80度<C<90度
X軸:屈折率楕円体の面内最大屈折率の方向
Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z軸:屈折率楕円体の屈折率最小方向
A:X軸とZ軸のなす角度
B:X軸とY軸のなす角度
C:Y軸とZ軸のなす角度
β:フィルムの法線方向とZ軸のなす角度
2.前記位相差フィルムがセルロースエステルフィルムであることを特徴とする前記1記載の位相差フィルム。
(1)0≦β≦50度
(2)B=90度
(3)A≠90度
(4)C≠90度
(5)80度<A<90度
(6)80度<C<90度
X軸:屈折率楕円体の面内最大屈折率の方向
Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z軸:屈折率楕円体の屈折率最小方向
A:X軸とZ軸のなす角度
B:X軸とY軸のなす角度
C:Y軸とZ軸のなす角度
β:フィルムの法線方向とZ軸のなす角度
2.前記位相差フィルムがセルロースエステルフィルムであることを特徴とする前記1記載の位相差フィルム。
本発明によれば、長期間連続生産しても、傾斜角度のバラツキが小さい位相差フィルムを提供することができる。
以下本発明を実施するための最良の形態について詳細に説明するが、本発明はこれらに限定されるものではない。
本発明の位相差フィルムは、製膜されたフィルムそのものであり、液晶や光学異方性化合物などを塗布加工することなく、フィルムそのものに光学的な機能である位相差を付与したフィルムであり、位相差機能として必要な部分が1回の製膜工程で出来上がったフィルムである。
つまり、厚さ方向の成分・組成が異なる層を積層した場合でも、1回の製膜で作製された位相差フィルムであれば、本発明の位相差フィルムに属するものである。
例えば、共流延や共押し出し等で滑り製を付与する層を設けたりするものは本発明の位相差フィルムに属する。
本発明の位相差フィルムは、光学的に2軸の位相差フィルムであってX軸、Y軸、Z軸、A、B、C、βを下記のように定義するとき、次の(1)~(6)の関係を全て有することを特徴とする。
(1)0≦β≦50度
(2)B=90度
(3)A≠90度
(4)C≠90度
(5)80度<A<90度
(6)80度<C<90度
X軸:屈折率楕円体の面内最大屈折率の方向
Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z軸:屈折率楕円体の屈折率最小方向
A:X軸とZ軸のなす角度
B:X軸とY軸のなす角度
C:Y軸とZ軸のなす角度
β:フィルムの法線方向とZ軸のなす角度(傾斜角度)
これらの軸および角度は傾斜測定が可能な複屈折計(例えば王子計測機器製KOBRA-31WR、オプトサイエンス社製Axoscan、株式会社溝尻光学工業所製分光エリプソメータDVA36VW)を用いて測定、あるいは算出することができる。
(1)0≦β≦50度
(2)B=90度
(3)A≠90度
(4)C≠90度
(5)80度<A<90度
(6)80度<C<90度
X軸:屈折率楕円体の面内最大屈折率の方向
Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z軸:屈折率楕円体の屈折率最小方向
A:X軸とZ軸のなす角度
B:X軸とY軸のなす角度
C:Y軸とZ軸のなす角度
β:フィルムの法線方向とZ軸のなす角度(傾斜角度)
これらの軸および角度は傾斜測定が可能な複屈折計(例えば王子計測機器製KOBRA-31WR、オプトサイエンス社製Axoscan、株式会社溝尻光学工業所製分光エリプソメータDVA36VW)を用いて測定、あるいは算出することができる。
βは例えば王子計測機器製KOBRA31WRにより測定できる。屈折率楕円体の三軸X、Y、Zは、エリプソメータ(例えば株式会社溝尻光学工業所製分光エリプソメータDVA36VW)などで測定し、それぞれの軸のなす角度となる。
Z方向が屈折率楕円体の面法線方向からの傾斜の有無は簡易的に複屈折計による確認も可能である。Zが法線より傾いていれば、A≠90度、C≠90度となる。屈折率楕円体の面内進相軸を傾斜軸として測定を行い、位相差がゼロとなる角度(すなわち光軸)を測定し、その中心(平均)角度を算出することでZ軸を算出することができる。
本発明と特許文献2~4に記載の技術との最も大きな違いは、これまでの傾斜フィルムがA=B=C=90度であったのに対し、本発明の傾斜フィルムはB=90度であるものの、A≠90度、C≠90度であることを特徴としている。
このことが、中間調の特に下方向の反転を抑制しながらコントラスト視野角を広げるという本発明の効果を顕著にしている。
本発明の位相差フィルムの傾斜角度βは0≦β≦50度の範囲であるが、15≦β≦45度であることがより好ましい。
本発明の傾斜フィルムは、セルロースエステルからなる以下に説明するセルロースエステルフィルムであることが好ましい。
<本発明のセルロースエステルフィルム>
本発明の光学フィルムは、以下に述べるセルロースエステルフィルムであることが好ましく、下記一般式(I)に示す芳香族末端ポリエステル系化合物、
一般式(I) B-(G-A)n-G-B
(式中、Bはアリールカルボン酸残基、Gは炭素数2~12のアルキレングリコール残基または炭素数6~12のアリールグリコール残基または炭素数が4~12のオキシアルキレングリコール残基、Aは炭素数4~12のアルキレンジカルボン酸残基または炭素数6~12のアリールジカルボン酸残基を表し、またnは1以上の整数を表す。)
およびピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下有し、その構造のOH基のすべてもしくは一部をエステル化したエステル化合物、から選択される少なくとも1種の化合物を含有するセルロースエステルフィルムであることが好ましい。
<本発明のセルロースエステルフィルム>
本発明の光学フィルムは、以下に述べるセルロースエステルフィルムであることが好ましく、下記一般式(I)に示す芳香族末端ポリエステル系化合物、
一般式(I) B-(G-A)n-G-B
(式中、Bはアリールカルボン酸残基、Gは炭素数2~12のアルキレングリコール残基または炭素数6~12のアリールグリコール残基または炭素数が4~12のオキシアルキレングリコール残基、Aは炭素数4~12のアルキレンジカルボン酸残基または炭素数6~12のアリールジカルボン酸残基を表し、またnは1以上の整数を表す。)
およびピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下有し、その構造のOH基のすべてもしくは一部をエステル化したエステル化合物、から選択される少なくとも1種の化合物を含有するセルロースエステルフィルムであることが好ましい。
〈セルロースエステル〉
本発明のセルロースエステルとしては特に限定はないが、セルロースエステルとして炭素数2~22程度のカルボン酸エステルであり、芳香族カルボン酸のエステルでもよく、特に炭素数が6以下の低級脂肪酸エステルであることが好ましい。
本発明のセルロースエステルとしては特に限定はないが、セルロースエステルとして炭素数2~22程度のカルボン酸エステルであり、芳香族カルボン酸のエステルでもよく、特に炭素数が6以下の低級脂肪酸エステルであることが好ましい。
水酸基に結合するアシル基は、直鎖であっても分岐してもよく、また環を形成してもよい。更に別の置換基が置換してもよい。同じ置換度である場合、前記炭素数が多いと複屈折性が低下するため、炭素数としては炭素数2~6のアシル基の中で選択することが好ましい。前記セルロースエステルとしての炭素数が2~4であることが好ましく、炭素数が2~3であることがより好ましい。
具体的には、セルロースエステルとしては、セルロースアセテート、セルロースアセテートプロピオネート、セルロースアセテートブチレート、またはセルロースアセテートプロピオネートブチレートのようなアセチル基の他にプロピオネート基またはブチレート基が結合したセルロースの混合脂肪酸エステルを用いることができる。
なお、ブチレートを形成するブチリル基としては、直鎖状でも分岐していてもよい。本発明において好ましく用いられるセルロースエステルとしては、特にセルロースアセテート、セルロースアセテートブチレート、セルロースアセテートプロピオネート、セルロースアセテートフタレートが好ましく用いられる。
本発明に好ましいセルロースアセテートフタレート以外のセルロースエステルとしては、下記式(1)および(2)を同時に満足するものが好ましい。
式(1) 2.0≦X+Y≦3.0
式(2) 0≦Y≦1.5
式中、Xはアセチル基の置換度、Yはプロピオニル基またはブチリル基、もしくはその混合物の置換度である。
式(2) 0≦Y≦1.5
式中、Xはアセチル基の置換度、Yはプロピオニル基またはブチリル基、もしくはその混合物の置換度である。
また、目的に叶う光学特性を得るために置換度の異なる樹脂を混合して用いても良い。混合比としては10:90~90:10(質量比)が好ましい。
この中で特にセルロースアセテートプロピオネートが好ましく用いられる。セルロースアセテートプロピオネートでは、1.0≦X≦2.5であり、0.1≦Y≦1.5、2.0≦X+Y≦3.0であることが好ましい。アシル基の置換度の測定方法はASTM-D817-96に準じて測定することができる。
本発明に用いられるセルロースエステルの数平均分子量は、60000~300000の範囲が、得られるフィルムの機械的強度が強く好ましい。更に70000~200000のものが好ましく用いられる。
セルロースエステルの重量平均分子量Mw、数平均分子量Mnは、ゲルパーミエーションクロマトグラフィー(GPC)を用いて測定した。
測定条件は以下の通りである。
溶媒: メチレンクロライド
カラム: Shodex K806、K805、K803G(昭和電工(株)製を3本接続して使用した)
カラム温度:25℃
試料濃度: 0.1質量%
検出器: RI Model 504(GLサイエンス社製)
ポンプ: L6000(日立製作所(株)製)
流量: 1.0ml/min
校正曲線: 標準ポリスチレンSTK standard ポリスチレン(東ソー(株)製)Mw=1000000~500の13サンプルによる校正曲線を使用した。13サンプルは、ほぼ等間隔に用いる。
カラム: Shodex K806、K805、K803G(昭和電工(株)製を3本接続して使用した)
カラム温度:25℃
試料濃度: 0.1質量%
検出器: RI Model 504(GLサイエンス社製)
ポンプ: L6000(日立製作所(株)製)
流量: 1.0ml/min
校正曲線: 標準ポリスチレンSTK standard ポリスチレン(東ソー(株)製)Mw=1000000~500の13サンプルによる校正曲線を使用した。13サンプルは、ほぼ等間隔に用いる。
本発明に用いられる、セルロースエステルの原料のセルロースとしては、特に限定はないが、綿花リンター、木材パルプ、ケナフなどを挙げることができる。またそれらから得られたセルロースエステルはそれぞれ任意の割合で混合使用することができる。
本発明のセルロースアセテートフタレート等のセルロースエステルは、公知の方法により製造することができる。具体的には特開平10-45804号に記載の方法を参考にして合成することができる。
〈一般式(I)に示す芳香族末端ポリエステル系化合物〉
本発明では、下記一般式(I)で表せる芳香族末端ポリエステル系化合物を使用する。
本発明では、下記一般式(I)で表せる芳香族末端ポリエステル系化合物を使用する。
一般式(I) B-(G-A)n-G-B
(式中、Bはアリールカルボン酸残基、Gは炭素数2~12のアルキレングリコール残基または炭素数6~12のアリールグリコール残基または炭素数が4~12のオキシアルキレングリコール残基、Aは炭素数4~12のアルキレンジカルボン酸残基または炭素数6~12のアリールジカルボン酸残基を表し、またnは1以上の整数を表す。)
一般式(I)中、Bで示されるアリールカルボン酸残基とGで示されるアルキレングリコール残基またはオキシアルキレングリコール残基またはアリールグリコール残基、Aで示されるアルキレンジカルボン酸残基またはアリールジカルボン酸残基とから構成されるものであり、通常のポリエステル系化合物と同様の反応により得られる。
(式中、Bはアリールカルボン酸残基、Gは炭素数2~12のアルキレングリコール残基または炭素数6~12のアリールグリコール残基または炭素数が4~12のオキシアルキレングリコール残基、Aは炭素数4~12のアルキレンジカルボン酸残基または炭素数6~12のアリールジカルボン酸残基を表し、またnは1以上の整数を表す。)
一般式(I)中、Bで示されるアリールカルボン酸残基とGで示されるアルキレングリコール残基またはオキシアルキレングリコール残基またはアリールグリコール残基、Aで示されるアルキレンジカルボン酸残基またはアリールジカルボン酸残基とから構成されるものであり、通常のポリエステル系化合物と同様の反応により得られる。
本発明で使用される芳香族末端ポリエステル系化合物のアリールカルボン酸成分としては、例えば、安息香酸、パラターシャリブチル安息香酸、オルソトルイル酸、メタトルイル酸、パラトルイル酸、ジメチル安息香酸、エチル安息香酸、ノルマルプロピル安息香酸、アミノ安息香酸、アセトキシ安息香酸等があり、これらはそれぞれ1種または2種以上の混合物として使用することができる。
本発明に用いることのできる芳香族末端ポリエステル系化合物の炭素数2~12のアルキレングリコール成分としては、エチレングリコール、1,2-プロピレングリコール、1,3-プロピレングリコール、1,2-ブタンジオール、1,3-ブタンジオール、1,2-プロパンジオール、2-メチル1,3-プロパンジオール、1,4-ブタンジオール、1,5-ペンタンジオール、2,2-ジメチル-1,3-プロパンジオール(ネオペンチルグリコール)、2,2-ジエチル-1,3-プロパンジオール(3,3-ジメチロールペンタン)、2-n-ブチル-2-エチル-1,3プロパンジオール(3,3-ジメチロールヘプタン)、3-メチル-1,5-ペンタンジオール1,6-ヘキサンジオール、2,2,4-トリメチル1,3-ペンタンジオール、2-エチル1,3-ヘキサンジオール、2-メチル1,8-オクタンジオール、1,9-ノナンジオール、1,10-デカンジオール、1,12-オクタデカンジオール等があり、これらのグリコールは、1種または2種以上の混合物として使用される。
特に炭素数2~12のアルキレングリコールがセルロースエステルとの相溶性に優れているため、特に好ましい。
また、上記芳香族末端ポリエステル系化合物の炭素数4~12のオキシアルキレングリコール成分としては、例えば、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、ジプロピレングリコール、トリプロピレングリコール等があり、これらのグリコールは、1種または2種以上の混合物として使用できる。
芳香族末端ポリエステル系化合物の炭素数4~12のアルキレンジカルボン酸成分としては、例えば、コハク酸、マレイン酸、フマール酸、グルタール酸、アジピン酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸等があり、これらは、それぞれ1種または2種以上の混合物として使用される。
炭素数6~12のアリーレンジカルボン酸成分としては、フタル酸、テレフタル酸、イソフタル酸、1,5ナフタレンジカルボン酸、1,4ナフタレンジカルボン酸等がある。
本発明で使用される芳香族末端ポリエステル系化合物は、nが1以上100以下であることが好ましく、数平均分子量が、好ましくは300~1500、より好ましくは400~1000の範囲が好適である。
