3-Cvanoalkyl- und 3-HydroxyalkvI-Indole und ihre Verwendung
Die vorliegende Anmeldung betrifft neue, 3-Cyanoalkyl- und 3-Hydroxyalkyl-substituierte Indol- Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prävention von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prävention von Krankheiten, insbesondere zur Behandlung und/oder Prävention von kardiovaskulären Erkrankungen.
Aldosteron spielt eine Schlüsselrolle in der Aufrechterhaltung der Flüssigkeits- und Elektrolythomöostase, indem es im Epithel des distalen Nephrons die Natriumretention und Kaliumsekretion fördert, was zur Konstanthaltung des Extrazellulärvolumens und damit zur Blutdruckregulation beiträgt. Daneben entfaltet Aldosteron direkte Effekte auf die Struktur und Funktion des Herz- und Gefäßsystems, wobei die dafür zugrunde liegenden Mechanismen noch nicht erschöpfend geklärt sind [R.E. Booth, J.P. Johnson, J.D. Stockand, Adv. Physiol. Educ. 26 (1), 8-20 (2002)].
Aldosteron ist ein Steroidhormon, das in der Nebennierenrinde gebildet wird. Seine Produktion wird indirekt ganz wesentlich in Abhängigkeit von der Nierendurchblutung reguliert. Jede Ab- nähme der Nierendurchblutung führt in der Niere zu einer Ausschüttung des Enzyms Renin in den Blutkreislauf. Dieses wiederum aktiviert die Bildung von Angiotensin II, das einerseits verengend auf die arteriellen Blutgefäße wirkt, andererseits aber auch die Bildung von Aldosteron in der Nebennierenrinde stimuliert. Somit fungiert die Niere als Blutdruck- und damit indirekter Volumen-Sensor im Blutkreislauf und wirkt über das Renin-Angiotensin-Aldosteron-System kri- tischen Volumenverlusten entgegen, indem einerseits der Blutdruck gesteigert wird (Angiotensin U- Wirkung), andererseits durch verstärkte Rückresorption von Natrium und Wasser in der Niere der Füllungszustand des Gefäßsystems wieder ausgeglichen wird (Aldosteron- Wirkung).
Dieses Regelsystem kann in vielfältiger Weise krankhaft gestört sein. So führt eine chronische Minderdurchblutung der Nieren (z.B. infolge einer Herzinsuffizienz und des hierdurch verursach- ten Blutrückstaus im Venensystem) zu einer chronisch überhöhten Ausschüttung von Aldosteron. Diese wiederum zieht eine Expansion des Blutvolumens nach sich und verstärkt hiermit die Herzschwäche durch ein Volumenüberangebot an das Herz. Eine Stauung von Blut in den Lungen mit Atemnot und Ödembildung in den Extremitäten sowie Aszites und Pleuraergüsse können die Folge sein; die Nierendurchblutung sinkt weiter ab. Überdies führt die übersteigerte Aldosteron- Wirkung zu einer Minderung der Kalium-Konzentration im Blut und in der Extrazellulärflüssigkeit. In ohnehin vorgeschädigten Herzmuskeln kann die Unterschreitung eines kritischen Mindestwertes tödlich endende Herzrhythmusstörungen auslösen. Hierin dürfte eine der Hauptursachen des häufig bei Patienten mit Herzinsuffizienz eintretenden plötzlichen Herztodes TU suchen sein.
Zusätzlich wird Aldosteron auch für eine Reihe der typischerweise bei Herzinsuffizienten zu beobachtenden Umbauprozesse des Herzmuskels verantwortlich gemacht. Somit ist der Hyperaldo- steronismus eine entscheidende Komponente in der Pathogenese und Prognose der Herzinsuffizienz, die ursprünglich durch unterschiedliche Schädigungen, wie z.B. einen Herzinfarkt, eine Herzmuskelentzündung oder Bluthochdruck, ausgelöst werden kann. Diese Annahme wird durch die Tatsache erhärtet, dass in umfangreichen klinischen Studien in Patientenkollektiven mit chronischer Herzinsuffizienz bzw. nach akutem Myokardinfarkt durch Anwendung von Aldosteron-Antagonisten die Gesamtmortalität deutlich gesenkt wurde [B. Pitt, F. Zannad, WJ. Remme et al., N. Engl. J. Med. 341, 709-717 (1999); B. Pitt, W. Remme, F. Zannad et al., N. Engl. J. Med. 348, 1309-1321 (2003)]. Dies konnte unter anderem durch eine Senkung der Inzidenz des plötzlichen Herztodes erreicht werden.
Neueren Studien zufolge findet man auch bei einem nicht unerheblichen Teil der Patienten, die unter einer essentiellen Hypertonie leiden, eine sogenannte normokaliämische Variante des primären Hyperaldosteronismus [Prävalenz bis 11% aller Hypertoniker: L. Seiler und M. Reincke, Der Aldosteron-Renin-Quotient bei sekundärer Hypertonie, Herz 28, 686-691 (2003)]. Als beste Diagnostikmethode dient beim normokaliämischen Hyperaldosteronismus der Aldosteron/Renin- Quotient der entsprechenden Plasmakonzentrationen, so dass auch relative Aldosteron-Erhöhungen in Bezug auf die Renin-Plasmakonzentrationen diagnostizierbar und letztlich therapierbar werden. Deshalb ist ein im Zusammenhang mit einer essentiellen Hypertonie diagnostizierter Hyperaldo- steronismus ein Ansatzpunkt für eine kausale und prophylaktisch sinnvolle Therapie.
Weitaus seltener als die oben aufgeführten Formen des Hyperaldosteronismus sind solche Krankheitsbilder, bei denen die Störung entweder in den Hormon-produzierenden Zellen der Nebenniere selbst zu finden ist oder deren Anzahl oder Masse durch eine Hyperplasie oder Wucherung vermehrt ist. Adenome oder diffuse Hyperplasien der Nebennierenrinde sind die häufigste Ursache des auch als Conn-Syndrom bezeichneten primären Hyperaldosteronismus, dessen Leitsymptome Hypertonie und hypokaliämische Alkalose sind. Auch hier steht neben der chirurgischen Entfernung des krankhaften Gewebes die medikamentöse Therapie mit Aldosteron-Antagonisten im Vordergrund [H.A. Kühn und J. Schirmeister (Hrsg.), Innere Medizin, 4. Aufl., Springer Verlag, Berlin, 1982].
Ein anderes typischerweise mit Erhöhung der Aldosteron-Konzentration im Plasma einhergehendes Krankheitsbild ist die fortgeschrittene Leberzirrhose. Die Ursache der Aldosteron- erhöhung liegt hier vorwiegend im infolge der Leberfunktionsstörung eingeschränkten Abbau des Aldosterons. Volumenüberlastung, Ödeme und Hypokaliämie sind die typischen Folgen, die in der klinischen Praxis durch Aldosteron-Antagonisten erfolgreich gelindert werden können.
Die Wirkungen von Aldosteron werden über den in den Zielzellen intrazellulär lokalisierten Mineralokorticoid-Rezeptor vermittelt. Die bislang verfügbaren Aldosteron-Antagonisten besitzen wie Aldosteron selbst eine Steroid-Grundstruktur. Begrenzt wird die Anwendbarkeit derartiger steroidaler Antagonisten durch ihre Wechselwirkungen mit den Rezeptoren anderer Steroidhor- mone, die zum Teil zu erheblichen Nebenwirkungen wie Gynäkomastie und Impotenz und zum Abbrechen der Therapie fuhren [M.A. Zaman, S. Oparil, D.A. Calhoun, Nature Rev. DrugDisc. 1, 621-636 (2002)].
Die Identifizierung wirkstarker, nicht-steroidaler und für den Mineralokorticoid-Rezeptor selektiver Antagonisten eröffnet die Möglichkeit, dieses Nebenwirkungsprofil zu umgehen und dadurch einen deutlichen Therapievorteil zu erzielen [vgl. MJ. Meyers und X. Hu, Expert Opin. Ther. Patents JJ (1), 17-23 (2007)].
Aufgabe der vorliegenden Erfindung ist daher die Bereitstellung neuer Verbindungen, die als potente und selektive Mineralokorticoid-Rezeptor-Antagonisten wirken und so für die Behandlung von Erkrankungen, insbesondere von kardiovaskulären Erkrankungen, eingesetzt werden können.
In WO 2004/067529, WO 2005/092854 und M. G. Bell, J. Med. Chem. 2007, 50 (26), 6443-6445 werden verschiedene in 3-Position substituierte Indol-Derivate als Modulatoren von Steroidhormon-Rezeptoren beschrieben. Indol-3-yl(phenyl)essigsäure-Derivate als Endothelin- Rezeptorantagonisten werden in WO 97/43260 und α-Amino(indol-3-yl)essigsäure-Derivate mit anti-diabetischer Wirkung werden in WO 90/05721 offenbart. In WO 2007/062994 und WO 2005/118539 werden 3-(3-Amino-l-arylpropyl)-Indole zur Behandlung von Depression und Angstzuständen beansprucht. 3-(Indol-3-yl)-3-phenylpropionitril-Derivate sind unter anderem in US 2 752 358, US 2 765 320 und US 2 778 819 beschrieben. Die Herstellung von 2- unsubstituierten Indolen wird in WO 98/06725 und US 5,808,064 offenbart. Über die Herstellung von 2-(Indol-3-yl)-2-phenylethanol-Derivaten wird unter anderem in M.L. Kantam et al., Tetrahedron Lett. 47 (35), 6213-6216 (2006) berichtet. In EP 0 778 277-A1 werden verschiedene azabicyclische Verbindungen als CRF-Antagonisten offenbart. WO 2007/040166 beansprucht annelierte Pyrrol-Derivate als Glucocorticoid-Rezeptor Modulatoren, die anti-inflammatorisch und anti-diabetisch wirken. In WO 2007/070892 werden substituierte Indole zur Behandlung von Angst, Schmerz und kognitiven Störungen beschrieben.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
in welcher
A für C-R5 oder N steht,
wobei
R5 für Wasserstoff, Fluor, Chlor oder (CrC4)-Alkyl steht,
R1 für Halogen, Cyano, Nitro, (C,-C6)-Alkyl, (CrC6)-Alkoxy, Amino, Mono-CQ-C^-alkyl- amino, Di-(Ci-C6)-alkylamino oder eine Gruppe der Formel -(CH2)P-NR6-SO2-R7 steht,
wobei (Ci-C6)-Alkyl und (Ci-Co)-AIkOXy mit 1 bis 3 Substituenten Fluor substituiert sein können,
wobei (Ci-C6)-Alkyl und (Ci-C6)-Alkoxy mit einem Substituenten ausgewählt aus der
Gruppe Hydroxy und (Ci-C4)-Alkoxy substituiert sein können
und wobei
p für die Zahl 0, 1 oder 2 steht,
R6 für Wasserstoff oder (CrC4)-Alkyl steht,
und
R7 für (Ci-C6)-Alkyl, (C3-C7)-Cycloalkyl, Phenyl, Benzyl oder 5- oder 6-gliedriges Heteroaryl steht,
worin Phenyl, Benzyl und 5- oder 6-gliedriges Heteroaryl mit 1 bis 3 Substituentenn unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Nitro, (Ci-C4)-Alkyl, Trifiuormethyl, Hydroxy, (CrC4)-Alkoxy, Trifluor- methoxy und Amino substituiert sein können,
R2 für Wasserstoff, Fluor, Chlor oder (CrC4)-Alkyl steht,
R3 für Phenyl oder Naphthyl steht,
wobei Phenyl und Naphthyl mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen, Cyano, Nitro, (Ci-C4)-Alkyl, Trifluormethyl, (Ci-C4)-Alkoxy, Trifluormethoxy, Mono-(Ci-C4)-alkylamino, Di-(C i-C4)-alkylamino, Aminocarbonyl, Mono-(Ci-C4)-alkylaminocarbonyl und Di-(Ci-C4)-alkylaminocarbonyl substituiert sein können,
n für die Zahl 2 oder 3 steht,
R4A für Wasserstoff, Fluor oder (CrC4)-Alkyl steht,
R4B für Wasserstoff, Fluor oder (CrC4)-Alkyl steht,
und
Z für Hydroxy oder Cyano steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereo- isomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasser- stoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsaure, Propionsäure, Milchsäure, Weinsäure, Apfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfϊndungsgemäßen Verbin- düngen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbin- düngen. Der Begriff "Prodrugs" umfasst Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein linearer oder verzweigter Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, iso-Butyl, sec.-Butyl, tert.-Butyl, 1-Ethylpropyl, n-Pentyl und n-Hexyl.
Cvcloalkyl steht im Rahmen der Erfindung für einen monocyclischen, gesättigten Carbocyclus mit 3 bis 7 bzw. 3 bis 6 Ring-Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
Alkoxy steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein linearer oder verzweigter Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, tert.-Butoxy, n-Pentoxy und n-Hexoxy.
Mono-alkylamino steht im Rahmen der Erfindung für eine Amino-Gruppe mit einem linearen oder verzweigten Alkylsubstituenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweist. Bevorzugt ist ein linearer oder verzweigter Monoalkylamino-Rest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methylamino, Ethylamino, n-Propylamino, Isopropylamino, n- Butylamino, tert.-Butylamino, n-Pentylamino und n-Hexylamino.
Di-alkylamino steht im Rahmen der Erfindung für eine Amino-Gruppe mit zwei gleichen oder verschiedenen linearen oder verzweigten Alkylsubstituenten, die jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweisen. Bevorzugt sind lineare oder verzweigte Dialkylamino-Reste mit jeweils 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: NN- Dimethylamino, NN-Diethylamino, N-Ethyl-N-methylamino, N-Methyl-N-n-propylamino, N-Iso- propyl-N-n-propylamino, NN-Diisopropylamino, N-n-Butyl-N-methylamino, N-tert.-Butyl-N- methylamino, N-Ethyl-N-n-pentylamino und N-n-Hexyl-N-methylamino.
Mono-alkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die einen linearen oder verzweigten Alkylsubstituenten mit 1 bis 4 Kohlenstoffatomen aufweist. Beispielhaft und vorzugsweise seien genannt: Methyl- aminocarbonyl, Ethylaminocarbonyl, /j-Propylaminocarbonyl, Isopropylaminocarbonyl, n- Butylaminocarbonyl, tert.-Butylaminocarbonyl, n-Pentylaminocarbonyl und n- Hexylaminocarbonyl .
Di-alkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die zwei gleiche oder verschiedene lineare oder verzweigte Alkylsubstituenten mit jeweils 1 bis 4 Kohlenstoffatomen aufweist. Beispielhaft und vorzugsweise seien genannt: NN-Dimethylaminocarbonyl, NN-Diethylaminocarbonyl, N-Ethyl-N-methylamino- carbonyl, N-Methyl-N-n-propylaminocarbonyl, N-«-Butyl-N-methylaminocarbonyl, N-tert.-Butyl- N-methylaminocarbonyl, N-n-Pentyl-N-methylaminocarbonyl und N-n-Hexyl-N- methy laminocarbonyl .
