WO2007095976A2 - Adjuvanz in form einer lipid-modifizierten nukleinsäure - Google Patents
Adjuvanz in form einer lipid-modifizierten nukleinsäure Download PDFInfo
- Publication number
- WO2007095976A2 WO2007095976A2 PCT/EP2006/008321 EP2006008321W WO2007095976A2 WO 2007095976 A2 WO2007095976 A2 WO 2007095976A2 EP 2006008321 W EP2006008321 W EP 2006008321W WO 2007095976 A2 WO2007095976 A2 WO 2007095976A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nucleic acid
- lipid
- cancer
- rna
- adjuvant according
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
- A61K31/355—Tocopherols, e.g. vitamin E
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55561—CpG containing adjuvants; Oligonucleotide containing adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55572—Lipopolysaccharides; Lipid A; Monophosphoryl lipid A
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to an immunostimulatory adjuvant in the form of a lipid-modified nucleic acid, optionally in combination with other adjuvants.
- the invention further relates to a pharmaceutical composition and a vaccine, each containing an immunostimulatory adjuvant according to the invention, at least one active ingredient and optionally a pharmaceutically suitable carrier and / or further auxiliaries and additives and / or further adjuvants.
- the present invention also relates to the use of the pharmaceutical composition according to the invention and the vaccine according to the invention for the treatment of infectious diseases or cancers.
- the present invention comprises the use of the immunostimulatory adjuvant according to the invention for the preparation of a pharmaceutical composition for the treatment of cancers or infectious diseases.
- adjuvants ie substances or compositions which can increase and / or influence an immune response, eg against an antigen.
- adjuvant usually refers to a compound or composition which serves as a binder, carrier or excipient for immunogens and / or other pharmaceutically active compounds.
- adjuvants e.g. Freund's adjuvant, metal oxides (aluminum hydroxide, etc.), alum, inorganic chelates or salts thereof, various paraffin-like oils, synthesized resins, alginates, mucoids, polysaccharide compounds, caseinates, and compounds isolated from blood and / or blood clots such as Fibrin derivatives, etc .
- adjuvants usually produce unwanted side effects, for example. Very painful irritation and inflammation at the site of the application. Furthermore, toxic side effects, especially tissue necrosis, are also observed. Finally, in most cases, these known adjuvants cause only insufficient stimulation of the cellular immune response since only B cells are activated.
- Kidney, liver and / or spleen cells significantly disrupt. Also, the property of gelatin to swell under parenteral administration may result in unpleasant side effects, such as hypersensitivity, in e.g. Swelling, especially at the site of application and cause discomfort.
- Fibrin derivatives For compounds isolated from blood and / or blood clots, e.g. Fibrin derivatives, etc. have typically been found to have immunostimulatory effects. However, most of these compounds are presently inappropriate as adjuvants due to their (in parallel with the quite desirable immunogenic properties occurring) side effects on the immune system. For example, many of these compounds are classified as allergenic and may cause an overreaction of the immune system far beyond the desired level. These compounds are therefore also unsuitable for the reasons mentioned as an adjuvant for immune stimulation.
- immune responses can also be generated directly with nucleic acids as an adjuvant.
- DNA plays a central role in the generation of immune responses Role.
- CpG DNA has an immunostimulating effect due to the presence of unmethylated CG motifs, which is why such CpG DNA has been proposed as an immunostimulating agent and adjuvant for vaccines (see US Patent 5,663,153).
- This immunostimulatory property of DNA can also be achieved by DNA oligonucleotides stabilized by phosphorothioate modification (US Patent 6,239,116).
- U.S. Patent 6,406,705 discloses adjuvant compositions containing a synergistic combination of a CpG oligodeoxyribonucleotide and a non-nucleic acid adjuvant.
- DNA is broken down relatively slowly in the bloodstream, so that the use of immunostimulatory (foreign) DNA can lead to the formation of anti-DNA antibodies, which has been confirmed in the animal model in the mouse (Gilkeson et al., J Clin Invest 1995, 95: 1398-1402).
- the possible persistence of (foreign) DNA in the organism can thus lead to overactivation of the immune system, which is known to result in splenomegaly in mice (Montheith et al., Anticancer Drug Res. 1997, 12 (5): 421-432).
- (foreign) DNA can interact with the host genome, and in particular cause mutations by integration into the host genome.
- insertion of the introduced (foreign) DNA into an intact gene can occur, which represents a mutation that can hinder the function of the endogenous gene or even completely switch it off.
- integration events can destroy vital enzyme systems for the cell; on the other hand, the risk of transformation of the cell thus altered into a degenerate state, if the integration of the (foreign) DNA, a gene crucial for the regulation of cell growth is changed. Therefore, in previous methods when using (foreign) DNA as an immunostimulating agent, a risk of cancer formation may not be excluded.
- RNA More advantageous for the generation of such immune responses is therefore generally the use of RNA as an adjuvant, since RNA has a much lower half-life in vivo compared to DNA. Nevertheless, there are also limitations in the use of RNA as adjuvant.
- hitherto disclosed in the prior art RNA sequences in vivo only limited cell permeability. This can turn one Increased amount of RNA for immunostimulation require, which, despite the increased cost due to increased amounts of RNA to be applied, the risk of the generally previously described mostly undesirable side effects harbors, for example. Very painful irritation and inflammation at the site of application. Also toxic side effects can not be excluded with high doses of the administered immunostimulant.
- an immunostimulatory adjuvant according to the invention in the form of a lipid-modified nucleic acid.
- this lipid-modified nucleic acid consists of a nucleic acid, at least one linker covalently linked to this nucleic acid and at least one lipid covalently linked to the respective linker.
- the lipid-modified nucleic acid according to the invention consists of (at least) one nucleic acid and at least one (bifunctional) lipid covalently linked to this nucleic acid (without linker).
- the lipid-modified nucleic acid according to the invention consists of a nucleic acid, at least one linker covalently linked to this nucleic acid and at least one lipid covalently linked to the respective linker and at least one (bifunctional) lipid covalently linked to this nucleic acid (without linker).
- An “immunostimulatory” adjuvant according to the present invention is preferably capable of eliciting an immune response
- An immune response can generally be induced in a variety of ways.
- An essential factor for a suitable immune response is the stimulation of different T cell subpopulations. Lymphocytes typically differentiate into two subpopulations, the T helper 1 (ThI) and T helper 2 (Th2) cells with which the immune system is capable of intracellular (Th!) And extracellular (Th2) pathogens (eg, antigens ) to destroy.
- the two Th cell populations differ in the pattern of the effector proteins (cytokines) they produce.
- ThI cells support the cellular immune response by activating macrophages and cytotoxic T cells.
- Th2 cells promote the humoral immune response by stimulating the B cells to transform into plasma cells and by producing antibodies (eg against antigens).
- the Th1 / Th2 ratio is therefore of great importance in the immune response.
- the adjuvant according to the invention preferably shifts the Th1 / Th2 ratio of the immune response in the direction of the cellular response (Th1 response) and thus induces a cellular immune response.
- the nucleic acid used according to the invention for the lipid-modified nucleic acid (adjuvant) may be an RNA or DNA (for example a cDNA), an RNA or DNA oligonucleotide, an RNA or DNA homopolymer, a CpG nucleic acid, etc. It may be single-stranded or double-stranded, homo- or heteroduplex, and linear or circular.
- the nucleic acid used according to the invention for the lipid-modified nucleic acid (adjuvant) is particularly preferably present as single-stranded RNA.
- the lipid-modified nucleic acid is relatively short nucleic acid molecules, for example, from about 2 to about 1000 nucleotides, preferably from about 5 to 200, 6 to about 200 nucleotides, and more preferably from 6 to about 40 or 6 to about 31 nucleotides.
- Nucleotides are in this context preferably all naturally occurring nucleotides and their analogs, such as ribonucleotides and / or deoxyribonucleotides and include, but are not limited to, for example, purines (adenine (A), guanine (G)) or pyrimidines (thymine (T)).
- the lipid-modified nucleic acid may comprise any naturally occurring nucleic acid sequence, its complement or a fragment thereof.
- a fragment of such a nucleic acid sequence in this context preferably has a length of preferably about 5 to 200, 6 to about 200 nucleotides, and more preferably from 6 to about 40 or 6 to about 31 nucleotides.
- the lipid-modified nucleic acid may be partially or wholly synthetic.
- CpG nucleic acid is used in the lipid-modified nucleic acid, in particular CpG RNA or CpG DNA.
- a CpG RNA or CpG DNA used according to the invention may be a single-stranded CpG DNA (see also CpG-DNA), a double-stranded CpG-DNA (dsDNA), a single-stranded CpG-RNA (see also CpG-RNA) or a double-stranded CpG-RNA ( ds CpG RNA).
- the CpG nucleic acid used according to the invention is preferably present as CpG RNA, more preferably as single-stranded CpG RNA (see also CpG RNA). Also preferably, such CpG nucleic acids have a length as described above.
- the CpG nucleic acid used according to the invention preferably contains at least one or more (mitogenic) cytosine / guanine dinucleotide sequence (s) (CpG motif (s)) which are represented by the generic formulas 5'-X 1 X 2 CGX 3 X 4 ⁇ '("Hexamer", SEQ ID NO: 1) or 5'-X, X 2 X 3 CGX 4 X 5 X 6 -3'("octamer", SEQ ID NO: 2).
- At least one CpG motif contained in these hexamer or octamer sequences ie the C (cytosine) and the G (guanine) of this CpG motif, is unmethylated. All other cytosines or guanines optionally contained in the hexamer or octamer sequences can either be methylated or non-methylated. methylated. However, according to another preferred alternative, the C (cytosine) and the G (guanine) of the CpG motif may also be methylated.
- X 1 , X 2 , X 3 , X 4 , X 5 and X 6 are preferably nucleotides which can be selected independently of each other or together from all naturally occurring nucleotides and their analogs, as previously described generally for nucleic acids used herein.
- the CpG nucleic acid used according to the invention contains as CpG motif at least one or more octamers selected from the group consisting of: GACGTTCC (SEQ ID NO: 3); GACGCTCC (SEQ ID NO: 4); GACGTCCC (SEQ ID NO: 5); GACGCCCC (SEQ ID NO: 6); AGCGTTCC (SEQ ID NO: 7); AGCGCTCC (SEQ ID NO: 8); AGCGTCCC (SEQ ID NO: 9); AGCGCCCC (SEQ ID NO: 10); AACGTTCC (SEQ ID NO: 11); AACGCTCC (SEQ ID NO: 12); AACGTCCC (SEQ ID NO: 13); AACGCCCC (SEQ ID NO: 14); GGCGTTCC (SEQ ID NO: 15); GGCGCTCC (SEQ ID NO: 16); GGCGTCCC (SEQ ID NO: 17); GGCGCCCC (SEQ ID NO: 18); GACGTTCG (SEQ ID NO:
- the CpG nucleic acid used according to the invention contains as CpG motif at least one or more octamers selected from the group consisting of: GACGTTCC (SEQ ID NO: 3), AACGTTCC (SEQ ID NO: 1 1), GACGTTCG (SEQ ID NO : 19) and AACGTTCG (SEQ ID NO: 23). Also included are those sequences which have an identity to one of the foregoing sequences of at least 60%, more preferably 70 or 80%, and most preferably 90 or 95%. To determine the percent identity of two nucleic acid sequences to each other, the sequences can be aligned to be compared below.
- gaps in the sequence of the first nucleic acid sequence can be introduced and the nucleotides can be compared at the corresponding position of the second nucleic acid sequence. If a position in the first nucleic acid sequence is occupied by the same nucleotide as it is at a position in the second sequence, then both sequences are at it Identical position.
- the determination of the percentage identity of two sequences can be carried out using a mathematical algorithm.
- a preferred, but not limiting, example of a mathematical algorithm that can be used to compare two sequences is the algorithm of Karlin et al. (1993), PNAS USA, 90: 5873-5877. Such an algorithm is integrated into the NBLAST program which can identify sequences having a desired identity to the sequences of the present invention. To obtain a gapped alignment as described above, the gapped BLAST program can be used as described in Altschul et al. (1997) Nucleic Acids Res. 25: 3389-3402.
- the mitogenic CpG motif contained in the CpG nucleic acid used according to the invention preferably occurs at least once in the CpG nucleic acid. Most preferably, at the 5 'and 3' ends or near the 5 'and 3' ends of the used CpG nucleic acid, e.g. in a range of 1 to 3 or 1 to 6 nucleotides, no GCG trinucleotide.
- the hexamer or octamer sequences according to one of the sequences SEQ ID NO: 1 to 34 can occur at least once in the CpG nucleic acid used according to the invention, i. at least one hexamer and / or octamer sequence according to one of the sequences SEQ ID NO: 1 to 34 is contained.
- the hexamer or octamer sequences according to any one of the sequences SEQ ID NOS: 1 to 34 in the CpG nucleic acid used according to the invention may occur as a multimer, e.g.
- the individual hexamer or octamer sequences according to SEQ ID NO: 1 to 34 can be separated from one another by 1-30, preferably 1-20, more preferably 1-10, of the abovementioned nucleotides or alternatively directly follow one another without intervening nucleotides.
- the CpG nucleic acid used according to the invention can furthermore be present as a "stabilized oligonucleotide", ie as an oligoribo- or oligodeoxyribonucleotide which is resistant to in vivo degradation (for example by an exo- or endonuclease) Stabilization can be carried out, for example, by a modified phosphate backbone of the CpG nucleic acid used according to the invention.
- Nucleotides preferably used in this context contain a phosphorothioate-modified phosphate backbone, wherein preferably at least one of the phosphate oxygens contained in the phosphate backbone is replaced by a sulfur atom.
- oligonucleotides include, for example, nonionic analogs, such as alkyl and aryl phosphonates, in which the charged phosphonate oxygen is replaced by an alkyl or aryl group, or phosphodiester and alkyl phosphotriester in which the charged oxygen Radical alkylated.
- nonionic analogs such as alkyl and aryl phosphonates, in which the charged phosphonate oxygen is replaced by an alkyl or aryl group, or phosphodiester and alkyl phosphotriester in which the charged oxygen Radical alkylated.
- the nucleic acid used for the lipid-modified nucleic acid is present as RNA or DNA homopolymer, more preferably as RNA homopolymer.
- RNA or DNA homopolymer typically comprises single-stranded or double-stranded, preferably single-stranded, polynucleotides such as, for example, polyinosinic acid (I), polyadenic acid (A), polyuridic acid (U), polyxanthenic acid (X) or polyguaninic acid (G).
- the RNA or DNA homopolymers used according to the invention can occur as single-stranded RNA or DNA homopolymers.
- RNA or DNA homopolymers are well known in the art and typically do not have a uniform molecular weight.
- Molecular weights for double-stranded complexes of copolymers have been determined, for example, in a range of about 1 ⁇ 10 5 to 1.5 ⁇ 10 6 .
- RNA or DNA homopolymers comprises single-stranded RNA or DNA homopolymers.
- Such single-stranded RNA or DNA homopolymers typically contain a ribo or deoxyribonucleotide as defined above in n-fold repetition, where n is preferably equal to the length of the nucleic acids described above according to the invention and in a range from 2 to about 1000, preferably 5 to 200, more preferably 6 to about 200, and most preferably 6 to about 40 or 6 to about 31.
- RNA or DNA homopolymers include, but are not limited to, the following sequences: 5'-AAAAAAAAAAAAAA-3 '(SEQ ID NO: 35), 5'-UUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUU-3' (SEQ ID NO: 36), 5 ' GGGGGGGGGGGGGGGGGG-3 '(SEQ ID NO: 37), 5'-
- sequences which have an identity to any of the foregoing sequences of at least 60%, more preferably at least 70 or 80%, and most preferably at least 90 or 95%.
- Chemically modified polynucleotides represent a second preferred alternative of the DNA or RNA homopolymers.
- Chemically modified polynucleotides according to the present invention may be as described above DNA or RNA polymers having in their sequence at least one nucleotide, for example an analog or derivative of purines (adenine (A), guanine (G)) or pyrimidines (thymine (T), cytosine (C), uracil (U)) as previously described.
- such chemically altered polynucleotides have a proportion of analogues and derivatives of 1 to 100%, eg 1 - 20, 10 - 30, 20 - 40, 30 - 50, 40 - 60, 50 - 70, 60 - 80, 70 - 90 or 80 - 100% on.
- Such chemically altered polynucleotides can also be prepared by methods known in the art (see Methods of Making Complexes of Homopolymers).
- Exemplary chemically altered polynucleotides include, but are not limited to, compounds such as poly-N 1 -methyladenylate, poly "6-methyladenylate", poly-N7-methylinosate, poly-N7-methylguanylate, poly-5-methyluridylate, poly 5-fluorouridylate, poly-5-bromouridylate, poly-5-bromocytidylate and poly-5-iodocytidylate, etc.
- combinations of the RNA or DNA homopolymers described above can also be used as RNA or DNA homopolymers.
- Such combinations preferably include nucleic acid sequences containing at least two of the above alternatives of the RNA or DNA homopolymers or multimers thereof described herein, eg, a sequence of 2 to 5, 5 to 10, 10 to 15, 15 to 20, 20 to 30 , 30 to 40, 40 to 50 or 50 to 100 of one or more of the above-described RNA or DNA homopolymers, more preferably according to one of SEQ ID NO: 35 to 39.
- RNA or DNA homopolymers can by 1 -30, preferably 1 -20, more preferably 1 to 10 of the nucleotides previously described herein are separated from each other, or alternatively immediately follow one another without intervening nucleotides.
- the lipid-modified nucleic acid those nucleic acids which are not assigned to any of the abovementioned classes of nucleic acid and which are already known as immunogenic in the prior art or which are not yet known in the prior art but have immunogenic properties ,
- nucleic acids according to this alternative are present as RNA or DNA, more preferably as RNA.
- these nucleic acids have a length as described above and contain nucleotides, eg ribo- or deoxyribonucleotides, as disclosed herein before.
- nucleic acids can encode, for example, antigens.
- nucleic acids can encode epitopes (of proteins).
- such nucleic acids then have an ATG as a start signal, which marks the start of translation of the encoded RNA.
- the nucleic acid sequence of the lipid-modified nucleic acid has at least one sequence according to one of SEQ ID NOs: 40-67, as listed below: 5'-GCCCGUCUGUUGUGUGACUC-S '(SEQ ID NO: 40, also as RNA 40), 5'-GGUAAGUGUAAGGUGUAAGG-S '(SEQ ID NO: 41, also referred to as RNA CV1), 5'-AAUGGAUAUGGAAUAUGGAA-S' (SEQ ID NO: 42, also referred to as RNA CV2), 5 1 -UCCAUGACGUUCCUGACGUU -3 1 (SEQ ID NO: 43), 5'-UCCAGGACUUCUCUCAGGUU-S 1 (SEQ ID NO: 44), 5'-
- nucleic acids which are a multimer of one or more of the nucleic acids described above, e.g. a sequence of 2 to 5, 5 to 10, 10 to 15, 15 to 20, 20 to 30, 30 to 40, 40 to 50 or 50 to 100 of the above-described nucleic acids, particularly preferably according to SEQ ID NO: 1 to 67, exhibit.
- the order of the nucleic acids can be chosen arbitrarily.
- the individual nucleic acids / nucleic acid units of the multimer can be separated from one another by 1-30, preferably 1-20, more preferably 1-10, of the nucleotides described above, or alternatively directly follow one another without intervening nucleotides.
- the nucleic acid used for the lipid-modified nucleic acid according to the invention may additionally have at least one chemical modification in addition to the lipid modification.
- chemical modifications can typically be introduced at the 5' end and / or within the sequence of the nucleic acid used in the invention.
- the lipid modification is present at the 5 'end of the nucleic acid used according to the invention, chemical modifications can typically be introduced at the 3' end and / or within the sequence of the nucleic acid used according to the invention. If, on the other hand, the lipid modification is present at the 3 'end and at the 5' end of the nucleic acid used according to the invention, the chemical modifications become preferably introduced within the sequence of the nucleic acid used according to the invention.
- the chemical modification of the lipid-modified nucleic acid according to the invention is preferably such that the nucleic acid used for this, preferably RNA, contains at least one analogous naturally occurring nucleotide.
- Such analogs include the nucleotides and their analogs described above.
- all of the aforementioned nucleotides and their analogs may be replaced by e.g. acetylation,
- the nucleic acid or the lipid-modified nucleic acid itself used for the lipid-modified nucleic acid itself can furthermore be stabilized.
- any nucleic acid can be used in principle for the lipid-modified nucleic acid.
- the use of RNA for such a nucleic acid is preferred.
- RNA does not involve the risk of being stably integrated into the genome of the transfected cell.
- RNA is much easier to degrade in vivo. Likewise, probably due to the relatively short DNA half-life of RNA in the bloodstream, so far no anti-RNA antibodies were detected.
- RNA in solution is much more unstable, for which essentially RNA-degrading enzymes, so-called RNAases (ribonucleases), are responsible. Even the smallest impurities of ribonucleases are sufficient to completely degrade RNA in solution. Such RNase contaminants can generally only be eliminated by special treatments, in particular diethylpyrocarbonate (DEPC).
- DEPC diethylpyrocarbonate
- the natural degradation of mRNA in the cytoplasm of cells is very finely regulated. In this regard, several mechanisms are known in the art. Thus, for a RNA in vivo, typically the terminal structure is critically important.
- RNA At the 5 'end of naturally occurring RNAs is usually a so-called “cap structure” (a modified guanosine nucleotide) and at the 3' end a sequence of up to 200 adenosine nucleotides (the so-called poly-A-tail) , Therefore, the nucleic acid of the lipid-modified nucleic acid, if present as RNA, can be stabilized by the addition of a so-called "5'-cap” structure to degradation by RNases.
- cap structure a modified guanosine nucleotide
- a m7G (5 ') ppp (5' (A, G (5 ') ppp (5') A or G (5 ') ppp (5') G is particularly preferred in this context.
- a modification for example a lipid modification, has not already been introduced at the 5 'end of the nucleic acid used according to the invention.
- the 3 'end of the nucleic acid of the lipid-modified nucleic acid may be defined by a sequence of at least 50 adenosine nucleotides, preferably at least 70 adenosine nucleotides, more preferably at least 100
- Adenosine nucleotides more preferably at least 200 adenosine nucleotides are modified. Analogously, such a modification can only be introduced if the 3'-end of the nucleic acid used according to the invention does not already have a modification, e.g. a lipid modification was introduced.
- the lipid contained in the lipid-modified nucleic acid according to the invention is typically a lipid or a lipophilic residue, which is preferably biologically active per se.
- lipids preferably include natural products or compounds such as vitamins, eg ⁇ -tocopherol (vitamin E), including RRR- ⁇ -tocopherol (formerly D- ⁇ -tocopherol), L- ⁇ -tocopherol, the racemate D, L- ⁇ -tocopherol , Vitamin E succinate (VES), or vitamin A and its derivatives, eg retinoic acid, retinol, vitamin D and its derivatives, eg vitamin D and its ergosterol precursors, vitamin E and its derivatives, vitamin K and its derivatives, eg vitamin K and related quinone or phytol compounds, or steroids, such as bile acids, for example, cholic acid, deoxycholic acid, dehydrocholic acid, cortisone, digoxygenin, testosterone, cholesterol or thiocholesterol.
- lipids or lipophilic radicals for the purposes of the present invention include, but are not limited to, polyalkylene glycols (Oberhauser et al., Nucl. Acids Res., 1992, 20, 533), aliphatic groups such as C, -C 20 alkanes C 1 -C 20 -alkenes, or C 1 -C 20 -alkanol compounds, etc., such as, for example, dodecanediol, hexadecanol or undecyl radicals (Saison-Behmoaras et al., EMBO J, 1991, 10, 11 Kabanov et al., FEBS Lett., 1990, 259, 327, Svinarchuk et al., Biochimie, 1993, 75, 49), phospholipids such as phosphatidylglycerol, diacylphosphatidylglycerol, Phosphatidylcholine, dipalmitoylphosphatidy
- the linkage between the lipid and the nucleic acid used according to the invention can in principle take place at each nucleotide, at the base or the sugar moiety of each nucleotide, at the 3 'and / or 5' end, and / or at the phosphate backbone of the nucleic acid used according to the invention.
- a terminal lipid modification of the nucleic acid used according to the invention at its 3 'and / or 5' end is particularly preferred. Terminal modification has several advantages over sequence-internal modifications. On the one hand, sequence-internal modifications may influence the hybridization behavior, which may negatively affect sterically demanding residues.
- nucleic acid sequence in a synthetic preparation of a lipid-modified nucleic acid according to the invention which is exclusively terminally modified, the synthesis of the nucleic acid sequence can be carried out with commercially available, widely available monomers and synthesis protocols known in the art can be used.
- the link between the nucleic acid used according to the invention and at least one lipid used is via a linker (covalently linked to the nucleic acid).
- Linkers in the sense of the present invention typically have at least two and optionally 3, 4, 5, 6 , 7, 8, 9, 10, 10-20, 20-30 or more reactive groups each selected from, for example, a hydroxy group, an amino group, an alkoxy group, etc.
- a reactive group is used to bond the above-described invention used in the present invention
- Nucleic acid for example of an RNA oligonucleotide
- This reactive group may be in a protected form, eg as DMT group (dimethoxytrityl chloride), as Fmoc group, as MMT (monomethoxytrityl) group, as TFA (trifluoroacetic acid) group, etc.
- sulfur groups can be replaced by disulfides, eg alkylthiols such as, for example, Thiopropanol, or protected with activated components such as 2-thiopyridine.
- One or more further reactive groups are used according to the invention for the covalent binding of one or more lipids.
- a nucleic acid used according to the invention may therefore bind at least one lipid via the covalently bound linker, for example 1, 2, 3, 4, 5, 5-10, 10-20, 20-30 or more lipid (s), more preferably at least 3-8 or more lipid (s) per nucleic acid.
- the bound lipids can be bound separately to different positions of the nucleic acid, but also present as a complex at one or more positions of the nucleic acid.
- An additional reactive group of the linker may be used for direct or indirect (cleavable) attachment to a support material, eg a solid phase.
- Preferred linkers according to the present invention are, for example, glycol, glycerol and glycerol derivatives, 2-aminobutyl-1, 3-propanediol and 2-aminobutyl-1,3-propanediol derivatives / skeleton, pyrrolidine-linker or pyrrolidine-containing organic molecules (in particular for a modification at the 3'-end), etc .
- Particularly preferred according to the invention as linker glycerol or glycerol derivatives (C 3 -Anker) or a 2-aminobutyl-1, 3-propanediol derivative / scaffold (C 7 -Anker) is used.
- a glycerol derivative (C 3 anchor) as a linker is particularly preferred when the lipid modification can be introduced via an ether linkage. If the lipid modification is to be introduced, for example, via an amide or a urethane bond, for example a 2-aminobutyl-1,3-propanediol skeleton (C 7 acceptor) is preferred.
- the bond formed between the linker and the nucleic acid used according to the invention is preferably such that it is compatible with the conditions and chemicals of amido-chemistry, that is, it is preferably neither acid nor base labile.
- bonds are preferred that are synthetically readily accessible and are not hydrolyzed by the ammoniacal cleavage procedure of a nucleic acid synthesis method.
- Suitable bonds are in principle all correspondingly suitable bonds, preferably ester bonds, amide bonds, urethane and ether bonds.
- the ether bond is particularly preferred because of their relatively high biological stability to enzymatic hydrolysis.
- the linkage (at least one) of the nucleic acid used according to the invention is carried out directly with at least one (bifunctional) lipid as described above, ie without the use of a linker as described above.
- the (bifunctional) lipid used according to the invention preferably has at least two reactive groups, or optionally 3, 4, 5, 6, 7, 8, 9, 10, or more reactive groups, wherein a first reactive group to the direct or indirect binding of the lipid to a carrier material described herein and at least one further reactive group for binding a nucleic acid used according to the invention is used.
- a nucleic acid used according to the invention may therefore preferably bind at least one lipid (directly without linker), for example 1, 2, 3, 4, 5, 5-10, 10-20, 20-30 or more lipid (s), more preferably at least 3-8 or more lipid (s) per nucleic acid.
- the bound lipids can be bound separately to different positions of the nucleic acid, but also present as a complex at one or more positions of the nucleic acid.
- at least one nucleic acid can be bound to a lipid as described above via its reactive groups, for example optionally 3, 4, 5, 6, 7, 8, 9, 10, 10-20, 20-30 or more nucleic acids .
- Particularly suitable lipids for this second embodiment include those (bifunctional) lipids which allow coupling (preferably at their termini or optionally intramolecularly), such as polyethylene glycol (PEG) and derivatives thereof, hexaethylene glycol (HEG) and derivatives thereof, alkanediols, Aminoalkane, thioalkanols, etc.
- PEG polyethylene glycol
- HEG hexaethylene glycol
- alkanediols alkanediols
- Aminoalkane thioalkanols, etc.
- the linkage between the nucleic acid used according to the invention and at least one lipid as described above can take place simultaneously via both of the abovementioned embodiments.
- the nucleic acid at one position of the nucleic acid can be linked to at least one lipid via a linker (analogous to the first embodiment) and at another position of the nucleic acid directly with at least one lipid without use a linker (analog second embodiment).
- At least one lipid as described above can be covalently linked to the nucleic acid via a linker at the 3 'end of a nucleic acid used according to the invention, and at the 5' end of the nucleic acid a lipid as described above can be covalently linked to the nucleic acid without a linker.
- at least one lipid as described above can be covalently linked to the nucleic acid via a linker at the 5 'end of a nucleic acid used according to the invention, and at the 3' end of the nucleic acid a lipid as described above can be covalently linked to the nucleic acid without a linker.
- covalent linkages can take place not only at the termini of the nucleic acid but also intramolecularly, as described above, eg at the 3'-end and intramolecularly, at the 5'-end and intramolecularly, at the 3'- and 5'-end and intramolecularly, exclusively intramolecular, etc.
- the lipid-modified nucleic acid (s) used as the adjuvant of the invention can be obtained by various methods.
- the lipid modification can in principle - as defined above - be introduced at any position of the nucleic acid sequence used, for example at the 3 'and / or 5 ' ends or on the phosphate backbone of the nucleic acid sequence used and / or at each base or on the sugar each nucleotide of the nucleic acid sequence used.
- terminal lipid modifications at the 3 'and / or 5' ends of the nucleic acids used are preferred.
- a large number of differently derivatized nucleic acids can be obtained according to the invention. Exemplary and inventively encompassed variants are shown in Figure 1.
- the method for producing such lipid-modified nucleic acids is preferably selected depending on the position of the lipid modification.
- the lipid modification typically takes place either before or after provision of the nucleic acid used according to the invention.
- the provision of the nucleic acid used in accordance with the invention can be carried out by direct synthesis of the nucleic acid or by addition of an already completely synthesized (eg commercially available) or nucleic acid isolated from samples.
- the nucleic acid of a 3'-lipid-modified nucleic acid according to the invention is synthesized directly prior to the introduction of the lipid, typically by methods known in the art for the synthesis of nucleic acids.
- a starting nucleoside for example via a coupling molecule, for example a succinyl radical, is preferably bound to a solid phase and the nucleic acid is synthesized, for example by the process of amidite chemistry.
- a linker as described above is preferably covalently bound to the 3 'end of the nucleic acid via a first reactive group of the linker.
- a second reactive group of the linker can then be covalently linked to the linker as described above a lipid.
- the linker may be covalently linked to the lipid prior to binding to the 3 'end of the nucleic acid.
- the nucleic acid can be cleaved off from the solid phase and deprotected. If the synthesis was carried out in solution, after the synthesis of the lipid-modified nucleic acid (and, if appropriate, before the removal from the carrier material), a washing and purification step can be carried out to remove unreacted reactants and also solvents and unwanted by-products.
- the nucleic acid of a 3'-lipid-modified nucleic acid according to the invention is synthesized after introduction of the lipid to a reactive group of the linker or bound as ready-synthesized or isolated from samples (commercially available) nucleic acid to the reactive group of the linker (see eg Fig. 2).
- a first reactive group of a linker as described above can be reacted with a lipid as described above.
- a second reactive group of the linker is preferably provided with an acid-stable protective group, for example DMT, Fmoc, etc., in order to enable subsequent binding of the nucleic acid to this reactive group.
- the linker can be bound directly or indirectly to a solid phase.
- Indirect binding is possible, for example, via a (coupling) molecule, which can be covalently bound both to the linker and to the solid phase.
- a (coupling) molecule is, for example, a succinyl radical, etc. as described hereinafter.
- the deprotection of the third reactive group of the linker is usually carried out and the binding or synthesis of a nucleic acid on the now accessible reactive group.
- the cleavage of the lipid-modified nucleic acid from the carrier material (and optionally the removal of the protective groups on the nucleic acid) is typically carried out.
- another lipid can also be coupled to the 3 'end of the coupled nucleic acid, preferably according to one of the previously described steps.
- a linker as described above can be bound directly or indirectly to a solid phase via a first reactive group.
- An acid-stable protective group is then first bound to a second reactive group of the linker.
- a third reactive group of the linker may first be attached to a lipid as described above. Subsequently, the removal of the protective group on the third reactive group of the linker, the binding or synthesis of a nucleic acid on the now accessible reactive group as well as the cleavage of the lipid-modified nucleic acid from the carrier material (and optionally the removal of the protective groups on the nucleic acid) preferably likewise take place.
- the 3'-lipid modification, a lipid-modified nucleic acid can be via a linker having three reactive groups (a trifunctional anchor connection) based on a glycerol base body (C3 anchor) and having a monofunctional lipid, such as a palmityl residue, cholesterol or tocopherol.
- a linker having three reactive groups (a trifunctional anchor connection) based on a glycerol base body (C3 anchor) and having a monofunctional lipid, such as a palmityl residue, cholesterol or tocopherol.
- ß-isopropylidene-glycerol a glycerol containing a ketal protecting group
- ß-isopropylidene-glycerol a glycerol containing a ketal protecting group
- the ether linkage in the first step may be linked by another method, eg, by forming a tosylate of the ⁇ , ⁇ -isopropylidene glycerol, and reacting the tosylate with the reactive group of a lipid, eg, an acidic proton, to the corresponding ether.
- the ketal protecting group can be removed with an acid, eg, acetic acid, dilute hydrochloric acid, etc., and then the primary hydroxy group of the diol selectively protected by dimethoxytrityl chloride (DMT-CI).
- a lipid-modified nucleic acid can be obtained by using a (bifunctional) lipid, e.g. Polyethylene glycol (PEG) or hexaethylene glycol (HEG) without using a linker as described above.
- a bifunctional lipid typically have two functional groups as described above, preferably one end of the bifunctional lipid via a (coupling) molecule, e.g. a base-labile succinic anchor, etc., as described herein, can be attached to the support material and the nucleic acid can be synthesized onto the other end of the bifunctional lipid (E. Bayer, M. Maier, K. Bleicher, H.-J. Gauss.
- the bifunctional lipid used in the invention e.g. Polyethylene glycol
- a protecting group e.g. DMT
- esterification of the lipid protected with a reactive group with succinic anhydride under catalysis of DMAP to the succinate is usually carried out.
- the bifunctional lipid can be coupled to a support material and deprotected, followed by the synthesis of the nucleic acid according to a method as described above in a fourth step.
- the deprotection of the nucleic acid and the cleavage of the lipid-modified nucleic acid from the carrier material are optionally, after the deprotection of the nucleic acid and the cleavage of the lipid-modified nucleic acid from the carrier material.
- the lipid modification takes place at the 5 'end of the nucleic acid.
- the lipid modification typically takes place either after provision or after synthesis of the nucleic acid used according to the invention.
- the provision of the nucleic acid according to the invention can thereby - as defined above - be carried out via a direct synthesis of the nucleic acid or via the addition of an already completely synthesized or isolated from samples or commercially available nucleic acid.
- a synthesis of the nucleic acid is carried out, preferably analogously to the above, by methods known in the art for Nucleic acid synthesis, even more preferably by the phosphoramidite method (see, eg, FIG. 4).
- the lipid modification takes place at the 5 'end of the nucleic acid used according to the invention by means of particularly modified phosphoramidites following a phosphoramidite method for the synthesis of the nucleic acid.
- particularly modified phosphoramidites following a phosphoramidite method for the synthesis of the nucleic acid.
- Such relatively readily synthetically available amidites are usually coupled as the last monomer to a commercially available or to a finished synthesized nucleic acid. These reactions are characterized by a relatively fast reaction kinetics and very high coupling yields.
- the synthesis of the modified amidites is preferably accomplished by the reaction of a phosphoramidite, e.g.
- ⁇ -cyanoethylmonochlorophosphoramidite phosphorous acid mono- (2-cyanoethyl ester) -diisopropyl-amide chloride
- a suitable solvent e.g. in absolute dichloromethane, dissolved alcohol of a lipid as defined above, e.g. a lipid alcohol of tocopherol, cholesterol, hexadecanol, DMT-PEG, etc.
- the reaction solution is added as an acid scavenger DIPEA.
- phosphoramidites used for the synthesis of the 5'-lipid-modified nucleic acids according to the invention are relatively resistant to hydrolysis and can be purified by chromatography (before synthesis) with silica gel.
- the eluent is typically given a small amount of a weak base, e.g. Triethylamine added to avoid decomposition of the amidite. It is important that this base is completely removed from the product again to avoid poor coupling yields. This can e.g. by simple drying in vacuo, but preferably by purification of the phosphoramidites by their precipitation from tert-butyl I metyl ether with pentane.
