WO2009097995A1 - Neue phenyl-substituierte imidazolidine, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung - Google Patents
Neue phenyl-substituierte imidazolidine, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung Download PDFInfo
- Publication number
- WO2009097995A1 WO2009097995A1 PCT/EP2009/000588 EP2009000588W WO2009097995A1 WO 2009097995 A1 WO2009097995 A1 WO 2009097995A1 EP 2009000588 W EP2009000588 W EP 2009000588W WO 2009097995 A1 WO2009097995 A1 WO 2009097995A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- cycloalkyl
- aryl
- inhibitors
- formula
- Prior art date
Links
- 0 CN(C1)CC1C(*)=O Chemical compound CN(C1)CC1C(*)=O 0.000 description 7
- DDBAVZIFAIWSOK-UHFFFAOYSA-N CC(C)(C(N1c(cc2)cc(C(F)(F)F)c2C#N)=O)N(Cc(cccc2)c2Cl)C1=O Chemical compound CC(C)(C(N1c(cc2)cc(C(F)(F)F)c2C#N)=O)N(Cc(cccc2)c2Cl)C1=O DDBAVZIFAIWSOK-UHFFFAOYSA-N 0.000 description 1
- MBWRUQSAXPZHDG-UHFFFAOYSA-N CC(C)(C(N1c(cc2Cl)ccc2Cl)=O)N(Cc2cc(C(F)(F)F)cc(C(F)(F)F)c2)C1=O Chemical compound CC(C)(C(N1c(cc2Cl)ccc2Cl)=O)N(Cc2cc(C(F)(F)F)cc(C(F)(F)F)c2)C1=O MBWRUQSAXPZHDG-UHFFFAOYSA-N 0.000 description 1
- PMGZJNCIQHGNLT-UHFFFAOYSA-N CC(C)(C)C(OCOP(C(CCCc1cc(Oc2ccccc2)ccc1)S(O)(=O)=O)(OCOC(C(C)(C)C)=O)=O)=O Chemical compound CC(C)(C)C(OCOP(C(CCCc1cc(Oc2ccccc2)ccc1)S(O)(=O)=O)(OCOC(C(C)(C)C)=O)=O)=O PMGZJNCIQHGNLT-UHFFFAOYSA-N 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N CCC(CC)CC1(CCCCC1)C(Nc(cccc1)c1SC(C(C)C)=O)=O Chemical compound CCC(CC)CC1(CCCCC1)C(Nc(cccc1)c1SC(C(C)C)=O)=O YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- MVCQKIKWYUURMU-UHFFFAOYSA-N CCCCCCCCCCCCCCCCOC(O1)=Nc2ccc(C)cc2C1=O Chemical compound CCCCCCCCCCCCCCCCOC(O1)=Nc2ccc(C)cc2C1=O MVCQKIKWYUURMU-UHFFFAOYSA-N 0.000 description 1
- UNAZAADNBYXMIV-UHFFFAOYSA-N CCNC(CC1)(CCN1c1ncnc2c1nc(-c(cccc1)c1Cl)[n]2-c(cc1)ccc1Cl)C(N)=O Chemical compound CCNC(CC1)(CCN1c1ncnc2c1nc(-c(cccc1)c1Cl)[n]2-c(cc1)ccc1Cl)C(N)=O UNAZAADNBYXMIV-UHFFFAOYSA-N 0.000 description 1
- KPRTURMJVWXURQ-UHFFFAOYSA-N CCOP(Cc(cc1)ccc1C(Nc(c(C#N)c1)ccc1Br)=O)(OCC)=O Chemical compound CCOP(Cc(cc1)ccc1C(Nc(c(C#N)c1)ccc1Br)=O)(OCC)=O KPRTURMJVWXURQ-UHFFFAOYSA-N 0.000 description 1
- OMUQJPMCQJZXMX-NNJIEVJOSA-N CN(C(N(Cc1ccccc1C#N)C(N(CCC1)C[C@@H]1N)=C1)O)C1=O Chemical compound CN(C(N(Cc1ccccc1C#N)C(N(CCC1)C[C@@H]1N)=C1)O)C1=O OMUQJPMCQJZXMX-NNJIEVJOSA-N 0.000 description 1
- NXFFJDQHYLNEJK-CYBMUJFWSA-N CS(c1c2[n](Cc(cc3)ccc3Cl)c([C@@H](CC(O)=O)CC3)c3c2cc(F)c1)(=O)=O Chemical compound CS(c1c2[n](Cc(cc3)ccc3Cl)c([C@@H](CC(O)=O)CC3)c3c2cc(F)c1)(=O)=O NXFFJDQHYLNEJK-CYBMUJFWSA-N 0.000 description 1
- HPTQTAOCTUDADS-LERMDLETSA-N C[C@@H]([C@@H](C[C@@]1(CNc2ncc[s]2)c(cc2)ccc2S(C)(=O)=O)C2CCCCC2)C1=O Chemical compound C[C@@H]([C@@H](C[C@@]1(CNc2ncc[s]2)c(cc2)ccc2S(C)(=O)=O)C2CCCCC2)C1=O HPTQTAOCTUDADS-LERMDLETSA-N 0.000 description 1
- LESOJAIAPGUSGB-UHFFFAOYSA-N Cc1cc(C)c(C)c(C[n](cc2)c3c2c(CNNC(c(cc2)cc(C#N)c2O)=O)ccc3)c1C Chemical compound Cc1cc(C)c(C)c(C[n](cc2)c3c2c(CNNC(c(cc2)cc(C#N)c2O)=O)ccc3)c1C LESOJAIAPGUSGB-UHFFFAOYSA-N 0.000 description 1
- IASPBORHOMBZMY-UHFFFAOYSA-N O=C(c1nc(cccc2)c2nc1)Nc1ccccc1-c1c[n](c(CN2CCNCC2)c[s]2)c2n1 Chemical compound O=C(c1nc(cccc2)c2nc1)Nc1ccccc1-c1c[n](c(CN2CCNCC2)c[s]2)c2n1 IASPBORHOMBZMY-UHFFFAOYSA-N 0.000 description 1
- NMRWDFUZLLQSBN-UHFFFAOYSA-N O=S(c(ccc(Cl)c1)c1Cl)(Nc(cc1Cl)cc(Cl)c1Oc1cc(cccc2)c2nc1)=O Chemical compound O=S(c(ccc(Cl)c1)c1Cl)(Nc(cc1Cl)cc(Cl)c1Oc1cc(cccc2)c2nc1)=O NMRWDFUZLLQSBN-UHFFFAOYSA-N 0.000 description 1
- VJLMRHSHSNLOGC-NOPZTHQXSA-N OC(C[C@@H]([C@H](C(O)=O)OC(/C=C/c(cc1)ccc1O)=O)OC(/C=C/c(cc1)ccc1O)=O)=O Chemical compound OC(C[C@@H]([C@H](C(O)=O)OC(/C=C/c(cc1)ccc1O)=O)OC(/C=C/c(cc1)ccc1O)=O)=O VJLMRHSHSNLOGC-NOPZTHQXSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N OC(c1ccccc1)=O Chemical compound OC(c1ccccc1)=O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N OC(c1cccnc1)=O Chemical compound OC(c1cccnc1)=O PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N OS(c1ccccc1)(=O)=O Chemical compound OS(c1ccccc1)(=O)=O SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- DPVGVPDHEWJQMW-UHFFFAOYSA-N Oc1ccccc1C(Oc(cccc1)c1O)=O Chemical compound Oc1ccccc1C(Oc(cccc1)c1O)=O DPVGVPDHEWJQMW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Abstract
Die Erfindung betrifft Verbindungen der Formel (I) worin die Reste die angegebenen Bedeutungen haben, sowie deren physiologisch verträgliche Salze. Die Verbindungen eignen sich z.B. als Antiadiposita und zur Behandlung des cardio-metabolischen Syndroms.
Description
Neue Phenyl-substituierte Imidazolidine, Verfahren zu deren Herstellung, diese Verbindungen enthaltende Arzneimittel und deren Verwendung
Die Erfindung betrifft Imidazolidindione, die am Imidstickstoff (N3) des Imidazolidin-2,4- dionsystems mit substituiertem Phenyl substituiert sind und ihre physiologisch verträglichen Salze.
Es sind bereits Imidazolidin-2,4-dione mit anti-androgener Wirkung sowie deren Verwendung zur Behandlung von Neoplasien der Prostata beschrieben worden (US 5,411,981).
Der Erfindung lag die Aufgabe zugrunde, Verbindungen zur Verfügung zu stellen, die eine therapeutisch verwertbare Wirkung entfalten. Insbesonders bestand die Aufgabe darin, neue Verbindungen zu finden, die zur Behandlung des metabolischen Syndroms, des Diabetes Typ II und der Adipositas geeignet sind.
Die Erfindung betrifft daher Verbindungen der Formel I,

worin bedeuten
Rl CN, NO2 oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (CrC12)-Alkyl, ((C6-C 12)-Aryl, (C]-Ci2)- Alkyien-(C6-Ci2)-Aryl, wobei (Ci-Ci2)-Alkyl, (C6-C12)-Aryl, (d-C12)-Alkylen- (C6-Ci 2)- Aryl bis zu dreifach unabhängig voneinander substituiert sein können mit Halogen, CN, CF3;
R5, R6, R7 unabhängig voneinander H, F, Cl, Br, CN, CF3, SF5, OCF3, NO2, S(O)J(C1-C6)- Alkyl], S(O)m[(C3-C9)-Cycloalkyl], S(O)mCF3, (C ,-Q)-AhCyI, (C1-Ce)-AIk0Xy, (C2-C6)-Alkenyl, (C2-C6)-Alkenyloxy, (C2-C6)- Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(CrC12)-Alkyl], -O(C=O)-(C6-Ci2)-Aryl, W-COOH, W-
CONH2, W-CO-NH[(d-C6)-Alkyl], W-CO-N[(Ci-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl], W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHR8-CO-R9 W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(d-C6)-Alkyl], W-CO-NH-C(=NH)N[(Ci-C6)-Alkyl]2, (CrC8)-Acyl, (C,-C7)-Acyloxy, W-C(=NH)NH2, W-C(=NH)NH0H, W-C(^N-SO2-NH2)NH2, W-C(=N-SO2-CF3)NH2, W-C[=N-SO2-(C1-C6)-Alkyl]NH2, W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2, NH2, NH-(Ci-Ci2)-Alkyl, N-[(Ci-Ci2)-Alkyl]2, W-NH-C(=NH)NH2, W-NH-C(=NH)NH[(Ci-C6)-Alkyl], W-NH-C(=NH)N[(Ci-C6)-Alkyl]2, W-NH-CO-NH2, W-NH-CO-NH[(Ci-C6)-Alkyl], W-NH-CO-N[(Ci-C6)- Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl], W-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-O-[(Ci-C6)-Alkyl], W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(Ci-C6)-Alkyl, W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(Ci-C6)-Alkyl, W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl], W-NH-C(=NH)-NH-C(=NH)-NH2, W-NH-C(=NH)-NH-C(=NH)-NH[(C,-C6)-Alkyl], W-NH-C(=NH)-NH-C(=NH)-N[(C , -C6)-Alkyl]2, W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(Ci-C6)-Alkyl], W-NH- W-SO2-N[(Ci-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl],
W-NH-W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[CC1-C6)- Alkyl], W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2, W-SO2-NH2, W-SO2-NH[(C1-C6)-Alkyl], W-SO2-N[(C1-C6)-Alkyl]2, W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2, W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NHt(C1-C6)- Alkyl], W-SO2-NH-CO-N[(C1-C6)-Alkyl]2, W-SO2-NH-CO-NH[CC3-C9)- Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C1-C6)- Alkyl], W-P(O)[O-(C1-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O- CH2-Aryl)2, W-P(O)(OH)2, (C6-C 12)-Aryl, O-(C6-C12)-Aryl, O-td-C^-Alkylen- (C6-C 12)-Aryl, S(O)m-(C6-C12)-Aryl, Tri(C1-C12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
m 0, 1, 2;
W eine Bindung oder (C1 -C6)- Alkyl;
R8 H, (C1-C6)-Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(=NH)NH2, COOH, CO-O(C1-C6)-Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(Ci-Ci2)-Alkyl, N[(C1-C12)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, N[(C3-
C9)-Cycloalkyl]2;
RIO NH-(d-C6)-Alkyl-SO3H, NH-(C!-C6)-Alkyl-SO2NH2, NH-(C1-C6)-Alkyl-SO2-(C1-
C6)-Alkyl, NH-(C1-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(C1-C6)-Alkyl-SO2-
.

sowie deren physiologisch verträgliche Salze.
Bevorzugt sind Verbindungen der Formel I, worin ein oder mehrere Reste die folgenden Bedeutungen haben:
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C1-Ci2)-Alkyl, ((C6-C i2)-Aryl, (C1-C12)- Alkylen-(C6-C12)-Aryl, wobei (d-C12)-Alkyl, (C6-C12)-Aryl, (d-C12)-Alkylen- (C6-C 12)- Aryl bis zu dreifach unabhängig voneinander substituiert sein können mit Halogen, CN, CF3;
R5 F, Cl, Br, CN, CF3, SF5, OCF3, NO2, S(O)ra[(C1-C6)-Alkyl], S(OU(C3-C9)-
Cycloalkyl], S(O)01CF3, (Ci-C6)-Alkyl, (C1-Ce)-AIk0Xy, (C2-C6)-Alkenyl, (C2-C6)-
Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(d-C12)-Alkyl],
-O(C=O)-(C6-C12)-Aryl, W-COOH, W-CONH2, W-CO-NH[(Ci-C6)-Alkyl],
W-CO-N[(Ci-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl],
W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHR8-CO-R9,
W-CO-RlO, W-C0-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(C1-C6)-Alkyl],
W-CO-NH-C(=NH)N[(CrC6)-Alkyl]2, (C1-Cg)-ACyI, (Ci-C7)-Acyloxy,
W-C(^NH)NH2, W-C(=NH)NH0H, W-C(=N-SO2-NH2)NH2,
W-C(=N-SO2-CF3)NH2, W-C[=N-SO2-(Ci-C6)-Alkyl]NH2,
W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2,
NH2, NH-(d-Ci2)-Alkyl, N-[(C1-C12)-Alkyl]2, W-NH-C(=NH)NH2,
W-NH-C(=NH)NH[(C1-C6)-Alkyl], W-NH-C(=NH)N[(C1-C6)-Alkyl]2,
W-NH-CO-NH2, W-NH-CO-NH[(d-C6)-Alkyl],
W-NH-C0-N[(C ! -C6)-Alkyl]2, W-NH-CO-NH [(C3-C9)-Cycloalkyl] ,
W-NH-CO-N[(C3-C9)-Cycloalkyl]2,
W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-O-[(d-C6)-Alkyl],
W-NH-CO-NH-[(C1-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(Ci-C6)-Alkyl,
W-NH-CO-NH-Sθ2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(Ci-C6)-Alkyl,
W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl], W-NH-C(=NH)-NH-C(=NH)-NH2,
W-NH-C(=NH)-NH-C(=NH)-NH[(CrC6)-Alkyl],
W-NH-C(=NH)-NH-C(=NH)-N[(C1-C6)-Alkyl]2,
W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(Ci-C6)-Alkyl],
W-NH- W-SO2-N[(d-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl],
W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[(d-C6)-Alkyl],
W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2,
W-SO2-NH2, W-SO2-NH[(C1-C6)-Alkyl], W-SO2-N[(C1-C6)-Alkyl]2,
W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2,
W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NHt(C1-C6)-
Alkyl], W-SO2-NH-CO-Nt(C1 -C6)- Alkyl]2, W-SO2-NH-CO-NH[(C3-C9)-
Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C1-C6)-
Alkyl], W-P(O)[O-(CrC6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O-
CH2-Aryl)2, W-P(O)(OH)2, (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(d-C12)-Alkylen-
(C6-C I2)-Aryl, S(O)01-(C6-C 12)-Aryl, Tri(C1-Ci2)-alkylsilyl, wobei das Alkyl 1 bis 6
Kohlenstoffatome aufweist;
R6, R7 unabhängig voneinander H, Halogen, CN, CF3, SF5, OCF3, S (O)n, [(C1 -C6)- Alkyl], S(O)m[(C3-C9)-Cycloalkyl], NO2, S(O)mCF3, (C ,-C6)- Alkyl, (C1-Q)-AIkOXy, (C2- C6)-Alkenyl, (C2-C6)- Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(d-C12)-Alkyl], -O(C=O)-(C6-C12)-Aryl, W-COOH, W-CONH2, W-CO-NH[(Ci-C6)-Alkyl], W-CO-N[(Ci-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl], W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHR8-CO-R9, W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(d-C6)-Alkyl], W-CO-NH-C(=NH)N[(d-C6)-Alkyl]2, (d-C8)-Acyl, (d-C7)-Acyloxy, W-C(=NH)NH2, W-C(=NH)NH0H, W-C(=N-SO2-NH2)NH2, W-C(^N-SO2-CF3)NH2, W-C[=N-SO2-(Ci-C6)-Alkyl]NH2, W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2, NH2, NH-(Ci-Ci2)-Alkyl, N- [(C 1-C12)- Alkyl] 2, W-NH-C(=NH)NH2,
W-NH-C(=NH)NH[(C1-C6)-Alkyl], W-NH-C(=NH)N[(Cι-C6)-Alkyl]2, W-NH-CO-NH2, W->ffl-CO-NH[(Ci-C6)-Alkyl], W-NH-CO-N[(Ci-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl], W-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-NH-CO-NH-[(C1-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], W-NH-CO-NH-[(C1-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(C1-C6)-Alkyl, W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(C1 -C6)- Alkyl, W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl] , W-NH-C(=NH)-NH-C(=NH)-NH2, W-NH-C(=NH)-NH-C(=NH)-NH[(Cj-C6)-Alkyl], W-NH-C(=NH)-NH-C(=NH)-N[(Ci-C6)-Alkyl]2, W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(Ci-C6)-Alkyl], W-NH-W-SO2-Nt(C1 -C6)- Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl], W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[(C1-C6)-Alkyl], W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2, W-SO2-NH2, W-SO2-NH[(d-C6)-Alkyl], W-SO2-N[(Ci-C6)-Alkyl]2, W-SO2-NH[(C3-C9)-Cycloalkyl] , W-SO2-N[(C3-C9)-Cycloalkyl]2, W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NH[(Cj-C6)- Alkyl], W-SO2-NH-CO-N[(C1-C6)-Alkyl]2, W-SO2-NH-CO-NH[(C3-C9)- Cycloalkyl] , W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C , -C6)- Alkyl], W-P(O)[O-(C1-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O- CH2-Aryl)2, W-P(O)(OH)2, (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(Cj-C12)-Alkylen- (C6-C I2)- Aryl, S(O)m-(C6-Ci2)-Aryl, Tri(Ci-C12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
m 0, 1, 2;
W eine Bindung oder (C J-C6)- Alkyl;
R8 H, (C 1-C6)- Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(=NH)NH2, COOH, CO-O(C J-C6)- Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(C,-C12)-Alkyl, N[(Ci-C12)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, Nf(C3-
C9)-Cycloalkyi]2;
RIO NH-(d-C6)-Alkyl-SO3H, NH-(CrC6)-Alkyl-SO2NH2, NH-(C1-C6)-Alkyl-SO2-(C1-
C6)-Alkyl, NH-(C1-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(C1-C6)-Alkyl-SO2-
.

sowie deren physiologisch verträgliche Salze.
Besonders bevorzugt sind Verbindungen der Formel I, worin ein oder mehrere Reste die folgenden Bedeutungen haben:
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C1-C12)-AUcylen-(C6-C12)-Aryl;
R5 F, Cl, Br, CF3, SF5, OCF3, S(O)2[(d-C6)-Alkyl], (C1 -C6)- Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[(C1-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[CC1-C6)- Alkyl], (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(CrC12)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CN, CF3, SF5, OCF3, S(O)01 [(Ci -C6)- Alkyl], S(O)m[(C3-C9)-Cycloalkyl], S(O)mCF3, (C ,-C6)- Alkyl, (C1-Ce)-AIk0Xy, (C2-C6)- Alkenyl, (C2-C6)- Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(Ci-Ci2)-Alkyl],
-O(C=O)-(C6-C12)-Aryl, W-COOH, W-CONH2, W-CO-NH [(Ci -C6)- Alkyl],
W-CO-N[(C1-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl],
W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHR8-CO-R9,
W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(C,-C6)- Alkyl],
W-CO-NH-C(=NH)N[(CrC6)-Alkyl]2, (Ci-C8)-Acyl, (d-C^-Acyloxy,
W-C(=NH)NH2, W-C(=NH)NHOH, W-C(=N-SO2-NH2)NH2,
W-C(=N-SO2-CF3)NH2, W-C[=N-SO2-(C1-C6)-Alkyl]NH2,
W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2,
NH2, NH-(d-Ci2)-Alkyl, N-[(Ci-C12)-Alkyl]2, W-NH-C(=NH)NH2,
W-NH-C(=NH)NH[(C1-C6)-Alkyl], W-NH-C(=NH)N[(Ci-C6)-Alkyl]2,
W-NH-CO-NH2, W-NH-CO-NH[(Ci-C6)- Alkyl],
W-NH-CO-N[(Cj-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl],
W-NH-CO-N[(C3-C9)-Cycloalkyl]2,
W-NH-CO-NH-KC1-C6)- Alkyl]-CO-O-[(C!-C6)- Alkyl],
W-NH-CO-NH-[(C1-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(C1-C6)-Alkyl,
W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(C1-Ce)-AIlCyI,
W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl] , W-NH-C(=NH)-NH-C(=NH)-NH2,
W-NH-C(=NH)-NH-C(=NH)-NH[(Ci-C6)-Alkyl],
W-NH-C(=NH)-NH-C(=NH)-N[(C1-C6)-Alkyl]2,
W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(d-C6)-Alkyl],
W-NH- W-SO2-N[(Ci-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl],
W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[(CrC6)-Alkyl],
W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2,
W-SO2-NH2, W-SO2-NH[(d-C6)-Alkyl], W-SO2-N[(Ci-C6)-Alkyl]2,
W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2,
W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NH[(C J-C6)-
Alkyl], W-SO2-NH-CO-Nt(C1-Ce)-AIlCyI]2, W-SO2-NH-CO-NH[(C3-C9)-
Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C-C6)-
Alkyl], W-P(O)[O-(d-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O-CH2-
Aryl)2, W-P(O)(OH)2, (C6-C 12)-Aryl, O-(C6-C,2)-Aryl, O-(C1-C12)-Alkylen-(C6-
C12)-Aryl, S(O)m-(C6-Ci2)-Aryl, Tri(CrC12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
m 0, 1, 2;
W eine Bindung oder (C1 -C6)- Alkyl;
R8 H, (Ci-C6)-Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(=NH)NH2, COOH, CO-O(C ,-C6)- Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(C,-Ci2)-Alkyl, N[(C1-C12)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, Nf(C3-
C9)-Cycloalkyl]2;
RIO NH-(Ci -C6)- Alkyl-SO3H, NH-(Ci -C6)- Alkyl-SO2NH2, NH-(C,-C6)-Alkyl-SO2-(C1-
C6)-Alkyl, NH-(C,-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(Ci-C6)-Alkyl-SO2-