Heteroaryl steht im Rahmen der Erfindung für einen monocyclischen aromatischen Heterocyclus (Heteroaromaten) mit insgesamt 5 oder 6 Ringatomen, der bis zu drei gleiche oder verschiedene Ring-Heteroatome aus der Reihe Ν, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls über ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Furyl,
Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl. Bevorzugt sind monocyclische 5- oder 6-gliedrige Heteroaryl-Reste mit bis zu zwei Ring-Heteroatomen aus der Reihe N, O und/oder S wie beispielsweise Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl, Isoxazolyl, Pyrazolyl, Imidazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor oder Fluor.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Er- findung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit einem oder zwei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt sind Verbindungen der Formel (I), in welcher
A für C-R5 steht,
wobei
R5 für Wasserstoff steht,
R1 für Chlor, Brom, Cyano, Nitro, (Ci-C4)-Alkyl, (C1-C4)-Alkoxy, Amino, Mono-(CrC4)- alkylamino, Di-(C i-C4)-alkylamino oder eine Gruppe der Formel -(CH2)P-NR6-Sθ2-R7 steht,
wobei (Ci-C4)-Alkyl und (Ci-C4)-Alkoxy mit 1 bis 3 Substituenten Fluor substituiert sein können,
wobei (Ci-C4)-Alkyl und (Ci-C4)-Alkoxy mit einem Substituenten ausgewählt aus der Gruppe Hydroxy und (Ci-C4)-Alkoxy substituiert sein können
und wobei
p für die Zahl 0 oder 1 steht,
R6 für Wasserstoff oder Methyl steht,
und
R7 für (CrC6)-Alkyl, (C3-C6)-Cycloalkyl, Phenyl, Benzyl oder 5- oder 6-gliedriges Heteroaryl steht,
worin Phenyl, Benzyl und 5- oder 6-gliedriges Heteroaryl mit 1 oder 2 Substituentenn unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Nitro, (CrC4)-Alkyl, Trifluormethyl, Hydroxy, (CrC4)-Alkoxy,
Trifluormethoxy und Amino substituiert sein können,
R2 für Wasserstoff, Fluor oder Methyl steht,
R3 für Phenyl oder Naphthyl steht,
wobei Phenyl und Naphthyl mit 1 oder 2 Substiruenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Brom, Cyano, Nitro, (Ci-C4)-Alkyl,
Trifluormethyl, (Ci-C4)-Alkoxy und Trifluormethoxy substituiert sein können,
n für die Zahl 2 oder 3 steht,
R4A für Wasserstoff, Fluor oder Methyl steht,
R4B für Wasserstoff, Fluor oder Methyl steht,
und
Z für Hydroxy oder Cyano steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt sind Verbindungen der Formel (I), in welcher
A für C-R5 steht,
wobei
R5 für Wasserstoff steht,
R1 für Brom, Cyano, Methyl, Ethyl, Trifluormethyl oder eine Gruppe der Formel -(CH2)P- NR6-SO2-R7 steht,
und wobei
p für die Zahl 0 steht,
R6 für Wasserstoff steht,
und
R7 für Methyl oder Ethyl steht,
R2 für Wasserstoff oder Fluorsteht,
R3 für Phenyl oder Naphthyl steht,
wobei Phenyl mit 1 oder 2 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Fluor, Chlor, Methyl oder Trifluormethyl substituiert sein kann,
n für die Zahl 2 oder 3 steht,
R4A für Wasserstoff steht,
R4B für Wasserstoff steht,
und
Z für Hydroxy oder Cyano steht,
sowie ihre Salze, Solvate und Solvate der Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Ver- bindungen der Formel (I), dadurch gekennzeichnet, dass man
[A] zunächst ein Indol-Derivat der Formel (II)
(H),
in welcher A, R
1 und R
2 die oben angegebenen Bedeutungen haben,
in einem inerten Lösungsmittel, gegebenenfalls in Gegenwart einer Säure und/oder Base, mit einem Benzaldehyd der Formel (HI)
in welcher R3 die oben angegebene Bedeutung hat,
und einem Malonsäureester der Formel (IV)
in welcher
T1 und T2 gleich oder verschieden sind und für (Ci-C4)-Alkyl stehen oder beide gemeinsam eine >C(CH3)2-Brücke bilden,
zu einer Verbindung der Formel (V)
in welcher A, R1, R2, R3, T1 und T2 jeweils die oben angegebenen Bedeutungen haben,
kondensiert, anschließend den Diester unter Decarboxylierung zu einer Verbindung der Formel (VI)
in welcher A, R1, R2 und R3 jeweils die oben angegebenen Bedeutungen haben
und
T3 für Wasserstoff oder (Q -C4)-Alkyl steht,
spaltet und diese dann in einem inerten Lösungsmittel mit einem geeigneten Reduktionsmittel, wie beispielsweise Lithiumaluminiumhydrid, in die erfindungsgemäße Verbindung der Formel (I- 1)
in welcher A, R , R und R jeweils die oben angegebenen Bedeutungen haben,
überführt,
[B] die Verbindung der Formel (I- 1) ihrerseits nach Standardmethoden über eine Verbindung der Formel (VII)
in welcher A, R , R und R jeweils die oben angegebenen Bedeutungen haben
und
X für eine geeignete Abgangsgruppe wie beispielsweise Halogen, Mesylat, Tosylat oder Triflat steht,
und nachfolgende Substitutionsreaktion mit einem Alkalicyanid zur erfϊndungsgemäßen Verbindung der Formel (1-2)
in welcher A, R > 1 , r R> 2 und Λ R D3 jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
[C] die Verbindung der Formel (1-2) ihrerseits zunächst zur Carbonsäure der Formel (VEl)
in welcher A, R , R und R jeweils die oben angegebenen Bedeutungen haben,
hydrolysiert und diese dann in einem inerten Lösungsmittel mit einem geeigneten Reduktionsmittel, wie beispielsweise Lithiumaluminiumhydrid, in die erfindungsgemäße Verbindung der Formel (1-3)
in welcher A, R > 1 , D R
2
r R>3 j :eweils die oben angegebenen Bedeutungen haben,
überführt
und
[D] die Verbindung der Formel (1-3) wiederum nach Standardmethoden über eine Verbindung der Formel (DC)
in welcher A, R1, R2 und R3 jeweils die oben angegebenen Bedeutungen haben
und
X für eine geeignete Abgangsgruppe wie beispielsweise Halogen, Mesylat, Tosylat oder Triflat steht,
und nachfolgende Substitutionsreaktion mit einem Alkalicyanid zur erfindungsgemäßen Verbindung der Formel (1-4)
in welcher A, R1, R2 und R3 jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
und gegebenenfalls die resultierenden Verbindungen der Formel (I- 1), (1-2), (1-3) bzw. (1-4) nach dem Fachmann bekannten Methoden in ihre Enantiomere und/oder Diastereomere trennt und/oder mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Weitere erfϊndungsgemäße Verbindungen können gegebenenfalls auch hergestellt werden durch Umwandlungen von funktionellen Gruppen einzelner Substituenten, insbesondere den zu R1 und R3 aufgeführten, ausgehend von den nach obigen Verfahren erhaltenen Verbindungen der Formel (I). Diese Umwandlungen werden nach üblichen, dem Fachmann bekannten Methoden durch- geführt und umfassen beispielsweise Reaktionen wie nukleophile, elektrophile oder Übergangs- metall-katalysierte Substitutionsreaktionen, Oxidation, Reduktion, Hydrierung, Alkylierung, Acylierung, Aminierung, Veresterung, Esterspaltung, Veretherung, Etherspaltung, Bildung von Carbonamiden und Sulfonamiden, sowie die Einführung und Entfernung temporärer Schutzgruppen [vgl. auch nachfolgende Syntheseschemata 2-7].
Erfindungsgemäße Verbindungen der Formel (I), worin einzelne Reste R4A und/oder R4B für Fluor oder (Ci-C4)-Alkyl stehen, können nach bekannten Methoden zur Fluorierung bzw. Alkylierung von Carbonylverbindungen ausgehend von den oben beschriebenen Verbindungen der Formeln (VI), (VHI), (1-2) oder (1-4) hergestellt werden [vgl. z.B. Z. Xu et al., J. Fluorine Chem. 58 (1), 71- 79 (1992); A. Malabarba et al., Farmaco Ed. Sei. 39 (12), 1050-1060 (1984)].
Der Verfahrensschritt (ET) + (ET) + (IV) — » (V) kann einstufig als 3 -Komponenten-Reaktion durchgeführt werden oder auch zweistufig, indem man zunächst den Benzaldehyd der Formel (DI) mit dem Malonsäureester der Formel (FV) nach Standardmethoden zu einer Benzyliden- Verbindung der Formel (X)
in welcher R3, T1 und T2 jeweils die oben angegebenen Bedeutungen haben,
kondensiert und diese dann in einem separaten Reaktionsschritt mit dem Indol der Formel (II) umsetzt.
Bei der einstufigen Reaktionsführung (IT) + (DI) + (IV) — > (V) wird als Malonester-Komponente (IV) bevorzugt Meldrum-Säure (cycl.-Isopropylidenmalonat) verwendet. Das hierbei resultierende Produkt der Formel (Va)
in welcher A, R1, R2 und R3 jeweils die oben angegebenen Bedeutungen haben,
wird anschließend durch Solvolyse mit Methanol oder Ethanol in Gegenwart von Pyridin und Kupfer-Pulver in einen Ester der Formel (VI) [T3 = Methyl bzw. Ethyl] überfuhrt [vgl. Y. Oikawa et al., Tetrahedron Lett., 1759-1762 (1978)].
Die einstufige Verfahrensvariante (IT) + (HI) + (IV) — > (V) sowie - bei zweistufiger Reaktionsführung - die Kondensation (IH) + (IV) — » (X) werden bevorzugt in Gegenwart eines Säure/Base- Katalysators, wie beispielsweise D,L-Prolin oder Piperidiniumacetat, durchgeführt. Die Umsetzung (X) + (E) — > (V) kann gegebenenfalls vorteilhaft mit Hilfe einer Aminbase wie Triethylamin oder einer Lewis-Säure wie Kupfer(II)- oder Ytterbium-trifluormethansulfonat erfolgen.
Als Lösungsmittel für die Verfahrensschritte (II) + (Iu) + (IV) → (V) und (X) + (II) → (V) eignen sich alle organischen Lösungsmittel, die unter den Reaktionsbedingungen inert sind. Hierzu gehören acyclische und cyclische Ether wie Diethylether, Methyl-tert.-butylether, 1 ,2-Dimethoxy- ethan, Tetrahydrofuran und Dioxan, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan und Cyclohexan, chlorierte Kohlenwasserstoffe wie Dichlormethan, Trichlormethan und Chlorbenzol, oder dipolar-aprotische Lösungsmittel wie Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), N-Methylpyrrolidinon (ΝMP) und Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Acetonitril verwendet.
Die Reaktionen erfolgen im Allgemeinen in einem Temperaturbereich von 00C bis +1200C, bevor- zugt bei 00C bis +600C. Die Umsetzungen können bei normalem, erhöhtem oder erniedrigtem Druck durchgeführt werden (z.B. im Bereich von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Als Reduktionsmittel in den Verfahrensschritten (VI) — > (I- 1) und (VHI) → (1-3) ist insbesondere
Lithiumaluminiumhydrid oder Lithiumborhydrid geeignet. Im Falle der Carbonsäuren (VIE) und (VI) [T3 = H] können alternativ auch Diboran oder Boran-Komplexe verwendet werden. Die
Umsetzungen werden bevorzugt in einem Ether wie Diethylether oder Tetrahydrofuran als inertem Lösungsmittel in einem Temperaturbereich von 00C bis +800C durchgeführt.
Für die Verfahrensschritte (VII) — » (1-2) und (EX) — > (1-4) eignen sich insbesondere Ether, wie Diethylether, Methyl-terf.-butylether, 1 ,2-Dimethoxyethan, Tetrahydrofuran und Dioxan, oder dipolar-aprotische Lösungsmittel, wie Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), N- Methylpyrrolidinon (ΝMP) und Acetonitril, als inerte Lösungsmittel. Ebenso ist es möglich, Gemische dieser Lösungsmittel einzusetzen. Bevorzugt wird Dimethylformamid verwendet. Die Reaktionen erfolgen im Allgemeinen in einem Temperaturbereich von +200C bis +1500C, bevorzugt bei +400C bis +1000C.
Die Hydrolyse der Νitrile (1-2) zu den Carbonsäuren (Vm) wird bevorzugt mit wässrigen Lösungen von Alkali- oder Erdalkali-Hydroxiden wie Lithium-, Natrium-, Kalium-, Calcium- oder Bariumhydroxid durchgeführt. Als Kosolventien eignen sich Alkohole wie Methanol, Ethanol, n- Propanol, Isopropanol, n-Butanol oder tert. -Butanol, Ether wie Diethylether, Tetrahydrofuran, Dioxan oder 1 ,2-Dimethoxyethan, andere Lösungsmittel wie Aceton, Dimethylformamid (DMF) oder Dimethylsulfoxid (DMSO), oder Gemische dieser Lösungsmittel. Die Hydrolyse erfolgt im Allgemeinen in einem Temperaturbereich von +500C bis +1500C, bevorzugt bei +600C bis +1000C.
Die Verbindungen der Formeln (II), (HI) und (IV) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch die folgenden Syntheseschemata veranschaulicht werden:
Schema 1
[a): cat. D,L-Prolin, Acetonitril, RT; b): cat. Cu-Pulver, EtOH/Pyridin, Rückfluss; c): LiAlH4, Et2O, O0C → RT; d): MsCl, Et3N, DMAP, CH2Cl2, RT; e): KCN, DMF, 800C; f): wässr. KOH, EtOH, 800C; g): LiAlH4, THF, 600C; h): MsCl, Et3N, DMAP, CH2Cl2, RT; i): KCN, DMF, 800C].
Schema 2
[a): Zn(CN)2, cat. Pd(PPh3)4, DMF, Mikrowelle, 2000C].
Schema 3
[a): H2, Pd/C, EtOH/THF, RT, Normaldruck; b): LiAlH4, THF, 600C; c): MsCl, Pyridin, THF, RT; d): MsCl, NEt3, DMAP, CH2Cl2, RT; e): KCN, DMF, 800C].
Schema 4
[a): SOj Pyridin, Et3N, DMSO/CH2C12, RT; b): KCN, /J-Bu3BnN+Cl", H2O/EtOAc, RT; c): DAST, CH2Cl2, RT].
Schema 5
[a): KOH, EtOH, Rückfluss; b): Kupferchromoxid, Chinolin, 205°C; c): 2,2-Dimethyl-5-[4-(tri- fluormethyl)benzyliden]-l,3-dioxan-4,6-dion, Acetonitril, Rückfluss; d): SOCl2, Et2O, RT; EtOH, RT; e): LiBH4, THF, RT; f): H2, Pd/C, RT; MsCl, Et3N, DMAP, CH2Cl2, RT; KCN, DMF, 800C].
Die erfindungsgemäßen Verbindungen sind potente und selektive Antagonisten des Mineralokorti- coid-Rezeptors und zeigen ein nicht vorhersehbares, wertvolles pharmakologisches Wirkspektrum. Sie eignen sich daher zur Verwendung als Arzneimittel zur Behandlung und/oder Prophylaxe von Krankheiten bei Menschen und Tieren.
Die erfϊndungsgemäßen Verbindungen sind geeignet für die Prophylaxe und/oder Behandlung von verschiedenen Erkrankungen und krankheitsbedingten Zuständen, insbesondere von Erkrankungen, die entweder durch eine Erhöhung der Aldosteron-Konzentration im Plasma oder durch eine Veränderung der Aldosteron-Plasmakonzentration relativ zur Renin-Plasmakonzentration gekennzeichnet sind oder mit diesen Veränderungen einhergehen. Beispielsweise seien genannt: idio- pathischer primärer Hyperaldosteronismus, Hyperaldosteronismus bei Nebennierenhyperplasie, Nebennierenadenomen und/oder Nebennierencarzinomen, Hyperaldosteronismus bei Leberzirrho-
se, Hyperaldosteronismus bei Herzinsuffizienz sowie (relativer) Hyperaldosteronismus bei essentieller Hypertonie.
Die erfindungsgemäßen Verbindungen sind aufgrund ihres Wirkmechanismus ferner geeignet für die Prophylaxe des plötzlichen Herztodes bei Patienten, die unter einem erhöhten Risiko stehen, an einem plötzlichen Herztod zu versterben. Dies sind insbesondere Patienten, die z.B. an einer der folgenden Erkrankungen leiden: primäre und sekundäre Hypertonie, hypertensive Herzkrankheit mit oder ohne kongestive Herzinsuffizienz, therapieresistente Hypertonie, akute und chronische Herzinsuffizienz, koronare Herzerkrankung, stabile und instabile Angina pectoris, myokardiale Ischämie, Myokardinfarkt, dilatative Kardiomyopathien, angeborene primäre Kardiomyopathien wie z.B. Brugada-Syndrom, durch die Chagas-Erkrankung hervorgerufene Kardiomyopathien, Schock, Arteriosklerose, atriale und ventrikuläre Arrhythmie, transitorische und ischämische Attacken, Hirnschlag, entzündliche kardiovaskuläre Erkrankungen, periphere und kardiale Gefäßerkrankungen, periphere Durchblutungsstörungen, arterielle Verschlusskrankheiten wie Claudi- catio intermittens, asymptomatische linksventrikuläre Dysfunktion, Myokarditis, hypertrophe Ver- änderungen des Herzens, pulmonale Hypertonie, Spasmen der Koronararterien und peripherer Arterien, Thrombosen, thromboembolische Erkrankungen sowie Vaskulitis.