- the lipid-modified amidites used have a very high viscosity, e.g. As a viscous oil, a (rapid) column chromatography can be carried out, which makes it possible to dispense with triethylamine as a base.
- a (rapid) column chromatography can be carried out, which makes it possible to dispense with triethylamine as a base.
- purification is typically not performed on PEG-modified amidites since they contain the acid-labile DMT protecting group.
- a solution of chlorinated hydrocarbons is therefore preferably used for the coupling reactions, for example a 0.1 M solution in (absolute) dichloromethane.
- dichloromethane requires some changes in the standard protocol of the synthesis cycle.
- lipid-modified amidites typically results in high coupling yields comparable to the coupling yield of amidites commonly used in the art.
- the kinetics of the reaction of lipid-modified amidites is generally slower.
- the coupling times are preferably (significantly) prolonged when using lipid-modified amidites.
- Such coupling times can be readily determined by one skilled in the art. Since it is possible to dispense with a capping step after the coupling, it is also possible, if necessary, to carry out a further synthesis cycle with the same lipid-modified amidite in order to increase the overall yield of the reaction. In this case, usually, e.g. in DMT-modified lipids such as DMT-PEG, the Detrityl michs Colour not executed.
- the phosphite triester via which the lipid is bound to the nucleic acid, can be oxidized by a sulfurizing agent.
- a sulfurizing agent for this purpose, preference is given to using such a sulfurizing agent as completely as possible to achieve the oxidation of the phosphotriester.
- the sulfurization reaction for example, for steric reasons, so incomplete that after ammoniacal cleavage and deprotection of the MON little or no product is obtained.
- the oxidation is carried out with iodine.
- a phosphodiester bond is introduced by the neighborhood of the However, lipid residue is not expected to be recognized as a substrate of nucleases.
- the linkers or (bifunctional) lipids present in the lipid-modified nucleic acid used according to the invention or optionally the nucleic acids used can, as described above, be coupled directly or indirectly to a carrier material.
- a direct coupling is preferably carried out directly with the carrier material, while an indirect coupling to the carrier material is typically carried out via a further (coupling) molecule.
- the bond formed by the coupling to a carrier material preferably has a (cleavable) covalent bond with the linker or bifunctional lipid and / or a (cleavable) covalent bond with the solid phase.
- Linkers, (bifunctional) lipids or optionally used nucleic acids, such as aminoalkyl residues (for example aminopropyl) or Aminohexanylreste carry a free amino function, can be bound via a Phtalimid linker to the carrier material.
- Thiol-containing linkers, (bifunctional) lipids or optionally used nucleic acids can be bound as a disulfide to the carrier material.
- the succinates of the described linkers or bifunctional lipids used according to the invention may be coupled to a support material with TBTU / NMM (1H-benzotriazol-1-yl-1,1,3,3-tetramethyluronium tetrafluoroborate / N-methylmorpholine).
- PS-PEG support materials in the 1 ⁇ mol scale usually used achieve the best results with loadings of between 50 and 100 ⁇ mol / g (E. Bayer, K. Bleicher, M. Maier Z. Naturforsch., 50b (1995) 1096).
- the loading of the support materials is preferably as high as possible (> 100 ⁇ mol). According to the invention, such a process also leads to good coupling yields (M. Gerster, M. Maier, N. Clausen, J. Schewitz, E. Bayer Z.
- support materials such as resins with up to 138 .mu.mol / g loading or possibly more with good Synthesis yields are used. Since the coupling yields with the linkers or bifunctional lipids described above are approximately 100%, the stoichiometry of these compounds allows the loading of the carrier material to be adjusted relatively precisely.
- the control of the loading is preferably carried out by the spectroscopic quantification of the cleaved DMT protective group (see experimental part).
- the residual amino functions still present on the support material can be capped with acetic anhydride. This capping is usually carried out after the occupancy of the carrier material, but can also be carried out directly in the nucleic acid synthesis, for example on the DNA synthesizer.
- Dioxane / water 9: 1) after the usually necessary oxidation step (to oxidize the phosphite triester) during the amidite process to remove traces of iodine.
- the immunostimulatory adjuvant of the invention may be combined with other adjuvants known in the art.
- the present invention also relates to pharmaceutical compositions comprising an immunostimulatory adjuvant as described above, at least one active ingredient and optionally a pharmaceutically suitable carrier and / or further auxiliaries and additives and / or adjuvants.
- compositions according to the present invention typically comprise a safe and effective amount of the immunostimulatory adjuvant of the invention.
- safe and effective amount means an amount of a compound that is sufficient to significantly induce a positive modification of a condition to be treated, eg, a tumor or infectious disease.
- a "safe and effective amount” is small enough to avoid serious side effects, allowing a reasonable balance of benefit and risk. The definition of these limits is typically within the scope of reasonable medical judgment.
- the term "safe and effective amount” preferably means that amount which is capable of stimulating the immune system in such a way that no excessive or detrimental immune responses are obtained, but preferably also no such immune reactions Below a measurable level
- a "safe and effective amount" of an adjuvant of the invention will vary depending on the particular condition being treated, the age and physical condition of the patient to be treated, the severity of the condition, duration the treatment, the nature of the accompanying therapy, the used particular pharmaceutically-acceptable carrier and like factors within the knowledge and experience of the attendant physician.
- the pharmaceutical compositions of the invention can be used according to the invention for human and veterinary purposes.
- the pharmaceutical composition according to the invention preferably contains at least one active substance.
- An active ingredient in this context is a compound which has a therapeutic effect against a particular indication, preferably cancer or infectious diseases.
- Such compounds include, but are not limited to, peptides, proteins, nucleic acids, (therapeutically-effective) low molecular weight organic or inorganic compounds (molecular weight less than 5,000, preferably less than 1,000), sugars, antigens or antibodies already known in the art Therapeutics, etc.
- the immunostimulatory adjuvant according to the invention described above may itself be an active ingredient.
- the lipid of the lipid-modified nucleic acid is a therapeutically active molecule, e.g. a vitamin as described above, or steroid, for example ⁇ -tocopherol (vitamin E), D- ⁇ -tocopherol, L- ⁇ -tocopherol, D, L- ⁇ -tocopherol, vitamin E succinate (VES), vitamins A and its derivatives, vitamin D and its derivatives, vitamin K and its derivatives, etc .
- the active ingredient contained in the pharmaceutical composition according to the invention is present as an antigen or as an immunogen.
- An "antigen” as well as an “immunogen” is understood to mean any structure which can cause the formation of antibodies and / or the activation of a cellular immune response. Therefore, according to the invention, the terms “antigen” and “immunogen” are used interchangeably.
- antigens are peptides, polypeptides, including proteins, cells, cell extracts, polysaccharides, polysaccharide conjugates, lipids, glycolipids and carbohydrates.
- antigens are, for example, tumor antigens, viral, bacterial, fungal and protozoological antigens into consideration.
- the antigen can for example in a vaccine according to the invention, also in the form of a hapten coupled to a suitable carrier.
- the active ingredient contained in the pharmaceutical composition according to the invention may be present according to a second embodiment as an antibody.
- any therapeutically suitable antibody can be used.
- Particularly preferred according to the invention is an antibody which is directed against antigens, proteins or nucleic acids which have an essential role in cancers or infectious diseases, e.g. Cell surface proteins, tumor suppressor genes or their inhibitors, growth and elongation factors, apoptosis-relevant proteins, tumor antigens or antigens as described above, etc.
- the active ingredient contained in the pharmaceutical composition according to the invention is present as nucleic acid.
- nucleic acid may be single-stranded or double-stranded, and as homo- or
- a nucleic acid contained as an active ingredient in the pharmaceutical composition is not limited in its length and may comprise any naturally occurring nucleic acid sequence or its complement or a fragment thereof. Also in this
- nucleic acid be partially or completely synthetic nature.
- nucleic acid may comprise such a nucleic acid which comprises
- an antigen is preferably an antigen as described above.
- the nucleic acid contained as active ingredient in the pharmaceutical composition according to the invention is present as mRNA.
- mRNA can be added to the pharmaceutical composition according to the invention in its naked form or in a stabilized form which reduces or even prevents the degradation of the nucleic acid in vivo, for example by exo- and / or endonucleases.
- the mRNA contained as an active ingredient in the pharmaceutical composition according to the invention with a 5'-cap and / or a poly-A tail as defined above at the 3'-end of at least 50 nucleotides, preferably at least 70 nucleotides, more preferably at least 100 nucleotides , more preferably at least 200 nucleotides are stabilized.
- the terminal structure is crucial in vivo. Through these structures, the RNA is recognized as mRNA and regulated the degradation.
- there are other processes that stabilize or destabilize RNA Many of these processes are still unknown, but often an interaction between RNA and proteins seems to be crucial. For example. Recently, an "mRNA surveillance system" has been described (Hellerin and Parker, Ann. Rev. Genet., 1999, 33: 229-260), in which incomplete or nonsense mRNA is recognized by certain feedback protein interactions in the cytosol, and the Degradation, with a major portion of these processes being performed by exonucleases.
- the stabilization of the mRNA contained in the pharmaceutical composition according to the invention by the fact that the mRNA with a cationic compound, in particular a polycationic compound, for example.
- a cationic compound in particular a polycationic compound, for example.
- a (poly) cationic peptide or protein is associated or complexed or bound thereto.
- protamine, nucleolin, spermine or spermidine as a polycationic, nucleic acid-binding protein is particularly effective.
- other cationic peptides or proteins such as poly-L-lysine or histones, is also possible. This procedure for the stabilization of mRNA is described in EP-A-1083232, the relevant disclosure content of which is fully included in the present invention.
- cationic polysaccharides eg chitosan, polybrene, polyethylenimine (PEI) or poly-L-lysine (PLL), etc.
- PKI polyethylenimine
- PLL poly-L-lysine
- Another possibility for stabilizing mRNA which may be present as an active ingredient in the pharmaceutical composition according to the invention, is the targeted modification of the sequence of the mRNA by removing or altering so-called destabilizing sequence elements (DSE).
- DSE destabilizing sequence elements
- Signaling proteins can bind to these destabilizing sequence elements (DSE), which occur in particular in the case of eukaryotic mRNA, and regulate the enzymatic degradation of the mRNA in vivo. Therefore, for further stabilization of the mRNA contained as active ingredient, one or more changes are preferably made to the corresponding region of the wild-type mRNA, so that no destabilizing sequence elements are contained.
- AURES AU-rich sequences
- the mRNA used as the active ingredient is therefore preferably modified in such a way with respect to the wild-type mRNA that it has no such destabilizing sequences.
- sequence motifs which are recognized by possible endonucleases, for example the sequence GAACAAG, which is contained in the 3 'UTR segment of the gene coding for the transferin receptor (Binder et al., EMBO J. 1994, 13: 1969 to 1980).
- sequence motifs are also eliminated from the lipid-modified nucleic acid according to the invention.
- the mRNA optionally present as an active ingredient in the pharmaceutical composition according to the invention may further be e.g. for an optionally desired efficient translation, be changed so that an effective binding of the ribosomes to the ribosome binding site (Kozak sequence: GCCGCCACCAUGG, the AUG forms the start codon) takes place.
- Kozak sequence: GCCGCCACCAUGG the AUG forms the start codon
- IRES internal ribosomal entry side
- An IRES can thus act as the sole ribosome binding site, but it can also serve to provide an mRNA encoding several peptides or polypeptides that are to be translated independently by the ribosomes ("multicistronic mRNA").
- IRES sequences which can be used according to the invention are those from picornaviruses (eg FMDV), pestiviruses (CFFV), polioviruses (PV), encephalococytitis viruses (ECMV), foot-and-mouth disease viruses (FMDV), hepatitis C viruses ( HCV), classical swine fever virus (CSFV), murine leucoma virus (MLV), simean immunodeficiency virus (SIV) or cricket paralysis virus (CrPV).
- picornaviruses eg FMDV
- CFFV pestiviruses
- PV polioviruses
- ECMV encephalococytitis viruses
- FMDV foot-and-mouth disease viruses
- HCV hepatitis C viruses
- CSFV classical swine fever virus
- MMV murine leucoma virus
- SIV simean immunodeficiency virus
- CrPV cricket paralysis virus
- the mRNA optionally used as the active ingredient in the pharmaceutical composition according to the invention may also have stabilizing sequences in its 5 'and / or 3' untranslated regions which are capable of increasing the half-life of the mRNA in the cytosol.
- These stabilizing sequences may have 100% sequence homology to naturally occurring sequences found in viruses, bacteria and eukaryotes, but may also be partially or wholly synthetic.
- the untranslated sequences (UTR) of the ⁇ -globin gene for example of Homo sapiens or Xenopus laevis, may be mentioned.
- a stabilization sequence has the general formula (C / U) CCAN x CCC (U / A) Py x UC (C / U) CC contained in the 3 1 UTR of the very stable mRNA coding for ⁇ -globin , ⁇ - (I) collagen, 15-lipoxygenase or tyrosine hydroxylase (see Holcik et al., Proc. Natl. Acad., USA, 1997, 94: 2410-2414).
- stabilizing sequences may be used alone or in combination with each other as well as in combination with other stabilizing sequences known to one skilled in the art.
- the mRNA used as the active ingredient may have the following modifications to a corresponding wild-type mRNA, which may be present either alternatively or in combination.
- the G / C content of the peptide or polypeptide coding region of the modified mRNA may be greater than the G / C content of the coding region of the wild-type mRNA encoding the peptide or polypeptide, the encoded amino acid sequence being wild-type unchanged.
- This modification is based on the fact that the sequence of sequence of the mRNA to be translated is essential for efficient translation of an mRNA.
- the composition and the sequence of the various nucleotides play a major role here.
- sequences with elevated G (guanosine) / C (cytosine) content more stable than sequences with increased A (adenosine) / U (uracil) content. Therefore, according to the invention, while maintaining the translated amino acid sequence, the codons are varied with respect to the wild-type mRNA in such a way that they increasingly contain G / C nucleotides. Since several codons code for one and the same amino acid (degeneracy of the genetic code), the most favorable for the stability codons can be determined (alternative codon usage, English, "codon usage"). Depending on the amino acid to be coded by the mRNA, different possibilities for modifying the mRNA sequence compared to the wild-type sequence are possible.
- codons containing only G or C nucleotides no modification of the codon is required.
- the codons for Pro (CCC or CCG), Arg (CGC or CGG), Ala (GCC or GCG), and Gly (GGC or GGG) require no change since there is no A or U present.
- the codons containing A and / or U nucleotides are altered by substituting other codons which encode the same amino acids but do not contain A and / or U.
- the codons for Pro can be changed from CCU or CCA to CCC or CCG; the codons for Arg can be changed from CGU or CGA or AGA or AGG to CGC or CGG; the codons for AIa can be changed from GCU or GCA to GCC or GCG; the codons for GIy can be changed from GGU or GGA to GGC or GGG. While in other cases A or U nucleotides can not be eliminated from the codons, it is possible to reduce the A and U content by using codons containing less A and / or U nucleotides.
- the codons for Phe can be changed from UUU to UUC; the codons for Leu can be changed from UUA, CUU or CUA to CUC or CUG; the codons for Ser can be changed from UCU or UCA or AGU to UCC, UCG or AGC; the codon for Tyr can be changed from UAU to UAC; the stop codon UAA can be changed to UAG or UGA; the codon for Cys can be changed from UGU to UGC; the codon His can be changed from CAU to CAC; the codon for GIn can be changed from CAA to CAG; the codons for He can be changed from AUU or AUA to AUC; the codons for Thr can be changed from ACU or ACA to ACC or ACG; the codon for Asn can be changed from AAU to AAC; the codon for Lys can be changed from AAA to AAG; the codons for VaI can be changed from GUU or GUA to GUC or GUG; the codon for VaI
- the G / C content of the peptide or polypeptide coding region (or any other optional portion) of the mRNA is at least 7%, more preferably at least 15%, more preferred increased by at least 20% points over the G / C content of the encoded region of the wild-type mRNA encoding the corresponding peptide or polypeptide. It is particularly preferred in this Related to maximally increase the G / C content of the thus modified mRNA compared to the wild-type sequence.
- Another preferred modification of an mRNA used as an active ingredient in the pharmaceutical composition is based on the finding that the translation efficiency is also determined by a different frequency in the occurrence of tRNAs in cells. Therefore, if so-called "rare" codons are increasingly present in an RNA sequence, the corresponding mRNA is significantly worse translated than in the case where codons coding for relatively "frequent" tRNAs are present.
- the coding region is changed relative to the corresponding region of the wild-type mRNA in such a way that at least one codon of the wild-type sequence which codes for a relatively rare tRNA in the cell is exchanged for a codon, which codes for a relatively frequent in the cell tRNA, which carries the same amino acid as the relatively rare tRNA.
- This modification modifies the RNA sequences to insert codons for which common tRNAs are available. Which tRNAs occur relatively frequently in the cell and which, in contrast, are relatively rare, is known to a person skilled in the art; see. eg Akashi, Curr. Opin. Genet. Dev.
- all codons of the wild-type sequence which code for a relatively rare tRNA in the cell can each be exchanged for a codon which codes for a relatively frequent tRNA in the cell, which carries the same amino acid as the relative one rare tRNA. It is particularly preferred to link the, in particular the maximum, sequential G / C portion which is increased in the above-described mRNA with the "frequent" codons, without the amino acid sequence of an antigenic peptide or polypeptide encoded by the coding region of the mRNA. one or more).
- the nucleic acid contained as active ingredient in the pharmaceutical composition according to the invention is present as dsRNA, preferably as siRNA.
- dsRNA especially an siRNA
- a dsRNA, especially an siRNA is of particular interest in connection with the phenomenon of RNA interference. The phenomenon of RNA interference was investigated in the course of immunological analysis Research carefully.
- RNA-mediated virus resistance in plants RNA-mediated virus resistance in plants
- PTGS post-transcriptional gene silencing
- RNA interference in eukaryotes are therefore based on a common mode of operation
- RNA interference is based on double-stranded RNA molecules (dsRNA) that trigger the sequence-specific suppression of gene expression (Zamore (2001) Nat. Struct. Biol. 9: 746-750; Sharp (2001) Genes Dev.
- dsRNA molecules have also been used in vivo (McCaffrey et al (2002), Nature 418: 38-39, Xia et al (2002) ), Nature Biotech 20: 1006-1010, Brummelkamp et al (2002), Cancer Cell 2: 243-247).
- the double-stranded RNA (dsRNA) used as the active ingredient therefore preferably contains a sequence with the general structure 5 '- (N 17, 29 ) -3', where N is any base and represents nucleotides.
- the general structure is composed of a double-stranded RNA with a macromolecule composed of ribonucleotides, the ribonucleotide consisting of a pentose (ribose), an organic base and a phosphate.
- the organic bases in the RNA consist of the purine bases adenine (A) and guanine (G) as well as the pyrimidine bases cytosine (C) and uracil (U).
- the dsRNA used according to the invention as active ingredient contains such nucleotides or nucleotide analogues with a directed structure.
- dsRNA the general structure 5 '- (. N 19 2s) -3', more preferably 5 '- (. N 19 24) -3', even more preferably 5 '- (N 21 23). -3 ', where N is any base.
- N is any base.
- 90% complementary means that, given a length of a dsRNA used according to the invention of, for example, 20 nucleotides, it has at most 2 nucleotides without corresponding complementarity with the corresponding segment on the (m) RNA.
- the sequence of the double-stranded RNA used according to the invention with its general structure is preferably but completely complementary to a section of the (m) RNA of a previously described as active ingredient protein or antigen.
- dsRNAs used as active ingredient may also be directed against nucleotide sequences of a previously (as active ingredient) described (therapeutically relevant) protein or antigen that is not in the coding region, in particular in the non-coding 5 'region of the (m) RNA, eg ie against non-coding regions of the (m) RNA with regulatory function.
- the target sequence of the dsRNA used as an active ingredient of a previously described protein or antigen can thus lie in the translated and untranslated region of the (m) RNA and / or in the region of the control elements.
- the target sequence of a dsRNA used as the drug may be in the overlap region of untranslated and translated sequence, more particularly, the target sequence may comprise at least one nucleotide upstream of the starting triplet of the coding region of the (m) RNA.
- a modified nucleotide in the case of a dsRNA contained as active ingredient in the pharmaceutical composition according to the invention, a modified nucleotide can preferably occur.
- the term "modified nucleotide” means that the particular nucleotide is chemically modified.
- the term “chemical modification” means that the modified nucleotide is replaced, added or removed by one or more atoms or atomic groups in comparison to naturally occurring nucleotides is changed.
- At least one modified nucleotide in dsRNA used according to the invention serves, on the one hand, for stability and, on the other hand, for preventing dissociation.
- between 2 and 10 preferably between 2 and 5 nucleotides are modified in a dsRNA used according to the invention.
- At least one 2'-hydroxy group of the nucleotides of the dsRNA in the double-stranded structure is replaced by a chemical group, preferably a 2'-amino or a 2'-methyl group.
- At least one nucleotide in at least one strand of the double-stranded structure may also be a so-called "locked nucleotide” having a chemically modified sugar ring, preferably by a 2'-O, 4'-C-methylene bridge
- several nucleotides of the dsRNA used according to the invention are " locked nucleotides ".
- by modifying the backbone of a dsRNA used according to the invention premature degradation thereof can be prevented.
- dsRNA which is modified in the form of phosphorothioate, 2'-O-methyl RNA, LNA, LNA / DNA gapers, etc. and therefore has a longer half-life in vivo.
- the ends of the double-stranded RNA (dsRNA) employed as the active ingredient in the pharmaceutical composition of the invention may be modified to counteract degradation in the cell or dissociation into the single strands, particularly to avoid premature degradation by nucleases.
- a regularly unwanted dissociation of the single strands of dsRNA occurs especially when using low concentrations of the same or short chain lengths.
- the cohesion of the double-stranded structure of dsRNA used by the nucleotide pairs can be increased by at least one, preferably more chemical linkage (s).
- a dsRNA used as an active ingredient according to the invention whose dissociation is reduced, has a higher stability against enzymatic and chemical degradation in the cell or in the organism ⁇ in vivo) or ex vivo and therefore has a higher half-life.
- a further possibility for preventing premature dissociation of dsRNA used according to the invention in the cell is that hairpin loops (n) can be formed at the ends of the strands.
- a dsRNA used according to the invention therefore has a hairpin structure in order to slow down the dissociation kinetics.
- a loop structure is preferably formed at the 5 'and / or 3' end.
- Such a loop structure does not have hydrogen bonds, typically no complementarity, between nucleotide bases.
- such a "loop” has a length of at least 5, preferably at least 7 nucleotides and connects in this way the two complementary single strands of a dsRNA used in the invention.
- the nucleotides of the two strands of the dsRNA used according to the invention in order to prevent dissociation of the strands, may also be modified in such a way that amplification of the hydrogen bond is achieved, for example by increasing the hydrogen bonding capacity between the bases by optionally modified nucleotides. This increases the stability of the interaction between the strands and protects the dsRNA against attack by RNAses.
- the dsRNA used as active ingredient in the pharmaceutical composition according to the invention is directed against the (m) RNA of a protein or antigen as described above.
- the dsRNA used herein suppresses translation of a previously described protein or antigen in a cell to at least 50%, more preferably 60%, even more preferably 70%, and most preferably at least 90%, i. the cell preferably contains at most half of the native cellular concentration of a previously described protein or antigen (without treatment with dsRNA used according to the invention).
- the suppression of the translation of these proteins or antigens into cells after the addition of dsRNA molecules used according to the invention is based on the phenomenon of RNA interference induced by such molecules.
- the dsRNA used according to the invention is then so-called siRNA, which can trigger the phenomenon of RNA interference and bind the (m) RNA of a previously described protein or antigen.
- the measurement or detection of translational suppression in cells triggered by the dsRNA used according to the invention can be carried out by Northern blot, quantitative real-time PCR or at the protein level with specific antibodies to a previously described protein or antigen.
- the dsRNA optionally used as active ingredient in the pharmaceutical composition according to the invention and a corresponding siRNA can be prepared by methods known to the person skilled in the art.
- the pharmaceutical composition of the invention may additionally contain one or more excipients.
- excipients preferably a synergistic effect of the immunostimulatory adjuvant according to the invention and optionally in addition in the pharmaceutical composition contained adjuvant and / or optionally a drug as described above.
- various mechanisms may be considered. For example. Compounds which allow the maturation of dendritic cells (DC), for example lipopolysaccharides, TNF- ⁇ or CD40 ligand, form a first class of suitable excipients.
- DC dendritic cells
- any "danger signal” (LPS, GP96, etc.) type of agent which affects the immune system or cytokines, such as GM-CFS, can be used as adjuvant, which make it possible to intensify an immune response generated by the immunostimulatory adjuvant according to the invention and / or directed to influence.
- cytokines such as monokines, lymphokines, interleukins or chemokines, eg IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL- 9, IL-I O 7 IL-12, INF- ⁇ , INF- ⁇ , GM-CFS, M-CSF, G-CSF, LT- ⁇ , TNF- ⁇ , or interferons, for example IFN- ⁇ , or growth factors , eg hGH.
- cytokines such as monokines, lymphokines, interleukins or chemokines, eg IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL- 9, IL-I O 7 IL-12, INF- ⁇ , INF- ⁇ , GM-CFS, M-CSF, G-CSF, LT- ⁇ , TNF- ⁇ , or interferons, for example
- the pharmaceutical composition of the invention may further contain an adjuvant known in the art.
- adjuvants known in the art include, but are not limited to, aluminum hydroxide, Freund's (complete or incomplete) adjuvant, and stabilizing cationic peptides or polypeptides such as protamine, nucleolin, spermine or spermidine previously described and cationic polysaccharides, especially chitosan, TDM, MDP, muramyl dipeptide, alum solution, Pluronics, etc.
- lipopeptides, such as Pam3Cys are also particularly suitable to be combined with the immunostimulatory adjuvant of the invention (see Deres et al, Nature 1989, 342: 561-564).
- the pharmaceutical composition of the invention may contain a pharmaceutically acceptable carrier.
- pharmaceutically acceptable carrier as used herein preferably includes one or more compatible solid or liquid fillers, diluents or encapsulating compounds which are suitable for administration to a subject.
- compatible as used herein means that the components of the composition are capable of being mixed together with the active ingredient, the adjuvant itself, and each other in such a manner that no interaction will occur which would significantly reduce the pharmaceutical effectiveness of the composition under ordinary conditions of use.
- pharmaceutically acceptable carriers must have sufficiently high purity and sufficiently low toxicity to render them suitable for administration to a subject to be treated.
- Some examples of compounds which may serve as pharmaceutically acceptable carriers or components thereof are sugars such as lactose, glucose and sucrose; Starches such as corn starch or potato starch; Cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose, cellulose acetate; powdered tragacanth; Malt; Gelatin; Tallow; solid lubricants such as stearic acid, magnesium stearate; Calcium sulfate; vegetable oils such as peanut oil, cottonseed oil, sesame oil, olive oil, corn oil and oil from theobromine; Polyols such as polypropylene glycol, glycerine, sorbitol, mannitol and polyethylene glycol; alginic acid; Emulsifiers, such as Tween ®; Wetting agents, such as sodium lauryl sulfate; coloring agents; taste-promoting agents, excipients; tablet-forming agents; stabilizers; antioxidants; Preservatives; pyrogen-free water
- a pharmaceutically acceptable carrier is principally determined by the manner in which the pharmaceutical compositions of the invention are administered.
- the pharmaceutical compositions according to the invention can be administered systemically, for example. Routes for administration include, for example, transdermal, oral, parenteral, including subcutaneous or intravenous injections, topical and / or intranasal routes.
- the appropriate amount of the pharmaceutical composition to be used can be determined by routine experimentation with animal models. Such models include, but are not limited to, models of rabbit, sheep, mouse, rat, dog and non-human primate models.
- Preferred unit dosage forms for injection include sterile solutions of water, physiological saline or mixtures thereof. The pH of such solutions should be adjusted to about 7.4.
- Suitable carriers for injection include hydrogels, controlled or sustained release devices, polylactic acid, and collagen matrices.
- Suitable pharmaceutically acceptable carriers for topical use include those suitable for use in lotions, creams, gels and the like are suitable. If the compound is to be administered orally, tablets, capsules and the like are the preferred unit dosage form.
- the pharmaceutically acceptable carriers for the preparation of unit dosage forms useful for oral administration are well known in the art. Their selection will depend on secondary considerations such as taste, cost and shelf life, which are not critical to the purposes of the present invention, and can be readily made by one skilled in the art.
- the pharmaceutical composition may also be present as a vaccine.
- vaccines according to the invention typically comprise a composition as described above for pharmaceutical composition, the composition of such vaccines according to the invention being determined, in particular, by the way in which they are administered.
- vaccines according to the invention are administered systemically.
- Routes for administering such vaccines typically include transdermal, oral, parenteral, including subcutaneous or intravenous injections, topical and / or intranasal routes. Therefore, vaccines are preferably formulated in liquid or solid form. It is also possible, if appropriate, for further excipients to be incorporated into a vaccine according to the invention which can further increase the immunogenicity of the vaccine.
- one or more other such adjuvants as previously defined will have to be selected depending on the immunogenicity and other properties of the active ingredient in the vaccine according to the invention.
- the pharmaceutical compositions according to the invention are used for the treatment of exemplary indications mentioned below.
- Pharmaceutical compositions according to the invention can be used to treat, for example, those diseases or conditions which are linked to various pathologically absent immune responses or which require an immune response, preferably an enhanced immune response, in the context of a therapy. Preference is given to using pharmaceutical compositions or vaccines according to the invention, to trigger tumor-specific or pathogen-specific immune responses.
- Particularly preferred such pharmaceutical compositions or vaccines of the present invention can be used to enhance immune responses of antigen presenting cells (APC).
- APC antigen presenting cells
- the pharmaceutical compositions or vaccines according to the invention can be used for the treatment of cancer or tumor diseases, preferably selected from colon carcinomas, melanomas, renal carcinomas, lymphomas, acute myeloid leukemia (AML), acute lymphoid leukemia (ALL), chronic myeloid leukemia (CML ), chronic lymphocytic leukemia (CLL), gastrointestinal tumors, lung carcinomas, gliomas, thyroid tumors, breast carcinomas, prostate tumors, hepatomas, various virus-induced tumors such as papillomavirus-induced carcinomas (eg cervical carcinomas), adenocarcinomas, herpesvirus-induced tumors (eg Burkitt's Lymphoma, EBV-induced B cell lymphoma), hepatitis B-induced tumors (hepatocellular carcinoma), HTLV-1 and HTLV-2 induced lymphoma, acoustic neuroma, cervical cancer, lung cancer, pharyngeal cancer, anal carcinoma
- Thyroid carcinoma Bladder cancer, Hodgkin syndrome, Meningioma, Schneeberger disease, Bronchial carcinoma, Pituitary tumor, Mycosis fungoides, Esophageal cancer, Breast cancer, Carcinoids, Neurinomas, Spinaliomas, Burkitt's lymphoma, Laryngeal cancer, Kidney cancer, Thymoma, Corpus carcinoma, Bone cancer, Non-Hodgkin's lymphoma, Urethral cancer, CUP, head and neck, oligodendroglioma, vulvar, colon, colon, oesophageal, warts, small intestine, craniopharyngioma, ovarian, soft tissue, ovarian, liver, pancreatic, cervical,
- ⁇ -tocopherol (vitamin E)
- D- ⁇ -tocopherol, L- ⁇ -tocopherol, D, L- ⁇ -tocopherol or vitamin E succinate (VES)
- ⁇ -tocopherol (vitamin E) is low in toxicity and has a potent anti-tumor effect (A. Bendich, LJ Machlin Am J. Clin Nutr 48 (1988) 612), what of its in cancer therapy looks promising.
- vitamin E is a potent antioxidant and a good free radical scavenger (C. Borek Ann, NY Acad., 570 (1990) 417
- it can prevent tumor growth by stimulating the immune response (G. Shklar, J. Schwartz, DP Trickler, S. Reid J. Oral Pathol., Med. 19 (1990) 60).
- an association between the expression of the tumor suppressor gene p53 in tumor cells (oral squamous cance) and a treatment with vitamin E succinate (VES) was furthermore found (J Schwartz, G. Shklar, D. Trickler Oral Oncol. Europ.
- vitamin E is the RRR- ⁇ -tocopherol (formerly D- ⁇ -tocopherol), but nowadays the racemate (D, L- ⁇ -tocopherol) is predominantly used. All forms of vitamin E mentioned hereinbefore are likewise included as lipid in the sense of the present invention.
- infectious diseases are preferably selected from influenza, malaria, SARS, yellow fever, AIDS, Lyme disease, Leishmaniasis, anthrax, meningitis, viral infectious diseases such as AIDS, condyloma acuminata, mollusc warts, dengue fever, three-day fever, Ebola virus, common cold, tick-borne encephalitis (TBE), influenza, shingles, hepatitis, herpes simplex Type I, Herpes simplex type II, Herpes zoster, Influenza, Japanese encephalitis, Lassa fever, Marburg virus, measles, foot-and-mouth disease, mononucleosis, mumps, Norwalk virus infection, Pfeifer's glandular fever, smallpox, polio (polio) , Pseudo-croup, ringworm, rabies, warts, West Nile fever, chickenpo
- Another object of the invention relates to the use of an immunostimulatory adjuvant according to the invention for the preparation of a pharmaceutical composition according to the invention or a vaccine according to the invention for the treatment of previously described indications, for example for the treatment of said tumor or infectious diseases.
- the invention includes the (therapeutic) Use of an immunostimulatory adjuvant according to the invention for the treatment of tumor or infectious diseases, as described above.
- kits comprising an immunostimulatory adjuvant according to the invention and / or a pharmaceutical composition according to the invention and / or a vaccine according to the invention and optionally a technical instruction manual with instructions for administration and
- Fig. 1 shows various possibilities according to the invention of the final modification of
- Nucleic acids with lipids Shown in particular are the lipid-modified linker or bifunctional peptides which are used for coupling or
- Synthesis with nucleic acid sequences (in short: ODN sequence) can be used.
- Figure 2 exemplifies a synthetic route for (trifunctional) lipid-modified linkers with e.g. a tocopherol modification to the 3 'end of a
- Nucleic acid can be introduced. Such exemplified compounds represent an intermediate in the preparation of the 5'- or 3'-lipid-modified nucleic acids according to the invention and the adjuvants according to the invention.
- Fig. 3 shows by way of example a bifunctional lipid with a succinyl anchor, the one
- Fig. 5A, B describe the stimulation of human PBMCs with immunostimulatory adjuvants according to the invention as well as with different RNA oligonucleotides. A) It is especially in the release of cytokines
- IL-G immunostimulatory adjuvants according to the invention without the addition of protamine have a more than 5-fold increase in cytokine secretion (IL-6) against the medium as well as in Addition of protamine to ⁇ -galactosidase and RNA oligo 40 alone (SEQ ID NO: 40) slightly improved release of IL-6.
- FIG. 6 shows the release of TNF- ⁇ by human PBMC cells after stimulation with RNA oligonucleotides used according to the invention as well as immunostimulatory adjuvants according to the invention.
- Fig. 6 shows in particular that immunostimulatory adjuvants according to the invention in
- RNA Oligo 40 Form of a lipid-modified nucleic acid containing z, B; one of the sequences SEQ ID NO: 40, 41 or 42, compared to eg an unmodified RNA oligonucleotide of the sequence according to SEQ ID NO: 40 (RNA Oligo 40) a significantly improved release of TNF- ⁇ and thus a significantly improved immune stimulation exhibit.
- RNA Oligo 40 a significantly improved release of TNF- ⁇ and thus a significantly improved immune stimulation exhibit.
- the best results with an increase in the immune stimulation by more than 10 times compared to the unmodified RNA oligonucleotide could be achieved with a tocopherol-modified sequence according to SEQ ID NO: 42 (RNA Oligo Toc CV2).
- PEG can also be detected with Dragendorff Citizen Spray Reagent.
- the solvent is removed and the product taken up in 50 ml of DCM.
- the organic phase is washed twice with 25 ml of 5% NaHCOj solution and twice with 25 ml H 2 O each time.
- the phase separation between aqueous and organic phase is tedious, since PEG has both hydrophobic and hydrophilic character.
- the following procedure is applicable to both DMT-PEG 1500 and DMT-HEG.
- reaction mixture is allowed to stir at RT for 1 h before the solution is diluted with 50 ml of ethyl acetate / TEA (20: 1).
- the organic phase is washed twice with 15 ml of a 10% NaHCO 3 solution and twice with a saturated NaCl solution and then dried over Na 2 SO 4 .
- the solvent is removed and the crude product by column chromatography on silica gel with
- Dimethoxytrityl chloride (dissolved in 50 ml of pyridine) slowly added dropwise and stirred for 24 h at RT. Thereafter, the reaction is stopped with 5 ml of methanol and the
- Nucleic acid containing a sequence according to SEQ ID NO: 40, 41 or 42 co-incubated with human cells.