sowie deren physiologisch verträgliche Salze.
Ganz besonders bevorzugt sind Verbindungen der Formel I, worin ein oder mehrere Reste die folgenden Bedeutungen haben:
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (Ci-C12)-Alkylen-(C6-C12)-Aryl;
R5 F, Cl, Br, CF3, SF5, OCF3, S(O)2[(Ci-C6)-Alkyl], (CrC6)-Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[(C1-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(Ci-C6)-Alkyl], (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(C1-C12)-Alkylen-(C6-C,2)-Aryl, S(O)m-(C6-C12)-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CF3, SF5, OCF3, S(O)2[(Ci-C6)-Alkyl], (Cj- C6)-Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[CC1-C6)- AlkylJ-CO-O-Kd-C^-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(d-C6)-Alkyl], (C6-C12)-Aryl, 0-(C6-C J2)- Aryl, O-(C1-C12)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl;
sowie deren physiologisch verträgliche Salze.
Weiter ganz besonders bevorzugt sind Verbindungen der Formel I, worin ein oder mehrere Reste die folgenden Bedeutungen haben:
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A CH;
B CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C1-C12)-Alkylen-(C6-C12)-Aryl;
R5 SF5, OCF3, S(O)2[(Ci -C6)-Alkyl], -NH-CO-NH- [(C1 -C6)- Alkyl] -CO-O- [(Ci -C6)-
Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(C1-C6)-Alkyl],
O-CCrC^-Alkylen-CQ-C^-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CF3, SF5, OCF3, S(O)2 [(Cj -C6)- Alkyl], (C1- C6)-Alkyl, OH5 -COOH,
NH2, -NH-CO-NH-[(C,-C6)-Alkyl]-CO-O-[(Ci-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(d-C6)-Alkyl], (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(C1-C12)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl;
sowie deren physiologisch verträgliche Salze.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen Rl gleich CN ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen Rl gleich NO2 ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen Rl gleich Halogen ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen R2 gleich CF3 ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen Rl gleich Halogen ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen A gleich CH ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen A gleich N ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen B gleich CH ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen B gleich N ist.
In einer Ausfuhrungsform sind Verbindungen der Formel I bevorzugt, in denen A und B gleich CH sind.
In einer Ausführungsform sind Verbindungen der Formel I bevorzugt, in denen A gleich N und B gleich CH ist.
In einer Ausführungsform sind Verbindungen der Formel I bevorzugt, in denen R5 ungleich H ist.
In einer Ausführungsform sind Verbindungen der Formel I bevorzugt, in denen R5 und R6 ungleich H sind.
In einer Ausführungsform sind Verbindungen der Formel I bevorzugt, in denen R5 gleich OCF3 ist.
In einer Ausführungsform sind Verbindungen der Formel I bevorzugt, in denen R5 gleich SF5 ist.
Gegenstand der Erfindung sind weiterhin sowohl Stereoisomerengemische der Formel I als auch die reinen Stereoisomere der Formel I, sowie Diastereoisomerengemische der Formel I als auch die reinen Diastereoisomere. Die Trennung der Gemische erfolgt z. B. auf chromatographischem Weg.
Die Erfindung bezieht sich auf Verbindungen der Formel I, in Form ihrer Tautomere, Racemate, racemischen Mischungen, Stereoisomerengemische, reinen Stereoisomere, Diastereoisomerengemische, reinen Diastereoisomere. Die Trennung der Gemische erfolgt z. B. auf chromatographischem Weg.
Pharmazeutisch verträgliche Salze sind aufgrund ihrer höheren Wasserlöslichkeit gegenüber den Ausgangs- bzw. Basisverbindungen besonders geeignet für medizinische Anwendungen. Diese Salze müssen ein pharmazeutisch verträgliches Anion oder Kation aufweisen. Geeignete pharmazeutisch verträgliche Säureadditionssalze der erfindungsgemäßen Verbindungen sind Salze anorganischer Säuren, wie Salzsäure, Bromwasserstoff-, Phosphor-, Metaphosphor-, Salpeter- und Schwefelsäure sowie organischer Säuren, wie z.B. Essigsäure, Benzolsulfon-, Benzoe-, Zitronen-, Ethansulfon-, Fumar-, Glucon-, Glykol-, Isethion-, Milch-, Lactobion-, Malein-, Äpfel-, Methansulfon-, Bernstein-, p-Toluolsulfon- und Weinsäure. Geeignete pharmazeutisch verträgliche basische Salze sind Ammoniumsalze, Alkalimetallsalze (wie Natrium- und Kaliumsalze), Erdalkalisalze (wie Magnesium- und Calciumsalze), Trometamol (2-Amino-2-hydroxymethyl-l,3-propandiol), Diethanolamin, Lysin oder Ethylendiamin.
Salze mit einem nicht pharmazeutisch verträglichen Anion, wie zum Beispiel Trifluoracetat, gehören ebenfalls in den Rahmen der Erfindung als nützliche Zwischenprodukte für die Herstellung oder Reinigung pharmazeutisch verträglicher Salze und/oder für die Verwendung in nicht-therapeutischen, zum Beispiel in-vitro- Anwendungen.
Die erfindungsgemäßen Verbindungen können auch in verschiedenen polymorphen Formen vorliegen, z.B. als amorphe und kristalline polymorphe Formen. Alle polymorphen Formen der erfindungsgemäßen Verbindungen gehören in den Rahmen der Erfindung und sind ein weiterer Aspekt der Erfindung.
Nachfolgend beziehen sich alle Verweise auf "Verbindung(en) gemäß Formel I" auf Verbindung(en) der Formel I wie vorstehend beschrieben, sowie ihre Salze und Solvate wie hierin beschrieben.
Unter (Ci-C12)-Alkyl wird eine geradkettige oder verzweigte Kohlenwasserstoffkette mit einem bis zwölf Kohlenstoffen verstanden, wie z.B. Methyl, Ethyl, iso-Propyl, tert.-Butyl, Hexyl, Dodecyl.
Unter Halogen wird F, Cl oder Br verstanden.
Unter einem Arylrest wird ein Phenyl, Naphthyl-, Biphenyl-, Tetrahydronaphthyl-, alpha- oder beta-Tetralon-, Indanyl- oder Indan-1-on-ylrest verstanden.
Die Arylreste können ein oder mehrfach mit geeigneten Gruppen wie oben beschrieben substituiert sein.
Unter Heteroarylrest werden aromatische Ringe und Ringsysteme verstanden, die außer Kohlenstoff noch Heteroatome, wie zum Beispiel Stickstoff, Sauerstoff oder Schwefel enthalten. Ferner gehören auch Ringsysteme zu dieser Definition, worin der Heteroarylrest mit Benzolkernen kondensiert ist. Ebenso fallen darunter Systeme, bei welchen eine oder mehrere CH-Gruppe(n) durch C=O oder C=S, vorzugsweise C=O, ersetzt ist (sind).
Geeignete Heteroarylreste sind z.B. Furyl, Imidazolyl, Benzimidazolyl, Indolyl, Indolinyl, Pyrimidinyl, Pyridyl, Pyrazinyl, Pyrrolyl, Thiazolyl, Oxazolyl, Thienyl, 1,2,3-Triazolyl, 1,2,4- Triazolyl, Tetrazolyl, Isoxazolyl, Pyridazinyl, 1,3,5-Triazinyl, 1,2,4-Triazinyl; das 2H- Pyridazin-3-on-, Dihydropyridazin-3,6-dion-, Imidazolidin-2-on-, l,3-Dihydro-imidazol-2-on-, Imidazolidin-2,5-dion-, Chinolin-, Isochinolin-, Chinoxalin-, Chinazolin-System.
Die Verknüpfung mit den Heteroarylresten kann an jedem der dafür in Frage kommenden Atome erfolgen; so kann z. B. Pyridyl sowohl für 2-, 3- als auch 4-Pyridyl stehen; Thienyl sowohl für 2- als auch 3 -Thienyl stehen; Furyl sowohl für 2- als auch 3 -Furyl stehen.
Umfasst sind weiterhin die entsprechenden N-Oxide dieser Verbindungen, also z.B. l-Oxy-2-, 3- oder 4-pyridyl.
Die Heteroarylreste können ein- oder mehrfach mit geeigneten Gruppen wie oben beschrieben substituiert sein.
Die Erfindung umfasst auch Solvate oder Hydrate der Verbindungen der Formel I.
Die Verbindungen der Formel I stellen Cannabinoid Rezeptor 1 (CBlR) Modulatoren dar und sind als solche beim Menschen und bei Tieren zur Behandlung oder zur Verhütung von Krankheiten geeignet, die auf einer Störung des Endocannabinoid-systems beruhen. Zum Beispiel, und nicht einschränkend, sind die Verbindungen der Formel I als psychotrope Medikamente nützlich, insbesondere zur Behandlung psychiatrischer Störungen, darunter Angstzustände, Depressionen, Gemütsstörungen, Schlaflosigkeit, Delirien, Zwangsneurosen, generelle Psychosen, Schizophrenie, Defizit der Aufmerksamkeit und Hyperaktivität (ADHS) bei hyperkinetischen Kindern, sowie zur Behandlung von Störungen in Zusammenhang mit dem Gebrauch psychotroper Substanzen, insbesondere in dem Fall eines Missbrauchs einer Substanz und/oder einer Abhängigkeit von einer solchen Substanz, darunter Alkoholabhängigkeit und Nikotinabhängigkeit aber auch Abhängigkeit von Kokain, Methamphetamin und Heroin (siehe z.B. Behavioural Pharmacology 2005, 16:275-296). Übersichten über CB IR- vermittelte therapeutische Eingriffsmöglichkeiten finden sich z. B. in Ken Mackie: Annu. Rev. Pharmacol. Toxicol. 46, 101-122 (2006), S. C. Black: Curr. Opin. Investig. Drugs 5, 389-394 (2004), V. Di Marzio et al.: Nat. Rev. Drug Discov. 3, 771-784 (2004), B. Le FoIl et al.: J. Pharmacol. Exp. Ther. 312, 875-883 (2005) oder L. Walter et al.: Br. J. Pharmacol. 141, 775-785 (2004).
Die erfindungsgemäßen Verbindungen der Formel I können als Medikamente zur Behandlung von Migräne, Stress, Krankheiten psychosomatischen Ursprungs, Panikattackenkrisen, Epilepsie, Bewegungsstörungen, insbesondere Dyskinesien oder Parkinsonsche Krankheit, Zittern und Dystonie verwendet werden.
Die erfindungsgemäßen Verbindungen der Formel I können weiterhin auch als Medikamente zur Behandlung von Gedächtnisstörungen, geistiger Defekte, insbesondere zur Behandlung der Altersdemenzen, der Alzheimer' sehen Krankheit sowie zur Behandlung verminderter Aufmerksamkeit oder Wachsamkeit verwendet werden.
Ferner können die Verbindungen der Formel I als Neuroprotektoren, zur Behandlung von Ischämie, Schädelverletzungen und Behandlung neurodegenerativer Krankheiten, darunter Chorea, Chorea Huntington, Tourette-Syndrom, verwendet werden.
Die erfindungsgemäßen Verbindungen der Formel I können ferner als Medikamente bei der Schmerzbehandlung verwendet werden; dazu zählen neuropathische Schmerzen, akute periphere Schmerzen, chronische Schmerzen entzündlicher Herkunft.
Die erfindungsgemäßen Verbindungen der Formel I können weiterhin als Medikamente zur Behandlung von Essstörungen (z. B. zwanghafte Essanfälle (hinge eating disorder), Anorexie und Bulimie), zur Behandlung der Sucht nach Süßigkeiten, Kohlenhydraten, Drogen, Alkohol oder anderen suchterzeugenden Substanzen dienen.
Die erfindungsgemäßen Verbindungen der Formel I sind besonders geeignet zur Behandlung der Adipositas oder der Bulimie sowie zur Behandlung von Diabetes Typ II wie auch zur Behandlung von Dyslipidämien und des metabolischen Syndroms. Die erfindungsgemäßen Verbindungen der Formel I sind daher zur Behandlung der Adipositas und der Gefahren in Zusammenhang mit Adipositas, insbesondere der kardiovaskulären Gefahren, nützlich. Ferner können die erfindungsgemäßen Verbindungen der Formel I als Medikamente zur Behandlung gastrointestinaler Störungen, zur Behandlung von Durchfällen, von Magen- Darmgeschwüren, von Erbrechen, von Blasenleiden und Störungen des Wasserlassens, von Störungen endokrinen Ursprungs, von kardiovaskulären Problemen, von niedrigem Blutdruck, des hämorrhagischen Schocks, des septischen Schocks, chronischer Leberzirrhose, Lebersteatose, der nicht alkoholischen Steatohepatitis, von Asthma, des Raynaudschen Syndroms, des Glaukoms, von Fruchtbarkeitsbeschwerden, Schwangerschaftsunterbrechung, Frühgeburt, Entzündungserscheinungen, Krankheiten des Immunsystems, insbesondere autoimmun- und neuroinflammatorische, wie zum Beispiel rheumatische Gelenkentzündung, reaktive Arthritis, von Krankheiten, die zu Demyelinisation fuhren, der multiplen Sklerose, von Infektionskrankheiten und viralen Erkrankungen, wie zum Beispiel von Enzephalitis, ischämischem Schlaganfall sowie als Medikamente zur Krebschemotherapie, zur Behandlung des Guillain-Barre-Syndroms und zur Behandlung der Osteoporose verwendet werden. Die erfindungsgemäßen Verbindungen der Formel I können weiterhin auch als Medikamente zur Behandlung des Syndroms der polycystischen Ovarien (PCOS, polycystic ovary Syndrome) Verwendung finden.
Gemäß der vorliegenden Erfindung sind die Verbindungen der Formel I besonders nützlich zur Behandlung psychotischer Beschwerden, insbesondere der Schizophrenie, verminderter Aufmerksamkeit und Hyperaktivität (ADHS) bei hyperkinetischen Kindern, zur Behandlung von Essstörungen und der Adipositas, zur Behandlung des Diabetes Typ II, zur Behandlung von Gedächtnisdefiziten und kognitiven Defiziten, zur Behandlung der Alkoholsucht, der Nikotinsucht, das heißt für die Alkohol- und Tabakentwöhnung.
Ganz besonders nützlich sind die erfindungsgemäßen Verbindungen der Formel I zur Behandlung und Verhütung von Essstörungen Appetitstörungen, metabolischen Störungen, gastrointestinalen Störungen, Entzündungserscheinungen, Erkrankungen des Immunsystems, psychotischen Störungen, der Alkoholsucht und der Nikotinsucht.
Gemäß einem ihrer Aspekte bezieht sich die Erfindung auf den Gebrauch einer Verbindung der Formel I, ihrer pharmazeutisch akzeptablen Salze und deren Solvate oder Hydrate zur Behandlung der oben angegebenen Störungen und Erkrankungen.
Die Verbindung(en) der Formel I können auch in Kombination mit weiteren Wirkstoffen verabreicht werden.
Die Menge einer Verbindung gemäß Formel I, die erforderlich ist, um den gewünschten biologischen Effekt zu erreichen, ist abhängig von einer Reihe von Faktoren, z.B. der gewählten spezifischen Verbindung, der beabsichtigten Verwendung, der Art der Verabreichung und dem klinischen Zustand des Patienten. Im allgemeinen liegt die Tagesdosis im Bereich von 0,3 mg bis 100 mg (typischerweise von 3 mg und 50 mg) pro Tag pro Kilogramm Körpergewicht, z.B. 3-10 mg/kg/Tag. Eine intravenöse Dosis kann z.B. im Bereich von 0,3 mg bis 1,0 mg/kg liegen, die geeigneterweise als Infusion von 10 ng bis 100 ng pro Kilogramm pro Minute verabreicht werden kann. Geeignete Infusionslösungen für diese Zwecke können z.B. von 0,1 ng bis 10 mg, typischerweise von 1 ng bis 10 mg pro Milliliter, enthalten. Einzeldosen können z.B. von 1 mg bis 10 g des Wirkstoffs enthalten. Somit können Ampullen für Injektionen beispielsweise von 1 mg bis 100 mg, und oral verabreichbare Einzeldosisformulierungen, wie zum Beispiel Tabletten oder Kapseln, können beispielsweise von 1,0 bis 1000 mg, typischerweise von 10 bis 600 mg enthalten. Zur Therapie der oben genannten Zustände können die Verbindungen gemäß Formel I selbst als Verbindung verwendet werden, vorzugsweise liegen sie jedoch mit einem verträglichen Träger in Form einer pharmazeutischen Zusammensetzung vor. Der Träger muss natürlich verträglich sein, in dem Sinne, dass er mit den anderen Bestandteilen der Zusammensetzung kompatibel ist und nicht gesundheitsschädlich für den Patienten ist. Der Träger kann ein Feststoff oder eine Flüssigkeit oder beides sein und wird vorzugsweise mit der Verbindung als Einzeldosis formuliert, beispielsweise als Tablette, die von 0,05% bis 95 Gew.- % des Wirkstoffs enthalten kann. Weitere pharmazeutisch aktive Substanzen können ebenfalls
vorhanden sein, einschließlich weiterer Verbindungen gemäß Formel I. Die erfindungsgemäßen pharmazeutischen Zusammensetzungen können nach einer der bekannten pharmazeutischen Methoden hergestellt werden, die im wesentlichen darin bestehen, dass die Bestandteile mit pharmakologisch verträglichen Träger- und/oder Hilfsstoffen gemischt werden.
Erfindungsgemäße pharmazeutische Zusammensetzungen sind solche, die für orale, rektale, topische, perorale (z.B. sublinguale) und parenterale (z.B. subkutane, intramuskuläre, intradermale oder intravenöse) Verabreichung geeignet sind, wenngleich die geeignetste Verabreichungsweise in jedem Einzelfall von der Art und Schwere des zu behandelnden Zustandes und von der Art der jeweils verwendeten Verbindung gemäß Formel I abhängig ist. Auch dragierte Formulierungen und dragierte Retardformulierungen gehören in den Rahmen der Erfindung. Bevorzugt sind säure- und magensaftresistente Formulierungen. Geeignete magensaftresistente Beschichtungen umfassen Celluloseacetatphthalat, Poylvinylacetatphthalat, Hydroxypropylmethylcellulosephthalat und anionische Polymere von Methacrylsäure und Methacrylsäuremethylester.
Geeignete pharmazeutische Verbindungen für die orale Verabreichung können in separaten Einheiten vorliegen, wie zum Beispiel Kapseln, Oblatenkapseln, Lutschtabletten oder Tabletten, die jeweils eine bestimmte Menge der Verbindung gemäß Formel I enthalten; als Pulver oder Granulate; als Lösung oder Suspension in einer wässrigen oder nicht-wässrigen Flüssigkeit; oder als eine Öl-in- Wasser- oder Wasser-in-Öl-Emulsion. Diese Zusammensetzungen können, wie bereits erwähnt, nach jeder geeigneten pharmazeutischen Methode zubereitet werden, die einen Schritt umfasst, bei dem der Wirkstoff und der Träger (der aus einem oder mehreren zusätzlichen Bestandteilen bestehen kann) in Kontakt gebracht werden. Im allgemeinen werden die Zusammensetzungen durch gleichmäßiges und homogenes Vermischen des Wirkstoffs mit einem flüssigen und/oder feinverteilten festen Träger hergestellt, wonach das Produkt, falls erforderlich, geformt wird. So kann beispielsweise eine Tablette hergestellt werden, indem ein Pulver oder Granulat der Verbindung verpresst oder geformt wird, gegebenenfalls mit einem oder mehreren zusätzlichen Bestandteilen. Gepresste Tabletten können durch tablettieren der Verbindung in frei fließender Form, wie beispielsweise einem Pulver oder Granulat, gegebenenfalls gemischt mit einem Bindemittel, Gleitmittel, inertem Verdünner und/oder einem (mehreren) oberflächenaktiven/dispergierenden Mittel in einer geeigneten Maschine hergestellt
werden. Geformte Tabletten können durch Formen der pul verförmigen, mit einem inerten flüssigen Verdünnungsmittel befeuchteten Verbindung in einer geeigneten Maschine hergestellt werden.
Pharmazeutische Zusammensetzungen, die für eine perorale (sublinguale) Verabreichung geeignet sind, umfassen Lutschtabletten, die eine Verbindung gemäß Formel I mit einem Geschmacksstoff enthalten, üblicherweise Saccharose und Gummi arabicum oder Tragant, und Pastillen, die die Verbindung in einer inerten Basis wie Gelatine und Glycerin oder Saccharose und Gummi arabicum umfassen.
Geeignete pharmazeutische Zusammensetzungen für die parenterale Verabreichung umfassen vorzugsweise sterile wässrige Zubereitungen einer Verbindung gemäß Formel I, die vorzugsweise isotonisch mit dem Blut des vorgesehenen Empfangers sind. Diese Zubereitungen werden vorzugsweise intravenös verabreicht, wenngleich die Verabreichung auch subkutan, intramuskulär oder intradermal als Injektion erfolgen kann. Diese Zubereitungen können vorzugsweise hergestellt werden, indem die Verbindung mit Wasser gemischt wird und die erhaltene Lösung steril und mit dem Blut isotonisch gemacht wird. Injizierbare erfindungsgemäße Zusammensetzungen enthalten im allgemeinen von 0,1 bis 5 Gew.-% der aktiven Verbindung.
Geeignete pharmazeutische Zusammensetzungen für die rektale Verabreichung liegen vorzugsweise als Einzeldosis-Zäpfchen vor. Diese können hergestellt werden, indem man eine Verbindung gemäß Formel I mit einem oder mehreren herkömmlichen festen Trägern, beispielsweise Kakaobutter, mischt und das entstehende Gemisch in Form bringt.
Geeignete pharmazeutische Zusammensetzungen für die topische Anwendung auf der Haut liegen vorzugsweise als Salbe, Creme, Lotion, Paste, Spray, Aerosol oder Öl vor. Als Träger können Vaseline, Lanolin, Polyethylenglykole, Alkohole und Kombinationen von zwei oder mehreren dieser Substanzen verwendet werden. Der Wirkstoff ist im allgemeinen in einer Konzentration von 0,1 bis 15 Gew.-% der Zusammensetzung vorhanden, beispielsweise von 0,5 bis 2%.
Auch eine transdermale Verabreichung ist möglich. Geeignete pharmazeutische Zusammensetzungen für transdermale Anwendungen können als einzelne Pflaster vorliegen, die für einen langzeitigen engen Kontakt mit der Epidermis des Patienten geeignet sind. Solche Pflaster enthalten geeigneterweise den Wirkstoff in einer gegebenenfalls gepufferten wässrigen Lösung, gelöst und/oder dispergiert in einem Haftmittel oder dispergiert in einem Polymer. Eine geeignete Wirkstoff-Konzentration beträgt ca. 1% bis 35%, vorzugsweise ca. 3% bis 15%. Als eine besondere Möglichkeit kann der Wirkstoff, wie beispielsweise in Pharmaceutical Research, 2(6): 318 (1986) beschrieben, durch Elektrotransport oder Iontophorese freigesetzt werden.
Als weitere Wirkstoffe für die Kombinationspräparate sind geeignet: Alle Antidiabetika, die in der Roten Liste 2007, Kapitel 12 genannt sind; alle Abmagerungsmittel/ Appetitzügler, die in der Roten Liste 2007, Kapitel 1 genannt sind; alle Diuretika, die in der Roten Liste 2007, Kapitel 36 genannt sind; alle Lipidsenker, die in der Roten Liste 2007, Kapitel 58 genannt sind. Sie können mit der erfindungsgemäßen Verbindung der Formel I insbesondere zur synergistischen Wirkungsverbesserung kombiniert werden. Die Verabreichung der Wirkstoffkombination kann entweder durch getrennte Gabe der Wirkstoffe an den Patienten oder in Form von Kombinationspräparaten, worin mehrere Wirkstoffe in einer pharmazeutischen Zubereitung vorliegen, erfolgen. Erfolgt die Gabe der Wirkstoffe durch getrennte Verabreichung der Wirkstoffe, so kann diese gleichzeitig oder nacheinander erfolgen. Die meisten der nachfolgend aufgeführten Wirkstoffe sind in USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2006, offenbart.
Antidiabetika umfassen Insulin und Insulinderivate, wie z.B. Lantus® (siehe www.lantus.com) oder HMR 1964 oder Levemir® (insulin detemir), Humalog^ (Insulin Lispro), Humulin^, VIAject™, SuliXen(R) oder solche, wie sie in WO2005005477 (Novo Nordisk) beschrieben sind, schnell wirkende Insuline (siehe US 6,221,633), inhalierbare Insuline, wie z. B. Exubera ® , Nasulin™, oder orale Insuline, wie z. B. IN- 105 (Nobex) oder Oral-lyn ™ (Generex Biotechnology) oder Technosphere^ Insulin (MannKind) oder Cobalamin™ orales Insulin oder Insuline, wie sie in WO2007128815, WO2007128817, WO2008034881, WO2008049711 beschrieben sind oder Insuline, die transdermal verabreicht werden können;
GLP-I -Derivate und GLP-I Agonisten wie z.B. Exenatide oder spezielle Zubereitungen davon, wie sie z.B. in WO2008061355 beschrieben sind, Liraglutide, Taspoglutide (R-1583), Albiglutide, Lixisenatide oder diejenigen die in WO 98/08871, WO2005027978, WO2006037811, WO2006037810 von Novo Nordisk A/S, in WO 01/04156 von Zealand oder in WO 00/34331 von Beaufour-Ipsen offenbart wurden, Pramlintide Acetat (Symlin; Amylin Pharmaceuticals), AVE-0010, BIM-51077 (R-1583, ITM-077), PC-DAC :Exendin-4 (ein Exendin-4 Analogon, welches kovalent an rekombinantes menschliches Albumin gebunden ist), CVX-73, CVX-98 und CVx-96 (GLP-I Analoga, welche kovalent an einen monoklonalen Antikörper gebunden sind, der spezifische Bindungsstellen für das GLP-I Peptid aufweist), CNTO-736 (ein GLP-I Analogon, welches an eine Domäne gebunden ist, welche den Fc-Teil eines Antikörpers beinhaltet), PGC-GLP-I (GLP-I gebunden an einen Nanocarri er), Agonisten wie sie z.B. bei D. Chen et al., Proc. Natl. Acad. Sei. USA 104 (2007) 943 beschrieben sind, solche wie sie in WO2006124529, WO2007124461, WO2008062457, WO2008082274, WO2008101017, WO2008081418, WO2008112939, WO2008112941, WO2008113601, WO2008116294, WO2008116648, WO2008119238 beschrieben sind, Peptide wie z.B. Obinepitide (TM-30338), Amylinrezeptor Agonisten, wie sie z.B. in WO2007104789 beschrieben sind, Analoga des humanen GLP-I, wie sie in WO2007120899, WO2008022015, WO2008056726 beschrieben sind, sowie oral wirksame hypoglykämische Wirkstoffe.
Antidiabetika umfassen auch Agonisten des Glukose-abhängigen insulinotropen Polypeptids (GIP) Rezeptors wie sie z.B. in WO2006121860 beschrieben sind.
Antidiabetika umfassen auch das Glukose-abhängige insulinotrope Polypeptid (GIP) wie auch analoge Verbindungen wie sie z.B. in WO2008021560 beschrieben sind.
Antidiabetika umfassen auch Analoga und Derivate des Fibroblastenwachstumsfaktors 21 (FGF-21, fibroblast growth factor 21).
Die oral wirksamen hypoglykämischen Wirkstoffe umfassen vorzugsweise Sulfonylharnstoffe,
Biguanidine,
Meglitinide,
Oxadiazolidindione,
Thiazolidindione,
PPAR- und RXR-Modulatoren,
Glukosidase-Inhibitoren.
Hemmstoffe der Glykogenphosphorylase,
Glukagonrezeptor- Antagonisten,
Glukokinaseaktivatoren,
Inhibitoren der Fructose-l,6-bisphosphatase,
Modulatoren des Glukosetransporters-4 (GLUT4),
Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase (GFAT),
GLP- 1 - Agonisten,
Kaliumkanalöffner, wie z.B. Pinacidil, Cromakalim, Diazoxid oder solche wie sie bei R. D.
Carr et al., Diabetes 52, 2003, 2513.2518, bei J. B. Hansen et al, Current Medicinal Chemistry
11, 2004, 1595-1615, bei T. M. Tagmose et al., J. Med. Chem. 47, 2004, 3202-3211 oder bei M.
J. Coghlan et al., J. Med. Chem. 44, 2001, 1627-1653 beschrieben sind, oder diejenigen, die in
WO 97/26265 und WO 99/03861 von Novo Nordisk A/S offenbart wurden,
Wirkstoffe, die auf den ATP-abhängigen Kaliumkanal der Betazellen wirken,
Inhibitoren der Dipeptidylpeptidase-IV (DPP-IV),
Insulin-Sensitizer,
Inhibitoren von Leberenzymen, die an der Stimulation der Glukoneogenese und/oder
Glykogenolyse beteiligt sind,
Modulatoren der Glukoseaufnahme, des Glukosetransports und der Glukoserückresorption,
Modulatoren der natrium-abhängigen Glukosetransporter 1 oder 2 (SGLTl, SGLT2),
Hemmstoffe der 11-beta-Hydroxysteroid-Dehydrogenase-l (l lß-HSDl),
Inhibitoren der Protein-Tyrosin-Phosphatase-1B (PTP-IB),
Nikotinsäurerezeptoragonisten,
Inhibitoren der hormon-sensitiven bzw. endothelialen Lipasen,
Hemmstoffen der Acetyl-CoA Carboxylase (ACCl und/oder ACC2) oder
Inhibitoren der GSK-3 beta.
Weiterhin sind umfasst den Fettstoffwechsel verändernde Verbindungen wie antihyperlipidämische Wirkstoffe und antilipidämische Wirkstoffe,
HMGCoA-Reduktase-Inhibitoren,
Farnesoid X Rezeptor (FXR) Modulatoren,
Fibrate,
Cholesterinresreptionsinhibitoren,
CETP-Inhibitoren,
Gallensäureresoφtionsinhibitoren,
MTP-Inhibitoren,
Agonisten des Estrogenrezeptors gamma (ERRD Agonisten),
Sigma-1 Rezeptorantagonisten,
Antagonisten des Somatostatin 5 Rezeptors (SST5 Rezeptor);
Verbindungen, die die Nahrungsmitteleinnahme verringern und
Verbindungen, die die Thermogenese erhöhen.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Insulin verabreicht.
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit einem Wirkstoff, der auf den ATP-abhängigen Kaliumkanal der Betazellen wirkt, z.B. Sulfonylharnstoffe, wie z.B. Tolbutamid, Glibenclamid, Glipizid, Gliclazide oder Glimepirid, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einer Tablette verabreicht, die sowohl Glimeprid enthält, welches schnell freigesetzt wird wie auch Metformin enthält, welches über einen längeren Zeitraum freigesetzt wird (wie z.B. in US2007264331, WO2008050987, WO2008062273 beschrieben).
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit einem Biguanid, wie z.B. Metformin, verabreicht.
Bei wieder einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit einem Meglitinid, wie z.B. Repaglinide, Nateglinid oder Mitiglinide verabreicht.
Bei einer weiteren Ausfuhrungsform wird die Verbindung der Formel I mit einer Kombination von Mitiglinide mit einem Glitazon, z.B. Pioglitazon Hydrochlorid, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I mit einer Kombination von Mitiglinide mit einem alpha-Glukosidaseinhibitor verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit antidiabetischen Verbindungen, wie sie in WO2007095462, WO2007101060, WO2007105650 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit antihypoglykämischen Verbindungen, wie sie in WO2007137008, WO2008020607 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Thiazolidindion, wie z.B. Troglitazon, Ciglitazon, Pioglitazon, Rosiglitazon oder den in WO 97/41097 von Dr. Reddy's Research Foundation offenbarten Verbindungen, insbesondere 5-[[4- [(3,4-Dihydro-3-methyl-4-oxo-2-chinazolinylmethoxy]phenyl]methyl]-2,4-thiazolidindion, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem PPAR gamma Agonisten, wie z.B. Rosiglitazon, Pioglitazon, JTT-501, Gl 262570, R-483, CS-OI l (Rivoglitazon), DRL-17564, DRF-2593 (Balaglitazon), INT-131, T-2384 oder solchen, wie sie in WO2005086904, WO2007060992, WO2007100027, WO2007103252, WO2007122970, WO2007138485, WO2008006319, WO2008006969, WO2008010238, WO2008017398, WO2008028188, WO2008066356, WO2008084303, WO2008089461- WO2008089464, WO2008093639, WO2008096769, WO2008096820, WO2008096829, US2008194617, WO2008099944, WO2008108602, WO2008109334, WO2008126731, WO2008126732 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Competact™, einer festen Kombination von Pioglitazon Hydrochlorid mit Metformin Hydrochlorid, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Tandemact™. einer festen Kombination von Pioglitazon mit Glimeprid, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Pioglitazon Hydrochlorid mit einem Angiotensin II Agonisten, wie z.B. TAK-536, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem PPAR alpha Agonisten bzw. gemischten PPAR alpha/PPAR delta Agonisten, wie z.B. GW9578, GW-590735, K-H l, LY-674, KRP-101, DRF-10945, LY-518674, CP-900691, BMS-687453, BMS-711939 oder solchen wie sie in WO2001040207, WO2002096894, WO2005097076, WO2007056771, WO2007087448, WO2007089667, WO2007089557, WO2007102515, WO2007103252, JP2007246474, WO2007118963, WO2007118964, WO2007126043, WO2008006043, WO2008006044, WO2008012470, WO2008035359, WO2008087365, WO2008087366, WO2008087367, WO2008117982 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem gemischten PPAR alpha/gamma Agonisten, wie z.B. Naveglitazar, LY-510929, ONO-5129, E-3030, AVE 8042, AVE 8134, AVE 0847, CKD-501 (Lobeglitazon Sulfat), MBX-213, KY-201 oder wie in WO 00/64888, WO 00/64876, WO03/020269, WO2004024726, WO2007099553, US2007276041, WO2007085135, WO2007085136, WO2007141423, WO2008016175, WO2008053331, WO2008109697, WO2008109700, WO2008108735 oder in J.P.Berger et al., TRENDS in Pharmacological Sciences 28(5), 244- 251, 2005 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem PPAR delta Agonisten, wie z.B. GW-501516 oder wie sie in WO2006059744, WO2006084176, WO2006029699, WO2007039172-WO2007039178, WO2007071766, WO2007101864, US2007244094, WO2007119887, WO2007141423, US2008004281, WO2008016175, WO2008066356, WO2008071311, WO2008084962, US2008176861 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem pan-SPPARM (selective PPAR modulator alpha, gamma, delta), wie z.B. GFT-505 oder solchen wie sie in WO2008035359 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Metaglidasen oder mit MBX-2044 oder anderen partiellen PPAR gamma Agonisten/ Antagonisten verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem α- Glukosidase-Inhibitor, wie z.B. Miglitol oder Acarbose oder solchen, wie sie z.B. in WO2007114532, WO2007140230, US2007287674, US2008103201, WO2008065796, WO2008082017 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Hemmstoff der Glykogenphosphorylase, wie z.B. PSN-357 oder FR-258900 oder solchen wie in WO2003084922, WO2004007455, WO2005073229-31, WO2005067932, WO2008062739, WO2008099000, WO2008113760 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Glukagon- Rezeptor-Antagonisten, wie z.B. A-770077 oder NNC-25-2504 oder wie in WO2004100875, WO2005065680, WO2006086488, WO2007047177, WO2007106181, WO2007111864, WO2007120270, WO2007120284, WO2007123581, WO2007136577, WO2008042223, WO2008098244 beschrieben, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einer Antisense-Verbindung, z.B. ISIS-325568, verabreicht, welche die Produktion des Glukagonrezeptors inhibiert.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Aktivatoren der Glukokinase, wie z. B. LY-2121260 (WO2004063179), PSN-105, PSN-110, GKA-50 oder solchen wie sie z. B. in WO2004072031, WO2004072066, WO2005080360, WO2005044801, WO2006016194, WO2006058923, WO2006112549, WO2006125972, WO2007017549, WO2007017649, WO2007007910, WO2007007040-42, WO2007006760-61, WO2007006814,
WO2007007886, WO2007028135, WO2007031739, WO2007041365, WO2007041366, WO2007037534, WO2007043638, WO2007053345, WO2007051846, WO2007051845, WO2007053765, WO2007051847, WO2007061923, WO2007075847, WO2007089512, WO2007104034, WO2007117381, WO2007122482, WO2007125103, WO2007125105, US2007281942, WO2008005914, WO2008005964, WO2008043701, WO2008044777, WO2008047821, US2008096877, WO2008050117, WO2008050101, WO2008059625, US2008146625, WO2008078674, WO2008079787, WO2008084043, , WO2008084044, WO2008084872, WO2008089892, WO2008091770, WO2008075073, WO2008084043, WO2008084044, WO2008084872, WO2008084873, WO2008089892, WO2008091770, JP2008189659, WO2008104994, WO2008111473, WO2008116107, WO2008118718, WO2008120754 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Glukoneogenese, wie sie z. B. in FR-225654, WO2008053446 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Fructose-l,6-bisphosphatase (FBPase) wie z.B. MB-07729, CS-917 (MB-06322) oder MB- 07803 oder solchen wie sie in WO2006023515, WO2006104030, WO2007014619, WO2007137962, WO2008019309, WO2008037628 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des Glukosetransporters-4 (GLUT4), wie z. B. KST-48 (D. -O. Lee et al.: Arzneim. -Forsch. Drug Res. 54 (12), 835 (2004)), verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase (GFAT), wie sie z. B. in WO2004101528 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Dipeptidylpeptidase-IV (DPP-IV), wie z. B. Vildagliptin (LAF-237), Sitagliptin (MK- 0431), Sitagliptin Phosphat, Saxagliptin ((BMS-477118), GSK-823093, PSN-9301, SYR-322,
SYR-619, TA-6666, TS-021, GRC-8200 (Melogliptin), GW-825964X, KRP-104, DP-893, ABT-341, ABT-279 oder ein anderes Salz davon, S-40010, S-40755, PF-00734200, BI-1356, PHX-1149, Alogliptin Benzoat, Linagliptin, Melogliptin oder solchen Verbindungen wie sie in WO2003074500, WO2003106456, WO2004037169, WO200450658, WO2005037828, WO2005058901, WO2005012312, WO2005/012308, WO2006039325, WO2006058064, WO2006015691, WO2006015701, WO2006015699, WO2006015700, WO2006018117, WO2006099943, WO2006099941, JP2006160733, WO2006071752, WO2006065826, WO2006078676, WO2006073167, WO2006068163, WO2006085685, WO2006090915, WO2006104356, WO2006127530, WO2006111261, US2006890898, US2006803357, US2006303661, WO2007015767 (LY-2463665), WO2007024993, WO2007029086, WO2007063928, WO2007070434, WO2007071738, WO2007071576, WO2007077508, WO2007087231, WO2007097931, WO2007099385, WO2007100374, WO2007112347, WO2007112669, WO2007113226, WO2007113634, WO2007115821, WO2007116092, US2007259900, EP1852108, US2007270492, WO2007126745, WO2007136603, WO2007142253, WO2007148185, WO2008017670, US2008051452, WO2008027273, WO2008028662, WO2008029217, JP2008031064, JP2008063256, WO2008033851, WO2008040974, WO2008040995, WO2008060488, WO2008064107, WO2008066070, WO2008077597, JP2008156318, WO2008087560, WO2008089636, WO2008093960, WO2008096841, WO2008101953, WO2008118848, WO2008119005, WO2008119208, WO2008120813, WO2008121506 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Janumet™, einer festen Kombination von Sitagliptin Phosphat mit Metformin Hydrochlorid, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Eucreas(R), einer festen Kombination von Vildagliptin mit Metformin Hydrochlorid, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Alogliptin Benzoat mit Pioglitazone verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einer festen Kombination von eines Salzes von Sitagliptin mit Metformin Hydrochlorid, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einer Kombination eines DPP-IV-Inhibitors mit omega-3 -Fettsäuren oder omega-3 -Fettsäureestern, wie z.B. in WO2007128801 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einer festen Kombination von eines Salzes von Sitagliptin mit Metformin Hydrochlorid, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einer die Insulinsekretion verstärkende Substanz, wie z. B. KCP-265 (WO2003097064), oder solchen wie sie in WO2007026761, WO2008045484, US2008194617 beschrieben sind, verabreicht. Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Agonisten des glucose-abhängigen insulinotropischen Rezeptors (GDIR) wie z. B. APD-668 verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem ATP-Citrat-Lyase Inhibitor, wie z.B. SB-204990, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des natrium-abhängigen Glukosetransporters 1 oder 2 (SGLTl, SGLT2), wie z.B. KGA-2727, T-1095, SGL-0010, AVE 2268, SAR 7226, SGL-5083, SGL-5085, SGL-5094, ISIS-388626, Sergliflozin oder Dapagliflozin oder wie sie z. B. in WO2004007517, WO200452903, WO200452902, PCT/EP2005/005959, WO2005085237, JP2004359630, WO2005121161, WO2006018150, WO2006035796, WO2006062224, WO2006058597, WO2006073197, WO2006080577, WO2006087997, WO2006108842, WO2007000445, WO2007014895, WO2007080170, WO2007093610, WO2007126117, WO2007128480, WO2007129668, US2007275907, WO2007136116, WO2007143316, WO2007147478, WO2008001864, WO2008002824, WO2008013277, WO2008013280, WO2008013321, WO2008013322, WO2008016132, WO2008020011, JP2008031161, WO2008034859, WO2008042688, WO2008044762, WO2008046497, WO2008049923, WO2008055870, WO2008055940, WO2008069327, WO2008070609, WO2008071288, WO2008072726, WO2008083200, WO2008090209, WO2008090210, WO2008101586, WO2008101939, WO2008116179, WO2008116195, US2008242596 oder von A. L. Handion in Expert Opin. Ther. Patents (2005) 15(11), 1531-1540 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Hemmstoffen der 11 -beta-Hydroxysteroid-Dehydrogenase-1 (l lß-HSDl), wie z. B. BVT-2733, JNJ-25918646, INCB-13739, INCB-20817, DIO-92 ((-)-Ketoconazol) oder solche, wie sie z. B. in WO200190090-94, WO200343999, WO2004112782, WO200344000, WO200344009, WO2004112779, WO2004113310, WO2004103980, WO2004112784, WO2003065983, WO2003104207, WO2003104208, WO2004106294, WO2004011410, WO2004033427, WO2004041264, WO2004037251, WO2004056744, WO2004058730, WO2004065351, WO2004089367, WO2004089380, WO2004089470-71, WO2004089896, WO2005016877, WO2005063247, WO2005097759, WO2006010546, WO2006012227, WO2006012173, WO2006017542, WO2006034804, WO2006040329, WO2006051662, WO2006048750, WO2006049952, WO2006048331, WO2006050908, WO2006024627, WO2006040329, WO2006066109, WO2006074244, WO2006078006, WO2006106423, WO2006132436, WO2006134481, WO2006134467, WO2006135795, WO2006136502, WO2006138508, WO2006138695, WO2006133926, WO2007003521, WO2007007688, US2007066584, WO2007029021, WO2007047625, WO2007051811, WO2007051810, WO2007057768, WO2007058346, WO2007061661, WO2007068330, WO2007070506, WO2007087150, WO2007092435, WO2007089683, WO2007101270, WO2007105753, WO2007107470, WO2007107550, WO2007111921, US2007207985, US2007208001, WO2007115935, WO2007118185, WO2007122411, WO2007124329, WO2007124337, WO2007124254, WO2007127688, WO2007127693, WO2007127704, WO2007127726, WO2007127763, WO2007127765, WO2007127901, US2007270424, JP2007291075, WO2007130898, WO2007135427, WO2007139992, WO2007144394, WO2007145834. WO2007145835, WO2007146761, WO2008000950, WO2008000951, WO2008003611, WO2008005910, WO2008006702, WO2008006703, WO2008011453, WO2008012532, WO2008024497, WO2008024892, WO2008032164, WO2008034032, WO2008043544, WO2008044656, WO2008046758, WO2008052638, WO2008053194, WO2008071169, WO2008074384, WO2008076336, WO2008076862, WO2008078725, WO2008087654, WO2008088540, WO2008099145, WO2008101885, WO2008101886, WO2008101907, WO2008101914, WO2008106128, WO2008110196, WO2008119017, WO2008120655, WO2008127924 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der Protein-Tyrosin-Phosphatase-1B (PTP-IB), wie sie z. B. in WO200119830-31, WO200117516, WO2004506446, WO2005012295, WO2005116003, WO2005116003, WO2006007959, DE 10 2004 060542.4, WO2007009911, WO2007028145, WO2007067612- 615, WO2007081755, WO2007115058, US2008004325, WO2008033455, WO2008033931, WO2008033932, WO2008033934, WO2008089581 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Agonisten des GPRl 09 A (HM74A Rezeptor Agonisten; NAR-Agonisten (Nikotinsäurerezeptoragonisten)), wie z.B. Nicotinsäure oder „extended release niacin" in Verbindung mit MK-0524A (Laropiprant) oder MK-0524 oder solchen Verbindungen, wie sie in WO2004041274, WO2006045565, WO2006045564, WO2006069242, WO2006085108, WO2006085112, WO2006085113, WO2006124490, WO2006113150, WO2007017261, WO2007017262, WO2007017265, WO2007015744, WO2007027532, WO2007092364, WO2007120575, WO2007134986, WO2007150025, WO2007150026, WO2008016968, WO2008051403, WO2008086949, WO2008091338, WO2008097535, WO2008099448, US2008234277, WO2008127591 beschrieben sind, verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Niacin mit Simvastatin verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Nicotinsäure oder „extended release niacin" in Verbindung mit MK-0524A (Laropiprant) verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Nicotinsäure oder „extended release niacin" in Verbindung mit MK-0524A (Laropiprant) und mit Simvastatin verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Nicotinsäure oder einem anderen Nicotinsäurerezeptoragonisten und einem Prostaglandin
DP Rezeptorantagonisten, wie z.B. solchen wie sie in WO2008039882 beschrieben sind, verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Agonisten des GPRl 16, wie sie z.B. in WO2006067531, WO2006067532 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des GPR40, wie sie z.B. in WO2007013689, WO2007033002, WO2007106469, US2007265332, WO2007123225, WO2007131619, WO2007131620, WO2007131621, US2007265332, WO2007131622, WO2007136572, WO2008001931, WO2008030520, WO2008030618, WO2008054674, WO2008054675, WO2008066097, US2008176912 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des GPRl 19 (G-Protein-gekoppelter Glukose-abhängiger insulinotroper Rezeptor), wie z.B. PSN-119-1, PSN-821, PSN-119-2, MBX-2982 oder solchen wie sie z. B. in WO2004065380, WO2005061489 (PSN-632408), WO2006083491, WO2007003960-62 und WO2007003964, WO2007035355, WO2007116229, WO2007116230, WO2008005569, WO2008005576, WO2008008887, WO2008008895, WO2008025798, WO2008025799, WO2008025800, WO2008070692, WO2008076243, WO200807692, WO2008081204, WO2008081205, WO2008081206, WO2008081207, WO2008081208, WO2008083238, WO2008085316, WO2008109702 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des GPRl 20, wie sie z.B. in EP 1688138, WO2008066131, WO2008066131, WO2008103500, WO2008103501 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der hormon-sensitiven Lipase (HSL) und/oder Phospholipasen, wie z. B. in WO2005073199, WO2006074957, WO2006087309, WO2006111321, WO2007042178, WO2007119837, WO2008122352, WO2008122357 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der endothelialen Lipase, wie z. B. in WO2007110216 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Phospholipase A2 Inhibitor wie z.B. Darapladib oder A-002 oder solchen, wie sie in WO2008048866, WO20080488867 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Myricitrin, einem Lipase-Inhibitor (WO2007119827), verabreicht.