また、その酸価は、0.5mgKOH/g以下、水酸基価は25mgKOH/g以下、より好ましくは酸価0.3mgKOH/g以下、水酸基価は15mgKOH/g以下のものである。
本発明の一般式(I)に示す芳香族末端ポリエステル系化合物は、セルロースエステルに対して、0.5~30質量%含有させることが好ましい。
以下に、本発明に用いることのできる芳香族末端ポリエステル系化合物の具体的化合物を示すが、本発明はこれに限定されない。




〈ピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下有しその構造のOH基のすべてもしくは一部をエステル化したエステル化合物〉
本発明のセルロースエステルフィルムは、ピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下有しその構造のOH基のすべてもしくは一部をエステル化したエステル化合物を含むことを特徴とする。
本発明のセルロースエステルフィルムは、ピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下有しその構造のOH基のすべてもしくは一部をエステル化したエステル化合物を含むことを特徴とする。
エステル化の割合としては、ピラノース構造またはフラノース構造内に存在するOH基の70%以上であることが好ましい。
本発明においては、エステル化合物を総称して、糖エステル化合物とも称す。
本発明のエステル化合物の例としては、例えば以下のようなものを挙げることができるが、本発明はこれらに限定されるものではない。
グルコース、ガラクトース、マンノース、フルクトース、キシロース、あるいはアラビノース、ラクトース、スクロース、ニストース、1F-フラクトシルニストース、スタキオース、マルチトール、ラクチトール、ラクチュロース、セロビオース、マルトース、セロトリオース、マルトトリオース、ラフィノースあるいはケストース挙げられる。
この他、ゲンチオビオース、ゲンチオトリオース、ゲンチオテトラオース、キシロトリオース、ガラクトシルスクロースなども挙げられる。
これらの化合物の中で、特にピラノース構造とフラノース構造を両方有する化合物が好ましい。
例としてはスクロース、ケストース、ニストース、1F-フラクトシルニストース、スタキオースなどが好ましく、更に好ましくは、スクロースである。
本発明ピラノース構造またはフラノース構造中のOH基のすべてもしくは一部をエステル化するのに用いられるモノカルボン酸としては、特に制限はなく、公知の脂肪族モノカルボン酸、脂環族モノカルボン酸、芳香族モノカルボン酸等を用いることができる。用いられるカルボン酸は1種類でもよいし、2種以上の混合であってもよい。
好ましい脂肪族モノカルボン酸としては、酢酸、プロピオン酸、酪酸、イソ酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、2-エチル-ヘキサンカルボン酸、ウンデシル酸、ラウリン酸、トリデシル酸、ミリスチン酸、ペンタデシル酸、パルミチン酸、ヘプタデシル酸、ステアリン酸、ノナデカン酸、アラキン酸、ベヘン酸、リグノセリン酸、セロチン酸、ヘプタコサン酸、モンタン酸、メリシン酸、ラクセル酸等の飽和脂肪酸、ウンデシレン酸、オレイン酸、ソルビン酸、リノール酸、リノレン酸、アラキドン酸、オクテン酸等の不飽和脂肪酸等を挙げることができる。
好ましい脂環族モノカルボン酸の例としては、酢酸、シクロペンタンカルボン酸、シクロヘキサンカルボン酸、シクロオクタンカルボン酸、またはそれらの誘導体を挙げることができる。
好ましい芳香族モノカルボン酸の例としては、安息香酸、トルイル酸等の安息香酸のベンゼン環にアルキル基、アルコキシ基を導入した芳香族モノカルボン酸、ケイ皮酸、ベンジル酸、ビフェニルカルボン酸、ナフタリンカルボン酸、テトラリンカルボン酸等のベンゼン環を2個以上有する芳香族モノカルボン酸、またはそれらの誘導体を挙げることができ、より、具体的には、キシリル酸、ヘメリト酸、メシチレン酸、プレーニチル酸、γ-イソジュリル酸、ジュリル酸、メシト酸、α-イソジュリル酸、クミン酸、α-トルイル酸、ヒドロアトロパ酸、アトロパ酸、ヒドロケイ皮酸、サリチル酸、o-アニス酸、m-アニス酸、p-アニス酸、クレオソート酸、o-ホモサリチル酸、m-ホモサリチル酸、p-ホモサリチル酸、o-ピロカテク酸、β-レソルシル酸、バニリン酸、イソバニリン酸、ベラトルム酸、o-ベラトルム酸、没食子酸、アサロン酸、マンデル酸、ホモアニス酸、ホモバニリン酸、ホモベラトルム酸、o-ホモベラトルム酸、フタロン酸、p-クマル酸を挙げることができるが、特に安息香酸が好ましい。
オリゴ糖のエステル化合物を、本発明に係るピラノース構造またはフラノース構造の少なくとも1種を1~12個を有する化合物として適用できる。
オリゴ糖は、澱粉、ショ糖等にアミラーゼ等の酵素を作用させて製造されるもので、本発明に適用できるオリゴ糖としては、例えば、マルトオリゴ糖、イソマルトオリゴ糖、フラクトオリゴ糖、ガラクトオリゴ糖、キシロオリゴ糖が挙げられる。
また、前記エステル化合物は、下記一般式(A)で表されるピラノース構造またはフラノース構造の少なくとも1種を1個以上12個以下縮合した化合物である。ただし、R11~R15、R21~R25は、炭素数2~22のアシル基または水素原子を、m、nはそれぞれ0~12の整数、m+nは1~12の整数を表す。

R11~R15、R21~R25は、ベンゾイル基、水素原子であることが好ましい。ベンゾイル基は更に置換基R26(pは0~5)を有していてもよく、例えばアルキル基、アルケニル基、アルコキシル基、フェニル基が挙げられ、更にこれらのアルキル基、アルケニル基、フェニル基は置換基を有していてもよい。オリゴ糖も本発明のエステル化合物と同様な方法で製造することができる。
以下に、本発明に係るエステル化合物の具体例を挙げるが、本発明はこれに限定されるものではない。
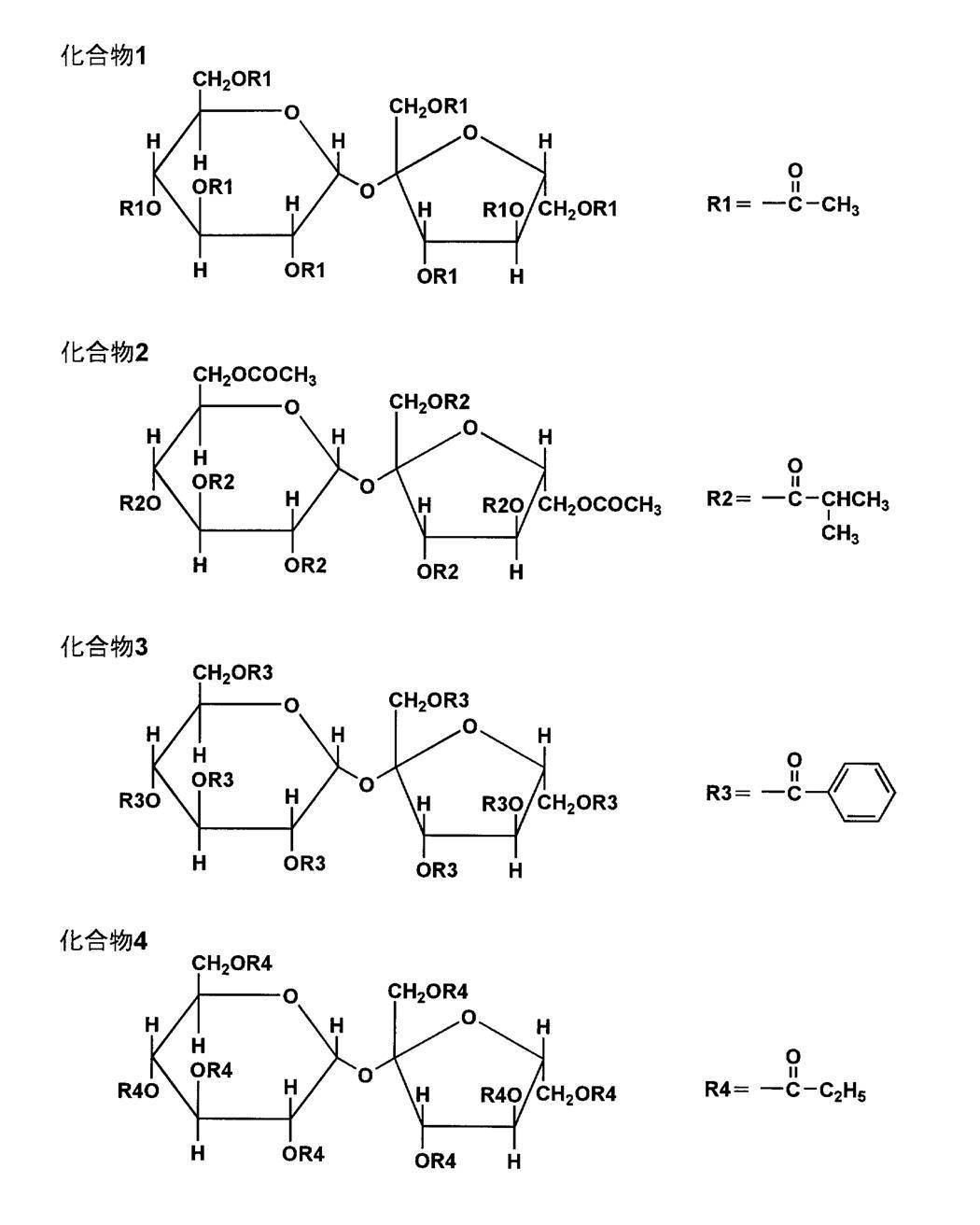







本発明のセルロースエステルフィルムは、位相差値の変動を抑制して、表示品位を安定化する為に、本発明の糖エステル化合物を、セルロースエステルフィルムの0.5~30質量%含むことが好ましく、特には、5~30質量%含むことが好ましい。
本発明の一般式(I)に示す芳香族末端ポリエステル系化合物と糖エステル化合物の含有量は、質量比で99:1~1:99の範囲で選択することができ、両化合物の全体量は、セルロースエステルに対して、1~40質量%であることが好ましい。
〈可塑剤〉
本発明のセルロースエステルフィルムは、本発明の効果を得る上で必要に応じて可塑剤を含有することができる。
本発明のセルロースエステルフィルムは、本発明の効果を得る上で必要に応じて可塑剤を含有することができる。
可塑剤は特に限定されないが、好ましくは、(メタ)アクリル系重合体可塑剤、多価カルボン酸エステル系可塑剤、グリコレート系可塑剤、フタル酸エステル系可塑剤、脂肪酸エステル系可塑剤および多価アルコールエステル系可塑剤、ポリエステル系可塑剤等から選択される。
そのうち、可塑剤を2種以上用いる場合は、少なくとも1種は多価アルコールエステル系可塑剤であることが好ましい。
〈(メタ)アクリル系重合体〉
本発明に用いられる(メタ)アクリル系重合体としては、光学補償フィルムに含有させた場合、機能として延伸方向に対して負の複屈折性を示すことが好ましく、特に構造が限定されるものではないが、エチレン性不飽和モノマーを重合して得られた重量平均分子量が500以上30000以下である重合体であることが好ましい。なお、複屈折性については、下記試験によりその存在を確認した。
本発明に用いられる(メタ)アクリル系重合体としては、光学補償フィルムに含有させた場合、機能として延伸方向に対して負の複屈折性を示すことが好ましく、特に構造が限定されるものではないが、エチレン性不飽和モノマーを重合して得られた重量平均分子量が500以上30000以下である重合体であることが好ましい。なお、複屈折性については、下記試験によりその存在を確認した。
((メタ)アクリル系重合体の複屈折性試験法)
(メタ)アクリル系重合体を溶媒に溶解しキャスト製膜した後、加熱乾燥し、透過率80%以上のフィルムについて複屈折性の評価を行った。
(メタ)アクリル系重合体を溶媒に溶解しキャスト製膜した後、加熱乾燥し、透過率80%以上のフィルムについて複屈折性の評価を行った。
アッベ屈折率計-4T((株)アタゴ製)に多波長光源を用いて屈折率測定を行った。延伸方向の屈折率nyおよび直交する面内方向の屈折率をnxとした。550nmの各々の屈折率について(ny-nx)<0であるフィルムについて、(メタ)アクリル系重合体は延伸方向に対して負の複屈折性であると判断する。
本発明に用いられる重量平均分子量が500以上30000以下である(メタ)アクリル系重合体は、芳香環を側鎖に有する(メタ)アクリル系重合体またはシクロヘキシル基を側鎖に有する(メタ)アクリル系重合体であってもよい。
該重合体の重量平均分子量が500以上30000以下のもので該重合体の組成を制御することにより、例えば光学補償フィルムが本発明において特に好ましいセルロースエステルフィルムである場合、該セルロースエステルと該重合体との相溶性を良好にすることができる。
芳香環を側鎖に有する(メタ)アクリル系重合体またはシクロヘキシル基を側鎖に有する(メタ)アクリル系重合体について、好ましくは重量平均分子量が500以上10000以下のものであれば、上記に加え、製膜後のセルロースエステルフィルムの透明性が優れ、透湿度も極めて低く、偏光板用保護フィルムとして優れた性能を示す。
該重合体は、重量平均分子量が500以上30000以下であるから、オリゴマーから低分子量重合体の間にあると考えられるものである。このような重合体を合成するには、通常の重合では分子量のコントロールが難しく、分子量を余り大きくしない方法でできるだけ分子量を揃えることのできる方法を用いることが望ましい。
特に、本発明の光学補償フィルムに用いられる(メタ)アクリル系重合体としては、分子内に芳香環と水酸基を有しないエチレン性不飽和モノマーXaと、分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーXbとXa、Xbを除く共重合可能なエチレン性不飽和モノマーとを共重合して得られた重量平均分子量2000以上30000以下の重合体X、または芳香環を有さないエチレン性不飽和モノマーYaと、Yaと共重合可能なエチレン性不飽和モノマーとを重合して得られた重量平均分子量500以上3000以下の重合体Yであることが好ましい。
[重合体X、重合体Y]
本発明に係る光学補償フィルムのRoおよびRtを調整する方法としては、分子内に芳香環と水酸基を有しないエチレン性不飽和モノマーXaと、分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーXbとXa、Xbを除く共重合可能なエチレン性不飽和モノマーとを共重合して得られた重量平均分子量2000以上30000以下の高分子量の重合体X、そして、より好ましくは、芳香環を有さないエチレン性不飽和モノマーYaと、Yaと共重合可能なエチレン性不飽和モノマーとを重合して得られた重量平均分子量500以上3000以下の低分子量の重合体Yを含有することが好ましい。
本発明に係る光学補償フィルムのRoおよびRtを調整する方法としては、分子内に芳香環と水酸基を有しないエチレン性不飽和モノマーXaと、分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーXbとXa、Xbを除く共重合可能なエチレン性不飽和モノマーとを共重合して得られた重量平均分子量2000以上30000以下の高分子量の重合体X、そして、より好ましくは、芳香環を有さないエチレン性不飽和モノマーYaと、Yaと共重合可能なエチレン性不飽和モノマーとを重合して得られた重量平均分子量500以上3000以下の低分子量の重合体Yを含有することが好ましい。
本発明に用いられる重合体Xは、分子内に芳香環と水酸基を有しないエチレン性不飽和モノマーXaと分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーXbとXa、Xbを除く共重合可能なエチレン性不飽和モノマーとを共重合して得られた重量平均分子量2000以上、30000以下の重合体である。
好ましくは、Xaは分子内に芳香環と水酸基を有しないアクリルまたはメタクリルモノマー、Xbは分子内に芳香環を有せず水酸基を有するアクリルまたはメタクリルモノマーである。
本発明に用いられる重合体Xは、下記一般式(X)で表される。
一般式(X)
-[Xa]m-[Xb]n-[Xc]p-
上記一般式(X)において、Xaは分子内に芳香環と水酸基とを有しないエチレン性不飽和モノマーを表し、Xbは分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーを表し、XcはXa、Xbを除く共重合可能なエチレン性不飽和モノマーを表す。m、nおよびpは、各々モル組成比を表す。ただし、m≠0、m+n+p=100である。
-[Xa]m-[Xb]n-[Xc]p-
上記一般式(X)において、Xaは分子内に芳香環と水酸基とを有しないエチレン性不飽和モノマーを表し、Xbは分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーを表し、XcはXa、Xbを除く共重合可能なエチレン性不飽和モノマーを表す。m、nおよびpは、各々モル組成比を表す。ただし、m≠0、m+n+p=100である。
更に、重合体Xとして好ましくは、下記一般式(X-1)で表される重合体である。
一般式(X-1)
-[CH2-C(-R1)(-CO2R2)]m-[CH2-C(-R3)(-CO2R4-OH)-]n-[Xc]p-
上記一般式(X-1)において、R1、R3は、それぞれ水素原子またはメチル基を表す。R2は炭素数1~12のアルキル基またはシクロアルキル基を表す。
-[CH2-C(-R1)(-CO2R2)]m-[CH2-C(-R3)(-CO2R4-OH)-]n-[Xc]p-
上記一般式(X-1)において、R1、R3は、それぞれ水素原子またはメチル基を表す。R2は炭素数1~12のアルキル基またはシクロアルキル基を表す。
R4は-CH2-、-C2H4-または-C3H6-を表す。Xcは、[CH2-C(-R1)(-CO2R2)]または[CH2-C(-R3)(-CO2R4-OH)-]に重合可能なモノマー単位を表す。m、nおよびpは、モル組成比を表す。ただしm≠0、m+n+p=100である。
本発明に係る重合体Xを構成するモノマー単位としてのモノマーを下記に挙げるが、これに限定されない。
Xにおいて、水酸基とは、水酸基のみならずエチレンオキシド連鎖を有する基をいう。
分子内に芳香環と水酸基を有しないエチレン性不飽和モノマーXaは、例えば、アクリル酸メチル、アクリル酸エチル、アクリル酸プロピル(i-、n-)、アクリル酸ブチル(n-、i-、s-、t-)、アクリル酸ペンチル(n-、i-、s-)、アクリル酸ヘキシル(n-、i-)、アクリル酸ヘプチル(n-、i-)、アクリル酸オクチル(n-、i-)、アクリル酸ノニル(n-、i-)、アクリル酸ミリスチル(n-、i-)、アクリル酸(2-エチルヘキシル)、アクリル酸(ε-カプロラクトン)、等、または上記アクリル酸エステルをメタクリル酸エステルに変えたものを挙げることができる。