- human PBMC cells were enriched with RNA in X-vivo15 medium (BioWhittaker) supplemented with 2 mM L-glutamine (BioWhittaker), 10 U / ml penicillin (BioWhittaker) and 10 ⁇ g / ml streptomycin 16h with 10 ⁇ g / ml RNA (mRNA encoding ⁇ -
- Galactosidase or phosphorothioate RNA oligonucleotide 40 5'GCCCGUCUGUUGUGUGACUC (SEQ ID NO: 40)) and optionally co-incubated with 10 ⁇ g / ml protamine. The supernatants were collected and analyzed by ELISA.
- cytokine release IL-6
- IL-6 cytokine release
- SEQ ID NO: 40 protamine ⁇ -galactosidase and RNA oligo 40 alone
- RNA oligonucleotides and adjuvants of the invention are provided.
- PBMC cells with 10 ug / ml RNA oligonucleotides in X-vivo15 medium (BioWhittaker), enriched with 2 mM L-glutamine (BioWhittaker), 10 U / ml penicillin (BioWhittaker) and 10 ug / ml streptomycin
- RNA oligos used here by way of example for the lipid modification carried the following sequences: RNA 40: 5 'GCCCGUGUGUGUGUGACUC (SEQ ID NO: 40), RNA CV1: GGUAAGUGUAAGGUGUAAGG (SEQ ID NO: 41), RNA CV2: AAGGGAUAUGGAAUAUGGAA (SEQ ID NO: 42); The supernatants were collected and analyzed by ELISA.
- each adjuvant in the form of a lipid-modified nucleic acid for example: a sequence according to SEQ ID NO: 40, 41 or 42, compared to eg an unmodified RNA OI oligonucleotide of the sequence according to SEQ ID NO : 40 (RNA Oligo 40) (or 41 or 42) significantly improved release of TNF- ⁇ and thus has a significantly improved immune stimulation.
- RNA Oligo 40 compared to eg an unmodified RNA OI oligonucleotide of the sequence according to SEQ ID NO : 40
- RNA Oligo 40 or 41 or 42
- the best results with an increase in the immune stimulation by more than 10 times compared with the unmodified RNA oligonucleotide could be achieved with a lipid-modified sequence according to SEQ ID NO: 42 (see FIG.
- the immunostimulatory adjuvant according to the invention in the form of a lipid-modified nucleic acid, in particular in the form of a 5'- and / or 3'-lipid-modified nucleic acid, on the one hand enables stabilization and better cell permeability of (pharmacological) active ingredients and a better distribution of these active ingredients within the cell.
- the immunostimulatory adjuvant according to the invention itself can produce an increase in the immune reaction as a biological effect. On the one hand, this can be achieved by supporting the actual antisense mechanism, e.g.
- the invention e.g. discloses a 3'-cholesterol-modified phosphodiester oligonucleotide as an immunostimulatory adjuvant which can be cytotoxic to specific tumor cells.
- inventive 3'-cholesterol-modified phosphodiester oligonucleotide can also act sequence-specific, but not by antisense effects.
- the receptor-mediated endocytosis described with cholesterol does not contribute significantly to the unusual immunostimulating effect found.
- the immunostimulatory adjuvant according to the invention exhibits an advantageous immunostimulating effect and at the same time increases the (cell) permeability of optionally additionally contained active substances.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Communicable Diseases (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Die vorliegende Erfindung betrifft ein immunstimulierendes Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure, optional in Kombination mit weiteren Adjuvanzien. Des weiteren betrifft die Erfindung eine pharmazeutische Zusammensetzung und eine Vakzine, jeweils enthaltend ein erfindungsgemäßes immunstimulierendes Adjuvanz, mindestens einen Wirkstoff und optional einen pharmazeutisch geeigneten Träger und/oder weitere Hilfs- und Zusatzstoffe und/oder weitere Adjuvanzien. Die vorliegende Erfindung betrifft ebenfalls die Verwendung der erfindungsgemäßen pharmazeutischen Zusammensetzung sowie der erfindungsgemäßen Vakzine zur Behandlung von Infektionserkrankungen oder Krebserkrankungen. Ebenso umfasst die vorliegende Erfindung die Verwendung des erfindungsgemäßen immunstimulierenden Adjuvanz zur Herstellung einer pharmazeutischen Zusammensetzung zur Behandlung von Krebserkrankungen oder Infektionserkrankungen.
Description
Anmelder:
CureVac GmbH
Paul-Ehrlich-Str. 15
72076 Tübingen
Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure
Die vorliegende Erfindung betrifft ein immunstimulierendes Adjuvanz in Form einer Lipid- modifizierten Nukleinsäure, optional in Kombination mit weiteren Adjuvanzien. Des weiteren betrifft die Erfindung eine pharmazeutische Zusammensetzung und eine Vakzine, jeweils enthaltend ein erfindungsgemäßes immunstimulierendes Adjuvanz, mindestens einen Wirkstoff und optional einen pharmazeutisch geeigneten Träger und/oder weitere Hilfs- und Zusatzstoffe und/oder weitere Adjuvanzien. Die vorliegende Erfindung betrifft ebenfalls die Verwendung der erfindungsgemäßen pharmazeutischen Zusammensetzung sowie der erfindungsgemäßen Vakzine zur Behandlung von Infektionserkrankungen oder Krebserkrankungen. Ebenso umfasst die vorliegende Erfindung die Verwendung des erfindungsgemäßen immunstimulierenden Adjuvanz zur Herstellung einer pharmazeutischen Zusammensetzung zur Behandlung von Krebserkrankungen oder Infektionserkrankungen.
Sowohl bei der klassischen als auch bei der genetischen Vakzinierung tritt häufig das Problem auf, dass nur eine geringe und damit häufig unzureichende Immunantwort im zu therapierenden bzw. zu impfenden Organismus hervorgerufen wird. Daher werden Vakzinen oder Wirkstoffen häufig sog. Adjuvanzien beigefügt, d.h. Substanzen oder Zusammensetzungen, die eine Immunantwort, z.B. gegen ein Antigen, erhöhen und/oder gerichtet beeinflussen können. So ist bspw. bekannt, dass die Effektivität einiger injizierbarer medizinischer Wirkstoffe in signifikanter Weise dadurch verbessert werden kann, dass der Wirkstoff mit einem Adjuvanz kombiniert wird, welcher in der Lage ist, die Freisetzung des Wirkstoffs in das Wirtszellsystem und gegebenenfalls die Aufnahme in die Wirtszellen zu beeinflussen. Auf diese Weise kann ein Effekt erzielt werden, der der periodischen Verabreichung vieler kleiner Dosen in regulären Intervallen vergleichbar ist. Der Begriff
„Adjuvanz" bezieht sich in diesem Zusammenhang üblicherweise auf eine Verbindung oder Zusammensetzung, die als Bindemittel, Träger oder Hilfsstoff für Immunogene und/oder andere pharmazeutisch wirksame Verbindungen dient.
Im Stand der Technik wurden eine Reihe von Verbindungen und Zusammensetzungen als Adjuvanz vorgeschlagen, z.B. das Freund'sche Adjuvanz, Metalloxide (Aluminiumhydroxid, etc.), Alaun, anorganische Chelate oder Salze davon, verschiedene Paraffin-artige Öle, synthetisierte Harze, Alginate, Mucoide, Polysaccharid-Verbindungen, Caseinate, sowie aus Blut und/oder Blutgeriήnseln isolierte Verbindungen wie z.B. Fibrin-Derivate, etc.. Derartige Adjuvanzien erzeugen jedoch zumeist unerwünschte Nebenwirkungen, bspw. sehr schmerzhafte Reizungen und Entzündungen am Ort der Applikation. Des weiteren werden auch toxische Nebenwirkungen, insbesondere Gewebsnekrosen, beobachtet. Schließlich bewirken diese bekannten Adjuvanzien in den meisten Fällen lediglich eine unzureichende Stimulation der zellulären Immunantwort, da ausschließlich B-Zellen aktiviert werden.
So wurden bspw. Alaune, Metalloxide und Chelate von Salzen mit der Erzeugung von sterilen Abszessen in Verbindung gebracht. Zudem wird in der Wissenschaftswelt bezweifelt, dass solche Verbindungen wieder vollständig ausgeschieden werden. Es wird eher angenommen, dass diese zu unerwünschten anorganischen Rückständen im Körper führen. Zwar weisen solche Verbindungen üblicherweise eine geringe Toxizität auf, jedoch besteht die Möglichkeit, dass diese durch die Zellen des reticulo-endothelialen Sytems (littorale und sinusoidale Zellen der Leber und der Milz) als Teil der unlöslichen Debris phagozytiert werden. Es gibt zudem Hinweise darauf, dass eine solche Debris schädlich auf die verschiedenen Filter-Mechanismen des Körpers, z.B. der Nieren, der Leber oder der Milz einwirken kann. Solche Rückstände stellen damit eine latente und ständig präsente Gefahrenquelle im Körper und allgemein für das Immunsystem dar.
Die als Adjuvanzien im Stand der Technik verwendeten synthetisierten Öle und Petroleum- Derivate führen ebenfalls zu nachteiligen Effekten. Diese Verbindungen sind jedoch insbesondere deshalb unerwünscht, weil sie im Körper schnell metabolisieren und in ihre aromatischen Kohlenwasserstoffverbindungen zerfallen. Von solchen aromatischen Kohlenwasserstoffverbindungen ist aber bekannt, dass sie in höchstem Maße kanzerogen
wirken können. Überdies wurde festgestellt, dass solche Verbindungen ebenfalls in Zusammenhang mit der Entstehung von sterilen Abszessen stehen und selten vollständig aus dem Körper wieder entfernt werden können.
Auch aus Tieren isolierte Verbindungen, wie z.B. Gelatine, sind häufig kaum als Adjuvanz zum Zwecke der Immunstimulation geeignet. Solche Verbindungen wirken zwar üblicherweise nicht zerstörerisch auf den Wirtsorganismus oder die jeweiligen Wirtszellen ein. Diese Verbindungen wandern jedoch typischerweise zu schnell von der Injektionsstelle in den Wirtsorganismus oder in die Wirtszellen, so dass die für ein Adjuvanz üblicherweise erwünschten Eigenschaften, wie z.B. eine verzögerte Freisetzung eines ggf. zusammen mit dem Adjuvanz injizierten Wirkstoffs, etc., selten erreicht werden. Einer solchen schnellen Verteilung kann z.T. mit Gerbstoffen oder anderen (anorganischen) Verbindungen entgegengewirkt werden. Der Metabolismus solcher zusätzlicher Verbindungen sowie deren Verbleib im Körper ist jedoch nicht vollständig geklärt. Es besteht daher auch hier die begründete Annahme, dass sich diese Verbindungen in der Debris ansammeln und so die Filtrationsmechanismen, z.B. der Nieren-, Leber und/oder Milzzellen, erheblich stören. Auch kann die Eigenschaft von Gelatine, bei parenteraler Verabreichung anzuschwellen, unter in ι//VoBedingungen zu unangenehmen Nebenwirkungen, wie z.B. Schwellungen, insbesondere an der Applikationsstelle und zu Unwohlheit, führen.
Bei aus Blut und/oder Blutgerinnseln isolierten Verbindungen wie z.B. Fibrin-Derivaten, etc. wurden typischerweise immunstimulatorische Effekte festgestellt. Jedoch sind die meisten dieser Verbindungen vorliegend als Adjuvanz aufgrund ihrer (parallel zu den durchaus erwünschten immunogenen Eigenschaften auftretenden) Nebenwirkungen auf das Immunsystem ungeeignet. So werden bspw. viele dieser Verbindungen als allergen eingestuft und bewirken unter Umständen eine weit über das gewünschte Maß hinausgehende Überreaktion des Immunsystems. Diese Verbindungen sind daher aus den genannten Gründen als Adjuvanz für die Immunstimmulation ebenfalls ungeeignet.
Immunantworten können darüber hinaus auch direkt mit Nukleinsäuren als Adjuvanz erzeugt werden. So spielt z.B. DNA bei der Erzeugung von Immunantworten eine zentrale
Rolle. Es ist bspw. von bakterieller DNA bekannt, dass sie aufgrund des Vorhandenseins von nicht-methylierten CG-Motiven immunstimulierend wirkt, weshalb derartige CpG-DNA als immunstimulierendes Mittel und als Adjuvanz für Vakzine vorgeschlagen wurde (vgl. US- Patent 5,663,153). Diese immunstimulierende Eigenschaft von DNA kann auch durch DNA-Oligonukleotide erreicht werden, die durch Phosphorthioat-Modifikation stabilisiert sind (US-Patent 6,239,1 16). Das US-Patent 6,406,705 offenbart schließlich Adjuvanz- Zusammensetzungen, welche eine synergistische Kombination aus einem CpG- Oligodesoxyribonukleotid und einem Nicht-Nukleinsäure-Adjuvanz enthalten.
Die Verwendung von DNA als Adjuvanz kann jedoch unter mehreren Gesichtspunkten weniger vorteilhaft sein. DNA wird in der Blutbahn nur relativ langsam abgebaut, so dass es bei der Verwendung von immunstimulierender (Fremd-)DNA zu einer Bildung von Anti- DNA-Antikörpern kommen kann, was im Tiermodell bei der Maus bestätigt wurde (Gilkeson et al., J. Clin. Invest. 1995, 95: 1398-1402). Die mögliche Persistenz von (Fremd-) DNA im Organismus kann so zu einer Überaktivierung des Immunsystems führen, welche bekanntermaßen bei Mäusen in einer Splenomegalie resultiert (Montheith et al., Anticancer Drug Res. 1997, 12(5): 421 -432). Des weiteren kann (Fremd-)DNA mit dem Wirtsgenom wechselwirken, und insbesondere durch Integration in das Wirtsgenom Mutationen hervorrufen. So kann es bspw. zu einer Insertion der eingebrachten (Fremd-)DNA in ein intaktes Gen kommen, welche eine Mutation darstellt, die die Funktion des endogenen Gens behindern oder gar vollkommen ausschalten kann. Durch solche Integrationsereignisse können einerseits für die Zelle lebenswichtige Enzymsysteme zerstört werden, andererseits besteht auch die Gefahr einer Transformation der so veränderten Zelle in einen entarteten Zustand, falls durch die Integration der (Fremd-) DNA ein für die Regulation des Zellwachstums entscheidendes Gen verändert wird. Daher kann bei bisherigen Verfahren bei der Verwendung von (Fremd-)DNA als immunstimulierendem Mittel ggf. ein Risiko der Krebsbildung nicht ausgeschlossen werden.
Vorteilhafter für die Erzeugung solcher Immunantworten ist daher allgemein die Verwendung von RNA als Adjuvanz, da RNA gegenüber DNA eine wesentlich geringere Halbwertszeit in vivo aufweist. Dennoch zeigt auch bei der Verwendung von RNA als Adjuvanz Beschränkungen. So weisen bisher im Stand der Technik offenbarte RNA- Sequenzen in vivo nur eine begrenzte Zellpermeabilität auf. Dies kann wiederum eine
erhöhte Menge an RNA zur Immunstimulation erfordern, was, ungeachtet der erhöhten Kosten aufgrund gesteigerter zu applizierender RNA-Mengen, die Gefahr der allgemein zuvor beschriebenen zumeist unerwünschten Nebenwirkungen birgt, bspw. sehr schmerzhaften Reizungen und Entzündungen am Ort der Applikation. Auch können toxische Nebenwirkungen bei hohen Gaben des verabreichten Immunstimulanzmittels nicht ausgeschlossen werden.
Trotz der bisher gezeigten Erfolge besteht daher ein erhöhter Bedarf sowie erhebliches Interesse an einer verbesserten Immunstimulation, insbesondere an Mitteln, die einerseits geeignet sind, eine effiziente Immunantwort im zu therapierenden bzw. zu impfenden Patienten hervorrufen, und andererseits effektiv die Aufnahme eines optional zusätzlich enthaltenen Wirkstoffs in den Körper bzw. Körperzellen unterstützen.
Diese Aufgabe wird gelöst durch ein erfindungsgemäßes immunstimulierendes Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure. Diese Lipid-modifizierte Nukleinsäure besteht erfindungsgemäß aus einer Nukleinsäure, mindestens einem mit dieser Nukleinsäure kovalent verknüpften Linker und mindestens einem mit dem jeweiligen Linker kovalent verknüpften Lipid. Alternativ besteht die Lipid-modifizierte Nukleinsäure erfindungsgemäß aus (mindestens) einer Nukleinsäure und mindestens einem mit dieser Nukleinsäure (ohne Linker) kovalent verknüpften (bifunktionellen) Lipid. Gemäß einer dritten Alternative besteht die Lipid-modifizierte Nukleinsäure erfindungsgemäß aus einer Nukleinsäure, mindestens einem mit dieser Nukleinsäure kovalent verknüpften Linker und mindestens einem mit dem jeweiligen Linker kovalent verknüpften Lipid sowie mindestens einem mit dieser Nukleinsäure (ohne Linker) kovalent verknüpften (bifunktionellen) Lipid.
Ein „immunstimulierendes" Adjuvanz gemäß der vorliegenden Erfindung ist bevorzugt in der Lage eine Immunreaktion auszulösen. Eine Immunreaktion kann allgemein auf verschiedene Art und Weise hervorgerufen werden. Ein wesentlicher Faktor für eine passende Immunantwort ist dabei die Stimulation unterschiedlicher T-ZeII- Subpopulationen. T-Lymphozyten differenzieren typischerweise in zwei Subpopulationen, die T-Helfer 1 (ThI )- und T-Helfer 2 (Th2) - Zellen, mit denen das Immunsystem in der Lage ist, intrazelluläre (Th! ) und extrazelluläre (Th2) Pathogene (z.B. Antigene) zu vernichten.
Die beiden Th-Zellpopulationen unterscheiden sich im Muster der von ihnen produzierten Effektorproteine (Zytokine). So unterstützen ThI -Zellen die zelluläre Immunantwort durch Aktivierung von Makrophagen und zytotoxischen T-Zellen. Th2-Zellen fördern dagegen die humorale Immunantwort durch Stimulierung der B-Zellen zur Verwandlung in Plasmazellen und durch Bildung von Antikörpern (z.B. gegen Antigene). Von hoher Wichtigkeit ist bei der Immunantwort daher das Th1/Th2-Verhältnis. Im Zusammenhang mit der vorliegenden Erfindung wird durch das erfindungsgemäße Adjuvanz das Th1/Th2-Verhältnis der Immunantwort bevorzugt in Richtung der zellulären Antwort (Th1 -Antwort) verschoben und damit eine zelluläre Immunantwort induziert.
Die erfindungsgemäß für die Lipid-modifizierte Nukleinsäure (Adjuvanz) verwendete Nukleinsäure, kann eine RNA oder DNA sein (bspw. eine cDNA), ein RNA- oder DNA- Oligonukleotid, ein RNA- oder DNA-Homopolymer, eine CpG-Nukleinsäure, etc.. Sie kann einzelsträngig oder doppelsträngig sein, als Homo- oder-Heteroduplex und linear oder zirkulär vorliegen. Besonders bevorzugt liegt die erfindungsgemäß für die Lipid-modifizierte Nukleinsäure (Adjuvanz) eingesetzte Nukleinsäure als einzelsträngige RNA vor.
Typischerweise handelt es sich bei der Lipid-modifizierten Nukleinsäure um relativ kurze Nukleinsäure-Moleküle, die bspw. aus etwa 2 bis etwa 1000 Nukleotiden, vorzugsweise aus etwa 5 bis 200, 6 bis etwa 200 Nukleotiden, und besonders bevorzugt aus 6 bis etwa 40 oder 6 bis etwa 31 Nukleotiden, bestehen. Nukleotide sind in diesem Zusammenhang bevorzugt alle natürlich auftretenden Nukleotide sowie deren Analoga, wie Ribonukleotide und/oder Desoxyribonukleotide und umfassen, ohne darauf beschränkt zu sein, bspw. Purine (Adenin (A), Guanin (G)) oder Pyrimidine (Thymin (T), Cytosin (C), Uracil (U)), sowie Analoge oder Derivate von Purinen und Pyrimidinen wie z.B. 1 -Methyl-Adenin, 2-Methyl- Adenin, 2-Methylthio-N6-isopentenyl-Adenin, N6-Methyl-Adenin, N6-Isopentenyl-Adenin, 2-Thio-Cytosin, 3-Methyl-Cytosin, 4-Acetyl-Cytosin, 5-Methyl-Cytosin, 2,6-Diaminopurin, 1-Methyl-Guanin, 2-Methyl-Guanin, 2,2-Dimethyl-Guanin, 7-Methyl-Guanin, Inosin, 1 - Methyl-Inosin, Dihydro-Uracil, 2-Thio-Uracil, 4-Thio-Uracil, 5- Carboxymethylaminomethyl^-thio-Uracil, 5-(Carboxyhydroxylmethyl)-Uracil, 5-Fluoro- Uracil, 5-Bromo-Uracil, 5-Carboxymethylaminomethyl-Uracil, 5-Methyl-2-Thio-Uracil, 5- Methyl-Uracil, N-Uracil-5-oxyessigsäuremethylester, 5-Methylaminomethyl-Uracil, 5- Methoxyaminomethyl-2-thio-Uracil, 5'-Methoxycarbonylmethyl-Uracil, 5-Methoxy-Uracil,
Uracil-5-oxyessigsäuremethylester, Uracil-5-oxyessigsäure (v), Pseudouracil, 1 -Methyl- Pseudouracil, Queosin, ß-D-Mannosyl-Queosin, Wybutoxosin, sowie Phosphoramidate, Phosphorthioate, Peptidnukleotide, Methylphosphonate, 7-Deazaguanosin, 5-Methylcytosin und Inosin. Die Herstellung derartiger Analoga sind einem Fachmann bspw. aus den US- Patenten 4,373,071, US 4,401 ,796, US 4,415,732, US 4,458,066, US 4,500,707, US 4,668,777, US 4,973,679, US 5,047,524, US 5,132,418, US 5,153,319, US 5,262,530 und 5,700,642 bekannt, deren Offenbarung durch Verweis hier vollumfänglich mit aufgenommen ist.
Die Lipid-modifizierte Nukleinsäure kann jede natürlich vorkommende Nukleinsäuresequenz, deren Komplement oder ein Fragment davon, umfassen. Ein Fragment einer solchen Nukleinsäuresequenz weist in diesem Zusammenhang bevorzugt eine Länge von vorzugsweise etwa 5 bis 200, 6 bis etwa 200 Nukleotiden, und besonders bevorzugt aus 6 bis etwa 40 oder 6 bis etwa 31 Nukleotiden, auf. Ebenfalls kann die Lipid- modifizierte Nukleinsäure teilweise oder vollständig synthetischer Natur sein.
Gemäß einer ersten bevorzugten Ausführungsform wird in der Lipid-modifizierte Nukleinsäure CpG-Nukleinsäure verwendet, insbesondere CpG-RNA oder CpG-DNA. Eine erfindungsgemäß verwendete CpG-RNA oder CpG-DNA kann eine einzelsträngige CpG- DNA (ss CpG-DNA), eine doppelsträngige CpG-DNA (dsDNA), eine einzelsträngige CpG- RNA (ss CpG-RNA) oder eine doppelsträngige CpG-RNA (ds CpG-RNA) sein. Bevorzugt liegt die erfindungsgemäß verwendete CpG-Nukleinsäure als CpG-RNA vor, noch bevorzugter als einzelsträngige CpG-RNA (ss CpG-RNA). Ebenfalls bevorzugt weisen solche CpG-Nukleinsäuren eine wie oben beschriebene Länge auf.
Bevorzugt enthält die erfindungsgemäß verwendete CpG-Nukleinsäure mindestens eine oder meherere (mitogene) Cytosin/Guanin-Dinukleotid-Sequenz(en) (CpG-Motiv(e)), welche durch die generischen Formeln 5'-X1X2CGX3X4^' („Hexamer", SEQ ID NO: 1 ) oder 5'- X,X2X3CGX4X5X6-3' („Oktamer", SEQ ID NO: 2) repräsentiert werden. Gemäß einer ersten bevorzugten Alternative ist dabei mindestens ein in diesen Hexamer- oder Oktamer- Sequenzen enthaltenes CpG-Motiv, d.h. das C (Cytosin) und das G (Guanin) dieses CpG- Motivs, nicht-methyliert. Alle weiteren, gegebenenfalls in den Hexamer- oder Oktamer- Sequenzen enthaltenen Cytosine oder Guanine können entweder methyliert oder nicht-
methyliert vorliegen. Gemäß einer weiteren bevorzugten Alternative können jedoch auch das C (Cytosin) und das G (Guanin) des CpG-Motivs methyliert vorliegen. Im Kontext der oben genannten Hexamer- oder Oktamer-Sequenzen stellen X1, X2, X3, X4, X5 und X6 bevorzugt Nukleotide dar, die unabhängig voneinander oder gemeinsam aus allen natürlich auftretenden Nukleotiden sowie deren Analoga ausgewählt werden können, wie zuvor allgemein für hier verwendete Nukleinsäuren beschrieben.
Gemäß einer bevorzugten Ausführungsform enthält die erfindungsgemäß verwendete CpG- Nukleinsäure als CpG-Motiv mindestens eines oder mehrere Oktamere ausgewählt aus der Gruppe bestehend aus: GACGTTCC (SEQ ID NO: 3); GACGCTCC (SEQ ID NO: 4); GACGTCCC (SEQ ID NO: 5); GACGCCCC (SEQ ID NO: 6); AGCGTTCC (SEQ ID NO: 7); AGCGCTCC (SEQ ID NO: 8); AGCGTCCC (SEQ ID NO: 9); AGCGCCCC (SEQ ID NO: 10); AACGTTCC (SEQ ID NO: 1 1 ); AACGCTCC (SEQ ID NO: 12); AACGTCCC (SEQ ID NO: 13); AACGCCCC (SEQ ID NO: 14); GGCGTTCC (SEQ ID NO: 15); GGCGCTCC (SEQ ID NO: 16); GGCGTCCC (SEQ ID NO: 17); GGCGCCCC (SEQ ID NO: 18); GACGTTCG (SEQ ID NO: 19); GACGCTCG (SEQ ID NO: 20); GACGTCCG (SEQ ID NO: 21 ); GACGCCCG (SEQ ID NO: 22); AGCGTTCG (SEQ ID NO: 23); AGCGCTCG (SEQ ID NO: 24); AGCGTCCG (SEQ ID NO: 25); AGCGCCCG (SEQ ID NO: 26); AACGTTCG (SEQ ID NO: 27); AACGCTCG (SEQ ID NO: 28); AACGTCCG (SEQ ID NO: 29); AACGCCCG (SEQ ID NO: 30); GGCGTTCG (SEQ ID NO: 31 ); GGCGCTCG (SEQ ID NO: 32); GGCGTCCG (SEQ ID NO: 33); GGCGCCCG (SEQ ID NO: 34). Am stärksten bevorzugt enthält die erfindungsgemäß verwendete CpG-Nukleinsäure als CpG-Motiv mindestens ein oder mehrere Oktamere ausgewählt aus der Gruppe bestehend aus: GACGTTCC (SEQ ID NO: 3), AACGTTCC (SEQ ID NO: 1 1), GACGTTCG (SEQ ID NO: 19) und AACGTTCG (SEQ ID NO: 23). Ebenfalls umfasst sind solche Sequenzen, die eine Identität zu einer der vorhergehenden Sequenzen von mindestens 60%, bevorzugter 70 oder 80%, und am stärksten bevorzugt von 90 oder 95 % aufweisen. Um die prozentuale Identität zweier Nukleinsäuresequenzen zueinander zu bestimmen, können die Sequenzen abgeglichen werden, um nachfolgend miteinander verglichen zu werden. Hierfür können z.B. Lücken in die Sequenz der ersten Nukleinsäuresequenz eingeführt werden und die Nukleotide an der entsprechenden Position der zweiten Nukleinsäuresequenz verglichen werden. Wenn eine Position in der ersten Nukleinsäuresequenz mit dem gleichen Nukleotid besetzt ist, wie es an einer Position in der zweiten Sequenz der Fall ist, dann sind beide Sequenzen an dieser
Position identisch. Die Bestimmung der prozentualen Identität zweier Sequenzen kann anhand eines mathematischen Algorithmus durchgeführt werden. Ein bevorzugtes, jedoch nicht beschränkendes, Beispiel eines mathematischen Algorithmus, der für den Vergleich zweier Sequenzen herangezogen werden kann, ist der Algorithmus von Karlin et al. (1993), PNAS USA, 90:5873-5877. Ein solcher Algorithmus ist in dem NBLAST-Programm integriert, mit dem Sequenzen identifiziert werden können, die eine gewünschte Identität zu den Sequenzen der vorliegenden Erfindung besitzen. Um einen Lücken-Abgleich (auch "gapped alignment"), wie oben beschrieben, zu erhalten, kann das "Gapped BLAST"- Programm verwendet werden, wie in Altschul et al. (1997), Nucleic Acids Res, 25:3389- 3402 beschrieben.
Das in der erfindungsgemäß eingesetzten CpG-Nukleinsäure enthaltene mitogene CpG- Motiv, bevorzugter ein Hexamer oder Octamer gemäß einer der Sequenzen SEQ ID NO: 1 bis 34, tritt bevorzugt mindestens einmal in der CpG-Nukleinsäure auf. Besonders bevorzugt tritt dabei am 5'- und am 3'-Ende oder nahe zum 5'- und 3'-Ende der verwendeten CpG-Nukleinsäure, z.B. in einem Bereich von 1 bis 3 oder 1 bis 6 Nukleotiden, kein GCG-Trinukleotid auf. Die Hexamer- oder Octamersequenzen gemäß einer der Sequenzen SEQ ID NO: 1 bis 34 können in der erfindungsgemäß verwendeten CpG-Nukleinsäure mindestens einmal auftreten, d.h. es ist mindestens eine Hexamer- und/oder Octamersequenz gemäß einer der Sequenzen SEQ ID NO: 1 bis 34 enthalten. Alternativ können die Hexamer- oder Octamersequenzen gemäß einer der Sequenzen SEQ ID NO: 1 bis 34 in der erfindungsgemäß verwendeten CpG-Nukleinsäure als Multimer auftreten z.B. als Abfolge von 2 bis 5, 5 bis 10, 10 bis 15, 15 bis 20, 20 bis 30, 30 bis 40 40 bis 50 oder 50 bis 100 der Sequenzen gemäß SEQ ID NO: 1 bis 34, wobei die Hexamer- und Octamersequenzen beliebig miteinander kombiniert werden können. Dabei können die einzelnen Hexamer- oder Octamersequenzen gemäß SEQ ID NO: 1 bis 34 durch 1 -30, bevorzugt 1 -20, stärker bevorzugt 1 bis 10 der zuvor genannten Nukleotide voneinander getrennt sein oder alternativ ohne dazwischenliegende Nukleotide unmittelbar aufeinander folgen.
Die erfindungsgemäß verwendete CpG-Nukleinsäure kann weiterhin als „stabilisiertes Oligonukleotid" vorliegen, d.h. als Oligoribo- oder Oligodesoxyribonukleotid, welches resistent gegenüber in vivo - Abbau (z.B. durch eine Exo- oder Endonuklease) ist. Ein solche
Stabilisierung kann z.B. durch ein modifiziertes Phosphat-Rückgrat (backbone) der erfindungsgemäß verwendeten CpG-Nukleinsäure erfolgen. In diesem Zusammenhang bevorzugt verwendete Nukleotide enthalten ein Phosphorthioat-modifiziertes Phosphat- Rückgrat (backbone), wobei bevorzugt mindestens einer der im Phosphat-Rückgrat enthaltenen Phosphat-Sauerstoffe durch ein Schwefelatom ersetzt wird. Andere stabilisierte Oligonukleotide schließen z.B. mit ein: nicht-ionische Analoga, wie z.B. Alkyl- und Aryl- Phophonate, bei welchen der geladene Phosphonat-Sauerstoff durch eine Alkyl- oder Aryl- Gruppe ersetzt wird, oder Phosphodiester und Alkylphosphotriester, bei welchen der geladene Sauerstoff-Rest alkyliert vorliegt.
Gemäß einer weiteren bevorzugten Ausführungsform liegt die für die Lipid-modifizierte Nukleinsäure verwendete Nukleinsäure als RNA- oder DNA-Homopolymer vor, stärker bevorzugt als RNA-Homopolymer. Ein solches DNA- oder RNA-Homopolymer umfasst typischerweise einzelsträngige oder doppelsträngige, bevorzugt einzelsträngige, Polynukleotide wie bspw. Polyinosinsäure (I), Polyadeninsäure (A), Polyuridinsäure (U), Polyxanthinsäure (X) oder Polyguaninsäure (G). Die erfindungsgemäß verwendeten RNA- oder DNA-Homopolymere können dabei als einzelsträngige RNA- oder DNA- Homopolymere auftreten. Solche RNA- oder DNA-Homopolymere sind im Stand der Technik gut bekannt und umfassen typischerweise kein einheitliches Molekulargewicht. Molekulargewichte bei doppelsträngigen Komplexen von Copolymeren wurden beispielsweise in einem Bereich von etwa 1 x 105 bis 1 ,5 x 106 bestimmt.
Eine erste bevorzugte Alternative der RNA- oder DNA-Homopolymere umfasst einzelsträngige RNA- oder DNA-Homopolymere. Solche einzelsträngigen RNA- oder DNA- Homopolymere enthalten typischerweise ein wie zuvor definiertes Ribo- oder Desoxyribonukleotid in n-facher Wiederholung, wobei n bevorzugt der Länge der zuvor beschriebenen erfindungsgemäß verwendeten Nukleinsäuren gleich ist und in einem Bereich von 2 bis etwa 1000, vorzugsweise 5 bis 200, stärker bevorzugt 6 bis etwa 200, und am stärksten bevorzugt 6 bis etwa 40 oder 6 bis etwa 31 liegt. Besonders bevorzugte einzelsträngige RNA- oder DNA-Homopolymere umfassen, ohne darauf beschränkt zu sein, folgende Sequenzen: 5'-AAAAAAAAAAAAAAAAAAAAAA-3' (SEQ ID NO: 35), 5'- UUUUUUUUUUUUUUUUUUUUUU-3' (SEQ ID NO: 36), 5'-
GGGGGGGGGGGGGGGGGGGGGG-3' (SEQ ID NO: 37), 5'-
CCCCCCCCCCCCCCCCCCCCCC-3' (SEQ ID NO: 38) und 5'- I M I I I I I I TTI I I I I I I I I I I - 3' (SEQ ID NO: 39). Ebenfalls υmfasst sind solche Sequenzen, die eine Identität zu einer der vorhergehenden Sequenzen von mindestens 60%, stärker bevorzugt mindestens 70 oder 80%, und am stärksten bevorzugt mindestens 90 oder 95 % aufweisen.
Chemisch veränderte Polynukleotide stellen eine zweite bevorzugte Alternative der DNA- oder RNA-Homopolymere dar. Chemisch veränderte Polynukleotide im Sinne der vorliegenden Erfindung können wie zuvor beschriebene DNA- oder RNA-Polymere sein, die in ihrer Sequenz mindestens ein Nukleotid, z.B. ein Analoges oder Derivat von Purinen (Adenin (A), Guanin (G)) oder Pyrimidinen (Thymin (T), Cytosin (C), Uracil (U)) aufweist, wie zuvor beschrieben. Stärker bevorzugt weisen solche chemisch veränderten Polynukleotide einen Anteil an Analogen und Derivaten von 1 bis 100%, z.B. 1 - 20, 10 - 30, 20 - 40, 30 - 50, 40 - 60, 50 - 70, 60 - 80, 70 - 90 oder 80 - 100 % auf. Solche chemisch veränderten Polynukleotide können ebenfalls nach im Stand der Technik bekannten Verfahren (siehe Verfahren zur Herstellung von Komplexen von Homopolymeren) hergestellt werden. Beispielhafte chemisch veränderte Polynukleotide umfassen, ohne darauf beschränkt zu sein, Verbindungen wie bspw. PoIy-N1- Methyladenylat, Poly-„6-Methyladenylat", Poly-N7-Methylinosat, Poly-N7-Methylguanylat, Poly-5-Methyluridylat, Poly-5-Fluorouridylat, Poly-5-Bromuridylat, Poly-5-Bromcytidylat und Poly-5-lodcytidylat, etc..
Gemäß einer sechsten Alternative können als RNA- oder DNA-Homopolymere auch Kombinationen der zuvor beschriebenen RNA- oder DNA-Homopolymere eingesetzt werden. Solche Kombinationen umfassen bevorzugt Nukleinsäuresequenzen, die mindestens zwei der oben genannten Alternativen der hier beschriebenen RNA- oder DNA- Homopolymere oder Multimere davon enthalten, z.B. eine Abfolge von 2 bis 5, 5 bis 10, 10 bis 15, 15 bis 20, 20 bis 30, 30 bis 40, 40 bis 50 oder 50 bis 100 einer oder mehrerer der zuvor beschriebenen RNA- oder DNA-Homopolymere, besonders bevorzugt gemäß einer der SEQ ID NO: 35 bis 39. Dabei können die einzelnen RNA- oder DNA-Homopolymere durch 1 -30, bevorzugt 1 -20, stärker bevorzugt 1 bis 10 der zuvor hier beschriebenen Nukleotide voneinander getrennt sein oder alternativ ohne dazwischenliegende Nukleotide unmittelbar aufeinander folgen.
Gemäß einer weiteren bevorzugten Ausführungsform können für die Lipid-modifizierte Nukleinsäure solche Nukleinsäuren eingesetzt werden, die keiner der oben benannte Nukleinsäureklassen zuzuordnen sind und im Stand der Technik bereits als immunogen bekannt sind oder die noch nicht im Stand der Technik bekannt sind, aber immunogene Eigenschaften aufweisen. Bevorzugt liegen Nukleinsäuren gemäß dieser Alternative als RNA oder DNA, noch bevorzugter als RNA vor. Ebenfalls bevorzugt weisen diese Nukleinsäuren eine wie oben beschriebene Länge auf und enthalten Nukleotide, z.B. Ribo- oder Desoxyribonukleotide, wie zuvor hier offenbart. Solche Nukleinsäuren können beispielsweise Antigene kodieren. Alternativ können solche Nukleinsäuren Epitope (von Proteinen) kodieren. Bevorzugt weisen solche Nukleinsäuren dann ein ATG als Startsignal auf, welches den Start der Translation der kodierten RNA markiert. Gemäß einer besonderen Ausführungsform weist z.B. die Nuklei nsäuresequenz der Lipid-modifizierten Nukleinsäure mindestens eine Sequenz gemäß einer der SEQ ID NOs: 40-67 auf, wie nachfolgend aufgeführt: 5'-GCCCGUCUGUUGUGUGACUC-S' (SEQ ID NO: 40, auch als RNA 40 bezeichnet), 5'-GGUAAGUGUAAGGUGUAAGG-S' (SEQ ID NO: 41 , auch als RNA CV1 bezeichnet), 5'-AAUGGAUAUGGAAUAUGGAA-S' (SEQ ID NO: 42, auch als RNA CV2 bezeichnet), 51-UCCAUGACGUUCCUGACGUU-31 (SEQ ID NO: 43), 5'- UCCAGGACUUCUCUCAGGUU-S1 (SEQ ID NO: 44), 5'-
UCCAUGACGUUCCUGAUGCU-S1 (SEQ ID NO: 45), 5'-
GCCCGUCUGUUGUGUGACUC-S1 (SEQ ID NO: 46), 5'-
GGUAAGUGUAAGGUGUAAGG-S' (SEQ ID NO: 47), 5'-
AAUGGAUAUGGAAUAUGGAA-S1 (SEQ ID NO: 48), 5'- CUCUGGAGGAAAAGAAAGUTT-S1 (SEQ ID NO: 49), 5'-
CAAUGCAACUCGCUUCUCGTT-3' (SEQ ID NO: 50), 5'-AGCUUAACCUGUCCUUCAA - 3' (SEQ ID NO: 51 ), 51-AAAAAAAACUGUCCUUCAA-31 (SEQ ID NO: 52), 5'- AAAAAAAAAUG UCCU UCAA-31 (SEQ ID NO: 53), 51-AAAAAAAAAAGUCCUUCAA-31 (SEQ ID NO: 54), 51-AAAAAAAAAAAUCCUUCAA-31 (SEQ ID NO: 55), 5'- AAAAAAAAAAAACCU UCAA-31 (SEQ ID NO: 56), 51-AGCUUAACCUGUCCUUAAA-31 (SEQ ID NO: 57), 5 '-AGCU UAACCUG UCCUAAAA-31 (SEQ ID NO: 58), 5'- AGCUUAACCUGUCCAAAAA-31 (SEQ ID NO: 59), 5l-AGCUUAACCUGAAAAAAAA-31 (SEQ ID NO: 60), 51-UGUCCUUCAAUGUCCUUCAA-31 (SEQ ID NO: 61 ) 5'-
AGCUUAACCUGUCCUUCAU-3' (SEQ ID NO: 62), 51-AGCUUAACCUGUCCUUCUU-3l (SEQ ID NO: 63), 5I-AGCUUAACCUGUCCUUCAACUACA-3I (SEQ ID NO: 64), 5'- CAAAUUGAAGGACAGGUUAAGCU-31 (SEQ ID NO: 65), 5'-UUAACCUGUCCUUCAA-3' (SEQ ID NO: 66), 5'-AACCUGUCCUUCA-S 1 (SEQ ID NO: 67). Ebenfalls umfasst sind solche Sequenzen, die eine Identität zu einer der vorhergehenden Sequenzen von mindestens 60%, bevorzugter 70 oder 80%, und am stärksten bevorzugt von 90 oder 95 % aufweisen.
Für die Lipid-modifizierte Nukleinsäure können gemäß einer weiteren Ausführungsform auch solche Nukleinsäuren eingesetzt werden, die ein Multimer aus einer oder mehreren der zuvor beschriebenen Nukleinsäuren darstellen, z.B. eine Abfolge von 2 bis 5, 5 bis 10, 10 bis 15, 15 bis 20, 20 bis 30, 30 bis 40, 40 bis 50 oder 50 bis 100 der oben beschriebenen Nukleinsäuren, besonders bevorzugt gemäß SEQ ID NO: 1 bis 67, aufweisen. Die Reihenfolge der Nukleinsäuren kann dabei beliebig gewählt werden. Dabei können die einzelnen Nukleinsäuren/Nukleinsäureeinheiten des Multimers durch 1 - 30, bevorzugt 1-20, stärker bevorzugt 1 bis 10 der zuvor beschriebenen Nukleotide voneinander getrennt sein oder alternativ ohne dazwischenliegende Nukleotide unmittelbar aufeinander folgen.
Die für die erfindungsgemäße Lipid-modifizierte Nukleinsäure verwendete Nukleinsäure, kann neben der Lipid-Modifizierung zusätzlich mindestens eine chemische Modifikation aufweisen. Dabei werden erfindungsgemäß vor allem solche chemischen Modifikationen bevorzugt, die die Immunogeniät des erfindungsgemäßen Adjuvanz steigern oder nicht mit der Lipid-Modifikation der erfindungsgemäß verwendeten Nukleinsäure interferieren. Beispielsweise können, falls die Lipid-Modifikation am 3' Ende der erfindungsgemäß verwendeten Nukleinsäure vorliegt, chemische Modifikationen typischerweise am 5'-Ende und/oder innerhalb der Sequenz der erfindungsgemäß verwendeten Nukleinsäure eingeführt werden. Falls die Lipid-Modifikation am 5' Ende der erfindungsgemäß verwendeten Nukleinsäure vorliegt, können typischerweise chemische Modifikationen am 3'-Ende und/oder innerhalb der Sequenz der erfindungsgemäß verwendeten Nukleinsäure eingeführt werden. Falls dagegen die Lipid-Modifikation am 3'-Ende und am 5'-Ende der erfindungsgemäß verwendeten Nukleinsäure vorliegt, wird die chemische Modifikationen
bevorzugt innerhalb der Sequenz der erfindungsgemäß verwendeten Nukleinsäure eingeführt.
Vorzugsweise ist die chemische Modifikation der erfindungsgemäßen Lipid-modifizierten Nukleinsäure derart ausgestaltet, dass die hierfür verwendete Nukleinsäure, bevorzugt RNA, mindestens ein Analoges natürlich vorkommender Nukleotide enthält. Solche Analoga schließen die zuvor beschriebenen Nukleotide und deren Analoga mit ein. Zusätzlich können alle zuvor genannten Nukleotide und deren Analoga, durch z.B. Acetylierung,
Methylierung, Hydroxylierung, etc. chemisch weiter modifiziert und erfindungsgemäß eingesetzt werden.
Die für die Lipid-modifizierte Nukleinsäure erfindungsgemäß verwendete Nukleinsäure bzw. die Lipid-modifizierte Nukleinsäure selbst, kann weiterhin stabilisiert werden. Wie zuvor ausgeführt, kann für die Lipid-modifizierte Nukleinsäure grundsätzlich jede Nukleinsäure verwendet werden. Unter dem Gesichtspunkt der Sicherheit ist der Einsatz von RNA für eine solche Nukleinsäure jedoch bevorzugt. Insbesondere bringt RNA nicht die Gefahr mit sich, stabil in das Genom der transfizierten Zelle integriert zu werden. Darüber hinaus wird RNA wesentlich einfacher in vivo abgebaut. Ebenso wurden, wohl aufgrund der gegenüber DNA relativ kurzen Halbwertszeit von RNA im Blutkreislauf, bisher keine anti-RNA-Antikörper nachgewiesen. Im Vergleich zu DNA ist RNA in Lösung allerdings wesentlich instabiler, wofür im wesentlichen RNA-abbauende Enzyme, sog. RNAasen (Ribonukleasen), verantwortlich sind. Selbst kleinste Verunreinigungen von Ribonukleasen reichen aus, um RNA in Lösung vollständig abzubauen. Derartige RNase- Verunreinigungen können allgemein nur durch besondere Behandlungen, insbesondere mit Diethylpyrocarbonat (DEPC), beseitigt werden. Der natürliche Abbau von mRNA im Cytoplasma von Zellen ist sehr fein reguliert. Diesbezüglich sind mehrere Mechanismen im Stand der Technik bekannt. So ist für eine RNA in vivo typischerweise die endständige Struktur von entscheidender Bedeutung. Am 5'-Ende natürlich auftretender RNAs befindet sich üblicherweise eine sogenannte "Cap-Struktur" (ein modifiziertes Guanosin-Nukleotid) und am 3 '-Ende eine Abfolge von bis zu 200 Adenosin-Nukleotiden (der sog. PoIy-A- Schwanz).
Daher kann die Nukleinsäure der Lipid-modifizierten Nukleinsäure, falls diese als RNA vorliegt, durch Anfügung einer sog. "5'-Cap"-Struktur gegenüber dem Abbau durch RNasen stabilisiert werden. Besonders bevorzugt wird in diesem Zusammenhang einer m7G(5')ppp (5'(A,G(5')ppp(5')A oder G(5')ppp(5')G als 5'-Cap"-Struktur. Eine solche Modifikation wird jedoch nur dann eingeführt, wenn am 5'-Ende der erfindungsgemäß verwendeten Nukleinsäure nicht bereits eine Modifikation, z.B. eine Lipid-Modifikation, eingeführt wurde.
Alternativ kann das 3'-Ende der Nukleinsäure der Lipid-modifizierten Nukleinsäure, falls diese als RNA vorliegt, durch eine Abfolge von mindestens 50 Adenosin-Nukleotiden, vorzugsweise mindestens 70 Adenosin-Nukleotiden, mehr bevorzugt mindestens 100
Adenosin-Nukleotiden, besonders bevorzugt mindestens 200 Adenosin-Nukleotiden modifiziert werden. Analog kann auch hier eine solche Modifikation nur dann eingeführt werden, wenn am 3'-Ende der erfindungsgemäß verwendeten Nukleinsäure nicht bereits eine Modifikation, z.B. eine Lipid-Modifikation, eingeführt wurde.
Das in der erfindungsgemäßen Lipid-modifizierten Nukleinsäure enthaltene Lipid ist typischerweise ein Lipid oder ein lipophiler Rest, der bevorzugt an sich biologisch aktiv ist. Solche Lipide umfassen bevorzugt Naturstoffe oder Verbindungen wie z.B. Vitamine, z.B. α-Tocopherol (Vitamin E), einschließlich RRR-α-Tocopherol (früher D-α-Tocopherol), L-α- Tocopherol, dem Racemat D,L-α-Tocopherol, Vitamin E-Succinat (VES), oder Vitamin A und dessen Derivate, z.B. Retinsäure, Retinol, Vitamin D und dessen Derivate, z.B. Vitamin D sowie dessen Ergosterol-Vorstufen, Vitamin E und dessen Derivate, Vitamin K und dessen Derivate, z.B. Vitamin K und verwandte Chinon bzw. Phytolverbindungen, oder Steroide, wie Gallensäuren, bspw. Cholinsäure, Desoxycholinsäure, Dehydrocholinsäure, Cortison, Digoxygenin, Testosteron, Cholesterol oder Thiocholesterol. Weitere Lipide oder lipophile Reste im Sinne der vorliegenden Erfindung umfassen, ohne darauf beschränkt zu sein, Polyalkylenglykole, (Oberhauser et al., Nucl. Acids Res., 1992, 20, 533), aliphatische Gruppen wie z.B. C,-C20-Alkane, C1-C20-Alkene, oder C1 -C20-Al kanol Verbindungen, etc. wie bspw. Dodecandiol, Hexadecanol oder Undecyl-Reste (Saison-Behmoaras et al., EMBO J, 1991 , 10, 1 1 1 ; Kabanov et al., FEBS Lett., 1990, 259, 327; Svinarchuk et al., Biochimie, 1993, 75, 49), Phospholipide wie z.B. Phosphatidylglycerol, Diacylphosphatidylglycerol,
Phosphatidylcholin, Dipalmitoylphosphatidylcholin, Distearoylphosphatidylcholin, Phosphatidylserin, Phosphatidylethanolamin, Di-hexadecyl-rac-glycerol, Sphingolipide, Cerebroside, Ganglioside, oder Triethylammonium 1 ,2-Di-O-hexadecyl-rac-glycero-3-H- phosphonat (Manoharan et al., Tetrahedron Lett., 1995, 36, 3651 ; Shea et al., Nucl. Acids Res., 1990, 18, 3777), Polyamine oder Polyalkylenglykole, wie z.B. Polyethylenglykol (PEG) (Manoharan et al., Nucleosides & Nucleotides, 1995, 14, 969), Hexaethylenglykol (HEG), Palmitin oder Palmityl-Reste (Mishra et al., Biochim. Biophys. Acta, 1995, 1264, 229), Octadecylamine oder Hexylamino-carbonyl-oxycholesterol-Reste (Crooke et al., J. Pharmacol. Exp. Ther., 1996, 277, 923), sowie Wachse, Terpene, alicyklische Kohlenwasserstoffe, gesättigte bzw. einfach- oder mehrfach ungesättigte Fettsäurereste, etc..
Die Verknüpfung zwischen dem Lipid und der erfindungsgemäß verwendeten Nukleinsäure kann grundsätzlich an jedem Nukleotid, an der Base oder dem Zuckerrest jedes Nukleotids, am 3'- und/oder 5'-Ende, und/oder am Phosphatrückgrat der erfindungsgemäß verwendeten Nukleinsäure stattfinden. Erfindungsgemäß wird eine terminale Lipid-Modifizierung der erfindungsgemäß verwendeten Nukleinsäure an deren 3'- und/oder 5'-Ende besonders bevorzugt. Eine terminale Modifizierung weist gegenüber sequenzinternen Modifikationen mehrere Vorteile auf. Zum einen können sequenzinterne Modifikationen das Hybridisierungsverhalten beeinflussen, was sich ggf. bei sterisch anspruchsvollen Resten negativ auswirkt. Andererseits kann bei einer synthetischen Herstellung einer erfindungsgemäßen Lipid-modifizierten Nukleinsäure, die ausschließlich terminal modifiziert ist, die Synthese der Nukleinsäuresequenz mit handelsüblichen, in großer Menge verfügbaren, Monomeren durchgeführt und im Stand der Technik bekannte Syntheseprotokolle verwendet werden.
Gemäß einer ersten bevorzugten Ausführungsform erfolgt die Verknüpfung zwischen der erfindungsgemäß verwendeten Nukleinsäure und mindestens einem verwendeten Lipid über einen (kovalent mit der Nukleinsäure verknüpften) „Linker". Linker im Sinne der vorliegenden Erfindung weisen typischerweise mindestens zwei und optional 3, 4, 5, 6, 7, 8, 9, 10, 10-20, 20-30 oder mehr reaktive Gruppen auf, jeweils ausgewählt aus z.B. einer Hydroxygruppe, einer Aminogruppe, einer Alkoxygruppe, etc.. Bevorzugt dient eine reaktive Gruppe zur Bindung der oben beschriebenen erfindungsgemäß verwendeten Nukleinsäure, z.B. eines RNA-Oligonukleotids. Diese reaktive Gruppe kann in einer
geschützten Form vorliegen, z.B. als DMT-Gruppe (Dimethoxytritylchlorid), als Fmoc- Gruppe, als MMT (Monomethoxytrityl-)-Gruppe, als TFA (Trifluoressigsäure)-Gruppe, etc. Weiterhin können Schwefelgruppen durch Disulfide, z.B. Alkylthiole wie bspw. 3- Thiopropanol, oder mit aktivierten Komponenten wie 2-Thiopyridin geschützt werden. Eine oder mehrere weitere reaktive Gruppen dienen erfindungsgemäß zur kovalenten Bindung eines oder mehrerer Lipide. Eine erfindungsgemäß verwendete Nukleinsäure kann daher gemäß der ersten Ausführungsform über den kovalent gebundenen Linker bevorzugt mindestens ein Lipid binden, z.B. 1 , 2, 3, 4, 5, 5-10, 10-20, 20-30 oder mehr Lipid(e), besonders bevorzugt mindestens 3-8 oder mehr Lipid(e) pro Nukleinsäure. Die gebundenen Lipide können dabei getrennt voneinander an verschiedene Positionen der Nukleinsäure gebunden werden, aber auch als Komplex an einer oder mehreren Positionen der Nukleinsäure vorliegen. Eine zusätzliche reaktive Gruppe des Linkers kann zur direkten oder indirekten (spaltbaren) Bindung an ein Trägermaterial, z.B. eine Festphase, verwendet werden. Bevorzugte Linker gemäß der vorliegenden Erfindung sind z.B. Glykol, Glycerin sowie Glycerinderivate, 2-Aminobutyl-1 ,3-propandiol sowie 2-Aminobutyl-1 ,3-propandiol- Derivate/Gerüst, Pyrrolidin-Linker bzw. Pyrrolidin enthaltende organische Moleküle (insbesondere für eine Modifikation am 3'-Ende), etc.. Besonders bevorzugt werden erfindungsgemäß als Linker Glycerin oder Glycerinderivate (C3-Anker) oder ein 2- Aminobutyl-1 ,3-propandiol-Derivat/Gerüst (C7-Anker) verwendet. Ein Glycerinderivat (C3- Anker) als Linker wird insbesondere dann bevorzugt, wenn die Lipid-Modifikation über eine Etherbindung eingeführt werden kann. Soll die Lipid-Modifikation z.B. über eine Amid- oder eine Urethanbindung eingeführt werden, wird z.B. ein 2-Aminobutyl-1 ,3-propandiol- Gerüst (C7-Anker) bevorzugt. In diesem Zusammenhang ist die zwischen dem Linker und der erfindungsgemäß verwendeten Nukleinsäure entstehende Bindung bevorzugt so beschaffen, dass sie mit den Bedingungen und Chemikalien der Amiditchemie kompatibel ist, das heißt sie ist bevorzugt weder säure- noch basenlabil. Insbesondere werden solche Bindungen bevorzugt, die synthetisch leicht zugänglich sind, und nicht durch die ammoniakalische Abspaltprozedur eines Nukleinsäuresyntheseverfahrens hydrolysiert werden. Als Bindungen kommen grundsätzlich alle entsprechend geeigneten Bindungen in Frage, bevorzugt Esterbindungen, Amidbindungen, Urethan- sowie Etherbindungen. Neben der guten Zugänglichkeit der Edukte (wenige Synthesestufen) ist dabei die Etherbindung aufgrund ihrer relativ hohen biologischen Stabilität gegenüber enzymatischer Hydrolyse besonders bevorzugt.
Gemäß einer zweiten bevorzugten Ausführungsform erfolgt die Verknüpfung (mindestens einer) der erfindungsgemäß verwendeten Nukleinsäure direkt mit mindestens einem wie oben beschriebenen (bifunktionellen) Lipid, d.h. ohne Verwendung eines wie oben beschriebenen Linkers. In diesem Fall weist das erfindungsgemäß verwendete (bifunktionelle) Lipid bevorzugt mindestens zwei reaktive Gruppen, oder optional 3, 4, 5, 6, 7, 8, 9, 10, oder mehr reaktive Gruppen, auf, wobei eine erste reaktive Gruppe zur direkten oder indirekten Bindung des Lipids an ein hier beschriebenes Trägermaterial und mindestens eine weitere reaktive Gruppe zur Bindung einer erfindungsgemäß verwendeten Nukleinsäure dient. Gemäß der zweiten Ausführungsform kann eine erfindungsgemäß verwendete Nukleinsäure daher bevorzugt mindestens ein Lipid (direkt ohne Linker) binden, z.B. 1 , 2, 3, 4, 5, 5-10, 10-20, 20-30 oder mehr Lipid(e), besonders bevorzugt mindestens 3- 8 oder mehr Lipid(e) pro Nukleinsäure. Die gebundenen Lipide können dabei getrennt voneinander an verschiedene Positionen der Nukleinsäure gebunden werden, aber auch als Komplex an einer oder mehreren Positionen der Nukleinsäure vorliegen. Alternativ kann gemäß der zweiten Ausführungsform an ein wie zuvor beschriebenes Lipid über dessen reaktive Gruppen mindestens eine Nukleinsäure gebunden werden, z.B. optional 3, 4, 5, 6, 7, 8, 9, 10, 10-20, 20-30 oder mehr Nukleinsäuren. Besonders bevorzugt umfassen für diese zweite Ausführungsform verwendbare Lipide solche (bifunktionellen) Lipide, die (bevorzugt an ihren Termini oder optional intramolekular) eine Kopplung ermöglichen, wie z.B. Polyethylenglykol (PEG) sowie Derivate davon,, Hexaethylenglykol (HEG) sowie Derivate davon, Alkandiole, Aminoalkan, Thioalkanole, etc.. Die Bindung zwischen einem (bifunktionellen) Lipid und einer wie zuvor beschriebenen erfindungsgemäß verwendeten Nukleinsäure ist bevorzugt so beschaffen, wie für die erste bevorzugte Ausführungsform beschrieben.
Gemäß einer dritten Ausführungsform kann die Verknüpfung zwischen der erfindungsgemäß verwendeten Nukleinsäure und mindestens einem wie oben beschriebenen Lipid über beide der zuvor genannten Ausführungsformen gleichzeitig erfolgen. So kann z.B: die Nukleinsäure an einer Position der Nukleinsäure mit mindestens einem Lipid über einen Linker verknüpft sein (analog 1 . Ausführungsform) und an einer anderen Position der Nukleinsäure direkt mit mindestens einem Lipid ohne Verwendung
eines Linkers (analog 2. Ausführungsform). Beispielsweise kann am 3'-Ende einer erfindungsgemäß verwendeten Nukleinsäure mindestens ein wie oben beschriebenes Lipid über einen Linker mit der Nukleinsäure kovalent verknüpft werden und am 5'-Ende der Nukleinsäure ein wie oben beschriebenen Lipid ohne einen Linker mit der Nukleinsäure kovalent verknüpft werden. Alternativ kann am 5'-Ende einer erfindungsgemäß verwendeten Nukleinsäure mindestens ein wie oben beschriebenes Lipid über einen Linker mit der Nukleinsäure kovalent verknüpft werden und am 3'-Ende der Nukleinsäure ein wie oben beschriebenen Lipid ohne einen Linker mit der Nukleinsäure kovalent verknüpft werden. Ebenso können kovalente Verknüpfungen nicht nur an den Termini der Nukleinsäure sondern auch intramolekular stattfinden, wie zuvor beschrieben, z.B. am 3'- Ende und intramolekular, am 5'-Ende und intramolekular, am 3'- und 5'-Ende und intramolekular, ausschließlich intramolekular, etc..
Bevorzugt kann/können die als erfindungsgemäßes Adjuvanz verwendeten Lipid- modifizierte(n) Nukleinsäure(n) durch verschiedene Verfahren erhalten werden. Die Lipid- Modifikation kann dabei grundsätzlich - wie oben definiert - an jeder Position der verwendeten Nukleinsäuresequenz eingeführt werden, z.B. an den 3'- und/oder 5 '-Enden oder am Phosphatrückgrat der verwendeten Nukleinsäuresequenz und/oder an jeder Base bzw. am Zucker jedes Nukleotids der verwendeten Nukleinsäuresequenz. Erfindungsgemäß werden terminale Lipid-Modifikationen an den 3'- und/oder 5 '-Enden der verwendeten Nukleinsäuren bevorzugt. Durch eine solche terminale chemische Modifizierung kann erfindungsgemäß eine große Zahl verschieden derivatisierter Nukleinsäuren erhalten werden. Beispielhafte und erfindungsgemäß mit umfasste Varianten sind in Figur 1 gezeigt. Das Verfahren zur Herstellung solcher Lipid-modifizierten Nukleinsäuren wird bevorzugt in Abhängigkeit der Position der Lipid-Modifizierung ausgewählt.
Findet bspw. die Lipid-Modifikation am 3'-Ende der Nukleinsäure statt, so erfolgt die Lipid- Modifikation typischerweise entweder vor oder nach Bereitstellung der erfindungsgemäß verwendeten Nukleinsäure. Die Bereitstellung der erfindungsgemäß verwendeten Nukleinsäure kann dabei über eine direkte Synthese der Nukleinsäure oder über Zugabe einer bereits fertig synthetisierten (z.B. kommerziell erhältlichen) oder aus Proben isolierten Nukleinsäure durchgeführt werden.
Gemäß einer ersten Alternative wird die Nukleinsäure einer erfindungsgemäßen 3'-Lipid- modifizierten Nukleinsäure vor der Einführung des Lipids direkt synthetisiert, typischerweise über im Stand der Technik bekannte Verfahren zur Synthese von Nukleinsäuren. Dazu wird bevorzugt ein Startnukleosid, z.B. über ein Kopplungsmolekül, z.B. ein Succinylrest, an eine Festphase gebunden sowie die Nukleinsäure, z.B. nach dem Verfahren der Amiditchemie, synthetisiert. Anschließend wird ein wie zuvor beschriebener Linker bevorzugt über eine erste reaktive Gruppe des Linkers kovalent an das 3'-Ende der Nukleinsäure gebunden. Über eine zweite reaktive Gruppe des Linkers kann danach ein wie zuvor beschriebenes Lipid kovalent mit dem Linker verknüpft werden. Alternativ kann der Linker bereits vor der Bindung an das 3'-Ende der Nukleinsäure mit dem Lipid kovalent verknüpft sein. In diesem Fall ist nur die Bindung einer ersten reaktiven Gruppe des Linkers mit dem 3'-Ende der Nukleinsäure erforderlich. Nach Synthese des Oligonukleotids bzw. nach Bindung des Lipids kann die Nukleinsäure von der Festphase abgespalten und entschützt werden. Falls die Synthese in Lösung durchgeführt wurde, kann nach der Synthese der Lipid-modifizierten Nukleinsäure ( und ggf. vor der Abspaltung von dem Trägermaterial) ein Wasch- und Aufreinigungsschritt zur Entfernung nicht umgesetzter Reaktanten sowie von Lösungsmitteln und unerwünschten Nebenprodukten durchgeführt werden.
Gemäß einer weiteren Alternative wird die Nukleinsäure einer erfindungsgemäßen 3'-Lipid- modifizierten Nukleinsäure nach der Einführung des Lipids an eine reaktive Gruppe des Linkers synthetisiert oder als fertig synthetisierte oder aus Proben isolierte (kommerziell erhältliche) Nukleinsäure an die reaktive Gruppe des Linkers gebunden (siehe z.B. Fig. 2). Dazu kann z.B. eine erste reaktive Gruppe eines wie oben beschriebenen Linkers mit einem wie zuvor beschriebenen Lipid umgesetzt werden. Danach wird bevorzugt in einem zweiten Schritt eine zweite reaktive Gruppe des Linkers mit einer säurestabilen Schutzgruppe versehen, z.B. DMT, Fmoc, etc., um eine spätere Bindung der Nukleinsäure an diese reaktive Gruppe zu ermöglichen. Danach kann über eine dritte reaktive Gruppe des Linkers der Linker direkt oder indirekt an eine Festphase gebunden werden. Eine indirekte Bindung ist z.B. über ein (Kopplungs-)Molekül möglich, das sich sowohl kovalent an den Linker wie auch an die Festphase binden lässt. Ein solches (Kopplungs-)Molekül ist z.B. ein Succinylrest, etc. wie hier im folgenden beschrieben. Anschließend erfolgt üblicherweise die Entfernung der Schutzgruppe an der dritten reaktiven Gruppe des Linkers
sowie die Bindung oder Synthese einer Nukleinsäure an der nun zugänglichen reaktiven Gruppe. Abschließend erfolgt typischerweise die Abspaltung der Lipid-modifizierten Nukleinsäure vom Trägermaterial (und ggf. die Entfernung der Schutzgruppen an der Nukleinsäure). Optional kann jedoch auch an das 3'-Ende der gekoppelten Nukleinsäure ein weiteres Lipid gekoppelt werden, bevorzugt gemäß einem der zuvor beschriebenen Schritte.
Entsprechend einer Variante dieser vorgenannten Alternative kann ein wie oben beschriebener Linker über eine erste reaktive Gruppe direkt oder indirekt an eine Festphase gebunden werden. An eine zweite reaktive Gruppe des Linkers wird dann zunächst eine säurestabile Schutzgruppe gebunden. Nach Bindung der Schutzgruppe an die zweite reaktive Gruppe kann an eine dritte reaktive Gruppe des Linkers zunächst ein wie oben beschriebenes Lipid gebunden werden. Anschließend erfolgt ebenfalls bevorzugt die Entfernung der Schutzgruppe an der dritten reaktiven Gruppe des Linkers, die Bindung oder Synthese einer Nukleinsäure an der nun zugänglichen reaktiven Gruppe sowie die Abspaltung der Lipid-modifizierten Nukleinsäure vom Trägermaterial (und ggf. die Entfernung der Schutzgruppen an der Nukleinsäure).
Gemäß einer besonders bevorzugten Ausführungsform der 3'-Lipid-Modifizierung kann eine Lipid-modifizierte Nukleinsäure über einen Linker mit drei reaktiven Gruppen (eine trifunktionelle Ankerverbindung) auf Basis eines Glycerin-Grundkörpers (C3-Anker) sowie mit einem monofunktionellen Lipid, wie z.B. einem Palmitylrest, Cholesterol oder Tocopherol, synthetisiert werden. Als Ausgangsstoff für die Synthese des Linkers kann z.B. α,ß-lsopropyliden-Glycerin (ein Glycerin enthaltend eine Ketalschutzgruppe) eingesetzt werden, das bevorzugt zunächst mit Natriumhydrid in das Alkoholat überführt und mit Hexadecylbromid und einem Lipid in einer Williamson-Synthese zum entsprechenden Ether umgesetzt wird. Alternativ kann die Etherbindung in der ersten Stufe mit einer anderen Methode geknüpft werden, z.B. durch Bildung eines Tosylats des α,ß-lsopropyliden- Glycerins, und der Umsetzung des Tosylats mit der reaktiven Gruppe eines Lipids, z.B. einem acidischen Proton, zum entsprechenden Ether. In einer zweiten Stufe kann die Ketal- Schutzgruppe mit einer Säure, z.B. Essigsäure, verdünnte Salzsäure, etc. entfernt und anschließend die primäre Hydroxygruppe des Diols selektiv durch Dimethoxytritylchlorid (DMT-CI) geschützt werden. In der letzten Stufe erfolgt bevorzugt die Umsetzung des aus
dem vorangegangenen Schritt erhaltenen Produkts mit Bernsteinsäureanhydrid zum Succinat mit DMAP als Katalysator. Ein solcher Linker ist z.B. zur Bindung von Palmitylresten oder Tocopherol als Lipid besonders geeignet (siehe z.B. Fig. 2).
Nach einer anderen Alternative der 3'-Lipid-Modifizierung kann eine Lipid-modifizierte Nukleinsäure über Verwendung eines (bifunktionellen) Lipids, wie z.B. Polyethylenglykol (PEG) oder Hexaethylenglykol (HEG), ohne einen wie oben beschriebenen Linker zu verwenden. Solche bifunktionelle Lipide weisen typischerweise zwei wie oben beschriebene funktionelle Gruppen auf, wobei bevorzugt ein Ende des bifunktionellen Lipids über ein (Kopplungs-)Molekül, z.B. einen basenlabilen Succinylanker, etc. wie hier beschrieben, an das Trägermaterial gebunden werden kann und auf das andere Ende des bifunktionellen Lipids die Nukleinsäure aufsynthetisiert werden kann (E. Bayer, M. Maier, K. Bleicher, H.-J. Gaus Z. Naturforsch. 50b (1995) 671). Durch das Wegfallen der dritten Funktionalisierung bzw. eines wie zuvor verwendeten Linkers vereinfacht sich die Synthese einer solchen erfindungsgemäßen Lipid-modifizierten Nukleinsäure (siehe z.B. Fig. 3). Zur Herstellung wird typischerweise das erfindungsgemäß verwendete bifunktionelle Lipid, z.B. Polyethylenglykol, zunächst mit einer Schutzgruppe, z.B. DMT, monosubstituiert. In einer zweiten Stufe erfolgt üblicherweise eine Veresterung des an einer reaktiven Gruppe geschützen Lipids mit Bernsteinsäureanhydrid unter Katalyse von DMAP zum Succinat. Danach kann in einer dritten Stufe das bifunktionelle Lipid an ein Trägermaterial gekoppelt und entschützt werden, worauf in einem vierten Schritt die Synthese der Nukleinsäure nach einem wie zuvor beschriebenen Verfahren erfolgt. Optional erfolgt danach die Entschützung der Nukleinsäure sowie die Abspaltung der Lipid-modifizierten Nukleinsäure vom Trägermaterial.
Entsprechend einer anderen bevorzugten Ausführungsform findet die Lipid-Modifikation am 5'-Ende der Nukleinsäure statt. Dabei erfolgt die Lipid-Modifikation typischerweise entweder nach Bereitstellung oder nach Synthese der erfindungsgemäß verwendeten Nukleinsäure. Die Bereitstellung der erfindungsgemäßen Nukleinsäure kann dabei - wie oben definiert - über eine direkte Synthese der Nukleinsäure oder über Zugabe einer bereits fertig synthetisierten oder aus Proben isolierten bzw. kommerziell erhältlichen Nukleinsäure durchgeführt werden. Eine Synthese der Nukleinsäure erfolgt dabei, bevorzugt analog zu dem zuvor Gesagten, nach im Stand der Technik bekannten Verfahren zur
Nuklei nsäuresynthese, noch stärker bevorzugt nach dem Phosphoramiditverfahren (siehe z.B. Fig. 4).
Gemäß einer besonders bevorzugten Ausfuhrungsform erfolgt die Lipid-Modifikation am 5'- Ende der erfindungsgemäß verwendeten Nukleinsäure durch besonders modifizierte Phosphoramidite im Anschluß an ein Phosphoramiditverfahren zur Synthese der Nukleinsäure. Solche relativ einfach synthetisch zugänglichen Amidite werden üblicherweise als letztes Monomer auf eine kommerziell erhältliche oder auf eine fertig synthetisierte Nukleinsäure aufgekuppelt. Diese Reaktionen zeichnen sich durch eine relativ schnelle Reaktionskinetik und sehr hohe Kupplungsausbeuten aus. Die Synthese der modifizierten Amidite erfolgt bevorzugt durch die Umsetzung eines Phosphoramidits, z.B. ß -Cyanoethyl-monochlorophosphoramidit (Phosphorigsäure-mono-(2-cyanoethylester)- diisopropyl-amid-chlorid), mit einem in einem geeigneten Lösungsmittel, z.B. in absolutem Dichlormethan, gelösten Alkohol eines wie oben definierten Lipids, z.B. einem Lipidalkohol von Tocopherol, Cholesterol, Hexadecanol, DMT-PEG, etc.. Ebenfalls bevorzugt wird der Reaktionslösung als Säurefänger DIPEA zugesetzt.
Diese für die Synthese der erfindungsgemäßen 5'-Lipid-modifizierten Nukleinsäuren verwendeten Phosphoramidite sind gegenüber Hydrolyse relativ beständig und können (vor der Synthese) mit Kieselgel chromatographisch gereinigt werden. Dafür wird dem Eluenten typischerweise eine kleine Menge einer schwachen Base wie z.B. Triethylamin zugesetzt, um eine Zersetzung des Amidits zu vermeiden. Wichtig ist dabei, dass diese Base wieder vollständig aus dem Produkt entfernt wird, um schlechte Kupplungsausbeuten zu vermeiden. Die kann z.B. durch einfaches Trocknen im Vakuum erfolgen, bevorzugt jedoch durch Reinigung der Phosphoramidite durch ihre Fällung aus tert-Buty I mety lether mit Pentan. Falls die verwendeten lipidmodifizierten Amidite eine sehr hohe Viskosität aufweisen, z.B. als zähes Öl vorliegen, kann auch eine (zügige) Säulenchromatographie erfolgen, welche es ermöglicht auf Triethylamin als Base zu verzichten. Eine solche Aufreinigung wird typischerweise jedoch nicht bei PEG-modifizierten Amiditen durchgeführt, da diese die säurelabile DMT-Schutzgruppe enthalten.
Für die Kupplungsreaktion der Lipid-modifizierten Phosphoramidite auf das 5'-Ende der erfindungsgemäß verwendeten Nukleinsäure werden bevorzugt solche Lösungsmittel
eingesetzt, in denen die verwendeten Amidite in ausreichender Weise löslich sind. So kann z.B. bedingt durch die hohe Lipophilie der erfindungsgemäß verwendeten Amidite deren Löslichkeit in Acetonitril begrenzt sein. Neben Acetonitril als typischerweise verwendetem Lösungsmittel wird daher für die Kupplungsreaktionen bevorzugt eine Lösung von chlorierten Kohlenwasserstoffen eingesetzt, z.B. eine 0,1 M Lösung in (absolutem) Dichlormethan . Die Verwendung von Dichlormethan erfordert jedoch einige Änderungen im Standardprotokoll des Synthesezyklus. So werden z.B., um das Ausfallen des Amidits in den Leitungen des Syntheseautomaten und auf dem Trägermaterial zu vermeiden, alle Ventile und Leitungen, die mit dem Amidit in Kontakt geraten, vor und nach dem eigentlichem Kupplungsschritt mit (absolutem) Dichlormethan gespült und trockengeblasen.
Bei Verwendung von Lipid-modifizierten Amiditen ergeben sich typischerweise hohe Kupplungsausbeuten, die mit der Kupplungsausbeute von üblicherweise im Stand der Technik verwendeten Amiditen vergleichbar sind. Dabei verläuft die Kinetik der Reaktion von Lipid-modifizierten Amiditen allgemein langsamer. Aus diesem Grund werden gegenüber Standardprotokollen die Kupplungszeiten bei Verwendung von Lipid- modifizierten Amiditen bevorzugt (deutlich) verlängert. Solche Kupplungszeiten können durch einen Fachmann einfach bestimmt werden. Da auf einen Capping-Schritt nach der Kupplung verzichtet werden kann, ist es ebenfalls möglich, bei Bedarf einen weiteren Synthesezyklus mit demselben Lipid-modifizierten Amiditen durchzuführen, um die Gesamtausbeute der Reaktion zu erhöhen. In diesem Fall wird üblicherweise, z.B. bei DMT-modifizierten Lipiden wie DMT-PEG, der Detritylierungsschritt nicht ausgeführt.
Bei der Synthese von erfindungsgemäßen 5 '-Lipid-modifizierten Nukleinsäuren kann der Phosphittriester, über den das Lipid an die Nukleinsäure gebunden wird, durch ein Sulfurierungsmittel oxidiert werden. Dazu wird bevorzugt ein solches Sulfurierungsmittel verwendet, das die Oxidation des Phosphotriesters möglichst vollständig erreicht. Andernfalls kann ggf. die Sulfurierungsreaktion, z.B. aus sterischen Gründen, so unvollständig ablaufen, dass nach der ammoniakalischen Abspaltung und Entschützung der MON nur wenig oder kein Produkt erhalten wird. Dieses Phänomen ist abhängig von der Art der Modifizierung, dem eingesetzten Sulfurierungsmittel und den Sulfurierungsbedingungen. Bevorzugt wird daher die Oxidation mit lod durchgeführt. Hierdurch wird zwar eine Phosphodiesterbindung eingeführt, durch die Nachbarschaft des
Lipidrestes ist jedoch nicht zu erwarten, dass diese Bindung als Substrat von Nukleasen erkannt wird.
Die in der erfindungsgemäß verwendeten Lipid-modifizierten Nukleinsäure enthaltenen Linker oder (bifunktionellen) Lipide oder ggf. die verwendeten Nukleinsäuren können, wie zuvor beschrieben, direkt oder indirekt an ein Trägermaterial gekoppelt werden. Eine direkte Kopplung erfolgt dabei bevorzugt unmittelbar mit dem Trägermaterial, während eine indirekte Kopplung an das Trägermaterial typischerweise über ein weiteres (Kopplungs- )Molekül erfolgt. Die durch die Kopplung an ein Trägermaterial entstehende Bindung weist bevorzugt eine (spaltbare) kovalente Bindung mit dem Linker oder bifunktionellen Lipid und/oder eine (spaltbare) kovalente Bindung mit der Festphase auf. Als (Kopplungs- )Molekül geeignete Verbindungen sind z.B. Dicarbonsäuren, bspw. Succinylreste (=Succinylanker), Oxalylreste (=Oxalylanker), etc.. Linker, (bifunktionelle) Lipide oder ggf. verwendete Nukleinsäuren, die wie z.B. Aminoalkylreste (bspw. Aminopropyl- oder Aminohexanylreste) eine freie Aminofunktion tragen, können über einen Phtalimid-Linker an das Trägermaterial gebunden werden. Thiolhaltige Linker, (bifunktionelle) Lipide oder ggf. verwendete Nukleinsäuren können als Disulfid an das Trägermaterial gebunden werden. Als Trägermaterial sind im Zusammenhang mit dieser Erfindung insbesondere Festphasen wie CPG, Tentagel®, aminofunktionalisiertes PS-PEG (Tentagel® S NH2) etc., bevorzugt Tentagel® oder aminofunktionalisiertes PS-PEG (Tentagel® S NH2), geeignet. Gemäß einer besonderen Ausführungsform können zur Kopplung an ein Trägermaterial z.B. die Succinate der beschriebenen erfindungsgemäß verwendeten Linker oder bifunktionellen Lipide bevorzugt mit TBTU/NMM (1 H-Benzotriazol-1 -yl-1 , 1 ,3,3- tetramethyluroniumtetrafluoroborat / N-Methylmorpholin) als Kupplungsreagenz auf aminofunktionalisiertem PS-PEG (Tentagel® S NH2) gekoppelt werden. Typischerweise werden bei PS-PEG Trägermaterialien im üblicherweise verwendeten 1 μmol-Maßstab die besten Resultate mit Beladungen zwischen 50 und 100 μmol/g erreicht (E. Bayer, K. Bleicher, M. Maier Z. Naturforsch. 50b (1995) 1096). Sollen erfindungsgemäß jedoch Nukleotide im größeren Maßstab (large scale) synthetisiert werden, ist die Beladung der Trägermaterialien bevorzugt möglichst hoch (> 100 μmol). Erfindungsgemäß führt ein solches Verfahren ebenfalls zu guten Kupplungsausbeuten (M. Gerster, M. Maier, N. Clausen, J. Schewitz, E. Bayer Z. Naturforsch. 52b (1997) 1 10). So können z.B. Trägermaterialien wie z.B. Harze mit bis zu 138 μmol/g Beladung oder ggf. mehr mit guten
Syntheseausbeuten verwendet werden. Da die Kupplungsausbeuten mit den zuvor beschriebenen Linkern oder bifunktionellen Lipiden annähernd 100% betragen, kann über die Stöchiometrie dieser Verbindungen relativ genau die Beladung des Trägermaterials eingestellt werden. Die Kontrolle der Beladung erfolgt bevorzugt über die spektroskopische Quantifizierung der abgespaltenen DMT-Schutzgruppe (siehe experimenteller Teil). Die auf dem Trägermaterial noch vorhandenen restlichen Aminofunktionen können mit Acetanhydrid gecappt werden. Dieses Capping wird normalerweise im Anschluss an die Belegung des Trägermaterials durchgeführt, kann aber auch direkt bei der Nukleinsäure- Synthese, z.B. am DNA-Synthesizer, ausgeführt werden. Für die Synthese von Lipid- modifizierten Nukleinsäuren auf den derivatisierten PS-PEG-Trägermaterialien werden bevorzugt speziell für Tentagel® entwickelte Synthesezyklen verwendet, die die charakteristischen Eigenschaften des Materials berücksichtigen (E. Bayer, M. Maier, K. Bleicher, H.-J. Gaus Z. Naturforsch. 50b (1995) 671 , E. Bayer, K. Bleicher, M. Maier Z Naturforsch. 50b (1995) 1096.). Bevorzugte Änderungen im Vergleich zum Standardprotokoll umfassen.
• Verlängerte Reaktionszeiten bei den Kupplungs-, Capping- und Oxidationsschritten;
• Erhöhung der Zahl der Detritylierungsschritte;
• Verlängerte Waschschritte nach jedem Schritt; • Verwendung einer ascorbinsäurehaltigen Waschlösung (0,1 M in
Dioxan/Wasser = 9:1 ) nach dem üblicherweise notwendigen Oxidationsschritt (zur Oxidation des Phosphittriesters) während des Amiditverfahrens, um Spuren von lod zu entfernen.
Dabei ist zu beachten, dass die Art der Modifizierung einen Einfluss auf die einzelnen Schritte des Synthesezyklus haben kann. Z.B. wird bei PEG,50o-derivatisierten Trägermaterialien eine stark verlangsamte Reaktionskinetik beobachtet, was eine nochmalige Erweiterung der Detritylierungsschritte und eine zusätzliche Verlängerung der Kupplungsdauer notwendig macht. Solche Änderungen und Anpassungen liegen im Bereich des normalen Könnens eines Fachmanns und können im Rahmen der vorliegenden Offenbarung jederzeit vorgenommen werden. Mit diesen derart veränderten Reaktionszyklen können sowohl Lipid-modifizierte Phosphordiester als auch Phosphorothioate synthetisiert werden. Die Kupplungsausbeuten von Amiditen auf
erfindungsgemäß verwendeten Linkern oder bifunktionellen Lipiden werden nicht durch die Lipidreste beeinträchtigt, sondern entsprechen üblichen Werten (97-99%). Die Möglichkeit der 5'-Derivatisierung sowie die Einführung weiterer Modifizierungen z.B. an Base, Zucker oder Phosphatrückgrat bleibt bei Verwendung solcher 3 '-Modifizierungen bestehen.
Gemäß einer weiteren Ausführungsform kann das erfindungsgemäße immunstimulierende Adjuvanz mit weiteren, im Stand der Technik bekannten Adjuvanzien kombiniert werden.
Die vorliegende Erfindung betrifft ebenfalls pharmazeutische Zusammensetzungen enthaltend ein wie oben beschriebenes immunstimulierendes Adjuvanz, mindestens einen Wirkstoff und optional einen pharmazeutisch geeigneten Träger und/oder weitere Hilfs- und Zusatzstoffe und/oder Adjuvanzien.
Die pharmazeutischen Zusammensetzungen gemäß der vorliegenden Erfindung umfassen typischerweise eine sichere und effektive Menge des erfindungsgemäßen immunstimulierenden Adjuvanz. Wie hier verwendet bedeutet "sichere und effektive Menge" eine solche Menge einer Verbindung, die ausreichend ist, um signifikant eine positive Modifikation eines zu behandelnden Zustands zu induzieren, z.B. einer Tumoroder Infektionserkrankung. Gleichzeiztig ist eine "sichere und effektive Menge" jedoch gering genug, um schwerwiegende Nebeneffekte zu vermeiden, also ein vernünftiges Verhältnis von Vorteil und Risiko zu ermöglichen. Die Festlegung dieser Grenzen liegen typischerweise innerhalb des Bereichs vernünftiger medizinischer Urteilskraft. In Bezug auf das erfindungsgemäße immunstimulierende Adjuvanz bedeutet der Begriff „sichere und effektive Menge" bevorzugt eine solche Menge, die geeignet ist, das Immunsystem in einer solchen Weise zu stimulieren, dass keine überschießenden bzw. schädlichen Immunreaktionen erzielt werden, jedoch bevorzugt auch keine solchen Immunreaktionen unterhalb eines messbaren Niveaus. Eine „sichere und effektive Menge" eines erfindungsgemäßen Adjuvanz bzw. eines erfindungsgemäßen Adjuvanz wird im Zusammenhang mit dem besonderen zu behandelnden Zustand variieren, sowie dem Alter und dem physischen Zustand des zu behandelnden Patienten, der Schwere des Zustandes, der Dauer der Behandlung, der Natur der begleitenden Therapie, des verwendeten
besonderen pharmazeutisch geeigneten Trägers und ähnlichen Faktoren innerhalb des Wissens und der Erfahrung des begleitenden Arztes. Die erfindungsgemäßen pharmazeutischen Zusammensetzungen können erfindungsgemäß für humane und wie auch für veterinärmedizinische Zwecke eingesetzt werden.
Zusätzlich zu dem erfindungsgemäßen immunstimulierenden Adjuvanz enthält die erfindungsgemäße pharmazeutische Zusammensetzung bevorzugt mindestens einen Wirkstoff. Ein Wirkstoff ist in diesem Zusammenhang eine Verbindung, die einen therapeutischen Effekt gegen eine bestimmte Indikation aufweist, bevorzugt Krebserkrankungen oder Infektionserkrankungen. Solche Verbindungen umfassen, ohne darauf beschränkt zu sein, Peptide, Proteine, Nukleinsäuren, (therapeutisch wirksame) niedermolekulare organische oder anorganische Verbindungen (Molekulargewicht kleiner als 5.000, bevorzugt kleiner als 1000), Zucker, Antigene, oder Antikörper, bereits im Stand der Technik bekannte Therapeutika, etc.. Gemäß einer besonderen Ausführungsform kann das zuvor beschriebene erfindungsgemäße immunstimulierende Adjuvanz selbst ein Wirkstoff sein. Dies ist insbesondere dann der Fall, wenn das Lipid der Lipid-modifizierten Nukleinsäure ein therapeutisch aktives Molekül darstellt, wie z.B. ein wie oben beschriebenes Vitamin, oder Steroid, bspw. α-Tocopherol (Vitamin E), D-α-Tocopherol, L- α-Tocopherol, D,L-α-Tocopherol, Vitamin E-Succinat (VES), Vitamin A und dessen Derivate, Vitamin D und dessen Derivate, Vitamin K und dessen Derivate, etc..
Gemäß einer ersten Ausführungsform liegt der in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthaltene Wirkstoff als Antigen bzw. als Immunogen vor. Unter einem „Antigen" wie auch einem „Immunogen" ist dabei jede Struktur zu verstehen, welche die Bildung von Antikörpern und/oder die Aktivierung einer zellulären Immunantwort hervorrufen kann. Erfindungsgemäß werden daher die Begriffe „Antigen" und „Immunogen" synonym verwendet. Beispiele von Antigenen sind Peptide, Polypeptide, also auch Proteine, Zellen, Zellextrakte, Polysaccharide, Polysaccharidkonjugate, Lipide, Glykolipide und Kohlenhydrate. Als Antigene kommen bspw. Tumorantigene, virale, bakterielle, Pilz- und protozoologische Antigene in Betracht. Bevorzugt sind dabei Oberflächenantigene von Tumorzellen und Oberflächenantigene, insbesondere sekretierte Formen, viraler, bakterieller, Pilz- oder protozoologischer Pathogene. Selbstverständlich kann das Antigen
z.B. in einer erfindungsgemäßen Vakzine, auch in Form eines an einen geeigneten Träger gekoppelten Haptens vorliegen.
Der in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthaltene Wirkstoff kann gemäß einer zweiten Ausführungsform als Antikörper vorliegen. In diesem Zusammenhang kann jeder therapeutisch geeignete Antikörper eingesetzt werden. Besonders bevorzugt ist erfindungsgemäß ein Antikörper, der gegen Antigene, Proteine oder Nukleinsäuren gerichtet ist, die eine wesentliche Rolle bei Krebserkrankungen oder Infektionserkrankungen aufweisen, z.B. Zelloberflächenproteine, Tumorsupressorgene oder deren Inhibitoren, Wachstums- und Elongationsfaktoren, Apoptoserelevante Proteine, Tumorantigene, oder Antigene, wie zuvor beschrieben, etc..
Gemäß einer dritten Ausführungsform liegt der in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthaltene Wirkstoff als Nukleinsäure vor. Eine solche Nukleinsäure kann einzelsträngig oder doppelsträngig sein, und als Homo- oder-
Heteroduplex sowie linear oder zirkulär vorliegen. Eine als Wirkstoff in der pharmazeutischen Zusammensetzung enthaltene Nukleinsäure ist dabei in ihrer Länge nicht beschränkt und kann jede natürlich vorkommende Nukleinsäuresequenz oder deren Komplement oder ein Fragment davon umfassen. Ebenfalls kann die in diesem
Zusammenhang eingesetzte Nukleinsäure teilweise oder vollständig synthetischer Natur sein. Beispielsweise kann die Nukleinsäure eine solche Nukleinsäure umfassen, die ein
(therapeutisch relevantes) Protein kodiert, und/oder die in der Lage ist, eine Immunreaktion hervorzurufen, z.B. ein Antigen oder eine ein Antigen kodierende Nukleinsäure. Ein Antigen ist dabei bevorzugt ein Antigen wie zuvor beschrieben.
Gemäß einer ersten bevorzugten Alternative der vorgenannten Ausführungsform liegt die als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung enthaltene Nukleinsäure als mRNA vor. Eine solche mRNA kann zur erfindungsgemäßen pharmazeutischen Zusammensetzung in ihrer nackten Form zugegeben werden oder in einer stabilisierten Form, die den Abbau der Nukleinsäure in vivo, z.B. durch Exo- und/oder Endonukleasen verringert oder gar verhindert.
Beispielsweise kann die in der erfindungsgemäßen pharmazeutischen Zusammensetzung als Wirkstoff enthaltene mRNA mit einem wie oben definierten 5'-Cap und/oder einem PoIy-A- Schwanz am 3'-Ende von mindestens 50 Nukleotiden, vorzugsweise mindestens 70 Nukleotiden, mehr bevorzugt mindestens 100 Nukleotiden, besonders bevorzugt mindestens 200 Nukleotiden stabilisiert werden. Wie bereits erwähnt, ist die endständige Struktur ist dabei in vivo von entscheidender Bedeutung. Über diese Strukturen wird die RNA als mRNA erkannt und der Abbau reguliert. Darüber hinaus gibt es jedoch weitere Prozesse, die RNA stabilisieren bzw. destabilisieren. Viele diese Prozesse sind noch unbekannt, oftmals scheint jedoch eine Wechselwirkung zwischen der RNA und Proteinen dafür maßgeblich zu sein. Bspw. wurde kürzlich ein „mRNA-Surveillance-System" beschrieben (Hellerin und Parker, Ann. Rev. Genet. 1999, 33: 229 bis 260), bei dem durch bestimmte Feedback-Protein-Wechselwirkungen im Cytosol unvollständige oder Nonsense- mRNA erkannt und dem Abbau zugänglich gemacht wird, wobei ein Hauptteil dieser Prozesse durch Exonucleasen vollzogen wird.
Ebenso kann die Stabilisierung der in der erfindungsgemäßen pharmazeutischen Zusammensetzung als Wirkstoff enthaltenen mRNA dadurch erfolgen, dass die mRNA mit einer kationischen Verbindung, insbesondere einer polykationischen Verbindung, bspw. einem (poly)kationischen Peptid oder Protein, assoziiert bzw. komplexiert oder daran gebunden ist. Insbesondere ist dabei die Verwendung von Protamin, Nukleolin, Spermin oder Spermidin als polykationischem, Nukleinsäure-bindenden Protein besonders wirksam. Des weiteren ist die Verwendung anderer kationischer Peptide oder Proteine, wie PoIy-L- Lysin oder Histonen, ebenfalls möglich. Diese Vorgehensweise zur Stabilisierung von mRNA ist in EP-A-1083232 beschrieben, deren diesbezüglicher Offenbarungsgehalt in die vorliegende Erfindung vollumfänglich eingeschlossen ist. Weitere bevorzugte kationische Substanzen, die zur Stabilisierung der als Wirkstoff enthaltnen mRNA verwendet werden können, schließen kationische Polysaccharide, bspw. Chitosan, Polybren, Polyethylenimin (PEI) oder Poly-L-Lysin (PLL), etc., ein. Die Assoziierung oder Komplexierung der mRNA mit kationischen Verbindungen erhöht bevorzugt neben der bereits vorteilhaften Wirkung der erfindungsgemäßen Lipid-modifizierten Nukleinsäure als Adjuvanz bei der Verbesserung der Zellpermeabilität den Transfer der als Wirkstoff enthaltenen mRNA in die zu behandelnden Zellen bzw. den zu behandelnden Organismus.
Eine weitere Möglichkeit zur Stablisierung von mRNA, die als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung vorliegen kann, ist die gezielte Veränderung der Sequenz der mRNA durch Entfernen oder Veränderung sogenannter destabilisierender Sequenzelemente (DSE). An diese destabilisierende Sequenzelemente (DSE), die insbesondere bei eukaryotischer mRNA auftreten, können Signalproteine binden und den enzymatischen Abbau der mRNA in vivo regulieren. Daher werden zur weiteren Stabilisierung der als Wirkstoff enthaltenen mRNA bevorzugt ein oder mehrere Veränderungen gegenüber dem entsprechenden Bereich der Wildtyp-mRNA vorgenommen, so dass keine destabilisierenden Sequenzelemente enthalten sind. Selbstverständlich ist es erfindungsgemäß ebenfalls bevorzugt, gegebenenfalls in den nicht-translatierten Bereichen (3'- und/oder 5'-UTR) vorhandene DSE aus der mRNA zu eliminieren. Beispiele der vorstehenden DSE sind AU-reiche Sequenzen ("AURES"), die in 3'-UTR-Abschnitten zahlreicher instabiler mRNA vorkommen (Caput et al., Proc. Natl. Acad. Sei. USA 1986, 83: 1670 bis 1674). Die als Wirkstoff eingesetzte mRNA ist daher vorzugsweise derart gegenüber der Wildtyp-mRNA verändert, dass sie keine derartigen destabilisierenden Sequenzen aufweist. Dies gilt auch für solche Sequenzmotive, die von möglichen Endonukleasen erkannt werden, bspw. die Sequenz GAACAAG, die im 3' UTR-Segment des für den Transferin-Rezeptor kodierenden Gens enthalten ist (Binder et al., EMBO J. 1994, 13: 1969 bis 1980). Vorzugsweise werden auch diese Sequenzmotive aus der erfindungsgemäßen Lipid-modifizierten Nukleinsäure eliminiert.
Die als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung gegebenenfalls vorliegende mRNA kann weiterhin z.B. für eine gegebenenfalls gewünschte effiziente Translation, so verändert werden, dass eine wirksame Bindung der Ribosomen an die Ribosomen-Bindungsstelle (Kozak-Sequenz: GCCGCCACCAUGG, das AUG bildet das Startcodon) erfolgt. Diesbezüglich ist festgestellt worden, dass ein erhöhter A/U-Gehalt um diese Stelle herum eine effizientere Ribosomen-Bindung an die mRNA ermöglicht.
Des weiteren ist es möglich, in die als Wirkstoff verwendete mRNA eine oder mehrere sog. IRES (engl, "internal ribosomal entry side") einzufügen. Eine IRES kann so als alleinige Ribosomen-Bindungsstelle fungieren, sie kann jedoch auch zur Bereitstellung einer mRNA dienen, die mehrere Peptide bzw. Polypeptide kodiert, die unabhängig voneinander durch die Ribosomen translatiert werden sollen ("multicistronische mRNA"). Beispiele
erfindungsgemäß verwendbarer IRES-Sequenzen sind diejenigen aus Picornaviren (z.B. FMDV), Pestviren (CFFV), Polioviren (PV), Enzephalo-Myocarditis-Viren (ECMV), Maul-und- Klauenseuche-Viren (FMDV), Hepatitis-C-Viren (HCV), Klassisches-Schweinefieber-Viren (CSFV), Murines-Leukoma-Virus (MLV), Simean-Immundefizienz-Viren (SIV) oder Cricket- Paralysis-Viren (CrPV).
Die als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung gegebenenfalls verwendete mRNA kann ebenfalls in ihren 5'- und/oder 3'-nicht translatierten Bereichen Stabilisierungssequenzen aufweisen, die befähigt sind, die Halbwertszeit der mRNA im Cytosol zu erhöhen. Diese Stabilisierungssequenzen können eine 100%ige Sequenzhomologie zu natürlich vorkommenden Sequenzen aufweisen, die in Viren, Bakterien und Eukaryoten auftreten, sie können aber auch teilweise oder vollständig synthetischer Natur sein. Als Beispiel für stabilisierende Sequenzen, die in der vorliegenden Erfindung verwendbar sind, können die nicht-translatierten Sequenzen (UTR) des ß- Globingens, bspw. von Homo sapiens oder Xenopus laevis, genannt werden. Ein anderes Beispiel einer Stabilisierungssequenz weist die allgemeine Formel (C/U)CCANxCCC(U/A)PyxUC(C/U)CC auf, die im 31UTR der sehr stabilen mRNA enthalten ist, die für α-Globin, α-(l)-Collagen, 15-Lipoxygenase oder für Tyrosin-Hydroxylase kodiert (vgl. Holcik et al., Proc. Natl. Acad. Sei. USA 1997, 94: 2410 bis 2414). Selbstverständlich können derartige Stabilisierungssequenzen einzeln oder in Kombination miteinander als auch in Kombination mit anderen, einem Fachmann bekannten, Stabilisierungssequenzen verwendet werden.
Zur weiteren Erhöhung einer gegebenenfalls gewünschten Translation kann die als Wirkstoff verwendete mRNA gegenüber einer entsprechenden Wildtyp-mRNA folgende Modifikationen aufweisen, die entweder alternativ oder in Kombination vorliegen können. Zum einen kann der G/C-Gehalt des für ein Peptid oder Polypeptid kodierenden Bereichs der modifizierten mRNA größer sein als der G/C-Gehalt des kodierenden Bereichs der für das Peptid oder Polypeptid kodierenden Wildtyp-mRNA, wobei die kodierte Aminosäuresequenz gegenüber dem Wildtyp unverändert ist. Diese Modifikation beruht auf der Tatsache, dass für eine effiziente Translation einer mRNA die Sequenzabfolge des zu translatierenden Bereichs der mRNA wesentlich ist. Dabei spielt die Zusammensetzung und die Abfolge der verschiedenen Nukleotide eine große Rolle. Insbesondere sind Sequenzen
mit erhöhtem G(Guanosin)/C(Cytosin)-Gehalt stabiler als Sequenzen mit einem erhöhten A(Adenosin)/U(Uracil)-Gehalt. Daher werden erfindungsgemäß unter Beibehaltung der translatierten Aminosäureabfolge die Codons gegenüber der Wildtyp-mRNA derart variiert, dass sie vermehrt G/C-Nukleotide beinhalten. Da mehrere Codons für ein und dieselbe Aminosäure kodieren (Degeneration des genetischen Codes), können die für die Stabilität günstigsten Codons ermittelt werden (alternative Codonverwendung, engl, "codon usage"). In Abhängigkeit von der durch die mRNA zu kodierenden Aminosäure sind unterschiedliche Möglichkeiten zur Modifikation der mRNA-Sequenz gegenüber der Wildtyp-Sequenz möglich. Im Fall von Aminosäuren, die durch Codons kodiert werden, die ausschließlich G- oder C- Nukleotide enthalten, ist keine Modifikation des Codons erforderlich. So erfordern die Codons für Pro (CCC oder CCG), Arg (CGC oder CGG), AIa (GCC oder GCG) und GIy (GGC oder GGG) keine Veränderung, da kein A oder U vorhanden ist. In folgenden Fällen werden die Codons, welche A-und/oder U-Nukleotide enthalten, durch Substituieren anderer Codons, welche die gleichen Aminosäuren kodieren, jedoch kein A und/oder U enthalten, verändert. Beispiele sind: Die Codons für Pro können von CCU oder CCA zu CCC oder CCG verändert werden; die Codons für Arg können von CGU oder CGA oder AGA oder AGG zu CGC oder CGG verändert werden; die Codons für AIa können von GCU oder GCA zu GCC oder GCG verändert werden; die Codons für GIy können von GGU oder GGA zu GGC oder GGG verändert werden. In anderen Fällen können A bzw. U-Nukleotide zwar nicht aus den Codons eliminiert werden, es ist jedoch möglich, den A- und U-Gehalt durch Verwendung von Codons zu vermindern, die weniger A- und/oder U-Nukleotide enthalten. Zum Beispiel: Die Codons für Phe können von UUU zu UUC verändert werden; die Codons für Leu können von UUA, CUU oder CUA zu CUC oder CUG verändert werden; die Codons für Ser können von UCU oder UCA oder AGU zu UCC, UCG oder AGC verändert werden; das Codon für Tyr kann von UAU zu UAC verändert werden; das Stop-Codon UAA kann zu UAG oder UGA verändert werden; das Codon für Cys kann von UGU zu UGC verändert werden; das Codon His kann von CAU zu CAC verändert werden; das Codon für GIn kann von CAA zu CAG verändert werden; die Codons für He können von AUU oder AUA zu AUC verändert werden; die Codons für Thr können von ACU oder ACA zu ACC oder ACG verändert werden; das Codon für Asn kann von AAU zu AAC verändert werden; das Codon für Lys kann von AAA zu AAG verändert werden; die Codons für VaI können von GUU oder GUA zu GUC oder GUG verändert werden; das Codon für Asp kann von GAU zu GAC verändert werden; das Codon für GIu
kann von GAA zu GAG verändert werden. Im Falle der Codons für Met (AUG) und Trp (UGG) besteht hingegen keine Möglichkeit der Sequenzmodifikation. Die vorstehend aufgeführten Substitutionen können selbstverständlich einzeln aber auch in allen möglichen Kombinationen zur Erhöhung des G/C-Gehalts der modifizierten mRNA gegenüber der ursprünglichen Sequenz verwendet werden. So können beispielsweise alle in der ursprünglichen (Wildtyp-) Sequenz auftretenden Codons für Thr zu ACC (oder ACG) verändert werden. Vorzugsweise werden jedoch Kombinationen der vorstehenden Substitutionsmöglichkeiten genutzt, z.B.: Substitution aller in der ursprünglichen Sequenz für Thr kodierenden Codons zu ACC (oder ACG) und Substitution aller ursprünglich für Ser kodierenden Codons zu UCC ( oder UCG oder AGC); Substitution aller in der ursprünglichen Sequenz für Me kodierenden Codons zu AUC und Substitution aller ursprünglich für Lys kodierenden Codons zu AAG und Substitution aller ursprünglich für Tyr kodierenden Codons zu UAC; Substitution aller in der ursprünglichen Sequenz für VaI kodierenden Codons zu GUC (oder GUG) und Substitution aller ursprünglich für GIu kodierenden Codons zu GAG und Substitution aller ursprünglich für AIa kodierenden Codons zu GCC (oder GCG) und Substitution aller ursprünglich für Arg kodierenden Codons zu CGC (oder CGG); Substitution aller in der ursprünglichen Sequenz für VaI kodierenden Codons zu GUC (oder GUG) und Substitution aller ursprünglich für GIu kodierenden Codons zu GAG und Substitution aller ursprünglich für AIa kodierenden Codons zu GCC (oder GCG) und Substitution aller ursprünglich für GIy kodierenden Codons zu GGC (oder GGG) und Substitution aller ursprünglich für Asn kodierenden Codons zu AAC; Substitution aller in der ursprünglichen Sequenz für VaI kodierenden Codons zu GUC (oder GUG) und Substitution aller usprünglich für Phe kodierenden Codons zu UUC und Substitution aller ursprünglich für Cys kodierenden Codons zu UGC und Substitution aller ursprünglich für Leu kodierenden Codons zu CUG (oder CUC) und Substitution aller ursprünglich für GIn kodierenden Codons zu CAG und Substitution aller ursprünglich für Pro kodierenden Codons zu CCC (oder CCG); usw.. Vorzugsweise wird der G/C-Gehalt des für das Peptid bzw. Polypeptid kodierenden Bereichs (bzw. jedes anderen gegebenenfalls vorhandenen weiteren Abschnitts) der mRNA um mindestens 7%-Punkte, mehr bevorzugt um mindestens 15%-Punkte, besonders bevorzugt um mindestens 20%- Punkte gegenüber dem G/C-Gehalt des kodierten Bereichs der für das entsprechende Peptid bzw. Polypeptid kodierenden Wildtyp-mRNA erhöht. Besonders bevorzugt ist es in diesem
Zusammenhang, den G/C-Gehalt der derart modifizierten mRNA im Vergleich zur Wildtyp- Sequenz maximal zu erhöhen.
Eine weitere bevorzugte Modifikation einer als Wirkstoff in der pharmazeutishen Zusammensetzung verwendeten mRNA beruht auf der Erkenntnis, dass die Translationseffizienz auch durch eine unterschiedliche Häufigkeit im Vorkommen von tRNAs in Zellen bestimmt wird. Sind daher in einer RNA-Sequenz vermehrt sogenannte "seltene" Codons vorhanden, so wird die entsprechende mRNA deutlich schlechter translatiert als in dem Fall, wenn für relativ "häufige" tRNAs kodierende Codons vorhanden sind. Somit wird erfindungsgemäß in der als Wirkstoff verwendeten mRNA, der kodierende Bereich gegenüber dem entsprechenden Bereich der Wildtyp-mRNA derart verändert, dass mindestens ein Codon der Wildtyp-Sequenz, das für eine in der Zelle relativ seltene tRNA kodiert, gegen ein Codon ausgetauscht wird, das für eine in der Zelle relativ häufige tRNA kodiert, welche die gleiche Aminosäure trägt wie die relativ seltene tRNA. Durch diese Modifikation werden die RNA-Sequenzen derart modifiziert, dass Codons eingefügt werden, für die häufig vorkommende tRNAs zur Verfügung stehen. Welche tRNAs relativ häufig in der Zelle vorkommen und welche demgegenüber relativ selten sind, ist einem Fachmann bekannt; vgl. bspw. Akashi, Curr. Opin. Genet. Dev. 2001, 1 1 (6): 660-666. Durch diese Modifikation können erfindungsgemäß alle Codons der Wildtyp-Sequenz, die für eine in der Zelle relativ seltene tRNA kodieren, jeweils gegen ein Codon ausgetauscht werden, das für eine in der Zelle relativ häufige tRNA kodiert, welche jeweils die gleiche Aminosäure trägt wie die relativ seltene tRNA. Besonders bevorzugt ist es, den in der wie vorstehend beschriebenen mRNA erhöhten, insbesondere maximalen, sequenziellen G/C-Anteil mit den "häufigen" Codons zu verknüpfen, ohne die Aminosäuresequenz eines durch den kodierenden Bereich des mRNA-kodierten antigenen Peptids bzw. Polypeptids (ein oder mehrere) zu verändern.
Gemäß einer zweiten bevorzugten Alternative der letztgenannten Ausführungsform liegt die in der erfindungsgemäßen pharmazeutischen Zusammensetzung als Wirkstoff enthaltene Nukleinsäure als dsRNA, bevorzugt als siRNA, vor. Eine dsRNA, v.a. eine siRNA, ist insbesondere im Zusammenhang mit dem Phänomen der RNA-I nterferenz von Interesse. Auf das Phänomen der RNA-I nterferenz wurde man im Zuge der immunologischen
Forschung aufmerksam. In den letzten Jahren wurde ein RNA-basierter Abwehrmechanismus entdeckt, der sowohl im Reich der Pilze, als auch im Pflanzen- und Tierreich vorkommt und wie ein „Immunsystem des Genoms" wirkt. Das System wurde ursprünglich unabhängig voneinander in verschiedenen Spezies, zuerst in C. elegans, beschrieben, ehe man die zugrundeliegenden Mechanismen der Vorgänge als identisch identifizieren konnte: RNA-vermittelte Virus-Resistenz in Pflanzen, PTGS („posttranscriptional gene silencing") bei Pflanzen, und RNA-I nterferenz bei Eukaryoten basieren demnach auf einer gemeinsamen Funktionsweise. Die in vitro Technik der RNA- Interferenz (RNAi) beruht auf doppelsträngigen RNA-Molekülen (dsRNA), welche die sequenzspezifische Suppression der Genexpression auslösen (Zamore (2001 ) Nat. Struct. Biol. 9: 746-750; Sharp (2001 ) Genes Dev. 5:485-490: Hannon (2002) Nature 41 : 244- 251 ). Die Aktivierung von Proteinkinase R und RNaseL bewirkt bei der Transfektion von Säugerzellen mit langer dsRNA unspezifische Effekte, wie z.B. eine Interferon-Antwort (Stark et. Al. (1998) Annu. Rev. Biochem. 67: 227-264; He und Katze (2002) Viral Immunol. 15: 95-1 19). Diese unspezifischen Effekte werden bei Verwendung von kürzerer, bspw. 21 - bis 23-merer, sog. siRNA (engl, „small interfering RNA") umgangen, da unspezifische Effekte durch siRNA, die kürzer als 30 bp ist, nicht ausgelöst werden (Elbashir et. al. (2001) Nature 411 : 494-498). Kürzlich wurden dsRNA-Moleküle auch in vivo zur Anwendung gebracht (McCaffrey et. al. (2002), Nature 418: 38-39; Xia et. al. (2002), Nature Biotech. 20: 1006- 1010; Brummelkamp et. al. (2002), Cancer Cell 2: 243-247).
Die als Wirkstoff verwendete doppelsträngige RNA (dsRNA) enthält daher bevorzugt eine Sequenz mit der allgemeinen Struktur 5'-(N17.29)-3', wobei N irgendeine Base ist und für Nukleotide steht. Die allgemeine Struktur setzt sich aus einer doppelsträngigen RNA mit einem aus Ribonukleotiden aufgebauten Makromolekül zusammen, wobei das Ribonukleotid aus einer Pentose (Ribose), einer organischen Base und einem Phosphat besteht. Hierbei bestehen die organischen Basen in der RNA aus den Purinbasen Adenin (A) und Guanin (G) sowie den Pyrimidinbasen Cytosin (C) und Uracil (U). Die erfindungsgemäß als Wirkstoff verwendete dsRNA enthält derartige Nukleotide oder Nukleotidanaloga mit einer gerichteten Struktur. Vorzugsweise weisen erfindungsgemäß als Wirkstoff verwendete dsRNAs die allgemeine Struktur 5'-(N19.2s)-3', stärker bevorzugt 5'-(N19.24)-3', noch stärker bevorzugt 5'-(N21.23)-3' auf, wobei N irgendeine Base ist. Hierbei werden bevorzugt zumindest 90%, vorzugsweise 95% und insbesondere 100% der Nukleotide einer als
Wirkstoff verwendeten dsRNA komplementär zu einem Abschnitt der (m)RNA-Sequenz eines zuvor (als Wirkstoff) beschriebenen (therapeutisch relevanten) Proteins oder Antigens sein. 90% komplementär bedeutet hierbei, dass bei einer Länge einer erfindungsgemäß verwendeten dsRNA von bspw. 20 Nukleotiden diese höchstens 2 Nukleotide ohne entsprechende Komplementarität mit dem entsprechenden Abschnitt auf der (m)RNA aufweist. Die Sequenz der erfindungsgemäß verwendeten doppelsträngigen RNA ist mit ihrer allgemeinen Struktur vorzugsweise aber vollständig komplementär zu einem Abschnitt der (m)RNA eines zuvor als Wirkstoff beschriebenen Proteins oder Antigens.
Grundsätzlich können alle im kodierenden Bereich der (m)RNA auftretenden 17 bis 29, vorzugsweise 19 bis 25 Basenpaare langen Abschnitte als Zielsequenz für eine erfindungsgemäß als Wirkstoff verwendete dsRNA dienen. Gleichwohl können als Wirkstoff verwendete dsRNAs auch gegen Nukleotidsequenzen eines zuvor (als Wirkstoff) beschriebenen (therapeutisch relevanten) Proteins oder Antigens, die nicht im kodierenden Bereich liegen, gerichtet sein, insbesondere im nicht-kodierenden 5'-Bereich der (m)RNA, bspw. also gegen nicht-kodierende Bereiche der (m)RNA mit Regulationsfunktion. Die Zielsequenz der als Wirkstoff eingesetzten dsRNA eines zuvor beschriebenen Proteins oder Antigens kann also im translatierten und nicht-translatierten Bereich der (m)RNA und/oder im Bereich der Steuerungselemente liegen. Auch kann die Zielsequenz einer als Wirkstoff verwendeten dsRNA im Überlappungsbereich von nicht-translatierter und translatierter Sequenz liegen, insbesondere kann die Zielsequenz mindestens ein Nukleotid stromaufwärts vom Starttriplett des kodierenden Bereichs der (m)RNA umfassen.
Bei einer in der erfindungsgemäßen pharmazeutischen Zusammensetzung als Wirkstoff enthaltenen dsRNA kann vorzugsweise ein modifiziertes Nukleotid auftreten. Der Begriff „modifiziertes Nukleotid" bedeutet erfindungsgemäß, dass das jeweilige Nukleotid chemisch modifiziert ist. Der Fachmann versteht unter dem Begriff „chemische Modifikation", dass das modifizierte Nukleotid durch Ersetzen, Anfügen oder Entfernen einzelner oder mehrerer Atome oder Atomgruppen im Vergleich zu natürlich vorkommenden Nukleotiden verändert ist. Mindestens ein modifiziertes Nukleotid in erfindungsgemäß verwendeter dsRNA dient einerseits der Stabilität und andererseits der Verhinderung der Dissoziation. Vorzugsweise sind zwischen 2 und 10, bevorzugt zwischen 2 und 5 Nukleotide in einer erfindungsgemäß verwendeten dsRNA modifiziert.
Vorteilhafterweise ist mindestens eine 2'-Hydroxygruppe der Nukleotide der dsRNA in der doppelsträngigen Struktur durch eine chemische Gruppe, vorzugsweise eine 2'-Amino- oder eine 2'-Methylgruppe, ersetzt. Mindestens ein Nukleotid in mindestens einem Strang der doppelsträngigen Struktur kann auch ein sogenanntes „locked nucleotide" mit einem, vorzugsweise durch eine 2'-O, 4'-C-Methylenbrücke, chemisch modifizierten Zuckerring sein. Vorteilhafterweise sind mehrere Nukleotide der erfindungsgemäß verwendeten dsRNA „locked nucleotides". Darüber hinaus kann durch Modifizierung des Rückgrates einer erfindungsgemäß verwendeten dsRNA ein vorzeitiger Abbau derselben verhindert werden. Insbesondere bevorzugt ist in diesem Zusammenhang eine dsRNA, die in Form von Phosphorthioat, 2'-O-Methyl-RNA, LNA, LNA/DNA-Gapmeren, etc. modifiziert ist und daher eine längere Halbwertszeit in-vivo aufweist.
Vorzugsweise können die Enden der als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung eingesetzten doppelsträngigen RNA (dsRNA) modifiziert werden, um einem Abbau in der Zelle oder einer Dissoziation in die Einzelstränge entgegenzuwirken, insbesondere um einen vorzeitigen Abbau durch Nukleasen zu umgehen. Eine regelmäßig nicht gewünschte Dissoziation der Einzelstränge von dsRNA tritt insbesondere bei Verwendung niedriger Konzentrationen derselben oder kurzer Kettenlängen auf. Zur besonders wirksamen Hemmung der Dissoziation kann der durch die Nukleotidpaare bewirkte Zusammenhalt der doppelsträngigen Struktur von erfindungsgemäß verwendeter dsRNA durch mindestens eine, vorzugsweise mehr chemische Verknüpfung/en erhöht werden. Eine als Wirkstoff erfindungsgemäß verwendete dsRNA, deren Dissoziation vermindert ist, weist eine höhere Stabilität gegen enzymatischen und chemischen Abbau in der Zelle bzw. im Organismus {in vivo) oder ex vivo auf und besitzt daher eine höhere Halbwertszeit. Eine weitere Möglichkeit zur Verhinderung frühzeitiger Dissoziation erfindungsgemäß verwendeter dsRNA in der Zelle besteht darin, dass an den Enden der Stränge jeweils Haarnadelschleife(n) ausgebildet sein können. In einer besonderen Ausführungsform weist eine erfindungsgemäß verwendete dsRNA daher eine Haarnadelstruktur auf, um die Dissoziationskinetik zu verlangsamen. Bei einer derartigen Struktur ist vorzugsweise am 5'- und/oder 3'-Ende eine Loop-Struktur ausgebildet. Eine derartige Loop-Struktur weist keine Wasserstoffbrücken, typischerweise also keine Komplementarität, zwischen Nukleotidbasen auf. Typischerweise weist ein derartiger „Loop" eine Länge von mindestens 5, vorzugsweise mindestens 7 Nukleotiden auf und
verbindet auf diese Weise die beiden komplementären Einzelstränge einer erfindungsgemäß verwendeten dsRNA. Bevorzugt können ebenfalls die Nukleotide der beiden Stränge der erfindungsgemäß verwendeten dsRNA, um eine Dissoziation der Stränge zu verhindern, so modifiziert sein, dass eine Verstärkung der Wasserstoffbrückenbindung erreicht wird, bspw. durch Erhöhung der Wasserstoffbrückenbindungskapazität zwischen den Basen durch ggf. modifizierte Nukleotide. Dadurch wird die Stabilität der Wechselwirkung zwischen den Strängen erhöht und die dsRNA gegen einen Angriff von RNAsen geschützt.
Gemäß einer besonders bevorzugten Ausführungsform ist die als Wirkstoff in der erfindungsgemäßen pharmazeutischen Zusammensetzung verwendete dsRNA gegen die (m)RNA eines wie zuvor beschriebenen Proteins oder Antigens gerichtet. Vorzugsweise unterdrückt dabei die verwendete dsRNA die Translation eines zuvor beschriebenen Proteins oder Antigens in einer Zelle mindestens zu 50%, stärker bevorzugt 60%, noch stärker bevorzugt 70%, und am stärksten bevorzugt zu mindestens 90%, d.h. die Zelle enthält vorzugsweise höchstens die Hälfte der nativ (ohne Behandlung mit erfindungsgemäß verwendeter dsRNA) auftretenden, zellulären Konzentration eines zuvor beschriebenen Proteins oder Antigens. Die Suppression der Translation dieser Proteine oder Antigene in Zellen nach Zugabe erfindungsgemäß verwendeter dsRNA-Moleküle beruht auf dem durch solche Moleküle hervorgerufenen Phänomen der RNA-I nterferenz. Bei der erfindungsgemäß verwendeten dsRNA handelt sich dann um sogenannte siRNA, die das Phänomen der RNA- Interferenz auslösen und die (m)RNA eines zuvor beschriebenen Proteins oder Antigens binden kann. Die Messung bzw. der Nachweis der durch die erfindungsgemäß verwendete dsRNA ausgelösten Translationssuppression in Zellen kann über Northern-Blot, quantitative Real-Time PCR oder auf Proteinebene mit spezifischen Antikörpern gegen eine zuvor beschriebenes Proteins oder Antigen erfolgen. Die in der erfindungsgemäßen pharmazeutischen Zusammensetzung ggf. als Wirkstoff eingesetzte dsRNA sowie eine entsprechende siRNA kann nach dem Fachmann bekannten Verfahren hergestellt werden.
Zur weiteren Erhöhung der Immunogenität kann die erfindungsgemäße pharmazeutische Zusammensetzung zusätzlich einen oder mehrere Hilfsstoffe enthalten. Hierbei wird vorzugsweise eine synergistische Wirkung des erfindungsgemäßen immunstimulatorischen Adjuvanz und eines gegebenenfalls zusätzlich in der pharmazeutischen Zusammensetzung
enthaltenen Hilfsstoffes und/oder ggf. eines wie oben beschriebenen Wirkstoffes erzielt. In Abhängigkeit der verschiedenen Arten von Hilfsstoffen können diesbezüglich verschiedene Mechanismen in Betracht kommen. Bspw. bilden Verbindungen, welche die Reifung von dendritischen Zellen (DC) erlauben, bspw. Lipopolysaccharide, TNF-α oder CD40-Ligand, eine erste Klasse geeigneter Hilfsstoffe. Allgemein kann jedes das Immunsystem beeinflussende Agenz von der Art eines "Gefahrsignals" (LPS, GP96 usw.) oder Cytokine, wie GM-CFS, als Hilfsstoff verwendet werden, welche es erlauben, eine durch das erfindungsgemäße immunstimulatorische Adjuvanz erzeugte Immunantwort zu verstärken und/oder gerichtet zu beeinflussen. Besonders bevorzugte Hilfsstoffe sind Cytokine, wie Monokine, Lymphokine, Interleukine oder Chemokine, z.B. IL-I , IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-I O7 IL-12, INF-γ, INF-ß, GM-CFS, M-CSF, G-CSF, LT-ß, TNF-α, oder Interferone, bspw. IFN-γ, oder Wachstumsfaktoren, bspw. hGH.
Die erfindungsgemäße pharmazeutische Zusammensetzung kann weiterhin zusätzlich ein im Stand der Technik bekanntes Adjuvanz enthalten. Im Zusammenhang mit der vorliegenden Erfindung schließen im Stand der Technik bekannte Adjuvanzien, ohne darauf beschränkt zu sein, Aluminiumhydroxid, das (komplette oder inkomplette) Freund'sche Adjuvanz sowie zuvor beschriebene stabilisierende kationische Peptide bzw. Polypeptide, wie Protamin, Nukleolin, Spermin oder Spermidin und kationische Polysaccharide, insbesondere Chitosan, TDM, MDP, Muramyldipeptid, Alaun-Lösung, Pluronics, etc. ein. Des weiteren sind Lipopeptide, wie Pam3Cys, ebenfalls besonders geeignet, um mit dem erfindungsgemäßen immunstimulierenden Adjuvanz kombiniert zu werden (vgl. Deres et al, Nature 1989, 342: 561-564).
Optional kann die erfindungsgemäße pharmazeutische Zusammensetzung einen pharmazeutisch geeigneten Träger enthalten. Der hier verwendete Begriff "pharmazeutisch geeigneter Träger" umfasst bevorzugt einen oder mehrere kompatible feste oder flüssige Füllstoffe, bzw. Verdünnungsmittel oder einkapselnde Verbindungen, welche für die Verabreichung an eine Person geeignet sind. Der Begriff "kompatibel", wie hier verwendet, bedeutet, dass die Bestandteile der Zusammensetzung in der Lage sind, mit dem Wirkstoff, dem Adjuvanz als solchem und miteinander in einer solchen Art zusammengemischt zu
werden, dass keine Interaktion auftritt, welche wesentlich die pharmazeutische Effektivität der Zusammensetzung unter gewöhnlichen Verwendungsbedingungen reduzieren würde. Pharmazeutisch geeignete Träger müssen selbstverständlich eine ausreichend hohe Reinheit und eine ausreichend geringe Toxizität aufweisen, um sie für die Verabreichung an eine zu behandelnde Person geeignet zu machen. Einige Beispiele von Verbindungen, welche als pharmazeutisch geeignete Träger oder Bestandteile davon dienen können, sind Zucker, wie beispielsweise Lactose, Glucose und Sukrose; Stärken wie beispielsweise Kornstärke oder Kartoffelstärke; Cellulose und seine Derivate, wie beispielsweise Natriumcarboxymethylcellulose, Ethylcellulose, Celluloseacetat; pulverisiertes Tragacanth; Malz; Gelatine; Talg; feste Gleitmittel, wie beispielsweise Stearinsäure, Magnesiumstearat; Kalziumsulfat; vegetabile Öle, wie beispielsweise Erdnussöl, Baumwollsamenöl, Sesamöl, Olivenöl, Kornöl und Öl aus Theobroma; Polyole, wie beispielsweise Polypropylenglycol, Glycerin, Sorbitol, Mannitol und Polyethylenglycol; Algininsäure; Emulgatoren, wie beispielsweise Tween®; Benetzungsmittel, wie beispielsweise Natriumlaurylsulfat; färbende Agenzien; geschmacksvermittelnde Agenzien, Arzneistoffträger; tablettenbildende Agenzien; Stabilisatoren; Antioxidantien; Konservierungsmittel; pyrogen-freies Wasser; isotonische Salzlösung und phosphatgepufferte Lösungen.
Die Wahl eines pharmazeutisch geeigneten Trägers wird grundsätzlich durch die Art bestimmt, durch die die erfindungsgemäßen pharmazeutischen Zusammensetzungen verabreicht werden. Die erfindungsgemäßen pharmazeutischen Zusammensetzungen können bspw. systemisch verabreicht werden. Routen zur Verabreichung schließen z.B. transdermale, orale, parenterale, einschließlich subkutane oder intravenöse Injektionen, topische und/oder intranasale Routen ein. Die geeignete Menge der zu verwendenen pharmazeutischen Zusammensetzung kann durch Routineexperimente mit Tiermodellen bestimmt werden. Solche Modelle schließen, ohne jedoch darauf begrenzt zu sein, Modelle von Kaninchen, Schaf, Maus, Ratte, Hund und nicht-humane Primatenmodelle mit ein. Bevorzugte Einheitsdosisformen zur Injektion schließen sterile Lösungen von Wasser, physiologischer Salzlösung oder Mischungen davon mit ein. Der pH solcher Lösungen sollte auf etwa 7,4 eingestellt werden. Geeignete Träger zur Injektion schließen Hydrogele, Vorrichtungen zur kontrollierten oder verzögerten Freigabe, polylaktische Säure und Collagenmatrizen mit ein. Geeignete pharmazeutisch-geeignete Träger zur topischen Anwendung schließen solche mit ein, die zur Verwendung in Lotionen, Cremes, Gels u.a.
geeignet sind. Falls die Verbindung peroral verabreicht werden soll, sind Tabletten, Kapseln u.a. die bevorzugte Einheitsdosisform. Die pharmazeutisch geeigneten Träger zur Herstellung von Einheitsdosisformen, die für die orale Verabreichung verwendbar sind, sind im Stand der Technik gut bekannt. Ihre Auswahl wird von sekundären Überlegungen wie Geschmack, Kosten und Lagerfähigkeit abhängen, welche für die Zwecke der vorliegenden Erfindung nicht kritisch sind, und ohne Schwierigkeiten durch einen Fachmann durchgeführt werden können.
Gemäß einer besonderen Ausführungsform kann die pharmazeutische Zusammensetzung auch als Vakzine vorliegen. Erfindungsgemäße Vakzine umfassen dabei typischerweise eine Zusammensetzung wie zuvor für pharmazeutische Zusammensetzung beschrieben, wobei die Zusammensetzung solcher erfindungsgemäßen Vakzine insbesondere durch die Art bestimmt wird, mit denen sie verabreicht werden. Bevorzugt werden erfindungsgemäße Vakzine systemisch verabreicht. Routen zur Verabreichung von solchen Vakzinen schließen typischerweise transdermale, orale, parenterale, einschließlich subkutane oder intravenöse Injektionen, topische und/oder intranasale Routen mit ein. Vakzine werden daher bevorzugt in flüssiger oder fester Form formuliert. Auch können ggf. in eine erfindungsgemäße Vakzine weitere Hilfsstoffe eingearbeitet sein, die die Immunogenität des Vakzins weiter steigern können. Vorteilhafterweise wird/werden ein oder mehr weitere solcher, wie zuvor definierte, Hilfsstoffe in Abhängigkeit der Immunogenität und anderen Eigenschaften des Wirkstoffes in der erfindungsgemäßen Vakzine zu wählen sein.
Gemäß einem weiteren bevorzugten Gegenstand der vorliegenden Erfindung werden die erfindungsgemäßen pharmazeutischen Zusammensetzungen, besonders bevorzugt die erfindungsgemäßen Vakzine, zur Behandlung von im folgenden genannten beispielhaften Indikationen verwendet. Mit erfindungsgemäßen pharmazeutischen Zusammensetzungen, besonders bevorzugt erfindungsgemäßen Vakzinen, können beispielsweise solche Erkrankungen oder Zustände behandelt werden, die mit diversen pathologisch unterbleibenden Immunantworten verknüpft sind oder die eine Immunantwort, bevorzugt eine gesteigerte Immunantwort, im Rahmen einer Therapie erfordern. Bevorzugt werden dabei erfindungsgemäße pharmazeutische Zusammensetzungen oder Vakzine eingesetzt,
um tumorspezifische oder pathogenspezifische Immunantworten auszulösen. Besonders bevorzugt können solche erfindungsgemäßen pharmazeutischen Zusammensetzungen oder Vakzine, zur Steigerung von Immunantworten antigenpräsentierender Zellen (APC) verwendet werden. Ebenfalls besonders bevorzugt können die erfindungsgemäßen pharmazeutischen Zusammensetzungen oder Vakzine zur Behandlung von Krebs bzw. Tumorerkrankungen eingesetzt werden, bevorzugt ausgewählt aus Kolonkarzinomen, Melanomen, Nierenkarzinomen, Lymphomen, akuter myeloider Leukämie (AML), akute lymphoider Leukämie (ALL), chronischer myeloider Leukämie (CML), chronischer lymphozytischer Leukämie (CLL), Gastroi ntestinalen Tumoren, Lungenkarzinomen, Gliomen, Schilddrüsentumoren, Mammakarzinomen, Prostatatumoren, Hepatomen, diversen virus-induzierten Tumoren wie z.B. Papillomvirus-induzierten Karzinomen (z.B. Cervixkarzinome), Adenokarzinomen, Herpesviren-induzierten Tumoren (z.B. Burkitt's Lymphom, EBV-induziertes B Zelllymphome), Hepatitis B-induzierten Tumoren (Hepatozellkarzinome), HTLV-1 und HTLV-2 induzierten Lymphomen, Akustikusneurinomen, Gebärmuttehalskrebs, Lungenkrebs, Rachenkrebs, Analkarzinomen, Glioblastomen, Lymphomen, Rektumkarzinomen, Astrozytomen, Hirntumoren, Magenkrebs, Retinoblastomen, Basaliomen, Hirnmetastasen, Medulloblastomen, Scheidenkrebs, Bauchspeicheldrüsenkrebs, Hodenkrebs, Melanomen,
Schilddrüsenkarzinomen, Blasenkrebs, Hodgkin-Syndrom, Meningeomen, Schneeberger Krankheit, Bronchialkarzinomen, Hypophysentumor, Mycosis fungoides, Speiseröhrenkrebs, Brustkrebs, Karzinoide, Neurinomen, Spinaliomen, Burkitt-Lymphomen, Kehlkopfkrebs, Nierenkrebs, Thymomen, Corpuskarzinomen, Knochenkrebs, Non-Hodgkin-Lymphomen, Urethrakrebs, CUP-Sysndrom, Kopf-Hals-Tumoren, Oligodendrogliomen, Vulvakrebs, Darmkrebs, Kolonkarzinomen, Ösphaguskarzinomen, Warzenbeteiligung, Dünndarmtumoren, Kraniopharyngeomen, Ovarial-Karzinomen, Weichteiltumoren, Eierstockkrebs, Leberkrebs, Pankreaskarzinomen, Zervixkarzinomen,
Endometriumkarzinomen, Lebermetastasen, Peniskrebs, Zungenkrebs, Gallenblasenkrebs, Leukämie, Plasmozytomen, Gebärmutterkrebs, Lidtumor, Prostata krebs, etc.. In diesem Zusammenhang ist es besonders bevorzugt, wenn als Lipid in der Lipid-modifizierten- Nukleinsäure oder als Wirkstoff α-Tocopherol (Vitamin E), D-α-Tocopherol, L-α- Tocopherol, D,L-α-Tocopherol oder Vitamin E-Succinat (VES) eingesetzt werden, α- Tocopherol (Vitamin E) ist wenig toxisch und zeigt eine potente Antitumor-Wirkung (A. Bendich, LJ. Machlin Am. J. Clin. Nutr. 48 (1988) 612), was dessen in der Krebstherapie
vielversprechend erscheinen lässt. Als Erklärung für die Inhibierung der Proliferation von Tumorzellen bzw. der zytotoxischen Wirkung auf sie sind v.a. zwei Mechanismen bekannt: Zum einen ist Vitamin E ein potentes Antioxidanz und ein guter Radikalfänger (C. Borek Ann. NY Acad. Sei. 570 (1990) 417), zum anderen kann es über eine Stimulierung der Immunantwort das Tumorwachstum verhindern (G. Shklar, J. Schwartz, D. P. Trickler, S. Reid J. Oral Pathol. Med. 19 (1990) 60). In neueren Arbeiten wurde weiterhin ein Zusammenhang zwischen der Expression des Tumorsuppressorgens p53 in Tumorzellen {oral squamous canceή und einer Behandlung mit Vitamin E-Succinat (VES) gefunden (J- Schwartz, G. Shklar, D. Trickler Oral Oncol. Europ. J. Cancer 29B (1993) 313). Dabei konnte sowohl eine Stimulierung der Produktion des wild type p53, welches als Tumorsuppressor gilt, beobachtet werden, als auch eine Reduktion von mutiertem p53, das onkogene Wirkung entfaltet. Interessanterweise ist die biologische Wirkung von VES auf diese Tumorzellen in zweifacher Hinsicht dosisabhängig: In physiologischen Dosen (0,001 - 50 μmol/l) ist ein ansteigendes Zellwachstum zu beobachten, in pharmakologischen Dosen (100-154 μmol/l) wird das Zellwachstum inhibiert. Dies wurde in Zellkultur gezeigt (T.M.A. Elattar, A.S. Virji Anticancer Res. 19 (1999) 365). Auch konnte in verschiedenen Brustkrebszelllinien durch Behandlung mit VES Apoptose induziert werden (W. Yu, K. Israel, Q.Y. Liao, C. M. Aldaz, B.G. Sanders, K. Kline Cancer Res. 59 (1999) 953). Die induzierte Apoptose wird dabei über eine Interaktion von Fas-Ligand und Fas-Rezeptor eingeleitet. Dies ist deshalb besonders hervorzuheben, weil bei den entsprechenden Zelllinien ein derartiger Mechanismus bisher nicht beobachtet werden konnte. Von Vitamin E existieren verschiedene Isomere, die sich in der Anzahl und Position der Methylgruppen am aromatischen Ring unterscheiden. In den beschriebenen Arbeiten wurde die biologisch wirksamste Form des natürlich vorkommenden Vitamin E, das α-Tocopherol, eingesetzt. Dieses kommt wiederum in verschiedenen Stereoisomeren vor, da das Molekül drei optisch aktive Zentren enthält. Die natürliche Form des Vitamin E ist das RRR-α- Tocopherol (früher D-α-Tocopherol), allerdings wird heutzutage überwiegend das Racemat (D,L-α-Tocopherol) eingesetzt. Alle hier zuvor genannten Formen von Vitamin E sind ebenfalls als Lipid im Sinne der vorliegenden Erfindung mit umfasst.
Ebenfalls besonders bevorzugt werden die erfindungsgemäßen pharmazeutischen Zusammensetzungen zur von Infektionserkrankungen eingesetzt. Ohne darauf beschränkt zu sein werden solche Infektionskrankheiten bevorzugt ausgewählt aus Influenza, Malaria,
SARS, Gelbfieber, AIDS, Lyme-Borreliose, Leischmaniasis, Anthrax, Meningitis, viralen Infektionskrankheiten wie AIDS, Condyloma acuminata, Dellwarze, Dengue-Fieber, Dreitagefieber, Ebolavirus, Erkältung, Frühsommermeningoenzephalitis (FSME), Grippe, Gürtelrose, Hepatitis, Herpes-Simplex Typ I, Herpes-Simplex Typ II, Herpes zoster, Influenza, Japanische Enzephalitis, Lassafieber, Marburg- Virus, Masern, Maul- und Klausenseuche, Mononukleose, Mumps, Norwalk-Virus-infektion, Pfeifersches- Drüsenfieber, Pocken, Polio (Kinderlähmung), Pseudokrupp, Ringelröteln, Tollwut, Warzen, West-Nil-Fieber, Windpocken, Zytomegalie-Virus (CMV), aus bakteriellen Infektionskrankheiten wie Abort (Prostataentzündung), Anthrax, Appendizitis (Blinddarmentzündung), Borreliose, Botulismus, Camphylobacter, Chlamydia trachomatis (Harnröhren-, Bindehautentzündung), Cholera, Diphterie, Diphterie, Donavanosis, Epiglottitis, Fleckfieber, Flecktyphys, Gasbrand, Gonorhoe, Hasenpest, Heliobakter pylori, Keuchhusten, klimatischer Bubo, Knochenmarksentzündung, Legionärskrankheit, Lepra, Listeriose, lungenentzündung, Meningitis, Bakterielle Gehirnhautentzündung, Milzbrand, Mittelohrentzündung, Mycoplasma hominis, Neugeborenensepsis (Chorioamnionitis), Noma, Paratyphus, Pest, Reitersyndrom, Rocky Mountain spotted fever, Salmonellen- Paratyphus, Salmonellen-Typhus, Scharlach, Syphilis, Tetanus, Tripper, Tsutsugamushi, Tuberkulose, Typhus, Vaginitis (Kolpitis), Weicher Schanker und aus durch Parasiten, Protozoen oder Pilze hervorgerufenen Inkektionskrankheiten wie Amöbenruhr, Bilharziose, Chagas-Krankheit, Fußpilz, Kleienpilzflechte, Krätze (Skabies), Malaria, Onchozerkose (Flussblindheit), oder Pilzerkrankungen, Toxoplasmose, Trichomoniasis, Trypanosomiasis (Schlafkrankheit), Viszerale Leishmaniose, Windeldermatitis, Schistosomiasis, Fischvergiftung (Ciguatera), Kandiose, Kutane Leishmaniose, Lamblienruhr (Giadiasis), oder Schlafkrankheit, oder aus Infektionskrankheiten hervorgerufen durch Echinococcus, Fischbandwurm, Fuchsbandwurm, Hundebandwurm, Läuse, Rinderbandwurm, Schweinebandwurm, Zwergbandwurm.
Ein weiterer Gegenstand der Erfindung betrifft die Verwendung eines erfindungsgemäßen immunstimulierenden Adjuvanz zur Herstellung einer erfindungsgemäßen pharmazeutischen Zusammensetzung oder eines erfindungsgemäßen Vakzins zur Behandlung von zuvor beschriebene Indikationen, z.B. zur Behandlung von den genannten Tumor- bzw. Infektionserkrankungen. Alternativ umfasst die Erfindung die (therapeutische)
Verwendung eines erfindungsgemäßen immunstimulierenden Adjuvanz zur Behandlung von Tumor- oder Infektionserkrankungen, wie zuvor beschrieben.
Ebenfalls umfasst werden von der vorliegenden Erfindung Kits, enthaltend ein erfindungsgemäßes immunstimulierendes Adjuvanz und/oder eine erfindungsgemäße pharmazeutische Zusammensetzung und/oder eine erfindungsgemäße Vakzine sowie optional eine technische Gebrauchsanweisung mit Angaben zur Verabreichung und
Dosierung des erfindungsgemäßen immunstimulierenden Adjuvanz und/oder der erfindungsgemäßen pharmazeutischen Zusammensetzung und/oder der erfindungsgemäßen Vakzine.
Die vorliegende Erfindung wird nachfolgend anhand von Figuren und Beispielen weiter illustriert, ohne dass diese dazu gedacht sind, die Gegenstände der vorliegenden Erfindung hierauf zu beschränken.
Figurenbeschreibung:
Fig. 1 : zeigt verschiedene erfindungsgemäße Möglichkeiten der Endmodifizierung von
Nukleinsäuren mit Lipiden. Dargestellt sind insbesondere die Lipid- modifizierten Linker bzw. bifunktionellen Peptide, die zur Kopplung bzw.
Synthese mit Nukleinsäuresequenzen (kurz: ODN-Sequenz) verwendet werden können.
Fig. 2: beschreibt beispielhaft einen Syntheseweg für (trifunktionale) Lipid-modifizierte Linker, mit der z.B. eine Tocopherolmodifizierung an das 3 '-Ende einer
Nukleinsäure eingebracht werden kann. Solche beispielhaft dargestellten Verbindungen stellen ein Zwischenprodukt bei der Herstellung der erfindungsgemäßen 5'- oder 3'-Lipid-modifizierten Nukleinsäuren sowie der erfindungsgemäßen Adjuvanzien dar.
Fig. 3: zeigt beispielhaft ein bifunktionelles Lipid mit einem Succinylanker, das eine
3 '-Modifizierung einer Nukleinsäure mit einem bifunktionellen Lipid, z.B. mit PEG, ermöglicht.
Fig. 4: stellt schematisch die Kopplung von Lipid-modifizierten Amiditen an das 5'-
Ende von Nukleinsäuren dar.
Fig. 5A,B: beschreiben die Stimulation humaner PBMCs mit erfindungsgemäßen immunstimulierenden Adjuvanzien sowie mit verschiedenen RNA- Oligonukleotiden. A) Es ist insbesondere bei der Ausschüttung von Zytokinen
(IL-G) zu beobachten, dass die erfindungsgemäßen immunstimulierenden Adjuvanzien ohne Zugabe von Protamin eine mehr als 5-fache Erhöhung der Zytokin-Ausschüttung (IL-6) gegenüber dem Medium aufweisen sowie bei
Zugabe von Protamin gegenüber ß-Galaktosidase und RNA Oligo 40 alleine (SEQ ID NO: 40) eine leicht verbesserte Freisetzung von IL-6. B) Bei der Bestimmung der TNFα-Ausschüttung lässt sich eine deutliche Stimulierung des Immunsystems feststellen, die der von ß-Galactosidase oder RNA mindestens gleichzusetzen ist.
Fig. 6: zeigt die Freisetzung von TNF-α durch humane PBMC-Zellen nach Stimulation mit erfindungsgemäß verwendeten RNA-Oligonukleotiden sowie erfindungsgemäßen immunstimulierenden Adjuvanzien. Fig. 6 zeigt insbesondere, dass immunstimulierende erfindungsgemäße Adjuvanzien in
Form einer Lipid-modifizierten Nukleinsäure, enthaltend z,B; eine der Sequenzen SEQ ID NO: 40, 41 oder 42, im Vergleich zu z.B. einem nicht modifizierten RNA-Oligonukleotid der Sequenz gemäß SEQ ID NO: 40 (RNA Oligo 40) eine deutlich verbesserte Freisetzung von TNF-α und damit eine deutlich verbesserte Immunstimulation aufweisen. Die besten Ergebnisse mit einer Steigerung der Immunstimulation um mehr als das 10-fache gegenüber dem nicht-modifizierten RNA-Oligonukleotid konnten mit einer Tocopherol- modifizierten Sequenz gemäß SEQ ID NO: 42 (RNA Oligo Toc CV2) erreicht werden.
Beispiele:
1. Synthese von l -(4,4"-Dimethoxytrityl)-polyethylenglykol (DMT-PEG15Ja)

30 ml absolutem Pyridin gelöst, das anschließend azeotrop abdestilliert wird. Das getrocknete Edukt wird in 35 ml abs. Pyridin gelöst. Zu dieser Lösung wird über einen Zeitraum von 30 min 4,7 g 4,4'-Dimethoxytritylchlorid (13,9 mmol) gelöst in 35 ml abs. Pyridin zugetropft. Es werden weitere 2 h bei RT gerührt, dabei wird der Reaktionsverlauf mittels DC kontrolliert. Neben des Nachweises der DMT-Gruppe mittels UV-Lampe können die DC-Platten in zwei Schritten entwickelt werden: 1. In HCl-gesättigter Atmosphäre zum Nachweis von DMT; 2. In der lodkammer zum Nachweis von PEG; PEG kann außerdem mit Dragendorff-Bürger-Sprühreagenz nachgewiesen werden. Nach Beendigung der Reaktion wird das Lösungsmittel entfernt und das Produkt in 50 ml DCM aufgenommen. Die organische Phase wird zwei mal mit je 25 ml 5% NaHCOj-Lösung sowie zwei mal mit je 25 ml H2O gewaschen. Die Phasentrennung zwischen wässriger und organischer Phase ist dabei langwierig, da PEG sowohl hydrophoben als auch hydrophilen Charakter besitzt. Nach Trocknen über Na2SO4 wird das Lösungsmittel entfernt und das Rohprodukt säulenchromatographisch über Kieselgel mit DCM/MeOH/TEA =
18:2:0,5 gereinigt. Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Man erhält ein gelbliches Öl, das nach gutem Trocknen im Hochvakuum zu einem wachsartigen Feststoff wird. Ausbeute: 18,3 g (72,5% der Theorie) DC (DCM/MeOH/TEA = 18:2:0,5): RrWert * 0,55 (durch die Molmassenverteilung von PEG verbreitertes Signal)
2. Synthese von 1 -(4.4'-Dimethoxytrityl)-hexaethylenglykol (DMT-HEG)
Arbeitsvorschrift: 10 g Hexaethylenglykol (35 mmol) werden durch Coevaporation mit zwei mal je 30 ml abs. Pyridin getrocknet und anschließend in 20 ml abs. Pyridin gelöst. Analog zur Vorschrift in Beispiel 1 wird das HEG mit 10 g
DMT-CI (29,5 mmol) gelöst in 50 ml abs. Pyridin umgesetzt. Die säulenchromatographische Aufreinigung erfolgt mit Ethylacetat/TEA = 95:5. Als getrocknetes Produkt erhält man ein zähes, gelbes Öl. Ausbeute: 12,5 g (60,5% der Theorie) DC (DCM/MeOH = 95:5): RrWert = 0,59
MS (FD): m/z 583,9 (M+)
1H-NMR (CDCI3): δ 3,21 (t, DMT-O-CH2-), 3,47-3,68 (m, -CH2-), 3,76 (s, -CH3), 6,77-7,46 (m, Aromaten)
3. Synthese von 1-(4.4"-Dimethoxytrityl)-polyethylenglvkolsuccinat (DMT-PEG-Suc)

Nachstehende Arbeitsvorschrift ist sowohl für DMT-PEG1500 als auch für DMT-HEG anwendbar.
Arbeitsvorschrift: 5 g DMT-PEG1500 (2,8 mmol) werden in 25 ml DCM/Pyridin = 5:1 gelöst und mit 420 mg Bernsteinsäureanhydrid (4,2 mmol, d.h. 1 ,5 eq) gelöst in 7 ml Pyridin sowie mit 170 mg DMAP (1 ,4 mmol, d.h. 0,5 eq) gelöst in 3 ml Pyridin versetzt. Nach 12 h Rühren bei RT werden die Lösungsmittel im Vakuum abgezogen und der Rückstand in DCM aufgenommen. Die organische Phase wird gründlich drei Mal mit NaHCO3-Lösung (10% in H2O) und zwei Mal mit gesättigter wässriger NaCI-Lösung gewaschen, um die überschüssige Bernsteinsäure abzutrennen. Nach
Trocknen über Na2SO4 wird das Lösungsmittel entfernt. Die Succinate können nach gründlicher Trocknung am Hochvakuum ohne weitere Aufarbeitung zur Kupplung auf aminomodifizierten Trägermaterialien eingesetzt werden. DC: DMT-PEG1500-SuC (DCM/MeOH/TEA = 18:2:0,5): RrWert = 0,41 DMT-HEG-Suc (DCM/MeOH = 9:2): RrWert = 0,70
4. Synthese von 1-Tosyl-2,3-isopropylidenglycerol

Arbeitsvorschrift: 200 mmol Isopropylidenglycerol (26,4 g) werden in 200 ml Acetonitril gelöst und mit 22,2 g Triethylamin (220 mmol) versetzt. 220 mmol p-Toluolsulfonsäurechlorid (41,9 g) werden in 250 ml Acetonitril gelöst und unter Rühren über 2 h zum Reaktionsgemisch zugetropft. Man lässt weitere 20 h bei RT rühren, wobei ein weißer Niederschlag ausfällt, welcher nach Beendigung der
Reaktion abfiltriert wird. Das Lösungsmittel wird entfernt und das Rohprodukt über Kieselgel säulenchromatographisch mit n-Hexan/Ethylacetat = 2:1 aufgereinigt. Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Das Produkt wird am Hochvakuum getrocknet. Es resultiert ein gelbliches Öl (L.N. Markovskii et al. / Org. Chem. UdSSRIb (1990) 2094.).
Ausbeute: 31,3 g (54,7% der Theorie) DC (n-Hexan/Ethylacetat = 2:1 ): RrWert = 0,2 MS (FD): m/z 272,0 (M+-CH) (286,3 berechnet) 1H-NMR (CDCI3): δ 1 ,31 (s); 1 ,34 (s); 2,45 (s); 3,74-3,79 (m); 3,90-4,08 (m); 4,23- 4,32 (m); 7,36 (d); 7,77 (d)
13C-NMR: δ 21 ,7; 25,2; 26,7 (Methyl-C); 66,2; 69,5; 72,9 (Glycerin-C); 1 10,1 (Methylen-C); 128,0; 129,9; 132,7; 145,1 (Aromaten-C)
5. Synthese von 2.3-lsopropyliden-1-D.L-α-tocopherolglycerol (Tod )

Arbeitsvorschrift: Zu 56 mmol D,L-cx-Tocopherol (24,06 g) in 280 ml DMSO werden 5,6 g pulverisiertes Kaliumhydroxid (100 mmol) zugegeben. Nach 2 h Rühren bei RT unter Lichtausschluss werden 56 mmol 1 -Tosyl-2,3- isopropylidenglycerol (16 g) gelöst in 20 ml DMSO zugetropft und weitere 12 h bei 600C gerührt. Danach wird das Reaktionsgemisch auf 1 I Eiswasser hydrolysiert und die wässrige Phase mit 1 ,5 I Toluol extrahiert. Nachdem über Natriumsulfat getrocknet wurde, wird das Lösungsmittel entfernt. Das Rohprodukt wird über Kieselgel säulenchromatographisch mit n-Hexan/Ethylacetat = 1 :1 aufgereinigt. Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Das Produkt wird im Hochvakuum getrocknet und unter Lichtausschluss aufbewahrt. Es resultiert ein gelbliches Öl (D.W. Will, T. Brown Tetrahedron Lett. 33 (1992) 2729.). Ausbeute: 24,2 g (79,2% der Theorie) DC (n-Hexan/Ethylacetat = 1 :1 ): RrWert = 0,69 MS (FD): m/z 544,6 (M+)
Synthese von 1 -D.L-α-Tocopherylglycerol (Toc2)

Arbeitsvorschrift: 16,9 mmol Tod (9,2 g) werden in 100 ml HCI(2 M)ATHF (1 :1) gelöst und 2 h bei RT unter Lichtausschluss gerührt. Anschließend wird das Lösungsmittel entfernt, der Rückstand zweimal mit je 50 ml absolutem Ethanol versetzt und das Gemisch wieder zur Trockene eingeengt. Das Rohprodukt wird mit
Diethylether/Toluol = 1 :1 über Kieselgel säulenchromatographisch aufgereinigt. Die prodυkthaltigen Fraktionen werden mit DC identifiziert, vereinigt und zur Trockene eingeengt. Das Produkt wird am Hochvakuum getrocknet und unter Lichtausschluss aufbewahrt. Es resultiert ein gelbes Öl.
Ausbeute: 6,6 g (77,5% der Theorie)
DC (Diethylether/Toluol = 1 :1 ): RrWert = 0,22
MS (FD): m/z 504,4 (M+)
Synthese von π -(4.4"-Dimethoxytrityl)l-3-D,L-α-tocopherylglycerol (Toc3)

Arbeitsvorschrift: 6,6 g Toc2 (13 mmol) werden zum Trocknen zweimal in 15 ml absolutem Pyridin gelöst, das wieder azeotrop abdestilliert wird. Das getrocknete Edukt wird in 50 ml absolutem Pyridin gelöst und mit 5,34 g DMT-Cl (15,8 mmol) versetzt. Nach 12 h Rühren bei RT unter Lichtausschluss wird die Reaktion durch die Zugabe von 50 ml Methanol beendet und das Reaktionsgemisch zur Trockene eingeengt. Der Rückstand wird in 500 ml Dichlormethan aufgenommen und zwei Mal mit je 150 ml wässriger, gesättigter NaCI-Lösung und anschließend einmal mit 150 ml Wasser gewaschen. Nachdem über Na2SO4 getrocknet wurde, wird das Lösungsmittel entfernt und der Rückstand säulenchromatographisch über Kieselgel gereinigt (n-
Hexan/Diethylether/Triethylamin = 40:60:1 ). Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Das Produkt wird am Hochvakuum getrocknet. Es resultiert ein gelbliches Öl, das unter Lichtausschluss aufbewahrt wird.
Ausbeute: 8,5 g (81 % der Theorie)
DC (n-Hexan/Diethylether/TEA = 40:60:1 ): RrWert = 0,43
MS (FD): m/z 807,2
8. Synthese von [1 -(4,4"-Dimethoxytrityl)l-2-succinyl-3-D.L-α-tocopherylglycerol (Toc4)

Arbeitsvorschrift: 1 ,45 g Toc3 (1,8 mmol) werden zur Trocknung zweimal in je 5 ml abs. Pyridin gelöst, das wieder azeotrop abdestilliert wird. Nachdem das Edukt in 8 ml abs. Pyridin gelöst wurde, gibt man im Argongegenstrom 140 mg DMAP (1 ,08 mmol) und 194,4 mg Bernsteinsäureanhydrid (1,8 mmol) zu. Anschließend wird 18 h unter Lichtausschluss bei RT gerührt. Zur Aufarbeitung wird die Reaktionslösung mit 45 ml DCM versetzt und vier Mal mit je 50 ml Wasser gewaschen. Nach Trocknung über Natriumsulfat wird das Lösungsmittel entfernt und das Rohprodukt säulenchromatographisch über Kieselgel mit Ethylacetat/Methanol/NH3(25% in H2O) = 5:1 :1 gereinigt. Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Nach Trocknung im Hochvakuum resultiert ein bräunliches, zähes Öl, das gekühlt und unter Lichtausschluss aufbewahrt wird. Ausbeute: 1 ,35 g (82,8% der Theorie) DC (EtOAc/NH3/MeOH = 5:1 :1 ): RrWert = 0,30 MS (FD): m/z 906,2 (M+), 604,2 (M+-DMT)
9. Synthese von D.L-α-Tocopheryl-ß-cvanoethvI-N.N-diisopropyl-phosphoramidit

Arbeitsvorschrift 5 g D,L-α-Tocopherol (11 ,6 mmol) werden zwei Mal mit je 25 ml Pyridin versetzt und durch azeotropes Schleppen getrocknet. Das Edukt wird in 40 ml DCMabs gelöst. Im Argongegenstrom werden 7,9 ml DIPEAabs (46,4 mmol) und 2,5 ml 2-Cyanoethyl-N,N-diisopropylphosphinchlorid (1 1 mmol) langsam zugetropft. Nachdem das Reaktionsgemisch 1 h bei RT unter Lichtausschluss gerührt hat, wird es mit 100 ml Ethylacetat/TEA (20:1 ) verdünnt und zwei Mal mit 25 ml 10% NaHCO3 sowie zwei Mal mit gesättigter NaCI-Lösung gewaschen. Anschließend wird die organische Phase über Na2SO4 getrocknet und das Lösungsmittel am Vakuum entfernt. Das Rohprodukt wird über Kieselgel säulenchromatographisch mit Ethy I acetat gereinigt. Ausbeute: 5,62 g (81 % der Theorie) DC (EtOAc): RrWert = 0,75 MS (FD): m/z 630,1 (M+) 31P-NMR: 152,24
10. Synthese von [1 -(4.4"-Dimethoxytrityl)1-(3-D.L-α-tocopheryl)-glycerol-2- phosphoramidit
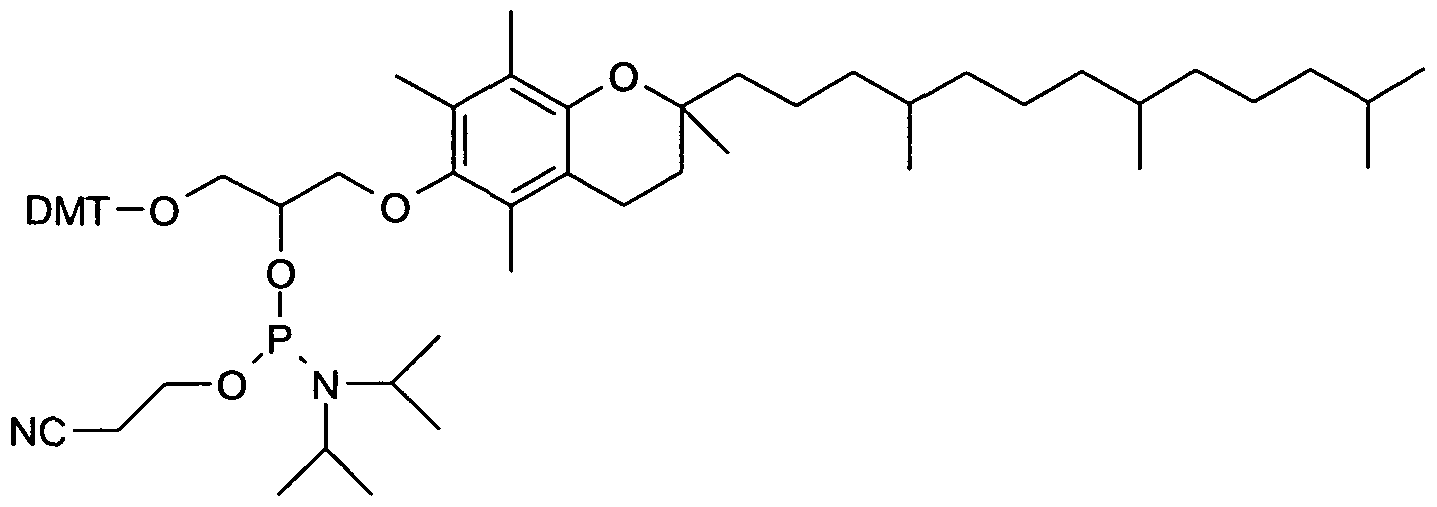
Man lässt das Reaktionsgemisch 1 h bei RT rühren, bevor die Lösung mit 50 ml Ethylacetat/TEA (20:1) verdünnt wird. Die organische Phase wird zwei Mal mit 15 ml einer 10%igen NaHCO3-Lösung und zwei Mal mit einer gesättigten NaCI- Lösung gewaschen und anschließend über Na2SO4 getrocknet. Das Lösungsmittel wird entfernt und das Rohprodukt säulenchromatographisch über Kieselgel mit
Ethylacetat/TEA (99:1 ) gereinigt. Die produkthaltigen Fraktionen werden mittels DC identifiziert, vereinigt und zur Trockene eingeengt. Nach Trocknung im Hochvakuum resultiert ein gelbbräunliches, sehr zähes Öl, das gekühlt und unter Lichtausschluss aufbewahrt wird. Ausbeute: 0, 76g (63,4% der Theorie)
DC (EtOAc, 1 % TEA): RrWert = 0,68
MS (FD): m/z 1006,4 (M+), 953,6 (M+-Cyanoethyl), 651,4 (M+-DMT,-Cyanoethyl), 603,1 (M+-DMT,-diisopropylamin), 303,1 (DMT+) 31P-NMR: 150,5
11. Synthese von 1 -Hexadecyl-2.3-isopropylidenglycerol (Pami)

Arbeits Vorschrift: Zu 0,1 mol D,L- , -Isopropyliden-Glycerin (12,4 ml) in 500 ml THFabs werden portionsweise unter Argongegenstrom 0,1 1 mol Natriumhydrid
(2,42 g) zugegeben. Nach 12 h Rühren bei RT wird zu dem entstandenen Alkoholat
0,1 1 mol 1 -Bromhexadecan (33,6 ml) in 80 ml THFabs zugetropft. Nach Zugabe von
0,5 mmol Tetrabutylammoniumiodid als Katalysator wird 12 h zum Sieden erhitzt.
Nach dem Abkühlen des Reaktionsgemisches wird das entstandene Natriumbromid abfiltriert und das Filtrat zur Trockene eingeengt. Der Rückstand wird in
Diethylether aufgenommen und die Etherphase drei Mal mit H2O ausgeschüttelt.
Nach dem Trocknen der organischen Phase über Na2SO4 wird zur Trockene eingeengt und der Rückstand über Kieselgel säulenchromatographisch (EtOAc/n- Hexan = 1 :9) gereinigt (S. Czernecki, C. Georgoulis, C. Provelenghiou Tetrahedron Lett. 39 (1976) 3535). Ausbeute: 18,5 g (52% der Theorie)
DC (EtOAc/n-Hexan = 1 :9): RrWert = 0,47 MS (FD): m/z 357,6 (M++1)
1H-NMR (CDCI3): 0,80 (t), 1 ,18 (s), 1,35 (s), 1 ,37 (s), 3,30-3,48 (m), 3,63-3,69 (dd), 3,96-4,01 (dd), 4,19 (q) 13C-NMR (CDCI3): 14,1 ; 22,7; 25,4; 26,1 ; 26,8; 29,4; 29,6; 29,7; 31 ,9 (Alkylkette),
67,0 (Alkyl-C-O-), 71 ,8; 71 ,9; 74,7 (Glycerin-C), 109,3 (Ketyl-C)
12. Synthese von 1 -Hexadecylglycerol (Pam2)
Arbeitsvorschrift: 18,5 g Pam1 (52 mmol) werden in 300 ml Essigsäure (65%)
24 h bei 400C gerührt. Der weiße Niederschlag wird abfiltriert und mehrfach mit n-
Hexan zur Trockene eingeengt. Zur Reinigung wird das Produkt dreimal mit n-
Hexan aufgeschlämmt und abfiltriert. Nicht entschütztes Edukt ist im Gegensatz zum Produkt in n-Hexan löslich und kann somit abgetrennt werden. Der verbleibende
Rückstand wird im Vakuum getrocknet. Die vereinigten Filtrate werden ebenfalls zur
Trockene eingeengt und erneut der Abspaltprozedur unterzogen (H. Paulsen, E.
Meinjohanns, F. Reck, I. Brockhausen Liebigs Ann. Chem. (1993) 721 ).
Ausbeute: 14,6 g (88% der Theorie) DC (EtOAc/n-Hexan = 1 :1 ): RrWert = 0,2
MS (FD): m/z 31 7,4 (M++1 )
1H-NMR (CDCI3): 0,86 (t), 1 ,23 (s), 3,30-3,48 (m), 3,41 -3,49 (m), 3,55-3,75 (m),
3,78-3,9 (m)
13C-NMR (CDCI3): 14,1 ; 22,7; 25,4; 26,1 ; 29,4; 29,5; 29,6; 31 ,9 (Alkylkette), 70,5 (Alkyl-C-O-), 64,2; 71 ,8; 72,4 (Glycerin-C)
13. Synthese von H -(4,4"-Dimethoxvtritvl)l-3-hexadecvlglvcerol (Pam3)

Arbeitsvorschrift: Zum Trocknen werden 10 mmol Pam2 (3,16 g) in 15 ml
Pyridinabs gelöst und das Lösungsmittel wieder abgezogen. Dieser Vorgang wird wiederholt. Zu einer Lösung des Diols in 100 ml Pyridinabs werden 12 mmol (4,06 g)
Dimethoxytrityl-chlorid (in 50 ml Pyridin gelöst) langsam zugetropft und 24 h bei RT gerührt. Danach wird die Reaktion mit 5 ml Methanol abgebrochen und das
Reaktionsgemisch zur Trockene eingeengt. Letzte Spuren von Pyridin werden durch azeotropes Schleppen mit Toluol entfernt. Der Rückstand wird in 300 ml DCM aufgenommen, mit gesättigter wässriger KCl-Lösung und H2O gewaschen und über
Na2SO4 getrocknet. Nach dem Entfernen des Lösungsmittels wird der Rückstand über Kieselgel chromatographiert (n-Hexan/Diethylether/TEA = 40:60:1 ) (R.A. Jones
"Oligonucleotide Synthesis: A Practical Approach" ed. MJ. Gait, IRL Press (1984)
23).
Ausbeute: 4,9 g (79,3% der Theorie)
DC (n-Hexan/Diethylether/TEA = 40:60:1 ): RrWert = 0,42
MS (FD): m/z 618,2 (M+), 303 (DMT+)
1H-NMR (CDCI3): 0,80 (t), 1 ,20 (s), 1 ,96 (s), 2,36 (d), 2,72 (s), 3,27 (d), 3,32-3,48
(m), 3,75 (m), 6,61 -6,76 (m), 7,08-7,37 (m)
14. Synthese von f1 -(4.4"-Dimethoxytrityl)1-2-succinyl-3-hexadecylglycerol (Pam4)

Arbeitsvorschrift: 1,26 mmol Pam3 (0,78 g) werden zwei Mal mit Pyridin getrocknet. Zu einer Lösung des Alkohols in 5 ml Pyridinabs werden 0,76 mmol DMAP (92 mg) und 1 ,26 mmol Bernsteinsäureanhydrid (126 mg) zugegeben. Nach 12 h Rühren bei RT wird die Reaktionslösung in 30 ml DCM aufgenommen, zwei Mal mit je 30 ml H2O gewaschen und über Na2SO4 getrocknet. Nach dem Entfernen des Lösungsmittels wird der Rückstand über Kieselgel chromatographiert (Ethylacetat/Methanol/NH3 (25% in H2O) = 5:1 :1 ) (D.W. Will, T. Brown Tetrahedron Lett. 33 (1992) 2729.). Ausbeute: 0,6 g (66,3%)
DC (EtOAc/MeOH/NH3(H2O) = 5:1 :1 ): RrWert = 0,32 MS (FD): m/z 718 (M+), 303 (DMT+), 1020,7 (M+DMT)+
15. Stimulation von humanen Zellen mit einem erfindungsgemäßem Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure
a) Zur Bestimmung der immunogenen Wirkung von erfindungsgemäßen Adjuvanzien wurden solche Adjuvanzien in Form einer Lipid-modifizierten
Nukleinsäure, die eine Sequenz gemäß SEQ ID NO: 40, 41 oder 42 enthalten, mit humanen Zellen koinkubiert. Dazu wurden bspw. humane PBMC-Zellen mit RNA in X-vivo15 medium (BioWhittaker), angereichert mit 2 mM L-Glutamine (BioWhittaker), 10 U/ml Penicillin (BioWhittaker) und 10 μg/ml Streptomycin 16h mit 10μg/ml RNA (mRNA kodierend ß-
Galactosidase oder Phosphorothioat RNA Oligonukleotid 40 5'GCCCGUCUGUUGUGUGACUC (SEQ ID NO: 40)) und optional mit 10 μg/ml Protamin koinkubiert. Die Überstände wurden abgenommen und mittels ELISA analysiert. Es ist insbesondere bei der Ausschüttung von Zytokinen (IL-6) zu beobachten, dass die erfindungsgemäßen Adjuvanzien ohne Zugabe von Protamin eine mehr als 5-fache Erhöhung der Zytokin- Ausschüttung (IL-6) gegenüber dem Medium aufweisen sowie bei Zugabe von Protamin gegenüber ß-Galaktosidase und RNA Oligo 40 alleine (SEQ ID NO: 40) eine leicht verbesserte Freisetzung von IL-6 (s. Figur 5A). Bei der Bestimmung der TNFα-Ausschüttung lässt sich eine deutliche Stimulierung des Immunsystems feststellen, die der von ß-Galactosidase oder RNA mindestens gleichzusetzen ist (s. Figur 5B).
b) In einem weiteren Experiment wurde die Freisetzung von TNF-α durch humane PBMC-Zellen nach Stimulation mit erfindungsgemäß verwendeten
RNA-Oligonukleotiden sowie erfindungsgemäßen Adjuvanzien bestimmt.
Dazu wurden humane PBMC-Zellen mit 10μg/ml RNA Oligonukleotiden in X-vivo15 medium (BioWhittaker), angereichert mit 2 mM L-Glutamin (BioWhittaker), 10 U/ml Penicillin (BioWhittaker) und 10 μg/ml Streptomycin
16h koinkubiert. Die hier beispielhaft für die Lipid-Modifikation verwendeten RNA-Oligos trugen die folgenden Sequenzen: RNA 40: 5' GCCCGUCUGUUGUGUGACUC (SEQ ID NO: 40), RNA CV1 :
GGUAAGUGUAAGGUGUAAGG (SEQ ID NO: 41 ), RNA CV2: AAUGGAUAUGGAAUAUGGAA (SEQ ID NO: 42); Die Überstände wurden abgenommen und mittels ELISA analysiert. Es zeigt sich deutlich, dass jedes Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure, das z.B: eine Sequenzen gemäß SEQ ID NO: 40, 41 oder 42, eine im Vergleich zu z.B. einem nicht modifizierten RNA-OI igonukleotid der Sequenz gemäß SEQ ID NO: 40 (RNA Oligo 40) (oder 41 oder 42) deutlich verbesserte Freisetzung von TNF-α und damit eine deutlich verbesserte Immunstimulation aufweist. Die besten Ergebnisse mit einer Steigerung der Immunstimulation um mehr als das 10-fache gegenüber dem nicht-modifizierten RNA-Oligonukleotid konnten mit einer Lipid-modifizierten Sequenz gemäß SEQ ID NO: 42 erreicht werden (s. Figur 6).
Vorteile der Erfindung:
Das erfindungsgemäße immunstimulierende Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure, insbesondere in Form einer 5'- und/oder 3 '-Lipid-modifizierten Nukleinsäure, ermöglicht einerseits eine Stabilisierung und eine bessere Zellgängigkeit von (pharmakologischen) Wirkstoffen sowie eine bessere Verteilung dieser Wirkstoffe innerhalb der Zelle. Darüber hinaus ist es von entscheidender Bedeutung, dass durch das erfindungsgemäße immunstimulierende Adjuvanz selbst als biologische Wirkung eine Steigerung der Immunreaktion hervorrufen kann. Dies kann einerseits durch eine Unterstützung des eigentlichen Antisense-Mechanismus, z.B. durch bessere Bindung der eingesetzten (Lipid-modifizierten) Nukleinsäuren aufgrund von Interkalation, geschehen, andererseits ist auch eine unabhängige Wirkung des erfindungsgemäßen immunstimulierenden Adjuvanz zur Steigerung der Stimulation denkbar. So wurde erfindungsgemäß z.B. ein 3'-Cholesterol-modifiziertes Phosphodiester-Oligonukleotid als immunstimulierendes Adjuvanz offenbart, das zytotoxisch auf spezielle Tumorzellen wirken kann. Zwar kann das erfindungsgemäße 3'-Cholesterol-modifiziertes Phosphodiester- Oligonukleotid auch sequenzspezifisch agieren, jedoch nicht durch Antisense-Effekte. Auch trägt die bei Cholesterol beschriebene rezeptorvermittelte Endozytose keinen wesentlichen Beitrag zur gefundenen ungewöhnlichen immunstimulierenden Wirkung. Dieser überraschende Effekt beruht daher im wesentlichen auf einer immunstimulierenden Wirkung der eingesetzten erfindungsgemäßen Lipid-modifizierten Nukleinsäure des erfindungsgemäßen Adjuvanz, wobei die Art der 3'- bzw. 5 '-Modifizierung (Lipid- Modifizierung) eine bedeutende Rolle spielt. Zusammenfassend zeigt daher das erfindungsgemäße immunstimulierende Adjuvanz eine vorteilhafte immunstimulierende Wirkung und erhöht zugleich die (Zell-)Permeabilität ggf. zusätzlich enthaltener Wirkstoffe.
Claims
1 . Immunstimulierendes Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure.
2. Immunstimulierendes Adjuvanz gemäß Anspruch 1, dadurch gekennzeichnet, dass die Lipid-modifizierte Nukleinsäure aus einer Nukleinsäure, mindestens einem mit dieser Nukleinsäure kovalent verknüpften Linker und einem mit dem jeweiligen Linker kovalent verknüpften Lipid besteht.
3. Immunstimulierendes Adjuvanz gemäß Anspruch 1 , dadurch gekennzeichnet, dass die Lipid-modifizierte Nukleinsäure aus einer mindestens einer Nukleinsäure, und mindestens einem mit dieser Nukleinsäure kovalent verknüpften (bifunktionellen) Lipid besteht.
4. Immunstimulierendes Adjuvanz gemäß Anspruch 1 , dadurch gekennzeichnet, dass die Lipid-modifizierte Nukleinsäure aus einer Nukleinsäure, mindestens einem mit dieser Nukleinsäure kovalent verknüpften Linker und einem mit dem jeweiligen Linker kovalent verknüpften Lipid sowie mindestens einem mit dieser Nukleinsäure kovalent verknüpften (bifunktionellen) Lipid besteht.
5. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, dass die Lipid-modifizierte Nukleinsäure mindestens 3-8 Lipide pro Nukleinsäure aufweist, wobei a) alle Lipide über einen Linker kovalent mit der Nukleinsäure verknüpft sind; b) alle Lipide direkt kovalent mit der Nukleinsäure verknüpft sind; c) ein Teil der Lipide über einen Linker kovalent mit der Nukleinsäure verknüpft sind und ein Teil der Lipide direkt kovalent mit der
Nukleinsäure verknüpft sind.
6. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine RNA oder DNA, ein RNA- oder DNA-Oligonukleotid, ein RNA- oder DNA- Homopolymer oder eine CpG-Nukleinsäure ist.
7. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 6, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure einzelsträngig oder doppelsträngig, als Homo- oder-Heteroduplex und/oder linear oder zirkulär vorliegt.
8. Immunstimulierendes Adjuvanz gemäß Anspruch 7, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine Länge von etwa 2 bis etwa 1000 Nukleotiden aufweist.
9. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 8, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine Länge von etwa 5 bis etwa 200 Nukleotiden aufweist.
10. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 9, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine
Länge von etwa 6 bis etwa 100 Nukleotiden aufweist.
11. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 10, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine Länge von etwa 6 bis etwa 40 Nukleotiden aufweist.
12. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 1 1 , dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure eine Länge von etwa 6 bis etwa 31 Nukleotiden aufweist.
13. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 12, dadurch gekennzeichnet, dass die Nukleinsäure ausgewählt wird aus einer der Sequenzen gemäß SEQ ID NO: 1 - 67.
14. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 12, dadurch gekennzeichnet, dass die Nukleinsäure ausgewählt wird aus einer Sequenz, die eine Identität zu einer der Sequenzen gemäß SEQ ID NO: 1 - 67 von mindestens 60 % aufweisen.
15. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 14, dadurch gekennzeichnet, dass das Lipid ausgewählt wird aus Lipiden oder lipophilen Resten umfassend Vitamine, einschließlich α-Tocopherol (Vitamin E), RRR-α-Tocopherol (D-α-Tocopherol), L-α-Tocopherol, Racemat D,L-α-Tocopherol, Vitamin A und dessen Derivate, einschließlich Retinsäure, Retinol, Vitamin D und dessen Derivate, einschließlich den Ergosterol-Vorstufen von Vitamin D, Vitamin E und dessen Derivate, einschließlich Vitamin E-Succinat (VES), Vitamin K und dessen Derivate, Chinon bzw. Phytolverbindungen, Steroide einschließlich Gallensäuren, ausgewählt aus Cholinsäure, Desoxycholinsäure, Dehydrocholinsäure, oder Cortison,
Digoxygenin, Testosteron, Cholesterol oder Thiocholesterol, oder Polyalkylenglykole, aliphatische Gruppen einschließlich C,-C20-Alkane, C1-C20- Alkene, oder C,-C20-Alkanolverbindungen, Dodecandiol, Hexadecanol oder Undecyl-Resten, oder Phospholipide einschließlich Phosphatidylglycerol, Diacylphosphatidylglycerol, Phosphatidylcholin, Dipalmitoylphosphatidylcholin,
Distearoylphosphatidylcholin, Phosphatidylserin, Phosphatidylethanolamin, oder Di-hexadecyl-rac-glycerol, Sphingolipide, Cerebroside, Ganglioside,
Triethylammonium 1 ^-Di-O-hexadecyl-rac-glycero-ß-H-phosphonat, Polyamine, Polyalkylenglykole, einschließlich Polyethylenglykol (PEG), Hexaethylenglykol (HEG), oder Palmitin und Palmityl-Reste, Octadecylamine oder Hexylamino- carbonyl-oxycholesterol-Reste, Wachse, Terpene, alicyklische Kohlenwasserstoffe, und gesättigte bzw. einfach- oder mehrfach ungesättigte Fettsäurereste.
16. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 2 und 4 bis 15, dadurch gekennzeichnet, dass der Linker ausgewählt wird aus einer Verbindung, die mindestens zwei, drei oder vier reaktive Gruppen aufweist, ausgewählt aus einer Hydroxygruppe, einer Aminogruppe und einer Alkoxygruppe.
17. Immunstimulierendes Adjuvanz gemäß Anspruch 16, dadurch gekennzeichnet, dass der Linker ausgewählt wird aus Glykol, Glycerin, Glycerinderivaten, 2-Aminobutyl-
1 ,3-propandiol, 2-Aminobutyl-1 ,3-propandiol-Derivaten oder einem 2-Aminobutyl-
1 ,3-propandiol-Gerüst, Pyrrolidin-Linkern bzw. Pyrrolidin enthaltenden organischen Molekülen.
18. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 1 7, dadurch gekennzeichnet, dass die Verknüpfung zwischen Nukleinsäure und Lipid oder zwischen Nukleinsäure und einem mit dem Lipid verknüpften Linker am 3'- und/oder 5'-Ende der Nukleinsäure stattfindet.
19. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 -18, dadurch gekennzeichnet, dass die Nukleinsäure der Lipid-modifizierten Nukleinsäure zusätzlich chemisch modifiziert ist.
20. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 -19, dadurch gekennzeichnet, dass die Nukleinsäure als RNA vorliegt und zusätzlich am 5'-Ende eine "Cap-Struktur" und/oder am 3'-Ende einen Poly-A-Schwanz aufweist.
21. Immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 -20, dadurch gekennzeichnet, dass das immunstimulierende Adjuvanz mit einem weiteren Adjuvanz kombiniert wird ausgewählt aus Aluminiumhydroxid, (komplettem oder inkomplettem) Freund'schen Adjuvanz, stabilisierenden kationischen Peptiden ausgewählt aus Polypeptiden, Protamin, Nukleolin, Spermin oder Spermidin, kationischen Polysacchariden, Chitosan, TDM, MDP, Muramyldipeptid, sowie
Alaun-Lösung, Pluronics und Lipopeptiden, einschließlich Pam3Cys.
22. Pharmazeutische Zusammensetzung enthaltend ein immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 21 , mindestens einen Wirkstoff und optional einen pharmazeutisch geeigneten Träger und/oder weitere Hilfs- und Zusatzstoffe und/oder Adjuvanzien.
23. Pharmazeutische Zusammensetzung gemäß Anspruch 22, dadurch gekennzeichnet, dass der Wirkstoff ausgewählt wird aus Peptiden, Proteinen, Nukleinsäuren, (therapeutisch wirksamen) niedermolekularen organischen oder anorganischen Verbindungen mit einem Molekulargewicht kleiner als 5.000, Zuckern, Antigenen, Antikörpern oder bereits im Stand der Technik bekannten Therapeutika.
24. Pharmazeutische Zusammensetzung gemäß Anspruch 23, dadurch gekennzeichnet, dass das weitere Adjuvanz wie in Anspruch 21 definiert ist.
25. Pharmazeutische Zusammensetzung gemäß einem der Ansprüche 22 bis 24, dadurch gekennzeichnet, dass die pharmazeutische Zusammensetzung als Vakzine vorliegt.
26. Verwendung eines immunstimulierenden Adjuvanz gemäß einem der Ansprüche 1 bis 21 zur Herstellung einer pharmazeutischen Zusammensetzung zur Behandlung von Krebserkrankungen oder Infektionserkrankungen.
27. Verwendung gemäß Anspruch 26, dadurch gekennzeichnet, dass Krebserkrankungen ausgewählt werden aus Kolonkarzinomen, Melanomen, Nierenkarzinomen, Lymphomen, akuter myeloider Leukämie (AML), akute lymphoider Leukämie (ALL), chronischer myeloider Leukämie (CML), chronischer lymphozytischer Leukämie (CLL), Gastroi ntestinalen Tumoren, Lungenkarzinomen, Gliomen, Schilddrüsentumoren, Mammakarzinomen, Prostatatumoren, Hepatomen, diversen virus-induzierten Tumoren wie z.B. Papillomvirus-induzierten Karzinomen (z.B. Cervixkarzinome), Adenokarzinomen, Herpesviren-induzierten Tumoren (z.B.
Burkitt's Lymphom, EBV-induziertes B Zelllymphome), Hepatitis B-induzierten Tumoren (Hepatozellkarzinome), HTLV-1 und HTLV-2 induzierten Lymphomen, Akustikusneurinomen, Gebärmuttehalskrebs, Lungenkrebs, Rachenkrebs, Analkarzinomen, Glioblastomen, Lymphomen, Rektumkarzinomen, Astrozytomen, Hirntumoren, Magenkrebs, Retinoblastomen, Basaliomen, Hirnmetastasen,
Medulloblastomen, Scheidenkrebs, Bauchspeicheldrüsenkrebs, Hodenkrebs, Melanomen, Schilddrüsenkarzinomen, Blasenkrebs, Hodgkin-Syndrom, Meningeomen, Schneeberger Krankheit, Bronchialkarzinomen, Hypophysentumor, Mycosis fungoides, Speiseröhrenkrebs, Brustkrebs, Karzinoide, Neurinomen, Spinaliomen, Burkitt-Lymphomen, Kehlkopfkrebs, Nierenkrebs, Thymomen, Corpuskarzinomen, Knochenkrebs, Non-Hodgkin-Lymphomen, Urethrakrebs, CUP- Sysndrom, Kopf-Hals-Tumoren, Oligodendrogliomen, Vulvakrebs, Darmkrebs, Kolonkarzinomen, Ösphaguskarzinomen, Warzenbeteiligung, Dünndarmtumoren,
Kraniopharyngeomen, Ovarial-Karzinomen, Weichteiltumoren, Eierstockkrebs, Leberkrebs, Pankreaskarzinomen, Zervixkarzinomen, Endometriumkarzinomen, Lebermetastasen, Peniskrebs, Zungenkrebs, Gallenblasenkrebs, Leukämie, Plasmozytomen, Gebärmutterkrebs, Lidtumor und Prostatakrebs.
28. Verwendung gemäß Anspruch 26, dadurch gekennzeichnet, dass Infektionserkrankungen ausgewählt werden aus Influenza, Malaria, SARS, Gelbfieber, AIDS, Lyme-Borreliose, Leischmaniasis, Anthrax, Meningitis, viralen Infektionskrankheiten wie AIDS, Condyloma acuminata, Dellwarze, Dengue-Fieber, Dreitagefieber, Ebolavirus, Erkältung, Frühsommermeningoenzephalitis (FSME),
Grippe, Gürtelrose, Hepatitis, Herpes-Simplex Typ I, Herpes-Simplex Typ II, Herpes zoster, Influenza, Japanische Enzephalitis, Lassafieber, Marburg-Virus, Masern, Maul- und Klausenseuche, Mononukleose, Mumps, Norwalk-Virus-infektion, Pfeifersches-Drüsenfieber, Pocken, Polio (Kinderlähmung), Pseudokrupp, Ringelröteln, Tollwut, Warzen, West-Nil-Fieber, Windpocken, Zytomegalie-Virus
(CMV), bakteriellen Infektionskrankheiten wie Abort (Prostataentzündung), Anthrax, Appendizitis (Blinddarmentzündung), Borreliose, Botulismus, Camphylobacter, Chlamydia trachomatis (Harnröhren-, Bindehautentzündung), Cholera, Diphterie, Diphterie, Donavanosis, Epiglottitis, Fleckfieber, Flecktyphys, Gasbrand, Gonorhoe, Hasenpest, Heliobakter pylori, Keuchhusten, klimatischer Bubo,
Knochenmarksentzündung, Legionärskrankheit, Lepra, Listeriose, lungenentzündung, Meningitis, Bakterielle Gehirnhautentzündung, Milzbrand, Mittelohrentzündung, Mycoplasma hominis, Neugeborenensepsis
(Chorioamnionitis), Noma, Paratyphus, Pest, Reitersyndrom, Rocky Mountain spotted fever, Salmonellen-Paratyphus, Salmonellen-Typhus, Scharlach, Syphilis,
Tetanus, Tripper, Tsutsugamushi, Tuberkulose, Typhus, Vaginitis (Kolpitis), Weicher Schanker und durch Parasiten, Protozoen oder Pilze hervorgerufene Inkektionskrankheiten wie Amöbenruhr, Bilharziose, Chagas-Krankheit, Fußpilz, Kleienpilzflechte, Krätze (Skabies), Malaria, Onchozerkose (Flussblindheit), oder Pilzerkrankungen, Toxoplasmose, Trichomoniasis, Trypanosomiasis
(Schlafkrankheit), Viszerale Leishmaniose, Windeldermatitis, Schistosomiasis, Fischvergiftung (Ciguatera), Kandiose, Kutane Leishmaniose, Lamblienruhr (Giadiasis), oder Schlafkrankheit, oder Infektionskrankheiten hervorgerufen durch
Echinococcus, Fischbandwurm, Fuchsbandwurm, Hundebandwurm, Läuse, Rinderbandwurm, Schweinebandwurm und Zwergbandwurm.
29. Kit, enthaltend ein immunstimulierendes Adjuvanz gemäß einem der Ansprüche 1 bis 21 und/oder eine pharmazeutische Zusammensetzung gemäß einem der
Ansprüche 22 bis 25, sowie optional eine technische Gebrauchsanweisung mit
Angaben zur Verabreichung und Dosierung des immunstimulierenden Adjuvanz oder der pharmazeutischen Zusammensetzung.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/748,181 US20070280929A1 (en) | 2006-02-17 | 2007-05-14 | Adjuvant in the form of a lipid-modified nucleic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102006007433.5 | 2006-02-17 | ||
| DE102006007433A DE102006007433A1 (de) | 2006-02-17 | 2006-02-17 | Adjuvanz in Form einer Lipid-modifizierten Nukleinsäure |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/748,181 Continuation-In-Part US20070280929A1 (en) | 2006-02-17 | 2007-05-14 | Adjuvant in the form of a lipid-modified nucleic acid |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007095976A2 true WO2007095976A2 (de) | 2007-08-30 |
| WO2007095976A3 WO2007095976A3 (de) | 2007-11-01 |
Family
ID=37891885
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/008321 WO2007095976A2 (de) | 2006-02-17 | 2006-08-24 | Adjuvanz in form einer lipid-modifizierten nukleinsäure |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20070280929A1 (de) |
| DE (1) | DE102006007433A1 (de) |
| WO (1) | WO2007095976A2 (de) |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102009034779A1 (de) | 2009-07-25 | 2011-02-03 | Emc Microcollections Gmbh | Synthetische Analoga bakterieller Lipopeptide und ihre Anwendung zur Therapie und Prophylaxe allergischer Erkrankungen |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| US8999380B2 (en) | 2012-04-02 | 2015-04-07 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9107886B2 (en) | 2012-04-02 | 2015-08-18 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding basic helix-loop-helix family member E41 |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9186372B2 (en) | 2011-12-16 | 2015-11-17 | Moderna Therapeutics, Inc. | Split dose administration |
| US9283287B2 (en) | 2012-04-02 | 2016-03-15 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of nuclear proteins |
| US9334328B2 (en) | 2010-10-01 | 2016-05-10 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9428535B2 (en) | 2011-10-03 | 2016-08-30 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9533047B2 (en) | 2011-03-31 | 2017-01-03 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9597380B2 (en) | 2012-11-26 | 2017-03-21 | Modernatx, Inc. | Terminally modified RNA |
| WO2017049245A2 (en) | 2015-09-17 | 2017-03-23 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| WO2017066781A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs with modified phosphate linkage |
| WO2017066793A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs and methods of mrna capping |
| WO2017066782A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Hydrophobic mrna cap analogs |
| WO2017066789A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs with modified sugar |
| WO2017066791A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Sugar substituted mrna cap analogs |
| WO2017112865A1 (en) | 2015-12-22 | 2017-06-29 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of agents |
| WO2017218704A1 (en) | 2016-06-14 | 2017-12-21 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| WO2018089540A1 (en) | 2016-11-08 | 2018-05-17 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| US10064934B2 (en) | 2015-10-22 | 2018-09-04 | Modernatx, Inc. | Combination PIV3/hMPV RNA vaccines |
| US10064935B2 (en) | 2015-10-22 | 2018-09-04 | Modernatx, Inc. | Human cytomegalovirus RNA vaccines |
| WO2018170306A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| WO2018170336A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Lipid nanoparticle formulation |
| US10124055B2 (en) | 2015-10-22 | 2018-11-13 | Modernatx, Inc. | Zika virus RNA vaccines |
| WO2018232120A1 (en) | 2017-06-14 | 2018-12-20 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of agents |
| WO2019036638A1 (en) | 2017-08-18 | 2019-02-21 | Modernatx, Inc. | METHODS FOR PREPARING MODIFIED RNA |
| WO2019046809A1 (en) | 2017-08-31 | 2019-03-07 | Modernatx, Inc. | METHODS OF MANUFACTURING LIPID NANOPARTICLES |
| US10273269B2 (en) | 2017-02-16 | 2019-04-30 | Modernatx, Inc. | High potency immunogenic zika virus compositions |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US10449244B2 (en) | 2015-07-21 | 2019-10-22 | Modernatx, Inc. | Zika RNA vaccines |
| WO2020061457A1 (en) | 2018-09-20 | 2020-03-26 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| WO2020061367A1 (en) | 2018-09-19 | 2020-03-26 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| US10653767B2 (en) | 2017-09-14 | 2020-05-19 | Modernatx, Inc. | Zika virus MRNA vaccines |
| US10695419B2 (en) | 2016-10-21 | 2020-06-30 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| WO2020160397A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| WO2020160430A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Vortex mixers and associated methods, systems, and apparatuses thereof |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| US11103578B2 (en) | 2016-12-08 | 2021-08-31 | Modernatx, Inc. | Respiratory virus nucleic acid vaccines |
| WO2021204175A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Lipid nanoparticle composition |
| WO2021204179A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Nucleic acid vaccines for coronavirus |
| WO2022002040A1 (en) | 2020-06-30 | 2022-01-06 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2022037652A1 (en) | 2020-08-20 | 2022-02-24 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| US11351242B1 (en) | 2019-02-12 | 2022-06-07 | Modernatx, Inc. | HMPV/hPIV3 mRNA vaccine composition |
| US11364292B2 (en) | 2015-07-21 | 2022-06-21 | Modernatx, Inc. | CHIKV RNA vaccines |
| WO2022152141A2 (en) | 2021-01-14 | 2022-07-21 | Suzhou Abogen Biosciences Co., Ltd. | Polymer conjugated lipid compounds and lipid nanoparticle compositions |
| WO2022152109A2 (en) | 2021-01-14 | 2022-07-21 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| EP4035659A1 (de) | 2016-11-29 | 2022-08-03 | PureTech LYT, Inc. | Exosome zur ausgabe von therapeutischen wirkstoffen |
| US11406703B2 (en) | 2020-08-25 | 2022-08-09 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| US11497807B2 (en) | 2017-03-17 | 2022-11-15 | Modernatx, Inc. | Zoonotic disease RNA vaccines |
| WO2022247755A1 (en) | 2021-05-24 | 2022-12-01 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023044343A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Acyclic lipids and methods of use thereof |
| WO2023044333A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| EP4162950A1 (de) | 2021-10-08 | 2023-04-12 | Suzhou Abogen Biosciences Co., Ltd. | Nukleinsäureimpfstoffe gegen coronavirus |
| WO2023056917A1 (en) | 2021-10-08 | 2023-04-13 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023056914A1 (en) | 2021-10-08 | 2023-04-13 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023116804A1 (zh) | 2021-12-23 | 2023-06-29 | 苏州艾博生物科技有限公司 | 脂质化合物和脂质纳米颗粒组合物 |
| WO2023122752A1 (en) | 2021-12-23 | 2023-06-29 | Renagade Therapeutics Management Inc. | Constrained lipids and methods of use thereof |
| WO2023196931A1 (en) | 2022-04-07 | 2023-10-12 | Renagade Therapeutics Management Inc. | Cyclic lipids and lipid nanoparticles (lnp) for the delivery of nucleic acids or peptides for use in vaccinating against infectious agents |
| US11866754B2 (en) | 2015-10-16 | 2024-01-09 | Modernatx, Inc. | Trinucleotide mRNA cap analogs |
| WO2024037578A1 (en) | 2022-08-18 | 2024-02-22 | Suzhou Abogen Biosciences Co., Ltd. | Composition of lipid nanoparticles |
| US12070495B2 (en) | 2019-03-15 | 2024-08-27 | Modernatx, Inc. | HIV RNA vaccines |
| WO2024192291A1 (en) | 2023-03-15 | 2024-09-19 | Renagade Therapeutics Management Inc. | Delivery of gene editing systems and methods of use thereof |
| WO2024192277A2 (en) | 2023-03-15 | 2024-09-19 | Renagade Therapeutics Management Inc. | Lipid nanoparticles comprising coding rna molecules for use in gene editing and as vaccines and therapeutic agents |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6207646B1 (en) | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US7956043B2 (en) | 2002-12-11 | 2011-06-07 | Coley Pharmaceutical Group, Inc. | 5′ CpG nucleic acids and methods of use |
| TR201909609T4 (tr) | 2005-08-23 | 2019-07-22 | Univ Pennsylvania | Modifiye edilmiş nükleosidleri içeren rna ve kullanım yöntemleri. |
| WO2008014979A2 (en) | 2006-07-31 | 2008-02-07 | Curevac Gmbh | NUCLEIC ACID OF FORMULA (I): GIXmGn, OR (II): CIXmCn, IN PARTICULAR AS AN IMMUNE-STIMULATING AGENT/ADJUVANT |
| DE102006035618A1 (de) * | 2006-07-31 | 2008-02-07 | Curevac Gmbh | Nukleinsäure der Formel (I): GlXmGn, insbesondere als immunstimulierendes Adjuvanz |
| NZ575437A (en) | 2006-09-27 | 2012-02-24 | Coley Pharm Gmbh | Cpg oligonucleotide analogs containing hydrophobic t analogs with enhanced immunostimulatory activity |
| WO2009030254A1 (en) * | 2007-09-04 | 2009-03-12 | Curevac Gmbh | Complexes of rna and cationic peptides for transfection and for immunostimulation |
| WO2009046739A1 (en) | 2007-10-09 | 2009-04-16 | Curevac Gmbh | Composition for treating prostate cancer (pca) |
| PL2176408T3 (pl) | 2008-01-31 | 2015-08-31 | Curevac Gmbh | Kwasy nukleinowe zawierające wzór (NuGiXmGnNv)a i ich pochodne jako środki immunostymulujące/ adiuwanty |
| US8728465B2 (en) * | 2008-06-17 | 2014-05-20 | Cedars-Sinai Medical Center | Use of toll-like receptor ligands as adjuvants to vaccination therapy for brain tumors |
| WO2010037408A1 (en) * | 2008-09-30 | 2010-04-08 | Curevac Gmbh | Composition comprising a complexed (m)rna and a naked mrna for providing or enhancing an immunostimulatory response in a mammal and uses thereof |
| WO2010075525A1 (en) * | 2008-12-24 | 2010-07-01 | Cedars-Sinai Medical Center | Method of using tumor cell debris to reduce brain tumor recurrence or growth |
| US20110053829A1 (en) | 2009-09-03 | 2011-03-03 | Curevac Gmbh | Disulfide-linked polyethyleneglycol/peptide conjugates for the transfection of nucleic acids |
| ES2558106T3 (es) | 2010-07-30 | 2016-02-02 | Curevac Ag | Formación de complejos de ácidos nucleicos con componentes catiónicos disulfuro-reticulados para la transfección e inmunoestimulación |
| WO2012019630A1 (en) | 2010-08-13 | 2012-02-16 | Curevac Gmbh | Nucleic acid comprising or coding for a histone stem-loop and a poly(a) sequence or a polyadenylation signal for increasing the expression of an encoded protein |
| WO2012116715A1 (en) | 2011-03-02 | 2012-09-07 | Curevac Gmbh | Vaccination in newborns and infants |
| WO2012113413A1 (en) | 2011-02-21 | 2012-08-30 | Curevac Gmbh | Vaccine composition comprising complexed immunostimulatory nucleic acids and antigens packaged with disulfide-linked polyethyleneglycol/peptide conjugates |
| WO2012116714A1 (en) | 2011-03-02 | 2012-09-07 | Curevac Gmbh | Vaccination in elderly patients |
| CN102351932A (zh) * | 2011-07-07 | 2012-02-15 | 中国人民解放军军事医学科学院放射与辐射医学研究所 | 一种抗流感病毒寡核苷酸的结构、制备方法和用途 |
| CN102295673A (zh) * | 2011-07-07 | 2011-12-28 | 中国人民解放军军事医学科学院放射与辐射医学研究所 | 一种脂肪链修饰的寡核苷酸的结构、制备方法及用途 |
| WO2013067203A1 (en) | 2011-11-02 | 2013-05-10 | The Regents Of The University Of California | Reprogramming of cellular adhesion |
| WO2013113326A1 (en) | 2012-01-31 | 2013-08-08 | Curevac Gmbh | Pharmaceutical composition comprising a polymeric carrier cargo complex and at least one protein or peptide antigen |
| WO2014160243A1 (en) | 2013-03-14 | 2014-10-02 | The Trustees Of The University Of Pennsylvania | Purification and purity assessment of rna molecules synthesized with modified nucleosides |
| AU2014310931B2 (en) | 2013-08-21 | 2019-12-19 | CureVac SE | Rabies vaccine |
| JP6896421B2 (ja) | 2013-08-21 | 2021-06-30 | キュアバック アーゲー | 呼吸器合胞体ウイルス(rsv)ワクチン |
| US10369216B2 (en) | 2014-04-01 | 2019-08-06 | Curevac Ag | Polymeric carrier cargo complex for use as an immunostimulating agent or as an adjuvant |
| EP3981437B1 (de) | 2014-04-23 | 2024-10-09 | ModernaTX, Inc. | Nukleinsäureimpfstoffe |
| EP3461904A1 (de) * | 2014-11-10 | 2019-04-03 | ModernaTX, Inc. | Alternative nukleinsäuremoleküle mit reduziertem uracilgehalt und verwendungen davon |
| EP3294885B1 (de) | 2015-05-08 | 2020-07-01 | CureVac Real Estate GmbH | Verfahren zur herstellung von rna |
| ES2960429T3 (es) | 2015-05-29 | 2024-03-04 | Curevac Mfg Gmbh | Método para producir y purificar ARN, que comprende al menos una etapa de filtración de flujo tangencial |
| TN2018000152A1 (en) | 2015-10-22 | 2019-10-04 | Modernatx Inc | Nucleic acid vaccines for varicella zoster virus (vzv) |
| EP3364982A4 (de) | 2015-10-22 | 2019-04-17 | ModernaTX, Inc. | Impfstoffe gegen geschlechtskrankheiten |
| WO2017081110A1 (en) | 2015-11-09 | 2017-05-18 | Curevac Ag | Rotavirus vaccines |
| EP3558356A2 (de) | 2016-12-23 | 2019-10-30 | CureVac AG | Mers-coronavirus-impfstoff |
| EP3595713A4 (de) | 2017-03-15 | 2021-01-13 | ModernaTX, Inc. | Impfstoff gegen respiratorisches synzytialvirus |
| WO2018170270A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Varicella zoster virus (vzv) vaccine |
| WO2018170256A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Herpes simplex virus vaccine |
| RU2020117611A (ru) * | 2017-11-02 | 2021-12-02 | Янссен Байофарма, Инк. | Олигонуклеотидные конструкты и их применение |
| TWI818943B (zh) * | 2018-01-04 | 2023-10-21 | 中央研究院 | 用於增加療效之可與細胞結合的免疫佐劑 |
| WO2019148101A1 (en) | 2018-01-29 | 2019-08-01 | Modernatx, Inc. | Rsv rna vaccines |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3906092A (en) * | 1971-11-26 | 1975-09-16 | Merck & Co Inc | Stimulation of antibody response |
| US6406705B1 (en) * | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| WO2003066649A1 (en) * | 2002-02-04 | 2003-08-14 | Biomira Inc. | Immunostimulatory, covalently lipidated oligonucleotides |
| US20030225016A1 (en) * | 2001-06-21 | 2003-12-04 | Fearon Karen L. | Chimeric immunomodulatory compounds and methods of using the same - III |
| WO2004004743A1 (de) * | 2002-07-03 | 2004-01-15 | Curevac Gmbh | Immunstimulation durch chemisch modifizierte rna |
| EP1393745A1 (de) * | 2002-07-29 | 2004-03-03 | Hybridon, Inc. | Modulierung der immunstimulierenden Eigenschaften von Oligonukleotiden und Analoga durch optimale Darstellung der 5'-Enden |
| WO2004058159A2 (en) * | 2002-12-23 | 2004-07-15 | Dynavax Technologies Corporation | Branched immunomodulatory compounds and methods of using the same |
| WO2005030259A2 (en) * | 2003-09-25 | 2005-04-07 | Coley Pharmaceutical Group, Inc. | Nucleic acid-lipophilic conjugates |
| WO2006116458A2 (en) * | 2005-04-26 | 2006-11-02 | Coley Pharmaceutical Gmbh | Modified oligoribonucleotide analogs with enhances immunostimulatory activity |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1196558A1 (de) * | 1999-06-08 | 2002-04-17 | Aventis Pasteur | Immunostimulatorisches oligonukleotid |
| WO2005001022A2 (en) * | 2003-04-10 | 2005-01-06 | 3M Innovative Properties Company | Methods and compositions for enhancing immune response |
-
2006
- 2006-02-17 DE DE102006007433A patent/DE102006007433A1/de not_active Withdrawn
- 2006-08-24 WO PCT/EP2006/008321 patent/WO2007095976A2/de active Application Filing
-
2007
- 2007-05-14 US US11/748,181 patent/US20070280929A1/en not_active Abandoned
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3906092A (en) * | 1971-11-26 | 1975-09-16 | Merck & Co Inc | Stimulation of antibody response |
| US6406705B1 (en) * | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| US20030225016A1 (en) * | 2001-06-21 | 2003-12-04 | Fearon Karen L. | Chimeric immunomodulatory compounds and methods of using the same - III |
| WO2003066649A1 (en) * | 2002-02-04 | 2003-08-14 | Biomira Inc. | Immunostimulatory, covalently lipidated oligonucleotides |
| WO2004004743A1 (de) * | 2002-07-03 | 2004-01-15 | Curevac Gmbh | Immunstimulation durch chemisch modifizierte rna |
| EP1393745A1 (de) * | 2002-07-29 | 2004-03-03 | Hybridon, Inc. | Modulierung der immunstimulierenden Eigenschaften von Oligonukleotiden und Analoga durch optimale Darstellung der 5'-Enden |
| WO2004058159A2 (en) * | 2002-12-23 | 2004-07-15 | Dynavax Technologies Corporation | Branched immunomodulatory compounds and methods of using the same |
| WO2005030259A2 (en) * | 2003-09-25 | 2005-04-07 | Coley Pharmaceutical Group, Inc. | Nucleic acid-lipophilic conjugates |
| WO2006116458A2 (en) * | 2005-04-26 | 2006-11-02 | Coley Pharmaceutical Gmbh | Modified oligoribonucleotide analogs with enhances immunostimulatory activity |
Non-Patent Citations (4)
| Title |
|---|
| BAYARD B ET AL: "ANTIVIRAL ACTIVITY IN L-1210 CELLS OF LIPOSOME-ENCAPSULATED 2'-5' OLIGOADENYLATE ANALOGS" EUROPEAN JOURNAL OF BIOCHEMISTRY, Bd. 151, Nr. 2, 1985, Seiten 319-326, XP002440943 ISSN: 0014-2956 * |
| HAUSCH F ET AL: "A Novel Carboxy-functionalized Photocleavable Dinucleotide Analog for the Selection of RNA Catalysts" TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, Bd. 39, Nr. 34, 20. August 1998 (1998-08-20), Seiten 6157-6158, XP004128903 ISSN: 0040-4039 * |
| KWIATKOWSKI, M. ET AL: "The 9-(4- octadecyloxyphenylxanthen )-9-yl-group. A new acid-labile hydroxyl protective group and its application in the preparative reverse-phase chromatographic separation of oligoribonucleotides" ACTA CHEMICA SCANDINAVICA, SERIES B: ORGANIC CHEMISTRY AND BIOCHEMISTRY , B38(8), 657-71 CODEN: ACBOCV; ISSN: 0302-4369, 1984, XP009086061 * |
| SHEA R G ET AL: "Synthesis, hybridization properties and antiviral activity of lipid-oligodeoxynucleotide conjugates" NUCLEIC ACIDS RESEARCH, OXFORD UNIVERSITY PRESS, SURREY, GB, Bd. 18, Nr. 13, 1990, Seiten 3777-3783, XP002244700 ISSN: 0305-1048 * |
Cited By (129)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011012240A2 (de) | 2009-07-25 | 2011-02-03 | Emc Microcollections Gmbh | Lipopeptide zur therapie und prophylaxe allergischer erkrankungen |
| DE102009034779A1 (de) | 2009-07-25 | 2011-02-03 | Emc Microcollections Gmbh | Synthetische Analoga bakterieller Lipopeptide und ihre Anwendung zur Therapie und Prophylaxe allergischer Erkrankungen |
| US9937233B2 (en) | 2010-08-06 | 2018-04-10 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9447164B2 (en) | 2010-08-06 | 2016-09-20 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9701965B2 (en) | 2010-10-01 | 2017-07-11 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9334328B2 (en) | 2010-10-01 | 2016-05-10 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US9533047B2 (en) | 2011-03-31 | 2017-01-03 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US10751386B2 (en) | 2011-09-12 | 2020-08-25 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9428535B2 (en) | 2011-10-03 | 2016-08-30 | Moderna Therapeutics, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9271996B2 (en) | 2011-12-16 | 2016-03-01 | Moderna Therapeutics, Inc. | Formulation and delivery of PLGA microspheres |
| US9186372B2 (en) | 2011-12-16 | 2015-11-17 | Moderna Therapeutics, Inc. | Split dose administration |
| US9295689B2 (en) | 2011-12-16 | 2016-03-29 | Moderna Therapeutics, Inc. | Formulation and delivery of PLGA microspheres |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US9107886B2 (en) | 2012-04-02 | 2015-08-18 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding basic helix-loop-helix family member E41 |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| US9233141B2 (en) | 2012-04-02 | 2016-01-12 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins associated with blood and lymphatic disorders |
| US9283287B2 (en) | 2012-04-02 | 2016-03-15 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of nuclear proteins |
| US9220792B2 (en) | 2012-04-02 | 2015-12-29 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding aquaporin-5 |
| US9303079B2 (en) | 2012-04-02 | 2016-04-05 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9301993B2 (en) | 2012-04-02 | 2016-04-05 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding apoptosis inducing factor 1 |
| US9220755B2 (en) | 2012-04-02 | 2015-12-29 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins associated with blood and lymphatic disorders |
| US9221891B2 (en) | 2012-04-02 | 2015-12-29 | Moderna Therapeutics, Inc. | In vivo production of proteins |
| US9216205B2 (en) | 2012-04-02 | 2015-12-22 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding granulysin |
| US9192651B2 (en) | 2012-04-02 | 2015-11-24 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of secreted proteins |
| US9149506B2 (en) | 2012-04-02 | 2015-10-06 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding septin-4 |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| US8999380B2 (en) | 2012-04-02 | 2015-04-07 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9050297B2 (en) | 2012-04-02 | 2015-06-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding aryl hydrocarbon receptor nuclear translocator |
| US9061059B2 (en) | 2012-04-02 | 2015-06-23 | Moderna Therapeutics, Inc. | Modified polynucleotides for treating protein deficiency |
| US9089604B2 (en) | 2012-04-02 | 2015-07-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for treating galactosylceramidase protein deficiency |
| US9095552B2 (en) | 2012-04-02 | 2015-08-04 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding copper metabolism (MURR1) domain containing 1 |
| US9878056B2 (en) | 2012-04-02 | 2018-01-30 | Modernatx, Inc. | Modified polynucleotides for the production of cosmetic proteins and peptides |
| US9828416B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US9114113B2 (en) | 2012-04-02 | 2015-08-25 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding citeD4 |
| US9675668B2 (en) | 2012-04-02 | 2017-06-13 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding hepatitis A virus cellular receptor 2 |
| US9827332B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of proteins |
| US9254311B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of proteins |
| US9782462B2 (en) | 2012-04-02 | 2017-10-10 | Modernatx, Inc. | Modified polynucleotides for the production of proteins associated with human disease |
| US9814760B2 (en) | 2012-04-02 | 2017-11-14 | Modernatx, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9597380B2 (en) | 2012-11-26 | 2017-03-21 | Modernatx, Inc. | Terminally modified RNA |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US10449244B2 (en) | 2015-07-21 | 2019-10-22 | Modernatx, Inc. | Zika RNA vaccines |
| US10702597B2 (en) | 2015-07-21 | 2020-07-07 | Modernatx, Inc. | CHIKV RNA vaccines |
| US11364292B2 (en) | 2015-07-21 | 2022-06-21 | Modernatx, Inc. | CHIKV RNA vaccines |
| US11007260B2 (en) | 2015-07-21 | 2021-05-18 | Modernatx, Inc. | Infectious disease vaccines |
| EP3736261A1 (de) | 2015-09-17 | 2020-11-11 | ModernaTX, Inc. | Verbindungen und zusammensetzungen zur intrazellulären verabreichung von therapeutischen wirkstoffen |
| EP4286012A2 (de) | 2015-09-17 | 2023-12-06 | ModernaTX, Inc. | Verbindungen und zusammensetzungen zur intrazellulären verabreichung von therapeutischen wirkstoffen |
| WO2017049245A2 (en) | 2015-09-17 | 2017-03-23 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| WO2017066793A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs and methods of mrna capping |
| WO2017066781A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs with modified phosphate linkage |
| EP4086269A1 (de) | 2015-10-16 | 2022-11-09 | ModernaTX, Inc. | Mrna-cap-analoga mit modifizierter phosphatverbindung |
| US11866754B2 (en) | 2015-10-16 | 2024-01-09 | Modernatx, Inc. | Trinucleotide mRNA cap analogs |
| US10570388B2 (en) | 2015-10-16 | 2020-02-25 | Modernatx, Inc. | Phosphate replacement MRNA cap analogs |
| US10563195B2 (en) | 2015-10-16 | 2020-02-18 | Modernatx, Inc. | Phosphate replacement mRNA cap analogs |
| US10428106B2 (en) | 2015-10-16 | 2019-10-01 | Modernatx, Inc. | Phosphate replacement mRNA cap analogs |
| WO2017066782A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Hydrophobic mrna cap analogs |
| WO2017066789A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Mrna cap analogs with modified sugar |
| WO2017066791A1 (en) | 2015-10-16 | 2017-04-20 | Modernatx, Inc. | Sugar substituted mrna cap analogs |
| US10272150B2 (en) | 2015-10-22 | 2019-04-30 | Modernatx, Inc. | Combination PIV3/hMPV RNA vaccines |
| US11278611B2 (en) | 2015-10-22 | 2022-03-22 | Modernatx, Inc. | Zika virus RNA vaccines |
| US11872278B2 (en) | 2015-10-22 | 2024-01-16 | Modernatx, Inc. | Combination HMPV/RSV RNA vaccines |
| US10238731B2 (en) | 2015-10-22 | 2019-03-26 | Modernatx, Inc. | Chikagunya virus RNA vaccines |
| US10517940B2 (en) | 2015-10-22 | 2019-12-31 | Modernatx, Inc. | Zika virus RNA vaccines |
| US10543269B2 (en) | 2015-10-22 | 2020-01-28 | Modernatx, Inc. | hMPV RNA vaccines |
| US11484590B2 (en) | 2015-10-22 | 2022-11-01 | Modernatx, Inc. | Human cytomegalovirus RNA vaccines |
| US10064934B2 (en) | 2015-10-22 | 2018-09-04 | Modernatx, Inc. | Combination PIV3/hMPV RNA vaccines |
| US10383937B2 (en) | 2015-10-22 | 2019-08-20 | Modernatx, Inc. | Human cytomegalovirus RNA vaccines |
| US11235052B2 (en) | 2015-10-22 | 2022-02-01 | Modernatx, Inc. | Chikungunya virus RNA vaccines |
| US10064935B2 (en) | 2015-10-22 | 2018-09-04 | Modernatx, Inc. | Human cytomegalovirus RNA vaccines |
| US10675342B2 (en) | 2015-10-22 | 2020-06-09 | Modernatx, Inc. | Chikungunya virus RNA vaccines |
| US10933127B2 (en) | 2015-10-22 | 2021-03-02 | Modernatx, Inc. | Betacoronavirus mRNA vaccine |
| US10124055B2 (en) | 2015-10-22 | 2018-11-13 | Modernatx, Inc. | Zika virus RNA vaccines |
| US10702600B1 (en) | 2015-10-22 | 2020-07-07 | Modernatx, Inc. | Betacoronavirus mRNA vaccine |
| US10702599B2 (en) | 2015-10-22 | 2020-07-07 | Modernatx, Inc. | HPIV3 RNA vaccines |
| US10716846B2 (en) | 2015-10-22 | 2020-07-21 | Modernatx, Inc. | Human cytomegalovirus RNA vaccines |
| EP4036079A2 (de) | 2015-12-22 | 2022-08-03 | ModernaTX, Inc. | Verbindungen und zusammensetzungen zur intrazellulären verabreichung von wirkstoffen |
| WO2017112865A1 (en) | 2015-12-22 | 2017-06-29 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of agents |
| WO2017218704A1 (en) | 2016-06-14 | 2017-12-21 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| US11197927B2 (en) | 2016-10-21 | 2021-12-14 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| US10695419B2 (en) | 2016-10-21 | 2020-06-30 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| US11541113B2 (en) | 2016-10-21 | 2023-01-03 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| WO2018089540A1 (en) | 2016-11-08 | 2018-05-17 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| EP4035659A1 (de) | 2016-11-29 | 2022-08-03 | PureTech LYT, Inc. | Exosome zur ausgabe von therapeutischen wirkstoffen |
| US11103578B2 (en) | 2016-12-08 | 2021-08-31 | Modernatx, Inc. | Respiratory virus nucleic acid vaccines |
| US10273269B2 (en) | 2017-02-16 | 2019-04-30 | Modernatx, Inc. | High potency immunogenic zika virus compositions |
| WO2018170306A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| EP4186888A1 (de) | 2017-03-15 | 2023-05-31 | ModernaTX, Inc. | Verbindung und zusammensetzungen zur intrazellulären verabreichung von therapeutischen wirkstoffen |
| WO2018170336A1 (en) | 2017-03-15 | 2018-09-20 | Modernatx, Inc. | Lipid nanoparticle formulation |
| US11497807B2 (en) | 2017-03-17 | 2022-11-15 | Modernatx, Inc. | Zoonotic disease RNA vaccines |
| WO2018232120A1 (en) | 2017-06-14 | 2018-12-20 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of agents |
| WO2019036638A1 (en) | 2017-08-18 | 2019-02-21 | Modernatx, Inc. | METHODS FOR PREPARING MODIFIED RNA |
| WO2019046809A1 (en) | 2017-08-31 | 2019-03-07 | Modernatx, Inc. | METHODS OF MANUFACTURING LIPID NANOPARTICLES |
| US11207398B2 (en) | 2017-09-14 | 2021-12-28 | Modernatx, Inc. | Zika virus mRNA vaccines |
| US10653767B2 (en) | 2017-09-14 | 2020-05-19 | Modernatx, Inc. | Zika virus MRNA vaccines |
| WO2020061367A1 (en) | 2018-09-19 | 2020-03-26 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| WO2020061457A1 (en) | 2018-09-20 | 2020-03-26 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| EP4427739A2 (de) | 2019-01-31 | 2024-09-11 | ModernaTX, Inc. | Verfahren zur herstellung von lipidnanopartikeln |
| WO2020160430A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Vortex mixers and associated methods, systems, and apparatuses thereof |
| WO2020160397A1 (en) | 2019-01-31 | 2020-08-06 | Modernatx, Inc. | Methods of preparing lipid nanoparticles |
| US11351242B1 (en) | 2019-02-12 | 2022-06-07 | Modernatx, Inc. | HMPV/hPIV3 mRNA vaccine composition |
| US12070495B2 (en) | 2019-03-15 | 2024-08-27 | Modernatx, Inc. | HIV RNA vaccines |
| WO2021204179A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Nucleic acid vaccines for coronavirus |
| WO2021204175A1 (en) | 2020-04-09 | 2021-10-14 | Suzhou Abogen Biosciences Co., Ltd. | Lipid nanoparticle composition |
| WO2022002040A1 (en) | 2020-06-30 | 2022-01-06 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2022037652A1 (en) | 2020-08-20 | 2022-02-24 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| US11406703B2 (en) | 2020-08-25 | 2022-08-09 | Modernatx, Inc. | Human cytomegalovirus vaccine |
| WO2022152141A2 (en) | 2021-01-14 | 2022-07-21 | Suzhou Abogen Biosciences Co., Ltd. | Polymer conjugated lipid compounds and lipid nanoparticle compositions |
| WO2022152109A2 (en) | 2021-01-14 | 2022-07-21 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2022247755A1 (en) | 2021-05-24 | 2022-12-01 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023044333A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| WO2023044343A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Acyclic lipids and methods of use thereof |
| EP4162950A1 (de) | 2021-10-08 | 2023-04-12 | Suzhou Abogen Biosciences Co., Ltd. | Nukleinsäureimpfstoffe gegen coronavirus |
| WO2023056914A1 (en) | 2021-10-08 | 2023-04-13 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023056917A1 (en) | 2021-10-08 | 2023-04-13 | Suzhou Abogen Biosciences Co., Ltd. | Lipid compounds and lipid nanoparticle compositions |
| WO2023122752A1 (en) | 2021-12-23 | 2023-06-29 | Renagade Therapeutics Management Inc. | Constrained lipids and methods of use thereof |
| WO2023116804A1 (zh) | 2021-12-23 | 2023-06-29 | 苏州艾博生物科技有限公司 | 脂质化合物和脂质纳米颗粒组合物 |
| WO2023196931A1 (en) | 2022-04-07 | 2023-10-12 | Renagade Therapeutics Management Inc. | Cyclic lipids and lipid nanoparticles (lnp) for the delivery of nucleic acids or peptides for use in vaccinating against infectious agents |
| WO2024037578A1 (en) | 2022-08-18 | 2024-02-22 | Suzhou Abogen Biosciences Co., Ltd. | Composition of lipid nanoparticles |
| WO2024192291A1 (en) | 2023-03-15 | 2024-09-19 | Renagade Therapeutics Management Inc. | Delivery of gene editing systems and methods of use thereof |
| WO2024192277A2 (en) | 2023-03-15 | 2024-09-19 | Renagade Therapeutics Management Inc. | Lipid nanoparticles comprising coding rna molecules for use in gene editing and as vaccines and therapeutic agents |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102006007433A1 (de) | 2007-08-23 |
| US20070280929A1 (en) | 2007-12-06 |
| WO2007095976A3 (de) | 2007-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007095976A2 (de) | Adjuvanz in form einer lipid-modifizierten nukleinsäure | |
| DE102006035618A1 (de) | Nukleinsäure der Formel (I): GlXmGn, insbesondere als immunstimulierendes Adjuvanz | |
| AU2007280690C1 (en) | Nucleic acid of formula (I): GIXmGn, or (II): CIXmCn, in particular as an immune-stimulating agent/adjuvant | |
| DE69008521T2 (de) | Kovalente konjugate von lipiden und oligonukleotiden. | |
| EP1797886B1 (de) | Immunstimulation durch chemisch modifizierte RNA | |
| TWI680767B (zh) | 用於增強的細胞攝取之化合物及方法 | |
| DE60132170T2 (de) | MODULIERUNG DER DURCH CpG-OLIGONUKLEOTIDE VERURSACHTEN IMMUNSTIMULIERUNG DURCH POSITIONSBEDINGTE VERÄNDERUNG VON NUKLEOSIDEN | |
| EP3087988A2 (de) | Verwendung von inhibitoren von toll-like-rezeptoren zur vorbeugung und behandlung von hypercholesterolämie und hyperlipidämie und damit zusammenhängenden krankheiten | |
| KR20180031025A (ko) | 약물 전달을 위한 다중 리간드 제제 | |
| US8153605B2 (en) | Modulation of toll-like receptor 3 expression by antisense oligonucleotides | |
| CN102459301A (zh) | 亲脂性多聚核苷酸缀合物 | |
| CN103415302A (zh) | 调控基于toll-样受体的免疫应答的免疫调节寡核苷酸(iro)化合物 | |
| JP2010536787A (ja) | Toll様受容体モジュレータ | |
| KR20110039382A (ko) | 안티센스 올리고누클레오티드에 의한 톨-유사 수용체 8 발현의 조절 | |
| JP2021512044A (ja) | オリゴヌクレオチドコンストラクト及びその使用 | |
| EP1204742A2 (de) | Oligonukleotide zur inhibierung der expression von humanem eg5 | |
| WO2001008707A2 (de) | Konjugate und verfahren zu deren herstellung sowie deren verwendung zum transport von molekülen über biologische membranen | |
| KR20110039381A (ko) | 안티센스 올리고누클레오티드에 의한 톨-유사 수용체 7 발현의 조절 | |
| WO2020044349A1 (en) | Compounds, cojugates and compositions for use in the methods for trans-membrane delivery of molecules | |
| JP2021509406A (ja) | 分子の膜貫通送達のための化合物および方法 | |
| TW202412845A (zh) | 用於遞送治療劑至cns組織之脂質結合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 11748181 Country of ref document: US |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 11748181 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06791641 Country of ref document: EP Kind code of ref document: A2 |