Bei einer Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Glykogen Synthase Kinase-3 beta (GSK-3 beta), wie z. B. in US2005222220, WO2005085230, WO2005111018, WO2003078403, WO2004022544, WO2003106410, WO2005058908, US2005038023, WO2005009997, US2005026984, WO2005000836, WO2004106343, EP1460075, WO2004014910, WO2003076442, WO2005087727, WO2004046117, WO2007073117, WO2007083978, WO2007120102, WO2007122634, WO2007125109, WO2007125110, US2007281949, WO2008002244, WO2008002245, WO2008016123, WO2008023239, WO2008044700, WO2008056266, WO2008057940, WO2008077138, EP1939191, EP1939192, WO2008078196, WO2008094992, WO2008112642, WO2008112651, WO2008113469, WO2008121063, WO2008121064 beschrieben.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Phosphoenolpyruvatcarboxykinase (PEPCK), wie z.B. solchen, wie in WO2004074288 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Phosphoinositidkinase-3 (PI3K), wie z.B. solchen, wie in WO2008027584, WO2008070150, WO2008125833, WO2008125835, WO2008125839 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Serum/Glucocorticoid regulierten Kinase (SGK), wie z. B. in WO2006072354, WO2007093264, WO2008009335, WO2008086854 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Modulator des Glucocorticoidrezeptors, wie z. B. in WO2008057855, WO2008057856, WO2008057857, WO2008057859, WO2008057862, WO2008059867, WO2008059866, WO2008059865, WO2008070507, WO2008124665, WO2008124745 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Modulator des Mineralocorticoidrezeptors (MR), wie z. B. Drospirenone, oder solchen wie sie in WO2008104306, WO2008119918 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Protein Kinase C beta (PKC beta), wie z. B. Ruboxistaurin, oder solchen wie sie in WO2008096260, WO2008125945 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Protein Kinase D, wie z. B. Doxazosin (WO2008088006), verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Aktivator der AMP-aktivierten Proteinkinase (AMPK), wie sie z. B. in WO2007062568, WO2008006432, WO2008016278, WO2008016730, WO2008083124 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Ceramidkinase, wie sie z. B. in WO2007112914, WO2007149865 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der MAPK-interagierenden Kinase 1 oder 2 (MNKl oder 2), wie sie z.B. in
WO2007104053, WO2007115822, WO2008008547, WO2008075741 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der „I-kappaB kinase" (IKK Inhibitoren), wie sie z. B. in WO2001000610, WO2001030774, WO2004022057, WO2004022553, WO2005097129, WO2005113544, US2007244140, WO2008099072, WO2008099073, WO2008099073, WO2008099074, WO2008099075 beschrieben sind, verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der NF-kappaB (NFKB) Aktivierung, wie sie z. B. Salsalate verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der ASK-I (apoptosis signal-regulating kinase 1), wie sie z. B. in WO2008016131 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindungen der Formel I in Kombination mit einem HMGCoA-Reduktase Inhibitor wie Simvastatin, Fluvastatin, Pravastatin, Lovastatin, Atorvastatin, Cerivastatin, Rosuvastatin, Pitavastatin, L-659699, BMS-644950 oder solchen, wie sie in US2007249583, WO2008083551 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Farnesoid X Rezeptor (FXR) Modulatoren, wie z.B. WAY-362450 oder solchen wie in WO2003099821, WO2005056554, WO2007052843, WO2007070796, WO2007092751, JP2007230909, WO2007095174, WO2007140174, WO2007140183, WO2008000643, WO2008002573, WO2008025539, WO2008025540, JP2008214222 beschrieben, verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Liganden des Leber X Rezeptors (liver X receptor; LXR), wie z.B. in WO2007092965, WO2008041003, WO2008049047, WO2008065754, WO2008073825, US2008242677 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Fibrat, wie z.B. Fenofibrat, Clofibrat, Bezafibrat, oder solchen wie sie in WO2008093655 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Fibraten, wie z.B. dem Cholinsalz von Fenofibrat (SLV-348), verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Fibraten, wie z.B. dem Cholinsalz von Fenofibrat und einem HMGCoA Reduktase Inhibitor, wie z.B. Rosuvastatin, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Bezafibrat und Diflunisal verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Fenofibrat oder einem Salz davon mit Simvastatin, Rosuvastatin, Fluvastatin, Lovastatin, Cerivastatin, Pravastatin, Pitavastatin oder Atorvastatin verabreicht.
Bei einer weiteren Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Synordia (R), einer festen Kombination von Fenofibrat mit Metformin, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Cholesterinresorptionsinhibitor, wie z.B. Ezetimibe, Tiqueside, Pamaqueside, FM- VP4 (sitostanol/campesterol ascorbyl phosphat; Forbes Medi-Tech, WO2005042692, WO2005005453), MD-0727 (Microbia Inc., WO2005021497, WO2005021495) oder mit Verbindungen, wie in WO2002066464, WO2005000353 (Kotobuki Pharmaceutical Co. Ltd.) oder WO2005044256 oder WO2005062824 (Merck & Co.) oder WO2005061451 und WO2005061452 (AstraZeneca AB) und WO2006017257 (Phenomix) oder WO2005033100 (Lipideon Biotechnology AG) oder wie in WO2002050060, WO2002050068, WO2004000803, WO2004000804, WO2004000805, WO2004087655, WO2004097655, WO2005047248,
WO2006086562, WO2006102674, WO2006116499, WO2006121861, WO2006122186, WO2006122216, WO2006127893, WO2006137794, WO2006137796, WO2006137782, WO2006137793, WO2006137797, WO2006137795, WO2006137792, WO2006138163, WO2007059871, US2007232688, WO2007126358, WO2008033431, WO2008033465, WO2008052658, WO2008057336, WO2008085300 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem NPC ILl -Antagonisten, wie z.B. solchen, wie sie in WO2008033464, WO2008033465 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Vytorin™, einer festen Kombination von Ezetimibe mit Simvastatin, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Ezetimibe mit Atorvastatin, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Ezetimibe mit Fenofibrat verabreicht.
Bei einer Ausführungsform der Erfindung ist der weitere Wirkstoff ein Diphenylazetidinonderivat, wie z.B. in US 6,992,067 oder US 7,205,290 beschrieben.
Bei einer weiteren Ausführungsform der Erfindung ist der weitere Wirkstoff ein Diphenylazetidinonderivat, wie z.B. in US 6,992,067 oder US 7,205,290 beschrieben, kombiniert mit einem Statin, wie z.B. Simvastatin, Fluvastatin, Pravastatin, Lovastatin, Cerivastatin, Atorvastatin, Pitavastatin oder Rosuvastatin.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Lapaquistat, einem Squalensynthase-Inhibitor, mit Atorvastatin verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem CETP-Inhibitor. wie z.B. Torcetrapib, Anacetrapib oder JTT-705 (Dalcetrapib) oder solchen wie sie in WO2006002342, WO2006010422, WO2006012093, WO2006073973, WO2006072362, WO2007088996, WO2007088999, US2007185058, US2007185113, US2007185154, US2007185182, WO2006097169, WO2007041494, WO2007090752, WO2007107243, WO2007120621, US2007265252, US2007265304, WO2007128568, WO2007132906, WO2008006257, WO2008009435, WO2008018529, WO2008058961, WO2008058967, WO2008059513, WO2008070496, WO2008115442, WO2008111604 beschrieben sind, verabreicht.
Bei einer Ausfiihrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Gallensäureresorptionsinhibitoren (Inhibitoren des intestinalen Gallensäuretransporters (IBAT)) (siehe z.B. US 6,245,744, US 6,221,897 oder WO00/61568), wie z.B. HMR 1741 oder solchen wie in DE 10 2005 033099.1 und DE 10 2005 033100.9, DE 10 2006 053635, DE 10 2006 053637, WO2007009655-56, WO2008058628, WO2008058629, WO2008058630, WO2008058631 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Agonisten des GPBARl (G-protein-coupled-bile-acid-receptor-l; TGR5), wie sie z.B. in US20060199795, WO2007110237, WO2007127505, WO2008009407, WO2008067219, WO2008067222, FR2908310, WO2008091540, WO2008097976 beschrieben sind, verabreicht.
Bei einer Ausfiihrungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren des TRPM5 Kanals (TRP-Cation-Channel-M5), wie sie z.B. in WO2008097504 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem polymeren Gallensäureadsorber, wie z.B. Cholestyramin, Colesevelam Hydrochlorid, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit Colesevelam Hydrochlorid und Metformin oder einem Sulfonylharnstoff oder Insulin verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Phytosterole enthaltenden Kaugummi (Reductol™) verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Inhibitor des mikrosomalen Triglycerid-Transfer-Proteins (MTP-Inhibitor), wie z.B. Implitapide , BMS-201038, R-103757, AS-1552133, SLx-4090, AEGR-733 oder solchen wie in WO2005085226, WO2005121091, WO2006010423, WO2006113910, WO2007143164, WO2008049806, WO2008049808, WO2008090198, WO2008100423 beschrieben, verabreicht.
Bei einer weiteren Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer Kombinbation eines Cholesterolabsorptionsinhibitors, wie z.B. Ezetimibe, und einem Inhibitor des Triglycerid-Transfer-Proteins (MTP-Inhibitor), wie z.B. Implitapide, wie in WO2008030382 oder in WO2008079398 beschrieben, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem antihypertriglyceridämischen Wirkstoff, wie z.B. solchen wie sie in WO2008032980 beschrieben sind, verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Antagonisten des Somatostatin 5 Rezeptors (SST5 Rezeptor), wie z.B. solchen wie sie in WO2006094682 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem ACAT-Inhibitor, wie z.B. Avasimibe, SMP-797 oder KY-382 oder solchen, wie sie in WO2008087029, WO2008087030, WO2008095189 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Leber-Carnitin Palmitoyltransferase-1 (L-CPTl), wie sie
z.B. in WO2007063012, WO2007096251 (ST-3473), WO2008015081, US2008103182, WO2008074692 beschrieben sind, verabreicht.
Bei einer weiteren Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Modulator der Serin-Palmitoyltransferase (SPT), wie sie z.B. in WO2008031032, WO2008046071, WO2008083280, WO2008084300 beschrieben sind, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Squalen Synthetase Inhibitor, wie z.B. BMS-188494, TAK-475 (Lapaquistat Acetat) oder wie in WO2005077907, JP2007022943, WO2008003424 beschrieben, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit ISIS-301012 (Mipomersen), einem Antisense-Oligonukleotid, welches in der Lage ist, das Apolipoprotein B Gen zu regulieren, verabreicht.
Bei einer Ausfuhrungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Stimulator des ApoA-1 Gens, wie er z.B. in WO2008092231 beschrieben ist, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem LDL-Rezeptorinducer (siehe US 6,342,512), wie z.B. HMRl 171, HMR1586, oder solchen wie in WO2005097738, WO2008020607 beschrieben, verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem HDL-Cholesterol-erhöhenden Agens, wie z.B. solchen wie sie in WO2008040651, WO2008099278 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem ABCAl Expressionsverstäker, wie sie z.B. in WO2006072393, WO2008062830 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipoprotein-Lipase Modulator, wie z.B. Ibrolipim (NO- 1886), verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipoprotein(a) antagonist, wie z.B. Gemcabene (CI- 1027) verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Lipase Inhibitor, wie z.B. Orlistat oder Cetilistat (ATL-962), verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Adenosin Al Rezeptor Agonisten (Adenosin Al R), wie sie z.B. in EP 1258247, EP1375508, WO2008028590, WO2008077050 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Adenosin A2B Rezeptor Agonisten (Adenosin A2B R) wie z.B. ATL-801 verabreicht.
Bei einer anderen Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Modulator der Adenosin A2A und/oder Adenosin A3 Rezeptoren, wie z.B. in WO2007111954, WO2007121918, WO2007121921, WO2007121923, WO2008070661 beschrieben, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Agonisten der Adenosin A1/A2B Rezeptoren, wie z.B. in WO2008064788, WO2008064789 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Adenosin A2B Rezeptor Antagonisten (Adenosin A2B R), wie sie in US2007270433, WO2008027585, WO2008080461 beschrieben sind, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Hemmstoffen der Acetyl-CoA Carboxylase (ACCl und/oder ACC2) wie z. B. solchen wie in
WO199946262, WO200372197, WO2003072197, WO2005044814, WO2005108370, JP2006131559, WO2007011809, WO2007011811, WO2007013691, WO2007095601-603, WO2007119833, WO2008065508, WO2008069500, WO2008070609, WO2008072850, WO2008079610, WO2008088688, WO2008088689, WO2008088692, US2008171761, WO2008090944, JP2008179621 , US2008200461, WO2008102749, WO2008103382, WO2008121592 beschrieben, verabreicht.
Bei einer anderen Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Modulatoren der mikrosomalen Acyl-CoA:Glycerol-3-Phosphat-Acyltransferase 3 (GP AT3, beschrieben in WO2007100789) oder mit Modulatoren der mikrosomalen Acyl-CoA:Glycerol- 3-Phosphat-Acyltransferase 4 (GP AT4, beschrieben in WO2007100833) verabreicht.
Bei einer weiteren Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Modulatoren der Xanthin-Oxidoreductase (XOR) verabreicht.
Bei einer anderen Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren der löslichen Epoxidhydrolase (sEH), wie sie z.B. in WO2008051873, WO2008051875, WO2008073623, WO2008094869, WO2008112022 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit CART-Modulatoren (siehe "Cocaine-amphetamine-regulated transcript influences energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al.: Hormone and Metabolie Research (2001), 33(9), 554-558);
NPY-Antagonisten wie z.B. Naphthalin-l-sulfonsäure-{4-[(4-amino-quinazolin-2-ylamino)- methyl]-cyclohexylmethyl}-amid Hydrochlorid (CGP 71683A) oder Velneperit;
NPY-5 Rezeptorantagonisten wie L-152804 oder die Verbindung „NPY-5-BY" der Firma Banyu oder wie sie z. B. in WO2006001318, WO2007103295, WO2007125952, WO2008026563, WO2008026564, WO2008052769, WO2008092887, WO2008092888, WO2008092891 beschrieben sind;
NPY-4-Rezeptorantagonisten wie sie z. B. in WO2007038942 beschrieben sind;
NPY-2-Rezeptorantagonisten wie sie z. B. in WO2007038943 beschrieben sind;
Peptid YY 3-36 (PYY3-36) oder analoge Verbindungen wie z. B. CJC- 1682 (PYY3-36 konjugiert mit humanem Serum Albumin über Cys34) oder CJC- 1643 (Derivat des PYY3-36, welches sich in vivo an Serum Albumin konjugiert) oder solche, wie sie in WO2005080424, WO2006095166, WO2008003947 beschrieben sind;
Derivaten des Peptids Obestatin wie sie WO2006096847 beschrieben sind;
CBlR (Cannabinoid Rezeptor 1) Antagonisten wie z.B. Rimonabant, Surinabant (SR147778), SLV-319 (Ibipinabant), AVE-1625, Taranabant (MK-0364) oder Salze davon, Otenabant (CP- 945,598), Rosonabant, V-24343 oder solche Verbindungen wie sie in z. B. EP 0656354, WO 00/15609, WO2001/64632-64634, WO 02/076949, WO2005080345, WO2005080328, WO2005080343, WO2005075450, WO2005080357, WO200170700, WO2003026647-48, WO200302776, WO2003040107, WO2003007887, WO2003027069, US6,509,367, WO200132663, WO2003086288, WO2003087037, WO2004048317, WO2004058145, WO2003084930, WO2003084943, WO2004058744, WO2004013120, WO2004029204, WO2004035566, WO2004058249, WO2004058255, WO2004058727, WO2004069838, US20040214837, US20040214855, US20040214856, WO2004096209, WO2004096763, WO2004096794, WO2005000809, WO2004099157, US20040266845, WO2004110453, WO2004108728, WO2004000817, WO2005000820, US20050009870, WO200500974, WO2004111033-34, WO200411038-39, WO2005016286, WO2005007111, WO2005007628, US20050054679, WO2005027837, WO2005028456, WO2005063761-62, WO2005061509, WO2005077897, WO2006018662, WO2006047516, WO2006060461, WO2006067428, WO2006067443, WO2006087480, WO2006087476, WO2006100208, WO2006106054, WO2006111849, WO2006113704, WO2007009705, WO2007017124, WO2007017126, WO2007018459, WO2007018460, WO2007016460, WO2007020502, WO2007026215, WO2007028849, WO2007031720, WO2007031721, WO2007036945, WO2007038045, WO2007039740, US20070015810, WO2007046548, WO2007047737, WO2007057687, WO2007062193, WO2007064272, WO2007079681, WO2007084319, WO2007084450,
WO2007086080, EP1816125, US2007213302, WO2007095513, WO2007096764, US2007254863, WO2007119001, WO2007120454, WO2007121687, WO2007123949, US2007259934, WO2007131219, WO2007133820, WO2007136571, WO2007136607, WO2007136571, US7297710, WO2007138050, WO2007139464, WO2007140385, WO2007140439, WO2007146761, WO2007148061, WO2007148062, US2007293509, WO2008004698, WO2008017381, US2008021031, WO2008024284, WO2008031734, WO2008032164, WO2008034032, WO2008035356, WO2008036021, WO2008036022, WO2008039023, WO2998043544, WO2008044111, WO2008048648, EP1921072-A1, WO2008053341, WO2008056377, WO2008059207, WO2008059335, WO2008062424, WO2008068423, WO2008068424, WO2008070305, WO2008070306, WO2008074816, WO2008074982, WO2008075012, WO2008075013, WO2008075019, WO2008075118, WO2008076754, WO2008081009, WO2008084057, EP 1944295, US2008090809, US2008090810, WO2008092816, WO2008094473, WO2008094476, WO2008099076, WO2008099139, WO2008101995, US2008207704, WO2008107179, WO2008109027, WO2008112674, WO2008115705, WO2008118414, WO2008119999, WO200812000, WO2008121257, WO2008127585 beschrieben sind;
Cannabinoid Rezeptor 1 / Cannabinoid Rezeptor 2 (CB1/CB2) modulierende Verbindungen wie z.B. delta-9-Tetrahydrocannabivarin oder solchen wie sie z.B. in WO2007001939, WO2007044215, WO2007047737, WO2007095513, WO2007096764, WO2007112399, WO2007112402, WO2008122618 beschrieben sind;
Modulatoren der FAAH (fatty acid amide hydrolase) wie sie z.B. in WO2007140005, WO2008019357, WO2008021625, WO2008023720, WO2008030532 beschrieben sind;
Inhibitoren der Fettsäuresynthase (fatty acid synthase; FAS), wie sie z.B. in WO2008057585, WO2008059214, WO2008075064, WO2008075070, WO2008075077 beschrieben sind;
Inhibitoren der LCE (long chain fatty acid elongase), wie sie z.B. in WO2008120653 beschrieben sind;
Vanilloid-1 -Rezeptor Modulatoren (Modulatoren des TRPVl), wie sie z.B. in WO2007091948, WO2007129188, WO2007133637, WO2008007780, WO2008010061, WO2008007211, WO2008010061, WO2008015335, WO2008018827, WO2008024433, WO2008024438, WO2008032204, WO2008050199, WO2008059339, WO2008059370, WO2008066664, WO2008075150, WO2008090382, WO2008090434, WO2008093024, WO2008107543, WO2008107544, WO2008110863 beschrieben sind;
Modulatoren, Antagonisten oder inverse Agonisten der Opioidrezeptoren, wie z.B. GSK-982 oder solche wie sie z.B. in WO2007047397, WO2008021849, WO2008021851, WO2008032156, WO2008059335 beschrieben sind;
Modulatoren des „orphan Opioid (ORL-I) receptor" wie sie z.B. in US2008249122, WO2008089201 beschrieben sind;
Agonisten des Prostaglandinrezeptors, wie z.B. Bimatoprost oder solchen Verbindungen wie sie in WO2007111806 beschrieben sind;
MC4-Rezeptor Agonisten (Melanocortin-4 Rezeptor Agonisten, MC4R Agonisten wie z.B. 1- Amino-l,2,3,4-tetrahydro-naphthalin-2-carbonsäure [2-(3a-benzyl-2-methyl-3-oxo- 2,3 ,3 a,4,6,7-hexahydro-pyrazolo [4,3 -c]pyridin-5 -yl)- 1 -(4-chloro-phenyl)-2-oxo-ethyl] -amid; (WO 01/91752)) oder LB53280, LB53279, LB53278 oder THIQ, MB243, RY764, CHIR-785, PT-141, MK-0493 oder solche wie sie in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720, US20050124652, WO2005051391, WO2004112793, WOUS20050222014, US20050176728, US20050164914, US20050124636, US20050130988, US20040167201, WO2004005324, WO2004037797, WO2005042516, WO2005040109, WO2005030797, US20040224901, WO200501921, WO200509184, WO2005000339, EP1460069, WO2005047253, WO2005047251, WO2005118573, EP1538159, WO2004072076, WO2004072077, WO2006021655-57, WO2007009894, WO2007015162, WO2007041061, WO2007041052, JP2007131570, EP-1842846, WO2007096186, WO2007096763, WO2007141343, WO2008007930, WO2008017852, WO2008039418, WO2008087186, WO2008087187, WO2008087189, WO2008087186- WO2008087190, WO2008090357 beschrieben sind;
Orexin-Rezeptor 1 Antagonisten (OXlR Antagonisten), Orexin-Rezeptor 2 Antagonisten (0X2R Antagonisten) oder gemischte OX1R/OX2R Antagonisten (z.B. l-(2-Methyl- benzoxazol-6-yl)-3-[l,5]naphthyridin-4-yl-hamstoff Hydrochlorid (SB-334867-A) oder solche, wie sie z. B. in WO200196302, WO200185693, WO2004085403, WO2005075458, WO2006067224, WO2007085718, WO2007088276, WO2007116374, WO2007122591, WO2007126934, WO2007126935, WO2008008517, WO2008008518, WO2008008551, WO2008020405, WO2008026149, WO2008038251, US2008132490, WO2008065626, WO2008078291, WO2008087611, WO2008081399, WO2008108991, WO2008107335, US2008249125 beschrieben sind);
Histamin H3 Rezeptor Antagonisten/inverse Agonisten (z. B. 3-Cyclohexyl-l-(4,4-dimethyl- l,4,6,7-tetrahydro-imidazo[4,5-c]pyridin-5-yl)-propan-l-on Oxalsäuresalz (WO 00/63208) oder solche, wie sie in WO200064884, WO2005082893, US2005171181 (z.B. PF-00389027), WO2006107661, WO2007003804, WO2007016496, WO2007020213, WO2007049798, WO2007055418, WO2007057329, WO2007065820, WO2007068620, WO2007068641, WO2007075629, WO2007080140, WO2007082840, WO2007088450, WO2007088462, WO2007094962, WO2007099423, WO2007100990, WO2007105053, WO2007106349, WO2007110364, WO2007115938, WO2007131907, WO2007133561, US2007270440, WO2007135111, WO2007137955, US2007281923, WO2007137968, WO2007138431, WO2007146122, WO2008005338, WO2008012010, WO2008015125, WO2008045371, EP1757594, WO2008068173, WO2008068174, US20080171753, WO2008072703, WO2008072724, US2008188484, US2008188486, US2008188487, WO2008109333, WO2008109336 beschrieben sind);
Histamin Hl / Histamin H3 Modulatoren, wie z. B. Betahistin bzw. seinem Dihydrochlorid;
Modulatoren des Histamin H3 Transporters oder der Histamin H3 / Serotonin Transporter wie sie z.B. in WO2008002816, WO2008002817, WO2008002818, WO2008002820 beschrieben sind;
Histamin H4 Modulatoren wie sie z.B. in WO2007117399 beschrieben sind;
CRF-Antagonisten (z.B. [2-Methyl-9-(2,4,6-trimethyl-phenyl)-9H-l ,3,9-triaza-fluoren-4-yl]- dipropyl-amin (WO 00/66585) oder solche CRFl -Antagonisten, wie sie in WO2007105113, WO2007133756, WO2008036541, WO2008036579, WO2008083070 beschrieben sind);
CRF BP-Antagonisten (z.B. Urocortin);
Urocortin-Agonisten;
Modulatoren des beta-3 Adrenoceptors wie z.B. l-(4-Chloro-3-methanesulfonylmethyl- phenyl)-2-[2-(2,3-dimethyl- 1 H-indol-6-yloxy)-ethylamino]-ethanol Hydrochlorid (WO 01/83451) oder Solabegron (GW-427353) oder N-5984 (KRP-204) oder solche, wie sie in JP2006111553, WO2002038543, WO2002038544, WO2007048840-843, WO2008015558, EP 1947103 beschrieben sind;
MSH (Melanocyt-stimulierendes Hormon)- Agonisten;
MCH (melanin-konzentrierendes Hormon) Rezeptor Antagonisten (wie z. B. NBI-845, A-761, A-665798, A-798, ATC-0175, T-226296, T-71 (AMG-071, AMG-076), GW-856464, NGD- 4715, ATC-0453, ATC-0759, GW-803430 oder solche Verbindungen, wie sie in WO2005085200, WO2005019240, WO2004011438, WO2004012648, WO2003015769, WO2004072025, WO2005070898, WO2005070925, WO2004039780, WO2004092181, WO2003033476, WO2002006245, WO2002089729, WO2002002744, WO2003004027, FR2868780, WO2006010446, WO2006038680, WO2006044293, WO2006044174, JP2006176443, WO2006018280, WO2006018279, WO2006118320, WO2006130075, WO2007018248, WO2007012661, WO2007029847, WO2007024004, WO2007039462, WO2007042660, WO2007042668, WO2007042669, US2007093508, US2007093509, WO2007048802, JP2007091649, WO2007092416; WO2007093363-366, WO2007114902, WO2007114916, WO2007141200, WO2007142217, US2007299062, WO2007146758, WO2007146759, WO2008001160, WO2008016811, WO2008020799, WO2008022979, WO2008038692, WO2008041090, WO2008044632, WO2008047544, WO2008061109, WO2008065021, WO2008068265, WO2008071646, WO2008076562, JP2008088120, WO2008086404, WO2008086409 beschrieben sind);
CCK-A (CCK-I) Agonisten/Modulatoren (wie z.B. {2-[4-(4-Chloro-2,5-dimethoxy-phenyl)-5- (2-cyclohexyl-ethyl)-thiazol-2-ylcarbamoyl]-5,7-ciimethyl-indol-l-yl}-essigsäure Trifluoressigsäuresalz (WO 99/15525) oder SR-146131 (WO 0244150) oder SSR-125180) oder solchen, wie sie in WO2005116034, WO2007120655, WO2007120688, WO2007120718, WO2008091631 beschrieben sind;
Serotonin- Wiederaufhahme-Inhibitoren (z.B. Dexfenfluramine) oder solchen wie sie in WO2007148341, WO2008034142, WO2008081477, WO2008120761 beschrieben sind;
gemischte Serotonin-/Dopamin- Wiederaufnahme-Inhibitoren (z.B. Bupropion) oder solche wie sie in WO2008063673 beschrieben sind oder feste Kombinationen von Bupropion mit Naltrexon oder Bupropion mit Zonisamid;
gemischte Wiederaufnahmeinhibitoren wie z.B. DOV-21947;
gemischte Sertonin- und noradrenerge Verbindungen (z.B. WO 00/71549);
5-HT-Rezeptor Agonisten z.B. l-(3-Ethyl-benzofuran-7-yl)-piperazin Oxalsäuresalz (WO 01/09111);
gemischte Dopamin/Norepinephrin/ Acetylcholin- Wiederaufnahme-Inhibitoren (z.B. Tesofensine) oder solchen wie sie z.B. in WO2006085118 beschrieben sind;
Dopaminantagonisten wie sie z.B. in WO2008079838, WO2008079839, WO2008079847, WO2008079848 beschrieben sind;
Norepinephrin- Wiederaufhahme-Inhibitoren wie sie z.B. in US2008076724 beschrieben sind;
5-HT2A Rezeptor Antagonisten wie sie z.B. in WO2007138343 beschrieben sind;
5-HT2C Rezeptor Agonisten (wie z.B. Lorcaserin Hydrochlorid (APD-356) oder BVT-933 oder solche, wie sie in WO200077010, WO200077001-02, WO2005019180, WO2003064423,
WO200242304, WO2005035533, WO2005082859, WO2006004937, US2006025601, WO2006028961, WO2006077025, WO2006103511, WO2007028132, WO2007084622, US2007249709; WO2007132841, WO2007140213, WO2008007661, WO2008007664, WO2008009125, WO2008010073, WO2008108445 beschrieben sind);
5-HT6 Rezeptor Modulatoren, wie z.B. E-6837, BVT-74316 oder PRX-07034 oder solche wie sie z.B. in WO2005058858, WO2007054257, WO2007107373, WO2007108569, WO2007108742-744, WO2008003703, WO2008027073, WO2008034815, WO2008054288, EP 1947085, WO2008084491, WO2008084492, WO2008092665, WO2008092666, WO2008101247, WO2008110598, WO2008116831, WO2008116833 beschrieben sind;
Agonisten des Estrogenrezeptors gamma (ERRγ Agonisten), wie sie z.B. in WO2007131005, WO2008052709 beschrieben sind;
Agonisten des Estrogenrezeptors alpha (ERRα / ERRl Agonisten), wie sie z.B. in WO2008109727 beschrieben sind;
Sigma-1 Rezeptorantagonisten, wie sie z.B. in WO2007098953, WO2007098961, WO2008015266, WO2008055932, WO2008055933 beschrieben sind;
Muscarin 3 Rezeptor (M3R) Antagonisten, wie sie z.B. in WO2007110782, WO2008041184 beschrieben sind;
Bombesin-Rezeptor Agonisten (BRS-3 Agonisten), wie sie z.B. in WO2008051404, WO2008051405, WO2008051406, WO2008073311 beschrieben sind;
Galanin-Rezeptor Antagonisten;
Wachstumshormon (z.B. humanes Wachstumshormon oder AOD-9604);
Wachstumshormon freisetzende Verbindungen (6-Benzyloxy-l-(2-diisopropylamino- ethylcarbamoyl)-3,4-dihydro-l H-isochinolin-2-carbonsäuretertiärbutylester (WO 01/85695));
Growth Hormone Secretagogue Receptor Antagonisten (Ghrelin Antagonisten) wie z. B. A- 778193 oder solchen, wie sie in WO2005030734, WO2007127457, WO2008008286 beschrieben sind;
Growth Hormone Secretagogue Receptor Modulatoren (Ghrelin-Modulatoren) wie z.B. JMV- 2959, JMV-3002, JMV-2810, JMV-2951 oder solchen, wie sie in WO2006012577 (z.B. YIL- 781 oder YIL-870), WO2007079239, WO2008092681 beschrieben sind;
TRH-Agonisten (siehe z.B. EP 0 462 884);
entkoppelnde Protein 2- oder 3 -Modulatoren;
chemische Entkoppler (z.B. WO2008059023, WO2008059024, WO2008059025, WO2008059026);
Leptinagonisten (siehe z.B. Lee, Daniel W.; Leinung, Matthew C; Rozhavskaya-Arena, Marina; Grasso, Patricia. Leptin agonists as a potential approach to the treatment of obesity. Drugs of the Future (2001), 26(9), 873-881);
DA-Agonisten (Bromocriptin, Doprexin);
Lipase/Amylase-Inhibitoren (z.B. WO 00/40569, WO2008107184);
Inhibitoren der Diacylglycerol O-Acyltransferasen (DGATs) wie z. B. BA Y- 74-4113 oder wie z. B. in US2004/0224997, WO2004094618, WO200058491, WO2005044250, WO2005072740, JP2005206492, WO2005013907, WO2006004200, WO2006019020, WO2006064189, WO2006082952, WO2006120125, WO2006113919, WO2006134317, WO2007016538, WO2007060140, JP2007131584, WO2007071966, WO2007126957, WO2007137103, WO2007137107, WO2007138304, WO2007138311, WO2007141502, WO2007141517, WO2007141538, WO2007141545, WO2007144571, WO2008011130, WO2008011131, WO2008039007, WO2008048991, WO2008067257, WO2008099221 beschrieben;
Inhibitoren der Monoacylglycerolacyltransferase (2-Acylglycerol-O-Acyltransferase; MGAT) wie sie z.B. in WO2008038768 beschrieben sind;
Inhibitoren der Fettsäuresynthase (FAS) wie z.B. C75 oder solchen, wie in WO2004005277, WO2008006113 beschrieben;
Inhibitoren der Stearoyl-CoA delta9 Desaturase (SCDl) wie sie z.B. in WO2007009236, WO2007044085, WO2007046867, WO2007046868, WO20070501124, WO2007056846, WO2007071023, WO2007130075, WO2007134457, WO2007136746, WO2007143597, WO2007143823, WO2007143824, WO2008003753, WO2008017161, WO2008024390, WO2008029266, WO2008036715, WO2008043087, WO2008044767, WO2008046226, WO2008056687, WO2008062276, WO2008064474, WO2008074824, WO2008074832, WO2008074833, WO2008074834, WO2008074835, WO2008089580, WO2008096746, WO2008104524, WO2008116898, US2008249100, WO2008120744, WO2008120759, WO2008123469, WO2008127349 beschrieben sind;
Inhibitoren der Fatty-Acid-Desaturase-1 (delta5 Desaturase) wie sie z.B. in WO2008089310 beschrieben sind;
hypoglykämische/hypertriglyceridämische Indolinverbindungen wie sie in WO2008039087 beschrieben sind;
Inhibitoren des „Adipocyte fatty acid-binding protein aP2" wie z.B. BMS-309403;
Aktivatoren der Adiponectinsekretion, wie z.B. in WO2006082978, WO2008105533 beschrieben;
Promotoren der Adiponectinproduktion, wie z.B. in WO2007125946, WO2008038712 beschrieben; modifizierte Adiponectine wie z.B. in WO2008121009 beschrieben;
Oxyntomodulin oder Analoga davon;
Oleoyl-Estron;
oder Agonisten oder partiellen Agonisten des Schilddrüsenhormonrezeptors (thyroid hormone receptor agonists) wie z. B: KB-2115 (Eprotirome), QRX-431 (Sobetirome) oder DITPA oder solche, wie in WO20058279, WO200172692, WO200194293, WO2003084915, WO2004018421, WO2005092316, WO2007003419, WO2007009913, WO2007039125, WO2007110225, WO2007110226, WO2007128492, WO2007132475, WO2007134864, WO2008001959, WO2008106213 beschrieben;
oder Agonisten des Schilddrüsenhormonrezeptors beta (TR-beta) wie z. B. MB-07811 oder MB-07344, oder solchen wie in WO2008062469 beschrieben, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einer Kombination von Eprotirome mit Ezetimibe verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Site-1 Protease (SlP), wie z.B. PF-429242, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Modulator des "Trace- Amine- Associated-Receptor-1" (TAARl), wie sie z.B. in US2008146523, WO2008092785 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Inhibitor des Growth-Factor-Receptor-Bound-Protein-2 (GRB2), wie z.B. in WO2008067270 beschrieben, verabreicht.
Bei einer weiteren Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem RNAi (siRNA) Therapeutikum, welches gegen PCSK9 (Proprotein Convertase Subtilisin/Kexin Typ 9) gerichtet ist, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Omacor® oder Lovaza™ (Omega-3 -Fettsäureester; hochkonzentrierte Ethyiester der Eicosapentaensäure und der Docosahexaensäure) verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Lycopin verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Antioxidans, wie z.B. OPC- 14117, AGI- 1067 (Succinobucol), Probucol, Tocopherol, Ascorbinsäure, ß-Caroten oder Selen verabreicht.
Bei einer Ausführungsform der Erfindung wird die Verbindung der Formel I in Kombination mit einem Vitamin, wie z. B. Vitamin B6 oder Vitamin B12 verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit mehr als einer der vorstehend genannten Verbindungen, z.B. in Kombination mit einem Sulfonylharnstoff und Metformin, einem Sulfonylharnstoff und Acarbose, Repaglinide und Metformin (PrandiMet (TM)), Insulin und einem Sulfonylharnstoff, Insulin und Metformin, Insulin und Troglitazon, Insulin und Lovastatin, etc. verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Inhibitor der Carboanhydrase Typ 2 (Carbonic anhydrase type 2), wie z.B. solchen, wie in WO2007065948 beschrieben, verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit Topiramat oder einem Derivat davon, wie es in WO2008027557 beschrieben ist, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einer festen Kombination von Topiramat mit Phentermin (Qnexa™) verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einer Antisense- Verbindung, z.B. ISIS-377131, verabreicht, welche die Produktion des Glukokortikoidrezeptors inhibiert.
Bei einer anderen Ausfuhrungsform wird die Verbindung der Formel I in Kombination mit einem Aldosteronsynthaseinhibitor und einem Antagonisten des Glucocorticoidrezeptors, einem Cortisolsyntheseinhibitor und/oder einem Antagonisten des Corticotropin-freisetzenden Faktors (corticotropin releasing factor), wie z.B. in EPl 886695, WO2008119744 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Agonisten des RUP3 Rezeptors, wie z. B. in WO2007035355, WO2008005576 beschrieben, verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Aktivator des Gens, welches für die Ataxia Telangiectasia Mutated (ATM) Proteinkinase kodiert, wie z. B. Chloroquin, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Tau- Protein-Kinase-1 -Inhibitor (TPKl Inhibitor), wie z. B. in WO2007119463 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem „c- Jun N-terminal kinase" Inhibitor (JNK-Inhibitor), wie z. B. in WO2007125405, WO2008028860, WO2008118626 beschrieben, verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Endothelin- A-Rezeptor Antagonisten, wie z. B. Avosentan (SPP-301), verabreicht.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Modulatoren des Glukokortikoidrezeptors (GR), wie z.B. KB-3305 oder solchen Verbindungen wie sie z. B.
in WO2005090336, WO2006071609, WO2006135826, WO2007105766, WO2008120661 beschrieben sind, verabreicht.
Bei einer Ausführungsform ist der weitere Wirkstoff Varenicline Tartrate, ein partieller Agonist des alpha 4-beta 2 nikotinischen Acetylcholinrezeptors.
Bei einer Ausführungsform ist der weitere Wirkstoff Trodusquemine.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Modulator des Enzyms SIRTl und/oder SIRT3 (einer NAD+-abhängigen Proteindeacetylase); dieser Wirkstoff kann z.B. Resveratrol in geeigneten Formulierungen sein, oder solche Verbindungen wie sie in WO2007019416 (z.B. SRT-1720), WO2008073451 genannt sind.
Bei einer Ausführungsform der Erfindung ist der weitere Wirkstoff DM-71 (N-Acetyl-L- Cy stein mit Bethanechol).
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit anti- hypercholesterolemisch wirkenden Verbindungen, wie sie z.B. in WO2007107587, WO2007111994, WO2008106600, WO2008113796 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit Inhibitoren des SREBP (sterol regulatory element-binding protein), wie sie z.B. in WO2008097835 beschrieben sind, verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit einem cyclischen Peptidagonisten des VPAC2 Rezeptors, wie sie z.B. in WO2007101146, WO2007133828 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einem Agonisten des Endothelinrezeptors, wie sie z.B. in WO2007112069 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit AKP-020 (Bis(ethylmaltolato)oxovanadium-IV) verabreicht.
Bei einer anderen Ausführungsform wird die Verbindung der Formel I in Kombination mit gewebe-selektiven Androgenrezeptor Modulatoren („tissue-selective androgen receptor modulators"; SARM), wie sie z.B. in WO2007099200, WO2007137874 beschrieben sind, verabreicht.
Bei einer weiteren Ausführungsform wird die Verbindung der Formel I in Kombination mit einem AGE (advanced glycation endproduct) Inhibitor, wie sie z.B. in JP2008024673 beschrieben sind, verabreicht.
Bei einer Ausführungsform der Erfindung ist der weitere Wirkstoff Leptin; siehe z.B. "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez-Ambrosi,
Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2(10), 1615-1622.
Bei einer anderen Ausführungsform der Erfindung ist der weitere Wirkstoff Metreleptin (rekombinantes Methionyl-Leptin) kombiniert mit Pramlintide.
Bei einer weiteren Ausführungsform der Erfindung ist der weitere Wirkstoff das Tetrapeptid ISF-402.
Bei einer Ausführungsform ist der weitere Wirkstoff Dexamphetamin oder Amphetamin.
Bei einer Ausführungsform ist der weitere Wirkstoff Fenfluramin oder Dexfenfluramin.
Bei noch einer Ausführungsform ist der weitere Wirkstoff Sibutramin oder solche Derivate wie sie in WO2008034142 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff Mazindol oder Phentermin.
Bei einer weiteren Ausführungsform ist der weitere Wirkstoff Geniposidinsäure (geniposidic acid; WO2007100104) oder Derivate davon (JP2008106008).
Bei einer Ausführungsform ist der weitere Wirkstoff ein nasal verabreichter Calciumkanalblocker wie z.B. Diltiazem oder solche, wie sie in US 7,138,107 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Inhibitor des Natrium-Calcium-Ionen- Austausches wie z.B. solche, wie sie in WO2008028958, WO2008085711 beschrieben sind.
Bei einer weiteren Ausführungsform ist der weitere Wirkstoff ein Blocker von Calciumkanälen wie z.B. des CaV3.2 oder CaV2.2 wie sie in WO2008033431, WO2008033447, WO2008033356, WO2008033460, WO2008033464, WO2008033465, WO2008033468, WO2008073461 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Modulator eines Calciumkanals wie z.B. solche, wie sie in WO2008073934, WO2008073936 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Blocker des „T-type calcium Channel" wie sie z.B. in WO2008033431, WO2008110008 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Inhibitor des KCNQ-Kaliumkanal-2 bzw. -3 wie z.B. solche, wie sie in US2008027049, US2008027090 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Inhibitor des Kalium KvI.3 Ionenkanals wie z.B. solchen, wie sie in WO2008040057, WO2008040058, WO2008046065 beschrieben sind.
Bei einer anderen Ausführungsform ist der weitere Wirkstoff ein Modulator des MCP-I Rezeptors (monocyte chemoattractant protein- 1 (MCP-I)) wie z.B. solche, wie sie in WO2008014360, WO2008014381 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Modulator des Somatostatinrezeptors 5 (SSTR5) wie z.B. solche, wie sie in WO2008019967, US2008064697, US2008249101, WO2008000692 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Modulator des Somatostatinrezeptors 2 (SSTR2) wie z.B. solche, wie sie in WO2008051272 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Erythropoietin-mimetisches Peptid, welches als Erythropoietin (EPO) Rezeptoragonist agiert. Solche Moleküle sind z.B. in WO2008042800 beschrieben.
Bei einer weiteren Ausführungsform ist der weitere Wirkstoff ein Anorektikum/eine hypoglykämische Verbindung wie z.B. solche, wie sie in WO2008035305, WO2008035306, WO2008035686 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Induktor der Liponsäuresynthetase wie z.B. solche, wie sie in WO2008036966, WO2008036967 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Stimulator der endothelialen Nitric- Oxid-Synthase (eNOS) wie z.B. solche, wie sie in WO2008058641, WO2008074413 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Modulator des Kohlenhydrat- und/oder Lipidstoffwechsels wie z.B. solche, wie sie in WO2008059023, WO2008059024, WO2008059025, WO2008059026 beschrieben sind.
Bei einer weiteren Ausführungsform ist der weitere Wirkstoff ein Angiotensin II Rezeptorantagonist wie z.B. solche, wie sie in WO2008062905, WO2008067378 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Agonist des Sphingosin-1- Phosphatrezeptors (SlP) wie z.B. solche, wie sie in WO2008064315, WO2008074820. WO2008074821 beschrieben sind.
Bei einer Ausführungsform ist der weitere Wirkstoff ein Mittel, welches die Magenentleerung retardiert wie z.B. 4-Hydroxyisoleucin (WO2008044770).
Bei einer Ausfuhrungsform ist der weitere Wirkstoff eine Muskel-relaxierende Substanz wie sie z.B. in WO2008090200 beschrieben ist.
Bei einer weiteren Ausführungsform ist der weitere Wirkstoff ein Inhibitor der Monoaminoxidase B (MAO-B) wie z.B. solche, wie sie in WO2008092091 beschrieben sind.
Bei einer anderen Ausführungsform ist der weitere Wirkstoff ein Inhibitor der Bindung von Cholesterol und/oder Triglyceriden an das SCP-2 Protein (sterol carrier protein-2) wie z.B. solche, wie sie in US2008194658 beschrieben sind.
Bei einer anderen Ausführungsform ist der weitere Wirkstoff Lisofylline, welcher Autoimmunschäden an insulinproduzierenden Zellen verhindert.
Bei einer Ausführungsform wird die Verbindung der Formel I in Kombination mit Ballaststoffen, vorzugsweise unlöslichen Ballaststoffen (siehe z.B. Carob/ Caromax® (Zunft H J; et al., Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct), 18(5), 230-6.) Caromax ist ein Carob enthaltendes Produkt der Fa. Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark Höchst, 65926 Frankfurt / Main)) verabreicht. Die Kombination mit Caromax® kann in einer Zubereitung erfolgen, oder durch getrennte Gabe von Verbindungen der Formel I und Caromax®. Caromax® kann dabei auch in Form von Lebensmitteln, wie z.B. in Backwaren oder Müsliriegeln, verabreicht werden.
Es versteht sich, dass jede geeignete Kombination der erfindungsgemäßen Verbindungen mit einer oder mehreren der vorstehend genannten Verbindungen und wahlweise einer oder mehreren weiteren pharmakologisch wirksamen Substanzen als unter den Schutzbereich der vorliegenden Erfindung fallend angesehen wird.