中でも、アクリル酸メチル、アクリル酸エチル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル(i-、n-)であることが好ましい。
分子内に芳香環を有せず、水酸基を有するエチレン性不飽和モノマーXbは、水酸基を有するモノマー単位として、アクリル酸またはメタクリル酸エステルが好ましく、例えば、アクリル酸(2-ヒドロキシエチル)、アクリル酸(2-ヒドロキシプロピル)、アクリル酸(3-ヒドロキシプロピル)、アクリル酸(4-ヒドロキシブチル)、アクリル酸(2-ヒドロキシブチル)、またはこれらアクリル酸をメタクリル酸に置き換えたものを挙げることができ、好ましくは、アクリル酸(2-ヒドロキシエチル)およびメタクリル酸(2-ヒドロキシエチル)、アクリル酸(2-ヒドロキシプロピル)、アクリル酸(3-ヒドロキシプロピル)である。
Xcとしては、Xa、Xb以外のモノマーで、かつ共重合可能なエチレン性不飽和モノマーであれば、特に制限はないが、芳香環を有していないものが好ましい。
XaおよびXbのモル組成比m:nは99:1~65:35の範囲が好ましく、更に好ましくは95:5~75:25の範囲である。Xcのpは0~10である。Xcは複数のモノマー単位であってもよい。
Xaのモル組成比が多いと、セルロースエステルとの相溶性が良化するがフィルム厚み方向のレターデーション値Rtが大きくなる。Xbのモル組成比が多いと上記相溶性が悪くなるが、Rtを低減させる効果が高い。
また、Xbのモル組成比が上記範囲を超えると製膜時にヘイズが出る傾向があり、これらの最適化を図りXa、Xbのモル組成比を決めることが好ましい。
高分子量の重合体Xの分子量は、重量平均分子量が5000以上30000以下であることがより好ましく、更に好ましくは8000以上25000以下である。
重量平均分子量を5000以上とすることにより、光学補償フィルムの高温高湿下における寸法変化が少ない、偏光板保護フィルムとしてカールが少ない等の利点が得られ好ましい。
重量平均分子量が30000以下とした場合は、セルロースエステルとの相溶性がより向上し、高温高湿下においてのブリードアウト、更に製膜直後でのヘイズの発生が抑制される。
本発明に係る重合体Xの重量平均分子量は、公知の分子量調節方法で調整することができる。そのような分子量調節方法としては、例えば、四塩化炭素、ラウリルメルカプタン、チオグリコール酸オクチル等の連鎖移動剤を添加する方法等が挙げられる。
また、重合温度は、通常、室温から130℃、好ましくは50℃から100℃で行われるが、この温度または重合反応時間を調整することで可能である。
重量平均分子量の測定方法は、下記の方法により求めることができる。
(平均分子量測定方法)
重量平均分子量Mw、数平均分子量Mnは、ゲルパーミエーションクロマトグラフィー(GPC)を用いて測定した。
重量平均分子量Mw、数平均分子量Mnは、ゲルパーミエーションクロマトグラフィー(GPC)を用いて測定した。
測定条件は以下の通りである。
溶媒: メチレンクロライド
カラム: Shodex K806、K805、K803G(昭和電工(株)製を3本接続して使用した)
カラム温度:25℃
試料濃度: 0.1質量%
検出器: RI Model 504(GLサイエンス社製)
ポンプ: L6000(日立製作所(株)製)
流量: 1.0ml/min
校正曲線: 標準ポリスチレンSTK standard ポリスチレン(東ソー(株)製)Mw=1000000~500の13サンプルによる校正曲線を使用した。13サンプルは、ほぼ等間隔に用いる。
カラム: Shodex K806、K805、K803G(昭和電工(株)製を3本接続して使用した)
カラム温度:25℃
試料濃度: 0.1質量%
検出器: RI Model 504(GLサイエンス社製)
ポンプ: L6000(日立製作所(株)製)
流量: 1.0ml/min
校正曲線: 標準ポリスチレンSTK standard ポリスチレン(東ソー(株)製)Mw=1000000~500の13サンプルによる校正曲線を使用した。13サンプルは、ほぼ等間隔に用いる。
本発明に用いられる低分子量の重合体Yは、芳香環を有さないエチレン性不飽和モノマーYaを重合して得られた重量平均分子量500以上3000以下の重合体である。重量平均分子量500以上であれば重合体の残存モノマーが減少し好ましい。
また、3000以下とすることは、レターデーション値Rt低下性能を維持するために好ましい。Yaは、好ましくは芳香環を有さないアクリルまたはメタクリルモノマーである。
本発明に用いられる重合体Yは、下記一般式(Y)で表される。
一般式(Y)
-[Ya]k-[Yb]q-
上記一般式(Y)において、Yaは芳香環を有しないエチレン性不飽和モノマーを表し、YbはYaと共重合可能なエチレン性不飽和モノマーを表す。kおよびqは、各々モル組成比を表す。ただし、k≠0、k+q=100である。
-[Ya]k-[Yb]q-
上記一般式(Y)において、Yaは芳香環を有しないエチレン性不飽和モノマーを表し、YbはYaと共重合可能なエチレン性不飽和モノマーを表す。kおよびqは、各々モル組成比を表す。ただし、k≠0、k+q=100である。
本発明に係る重合体Yにおいて、更に好ましくは下記一般式(Y-1)で表される重合体である。
一般式(Y-1)
-[CH2-C(-R5)(-CO2R6)]k-[Yb]q-
上記一般式(Y-1)において、R5は、それぞれ水素原子またはメチル基を表す。R6は炭素数1~12のアルキル基またはシクロアルキル基を表す。Ybは、[CH2-C(-R5)(-CO2R6)]と共重合可能なモノマー単位を表す。kおよびqは、それぞれモル組成比を表す。ただしk≠0、k+q=100である。
-[CH2-C(-R5)(-CO2R6)]k-[Yb]q-
上記一般式(Y-1)において、R5は、それぞれ水素原子またはメチル基を表す。R6は炭素数1~12のアルキル基またはシクロアルキル基を表す。Ybは、[CH2-C(-R5)(-CO2R6)]と共重合可能なモノマー単位を表す。kおよびqは、それぞれモル組成比を表す。ただしk≠0、k+q=100である。
Ybは、Yaである[CH2-C(-R5)(-CO2R6)]と共重合可能なエチレン性不飽和モノマーであれば特に制限はない。Ybは複数であってもよい。k+q=100、qは好ましくは1~30である。
芳香環を有さないエチレン性不飽和モノマーを重合して得られる重合体Yを構成するエチレン性不飽和モノマーYaは、アクリル酸エステルとして、例えば、アクリル酸メチル、アクリル酸エチル、アクリル酸プロピル(i-、n-)、アクリル酸ブチル(n-、i-、s-、t-)、アクリル酸ペンチル(n-、i-、s-)、アクリル酸ヘキシル(n-、i-)、アクリル酸ヘプチル(n-、i-)、アクリル酸オクチル(n-、i-)、アクリル酸ノニル(n-、i-)、アクリル酸ミリスチル(n-、i-)、アクリル酸シクロヘキシル、アクリル酸(2-エチルヘキシル)、アクリル酸(ε-カプロラクトン)、アクリル酸(2-ヒドロキシエチル)、アクリル酸(2-ヒドロキシプロピル)、アクリル酸(3-ヒドロキシプロピル)、アクリル酸(4-ヒドロキシブチル)、アクリル酸(2-ヒドロキシブチル)、メタクリル酸エステルとして、上記アクリル酸エステルをメタクリル酸エステルに変えたもの;不飽和酸として、例えば、アクリル酸、メタクリル酸、無水マレイン酸、クロトン酸、イタコン酸等を挙げることができる。
Ybは、Yaと共重合可能なエチレン性不飽和モノマーであれば特に制限はないが、ビニルエステルとして、例えば、酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、吉草酸ビニル、ピバリン酸ビニル、カプロン酸ビニル、カプリン酸ビニル、ラウリン酸ビニル、ミリスチン酸ビニル、パルミチン酸ビニル、ステアリン酸ビニル、シクロヘキサンカルボン酸ビニル、オクチル酸ビニル、メタクリル酸ビニル、クロトン酸ビニル、ソルビン酸ビニル、桂皮酸ビニル等が好ましい。Ybは複数であってもよい。
重合体X、Yを合成するには、通常の重合では分子量のコントロールが難しく、分子量を余り大きくしない方法で、かつできるだけ分子量を揃えることのできる方法を用いることが望ましい。
かかる重合方法としては、クメンペルオキシドやt-ブチルヒドロペルオキシドのような過酸化物重合開始剤を使用する方法、重合開始剤を通常の重合より多量に使用する方法、重合開始剤の他にメルカプト化合物や四塩化炭素等の連鎖移動剤を使用する方法、重合開始剤の他にベンゾキノンやジニトロベンゼンのような重合停止剤を使用する方法、更に特開2000-128911号または同2000-344823号公報にあるような一つのチオール基と2級の水酸基とを有する化合物、或いは、該化合物と有機金属化合物を併用した重合触媒を用いて塊状重合する方法等を挙げることができ、何れも本発明において好ましく用いられる。
特に、重合体Yは、分子中にチオール基と2級の水酸基とを有する化合物を連鎖移動剤として使用する重合方法が好ましい。この場合、重合体Yの末端には、重合触媒および連鎖移動剤に起因する水酸基、チオエーテルを有することとなる。この末端残基により、Yとセルロースエステルとの相溶性を調整することができる。
重合体XおよびYの水酸基価は、30~150[mgKOH/g]であることが好ましい。
(水酸基価の測定方法)
水酸基価の測定は、JIS K 0070(1992)に準ずる。この水酸基価は、試料1gをアセチル化させたとき、水酸基と結合した酢酸を中和するのに必要とする水酸化カリウムのmg数と定義される。
水酸基価の測定は、JIS K 0070(1992)に準ずる。この水酸基価は、試料1gをアセチル化させたとき、水酸基と結合した酢酸を中和するのに必要とする水酸化カリウムのmg数と定義される。
具体的には試料Xg(約1g)をフラスコに精秤し、これにアセチル化試薬(無水酢酸20mlにピリジンを加えて400mlにしたもの)20mlを正確に加える。フラスコの口に空気冷却管を装着し、95~100℃のグリセリン浴にて加熱する。1時間30分後、冷却し、空気冷却管から精製水1mlを加え、無水酢酸を酢酸に分解する。
次に電位差滴定装置を用いて0.5mol/L水酸化カリウムエタノール溶液で滴定を行い、得られた滴定曲線の変曲点を終点とする。
更に空試験として、試料を入れないで滴定し、滴定曲線の変曲点を求める。水酸基価は、次の式によって算出する。
水酸基価={(B-C)×f×28.05/X}+D
式中、Bは空試験に用いた0.5mol/Lの水酸化カリウムエタノール溶液の量(ml)、Cは滴定に用いた0.5mol/Lの水酸化カリウムエタノール溶液の量(ml)、fは0.5mol/L水酸化カリウムエタノール溶液のファクター、Dは酸価、また、28.05は水酸化カリウムの1mol量56.11の1/2を表す。
式中、Bは空試験に用いた0.5mol/Lの水酸化カリウムエタノール溶液の量(ml)、Cは滴定に用いた0.5mol/Lの水酸化カリウムエタノール溶液の量(ml)、fは0.5mol/L水酸化カリウムエタノール溶液のファクター、Dは酸価、また、28.05は水酸化カリウムの1mol量56.11の1/2を表す。
上述の重合体X、重合体Yは何れもセルロースエステルとの相溶性に優れ、蒸発や揮発もなく生産性に優れ、偏光板用保護フィルムとしての保留性がよく、透湿度が小さく、寸法安定性に優れている。
重合体Xと重合体Yの光学補償フィルム中での含有量は、下記式(i)、式(ii)を満足する範囲であることが好ましい。重合体Xの含有量をXg(質量%=(重合体Xの質量/セルロースエステルの質量)×100)、重合体Yの含有量をYg(質量%)とすると、
式(i) 5≦Xg+Yg≦35(質量%)
式(ii) 0.05≦Yg/(Xg+Yg)≦0.4
式(i)の(Xg+Yg)の好ましい範囲は、10~35質量%である。重合体Xと重合体Yは、セルロースエステル全質量に対し、総量として5質量%以上であれば、レターデーション値Rtの調整に十分な作用をする。また、総量として35質量%以下であれば、偏光子PVAとの接着性が良好である。
式(i) 5≦Xg+Yg≦35(質量%)
式(ii) 0.05≦Yg/(Xg+Yg)≦0.4
式(i)の(Xg+Yg)の好ましい範囲は、10~35質量%である。重合体Xと重合体Yは、セルロースエステル全質量に対し、総量として5質量%以上であれば、レターデーション値Rtの調整に十分な作用をする。また、総量として35質量%以下であれば、偏光子PVAとの接着性が良好である。
重合体Xと重合体Yは、後述するドープ液を構成する素材として直接添加、溶解するか、もしくはセルロースエステルを溶解する有機溶媒に予め溶解した後ドープ液に添加することができる。
多価アルコールエステル系可塑剤は2価以上の脂肪族多価アルコールとモノカルボン酸のエステルよりなる可塑剤であり、分子内に芳香環またはシクロアルキル環を有することが好ましい。好ましくは2~20価の脂肪族多価アルコールエステルである。
本発明に好ましく用いられる多価アルコールは次の一般式(a)で表される。
一般式(a) R1-(OH)n
但し、R1はn価の有機基、nは2以上の正の整数、OH基はアルコール性、および/またはフェノール性水酸基を表す。
但し、R1はn価の有機基、nは2以上の正の整数、OH基はアルコール性、および/またはフェノール性水酸基を表す。
好ましい多価アルコールの例としては、例えば以下のようなものを挙げることができるが、本発明はこれらに限定されるものではない。
アドニトール、アラビトール、エチレングリコール、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、1,2-プロパンジオール、1,3-プロパンジオール、ジプロピレングリコール、トリプロピレングリコール、1,2-ブタンジオール、1,3-ブタンジオール、1,4-ブタンジオール、ジブチレングリコール、1,2,4-ブタントリオール、1,5-ペンタンジオール、1,6-ヘキサンジオール、ヘキサントリオール、ガラクチトール、マンニトール、3-メチルペンタン-1,3,5-トリオール、ピナコール、ソルビトール、トリメチロールプロパン、トリメチロールエタン、キシリトール等を挙げることができる。
特に、トリエチレングリコール、テトラエチレングリコール、ジプロピレングリコール、トリプロピレングリコール、ソルビトール、トリメチロールプロパン、キシリトールが好ましい。
多価アルコールエステルに用いられるモノカルボン酸としては、特に制限はなく、公知の脂肪族モノカルボン酸、脂環族モノカルボン酸、芳香族モノカルボン酸等を用いることができる。脂環族モノカルボン酸、芳香族モノカルボン酸を用いると透湿性、保留性を向上させる点で好ましい。
好ましいモノカルボン酸の例としては以下のようなものを挙げることができるが、本発明はこれに限定されるものではない。
脂肪族モノカルボン酸としては、炭素数1~32の直鎖または側鎖を有する脂肪酸を好ましく用いることができる。炭素数は1~20であることが更に好ましく、1~10であることが特に好ましい。酢酸を含有させるとセルロースエステルとの相溶性が増すため好ましく、酢酸と他のモノカルボン酸を混合して用いることも好ましい。
好ましい脂肪族モノカルボン酸としては、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、2-エチル-ヘキサン酸、ウンデシル酸、ラウリン酸、トリデシル酸、ミリスチン酸、ペンタデシル酸、パルミチン酸、ヘプタデシル酸、ステアリン酸、ノナデカン酸、アラキン酸、ベヘン酸、リグノセリン酸、セロチン酸、ヘプタコサン酸、モンタン酸、メリシン酸、ラクセル酸等の飽和脂肪酸、ウンデシレン酸、オレイン酸、ソルビン酸、リノール酸、リノレン酸、アラキドン酸等の不飽和脂肪酸等を挙げることができる。