Die erfindungsgemäßen Verbindungen können ferner verwendet werden für die Prophylaxe und/ oder Behandlung von Ödembildung, wie zum Beispiel pulmonales Ödem, renales Ödem oder Herzinsuffizienz-bedingtes Ödem, und von Restenosen, wie nach Thrombolysetherapien, percutan- transluminalen Angioplastien (PTA) und Koronarangioplastien (PTCA), Herztransplantationen sowie Bypass-Operationen.
Außerdem können die erfindungsgemäßen Verbindungen eingesetzt werden für die Prophylaxe und/oder Behandlung von erektiler Dysfunktion.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Verwendung als kaliumsparendes Diuretikum und bei Elektrolytstörungen wie zum Beispiel Hyperkalzämie, Hypernatriämie oder Hypokaliämie, einschliesslich genetisch bedingter Formen wie das Gitelman oder Barrter Syndrom.
Die erfindungsgemäßen Verbindungen eignen sich ebenso zur Behandlung von Nierenerkrankungen, wie akutem und chronischem Nierenversagen, hypertensiver Nierenkrankheit, arteriosklero- tischer Nephritis (chronisch und interstitiell), Nephrosklerose, chronischer Niereninsuffizienz und zystischen Nierenerkrankungen, zur Verhinderung von Nierenschäden, die zum Beispiel durch Immunsuppressiva wie Cyclosporin A bei Organtransplantationen hervorgerufen werden können, sowie bei Nierenkrebs.
Außerdem können die erfϊndungsgemäßen Verbindungen eingesetzt werden für die Prophylaxe und/oder Behandlung von Diabetes mellitus und diabetischen Folgeerkrankungen wie z.B. Neuropathie, Nephropathie und Cardiomyopathie.
Weiterhin können die erfindungsgemäßen Verbindungen für die Prophylaxe und/oder Behandlung von Augenerkrankungen, insbesondere auf Angiogenese und Neovaskularisierung beruhende Formen wie z.B. Neugeborenenretinopathie, diabetischer Retinopathie, sowie altersbedingter Maculadegeneration und Glaukom eingesetzt werden.
Die erfϊndungsgemäßen Verbindungen können ferner verwendet werden für die Prophylaxe und/ oder Behandlung von Mikroalbuminurie, zum Beispiel bedingt durch Diabetes mellitus oder Blut- hochdruck, sowie der Proteinurie.
Die erfindungsgemäßen Verbindungen eignen sich auch für die Prophylaxe und/oder Behandlung von Erkrankungen, die entweder mit einer Erhöhung der Glukokortikoid-Konzentration im Plasma oder mit einer lokalen Konzentrationserhöhung von Glukokortikoiden im Gewebe (z.B. des Herzens) einhergehen. Beispielsweise seien genannt: Funktionsstörungen der Nebenniere, die zur Überproduktion von Glukokortikoiden führen (Cushing-Syndrom), Nebennierenrindentumore mit resultierender Überproduktion von Glukokortikoiden sowie Hypophysentumore, die autonom ACTH (adrenokortikotropes Hormon) produzieren und dadurch zu Nebennierenhyperplasien mit resultierendem Morbus Cushing führen.
Außerdem können die erfindungsgemäßen Verbindungen für die Prophylaxe und/oder Behandlung von Obesitas, des metabolischen Syndroms und der obstruktiven Schlaf-Apnoe eingesetzt werden.
Die erfindungsgemäßen Verbindungen können ferner verwendet werden für die Prophylaxe und/ oder Behandlung von entzündlichen Erkrankungen, die z.B. durch Viren, Spirochäten, Pilze, Bakterien oder Mykobakterien hervorgerufen werden, sowie von entzündlichen Erkrankungen unbekannter Ätiologie, wie der Polyarthritis, dem Lupus erythematodes, der Peri- oder Polyarteriitis, der Dermatomyositis, der Sklerodermie und der Sarkoidose.
Weiterhin können die erfindungsgemäßen Verbindungen eingesetzt werden für die Behandlung von zentralnervösen Erkrankungen wie Depressionen, Angstzuständen und chronischen Schmerzen, insbesondere Migräne, sowie bei neurodegenerativen Erkrankungen wie der Alzheimer'schen Krankheit und dem Parkinson-Syndrom.
Die erfindungsgemäßen Verbindungen sind auch geeignet für die Prophylaxe und/oder Behandlung von vaskulären Schäden, z.B. nach Interventionen wie percutan-transluminaler koronarer Angioplastie (PTCA), Implantationen von Stents, koronarer Angioskopie, Reokklusion oder
Restenose nach Bypass-Operationen, sowie bei endothelialer Dysfunktion, bei Morbus Raynaud, bei der Thrombangiitis obliterans (Buerger-Syndrom) und beim Tinnitus-Syndrom.
Die erfindungsgemäßen Verbindungen eignen sich auch für die Prophylaxe und/oder Behandlung von gynaekologischen Erkrankungen wie Endometriose, Leiomyomen des Uterus, dysfunktionelle Blutungen und der Dysmenorrhoe.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Ver- bindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Weiterer Gegenstand der vorliegenden Erfindung sind die erfindungsgemäßen Verbindungen zur Verwendung in einem Verfahren zur Behandlung und/oder Prophylaxe von Aldosteronismus, Bluthochdruck, akuter und chronischer Herzinsuffizienz, den Folgen eines Myokardinfarkts, Leberzirrhose, Niereninsuffizienz und Hirnschlag.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prävention der zuvor genannten Erkrankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der CaI- cium-Antagonisten, Angiotensin AH-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten,
Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker und Rho-Kinase-Inhibi- toren;
• Diuretika, insbesondere Schleifendiuretika sowie Thiazide und Thiazid-ähnliche Diuretika;
• antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Thrombo- zytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen;
• den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, CETP- Inhibitoren, MTP-Inhibitoren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Absorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Gallen- säure-Reabsoφtionshemmer und Lipoprotein(a)-Antagonisten;
• organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-I, sowie inhalatives NO;
• positiv-inotrop wirksame Verbindungen, wie beispielsweise Herzglycoside (Digoxin), beta- adrenerge und dopaminerge Agonisten wie Isoproterenol, Adrenalin, Noradrenalin, Dopamin und Dobutamin;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) und/oder cyclischem Adenosinmonophosphat (cAMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2, 3, 4 und/oder 5, insbesondere PDE 5-Inhibitoren wie Sildenafil, Vardenafil und Tadalafil, sowie PDE 3 -Inhibitoren wie Amrinone und Milrinone;
• natriuretische Peptide, wie z.B. "atrial natriuretic peptide" (ANP, Anaritide), "B-type natriuretic peptide" oder "brain natriuretic peptide" (BNP, Nesiritide), "C-type natriuretic peptide" (CNP) sowie Urodilatin;
• Calcium-Sensitizer, wie beispielhaft und vorzugsweise Levosimendan;
• NO- und Häm-unabhängige Aktivatoren der Guanylatcyclase, wie insbesondere Cinaciguat sowie die in WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 und WO 02/070510 beschriebenen Verbindungen;
• NO-unabhängige, jedoch Häm-abhängige Stimulatoren der Guanylatcyclase, wie insbesondere Riociguat sowie die in WO 00/06568, WO 00/06569, WO 02/42301 und WO 03/095451 beschriebenen Verbindungen;
• Modulatoren der Adenosin Rezeptoren, inbesondere Adenosin Al Antagonisten, wie KW- 3902, SLV-320 oder BG-9928 (Adentri);
• Vasopressin Rezeptor Antagonisten, wie beispielsweise Conivaptan (Vaprisol), Tolvaptan, Satavaptan, Lixivaptan, Relcovaptan, RWJ-339489 oder RWJ-351647.
• Inhibitoren der humanen neutrophilen Elastase (HNE), wie beispielsweise Sivelestat oder DX- 890 (Reltran);
• die Signaltransduktionskaskade inhibierende Verbindungen, wie beispielsweise Tyrosinkinase- Inhibitoren, insbesondere Sorafenib, Imatinib, Gefϊtinib und Erlotinib; und/oder
• den Energiestoffwechsel des Herzens beeinflussende Verbindungen, wie beispielhaft und vorzugsweise Etomoxir, Dichloracetat, Ranolazine oder Trimetazidine.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, Bumetanid, Torsemid, Bendroflumethiazid, Chlorthiazid, Hydrochlorthiazid, Hydroflumetbiazid, Methyclothiazid, Polythiazid, Trichlormethiazid, Chlorthalidon, Indapamid, Metolazon, Quineth- azon, Acetazolamid, Dichlorphenamid, Methazolamid, Glycerin, Isosorbid, Mannitol, Amilorid oder Triamteren, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin Aü-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-mhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Rho-Kinase-Inhibitoren sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin AH-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Candesartan, Valsartan, Telmisartan oder Embusartan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600, SPP-635, SPP-676, SPP-800 oder SPP-1148, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem alpha- 1 -Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Meti- pranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucin- dolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Rho-Kinase-Inhibitor, wie beispielhaft und vorzugsweise Fasu- dil, Y-27632, SLx-2119, BF-66851, BF-66852, BF-66853, KI-23095 oder BA-1049, verabreicht.
Unter antithrombotisch wirkenden Mitteln (Antithrombotika) werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrino- lytischen Substanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPÜb/ÜIa-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban (BAY 59-7939), DU-176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux,
Idraparinux, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibi- toren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Absorptions- hemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, Lipase-Inhibitoren sowie der Lipoprotein(a)-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Dalcetrapib, BAY 60-5521, Anacetrapib oder CETP-vaccine (CETi-I), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem PPAR-gamma-Agonisten, wie beispielhaft und vorzugsweise Pioglitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-delta-Agonisten, wie beispielhaft und vorzugsweise GW- 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin-Absorptionshemmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI-1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugs- weise Gemcabene calcium (CI- 1027) oder Nicotinsäure, verabreicht.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszuberei- tungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale und die intravenöse Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy-
ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körper- gewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindest- menge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Abkürzungen und Akronyme:
Ac Acetyl
Bn Benzyl
Bu Butyl cat. katalytisch
CI chemische Ionisation (bei MS)
DAST Diethylaminoschwefeltrifluorid
DMAP 4-N,N-Dimethylaminopyridin
DMF Dimethylformamid
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute)
EI Elektronenstoß-Ionisation (bei MS) eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
EtOAc Ethylacetat ges. gesättigt h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie konz. konzentriert
LC-MS Flüssigchromatographie-gekoppelte Massenspektrometrie
Me Methyl min Minute(n)
Ms Methansulfonyl (Mesyl)
MS Massenspektrometrie
NMR Kernresonanzspektrometrie
Pd/C Palladium auf Aktivkohle
Ph Phenyl
RT Raumtemperatur
Rt Retentionszeit (bei HPLC) THF Tetrahydrofuran
UV Ultraviolett-Spektrometrie v/v Volumen zu Volumen-Verhieltnis (einer Lösung)
wässr. wässrig, wässrige Lösung
LC-MS- und HPLC-Methoden:
Methode 1 CLC-MSV Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 2 (LC-MS): Gerätetyp MS: Waters ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith RP18e, 100 mm x 3 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2 min 65% A → 4.5 min 5% A → 6 min 5% A; Fluss: 2 ml/min; Ofen: 400C; UV-Detektion: 210 nm.
Methode 3 (LC-MS): Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Onyx Monolithic C 18, 100 mm x 3 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2 min 65% A → 4.5 min 5% A → 6 min 5% A; Fluss: 2 ml/min; Ofen: 4O0C; UV-Detektion: 208-400 nm.
Methode 4 (HPLC): Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml HClO4 (70%-ig) / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 9 min 90% B → 9.2 min 2% B -> 10 min 2% B; Fluss: 0.75 ml/min; Säulentemperatur: 300C; UV-Detektion: 210 nm.
Methode 5 (HPLC): Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml HClO4 (70%-ig) / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 6.5 min 90% B → 6.7 min 2% B → 7.5 min 2% B; Fluss: 0.75 ml/min; Säulentemperatur: 300C; UV-Detektion: 210 nm.
Methode 6 (LC-MS): Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A — » 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 7 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ MAX-RP 100A Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser +
0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient:
0.0 min 90% A → 0.1 min 90% A → 3.0 min 5% A → 4.0 min 5% A → 4.01 min 90% A; Fluss: 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 8 (LC-MS): Instrument: Micromass QuattroPremier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD 1.9μ 50 mm x 1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisen- säure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 0.1 min 90% A → 1.5 min 10% A → 2.2 min 10% A; Fluss: 0.33 ml/min; Ofen: 5O0C; UV-Detektion: 210 nm.
Methode 9 (LC-MS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Ausgangsverbindungen und Intermediate:
Beispiel IA
5-{(7-Ethyl-lH-indol-3-yl)[4-(trifluormethyl)phenyl]methyl}-2,2-dimethyl-l,3-dioxan-4,6-dion
Zu einer Lösung von 40.0 g 7-Ethylindol (275 mmol), 39.7 g Meldrum-Säure (275 mmol) und 48.0 g 4-Trifluormethylbenzaldehyd (275 mmol) in 400 ml Acetonitril wurden 1.6 g D,L-Prolin (14 mmol) gegeben. Die Mischung wurde über Nacht bei RT gerührt. Der ausgefallene Feststoff wurde danach abgesaugt, mit Acetonitril gewaschen und im Hochvakuum getrocknet. Man erhielt 115 g (94% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, DMSOd6): δ = 1.27 (t, 3H), 1.61 (s, 3H), 1.85 (s, 3H), 2.85 (q, 2H), 5.36 (d, IH), 5.46 (br. s, IH), 6.82-6.88 (m, 2H), 6.91 (d, IH), 7.05 (d, IH), 7.14 (m,., IH), 7.52 (d, 2H), 7.61 (d, 2H), 11.04 (s, IH).
LC-MS (Methode 1): Rt = 2.74 min; MS (ESIneg): m/z = 444.3 [M-H]".
Beispiel 2A
3-(7-Ethyl-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propionsäureethylester
Zu 25.0 g der Verbindung aus Beispiel IA (56.1 mmol) in 100 ml Pyridin und 20 ml Ethanol wurden 0.18 g Kupferpulver (-150 mesh, 2.1 mmol) gegeben. Das Gemisch wurde über Nacht unter Rückfluss erhitzt. Das Lösungsmittel wurde danach bei vermindertem Druck entfernt und der Rückstand über eine Kieselgelsäule chromatographisch gereinigte (Laufmittel: Cyclohexan/Ethylacetat 100:1 → 3:1). Es wurden 20.5 g (94% d. Th.) der Zielverbindung erhalten.
1H-NMR (200 MHz, DMSO-(I6): δ = 1.07 (t, 3H), 1.25 (t, 3H), 2.85 (q, 2H), 3.14 + 3.24 (AB- Signal, zusätzlich zum d aufgespalten, 2H), 4.00 (q, 2H), 4.75 (t, IH), 6.80-6.91 (m, 2H), 7.23 (dd, IH), 7.38 (d, IH), 7.61 (s, 4H), 11.0 (s, IH).
Beispiel 3A
3-(7-Ethyl-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propyhnethansulfonat
Zu 500 mg der Verbindung aus Beispiel 1 in 5 ml Dichlormethan wurden 0.341 ml Triethylamin (247 mg, 2.45 mmol) und 18 mg 4-N(N-Dimethylaminopyridin (0.14 mmol) gegeben. Man ließ 5 min bei RT rühren und gab dann 0.223 ml Methansulfonsäurechlorid (329 mg, 2.88 mmol) hinzu. Die Reaktionsmischung wurde über Nacht bei RT gerührt und dann mit 15 ml Essigsäureethyl- ester versetzt. Nach Extraktion mit je 20 ml 1 N Salzsäure, Wasser und ges. wässr. Natrium-
chlorid-Lösung wurde die organische Phase über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde ohne weitere Reinigung weiter eingesetzt.