LY-518674

KRP-101

LY-510929 GW-501516

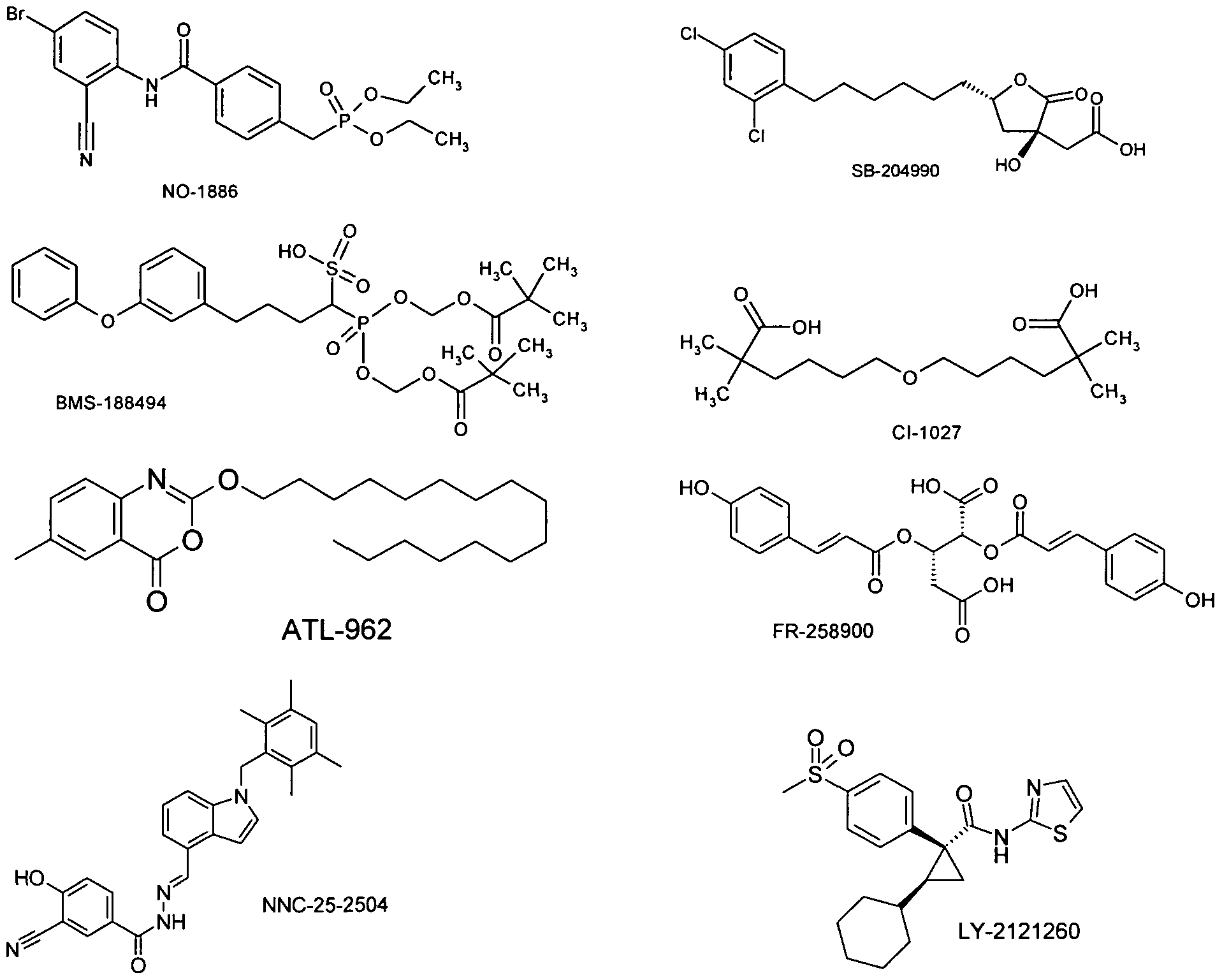

FR-225654







PSN-632408 SYR-322

DP-893 Varenicline Tartrat

Trodusquemine

Solabegron Lorcaserin Hydrochlorid



Leu — Tyr — Se r — S er — VaI — Asp — Se r
GIu — G Iy — GIn — AIa — AIa . ys — G Iu Trp — AIa ■ -He -Phe

 BIM-51077 TAK-536
BIM-51077 TAK-536
E-6837 Tesofensine

BVT-74316 ABT-341

Sergliflozin SLV-319
AVE 1625 (proposed INN: drinabant) TAK-475 (Lapaquistat Acetat)

AS-1552133 MB-07344

CKD-501 (Lobeglitazon Sulfat) MB-07811

JMV-2959 JMV-3002

JMV-2810 JMV-2951

BMS-309403 PSN-119-1

S-40755 LY-2463665

Dapagliflozin, BMS-512148 BI-1356

PF-429242 SLV-348

Balaglitazon


BMS-711939 BMS-687453

ST-3473 DOV-21947

DM-71 AEGR-733


PF-00389027 KB-3305

ISF-402 SRT- 1720
 darapladib A-002
darapladib A-002

DITPA DGAT-I Inhibitor aus WO2007137103





MB-07229 MB-07803

Succinobucol


T-2384 BMS-644950
 alogliptin benzoate Nicotinsäure / Laropiprant
alogliptin benzoate Nicotinsäure / Laropiprant
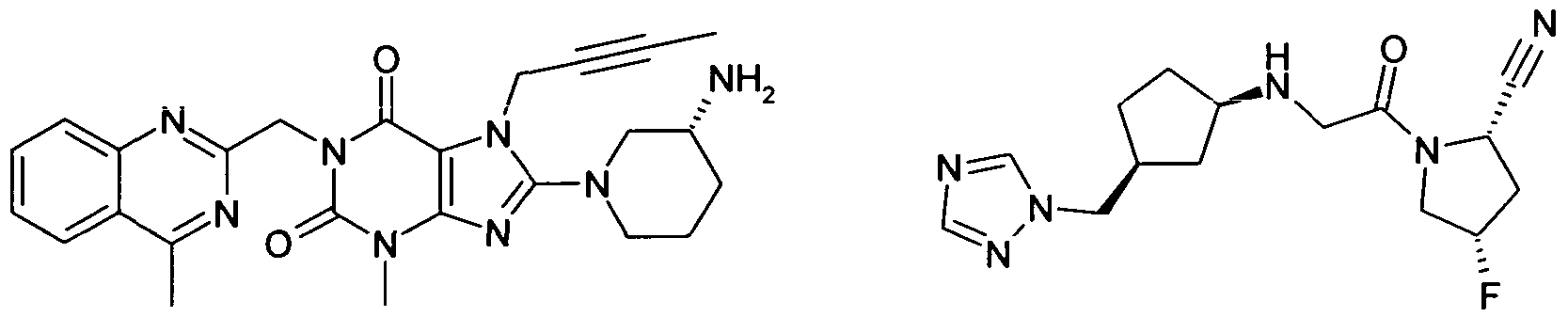 linagliptin melogliptin
linagliptin melogliptin


PSN-119-2 drospirenone

Weiterhin sind folgende Wirkstoffe für Kombinationspräparate geeignet:
Alle Antiepileptika, die in der Roten Liste 2007, Kapitel 15 genannt sind; alle Antihypertonika, die in der Roten Liste 2007, Kapitel 17 genannt sind; alle Hypotonika, die in der Roten Liste 2007, Kapitel 19 genannt sind; alle Antikoagulantia, die in der Roten Liste 2007, Kapitel 20 genannt sind; alle Arteriosklerosemittel, die in der Roten Liste 2007, Kapitel 25 genannt sind; alle Betarezeptoren-, Calciumkanalblocker und Hemmstoffe des Renin-Angiotensin-Systems, die in der Roten Liste 2007, Kapitel 27 genannt sind;
alle Diuretika und Durchblutungsfördernde Mittel, die in der Roten Liste 2007, Kapitel 36 und
37 genannt sind; alle Entwöhnungsmittel/Mittel zur Behandlung von Suchterkrankungen, die in der Roten Liste
2007, Kapitel 39 genannt sind; alle Koronarmittel und Magen-Darm-Mittel, die in der Roten Liste 2007, Kapitel 55 und 60 genannt sind; alle Migränemittel, Neuropathiepräparate und Parkinsonmittel, die in der Roten Liste 2007,
Kapitel 61, 66 und 70 genannt sind.
Ein erfindungsgemäßes Verfahren („A") zur Herstellung einer Verbindung der Formel I umfasst entweder das Umsetzen einer Verbindung der Formel

worin R1, R2 und X die vorstehenden Definitionen aufweisen, mit einer Verbindung der Formel
CH3 m
R3'- NH- C— CN CH3
in Gegenwart einer tertiären Base, um eine Verbindung der Formel


Verbindungen der Formel IV können gegebenenfalls einer oder mehrerer der folgenden Reaktionen in beliebiger Reihenfolge unterworfen werden: a) Hydrolysereaktion von C=NH zu einer Ketonfunktion oder Umwandlung von C=S in C=O b) Umwandlungsreaktion von C=O in C=S
c) Umsetzen der Produkte der Formel IV, worin R3 > Wasserstoff ist, und nach Hydrolyse von C=NH in ein Keton, mit einer Verbindung der Formel R3"-Halogen.
Ein weiteres Verfahren („B") zur Herstellung der Verbindungen der Formel I
Säure

Aktivierung


Verfahren „B"
besteht darin, dass ein geeignet substituiertes Anilin der Formel A, in welchem die Reste R1, R2 und R3' die weiter oben beschriebene Bedeutung haben, in ein Isocyanat der Formel B umgesetzt wird. Diese Umsetzung kann z. B. mit Phosgen in Toluol oder mit Diphosgen oder Triphosgen durchgeführt werden. Das Isocyanat B wird anschließend mit dem Methylester oder einem anderen Ester (z.B. tert.-Butyl) der Aminosäure J, in welcher R und R' die Bedeutung CH3 haben, oder einem Salz eines Esters der Aminosäure /unter Zugabe von Base (z. B. Triethylamin) zu einem Harnstoff der Formel K umgesetzt. Dieser Harnstoff kann unter basischen oder sauren Bedingungen, vorzugsweise sauren Bedingungen, zum Imidazolidin-2,4- dion der Formel L ringgeschlossen werden. Die weitere Umsetzung zu einer Verbindung der Formel M, in welcher der Rest R3 die oben beschriebene Bedeutung hat, kann z. B. so erfolgen, dass L mit einer geeignet substituierten Verbindung Q, wobei R3 die weiter oben beschriebenen Bedeutungen hat und V Halogen, bevorzugt Brom, bedeutet alkylierend umgesetzt wird.
In einem weiteren Verfahren („C") zur Herstellung der Verbindungen der Formel I