好ましい脂環族モノカルボン酸の例としては、シクロペンタンカルボン酸、シクロヘキサンカルボン酸、シクロオクタンカルボン酸、またはそれらの誘導体を挙げることができる。
好ましい芳香族モノカルボン酸の例としては、安息香酸、トルイル酸等の安息香酸のベンゼン環にアルキル基、メトキシ基あるいはエトキシ基などのアルコキシ基を1~3個を導入したもの、ビフェニルカルボン酸、ナフタリンカルボン酸、テトラリンカルボン酸等のベンゼン環を2個以上有する芳香族モノカルボン酸、またはそれらの誘導体を挙げることができる。特に安息香酸が好ましい。
多価アルコールエステルの分子量は特に制限はないが、300~1500であることが好ましく、350~750であることが更に好ましい。分子量が大きい方が揮発し難くなるため好ましく、透湿性、セルロースエステルとの相溶性の点では小さい方が好ましい。
多価アルコールエステルに用いられるカルボン酸は1種類でもよいし、2種以上の混合であってもよい。また、多価アルコール中のOH基は、全てエステル化してもよいし、一部をOH基のままで残してもよい。
以下に、多価アルコールエステルの具体的化合物を例示する。




グリコレート系可塑剤は特に限定されないが、アルキルフタリルアルキルグリコレート類が好ましく用いることができる。
アルキルフタリルアルキルグリコレート類としては、例えばメチルフタリルメチルグリコレート、エチルフタリルエチルグリコレート、プロピルフタリルプロピルグリコレート、ブチルフタリルブチルグリコレート、オクチルフタリルオクチルグリコレート、メチルフタリルエチルグリコレート、エチルフタリルメチルグリコレート、エチルフタリルプロピルグリコレート、メチルフタリルブチルグリコレート、エチルフタリルブチルグリコレート、ブチルフタリルメチルグリコレート、ブチルフタリルエチルグリコレート、プロピルフタリルブチルグリコレート、ブチルフタリルプロピルグリコレート、メチルフタリルオクチルグリコレート、エチルフタリルオクチルグリコレート、オクチルフタリルメチルグリコレート、オクチルフタリルエチルグリコレート等が挙げられる。
フタル酸エステル系可塑剤としては、ジエチルフタレート、ジメトキシエチルフタレート、ジメチルフタレート、ジオクチルフタレート、ジブチルフタレート、ジ-2-エチルヘキシルフタレート、ジオクチルフタレート、ジシクロヘキシルフタレート、ジシクロヘキシルテレフタレート等が挙げられる。
クエン酸エステル系可塑剤としては、クエン酸アセチルトリメチル、クエン酸アセチルトリエチル、クエン酸アセチルトリブチル等が挙げられる。
脂肪酸エステル系可塑剤として、オレイン酸ブチル、リシノール酸メチルアセチル、セバシン酸ジブチル等が挙げられる。
リン酸エステル系可塑剤としては、トリフェニルホスフェート、トリクレジルホスフェート、クレジルジフェニルホスフェート、オクチルジフェニルホスフェート、ジフェニルビフェニルホスフェート、トリオクチルホスフェート、トリブチルホスフェート等が挙げられる。
多価カルボン酸エステル化合物としては、2価以上、好ましくは2価~20価の多価カルボン酸とアルコールのエステルよりなる。また、脂肪族多価カルボン酸は2~20価であることが好ましく、芳香族多価カルボン酸、脂環式多価カルボン酸の場合は3価~20価であることが好ましい。
多価カルボン酸は次の一般式(b)で表される。
一般式(b) R2(COOH)m(OH)n
(但し、R2は(m+n)価の有機基、mは2以上の正の整数、nは0以上の整数、COOH基はカルボキシル基、OH基はアルコール性またはフェノール性水酸基を表す)
好ましい多価カルボン酸の例としては、例えば以下のようなものを挙げることができるが、本発明はこれらに限定されるものではない。
(但し、R2は(m+n)価の有機基、mは2以上の正の整数、nは0以上の整数、COOH基はカルボキシル基、OH基はアルコール性またはフェノール性水酸基を表す)
好ましい多価カルボン酸の例としては、例えば以下のようなものを挙げることができるが、本発明はこれらに限定されるものではない。
トリメリット酸、トリメシン酸、ピロメリット酸のような3価以上の芳香族多価カルボン酸またはその誘導体、コハク酸、アジピン酸、アゼライン酸、セバシン酸、シュウ酸、フマル酸、マレイン酸、テトラヒドロフタル酸のような脂肪族多価カルボン酸、酒石酸、タルトロン酸、リンゴ酸、クエン酸のようなオキシ多価カルボン酸などを好ましく用いることができる。特にオキシ多価カルボン酸を用いることが、保留性向上などの点で好ましい。
本発明に用いることのできる多価カルボン酸エステル化合物に用いられるアルコールとしては特に制限はなく公知のアルコール、フェノール類を用いることができる。
例えば炭素数1~32の直鎖または側鎖を持った脂肪族飽和アルコールまたは脂肪族不飽和アルコールを好ましく用いることができる。炭素数1~20であることが更に好ましく、炭素数1~10であることが特に好ましい。
また、シクロペンタノール、シクロヘキサノールなどの脂環式アルコールまたはその誘導体、ベンジルアルコール、シンナミルアルコールなどの芳香族アルコールまたはその誘導体なども好ましく用いることができる。
多価カルボン酸としてオキシ多価カルボン酸を用いる場合は、オキシ多価カルボン酸のアルコール性またはフェノール性の水酸基を、モノカルボン酸を用いてエステル化しても良い。好ましいモノカルボン酸の例としては以下のようなものを挙げることができるが、本発明はこれに限定されるものではない。
脂肪族モノカルボン酸としては炭素数1~32の直鎖または側鎖を持った脂肪酸を好ましく用いることができる。炭素数1~20であることが更に好ましく、炭素数1~10であることが特に好ましい。
好ましい脂肪族モノカルボン酸としては酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、2-エチル-ヘキサンカルボン酸、ウンデシル酸、ラウリン酸、トリデシル酸、ミリスチン酸、ペンタデシル酸、パルミチン酸、ヘプタデシル酸、ステアリン酸、ノナデカン酸、アラキン酸、ベヘン酸、リグノセリン酸、セロチン酸、ヘプタコサン酸、モンタン酸、メリシン酸、ラクセル酸などの飽和脂肪酸、ウンデシレン酸、オレイン酸、ソルビン酸、リノール酸、リノレン酸、アラキドン酸などの不飽和脂肪酸などを挙げることができる。
好ましい脂環族モノカルボン酸の例としては、シクロペンタンカルボン酸、シクロヘキサンカルボン酸、シクロオクタンカルボン酸、またはそれらの誘導体を挙げることができる。
好ましい芳香族モノカルボン酸の例としては、安息香酸、トルイル酸などの安息香酸のベンゼン環にアルキル基を導入したもの、ビフェニルカルボン酸、ナフタリンカルボン酸、テトラリンカルボン酸などのベンゼン環を2個以上持つ芳香族モノカルボン酸、またはそれらの誘導体を挙げることができる。特に酢酸、プロピオン酸、安息香酸であることが好ましい。
多価カルボン酸エステル化合物の分子量は特に制限はないが、分子量300~1000の範囲であることが好ましく、350~750の範囲であることが更に好ましい。保留性向上の点では大きい方が好ましく、透湿性、セルロースエステルとの相溶性の点では小さい方が好ましい。
本発明に用いることのできる多価カルボン酸エステルに用いられるアルコール類は一種類でも良いし、二種以上の混合であっても良い。
本発明に用いることのできる多価カルボン酸エステル化合物の酸価は1mgKOH/g以下であることが好ましく、0.2mgKOH/g以下であることが更に好ましい。酸価を上記範囲にすることによって、レターデーションの環境変動も抑制されるため好ましい。
なお、酸価とは、試料1g中に含まれる酸(試料中に存在するカルボキシル基)を中和するために必要な水酸化カリウムのミリグラム数をいう。酸価はJIS K0070に準拠して測定したものである。
特に好ましい多価カルボン酸エステル化合物の例を以下に示すが、本発明はこれに限定されるものではない。
例えば、トリエチルシトレート、トリブチルシトレート、アセチルトリエチルシトレート(ATEC)、アセチルトリブチルシトレート(ATBC)、ベンゾイルトリブチルシトレート、アセチルトリフェニルシトレート、アセチルトリベンジルシトレート、酒石酸ジブチル、酒石酸ジアセチルジブチル、トリメリット酸トリブチル、ピロメリット酸テトラブチル等が挙げられる。
本発明の位相差フィルムは、Roの範囲が10~70nm、Rtの範囲が70~400nmが好ましい。
なお、Ro=(nx-ny)×d
Rt=((nx+ny)/2-nz)×d
(式中、nxはセルロースエステルフィルムの面内の遅相軸方向の屈折率を、nyは面内で遅相軸に直交する方向の屈折率を、nzは厚み方向の屈折率を、dはセルロースエステルフィルムの厚み(nm)をそれぞれ表す。屈折率の測定波長は590nmである。)
上記屈折率は、例えばKOBRA-21ADH(王子計測機器(株))を用いて、23℃、55%RHの環境下で、波長が590nmで求めることができる。
<その他の添加剤>
以下、本発明の光学フィルムに添加することが可能な、他の一般的な添加剤について述べる。
Rt=((nx+ny)/2-nz)×d
(式中、nxはセルロースエステルフィルムの面内の遅相軸方向の屈折率を、nyは面内で遅相軸に直交する方向の屈折率を、nzは厚み方向の屈折率を、dはセルロースエステルフィルムの厚み(nm)をそれぞれ表す。屈折率の測定波長は590nmである。)
上記屈折率は、例えばKOBRA-21ADH(王子計測機器(株))を用いて、23℃、55%RHの環境下で、波長が590nmで求めることができる。
<その他の添加剤>
以下、本発明の光学フィルムに添加することが可能な、他の一般的な添加剤について述べる。
(紫外線吸収剤)
本発明に係るセルロースエステルフィルムBは、紫外線吸収剤を含有することもできる。紫外線吸収剤は400nm以下の紫外線を吸収することで、耐久性を向上させることを目的としており、特に波長370nmでの透過率が10%以下であることが好ましく、より好ましくは5%以下、更に好ましくは2%以下である。
本発明に係るセルロースエステルフィルムBは、紫外線吸収剤を含有することもできる。紫外線吸収剤は400nm以下の紫外線を吸収することで、耐久性を向上させることを目的としており、特に波長370nmでの透過率が10%以下であることが好ましく、より好ましくは5%以下、更に好ましくは2%以下である。
本発明に用いられる紫外線吸収剤は特に限定されないが、例えばオキシベンゾフェノン系化合物、ベンゾトリアゾール系化合物、サリチル酸エステル系化合物、ベンゾフェノン系化合物、シアノアクリレート系化合物、トリアジン系化合物、ニッケル錯塩系化合物、無機粉体等が挙げられる。
例えば、5-クロロ-2-(3,5-ジ-sec-ブチル-2-ヒドロキシルフェニル)-2H-ベンゾトリアゾール、(2-2H-ベンゾトリアゾール-2-イル)-6-(直鎖および側鎖ドデシル)-4-メチルフェノール、2-ヒドロキシ-4-ベンジルオキシベンゾフェノン、2,4-ベンジルオキシベンゾフェノン等があり、また、チヌビン109、チヌビン171、チヌビン234、チヌビン326、チヌビン327、チヌビン328等のチヌビン類があり、これらはいずれもチバ・ジャパン社製の市販品であり好ましく使用できる。
本発明で好ましく用いられる紫外線吸収剤は、ベンゾトリアゾール系紫外線吸収剤、ベンゾフェノン系紫外線吸収剤、トリアジン系紫外線吸収剤であり、特に好ましくはベンゾトリアゾール系紫外線吸収剤、ベンゾフェノン系紫外線吸収剤、である。
この他、1,3,5トリアジン環を有する化合物等の円盤状化合物も紫外線吸収剤として好ましく用いられる。
本発明に係わる偏光板保護フィルムは紫外線吸収剤を2種以上を含有することが好ましい。
また、紫外線吸収剤としては高分子紫外線吸収剤も好ましく用いることができ、特に特開平6-148430号記載のポリマータイプの紫外線吸収剤が好ましく用いられる。
紫外線吸収剤の添加方法は、メタノール、エタノール、ブタノール等のアルコールやメチレンクロライド、酢酸メチル、アセトン、ジオキソラン等の有機溶媒あるいはこれらの混合溶媒に紫外線吸収剤を溶解してからドープに添加するか、または直接ドープ組成中に添加してもよい。
無機粉体のように有機溶剤に溶解しないものは、有機溶剤とセルロースエステル中にディゾルバーやサンドミルを使用し、分散してからドープに添加する。
紫外線吸収剤の使用量は、紫外線吸収剤の種類、使用条件等により一様ではないが、偏光板保護フィルムの乾燥膜厚が30~200μmの場合は、偏光板保護フィルムに対して0.5~10質量%が好ましく、0.6~4質量%が更に好ましい。
(酸化防止剤)
酸化防止剤は劣化防止剤ともいわれる。高湿高温の状態に液晶画像表示装置などがおかれた場合には、セルロースエステルフィルムの劣化が起こる場合がある。
酸化防止剤は劣化防止剤ともいわれる。高湿高温の状態に液晶画像表示装置などがおかれた場合には、セルロースエステルフィルムの劣化が起こる場合がある。
酸化防止剤は、例えば、セルロースエステルフィルム中の残留溶媒量のハロゲンやリン酸系可塑剤のリン酸等によりセルロースエステルフィルムが分解するのを遅らせたり、防いだりする役割を有するので、前記セルロースエステルフィルム中に含有させるのが好ましい。
このような酸化防止剤としては、ヒンダードフェノール系の化合物が好ましく用いられ、例えば、2,6-ジ-t-ブチル-p-クレゾール、ペンタエリスリチル-テトラキス〔3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート〕、トリエチレングリコール-ビス〔3-(3-t-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート〕、1,6-ヘキサンジオール-ビス〔3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート〕、2,4-ビス-(n-オクチルチオ)-6-(4-ヒドロキシ-3,5-ジ-t-ブチルアニリノ)-1,3,5-トリアジン、2,2-チオ-ジエチレンビス〔3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート〕、オクタデシル-3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート、N,N′-ヘキサメチレンビス(3,5-ジ-t-ブチル-4-ヒドロキシ-ヒドロシンナマミド)、1,3,5-トリメチル-2,4,6-トリス(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)ベンゼン、トリス-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-イソシアヌレイト等を挙げることができる。
特に、2,6-ジ-t-ブチル-p-クレゾール、ペンタエリスリチル-テトラキス〔3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート〕、トリエチレングリコール-ビス〔3-(3-t-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート〕が好ましい。また、例えば、N,N′-ビス〔3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオニル〕ヒドラジン等のヒドラジン系の金属不活性剤やトリス(2,4-ジ-t-ブチルフェニル)フォスファイト等のリン系加工安定剤を併用してもよい。
これらの化合物の添加量は、セルロース誘導体に対して質量割合で1ppm~1.0%が好ましく、10~1000ppmが更に好ましい。