LC-MS (Methode 2): R, = 3.95 min; MS (ESIpos): m/z = 426.2 [M+H]+.
Beispiel 4A
4-(7-Ethyl-lH-indol-3-yl)-4-(4-trifluormethylphenyl)buttersäure
958 mg der Verbindung aus Beispiel 2 (2.69 mmol) wurden in 9 ml Ethanol vorgelegt. Es wurden 603 mg Kaliumhydroxid in 4.5 ml Wasser zugegeben und die Mischung 6 h bei 800C gerührt. Der Ansatz wurde danach abgekühlt, auf Eiswasser gegossen und mit 1 N Salzsäure auf pH 3 eingestellt. Es wurde zweimal mit 20 ml Essigsäureethylester extrahiert. Die vereinigteen organischen Phasen wurden mit 20 ml ges. wässr. Natriumchlorid-Lösung extrahiert, über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde über eine Kieselgelfritte gereinigte (Laufmittel: Cyclohexan/Essigsäureethylester 1 : 1). Es wurden 927 mg (90% d. Th.) der Zielverbindung erhalten.
1H-NMR (400 MHz, DMSOd6): δ = 1.23 (t, 3H), 2.10-2.30 (m, 3H), 2.35-2.46 (m, IH), 4.03 (q, 2H), 4.26 (t, IH), 6.79-6.88 (m, 2H), 7.19 (d, IH), 7.32 (d, IH), 7.54 (d, 2H), 7.62 (d, 2H), 10.93 (s, IH), 12.07 (s, IH).
MS (CIpos): m/z = 393.0 [M+NH4]"1".
Beispiel 5A
3-(7-Nitro-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propionsäureethylester
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Nitroindol analog zur Synthese der Verbindung aus Beispiel 2A.
1H-NMR (400 MHz, DMSO-d6): δ = 1.03 (t, 3H), 3.18 + 3.25 (AB-Signal, zusätzlich zum d auf- gespalten, 2H), 3.96 (q, 2H), 4.85 (t, IH), 7.16 (dd, IH), 7.60-7.66 (m, 5H), 7.97 (d, IH), 8.06 (d, IH), 11.85 (s, IH).
HPLC (Methode 4): R, = 5.20 min; MS (ESIneg): m/z = 405.2 [M-H]".
Beispiel 6A
3-(7-Amino-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propionsäureethylester
3.66 g der Verbindung aus Beispiel 5A (9.01 mmol) in 30 ml Ethanol und 60 ml TΗF wurden mit 400 mg Palladium auf Kohle versetzt und über Nacht unter Normaldruck bei RT hydriert. Es wurde über Celite filtriert, mit Ethanol nachgewaschen und das Filtrat bei vermindertem Druck eingeengt. Der Rückstand wurde über eine Kieselgelsäule chromatographisch gereinigte (Laufmittel: Dichlormethan → Dichlormethan/Methanol 100:1). Es wurden 3.32 g (98% d. Th.) der Zielverbindung erhalten.
1H-NMR (400 MHz, DMSO-(I6): δ = 1.05 (t, 3H), 3.08 + 3.16 (AB-Signal, zusätzlich zum d aufgespalten, 2H), 3.96 (q, 2H), 4.65 (t, IH), 4.96 (s, 2H), 6.26 (dd, IH), 6.58-6.65 (m, 2H), 7.26 (d, IH), 7.54 (d, 2H), 7.59 (d, 2H), 10.51 (s, IH).
HPLC (Methode 5): R, = 4.28 min.
Beispiel 7A
3-[4-(Trifluormethyl)phenyl]-3-{7-[(methylsulfonyl)amino]-lH-indol-3-yl}propylmethansulfonat
Zu 45.0 mg der Verbindung aus Beispiel 16 (0.109 mmol) in 0.5 ml Dichlormethan wurden 1.3 mg 4-NN- Dimethylaminopyridin (0.011 mmol) und 26 μl Triethylamin (19 mg, 0.19 mmol) gegeben. Man ließ 5 min rühren und gab dann 13 μl Methansulfonsäurechlorid (19 mg, 0.16 mmol) hinzu. Nach Rühren über Nacht bei RT wurden 5 ml Essigsäureethylester und 5 ml Wasser zugesetzt. Die organische Phase wurde mit je 5 ml 1 N Salzsäure, Wasser und ges. wässr. Natriumchlorid-Lösung extrahiert. Die organische Phase wurde über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde über eine Kieselgelsäule chromatogra- phisch gereinigte (Laufmittel: Dichlormethan/Methanol 100:1). Es wurden 44.9 mg (76% d. Th.) der Zielverbindung erhalten.
LC-MS (Methode 3): R, = 3.58 min; MS (ESIpos): m/z = 491.2 [M+Η]+.
Beispiel 8A
N-(3-{3-Oxo-l-[4-(trifluormethyl)phenyl]propyl}-lH-indol-7-yl)methansulfonamid
2.80 g Schwefeltrioxid-Pyridin-Komplex (17.6 mmol) wurden bei 00C in 12 ml DMSO/Dichlor- methan (1 : 1) gelöst. Nach 15 min Rühren wurden 1.45 g der Verbindung aus Beispiel 16 (3.52 mmol) und 5.9 ml Triethylamin (4.27 g, 42.2 mmol) hinzu gegeben, die Lösung innerhalb von 30 min auf RT aufgewärmt und 1 h bei RT nachgerührt. Anschließend wurden jeweils 20 ml
Dichlormethan und Wasser zugesetzt und die Phasen getrennt. Die organische Phase wurde zweimal mit Wasser gewaschen, über Natriumsulfat getrocknet und bei vermindertem Druck vom
Lösungsmittel befreit. Der Rückstand wurde mittels präparativer HPLC gereinigte (Eluent:
Acetonitril/Wasser, Gradient 30:70 → 98:2). Man erhielt 0.67 g (87% Reinheit, 40% d. Th.) der Zielverbindung.
LC-MS (Methode 7): R, = 1.96 min; MS (ESIneg): m/z = 409.3 [M-H]-.
Beispiel 9A
N- 1 H-Indol-7-ylmethansulfonamid
4.34 g (32.8 mmol) 7-Amino-lH-indol wurden in 120 ml Dichlormethan vorgelegt, 3.76 g (32.8 mmol) Methansulfonsäurechlorid und 2.60 g (32.8 mmol) Pyridin hinzu gegeben und der Ansatz drei Tage bei RT gerührt. Nach Einengen auf ein Drittel des Volumens wurde Essigsäureethylester hinzu gegeben und nacheinander mit 1 M Salzsäure, Wasser und gesättigter Natriumchlorid- Lösung gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet, filtriert und
eingeengt. Man reinigte den Rückstand mittels Flash-Chromatographie (Laufmittel: Toluol/Essigsäureethylester 9:1) und erhielt 5.63 g (82% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-Cl6): δ = 2.97 (s, 3H), 6.46 (t, IH), 6.98 (t, IH), 7.06 (d, IH), 7.36 (t, IH), 7.41 (d, IH), 9.31 (s, IH), 10.8 (s, IH).
LC-MS (Methode 8): R, = 0.81 min; MS (ESIpos): m/z = 211 [M+H]+.
Beispiel IQA
N-(3-{(2,2-Dimethyl-4,6-dioxo-l,3-dioxan-5-yl)[2-fluor-4-(trifluormethyl)phenyl]methyl}-lH- indol-7-yl)methansulfonamid
Die Herstellung der Titelverbindung erfolgte ausgehend von 1.00 g (4.76 mmol) der Verbindung aus Beispiel 9A analog zur Synthese der Verbindung aus Beispiel IA. Man reinigte das Rohprodukt zunächst mittels Flash-Chromatographie an Kieselgel (Laufmittel: Toluol/Essigsäureethylester-Gradient) und anschließend mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% Ameisensäure). Man erhielt 1.59 g (63% d. Th.) der Titelverbindung.
LC-MS (Methode 8): R, = 1.23 min; MS (ESIneg): m/z = 527 [M-H]".
Die in der folgenden Tabelle aufgeführten Verbindungen wurden analog zur Synthese der Verbindung aus Beispiel 10A hergestellt. Abweichend wurde bei der Reaktion zu Beispiel 16A zunächst 8 h bei 60 0C und anschließend zwei Tage bei RT gerührt:
(s,
Beispiel 17A
3-[2-Fluor-4-(trifluormethyl)phenyl]-3-{7-[(methylsulfonyl)amino]-lH-indol-3- yljpropansäureethylester
Zu 1.59 g (3.01 mmol) der Verbindung aus Beispiel 1OA in 21 ml Pyridin und 5.4 ml Ethanol wurden 2 mg (0.03 mmol) Kupferpulver gegeben. Das Gemisch wurde 1 h unter Rückfluss erhitzt. Das Lösungsmittel wurde danach bei vermindertem Druck entfernt, der Rückstand in Ethylacetat aufgenommen, mit 1 M Salzsäure gewaschen und die org. Phase über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wurde über eine Kieselgelsäule chromatographisch gereinigte (Laufmittel: Cyclohexan/Ethylacetat-Gradient). Es wurden 1.00 g (70% d. Th.) der Zielverbindung erhalten.
1H-NMR (400 MHz, DMSO-d6): δ = 1.04 (t, 3H), 2.96 (s, 3H), 3.22 (d, 2H), 3.97 (q, 2H), 4.98 (t, IH), 6.94 (t, IH), 7.04 (d, IH), 7.27 (d, IH), 7.38 (d, IH), 7.49 (d, IH), 7.59-7.68 (m, 2H), 9.30 (s, IH), 10.8 (s, IH).
LC-MS (Methode 8): Rt = 1.33 min; MS (ESIpos): m/z = 473 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen wurden analog zur Synthese der Verbindung aus Beispiel 17A hergestellt. Abweichend konnte bei der Reaktion auch 2 h unter Rückfluss erhitzt werden. Das Reaktionsgemisch konnte alternativ auch ohne weitere Aufarbeitung direkt mittels Flash-Chromatographie (Laufmittel: Cyclohexan/Essigsäureethylester-Gradient) und anschließender präparativer HPLC (RPl 8 -Säule; Laufmittel: Acetonitril/Wasser-Gradient) gereinigt werden.
Beispiel 24A
5 -Fluor-7-nitro- 1 H-indol-2-carbonsäure
1.85 g (6.15 mmol) 5-Fluor-7-nitro-lH-indol-2-carbonsäureethylester wurden in 20 ml Ethanol vorgelegt, mit 517 mg (9.22 mmol) Kaliumhydroxid versetzt und über Nacht unter Rückfluss erhitzt. Anschließend wurde Essigsäureethylester hinzugesetzt, mit 1 M Natronlauge extrahiert und der pH-Wert der wässrigen Phase mit Salzsäure auf pH 2 eingestellt. Es wurde mehrmals mit Essigsäureethylester extrahiert, die organische Phase wurde mit gesättigter Natriumchlorid-Lösung gewaschen und über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man erhielt 1.35 g (98% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSCVd6): δ = 7.37 (d, IH), 8.12 (dd, IH), 8.16 (dd, IH), 11.3 (s, IH), 13.7 (s, IH).
Beispiel 25A
5-Fluor-7-nitro-lH-indol
1.35 g (6.02 mmol) der Verbindung aus Beispiel 24A wurden in 13.5 ml Chinolin vorgelegt, mit 349 mg (1.51 mmol) Kupferchromoxid versetzt und 2 h bei 2050C gerührt. Nach dem Abkühlen wurde Essigsäureethylester hinzugesetzt und mit 1 M Salzsäure extrahiert. Die organische Phase wurde mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man reinigte den Rückstand mittels Flash-Chromatographie (Laufmittel: Cyclohexan/Dichlormethan 2:1) und erhielt 975 mg (90% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-dβ): δ = 6.74 (d, IH), 7.63 (d, IH), 7.92-8.01 (m, 2H), 12.0 (s, IH).
Beispiel 26A
3-(5 -Fluor-7-nitro- 1 H-indol-3 -yl)-3 -[4-(trifluormethyl)phenyl]propansäure
1.02 g (5.66 mmol) der Verbindung aus Beispiel 25 A wurden in 20 ml Acetonitril vorgelegt, mit 5.10 g (17.0 mmol) 2,2-Dimethyl-5-[4-(trifluormethyl)benzyliden]-l,3-dioxan-4,6-dion versetzt und zwei Tage unter Rückfluss erhitzt. Man engte danach ein und reinigte den Rückstand zunächst mittels Flash-Chromatographie (Laufmittel: Dichlormethan/Methanol 20:1) und dann mittels prä- parativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 1% Ameisensäure). Es wurden 1.33 g (54% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSO-Cl6): δ = 3.09 (dd, IH), 3.20 (dd, IH), 4.81 (t, IH), 7.60-7.71 (m, 5H), 7.89-7.95 (m, 2H), 11.9 (s, IH), 12.2 (s, IH).
LC-MS (Methode 9): R, = 2.65 min; MS (ESIpos): m/z = 397 [M+H]+.
Beispiel 27A
3-(5-Fluor-7-nitro-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propansäureethylester
1.07 g (2.70 mmol) der Verbindung aus Beispiel 26A wurden in 20 ml Diethylether vorgelegt, mit 842 mg (4.05 mmol) Thionylchlorid versetzt und 1 h bei RT gerührt. Anschließend wurden 10 ml Ethanol hinzu gegeben und das Reaktionsgemisch über Nacht bei RT gerührt. Der Ansatz wurde dann auf Wasser gegossen und mit Essigsäureethylester extrahiert. Die organische Phase wurde mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und
eingeengt. Man reinigte den Rückstand mittels Flash-Chromatographie (Laufmittel: Dichlormethan/ Cyclohexan 2: 1) und erhielt 560 mg (49% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-Cl6): δ = 1.03 (t, 3H), 3.20 (dd, IH), 3.29 (dd, IH), 3.96 (q, 2H), 4.84 (t, IH), 7.60-7.63 (m, 2H), 7.66-7.70 (m, 2H), 7.72 (s, IH), 7.91 (dd, IH), 7.95 (dd, IH), 11.9 (s, IH).
MS (ESIneg): m/z = 423 [M-H]".
Ausführungsbeispiele:
Beispiel 1
3-(7-Ethyl-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Zu 3.51 g Lithiumaluminiumhydrid (92.4 mmol) in 150 ml Diethylether gab man bei O0C langsam 12.0 g der Verbindung aus Beispiel 2A (30.8 mmol) hinzu. Man rührte über Nacht bei RT und beendete die Reaktion durch Zugabe von 10 ml Isopropanol bei 00C. Die Reaktionslösung wurde mit ges. wässr. Ammoniumchlorid-Lösung neutralisiert. Die wässrige Phase wurde mit 200 ml Diethylether extrahiert und die organische Phase mit 50 ml 1 N Salzsäure gewaschen. Die vereinigteen organischen Phasen wurden über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Als Rückstand erhielt man 10.1 g (95% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, CDCl3): δ = 1.35 (t, 3H), 2.22-2.33 + 2.44-2.54 (AB-Signal, 2m, 2H), 2.85 (q, 2H), 3.59- 3.74 (m, 2H), 4.48 (t, IH), 6.96-7.04 (m, 2H), 7.10 (d, IH), 7.27 (m, IH), 7.41 (d, 2H), 7.51 (d, 2H), 8.02 (s, IH).
Beispiel 2
4-(7-Ethyl-lH-indol-3-yl)-4-[4-(trifiuormethyl)phenyl]butannitril
Zu 1.45 g der Verbindung aus Beispiel 3A (3.41 mmol) in 14.5 ml DMF wurden 444 mg Kalium- cyanid (6.82 mmol) gegeben. Man erhitzte 3 h auf 800C und beendete dann die Reaktion durch Zugabe von je 20 ml Essigsäureethylester und Wasser. Die organische Phase wurde mit 30 ml ges. wässr. Natriumhydrogencarbonat-Lösung gewaschen, über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde über eine Kieselgelsäule chromatographisch gereinigte (Laufmittel: Dichlormethan/Cyclohexan 2:1). Es wurden 960 mg (78% d. Th.) der Zielverbindung erhalten.