C"
wird so vorgegangen, dass das Isocyanat B mit einem geeignet substituierten Aminosäureesterderivat C, wobei der im Schema gezeigte Methylester ein nicht limitierendes Beispiel für einen Ester ist, und wobei R3', R und R' die oben benannten Bedeutungen haben, unter Zugabe einer Base (z. B. Triethylamin) zu einem Harnstoff der Formel F umgesetzt wird. Das Aminosäureesterderivat C kann aus der Verbindung D, worin R3 die weiter oben
beschriebenen Bedeutungen hat und X die Bedeutung Halogen, bevorzugt Brom, hat, mit einem Aminosäureester der Formel E, worin R und R* die in Formei I genannten Bedeutungen haben, unter alkylierenden Bedingungen hergestellt werden. Alternativ kann die Verbindung der Formel C durch reduktive Aminierung des Aldehyds D, worin R3 die Bedeutung Aryl oder Heteroaryl und X die Bedeutung (C0-C1 i)-CHO hat, mit dem Aminosäurederivat E gewonnen werden. Der Harnstoff F kann unter basischen oder sauren Bedingungen, vorzugsweise sauren Bedingungen, zum Imidazolidin-2,4-dion der Formel G, worin R3 die weiter oben für Formel I beschriebenen Bedeutungen hat ringgeschlossen werden.
Die optionale Hydrolysereaktion von C=NH in C=O wird vorzugsweise mit einer Säure, wie wässriger Salzsäure unter Rückfluss, durchgeführt. Wenn die Hydrolyse von C=NH in C=O mit einem Molekül durchgeführt wird, das ebenfalls C=S enthält, kann das Letztere in eine C=O-Gruppe umgewandelt werden. Die Umwandlung der Gruppe C=O in C=S erfolgt mit einem Lawesson-Reagens der Formel

bei dem es sich um ein beispielsweise von Fluka vertriebenes kommerzielles Reagens handelt und das in Bull. Soc. Chim. BeIg., Bd. 87 Nr. 3 (1987), S. 229 beschrieben ist. Werden zwei C=O in C=S umgewandelt, wird die Umsetzung in einem Überschuss des Lawesson-Reagens durchgeführt. Das gleiche wird auch verwendet, wenn das Molekül sowohl C=S als auch C=O enthält und gewünscht ist, C=O in C=S umzuwandeln.
Wenn dagegen ein Teil des Moleküls zwei C=O enthält und gewünscht ist, ein Produkt mit nur einem C=S zu erhalten, wird ein Mangel an Lawesson-Reagens verwendet, um ein Gemisch von 3 Produkten zu erhalten, zwei Produkte mit jeweils einem C=O und C=S und ein Produkt mit zwei C=S. Diese Produkte können durch bekannte Verfahren, wie Chromatographie, getrennt werden.
Die nachfolgenden Beispiele dienen zur näheren Erläuterung der Erfindung, ohne dieselbe auf in den Beispielen beschriebene Produkte und Ausführungsformen einzuschränken.
Allgemeine experimentelle Verfahren:
1H-NMR:
Die 1H-NMR Spektren wurden in deuteriertem Dimethylsulfoxid an einem 500 MHz-Gerät
(DRX 500, Firma Bruker) bei 3000K gemessen. Angaben: δ in ppm, Multiplizität (s für
Singulett, d für Dublett, t für Triplett, q für Quartett, m für Multiplett, x H (Anzahl der
Wasserstoffatome)
HPLC/MS:
Die HPLC-MS-Messungen wurden an einem LCT-Gerät der Firma Waters durchgeführt.
Säule: YMC Jshere 33x2 4 μm; Gradient [A]: (Acetonitril + 0.05% Trifluoressigsäure) :
(Wasser + 0.05% Trifluoressigsäure) 5:95 (0 Minuten) nach 95:5 (3 Minuten); Gradient [B]:
(Acetonitril + 0.05% Trifluoressigsäure) : (Wasser + 0.05% Trifluoressigsäure) 5:95 (0
Minuten) nach 95:5 (2.5 Minuten) nach 95:5 (3.0 Minuten); Gradient [C]: (Acetonitril + 0.05%
Trifluoressigsäure) : (Wasser + 0.05% Trifluoressigsäure) 5:95 (0 Minuten) nach 95:5 (3.4
Minuten) nach 95:5 (4.4 Minuten); Gradient [D]: (Acetonitril + 0.05% Trifluoressigsäure) :
(Wasser + 0.05% Trifluoressigsäure) 5:95 (0 Minuten) nach 5:95 (0.3 Minuten) nach 95:5 (3.5
Minuten) nach 95:5 (4 Minuten); Gradient [E]: (Acetonitril + 0.05% Trifluoressigsäure) :
(Wasser + 0.05% Trifluoressigsäure) 2:98 (1 Minute) nach 95:5 (5 Minuten) nach 95:5 (6.25
Minuten); Detektor: Tecan-LCT.
Chromatographische Reinigungsmethoden:
[RPl]: Fluss: 30 ml/min; Gradient: Acetonitril/Wasser + 0,1% Trifluoressigsäure; 30 min.
Säule: XTerra Cl 8 5 μm 30x100 mm; Detektion: MS (ESI), UV (DAD).
[RP2]: Fluss: 150 ml/min; Gradient: Acetonitril/Wasser + 0,1% Trifluoressigsäure; 20 min.
Säule: XTerra Cl 8 10 μm 50x250 mm; Detektion: MS (ESI), UV (DAD).
Beispiel 1: 4-[3-(3,5-Bis-trifluoromethyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l- y 1] -2 -trifluoromethyl-benzonitril :

Die Verbindung 1.1 kann nach Verfahren "A" dargestellt werden.Eine Lösung von 10 g 4-Cyano-3-trifluormethylanilin (im europäischen Patent Nr. 0,002,892 beschrieben) in 30 ml Essigsäureethylester wurde bei 0 bis 5°C zu 33,6 ml einer Toluollösung mit 1,93 M/l Phosgen hinzugefügt, und nach Rühren bei 0 bis 5°C für 30 Minuten wurde die Temperatur auf 25 °C erhöht. Das Gemisch wurde destilliert, während frisches Toluol eingebracht wurde, das auf einer konstanten Höhe gehalten wurde, um das destillierte Volumen Toluol zu kompensieren, bis eine Temperatur von etwa 110°C erreicht war. Das Gemisch wurde bei Rückfluss gehalten, bis die Abgabe von Chlorwasserstoff abnahm (4 1/2 Stunden). Die Temperatur kehrte zu Raumtemperatur zurück, und der weiße Feststoff wurde über Natriumsulfat getrocknet und 3 Mal mit Toluol gespült. Die organische Phase wurde unter verringertem Druck bis zur Trockne verdampft, bei 6O0C für eine Stunde erhitzt und dann unter Argon abgekühlt, um 11,6 g 4-Isocyanato-2- trifluoromethylbenzonitril zu erhalten.
Eine Lösung von 6,6 g 4-Isocyanato-2-trifluoromethylbenzonitril in 10 ml Dichlorethan wurde bei 5°C zu einer Lösung von 2,63 g 2-Amino-2-cyanopropan und 36 ml Dichlorethan und 0,9 ml Triethylamin hinzugefügt, und nach Rühren für 16 Stunden bei Raumtemperatur wurde das Gemisch bis zur Trockne verdampft. Die 7,7 g Rückstand wurden auf Kieselgel chromatographiert und mit einem 85 / 15 - Methylenchlorid / Aceton-Gemisch eluiert, um 3,54 g des gewünschten Produkts zu erhalten, das bei 228°C schmilzt. Eine Analyseprobe wurde durch Kristallisieren von 300 mg aus Isopropanol hergestellt, um 267 mg des Produkts zu erhalten, das bei 228°C schmilzt.
Analyse: C13HnF3N4O; Molekülmasse = 296,25
% C % H % F % N
Berechnet: 52,71 3,74 19,24 18,91 Gefunden: 52,7 3,6 19,1 18,6
2) Herstellung von 4-(4,4-Dimethyl-2,5-dioxo-imidazolidin- 1 -yl)-2-trifluoromethyl- benzonitril (1.2):
Herstellung nach Verfahren „A": Eine Lösung von 2,76 g der Verbindung 1.1 und 60 ml 0,5 N Salzsäure wurde für 35 Minuten unter Rückfluss erhitzt und in 100 g Wasser und Eis gegossen. Das Gemisch wurde mit Essigsäureethylester extrahiert, und die organische Phase wurde mit Wasser gewaschen, getrocknet und unter verringertem Druck bis zur Trockne verdampft, um 2,70 g des gewünschten Produkts zu erhalten, das bei 210°C schmilzt. Eine Analyseprobe wurde durch Kristallisieren von 440 mg Produkt aus Isopropanol erhalten, um 383 mg Produkt zu erhalten, das bei 210°C bis 211°C schmilzt.
Herstellung nach Verfahren „B": 14,74 g (79,21 mMol) 4-Amino-2-trifluoromethyl- benzonitril wurden in 200 ml trockenem Acetonitril gelöst. Diese Lösung wurde unter Rühren zu einer auf 700C erwärmten 20%igen Lösung von Phosgen in Toluol zugetropft und anschließend 1 h gerührt. Die abgekühlte Reaktionslösung wurde im Vakuum eingeengt, der Rückstand mit Toluol aufgenommen und wieder im Vakuum eingeengt. Schließlich wurde der Rückstand in 150 ml trockenem Acetonitril gelöst und die Lösung unter Rühren mit 15,5 g (79,21 mMol) 2-Amino-2-methyl-propionsäure-tert.-butylester Hydrochlorid versetzt. Zu die Reaktionsmischung wurden langsam 12,02 g (118,8 mMol) Triethylamin zugetropft und anschließend 45 min. bei Raumtemperatur gerührt. Danach wurde die Mischung vorsichtig mit 50 ml konzentrierter Salzsäure versetzt und 1 h bei 700C gerührt. Die abgekühlte Reaktionsmischung wurde im Vakuum eingeengt und der Rückstand mit Essigsäureethylester und Wasser versetzt. Die organische Phase wurde abgetrennt, mit gesättigter Natriumhydrogencarbonatlösung und anschließend mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und im Vakuum eingeengt. Der Rückstand wurde chromatographisch über Kieselgel mit Heptan /
Essigsäureethylester 2:1 gereinigt. Man erhielt 21,2 g (90% Ausbeute) 4-(4,4-Dimethyl- 2,5-dioxo-imidazolidin-l-yl)-2-trifiuoromethyl-benzonitril i.2 mit dem Schmelzpunkt 208 - 2110C.
3) Herstellung von 4-[3-(3,5-Bis-trifluoromethyl-benzyl)-4,4-dimethyl-2,5-dioxo- imidazolidin-1 -yl]-2-trifluoromethyl-benzonitril (1):
100 mg der Verbindung 1.2 und 103 mg 3,5-Bis-(trifluoromethyl)-benzylbromid wurden in 2,5 ml trockenem Acetonitril gelöst, mit 110 mg Cäsiumcarbonat versetzt und 4 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung mit Essigsäureethylester und Wasser versetzt. Die organische Phase wurde abgetrennt, über Magnesiumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde chromatographisch (Methode [RPl]) gereinigt. Man erhielt 4-[3-(3,5-Bis- trifluoromethyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2-trifluoromethyl- benzonitril; 1H NMR: 8.35, d, IH; 8.23, s, IH; 8.19, s, 2H; 8.1, d, IH; 8.03, s, IH; 4.81, s, 2H; 1.42, s, 6H.
Beispiel 2: 4-[4,4-Dimethyl-2,5-dioxo-3-(4-trifluoromethoxy-benzyl)-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril :

Die Verbindung des Beispiels 2 wurde in analoger Weise durch Umsatz von 1.2 mit 4- (Trifluoromethoxy)-benzylbromid hergestellt. 1H NMR: 8.35, d, IH; 8.23, s, IH; 8.09, d, IH; 7.58, d, 2H; 7.32, d, 2H; 4.63, s, 2H; 1.41, s, 6H.
Beispiel 3: 4-[4,4-Dimethyl-2,5-dioxo-3-(3-trifluoromethyl-benzyl)-imidazolidin-l- yl] -2-trifluoromethyl-benzonitril :

Beispiel 4: 4-[4,4-Dimethyl-2,5-dioxo-3-(4-pentafluorosulfanyl-benzyl)-imidazolidin-l-yl]- 2-trifluoromethyl-benzonitril :

1) Herstellung von (4-Pentafluorosulfanyl-phenyl)-methanol (4.1):

10 g 4-(Pentafluorosulfanyl)-benzoesäure wurden in 100 ml trockenem Tetrahydrofuran gelöst und bei einer Temperatur von -5 bis 0°C innerhalb von 30 min tropfenweise mit 48,4 ml einer 1 M Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran versetzt. Danach ließ man langsam auf Raumtemperatur erwärmen und rührte noch eine Stunde bei Raumtemperatur. Zur Aufarbeitung wurde die Reaktionsmischung unter Kühlung vorsichtig mit 2 n Salzsäure auf pH 3 eingestellt. Die Mischung wurde filtriert, mit 300 ml Essigsäureethylester versetzt und ausgeschüttelt. Die organische Phase wurde abgetrennt, über Magnesiumsulfat getrocknet und chromatographisch (Kieselgel; n- Heptan / Essigsäureethylester 2/1) gereinigt. Man erhielt 7,66 g (81% Ausbeute) (4- Pentafluorosulfanyl-phenyl)-methanol als Hauptprodukt; 1H NMR: 7.86, d, 2H; 7.53, d, 2H; 5.45, t, IH; 4.6, d, 2H.
Als Nebenprodukt wurden 165 mg (4-Mercapto-phenyl)-methanol isoliert; 1H NMR: 7.25, d, 2H; 7.19, d, 2H; 5.3, s, IH; 5.11, t, IH; 4.41, d, 2H.
2) Herstellung von l-Brommethyl-4-pentafluorosulfanyl-benzol (4.2):
 6,23 g Triphenylphosphin und 1,93 g Imidazol wurden in 60 ml Dichlor-methan vorgelegt; zu dieser Mischung wurden unter Rühren bei 5°C 3,79 g Brom, gelöst in 10 ml Dichlormethan, zugetropft. Die Mischung wurde 10 min bei 5°C gerührt. Anschließend wurde bei 5°C eine Lösung von 5,3 g der Verbindung 4.1 in 60 ml Dichlormethan langsam unter Rühren zugetropft. Nach beendeter Zugabe wurde eine weitere Stunde bei 5°C gerührt; nach dem Erwärmen auf Raumtemperatur wurde die Mischung weitere 3 Stunden gerührt. Zur Aufarbeitung wurde die Mischung mit 60 ml 1 n Salzsäure versetzt; die organische Phase wurde abgetrennt, mit Wassr gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde mit 150 ml Diethylether versetzt, gerührt, filtriert und erneut eingeengt. Der Rückstand wurde dann chromatographisch (Kieselgel; n-Heptan / Essigsäureethylester 3 / 1) gereinigt. Man erhielt l-Brommethyl-4-pentafluorosulfanyl-benzol (4.2); 1H NMR: 7.91, d, 2H; 7.69, d, 2H; 4.8, s, 2H.
6,23 g Triphenylphosphin und 1,93 g Imidazol wurden in 60 ml Dichlor-methan vorgelegt; zu dieser Mischung wurden unter Rühren bei 5°C 3,79 g Brom, gelöst in 10 ml Dichlormethan, zugetropft. Die Mischung wurde 10 min bei 5°C gerührt. Anschließend wurde bei 5°C eine Lösung von 5,3 g der Verbindung 4.1 in 60 ml Dichlormethan langsam unter Rühren zugetropft. Nach beendeter Zugabe wurde eine weitere Stunde bei 5°C gerührt; nach dem Erwärmen auf Raumtemperatur wurde die Mischung weitere 3 Stunden gerührt. Zur Aufarbeitung wurde die Mischung mit 60 ml 1 n Salzsäure versetzt; die organische Phase wurde abgetrennt, mit Wassr gewaschen, über Magnesiumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wurde mit 150 ml Diethylether versetzt, gerührt, filtriert und erneut eingeengt. Der Rückstand wurde dann chromatographisch (Kieselgel; n-Heptan / Essigsäureethylester 3 / 1) gereinigt. Man erhielt l-Brommethyl-4-pentafluorosulfanyl-benzol (4.2); 1H NMR: 7.91, d, 2H; 7.69, d, 2H; 4.8, s, 2H.
3) Herstellung von 4-[4,4-Dimethyl-2,5-dioxo-3-(4-pentafluorosulfanyl-benzyl)- imidazolidin-l-yl]-2-trifluoromethyl-benzonitril:
Die Verbindung des Beispiels 4 wurde durch Umsatz von 1.2 mit 4.2 analog der Vorgehensweise bei der Herstellung der Verbindung des Beispiels 1 gewonnen. 1H NMR: 8.35, d, IH; 8.25, s, IH; 8.09, d, IH; 7.89, d, 2H; 7.7, d, 2H; 4.7, s, 2H; 1.42, s, 6H.
Beispiel 5 : 4- [3 -(6-Chlor-pyridin-3 -ylmethyl)-4,4-dimethyl-2,5 -dioxo-imidazolidin- 1 -yl] -2- trifiuoromethyl-benzonitril :
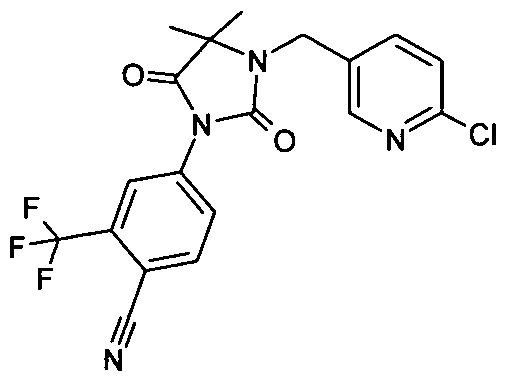
Die Verbindung des Beispiels 5 wurde durch Umsatz von 1.2 mit 2-Chlor-5- chlormethyl-pyridin ähnlich der Vorgehensweise bei der Herstellung der Verbindung
des Beispiels 1 gewonnen. Als Lösungsmittel fand Dimethylformamid und als Base Natriumhydrid Verwendung. 1H NMR: 8.51, s, IH; 8.33, d, IH; 8.23, s, IH; 8.07, d, IH; 7.94, d, IH; 4.67, s, 2H; 1.42, s, 6H.
Beispiel 6: 4-[4,4-Dimethyl-2,5-dioxo-3-(6-trifluoromethyl-pyridin-3-ylmethyl)- imidazolidin-l-yl]-2-trifluoromethyl-benzonitril:

Die Verbindung des Beispiels 6 wurde durch Umsatz von 1.2 mit 3-(Chlormethyl)-6- (trifluoromethyl)pyridin ähnlich der Vorgehensweise bei der Herstellung der Verbindung des Beispiels 1 gewonnen. Als Lösungsmittel fand Dimethylformamid und als Base Natriumhydrid Verwendung. 1H NMR: 8.87, s, IH; 8.33, d, IH; 8.23, s, IH; 8.09, d, IH; 7.9, d, IH; 4.79, s, 2H; 1.46, s, 6H.
Beispiel 7: 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl- 1 -(6-trifiuoromethyl- pyridin-3-ylmethyl)-imidazolidin-2,4-dion:

Die Verbindung des Beispiels 7 kann nach Verfahren „C" hergestellt werden: 1 ) Herstellung von N-Memoxy-N-methyl-ό-trifluoromethyl-nicotinamid (7.1):

Zur Aufarbeitung wurde die Reaktionsmischung im Vakuum eingeengt, der Rückstand in 50 ml Essigsäureethylester aufgenommen und zweimal mit jeweils 25 ml Natriumhydrogensulfatlösung und zweimal mit jeweils 25 ml gesättigter Natriumcarbonatlösung ausgeschüttelt. Die organische Phase wurde abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt N- Methoxy-N-methyl-6-trifluoromethyl-nicotinamid (7.1). 1H NMR: 8.94, s, IH; 8.3, d, IH; 8.01, d, IH; 3.58, s, 3H; 3.34, s, 3H.
2) Herstellung von 6-Trifluoromethyl-pyridin-3-carbaldehyd (7.2):

1,62 g der Verbindung 7.1 wurden in 35 ml trockenem Tetrahydrofuran gelöst und bei - 60°C tropfenweise unter Rühren mit 6,9 ml einer 1 -molaren Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran versetzt. Nach beendeter Zugabe wurde noch eine Stunde bei -60°C gerührt; danach ließ man die Mischung sich auf Raumtemperatur erwärmen. Zur Aufarbeitung wurde die Reaktionsmsichung tropfenweise unter Rühren mit 40 ml kalter Kaliumhydrogensulfatlösung versetzt. Die Mischung wurde zweimal mit jeweils 50 ml Essigsäureethylester ausgeschüttelt; die organische Phase wurde mit gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt 6- Trifluoromethyl-pyridin-3-carbaldehyd (7.2). 1H NMR: 10.2, s, IH; 9.28, s, IH; 8.55, d, IH; 8.18, d, IH.
3) Herstellung von 2-Methyl-2-{[l-(6-trifluoromethyl-pyridin-3-yl)-methyliden]-amino}- propionsäuremethylester (7.3):

1 g 2-Amino-2-methyl-propionsäuremethylester Hydrochlorid wurde in 20 ml Dichlormethan suspendiert und unter Rühren tropfenweise mit 0,66 g Triethylamin versetzt. 15 Minuten nach beendeter Zugabe wurde die Mischung mit 1,57 g Magnesiumsulfat und 1,14 g der Verbindung 7.2 versetzt und 24 Stunden bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung filtriert, das Filtrat mit Wasser und gesättigter Kochsalzlösung ausgeschüttelt, die organische Phase abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt 2-Methyl-2- { [ 1 -(6-trifluoromethyl-pyridin-3-yl)-methyliden] -amino } - propionsäuremethylester (7.3). 1H NMR: 9.1, s, IH; 8.56, s, IH; 8.44, d, IH; 8.0, d, IH; 3.69, s, 3H; 1.5, s, 6H.
4) Herstellung von 2-Methyl-2-[(6-trifluoromethyl-pyridin-3-ylmethyl)-amino]- propionsäuremethylester (7.4):

1,7 g der Verbindung 7.3 wurden in einer Mischung aus 7,5 ml trockenem Dichlormethan und 7,5 ml trockenem Methanol gelöst, mit 66 mg Palladium-auf-Kohle (10%ig) versetzt und bei 1 bar bis zu beendeten Wasserstoffaufnahme hydriert. Zur Aufarbeitung wurde vom Katalysator abfiltriert, das Filtrat im Vakuum eingeengt und der Rückstand chromatographisch (Kieselgel; Dichlormethan / Methanol 10 / 1) gereinigt. Man erhielt 2-Methyl-2-[(6-trifluoromethyl-pyridin-3-ylmethyl)-amino]- propionsäuremethylester (7.4). Molekulargewicht 276,10 (C12Hi5F3N2O2); Retentionszeit R1 = 0.84 min. [C]; MS (ESI): 277,13 (MH+).
5) Herstellung von 3-(4-Fluoro-3-trifiuoromethyl-phenyl)-5,5-dimethyl-l-(6- trifluoromethyl-pyridin-3-ylmethyl)-imidazolidin-2,4-dion:
0,15 mmol des Aminosäureesters 7.4 wurden in 1 ml trockenem Acetonitril gelöst, mit 0,165 mmol l-Fluoro-4-isocyanato-2-trifluoromethyl-benzol versetzt und über Nacht bei Raumtemperatur unter Feuchtigkeits-ausschluss gerührt. Nach beendeter Reaktion wurde die Mischung mit 100 μl konzentrierter Salzsäure versetzt und 3 h bis zum vollständig erfolgten Ringschluss gerührt. Danach wurde das Lösungsmittel im Vakuum entfernt und der Rückstand chromatographisch (Methode [RPl]) gereinigt. Man erhielt 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(6-trifluoro-methyl-pyridin-3- ylmethyl)-imidazolidin-2,4-dion (7). Molekulargewicht 449,09 (C19H14F7N3O2); Retentionszeit R1 = 2.03 min. [B]; MS (ESI): 450,25 (MH+).
Beispiel 8: 3-(4-Chlor-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(6-trifluoro- methyl- pyridin-3-ylmethyl)-imidazolidin-2,4-dion:

Die Verbindung des Beispiels 8 wurde wie die Verbindung des Beispiels 7 hergestellt, mit dem Unterschied, dass die Verbindung 7.4 nicht mit l-Fluoro-4-isocyanato-2- trifluoromethyl-benzol sondern mit 1 -Chlor-4-isocyanato-2-trifluoromethyl-benzol umgesetzt wurde. Molekulargewicht 465,06 (C19H14ClF6N3O2); Retentionszeit R1 = 2.13 min. [B]; MS (ESI): 466,24 (MH+).
Die Verbindungen der Beispiele 21 (3-(3,4-Difluoro-phenyl)-5,5-dimethyl-l-(6-
trifluoromethyl-pyridin-3-ylmethyl)-imidazolidin-2,4-dion,
 , 1H NMR: 8.85, s, IH; 8.13, d, IH; 7.9, d, IH; 7.6, m, 2H; 7.4, m, IH; 4.76, s, 2H; 1.41, s, 6H) und 22 (3-(3,4- Dichlor-phenyl)-5,5-dimethyl-l-(6-trifluoromethyl-pyridin-3-ylmethyl)-imidazolidin-2,4-dion,
, 1H NMR: 8.85, s, IH; 8.13, d, IH; 7.9, d, IH; 7.6, m, 2H; 7.4, m, IH; 4.76, s, 2H; 1.41, s, 6H) und 22 (3-(3,4- Dichlor-phenyl)-5,5-dimethyl-l-(6-trifluoromethyl-pyridin-3-ylmethyl)-imidazolidin-2,4-dion,
 , 1H NMR: 8.85, s, IH; 8.16, d, IH; 7.9, d, IH; 7.8, m, 2H; 7.51, d, IH; 4.76, s, 2H; 1.41, s, 6H) wurden in analoger Weise durch Umsatz von 7.4 mit 1 ,2-Difluoro-4- isocyanato-benzol bzw. 1 ,2-Dichlor-4-isocyanato-benzol gewonnen.
, 1H NMR: 8.85, s, IH; 8.16, d, IH; 7.9, d, IH; 7.8, m, 2H; 7.51, d, IH; 4.76, s, 2H; 1.41, s, 6H) wurden in analoger Weise durch Umsatz von 7.4 mit 1 ,2-Difluoro-4- isocyanato-benzol bzw. 1 ,2-Dichlor-4-isocyanato-benzol gewonnen.
Beispiel 9 : 1 -(3 ,5-Bis-trifluoromethyl-benzyl)-3 -(4-fluoro-3 -trifluoromethyl-phenyl)-5 ,5 - dimethy l-imidazolidin-2 ,4-dion :

Die Verbindung des Beispiels 9 wurde über eine analoge Reaktionssequenz gewonnen: Reaktion von 3,5-Bis-trifluoromethyl-benzaldehyd mit 2-Amino-2-methyl- propionsäuremethylester Hydrochlorid lieferte 2-{[l-(3,5-Bis-trifluoromethyl-phenyl)- methyliden]-amino}-2-methyl-propionsäuremethylester (9.3; 1H NMR: 8.6, s, IH; 8.45, s, 2H; 8.25, s, IH; 3.69, s, 3H; 1.5, s, 6H). Dessen Hydrierung lieferte das Aminosäurederivat
2-(3,5-Bis-trifluoromethyl-benzylamino)-2-methyl-propionsäuremethylester (9.4; Molekulargewicht 343,10 (Ci4H15F6NO2); Retentionszeit Rt = 1.36 min. [C]; MS (ESI): 344,19 (MH+)). Der weitere Umsatz der Verbindung 9.4 mit l-Fluoro-4-isocyanato-2- trifluoromethyl-benzol ergab 1 -(3,5-Bis-trifluoromethyl-benzyl)-3-(4-fluoro-3- trifluoromethyl-phenyl)-5,5-dimethyl-imidazolidin-2,4-dion (9; 1H NMR: 8.18, s, 2H; 8.03, s, IH; 7.98, m, IH; 7.89, m, IH; 7.7, m, IH; 4.8, s, 2H; 1.42, s, 6H).
Beispiel 10: 1 -(3,5-Bis-trifluoromethyl-benzyl)-3-(4-chlor-3-trifluoromethyl-phenyl)-5,5- dimethyl-imidazolidin-2,4-dion:

In analoger Weise, jedoch durch Umsatz mit l-Chlor-4-isocyanato-2-trifiuoromethyl- benzol anstatt mit l-Fluoro-4-isocyanato-2-trifluoromethyl-benzol, wurde 10 aus 9.4 gewonnen. 1H NMR: 8.17, s, 2H; 8.08, s, IH; 8.02, m, IH; 7.88, m, 2H; 4.8, s, 2H; 1.42, s, 6H).
Beispiel 11: 4-[4,4-Dimethyl-2,5-dioxo-3-(3-pentafluorosulfanyl-benzyl)-imidazolidin-l-yl]- 2-trifluoromethyl-benzonitril:

Bei der Herstellung der Verbindung des Beispiels 11 wurde so vor-gegangen wie es für die Verbindung 4 weiter oben beschrieben ist:
(3-Pentafluorosulfanyl-phenyl)-methanol (11.1; 1H NMR: 7.83, s, IH; 7.78, d, IH; 7.59, m, 2H; 5.5, t, IH; 4.6, d, 2H) wurde durch Lithiumaluminium-hydridreduktion von 3- (Pentafluorosulfanyl)-benzoesäure erhalten. 1 -Bromrnethyl-3-pentafluorosulfanyl- benzol (11.2; 1H NMR: 8.02, s, IH; 7.87, d, IH; 7.78, d, IH; 7.63, m, IH; 4.83, s, 2H) wurde aus 11.1 durch Reaktion mit Phosphortribromid in Dichlormethan gewonnen. Die alkylierende Umsetzung von 1.2 mit 11.2 lieferte 4-[4,4-Dimethyl-2,5-dioxo-3-(3- pentafluorosulfanyl-benzyl)-imidazolidin-l-yl]-2-trifluoro-methyl-benzonitril (11; Molekulargewicht 513,07 (C20H15F8N3O2S); Retentionszeit R, = 2.19 min. [B]; MS (ESI)^H5IO (MH+)).
Beispiel 12: 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(3-pentafluorosulfanyl- bεnzyl)-iniidazolidin-2,4-dion:

1) Herstellung von 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-imidazolidin-2,4- dion (12.1):
Die Verbindung 12.1 kann nach Verfahren „B" dargestellt werden. Dazu wurden 1,5 g (9,76 mMol) 2-Amino-2-methyl-propionsäuremethylester Hydrochlorid in 20 ml trockenem Tetrahydrofuran suspendiert, mit 1,38 ml (9,76 mMol) Triethylamin und 2 g (9,76 mMol) l-Fluoro-4-isocyanato-2-trifluoromethyl-benzol versetzt. Die Mischung wurde 1 h bei 700C gerührt; danach ließ man etwas abkühlen, fügte 10 ml konzentrierte Salzsäure zu und rührte für 2 h bei 700C. Die abgekühlte Reaktionsmischung wurde mit Essigsäureethylester und Wasser versetzt; die organische Phase wurde abgetrennt, über Natriumsulfat getrocknet, filtriert und im Vakuum eingeengt. Der Rückstand wurde chromatographisch gereinigt (Methode [RP2]) und wurde nach Lösen in Essigsäureethylester, Trocknen der Lösung, Einengen im Vakuum und erneutem Lösen in Dichlormethan mit n-Heptan zur Kristallisation gebracht. Man erhielt 2,8 g 3-(4- Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-imidazolidine-2,4-dion (12.1) mit dem Schmelzpunkt 111 - 1140C. Molekulargewicht 290,06 (Ci2H10F4N2O2); Retentionszeit Rt = 1.55 min. [B]; MS (ESI): 291,27 (MH+).
2) Herstellung von 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(3- pentafluorosulfanyl-benzyl)-imidazolidin-2,4-dion (12):
Die alkylierende Umsetzung (Acetonitril, Cäsiumcarbonat, 90 Minuten, 70°C) von 12.1 mit 11.2 lieferte 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(3- pentafluorosulfanyl-benzyl)-imidazolidin-2,4-dion (12; Molekulargewicht 506,07 (C19Hi5F9N2O2S); Retentionszeit R1 = 2.24 min. [B]; MS (ESI): 507,15 (MH+)).
Beispiel 13: 4-[3-(3-Chlor-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2-tri- fluoromεthyl-bεnzonitril :

Der alkylierende Umsatz der Verbindung 1.2 mit l-Brommethyl-3-chlor- benzol lieferte 4-[3-(3-Chlor-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l- yl]-2-trifluoromethyl-benzonitril 13. 1H NMR (300 MHz): 8.33, d, IH; 8.26, d, IH; 8.10, dd, IH; 7.53, t, IH; 7.47 - 7.31, m, 3H; 4.62, s, 2H; 1.41, s, 6H.
Beispiel 14 : 3 -(4-Fluoro-3 -trifluoromethyl-phenyl)-5 ,5 -dimethyl- 1 -(4-trifluoro-methoxy- benzyl)-imidazolidin-2,4-dion:

Die Verbindung des Beispiels 14 wurde analog der Vorgehensweise, wie sie für die Verbindung des Beispiels 7 beschrieben wurde, nach Verfahren „C" hergestellt:
1 ) Herstellung von 2-Methyl-2- { [ 1 -(4-trifluoromethoxy-phenyl)-methyliden] -amino } - propionsäuremethylester (14.1):

2) Herstellung von 2-Memyl-2-(4-trifiuoromethoxy-benzylamino)-propionsäure- methylester (14.2):

Die Verbindung 14.1 wurde in einer Mischung aus trockenem Dichlormethan und trockenem Methanol gelöst, mit Palladium-auf-Kohle (10%ig) versetzt und bei 1 bar bis zu beendeten Wasserstoffaufnahme hydriert. Man erhielt 2-Methyl-2-(4- trifluoromethoxy-benzylamino)-propionsäure-methylester (14.2, 1H NMR: 7.43, d, 2H; 7.28, d, 2H; 3.63, s, 3H; 3.6, d, 2H; 2.51, t, IH; 1.28, s, 6H).
3) Herstellung von 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(4-trifluoro- methoxy-benzyl)-imidazolidin-2,4-dion:
Der Aminosäureesters 14.2 wurde in trockenem Acetonitril gelöst, mit l-Fluoro-4- isocyanato-2-trifluoromethyl-benzol versetzt und über Nacht bei Raumtemperatur unter Feuchtigkeits-ausschluss gerührt. Nach beendeter Reaktion wurde die Mischung mit konzentrierter Salzsäure versetzt und 3 h bis zum vollständig erfolgten Ringschluss gerührt. Man erhielt 3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(4-trifluoro- methoxy-benzyl)-imidazolidin-2,4-dion (14; 1H NMR: 7.98, m, IH; 7.88, m, IH; 7.68, t, IH; 7.58, d, 2H; 7.33, d, 2H; 4.63, s, 2H; 1.4, s, 6H).
Beispiel 15: 3-(4-Chlor-3-trifluoromethyl-phenyl)-5,5-dimethyl- 1 -(4-trifluoro- methoxy-benzyl)-imidazolidin-2,4-dion:

Die Verbindung des Beispiels 15 wurde analog der Vorgehensweise wie sie für die Verbindung des Beispiels 14 beschrieben wurde nach Verfahren „C" hergestellt, wobei in der letzten Stufe l-Chlor-4-isocyanato-2-trifluoro-methyl-benzol anstelle von 1- Fluoro-4-isocyanato-2-trifluoromethyl-benzol eingesetzt wurde. 15: 1H NMR: 8.05, s, IH; 7.88, m, 2H; 7.58, d, 2H; 7.35, d, 2H; 4.62, s, 2H; 1.4, s, 6H.
Die Verbindungen der Beispiele 19 (3-(3,4-Difluoro-phenyl)-5,5-dimethyl-l-(4-
trifluoromethoxy-benzyl)-imidazolidin-2,4-dion,
 , H NMR: 7.6, m, 4H;
, H NMR: 7.6, m, 4H;
7.35, m, 3H; 4.62, s, 2H; 1.39, s, 6H) und 20 (3-(3,4-Dichlor-phenyl)-5,5-dimethyl-l-(4-
trifluoromethoxy-benzyl)-imidazolidin-2,4-ion,
 , H NMR: 7.83, s, IH;
, H NMR: 7.83, s, IH;
7.79, d, IH; 7.59, d, 2H; 7.51, d, IH; 7.35, d, 2H; 4.62, s, 2H; 1.39, s, 6H) wurden in analoger Weise durch Umsatz von 14.2 mit l,2-Difluoro-4-isocyanato-benzol bzw. 1 ,2-Dichlor-4- isocyanato-benzol gewonnen.
Beispiel 16: 4-[4,4-Dimethyl-2,5-dioxo-3-(2-trifluoromethyl-benzyl)-imidazolidin-l- yl] -2-trifluoromethyl-benzonitril :

Die Verbindung des Beispiels 16 wurde durch Umsatz von 1.2 mit l-Brom-methyl-2- trifluoromethyl-benzol gewonnen. 1H NMR: 8.36, d, IH; 8.27, s, IH; 8.11, m, IH; 7.77, m, 2H; 7.69, t, IH; 7.51, t, IH; 4.74, s, 2H; 1.4, s, 6H.
Die Verbindungen der Beispiele 25, 4-[3-(2-Chlor-benzyl)-4,4-dimethyl-2,5-dioxo-
imidazolidin-1-yl] -2-trifluoromethyl-benzonitril,
 (1H NMR: 8.35, d, IH;
(1H NMR: 8.35, d, IH;
8.24, s, IH; 8.1, d, IH; 7.6, m, IH; 7.49, m, IH; 7.32, m, 2H; 4.68, s, 2H; 1.41, s, 6H); 26, 4-[3- (3,5-Bis-methansulfonyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2-trifluoromethyl-
benzonitril,
 (1H NMR: 8.36, m, 4H; 8.25, s, IH; 8.1, d, IH; 4.88, s,
(1H NMR: 8.36, m, 4H; 8.25, s, IH; 8.1, d, IH; 4.88, s,
2H; 2.55, s, 6H; 1.47, s, 6H); 27, 4-[3-(2-Methansulfonyl-benzyl)-4,4-dimethyl-2,5-dioxo-
imidazolidin-l-yl]-2-trifluoromethyl-benzonitril,
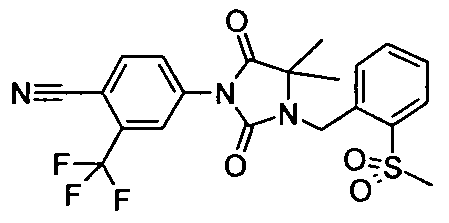 (1H NMR: 8.36, d, IH;
(1H NMR: 8.36, d, IH;
8.25, s, IH; 8.1, d, IH; 7.98, d, IH; 7.78, d, IH; 7.7, t, IH; 7.59, t, IH; 5.1, s, 2H; 2.55, s, 3H; 1.42, s, 6H); 28, 4-[3-(5-Fluoro-2-methansulfonyl-benzyl)-4,4-dimethyl-2,5-dioxo-
imidazolidin-l-yl]-2-trifluoromethyl-benzonitril,
 (1H NMR: 8.35, d, IH;
(1H NMR: 8.35, d, IH;
8.25, s, IH; 8.1, d, IH; 8.03, m, IH; 7.62, d, IH; 7.41, t, IH; 5.06, s, 2H; 2.55, s, 3H; 1.48, s, 6H) und 29, 4-[3-(2-Brombenzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2-trifluoromethyl-
benzonitril,
 (1H NMR: 8.35, d, IH; 8.25, s, IH; 8.1, d, IH; 7.67, d, IH;
7.59, d, IH; 7.4, t, IH; 7.26, t, IH; 4.62, s, 2H; 1.42, s, 6H) wurden, wie für das Beispiel 16 beschrieben, durch alkyüerεnden Umsatz von 1.2 mit 1 -Brommethyl-2-chlor-benzol (für 25), mit l-Brommethyl-3,5-bis-methansulfonyl-benzol (für 26; hergestellt aus dem entsprechenden Benzylalkohol durch Umsatz mit Phosphortribromid (1H NMR: 8.39, m, 3H; 4.91, s, 2H; 2.55, s, 6H), mit l-Brommethyl-2-methansulfonyl-benzol (für 27), mit 2-Brommethyl-4-fluoro-l- methansulfonyl-benzol (für 28) und mit l-Brom-2-brommethyl-benzol (für 29) gewonnen.
(1H NMR: 8.35, d, IH; 8.25, s, IH; 8.1, d, IH; 7.67, d, IH;
7.59, d, IH; 7.4, t, IH; 7.26, t, IH; 4.62, s, 2H; 1.42, s, 6H) wurden, wie für das Beispiel 16 beschrieben, durch alkyüerεnden Umsatz von 1.2 mit 1 -Brommethyl-2-chlor-benzol (für 25), mit l-Brommethyl-3,5-bis-methansulfonyl-benzol (für 26; hergestellt aus dem entsprechenden Benzylalkohol durch Umsatz mit Phosphortribromid (1H NMR: 8.39, m, 3H; 4.91, s, 2H; 2.55, s, 6H), mit l-Brommethyl-2-methansulfonyl-benzol (für 27), mit 2-Brommethyl-4-fluoro-l- methansulfonyl-benzol (für 28) und mit l-Brom-2-brommethyl-benzol (für 29) gewonnen.
Beispiel 17: l-(3,5-Bis-trifluoromethyl-benzyl)-3-(3,4-difluoro-phenyl)-5,5-di-methyl- imidazolidin-2,4-dion:

Die Verbindung des Beispiels 17 wurde analog der Vorgehensweise, wie sie für die Verbindung des Beispiels 7 beschrieben wurde, nach Verfahren „C" hergestellt:
Die Reaktion des 3,5-Bis(trifluoromethyl)-benzaldehyds mit 2-Amino-2-methyl- propionsäuremethylester Hydrochlorid und Triethylamin in Dichlormethan lieferte 2- {[l-(3,5-Bis-trifluoromethyl-phenyl)-methyliden]-amino}-2-methyl- propionsäuremethylester (17.1; 1H NMR: 8.59, s, IH; 8.45, s, 2H; 8.25, s, IH; 3.7, s, 3H), 1.5, s, 6H). Die Reduktion des Imins mit Wasserstoff und Palladium auf Kohle lieferte das Aminosäureester-derivat 17.2, 2-(3,5-Bis-trifluoromethyl-benzylamino)-2- methyl-propionsäuremethylester (1H NMR: 8.05, s, 2H; 7.94, s, IH; 3.8, d, 2H; 3.61, s, 3H; 2.98, t, IH; 1.28, s, 6H). Der Umsatz von 17.2 mit 1 ,2-Difluoro-4-isocyanato- benzol lieferte 1 -(3,5-Bis-trifluoromethyl-benzyl)-3-(3,4-difluoro-phenyl)-5,5-di- methyl-imidazolidin-2,4-dion (17; 1H NMR: 8.16, s, 2H; 8.03, s, IH; 7.62, m, 2H; 7.4, m, IH; 4.8, s, 2H; 1.41, s, 6H).
Die Verbindung des Beispiels 18 (l-(3,5-Bis-trifluoromethyl-benzyl)-
3-(3,4-dichlor-phenyl)-5,5-dimethyl-imidazolidin-2,4-dion;
 wurde in analoger Weise durch Umsatz von 17.2 mit 1 ,2-Dichlor-4-isocyanato-benzol gewonnen. 1H NMR: 8.16, s, 2H; 8.03, s, IH; 7.81, m, 2H; 7.53, d, IH; 4.8, s, 2H; 1.4, s, 6H.
wurde in analoger Weise durch Umsatz von 17.2 mit 1 ,2-Dichlor-4-isocyanato-benzol gewonnen. 1H NMR: 8.16, s, 2H; 8.03, s, IH; 7.81, m, 2H; 7.53, d, IH; 4.8, s, 2H; 1.4, s, 6H.
Beispiel 23: 3-(3,4-Difluoro-phenyl)-5,5-dimethyl-l-(4-pentafluorosulfanyl-benzyl)- imidazolidin-2,4-dion:

Die Verbindung des Beispiels 23 kann nach Verfahren „C" hergestellt werden: 1 ) Herstellung von N-Methoxy-N-methyl-4-pentafluorosulfanyl-benzamid (23.1):

1,25 g 4-Pentafluorosulfanyl-benzoesäure und 0,54 g N,O-Dimethylhydroxylamin Hydrochlorid wurden in 20 ml Dichlormethan gelöst, die Lösung mit 3,2 g 2,4,6- Tripropyl-[l,3,5,2,4,6]trioxatriphosphinan-2,4,6-trioxid und 1,01 g Triethylamin versetzt und anschließend 16 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung im Vakuum eingeengt, der Rückstand in 50 ml Essigsäureethylester aufgenommen und zweimal mit jeweils 25 ml Natriumhydrogensulfatlösung und zweimal mit jeweils 25 ml gesättigter Natriumcarbonatlösung ausgeschüttelt. Die organische Phase wurde abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt N- Methoxy-N-methyl-4-pentafluorosulfanyl-benzarnid (23.1), welches ohne weitere Reinigung in die nächste Stufe eingesetzt wurde.
2) Herstellung von 4-Pentafluorosulfanyl-benzaldehyd (23.2):

1,31 g der Verbindung 23.1 wurden in 35 ml trockenem Tetrahydrofuran gelöst und bei -60°C tropfenweise unter Rühren mit 4,95 ml einer 1 -molaren Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran versetzt. Nach beendeter Zugabe wurde noch eine Stunde bei -60°C gerührt; danach ließ man die Mischung sich auf Raumtemperatur erwärmen. Zur Aufarbeitung wurde die Reaktionsmsichung tropfenweise unter Rühren mit 40 ml kalter Kaliumhydrogensulfatlösung versetzt. Die Mischung wurde zweimal mit jeweils 50 ml Essigsäureethylester ausgeschüttelt; die organische Phase wurde mit gesättigter Kochsalzlösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt 4- Pentafluorosulfanyl-benzaldehyd (23.2). Der Aldehyd wurde ohne weitere Reinigung weiterverarbeitet.
3) Herstellung von 2-Methyl-2-{[l-(4-pentafluorosulfanyl-phenyl)-methyliden]-amino}- propionsäuremethylester (23.3):

0,53 g 2-Amino-2-methyl-propionsäuremethylester Hydrochlorid wurden in 20 ml Dichlormethan suspendiert und unter Rühren tropfenweise mit 0,35 g Triethylamin versetzt. 15 Minuten nach beendeter Zugabe wurde die Mischung mit 0,83 g Magnesiumsulfat und 0,8 g der Verbindung 23.2 versetzt und 24 Stunden bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung filtriert, das Filtrat mit Wasser und gesättigter Kochsalzlösung ausgeschüttelt, die organische Phase abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt 2-Methyl-2- { [ 1 -(4-pentafluorosulfanyl-phenyl)-methyliden]-amino} - propionsäuremethylester (23.3), welches als Rohprodukt weiter umgesetzt wurde.
4) Herstellung von 2-Methyl-2-(4-pentafluorosulfanyl-benzylamino)-propion- säuremethylester (23.4):

1,05 g der Verbindung 23.3 wurden in 20 ml trockenem Dichlormethan gelöst und bei Raumtemperatur portionsweise mit insgesamt 1,61 g Natriumtriacetoxyborhydrid versetzt. Die Mischung wurde über Nacht bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung mit 30 ml gesättigter Natriumhydrogencarbonatlösung und 50 ml Dichlormethan versetzt. Die organische Phase wurde abgetrennt, mit gesättigter Kochsalzlösung ausgeschüttelt und über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Der Rückstand wurde über Kieselgel mit n-Heptan / Essigsäure-ethylester 3 / 1 chromatographisch gereinigt. Man erhielt 2-Methyl-2-(4-penta-fluorosulfanyl-benzylamino)-propion-säuremethylester (23.4). 1H NMR: 7.8, d, 2H; 7.56, d, 2H; 3.69, d, 2H; 3.62, s, 3H; 2.7, t, IH; 1.29, s, 6H.
5) Herstellung von 3-(3,4-Difluoro-phenyl)-5,5-dimethyl-l-(4-pentafluoro-sulfanyl- benzyl)-imidazolidin-2,4-dion:
0,15 mmol des Aminosäureesters 23.4 wurden in 1 ml trockenem Acetonitril gelöst, mit 0,165 mmol l,2-Difluoro-4-isocyanato-benzol versetzt und über Nacht bei Raumtemperatur unter Feuchtigkeitsausschluss gerührt. Nach beendeter Reaktion wurde die Mischung mit 100 μl konzentrierter Salzsäure versetzt und 3 h bis zum vollständig erfolgten Ringschluss gerührt. Danach wurde das Lösungsmittel im Vakuum entfernt und der Rückstand chromatographisch (Methode [RPl]) gereinigt. Man erhielt 3-(3,4- Difluoro-phenyl)-5,5-dimethyl-l-(4-pentafluoro-sulfanyl-benzyl)-imidazolidin-2,4-dion (23). 1H NMR: 7.9, d, 2H; 7.62, m, 4H; 7.36, m, IH; 4.7, s, 2H; 1.4, s, 6H.
Beispiel 24: 3-(3,4-Dichlor-phenyl)-5,5-dimethyl-l-(4-pentafluorosulfanyl-benzyl)- imidazolidin-2,4-dion:
F
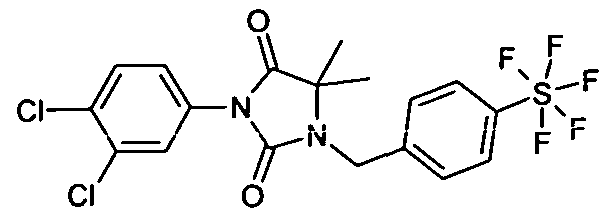
Die Verbindung des Beispiels 24 wurde in analoger Weise durch Umsatz von 23.4 mit l,2-Dichlor-4-isocyanato-benzol gewonnen. 1H NMR: 7.85, m, 4H; 7.67, d, 2H; 7.52, d, IH; 4.7, s, 2H; 1.4, s, 6H.
Die Verbindung 32 (4-[3-(2-Fluoro-3-methyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l- yl]-2-trifluoromethyl-benzonitril,

1H NMR: 8.34, d, IH; 8.21, s, IH; 8.19, d, IH; 7.35, t, IH; 7.21, t, IH; 7.09, t, IH; 4.62, s, 2H; 2.25, s, 3H; 1.4, s, 6H) wurde in analoger Weise, wie für die Verbindung des Beispiels 1 beschrieben, durch Umsatz von 1.2 mit l-Brommethyl-2-fluoro-3-methyl-benzol, erhalten.
Beispiel 33 : 4- [3 -(3 ,4-Bis-benzyloxy-benzyl)-4,4-dimethyl-2,5 -dioxo-imidazolidin- 1 -yl] -2- trifluoromethyl-benzonitril :

1) Herstellung von 2-(3,4-Bis-benzyloxy-benzylamino)-2-methyl-propionsäure- methylester 33.1:

2) 4-[3-(3,4-Bis-benzyloxy-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril 33:
Zu einer Lösung von 0,15 mMol der Verbindung 33.1 in 1 ml trockenem Acetonitril wurden 0,165 mMol 4-Isocyanato-2-trifiuoromethyl-benzonitril zugegeben und die Mischung über Nacht bei Raumtemperatur gerührt. Anschließend wurden 100 μl konzentrierte Salzsäure zugesetzt und die Mischung weitere 3 h zur Vervollständigung des Ringschlusses gerührt. Das Lösemittel wurde im Vakuum entfernt und der Rückstand wurde chromatographisch (Methode RPl) gereinigt. Man erhielt 4-[3-(3,4- Bis-benzyloxy-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2-trifluoro-methyl- benzonitril: 33. 1H NMR: 8.35, d, IH; 8.24, s, IH; 8.09, d, IH; 7.47 - 7.27, m, 10H; 7.1, s, IH; 7.0, m, 2H; 5.15, s, 2H; 5.11, s, 2H; 4.5, s, 2H; 1.3, s, 6H.
In analoger Weise (Tabelle 1) wurden die Verbindungen der Beispiele 36, 37, 39 und 40 hergestellt:
36 durch Umsatz von 33.1 mit l-Fluoro-4-isocyanato-2-trifluoromethyl-benzol;
37 durch Umsatz von 33.1 mit l-Chlor-4-isocyanato-2-trifluoromethyl-benzol;
39 durch Umsatz von 33.1 mit l,2-Difluoro-4-isocyanato-benzol; und
40 durch Umsatz von 33.1 mit l,2-Dichlor-4-isocyanato-benzol.
Tabelle 1


1) Herstellung von l-Brommethyl-2-phenoxy-benzol (34.2):

2,5 g (12,5 mMol) 2-Phenoxybenzylalkohol wurden in 45 ml Dichlormethan gelöst und bei 5° C tropfenweise mit einer Lösung von 1,35 g (5 mMol) Phosphortribromid in 5 ml Dichlormethan versetzt. Der Ansatz stand über Nacht bei Raumtemperatur. Danach wurde die Reaktionsmischung mit 5 ml gesättigter Natriumcarbonatlösung versetzt, die organische Phase abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Man erhielt 3,25 g (quantitativ) l-Brommethyl-2-phenoxy-benzol 34.2. 1H NMR: 7.56, d, IH; 7.4, m, 2H; 7.3, m, IH; 7.15, m, 2H; 7.01, d, 2H; 6.81, d, IH; 4.7, s, 2H. Molekulargewicht 261,99 (C13H11BrO).
2) Herstellung von 2-Methyl-2-(2-phenoxy-benzylamino)-propionsäure-tert-butyl ester (34.1):

Die Verbindung 34.1 kann nach Verfahren "C", dargestellt werden. Dazu wurden 3,21 g (76,7 mMol) Lithiumhydroxid-Hydrat in 125 ml trockenem Dimethylformamid vorgelegt, mit 20 g Molsieb 4 A versetzt und 30 Minutem bei Raumtemperatur gerührt. Danach wurden 7,5 g (38,3 mMol) 2-Amino-2- methylpropionsäure-tert.butyl-ester Hydrochlorid zugegeben und 15 Minuten bei Raumtemperatur gerührt, bevor 11,09 g (42,16 mMol) des Bromids 34.2 , gelöst in 25 ml trockenem Dimethylformamid bei Raumtemperatur zugetropft wurden. Der
Reaktionsansatz wurde 20 h bei Raumtemperatur gerührt. Die Reaktionsmischung wurde mit Wasser und Essigsäureethylester versetzt, die organische Phase abgetrennt, über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Der Rückstand wurde chromatographisch gereinigt (Kieselgel; n-Heptan/Essigsäureethylester 10/1) und lieferte 8,3 g (64% Ausbeute) 2-Methyl-2-(2-phenoxy-benzylamino)-propionsäure-tert- butylester 34.1. Molekulargewicht 341,19 (C2]H27NO3); Retentionszeit R, = 1.58 min. [B]; MS (ESI): 342,49 (MH+).
3) 4-[4,4-Dimethyl-2,5-dioxo-3-(2-phenoxy-benzyl)-imidazolidin-l-yl]-2-trifluoromethyl- benzonitril 34:
Wie im Beispiel 33 beschrieben, jedoch unter Einsatz von 2-Methyl-2-(2-phenoxy- benzylamino)-propionsäure-tert-butylester 33.1 und 4-Isocyanato-2-trifluoromethyl- benzonitril wurde 34 erhalten. Molekulargewicht 479,15 (C26H20F3N3O3); Retentionszeit R1 = 2.93 min. [C]; MS (ESI): 480,28 (MH+).
Die Verbindungen der Beispiele 43 (3-(3,4-Dichlor-phenyl)-5,5-dimethyl-l-(2-phenoxy- benzyl)-imidazolidin-2,4-dion),
44 (3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(2-phenoxy-benzyl)-imidazolidin-
2,4-dion) und 55 (3 -(3 ,4-Difluoro-phenyl)-5 ,5 -dimethyl- 1 -(2-phenoxy-benzyl)-imidazolidin-2,4-dion
(siehe Tabelle 2) wurden in ähnlicher Weise wie für die Herstellung der Verbindung 34 beschrieben durch
Umsatz von 33.1 mit 1 ,2-Dichlor-4-isocyanato-benzol (für 43), mit l-Fluoro-4-isocyanato-2-trifluoromethyl-benzol (für 44) und mit 1 ,2-Difluoro-4-isocyanato-benzol (für 55) gewonnen.
Tabelle 2

Beispiel 35: 4-(3-Benzhydryl-4,4-dimethyl-2,5-dioxo-imidazolidin- 1 -yl)-2-trifluoromethyl- benzonitril

100 mg der Verbindung 1.2 wurden mit 88 mg Bromdiphenylmethan in 2,5 ml trockenem Acetonitril gelöst, mit 110 mg Cäsiumcarbonat versetzt und 4 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung mit Essigsäureethylester und Wasser versetzt, die organische Phase abgetrennt, über
Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Die chromatographische Reinigung erfolgte mit der Methode [RP2]. Man erhielt 4-(3-Benzhydryl- 4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl)-2-trifluoromethyl-benzonitril 35. 1H NMR: 7.56, d, IH; 7.4, m, 2H; 7.3, m, IH; 7.15, m, 2H; 7.01, d, 2H; 6.81, d, IH; 4.7, s, 2H.
Die Verbindungen der Beispiele 38 (4-(3-Biphenyl-2-ylmethyl-4,4-dimethyl-2,5-dioxo- imidazolidin- 1 -yl)-2-trifluoromethyl-benzonitril),
41 (4-[3-(2-Isopropyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin- 1 -yl]-2-trifluoromethyl- benzonitril),
45 (3-[3-(4-Cyano-3-trifluoromethyl-phenyl)-5,5-dimethyl-2,4-dioxo-imidazolidin-l-ylmethyl]- benzoesäure),
46 (4-[3-(4-Benzolsulfonyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril),
48 (4-[4,4-Dimethyl-2,5-dioxo-3-(4-phenoxy-benzyl)-imidazolidin-l-yl]-2-trifluoromethyl- benzonitril),
49 (4-[3-(l ,3-Diphenyl-propyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l -yl]-2-trifluoromethyl- benzonitril),
50 (4-[4,4-Dimethyl-2,5-dioxo-3-(3-phenoxy-benzyl)-imidazolidin-l-yl]-2-trifluoromethyl- benzonitril),
53 (4-(4,4-Dimethyl-2,5-dioxo-3-pyridin-4-ylmethyl-imidazolidin- 1 -yl)-2-trifluoromethyl- benzonitril) und
54 (4-[3-(3-Hydroxy-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin- 1 -yl]-2-trifluoromethyl- benzonitril), (siehe Tabelle 3) wurden in analoger Weise durch Reaktion von 1.2 mit 2-Brommethyl-biphenyl (für 38), mit l-Brommethyl-2-isopropyl-benzol (41.1 für 41; 41.1 wurde über die Reaktions-sequenz 2- Isopropyl-benzoesäuremethylester -> (2-Isopropyl-phenyl)-methanol (41.2, durch Reduktion mit Lithiumaluminiumhydrid; 1H NMR: 7.32, d, IH; 7.27, d, IH; 7.21, t, IH; 7.15, t, IH; 5.03, t, IH; 4.55, d, 2H; 3.19, p, IH; 1.19, d, 6H) -» l-Brommethyl-2-isopropyl-benzol (41.1, durch Umsatz von 41.2 mit Phosphortribromid; 1H NMR: 7.4 - 7.3, m, 3H; 7.18, t, IH; 4.78, s, 2H; 3.3, p, IH; 1.22, d, 6H) erhalten),
mit (2-Brom-ethoxymethyl)-benzol (für 42; in diesem Falle wurde Natriumhydrid als Base und
Dimεthylformamid als Lösemittel verwendet), mit l-Benzolsulfonyl-4-brommethyl-benzol (46.1 für 46; 46.1 (1H NMR: 7.98, m, 4H; 7.7 -
7.6, m, 5H; 4.74, s, 2H) wurde durch Umsatz von (4-Benzolsulfonyl-phenyl)-methanol (46.2;
1H NMR: 7.93, m, 4H; 7.7 - 7.5, m, 5H; 5.41, t, IH; 4.55, d, 2H) mit Phosphortribromid gewonnen; der Alkohol 46.2 seinerseits war durch Reduktion von 4-Benzolsulfonyl- benzoesäure mit Lithiumaluminiumhydrid hegestellt worden), mit l-Brommethyl-4-phenoxy-benzol (für 48), mit l-Brom-l,3-diphenyl-propan (49.1 für 49; 49.1 wurde aus dem entsprechenden Alkohol durch Umsatz mit Phosphortribromid hergestellt: 1H NMR: 7.6, m, 2H; 7.4 - 7.19, m, 8H; 5.2, t, IH; 2.75, m, IH; 2.55 m, 2H; 2.4, m, IH), mit l-Brommethyl-3-phenoxy-benzol (50.1 für 50; die Verbindung 50.1 wurde durch Reaktion von (3-Phenoxy-phenyl)-methanol mit Phosphortribromid hergestellt: 1H NMR: 7.4, m, 3H;
7.2, m, 2H; 7.1, m, IH; 7.03, m, 2H; 6.93, m, IH; 4.7, s, 2H), mit 4-Brommethyl-pyridin (für 53) und mit (3-Brommethyl-phenoxy)-trimethylsilan (mit nachfolgender Schutzgruppenabspaltung für 54) gewonnen.
Tabelle 3



Beispiel 51: 2-(3-{5-Fluoro-2-[3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-2,4- dioxo-imidazolidin- 1 -ylmethyl] -phenyl } -ureido)-2-methyl- propionsäuremethylester

1) Herstellung von l-Brommethyl-4-fluoro-2-nitro-benzol (51.3):

0,776 g 4-Fluoro-2-nitro-toluolwuτden bei Raumtemperatur in 10 ml trockenem Chlorbenzol gelöst und auf 1200C erhitzt. Anschließend wurden innerhalb 1 h portionsweise insgesamt 1,07 g N-Bromsuccinimid und 0,12 g Benzoylperoxid gut vermischt zugegeben. Nach beendeter Zugabe wurde die Mischung eine weitere Stunde bei 120°C gerührt. Zur Aufarbeitung wurde die abgekühlte Reaktionsmischung im Vakuum eingeengt und der Rückstand mit Methyl-tert,-butylether aufgenommen. Die etherische Lösung wurde mit 1 N Natronlauge und danach mit gesättigter Kochsalzlösung gewaschen, die organische Phase über Magnesiumsulfat getrocknet, filtriert und im Vakuum eingeengt. Nach chromatographischer Reinigung (Kieselgel; n- Heptan/Essigsäureethylester 90/10 -> n-Heptan/Essigsäureethylester 80/20 in 35 min.)
erhielt man l-Brommethyl-4-fluoro-2-nitro-benzol (51.3). 1H NMR: 8.01, d, IH; 7.83, t, IH; 7.69, t, IH; 4.9, s, 2H.
2) Herstellung von l-(4-Fluoro-2-nitro-benzyl)-3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5- dimethyl-imidazolidin-2,4-dion (51.2):