(微粒子)
本発明に係るセルロースエステルフィルムは、微粒子を含有することが好ましい。
本発明に係るセルロースエステルフィルムは、微粒子を含有することが好ましい。
本発明に使用される微粒子としては、無機化合物の例として、二酸化珪素、二酸化チタン、酸化アルミニウム、酸化ジルコニウム、炭酸カルシウム、炭酸カルシウム、タルク、クレイ、焼成カオリン、焼成ケイ酸カルシウム、水和ケイ酸カルシウム、ケイ酸アルミニウム、ケイ酸マグネシウムおよびリン酸カルシウムを挙げることができる。また、有機化合物の微粒子も好ましく使用することができる。
有機化合物の例としてはポリテトラフルオロエチレン、セルロースアセテート、ポリスチレン、ポリメチルメタクリレート、ポリプピルメタクリレート、ポリメチルアクリレート、ポリエチレンカーボネート、アクリルスチレン系樹脂、シリコーン系樹脂、ポリカーボネート樹脂、ベンゾグアナミン系樹脂、メラミン系樹脂、ポリオレフィン系粉末、ポリエステル系樹脂、ポリアミド系樹脂、ポリイミド系樹脂、あるいはポリ弗化エチレン系樹脂、澱粉等の有機高分子化合物の粉砕分級物もあげられる。あるいは又懸濁重合法で合成した高分子化合物、スプレードライ法あるいは分散法等により球型にした高分子化合物、または無機化合物を用いることができる。
微粒子は珪素を含むものが濁度が低くなる点で好ましく、特に二酸化珪素が好ましい。
微粒子の一次粒子の平均粒径は5~400nmが好ましく、更に好ましいのは10~300nmである。
これらは主に粒径0.05~0.3μmの二次凝集体として含有されていてもよく、平均粒径100~400nmの粒子であれば凝集せずに一次粒子として含まれていることも好ましい。
偏光板保護フィルム中のこれらの微粒子の含有量は0.01~1質量%であることが好ましく、特に0.05~0.5質量%が好ましい。共流延法による多層構成の偏光板保護フィルムの場合は、表面にこの添加量の微粒子を含有することが好ましい。
二酸化珪素の微粒子は、例えば、アエロジルR972、R972V、R974、R812、200、200V、300、R202、OX50、TT600(以上日本アエロジル(株)製)の商品名で市販されており、使用することができる。
酸化ジルコニウムの微粒子は、例えば、アエロジルR976およびR811(以上日本アエロジル(株)製)の商品名で市販されており、使用することができる。
ポリマーの例として、シリコーン樹脂、フッ素樹脂およびアクリル樹脂を挙げることができる。シリコーン樹脂が好ましく、特に三次元の網状構造を有するものが好ましく、例えば、トスパール103、同105、同108、同120、同145、同3120および同240(以上東芝シリコーン(株)製)の商品名で市販されており、使用することができる。
これらの中でもアエロジル200V、アエロジルR972Vが偏光板保護フィルムの濁度を低く保ちながら、摩擦係数を下げる効果が大きいため特に好ましく用いられる。本発明で用いられる偏光板保護フィルムにおいては、少なくとも一方の面の動摩擦係数が0.2~1.0であることが好ましい。
各種添加剤は製膜前のセルロースエステル含有溶液であるドープにバッチ添加してもよいし、添加剤溶解液を別途用意してインライン添加してもよい。特に微粒子は濾過材への負荷を減らすために、一部または全量をインライン添加することが好ましい。
添加剤溶解液をインライン添加する場合は、ドープとの混合性をよくするため、少量のセルロースエステルを溶解するのが好ましい。好ましいセルロースエステルの量は、溶剤100質量部に対して1~10質量部で、より好ましくは、3~5質量部である。
本発明においてインライン添加、混合を行うためには、例えば、スタチックミキサー(東レエンジニアリング製)、SWJ(東レ静止型管内混合器 Hi-Mixer)等のインラインミキサー等が好ましく用いられる。
<本発明のセルロースエステルフィルムの製造方法-フィルム化工程->
〈セルロースエステルフィルムの製造方法〉
次に、本発明のセルロースエステルフィルムの製造方法について説明する。
<本発明のセルロースエステルフィルムの製造方法-フィルム化工程->
〈セルロースエステルフィルムの製造方法〉
次に、本発明のセルロースエステルフィルムの製造方法について説明する。
本発明に係るセルロースエステルフィルムは溶液流延法で製造されたフィルムであっても溶融流延法で製造されたフィルムであっても好ましく用いることができる。
本発明のセルロースエステルフィルムの製造は、セルロースエステルおよび添加剤を溶剤に溶解させてドープを調製する工程、ドープを無限に移行する無端の金属支持体上に流延する工程、流延したドープをウェブとして乾燥する工程、金属支持体から剥離する工程、延伸または幅保持する工程、更に乾燥する工程、仕上がったフィルムを巻取る工程により行われる。
ドープを調製する工程について述べる。ドープ中のセルロースエステルの濃度は、濃い方が金属支持体に流延した後の乾燥負荷が低減できて好ましいが、セルロースエステルの濃度が濃過ぎると濾過時の負荷が増えて、濾過精度が悪くなる。これらを両立する濃度としては、10~35質量%が好ましく、更に好ましくは、15~25質量%である。
ドープで用いられる溶剤は、単独で用いても2種以上を併用してもよいが、セルロースエステルの良溶剤と貧溶剤を混合して使用することが生産効率の点で好ましく、良溶剤が多い方がセルロースエステルの溶解性の点で好ましい。
良溶剤と貧溶剤の混合比率の好ましい範囲は、良溶剤が70~98質量%であり、貧溶剤が2~30質量%である。良溶剤、貧溶剤とは、使用するセルロースエステルを単独で溶解するものを良溶剤、単独で膨潤するかまたは溶解しないものを貧溶剤と定義している。
そのため、セルロースエステルの平均酢化度(アセチル基置換度)によっては、良溶剤、貧溶剤が変わり、例えばアセトンを溶剤として用いる時には、セルロースエステルの酢酸エステル(アセチル基置換度2.4)、セルロースアセテートプロピオネートでは良溶剤になり、セルロースの酢酸エステル(アセチル基置換度2.8)では貧溶剤となる。
本発明に用いられる良溶剤は特に限定されないが、メチレンクロライド等の有機ハロゲン化合物やジオキソラン類、アセトン、酢酸メチル、アセト酢酸メチル等が挙げられる。特に好ましくはメチレンクロライドまたは酢酸メチルが挙げられる。
また、本発明に用いられる貧溶剤は特に限定されないが、例えば、メタノール、エタノール、n-ブタノール、シクロヘキサン、シクロヘキサノン等が好ましく用いられる。また、ドープ中には水が0.01~2質量%含有していることが好ましい。
また、セルロースエステルの溶解に用いられる溶媒は、フィルム製膜工程で乾燥によりフィルムから除去された溶媒を回収し、これを再利用して用いられる。
回収溶剤中に、セルロースエステルに添加されている添加剤、例えば可塑剤、紫外線吸収剤、ポリマー、モノマー成分などが微量含有されていることもあるが、これらが含まれていても好ましく再利用することができるし、必要であれば精製して再利用することもできる。
上記記載のドープを調製する時の、セルロースエステルの溶解方法としては、一般的な方法を用いることができる。加熱と加圧を組み合わせると常圧における沸点以上に加熱できる。
溶剤の常圧での沸点以上でかつ加圧下で溶剤が沸騰しない範囲の温度で加熱しながら攪拌溶解すると、ゲルやママコと呼ばれる塊状未溶解物の発生を防止するため好ましい。
また、セルロースエステルを貧溶剤と混合して湿潤あるいは膨潤させた後、更に良溶剤を添加して溶解する方法も好ましく用いられる。
加圧は窒素ガス等の不活性気体を圧入する方法や、加熱によって溶剤の蒸気圧を上昇させる方法によって行ってもよい。加熱は外部から行うことが好ましく、例えばジャケットタイプのものは温度コントロールが容易で好ましい。
溶剤を添加しての加熱温度は、高い方がセルロースエステルの溶解性の観点から好ましいが、加熱温度が高過ぎると必要とされる圧力が大きくなり生産性が悪くなる。
好ましい加熱温度は45~120℃であり、60~110℃がより好ましく、70℃~105℃が更に好ましい。また、圧力は設定温度で溶剤が沸騰しないように調整される。
もしくは冷却溶解法も好ましく用いられ、これによって酢酸メチルなどの溶媒にセルロースエステルを溶解させることができる。
次に、このセルロースエステル溶液を濾紙等の適当な濾過材を用いて濾過する。濾過材としては、不溶物等を除去するために絶対濾過精度が小さい方が好ましいが、絶対濾過精度が小さ過ぎると濾過材の目詰まりが発生し易いという問題がある。
このため絶対濾過精度0.008mm以下の濾材が好ましく、0.001~0.008mmの濾材がより好ましく、0.003~0.006mmの濾材が更に好ましい。
濾材の材質は特に制限はなく、通常の濾材を使用することができるが、ポリプロピレン、テフロン(登録商標)等のプラスチック製の濾材や、ステンレススティール等の金属製の濾材が繊維の脱落等がなく好ましい。
濾過により、原料のセルロースエステルに含まれていた不純物、特に輝点異物を除去、低減することが好ましい。
輝点異物とは、2枚の偏光板をクロスニコル状態にして配置し、その間に光学フィルム等を置き、一方の偏光板の側から光を当てて、他方の偏光板の側から観察した時に反対側からの光が漏れて見える点(異物)のことであり、径が0.01mm以上である輝点数が200個/cm2以下であることが好ましい。
より好ましくは100個/cm2以下であり、更に好ましくは50個/m2以下であり、更に好ましくは0~10個/cm2以下である。また、0.01mm以下の輝点も少ない方が好ましい。
ドープの濾過は通常の方法で行うことができるが、溶剤の常圧での沸点以上で、かつ加圧下で溶剤が沸騰しない範囲の温度で加熱しながら濾過する方法が、濾過前後の濾圧の差(差圧という)の上昇が小さく、好ましい。
好ましい温度は45~120℃であり、45~70℃がより好ましく、45~55℃であることが更に好ましい。
濾圧は小さい方が好ましい。濾圧は1.6MPa以下であることが好ましく、1.2MPa以下であることがより好ましく、1.0MPa以下であることが更に好ましい。
ここで、ドープの流延について説明する。
流延(キャスト)工程における金属支持体は、表面を鏡面仕上げしたものが好ましく、金属支持体としては、ステンレススティールベルトもしくは鋳物で表面をメッキ仕上げしたドラムが好ましく用いられる。
キャストの幅は1~4mとすることができる。流延工程の金属支持体の表面温度は-50℃~溶剤の沸点未満の温度で、温度が高い方がウェブの乾燥速度が速くできるので好ましいが、余り高過ぎるとウェブが発泡したり、平面性が劣化する場合がある。
好ましい支持体温度は0~55℃であり、25~50℃が更に好ましい。あるいは、冷却することによってウェブをゲル化させて残留溶媒を多く含んだ状態でドラムから剥離することも好ましい方法である。
金属支持体の温度を制御する方法は特に制限されないが、温風または冷風を吹きかける方法や、温水を金属支持体の裏側に接触させる方法がある。温水を用いる方が熱の伝達が効率的に行われるため、金属支持体の温度が一定になるまでの時間が短く好ましい。温風を用いる場合は目的の温度よりも高い温度の風を使う場合がある。
セルロースエステルフィルムが良好な平面性を示すためには、金属支持体からウェブを剥離する際の残留溶媒量は10~150質量%が好ましく、更に好ましくは20~40質量%または60~130質量%であり、特に好ましくは、20~30質量%または70~120質量%である。
本発明においては、残留溶媒量は下記式で定義される。
残留溶媒量(質量%)={(M-N)/N}×100
なお、Mはウェブまたはフィルムを製造中または製造後の任意の時点で採取した試料の質量で、NはMを114℃で1時間の加熱後の質量である。
なお、Mはウェブまたはフィルムを製造中または製造後の任意の時点で採取した試料の質量で、NはMを114℃で1時間の加熱後の質量である。
また、セルロースエステルフィルムの乾燥工程においては、ウェブを金属支持体より剥離し、更に乾燥し、残留溶媒量を1質量%以下にすることが好ましく、更に好ましくは0.1質量%以下であり、特に好ましくは0~0.01質量%以下である。
フィルム乾燥工程では一般にロール乾燥方式(上下に配置した多数のロールにウェブを交互に通し乾燥させる方式)やテンター方式でウェブを搬送させながら乾燥する方式が採られる。
本発明のセルロースエステルフィルムを作製するためには、ウェブの両端をクリップ等で把持するテンター方式で幅方向(横方向)に延伸を行うことが特に好ましい。剥離張力は300N/m以下で剥離することが好ましい。
ウェブを乾燥させる手段は特に制限なく、一般的に熱風、赤外線、加熱ロール、マイクロ波等で行うことができるが、簡便さの点で熱風で行うことが好ましい。
ウェブの乾燥工程における乾燥温度は40~200℃で段階的に高くしていくことが好ましい。
セルロースエステルフィルムの膜厚は、特に限定はされないが10~200μmが用いられる。特に膜厚は10~100μmであることが特に好ましい。更に好ましくは20~60μmである。
本発明のセルロースエステルフィルムは、幅1~4mのものが用いられる。特に幅1.4~4mのものが好ましく用いられる。4mを超えると搬送が困難となる。
(本発明のセルロースエステルフィルムの傾斜処理工程)
本発明の位相差フィルムは光学的に軸が歪んだ形の二軸性の位相差フィルムであり、屈折率が最小もしくは最大の方向を傾斜させたものであり、フィルム面に剪断応力を発生させて圧延処理(剪断圧延処理、剪断処理ともいう)をすることなどでも作製することができる。
本発明の位相差フィルムは光学的に軸が歪んだ形の二軸性の位相差フィルムであり、屈折率が最小もしくは最大の方向を傾斜させたものであり、フィルム面に剪断応力を発生させて圧延処理(剪断圧延処理、剪断処理ともいう)をすることなどでも作製することができる。
より具体的には、等速で荷重を変えた2つ以上のローラに挟んで処理する方法や、周速の異なるローラ(図1に記載の駆動ローラおよび追随ローラ)やベルトに挟んで処理する方法、あるいはフィルムを製膜時に基材(ドラム、ベルトなど)から剥離する際の剥離角度と剥離張力を制御する方法、あるいは文献4に記載の風による軸傾斜処理やフィルムをローラ搬送する際にローラの反対側を回転・駆動しないものを用いて剪断圧延処理によって作製することも可能である。
ここで、追随ローラとは、図1に示すように、搬送されるフィルムとの接触圧力によって自由回転又は強制回転する回転ローラであって、駆動ローラによってフィルムを搬送させる力が掛るフィルム面と反対のフィルム面でブレーキが掛るように反対方向に力を作用させるために使用する回転ローラをいう。回転に要する負荷は、各種のブレーキを使用することができる。ポイントとしては、負荷トルクが変動しない構造とすることが重要であり、駆動ローラを含めて、一定のトルクとなるような制御が必要である。
これらの処理は、一般にフィルムの処理温度をTg(フィルムのガラス転移温度)-50℃~Tg+40℃の範囲で実施することが好ましい。しかしこれらの方法で本発明の位相差フィルムを作製するにはバラツキが発生してしまい、安定して製造するためには更に工程を工夫する必要がある。
本発明の位相差フィルムをさらに安定して製造するには、例えば搬送方向への剪断応力を発生させる圧延処理の後、斜め方向の剪断応力を発生させる工程を幅手の一方の側ともう一報の側とから実施する、あるいは一度異周速ローラ間での処理を行った後、反対方向に剪断応力を発生させるローラと搬送方向に回転するローラでフィルムを圧延処理を実施する、フィルムの片面ともう片面とで残留溶媒差をつけた状態で斜め方向に剪断処理を行う、などの方法で作製することができている。