1H-NMR (400 MHz, DMSO-Cl6): δ = 1.23 (t, 3H), 2.31-2.39 (m, IH), 2.40-2.45 (m, 2H), 2.82 (q, 2H), 4.33 (t, IH), 6.82-6.89 (m, 2H), 7.22 (dd, IH), 7.40 (d, IH), 7.58 (d, 2H), 7.63 (d, 2H), 11.00 (s, IH).
HPLC (Methode 4): R, = 5.15 min; MS (ESIneg): m/z = 355.2 [M-H]".
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 15 ml/min; Temperatur: 400C; UV-Detektion: 220 nm] wurden die Enantiomere getrennt:
Enantiomer 2-1:
R, = 6.43 min [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 1.0 ml/min; Temperatur: 250C; UV-Detektion: 210 nm];
Enantiomer 2-2:
Rt = 8.40 min [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 1.0 ml/min; Temperatur: 250C; UV-Detektion: 210 nm].
Beispiel 3
4-(7-Ethyl-lH-indol-3-yl)-4-[4-(trifluormethyl)phenyl]butan-l-ol
Zu 9.78 g der Verbindung aus Beispiel 4A (26.0 mmol) in 100 ml TΗF wurden 2.47 g Lithium- aluminiumhydrid (65.0 mmol) in 30 ml TΗF gegeben und die Reaktionsmischung über Nacht bei 6O0C gerührt. Nach dem Abkühlen wurden erst 100 ml Isopropanol, dann 100 ml 1 N Salzsäure zugesetzt. Nach Filtration der Mischung über eine Kieseigelfritte und Nachwaschen mit Essigsäure- ethylester wurden die Phasen des Filtrats getrennt. Die wässrige Phase wurde mit 100 ml Essig- säureethylester extrahiert. Die organische Phase wurde mit je 100 ml Wasser, ges. wässr. Natrium- hydrogencarbonat-Lösung und ges. wässr. Natriumchlorid-Lösung gewaschen. Die vereinigteen organischen Phasen wurden über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde über eine Kieselgelsäule chromatographisch gereinigte (Laufmittel: Dichlormethan). Es wurden 9.05 g (91% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSO-(I6): δ = 1.23 (t, 3H), 1.28-1.40 + 1.40-1.52 (AB-Signal, 2m, 2H), 1.97-2.07 + 2.12-2.23 (AB-Signal, 2m, 2H), 2.81 (q, 2H), 3.24 (td, IH), 4.22 (t, IH), 4.37 (t, IH), 6.79-6.86 (m, 2H), 7.20 (dd, IH), 7.29 (d, IH), 7.54 (d, 2H), 7.60 (d, 2H), 10.89 (s, IH).
HPLC (Methode 5): R, = 4.82 min; MS (ESIpos): m/z = 362.3 [M+H]+.
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralpak OD-H, 250 mm x 20 mm; Eluent: Isopropanol/Isohexan 20:80; Fluss: 20 ml/min; Temperatur: 24°C; UV-Detektion: 230 nm] wurden die Enantiomere getrennt:
Enantiomer 3-1:
Rt = 6.27 min [Säule: Daicel Chiralpak OD-H, 250 mm x 4 mm; Eluent: Isopropanol/Isohexan 20:80; Fluss: 1 ml/min; UV-Detektion: 230 nm];
Enantiomer 3-2:
R, = 8.67 min [Säule: Daicel Chiralpak OD-H, 250 mm x 4 mm; Eluent: Isopropanol/Isohexan 20:80; Fluss: 1 ml/min; UV-Detektion: 230 nm].
Beispiel 4
5-(7-Ethyl-lH-indol-3-yl)-5-[4-(trifluormethyl)phenyl]pentannitril
Zu 2.20 g der Verbindung aus Beispiel 3 (5.26 mmol) in 28 ml Dichlormethan wurden 1.34 ml Tri- ethylamin (974 mg, 9.62 mmol) und 69 mg 4-N,N-Dimethylaminopyridin (0.57 mmol) gegeben. Man ließ 10 min rühren und gab dann 657 μl Methansulfonsäurechlorid (973 mg, 8.49 mmol) bei 00C hinzu. Nach 40 min Rühren bei RT wurde die Mischung mit 100 ml Diethylether verdünnt. Man extrahierte nacheinander mit je 20 ml Wasser, 1 N Salzsäure, Wasser, ges. wässr. Natrium- hydrogencarbonat-Lösung, Wasser und ges. wässr. Natriumchlorid-Lösung. Die organische Phase wurde über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit.
Der Rückstand wurde in 28 ml DMF gelöst, mit 728 mg Kaliumcyanid (11.2 mmol) versetzt und über Nacht bei 800C gerührt. Nach dem Abkühlen wurden je 30 ml Wasser und Diethylether zugesetzt. Die organische Phase wurde zweimal mit je 20 ml Wasser und ges. wässr. Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde aus Ethanol umkristallisiert. Man erhielt 1.25 g (60% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, CDCl3): δ = 1.36 (t, 3H), 1.60-1.82 (m, 2H), 2.13-2.24 + 2.32-2.45 (AB- Signal, 2m, 2H), 2.37 (t, 2H), 2.85 (q, 2H), 4.25 (t, IH), 6.97-7.04 (m, 2H), 7.10 (d, IH), 7.24 (dd, IH), 7.41 (d, 2H), 7.53 (d, 2H), 8.04 (s, IH).
MS (CIpos): m/z = 388.0 [M+NH,]*.
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isohexan/Isopropanol 4:1; Fluss: 15 ml/min; Temperatur: 24°C; UV-Detektion: 240 nm] wurden die Enantiomere getrennt:
Enantiomer 4-1:
Rt = 4.19 min [Säule: Daicel Chiralcel AD-H, 250 mm x 4.6 mm; Eluent: Isopropanol/Isohexan 30:70; Fluss: 1 ml/min; UV-Detektion: 220 nm];
Enantiomer 4-2:
R, = 4.80 min [Säule: Daicel Chiralcel AD-H, 250 mm x 4.6 mm; Eluent: Isopropanol/Isohexan 30:70; Fluss: 1 ml/min; UV-Detektion: 220 nm].
Beispiel 5
3-(7-Methyl-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Methylindol analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, CDCl3): δ = 1.31 (t, IH), 2.22-2.32 + 2.44-2.54 (AB-Signal, 2m, 2H), 2.61 (s, 3H), 3.67 (Hi0, 2H), 4.48 (t, IH), 6.93-7.00 (m, 2H), 7.11 (d, IH), 7.26 (d, IH), 7.43 (d, 2H), 7.51 (d, 2H), 7.99 (s, IH).
MS (CIpos): m/z = 334.3 [M+H]+.
Beispiel 6
4-(7-Methyl-lH-indol-3-yl)-4-[4-(trifluormethyl)phenyl]butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 5 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, CDCl3): δ = 2.33 (t, 2H), 2.33-2.43 + 2.54-2.62 (AB-Signal, 2m, 2H), 2.49 (s, 3H), 4.41 (t, IH), 6.95-7.02 (m, 2H), 7.11 (d, 2H), 7.25 (d, IH), 7.44 (d, 2H), 7.55 (d, 2H), 8.05 (s, IH).
MS (CIpos): m/z = 360.4 [M+NH,]*.
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 15 ml/min; Temperatur: 40°C; UV-Detektion: 220 nm] wurden die Enantiomere getrennt:
Enantiomer 6-1 :
Rt = 6.40 min [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 1.0 ml/min; Temperatur: 25°C; UV-Detektion: 210 nm];
Enantiomer 6-2:
R, = 8.47 min [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 1.0 ml/min; Temperatur: 25°C; UV-Detektion: 210 nm].
Beispiel 7
4-(7-Methyl-lH-indol-3-yl)-4-[4-(trifluormethyl)phenyl]butan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 6 analog zur Synthese der Verbindung aus Beispiel 3.
1H-NMR (400 MHz, CDCl3): δ = 1.21 (br. s, IH), 1.50-1.72 (m, 2H), 2.04-2.16 + 2.25-2.36 (AB- Signal, 2m, 2H), 2.47 (s, 3H), 3.68 (td, 2H), 4.24 (t, IH), 2.92-2.99 (m, 2H), 7.10 (d, IH), 7.24 (d, IH), 7.41 (d, 2H), 7.51 (d, 2H), 7.96 (s, IH).
Beispiel 8
5 -(7-Methyl- 1 H-indol-3 -yl)-5 -[4-(trifluormethyl)phenyl]pentannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 7 analog zur Synthese der Verbindung aus Beispiel 4.
1H-NMR (400 MHz, CDCl3): δ = 1.59-1.82 (m, 2H), 2.12-2.24 + 2.31-2.43 (AB-Signal, 2m, 2H), 2.36 (t, 2H), 2.48 (s, 3H), 4.25 (t, IH), 6.93-7.00 (m, 2H), 7.11 (d, IH), 7.23 (d, IH), 7.43 (d, 2H), 7.52 (d, 2H), 8.01 (s, IH).
MS (CIpos): m/z = 374.6 [MfNHJ+.
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isohexan/Isopropanol 3:1; Fluss: 15 ml/min; Temperatur: 240C; UV-Detektion: 240 nm] wurden die Enantiomere getrennt:
Enantiomer 8-1 :
R, = 4.53 min [Säule: Daicel Chiralcel OD-H, 250 mm x 4.6 mm; Eluent: Isopropanol/Isohexan 50:50; Fluss: 1 ml/min; UV-Detektion: 220 nm];
Enantiomer 8-2:
Rt = 5.76 min [Säule: Daicel Chiralcel OD-H, 250 mm x 4.6 mm; Eluent: Isopropanol/Isohexan 50:50; Fluss: 1 ml/min; UV-Detektion: 220 nm].
Beispiel 9
3-(7-Brom-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Bromindol analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, CDCl3): δ = 1.29 (t, IH), 2.22-2.32 + 2.43-2.53 (2m, AB-Signal, 2H), 3.59- 3.74 (m, 2H), 4.48 (t, IH), 6.91 (dd, IH), 7.18 (d, IH), 7.32 (d, IH), 7.33 (d, IH), 7.41 (d, 2H), 7.54 (d, 2H), 8.24 (s, IH).
Beispiel 10
4-(7-Brom-lH-indol-3-yl)-4-[4-(trifluormethyl)phenyl]butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 9 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, CDCl3): δ = 2.26-2.45 (m, 3H), 2.50-2.61 (m, IH), 4.40 (t, IH), 6.94 (dd, IH), 7.18 (d, IH), 7.31 (d, IH), 7.35 (d, IH), 7.42 (d, 2H), 7.57 (d, 2H), 8.30 (s, IH).
LC-MS (Methode 1): R, = 2.71 min; MS (ESIpos): m/z = 407.1 [M+H]+.
Beispiel 11
4-(7-Brom- 1 H-indol-3 -yl)-4-[4-(trifluormethyl)phenyl]butan- 1 -ol
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 10 analog zur Synthese der Verbindung aus Beispiel 3.
1H-NMR (400 MHz, CDCl3): δ = 1.23 (br. s, IH), 1.50-1.70 (m, 2H), 2.05-2.16 + 2.25-2.35 (AB- Signal, 2m, 2H), 3.68 (t, 2H), 4.22 (t, IH), 6.90 (dd, IH), 7.18 (d, IH), 7.30 (d, IH), 7.31 (d, IH), 7.39 (d, IH), 7.52 (d, 2H), 8.22 (s, IH).
Beispiel 12
5-(7-Brom-lH-indol-3-yl)-5-[4-(trifluormethyl)phenyl]pentannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 11 analog zur Synthese der Verbindung aus Beispiel 4.
1H-NMR (400 MHz, CDCl3): δ = 1.61-1.80 (m, 2H), 2.14-2.23 + 2.32-2.43 (AB-Signal, 2m, 2H), 2.38 (m, 2H), 4.23 (t, IH), 6.92 (dd, IH), 7.19 (d, IH), 7.30 (d, IH), 7.33 (d, IH), 7.39 (d, 2H), 7.54 (d, 2H), 8.26 (s, IH).
MS (CIpos): m/z = 438.0 [M+NH,]*.
Beispiel 13
3- { 3 -Cyano- 1 -[4-(trifluormethyl)phenyl]propyl } - 1 H-indol-7-carbonitril
Durch eine Lösung von 50.0 mg der Verbindung aus Beispiel 10 (0.123 mmol) in 1.2 ml trockenem DMF ließ man für 10 min Argon-Gas perlen und gab dann 15.9 mg Zinkcyanid (0.135 mmol) sowie 14 mg Tetrakis(triphenylphosphin)palladium (0.012 mmol) hinzu. Der Ansatz wurde im Mikrowellenreaktor 30 Sekunden bei RT und dann 15 min bei 2000C gerührt. Nach Abkühlen wurde über Kieselgur filtriert und mit DMF nachgewaschen. Das Lösungsmittel des Filtrats wurde bei vermindertem Druck entfernt. Zum Rückstand wurden je 10 ml Wasser und Methyl-te/Y.-butyl- ether gegeben. Die organische Phase wurde mit je 10 ml Wasser und ges. wässr. Natriumchlorid- Lösung gewaschen, durch Extrelut filtriert und bei vermindertem Druck vom Lösungsmittel
befreit. Der Rückstand wurde mittels präparativer HPLC gereinigte (Eluent: Acetonitril/Wasser mit 0.1% Ameisensäure, Gradient 10:90 → 95:5). Man erhielt 33 mg (76% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, CDCl3): δ = 2.25-2.44 (m, 3H), 2.51-2.62 (m, IH), 4.44 (t, IH), 7.11 (d, IH), 7.25 (d, IH), 7.42 (d, 2H), 7.52 (d, IH), 7.59 (d, 2H), 8.73 (s, IH).
MS (CIpos): m/z = 371.1 [M+NEUf.
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralcel OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isohexan/Isopropanol 70:30; Fluss: 20 ml/min; Temperatur: 24°C; UV-Detektion: 230 nm] wurden die Enantiomere getrennt:
Enantiomer 13-1:
R, = 5.94 min [Säule: Daicel Chiralcel OD-H, 250 mm x 4 mm, Eluent: Isopropanol/Isohexan 30:70; Fluss: 1 ml/min; UV-Detektion: 225 nm];
Enantiomer 13-2:
Rt = 10.04 min [Säule: Daicel Chiralcel OD-H, 250 mm x 4 mm, Eluent: Isopropanol/Isohexan 30:70; Fluss: 1 ml/min; UV-Detektion: 225 nm].
Beispiel 14
3-{4-Cyano-l-[4-(trifluormethyl)phenyl]butyl}-lH-indol-7-carbonitril
Durch eine Lösung von 55.0 mg der Verbindung aus Beispiel 12 (0.131 mmol) in 1.3 ml trockenem DMF ließ man für 10 min Argon-Gas perlen und gab dann 16.9 mg Zinkcyanid (0.144 mmol) sowie 15 mg Tetrakis(triphenylphosphin)palladium (0.013 mmol) hinzu. Der Ansatz wurde im Mikrowellenreaktor 30 Sekunden bei RT und dann 15 min bei 2000C gerührt. Nach Abkühlen
wurde über Kieselgur filtriert und mit DMF nachgewaschen. Das Lösungsmittel des Filtrats wurde bei vermindertem Druck entfernt. Zum Rückstand wurden je 10 ml Wasser und Methyl-tert.-butyl- ether gegeben. Die organische Phase wurde mit je 10 ml Wasser und ges. wässr. Natriumchlorid- Lösung gewaschen, durch Extrelut filtriert und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde mittels präparativer HPLC gereinigte (Eluent: Acetonitril/Wasser mit 0.1% Ameisensäure, Gradient 30:70 → 95:5). Man erhielt 36 mg (75% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, CDCl3): δ = 1.59-1.80 (m, 2H), 2.14-2.25 (m, IH), 2.30-2.46 (m, 2H), 4.26 (t, 2H), 7.08 (dd, IH), 7.38 (d, 2H), 7.50 (d, IH), 7.56 (d, 2H), 7.57 (d, IH), 8.66 (s, IH).
MS (ESIneg): m/z = 366.1 [M-H]-.
Beispiel 15
3-(7-Amino-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Zu 61.8 mg Lithiumaluminiumhydrid (1.63 mmol) in 5 ml TΗF gab man langsam 175 mg der Ver- bindung aus Beispiel 6A (0.465 mmol) hinzu. Man rührte über Nacht bei 600C und beendete die
Reaktion durch Zugabe von 20 ml Wasser. Es wurde über Celite filtriert und mit Essigsäureethyl- ester und Wasser nachgewaschen. Das Filtrat wurde mit 25 ml Essigsäureethylester extrahiert. Die organische Phase wurde über Magnesiumsulfat getrocknet und bei vermindertem Druck vom
Lösungsmittel befreit. Der Rückstand wurde mittels präparativer ΗPLC gereinigte (Eluent: Aceto- nitril/Wasser mit 0.1% Ameisensäure, Gradient 10:90 → 95:5). Man erhielt 112 mg (72% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, DMSO-Cl6): δ = 2.05-2.16 + 2.25-2.38 (AB-Signal, 2m, 2H), 3.30-3.42 (m, 2H), 4.34 (t, IH), 4.48 (t, IH), 4.96 (s, 2H), 6.22-6.28 (m, IH), 6.58-6.62 (m, 2H), 7.24 (d, IH), 7.50 (d, 2H), 7.59 (d, 2H), 10.47 (s, IH).
HPLC (Methode 5): R, = 3.81 min; MS (ESIneg): m/z = 333.1 [M-H]".
Beispiel 16
N-(3-{3-Hydroxy-l-[4-(trifluormethyl)phenyl]propyl}-lH-indol-7-yl)methansulfonamid
50.0 mg der Verbindung aus Beispiel 15 (0.150 mmol) wurden in 1 ml TΗF vorgelegt. Es wurden bei 00C 13 μl Pyridin (13 mg, 0.17 mmol) und 5 min danach 13 μl Methansulf onsäurechlorid (19 mg, 0.17 mmol) zugegeben. Man ließ anschließend über Nacht bei RT rühren. Das Lösungsmittel wurde dann bei vermindertem Druck entfernt und der Rückstand mittels präparativer ΗPLC gereinigte (Eluent: Acetonitril/Wasser mit 0.1% Ameisensäure, Gradient 10:90 -» 95:5). Man erhielt 53.5 mg (87% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, DMSOd6): δ = 2.08-2.19 + 2.27-2.38 (AB-Signal, 2m, 2H), 2.96 (s, 3H), 3.29-3.42 (m, 2H), 4.43 (t, IH), 4.51 (t, IH), 6.69 (dd, IH), 7.01 (d, IH), 7.24 (d, IH), 7.35 (d, IH), 7.54 (d, 2H), 7.61 (d, 2H), 9.27 (s, IH), 10.69 (s, IH).
HPLC (Methode 5): R, = 4.07 min; MS (CIpos): m/z = 430.1 [M+NK,]*.
Beispiel 17
N-(3-{4-Cyano-l-[4-(trifluormethyl)phenyl]butyl}-lH-indol-7-yl)methansulfonamid
Zu 37.8 mg der Verbindung aus Beispiel 7A (0.0771 mmol) in 1 ml DMF wurden 10.0 mg Kalium- cyanid (0.154 mmol) gegeben und die Mischung 3 h bei 800C gerührt. Danach wurden 5 ml Essigsäureethylester zugesetzt. Man extrahierte mit je 5 ml Wasser und ges. wässr. Natriumhydro- gencarbonat-Lösung. Die organische Phase wurde über Magnesiumsulfat getrocknet und bei vermindertem Druck vom Lösungsmittel befreit. Der Rückstand wurde mittels präparativer HPLC gereinigte (Eluent: Acetonitril/Wasser mit 0.1% Ameisensäure, Gradient 10:90 -» 95:5). Man erhielt 24 mg (73% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, DMSOd6): δ = 2.30-2.40 (m, 2H), 2.43 (t, 2H), 2.96 (s, 3H), 4.35 (t, IH), 6.92 (dd, IH), 7.03 (d, IH), 7.28 (d, IH), 7.44 (d, IH), 7.59 (d, 2H), 7.64 (d, 2H), 9.29 (s, IH), 10.80 (s, IH).
HPLC (Methode 5): R, = 4.57 min; MS (ESIneg): m/z = 420.1 [M-H]".
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralpak OD-H, 5 μm, 250 mm x 20 mm; Eluent: Isopropanol/Isohexan 50:50; Fluss: 20 ml/min; Temperatur: 250C; UV-Detektion: 230 nm] wurden die Enantiomere getrennt:
Enantiomer 17-1:
R, = 10.70 min [Säule: Daicel Chiralpak OD-H, 250 mm x 4 mm; Eluent: Isopropanol/Isohexan 50:50; Fluss: 1 ml/min; UV-Detektion: 230 nm];
Enantiomer 17-2:
Rt = 12.47 min [Säule: Daicel Chiralpak OD-H, 250 mm x 4 mm; Eluent: Isopropanol/Isohexan 50:50; Fluss: 1 ml/min; UV-Detektion: 230 nm].
Beispiel 18
N-(3 - {4-Hydroxy- 1 -[4-(trifluormethyl)phenyl]butyl } - 1 H-indol-7-yl)methansulfonamid
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 17 analog zur Synthese der Verbindung aus Beispiel 3.
1H-NMR (400 MHz, DMSO-d6): δ = 1.28-1.39 + 1.39-1.51 (AB-Signal, 2m, 2H), 1.96-2.07 + 2.12- 2.22 (AB-Signal, 2m, 2H), 2.96 (s, 3H), 3.42 (td, 2H), 4.25 (t, IH), 4.38 (t, IH), 6.89 (dd, IH), 7.01 (d, IH), 7.25 (d, IH), 7.34 (d, IH), 7.55 (d, 2H), 7.61 (d, 2H), 9.27 (s, IH), 10.70 (s, IH).
HPLC (Methode 5): R, = 4.28 min; MS (ESIneg): m/z = 425.1 [M-H]".
Beispiel 19
N-(3-{4-Cyano-l-[4-(trifluormethyl)phenyl]butyl}-lH-indol-7-yl)methansulfonamid
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 18 analog zur Synthese der Verbindung aus Beispiel 4.
1H-NMR (400 MHz, DMSO-Cl6): δ = 1.41-1.64 (m, 2H), 2.05-2.16 + 2.19-2.30 (AB-Signal, 2m, 2H), 2.55 (t, 2H), 2.96 (s, 3H), 4.33 (t, IH), 6.90 (dd, IH), 7.02 (d, IH), 7.27 (d, IH), 7.38 (d, IH), 7.57 (d, 2H), 7.63 (d, 2H), 9.28 (s, IH), 10.74 (s, IH).
LC-MS (Methode 2): R, = 3.53 min; MS (ESIpos): m/z = 436.1 [M+H]+.
Beispiel 20
3-(7-Ethyl-lH-indol-3-yl)-3-(4-fluorphenyl)propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 4-Fluorbenzaldehyd analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, DMSO-d6): δ = 1.23 (t, 3H), 2.03-2.14 + 2.24-2.34 (2m, AB-Signal, 2H), 2.81 (q, 2H), 3.30-3.40 (m, 2H), 4.30 (t, IH), 4.45 (t, IH), 6.78-6.86 (m, 2H), 7.02-7.07 (m, 2H), 7.18 (d, IH), 7.23 (d, IH), 7.30-7.35 (m, 2H), 10.82 (s, IH).
HPLC (Methode 5): R, = 4.44 min; MS (CIpos): m/z = 315.1 [M+NHJ*.
Beispiel 21
4-(7-Ethyl-lH-indol-3-yl)-4-(4-fluorphenyl)butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 20 analog zur Synthese der Verbindung 2.
1H-NMR (400 MHz, DMSO-d6): δ = 1.23 (t, 3H), 2.24-2.55 (m, 4H), 2.81 (q, 2H), 4.22 (t, IH), 6.81-6.88 (m, IH), 7.09 (nie, 2H), 7.21 (dd, IH), 7.32-7.40 (m, 3H), 10.94 (s, IH).
HPLC (Methode 5): R, = 4.97 min; MS (ESIneg): m/z = 305.2 [M-H]".
Beispiel 22
3-[7-(Trifluormethyl)-lH-indol-3-yl]-3-[4-(trifluormethyl)phenyl]propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Trifluormethylindol [Y. Murakami et al., Chem. Pharm. Bull. 41 (11), 1910-1919 (1993)] analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, DMSO-d6): δ = 2.12-2.21 + 2.30-2.40 (2m, AB-Signal, 2H), 3.30-3.43 (m, 2H), 4.51 (t, IH), 4.53 (t, IH), 7.07 (dd, IH), 7.39 (d, IH), 7.47 (d, IH), 7.56 (d, 2H), 7.61 (d, 2H), 7.69 (d, IH), 11.36 (s, IH).
LC-MS (Methode 3): R, = 3.90 min; MS (ESIpos): m/z = 388.2 [M+H]+.
Beispiel 23
4-[7-(Trifluormethyl)-lH-indol-3-yl]-4-[4-(trifluormethyl)phenyl]butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 22 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, DMSO-d6): δ = 2.31-2.60 (m, 4H), 4.42 (t, IH), 7.09 (dd, IH), 7.41 (d, IH), 7.58 (d, IH), 7.61 (d, 2H), 7.64 (d, 2H), 7.73 (d, IH), 11.45 (s, IH).
LC-MS (Methode 3): R1 = 4.17 min; MS (CIpos): m/z = 397.2 [M+NHJ*.
Beispiel 24
3-(7-Methoxy-lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Methoxyindol analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, CDCl3): δ = 1.28 (t, IH), 2.22-2.32 + 2.43-2.53 (2m, AB-Signal, 2H), 3.60- 3.73 (m, 2H), 3.94 (s, 3H), 4.47 (t, IH), 6.62 (d, IH), 6.94 (dd, IH), 7.01 (d, IH), 7.07 (d, IH), 7.43 (d, 2H), 7.51 (d, 2H), 8.26 (s, IH).
Beispiel 25
4-(7-Methoxy-lH-indol-3-yl)-4-[4-(trifluormethyl)phenyl]butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 24 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, CDCl3): δ = 2.30-2.43 (m, 3H), 2.51-2.63 (m, IH), 3.95 (s, 3H), 4.39 (t, IH), 6.64 (dd, IH), 6.94-7.00 (m, 2H), 7.07 (d, IH), 7.44 (d, 2H), 7.55 (d, 2H), 8.31 (s, IH).
MS (CIpos): m/z = 376.0 [M+NKJ*.
Die in der folgenden Tabelle aufgeführten Verbindungen wurden analog zur Synthese der Verbindung aus Beispiel 1 hergestellt:
'die Reduktion des Carbonsäureethylesters zum primären Alkohol (vgl. Beispiel 2A — » Beispiel 1) erfolgte hier mit 2 eq. Lithiumborhydrid in THF (statt Lithiumaluminiumhydrid in Diethylether).
Beispiel 32
N-(3 - { 3 -Cyano-3 -fluor- 1 -[4-(trifluormethyl)phenyl]propyl } - lH-indol-7-yl)methansulfonamid
Zu 100 mg der Verbindung aus Beispiel 8A (0.21 mmol, 87% Reinheit) in 3 ml Essigsäureethyl- ester wurden 33 mg Benzyltri-«-butylammoniumchlorid (0.11 mmol), 15 mg Kaliumcyanid (0.22 mmol) und 6 ml Wasser gegeben. Nach 1 h Rühren bei RT wurden 5 ml Essigsäureethylester hin- zugefügt, und die Lösung wurde über Natriumsulfat getrocknet. Der Feststoff wurde abfiltriert und das Lösungsmittel unter vermindertem Druck entfernt. Der Rückstand wurde in 2 ml Dichlormethan gelöst, bei 00C mit 38 mg Diethylaminoschwefeltrifluorid (0.23 mmol) versetzt und die Lösung anschließend 16 h bei RT gerührt. Nach Zugabe von 1 ml Wasser wurde das Rohprodukt zunächst mittels präparativer HPLC vorgereinigte (Eluent: Acetonitril/Wasser- Gradient 30:70 — > 98:2). Von den produkthaltigen Fraktionen wurde das Lösungsmittel im Vakuum entfernt, und der Rückstand wurde säulenchromatographisch an Kieselgel weiter gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 1:1). Man erhielt 9 mg (13% d. Th.) der Zielverbindung.
1H-NMR (400 MHz, DMSOd6): δ = 2.72-3.05 (m, 5H), 4.48-4.57 (m, IH), 5.35-5.63 (m, IH), 6.89-6.95 (m, IH), 7.01-7.05 (m, IH), 7.29-7.37 (m, IH), 7.48-7.56 (dd, IH), 7.62-7.66 (m, 4H), 9.28 (s, IH), 10.83 (d, IH).
LC-MS (Methode 8): R, = 1.30 min; MS (ESIpos): m/z = 440.1 [M+H]+.
Beispiel 33
3-(7-Ethyl-lH-indol-3-yl)-3-naphth-2-ylpropan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 2-Naphthaldehyd analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, DMSO-de): δ = 1.23 (t, 3H), 2.17-2.29 (m, IH), 2.32-2.43 (m, IH), 2.81 (q, 2H), 3.34-3.46 (m, 2H), 4.42-4.51 (m, 2H), 6.74-6.85 (m, 2H), 7.23 (d, IH), 7.30 (s, IH), 7.38-7.49 (m, 3H), 7.73-7.88 (m, 4H), 10.85 (s, IH).
HPLC (Methode 5): R, = 4.76 min; MS (CIpos): m/z = 347.2 [M+NHJ*.
Beispiel 34
3-(7-Ethyl-lH-indol-3-yl)-3-naphth-2-ylbutannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 33 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, DMSOd6): δ = 1.23 (t, 3H), 2.40-2.52 (m, 4H), 2.81 (q, 2H), 4.35-4.41 (m, IH), 6.78-6.87 (m, 2H), 7.25 (d, IH), 7.38-7.51 (m, 4H), 7.77-7.93 (m, 4H), 10.95 (s, IH).
LC-MS (Methode 7): R, = 2.43 min; MS (ESIpos): m/z = 339.3 [M+H]+.
Beispiel 35
3-(7-Brom-lH-pyrrolo[2,3-c]pyridin-3-yl)-3-[4-(trifluormethyl)phenyl]propan-l-ol
Die Herstellung der Titelverbindung erfolgte ausgehend von 7-Brom-lH-pyrrolo[2,3-c]pyridin analog zur Synthese der Verbindung aus Beispiel 1.
1H-NMR (400 MHz, DMSO-d6): δ = 2.14-2.23 (m, IH), 2.29-2.38 (m, IH), 3.26-3.43 (m, 2H), 4.47 (t, IH), 4.51 (t, IH), 7.43 (d, IH), 7.56 (d, 2H), 7.61 (d, 2H), 7.67 (d, IH), 7.79 (d, IH), 11.75 (s, IH).
HPLC (Methode 4): Rt = 3.82 min; MS (CIpos): m/z = 399.0 [M+H]+.
Beispiel 36
3-(7-Brom-lH-pyrrolo[2,3-c]pyridin-3-yl)-3-[4-(trifluormethyl)phenyl]butannitril
Die Herstellung der Titelverbindung erfolgte ausgehend von der Verbindung aus Beispiel 35 analog zur Synthese der Verbindung aus Beispiel 2.
1H-NMR (400 MHz, DMSOd6): δ = 2.34-2.59 (m, 4H), 4.35-4.42 (m, IH), 7.48 (d, IH), 7.61 (d, 2H), 7.65 (d, 2H), 7.79 (s, IH), 7.81 (d, IH), 11.87 (s, IH).
HPLC (Methode 4): R, = 4.02 min; MS (CIpos): m/z = 408.0 [M+H]+.
Beispiel 37
N-(3- { 1 -[2-Fluor-4-(trifluormethyl)phenyl]-3-hydroxypropyl} -lH-indol-7-yl)methansulfonamid
Die Herstellung der Titelverbindung erfolgte ausgehend von 985 mg (2.09 mmol) der Verbindung aus Beispiel 17A analog zur Synthese der Verbindung aus Beispiel 1. Es wurde jedoch Tetrahydrofuran als Lösungsmittel verwendet und 2 h bei 60 0C gerührt. Man reinigte das Rohprodukt mittels präparativer HPLC (RP18-Säule; Laufimittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% Ameisensäure) und erhielt 630 mg (70% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-(I6): δ = 2.13-2.23 (m, IH), 2.30-2.40 (m, IH), 2.96 (s, 3H), 3.33-3.46 (m, 2H), 4.53 (t, IH), 4.71 (t, IH), 6.92 (t, IH), 7.03 (d, IH), 7.25 (d, IH), 7.33 (d, IH), 7.49 (d, IH), 7.57-7.63 (m, 2H), 9.29 (s, IH), 10.7 (s, IH).
LC-MS (Methode 9): R, = 2.26 min; MS (ESIpos): m/z = 431 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen wurden analog zur Synthese der Verbindung aus Beispiel 37 hergestellt. Alternativ konnte auch nur 1 h bei 60
0C gerührt werden und die Aufarbeitung durch Quenchen mit Wasser und 1 M Salzsäure, mehrmaliger Extraktion der wässrigen Phase mit Wasser, Waschen der organischen Phasen mit gesättigter wässriger Natriumchlorid-Lösung, Trocknen mit Natriumsulfat, Filtration und Einengen erfolgen.
Beispiel 44
N-(3 - { 3-Cyan- 1 -[2-fluor-4-(trifluormethyl)phenyl]propyl } - 1 H-indol-7-yl)methansulfonamid
Zu 630 mg (1.46 mmol) der Verbindung aus Beispiel 37 in 30 ml Dichlormethan wurden 18 mg (0.15 mmol) 4-NN-Dimethylaminopyridin und 0.35 ml (2.45 mmol) Triethylamin gegeben. Man ließ 5 min rühren und gab dann 0.17 ml (2.20 mmol) Methansulfonsäurechlorid hinzu. Nach Rühren über Nacht bei RT wurde Dichlormethan hinzu gegeben und die Lösung mit IM Salzsäure, Wasser und ges. Natriumchlorid-Lösung gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 1% Ameisensäure) gereinigt. Das Zwischenprodukt wurde in DMF gelöst, mit 160 mg (2.46 mmol) Kaliumcyanid versetzt und die Lösung 4 h bei 80 0C gerührt. Die Reaktionsmischung wurde eingeengt, der Rückstand in Ethylacetat aufgenommen und nacheinander mit ges. Natriumhydrogencarbonat- Lösung, Wasser und ges. Natriumchlorid-Lösung gewaschen. Die organische Phase wurde über Magnesiumsulfat getrocknet, filtriert und eingeengt und der Rückstand mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 1% Ameisensäure) gereinigt. Man erhielt 439 mg (23% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-d6): δ = 2.32-2.42 (m, IH), 2.47-2.58 (m, 3H), 2.97 (s, 3H), 4.60-4.66 (m, IH), 6.95 (t, IH), 7.05 (d, IH), 7.28 (d, IH), 7.43 (d, IH), 7.52 (d, IH), 7.62-7.67 (m, 2H), 9.31 (s, IH), 10.9 (s, IH).
LC-MS (Methode 7): R, = 2.05 min; MS (ESIpos): m/z = 440 [M+H]+.
Die in der folgenden Tabelle aufgeführten Verbindungen wurden analog zur Synthese der Verbindung aus Beispiel 44 hergestellt. Alternativ konnten die Aufreinigungen der Rohprodukte mittels präparativer HPLC auch ohne vorherige Aufarbeitung erfolgen:
bei der zweiten Teilreaktion (Umsetzung des Mesylates zum Cyanid) wurde abweichend 2 h bei 100
0C in DMF gerührt.
Beispiel 51
3-(5-Fluor-7-nitro- lH-indol-3-yl)-3-[4-(trifluormethyl)phenyl]propan- 1 -ol
544 mg (1.28 mmol) der Verbindung aus Beispiel 26A wurden in 10 ml Tetrahydrofuran vorgelegt, bei 00C mit 55.9 mg (2.56 mmol) Lithiumborhydrid versetzt und über Nacht bei RT gerührt. Danach wurden weitere 28.0 mg (1.28 mmol) Lithiumborhydrid zugegeben und die Reaktions- mischung wiederum über Nacht bei RT gerührt. Anschließend wurde Wasser zugesetzt und mit Essigsäureethylester extrahiert. Die organische Phase wurde mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man reinigte den Rückstand mittels Flash-Chromatographie (Laufmittel: Dichlormethan/Methanol 100:1) und erhielt 194 mg (40% d. Th.) der Titelverbindung.
1H-NMR (400 MHz, DMSO-Cl6): δ = 2.13-2.24 (m, IH), 2.29-2.39 (m, IH), 3.30-3.42 (m, 2H), 4.50-4.56 (m, 2H), 6.59-7.65 (m, 5H), 7.84 (dd, IH), 7.90 (dd, IH), 11.9 (s, IH).
LC-MS (Methode 9): R, = 2.70 min; MS (ESIpos): m/z = 383 [M+H]+.
Beispiel 52
N-(3-{3-Cyano-l-[4-(trifluormethyl)phenyl]propyl}-5-fluor-lH-indol-7-yl)methansulfonamid
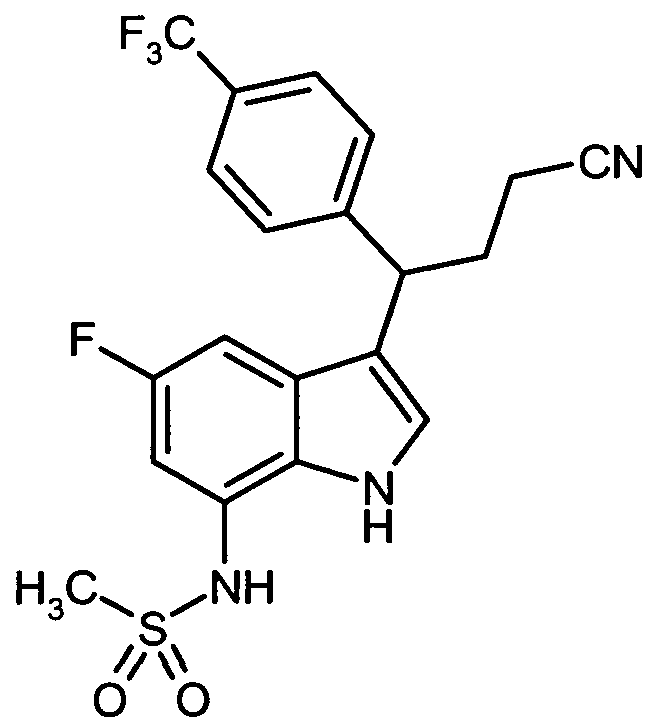
176 mg (460 μmol) der Verbindung aus Beispiel 51 wurden in 10 ml Ethanol vorgelegt, mit 17.5 mg Palladium auf Kohle (10%-ig) versetzt und über Nacht bei Normaldruck und RT hydriert. Man filtrierte danach über Celite, wusch mit Ethanol nach, engte das Filtrat ein und erhielt 160 mg des rohen Zwischenprodukts. 153 mg (433 μmol) dieses Zwischenprodukts wurden unter Argon in Dichlormethan vorgelegt, mit 5.3 mg (43 μmol) 4-N,N-Dimethylaminopyridin, 149 mg (1.47 mmol) Triethylamin sowie 149 mg (1.30 mmol) Methansulfonsäurechlorid versetzt und über Nacht bei RT gerührt. Anschließend wurde Essigsäureethylester hinzugegeben, nacheinander mit 1 M Salzsäure, Wasser und gesättigter wässriger Natriumchlorid-Lösung gewaschen, die organische Phase über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man reinigte den Rückstand mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient) und erhielt 207 mg des zweiten Zwischenprodukts. 149 mg dieses Zwischenprodukts wurden in 5 ml Di- methylformamid vorgelegt, mit 33.1 mg (508 μmol) Kaliumcyanid versetzt und über Nacht bei 8O
0C gerührt. Man reinigte dann die Reaktionsmischung direkt mittels präparativer HPLC (RP 18- Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 1% Ameisensäure) und erhielt 63 mg der Titelverbindung.
1H-NMR (400 MHz, DMSO-d6): δ = 2.29-2.38 (m, IH), 2.39-2.45 (m, 2H), 2.46-2.55 (m, IH), 3.03 (s, 3H), 4.31 (t, IH), 6.89 (dd, IH), 7.06 (dd, IH), 7.53 (d, IH), 7.58-7.67 (m, 4H), 9.56 (s, IH), 10.9 (s, IH).
MS (CIpos): m/z = 457 [Mt-NH4]".
Durch präparative HPLC an chiraler Phase [Säule: Daicel Chiralpak AD-H, 5 μm, 250 mm x 20 mm; Eluent: Ethanol; Fluss: 10 ml/min; Temperatur: 400C; UV-Detektion: 220 nm] wurden die Enantiomere getrennt:
Enantiomer 52-1:
Rt = 4.80 min [Säule: Daicel Chiralpak AD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Ethanol; Fluss: 0.8 ml/min; Temperatur: 45°C; UV-Detektion: 220 nm];
Enantiomer 52-2:
R1 = 8.37 min [Säule: Daicel Chiralpak AD-H, 5 μm, 250 mm x 4.6 mm; Eluent: Ethanol; Fluss: 0.8 ml/min; Temperatur: 45°C; UV-Detektion: 220 nm].
Beispiel 53
N-(3-{3 -Cyano- 1 -[4-(trifluormethyl)phenyl]propyl } - 1 H-indol-7-yl)ethansulfonamid
80.0 mg (239 μmol) der Verbindung aus Beispiel 15 wurden in 1.5 ml Tetrahydrofuran vorgelegt und bei 00C mit 48 μl (598 μmol) Pyridin und 57 μl (598 μmol) Ethansulfonylchlorid versetzt. Die Reaktionsmischung wurde über Nacht bei RT und anschließend 4 h bei 500C gerührt. Man gab da- nach weitere 23 μl (239 μmol) Ethansulfonylchlorid und 19 μl (239 μmol) Pyridin hinzu und rührte wiederum über Nacht bei RT. Anschließend wurden nochmals 35 μl (359 μmol) Ethansulfonylchlorid und 29 μl (359 μmol) Pyridin hinzugegeben und die Mischung erneut über Nacht bei RT gerührt. Die letztere Prozedur wurde noch einmal wiederholt. Schließlich wurde Essigsäureethylester zugesetzt und die Mischung nacheinander mit 1 M Salzsäure, Wasser und gesättigter wässriger Natriumchlorid-Lösung extrahiert. Die organische Phase wurde über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man reinigte den Rückstand mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 0.1% Ameisensäure) und erhielt 48.7 mg des Zwischenprodukts. 45 mg dieses Zwischenprodukts wurden in 1 ml Dimethylformamid vorgelegt, mit 11.3 mg (174 μmol) Kaliumcyanid versetzt und 3 h bei 800C gerührt. Anschließend wurde Essigsäureethylester hinzugegeben, mit Wasser und gesättigter Natriumhydrogencarbonat-Lösung extrahiert, die organische Phase über Magnesiumsulfat getrocknet, filtriert und eingeengt. Man reinigte das Rohprodukt mittels präparativer HPLC (RP18-Säule; Laufmittel: Acetonitril/Wasser-Gradient unter Zusatz von 1% Ameisensäure) und erhielt so 30.4 mg der Titelverbindung.
1H-NMR (400 MHz, DMSOd6): δ = 1.19 (t, 3H), 2.31-2.39 (m, IH), 2.40-2.46 (m, 2H), 2.46-2.52 (m, IH), 3.07 (q, 2H), 4.34 (t, IH), 6.90 (t, IH), 7.03 (d, IH), 7.25 (d, IH), 7.43-7.45 (m, IH), 7.56-7.61 (m, 2H), 7.62-7.67 (m, 2H), 9.33 (s, IH), 10.7 (s, IH).
MS (ESIneg): m/z = 434 [M-H]".
Durch präparative HPLC an chiraler Phase [Säule: chirale Kieselgel-Phase basierend auf dem Selektor Poly(N-methacryloyl-Z-leucin-Z)-menthylamid), 5 μm, 250 mm x 30 mm; Eluent: Ethyl-
acetat; Fluss: 50 ml/min; Temperatur: 24°C; UV-Detektion: 260 nm] wurden die Enantiomere getrennt:
Enantiomer 53-1:
Rt = 3.68 min [Säule: chirale Kieselgel-Phase basierend auf dem Selektor Poly(N-methacryloyl-Z- leucin-Z>-menthylamid), 5 μm, 250 mm x 4.6 mm; Eluent: Ethylacetat; Fluss: 2 ml/min; Temperatur: 24°C; UV-Detektion: 260 nm];
Enantiomer 53-2:
Rt = 4.84 min [Säule: chirale Kieselgel-Phase basierend auf dem Selektor Poly(N-methacryloyl-Z- leucin-Z)-menthylamid), 5 μm, 250 mm x 4.6 mm; Eluent: Ethylacetat; Fluss: 2 ml/min; Tempera- tur: 24°C; UV-Detektion: 260 nm].
- O CQO -
B. Bewertung der pharmakologischen Wirksamkeit
Abkürzungen:
DMEM Dulbecco's Modifϊed Eagle Medium
DNA Desoxyribo Nucleic Acid
FCS Fetal CaIf Serum
HEPES 4-(2-Hydroxyethyl)- 1 -piperazin-ethansulfonsäure
PCR Polymerase Chain Reaction
Tris Tris-(hydroxymethyl)-methylamin
Die vorteilhaften pharmakologischen Eigenschaften der erfindungsgemäßen Verbindungen können in folgenden Assays gezeigt werden:
1. Zellulärer in vtϊro-Test zur Bestimmung der inhibitorischen MR-Aktivität und MR- Selektivität gegenüber anderen Steroidhormon-Rezeptoren
Die Identifizierung von Antagonisten des humanen Mineralokorticoid-Rezeptors (MR) sowie die Quantifizierung der Wirksamkeit der hier beschriebenen Verbindungen erfolgt mit Hilfe einer rekombinanten Zelllinie. Die Zelle leitet sich ursprünglich von einer Ovarepithelzelle des Hamsters ab (Chinese Hamster Ovary, CHO Kl, ATCC: American Type Culture Collection, VA 20108, USA).
In dieser CHO Kl -Zelllinie wird ein etabliertes Chimärensystem verwendet, in dem die Liganden- Bindungsdomänen humaner Steroidhormon-Rezeptoren an die DNA-Bindungsdomäne des Hefe- Transkriptionsfaktors GAL4 fusioniert werden. Die so entstehenden GAL4-Steroidhormonrezep- tor-Chimären werden in den CHO-Zellen mit einem Reporterkonstrukt co-transfiziert und stabil exprimiert.
Klonierungen:
Zur Generierung der GAL4-Steroidhormonrezeptor-Chimären wird die GAL4-DNA-Bindungs- domäne (Aminosäuren 1-147) aus dem Vektor pFC2-dbd (Fa. Stratagene) mit den PCR-amplifi- zierten Liganden-Bindungsdomänen des Mineralokorticoid-Rezeptors (MR, Aminosäuren 734-
985), des Glucokorticoid-Rezeptors (GR, Aminosäuren 443-777), des Progesteron-Rezeptors (PR,
Aminosäuren 680-933) und des Androgen-Rezeptors (AR, Aminosäuren 667-919) in den Vektor pIRES2 (Fa. Clontech) kloniert. Das Reporterkonstrukt, welches fünf Kopien der GAL4-Binde- stelle, vorgeschaltet vor einem Thymidinkinase-Promotor enthält, fuhrt zur Expression der Firefly-
Luciferase (Photinus pyralis) nach Aktivierung und Bindung der GAL4-Steroidhormonrezeptor- Chimären durch die jeweiligen spezifischen Agonisten Aldosteron (MR), Dexamethason (GR), Progesteron (PR) und Dihydrotestosteron (AR).
Testablauf:
Die MR-, GR-, PR- und AR-Zellen werden am Tag vor dem Test in Medium (Optimem, 2.5% FCS, 2 mM Glutamin, 10 mM HEPES) in 96- (oder 384- bzw. 1536-) Loch-Mikrotiterplatten ausplattiert und in einem Zellinkubator (96% Luftfeuchtigkeit, 5% v/v CO2, 37°C) gehalten. Am Testtag werden die zu prüfenden Substanzen in oben genanntem Medium aufgenommen und zu den Zellen hinzugegeben. Etwa 10 bis 30 Minuten nach Zugabe der Testsubstanzen werden die jeweili- gen spezifischen Agonisten der Steroidhormon-Rezeptoren hinzugesetzt. Nach einer weiteren Inkubationszeit von 5 bis 6 Stunden wird die Luciferaseaktivität mit Hilfe einer Videokamera gemessen. Die gemessenen relativen Lichteinheiten ergeben in Abhängigkeit von der Substanzkonzentration eine sigmoide Stimulationskurve. Die Berechnung der IC50- Werte erfolgt mit Hilfe des Computerprogramms GraphPad PRISM (Version 3.02).
Tabelle A zeigt die IC50- Werte repräsentativer Beispielverbindungen:
Tabelle A
2. In vi'vo-Test zum Nachweis der kardiovaskulären Wirkung: Diurese-Untersuchungen an wachen Ratten in Stoffwechselkäfigen
Wistar-Ratten (250-350 g Körpergwicht) werden mit freiem Zugang zu Futter (Altromin) und Trinkwasser gehalten. Ab ca. 72 Stunden vor Versuchsbeginn erhalten die Tiere anstelle des nor-
malen Futters ausschließlich Kochsalz-reduziertes Futter mit einem Gehalt von 0.02% Natriumchlorid (ssniff R/M-H, 10 mm mit 0.02% Na, S0602-E081, Fa. ssniff Spezialdiäten GmbH, D- 59494 Soest). Während des Versuches werden die Tiere für ca. 24 Stunden einzeln in für Ratten dieser Gewichtsklasse geeigneten Stoffwechselkäfigen (Fa. Tecniplast Deutschland GmbH, D- 82383 Hohenpeißenberg) mit freiem Zugang zu Kochsalz-reduziertem Futter und Trinkwasser gehalten. Am Versuchsbeginn wird den Tieren die zu prüfende Substanz in einem Volumen von 0.5 ml/kg Körpergewicht eines geeigneten Lösemittels mittels einer Schlundsonde in den Magen verabreicht. Als Kontrolle dienende Tiere erhalten nur Lösemittel. Kontrollen und Substanz- testungen werden am selben Tag parallel durchgeführt. Kontrollgruppen und Substanz-Dosis- gruppen bestehen aus jeweils 6 bis 8 Tieren. Während des Versuchs wird der von den Tieren ausgeschiedene Urin kontinuierlich in einem Auffangbehälter am Käfigboden gesammelt. Für jedes Tier wird gesondert das Urinvolumen pro Zeiteinheit bestimmt und die Konzentration der im Urin ausgeschiedenen Natrium- bzw. Kalium-Ionen mittels flammenphotometrischer Standardmethoden gemessen. Aus den Messwerten wird der Natrium/Kalium-Quotient als ein Maß für die Substanzwirkung berechnet. Die Messintervalle betragen typischerweise den Zeitraum bis zu 8 Stunden nach Versuchsbeginn (Tagintervall) und den Zeitraum von 8 bis 24 Stunden nach Versuchsbeginn (Nachtintervall). In einer abgewandelten Versuchsanordnung wird der Urin während des Tagintervalls im Abstand von zwei Stunden gesammelt und gemessen. Um eine hierfür ausreichende Urinmenge zu erhalten, wird den Tieren zu Versuchsbeginn und dann im Abstand von zwei Stunden per Schlundsonde eine definierte Menge Wasser zugeführt.
3. DOCA/Salz-Modell
Die Verabreichung von Desoxycorticosteronacetat (DOCA) in Kombination mit einer Hochsalzdiät und einseitiger Nierenentfernung induziert bei der Ratte einen Hypertonus, der durch relativ niedrige Reninspiegel charakterisiert ist. Als Folge dieser endokrinen Hypertonie (DOCA ist eine direkte Vorstufe von Aldosteron) kommt es in Abhängigkeit von der gewählten DOCA-Konzen- tration zu einer Hypertrophie des Herzens und weiteren Endorgan-Schäden, z.B. der Niere, die u.a. durch Proteinurie und Glomerulosklerose charakterisiert sind. In diesem Rattenmodell lassen sich somit Testsubstanzen auf vorhandene antihypertrophe und Endorgan-schützende Wirkung hin untersuchen.
Etwa 8 Wochen alte (Körpergewicht zwischen 250 und 300 Gramm), männliche Sprague Dawley (SD)-Ratten werden linksseitig uninephrektomiert. Dazu werden die Ratten mit 1.5-2%-igem Iso- fluran in einer Mischung aus 66% N2O und 33% O2 anästhesiert und die Niere über einen Flankenschnitt entfernt. Als spätere Kontrolltiere dienen sogenannte sham-operierte Tiere, denen keine Niere entfernt wird.
Uninephrektomierte SD-Ratten erhalten 1% Natriumchlorid im Trinkwasser und einmal wöchentlich eine subkutane Injektion von Desoxycorticosteronacetat (gelöst in Sesamöl; Fa. Sigma) zwischen die Schulterblätter gespritzt (Hochdosis: 100 mg/kg/Woche s.c; Normaldosis: 30 mg/kg/Woche s.c).
Die Substanzen, die auf ihre protektive Wirkung in vivo untersucht werden sollen, werden per Gavage oder über das Futter (Fa. Ssniff) verabreicht. Die Tiere werden einen Tag vor Versuchsbeginn randomisiert und Gruppen mit gleicher Tierzahl, in der Regel n = 10, zugeordnet. Während des gesamten Versuchs steht den Tieren Trinkwasser und Futter ad libitum zur Verfügung. Die Substanzen werden einmal täglich 4-8 Wochen lang per Gavage oder per Futter verabreicht. Als Plazebogruppe dienen Tiere, die genauso behandelt werden, aber entweder nur das Lösungsmittel oder das Futter ohne Testsubstanz erhalten.
Die Wirkung der Testsubstanzen wird durch Messung hämodynamischer Parameter [Blutdruck, Herzfrequenz, Inotropie (dp/dt), Relaxationszeit (tau), maximaler linksventrikulärer Druck, links- ventrikulärer enddiastolischer Druck (LVEDP)], Gewichtsbestimmung von Herz, Niere und Lunge, Messung der Proteinausscheidung sowie durch Messung der Genexpression von Bio- markern (z.B. ANP, Atrial Natriuretic Peptide, und BNP, Brain Natriuretic Peptide) mittels RT/TaqMan-PCR nach RNA-Isolation aus kardialem Gewebe bestimmt.
Die statistische Auswertung erfolgt mit Student's t-Test nach vorheriger Überprüfung der Varianzen auf Homogenität.
4. In vivo-Test zum Nachweis von anti-mineralokortikoider Wirksamkeit an wachen Hunden
Primäres Versuchsziel ist die Untersuchung des Einflusses von Testverbindungen mit anti- mineralokortikoider Rezeptorwirksamkeit auf die Aldosteron-induzierte Natrium-Retention. Hierbei wird analog einer publizierten Methode vorgegangen (Rosenthale,M.E., Schneider,F., Kassarich,J. & Datko,L. (1965). Determination of antialdosterone activity in normal dogs. Proc Soc Exp Biol Med, 118, 806-809. et al, 1965).
Männliche oder weibliche Beagle mit einem Gewicht zwischen 8 und 20 Kilogramm erhalten eine Standarddiät und haben freien Zugang zu Trinkwasser. An den Versuchstagen sind die Hunde nüchtern. Mit Propofol (4-6 mg/kg intravenös; Propofol 1% Parke-Davis®, Gödecke, Deutschland) wird eine Kurzzeitnarkose induziert, um mit einem Blasenkatheter wird ein Aliquot an Urin (als Ausgangswert Tag 1) zu gewinnen.
Am Tag 2 erhalten alle Hunde um 16 Uhr 0.3 mg Astonin, ein metabolisch stabiles Aldosteron- derivat (Astonin H, Merck, Deutschland).
Am folgenden Morgen (Tag 3) wird den Hunden die Testsubstanz oral in einer Gelatine-Kapsel verabreicht. 5 Stunden später wird den Hunden Blut zur Bestimmung der Plasmakonzentration der Substanz entnommen. Anschliessend wird erneut in einer Kurzzeitnarkose über einen Blasenkatheter Urin gewonnen.
Die Behandlung mit den Testsubstanzen fuhrt nach 5 Stunden zu einer Zunahme des Natrium- Kalium-Verhältnisses im Urin (die Natrium- und Kalium-Bestimmung erfolgt flammenphotometrisch). Als Positivkontrolle dient Spironolakton, das ebenso das Natrium/Kalium- Verhältnis im Urin dosisabhängig erhöht, als Negativkontrolle dient die Behandlung mit einer Leerkapsel.
Die Auswertung erfolgt über den Vergleich des Natrium/Kaliumverhältnisses im Urin zwischen Tag 1 und 3. Alternativ kann auch das Natrium/Kaliumverhältnisses zwischen Plazebo und Substanz an Tag 3 verglichen werden.
5. Chronisches Mvokardinfarkt-Modell der wachen Ratte
Männliche Wistar-Ratten (280-300 g Körpergewicht; Harlan- Winkelmann) werden mit 5% Isoflu- ran im Narkosekäfig narkotisiert, intubiert, an eine Beatmungspumpe (ugo basile 7025 rodent, 50 Hübe/min, 7 ml) angeschlossen und mit 2% Isofluran/N2O/O2 beatmet. Die Körpertemperatur wird durch eine Wärmematte auf 37-38°C gehalten. Als Schmerzmittel werden 0.05 mg/kg Temgesic subkutan gegeben. Der Brustkorb wird zwischen der dritten und vierten Rippe seitlich geöffnet und das Herz freigelegt. Die Herzkranzarterie des linken Ventrikels (LAD) wird mit einem Okklu- sionsfaden (Prolene 1 metric 5-0 EthiconlH) kurz unterhalb ihres Ursprungs (unterhalb des linken Atriums) unterstochen und permanent abgebunden. Das Auftreten eines Myokardinfarkts wird durch eine EKG-Messung (Cardioline, Remco, Italien) überwacht. Der Thorax wird wieder ge- schlössen und die Muskelschichten mit Ethibond excel 1 metric 5/0 695 IH und die Oberhaut mit Ethibond excel 3/0 6558H zugenäht. Die OP-Naht wird mit Sprühpflaster benetzt (z.B. Nebace- tin®N Sprühverband, Wirkstoff: Neomycinsulfat) und anschließend die Narkose beendet.
Eine Woche nach der Okklusion der LAD wird die Größe des Myokardinfarkts durch Echokardiographie (Sequoia 512, Acuson) abgeschätzt. Die Tiere werden randomisiert und in einzelne Be- handlungsgruppen sowie eine Kontrollgruppe ohne Substanzbehandlung aufgeteilt. Als weitere Kontrolle wird eine "sham"-Gruppe, bei der nur der Operationsvorgang, nicht aber die LAD- Okklusion durchgeführt wurde, mitgeführt.
Die Substanzbehandlung erfolgt über 8 Wochen per Gavage oder durch Zusatz der Testverbindung zum Futter oder Trinkwasser. Die Tiere werden wöchentlich gewogen und der Wasser- bzw. Futterverbrauch alle 14 Tage bestimmt.
Nach 8 Wochen Behandlung werden die Tiere erneut narkotisiert (2% Isofluran/N2θ/Luft) und ein Druckkatheter (Miliar SPR-320 2F) über die A. carotis in den linken Ventrikel eingeführt. Dort werden Herzfrequenz, linksventrikulärer Druck (LVP), linksventrikulärer enddiastolischer Druck (LVEDP), Kontraktilität (dp/dt) und Relaxationsgeschwindigkeit (τ) mit Hilfe des Powerlab- Systems (AD Instruments, ADI-PWLB-4SP) und der Chart 5-Software (SN 425-0586) erfasst und ausgewertet. Anschließend wird eine Blutprobe zur Bestimmung der Substanz-Plasmaspiegel und Plasmabiomarker entnommen und die Tiere abgetötet. Herz (Herzkammern, linker Ventrikel mit Septum, rechter Ventrikel), Leber, Lunge und Niere werden entnommen und gewogen.
6. Modell der stroke-prone spontan-hypertensiven Ratte
Die Verabreichung von Kochsalz bei der sogenannten stroke-prone spontan-hypertensiven Ratte (SP-SHR) führt in diesem Modell paradoxerweise nach wenigen Tagen zu einer Aufhebung der physiologischen salzinduzierten Repression der Renin- und Angiotensin-Freisetzung. Somit ist der Hypertonus in den SP-SHR-Tieren charakterisiert durch einen relativ hohen Reninspiegel. Als Folge des sich entwickelnden Hypertonus kommt es zu ausgeprägten Endorganschäden des Herzens und der Niere, die u.a. durch Proteinurie und Glomerulosklerose charakterisiert sind, sowie zu allgemeinen vaskulären Veränderungen. So können sich vor allem durch cerebrovaskuläre Läsionen im weiteren Verlauf Schlaganfälle bilden ("stroke-prone"), die zu einer hohen Mortalität der unbehandelten Tiere führen. In diesem Rattenmodell lassen sich somit Testsubstanzen auf Blutdruck-senkende und Endorgan-schützende Wirkung hin untersuchen.
Etwa 10 Wochen alte, männliche SP-SH-Ratten (Körpergewicht zwischen 190 und 220 g) werden einen Tag vor Versuchsbeginn randomisiert und Gruppen mit gleicher Tierzahl, in der Regel n = 12-14, zugeordnet. Während des gesamten Versuchs steht den Tieren kochsalzhaltiges Trinkwasser (2% NaCl) und Futter ad libitum zur Verfügung. Die Substanzen werden einmal täglich 6-8 Wochen lang per Gavage oder per Futter verabreicht (Ssniff, Germany). Als Plazebogruppe dienen Tiere, die genauso behandelt werden, aber entweder nur das Lösungsmittel oder das Futter ohne Testsubstanz erhalten. Im Rahmen einer Mortalitätsstudie wird der Versuch beendet, wenn ca. 50% der Plazebo-behandelten Tiere verstorben sind.
Die Wirkung der Testsubstanzen wird durch Verlaufsmessungen des systolischen Blutdrucks (über eine Schwanzmanschette) sowie der Proteinausscheidung im Urin verfolgt. Post mortem erfolgen Gewichtsbestimmungen von Herz, Niere und Lunge, sowie histopathologische Analysen von Herz,
Niere und Gehirn mit semi-quantitativem Scoring der histologischen Veränderungen. Verschiedene Biomarker (z.B. ANP, Atrial Natriuretic Peptide, und BNP, Brain Natriuretic Peptide, KIM-I, kidney induced molecule 1, Osteopontin-1) werden mittels RT/TaqMan-PCR nach RNA-Isolation aus Herz- und Nierengewebe bzw. Serum oder Plasma bestimmt.
Die statistische Auswertung erfolgt mit Student's t-Test nach vorheriger Überprüfung der Varianzen auf Homogenität.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette:
Zusammensetzung:
100 mg der erfmdungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfmdungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfmdungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfϊndungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.


































































