0,73 g der Verbindung des Beispiels 47 wurden bei Raumtemperatur in 20 ml trockenem Acetonitril gelöst, mit 0,7 g der Verbindung 51.3 und 0,9 g Cäsiumcarbonat versetzt und 24 h bei Raumtemperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung filtriert; das Filtrat wurde im Vakuum eingeengt, der Rückstand mit Wasser verrührt, abgesaugt, mit Wasser gewaschen und getrocknet. Man erhielt 1- (4-Fluoro-2-nitro-benzyl)-3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl- imidazolidin-2,4-dion (51.2). Molekulargewicht 443,09 (Ci9H]4F5N3O4); Retentionszeit R1 = 3.56 min. [D]; MS (ESI): 444,08 (MH+).
3) Herstellung von l-(2-Amino-4-fluoro-benzyl)-3-(4-fluoro-3-trifluoromethyl-phenyl)- 5,5-dimethyl-imidazolidin-2,4-dion (51.1):

1 ,03 g der Verbindung 51.2 wurden bei Raumtemperatur in 20 ml trockenem Methanol gelöst und unter einer Argonatmosphäre wurden 16 mg Palladiumhydroxid-auf-Kohle und danach 0,20 g Trimethylamin-Boran-Komplex zugegeben und die Reaktionsmischung 4 h unter Rückfluss gerührt. Nach Zugabe von weiteren 0,1 g Trimethylamin-Boran-Komplex und nochmaligem 4-stündigen Erhitzen war die Reaktion beendet. Zur Aufarbeitung wurde die abgekühlte Reaktionsmischung über ein Faltenfilter filtriert, das Filtrat im Vakuum eingeengt und der Rückstand mit Cyclohexan verrührt, abgesaugt, mit Cyclohexan gewaschen und getrocknet. Man
erhielt 1 -(2- Amino-4-fluoro-benzyl)-3 -(4-fluoro-3 -trifluoromethyl-phenyl)-5 ,5 - dimethyl-imidazolidin-2,4-dion (51.1). Molekulargewicht 413,11 (C19H]6F5N3O2); Retentionszeit R1 = 3.78 min. [E]; MS (ESI): 414,06 (MH+).
4) 2-(3 - { 5 -Fluoro-2- [3 -(4-fluoro-3 -trifluoromethyl-phenyl)-5 ,5 -dimethyl-2,4-dioxo- imidazolidin- 1 -ylmethyl] -phenyl } -ureido)-2-methyl-propionsäure-methylester :
0,21 g der Verbindung des Beispiels 51.1 wurden bei Raumtemperatur in 5 ml trockenem Pyridin gelöst, mit 0,14 g 2-Isocyanoto-2-Methyl-propion-säuremethylester versehen und 24 h bei Raumteperatur gerührt. Zur Aufarbeitung wurde die Reaktionsmischung im Vakuum eingeengt und der Rückstand chromatographisch (Methode [RPl] gereinigt. Man erhielt 2-(3-{5-Fluoro-2-[3-(4-fluoro-3-trifluoromethyl- phenyl)-5 ,5 -dimethyl-2,4-dioxo-imidazolidin- 1 -ylmethyl] -phenyl } -ureido)-2-methyl- propionsäure-methylester 51. 1H NMR: 8.1, s, IH; 8.9, m, IH; 7.9, m, IH; 7.7, t, IH; 7.54, d, IH; 7.4, t, IH; 7.0, s, IH; 6.82, t, IH; 4.48, s, 2H; 3.6, s, 3H; 1.45, s, 3H; 1.4, s, 3H.
Beispiel 52: N-{5-Fluoro-2-[3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-2,4-dioxo- imidazolidin- 1 -ylmethyl] -phenyl } -sulfamid

1) Herstellung von N-{5-Fluoro-2-[3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-
2,4-dioxo-imidazolidin-l-ylmethyl]-phenyl}-N'-tert.-Butyloxycarbonyl-sulfamid (52.1):

2) N- { 5 -Fluoro-2- [3 -(4-fluoro-3 -trifluoromethyl-phenyl)-5 ,5 -dimethyl-2,4-dioxo- imidazolidin- 1 -ylmethyl]-phenyl } -sulfamid:
0,69 g der Verbindung 52.1 wurden bei Raumtemperatur in 15 ml trockenem Dichlormethan gelöst, mit 1 ,79 ml Trifluoroessigsäure und 0, 18 ml Wasser versetzt und 4 h bei Raumtemperatur gerührt und dann über Nacht stehen gelassen. Zur Aufarbeitung wurde die Reaktionsmischung im Vakuum eingeengt, der Rückstand mit Toluol versetzt und erneut eingeengt. Schließlich wurde der Rückstand in Dichlormethan gelöst, die organische Phase mit gesättigter Natriumhydrogencarbonatlösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und das Filtrat im Vakuum eingeengt. Man erhielt N-{5-Fluoro-2-[3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-2,4-dioxo- imidazolidin-l-ylmethyl]-phenyl} -sulfamid 52. Molekulargewicht 492,08 (Ci9H17F5N4O6S); Retentionszeit R1 = 3.24 min. [D]; MS (ESI): 493,10 (MH+).
Die erfindungsgemäßen Verbindungen der Formel I zeigen eine hohe Affinität gegenüber dem humanen Cannabinoid Rezeptor 1 (hCBlR). Diese Affinität ist der verglichen mit der an dem humanen Androgenrezeptor (hAR) deutlich ausgeprägter. So ist die Selektivität etwa um den Faktor 5 größer als sie für Beispiele der in der in der Anmeldung US 5,411,981 beschriebenen Verbindungen gefunden wurde.
Pharmakologische Prüfungen:
Bindung an den humanen Cannabinoid Rezeptor 1 (hCBlR):
Testverbindungen: Die Verbindungen (3 μl, 10 mM, 100% DMSO), einpipettiert in 96- well PP
Mikrotiterplatten, wurden mit 27 μl 100 % DMSO (Dimethylsulfoxid) verdünnt. Ausgehend von dieser Lösung wurden weitere 3-fach Verdünnungsschritte vorgenommen, indem jeweils
10 μl auf einer neue PP Mikrotiterplatte überführt und weitere 20 μl 100 % DMSO zugefügt wurden. Jeweils 6 μl dieser Lösungen wurden in neue 96-well PP Mikrotiterplatten transferiert und mit 144 μl Assaypuffer aufgefüllt. Die Endkonzentrationen reichten von 10 μM bis 0.005 μM.
Negativkontrolle: AM 251, gelöst in Assaypuffer mit 1% DMSO, wurde zu den
Verdünnungsreihen in den Mikrotiterplatten als Kontrolle mitgeführt. Die Endkonzentration betrug 1 μM.
Leerkontrolle: Assaypuffer mit 1 % DMSO wurde in den Verdünnungsreihen der
Mikrotiterplatten als Leerkontrolle mitgeführt.
Zusammenfassung der Assayparameter:

Hohe Kontrolle: 3H Bindung ohne Zugabe der Verbindung Niedrige Kontrolle: 3H Bindung in Gegenwart von 1 μM AM 251 Die Werte wurden über die korrigierten Rohdaten berechnet.
, •■ • , • . , • . ,n, ^ 1 ΛΛ * (Λ {sample -loweontrol ) \
Inhibierung der Ligandenbindung (%) = 1 UU * ^l - {highcontro ^lowcontrol ) )
Die aufgeführten Werte wurden als Durchschnittswerte einer Doppelbestimmung gewonnen. Die IC50 Werte wurden aus den Messwerten mit dem Programm Xlfit, Formel 205, berechnet. Ki- Werte wurden aus den IC50- and Kd- Werten unter Benutzung der Cheng-Prusoff Gleichung erhalten:
/C50
Ki = -
(C= Konzentration des Radio-liganden)
1 + c_
Kd
Literatur: Cheng, Y.-C, und Prusoff, W.H. (1973) Biochem. Pharmacol 22, 3099-3108
Ergebnisse: Kj- Werte von Beispielverbindungen; Tabelle 4:


Aus dem Messdaten ist abzulesen, dass die erfindungsgemäßen Verbindungen der Formel I mit hoher Affinität an den hCBlR binden und daher gut zur Behandlung des metabolischen Syndroms, des Diabetes Typ II und der Adipositas geeignet sind.
Bindungsassay mit humanem Androgenrezeptor:
Der Bindungsassays am Androgenrezeptor wurden gemäss der Vorschrift von D. T. Zava et al. (1979) durchgeführt ("Androgen Receptor Assay with [3H]Methyltrienolone (Rl 881) in the Presence of Progesterone Receptor", Endocrinology, 104, 1007-1012). Als radioaktiver Ligand für die Bindungsmessung diente [3H] Methyltrienolon und Referenzsubstanz war dieselbe Verbindung in nichtmarkierter (= nichtradioaktiver) Form. Zur Bestimmung des unspezifischen Anteils der Bindung wurde 1 μM Miboleron verwendet. In Abweichung von der zitierten Vorschrift wurde für die Präparation von cytosolischem Rezeptorprotein die androgen-sensitive humane Prostataadenokarzinom-zellinie LNCaP verwendet. Zur Messung wurden Aliquote einer Zellzytosolfraktion (ausgehend von 106 Zellen pro Messpunkt) für 24 Stunden bei 4°C mit 0,5 nM [ H] Methyltrienolon in Gegenwart oder Abwesenheit von Prüfsubstanz in einem Puffer inkubiert (25 mM HEPES/Tris, 1 mM EDTA, 10 mM Na2MoO4, 2 mM DTT, 10% Glycerol; pH 7.4). Die Proben wurden dann mit je 400 μl einer Aktivkohlesuspension gemischt und die Mischungen zentrifugiert (10 Minuten, 8000 x g). Überstände wurden entnommen, mit je 5 ml Szintillationscocktail gemischt und die Radioaktivität der Proben im Szintillationszähler gemessen. Spezifische Ligandbindung zu den Rezeptoren wurde errechnet als die Differenz zwischen totaler Bindung und der unspezifischen Bindung in Gegenwart eines Überschusses an nichtmarkiertem Ligand. Endergebnisse wurden dargestellt als Prozent spezifische Bindung im Vergleich zur Bindung von Kontrollsubstanz.
Die Stärke der Bindung an den humanen Androgenrezeptor wird ausgedrückt als Prozent Inhibierung der Bindung von [ H] Methyltrienolon an den humanen Androgenrezeptor. Dabei beträgt die Konzentration der zu untersuchenden Verbindungen 1 μM bzw. 10 μM. Je größer
der Zahlenwert der „Prozent Inhibierung bei 1 bzw. 10 μM" ausfallt, umso stärker ist die Bindung der Testsubstanz an den humanen Androgenrezeptor. Alternativ ist der Ki- Wert angegeben; je größer dieser Wert im Vergleich zum Ki- Wert bezüglich der Bindung an den hCBlR ist, umso geringer ist die Bindung an den hAR.
Ergebnisse: Prozent Inhibierung der Bindung von [3H]Methyltrienolon an den humanen Androgenrezeptor (hAR) durch Beispielverbindungen bei 1 bzw. 10 μM; Tabelle 5:

Die Verbindung des Beispiels 29 (4-[4,4-Dimethyl-2,5-dioxo-3-(4-trifluoro- methyl-benzyl)-imidazolidin-l-yl]-2-trifluoromethyl-benzonitril) der Anmeldung US 5,411,981 weist einen Kj- Wert von 76 nM bezüglich ihrer Bindung an den humanen Cannabinoid
Rezeptor 1 auf und einen solchen von 96 nM bezüglich ihrer Bindung an den humanen Androgenrezeptor.
Aus den Messdaten der Tabellen 4 und 5 ist abzulesen, dass die erfindungsgemäßen Verbindungen der Formel I gegenüber dem humanen Androgenrezeptor eine deutlich bzw. sehr deutlich verminderte Affinität aufweisen und die Selektivität gegenüber dem humanen Cannabinoid Rezeptor 1 erhöht ist.
Claims
1. Verbindungen der Formel I,

worin bedeuten
Rl CN, NO2 oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (d-C12)-Alkyl, ((C6-Ci2)-Aryl, (Ci-C12)- Alkylen-(C6-Ci2)-Aryl, wobei (C rC^-Alkyl, (C6-C ,2)-Aryl, (C1-C12)-Alkylen- (C6-C J2)- Aryl bis zu dreifach unabhängig voneinander substituiert sein können mit Halogen, CN, CF3;
R5, R6, R7 unabhängig voneinander H, F, Cl, Br, CN, CF3, SF5, OCF3, NO2, S(O)m[(Ci-C6)- Alkyl], S(O)m[(C3-C9)-Cycloalkyl], S(O)1nCF3, (C,-C6)-Alkyl, (C1-Q)-AIk0Xy, (C2-C6)-Alkenyl, (C2-C6)-Alkenyloxy, (C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(C!-C12)-Alkyl], -0(C=O)-(C6-C, 2)-Aryl, W-C00H, W-
CONH2, W-CO-NH[(CrC6)-Alkyl], W-CO-N[(C,-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl], W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHRS-CO-RS W-CO-Rl 0, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(Ci-C6)-Alkyl], W-CO-NH-C(=NH)N[(Ci-C6)-Alkyl]2, (d-C8)-Acyl, (d-C7)-Acyloxy, W-C(=NH)NH2, W-CC=NH)NHOH, W-CC=N-SO2-NH2)NH2, W-CC=N-SO2-CF3)NH2, W-C[=N-SO2-(Ci-C6)-Alkyl]NH2, W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2, NH2, NH-Cd-d^-Alkyl, N-[(d-C12)-Alkyl]2, W-NH-C(=NH)NH2, W-NH-CC=NH)NH[CC1-C6)- Alkyl], W-NH-C(=NH)N[(d-C6)-Alkyl]2, W-NH-CO-NH2, W-NH-CO-NH[(d -C6)-Alkyl], W-NH-CO-N[(d-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl], W-NH-CO-N[CC3-C9)-Cycloalkyl]2, W-NH-CO-NH-[(C,-C6)-Alkyl]-CO-O-[(d-C6)- Alkyl], W-NH-CO-NH-[CC1-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(Ci-C6)-Alkyl, W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-CC1 -C6)- Alkyl, W-NH-CO-NH-CO-CC3-C9)-Cycloalkyl], W-NH-C(=NH)-NH-C(=NH)-NH2, W-NH-C(=NH)-NH-C(=NH)-NH[(d-C6)-Alkyl], W-NH-CC=NH)-NH-C(=NH)-N[Cd-C6)-Alkyl]2, W-NH-W-SO2-NH2, W-NH- W-SO2-NH[CCrC6)-Alkyl], W-NH- W-SO2-N[(Ci-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl], W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[CCi-C6)-Alkyl], W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2, W-SO2-NH2, W-SO2-NH[(d-C6)-Alkyl], W-SO2-N[(d-C6)-Alkyl]2, W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2, W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NH[CC1-C6)- Alkyl], W-SO2-NH-CO-N[(C,-C6)-Alkyl]2, W-SO2-NH-CO-NH[CC3-C9)- Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(Ci-C6)- Alkyl], W-P(O)[O-(d-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O- CH2-Aryl)2, W-P(O)(OH)2, (C6-C I2)-Aryl, O-(C6-C12)-Aryl, O-(d-C12)-Alkylen- (C6-C J2)- Aryl, S(O)m-(C6-C12)-Aryl, Tri(d-Ci2)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
0, 1, 2; W eine Bindung oder (Ci-C6)-Alkyl;
R8 H, (CrC6)-Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(=NH)NH2, COOH, CO-O(C1 -C6)- Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(C1-C12)-Alkyl, N[(Ci-C]2)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, N[(C3-
C9)-Cycloalkyl]2;
RIO NH-(CrC6)-Alkyl-SO3H, NH-(C!-C6)-Alkyl-SO2NH2, NH-(Ci-C6)-Alkyl-SO2-(d-
C6)-Alkyl, NH-(C1-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(C1-C6)-Alkyl-SO2-

sowie deren physiologisch verträgliche Salze.
2. Verbindungen der Formel I, gemäß Anspruch 1, dadurch gekennzeichnet, dass darin bedeuten
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (Cj-Ci2)-Alkyl, ((C6-C12)- Aryl, (Ci-C12)- Alkylen-(C6-C12)-Aryl, wobei (CrCi2)-Alkyl, (C6-Ci2)-Aryl, (Ci-C12)-Alkylen- (C6-C J2)- Aryl bis zu dreifach unabhängig voneinander substituiert sein können mit Halogen, CN, CF3; F, Cl, Br, CN, CF3, SF5, OCF3, NO2, S(O)m[(C,-C6)-Alkyl], S(O)m[(C3-C9)-
Cycloalkyl], S(O)111CF3, (C,-C6)-Alkyl, (C,-C6)-Alkoxy, (C2-C6)-Alkenyl, (C2-C6)-
Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(d-Ci2)-Alkyl],
-0(C=O)-(C6-Ci2)- Aryl, W-COOH, W-CONH2, W-CO-NH[(Ci-C6)-Alkyl],
W-CO-N[(C1-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl],
W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHRδ-CO-RQ,
W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(C1-C6)-Alkyl],
W-CO-NH-C(=NH)N[(C1-C6)-Alkyl]2, (d-C8)-Acyl, (d-C7)-Acyloxy,
W-C(=NH)NH2, W-C(=NH)NH0H, W-C(=N-S O2-NH2)NH2,
W-C(=N-SO2-CF3)NH2, W-C[=N-SO2-(Ci-C6)-Alkyl]NH2,
W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2,
NH2, NH-(C1-Ci2)-Alkyl, N-[(Ci-C12)-Alkyl]2, W-NH-C(^NH)NH2,
W-NH-C(=NH)NH[(C1-C6)-Alkyl], W-NH-C(=NH)N[(C)-C6)-Alkyl]2,
W-NH-CO-NH2, W-NH-CO-NH[(d-C6)-Alkyl],
W-NH-CO-N[(Ci-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl],
W-NH-CO-N[(C3-C9)-Cycloalkyl]2,
W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl],
W-NH-CO-NH-[(C,-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(C1-C6)-Alkyl,
W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(Ci-C6)-Alkyl,
W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl], W-NH-C(=NH)-NH-C(=NH)-NH2,
W-NH-C(=NH)-NH-C(=NH)-NH[(d-C6)-Alkyl],
W-NH-C(=NH)-NH-C(=NH)-N[(C,-C6)-Alkyl]2,
W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(d-C6)-Alkyl],
W-NH- W-SO2-N[(d-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl],
W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[(d-C6)-Alkyl],
W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2,
W-SO2-NH2, W-SO2-NH[(d-C6)-Alkyl], W-SO2-N[(C,-C6)-Alkyl]2,
W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2,
W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NHf(C1-C6)-
Alkyl], W-SO2-NH-CO-N[(d-C6)-Alkyl]2, W-SO2-NH-CO-NH[(C3-C9)-
Cycloalkyl], W-S02-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C1-C6)- Alkyl], W-P(O)[O-(Ci-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O- CH2-Aryl)2, W-P(O)(OH)2, (C6-C12)-Aryl, O-(C6-d2)-Aryl, 0-(C J-C12)- Aikylen- (C6-Ci2)-Aryl, S(O)m-(C6-Ci2)-Aryl, Tri(Ci-C12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
R6, R7 unabhängig voneinander H, Halogen, CN, CF3, SF5, OCF3, S(O)m[(C1-C6)-Alkyl], S(O)m[(C3-C9)-Cycloalkyl], NO2, S(O)01CF3, (C ,-C6)- Alkyl, (C1-Ce)-AIk0Xy, (C2- C6)-Alkenyl, (C2-C6)-Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-COO-[(CrC12)-Alkyl], -O(C=O)-(C6-Ci2)-Aryl, W-C00H, W-CONH2, W-CO-NH[(CrC6)-Alkyl], W-CO-Nf(C1-C6)- Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl], W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHRδ-CO-R^ W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(CrC6)-Alkyl], W-CO-NH-C(=NH)N[(d-C6)-Alkyl]2, (C1-Q)-ACyI, (d-C7)-Acyloxy, W-C(=NH)NH2, W-C(=NH)NH0H, W-C(=N-SO2-NH2)NH2, W-Q=N-SO2-CF3)NH2, W-C[=N-SO2-(d-C6)-Alkyl]NH2, W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2, NH2, NH-(d-C12)-Alkyl, N-[(C1-C,2)-Alkyl]2, W-NH-C(=NH)NH2, W-NH-C(=NH)NH[(C1-C6)-Alkyl], W-NH-C(=NH)N[(d-C6)-Alkyl]2, W-NH-CO-NH2, W-NH-CO-NH[(d-C6)-Alkyl], W-NH-CO-N[(d-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl], W-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-NH-CO-NH-[(d-C6)-Alkyl]-CO-O-[(d-C6)-Alkyl], W-NH-CO-NH-[(d-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(d-C6)-Alkyl, W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(C1-C6)- Alkyl, W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl], W-NH-C(=NH)-NH-C(=NH)-NH2, W-NH-C(=NH)-NH-C(=NH)-NH[(d-C6)-Alkyl], W-NH-C(=NH)-NH-C(=NH)-N[(C j -C6)-Alkyl]2, W-NH-W-SO2-NH2, W-NH- W-SO2-NH [(C1 -C6)- Alkyl], W-NH- W-SO2-N[(d-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl], W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O[(C1-C6)-Alkyl], W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2, W-SO2-NH2, W-SO2-NHt(C, -C6)-Alkyl], W-SO2-Nt(C1-C6)- Alkyl]2, W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2, W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NHt(C1-C6)- Alkyl], W-SO2-NH-CO-N[(d-C6)-Alkyl]2, W-SO2-NH-CO-NH [(C3-C9)- Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(C1-C6)- Alkyl], W-P(O)[O-(C1-C6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O- CH2-Aryl)2, W-P(O)(OH)2, (C6-C 12)-Aryl, O-(C6-C12)-Aryl, O-(C1-C12)-Alkylen- (C6-C 12)-Aryl, S(O)ra-(C6-C12)-Aryl, Tri(C1-C12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
m 0, 1, 2;
W eine Bindung oder (CrC6)-Alkyl;
R8 H, (C 1-C6)- Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(=NH)NH2, COOH, CO-O(Ci -C6)- Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(C1-C12)-Alkyl, N[(d-C12)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, N[(C3-
C9)-Cycloalkyl]2;
RIO NH-(d-C6)-Alkyl-SO3H, NH-(C1 -C6)- Alkyl-SO2NH2, NH-(Ci-C6)-Alkyl-SO2-(d-
C6)-Alkyl, NH-(Ci-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(Ci-C6)- Alkyl-SO2-

sowie deren physiologisch verträgliche Salze.
3. Verbindungen der Formel I, gemäß Anspruch 1 oder 2, dadurch gekennzeichnet, dass darin bedeuten
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C1-C12)-Alkylen-(C6-C12)-Aryl;
R5 F, Cl, Br, CF3, SF5, OCF3, S(O)2 [(Ci -C6)- Alkyl], (Ci-C6)-Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[(C1-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-Ot(C1-C6)- Alkyl], (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(C1-C12)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CN, CF3, SF5, OCF3, S(O)m[(C1-C6)-Alkyl], S(O)m[(C3-C9)-Cycloalkyl], S(O)mCF3, (C ,-C6)- Alkyl, (C,-C6)-Alkoxy, (C2-C6)- Alkenyl, (C2-C6)-Alkenyloxy,
(C2-C6)-Alkinyl, (C2-C6)-Alkinyloxy, OH, SH, W-C00-[(C1-d2)-Alkyl], -0(C=O)-(C6-C 12)-Aryl, W-C00H, W-CONH2, W-CO-NH[(Ci-C6)-Alkyl], W-CO-N[(C!-C6)-Alkyl]2, W-CO-NH[(C3-C9)-Cycloalkyl], W-CO-N[(C3-C9)-Cycloalkyl]2, W-CO-NH-CN, W-CO-NH-CHR8-CO-R9, W-CO-RlO, W-CO-NH-C(=NH)NH2, W-CO-NH-C(=NH)NH[(C1-C6)-Alkyl], W-CO-NH-C(=NH)N[(Ci-C6)-Alkyl]2, (C1-Q)-ACyI, (d-C7)-Acyloxy, W-C(=NH)NH2, W-C(=NH)NH0H, W-C(=N-SO2-NH2)NH2, W-C(^N-SO2-CF3)NH2, W-C[=N-SO2-(Ci-C6)-Alkyl]NH2, W-C[=N-SO2-(C3-C9)-Cycloalkyl]NH2, W-C(=N-SO2-Aryl)NH2, NH2, NH-^rd^-Alkyl, N-[(Ci-C12)-Alkyl]2, W-NH-C(=NH)NH2, W-NH-C(=NH)NH[(C,-C6)-Alkyl], W-NH-C(^NH)N [(C1 -C6)-Alkyl]2, W-NH-CO-NH2, W-NH-CO-NH[(C1-C6)-Alkyl], W-NH-CO-N[(Ci-C6)-Alkyl]2, W-NH-CO-NH[(C3-C9)-Cycloalkyl], W-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], W-NH-CO-NH-[(Ci-C6)-Alkyl]-CO-NH2, W-NH-CO-NH-SO2-(C1 -C6)-Alkyl, W-NH-CO-NH-SO2-(C3-C9)-Cycloalkyl], W-NH-CO-NH-CO-(C i-C6)-Alkyl, W-NH-CO-NH-CO-(C3-C9)-Cycloalkyl], W-NH-C(-NH)-NH-C(=NH)-NH2, W-NH-C(=NH)-NH-C(=NH)-NH[(d-C6)-Alkyl], W-NH-C(=NH)-NH-C(=NH)-N[(Ci-C6)-Alkyl]2, W-NH-W-SO2-NH2, W-NH- W-SO2-NH[(C!-C6)-Alkyl], W-NH- W-SO2-N[(C1-C6)-Alkyl]2, W-NH- W-SO2-NH[(C3-C9)-Cycloalkyl], W-NH- W-SO2-N[(C3-C9)-Cycloalkyl]2, W-NH- W-SO2-NH-CO-O [(Ci -C6)- Alkyl], W-NH-W-SO2-NH-CO-NH2, W-O-SO2-NH2, W-O-W-COOH, W-O-W-CONH2, W-SO2-NH2, W-SO2-NH[(C rC6)- Alkyl], W-SO2-N[(Ci-C6)-Alkyl]2, W-SO2-NH[(C3-C9)-Cycloalkyl], W-SO2-N[(C3-C9)-Cycloalkyl]2, W-SO3H, W-NH-W-SO3H, W-SO2-NH-CO-NH2, W-SO2-NH-CO-NHt(C1-C6)- Alkyl], W-SO2-NH-CO-N[(d-C6)-Alkyl]2, W-SO2-NH-CO-NH[(C3-C9)- Cycloalkyl], W-SO2-NH-CO-N[(C3-C9)-Cycloalkyl]2, W-P(O)(OH)[O-(Ci-C6)- Alkyl], W-P(O)[O-(CrC6)-Alkyl]2, W-P(O)(OH)(O-CH2-Aryl), W-P(O)(O-CH2- Aryl)2, W-P(O)(OH)2, (C6-Ci2)-Aryl, O-(C6-Ci2)-Aryl, O-(Ci-Ci2)-Alkylen-(C6- C12)-Aryl, S(O)m-(C6-C12)-Aryl, Tri(C1-C12)-alkylsilyl, wobei das Alkyl 1 bis 6 Kohlenstoffatome aufweist;
m 0, 1, 2;
W eine Bindung oder (Ci-C6)-Alkyl;
R8 H, (C 1-C6)- Alkyl, wobei die Alkylgruppe mit OH, SH. SCH3, Aryl, 4-Hydroxyaryl,
Heteroaryl, NH2, NH-C(^NH)NH2, COOH, CO-O(Ci -C6)- Alkyl, CONH2 substituiert sein kann;
R9 OH, NH2, NH-(Ci-Ci2)-Alkyl, N[(Ci-Ci2)-Alkyl]2, NH-(C3-C9)-Cycloalkyl, N[(C3-
C9)-Cycloalkyl]2; RIO NH-(Ci-C6)-Alkyl-SO3H, NH-(C1-C6)-Alkyl-SO2NH2, NH-(C1-C6)-Alkyl-SO2-(C1-
C6)-Alkyl, NI-I-(C1-C6)-Alkyl-SO2-(C3-C9)-Cycloalkyl, NH-(C1 -C6)- Alky 1-SO2-