本発明に記載の位相差フィルムを得るには、剪断処理を行う際にフィルムの一方の面ともう一方の面とで差がつくことが必要であると思われるが、直接効果が見られる物性は完全に把握されているわけではなく、フィルム片面ともう片面との温度差、残溶差、表面粗さの差、ガラス転移点差、同じガラス転移点であればその時の硬度差などをつけることで効果が出てくる。膜厚方向で可塑剤、位相差発現剤などの添加剤の濃度勾配を持たせることも効果がある。
本発明の位相差フィルムは、フィルムの片面ともう片方の面との速度差あるいは経路長の差をつけることで、剪断応力が発生して変形することで必要な機能を発現するものであるが、この処理をローラやドラム、あるいはベルトでフィルムを挟んで行う場合、事前にフィルム端部にエンボス処理などのナーリング加工を行っている場合には、処理幅をナーリング部より内側のみで実施することができる。処理後に端部をスリットし、再度ナーリング加工をしても良い。
本発明の位相差フィルムは、複数のローラもしくはドラムによる処理を行うことができるが、これらは必ず樹脂製もしくは弾性金属もしくはゴム被覆金属もしくは金属被覆されたゴムなど、表面硬度か弾性率の異なるものを組み合わせることが好ましい。
その場合、硬い側のローラ(もしくはドラム)の表面材質としては、ハードクロムメッキされたもの、ニッケルメッキなどを施されたもの、セラミックスの超硬材料である、タングステン-カーバイドなどが溶射されたものなどで、剥離性がよく、かつ鏡面に研磨できる材質であることが好ましい。表面粗度は、0.2s以下で、特に好ましくは、0.1s以下である。
本発明の位相差フィルムは、処理を行う上下のローラ(もしくはドラム)の温度を異なる温度にして処理を行っても良く、例えば150℃程度の差をつけても良い。表面硬度の異なるローラ(もしくはドラム)を用いる場合で、ローラに温度差をつける場合、高硬度側を高温にする方が制御の面から安定化させやすく好ましい。
また、ローラの弾性が異なる場合で、周速差をつける場合はより低弾性側のローラを低速駆動かもしくは連れ周りにする方が好ましい。
また、これらの処理を行う場合、ウェブに溶媒が残った状態で行っても良い。
ローラ周速は、2つのローラの周速比が重要である。この場合、接触距離に夜が、独立駆動で実施する場合は、(高速側の速度/低速側の速度)の値は、1.05~1.80にすることが好ましく、1.60以下がより好ましい。
これらは、屈折率楕円体の屈折率最小の方向を傾斜させるために必要な処理と考えられるが、さらに軸を安定に扁平させるためには他の処理を追加しても良い。
二つのローラの圧着力は、0.01~10MPaが好ましく、0.03~5MPaがより好ましい。圧着力が大きすぎると傾斜が発生しにくくなり好ましくなく、できるだけ低い圧着力で実施することが重要である。この際の圧着力は、ローラとウェブが滑らない最低限の力で、押しつぶす方向の変形は極力抑えることが必要である。
本発明の処理を行う温度は、用いる材料のガラス転移点以上で行うことが好ましい。ガラス転移点かそれ以下の温度では、処理を行う際にバラツキが生じやすくなり、安定性の確保が困難になる。
特に、処理の際に圧着力を低く抑えることが安定性に効果があるため、複屈折の発現性が必要量確保できる状態で、可能な限り高温で処理することが好ましい。
本発明の実施には金属ベルトを用いて処理しても良い。
使用する金属ベルトの厚みは、スリップしないために必要な圧着力を確保するために0.2mm以上が好ましい。
フィルムの膜厚は、フィルムの片面ともう片方の面とで速度差をつける処理を実施する場合、処理前で150μ以下が好ましいが、複屈折が小さい場合はこの限りではない。150μを超えると巻き癖がつきやすくなる他、位相差や光軸のムラが発生しやすくなるなど、安定性が確保できなくなる場合がある。
ムラ発生しやすさの詳細な原因は不明であるが、厚み方向に力が伝わる際に厚み方向の材料の分布などによる違いをより顕著に拾ってしまうため、と推察される。
そのような観点で考えた場合、厚みの分布はできるだけ小さく抑える必要がある。平均値に対して、通常、±10%以内、好ましくは±5%以内、さらに好ましくは±1%以内、特に好ましくは±0.5%以内である。
また、1cmあたりの厚みの変動は、通常は1%以下、好ましくは0.1%以下、さらに好ましくは0.01%以下、特に好ましくは0.005%以下であることが望ましい。光学フィルムの厚み分布を上記の範囲内に制御することにより、場合により延伸加工処理を行う際に、位相差ムラや光軸の配向ムラが発生することを防止することができる。
(延伸工程)
本発明の位相差フィルムを製造するために、場合により延伸加工を施しても良い。具体的には、公知の一軸延伸法または二軸延伸法を挙げることができる。
本発明の位相差フィルムを製造するために、場合により延伸加工を施しても良い。具体的には、公知の一軸延伸法または二軸延伸法を挙げることができる。
すなわち、テンター法による横一軸延伸法、ローラ間圧縮延伸法、円周の異なる二組のローラを利用する縦一軸延伸法など、あるいは横一軸と縦一軸を組合わせた二軸延伸法による延伸法などを用いることができる。
一軸延伸法の場合、延伸速度は、通常、0.1~200%/秒であり、好ましくは0.1~100%/秒である。
二軸延伸法の場合、同時2方向に延伸を行う場合や一軸延伸後に最初の延伸方向と異なる方向に延伸処理する場合がある。
この時、延伸後のフィルムの屈折率楕円体の形状を制御するための2つの延伸軸の交わり角度は、所望の特性により決定されるため特に限定はされないが、通常、45~120度の範囲である。
また、延伸速度は各延伸方向で同じであってもよく、異なっていてもよく0.1~200%/秒が好ましい。延伸倍率は、所望の特性により決定されるため特に限定はされないが、通常、1.01~10倍、好ましくは1.03~5倍、さらに好ましくは1.03~3倍である。延伸倍率が10倍を超えると、位相差の制御が困難になる場合がある。
延伸したフィルムは、そのまま冷却してもよいが、Tg-20℃~Tgの温度雰囲気下に1秒以上あることが好ましい。
これらの延伸処理はどの段階で行っても良いが、最終製品の仕上がりの観点では、ローラなどによる処理の後に実施することが好ましい。特に平面性や膜厚分布などをより均一にするためにはローラなどによる処理の後に実施することが好ましい。
(最終工程)
製膜の最終工程として、フィルムの端部をスリッターにより製品となる幅にスリットして裁ち落とした後、エンボスリングおよびバックロールよりなるナール加工装置によりナール加工(エンボッシング加工)をフィルム両端部に施し、巻取り機によって巻き取ることにより、セルロースエステルフィルム(元巻き)の貼り付きや、すり傷の発生を防止する。
製膜の最終工程として、フィルムの端部をスリッターにより製品となる幅にスリットして裁ち落とした後、エンボスリングおよびバックロールよりなるナール加工装置によりナール加工(エンボッシング加工)をフィルム両端部に施し、巻取り機によって巻き取ることにより、セルロースエステルフィルム(元巻き)の貼り付きや、すり傷の発生を防止する。
ナール加工の方法は、凸凹のパターンを側面に有する金属リングを加熱や加圧により加工することができる。なお、フィルム両端部のクリップの把持部分は通常、変形しており、フィルム製品として使用できないので、切除されて、原料として再利用される。
一般的に、溶融押出しでは流延ダイの形状により、端部側の滞留時間が長くなる傾向が知られており、それによりフィルム端部の着色が促進されると考えられる。
本発明では溶融押出し直後のフィルム幅手方向の端部のイエローインデックスYeと、フィルム中央部分のイエローインデックスYcは下式を満たすことが好ましく、より好ましくはYe/Ycが3.0以下である。Ye/Ycが5.0より大きいと、フィルム端部を切除して、原料として再利用した際に、生産したフィルムの着色が増加する。
なお、本発明で端部のイエローインデックスとはフィルム幅手方向の両端部から30mm以内での最大値と定義する。
式 1.0≦Ye/Yc≦5.0
光学フィルム用途の場合、該フィルムの厚さは、10~500μmが好ましい。特に、下限は20μm以上、好ましくは30μm以上である。上限は150μm以下、好ましくは120μm以下である。特に好ましい範囲は25~90μmである。
光学フィルム用途の場合、該フィルムの厚さは、10~500μmが好ましい。特に、下限は20μm以上、好ましくは30μm以上である。上限は150μm以下、好ましくは120μm以下である。特に好ましい範囲は25~90μmである。
本発明のセルロースエステルフィルムの膜厚変動は、±3%、さらに±1%の範囲とすることが好ましい。
なお、ここでいうガラス転移温度Tgとは、示差走査熱量測定器(Perkin Elmer社製DSC-7型)を用いて、10℃/分で昇温、降温を行った後にもう一度昇温処理を行い、その際のTgを測定した測定し、JIS K7121(1987)に従い求めた中間点ガラス転移温度(Tmg)とする。
(機能性層の形成)
本発明のセルロースエステルフィルム製造に際し、延伸の前および/または後で透明導電層、ハードコート層、反射防止層、易滑性層、易接着層、防眩層、バリアー層、光学補償層等の機能性層を塗設してもよい。
本発明のセルロースエステルフィルム製造に際し、延伸の前および/または後で透明導電層、ハードコート層、反射防止層、易滑性層、易接着層、防眩層、バリアー層、光学補償層等の機能性層を塗設してもよい。
特に、透明導電層、ハードコート層、反射防止層、易接着層、防眩層および光学補償層から選ばれる少なくとも1層を設けることが好ましい。この際、コロナ放電処理、プラズマ処理、薬液処理等の各種表面処理を必要に応じて施すことができる。
<偏光板>
偏光板は一般的な方法で作製することができる。本発明のセルロースエステルフィルムをアルカリ鹸化処理し、処理したフィルムを、ヨウ素溶液中に浸漬延伸して作製した偏光子の少なくとも一方の面に、完全鹸化型ポリビニルアルコール水溶液を用いて貼り合わせることが好ましい。もう一方の面にも本発明のセルロースエステルフィルムを用いても、別の光学フィルムや偏光板保護フィルムを用いてもよい。
<偏光板>
偏光板は一般的な方法で作製することができる。本発明のセルロースエステルフィルムをアルカリ鹸化処理し、処理したフィルムを、ヨウ素溶液中に浸漬延伸して作製した偏光子の少なくとも一方の面に、完全鹸化型ポリビニルアルコール水溶液を用いて貼り合わせることが好ましい。もう一方の面にも本発明のセルロースエステルフィルムを用いても、別の光学フィルムや偏光板保護フィルムを用いてもよい。
本発明のセルロースエステルフィルムに対して、もう一方の面に用いられる光学フィルムや偏光板保護フィルムは市販のセルロースエステルフィルムを用いることができる。例えば、市販のセルロースエステルフィルムとして、KC8UX、KC4UX、KC5UX、KC8UCR3、KC8UCR4、KC8UCR5、KC8UY、KC4UY、KC10UDR、KC4FR、KC4UE、KC8UE、KC8UY-HA、KC8UX-RHA、KC8UXW-RHA-C、KC8UXW-RHA-NC、KC4UXW-RHA-NC(以上、コニカミノルタオプト(株)製)等が好ましく用いられる。
あるいはさらにディスコチック液晶、棒状液晶、コレステリック液晶などの液晶化合物を配向させて形成した光学異方層を有している光学補償フィルムを兼ねる光学フィルムを用いることも好ましい。
例えば、特開2003-98348号記載の方法で光学異方性層を形成することができる。本発明のセルロースエステルフィルムと組み合わせて使用することによって、平面性に優れ、安定した視野角拡大効果を有する偏光板を得ることができる。
偏光板の主たる構成要素である偏光子とは、一定方向の偏波面の光だけを通す素子であり、現在知られている代表的な偏光子は、ポリビニルアルコール系偏光フィルムで、これはポリビニルアルコール系フィルムにヨウ素を染色させたものと二色性染料を染色させたものがある。
偏光子は、ポリビニルアルコール水溶液を製膜し、これを一軸延伸させて染色するか、染色した後一軸延伸してから、好ましくはホウ素化合物で耐久性処理を行ったものが用いられている。該偏光子の面上に、本発明のセルロースエステルフィルムの片面を貼り合わせて偏光板を形成する。好ましくは完全鹸化ポリビニルアルコール等を主成分とする水系の接着剤によって貼り合わせる。
本発明に従い溶融流延製膜方法により製造される長尺状セルロースエステルフィルムは、長尺状の偏光子(偏光フィルム)とアルカリケン化処理を施して貼合することができるため、特に100m以上の長尺で生産的効果が得られ、1500m、2500m、5000mとより長尺化する程偏光板製造の生産的効果が高まる。
また、本発明のセルロースエステルフィルムを用いた偏光板はリワーク性に優れるため、偏光板収率が向上するという効果も得ることができる。
<液晶表示装置>
本発明のセルロースエステルフィルムを含む偏光板は、通常の偏光板と比較して高い表示品質を発現させることができる。
<液晶表示装置>
本発明のセルロースエステルフィルムを含む偏光板は、通常の偏光板と比較して高い表示品質を発現させることができる。
本発明の偏光板は、TNモード、OCB(Optical Compensated Bend)モード、IPS(In-Plane Switching)モード等に用いることができる。
液晶表示装置はカラー化および動画表示用の装置として応用され、本発明により表示品質が改良され、コントラストの改善や偏光板の耐性が向上したことにより、疲れにくく忠実な動画像表示が可能となる。
以下に実施例を挙げて本発明を具体的に説明する。
実施例1
<位相差フィルムの作製>
以下、本発明について実施例を挙げて説明するが、本発明はこれらに限定されるものではない。なお、剪断処理を行う条件は全ての位相差フィルムの作製においてTg-30℃~Tg+100℃の範囲とした。使用したセルロースエステルは表1に記載した。
<セルロースエステルフィルム101の作製>
〈微粒子分散液1〉
微粒子(アエロジル R812 日本アエロジル(株)製) 11質量部
エタノール 89質量部
以上をディゾルバーで50分間攪拌混合した後、マントンゴーリンで分散を行った。
実施例1
<位相差フィルムの作製>
以下、本発明について実施例を挙げて説明するが、本発明はこれらに限定されるものではない。なお、剪断処理を行う条件は全ての位相差フィルムの作製においてTg-30℃~Tg+100℃の範囲とした。使用したセルロースエステルは表1に記載した。
<セルロースエステルフィルム101の作製>
〈微粒子分散液1〉
微粒子(アエロジル R812 日本アエロジル(株)製) 11質量部
エタノール 89質量部
以上をディゾルバーで50分間攪拌混合した後、マントンゴーリンで分散を行った。
〈微粒子添加液1〉
メチレンクロライドを入れた溶解タンクに十分攪拌しながら、微粒子分散液1をゆっくりと添加した。更に、二次粒子の粒径が所定の大きさとなるようにアトライターにて分散を行った。これを日本精線(株)製のファインメットNFで濾過し、微粒子添加液1を調製した。
メチレンクロライドを入れた溶解タンクに十分攪拌しながら、微粒子分散液1をゆっくりと添加した。更に、二次粒子の粒径が所定の大きさとなるようにアトライターにて分散を行った。これを日本精線(株)製のファインメットNFで濾過し、微粒子添加液1を調製した。
メチレンクロライド 99質量部
微粒子分散液1 5質量部
下記組成の主ドープ液を調製した。まず加圧溶解タンクにメチレンクロライドとエタノールを添加した。溶剤の入った加圧溶解タンクにセルロースエステルA、Bを攪拌しながら投入した。これを加熱し、攪拌しながら、完全に溶解し。これを安積濾紙(株)製の安積濾紙No.244を使用して濾過し、主ドープ液を調製した。
微粒子分散液1 5質量部
下記組成の主ドープ液を調製した。まず加圧溶解タンクにメチレンクロライドとエタノールを添加した。溶剤の入った加圧溶解タンクにセルロースエステルA、Bを攪拌しながら投入した。これを加熱し、攪拌しながら、完全に溶解し。これを安積濾紙(株)製の安積濾紙No.244を使用して濾過し、主ドープ液を調製した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE1 100質量部
本発明のポリエステル系化合物14 6.5質量部
本発明の糖エステル化合物3 6.0質量部
微粒子添加液1 1質量部
以上を密閉容器に投入し、攪拌しながら溶解してドープ液を調製した。次いで、無端ベルト流延装置を用い、ドープ液を温度33℃、1500mm幅でステンレスベルト支持体上に均一に流延した。ステンレスベルトの温度は30℃に制御した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE1 100質量部
本発明のポリエステル系化合物14 6.5質量部
本発明の糖エステル化合物3 6.0質量部
微粒子添加液1 1質量部
以上を密閉容器に投入し、攪拌しながら溶解してドープ液を調製した。次いで、無端ベルト流延装置を用い、ドープ液を温度33℃、1500mm幅でステンレスベルト支持体上に均一に流延した。ステンレスベルトの温度は30℃に制御した。
ステンレスバンド支持体で、残留溶剤量が105%になるまで溶媒を蒸発させ、剥離張力162N/mでステンレスバンド支持体上から剥離した。
その後120℃で乾燥させた後、毎分85mでウェブを搬送し、直径35cmの金属ローラと弾性金属ローラにてウェブを、2.1MPa(接触幅が6mm)の圧着力となるように設定して挟み、弾性金属ローラの速度を搬送速度と同一にして、金属ローラの速度を毎分98mとなるように設定し1回目の剪断処理を施した。このローラを通す際の残留溶媒は1.2質量%であった。
続いて、同じ設定で、回転方向が搬送方向と50度方向となる金属ローラ(駆動ローラ)/弾性金属ローラ(追随ローラ)による2回目の剪断処理、さらに、搬送方向と-50度方向となる金属ローラ/弾性金属ローラによって3回目の剪断処理を行った。2回目の剪断処理の際、金属ローラの速度は毎分99m、3回目の剪断処理の際、金属ローラの回転速度は毎分98.5mに設定した。金属ローラの設定温度は全て185℃とした。
この後、テンターにて搬送方向と直交する方向に延伸処理を行った。延伸温度は155℃に設定し、延伸倍率は1.08倍、延伸速度は15%/秒とした。
テンターで延伸後130℃で幅手張力を緩和して幅保持を開放した後、120℃、130℃の乾燥ゾーンを多数のロールで搬送させながら乾燥を終了させ、1.75m幅にスリットし、フィルム両端に幅10mm高さ7μmのナーリング加工を施し、初期張力220N/m、終張力110N/mで内径6インチコアに巻き取り、膜厚42μmの位相差フィルム101を得た。

<位相差フィルム102>
位相差フィルム101の作製において、剪断処理2回目と3回目の金属ローラの速度をいずれも毎分104mとして、テンターでの延伸を1.2倍(延伸速度は40%/秒)にした他は位相差フィルム101と同様にして膜厚34μmの位相差フィルム102を作製した。
<位相差フィルム103、104、105、106>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
位相差フィルム101の作製において、剪断処理2回目と3回目の金属ローラの速度をいずれも毎分104mとして、テンターでの延伸を1.2倍(延伸速度は40%/秒)にした他は位相差フィルム101と同様にして膜厚34μmの位相差フィルム102を作製した。
<位相差フィルム103、104、105、106>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
なお、剪断処理1回目、2回目、3回目の金属ローラの速度を毎分85.6m、テンターでの延伸倍率を1.20倍、延伸温度を165℃とし膜厚50μmの位相差フィルム103を、金属ローラの温度を195℃、テンターでの延伸倍率を1.15倍、延伸温度を145℃にし膜厚60μmの位相差フィルム104を作製した。金属ローラ処理時の残留溶媒はどちらも2.2質量%であった。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
なお、剪断処理1回目、2回目、3回目の金属ローラの速度を毎分85.6m、テンターでの延伸倍率を1.20倍、延伸温度を165℃とし膜厚50μmの位相差フィルム103を、金属ローラの温度を195℃、テンターでの延伸倍率を1.15倍、延伸温度を145℃にし膜厚60μmの位相差フィルム104を作製した。金属ローラ処理時の残留溶媒はどちらも2.2質量%であった。
位相差フィルム103のテンターでの延伸条件を175℃で1.55倍として膜厚25μmの位相差フィルム105を、位相差フィルム103の2回目の金属ローラ温度を195℃、3回目の温度を175℃にして膜厚70μmの位相差フィルム106を作製した。
<位相差フィルム107の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
<位相差フィルム107の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理2回目の金属ローラでの処理角度を搬送方向と5度の角度で、金属ローラの速度を毎分121mとした他は位相差フィルム102と同様にして膜厚45μの位相差フィルム107を作製した。
<位相差フィルム108、109の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理2回目の金属ローラでの処理角度を搬送方向と5度の角度で、金属ローラの速度を毎分121mとした他は位相差フィルム102と同様にして膜厚45μの位相差フィルム107を作製した。
<位相差フィルム108、109の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.8MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を200℃として処理を行い、テンターでの延伸温度を176℃、延伸倍率を1.68倍とした他は位相差フィルム101と同じようにして膜厚38μmの位相差フィルム108を、金属ベルトの速度を毎分86.5m、テンターの延伸倍率を1.9倍にした他は位相差フィルム101と同じようにして膜厚34μmの位相差フィルム109を作製した。
<位相差フィルム110の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.8MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を200℃として処理を行い、テンターでの延伸温度を176℃、延伸倍率を1.68倍とした他は位相差フィルム101と同じようにして膜厚38μmの位相差フィルム108を、金属ベルトの速度を毎分86.5m、テンターの延伸倍率を1.9倍にした他は位相差フィルム101と同じようにして膜厚34μmの位相差フィルム109を作製した。
<位相差フィルム110の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.8MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を200℃として処理を行い、テンターでの延伸温度を175℃、延伸倍率を1.70倍とした他は位相差フィルム101と同じようにして膜厚40μmの位相差フィルム110を作製した。
<位相差フィルム111の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.8MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を200℃として処理を行い、テンターでの延伸温度を175℃、延伸倍率を1.70倍とした他は位相差フィルム101と同じようにして膜厚40μmの位相差フィルム110を作製した。
<位相差フィルム111の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.6MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を186℃として処理を行い、テンターでの延伸温度を158℃、延伸倍率を1.50倍とした他は位相差フィルム101と同じようにして膜厚48μmの位相差フィルム111を作製した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 9.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.6MPa、接触幅が15cmとなるように設定し、金属ローラの速度を毎分88mとして、雰囲気温度を186℃として処理を行い、テンターでの延伸温度を158℃、延伸倍率を1.50倍とした他は位相差フィルム101と同じようにして膜厚48μmの位相差フィルム111を作製した。
(偏光板101~111の作製)
上記作製したセルロースエステルフィルム101~111を使って、下記に記載するアルカリケン化処理後、偏光板の作製を行った。
上記作製したセルロースエステルフィルム101~111を使って、下記に記載するアルカリケン化処理後、偏光板の作製を行った。
〈アルカリケン化処理〉
ケン化工程 2M-NaOH 50℃ 90秒
水洗工程 水 30℃ 45秒
中和工程 10質量%HCl 30℃ 45秒
水洗工程 水 30℃ 45秒
ケン化処理後、水洗、中和、水洗の順に行い、次いで80℃で乾燥を行った。
ケン化工程 2M-NaOH 50℃ 90秒
水洗工程 水 30℃ 45秒
中和工程 10質量%HCl 30℃ 45秒
水洗工程 水 30℃ 45秒
ケン化処理後、水洗、中和、水洗の順に行い、次いで80℃で乾燥を行った。
〈偏光子の作製〉
厚さ120μmの長尺ロールポリビニルアルコールフィルムを沃素1質量部、ホウ酸4質量部を含む水溶液100質量部に浸漬し、50℃で6倍に搬送方向に延伸して偏光子を作った。
厚さ120μmの長尺ロールポリビニルアルコールフィルムを沃素1質量部、ホウ酸4質量部を含む水溶液100質量部に浸漬し、50℃で6倍に搬送方向に延伸して偏光子を作った。
上記偏光子の片面に同様にケン化処理したコニカミノルタオプト(株)製KC8UX、その反対面側に前記アルカリケン化処理した本発明のセルロースエステルフィルムを完全ケン化型ポリビニルアルコール5%水溶液を接着剤として、偏光子の透過軸とフィルムの面内遅相軸が平行になるように各々貼り合わせ、乾燥した。
なお、偏光板107は、位相差フィルム107を90度回転させて、位相差フィルムの面内遅相軸と偏光子の透過軸が直交するように作製した。
<位相差フィルム112の作製>
ゼオノアフィルムZF-14(オプテス(株)製)を、直径35cmの金属ローラと弾性金属ローラにてウェブを毎分2.2mで搬送し、8.8MPa(接触幅が6mm)の圧着力となるように設定して挟み、弾性金属ローラの速度を搬送速度と同一にして、金属ローラの速度を毎分2.5mとなるように剪断処理した。
<位相差フィルム112の作製>
ゼオノアフィルムZF-14(オプテス(株)製)を、直径35cmの金属ローラと弾性金属ローラにてウェブを毎分2.2mで搬送し、8.8MPa(接触幅が6mm)の圧着力となるように設定して挟み、弾性金属ローラの速度を搬送速度と同一にして、金属ローラの速度を毎分2.5mとなるように剪断処理した。
続いて、同じ設定で、回転方向が搬送方向と50度方向となる金属ローラ/弾性金属ローラによる2回目の処理、さらに、搬送方向と-50度方向となる金属ローラ/弾性金属ローラによって3回目の処理を行った。2回目の処理の際、金属ローラの速度は毎分2.7m、3回目の処理の際、金属ローラの回転速度は毎分2.75mに設定した。金属ローラの設定温度は全て165℃とした。
この後、テンターにて搬送方向と直交する方向に延伸処理を行った。延伸温度は148℃に設定し、延伸倍率は1.15倍、延伸速度は15%/秒として位相差フィルム112を作製した。
<位相差フィルム113の作製>
位相差フィルム112と同じように製膜を行い、直径35cmの金属ローラと弾性金属ローラにてウェブを毎分2.2mで搬送し、8.8MPa(接触幅が6mm)の圧着力となるように設定して挟み、弾性金属ローラの速度を搬送速度と同一にして、金属ローラの速度を毎分3.2mとなるように設定した。
<位相差フィルム113の作製>
位相差フィルム112と同じように製膜を行い、直径35cmの金属ローラと弾性金属ローラにてウェブを毎分2.2mで搬送し、8.8MPa(接触幅が6mm)の圧着力となるように設定して挟み、弾性金属ローラの速度を搬送速度と同一にして、金属ローラの速度を毎分3.2mとなるように設定した。
続いて、同じ設定で、回転方向が搬送方向と50度方向となる金属ローラ/弾性金属ローラによる2回目の処理、さらに、搬送方向と-50度方向となる金属ローラ/弾性金属ローラによって3回目の処理を行った。2回目の処理の際、金属ローラの速度は毎分2.9m、3回目の処理の際、金属ローラの回転速度は毎分2.85mに設定した。金属ローラの設定温度は全て155℃とした。
この後、テンターにて搬送方向と直交する方向に延伸処理を行った。延伸温度は148℃に設定し、延伸倍率は1.3倍、延伸速度は20%/秒として位相差フィルム113を作製した。
<位相差フィルム201、202の作製>
位相差フィルム112と同様に製膜を行い、2回目、3回目のローラ処理を行わなかった以外は位相差フィルム112と同じ様にして位相差フィルム201を作製した。
<位相差フィルム201、202の作製>
位相差フィルム112と同様に製膜を行い、2回目、3回目のローラ処理を行わなかった以外は位相差フィルム112と同じ様にして位相差フィルム201を作製した。
位相差フィルム113と同様に製膜を行い、2回目、3回目のローラ処理を行わずに148℃で、延伸倍率は1.28倍、延伸速度は19%/秒として位相差フィルム202を作製した。
(偏光板112、113、201、202の作製)
上記作製した位相差フィルムを使って、アクリル系接着剤(住友スリーエム製、製品名「DP-8005クリア」)を介して次のように作製した偏光子と貼合した。
上記作製した位相差フィルムを使って、アクリル系接着剤(住友スリーエム製、製品名「DP-8005クリア」)を介して次のように作製した偏光子と貼合した。
〈偏光子の作製〉
厚さ120μmの長尺ロールポリビニルアルコールフィルムを沃素1質量部、ホウ酸4質量部を含む水溶液100質量部に浸漬し、50℃で6倍に搬送方向に延伸して偏光子を作った。
厚さ120μmの長尺ロールポリビニルアルコールフィルムを沃素1質量部、ホウ酸4質量部を含む水溶液100質量部に浸漬し、50℃で6倍に搬送方向に延伸して偏光子を作った。
上記位相差フィルムを貼合した偏光子の反対面に、上記ケン化処理したコニカミノルタオプト(株)製KC8UXを完全ケン化型ポリビニルアルコール5%水溶液を接着剤として、偏光子の透過軸とフィルムの面内遅相軸が平行になるように各々貼り合わせ、乾燥した。
<位相差フィルム203の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
<位相差フィルム203の作製>
位相差フィルム101の作製において、主ドープ液の組成を下記に変更したほかは同様にドープを作製、製膜した。
〈主ドープ液の組成〉
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.2MPa、搬送速度を毎分14m、接触幅が15cmとなるように設定し、金属ローラの速度を毎分15mとして、雰囲気温度を210℃として処理を行い、テンターでの延伸温度を178℃、延伸倍率を2.1倍に設定して位相差フィルム203を作製した。
<位相差フィルム204、205の作製>
位相差フィルム101と同様に製膜し、剪断処理2回目と3回目のローラ処理を実施せずに、テンターでの延伸条件をそれぞれ延伸温度130℃、延伸倍率1.21倍、114℃1.2倍で位相差フィルム204、205を作製した。
<位相差フィルム206の作製>
特開2000-155216号実施例1記載の光学補償フィルムに準じて作製した。
<位相差フィルム207の作製>
特開平7-98412号実施例1記載の光学補償フィルムに準じて作製した。
メチレンクロライド 340質量部
エタノール 64質量部
本発明のセルロースエステルCE2 100質量部
本発明のポリエステル系化合物14 8.0質量部
本発明の糖エステル化合物3 5.0質量部
微粒子添加液1 1質量部
位相差フィルム101の作製において、剪断処理1回目の処理を金属ローラの替わりにSUS製2.8mmの金属ベルトを用い、圧力が2.2MPa、搬送速度を毎分14m、接触幅が15cmとなるように設定し、金属ローラの速度を毎分15mとして、雰囲気温度を210℃として処理を行い、テンターでの延伸温度を178℃、延伸倍率を2.1倍に設定して位相差フィルム203を作製した。
<位相差フィルム204、205の作製>
位相差フィルム101と同様に製膜し、剪断処理2回目と3回目のローラ処理を実施せずに、テンターでの延伸条件をそれぞれ延伸温度130℃、延伸倍率1.21倍、114℃1.2倍で位相差フィルム204、205を作製した。
<位相差フィルム206の作製>
特開2000-155216号実施例1記載の光学補償フィルムに準じて作製した。
<位相差フィルム207の作製>
特開平7-98412号実施例1記載の光学補償フィルムに準じて作製した。
(偏光板203~207の作製)
位相差フィルム203~207を上記の条件でケン化処理を行い偏光子と接着し偏光板203~207を作製した。
位相差フィルム203~207を上記の条件でケン化処理を行い偏光子と接着し偏光板203~207を作製した。
偏光板207は、コニカミノルタオプト製KC8UXを用いた偏光板に粘着層を介して、遅相軸と偏光子の透過軸が平行になるように偏光板を作製した。
〈液晶表示装置の作製〉
(液晶表示装置の作製)
得られた偏光板は、TN型液晶表示装置であるNEC製17型液晶ディスプレイLCD72Vにあらかじめ貼合されていた偏光板を注意深く剥がし、もともと貼ってあった偏光板の透過軸にあわせて、粘着剤を介して作製した偏光板を貼り付け液晶表示装置を作製した。
(液晶表示装置の作製)
得られた偏光板は、TN型液晶表示装置であるNEC製17型液晶ディスプレイLCD72Vにあらかじめ貼合されていた偏光板を注意深く剥がし、もともと貼ってあった偏光板の透過軸にあわせて、粘着剤を介して作製した偏光板を貼り付け液晶表示装置を作製した。
(評価)
〈位相差(Ro,Nz)、傾斜角度βの測定〉
王子計測機器製KOBRA-31WRと溝尻工学社製DVA36VWを併用して、23℃55%RHの雰囲気下、波長590nmで測定した。
〈位相差(Ro,Nz)、傾斜角度βの測定〉
王子計測機器製KOBRA-31WRと溝尻工学社製DVA36VWを併用して、23℃55%RHの雰囲気下、波長590nmで測定した。
〈視野角評価〉
ELDIM社製EZ-Contrast160Dを用いて、白、黒の輝度測定よりコントラスト10が得られる角度を視野角とした。階調反転とカラーシフトは20人で目視による官能評価を行った。
ELDIM社製EZ-Contrast160Dを用いて、白、黒の輝度測定よりコントラスト10が得られる角度を視野角とした。階調反転とカラーシフトは20人で目視による官能評価を行った。
〈階調反転〉
白から黒までを8段階で表示したウェッジを用い、輝度が変化し始める角度を上、下、横、斜め下で観察し全方向で30度以上を○、下方向以外が30度以上で下方向は20~30度のものを△、どこか1方向でも20度以内のものを×とした。
白から黒までを8段階で表示したウェッジを用い、輝度が変化し始める角度を上、下、横、斜め下で観察し全方向で30度以上を○、下方向以外が30度以上で下方向は20~30度のものを△、どこか1方向でも20度以内のものを×とした。
〈カラーシフト〉
赤、青、緑、白の4色を上、下、斜め下、横方向で観察し、色変化が30度以上で気にならないものを○、20~30度でどこか一方向の色変化がはっきり認識できるものを△、2方向で20~30度、もしくは一方向でも10度以内ではっきり認識できるものを×とした。さらに白表示の際に、横方向で倒れ角60度における色味を確認した。
赤、青、緑、白の4色を上、下、斜め下、横方向で観察し、色変化が30度以上で気にならないものを○、20~30度でどこか一方向の色変化がはっきり認識できるものを△、2方向で20~30度、もしくは一方向でも10度以内ではっきり認識できるものを×とした。さらに白表示の際に、横方向で倒れ角60度における色味を確認した。
〈額縁ムラ〉
表示装置を50℃90%RHで200時間処理し、取り出した直後に黒表示にして室温で2時間放置して、全面および額縁状に発生したムラの大小・強弱で判断した。目だったムラがないか実用上問題ないムラが1箇所発生しているものを○、実用上問題にならないムラが2箇所まで発生しているものを△、1箇所でも目だったムラが発生しているものを×とした。
表示装置を50℃90%RHで200時間処理し、取り出した直後に黒表示にして室温で2時間放置して、全面および額縁状に発生したムラの大小・強弱で判断した。目だったムラがないか実用上問題ないムラが1箇所発生しているものを○、実用上問題にならないムラが2箇所まで発生しているものを△、1箇所でも目だったムラが発生しているものを×とした。
結果を表2に示す。

表2より、本発明はコントラスト視野角が広く、位相差ムラの小さいことが明確である。
1R 駆動ローラ
2R 追随ローラ
MD フィルム搬送方向
F フィルム
X 屈折率楕円体の面内最大屈折率の方向
Y 屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z 屈折率楕円体の屈折率最小方向
A X軸とZ軸のなす角度
B X軸とY軸のなす角度
C Y軸とZ軸のなす角度
β フィルムの法線方向とZ軸のなす角度
2R 追随ローラ
MD フィルム搬送方向
F フィルム
X 屈折率楕円体の面内最大屈折率の方向
Y 屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z 屈折率楕円体の屈折率最小方向
A X軸とZ軸のなす角度
B X軸とY軸のなす角度
C Y軸とZ軸のなす角度
β フィルムの法線方向とZ軸のなす角度
Claims (2)
- 光学的に2軸の位相差フィルムであってX軸、Y軸、Z軸、A、B、C、βを下記のように定義するとき、次の(1)~(6)の関係を全て有することを特徴とする位相差フィルム。
(1)0≦β≦50度
(2)B=90度
(3)A≠90度
(4)C≠90度
(5)80度<A<90度
(6)80度<C<90度
X軸:屈折率楕円体の面内最大屈折率の方向
Y軸:屈折率楕円体の屈折率楕円体の面内方向であって面内最大屈折率と直交する方向
Z軸:屈折率楕円体の屈折率最小方向
A:X軸とZ軸のなす角度
B:X軸とY軸のなす角度
C:Y軸とZ軸のなす角度
β:フィルムの法線方向とZ軸のなす角度 - 前記位相差フィルムがセルロースエステルフィルムであることを特徴とする請求項1記載の位相差フィルム。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011502694A JPWO2010100993A1 (ja) | 2009-03-05 | 2010-02-04 | 位相差フィルム |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-051832 | 2009-03-05 | ||
| JP2009051832 | 2009-03-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010100993A1 true WO2010100993A1 (ja) | 2010-09-10 |
Family
ID=42709555
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/051600 WO2010100993A1 (ja) | 2009-03-05 | 2010-02-04 | 位相差フィルム |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2010100993A1 (ja) |
| WO (1) | WO2010100993A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000121831A (ja) * | 1998-10-20 | 2000-04-28 | Sumitomo Chem Co Ltd | 位相差フィルム |
| JP2003075639A (ja) * | 2001-09-07 | 2003-03-12 | Hayashi Telempu Co Ltd | 位相差フィルムとその製造方法、および位相差フィルムを用いた液晶表示装置 |
| JP2003307617A (ja) * | 2002-04-17 | 2003-10-31 | Hayashi Telempu Co Ltd | 位相差フィルムとその製造方法、およびこの位相差フィルムを装着した液晶表示装置 |
| JP2007038646A (ja) * | 2005-06-28 | 2007-02-15 | Jsr Corp | 光学フィルムの製造方法、光学フィルムおよび偏光板 |
-
2010
- 2010-02-04 WO PCT/JP2010/051600 patent/WO2010100993A1/ja active Application Filing
- 2010-02-04 JP JP2011502694A patent/JPWO2010100993A1/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000121831A (ja) * | 1998-10-20 | 2000-04-28 | Sumitomo Chem Co Ltd | 位相差フィルム |
| JP2003075639A (ja) * | 2001-09-07 | 2003-03-12 | Hayashi Telempu Co Ltd | 位相差フィルムとその製造方法、および位相差フィルムを用いた液晶表示装置 |
| JP2003307617A (ja) * | 2002-04-17 | 2003-10-31 | Hayashi Telempu Co Ltd | 位相差フィルムとその製造方法、およびこの位相差フィルムを装着した液晶表示装置 |
| JP2007038646A (ja) * | 2005-06-28 | 2007-02-15 | Jsr Corp | 光学フィルムの製造方法、光学フィルムおよび偏光板 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2010100993A1 (ja) | 2012-09-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5299270B2 (ja) | 位相差フィルム、偏光板、液晶表示装置および位相差フィルムの製造方法 | |
| JP5201135B2 (ja) | 光学補償フィルムとそれを用いた偏光板及び液晶表示装置 | |
| JP5754445B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| JP2009210777A (ja) | 光学フィルム、及びそれを用いた偏光板 | |
| JP5671832B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| JP2009001696A (ja) | セルロースエステルフィルム、それを用いた偏光板及び液晶表示装置 | |
| JP5741850B2 (ja) | 光学フィルム、それを含む偏光板および液晶表示装置 | |
| WO2009119142A1 (ja) | セルロースエステルフィルム | |
| JPWO2008050603A1 (ja) | Ips型液晶表示装置及びips型液晶表示装置の製造方法 | |
| JP5737287B2 (ja) | 位相差フィルム、それを用いた偏光板、及び液晶表示装置 | |
| JP5428045B2 (ja) | 液晶表示装置 | |
| JP5170093B2 (ja) | 液晶表示装置 | |
| JPWO2011114764A1 (ja) | 位相差フィルム及びそれが備えられた偏光板 | |
| WO2009101839A1 (ja) | 位相差フィルム | |
| JP5549397B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| JP5233935B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置並びにリターデーション発現剤 | |
| JP6330870B2 (ja) | 偏光板およびこれを用いた液晶表示装置 | |
| WO2010041514A1 (ja) | 光学フィルム、及びそれを用いた偏光板 | |
| WO2010100993A1 (ja) | 位相差フィルム | |
| JP5446881B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| WO2011046027A1 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| JP2012072223A (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| WO2010106854A1 (ja) | 光学フィルム、光学フィルムの製造方法 | |
| JP5282310B2 (ja) | 光学フィルム、及びそれを用いた偏光板、液晶表示装置 | |
| JP5772680B2 (ja) | セルロースアシレート積層フィルムおよびその製造方法、並びにそれを用いた偏光板および液晶表示装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10748590 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011502694 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10748590 Country of ref document: EP Kind code of ref document: A1 |