sowie deren physiologisch verträgliche Salze.
4. Verbindungen der Formel I, gemäß einem oder mehreren der Ansprüche 1 bis 3 dadurch gekennzeichnet, dass darin bedeuten
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A, B unabhängig voneinander CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C!-C12)-Alkylen-(C6-Ci2)-Aryl;
R5 F, Cl, Br, CF3, SF5, OCF3, S(O)2 [(C1-Co)-AUCyI], (Ci-C6)-Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[(Ci-C6)-Alkyl]-CO-O-[(Ci-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(C1-C6)-Alkyl], (C6-C12)-Aryl, O-(C6-C,2)-Aryl, O-(C1-C12)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CF3, SF5, OCF3, S(O)2[(d-C6)-Alkyl], (C1- C6)-Alkyl, OH, -COOH,
NH2, -NH-CO-NH-[(C,-C6)-Alkyl]-CO-O-[(C1-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(Ci-C6)-Alkyl], (C6-C12)-Aryl, O-(C6-C12)-Aryl, O-(Ci-Ci2)-Alkylen-(C6-C12)-Aryl, S(O)m-(C6-C12)-Aryl; sowie deren physiologisch verträgliche Salze.
5. Verbindungen der Formel I, gemäß einem oder mehreren der Ansprüche 1 bis 4 dadurch gekennzeichnet, dass darin bedeuten
Rl CN oder Halogen;
R2 CF3 oder Halogen;
A CH;
B CH, N;
R3, R4 unabhängig voneinander Wasserstoff, (C!-Ci2)-Alkylen-(C6-Ci2)-Aryl;
R5 SF5, OCF3, S(O)2[(Ci-C6)-Alkyl], -NH-CO-NH- [(C !-C6)- Alkyl] -CO-O- [(C !-C6)-
Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(C1-C6)-Alkyl], O-(C1-Ci2)-Alkylen-(C6-C12)-Aryl;
R6, R7 unabhängig voneinander H, Halogen, CF3, SF5, OCF3, S(O)2[(CrC6)-Alkyl], (Cr C6)-Alkyl, OH5 -COOH,
NH2, -NH-CO-NH-[(C1-C6)-Alkyl]-CO-O-[(Ci-C6)-Alkyl], -NH-SO2-NH2, -NH-SO2-NH-CO-O[(C,-C6)-Alkyl], (C6-Ci2)-Aryl, O-(C6-C12)-Aryl,  S(O)m-(C6-C12)-Aryl;
S(O)m-(C6-C12)-Aryl;
sowie deren physiologisch verträgliche Salze.
6. Verbindungen der Formeln
4-[3-(3,5-Bis-trifluoromethyl-benzyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril, 4- [4,4-Dimethyl-2,5 -dioxo-3 -(4-trifluoromethoxy-benzyl)-imidazolidin- 1 -yl] -2- trifluoromethyl-benzonitril,
4-[4,4-Dimethyl-2,5-dioxo-3-(3-trifluoromethyl-benzyl)-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril,
4-[3-(6-Chlor-pyridin-3-ylmethyl)-4,4-dimethyl-2,5-dioxo-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril,
4-[4,4-Dimethyl-2,5-dioxo-3-(6-trifluoromethyl-pyridin-3-ylmethyl)-imidazolidin-l-yl]-
2-trifluoromethyl-benzonitril,
3 -(4-Fluoro-3 -trifluoromethyl-phenyl)-5 ,5 -dimethyl- 1 -(6-trifluoromethyl-pyridin-3 - ylmethyl)-imidazolidin-2,4-dion,
3-(4-Chlor-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(6-trifluoro- methyl-pyridin-3- ylmethyl)-imidazolidin-2,4-dion,
l-(3,5-Bis-trifluoromethyl-benzyl)-3-(4-fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl- imidazolidin-2,4-dion,
1 -(3 ,5-Bis-trifluoromethyl-benzyl)-3 -(4-chlor-3 -trifluoromethyl-phenyl)-5 ,5 -dimethyl- imidazolidin-2,4-dion,
4-[4,4-Dimethyl-2,5-dioxo-3-(3-pentafluorosulfanyl-benzyl)-imidazolidin-l-yl]-2- trifluoromethyl-benzonitril und
3-(4-Fluoro-3-trifluoromethyl-phenyl)-5,5-dimethyl-l-(3-pentafluorosulfanyl-benzyl)- imidazolidin-2,4-dion.
7. Arzneimittel enthaltend eine oder mehrere der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6.
8. Arzneimittel enthaltend eine oder mehrere der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 und mindestens einen weiteren Wirkstoff.
9. Arzneimittel, gemäß Anspruch 8, dadurch gekennzeichnet, dass es als weiteren Wirkstoff eine oder mehrere Antidiabetika, hypoglykämischen Wirkstoffe, HMGCoA- Reduktase Inhibitoren, Cholesterinresorptionsinhibitoren, PPAR gamma Agonisten, PPAR alpha Agonisten, PPAR alpha/gamma Agonisten, PPAR delta Agonisten, Fibrate, MTP-Inhibitoren, Gallensäureresorptionsinhibitoren, MTP-Inhibitoren, CETP- Inhibitoren, polymere Gallensäureadsorber, LDL-Rezeptorinducer, ACAT-Inhibitoren, Antioxidantien, Lipoprotein-Lipase Inhibitoren, ATP-Citrat-Lyase Inhibitoren, Squalen Synthetase Inhibitoren, Lipoprotein(a) Antagonisten, HM74A Rezeptor Agonisten, Lipase Inhibitoren, Insuline, Sulfonylharnstoffe, Biguanide, Meglitinide, Thiazolidindione, α-Glukosidase-Inhibitoren, auf den ATP-abhängigen Kaliumkanal der Betazellen wirkende Wirkstoffe, Glykogen Phosphorylase Inhibitoren, Glukagon- Rezeptor- Antagonisten, Aktivatoren der Glukokinase, Inhibitoren der Glukoneogenese, Inhibitoren der Fructose-l,6-biphosphatase, Modulatoren des Glukosetransporters-4, Inhibitoren der Glutamin-Fructose-6-Phosphat-Amidotransferase, Inhibitoren der Dipeptidylpeptidase-IV, Hemmstoffe der 11-beta-Hydroxysteroid-Dehydrogenase-l, Inhibitoren der Protein-Tyrosin-Phosphatase-1B, Modulatoren des natrium-abhängigen Glukosetransporters 1 oder 2, Modulatoren des GPR40, Inhibitoren der hormonsensitiven Lipase, Hemmstoffe der Acetyl-CoA Carboxylase, Inhibitoren der Phosphoenolpyruvatcarboxykinase, Inhibitoren der Glykogen Synthase Kinase-3 beta, Inhibitoren der Protein Kinase C beta, Endothelin- A-Rezeptor Antagonisten, Inhibitoren der I kappaB Kinase, Modulatoren des Glukocorticoidrezeptors, CART-Agonisten, NP Y- Antagonisten, MC4- Agonisten, Orexin- Antagonisten, H3 -Antagonisten, TNF- Agonisten, CRF- Antagonisten, CRF BP- Antagonisten, Urocortin- Agonisten, ß3- Agonisten, CBl -Rezeptor Antagonisten, MSH (Melanocyt-stimulierendes Hormon)- Agonisten, MCH-Antagonisten, CCK-Agonisten, Serotonin- Wiederaufnahme- Inhibitoren, gemischte Sertonin- und noradrenerge Verbindungen, 5HT-Modulatoren, Bombesin- Agonisten, Galanin-Antagonisten, Wachstumshormone, Wachstumshormon freisetzende Verbindungen, TRH- Agonisten, entkoppelnde Protein 2- oder 3- Modulatoren, Leptinagonisten, DA-Agonisten (Bromocriptin, Doprexin), Lipase/ Amylase-Inhibitoren, PPAR-Modulatoren, RXR-Modulatoren oder TR-ß- Agonisten oder Amphetamine enthält.
10. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung des metabolischen Syndroms.
11. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung des Diabetes.
12. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von Adipositas.
13. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Gewichtsreduktion.
14. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von Nikotinabhängigkeit.
15. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von Alkoholabhängigkeit.
16. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von ZNS-Erkrankungen.
17. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von Schizophrenie.
18. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung von Alzheimer.
19. Verwendung der Verbindungen gemäß einem oder mehreren der Ansprüche 1 bis 6 zur Herstellung eines Medikamentes zur Behandlung des Syndroms der polycystischen Ovarien (PCOS, polycystic ovary Syndrome).
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09707223A EP2242745A1 (de) | 2008-02-07 | 2009-01-30 | Neue phenyl-substituierte imidazolidine, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| US12/852,038 US20110178134A1 (en) | 2008-02-07 | 2010-08-06 | Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08290133 | 2008-02-07 | ||
| EP08290133.1 | 2008-02-07 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/852,038 Continuation US20110178134A1 (en) | 2008-02-07 | 2010-08-06 | Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009097995A1 true WO2009097995A1 (de) | 2009-08-13 |
Family
ID=39506661
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/000588 WO2009097995A1 (de) | 2008-02-07 | 2009-01-30 | Neue phenyl-substituierte imidazolidine, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20110178134A1 (de) |
| EP (1) | EP2242745A1 (de) |
| AR (1) | AR070578A1 (de) |
| CL (1) | CL2009000256A1 (de) |
| TW (1) | TW200946510A (de) |
| UY (1) | UY31645A1 (de) |
| WO (1) | WO2009097995A1 (de) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009286698A (ja) * | 2008-05-27 | 2009-12-10 | Ube Ind Ltd | ハロゲノメチルペンタフルオロスルファニルベンゼン化合物及びその製造方法 |
| WO2011029537A1 (en) * | 2009-09-11 | 2011-03-17 | Bayer Schering Pharma Aktiengesellschaft | Sustituted ( heteroarylmethyl ) thiohydantoins as anticancer drugs |
| WO2012030165A2 (ko) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | P P A R δ 활성물질의 태자재프로그래밍용도 |
| EP2746260A1 (de) | 2012-12-21 | 2014-06-25 | Basf Se | Substituierte [1,2,4]Triazol- und Imidazolverbindungen |
| EP2746259A1 (de) | 2012-12-21 | 2014-06-25 | Basf Se | Substituierte [1,2,4]Triazol- und Imidazolverbindungen |
| US9346769B2 (en) | 2010-05-05 | 2016-05-24 | Infinity Pharmaceuticals, Inc. | Tetrazolones as inhibitors of fatty acid synthase |
| CN112759581A (zh) * | 2020-11-16 | 2021-05-07 | 贵州大学 | 一种含苯并咪唑磺酰胺杨梅素衍生物、制备方法及用途 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016517883A (ja) * | 2013-05-03 | 2016-06-20 | オレゴン ヘルス アンド サイエンス ユニバーシティー | X連鎖副腎白質ジストロフィーの処置におけるソベチロムの使用 |
| WO2016079522A1 (en) * | 2014-11-20 | 2016-05-26 | University College Cardiff Consultants Limited | Androgen receptor modulators and their use as anti-cancer agents |
| CN113423684A (zh) | 2018-12-12 | 2021-09-21 | 速通医疗公司 | 新型拟甲状腺素药 |
| CA3130371A1 (en) | 2019-03-01 | 2020-09-10 | Autobahn Therapeutics, Inc. | Novel thyromimetics |
| CN113801064A (zh) * | 2021-09-26 | 2021-12-17 | 甘肃省化工研究院有限责任公司 | 一种[3+2]环加成反应构建苯基乙内酰脲的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0494819A1 (de) * | 1991-01-09 | 1992-07-15 | Roussel Uclaf | Phenylimidazolidine, Verfahren zu deren Herstellung, deren Anwendung als Arzneimittel und diese enthaltende pharmazeutische Zusammenstellungen |
Family Cites Families (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5411981A (en) * | 1991-01-09 | 1995-05-02 | Roussel Uclaf | Phenylimidazolidines having antiandrogenic activity |
| US7396814B2 (en) * | 1995-06-07 | 2008-07-08 | Palatin Technologies, Inc. | Metallopeptide compositions for treatment of sexual dysfunction |
| DE19726167B4 (de) * | 1997-06-20 | 2008-01-24 | Sanofi-Aventis Deutschland Gmbh | Insulin, Verfahren zu seiner Herstellung und es enthaltende pharmazeutische Zubereitung |
| US6221897B1 (en) * | 1998-06-10 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Benzothiepine 1,1-dioxide derivatives, a process for their preparation, pharmaceuticals comprising these compounds, and their use |
| DE19845405C2 (de) * | 1998-10-02 | 2000-07-13 | Aventis Pharma Gmbh | Arylsubstituierte Propanolaminderivate und deren Verwendung |
| EE200200095A (et) * | 1999-09-01 | 2003-04-15 | Aventis Pharma Deutschland Gmbh | Sulfonüülkarboksamiidi derivaadid, nende kasutamine, ravim ja nende valmistamismeetod |
| US20030105090A1 (en) * | 2000-12-21 | 2003-06-05 | David Bebbington | Pyrazole compounds useful as protein kinase inhibitors |
| DK1345895T3 (da) * | 2000-12-21 | 2007-05-07 | Sanofi Aventis Deutschland | Hidtil ukendt diphenylazetidioner, fremgangsmåde til deres fremstilling, lægemidler indeholdende disse forbindelser og deres anvendelse til behandling af lipidstofskifteforstyrrelser |
| US7718802B2 (en) * | 2001-08-10 | 2010-05-18 | Palatin Technologies, Inc. | Substituted melanocortin receptor-specific piperazine compounds |
| US7732451B2 (en) * | 2001-08-10 | 2010-06-08 | Palatin Technologies, Inc. | Naphthalene-containing melanocortin receptor-specific small molecule |
| CA2462200A1 (en) * | 2001-08-10 | 2003-02-20 | Palatin Technologies, Inc. | Peptidomimetics of biologically active metallopeptides |
| US7655658B2 (en) * | 2001-08-10 | 2010-02-02 | Palatin Technologies, Inc. | Thieno [2,3-D]pyrimidine-2,4-dione melanocortin-specific compounds |
| US7109216B2 (en) * | 2001-09-21 | 2006-09-19 | Solvay Pharmaceuticals B.V. | 1H-imidazole derivatives having CB1 agonistic, CB1 partial agonistic or CB1-antagonistic activity |
| US6509367B1 (en) * | 2001-09-22 | 2003-01-21 | Virginia Commonwealth University | Pyrazole cannabinoid agonist and antagonists |
| US20050124652A1 (en) * | 2002-02-04 | 2005-06-09 | Rustum Boyce | Guanidino compounds |
| US7138107B2 (en) * | 2003-02-18 | 2006-11-21 | Compellis Pharmaceuticals | Inhibition of olfactory neurosensory function to treat eating disorders and obesity |
| EP1591120A4 (de) * | 2003-01-28 | 2009-06-10 | Takeda Chemical Industries Ltd | Rezeptor-agonisten |
| US7145012B2 (en) * | 2003-04-23 | 2006-12-05 | Pfizer Inc. | Cannabinoid receptor ligands and uses thereof |
| US20040214856A1 (en) * | 2003-04-23 | 2004-10-28 | Pfizer Inc | Cannabinoid receptor ligands and uses thereof |
| US7268133B2 (en) * | 2003-04-23 | 2007-09-11 | Pfizer, Inc. Patent Department | Cannabinoid receptor ligands and uses thereof |
| US7049323B2 (en) * | 2003-04-25 | 2006-05-23 | Bristol-Myers Squibb Company | Amidoheterocycles as modulators of the melanocortin-4 receptor |
| AR044152A1 (es) * | 2003-05-09 | 2005-08-24 | Bayer Corp | Derivados de alquilarilo, metodo de preparacion y uso para el tratamiento de la obesidad |
| DE602004012858T2 (de) * | 2003-06-20 | 2009-05-14 | F. Hoffmann-La Roche Ag | 2-aminobenzothiazole als cb1 rezeptor inverse agonisten |
| US7276608B2 (en) * | 2003-07-11 | 2007-10-02 | Bristol-Myers Squibb Company | Tetrahydroquinoline derivatives as cannabinoid receptor modulators |
| US20050026984A1 (en) * | 2003-07-29 | 2005-02-03 | Aventis Pharma S.A. | Substituted thieno [2,3-c] pyrazoles and their use as medicinal products |
| BRPI0507374A (pt) * | 2004-02-02 | 2007-07-10 | Pfizer Prod Inc | moduladores do receptor de histamina-3 |
| EP1586318A1 (de) * | 2004-04-05 | 2005-10-19 | Neuropharma S.A.U. | Thiadiazolidinone als GSK3-Inhibitoren |
| NZ552806A (en) * | 2004-07-29 | 2009-09-25 | Athersys Inc | Tricyclic indeno-pyrrole derivatives as serotonin receptor modulators |
| DE602005014124D1 (de) * | 2004-09-07 | 2009-06-04 | Banyu Pharma Co Ltd | Carbamoylsubstituiertes spiro-derivat |
| EP1757587A1 (de) * | 2005-07-15 | 2007-02-28 | Laboratorios Del Dr. Esteve, S.A. | Substituierte Pyrazolinderivate, ihre Herstellung und Verwendung als Arzneimittel |
| MXPA05009633A (es) * | 2005-09-08 | 2007-03-07 | Silanes Sa De Cv Lab | Composicion farmaceutica estable de glimepirida de liberacion inmediata y metformina de liberacion prolongada. |
| EP1931652A2 (de) * | 2005-09-21 | 2008-06-18 | Incyte Corporation | Amidverbindungen und ihre verwendung als arzneimittel |
| AR056155A1 (es) * | 2005-10-26 | 2007-09-19 | Bristol Myers Squibb Co | Antagonistas del receptor 1 de la hormona de concentracion de melanina no basica |
| US7745447B2 (en) * | 2005-10-26 | 2010-06-29 | Bristol-Myers Squibb Company | Substituted thieno[3,2-D]pyrimidines as non-basic melanin concentrating hormone receptor-1 antagonists |
| US7691876B2 (en) * | 2006-02-07 | 2010-04-06 | Hoffmann-La Roche Inc. | Heterobicyclic amide compounds |
| US7745477B2 (en) * | 2006-02-07 | 2010-06-29 | Hoffman-La Roche Inc. | Heteroaryl and benzyl amide compounds |
| US7674815B2 (en) * | 2006-02-07 | 2010-03-09 | Hoffmann-La Roche Inc. | Heteroaryl and benzyl amide compounds |
| US7572823B2 (en) * | 2006-02-07 | 2009-08-11 | Hoffmann-La Roche Inc. | Heteroaryl carboxamide compounds |
| US7834178B2 (en) * | 2006-03-01 | 2010-11-16 | Bristol-Myers Squibb Company | Triazine 11-beta hydroxysteroid dehydrogenase type 1 inhibitors |
| US20070208001A1 (en) * | 2006-03-03 | 2007-09-06 | Jincong Zhuo | Modulators of 11- beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US20070270492A1 (en) * | 2006-03-06 | 2007-11-22 | Avestha Gengraine Technologies Pvt. Ltd. | Nananoic acid derivatives as dipeptidyl peptidase inhibitors |
| US20080051452A1 (en) * | 2006-03-06 | 2008-02-28 | Avestha Gengraine Technologies Pvt. Ltd. | Hexanoic acid derivatives as dipeptidyl peptidase inhibitors |
| AU2007226673A1 (en) * | 2006-03-10 | 2007-09-20 | Jenrin Discovery | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| US7645748B2 (en) * | 2006-04-03 | 2010-01-12 | Forbes Medi-Tech Inc. | Sterol/stanol phosphorylnitroderivatives and use thereof |
| CN101460491A (zh) * | 2006-04-12 | 2009-06-17 | 惠氏公司 | 苯胺基嘧啶苯基和苯并噻吩类似物 |
| UY30288A1 (es) * | 2006-04-18 | 2007-08-31 | Janssen Pharmaceutica Nv | Derivados del ácido benzoazepin-oxi-acético como agonistas de ppar-delta usados para aumentar hdl-c. reducir ldl-c y reducir colesterol |
| US20070249709A1 (en) * | 2006-04-18 | 2007-10-25 | Wyeth | Crystal forms of (S)-(8(2,6-dichlorophenyl)-6-fluoro-2,3-dihydro benzo[b][1,4]dioxin-2-yl)methanamine hydrochloride salt |
| US7659281B2 (en) * | 2006-04-25 | 2010-02-09 | Bristol-Myers Squibb Company | HMG-CoA reductase inhibitors |
| US20070254863A1 (en) * | 2006-04-27 | 2007-11-01 | Jochen Antel | Use of CBx cannabinoid receptor modulators as potassium channel modulators |
| EA030606B1 (ru) * | 2006-05-04 | 2018-08-31 | Бёрингер Ингельхайм Интернациональ Гмбх | Способы приготовления лекарственного средства, содержащего полиморфы |
| WO2007131219A2 (en) * | 2006-05-05 | 2007-11-15 | Jenrin Discovery | Cannabinoid receptor antagonists/inverse agonists |
| WO2007134149A2 (en) * | 2006-05-11 | 2007-11-22 | Janssen Pharmaceutica N.V. | 3,4-dihydro-2h-benzo[1,4]oxazine and thiazine derivatives as cetp inhibitors |
| JP2009536953A (ja) * | 2006-05-11 | 2009-10-22 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Cetp阻害剤としての1,2,3,4−テトラヒドロ−キノリン誘導体 |
| US7851468B2 (en) * | 2006-05-15 | 2010-12-14 | Cephalon, Inc. | Substituted pyrazolo[3,4-d]pyrimidines |
| AU2007254325B2 (en) * | 2006-05-15 | 2012-05-10 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| CA2652375A1 (en) * | 2006-05-17 | 2007-11-29 | Incyte Corporation | Heterocyclic inhibitors of 11-.beta. hydroxyl steroid dehydrogenase type i and methods of using the same |
| BRPI0712516A2 (pt) * | 2006-05-18 | 2016-08-23 | Hoffmann La Roche | compostos, composições farmacêuticas, antagonista receptor de a2b, método para uso terapêutico e/ou tratamento profilático e uso de antagonista receptor de a2b |
| CN101432261A (zh) * | 2006-05-19 | 2009-05-13 | 惠氏公司 | 作为组织胺-3拮抗剂的n-苯甲酰基-吡咯烷-3-基胺和n-苄基-吡咯烷-3-基胺 |
| EP2019679B1 (de) * | 2006-05-23 | 2018-06-20 | Theracos, Inc. | Glucosetransporthemmer und anwendungsverfahren |
| JP5105297B2 (ja) * | 2006-05-25 | 2012-12-26 | 味の素株式会社 | Ppar活性調節剤 |
| WO2007143422A2 (en) * | 2006-05-30 | 2007-12-13 | Janssen Pharmaceutica N.V. | Substituted pyridyl amide compounds as modulators of the histamine h3 receptor |
| US8008332B2 (en) * | 2006-05-31 | 2011-08-30 | Takeda San Diego, Inc. | Substituted indazoles as glucokinase activators |
| US20070287674A1 (en) * | 2006-06-08 | 2007-12-13 | Hej Research Institute Of Chemistry | New treatment of diabetes mellitus |
| US7629346B2 (en) * | 2006-06-19 | 2009-12-08 | Hoffmann-La Roche Inc. | Pyrazinecarboxamide derivatives as CB1 antagonists |
| WO2008002575A1 (en) * | 2006-06-26 | 2008-01-03 | The Procter & Gamble Company | Melanin concentrating hormone antagonists |
| US20080004281A1 (en) * | 2006-06-28 | 2008-01-03 | Kalypsys, Inc. | Methods for the modulation of crp by the selective modulation of ppar delta |
| US20080004325A1 (en) * | 2006-06-29 | 2008-01-03 | Wyeth | PTP1B inhibitors |
| US7803799B2 (en) * | 2006-07-07 | 2010-09-28 | National Health Research Institutes | Selenophene compounds |
| US7297710B1 (en) * | 2006-07-12 | 2007-11-20 | Sanofi-Aventis | Derivatives of N-[(1,5-diphenyl-1H-pyrazol-3-yl)methyl]sulfonamide, their preparation and their application in therapeutics |
| US7601856B2 (en) * | 2006-07-27 | 2009-10-13 | Wyeth | Benzofurans as potassium ion channel modulators |
| US7662831B2 (en) * | 2006-07-27 | 2010-02-16 | Wyeth Llc | Tetracyclic indoles as potassium channel modulators |
| AU2007283113A1 (en) * | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US7939661B2 (en) * | 2006-09-11 | 2011-05-10 | Hoffman-La Roche Inc. | Pyridine, quinoline and pyrimidine derivatives |
| US7531519B2 (en) * | 2006-09-21 | 2009-05-12 | Medical And Pharmaceutical Industry Technology And Development Center | Polygalatenosides and use thereof as an antidepressant agent |
| TWI339205B (en) * | 2006-10-02 | 2011-03-21 | Nat Health Research Institutes | Pyrazole compounds and pharmaceutical composition |
| TWI408136B (zh) * | 2006-10-02 | 2013-09-11 | Nat Health Research Institutes | 噻吩化合物及其醫藥組成物 |
| KR101444486B1 (ko) * | 2006-10-19 | 2014-09-24 | 다케다 야쿠힌 고교 가부시키가이샤 | 인돌 화합물 |
| US20080103201A1 (en) * | 2006-10-26 | 2008-05-01 | Wijayabandara Mirihanage Don J | Novel alpha-Glucosidase inhibitor from Tabernaemontana dichotoma |
| US7858645B2 (en) * | 2006-11-01 | 2010-12-28 | Hoffmann-La Roche Inc. | Indazole derivatives |
| US7750048B2 (en) * | 2006-11-15 | 2010-07-06 | Janssen Pharmaceutica Nv | GPR40 agonists |
| PE20081229A1 (es) * | 2006-12-01 | 2008-08-28 | Merck & Co Inc | Antagonistas de receptor de orexina de diazepam sustituido |
| US20080194658A1 (en) * | 2006-12-11 | 2008-08-14 | Que Lan | Sterol Carrier Protein-2 Inhibitors for Lowering Cholesterol and Triglyceride Levels in Mammals |
| US7902248B2 (en) * | 2006-12-14 | 2011-03-08 | Hoffmann-La Roche Inc. | Oxime glucokinase activators |
| US20080146523A1 (en) * | 2006-12-18 | 2008-06-19 | Guido Galley | Imidazole derivatives |
| PE20081559A1 (es) * | 2007-01-12 | 2008-11-20 | Merck & Co Inc | DERIVADOS DE ESPIROCROMANONA SUSTITUIDOS COMO INHIBIDORES DE ACETIL CoA CARBOXILASA |
| US20080176861A1 (en) * | 2007-01-23 | 2008-07-24 | Kalypsys, Inc. | Sulfonyl-substituted bicyclic compounds as ppar modulators for the treatment of non-alcoholic steatohepatitis |
| US7648979B2 (en) * | 2007-02-07 | 2010-01-19 | Hoffmann-La Roche Inc. | 5-amido-(1H-indol-2-yl)-piperazin-1-yl-methanone derivatives |
| US7557108B2 (en) * | 2007-02-07 | 2009-07-07 | Hoffmann-La Roche Inc. | (Indol-4-yl) or (indol-5-yl)-piperazinylmethanones |
| US7507736B2 (en) * | 2007-02-07 | 2009-03-24 | Hoffmann-La Roche Inc. | Indol-2-yl-piperazin-1-yl-methanone derivatives |
| EA200970746A1 (ru) * | 2007-02-09 | 2010-02-26 | Такеда Фармасьютикал Компани Лимитед | Конденсированные циклические соединения в качестве частичных агонистов ppar-гамма |
| WO2008103354A2 (en) * | 2007-02-20 | 2008-08-28 | Cropsolution, Inc. | Modulators of acetyl-coenzyme a carboxylase and methods of use thereof |
| US20080207704A1 (en) * | 2007-02-27 | 2008-08-28 | The Green Cross Corporation | Heteroaryl-imidazole derivatives as cannabinoid cb1 receptor antagonists |
| WO2008116742A1 (en) * | 2007-03-23 | 2008-10-02 | F. Hoffmann-La Roche Ag | Aza-pyridopyrimidinone derivatives |
| KR101171433B1 (ko) * | 2007-03-30 | 2012-08-06 | 에프. 호프만-라 로슈 아게 | 이미다졸리딘온 유도체 |
| AR065913A1 (es) * | 2007-04-02 | 2009-07-08 | Theracos Inc | Derivados de glicosido bencilico, composicion y combinacion farmaceutica y uso |
| WO2008122513A1 (en) * | 2007-04-04 | 2008-10-16 | F. Hoffmann-La Roche Ag | Heterocycles as orexin antagonists |
| US7799806B2 (en) * | 2007-04-04 | 2010-09-21 | Hoffmann-La Roche Inc. | Substituted n-benzyl piperidines as somatostatin receptor modulators |
| US7662819B2 (en) * | 2007-04-09 | 2010-02-16 | Gilead Palo Alto, Inc. | Pteridinone derivatives for use as stearoyl CoA desaturase inhibitors |
| US8741916B2 (en) * | 2007-04-09 | 2014-06-03 | Janssen Pharmaceutica Nv | 1,3,8-trisubstituted-1,3,8-triaza-spiro[4.5]decan-4-one derivatives as ligands of the ORL-1 receptor |
-
2009
- 2009-01-30 EP EP09707223A patent/EP2242745A1/de not_active Withdrawn
- 2009-01-30 WO PCT/EP2009/000588 patent/WO2009097995A1/de active Application Filing
- 2009-02-05 TW TW098103593A patent/TW200946510A/zh unknown
- 2009-02-05 AR ARP090100398A patent/AR070578A1/es unknown
- 2009-02-05 CL CL2009000256A patent/CL2009000256A1/es unknown
- 2009-02-06 UY UY031645A patent/UY31645A1/es not_active Application Discontinuation
-
2010
- 2010-08-06 US US12/852,038 patent/US20110178134A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0494819A1 (de) * | 1991-01-09 | 1992-07-15 | Roussel Uclaf | Phenylimidazolidine, Verfahren zu deren Herstellung, deren Anwendung als Arzneimittel und diese enthaltende pharmazeutische Zusammenstellungen |
Non-Patent Citations (2)
| Title |
|---|
| SOEDERHOLM, ANNU A. ET AL: "Three-Dimensional Structure-Activity Relationships of Nonsteroidal Ligands in Complex with Androgen Receptor Ligand-Binding Domain", JOURNAL OF MEDICINAL CHEMISTRY , 48(4), 917-925 CODEN: JMCMAR; ISSN: 0022-2623, 2005, XP009101948 * |
| VAN DORT, M. E. ET AL: "Synthesis and structure-activity studies of side-chain derivatized arylhydantoins for investigation as androgen receptor radioligands", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 11(8), 1045-1047 CODEN: BMCLE8; ISSN: 0960-894X, 2001, XP009101945 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009286698A (ja) * | 2008-05-27 | 2009-12-10 | Ube Ind Ltd | ハロゲノメチルペンタフルオロスルファニルベンゼン化合物及びその製造方法 |
| WO2011029537A1 (en) * | 2009-09-11 | 2011-03-17 | Bayer Schering Pharma Aktiengesellschaft | Sustituted ( heteroarylmethyl ) thiohydantoins as anticancer drugs |
| CN102639523A (zh) * | 2009-09-11 | 2012-08-15 | 拜耳医药股份有限公司 | 用作抗癌药的取代的(杂芳基甲基)乙内酰硫脲 |
| US9346769B2 (en) | 2010-05-05 | 2016-05-24 | Infinity Pharmaceuticals, Inc. | Tetrazolones as inhibitors of fatty acid synthase |
| WO2012030165A2 (ko) | 2010-08-31 | 2012-03-08 | 서울대학교산학협력단 | P P A R δ 활성물질의 태자재프로그래밍용도 |
| EP2746260A1 (de) | 2012-12-21 | 2014-06-25 | Basf Se | Substituierte [1,2,4]Triazol- und Imidazolverbindungen |
| EP2746259A1 (de) | 2012-12-21 | 2014-06-25 | Basf Se | Substituierte [1,2,4]Triazol- und Imidazolverbindungen |
| CN112759581A (zh) * | 2020-11-16 | 2021-05-07 | 贵州大学 | 一种含苯并咪唑磺酰胺杨梅素衍生物、制备方法及用途 |
| CN112759581B (zh) * | 2020-11-16 | 2022-08-02 | 贵州大学 | 一种含苯并咪唑磺酰胺杨梅素衍生物、制备方法及用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| UY31645A1 (es) | 2009-08-31 |
| CL2009000256A1 (es) | 2009-06-26 |
| AR070578A1 (es) | 2010-04-21 |
| EP2242745A1 (de) | 2010-10-27 |
| TW200946510A (en) | 2009-11-16 |
| US20110178134A1 (en) | 2011-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8530413B2 (en) | Heterocyclically substituted methoxyphenyl derivatives with an oxo group, processes for preparation thereof and use thereof as medicaments | |
| EP2061767B1 (de) | Arylaminoaryl-alkyl-substituierte Imidazolidin-2,4-dione, Verfahren zu ihrer Herstellung, diese Verbindungen enthaltende Arzneimittel und ihre Verwendung | |
| EP2590930B1 (de) | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| US8648038B2 (en) | (2-aryloxyacetylamino)phenylpropionic acid derivatives, processes for preparation thereof and use thereof as medicaments | |
| US20110112097A1 (en) | Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof | |
| US20110178134A1 (en) | Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof | |
| US20110059910A1 (en) | Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof | |
| WO2009039942A1 (de) | (cyclopropyl-phenyl)-phenyl-oxalamide, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2203416A1 (de) | (carboxylalkylen-phenyl)-phenyl-oxalamide, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2590929A1 (de) | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2091915A1 (de) | Neues mit piperazin-1-sulfonsäure substituiertes diphenylazetidinon mit verbesserten pharmakologischen eigenschaften | |
| EP2220040B1 (de) | Neue kristalline diphenylazetidinonhydrate, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| US20110046185A1 (en) | Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof | |
| US20130172248A1 (en) | 3-[4-(phenylaminooxalylamino)phenyl]hex-4-ynoic acids, process for preparation thereof and use thereof as a medicament | |
| EP2252591B1 (de) | Aryl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung | |
| WO2009097996A1 (de) | Verwendung von substituierten phenylimidazolidinen zur herstellung von arzneimitteln zur behandlung des metabolischen syndroms | |
| US20110046105A1 (en) | Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof | |
| WO2011107494A1 (de) | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2683704B1 (de) | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung | |
| DE102010015123A1 (de) | Benzylamidische Diphenylazetidinone, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09707223 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009707223 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |