WO2006090931A1 - スルホンアミド化合物の抗癌剤との新規併用 - Google Patents
スルホンアミド化合物の抗癌剤との新規併用 Download PDFInfo
- Publication number
- WO2006090931A1 WO2006090931A1 PCT/JP2006/304219 JP2006304219W WO2006090931A1 WO 2006090931 A1 WO2006090931 A1 WO 2006090931A1 JP 2006304219 W JP2006304219 W JP 2006304219W WO 2006090931 A1 WO2006090931 A1 WO 2006090931A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituent
- hydrogen atom
- optionally substituted
- halogen atom
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/498—Pyrazines or piperazines ortho- and peri-condensed with carbocyclic ring systems, e.g. quinoxaline, phenazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/343—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide condensed with a carbocyclic ring, e.g. coumaran, bufuralol, befunolol, clobenfurol, amiodarone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4741—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having oxygen as a ring hetero atom, e.g. tubocuraran derivatives, noscapine, bicuculline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/243—Platinum; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to a sulfonamide compound, a platinum complex substance such as Oxaliplatin or Cisplatin, a DNA-topoisomerase I inhibitor such as CPT-11, an antimetabolite such as Gemcitabine or Methotrexate, and a microtubule inhibitor such as Paclitaxel.
- a novel pharmaceutical composition and kit comprising a combination of at least one substance selected from the group consisting of antibiotics such as Doxorubicin, and a method for treating cancer and a method for inhibiting cancer or angiogenesis. is there.
- Conventional cancer chemotherapeutic agents include the alkylating agent cyclophosphamide, the antimetabolite methotrexate, fluorouracil, the antibiotics adriamycin, mitomycin, bleomycin, plant-derived taxol, vincristine, etoposide
- metal complexes such as cisplatin, but none of them have sufficient antitumor effects, and the development of new antitumor agents has been eagerly desired.
- the present invention has been made in view of such a situation, and the problem to be solved is a pharmaceutical composition and kit having excellent antitumor activity and / or angiogenesis inhibitory activity, and cancer. It is to find a method of treatment and / or a method of inhibiting angiogenesis.
- E7820 is a taxol (Paclitaxel), SN38 (CPT-11 activator), in vascular endothelial cell proliferation assay (in vitro), When combined with methotrexate, cisplatin, gemcitabine, and oxorubicin, it is statistically (combination index) for cell growth inhibition. It became clear that it showed a significant synergistic effect.
- E7820 is a combination of Paclitaxel, SN38 (CPT'll active substance), Cisplatin, Gemcitabine, and Doxorubicin in combination with vascular endothelial cell tube formation (in vitro).
- E7820 is statistically significant (two-way ANOVA) for antitumor effects when used in combination with Oxaliplatin or CPT-11. It became clear that In addition, in a human knee cancer cell line subcutaneous transplantation model (in vivo), E7820 shows a statistically significant (two-way ANOVA) synergistic effect on antitumor effects when used in combination with Gemcitabine. It was revealed.
- E7820 is used in combination with at least one compound selected from the group consisting of Oxaliplatin, CPT-11 and Gemcitabine, so that at least one selected from the group consisting of Oxaliplatin, CPT-11 and Gemcitabine is used.
- E7070 in this specification, “N— (3-chloro-1, 1 H-indole-7-yl) 4-sulfamoylbenzenesulfonamide”). It was found that the gene variation pattern and cytostatic activity by E7820, LY186641, LY295501, LY573636 or CQS or a combination thereof showed high correlation. In addition, in an assay for measuring cytostatic activity, it was found that the cancer cell lines E7820, LY186641, LY295501, LY-ASAP, LY573636, or CQS that are resistant to E7070 are cross-resistant.
- E7820, LY186641, LY295501, LY-ASAP, LY573636, or CQS or the combination of these yarns, Oxaliplatin, Cisplatin, CPT-11, Gemcitabine, Methotrexate Paclitaxel and Doxorubicin force
- Oxaliplatin, Cisplatin, CPT-11, Gemcitabine Methotrexate Paclitaxel and Doxorubicin force
- sulfoneamide compounds preferably E7820, LY186641, LY295501, LY-ASAP, LY573636
- the present invention relates to the following.
- Sulfonamide compound and at least one substance selected from the group consisting of a platinum complex substance, a DNA-topoisomerase I inhibitor, an antimetabolite, a microtubule inhibitor and an antibiotic, or a pharmacologically acceptable substance thereof Or a pharmaceutical composition comprising a combination of these salts and solvates thereof.
- a preparation comprising a sulfonamide compound and at least one substance selected from the group consisting of a platinum complex substance, a DNA-topoisomerase I inhibitor, an antimetabolite, a microtubule inhibitor and an antibiotic, or a substance thereof
- a kit comprising a pharmacologically acceptable salt or a preparation containing a solvate thereof as a set.
- sulfonamide compound for the production of a pharmaceutical composition in combination with a pharmacologically acceptable salt thereof or a solvate thereof.
- Sulfonamide compound and at least one substance selected from the group consisting of platinum complex substances, DNA-topoisomerase I inhibitors, antimetabolites, microtubule inhibitors and antibiotics, or pharmacologically acceptable A method for treating cancer, and a method for inhibiting Z or angiogenesis, comprising administering a salt or a solvate thereof to a patient.
- Sulfonamide compounds for coadministration to patients together with at least one substance selected from the group consisting of platinum complex substances, DNA-topoisomerase I inhibitors, antimetabolites, microtubule inhibitors and opi antibiotics.
- a pharmaceutical composition comprising
- the sulfonamide compound is N- (3-cyanol 4-methinole 1 H-indole 1-7 yl) -3 syananobenzenesolephone amide or a pharmacology thereof.
- the sulfonamide compound has the general formula (I)
- E is 1 O—, 1 N (CH 3 ) 1, 1 CH 2 —, _CH 2 CH 2 — or 1 CH 2 O1, D is 1 CH 2 — or 1 O—, Ria means a hydrogen atom or a halogen atom, and R 2a means a halogen atom or a trifluoromethyl group.
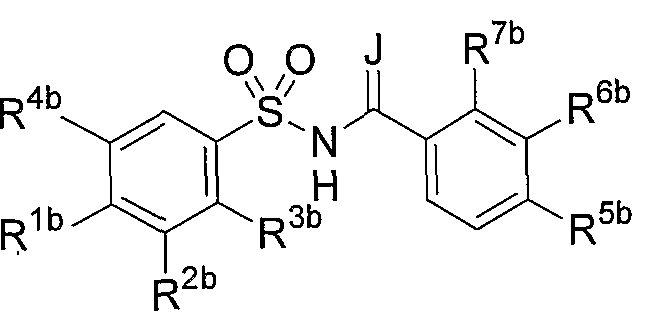
- Rib represents a hydrogen atom, a halogen atom, an optionally substituted 1 C 6 alkyl group, or an optionally substituted C i_C 4 alkoxy group, optionally substituted Ci—C 4 alkylthio group, 1 CF 3 , 10 CF 3 , — S CF 3 , optionally substituted C1—C4 alkoxycarbonyl Group, nitro group, azide group, 1 O (S 0 2 ) CH 3 , — N (CH 3 ) 2, hydroxyl group, phenyl group, substituted phenyl group, pyridinyl group, chenyl group, furyl group A quinolinyl group or a triazole group, R 2b is a hydrogen atom, a nitrogen atom, a cyano group, one CF 3 , an optionally substituted d-C 6 alkyl group, or a substituted group.
- R 3b is a hydrogen atom or an optionally substituted Ci-C 4 alkoxy group
- R 4b represents a hydrogen atom or An optionally substituted C—Ce alkyl group (wherein at least one of R 3 b and R 4 b is a hydrogen atom)
- R 5 b is a hydrogen atom, a halogen atom Ci_C 6 alkyl group, mono-CF 3 or nitro group which may have a substituent
- R 6b Ci-C 6 alkyl group which may have a hydrogen atom, a halogen atom or a substituent ( Provided that when R 6b is an optionally substituted Ci—C 6 alkyl group, R 5b is a hydrogen atom and R 7b is a halogen atom), R 7b is
- b is If have a have a substituent is also may CI- C 6 alkyl group force ⁇ or R? B force halogen atom or a substituent is also optionally Ci _C 6 alkyl group, R or RGb Each of which is a hydrogen atom).
- the group consisting of a platinum complex substance, a DNA-topoisomerase I inhibitor, an antimetabolite, a microtubule inhibitor and an antibiotic is Oxaliplatin ⁇ Cisp on atin, CPT-11, Mention may be made of the group consisting of Gemcitabine, Methotrexate Paclitaxel and Doxorubicin.
- the present invention provides a pharmaceutical composition and kit exhibiting excellent antitumor activity and / or angiogenesis inhibitory activity, as well as a method for treating cancer and / or a method for inhibiting angiogenesis.
- a sulfonamide compound that is, (A) E7820, (B) a compound represented by general formula (I), preferably LY186641 or LY295501, (C) represented by general formula (II) A compound, preferably LY-ASAP, (D) LY573636 and (E) at least one compound selected from CQS, and (i) a platinum complex substance, preferably Oxaliplatin or Cisplatin, (ii) DNA-topoisomerase I inhibition A substance, preferably CPT-11, (iii) an antimetabolite, preferably Gemcitabine or Methotrexate, (iv) a microtubule inhibitor, preferably Paclitaxel, and (v) an antibiotic, preferably at least one selected from Doxorubicin Combined with two substances, it has excellent antitumor activity and / or antiangiogenic activity.
- Pharmaceutical compositions and kits shown, as well as methods of cancer treatment and methods of inhibiting Z or angiogenesis have been provided that can be used to be
- Figure 1 shows the combined effect of E7820 and oxaliplatin on tumor growth inhibition in a human colon cancer cell line (Colo320DM) subcutaneous transplant model (in vivo).
- Colo320DM human colon cancer cell line
- * indicates a statistically significant synergistic effect with a risk factor of less than 0.05.
- days # indicate the number of days when the start of administration ⁇ is dayl.
- Figure 2 shows the combined effect of E7820 and CPT-11 on tumor growth inhibition in a human colon cancer cell line (Colo320DM) subcutaneous transplant model (in vivo).
- Colo320DM human colon cancer cell line
- * indicates that there was a statistically significant synergistic effect with a risk factor of less than 0.01.
- the number of days # indicates the number of days when the start date of administration is dayl.
- Figure 3 shows the combined effect of E7820 and Gemcitabine on tumor growth inhibition in a human knee cancer cell line (KP-1) subcutaneous transplant model (in vivo).
- KP-1 human knee cancer cell line
- * indicates a statistically significant synergistic effect with a risk factor of less than 0.01.
- the number of days # indicates the number of days when the start date of administration is dayl.
- FIG. 4 shows the results of hierarchical clustering analysis on the DNA microarray in Example 7.
- FIG. 5 shows the correlation coefficient in the DNA microarray in Example 8.
- FIG. 6 shows the results of hierarchical clustering analysis on the DNA microarray in Example 8.
- FIG. 7 shows the correlation coefficient in the DNA microarray in Example 8.
- FIG. 8 shows the result of hierarchical clustering analysis in the DNA microarray in Example 8.
- Fig. 9 shows HCT116-C9, which is used to measure cytostatic activity.
- FIG. 10 shows the growth inhibitory action of E7070 and LY573636 on HCT116-C9, HCT116-C9-C1 and HCT116.C9-C4 in the assay measuring cytostatic activity.
- the pharmaceutical composition Oppi Z or kit of the present invention, and the cancer treatment method Oppi Z or the method of inhibiting angiogenesis comprise a sulfonamide compound.
- the sulfonamide compound includes a compound represented by the following formula (V).
- E7820 The compound represented by the formula (V) is N- (3-cyanol-4-monomethyl-1H-india-llu 7-yl) 1-3-cyanobenzenesulfonamide and is called E7820. There is a case.
- E7820 can be produced by a known method.
- E7820 can be produced by the method described in International Publication No. 00/50395 pamphlet (WO00 / 50395).
- the sulfone compound includes a compound represented by the following general formula (I).
- E is 1 O—, 1 N (CH 3 ) —, 1 CH 2 , 1 CH 2 CH 2 — or 1 CH 2 0—, and D is 1 CH 2 — or 1 O—
- R la represents a hydrogen atom or a halogen atom (for example, a fluorine atom, a chlorine atom, a bromine atom, an iodine atom)
- R 2a represents a neurogen atom or a trifluoromethyl group, respectively.
- the compound represented by the general formula (I) of the present invention can be produced by a known method. For example, it can be produced by a method described in EP 0222475A1 (EP0222475A1).
- preferred compounds are LY186641 or LY295501.
- LY186641 refers to N — [[(4-Chromium-Fuel) amino] carbonyl] —2,3-Dihydro D 1 H—Indene 5—Sulfonamide, whose structural formula is represented by the following formula (VI): Show.
- LY186641 can be produced by a known method.
- LY186641 can be produced by the method described in European Patent Application Publication No. 0222475A1 (EP0222475A1).
- LY295501 refers to N — [[((3,4-dichlorophole) amino) carbol] —2,3-dihydrobenzofuran-5-sulfonamide, whose structural formula is It is shown in the following formula (VII).
- LY295501 can be produced by a known method, for example, the method described in European Patent Application Publication No. 0222475A1 (EP0222475A1) and Z or European Patent Application Publication No. 0555036A2 (EP0555036A2).
- the sulfonamide compound includes a compound represented by the following general formula (II).
- J represents 1 O— or 1 NH—
- Rib represents a hydrogen atom, a halogen atom, an optionally substituted d—C 6 alkyl group, or a substituent.
- Ci 1 C 4 alkoxy group which may have, Ci 1 C4 alkylthio group which may have a substituent, — CF 3 , 1 OCF 3 , 1 S CF 3 , which may have a substituent Good Ci —C 4 alkoxycarbonyl group, nitro group, azide group, — O (S0 2 ) CH 3 , —N (CH 3 ) 2, hydroxyl group, phenyl group, substituted furoyl group, pyridinyl group, chenyl A group, a furyl group, a quinolinyl group or a triazole group
- R 2 b is a hydrogen atom, a halogen atom, a cyan group, one CF 3 , an optionally substituted Ci 1 C
- R 5b is a hydrogen atom
- a R? B is a halogen atom
- R? B is a halogen atom
- a substituent D-Ce alkyl group or one CF 3 provided that either R 5b or R 7b is an optionally substituted Ci i C 6 alkyl group
- R? B force S a halogen atom or an optionally substituted d—C 6 alkyl group, either R 5b or R 6b is a hydrogen atom
- the “halogen atom” is preferably a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom.
- Ji 1 - ⁇ 6 Arukiru group includes straight Kusariwaka properly from 1 to 6 carbon atoms means a branched alkyl group, for example, but is not particularly limited Is a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a sec-butyl group, a tert-butyl group, an n-pentyl group (amyl group), an isopentyl group, or a neopentyl group.
- Tert-pentyl group 1-methylbutyl group, 2-methylbutyl group, 1,2-dimethylpropyl group, n-hexyl group, isohexyl group, 1-methylpentinole group, 2-methylpentyl group, 3-Methylenopentinole group, 1-ethylpropyl group, 1,1-dimethylbutyl group, 1,2-dimethylbutyl group, 2,2-dimethylbutyl group, 1,3-dimethylbutyl group, 2,3- Dimethylbutyl group, 3,3-dimethylbutyl 1-ethylbutyl group, 2-ethylbutyl group, 1,1,2-trimethylpropyl group, 1,2,2-trimethylpropyl group, 1-ethyl-1-methylpropyl group, 1-ethyl-2-methylpropyl group, etc.
- Suitable groups are methyl group, ethyl group, n-propyl group, isopyl pill group, n-butynole group, isobutyl group, sec-butyl group, tert-butyl group, n-pentyl group, n —A hexyl group can be exemplified.
- Ci 1 C 4 alkoxy group means an alkoxy group having 1 to 4 carbon atoms, and is not particularly limited, but is preferably a methoxy group, an ethoxy group. ⁇ -propoxy group, isopropoxy group, ⁇ -butoxy group, isobutoxy group, sec-butoxy group, tert-butoxy group and the like.
- the alkyl group is not particularly limited, but methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl and the like can be mentioned.
- examples of “C i—C 4 alkoxycarbonyl group” are not particularly limited, but include methoxycarbonyl group, ethoxycarbonyl group, n-propoxycarbonyl group, Examples thereof include a propoxycarbonyl group, an n-butoxycarbonyl group, an isobutoxycarbonyl group, a sec-butoxycarbonyl group, and a tert-butoxycarbonyl group.
- examples of the substituent to be introduced is not particularly limited, for example, C i _ C 6 alkyl group (e.g., methyl group, Echiru group, n- propyl group, an isopropyl group , n- butyl group, Isopuchiru group, sec- butyl group, tert one butyl group), d-C 4 alkoxy groups (e.g., main butoxy group, an ethoxy group, n - propoxy group, isopropoxy group, n- butoxy group , Isobutoxy group, sec-butoxy group, tert-butoxy group, etc.), amino group, hydroxyl group, halogen atom (for example, fluorine atom, chlorine atom, bromine atom, iodine atom) or silyl group .
- C i _ C 6 alkyl group e.g., methyl group, Echiru group, n- propyl group, an isopropyl group
- the compound represented by the general formula ( ⁇ ) of the present invention can be produced by a known method. For example, it can be produced by the method described in WO 02/098848 (Pamphlet of WO 02/098848). .
- the preferred compound is LY-ASAP.
- LY-ASAP refers to N- (2,4-dichlorobenzoyl) 14-cyclohexane sulfonamide, and its structural formula is shown in the following formula (VIII).
- LY-ASAP can be produced by a known method, for example, by the method described in International Publication No. 02/098848 (WO02 / 098848).
- examples of the sulfonamide compound include LY573636.
- LY573636 refers to N- (2,4-dichlorobenzoyl) 15-promothiophene-2-sulfonamide, and its structural formula is shown in the following formula ( ⁇ ).
- LY573636 is preferably a sodium salt.
- LY573636 can be produced by a known method.
- LY573636 can be produced in the same manner as described in International Publication No. 02/098848 (WO02 / 098848), and commercially available 5-bromothiophene 2-sulfonyl chloride. , 4-Dichlorodibenzoic acid.
- LY573636 can be produced by the method described in Example 63 in WO 03/035629 (WO03 / 035629).
- examples of the sulfonamide compound include CQS.
- CQS refers to 2-sulfanylamide 5-chloroquinoline, and the structural formula is shown in the following formula (IV).
- CQS can be produced by a known method, for example, by the method of (J. Am. Chem. Soc, 1947, 71, 6-10).
- Sulfonamide compounds may form pharmacologically acceptable salts with acids or bases.
- the sulfonamide compound in the present invention includes these pharmacologically acceptable salts.
- salts with acids include inorganic acid salts such as hydrochloride, hydrobromide, sulfate, phosphate, formic acid, acetic acid, lactic acid, succinic acid, fumaric acid, maleic acid.
- salts with organic acids such as acid, citrate, tartaric acid, benzoic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, and trifluoroacetic acid.
- alkali metal salts such as sodium salt and potassium salt
- alkaline earth metal salts such as calcium salt and magnesium salt
- the sulfonamide compound may be an anhydride or may form a solvate such as a hydrate.
- the solvate may be either a hydrate or a non-hydrate, but a hydrate is preferred.
- water, alcohol (for example, methanol, ethanol, n-propanol), dimethylformamide, or the like can be used.
- the sulfonamide compounds in the present invention include those solvates and / or optical isomers.
- the sulfonamide compound in the present invention also includes a sulfonamide compound that undergoes metabolism such as oxidation, reduction, hydrolysis, and conjugation in vivo.
- the sulfonamide compound in the present invention also includes a compound that generates a sulfonamide compound by undergoing metabolism such as oxidation, reduction, and hydrolysis in vivo.
- the pharmaceutical composition and kit or kit of the present invention, and the cancer treatment method and the Z or angiogenesis inhibition method include platinum complex substance, DNA-topoisomei'ase I inhibitor, metabolite, microtubule inhibition It contains at least one substance selected from the group consisting of substances and antibiotics.
- the platinum complex material may be a complex having platinum as the central metal (platinum complex), and depends on the degree of bonding between the central metal and the ligand, the charge of the complex, and the bridge structure of the ligand. It is not limited to an increase in the number of central metals.
- the platinum complex substance may be a platinum preparation in which a platinum complex is formulated. In the present invention, the platinum complex substance is sometimes referred to as a platinum preparation.
- platinum complex substances include Oxaliplatin, Carboplatin, Cisplatin (CDDP), Lobaplatin, AR_726, Miriplatin, Picoplatin, PLD'147, Satraplatin, Thioplatin, Triplatin, and preferably Oxaliplatin or Cisplatin. Oxaliplatin is more preferable.
- the platinum complex material can be produced by a known method or can be purchased.
- Oxaliplatin is oxalato (1R, 2R-cyclohexanediamme) platinum, which is a compound represented by the formula (IX).
- Oxaliplatin can be produced by a known method. Oxaliplatin can also be obtained by purchasing Eloxatin (registered trademark) from Sanofi Aventis.
- Carboplatin is cis-diammine (l, l-cyclobutanedicarboxylate) platinum.
- Carboplatin can be obtained by purchasing Paraplatin (Bristol Pharmaceutical Co., Ltd.).
- the DNA-topoisomerase I inhibitor means a substance having an action of inhibiting DNA-topoisomerase I.
- the DNA-topoisomerase I inhibitor includes CPT_11, Topotecan hydrochloride N Exatecan, Rubitecan, 9-amino-camptothecin, Lurtotecan dihydrochloride, Gimatecan, Edotecarin, etc., preferably CPT-11.
- the DNA-topoisomerase I inhibitor can be produced by a known method or can be purchased.
- CPT-11 means irinotecan hydrochloride trihydrate ([1,4'-bipiperidine] -l'-carboxylic acid (S) -4, ll-aiethyl-3,4,12, 14 -tetra ydro-4 'hydroxy- 3, 14-dioxo- lH-pyrano- [3', 4 ': 6,7] -mdolizmo [l, 2-b] quinoiin-9-yl ester Hydrochloride trihydrate)
- S -4, ll-aiethyl-3,4,12, 14 -tetra ydro-4 'hydroxy- 3, 14-dioxo- lH-pyrano- [3', 4 ': 6,7] -mdolizmo [l, 2-b] quinoiin-9-yl ester Hydrochloride trihydrate
- the formula is a compound represented by
- CPT-11 can be produced by a known method.
- CPT-11 can be obtained by purchasing Topotecin (registered trademark) from Daiichi Pharmaceutical Co., Ltd.
- SN38 which is a CPT'll active form, can be used as a DNA-topoisomerase I inhibitor.
- SN38 refers to 7-ethyl-10-hydro- (20) S-camptothecin.
- SN38 can be purchased from ABATRA.
- Topotecan hydrochloride is (4S) _10- [(dimethylammo) methyl] -4-ethyl-4, 9-di ydroxylH-pyranoL3 ', 4': 6, 7] indolizino [1, 2-b] qumoline-3, 14 (4H, 12H) -dione hydrochloride is a compound represented by formula (XI).
- Topotecan hydrochloride can be produced by a known method (US Pat. No. 5,004,858 (US5004758)). Topotecan hydrochloride can also be obtained by purchasing Hicamtin (registered trademark) from Nippon Kayaku.
- Exatecan means (IS, 9S) -l-amino-9-ethyl-5-fluoro-l, 2, 3 9, 12, 15-hexahvdro-9-ny roxy4-methyl-10H, 13H- benzo [de] pyrano [3 ', 4': 6 7] indolizino [l, 2-b] quinoline-10, 13-dione is a compound represented by the formula (XII).
- Exatecan can be produced by a known method (Japanese Patent Laid-Open No. 05-59061 (JP93-59061)).
- Rubitecan means (4S) -ethyl-4-hydroxy-10-nitro-l, 12 'dihydro-14H-pyrano [3', 4 ': 6, 7] -indolizino [l, 2-b ] quinoline-3, 14 (4H, 12H) -dione is a compound represented by the formula (XIII).
- Rubitecan can be produced by a known method (Journal of Medicinal Chemistry (1986), 29 (11), 2358.63, JP 59-051288).
- 9-amino-camptotliecin is (4S) -ethyl-4-hydroxy-10-amino-1, 12-dihydro-14H-pvrano [3 ', 4': 6, 7] -indolizmo [l , 2-b] qumoline-3, 14 (4H, 12H) -dione, a compound represented by the formula (XIV).
- 9-amino-camptothecin can be produced by a known method (Japanese Patent Laid-Open No. 59/0551289).
- Lurtotecan dihydrochloride is 7_ (4-methylpiperazinomethylene) -10, ll-ethylene ioxy20 (s) -camptothecin dihydrochloride, which is a compound represented by the formula (XV).
- Lurtotecan dihydrochloride can be produced by a known method (WO95 / 29919).
- Gimatecan is (4S) -11 _ [(E)-[[1, 1-aiinethylethoxy] iniino] methyl] -4-ethyl-4-hydroxyl, 12-dihydro-14H- pyrano [3 ', 4': 6, 7] -indolizino [l, 2-b] quinoline-3, 14 (4H) -dione is a compound represented by the formula (XVI).
- Gimatecan can be produced by a known method (WO00 / 053607).
- Edotecarin is 12-6-D-glucopyranosyl "2, 10-didydroxy-6-[[2-hydroxy-l- (hydroxymethyl) ethyl] ammo] -12, 13-dihydro-6H- Indolo [2,3-a] pyrrolo [3,4-c] carbazole-5,7-dione is a compound represented by the formula (XVII).
- Edotecarin can be produced by a known method (WO95 / 30682).
- the antimetabolite is a compound having a structure similar to that required for cell metabolism such as nucleic acid synthesis and protein synthesis, and inhibits cell metabolism due to its structural similarity.
- examples of the antimetabolite include Cytidine derivatives. Specific examples include Gemcitabine N Cytarabine (araC), Enocitabine, Citarabine ocfosfate ⁇ 5-azacytidine, CNDAC, and preferably Gemcitabine.
- the antimetabolite includes a folic acid antagonist, and specifically includes methotrexate. Folic acid antagonists inhibit nucleic acid synthesis by inhibiting dihydrofolate reductase.
- Antimetabolites can be produced by known methods and can be purchased.
- Gemcitabine refers to gemcitabine hydrochloride (2′-deoxy-2 ′, 2′_difluoro-cytidine hydrochloride), which is a compound represented by the formula (XVIII).
- Gemcitabine can be produced by a known method. Gemcitabine can also be obtained by purchasing GEMZAR (registered trademark) from Nippon Iri Lily.
- Cytarabine (araC) can be obtained by purchasing kiloside (Nippon Shinyaku) or kiloside N (Nippon Shinyaku).
- Enocitabine (BH-AC) can be obtained by purchasing sanrabin (Asahi Kasei).
- Citarabine ocfosfate can be obtained by purchasing Staraside (Nippon Glaze).
- 5-azacytidine can be produced by a known method, or can be obtained by purchasing from Nakarai Testa Co., Ltd.
- CNDAC refers to l- (2-C-cyano-2-deoxy-6-D-arabino-pentofuranosyl) -N-palinitoylcytosine, which is a compound represented by the formula (XIX).
- CNDAC can be produced by a known method (EP536936).
- methotrexate is N- ⁇ 4- [N- (2,4-diaminopteridin-6-ylmethyl) -N-methylamino] benzoyl ⁇ -L-glutmic acid, which is represented by the formula (XXI): It is a compound.
- Methotrexate can be obtained by purchasing Mesotrexate (registered trademark) or Rheumatrex (registered trademark) from Weiss Ichitakeda.
- the microtubule inhibitor means a substance having an action of inhibiting the function of microtubules such as a spindle formation function or a substance transport function in cell division.
- a microtubule inhibitor acts on a microtubule such as a cell or nerve cell in which cell division is active, and exhibits an antitumor effect.
- examples of the microtubule inhibitor include Paclitaxel, Docetaxel, etc., preferably Paclitaxel.
- the microtubule inhibitor can be produced by a known method or can be purchased.
- Paclitaxel is (-)-(lS, 2R, 3S, 4S, 5R, 7S, 8S, Jian,
- Paclitaxel can be obtained by purchasing Taxol from Bristol Pharmaceutical Co., Ltd.
- Docetaxel can be obtained by purchasing Taxotere (Sanofi-Aventis).
- the antibiotic is preferably an antitumor antibiotic.
- Antitumor antibiotics have an action such as inhibition of DNA synthesis or DNA strand breakage in tumor cells, and exhibit antitumor action.
- the antibiotic Doxorubicin (A Doria clarithromycin), DaunoruDicin, j irarubicm, Epirubicm , Idarubicm, Aclarubicin, mentioned force s like Amrubicin Mitoxantrone, preferably Doxorubicin.
- Antibiotics can be produced by known methods and can be purchased.
- Doxorubicin is 10-[(3-amino-2,3,6-trideoxy- -L-lyxo-hexoDyranosyl) oxy] -7,8,9, 10-tetrahydro-6, 8, 1 l -trihydroxy-8- (hydroxyacetyl) -l-methoxy (8S-cis) -5, 12-naphthacenedione is a compound represented by the formula (XXIII).
- Doxorubicin can be obtained by purchasing Adriacin (registered trademark) (Kyowa Hakko).
- Daunorubicin can be obtained by purchasing Daunomycin (registered trademark) (Meiji Seika).
- Piparubicin can be obtained by purchasing Teralubicin (registered trademark) (Meiji Seika) or Pinorbin (registered trademark) (Mershan I Nippon Kayaku).
- Epirubicin can be obtained by purchasing Falmorubicin (registered trademark) (Fuiza-Kyowa Hakko).
- Idarubicin can be obtained by purchasing Idamycin (registered trademark).
- Aclarubicin can be obtained by purchasing Acracinon (registered trademark) (Mershan Ichiyamanouchi).
- Amurubicin can be obtained by purchasing Calcedo (registered trademark) (Sumitomo Pharma).
- Mitoxantrone can be obtained by purchasing Novantron (registered trademark) (Weiss Ichitakeda).
- Platinum complex substances, DNA-topoisomerase I inhibitors, antimetabolites, microtubule inhibitors and antibiotics may form pharmacologically acceptable salts with acids or bases.
- the compounds listed in (1) to (5) above may form pharmacologically acceptable salts different from the salts exemplified in (1) to (5).
- the aforementioned substances also include these pharmacologically acceptable salts.
- the salt with acid include inorganic acid salts such as hydrochloride, hydrobromide, sulfate, and phosphate, formic acid, acetic acid, lactic acid, succinic acid, fumaric acid, maleic acid, citrate, tartaric acid, and benzoic acid.
- salts with organic acids such as acids, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, trifluoroacetic acid.
- alkali metal salts such as sodium salt and potassium salt
- alkaline earth metal salts such as calcium salt and magnesium salt
- salts with organic bases such as lysine (organic amine salts), and ammonium salts.
- platinum complex substances DNA-topoisomei'ase I inhibitors, antimetabolites, microtubule inhibitors and antibiotics may be anhydrous or form solvates such as hydrates. Good.
- the solvate may be either a hydrate or a non-hydrate.
- water alcohol (for example, methanol, ethanol, n-propanol), dimethylformamide or the like can be used.
- solvates and / or optical isomers of these substances are present, the substances in the present invention include those solvates and / or optical isomers.
- the substance in the present invention includes platinum complex substances that undergo metabolism such as oxidation, reduction, hydrolysis, and conjugation in vivo, DNA-topoisomerase I inhibitor substances, metabolic antagonist substances, microtubule inhibitor substances, and antibiotic substances. Also includes at least one selected. Still further, the substance according to the present invention is obtained from a platinum complex substance, a DNA-topoisomerase I inhibitor, an antimetabolite, a microtubule inhibitor, and an antibiotic after undergoing metabolism such as oxidation, reduction, and hydrolysis in vivo. Also includes substances that produce at least one selected.
- the present invention provides a sulfonamide compound, (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V)
- the present invention relates to a pharmaceutical composition, a kit, a method for treating cancer, and a method for inhibiting angiogenesis, characterized in that it is combined with at least one substance selected from the group consisting of antibiotics.
- the sulfonamide compound is as described in “1. Sulfonamide Compound”.
- Sulfonamide Compound For example, (A) E7820 (formula (V)), (B) a compound represented by the general formula (I) , Preferably LY186641 or LY295501, (C) selected from compounds represented by general formula (II), preferably LY-ASAP, (D) LY573636 (formula (III)) and (E) CQS (formula (IV)) At least one compound selected from the group consisting of LY295501 and LY573636, particularly preferably a sodium salt of LY573636.
- the sulfonamide compound is preferably E7820.
- platinum complex substance, a DNA-topoisomerase I inhibitor, an antimetabolite, a microtubule inhibitor, and an antibiotic are described in “2.
- Platinum complex substance, DNA-topoisomerase I inhibitor, antimetabolite, microtubule inhibitor” platinum complex substances are preferably Oxaliplatin, (ii) DNA-topoisomerase I inhibitors are preferably CPT-11 and (iii) antimetabolites are preferred. Is Gemcitabine.
- the platinum complex is preferably Cisplatin,
- the antimetabolite is preferably Methotrexate (iv) the microtubule inhibitor is preferably Paclitaxel, and (v) the antibiotic is preferred.
- Doxorubicin is preferably Doxorubicin.
- the sulfonamide compound and the substances (i) to (V) include pharmacologically acceptable salts or solvates such as hydrates thereof.
- the pharmaceutical composition of the present invention comprises a sulfonamide compound, (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) A pharmaceutical composition comprising a combination of at least one substance selected from antibiotics.
- the pharmaceutical composition of the present invention is useful as a pharmaceutical composition for cancer treatment Oppi Z or a pharmaceutical composition for inhibiting angiogenesis.
- “combined” means a combination for use in combination of compounds, and includes both forms in which different substances are used together at the time of administration, and forms as a mixture.
- the pharmaceutical composition of the present invention comprises (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) an antibiotic.
- a pharmaceutical composition comprising a sulfonamide compound for coadministration to a patient together with at least one substance selected from. A sulfonamide compound, and (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) an antibiotic.
- “Simultaneous” means that they are administered at the same timing in one dosing schedule, and the time of administration does not have to be completely the same. “Separately” means that they are administered at different times in one dosing schedule.
- the kit of the present invention comprises a preparation comprising a sulfonamide compound, (i) a platinum complex substance, '(ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor.
- a kit comprising an agent comprising at least one substance selected from the group consisting of antibiotics.
- the preparation contained in the kit of the present invention is not particularly limited as long as it contains a sulfonamide compound or at least one of the above-mentioned (i) to (V).
- the kit of the present invention is useful as a cancer treatment kit and / or angiogenesis inhibition kit.
- kits of the present invention a preparation comprising a sulfonamide compound
- the preparation containing at least one substance of (i) to (V) may be mixed, or may be stored separately and packaged together, or administered simultaneously. Either one may be administered first.
- the pharmaceutical composition and / or kit of the present invention and the cancer treatment method or angiogenesis inhibition method may be further combined with one or more other anticancer agents.
- Other anticancer agents are not particularly limited as long as they are preparations having anticancer activity.
- Other anticancer drugs Examples include 5-Fluorouracil (5-FU), Holinate Calcium (oralcocobalin), Docetaxel (Taxotere (registered trademark)), Gefitinib
- the other anticancer agents include 5-fluorouracil, folinate calcium, gefitinib, erlotinib, cetuximab, and bevacizumab are particularly preferred when the cancer type targeted for the cancer treatment is colorectal cancer, and gefitinib, erlotinib, Cetuximap and bevacizumab are particularly preferred.
- the cancer type targeted for the cancer therapeutic agent is colorectal cancer
- the combinations shown in Table 1 Yes for knee cancer, for example, the combinations shown in Table 2.
- Table 1 shows preferred combinations in the present invention when the cancer type targeted for the cancer therapeutic agent is colorectal cancer. In the table, LV indicates folinate calcium. Table 2
- Table 2 shows preferable combinations in the present invention when the cancer type targeted for the cancer therapeutic agent is knee cancer.
- the pharmaceutical composition and Z or kit of the present invention can be used as a cancer therapeutic agent or an angiogenesis inhibitor.
- the treatment is to reduce the symptoms of the disease, suppress the progression of the symptoms of the disease, remove the symptoms of the disease, improve the prognosis of the disease, prevent the recurrence of the disease. Is also included.
- the cancer therapeutic agent means an agent containing an antitumor agent, a cancer prognosis improving agent, a cancer recurrence preventing agent, a cancer metastasis inhibitor, and the like.
- the effect of cancer treatment can be confirmed by findings such as radiographs and CT, histopathological diagnosis of biopsy, or by the value of tumor marker.
- the pharmaceutical composition composition or kit of the present invention can be administered to a mammal (eg, human, rat, rabbit, hidge, puta, ushi, cat, inu, monkey, etc.). it can.
- a mammal eg, human, rat, rabbit, hidge, puta, ushi, cat, inu, monkey, etc.
- cancer there are no particular limitations on the types of cancer that can be used as therapeutic agents for cancer, such as brain tumors, cervical cancer, esophageal cancer, tongue cancer, lung cancer, breast cancer, knee cancer, gastric cancer, small intestine and duodenal cancer, colon cancer (colon cancer) , Direct cancer), bladder cancer, kidney cancer, liver cancer, prostate cancer, uterine cancer, ovarian cancer, thyroid cancer, gallbladder cancer, pharyngeal cancer, sarcoma (eg, osteosarcoma, chondrosarcoma, force positive sarcoma, muscle tumor, blood vessel Sarcoma, fibrosarcoma, etc.), leukemia (eg, chronic myelogenous leukemia (CML)), acute myeloid leukemia Disease (AML), chronic lymphocytic leukemia (CLL) and acute lymphocytic leukemia (ALL), lymphoma, multiple myeloma (MM), etc.
- CML chronic myelogenous leuk
- the cancer type that is the target of the cancer therapeutic agent is preferably at least one selected from the group consisting of colon cancer and vaginal cancer.
- the pharmaceutical composition and / or kit of the present invention can be administered orally or parenterally.
- the dosage of the sulfonamide compound is as follows: symptom level, patient age, sex, body weight, sensitivity difference, administration method, administration timing, administration interval, pharmaceutical It depends on the nature of the preparation, formulation, type, type of active ingredient, etc., and is not particularly limited. Usually adult (body weight 60 kg) 10-6000 mg per day, preferably 50-4000 mg, more preferably 50-2000 This can be administered in 1 to 3 divided doses per day.
- platinum complex substance preferably Oxaliplatin or Cisplatin
- DNA-topoisomerase I inhibitor preferably CPT-11
- antimetabolite The dosage of at least one substance selected from the group consisting of substances, preferably Gemcitabine or Methotrexate,
- microtubule inhibitors preferably Paclitaxel
- antibiotics preferably Doxorubicin
- it is usually 10 to 6000 mg per day for an adult, preferably 50 to 4000 mg, more preferably 50 to 2000 mg, which can be administered usually in 1 to 3 divided doses per day.
- the amount of the sulfonamide compound to be used is not particularly limited.
- Platinum complex substance preferably Oxaliplatin or Cisnlatin
- DNA-topoisomerase I inhibitor preferably CPT-11
- Antimetabolite Preferably depending on individual combinations with at least one substance selected from the group consisting of Gemcitabine or Methotrexate,
- a microtubule inhibitor preferably Paclitaxel
- an antibiotic preferably Doxorubicin
- the pharmaceutical composition of the present invention can be made into various dosage forms, for example, oral solid preparations or parenteral preparations such as injections, suppositories, ointments, cataplasms, and the like.
- the sulfonamide compound contained in the kit of the present invention (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and ( V) At least one substance selected from the group consisting of antibiotics is a variety of dosage forms, for example solid oral preparations, or parenterals such as injections, suppositories, ointments, cataplasms, etc. It can be made into preparations such as preparations.
- a solid preparation for oral administration When preparing a solid preparation for oral administration, add excipients, and if necessary, binders, disintegrants, lubricants, coloring agents, flavoring agents, etc. to the active ingredient. Covered tablets, granules, fine granules, powders, capsules, etc. In addition, oral non-solid preparations such as syrups can be appropriately prepared.
- Excipients include, for example, lactose, corn starch, sucrose, glucose, sonorev, crystalline cellulose, silicon dioxide, etc.
- binders include, for example, polyvinylenoleolecol, ethinoresenorelose, methinoresenorelose, gum arabic Hydroxypropylcellulose, hydroxypropylmethylcellulose, etc.
- lubricants for example, magnesium stearate, tanolec, silica, etc. are permitted to be added to pharmaceuticals as coloring agents.
- the flavoring agent for example, cocoa powder, hearth brain, aromatic acid, horsepower oil, dragon brain, cinnamon powder and the like are used.
- these tablets and granules may be appropriately coated with sugar coating, gelatin coating, etc. if necessary.
- suspending agent examples include methyl cellulose, polysorbate 80, hydroxetyl cellulose, gum arabic, tragacanth powder, canoleoxy methino resell mouth natnatrium, polyoxyethylene sorbitan monolaurate, and the like.
- solubilizers include polyoxyethylene hydrogenated castor oil, polysorbate 80, nicotinic acid amide, polyoxyethylene sonolebitan monolaurate, tuna gol, and castor oil fatty acid ether ester. .
- stabilizers include sodium sulfite and sodium metasulfite
- preservatives include methyl paraoxybenzoate, ethyl parabenzoate, sonolevic acid, phenol, crezonole, and chlorotalezonore. Can do.
- the pharmaceutical composition and / or kit of the present invention comprises the above sulfonamide compound, and (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) a metabolic antagonist, (iv) a microtubule inhibitor
- a platinum complex substance e.g., a platinum complex substance, ii) a DNA-topoisomerase I inhibitor, (iii) a metabolic antagonist, (iv) a microtubule inhibitor
- it may contain packaging containers, instruction manuals, package inserts, and the like.
- Packaging containers, instruction manuals, package inserts, etc. can describe combinations to be used in combination with compounds.
- the dose can be described. The usage and dosage can be described with reference to the above.
- the kit of the present invention comprises (a) a sulfonamide compound, (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) at least one selected from the group consisting of packaging containers, instruction manuals and package inserts, describing the use in combination with at least one substance selected from the group consisting of antibiotics; (b) It may be an embodiment containing a pharmaceutical composition containing a sulfonamide compound.
- the kit is useful as a cancer treatment kit and z or angiogenesis inhibition kit.
- the pharmaceutical composition containing the sulfonamide compound is useful as a pharmaceutical composition for treating cancer and / or a pharmaceutical composition for inhibiting angiogenesis.
- Packaging containers, instruction manuals, package inserts, etc. can indicate that a sulfonamide compound is used in combination with at least one substance selected from the above (i) to (V).
- Usage, dosage, etc. can be described in the form of a combination of these substances at the time of administration or in the form of a mixture. The usage and dose can be determined by referring to the description of the pharmaceutical composition / kit.
- the present invention includes a compound selected from the group consisting of (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) an antibiotic. Also included is the use of a sulfonamide compound for the manufacture of a pharmaceutical composition in combination with at least one of the substances described above. In the use of the present invention, the pharmaceutical composition is useful as a pharmaceutical composition for treating cancer and a pharmaceutical composition for inhibiting angiogenesis.
- the present invention also includes a sulfonamide compound, (i) a platinum complex substance, (ii) a DNA-topoisomerase I inhibitor, (iii) an antimetabolite, (iv) a microtubule inhibitor, and (V) It includes a method for treating cancer and / or a method for inhibiting angiogenesis, wherein at least one substance selected from the group consisting of antibiotics is administered to a patient simultaneously or separately.
- the administration route and administration method of the sulfonamide compound and at least one substance selected from the above (i) to (V) are not particularly limited. Reference can be made to the above description of the pharmaceutical composition of the present invention.
- Example 1 Combined effect of E7820 and anticancer drugs Paclitaxel, SN38 (CPT-11 active substance), Methotrexate, Cisplatin, Gemcitabine, Doxorubicin on cell proliferation in vascular endothelial cell proliferation assembly (in vitro)
- Human umbilical vein endothelial cells are suspended in Brett Kit EGM-2 (Cambrex) as a culture medium, adjusted to 1 X 10 4 cells / ml, and 100 ⁇ l of this solution is added to each well of a 96-well plate. The cells were cultured at 37 ° C in a 5% carbon dioxide incubator. On the next day, a solution containing E7820, a solution containing a combination anticancer agent, and a solution containing both compounds of E7820 and a combination anticancer agent were each diluted in a culture medium. Then, the diluted solution was added to each well in the culture at 100 ⁇ 1 / well and the culture was continued.
- E7820 is E7820, Paclitaxel, SN38 (CPT'll active form), Methotrexate Cisplatin, Gemcitabine or Doxorubicin alone. Compared with this, it showed a stronger cell growth inhibitory effect.
- E7820 is a combination index (CI) force S 1 or less when Doxorubicin is used in combination with Paclitaxel, SN38 (CPT-11 active substance), Methotrexate, Cisplatin, and Gemcitabine.
- CI combination index
- Table 3 shows the synergistic effect of E7820 and anticancer agents on cell growth inhibition in vascular endothelial cell proliferation assay (in vitro).
- Example 2 Combined E7820 with anticancer drugs Paclitaxel, SN38 (CPT-11 activator), Cisplatin, Gemcitabine, Doxorubicin for tube formation in vascular endothelial cell tube formation (in vitro)
- Human umbilical cord A venous endothelial cell line (HUVEC) was suspended in a serum-free medium (Human endothelial-SFM Basal Growth Medium, manufactured by GIBCO BRL) to prepare a cell suspension of 5 ⁇ 10 ⁇ cells / ml. This suspension 2001 was seeded in each well containing type I collagen gel and serum-free medium and cultured overnight.
- a serum-free medium Human endothelial-SFM Basal Growth Medium, manufactured by GIBCO BRL
- type I collagen gel 400 ⁇ 1 was layered and gelled at 37 ° C in a CO 2 incubator for 3 hours, and then serum-free medium containing E7820, combined anticancer agent and both E7820 and combined anticancer agent (10 ng 1.5 ml of / ml EGF and 20 ng / ml VEGF (Genzyme Techne Corp.) were added, and the cells were further cultured in a CO 2 incubator at 37 ° C for 3 days.
- Table 4 shows the synergistic effect of E7820 and anticancer drugs on the inhibition of vascular endothelial cell tube formation.
- superior blood vessels can be obtained by combining E7820 with at least one substance selected from the group consisting of platinum complex substances, DNA-topoisomerase I inhibitors, antimetabolites, microtubule inhibitors and antibiotics.
- a pharmaceutical composition and kit showing angiogenesis inhibitory activity, and a method for inhibiting angiogenesis and the pharmaceutical composition, kit and method of the present invention can be used for cancer treatment and angiogenesis inhibition. It was.
- Example 3 Combination of E7820 and Oxaliplatin in human colon cancer cell line (Colo320DM) subcutaneous transplant model (in vivo)
- Human colon cancer cell line Colo320DM (purchased from Dainippon Pharmaceutical Co., Ltd.) is cultured at 37 ° C in a 5% carbon dioxide incubator with RPMI1640 (including 10% FBS) until it becomes about 80% confluent. To collect the cells. With 50% matrigel containing phosphate buffer, prepared 7.5xl0 7 cells / mL suspension, resulting cells suspension was transplanted into nude mice side subcutaneously by 0.1 mL.
- Tumor volume TV Tumor major axis (mm) X Tumor minor axis 2 (mm 2 ) / 2
- Specific tumor volume RTV tumor volume on the measurement day / tumor volume on the administration start day
- Table 5 shows the antitumor effects of E7820, Oxaliplatin, and combinations of E7820 and Oxaliplatin in the Colo320DM nude mouse subcutaneous transplant model. The day of administration was Dayl.
- Human colon cancer cell line Colo320DM (purchased from Dainippon Pharmaceutical Co., Ltd.) is cultured at 37 ° C in a 5% carbon dioxide incubator with RPMI1640 (including 10% FBS) until it becomes about 80% confluent. To collect the cells. With 50% matrigel containing phosphate buffer, prepared 7.5xl0 7 cells / mL suspension, resulting cells suspension was Utsurikusunoki into nude mice side subcutaneously by 0.1 mL.
- E7820 50 mg / kg, twice a day for 2 weeks, oral administration
- CPT-11 topotecin (registered trademark)
- Daiichi Pharmaceutical 100 mg / kg, 4 days
- Tumor volume TV tumor major axis (mm) X tumor minor axis 2 ( mm 2) 12
- Specific tumor volume RTV Tumor volume on the measurement day Z Tumor volume on the administration start day
- Table 6 shows the antitumor effects of E7820, CPT-11, and combinations of E7820 and CPT-11 in the Colo320DM nude mouse subcutaneous transplant model. Day 1 of administration was Dayl.
- t Knee cancer cell line KP-1 (obtained from Kyushu Cancer Center) is cultured at 37 ° C in a 5% carbon dioxide incubator with RPMI1640 (including 10% FBS) until approximately 80% confluent.
- Cells were collected by EDTA.
- An lxlO 8 cells / mL suspension was prepared with a phosphate buffer, and the resulting cell suspension was transplanted subcutaneously into the nude mouse side in 0.1 mL increments.
- Tumor volume TV Tumor major axis (mm) X Tumor minor axis 2 (mm 2 ) 12
- Specific tumor volume RTV Tumor volume on the measurement day Z Tumor volume on the administration start day
- Table 7 shows the antitumor effects of E7820, Gemcitabine and the combination of E7820 and Gemcitabine in the KP-1 nude mouse subcutaneous transplantation model.
- the administration start date was Day 1.
- the cells in ISOGEN (Wako Pure Chemical) 3 ml of medium was removed by dissolving pressurized forte, after stirred with an equal volume of CHC1 3, and extract the aqueous phase.
- half the amount of isopropanol was added, and the mixture was allowed to stand for 5 minutes, and then the precipitate was collected by centrifugation.
- the precipitate was washed with 70% ethanol, sterilized water was added and dissolved, and the amount of RNA was quantified by measuring the absorbance at 260 nm with a spectrophotometer.
- RT-PCR reaction was performed using TaqMan Gold RT PCR kit (Applied Biosystems). That is, RNA 0.1 / g was added to the reaction solution 50 / l and reacted at 25 ° C for 10 minutes, 48 ° C for 30 minutes, and 95 ° C for 5 minutes to prepare cDNA.
- PCR reaction was performed with a primer for measuring DNA-topoisomerase II mRNA (ABI Taqman probe Hs00172214 ml), and the amount of RNA was quantified with ABI7700 (manufactured by Applied Biosystems).
- the amount of DNA-topoisomerase II mRNA was 4.4 / z g / ml in the untreated group, whereas it was 1.6 ⁇ g / ml in the E7820 treated group.
- human colon cancer cell line HCT116 American Type Culture Collection, Manassas, VA, USA
- human leukemia cell line MOLT-4 American Type Culture Collection, Manassas, VA, USA
- HCT116 cells and MOLT-4 cells were seeded at a rate of 2.0 ⁇ 10 6 cells in a 10 cm diameter cell culture dish, and the following compound treatment was performed after 24 hours of culture.
- E7820 (0 8 ⁇ ⁇ ), ⁇ 7070 (0.8 ⁇ ), LY295501 (30 ⁇ ), CQS (8 ⁇ ) ⁇ adriamycin (0.2 ⁇ ), daunomycin (0.2 ⁇ ), ICRF154 Twelve compounds (80 ⁇ ), ICRF159 (80 ⁇ ), kenpaullone (10 ⁇ ), alsterpuUone (10 ⁇ ), trichostatin A (0.1 ⁇ ) rapamycin (80 ⁇ ) were evaluated. On the other hand, for MOLT-4 cells, ⁇ 7070 (0.8 ⁇ ) was evaluated. It was.
- adriamycin and daunomycin are DNA-topoisonie se II inhibitors that intercalate with DNA
- ICRF154 and ICRF159 are catalvtic tvoe DNA-toOOisomerase II inhibitors i, Kenpaullone and aisterpullone ⁇ 3 ⁇ Cyclm-dependent kinases (CDKs) fe lingualism (J, tncnostatm A is an istone deacetylase inhibitor
- rapamycin is a known compound as an mTOR / FRAP inhibitor.
- the compound treatment concentration is HCT116 for each compound.
- the concentration is set to 3 to 5 times the reference, and is shown in parentheses following the above compound name. Cells were collected after 24 hours of treatment at the set concentration, and cells cultured for 24 hours without addition of compounds were also collected.
- RNA was dissolved in 100 / I diethylpyrocarbonate (DEPC) -sterilized water and further purified using an RNeasy column (QIAGEN). Superscript Choice System (Invitrogen) and T7_d (T) 2 2. Using 4 primers, synthesized double-stranded cDNA.
- cDNA was purified with phenol / cloform form, and labeled with biotin UTP and CTP according to the attached operation method using RNA Transcript Labeling Kit (Enzo Diagnostics). HNeasy force of reaction product After purification with ram, cRNA was fragmented by heating at 94 ° C for 35 minutes in 200 mM trisacetic acid pH 8.1, 150 mM magnesium acetate, 50 mM acetate.
- Fragmented cRNA was added in 100 mM MES, 1 M sodium salt, 20 mM EDTA, 0.01% Tween 20, 45. Hybridize to GeneChip (Affymetrix) Human Focus array for 16 hours in C. After hybridization, GeneChi was washed and stained according to the protocol Midi_euk2 attached to the Affymetrix fluidics station. For the staining, streptavidin-phycoerythrin and biotinylated anti-streptavidin antibody were used. The stained GeneChip was scanned using an HP Argon laser confocal microscope (Hewlett Packard), and the fluorescence intensity was measured. Measurements were carried out with excitation at a wavelength of 488 nm and fr with emission at a wavelength of 570 nm.
- HCT116 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 units / ml penicillin, 100 g / ml streptomycin. Hereinafter, culture and compound treatment, 5% C0 2, 37 ° line in an incubator adjusted to C ivy. HCT116 cells were seeded in a 10 cm diameter cell culture dish at a ratio of 2.0 ⁇ 10 6 cells Z dish, and the following compound treatment was performed after 24 hours of culture.
- MST16 induces the formation of a catalytic type of DNA-topoisomerase II inhibitor
- etoposide induces the formation of cleavable comple.
- DNA-topoisomerase II inhibitory U ethoxzolamide is carbonic anhydrase] 3 ⁇ 4 ⁇ authoritative agent
- capsaicin is tumor-specific plasma membrane NADH oxidase inhibition [J, trichostatin A is histone deacetylase inhibition U, kenpaullone is a known compound as cyclin-dependent kinases (CDKs) P and Ij.
- CDKs cyclin-dependent kinases
- the compound treatment concentration was determined as a concentration twice that of the 50% growth inhibitory concentration of each compound on HCT116 cells (based on the 3-day cytostatic activity using MTT). Cells were collected after 24 hours of treatment at the set concentration indicated in parentheses following the compound name. In addition, cells cultured for 24 hours without compound addition were also collected.
- Gene expression analysis using a DNA microarray was performed in the same manner as “(2) Gene expression analysis using DNA microarray” in Example 7.
- this example was duplicated for each sample (for convenience of experiments, branch numbers were assigned in the manner of control-l, conti'ol_2, E7070_l, E7070-2 so that each sample could be distinguished) .
- the RMA method (robust multi-array average method (Biostatistics (2003), 4, 249-264)) was applied to 26 cels (13 samples X2 of control + 12 compounds) obtained in this example, After normal distribution at the probe level, the log value of the signal intensity at the gene level was calculated. Subsequently, the log value of the signal intensity in the compound-untreated group (control-1) is subtracted from the log value of the signal intensity in the compound-treated group of each gene, and the log value of the signal ratio of the compound-treated group to control-1 is obtained. Obtained. The cosine correlation coefficient was calculated and used as the correlation coefficient between experiments (Fig. 5).
- LYlj is LY186641,“ LY2 ”is LY295501,“ LY5 ”is LY573636,“ CAI ”is ethoxzolamide,“ Cap ”is capsaicin,“ MST ”is MST16,“ Etop ” Indicates etoposide, “TSA” indicates trichosta.tin A, and “Kenp” indicates kenpaulkme.
- TSA indicates trichosta.tin A
- Kenp indicates kenpaulkme.
- De hclust (*,“ average ”) is a command used for statistical angle analysis, and indicates that clustering analysis by R was performed using the average value of dupulicate experimental data.
- the cancer cell lines used were DLD-1, HCT15, HCT116, HT29, SW480, SW620, WiDr (above human colon cancer cell line), A427, A549, LX-1, NCI-H460, NCI-H522, PC -9, PC-10 (above, human lung cancer cell line), GT3TKB, HGC27, MKN1, MKN7, MKN28, MKN74 (above, human stomach cancer cell line), AsPC-1, KP-1, KP-4, MiaPaCall , PANC-1, SUIT-2 (above, human spleen cancer cell line), BSY-1, HBC5, MCF-7, MDA-MB-231, MDA-MB-435, MDA-MB-468 (above, Human breast cancer cell line), CCRF-CEM, HL
- DLD-1 (1250 / weIl, 16.8 h) GT3TKB (2000 / well, 21.1 h) BSY-l (2000 / well, 46.1 h)
- HCT15 (1500 / well, 14.5 h)
- HGC27 (1500 / well, 14.6 h)
- HBC5 2000 / well, 31.8 h)
- Table 8 shows the types of human cancer cell lines, the number of sprinkled cells and the doubling time in the human cancer cell line panel.
- Table 9 shows the correlation coefficients between compounds (E7070, E7820, CQS, LY186641 and LY295501) in a human cancer cell line panel.
- Example 1 0 Cross resistance in E7070 resistant strains
- HCT116-C9 is a sub-strain isolated from human colorectal cancer-derived HCT116 (American Type Culture Collection, Manassas, VA, USA). This HCT116-C9 was cultured in the presence of E7070, and the E7070 concentration was gradually increased.
- the E7070-resistant substrains obtained by raising are HCT116-C9-C1 and HCT116-C9-C4 (Molecular Cancer Therapeutics, 2002, 1, 275-286) o
- HCT116-C9, HCT116-C9-C1 and HCT116-C9-C4 were spread on a 96-well microphone plate (flat bottom) at 3000 cells / well each (SO ⁇ l / well) and tripled after 24 hours. It was added compound of the rare ⁇ series (50 il / W ell). Furthermore, 72 hours later, the cells were analyzed by MTT method (Mossmann T., J. Immunol. Methods, 1983, 65, 55-63). Growth inhibition activity was evaluated. The 50% growth inhibitory inhibitory concentration for each cancer cell was determined by the least square method.
- the cell growth inhibitory activity of E7070 was 0.127 ⁇ M for HCT116-C9 (C9).
- HCT116-C9-C1 and HCT116-C9-C4 are activity against HCT116-C9-C1 and HCT116-C9-C4 .
- IC50 51.2 / ⁇ , 634 / ⁇ , 134 ⁇ , 111 ⁇ ⁇ ⁇ ⁇ ⁇ , 113 ⁇ ⁇ ⁇ ⁇ for HCT116-C9-C4,
- HCT116-C9-C4 IC50 52 ⁇ 8 / ⁇ M, 517 / iM, 138; uM, 110 / M, 90.3 / M, respectively.
- Example 10 the cell growth inhibitory activity of LY573636 was evaluated at the same time as E7070 using an E7070 resistant strain.
- a sulfonamide compound preferably E7070, LY186641, LY295501, LY_ASAP, LY573636 or CQS or a combination thereof, (i) a platinum complex material, preferably Oxaliplatin.
- Cisplatin (ii) DNA-topoisomerase I inhibitor, preferably CPT-11, (iii) antimetabolite, preferably Gemcitabine or Methotrexate, (iv) microtubule inhibitor, preferably Paclitaxel, and (v) antibiotics It has been shown that when used in combination with at least one compound selected from the group consisting of substances, preferably Doxorubicin, it exhibits excellent antitumor activity and angiogenesis inhibitory activity. Industrial applicability
- the present invention provides a pharmaceutical composition and kit exhibiting excellent antitumor activity and / or angiogenesis inhibitory activity, as well as a method for treating cancer and / or a method for inhibiting angiogenesis.
- a sulfonamide compound that is, (A) E7820, (B) a compound represented by the general formula (I), preferably LY186641 or LY295501, (C) a compound represented by the general formula (II) At least one compound selected from LY-ASAP, (D) LY573636, and (E) CQS, and (i) a platinum complex, preferably Oxaliplatin or Cisplatin, (ii) a DNA-topoisomerase I inhibitor , Preferably CPT-11, (iii) antimetabolite, preferably Gemcitabine or Methotrexate, (iv) a microtubule inhibitor, preferably Paclitaxel, and (v) Pharmaceutical compositions and kits that exhibit excellent anti-tumor activity and Z or angiogenesis inhibitory activity by combining with a biomaterial, preferably at least one substance selected from the group consisting of Doxorubicin, and methods for treating cancer and Z Or a method of inhibiting angiogenesis is provided
Abstract
本発明は、スルホンアミド化合物と、白金錯体物質、DNA-topoisomerase I阻害物質、代謝拮抗物質、微小管阻害物質または抗生物質とを組み合わせてなることを特徴とする医薬組成物およびキット、ならびに癌の治療方法および/または血管新生の阻害方法に関する。
Description
明細書 スルホンアミ ド化合物の抗癌剤との新規併用 技術分野
本発明は、 スルホンアミ ド化合物と、 Oxaliplatinまたは Cisplatinなどの白 金錯体物質、 CPT-11などの DNA-topoisomerase I阻害物質、 Gemcitabineま' たは Methotrexateなどの代謝拮抗物質、 Paclitaxelなどの微小管阻害物質、 お よび Doxorubicin などの抗生物質からなる群から選択される少なくとも一つの 物質とを組み合わせてなる新規な医薬組成物およびキット、 ならびに癌の治療方 法およひブまたは血管新生阻害方法に関するものである。 背景技術
癌の化学療法剤として従来用いられているものには、 アルキル化剤のサイクロ フォスフアミ ド、 代謝拮抗剤のメ トトレキセート、 フルォロウラシル、 抗生物質 のアドリアマイシン、 マイ トマイシン、 ブレオマイシン、 植物由来のタキソール、 ビンクリスチン、 エトポシド、 金属錯体のシスプラチンなどがあるが、 いずれも その抗腫瘍効果は十分であるとは言えず、 新しい抗腫瘍剤の開発が切望されてい た。
近年、 有用な抗腫瘍剤として、 スルホンアミ ド化合物が報告されている(1一 4 )。 特に、 N— ( 3—シァノー 4ーメチルー 1 H—インドールー 7—ィル) —3 シ ァノベンゼンスルホンアミ ド (以下、 「E7820」 と称する場合がある) 、 N— [ [ ( 4一クロ口フエニル) ァミノ] 力ルポ二ル] — 2 , 3—ジヒ ドロ一 1 H— インデンー 5—スルホンアミド (以下、 「LY186641」 と称する場合がある) 、 N— [ [ ( 3 , 4—ジクロロフエニル) ァミノ] カルボ-ル] 一 2, 3—ジヒ ド 口べンゾフラン一 5—スルホンアミ ド (以下、 「LY295501」 と称する場合があ る) 、 N— (2 , 4—ジクロ口べンゾィノレ) 一 4—クロ口フエニノレスルホンアミ ド (以下、 「LY'ASAPJ と称する場合がある) 、 N— (2 , 4—ジクロ口ベン
ゾィル) 一 5—ブロモチォフェン一 2—スルホンアミ ド (以下、 「LY573636」 と称する場合がある) 、 2—スルファニルアミ ドー 5—クロ口キノキサリン (以 下、' 「CQS」 と称する場合がある) などは、 種々のタイプの腫瘍に活性を示し 非常に有用である。
しかしながら、 これらの化合物の組み合わせにより、 いかなる効果を示すか否 かについては、 これまで報告されていない。
近年、 種々の DNAマイクロアレイを用い、 多数の遺伝子の発現量を同時に検 出する方法が確立され、 DNAマイクロアレイは、 幅広い目的に応用されている ( 5および 6 ) 。 また、 DNAマイクロアレイ (一部メレブランフィルターを用いたマ クロアレイ) を用いて、 腫瘍細胞に抗癌剤を作用させた際に起こる遺伝子発現変 化を検討した報告もいくつか成されている (7— 9 )。 これらの報告は、 遺伝子発現 の変動解析が、 複数の細胞集団の特性比較や、 薬剤の処理等により細胞に引き起 こされる生物学的な変化を、 分子レベルで包括的に研究するために極めて有用で あることを示している。
また、 米国 National Cancer Instituteの 60種類の癌細胞株パネルについて 遺伝子発現プロファイルを解析することにより、 これら細胞株を再分類し、 その 特性を検討した報告(1 G )、 さらに、 この 60種類の癌細胞株パネルの遺伝子発 現プロファイルと、 各細胞株の各種抗癌剤に対する感受性との間の関連について 考察した報告(m等がなされている。
参考文献
( 1 ) 国際公開第 0 0 Z 5 0 3 9 5号パンフレッ ト
( 2 ) 欧州特許出願公開第 0 2 2 2 4 7 5号明細書
( 3 ) 国際公開第 0 2 Z 0 9 8 8 4 8号パンフレツト
( 4 ) 国際公開第 0 3 Z 0 3 5 6 2 9号パンフレッ ト
( 5 ) Schena M, Shalon D, Davis RW, Brown PO. Science 1995, 270, 467-70.
( 6 ) Lockhart, D.J., Dong, H., Byrne, M.C., Follettie, M.T., Gallo, M.V., Ghee, M.S., Mittinann, Μ·, Wang C, Kobayashi, M.,
Horton, H. Brown, E.L., Nature Biotechnology, 1996, 14, 1675- 1680.
( 7 ) Rhee CH, Ruan S, Chen S, Chenchik A, Levin VA, Yung AW,
Fuller GN, Zhang W, Oncol Rep, 1999, 6, 393-401.
( 8 ) Zimmermann J, Erdmann D, Lalande I, Grossenbacher R,
Noorani M, Furst P, Oncogene, 2000, 19, 2913-20.
( 9 ) Kudoh K, Ramanna M, Ravatn R, Elkahloun AG, Bittner ML,
Meltzer PS, Trent JM, Dalton WS, Chin KV, Cancer Res, 2000,
4161-6.
( 1 0 ) Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman
P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO, Nat Genet, 2000, 24, 227- 35.
( 1 1 ) Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe
L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, Scudiero DA, Eisen MB, Sausville EA, Pommier Y, Botstein D, Brown PO: Weinstein JN, Nat Genet, 2000, 24, 236-44. 発明の開示
本発明は、 このような状況に鑑みてなされたものであり、 その解決しようとす る課題は、' 優れた抗腫瘍活性および/または血管新生阻害活性を有する医薬組成 物およびキット、 ならびに癌の治療方法および/または血管新生阻害方法を見出 すことにある。
本発明者らは、 .上記課題を解決するため、 鋭意検討を重ねた結果、 血管内皮細 胞増殖アツセィ (in vitro) において、 E7820は、 Taxol (Paclitaxel) 、 SN38 (CPT- 11 活性体) 、 Methotrexate, Cisplatin、 Gemcitabine , Doxorubicin と併用することにより、 細胞増殖抑制に対する統計的 (combination index) に
有意な相乗効果を示すことが明らかになった。 また、 血管内皮細胞管腔形成アツ セィ (in vitro) において、 E7820 は、 Paclitaxel、 SN38 (CPT' ll 活性体) 、 Cisplatin, Gemcitabine、 Doxorubicin と併用することにより、 管腔形成抑制 に対する統計的 (combination index) に有意な相乗効果を示すことが明らかに なった。 さらには、 ヒ ト大腸癌細胞株皮下移植モデル (in vivo) において、 E7820 は、 Oxaliplatin または CPT-11 と併用することにより、 抗腫瘍効果に 対する統計的 (two-way ANOVA) に有意な相乗効果を示すことが明らかにな つた。 また、 ヒ ト膝癌細胞株皮下移植モデル (in vivo) において、 E7820 は、 Gemcitabine と併用することによ り、 抗腫瘍効果に対する統計的 (two-way ANOVA) に有意な相乗効果を示すことが明らかになった。 さらに、 E7820 は、 Oxaliplatin, CPT-11および Gemcitabineからなる群から選択される少なくと も一つの化合物と併用するこ とによ り 、 Oxaliplatin、 CPT- 11 および Gemcitabine からなる群から選択される少なくとも一つの化合物単独では示す ごとができないような優れた抗腫瘍効果が認められた。
また、 DNAマイクロアレイおよび癌細胞株パネルの実験において、 E7070 (本明細書において、 「N— ( 3—クロ口一 1 H—インドールー 7—ィル) 一 4 ースルファモイルベンゼンスルホンアミ ド」 を意味する) 、 E7820、 LY186641、 LY295501、 LY573636 もしくは CQSまたはこれらの組み合わせによる遺伝子 変動パターンおよび細胞増殖抑制活性が、 高い相関を示すことを見出した。 また、 細胞増殖抑制活性を測定するアツセィにおいて、 E7070 に耐性を示す癌細胞株 力 E7820、 LY186641、 LY295501、 LY-ASAP、 LY573636もしくは CQSに 交叉耐性を示すことを見出した。 本発明者は、 これらの結果から、 E7070、 E7820、 LY186641、 LY295501, LY'ASAP、 LY573636もしくは CQSまたは これらの組み合わせは、 同一または類似の作用機序を有し、 同一または類似の遺 伝子変化および効果をもたらすという知見を得た。
よって、 E7820、 LY186641 , LY295501, LY-ASAP, LY573636 もしくは CQS またはこれらの糸且み合わせは、 Oxaliplatin、 Cisplatin、 CPT- 11、 Gemcitabine, Methotrexate Paclitaxelおよぴ Doxorubicin力 ら る群力 ら
選択される少なくとも一つの化合物と併用することにより、 すぐれた抗腫瘍活性 および/または血管新生阻害活性を示すと考えられ、 スルホンアミ ド化合物、 好 ましくは E7820、 LY186641 , LY295501 , LY-ASAP , LY573636 もしく(ま CQS またはこれらの組み合わせと、 Oxaliplatin、 Cisplatin、 CPT_ 11、 Gemcitabine、 Methotrexate, Pac丄 itaxelおよひ Doxorubicin T らなる群力 ら 選択される少なくと.も一つの物質との組み合わせは、 有用な医薬組成物およびキ ットとして使用し得ること、 ならびに癌の治療および Zまたは血管新生の阻害に 使用し得ることを見出した。
すなわち本発明は、 以下に関する。
( 1 ) スルホンアミ ド化合物と、 白金錯体物質、 DNA-topoisomerase I 阻害物 質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択さ れる少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 また はそれらの溶媒和物とを組み合わせてなる医薬組成物。
( 2 ) (a) スルホンアミ ド化合物と、 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から 選択される少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物とを併用することを記載した、 包装容器、 取扱説 明書および添付文書からなる群から選択される少なくとも一つと、
(b) スルホンアミ ド化合物を含む医薬組成物と、
を含有するキット。
( 3 ) スルホンァ ミ ド化合物を含んでなる製剤と、 白金錯体物質、 DNA- topoisomerase I阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質 からなる群から選択される少なくとも一つの物質、 もしくはその薬理学的 に許容される塩、 またはそれらの溶媒和物を含んでなる製剤とをセットに したことを特徴とするキット。
( 4 ) 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質おょぴ抗生物質からなる群から選択される少なくとも一つの物質、
もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物と組み合 わせてなる医薬組成物の製造のためのスルホンアミ ド化合物の使用。
(5) スルホンアミ ド化合物と、 白金錯体物質、 DNA-topoisomerase I 阻害物 質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択さ れる少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 また はそれらの溶媒和物とを患者に投与することを特徴とする癌の治療方法お よび Zまたは血管新生の阻害方法。
(6) 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質おょぴ抗生物質からなる群から選択される少なくとも一つの物質 とともに患者に併用投与するための、 スルホンアミ ド化合物を含む医薬組 成物。
上記 (1) 〜 (6) において、 前記スルホンアミ ド化合物は、 N— (3—シァ ノー 4—メチノレー 1 H—ィンドール一 7一ィル) ― 3一シァノベンゼンスノレホン アミ ドもしくはその薬理学的に許容される塩、 またはそれらの溶媒和物を挙げる ことができる。
また、 上記 (1) 〜 (6) において、 前記スルホンアミ ド化合物は、 一般式 (I)

[式中、 Eは、 一O—、 一 N (CH3) 一、 一 CH2—、 _CH2CH2—または一 CH2〇一を、 Dは、 一 CH2—または一 O—を、 Riaは、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 (II)
[式中、 Jは、 一 O—または一 NH_を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有していてもよい 一 C6アルキル基、 置換基を有していてもよい C i_C4アルコキシ基、 置換基を有していてもよい Ci—C4アルキルチオ基、 一 CF3、 一〇CF3、 — S CF3、 置換基を有していてもよい C1—C4アルコキ シカルボニル基、 ニトロ基、 アジド基、 一 O (S 02) CH3、 — N (CH3) 2、 水酸基、 フエ二ル基、 置換基を有するフエ二ル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ノヽ ロゲン原子、 シァノ基、 一 CF3、 置換基を有していてもよい d— C6アルキ ル基、 置換基を有していてもよい Ci一 C4アルコキシカルボニル基、 置換基を 有していてもよい Ci— C4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci—C4アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい C —Ceアルキル基 (伹し、 R3bおよび R4bの少な く とも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい Ci_C6アルキル基、 一 C F3またはニトロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい Ci一 C6アルキ ル基 (但し、 R6bが置換基を有していてもよい Ci— C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい Ci—C6アルキル基または _C F3 (但し、 R5bま たは R?bのいずれか一方が、 置換基を有していてもよい Ci— C6アルキル基で ある力 \ あるいは R?b力 ハロゲン原子または置換基を有していてもよい Ci _C6アルキル基である場合には、 R または RGbのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物および
式 (IV)
(IV)

より具体的には、 スルホンアミ ド化合物、 すなわち、 (A) E7820, (B) 一 般式 (I) で表される化合物、 好ましくは LY186641または LY295501、 (C) 一般式 (II) で表される化合物、 好ましくは LY-ASAP、 (D) LY573636およ ぴ (E) CQS から選択される少なくとも一つの化合物と、 (i) 白金錯体物質、 好ましくは Oxaliplatinまたは Cisplatin、 (ii) DNA-topoisomerase I阻害物質、 好ましくは CPT- 11、 (iii) 代謝拮抗物質、 好ましくは Gemcitabine または Methotrexate, (iv) 微小管阻害物質、 好ましくは Paclitaxel, および (v) 抗 生物質、 好ましくは Doxorubicinから選択される少なくとも一つの物質とを組 み合わせることにより、 すぐれた抗腫瘍活性および/または血管新生阻害活性を
示す医薬組成物およびキット、 ならびに癌の治療方法および Zまたは血管新生阻 害方法が提供され、 癌の治療または血管新生の阻害に用いることが可能となった, 図面の簡単な説明
図 1は、 ヒ ト大腸癌細胞株 (Colo320DM) 皮下移植モデル (in vivo) にお ける腫瘍増殖抑制に対する E7820 と oxaliplatin との併用効果を示す。 図 1中、 *は、 危険率 0.05未満で統計的に有意な相乗効果があったことを示す。 図 1中、 日数 #は、 投与開始 Θを daylとした日数を示す。
図 2は、 ヒ ト大腸癌細胞株 (Colo320DM) 皮下移植モデル (in vivo) にお ける腫瘍増殖抑制に対する E7820 と CPT-11 との併用効果を示す。 図 2中、 * は、 危険率 0.01 未満で統計的に有意な相乗効果があったことを示す。 図 2中、 日数 #は、 投与開始日を daylとした日数を示す。
図 3は、 ヒ ト膝癌細胞株 (KP-1) 皮下移植モデル (in vivo) における腫瘍 増殖抑制に対する E7820 と Gemcitabine との併用効果を示す。 図 3中、 *は、 危険率 0.01未満で統計的に有意な相乗効果があったことを示す。 図 3中、 日数 #は、 投与開始日を daylとした日数を示す。
図 4は、 実施例 7における DNAマイクロアレイにおける階層的クラスター リング解析の結果を示す。
図 5は、 実施例 8における DNAマイクロアレイにおける相関係数を示す。 図 6は、 実施例 8における DNAマイクロアレイにおける階層的クラスター リング解析の結果を示す。
図 7は、 実施例 8における DNAマイクロアレイにおける相関係数を示す。 図 8は、 実施例 8における DNAマイクロアレイにおける階層的クラスター リング解析の結果を示す。
図 9は、 細胞増殖抑制活性を測定するアツセィにおける、 HCT116-C9、
HCT116-C9-C1 および HCT116-C9-C4 に対する E7070、 E7820、 CQS、 LY186641、 LY295501および LY-ASAPの増殖抑制作用を示したものである。
図 1 0は、 細胞増殖抑制活性を測定するアツセィにおける、 HCT116-C9、 HCT116-C9-C1および HCT116.C9-C4に対する E7070および LY573636の 増殖抑制作用を示したものである。 発明を実施するための最良の形態
以下に本発明の実施の形態について説明する。 以下の実施の形態は、 本発明を 説明するための例示であり、 本発明をこの実施の形態にのみ限定する趣旨ではな レ、。 本発明は、 その要旨を逸脱しない限り、 さまざまな形態で実施をすることが できる。
なお、 本明細書において引用した文献、 および公開公報、 特許公報その他の特 許文献は、 参照として本明細書に組み込むものとする。
1 . スルホンァミ ド化合物
本発明の医薬組成物おょぴ Zまたはキット、 ならびに癌の治療方法おょぴ Zま たは血管新生阻害方法は、 スルホンアミ ド化合物を含むものである。
本発明において、 スルホンアミ ド化合物は、 下記の式 (V) で表される化合物 を含む。

E7820 式 (V) で表される化合物は、 N— (3—シァノ一 4一メチル—1 H—インド 一ルー 7—ィル) 一 3—シァノベンゼンスルホンアミ ドであり、 E7820 と称す る場合がある。
E7820 は、 公知の方法で製造でき、 例えば、 国際公開第 00/50395 号パンフ レッ ト (WO00/50395) に記載された方法によって製造することができる。 本発明において、 スルホン ミ ド化合物は、 下記の一般式 (I) で表される化 合物を含む。
 上記一般式 (I) において、 式中、 Eは、 一 O—、 一 N ( C H3) ―、 一 C H2 一、 一 C H2 C H2—または一 C H20—を、 Dは、 一 C H2—または一 O—を、 R laは、 水素原子またはハロゲン原子 (例えば、 フッ素原子、 塩素原子、 臭素原子、 ヨウ素原子) を、 R2aは、 ノヽロゲン原子またはトリフルォロメチル基をそれぞれ 意味する。
上記一般式 (I) において、 式中、 Eは、 一 O—、 一 N ( C H3) ―、 一 C H2 一、 一 C H2 C H2—または一 C H20—を、 Dは、 一 C H2—または一 O—を、 R laは、 水素原子またはハロゲン原子 (例えば、 フッ素原子、 塩素原子、 臭素原子、 ヨウ素原子) を、 R2aは、 ノヽロゲン原子またはトリフルォロメチル基をそれぞれ 意味する。
本発明の一般式 (I) で表される化合物は、 公知の方法により製造でき、 例え ば、 欧州特許出願公開第 0222475A1 号明細書 (EP0222475A1) に記載の方法 によって製造することができる。
一般式 (I) において、 好ましい化合物は、 LY186641または LY295501であ る。
LY186641 とは、 N— [ [ ( 4一クロ口フエ-ル) ァミノ] カルボニル] ― 2, 3—ジヒ ドロー 1 H—インデンー 5—スルホンアミ ドをいい、 その構造式を 以下の式 (VI) に示す。

LY186641
LY186641 は、 公知の方法で製造でき、 例えば、 欧州特許出願公開第 0222475A1 号明細書 (EP0222475A1) に記載の方法で製造することができる。 本発明において、 LY295501 とは、 N— [ [ ( 3 , 4—ジクロ口フエ-ル) ァミノ] カルボ-ル] —2 , 3—ジヒ ドロべンゾフラン一 5—スルホンアミ ドを いい、 その構造式を以下の式 (VII) に示す。

LY295501
LY295501 は、 公知の方法で製造でき、 例えば、 欧州特許出願公開第 0222475A1 号明細書 (EP0222475A1) および Zまたは欧州特許出願公開第 0555036A2 号明細書 (EP0555036A2) に記載の方法で製造することができる。 また、 本発明において、 スルホンアミ ド化合物は、 下記の一般式 (II) で表さ れる化合物を含む。

一般式 (Π) において、 式中、 Jは、 一 O—または一 NH—を、 Ribは、 水素 原子、 ハロゲン原子、 置換基を有していてもよい d— C6アルキル基、 置換基を 有していてもよい Ci一 C4アルコキシ基、 置換基を有していてもよい Ci一 C4ァ ルキルチオ基、 — CF3、 一 OCF3、 一 S CF3、 置換基を有していてもよい Ci —C4アルコキシカルボニル基、 ニトロ基、 アジド基、 — O (S02) CH3、 - N (CH3) 2、 水酸基、 フエニル基、 置換基を有するフユ-ル基、 ピリジニル基、 チェニル基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原 子、 ハロゲン原子、 シァノ基、 一 CF3、 置換基を有していてもよい Ci一 C6ァ ルキル基、 置換基を有していてもよい Cl_C4アルコキシカルボニル基、 置換基 を有していてもよい Ci—C4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または置 換基を有していてもよい Ci—C アルコキシ基を、 R4bは、 水素原子または置換 基を有していてもよい d— C6アルキル基 (但し、 R3bおよび R4bの少なくとも 一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換基を有し ていてもよい d—Csアルキル基、 一 CF3またはニトロ基を、 R6bは、 水素原 子、 ハロゲン原子または置換基を有していてもよい Ci—C6アルキル基 (但し、
2006/304219
Rebが置換基を有していてもよい C i一 C 6アルキル基のとき、 R 5bは水素原子で あり、 R?bはハロゲン原子である) を、 R?bは、 ハロゲン原子、 置換基を有して いてもよい d—C eアルキル基または一 C F 3 (但し、 R 5bまたは R 7bのいずれ か一方が、 置換基を有していてもよい C i一 C 6アルキル基である力 \ あるいは R ?b力 S、 ハロゲン原子または置換基を有していてもよい d— C 6アルキル基である 場合には、 R 5bまたは R 6bのいずれか一方が、 水素原子である) をそれぞれ意味 する。
一般式 (II) において、 「ハロゲン原子」 は、 フッ素原子、 塩素原子、 臭素原 子、 ヨウ素原子であることが好ましい。
—般式 (II) において、 「じ1ー〇6ァルキル基」 は、 炭素数が 1〜 6の直鎖若 しくは分枝状のアルキル基を意味し、 例えば、 特に限定されるわけではないが、 メチル基、 ェチル基、 n—プロピル基、 イソプロピル基、 n—ブチル基、 イソプ チル基、 sec—ブチル基、 tert—ブチル基、 n—ペンチル基 (ァミル基) 、 イソ ペンチル基、 ネオペンチル基、 tert—ペンチル基、 1一メチルブチル基、 2—メ チルブチル基、 1, 2—ジメチルプロピル基、 n—へキ ル基、 イソへキシル基、 1一メチルペンチノレ基、 2—メチルペンチル基、 3—メチノレペンチノレ基、 1—ェ チルプロピル基、 1, 1—ジメチルブチル基、 1 , 2—ジメチルブチル基、 2, 2—ジメチルプチル基、 1 , 3—ジメチルブチル基、 2, 3—ジメチルブチル基、 3 , 3—ジメチルブチル基、 1一ェチルブチル基、 2—ェチルブチル基、 1, 1, 2—トリメチルプロピル基、 1, 2, 2—トリメチルプロピル基、 1ーェチルー 1一メチルプロピル基、 1一ェチル— 2 _メチルプロピル基などを意味する。 こ れらのうち好ましい基としては、 メチル基、 ェチル基、 n—プロピル基、 イソプ 口ピル基、 n—ブチノレ基、 イソブチル基、 sec—ブチル基、 tert—ブチル基、 n 一ペンチル基、 n—へキシル基等を挙げることができる。
一般式 (II) において、 「C i一 C 4アルコキシ基」 は、 炭素数が 1〜4のアル コキシ基を意味し、 特に限定されるわけではないが、 好ましくは、 メ トキシ基、 エトキシ基、 η—プロポキシ基、 イソプロポキシ基、 η—ブトキシ基、 イソブト キシ基、 sec—ブトキシ基、 tert—ブトキシ基等が挙げられる。
一般式 (II) において、 「C i一 C 4アルキルチオ基」 において、 アルキル基は 特に限定されるわけではないが、 メチル、 ェチル、 n—プロピル、 イソプロピル、 n—プチル、 ィソブチル、 sec—ブチル、 tert—ブチル等を挙げることができる。 一般式 (Π) において、 「C i— C4アルコキシカルボニル基」 の例としては、 特に限定されるわけではないが、 メ トキシカルポニル基、 エトキシカルボ二ル基、 n一プロポキシカルボニル基、 ィソプロボキシカルボニル基、 n—ブトキシカノレ ボニル基、 イソブトキシカルボニル基、 sec—ブトキシカルボニル基、 tert—ブ トキシカルボ二ル基等を挙げることができる。
一般式 (II) において、 導入される置換基としては、 特に限定されるわけでは ないが、 例えば、 C i _ C 6アルキル基 (例えば、 メチル基、 ェチル基、 n—プロ ピル基、 イソプロピル基、 n—ブチル基、 イソプチル基、 sec—ブチル基、 tert 一ブチル基等) 、 d— C 4アルコキシ基 (例えば、 メ トキシ基、 エトキシ基、 n —プロポキシ基、 イソプロポキシ基、 n—ブトキシ基、 イソブトキシ基、 sec— ブトキシ基、 tert—ブトキシ基等) 、 アミノ基、 水酸基、 ハロゲン原子 (例えば、 フッ素原子、 塩素原子、 臭素原子、 ヨウ素原子) またはシリル基などの置換基を 挙げることができる。
本発明の一般式 (Π) で表される化合物は、 公知の方法により製造でき、 例え ば、 国際公開第 02/098848 号パンフレツ ト (WO02/098848) に記載の方法に よって製造することができる。
一般式 (II) において、 好ましい化合物は LY- ASAPである。
LY-ASAP とは、 N— ( 2, 4—ジクロロべンゾィル) 一 4一クロ口フエ-ル スルホンアミ ドをいい、 その構造式を以下の式 (VIII) に示す。
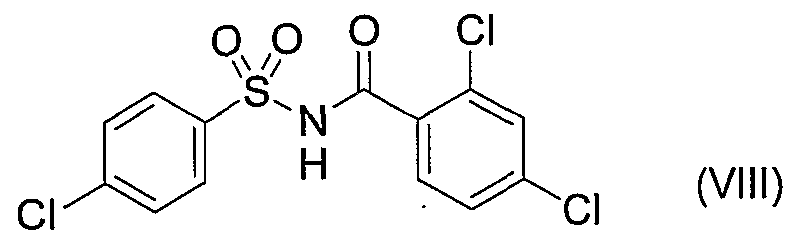
LY-ASAP
LY-ASAP は、 公知の方法で製造でき、 例えば、 国際公開第 02/098848 号パ ンフレッ ト (WO02/098848) に記載の方法で製造することができる。
本発明において、 スルホンアミ ド化合物には、 LY573636 を挙げることがで きる。 本発明において、 LY573636 とは、 N— (2, 4—ジクロロべンゾィ ル) 一 5—プロモチォフェン一 2—スルホンアミ ドをいい、 その構造式を以下の 式 (ΙΠ) に示す。

LY573636
LY573636は、 ナトリゥム塩であることが好ましい。
LY573636 は、 公知の方法で製造でき、 例えば、 国際公開第 02/098848号パ ンフレッ ト (WO02/098848) に記載の方法と同様にして、 市販の 5—ブロモチ ォフェン一 2ースルフォニルク口ライ ドと 2 , 4—ジクロ口安息香酸より製造す ることができる。
ま た 、 LY573636 は、 国際公開第 03/035629 号パ ンフ レ ツ ト (WO03/035629) における実施例 6 3に記載の方法で製造することができる。 本発明において、 スルホンアミ ド化合物には、 CQS を挙げることができる。 本発明において、 CQS とは、 2—スルファニルアミ ドー 5—クロ口キノキサリ ンをいい、 その構造式を以下の式 (IV) に示す。
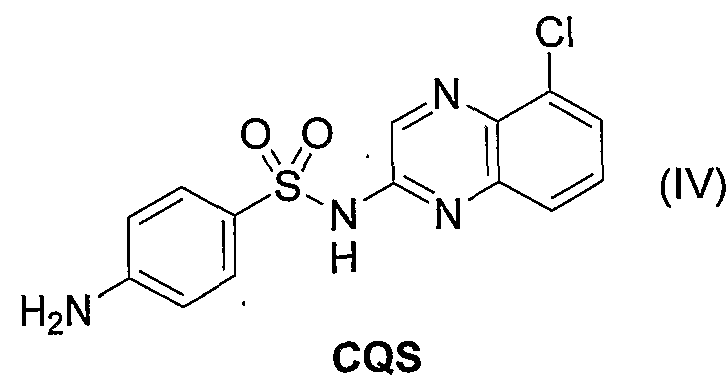
CQSは、 公知の方法で製造でき、 例えば、 (J. Am. Chem. Soc, 1947, 71, 6-10) の方法で製造することができる。
スルホンアミ ド化合物は、 酸または塩基と薬理学的に許容される塩を形成する 場合もある。 本発明におけるスルホンアミ ド化合物は、 これらの薬理学的に許容 される塩をも包含する。 酸との塩としては、 例えば、 塩酸塩、 臭化水素酸塩、 硫 酸塩、 燐酸塩等の無機酸塩や蟻酸、 酢酸、 乳酸、 コハク酸、 フマル酸、 マレイン
酸、 クェン酸、 酒石酸、 安息香酸、 メタンスルホン酸、 ベンゼンスルホン酸、 p 一トルエンスルホン酸、 トリフルォロ酢酸などの有機酸との塩を挙げることがで きる。 また、 塩基との塩としては、 ナトリウム塩、 カリウム塩などのアルカリ金 属塩、 カルシウム塩、 マグネシウム塩のようなアルカリ土類金属塩、 トリメチル ァミン、 トリェチルァミン、 ピリジン、 ピコリ ン、 ジシクロへキシルァミン、 N , N ' —ジベンジルエチレンジァミン、 アルギニン、 リジン等の有機塩基との塩 (有機アミン塩) 、 アンモニゥム塩を挙げることができる。
また、 スルホンアミ ド化合物は、 無水物であってもよく、 水和物などの溶媒和 物を形成していてもよい。 溶媒和物は水和物または非水和物のいずれであっても よいが、 水和物が好ましい。 溶媒は水、 アルコール (例えば、 メタノール、 エタ ノール、 n—プロパノール) 、 ジメチルホルムアミ ドなどを使用することができ る。
また、 これら化合物の溶媒和物および/または光学異性体が存在する場合には、 本発明におけるスルホンアミ ド化合物は、 それらの溶媒和物および/または光学 異性体が含まれる。 また、 本発明におけるスルホンアミ ド化合物は、 生体内で酸 ィ匕、 還元、 加水分解、 抱合などの代謝を受けるスルホンアミ ド化合物をも包含す る。 またさらに、 本発明におけるスルホンアミ ド化合物は、 生体内で酸化、 還元、 加水分解などの代謝を受けてスルホンアミ ド化合物を生成する化合物をも包含す る。
2 . 白金錯体物質、 DNA-topoisomei'ase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質
本発明の医薬組成物およひブまたはキット、 ならびに癌の治療方法および Zま たは血管新生阻害方法は、 白金錯体物質、 DNA-topoisomei'ase I 阻害物質、 代 謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択される少なくと も一^ 3の物質を含むものである。
( 1 ) 白金錯体物質
本発明において、 白金錯体物質は中心金属を白金とする錯体 (白金錯体) であ ればよく、 中心金属と配位子間との結合の程度、 錯体の荷電、 配位子のブリッジ ド構造による中心金属の数の増加などに限定されるものではない。 また、 本発明 において、 白金錯体物質は白金錯体を製剤化した白金製剤であってもよい。 本発 明において、 白金錯体物質を白金製剤と称する場合もある。
本発明において、 白金錯体物質には、 Oxaliplatin、 Carboplatin、 Cisplatin(CDDP), Lobaplatin、 AR_726、 Miriplatin、 Picoplatin, PLD'147、 Satraplatin、 Thioplatin、 Triplatin な どが挙げられ、 好ま し く は、 Oxaliplatinまたは Cisplatinであり、 より好ましくは Oxaliplatinである。 白 金錯体物質は、 公知の方法で製造でき、 また、 購入することができる。
本発 にぉレヽて、 Oxaliplatin とは、 oxalato (1R, 2R-cyclohexanediamme) platinumをいい、 式 (IX) で表される化合物である。

Oxaliplatin は、 公知の方法で製造できる。 また、 Oxaliplatin は、 Sanofi Aventis 社から Eloxatin (登録商標) を購入することによって、 入手すること ができる。
本 発 明 に ぉ レヽ て 、 Carboplatin と は 、 cis-diammine(l, l- cyclobutanedicarboxylate) platinumをレヽつ。
本発明において、 Carboplatinは、 パラプラチン (ブリス トル製薬株式会社) を購入することによって、 入手することができる。
本発 におレ、て、 Cisplatin(CDDP) fま、 cis-diamminedichloroplatinum II をいい、 式 (XX) で表される化合物である。
 0
0
1

1〇
( 2 ) DNA-topoisomerase I阻害物質
本発明において、 DNA-topoisomerase I阻害物質とは、 DNA-topoisomerase I 阻害する作用を有する物質を意味する。
本発明において、 DNA-topoisomerase I阻害物質には、 CPT_11、 Topotecan hydrochloride N Exatecan、 Rubitecan、 9-amino-camptothecin、 Lurtotecan dihydrochloride , Gimatecan , Edotecarin などが挙げられ、 好ましくは、 CPT-11である。
DNA-topoisomerase I 阻害物質は、 公知の方法で製造でき、 また、 購入する ことができる。
本発明において、 CPT- 11 とは、 塩酸イ リ ノテカン三水和物 ([1,4'- Bipiperidine] -l'-carboxylic acid (S)-4, ll-aiethyl-3,4,12, 14-tetra ydro-4' hydroxy- 3, 14-dioxo- lH-pyrano- [3',4':6,7]-mdolizmo[l, 2-b] quinoiin-9-yl ester Hydrochloride trihydrate) をいい、 式 は) で表される化合物である。

CPT-11 は、 公知の方法で製造できる。 また、 CPT-11 は、 第一製薬株式会社 からトポテシン (登録商標) を購入することによって、 入手することができる。 本発明において、 DNA-topoisomerase I阻害物質には CPT' ll活性体である SN38 を用レヽる こ と もでき る。 SN38 と は、 7-ethyl-10-hydro-(20)S- Camptothecinをいう。 SN38は ABATRA社から購入して入手することができ る。
本 発 明 に お い て 、 Topotecan hydrochloride と は 、 (4S)_10- [(dimethylammo)methyl]-4-ethyl-4, 9-di ydroxylH-pyranoL3', 4':6, 7] indolizino [1, 2-b] qumoline-3, 14 (4H, 12H)-dione hydrochlorideをレヽレヽ、 式 (XI) で表される化合物である。
Topotecan hydrochloride は、 公知の方法で製造することができる (米国特 許第 5 0 0 4 7 5 8 号明細書 ( US5004758 ) ) 。 また、 Topotecan hydrochloride は、 日本化薬からハイカムチン (登録商標) を購入することによ つて、 入手することができる。
本発明において、 Exatecanとは、 (IS, 9S)-l-amino-9-ethyl-5-fluoro- l, 2, 3 9, 12, 15-hexahvdro-9-ny roxy4-methyl-10H, 13H-benzo[de]pyrano[3', 4':6 7]indolizino[l, 2-b]quinoline-10, 13-dione をいい、 式 (XII) で表される化合 物である。

Exatecan は、 公知の方法で製造することができる (特開平 0 5— 5 9 0 6 1 号公報 (JP93-59061) ) 。
本発明において、 Rubitecan とは、 (4S)-ethyl-4-hydroxy-10-nitro- l, 12' dihydro-14H-pyrano[3', 4': 6, 7]-indolizino[l, 2-b]quinoline-3, 14(4H, 12H)- dioneをいい、 式 (XIII) で表される化合物である。

本発明において、 9-amino-camptotliecin とは、 (4S)-ethyl-4-hydroxy-10- amino-1, 12-dihydro- 14H-pvrano[3', 4': 6, 7] -indolizmo[l, 2-b]qumoline-3, 14(4H, 12H)-dioneをいい、 式 (XIV) で表される化合物である。

9-amino-camptothecin は、 公知の方法で製造することができる (特開昭 59· 051289) 。
本 発 明 に ぉ レヽ て 、 Lurtotecan dihydrochloride と は 、 7_(4- methylpiperazinomethylene)-10, ll-ethylene ioxy20(s)-camptothecin dihydrochlorideをいい、 式 (XV) で表される化合物である。

Lurtotecan dihydrochloride は、 公知の方法で製造するこ とができる (WO95/29919) 。
本 発 明 に お い て 、 Gimatecan と は 、 (4S)-11_[(E)- [[1, 1- aiinethylethoxy]iniino]methyl]-4-ethyl-4-hydroxy l, 12-dihydro- 14H- pyrano [3', 4': 6, 7]-indolizino[l, 2-b]quinoline-3, 14(4H)-dione をいい、 式 (XVI) で表される化合物である。

Gimatecanは、 公知の方法で製造することができる (WO00/053607) 。
本発明において、 Edotecarin と は、 12-6-D-glucopyranosyl"2, 10- di ydroxy-6-[[2-hydroxy-l-(hydroxymet yl)ethyl]ammo]- 12, 13-dihydro- 6H-indolo[2, 3-a]pyrrolo[3, 4-c]carbazole-5, 7-dioneをいい、 式 (XVII) で表 される化合物である。

Edotecarinは、 公知の方法で製造することができる (WO95/30682) 。
( 3 ) 代謝拮抗物質
本発明において、 代謝拮抗物質は、 核酸合成やタンパク質合成などの細胞の代 謝において必要な物質と類似の構造をもつ化合物であり、 その構造類似性により、 細胞の代謝を阻害するものである。
本発明において、 代謝拮抗物質には、 Cytidine誘導体が挙げられ、 具体的に は、 Gemcitabine N Cytarabine (araC)、 Enocitabine、 Citarabine ocfosfateヽ 5-azacytidine, CNDACなどが挙げられ、 好ましくは、 Gemcitabineである。
また、 本発明において、 代謝拮抗物質には、 葉酸拮抗剤が挙げられ、 具体的に は、 Methotrexate が挙げられる。 葉酸拮抗剤は、 ジヒ ドロ葉酸レダクターゼを 阻害することにより、 核酸合成を阻害するものである。
代謝拮抗剤は、 公知の方法で製造でき、 また、 購入することができる。
本発明において、 Gemcitabine とは、 塩酸ゲムシタビン (2'-deoxy-2',2'_ difluoro-cytidine hydrochloride) をいい、 式 (XVIII) で表される化合物であ る。

Gemcitabine は、 公知の方法で製造できる。 また、 Gemcitabine は、 日本ィ 一ライリ リー社から GEMZAR (登録商標) を購入することによって、 入手する ことができる。
Cytarabine (araC) は、 キロサイ ド (日本新薬) またはキロサイ ド N (日本 新薬) を購入することによって、 入手することができる。
Enocitabine (BH-AC) は、 サンラビン (旭化成) を購入することによって、 入手することができる。
Citarabine ocfosfate (SPAC) は、 スタラシド (日本ィ匕薬) を購入すること によって、 入手することができる。
5-azacytidine は、 公知の方法で製造することができ、 また、 ナカライテスタ 株式会社から購入することによって、 入手することができる。
本発明において、 CNDAC ,と は、 l-(2-C-cyano-2-deoxy-6-D-arabino- pentofuranosyl)-N-palinitoylcytosine をいい、 式 (XIX) で表される化合物で ある。

CNDACは、 公知の方法で製造することができる (EP536936) 。
本発明において、 Methotrexate とは、 N-{4- [N-(2,4-diaminopteridin-6- ylmethyl)-N-methylamino]benzoyl}-L- glutamic acidをレヽレヽ、 式 (XXI) 、表 される化合物である。

本発明において、 Methotrexate は、 ワイス一武田からメソトレキセート (登 録商標) またはリウマトレックス (登録商標) を購入することによって、 入手す ることができる。
( 4 ) 微小管阻害物質
本発明において、 微小管阻害物質は、 細胞分裂における紡錘体形成機能または 物質輸送機能などの微小管の機能を阻害する作用を有する物質を意味する。 例え ば、 微小管阻害物質は、 細胞分裂が盛んな細胞または神経細胞などの微小管に作 用して抗腫瘍効果を示す。
本発明において、 微小管阻害物質には、 Paclitaxel、 Docetaxel などが挙げら れ、 好ましくは Paclitaxelである。
微小管阻害物質は、 公知の方法で製造でき、 また、 購入することができる。 本発明において、 Paclitaxel とは、 (-)-(lS, 2R, 3S, 4S, 5R, 7S, 8S, 建,
.13S)-4, 10-diacetoxy-2-benzoyloxy-5,20-epoxy l,7-dihydroxy-9-oxotax- ll-
en- 13-yl (2S, 3S) -3-benzoylamino-2-hydroxy 3-phenylpropionateをレヽレヽ、 式
(XXI I ) で表される化合物である。

Paclitaxel は、 ブリス トル製薬株式会社からタキソール (Taxol) を購入する ことによって、 入手することができる。
Docetaxelは、 タキソテール (登録商標) (サノフィ ·アベンテイス) を購入 することによって、 入手することができる。
( 5 ) 抗生物質
本発明において、 抗生物質は、 好ましくは抗腫瘍性抗生物質である。 抗腫瘍性 抗生物質は、 腫瘍細胞中の DNA合成抑制または DNA鎖切断などの作用を有し、 抗腫瘍作用を示すものである。
本発明において、 抗生物質には、 Doxorubicin (ア ドリアマイシン) 、 DaunoruDicin、 j irarubicm、 Epirubicm、 Idarubicm、 Aclarubicin、 Amrubicin Mitoxantrone など力 s挙げられ、 好ましくは Doxorubicin である。 抗生物質は、 公知の方法で製造でき、 また、 購入することができる。
本発明において、 Doxorubicin とは、 10- [(3-amino-2,3,6-trideoxy- -L- lyxo-hexoDyranosyl)oxy] -7,8,9, 10-tetrahydro-6, 8, 1 l-trihydroxy-8- (hydroxyacetyl)-l-methoxy(8S-cis)-5, 12-naphthacenedione を い い 、 式 (XXIII) で表される化合物である。

Doxorubicin は、 アドリアシン (登録商標) (協和発酵) を購入することに よって、 入手することができる。
Daunorubicin は、 ダウノマイシン (登録商標) (明治製菓) を購入すること によって、 入手することができる。
Piparubicin は、 テラルビシン (登録商標) (明治製菓) またはピノルビン (登録商標) (メルシャン一日本化薬) を購入することによって、 入手すること ができる。
Epirubicin は、 フアルモルビシン (登録商標) (フアイザ——協和発酵) を 購入することによって、 入手することができる。
Idarubicin は、 イダマイシン (登録商標) (フアイザ一) を購入することに よって、 入手することができる。
Aclarubicin は、 ァクラシノン (登録商標) (メルシャン一山之内) を購入す ることによって、 入手することができる。
Amurubicinは、 カルセド (登録商標) (住友製薬) を購入することによって、 入手することができる。
Mitoxantrone は、 ノバントロン (登録商標) (ワイス一武田) を購入するこ とによって、 入手することができる。
( 6 ) 塩、 溶媒和物
白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害 物質および抗生物質は、 酸または塩基と薬理学的に許容される塩を形成する場合 もある。 また、 上記 (1 ) 〜 (5 ) にあげた化合物は、 (1 ) 〜 (5 ) で例示し た塩とは異なる薬理学的に許容される塩を形成する場合もある。 本発明における
前記の物質は、 これらの薬理学的に許容される塩をも包含する。 酸との塩として は、 例えば、 塩酸塩、 臭化水素酸塩、 硫酸塩、 燐酸塩等の無機酸塩や蟻酸、 酢酸、 乳酸、 コハク酸、 フマル酸、 マレイン酸、 クェン酸、 酒石酸、 安息香酸、 メタン スルホン酸、 ベンゼンスルホン酸、 p — トルエンスルホン酸、 トリフルォロ酢酸 などの有機酸との塩を挙げることができる。 また、 塩基との塩としては、 ナトリ ゥム塩、 カリウム塩などのアルカリ金属塩、 カルシウム塩、 マグネシウム塩のよ うなアルカリ土類金属塩、 トリメチルァミン、 トリェチルァミン、 ピリジン、 ピ コリン、 ジシクロへキシノレアミン、 N , N ' —ジベンジルエチレンジァミン、 了 ルギニン、 リジン等の有機塩基との塩 (有機アミン塩) 、 アンモニゥム塩を挙げ ることができる。
また、 白金錯体物質、 DNA-topoisomei'ase I阻害物質、 代謝拮抗物質、 微小 管阻害物質および抗生物質は、 無水物であってもよく、 水和物などの溶媒和物を 形成していてもよい。 溶媒和物は水和物または非水和物のいずれであってもよい 力 水和物が好ましい。 溶媒は水、 アルコール (例えば、 メタノール、 エタノー ル、 n—プロパノール) 、 ジメチルホルムアミ ドなどを使用することができる。 また、 これら物質の溶媒和物および/または光学異性体が存在する場合には、 本発明における前記物質には、 それらの溶媒和物および/または光学異性体が含 まれる。 また、 本発明における前記物質は、 生体内で酸化、 還元、 加水分解、 抱 合などの代謝を受ける白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮 抗物質、 微小管阻害物質および抗生物質から選択される少なくとも一つをも包含 する。 またさらに、 本発明における前記物質は、 生体内で酸化、 還元、 加水分解 などの代謝を受けて白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗 物質、 微小管阻害物質おょぴ抗生物質から選択される少なくとも一つを生成する 物質をも包含する。
3 . 医薬組成物、 キット、 癌の治療方法、 血管新生阻害方法
本発明は、 スルホンアミ ド化合物と、 (i ) 白金錯体物質、 (ii) DNA- topoisomerase I阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 およ
ぴ (V) 抗生物質からなる群から選択される少なくとも一つの物質とを組み合わ せる点に特徴を有する医薬組成物、 キット、 癌の治療方法および血管新生阻害方 法に関するものである。
本発明において、 スルホンアミ ド化合物は、 「1 . スルホンアミ ド化合物」 で 記載したとおりであるが、 例えば、 (A) E7820 (式 (V) ) 、 (B) 一般式 (I) で表される化合物、 好ましくは LY186641または LY295501、 (C) 一般 式 (II ) で表される化合物、 好ましくは LY-ASAP、 ( D ) LY573636 (式 (III) ) および (E) CQS (式 (IV) ) から選択される少なく とも一つの化合 物であり、 より好ましくは LY295501および LY573636から選択される少なく とも一つの化合物であり、 特に好ましくは LY573636のナトリウム塩である。 他方、 本発明において、 スルホンアミ ド化合物は、 好ましくは E7820 である。 本発明において、 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗 物質、 微小管阻害物質および抗生物質は、 「 2 . 白金錯体物質、 DNA- topoisomerase I阻害物質、 代謝拮抗物質、 微小管阻害物質おょぴ抗生物質」 で 記載したとおりであるが、 (i) 白金錯体物質は好ましくは Oxaliplatin、 (ii) DNA-topoisomerase I阻害物質は好ましくは CPT- 11および (iii) 代謝拮抗物 質は好ましくは Gemcitabineである。 他方、 本発明において、 (i) 白金錯体物 質は好ましくは Cisplatin、 (iii) 代謝拮抗物質は好ましくは Methotrexate (iv) 微小管阻害物質は好ましくは Paclitaxel、 および (v) 抗生物質は好まし く f Doxorubicinである。
本発明において、 上記スルホンアミ ド化合物および上記 (i) 〜 (V) の物質に は、 その薬理学的に許容される塩、 またはそれらの水和物などの溶媒和物も包含 される。
本発明の医薬組成物は、 スルホンアミ ド化合物と、 (i) 白金錯体物質、 (ii) DNA-topoisomerase I 阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻 害物質、 および (V) 抗生物質から選ばれる少なくとも一つの物質とを組み合わ せてなる医薬組成物である。 本発明の医薬組成物は、 癌治療用医薬組成物おょぴ Zまたは血管新生阻害用医薬組成物として有用である。
本発明において、 「組み合わせてなる」 とは、 化合物を併用して用いるための 組み合わせを意味し、 別々の物質を投与時に併用する形態、 および混合物として の形態の両方を含む。
また、 本発明の医薬組成物は、 ( i ) 白金錯体物質、 ( ii ) DNA- topoisomerase I阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 およ び (V) 抗生物質から選ばれる少なくとも一つの物質とともに患者に併用投与す るためのスルホンアミ ド化合物を含む医薬組成物の態様でも提供される。 スルホ ンアミド化合物、 および (i) 白金錯体物質、 (ii) DNA-topoisomerase I 阻害 物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 および (V) 抗生物質から 選ばれる少なくとも一つの物質は、 同時または別々に投与され得る。 「同時」 と は、 一つの投与スケジュールにおいて同一のタイミングで投与されることを意味 し、 投与の時分が完全に同一である必要はない。 「別々」 とは、 一つの投与スケ ジュールにおいて異なるタイミングで投与されることを意味する。
また、 本発明のキットは、 スルホンアミ ド化合物を含んでなる製剤と、 (i) 白金錯体物質、' (ii) DNA-topoisomerase I 阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 および (V) 抗生物質からなる群から選択される少なく とも一つの物質を含んでなる 剤とをセットにしたことを特徴とするキットであ る。 本発明のキットに含まれる製剤は、 スルホンアミ ド化合物または上記 (i) 〜 (V) の少なくとも一つの物質を含む限り、 その剤形は特に限定されない。 本 発明のキットは、 癌治療用キットおよび/または血管新生阻害用キットとして有 用である。
本発明のキットにおいて、 スルホンアミ ド化合物を含んでなる製剤と、 上記
(i) 〜 (V) の少なくとも一つの物質を含む製剤とは、 混合されていてもよいし、 あるいは、 別個に収納されて一体に包装されていてもよく、 また、 同時に投与さ れてもよいし、 いずれか一方を先に投与してもよい。
本発明の医薬組成物および/またはキットならびに癌の治療方法おょぴ また は血管新生阻害方法は、 さらに一または複数の他の抗癌剤を組み合わせてもよレ、。 他の抗癌剤は、 抗癌作用を有する製剤であれば、 特に限定されない。 他の抗癌剤
としては、 例えば、 5-フルォロゥラシル (5-FU) 、 ホリナートカルシウム (口 ィコボリン) 、 ドセタキセル (タキソテール (登録商標) ) 、 ゲフイチニブ
(Iressa (登録商標) ) 、 エル口チニブ (Tarceva (登録商標) ) 、 セツキシマ ブ (Erbitux (登録商標) 、 べバシズマブ (Avastin (登録商標) ) などが挙げ られる。 また、 前記他の抗癌剤としては、 癌治療剤の対象となる癌種が、 大腸癌 である場合には、 5-フルォロゥラシル、 ホリナートカルシウム、 ゲフイチニブ、 エルロチニブ、 セツキシマブ、 ベバシズマブが特に好ましく、 膝癌である場合に は、 ゲフイチニブ、 エルロチニブ、 セツキシマプ、 ベバシズマブが特に好ましい, さらに、 本発明において、 化合物の特に好ましい組み合わせとしては、 癌治療 剤の対象となる癌種が、 大腸癌である場合には、 例えば、 表 1に示した組み合わ せであり、 膝癌である場合には、 例えば、 表 2に示した組み合わせである。
表 1 組み合わせ化合物
1 E7070 oxaliplatin 5- -FU LV ゲフイチニブ
2 E7820 oxaliplatin 5- FU し V ゲフイチニブ
3 E7070 oxaliplatin 5- -FU LV ェ レ口チニブ
4 E7820 oxaliplatin 5- -FU LV エノレロチニブ
5 E7070 oxaliplatin 5- -FU LV セツキシマブ
6 E7820 oxaliplatin 5- -FU LV セツキシマブ
7 E7070 oxaliplatin 5- -FU LV ゲフイチニブ べバシズマブ
8 E7820 oxaliplatin 5- -FU LV ゲフイチニブ ベ/くシズマブ
9 E7070 oxaliplatin 5- -FU LV エレロチニブべパシズマブ
10 E7820 oxaliplatin 5- -FU LV エレロチニブべバシズマブ
1 1 E7070 oxaliplatin 5- -FU LV セツキシマブべバシズマブ
12 E7820 oxaliplatin 5- -FU LV セツキシマブべバシズマブ
13 E7070 CPT-1 1 5- -FU LV ゲフイチニブ
14 E7820 CPT-11 5- -FU LV ゲフイチニブ
15 E7070 CPT-11 5- -FU LV エノレロチニブ
16 E7820 CPT-1 1 5- -FU LV エレロチニブ
17 E7070 CPT-11 5- -FU LV セツキシマブ
18 E7820 CPT-1 1 5- -FU LV セツキシマブ
19 E7070 CPT-1 1 5- -FU LV ゲフイチニブ べバシズマブ
20 E7820 CPT-1 1 5- -FU LV ゲフイチニブ べノ シズマブ
21 E7070 CPT-11 5- -FU LV エル口チニブべパシズマブ
22 E7820 CPT-11 5- -FU LV エレロチニブべバシズマブ
23 E7070 CPT-1 1 5- -FU LV セツキシマブベ/くシズマブ
24 E7820 CPT-11 5- -FU LV セツキシマブべパシズマブ 表 1は、 本発明において、 癌治療剤の対象となる癌種が、 大腸癌である場合に おける好ましい組み合わせを示す。 表中、 LV は、 ホリナートカルシウムを示す。
表 2
組み合わせ化合物
1 E7070 Gemcitabine ゲフイチニブ
2 E7820 Gemcitabine ゲフイチニブ
3 E7070 Gemcitabine エル口チニブ
4 E7820 Gemcitabine エル口チニブ
5 E7070 Gemcitabine セツキシマブ
6 E7820 Gemcitabine セツキシマブ
7 E7070 Gemcitabine ゲフイチニブ べバシズマブ
8 E7820 Gemcitabine ゲフイチニブ べバシズマブ
9 E7070 Gemcitabine エル口チニブベ <^シズマブ
10 E7820 Gemcitabine エル口チニブべパ、シズマブ
1 1 E7070 Gemcitabine セツキシマブべバシズマブ
12 E7820 Gemcitabine セツキシマブべバシズマブ
表 2は、 本発明において、 癌治療剤の対象となる癌種が、 膝癌である場合にお ける好ましい組み合わせを示す。
本発明の医薬組成物および Zまたはキットは、 癌治療剤または血管新生阻害剤 として使用することができる。
本発明において、 治療は、 疾患の症状を軽減すること、 疾患の症状の進行を抑 制すること、 疾患の症状を除去すること、 疾患の予後を改善すること、 疾患の再 発を予防することも包含する。
本発明において、 癌治療剤とは、 抗腫瘍剤、 癌予後改善剤、 癌再発予防剤、 癌 転移抑制剤などを含むものをいう。
癌治療の効果は、 レントゲン写真、 CT等の所見や生検の病理組織診断により、 あるいは腫瘍マーカーの値により確認することができる。
本発明の医薬組'成物おょぴ またはキットは、 哺乳動物 (例、 ヒ ト、 ラット、 ゥサギ、 ヒッジ、 プタ、 ゥシ、 ネコ、 ィヌ、 サルなど) に対して、 投与すること ができる。
癌治療剤の対象となる癌種は、 特に限定されず、 例えば、 脳腫瘍、 頸癌、 食道 癌、 舌癌、 肺癌、'乳癌、 膝癌、 胃癌、 小腸や十二指腸の癌、 大腸癌 (結腸癌、 直 腸癌) 、 膀胱癌、 腎癌、 肝癌、 前立腺癌、 子宮癌、 卵巣癌、 甲状腺癌、 胆嚢癌、 咽頭癌、 肉腫 (例えば、 骨肉腫、 軟骨肉腫、 力ポジ肉腫、 筋肉腫、 血管肉腫、 線 維肉腫など) 、 白血病 (例えば、 慢性骨髄性白血病 (CML) 、 急性骨髄性白血
病 (AML) 、 慢性リ ンパ球性白血病 (CLL) 及び急性リンパ性白血病 (ALL) 、 リンパ腫、 多発性骨髄腫 (MM) など) およびメラノ一マからなる群から選択さ れる少なくとも一つなどがあげられる。 また、 癌治療剤の対象となる癌種は、 好 ましくは大腸癌および臈癌からなる群から選択される少なくとも一つである。 本発明の医薬組成物および/またはキットを使用する場合には、 経口もしくは 非経口的に投与することができる。
本発明の医薬組成物およびノまたはキットを使用する場合、 スルホンアミ ド化 合物の投与量は、 症状の程度、 患者の年齢、 性別、 体重、 感受性差、 投与方法、 投与時期、 投与間隔、 医薬製剤の性質、 調剤、 種類、 有効成分の種類等によって 異なり、 特に限定されないが、 通常成人 (体重 6 0 Kg) 1 日あたり 10〜6000 mg、 好ましくは 50〜4000 mg、 さらに好ましくは 50〜2000 mgであり、 これ を通常 1 日 1〜 3回に分けて投与することができる。
本発明の医薬組成物および/またはキットを使用する場合、 (i) 白金錯体物 質、 好ましくは Oxaliplatinまたは Cisplatin, (ii) DNA-topoisomerase I阻害 物質、 好ましくは CPT- 11、 (iii) 代謝拮抗物質、 好ましくは Gemcitabine ま たは Methotrexate , (iv) 微小管阻害物質、 好ましくは Paclitaxel、 および (V) 抗生物質、 好ましくは Doxorubicinからなる群から選択される少なくとも 一つの物質の投与量は、 特に限定されないが、 通常成人 1 日あたり 10〜6000 mg、 好ましくは 50〜4000 mg、 さらに好ましくは 50〜2000 mgであり、 これ を通常 1日 1〜 3回に分けて投与することができる。
使用するスルホンアミ ド化合物の量は、 特に限定されず、 (i) 白金錯体物質、 好ましくは Oxaliplatinまたは Cisnlatin、 (ii) DNA-topoisomerase I阻害物質、 好ましくは CPT-11、 . (iii) 代謝拮抗物質、 好ましくは Gemcitabine または Methotrexate, (iv) 微小管阻害物質、 好ましくは Paclitaxel、 および (v) 抗 生物質、 好ましくは Doxorubicinからなる群から選択される少なくとも一つの 物質との個々の組み合わせによって異なるが、 例えば、 (i) 〜 (V) から選択さ れる少なくとも一つの物質の約 0·01〜100倍 (重量比) である。 さらに好まし くは約 0.1〜10倍 (重量比) である。
本発明の医薬組成物は、 種々の剤形、 例えば、 経口用固形製剤、 または注射剤、 坐剤、 軟膏剤、 パップ剤などの非経口用製剤などにすることができる。
また、 本発明のキッ トに含まれるスルホンアミ ド化合物と、 (i) 白金錯体物 質、 (ii) DNA-topoisomerase I 阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管 阻害物質、 および (V) 抗生物質からなる群から選択される少なく とも一つの物 質とは、 それぞれ種々の剤形、 例えば、 経口用固形製剤、 または注射剤、 坐剤、 軟膏剤、 パップ剤などの非経口用製剤などの製剤にすることができる。
経口用固形製剤を調製する場合には、 主薬に賦形剤、 さらに必要に応じて結合 剤、 崩壊剤、 滑沢剤、 着色剤、 矯味矯臭剤などを加えた後、 常法により錠剤、 被 覆錠剤、 顆粒剤、 細粒剤、 散剤、 カプセル剤等とすることができる。 また、 シロ ップ剤等の経口用非固形製剤'も適宜調製することができる。
賦形剤としては、 例えば、 乳糖、 コーンスターチ、 白糖、 ぶどう糖、 ソノレビッ ト、 結晶セルロース、 二酸化ケイ素などが、 結合剤としては、 例えば、 ポリビニ ノレアノレコール、 ェチノレセノレロース、 メチノレセノレロース、 アラビアゴム、 ヒ ドロキ シプロピルセルロース、 ヒ ドロキシプロピルメチルセルロース等が、 滑沢剤とし ては、 例えば、 ステアリン酸マグネシウム、 タノレク、 シリカ等が、 着色剤として は医薬品に添加することが許可されているものが、 矯味矯臭剤としては、 例えば、 ココア末、 ハツ力脳、 芳香酸、 ハツ力油、 龍脳、 桂皮末等が用いられる。 これら の錠剤、 顆粒剤には糖衣、 ゼラチン衣、 その他必要により適宜コーティングする ことは勿論差し支えない。
注射剤を調製する場合には、 必要により主薬に pH調整剤、 緩衝剤、 懸濁化剤、 溶解補助剤、 安定化剤、 等張化剤、 保存剤などを添加し、 常法により静脈、 皮下、 筋肉内注射剤、 点滴静注剤とすることができる。 その際必要により、 常法により 凍結乾燥物とすることもできる。
懸濁化剤としては、 例えば、 メチルセルロース、 ポリソルベート 80、 ヒ ドロ キシェチルセルロース、 アラビアゴム、 トラガント末、 カノレボキシメチノレセル口 ースナトリゥム、 ポリオキシエチレンソルビタンモノラウレートなどを挙げるこ とができる。
'溶解補助剤としては、 例えば、 ポリオキシェチレン硬化ヒマシ油、 ポリソルべ ート 80、 ニコチン酸アミ ド、 ポリオキシエチレンソノレビタンモノラウレート、 マグロゴール、 ヒマシ油脂肪酸ェチルェステルなどを挙げることができる。
また安定化剤としては、 例えば、 亜硫酸ナトリウム、 メタ亜硫酸ナトリウム等 を、 保存剤としては、 例えば、 パラォキシ安息香酸メチル、 パラォキシ安息香酸 ェチル、 ソノレビン酸、 フエノール、 クレゾーノレ、 クロロタレゾーノレなどを挙げる ことができる。
本発明の医薬組成物および/またはキットは、 上記のスルホンアミド化合物、 および (i) 白金錯体物質、 (ii) DNA-topoisomerase I阻害物質、 (iii) 代謝拮 抗物質、 (iv) 微小管阻害物質、 および (V) 抗生物質からなる群から選択され る少なくとも一つの物質の他に、 包装容器、 取扱説明書、 添付文書等を含んでい てもよい。 包装容器、 取扱説明書、 添付文書等には、 化合物を併用して用いるた めの組み合わせを記載することができ、 また、 別々の物質を投与時に併用する形 態または混合物としての形態について、 用法、 用量などを記載することができる。 用法、 用量は、 上記を参照して記載することができる。
また、 本発明のキットは、 (a) スルホンアミド化合物と、 (i) 白金錯体物質、 (ii) DNA-topoisomerase I 阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻 害物質、 および (V) 抗生物質からなる群から選択される少なくとも一つの物質 とを併用して用いることを記載した、 包装容器、 取扱説明書および添付文書から なる群から選択される少なくとも一つと、 (b ) スルホンアミ ド化合物を含む医 薬組成物とを含有する態様であってもよい。 当該キットは、 癌治療用キットおよ び zまたは血管新生阻害用キットとして有用である。 前記スルホンアミ ド化合物 を含む医薬組成物は、 癌治療用医薬組成物および/または血管新生阻害用医薬組 成物として有用である。 包装容器、 取扱説明書、 添付文書等には、 スルホンアミ ド化合物と上記 (i) 〜 (V) から選択される少なくとも 1つの物質とを併用して 用いることを記載することができ、 また、 別々の物質を投与時に併用する形態ま たは混合物としての形態について、 用法、 用量などを記載することができる。 用 法、 用量は、 上記医薬組成物/キットの記載を参照して設定することができる。
さらに、 本発明には、 (i) 白金錯体物質、 (ii) DNA-topoisomerase I 阻害 物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 および (V) 抗生物質から なる群から選択される少なくとも一つの物質と組み合わせてなる医薬組成物の製 造のためのスルホンアミ ド化合物の使用も含まれる。 本発明の使用において、 上 記医薬組成物は、 癌治療用医薬組成物およびノまたは血管新生阻害用医薬組成物 として有用である。
また、 本発明には、 スルホンアミド化合物と、 (i) 白金錯体物質、 (ii) DNA- topoisomerase I阻害物質、 (iii) 代謝拮抗物質、 (iv) 微小管阻害物質、 およ び (V) 抗生物質からなる群から選択される少なくとも一つの物質とを、 同時ま たは別々に患者に投与する癌の治療方法および/または血管新生阻害方法をも含 むものである。 本発明の癌の治療方法およぴ Zまたは血管新生阻害方法において、 スルホンアミ ド化合物および上記 (i) 〜 (V) から選択される少なくとも一つの 物質の投与経路および投与方法は特に限定されないが、 上記本発明の医薬組成物 の記載を参照することができる。 以下に、 具体的な例をもって本発明を示すが、 本発明はこれに限られるもので はない。 実施例 1 血管内皮細胞増殖アツセィ (in vitro) における細胞増殖に対す る E7820 と抗癌剤 Paclitaxel、 SN38 (CPT-11 活性体) 、 Methotrexate, Cisplatin、 Gemcitabine、 Doxorubicin との併用効果
ヒ ト臍帯静脈内皮細胞を、 培養培地としてブレツトキット EGM-2 (Cambrex) に懸濁し、 1 X 104 cells/mlに調製して、 100 μ 1の本溶液を 96 well plateの各 well に加え、 37°C下、 5 %炭酸ガスインキュベータ一にて培養した。 翌日、 E7820を含む溶液、 併用抗癌剤を含む溶液並びに E7820および併用抗癌剤の両 化合物を含む溶液を、 培養培地にてそれぞれ希釈した。 そして、 当該希釈液を前 記培養中の各 wellに 100 μ 1 /well加えて培養を続けた。
3 日後、 Cell Counting Kit_8溶液 (Cell Counting Kit_8、 和光純薬) 10 μ 1 を添加し、 37°C下で 2〜3 時間培養後、 プレートリーダー (コロナ電気株式会 社) によって 450 nm の吸光度を測定した。 併用効果は、 Chou ら(Adv. Enzyme Regul., 22, 27.55, 1984)の計算式に従い算出した。
その結果、 E7820 と Paclitaxel、 SN38 (CPT' ll 活性体) 、 Methotrexate Cisplatin , Gemcitabine または Doxorubicin との組み合わせは、 E7820、 Paclitaxel、 SN38 ( CPT-11 活十生体) 、 Methotrexate 、 Cisplatin、 Gemcitabineまたは Doxorubicin単独に比べて、 より強い細胞増殖抑制作用を 示 した。 ま た、 E7820 と Paclitaxel、 SN38 ( CPT-11 活性体) 、 Methotrexate, Cisplatin, Gemcitabine には Doxorubicin とを併用しに場 合の combination index ( CI ) 力 S 1以下となったこと力 ら、 E7820 は、 Paclitaxel、 SN38 ( CPT-11 活性体) 、 Methotrexate、 Cisplatin、 Gemcitabineまたは Doxorubicin と併用することにより細胞增殖抑制に対する 相乗効果 (Synergistic) を示すことが明らかになった (表 3 ) 。 当該効果は、 併用により一般的に認められる効果に比べて、 顕著な効果であり、 当業者にとつ てまったく予想し得ないものであった。
表 3

24 ゥエルプレート (FALCON社製) に type I collagen gel (新田ゼラチ ン) 400 μ 1を分注し、 37°C、 C02インキュベーターに 40 minおいてゲル化し た。 20 ng/ml EGF ( GIBCO BRL 社製) を含む無血清培地 (Human endothelial-SFM Basal Growth Medium, インビトロジェン社製) を調製し、 この溶液 200 を type I collagen gelの入った各 wellに分注した。 ヒ ト臍帯
静脈内皮細胞株 (HUVEC) を、 無血清培地 (Human endothelial-SFM Basal Growth Medium, GIBCO BRL社製) に懸濁し、 5 x 10^ cells/ml細胞懸濁液 を調製した。 この懸濁液 200 1を type I collagen gelおよび無血清培地の入つ た各 wellにまきこみ一晩培養した。
翌日、 type I collagen gel 400 ^ 1を重層して 37°C、 CO2インキュベーターに 3 時間置いてゲル化させた後、 E7820、 併用抗癌剤並びに E7820 と併用抗癌剤 の両方を含む無血清培地 (10 ng/ml EGFおよび 20 ng/ml VEGF (Genzyme Techne Corp.) を含む) を 1.5 mlを加え、 更に 37°C、 CO2インキュベーター にて 3日間培養した。
培養後、 MTT (SIGMA社製) 3.3 mg/ml溶液 400 μ ΐを各 wellに分注し、 37 °C、 CO2インキュベータ一にて 3 時間反応させて細胞を発色させた。 HUVECが形成する管腔の画像を顕微鏡下 (SZX12、 OLYMPUS社製) で取り 込んだ (M-3204C、 OLYMPUS 社製) 。 取り込んだ画像を画像解析ソフ ト Mac SCOPE 2,56 (MITANI社製) を用いて管腔の長さを測定することにより HUVEC の管腔形成を定量した。 併用効果は Chou ら (Adv. Enzyme ReguL, 22, 27-55, 1984)の計算式に従い算出した。
その結果、 E7820 と Paclitaxel、 SN38 ( CPT- 11 活性体) 、 Cisplatin、 Gemcitabine または Doxorubicin との糸且み合わせは、 E7820、 Paclitaxel, SN38 (CPT-11活性体) 、 Cisplatin、 Gemcitabineまたは Doxorubicin単独 に比べて、 より強い管腔形成抑制作用を示した。 また、 E7820 と Paclitaxel、 SN38 (CPT-11活性体) 、 Cisplatin、 Gemcitabineまたは Doxorubicin とを 併用した場合の combination index (CI) が 1 ·以下となったことから、 E7320 は、 Paclitaxel、 SN38 (CPT-11 活性体) 、 Cisplatin、 Gemcitabine または Doxorubicin と併用するこ と によ り 管腔形成抑制に対する相乗効果 (Synergistic) を示すことが明らかになった (表 4 ) 。 当該効果は、 併用によ り一般的に認められる効果に比べて、 顕著な効果であり、 当業者にとってまった く予想し得ないものであった。
表 4

以上の結果から、 E7820と白金錯体物質、 DNA-topoisomerase I阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択される少なく とも一つの物質とを組み合わせることにより、 優れた血管新生阻害活性を示す医 薬組成物およびキット、 ならびに血管新生の阻害方法が提供され、 本発明の医薬 組成物、 キットおよび方法は、 癌の治療および血管新生の阻害に用いることが可 能となった。 実施例 3 ヒ ト大腸癌細胞株 (Colo320DM) 皮下移植モデル (in vivo) に おける E7820と Oxaliplatinとの併用
ヒ ト大腸癌細胞株 Colo320DM (大日本製薬より購入) を 37°C下、 5%炭酸ガ スインキュベーター内において RPMI1640 (10% FBS含) で約 80%コンフル ェントとなるまで培養し、 トリプシン一 EDTA により、 細胞を回収した。 50% マトリゲル含有リン酸緩衝液で、 7.5xl07cells/mL懸濁液を調製し、 得られた細 胞懸濁液を 0.1 mLずつヌードマウス体側皮下に移植した。 移植後 6日目より、 E7820 (50 mg/kg、 1 日 2 回 · 2 週間 ·経口投与) 、 Oxaliplatin (Eloxatin (登録商標) 、 Sanofi Aventis社) (10 mg/kg, 4日に 1回、 計 3回、 静脈内 投与) の単独投与あるいは併用投与を開始した。 腫瘍長径 ·短径をデジマチック キヤリパ (Mitsutoyo) で測定し、 以下の式で腫瘍体積、 比腫瘍体積を算出した。 腫瘍体積 TV=腫瘍長径 (mm) X腫瘍短径 2 (mm2) /2
比腫瘍体積 RTV=測定日の腫瘍体積/投与開始日の腫瘍体積
併用群において、 two-way ANOVA で統計的有意な相互作用が認められた場 合、 E7820と Oxaliplatinとの間に相乗効果を有すると判定した。
その結果、 E7820は、 Oxaliplatinと併用することにより、 相乗効果が認めら れ、 E7820 または Oxaliplatin単独の効果に比べ、 すぐれた抗腫瘍効果を示し た (表 5および図 1 ) 。 また、 E7820は、 Oxaliplatinと併用することにより、
Oxalip latin単独では示すことができないような優れた抗腫瘍効果が認められた (表 5および図 1 ) 。
表 5

以上の結果から、 E7820 と Oxaliplatin とを組み合わせることにより、 すぐ れた抗腫瘍活性を示す医薬組成物およびキット、 ならびに癌の治療方法が提供さ れ、 本発明の医薬組成物、 キットおよび方法は癌の治療に用いることが可能とな つ 7こ。 実施例 4 ヒ ト大腸癌細胞株 (Colo320DM) 皮下移植モデル (in vivo) .に おける E7820と CPT- 11との併用
ヒ ト大腸癌細胞株 Colo320DM (大日本製薬より購入) を 37°C下、 5%炭酸ガ スインキュベーター内において RPMI1640 (10% FBS含) で約 80%コンフル ェントとなるまで培養し、 トリプシン一 EDTA により、 細胞を回収した。 50% マトリゲル含有リン酸緩衝液で、 7.5xl07cells/mL懸濁液を調製し、 得られた細 胞懸濁液を 0.1 mLずつヌードマウス体側皮下に移楠した。 移植後 6日目より、 E7820 (50 mg/kg, 1 日 2回、 2週間、 経口投与) 、 CPT-11 (トポテシン (登 録商標) 、 第一製薬) (100 mg/kg, 4 日に 1回、 計 3回、 静脈内投与) の単独
投与あるいは併用投与を開始した。 腫瘍長径 ·短径をデジマチックキヤリパ
(Mistsutoyo) で測定し、 以下の式で腫瘍体積、 比腫瘍体積を算出した。
艟瘍体積 TV=腫瘍長径 (mm) X腫瘍短径 2 (mm2) 12
比腫瘍体積 RTV=測定日の腫瘍体積 Z投与開始日の腫瘍体積
併用群において、 two-way ANOVAで統計的有意な相互作用が認められた場 合、 E7820と CPT-11との間に相乗効果を有すると判定した。
その結果、 E7820 は、 CPT-11 と併用することにより、 相乗効果が認められ、 E7820 または CPT-11 単独の効果に比べ、 すぐれた抗腫瘍効果を示した (表 6 および図 2 ) 。 また、 E7820は、 CPT-11と併用することにより、 CPT-11単独 では示すことができないような優れた抗腫瘍効果が認められた (表 6および図 2 ) 。
表 6

以上の結果から、 E7820 と CPT-11 とを組み合わせることにより、 すぐれた 抗腫瘍活性を示す医薬組成物およびキット、 ならびに癌の治療方法が提供され、 本発明の医薬組成物、 キットおよび方法は癌の治療に用いることが可能となった。
実施例 5 ヒ ト鸱癌細胞株 (KP-1) 皮下移植モデル (in vivo) における E7820と Gemcitabineとの併用
t ト膝癌細胞株 KP- 1 (九州がんセンターより入手) を 37°C下、 5%炭酸ガス インキュベーター内において RPMI1640 (10% FBS含) で約 80%コンフルェ ントとなるまで培養し、 トリプシン一 EDTA により、 細胞を回収した。 リン酸 緩衝液で、 lxlO8 cells/mL懸濁液を調製し、 得られた細胞懸濁液を 0.1 mLず つヌードマウス体側皮下に移植した。 移植後 11 日目より、 E7820 (50 mg/kg, 1 日 2回、 3週間、 経口投与) 、 および Gemcitabine (GEMZAR (登録商標) 、 日本ィーライリ リー社) (200 mg/kg、 3日に 1回、 計 4回、 静脈内投与) の単 独投与あるいは併用投与を開始した。 腫瘍長径 '短径をデジマチックキヤリバ (Mitsutoyo) で測定し、 以下の式で腫瘍体積、 比腫瘍体積を算出した。
腫瘍体積 TV=腫瘍長径 (mm) X腫瘍短径 2 (mm2) 12
比腫瘍体積 RTV=測定日の腫瘍体積 Z投与開始日の腫瘍体積
併用群において、 two-way ANOVA で統計的有意な相互作用が認められた場 合、 E7820と Gemcitabine との間に相乗効果を有すると判定した。
その結果、 E7820 は、 Gemcitabine と併用することにより、 相乗効果が認め られ、 E7820または Gemcitabine単独の効果に比べ、 すぐれた抗腫瘍効果を示 した (表 7およぴ図 3 ) 。 また、 E7820 は、 Gemcitabine と併用することによ り、 Gemcitabine 単独では示すことができないような優れた抗腫瘍効果が認め られた (表 7および図 3 ) 。
表 7
Day22における比腫瘍体積 Two-way 検体投与 平均土標準偏差 ANOVA コントロール (無処置) 14.8 ±2.28
E7820 50 mg/kg 8.81 ±2.57
Gemcitabine 200 mg/kg 4.74± 1.50
E7820 50 mg/kg p < 0.01
0.96 ± 0.31
+ Gemcitabine 200 mg/kg 相乗効果
表 7は、 KP-1 ヌードマウス皮下移植モデルにおける、 E7820、 Gemcitabine および E7820 と Gemcitabine との組み合わせによる抗腫瘍効果を示す。 投与 開始日を D ay 1とした。
以上の結果から、 E7820と Gemcitabine とを組み合わせることにより、 すぐ れた抗腫瘍活性を示す医薬組成物およびキット、 ならびに癌の治療方法が提供さ れ、 本発明の医薬組成物、 キットおょぴ方法は癌の治療に用いることが可能とな ' つた。 実施例 6 DNA-topoisomerase II mRNAの定量
ヒト臍帯静脈内皮細胞 2 X 106個を 25 cm2細胞培養用ボトルに蒔き込んだ後、 EGM-2培地 (三光純薬) を用いて 37°C下、 C02インキュベータ一にてー晚培 養した。 翌日、 E7820を 1 Μになるように添加し、 6時間後に細胞から RNA を調製した。 すなわち、 培地を除去した細胞に ISOGEN (和光純薬) 3 ml を加 えて溶解し、 等量の CHC13を加えて攪拌後、 水相を抽出した。 次に、 半量の isopropanol を加えて 5 分間静置後、 遠心によって沈殿を回収した。 沈殿を 70%エタノールで洗浄後、 滅菌水を加えて溶解し、 分光光度計により 260 nm の吸光度を測定することにより RNA量を定量した。
次に、 TaqMan Gold RT PCR kit (アプライド バイオシステムズ社製) を 用いて RT-PCR反応を行った。 すなわち、 RNA 0.1 / gを反応液 50 / lへ加え、 25°C 10分間、 48°C 30分間、 95°C 5分間の反応を行い、 cDNAを調製した。 次 に、 DNA-topoisomerase II mRNA測定用プライマー (ABI Taqman probe Hs00172214ml) により PCR反応を行い、 ABI7700 (アプライド バイオシ ステムズ社製) によって RNA量を定量した。
その結果、 DNA-topoisomerase II mRNA量は、 未処理群では 4.4/z g/mlで あつたのに対し、. E7820処理群では 1.6 μ g/mlであった。
ところで、 抗癌剤による DNA-topoisomerase I 阻害により、 腫瘍における DNA-topoisomerase II が上昇する こ とが報告されている ( Kim R,
Hirabayashi N, Nishiyama M, et al. Experimental studies on biochemical modulation targeting topoisomerase I and II in human tumor xenografts in nude mice. Int J Cancer. 1992; 50: 760-6., Whitacre CM, Zborowska E, Gordon NH, et al. Topotecan increases to oisomerase Ilalpha levels and sensitivity to treatment with etoposide in schedule -dependent process. Cancer Res. 1997; 57: 1425-8.) 。 このため、 E7820 ίま、 DNA-topoisomerase IIの発現阻害作用に基づき、 DNA-topoisomerase I阻害剤との組み合わせによ り相乗的な抗腫瘍効果を示すと考える。
したがって、 スルホンアミ ド化合物は、 CPT- 11のみならず、 その他の DNA- topoisomerase I阻害剤との組み合わせにより相乗的な抗腫瘍効果を示すことが 強く示唆された。 実施例 7 DNAマイクロアレイ解析
( 1 ) 細胞培養、 化合物処理、 および RNAの抽出
化合物により誘導される遺伝子発現変化を DNAマイクロアレイ解析によって 調べる目的で、 ヒ ト大腸癌由来細胞株 HCT116 (American Type Culture Collection, Manassas, VA, U.S.A. ) およびヒ ト白血病由来細胞株 MOLT-4 (American Type Culture Collection, Manassas, VA, U.S.A.) を、 10 %の胎 児牛血清、 100 units/ml のペニシリン、 100 μ g/ml のス トレプトマイシンを添 加した RPMI-1640培地中で培養した。 以下、 培養および化合物処理は 5%CO2、 37°Cに調擎されたインキュベーター内で行った。 10 cm径の細胞培養ディッシ ュに 2.0X106細胞 ディッシュの割合で HCT116細胞および MOLT-4細胞を蒔 き、 24時間培養後に以下の化合物処理を行った。
HCT116 細胞に関しては、 E7820 (0·8 μ Μ)、 Ε7070 (0.8 μ Μ)、 LY295501 (30 μ Μ)、 CQS (8 μ Μ)ヽ adriamycin (0.2 μ Μ)、 daunomycin (0.2 μ Μ)、 ICRF154 (80 μ Μ) 、 ICRF159 (80 μ Μ) 、 kenpaullone (10 μ Μ) 、 alsterpuUone (10 μ Μ)、 trichostatin A (0.1 μ Μ) rapamycin (80 Μ)の 12 化合物を評価した。 一方、 MOLT-4細胞に関しては、 Ε7070 (0.8 μ Μ)を評価し
た。 ここで、 adriamycinおよぴ daunomycin は、 DNAにインターカレーショ ンする型の DNA-topoisonie se II 阻害剤、 ICRF154 および ICRF159 は、 catalvtic tvoe の DNA-toOOisomerase II 阻 · i 、 Kenpaullone わ.よび aisterpullone Γ3~、 cyclm-dependent kinases (CDKs) fe舌斉 (J、 tncnostatm A は、 istone deacetylase阻害剤、 rapamycinは、 mTOR/FRAP阻害剤として、 それぞれ公知の化合物である。 化合物処理濃度は、 各々の化合物の HCT116 細 胞に対する 50%増殖阻害濃度 (WST-8を用いた 3日間の細胞増殖抑制活性に基' づく) を基準にその 3〜5倍の濃度として設定し、 上記化合物名に続く括弧内に 示した設定濃度で 24 時間処理後に細胞を回収した。 また、 化合物を加えずに 24時間培養した細胞も同様に回収した。
回収した細胞からの全 RNA の抽出は、 TRIZOL試薬 (インビトロジヱン社 製) を用いて添付の操作法に従って行った。
( 2 ) DNAマイクロアレイによる遺伝子発現解析
得られた RNAを 100 / Iの diethylpyrocarbonate (DEPC) 処理をした滅菌 水に溶解し、 さ らに RNeasy カラム (QIAGEN ) を用いて精製し、 Superscript Choice System (インビトロジェン社製) および T7_d(T)24プライ マーを用いて 2.本鎖の cDNAを合成した。
まず 10 ^ g の RNA に 5 μ Μ の T7-d(T)24プライマー、 lx First strand buffer、 10 mM DTT、 500 μ M の dNTP mix、 および 20 units/ μ 1 の Superscript II Reverse Transcriptase を加え、 42°Cにて 1時間反応させ、 1 本鎖 DNAを合成した。 続いて、 lx Second strand buffer, 200 Mの dNTP mix, 67 U/ml DNA ligase, 270 U/ml DNA polymerase I、 および 13 U/ml RNase Hを添加して、 16°Cにて 2時間反応させ 2本鎖 cDNAを合成した。 さ らに、 67 U/ml T4 DNA polymerase Iを添加して、 16°Cにて 5分間反応させた 後、 10 z lの 0.5 M EDTAを加え反応を停止した。
得られた cDNA をフエノール/クロ口ホルムにて精製し、 RNA Transcript Labeling Kit (Enzo Diagnostics) を用レ、、 添付の操作法に従って、 ビォチン ィ匕 UTPならびに CTPによるラベル化反応を行った。 反応生成物を HNeasy力
ラムにて精製後、 200 mM トリス酢酸 pH8.1、 150 mM酢酸マグネシウム、 50 mM酢酸力リゥム中で 94°Cにて 35分間加熱して cRNAを断片化した。
断片化した cRNAを、 100 mM MES、 1 M sodium salt, 20 mM EDTA、 0.01% Tween 20 中、 45。Cにて 16 時間、 GeneChip (Affymetrix) Human Focus array にハイブリダィズさせた。 ハイブリダイズ後、 GeneChi は Affymetrix fluidics stationに添付のプロトコール Midi_euk2に従い洗浄およ ぴ染色した。 染色にはストレプトアビジン-フィコエリ トリンとビォチン化抗ス トレプトアビジンャギ抗体を用いた。 染色後の GeneChip を HP アルゴンィォ ンレーザー共焦点顕微鏡 (Hewlett Packard) を用いてスキャンし、 蛍光強度 を測定した。 測定は、 488 nm の波長で excitation を行い、 570 nm の波長の emissionで frつた。
定量的データ解析は全て GeneChip software (Affymetrix) ならびに Gene Spring (Silicongenetics) を用!/、て frつ 7こ。 GeneChip softwareを用レヽてィ匕合 物による遺伝子発現変化を評価する際には、 化合物処理群と未処理群の 2つの条 件間で RNAの定量値が 2倍以上解離している場合につき、 その遺伝子の発現が 有意に 「増加」 あるいは 「減少」 したと判断した。 Gene Spring を用いて、.各 化合物が誘導する遺伝子発現変化の類似性を評価する際には、 Human Focus Array に載っている全遺伝子の発現変化をもとに階層的クラスターリング解析 を行った。
HCT116細胞の階層的クラスターリング解析の結果を図 4に示した。
解析の转果、 同一の作用機序を有する adriamycin および daunomycir ICRF154および ICRF159、 Kenpaulloneおよび alsterpullone は、 それぞれ 類似の遺伝子変化を引き起こした (図 4 ) 。 よって、 同一の作用機序を有する化 合物が、 互いに類似の遺伝子変化を引き起こすことが確認された。
E7070、 E7820、 LY295501および CQSは、 類似の遺伝子変化を引き起こし た (図 4 ) 。 よって、 本解析により、 E7070、 E7820、 LY295501 および CQS は、 同一または類似の作用機序を有すると考えられ、 同一または類似の遺伝子変 化おょぴ効果をもたらすことが強く示唆された。
実施例 8 DNAマイクロアレイ解析
HCT116 細胞を、 10 %の胎児牛血清、 100 units/ml のペニシリン、 100 g/ml のストレプトマイシンを添加した RPMI- 1640培地中で培養した。 以下、 培養および化合物処理は、 5%C02、 37°Cに調整されたインキュベーター内で行 つた。 10 cm 径の細胞培養ディッシュに 2.0X106細胞 Zディッシュの割合で HCT116細胞を蒔き、 24時間培養後に以下の化合物処理を行った。
本実施例では HCT116細胞を用いて、 E 7820 (0.16 / M) , E 7070 (0.26 μ Μ)、 LY186641 (59 μ Μ)、 LY295501 (24 μ Μ)、 LY-573636 (9.6 μ Μ) , CQS (4.0 μ Μ)、 MST16 (100 μ Μ) , etoposide (3.6 μ Μ)、 ethoxzolamide (410 μ Μ)、 capsaicin (280 μ Μ)ヽ trichostatin Α (0. 16 μ Μ)、 kenpaullone (7.1 μ Μ)の 12 化合物で処理したときの遺伝子発現変化を調べた。
ここで、 MST16 は、 catalytic type の DNA-topoisomerase II 阻害剤、 etoposideは cleavable comple の形成を誘導する .DNA-topoisomerase II阻害 斉 U、 ethoxzolamide は carbonic anhydrase ]¾■著剤、 capsaicin は tumor- specific plasma membrane NADH oxidase阻 斉 [J、 trichostatin Aは histone deacetylase阻害斉 U、 kenpaulloneは cyclin-dependent kinases (CDKs)P且著斉 Ij として、 それぞれ公知の化合物である。
化合物処理濃度は、 各々の化合物の HCT116細胞に対する 50%増殖阻害濃度 (MTTを用いた 3日間の細胞増殖抑制活性に基づく) を基準に、 その 2倍の濃 度として ^定した。 上記化合物名に続く括弧内に示した設定濃度で 24時間処理 後に細胞を回収した。 また、 化合物を加えずに 24時間培養した細胞も同様に回 収した。
回収した細胞からの全 RNAの抽出は、 TRIZOL試薬 (インビトロジェン社 製) を用いて添付の操作法に従って行った。
DNA マイクロアレイによる遺伝子発現解析は、 実施例 7中の 「 (2 ) DNA マイクロアレイによる遺伝子発現解析」 と同様に行つた。
また、 本実施例は、 各サンプルについて duplicateで行った (実験の便宜上、 それぞれのサンプルは区別できるように control-l、 conti'ol_2、 E7070_ l、 E7070-2 の要領で枝番号を付した) 。 そして、 GeneChip ( Affymetrix ) system (Human Focus array)を用いて各化合物の誘導する遺伝子発現変化を解 祈した。
本実施例で得られた 26個 (control+ 12化合物の 13サンプル X2) の cel」 フアイルに対し RMA法 (robust multi-array average法(Biostatistics(2003), 4, 249-264)) を適用し、 プローブレベルでの正規分布化を行った後、 遺伝子レ ベルでのシグナル強度のログ値を算出した。 続いて、 各遺伝子の化合物処理群に おけるシグナル強度のログ値から化合物未処理群 (control-1) におけるシグナ ル強度のログ値を引き、 control-1 に対する化合物処理群のシグナル比のログ値 を得た。 そして、 コサイン相関係数を計算し、 実験間の相関係数とした (図 5 )。 この相関係数をもとに、 UPGMA法 (Unweighted Pair Group Method with Arithmetic mean法) により階層的クラスターリング解析した(図 6 )。 control- 2 についても、 同様の計算を行った(図 7および図 8 )。 使用したソフトウェアは R, 2.0. l(http ://www. r-project.org/) 、 affy package 1.5.8(http:〃 www. bioconductor.org)である。
図 5〜8において、 「LYlj は LY186641 を、 「LY2」 は LY295501 を、 「 LY5」 は LY573636 を、 「 CAI」 は ethoxzolamide を、 「 Cap」 は capsaicin を、 「MST」 は MST16 を、 「Etop」 は etoposide を、 「TSA」 は trichosta.tin Aを、 「Kenp」 は kenpaulkmeを示す。 図 6およぴ図 8において、
「 de hclust (*, "average")」 は、 統計角军析を行う時のコマン ドであり 、 dupulicate の実験データの平均値を用いて Rによるクラスターリング分析を行 つたことを示す。
解析の結果、 E7070、 E7820、 LY186641、 LY295501、 LY573636 および CQS は、 HCT_116 細胞に対して引き起こす遺伝子変化は非常に高い類似性を示 し、 他のどのィ匕合物 ( MST16、 etoposide、 ethoxzolamide、 capsaicin、 trichostatin A、 kenpaullone) のプロフアイノレとも異なることが明らかとなつ
た (図 5〜図 8 ) 。 よって、 本解析により、 E7070、 E7820、 LY186641 , LY295501, LY573636および CQSは、 同一または類似の作用機序を有すると 考えられ、 同一または類似の遺伝子変化およぴ効果をもたらすことが強く示唆さ れた。 実施例 9 癌細胞株パネル実験
36株のヒ ト癌細胞パネルを用いて、 E7820、 E7070、 CQS、 LY186641 , LY295501 の細胞増殖抑制活性の相関を調べた。 用いた癌細胞株は、 DLD-1, HCT15, HCT116, HT29, SW480, SW620, WiDr (以上、 ヒ ト大腸癌細胞株) 、 A427, A549, LX-1, NCI-H460, NCI-H522, PC-9, PC-10 (以上、 ヒ ト肺癌細胞 株) 、 GT3TKB, HGC27, MKN1, MKN7, MKN28, MKN74 (以上、 ヒ ト胃癌 細胞株) 、 AsPC-1, KP-1, KP-4, MiaPaCall, PANC-1, SUIT-2 (以上、 ヒ ト 陴臓癌細胞株) 、 BSY-1, HBC5, MCF-7, MDA-MB-231, MDA-MB-435, MDA-MB-468 (以上、 ヒ ト乳癌細胞株) 、 CCRF-CEM, HL60, K562, MOLT- 4 (以上、 ヒト白血病細胞株) の 3 6種類であり、 全ての細胞は 10%の胎児牛血 清、 100 units/ml のぺニシリン、 100 g/ml のス トレプトマイシンを添加した RPMI-1640培地を用いて 5% CO2条件下 37 °Cにて培養した (表 8 ) 。
表 8
36 human cancer cell lines tested in 3-day MTT assays
Colon Stomach Breast
DLD-1 (1250/weIl, 16.8 h) GT3TKB (2000/well, 21.1 h) BSY-l (2000/well, 46.1 h)
HCT15 (1500/well, 14.5 h) HGC27 (1500/well, 14.6 h) HBC5 (2000/well, 31.8 h)
HCT116 (1250/weII, 13.4 h) MKN1 (4000/well, 35.9 h) MCF-7 (3000/well, 29.5 h)
HT29 (2500/well, 19.8 h) MKN7 (3000/weII, 37.4 h) MDA-MB231 (2000/well, 21.6 h) SW480 (3000/weII, 19.5 h) MKN28 (2000/well, 22.7 h) MDA-MB-435 (3000/well, 24.4 h) SW620 (2500/well, 17.3 h) MK 74 (4000/well, 24.8 h) MDA-MB-468 (3000/well, 34.2 h) WiDr (2000/well, 18.9 h)
Pancreas Leukemia
Lung AsPC-1 (2500/well, 28.4 h) CCRF-CEM (1500/well, 27.2 h)
A427 (2500/well, 32.4 h) KP-1 (2000/well, 24.8 h) HL60 (1500/well, 29.5 h)
A549 (1250/well, 18.9 h) KP-4 (2000/well, 16.7 h) K562 (1500/well, 20.6 h)
LX-1 (2000/well, 17.2 h) MiaPaCall (2500/well, 19.1 h) MOLT-4 (1500/well, 22.3 h) NCI-H460 (1000/weIl, 13.6 h) PANC-1 (2500/well, 27.9 h)
NCI-H522 (4000/well, 80.4 h) SUIT-2 (2000/well, 15.6 h)
PC-9 (2000/well, 23.7 h)
PC-10 (2000/well, 24.0 h)
Cell line (initial cell number, doubling time)
表 8は、 ヒ ト癌細胞株パネルにおけるヒト癌細胞株の種類、 蒔きこみ細胞数お よび倍化時間を示す。
表 8に記載の細胞数で 96 ウェルマイク口プレート (平底) に蒔き (50 z 1/well) 、 24時間後に 3倍希釈系列の化合物を添加した (δθ μ ΐ/well) 。 さらに 72時間鋒に WST-8 (10 / 1/well) を添加し、 450 nm の吸光度を測定した。'最 小二乗法により全 36株の癌細胞に対する 50 %増殖抑制阻害濃度を求め、 その パターンを各化合物間で比較した。 相関の指標と しては、 Pearson's correlation coefficientsを用レヽた (Paull, K. D. et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088-1092; Monks, A. et al. Feasibility of a high-flux
anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757-766.) 。
その結果、 E7070, E7820, LY186641, LY295501および CQSは、 各癌細 胞株に対する増殖抑制活性において、 高い相関係数を示した (表 9 ) 。 よって、 本解析により、 E7070、 E7820、 LY186641、 LY295501および CQSは、 同一 または類似の作用機序を有すると考えられ、 同一または類似の遺伝子変化および 効果をもたらすことが強く示唆された。
表 9
E7070 E7820 CQS LY186641 LY295501
E7070 1.00 0.98 0.97 0.93 0.80
E7820 0.98 1.00 0.96 0.95 0.82
CQS 0.97 0.96 1.00 0.92 0.82
LY186641 0.93 0.95 0.92 1.00 0.81
LY295501 0.80 0.82 0.82 0.81 1.00 表 9は、 ヒ ト癌細胞株パネルにおける化合物間 (E7070、 E7820、 CQS , LY186641および LY295501) の相関係数を示す。 実施例 1 0 E7070耐性株における交差耐性
E7070耐性株を用レヽて、 E7820, LY186641, LY295501, LY-ASAPおよび CQS の細胞増殖抑制活性を評価した。 HCT116-C9 は、 ヒ ト大腸癌由来 HCT116 (American Type Culture Collection, Manassas, VA, U.S.A.) から 分離した亜株であり、 この HCT116-C9を E7070存在下で培養し、 E7070濃度 を漸次的 (こ上昇させることにより得た E7070耐性亜株が HCT116-C9-C1およ ぴ HCT116-C9-C4 である (Molecular Cancer Therapeutics, 2002, 1, 275- 286) o
HCT116-C9 , HCT116-C9-C1 , HCT116-C9-C4 の 3 細胞株を各々 3000 cells/wellで 96 ウェルマイク口プレート (平底) に蒔き (SO ^ l/well) 、 24時 間後に 3倍希^系列の化合物を添加した (50 i l/Well) 。 さらに、 72 時間後に MTT法 (Mossmann T., J. Immunol. Methods, 1983, 65, 55-63) により細胞
増殖抑制活性を評価した。 最小二乗法により各癌細胞に対する 50 %増殖抑制阻 害濃度を求めた。
その結果、 E7070の細胞増殖抑制活性は、 HCT116-C9 (C9) に対して IC50 は 0.127 μ M であった。 これに対し、 HCT116-C9-C1 ( C9C1 ) および HCT116-C9-C4 (C9C4) に対する活性はそれぞれ IC50 = 31.9μ Μ および 26.9 μ Μであり、 Ε7070の C9C1および C9C4に対する細胞増殖抑制活性が顕 著に低下することが確認された (図 9) 。 また、 E7820、 CQS、 LY186641、 LY295501, LY-ASAP の細胞増殖抑制活性については、 HCT116-C9 に対する 活性がそれぞれ IC50 = 0.080 μΜ、 1.73 Μ, 33.6 Μ、 10·9μΜ、 1.63 μΜ であったのに対し、 HCT116-C9-C1および HCT116-C9-C4 に対する活性は、 HCT116-C9-C1 について、 それぞれ IC50 = 51.2/ Μ, 634 /ζΜ、 134 Μ、 111μΜ、 113 Μであり、 HCT116-C9-C4について、 それぞれ IC50 = 52·8/ζ M、 517/iM、 138;uM、 110/ M、 90.3/ M であった。 したがって、 E7820、 CQS、 LY186641、 LY295501, LY-ASAPの細胞増殖抑制活性については、 C9 に対する活性に比べ、 C9C1 および C9C4 に対する活性が顕著に低下していた (図 9) 。 よって、 E7070、 E7820、 LY186641、 LY295501, LY-ASAP およ び CQS は、 同一または類似の作用機序を有すると考えられ、 同一または類似の 遺伝子変化および効果をもたらすことが強く示唆された。 実施例 1 1 E7070耐性株における交差耐性
実施例 10と全く同様にして、 E7070耐性株を用いて LY573636の細胞増殖 抑制活性を E7070と同時に評価した。
その結果、 E7070 の細胞増殖抑制活性は、 HCT116-C9 に対する活性に比べ (IC50 = 0.127 M) 、 HCT116-C9-C1および HCT116-C9-C4に対する活性 (それぞれ IC50 = 32.7 μΜ、 28.0 ^Μ) が顕著に低下することが再度確認され た (図 10) 。 また、 LY573636の細胞増殖抑制活性も、 HCT116-C9に対する 活性に比べ (IC50 = 5.11μΜ) 、 HCT116-C9-C1および HCT116-C9-C4に対 する活性 (それぞれ IC50 = 264 Μ、 240 μΜ) が顕著に低下していた (図 1
0 ) 。 よって、 LY573636 は、 E7070 と同一または類似の作用機序を有すると 考えられ、 同一または類似の遺伝子変化および効果をもたらすことが強く示唆さ れた。
これらの結果 (実施例 7— 1 1 ) 力 ら、 E7070、 E7820、 LY186641、 LY295501、 LY-ASAP、 LY573636 もしくは CQS またはこれらの組み合わせ 力 同一または類似の遺伝子変化ならびに同一または類似の作用および効果をも たらすことが明らかとなった。
よって、 E7820 と同様に (実施例 1一 6 ) 、 スルホンアミ ド化合物、 好まし くは E7070、 LY186641, LY295501, LY_ASAP、 LY573636 もしくは CQS またはこれらの組み合わせが、 (i) 白金錯体物質、 好ましくは Oxaliplatin ま たは Cisplatin、 (ii) DNA-topoisomerase I阻害物質、 好ましくは CPT-11、 (iii) 代謝拮抗物質、 好ましくは Gemcitabine または Methotrexate、 (iv) 微小管阻害物質、 好ましくは Paclitaxel、 および (v) 抗生物質、 好ましくは Doxorubicin からなる群から選択される少なくとも一つの化合物と併用するこ とにより、 すぐれた抗腫瘍活性および血管新生阻害活性を示すことが明らかにな つた。 産業上の利用可能性
本発明により、 すぐれた抗腫瘍活性および/または血管新生阻害活性を示す医 薬組成物およびキット、 ならびに癌の治療方法および/または血管新生阻害方法 が提供される。
より具体的には、 スルホンアミ ド化合物、 すなわち (A) E7820、 (B) 一般 式 (I) で表される化合物、 好ましくは LY186641または LY295501、 (C) 一 般式 (II) で表される化合物、 好ましくは LY-ASAP、 (D) LY573636 および (E) CQS から選択される少なくとも一つの化合物と、 (i) 白金錯体物質、 好 ましくは Oxaliplatinまたは Cisplatin、 (ii) DNA-topoisomerase I阻害物質、 好ましくは CPT-11、 (iii) 代謝拮抗物質、 好ましくは Gemcitabine または Methotrexate, (iv) 微小管阻害物質、 好ましくは Paclitaxel、 および (v) 抗
生物質、 好ましくは Doxorubicin からなる群から選択される少なくとも一つの 物質とを組み合わせることにより、 すぐれた抗腫瘍活性および Zまたは血管新生 阻害活性を示す医薬組成物およびキット、 ならびに癌の治療方法および Zまたは 血管新生阻害方法が提供される。 本発明の医薬組成物、 キットおよび方法は、 癌 の治療または血管新生の阻害に有用である。
Claims
請求の範囲 . N— ( 3—シァノー 4—メチルー 1 H—インドールー 7—ィル) 一 3—シァ ノベンゼンスルホンアミ ド、 もしくはその薬理学的に許容される塩、 また はそれらの溶媒和物と、 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択される 少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物とを組み合わせてなる医薬組成物。
. 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻 害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin, CPT- 11、 uemcitabine、 Methotrexate、 Paciitaxelおよび Doxorubicin力 り る群 である、 請求項 1に記載の医薬組成物。
. スルホンアミ ド化合物と、 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択される 少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物とを組み合わせてなる医薬組成物であって、
前記スルホンアミ ド化合物が、
一般式 (I)

で表わされる化合物、
一般式 (II)

[式中、 Jは、 一 O—または一 NH—を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有していてもよい 一 C6アルキル基、 置換基を有していてもよい C 1一 C4アルコキシ基、 置換基を有していてもよい Ci一 C4アルキルチオ基、 ― C F3、 _OCF3、 一 S CF3、 置換基を有していてもよい Ci—C4アルコキ シカルボ-ル基、 ニトロ基、 アジド基、 一 O (S02) CH3、 一 N (CH3) 2、 水酸基、 フエ二ル基、 置換基を有するフエニル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ノヽ ロゲン原子、 シァノ基、 一 CF3、 置換基を有していてもよい Ci_C6アルキ ル基、 置換基を有していてもよい d— C4アルコキシカルボニル基、 置換基を 有していてもよい Ci— C4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci—C アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい Ci—Csアルキル基 (但し、 R3bおよび R4bの少な く とも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい Ci—C6アルキル基、 一C F3または-トロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい C]_—C6アルキ ル基 (但し、 Rebが置換基を有していてもよい Ci—C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい Ci一 C6アルキル基または一 C F3 (伹し、 R5bま たは R7bのいずれか一方が、 置換基を有していてもよい Ci— C6アルキル基で ある力 、 あるいは R?b力 ハロゲン原子または置換基を有していてもよい Ci 一 C6アルキル基である場合には、 R5bまたは R6bのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物および
式 (IV)
(IV)

4 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻 害物質および抗生物質からなる群が、 Oxaliplatin, Cisplatin, CPT-11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 らなな群 である、 請求項 3に記載の医薬組成物。
5 . スルホンアミ ド化合物が、 N— [ [ ( 4ークロロフヱ-ル) ァミノ] カルボ ニル] 一 2 , 3—ジヒ ドロー 1 H—ィンデン一 5—スルホンァミ ド、 N—
[ [ ( 3, 4ージクロロフヱ-ル) ァミノ] カルボニル] _ 2, 3—ジヒ ドロべンゾフラン一 5—スルホンアミ ド、 N— (2 , 4—ジクロ口べンゾ ィノレ) 一 4—クロ口フエニノレスノレホンアミ ド、 N— (2, 4ージクロ口べ ンゾィノレ) 一 5—プロモチォフェン一 2—スノレホンアミ ドおよび 2—スル 'ファニルアミ ドー 5—クロ口キノキサリンからなる群から選択される少な く とも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそれ らの溶媒和物である、 請求項 3または 4に記載の医薬組成物。
6 . スルホンアミ ド化合物が、 N— [ [ ( 3 , 4—ジクロロフエニル) ァミノ] カノレボニ Λ^] — 2 , 3—ジヒ ドロべンゾフラン一 5—スノレホンアミ ドおよ び Ν— ( 2 , 4—ジクロ口べンゾィノレ) 一 5—ブロモチォフェン一 2—ス
ルホンアミ ドからなる群から選択される少なくとも一つの化合物、 もしく はその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請求項
3または 4に記載の医薬組成物。
スルホンアミ ド化合物が、 N— ( 2 , 4ージクロ口べンゾィル) 一 5—ブロ モチォフェン一 2—スルホンァミ ドのナトリ ゥム塩である、 請求項 3また は 4に記載の医薬組成物。
医薬組成物が、 癌治療用医薬組成物である、 請求項 1〜 7のいずれか一項に 記載の医薬組成物。
医薬組成物が、 血管新生阻害用医薬組成物である、 請求項 1〜 7のいずれか 一項に記載の医薬組成物。
. (a) N— ( 3—シァノ一 4ーメチルー 1 H—インドール一 7—ィル) 一 3—シァノベンゼンスルホンアミ ド、 もしくはその薬理学的に許容される 塩、 またはそれらの溶媒和物と、 白金錯体物質、 DNA-topoisomerase I 阻 害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選 択される少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物とを併用することを記載した、 包装容器、 取搀説 明書および添付文書からなる群から選択される少なくとも一つと、
(b) N— ( 3一シァノ一 4—メチルー 1 H—インドール一 7—ィル) - 3 - シァノベンゼンスルホンアミ ド、 もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物を含む医薬組成物と、
を含有するキット。
. 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT_11、 Gemcitabine、 Methotrexate ^ Paclitaxelぉょぴ Doxorubicin力 らなるお羊 である、 請求項 1 0に記載のキット。
. (a) ス レホンアミ ド化合物と、 白金錯体物質、 D A-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から 選択される少なくとも一つの物質、 もしくはその薬理学的に許容される塩、
またはそれらの溶媒和物とを併用することを記載した、 包装容器、 明書および添付文書からなる群から選択される少なくとも一つと、
(b) スルホンアミ ド化合物を含む医薬組成物と、
を含有するキットであって、
前記スルホンアミ ド化合物が、
一般式 (I)

[式中、 Eは、 一 O—、 一 N (CH3) ―、 一CH2—、 一 CH2CH2—または一 CH20—を、 Dは、 _CH2—または—〇一を、 は、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 (II) '

[式中、 Jは、 一 0—または一 NH—を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有していてもよい Ci一 C6アルキル基、 置換基を有していてもよい C ェ— C4アルコキシ基、 置換基を有していてもよい Ci— C4アルキルチオ基、 一
CF3、 一 OCF3、 一 SCF3、 置換基を有していてもよい Ci一 C4アルコキ シカルボ二ノレ基 トロ基、 アジド基、 一O (S02) CH3、 N (CH3) 2、 水酸基、 フエニル基、 置換基を有するフエ-ル基、 ピリジニル基、 チェ-ル 基、 フリル基.、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ハ ロゲン原子、 シァノ基、 —CF3、 置換基を有していてもよい d—Ceアルキ ル基、 置換基を有していてもよい d— C4アルコキシカルボ-ル基、 置換基を
有していてもよい Ci一 C4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci一 C4アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい 一 C6アルキル基 (但し、 R3bおよび R4bの少な くとも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい C —CGアルキル基、 一 C F3またはニトロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい Ci一 C6アルキ ル基 (但し、 R6bが置換基を有していてもよい Ci一 C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい Ci— C6アルキル基または一 C F3 (但し、 R5bま たは R?bのいずれか一方が、 置換基を有していてもよい d—C6アルキル基で ある力 \ あるレ、は R7b力 S、 ハロゲン原子または置換基を有していてもよい d
_C6アルキル基である場合には、 R5bまたは RSbのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物および
式 (IV).
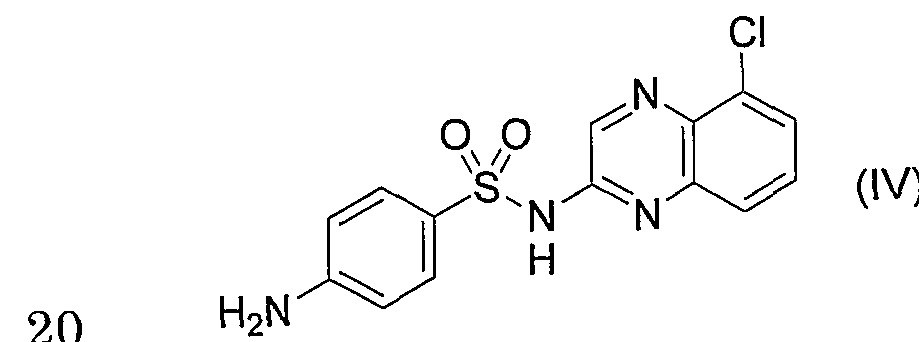
で表される化.合物からなる群から選択される少なく とも一つの化合物、 もし くはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 前記キ ッ卜。
1 3. 白金錯体物質、 DNA-topoisomei'ase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT_11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 り る群 である、 請求項 1 2に記載のキット。
14. スルホンァミ ド化合物が、 N— [ [ (4一クロロブヱニル) ァミノ] カル ボニル] — 2, 3—ジヒ ドロー 1 H—インデン一 5—スルホンアミ ド、 N 一 [ [ (3, 4—ジクロ口フエニル) ァミノ] カルボニル] — 2, 3—ジ ヒ ドロべンゾフラン一 5—スノレホンアミ ド、 N— (2, 4—ジクロ口ベン ゾィノレ) 一 4—クロ口 フエニノレスノレホンア ミ ド、 N— (2, 4—ジクロ ロ ベンゾィノレ) 一 5—ブロモチォフェン一 2—スノレホンアミ ドおよび 2—ス ルファニルアミ ドー 5—クロ口キノキサリンからなる群から選択される少 なく とも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物である、 請求項 1 2または 1 3に記載のキット。
1 5. スルホンアミ ド化合物が、 N— [ [ (3, 4—ジクロロフヱニル) アミ ノ] カルボニル] — 2, 3—ジヒ ドロべンゾフラン一 5—スルホンアミ ド およぴ N— (2, 4—ジクロロべンゾィノレ) 一 5—ブロモチォフェン一 2 ースルホンアミ ドからなる群から選択される少なくとも一つの化合物、 も しくはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請 求項 1 2または 1 3に記載のキット。
16. スノレホンアミ ド化合物が、 N— (2, 4—ジクロロべンゾィノレ) 一 5—ブ ロモチオフヱン一 2—スルホンァミ ドのナトリゥム.塩である、 請求項 1 2 または 1 3に記載のキット。
1 7. キットが、 癌治療用キットである、 請求項 1 0〜1 6のいずれか一項に記 載のキット。
1 8. キットが、 血管新生阻害用キットである、 請求項 1 0〜1 6のいずれか一 項に記載の.キット。
1 9. N— ( 3—シァノ一 4—メチルー 1 H—インドール一 7—ィル) 一 3—シ ァノベンゼンスルホンアミ ド、 もしくはその薬理学的に許容される塩、 ま
たはそれらの溶媒和物を含んでなる製剤と、 白金錯体物質、 DNA- 般 topoisomerase I阻害物質、 代謝拮抗物質、 微小管阻害物質および抗生物質 式.
からなる群から選択される少なくとも一つの物質、 もしくはその薬理学的
I
ノ 、 }
に許容される塩、 またはそれらの溶媒和物を含んでなる製剤とをセットに したことを特徴とするキット。
2 0 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT_11、 Gemcitabine、 Methotrexate、 Paclitaxelおよぴ Doxorubicin力51りなる群 である、 請求項 1 9に記載のキット。
2 1 . スルホンアミ ド化合物を含んでなる製剤と、 白金錯体物質、 DNA- topoisomerase I阻害物質、 代謝拮抗物質、 微小管阻害物質おょぴ抗生物質 からなる群から選択される少なく とも一つの物質、 もしくはその薬理学的 に許容される塩、 またはそれらの溶媒和物を含んでなる製剤とをセットに したことを特徴とするキットであって、
前記スルホンアミ ド化合物が、

[式中、. Eは、 一 O—、 一N ( C H3) ―、 一 C H2_、 一 C H2 C H2—または一 C H20 _を、 Dは、 一 C H2—または一 O—を、 R laは、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 (II) .

[式中、 Jは、 一〇一または一 NH—を、 は、 水素原子、 ハロゲン原子、 置換基を有していてもよい Ci一 C6アルキル基、 置換基を有していてもよい C 1一 C4アルコキシ基、 置換基を有していてもよい Cl_ C4アルキルチオ基、 一 CF3、 一〇C F3、 一 S CF3、 置換基を有していてもよい Ci_C4アルコキ シカルボニル基、 ニトロ基、 アジド基、 一 O (S02) CH3、 一 N (CH3) 2、 水酸基、 フエニル基、 置換基を有するフエ二ル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ハ ロゲン原子、 シァノ基、 一CF3、 置換基を有していてもよい Ci一 C6アルキ ル基、 置換基を有していてもよい Ci一 C4アルコキシカルボニル基、 置換基を 有していてもよい Ci一 C4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリ -ル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci一 C4アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい Ci—C6アルキル基 (但し、 R3bおよび R4bの少な くとも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい Ci—C6アルキル基、 — C F3またはニトロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい Ci一 C6アルキ ル基 (伹し、 R6bが置換基を有していてもよい d—Ceアルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい d—Csアルキル基または一 CF3 (但し、 R5bま たは R?bのいずれか一方が、 置換基を有していてもよい d— C6アルキル基で ある力 \ あるいは R7b力 ハロゲン原子または置換基を有していてもよい d 一 C6アルキル基である場合には、 R5bまたは RGbのいずれか一方が、 水素原 子である) を.それぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物および
式 (IV)
(IV)

2 2 . 白金錯体物質、 DNA'topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin, CPT-11、 Gemcitabine、 Methotrexate、 Paclitaxelおよぴ Doxorubicin力 らなる群 である、 請求項 2 1に記載のキット。
2 3 . スルホンアミ ド化合物が、 N— [ [ ( 4—クロロフヱニル) ァミノ] カル ボニノレ] 一 2, 3—ジヒ ドロー 1 H—インデンー 5—スノレホンアミ ド、 N 一 [ [ ( 3 , 4—ジクロロフヱニル) ァミノ] カルボ-ル] 一 2 , 3—ジ ヒ ドロべンゾフラン一 5—スノレホンアミ ド、 N— ( 2, 4ージクロ口ベン ゾイ レ) 一 4一クロ口フエニノレスノレホンアミ ド、 N— (2 , 4—ジクロ口 ベンゾィノレ) 一 5—ブロモチォフェン一 2—スノレホンアミ ドおよび 2—ス ルファ -ルァミ ドー 5—クロ口キノキサリンからなる群から選択される少 なく とも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物である、 請求項 2 1または 2 2に記載のキット。
2 4 . スルホ アミ ド化合物が、 N— [ [ ( 3, 4ージクロ口フエニル) アミ ノ] カノレポ二 Λ^] — 2, 3—ジヒ ドロべンゾフラン一 5—スルホンアミ ド および Ν— (2, 4—ジクロ口べンゾィル) 一 5—プロモチォフェン一 2
ースルホンアミ ドからなる群から選択される少なく とも一つの化合物、 も しくはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請 求項 2 1または 2 2に記載のキット。
5 · スルホンアミ ド化合物が、 N— ( 2 , 4—ジクロロべンゾィル) — 5—ブ 口モチォフェン一 2ースルホンァミ ドのナトリゥム塩である、 請求項 2 1 または 2 2に記載のキット。
6 . キットが、 癌治療用キットである、 請求項 1 9〜2 5のいずれか 1項に記 載のキット。
7 . キットが、 血管新生阻害用キットである、 請求項 1 9〜2 5のいずれか 1 項に記載のキット。
8 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群から選択される少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物と組み合 わせてなる医薬組成物の製造のための N— ( 3—シァノ一 4ーメチルー 1 H—イン ドーノレ一 7—ィノレ) 一 3—シァノベンゼンスルホンアミ ド、 もし くはその薬理学的に許容される塩、 またはそれらの溶媒和物の使用。 . 9 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質おょぴ抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT' 11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 らなる群 である、 請求項 2 8に記載の使用。
0 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群から選択される少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそれらの溶媒和物と組み合 わせてなる医薬組成物の製造のためのスルホンアミ ド化合物の使用であつ て、
前記スルホン.アミ ド化合物が、
一般式 (I)

[式中、 Eは、 一 O—、 _N (CH3) 一、 一CH2—、 一 CH2CH2—または— CH20—を、 Dは、 一 CH2—または一〇一を、 Rlaは、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 (II)

[式中、 Jは、 一 O—または一 NH—を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有していてもよい Ci—C6アルキル基、 置換基を有して.いてもよい C i—C4アルコキシ基、 置換基を有していてもよい Ci一 C4アルキルチオ基、. ― CF3、 _OCF3、 一 S CF3、 置換基を有していてもよい Ci一 C4アルコキ シカルボニル基、 ニトロ基、 アジド基、 一 O (S02) CH3、 一 N (CH3) 2、 水酸基、 フエニル基、 置換基を有するフエニル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ハ ロゲン原子、 シァノ基、 一 C F3、 置換基を有していてもよい Ci_C6アルキ ル基、 置換基を有していてもよい d—C4アルコキシカルボ二ル基、 置換基を 有していてもよい Ci— C4アルコキシ基、 置換基を有していてもよいフエ-ル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci—C4アルコキシ基を、 R4bは、 水素原子または 置換基を有レていてもよい Ci—C6アルキル基 (但し、 R3bおよび R4bの少な く とも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい Ci—C6アルキル基、 _C F3またはニトロ基を、 R6b
は、 水素原子、 ハロゲン原子または置換基を有していてもよい d—Ceアルキ ル基 (但し、 R6bが置換基を有していてもよい d—C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい Ci—Ceアルキル基または一 CF3 (但し、 R5bま たは R7bのいずれか一方が、 置換基を有していてもよい Ci_C6アルキル基で ある力 あるいは R7b力 ハロゲン原子または置換基を有していてもよい d 一 C6アルキル基である場合には、 R5bまたは RGbのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物およぴ
式 (IV)

3 1. 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質おょぴ抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT-11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 らなる; e である、 請求項 30に記載の使用。
32. スルホンアミ ド化合物が、 N— [ [ (4—クロロフヱ-ル) ァミノ] カル ボニル] — 2, 3—ジヒ ドロ— 1 H—インデン一 5—スルホンアミ ド、 N
— [ [ ( 3, 4—ジクロロフエニル) ァミノ] カルボニル] — 2 , 3—ジ ヒ ドロべンゾフラン一 5—スノレホンアミ ド、 N— ( 2, 4ージクロ口ベン ゾィノレ) 一 4—クロ口フエニノレスノレホンアミ ド、 N— ( 2 , 4—ジクロロ ベンゾィノレ) 一 5—プロモチォフェン一 2—スルホンアミ ドおよび 2—ス ルファニルアミ ドー 5—クロ口キノキサリンからなる群から選択される少 なくとも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物である、 請求項 3 0または 3 1に記載の使用。
3 3 . スルホンァミ ド化合物が、 N— [ [ ( 3, 4ージクロ口フエニル) 了ミ ノ] カノレボニノレ] — 2, 3—ジヒ ドロべンゾフラン一 5—スノレホンア ミ ド および N— ( 2 , 4—ジクロ ロべンゾィノレ) 一 5—プロモチォフェン一 2 ースルホンァミ ドからなる群から選択される少なくとも一つの化合物、 も しくはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請 求項 3 0または 3 1に記載の使用。
3 4 . スルホンアミ ド化合物が、 N— ( 2, 4—ジクロロべンゾィル) —5—ブ 口モチォフェン一 2—スルホンァミ ドのナトリゥム塩である、 請求項 3 0 または 3 1に記載の使用。
3 5 . 医薬組成物が、 癌治療用医薬組成物である、 請求項 2 8〜3 4のいずれか 一項に記載の使用。
3 6 . 医薬組成物が、 血管新生阻害用医薬組成物である、 請求項 2 8〜3 4のい ずれか一項に記載の使用。
3 7 . N - ( 3—シァノー 4—メチル一 1 H—インドール一 7—ィル) —3—シ ァノベンゼンスルホンアミ ド、 もしくはその薬理学的に許容される塩、 ま たはそれらの溶媒和物と、 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管阻害物質およぴ抗生物質からなる群から選択される 少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物とを患者に投与することを特徴とする癌の治療方法おょぴ
/または血管新生の阻害方法。
38. 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT-11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 らなる?^ である、 請求項 3 7に記載の方法。
39. スルホンアミ ド化合物と、 白金錯体物質、 DNA-topoisomerase I 阻害物 質、 代謝拮抗物質、 微小管阻害物質および抗生物質からなる群から選択さ れる少なくとも一つの物質、 もしくはその薬理学的に許容される塩、 また' はそれらの溶媒和物とを患者に投与することを特徴とする癌の治療方法お よび zまたは血管新生の阻害方法であって、
前記スルホンアミ ド化合物が、
一般式 (I)

[式中、 Eは、 一 O—、 一 N (CH3) 一、 _CH2—、 _CH2CH2_または一 CH20—を、 Dは、 一 CH2—または _〇一を、 Riaは、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 .(Π)

[式中、 Jは、 一 O—または一NH—を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有し Tいてもよい Ci一 C6アルキル基、 置換基を有していてもよい C i—C4アルコキシ基、 置換基を有していてもよい Ci—C4アルキルチオ基、 一 CF3、 一 OCF3、 —S CF3、 置換基を有していてもよい d— C4アルコキ
シカルボニル基、 ニトロ基、 アジド基、 — O (S02) CH3、 -N (CH3) 2、 水酸基、 フ ニル基、 置換基を有するフエニル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ノヽ ロゲン原子、 シァノ基、 一 CF3、 置換基を有していてもよい Ci一 C6アルキ ル基、 置換基を有していてもよい Ci一 C4アルコキシカルボニル基、 置換基を 有していてもよい d—C 4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci—C4アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい Ci—Ceアルキル基 (但し、 R3bおよび R4bの少な くとも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい d—C6アルキル基、 一 C F3またはニトロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい d—Csアルキ ル基 (但し、 R6bが置換基を有していてもよい Ci— C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい 一 C6アルキル基または一CF3 (但し、 R5bま たは R?bのいずれか一方が、 置換基を有していてもよい Ci一 C6アルキル基で あるカ、 あるいは R?b力 ハロゲン原子または置換基を有していてもよい d —C6アルキル基である場合には、 R5bまたは RGbのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物および
式 (IV)
 で表される化合物からなる群から選択される少なくとも一つの化合物、 もし くはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 前記方 法。
で表される化合物からなる群から選択される少なくとも一つの化合物、 もし くはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 前記方 法。
4 0 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin, CPT_11、 emcitabine、 Methotrexate ^ Paclitaxelおよび Doxorubicin力 り 7 る;^ である、 請求項 3 9に記載の方法。
4 1 . スルホンアミ ド化合物が、 N— [ [ ( 4 _クロ口フエニル) ァミノ] カル ボニル] —2 , 3—ジヒ ドロ _ 1 H—インデン一 5—スルホンアミ ド、 N
- [ [ ( 3 , 4ージクロ口フエニル) ァミノ] カルボニル] — 2, 3—ジ ヒ ドロべンゾフラン一 5—スノレホンアミ ド、 N一 ( 2, 4ージク口口ベン ゾィノレ) 一 4一クロ口フエ-ノレスルホンアミ ド、 N— ( 2 , 4—ジクロ口 ベンゾィル) — 5—ブロモチォフェン一 2—スノレホンアミ ドおよび 2—ス ルファニルアミ ドー 5—クロ口キノキサリ ンからなる群から選択される少 なくとも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物である、 請求項 3 9または 4 0 3 6に記載の方法。
4 2 . スルホンアミ ド化合物が、 N— [ [ ( 3, 4ージクロ口フエニル) アミ ノ] カノレポニル] 一 2, 3—ジヒ ドロべンゾフラン一 5—スノレホンアミ ド および N— (2 , 4—ジクロ口べンゾィル) 一 5—ブロモチォフェン一 2
—スルホンアミ ドからなる群から選択される少なくとも一つの化合物、 も しくはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請 求項 3 9または 4 0に記載の方法。
4 3 . スルホンアミ ド化合物が、 N— (2, 4—ジクロロべンゾィル) ー5—ブ 口モチォフェン一 2—スルホンァミ ドのナトリゥム塩である、 請求項 3 9 または 4 0に記載の方法。
4 4 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群から選択される少なくとも一つの物質 とともに患者に併用投与するための、 N— ( 3—シァノ一 4—メチルー 1 H—インドールー 7 _ィル) 一 3—シァノベンゼンスルホンアミ ド、 もし くはその薬理学的に許容される塩、 またはそれらの溶媒和物を含む医薬組 成物。
4 5 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin, Cisplatin, CPT' 11、 Gemcitabine、 Methotrexate、 Paclitaxelおよび Doxorubicin力 りなる群 である、 請求項 4 4に記載の医薬組成物。
4 6 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群から選択される少なく とも一つの物質 とともに患者に併用投与するための、 スルホンアミ ド化合物を含む医薬.組 成物であって、
前記スルホンアミ ド化合物が、
一般式 (I)

[式中、 Eは、 一O—、 一 N ( C H3) 一、 一 C H2—、 _ C H2 C H2—または一 C H2〇—を、 Dは、 _ C H2—または一O—を、 R laは、 水素原子またはハロ ゲン原子を、 R2aは、 ハロゲン原子またはトリフルォロメチル基をそれぞれ意 味する。 ]
で表わされる化合物、
一般式 (ID

[式中、 Jは、 一 O—または— NH—を、 Ribは、 水素原子、 ハロゲン原子、 置換基を有していてもよい en一 C6アルキル基、 置換基を有していてもよい C 1一 C4アルコキシ基、 置換基を有していてもよい C1— C4アルキルチオ基、 一 CF3、 一 OC F3、 一 S CF3、 置換基を有していてもよい C1— C4アルコキ シカルボニル基、 ニトロ基、 アジド基、 一 O (S02) CH3、 一 N (CH3) 2、 水酸基、 フエニル基、 置換基を有するフエニル基、 ピリジニル基、 チェニル 基、 フリル基、 キノリニル基またはトリァゾール基を、 R2bは、 水素原子、 ノヽ ロゲン原子、 シァノ基、 _CF3、 置換基を有していてもよい d— C6アルキ ル基、 置換基を有していてもよい d_ C4アルコキシカルボニル基、 置換基を 有していてもよい Cl一 C 4アルコキシ基、 置換基を有していてもよいフエニル 基または置換基を有していてもよいキノリニル基を、 R3bは、 水素原子または 置換基を有していてもよい Ci—C4アルコキシ基を、 R4bは、 水素原子または 置換基を有していてもよい Ci_C6アルキル基 (但し、 R3bおよび R4bの少な くとも一つは、 水素原子である) を、 R5bは、 水素原子、 ハロゲン原子、 置換 基を有していてもよい d—Ceアルキル基、 — C F3またはニトロ基を、 R6b は、 水素原子、 ハロゲン原子または置換基を有していてもよい Ci—Csアルキ ル基 (伹し、 R6bが置換基を有していてもよい Ci—C6アルキル基のとき、 R 5bは水素原子であり、 R7bはハロゲン原子である) を、 R7bは、 ハロゲン原子、 置換基を有していてもよい Ci—Csアルキル基または一 CF3 (但し、 R5bま たは R?bのいずれか一方が、 置換基を有していてもよい d—Ceアルキル基で ある力、 あるいは R?bが、 ハロゲン原子または置換基を有していてもよい d —C6アルキル基である場合には、 R5bまたは RGbのいずれか一方が、 水素原 子である) をそれぞれ意味する。 ]
で表わされる化合物、
式 (III)

で表わされる化合物およぴ
式 (IV)

4 7 . 白金錯体物質、 DNA-topoisomerase I 阻害物質、 代謝拮抗物質、 微小管 阻害物質および抗生物質からなる群が、 Oxaliplatin、 Cisplatin、 CPT- 11、 Gemcitabine、 Methotrexate ^ Paclitaxelお び Doxorubicin力 らなる群 である、 請求項 4 6に記載の医薬組成物。
4 8 . スルホンァミ ド化合物が、 N— [ [ ( 4一クロ口フエニル) ァミノ] '力ノレ ボニル] 一 2, 3—ジヒ ドロー 1 H—インデン一 5—スルホンアミ ド、 N _ [ [ ( 3 , 4—ジクロロフエニル) ァミノ] カルボエル] — 2 , 3—ジ ヒ ドロべンゾフラン一 5—スノレホンアミ ド、 N _ ( 2 , 4—ジクロ口ベン ゾィノレ) 一 4—クロ口フエニノレスルホンアミ ド、 N— ( 2 , 4—ジクロ口 ベンゾィノレ) - 5—プロモチォフェン一 2—スノレホンァミ ドおよび 2—ス ルファニルァミ ドー 5一クロ口キノキサリ ンからなる群から選択される少 なくとも一つの化合物、 もしくはその薬理学的に許容される塩、 またはそ れらの溶媒和物である、 請求項 4 6または 4 7に記載の医薬組成物。
4 9 . スルホンァミ ド化合物が、 N— [ [ ( 3, 4ージクロ口フエニル) ァミ ノ] カルボニル] 一 2, 3—ジヒ ドロべンゾフラン一 5—スルホンアミ ド
および N— ( 2, 4ージクロ口べンゾィル) — 5—ブロモチォフェン一 2
—スルホンアミ ドからなる群から選択される少なくとも一つの化合物、 も しくはその薬理学的に許容される塩、 またはそれらの溶媒和物である、 請 求項 4 6または 4 7に記載の医薬組成物。
. スルホンアミ ド化合物が、 N— ( 2 , 4ージクロロべンゾィル) 一 5—ブ 口モチォフェン一 2—スルホンァミ ドのナトリゥム塩である、 請求項 4 6 または 4 7に記載の医薬組成物。
. 医薬組成物が、 癌治療用医薬組成物である、 請求項 4 4〜 5 0のいずれか 一項に記載の医薬組成物。
. 医薬組成物が、 血管新生阻害用医薬組成物である、 請求項 4 4〜 5 0のい ずれか一項に記載の医薬組成物。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007504858A JP5106098B2 (ja) | 2005-02-28 | 2006-02-28 | スルホンアミド化合物の抗癌剤との新規併用 |
| US11/885,129 US20090047365A1 (en) | 2005-02-28 | 2006-02-28 | Novel Concomitant Use of Sulfonamide Compound with Anti-Cancer Agent |
| EP06715262A EP1859797A4 (en) | 2005-02-28 | 2006-02-28 | NEW SIMULTANEOUS USE OF A SULPHONAMIDE COMPOUND AND A MEDIUM AGAINST CANCER |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005055132 | 2005-02-28 | ||
| JP2005-055132 | 2005-02-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006090931A1 true WO2006090931A1 (ja) | 2006-08-31 |
Family
ID=36927548
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/304219 WO2006090931A1 (ja) | 2005-02-28 | 2006-02-28 | スルホンアミド化合物の抗癌剤との新規併用 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20090047365A1 (ja) |
| EP (1) | EP1859797A4 (ja) |
| JP (1) | JP5106098B2 (ja) |
| WO (1) | WO2006090931A1 (ja) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008088088A1 (ja) * | 2007-01-19 | 2008-07-24 | Eisai R & D Management Co., Ltd. | 膵癌治療用組成物 |
| WO2009060945A1 (ja) * | 2007-11-09 | 2009-05-14 | Eisai R & D Management Co., Ltd. | 血管新生阻害物質と抗腫瘍性白金錯体との併用 |
| US7973160B2 (en) | 2000-10-20 | 2011-07-05 | Eisai R&D Management Co., Ltd. | Nitrogen-containing aromatic derivatives |
| JP2011522814A (ja) * | 2008-06-09 | 2011-08-04 | サイクラセル リミテッド | スパシタビン(cndac)と、デシタビン及びプロカイン等のdnaメチルトランスフェラーゼ阻害剤との組合せ |
| US7994159B2 (en) | 2003-03-10 | 2011-08-09 | Eisai R&D Management Co., Ltd. | c-Kit kinase inhibitor |
| US8058474B2 (en) | 2003-11-11 | 2011-11-15 | Eisai R&D Management Co., Ltd. | Urea derivative and process for preparing the same |
| US8865737B2 (en) | 2006-08-28 | 2014-10-21 | Eisai R&D Management Co., Ltd. | Antitumor agent for undifferentiated gastric cancer |
| US8962655B2 (en) | 2007-01-29 | 2015-02-24 | Eisai R&D Management Co., Ltd. | Composition for treatment of undifferentiated gastric cancer |
| US8962650B2 (en) | 2011-04-18 | 2015-02-24 | Eisai R&D Management Co., Ltd. | Therapeutic agent for tumor |
| US8969379B2 (en) | 2004-09-17 | 2015-03-03 | Eisai R&D Management Co., Ltd. | Pharmaceutical compositions of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7=methoxy-6-quinolinecarboxide |
| US8969344B2 (en) | 2005-08-02 | 2015-03-03 | Eisai R&D Management Co., Ltd. | Method for assay on the effect of vascularization inhibitor |
| US9006256B2 (en) | 2006-05-18 | 2015-04-14 | Eisai R&D Management Co., Ltd. | Antitumor agent for thyroid cancer |
| US9012458B2 (en) | 2010-06-25 | 2015-04-21 | Eisai R&D Management Co., Ltd. | Antitumor agent using compounds having kinase inhibitory effect in combination |
| US9334239B2 (en) | 2012-12-21 | 2016-05-10 | Eisai R&D Management Co., Ltd. | Amorphous form of quinoline derivative, and method for producing same |
| US9945862B2 (en) | 2011-06-03 | 2018-04-17 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| US10226478B2 (en) | 2011-04-14 | 2019-03-12 | Cyclacel Limited | Dosage regimen for sapacitabine and decitabine in combination for treating acute myeloid leukemia |
| US10259791B2 (en) | 2014-08-28 | 2019-04-16 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10517861B2 (en) | 2013-05-14 | 2019-12-31 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of endometrial cancer subjects to lenvatinib compounds |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0011903D0 (en) * | 2000-05-18 | 2000-07-05 | Astrazeneca Ab | Combination chemotherapy |
| US20100105031A1 (en) * | 2005-08-01 | 2010-04-29 | Esai R & D Management Co., Ltd. | Method for prediction of the efficacy of vascularization inhibitor |
| EP1938842A4 (en) * | 2005-09-01 | 2013-01-09 | Eisai R&D Man Co Ltd | METHOD FOR PRODUCING A PHARMACEUTICAL COMPOSITION COMPRISING IMPROVED CRASHING CHARACTERISTICS |
| WO2007052849A1 (ja) * | 2005-11-07 | 2007-05-10 | Eisai R & D Management Co., Ltd. | 血管新生阻害物質とc-kitキナーゼ阻害物質との併用 |
| US20090247576A1 (en) * | 2005-11-22 | 2009-10-01 | Eisai R & D Management Co., Ltd. | Anti-tumor agent for multiple myeloma |
| JPWO2008001956A1 (ja) * | 2006-06-29 | 2009-12-03 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 肝線維症治療剤 |
| US8173686B2 (en) | 2006-11-06 | 2012-05-08 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8178564B2 (en) | 2006-11-06 | 2012-05-15 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8168661B2 (en) * | 2006-11-06 | 2012-05-01 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| US8168662B1 (en) | 2006-11-06 | 2012-05-01 | Poniard Pharmaceuticals, Inc. | Use of picoplatin to treat colorectal cancer |
| KR20090109130A (ko) * | 2007-02-09 | 2009-10-19 | 포니아드 파마슈티칼즈, 인크. | 캡슐화된 피코플라틴 |
| US20110033528A1 (en) * | 2009-08-05 | 2011-02-10 | Poniard Pharmaceuticals, Inc. | Stabilized picoplatin oral dosage form |
| US20100260832A1 (en) * | 2007-06-27 | 2010-10-14 | Poniard Pharmaceuticals, Inc. | Combination therapy for ovarian cancer |
| TW200916094A (en) * | 2007-06-27 | 2009-04-16 | Poniard Pharmaceuticals Inc | Stabilized picoplatin dosage form |
| WO2009011861A1 (en) * | 2007-07-16 | 2009-01-22 | Poniard Pharmaceuticals, Inc. | Oral formulations for picoplatin |
| AU2009210098B2 (en) * | 2008-01-29 | 2013-06-13 | Eisai R & D Management Co., Ltd. | Combined use of angiogenesis inhibitor and taxane |
| US20110052580A1 (en) * | 2008-02-08 | 2011-03-03 | Poniard Pharmaceuticals, Inc. | Use of picoplatin and bevacizumab to treat colorectal cancer |
| BRPI1009448A2 (pt) | 2009-03-11 | 2016-03-01 | Ardea Biosciences Inc | combinações farmacêuticas compreendendo edea119/bay 869766 para o tratamento de cânceres específicos |
| WO2011009059A2 (en) * | 2009-07-17 | 2011-01-20 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Method of treating or preventing cancer |
| KR20230017829A (ko) * | 2020-05-27 | 2023-02-06 | 코어셉트 쎄라퓨틱스 인코포레이티드 | 글루코코르티코이드 수용체 조절제 렐라코릴란트 및 cyp2c8과 cyp3a4의 이중 기질인 파클리탁셀의 병용 투여 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002036117A1 (fr) * | 2000-10-31 | 2002-05-10 | Eisai Co., Ltd. | Compositions medicinales destinees a etre utilise de maniere concomitante comme agent anticancereux |
| JP2002533416A (ja) * | 1998-12-23 | 2002-10-08 | ジー.ディー.サール & カンパニー | 新形成の治療における組み合わせ治療としてのシクロオキシゲナーゼ−2インヒビターおよび一つまたはそれ以上の抗腫瘍薬の使用方法 |
| WO2003074045A1 (fr) * | 2002-03-05 | 2003-09-12 | Eisai Co., Ltd. | Agent antitumoral comprenant une combinaison d'un compose heterocyclique contenant un sulfamide et d'un inhibiteur d'angiogenese |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5004758A (en) * | 1987-12-01 | 1991-04-02 | Smithkline Beecham Corporation | Water soluble camptothecin analogs useful for inhibiting the growth of animal tumor cells |
| IT1273391B (it) * | 1994-03-31 | 1997-07-08 | Boehringer Mannheim Italia | Platino complessi cationici trinucleari ad attivita' antitumorale e composizioni farmaceutiche che li contengono |
| CZ288912B6 (cs) * | 1998-05-27 | 2001-09-12 | Lachema, A. S. | Komplex platiny v oxidačním čísle IV, způsob přípravy tohoto komplexu, tento komplex jako léčivo a farmaceutická kompozice tento komplex obsahující |
| PL346643A1 (en) * | 1998-08-25 | 2002-02-25 | Deutsches Krebsforsch | Medicament containing platinum complex compounds and the use thereof |
| ATE382048T1 (de) * | 2002-09-19 | 2008-01-15 | Schering Corp | Pyrazolopyrimdine als hemmstoffe cyclin abhängiger kinasen |
| US8772269B2 (en) * | 2004-09-13 | 2014-07-08 | Eisai R&D Management Co., Ltd. | Use of sulfonamide-including compounds in combination with angiogenesis inhibitors |
| EP2364699A1 (en) * | 2004-09-13 | 2011-09-14 | Eisai R&D Management Co., Ltd. | Joint use of sulfonamide based compound with angiogenesis inhibitor |
| PL1859793T3 (pl) * | 2005-02-28 | 2011-09-30 | Eisai R&D Man Co Ltd | Nowe połączone zastosowanie związku sulfonamidowego w leczeniu choroby nowotworowej |
-
2006
- 2006-02-28 EP EP06715262A patent/EP1859797A4/en not_active Withdrawn
- 2006-02-28 WO PCT/JP2006/304219 patent/WO2006090931A1/ja active Application Filing
- 2006-02-28 US US11/885,129 patent/US20090047365A1/en not_active Abandoned
- 2006-02-28 JP JP2007504858A patent/JP5106098B2/ja not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002533416A (ja) * | 1998-12-23 | 2002-10-08 | ジー.ディー.サール & カンパニー | 新形成の治療における組み合わせ治療としてのシクロオキシゲナーゼ−2インヒビターおよび一つまたはそれ以上の抗腫瘍薬の使用方法 |
| WO2002036117A1 (fr) * | 2000-10-31 | 2002-05-10 | Eisai Co., Ltd. | Compositions medicinales destinees a etre utilise de maniere concomitante comme agent anticancereux |
| WO2003074045A1 (fr) * | 2002-03-05 | 2003-09-12 | Eisai Co., Ltd. | Agent antitumoral comprenant une combinaison d'un compose heterocyclique contenant un sulfamide et d'un inhibiteur d'angiogenese |
Non-Patent Citations (4)
| Title |
|---|
| KIM R; HIRABAYASHI N; NISHIYAMA M ET AL.: "Experimental studies on biochemical modulation targeting topoisomerase I and II in human tumor xenografts in nude mice", INT J CANCER, vol. 50, 1992, pages 760 - 6 |
| MOLECULAR CANCER THERAPEUTICS, vol. 1, 2002, pages 275 - 286 |
| MOSSMANN T., J. IMMUNOL. METHODS, vol. 65, 1983, pages 55 - 63 |
| See also references of EP1859797A4 |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7973160B2 (en) | 2000-10-20 | 2011-07-05 | Eisai R&D Management Co., Ltd. | Nitrogen-containing aromatic derivatives |
| US8372981B2 (en) | 2000-10-20 | 2013-02-12 | Eisai R&D Management Co., Ltd. | Nitrogen-containing aromatic derivatives |
| US7994159B2 (en) | 2003-03-10 | 2011-08-09 | Eisai R&D Management Co., Ltd. | c-Kit kinase inhibitor |
| US8058474B2 (en) | 2003-11-11 | 2011-11-15 | Eisai R&D Management Co., Ltd. | Urea derivative and process for preparing the same |
| US8969379B2 (en) | 2004-09-17 | 2015-03-03 | Eisai R&D Management Co., Ltd. | Pharmaceutical compositions of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7=methoxy-6-quinolinecarboxide |
| US9504746B2 (en) | 2004-09-17 | 2016-11-29 | Eisai R&D Management Co., Ltd. | Pharmaceutical compositions of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide |
| US9006240B2 (en) | 2005-08-02 | 2015-04-14 | Eisai R&D Management Co., Ltd. | Method for assay on the effect of vascularization inhibitor |
| US8969344B2 (en) | 2005-08-02 | 2015-03-03 | Eisai R&D Management Co., Ltd. | Method for assay on the effect of vascularization inhibitor |
| US9006256B2 (en) | 2006-05-18 | 2015-04-14 | Eisai R&D Management Co., Ltd. | Antitumor agent for thyroid cancer |
| US8865737B2 (en) | 2006-08-28 | 2014-10-21 | Eisai R&D Management Co., Ltd. | Antitumor agent for undifferentiated gastric cancer |
| WO2008088088A1 (ja) * | 2007-01-19 | 2008-07-24 | Eisai R & D Management Co., Ltd. | 膵癌治療用組成物 |
| US8962655B2 (en) | 2007-01-29 | 2015-02-24 | Eisai R&D Management Co., Ltd. | Composition for treatment of undifferentiated gastric cancer |
| JP5638244B2 (ja) * | 2007-11-09 | 2014-12-10 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 血管新生阻害物質と抗腫瘍性白金錯体との併用 |
| US8952035B2 (en) | 2007-11-09 | 2015-02-10 | Eisai R&D Management Co., Ltd. | Combination of anti-angiogenic substance and anti-tumor platinum complex |
| WO2009060945A1 (ja) * | 2007-11-09 | 2009-05-14 | Eisai R & D Management Co., Ltd. | 血管新生阻害物質と抗腫瘍性白金錯体との併用 |
| US8975239B2 (en) | 2008-06-09 | 2015-03-10 | Cyclacel Limited | Combinations of sapacitabine or CNDAC with DNA methyltransferase inhibitors such as decitabine and procaine |
| JP2011522814A (ja) * | 2008-06-09 | 2011-08-04 | サイクラセル リミテッド | スパシタビン(cndac)と、デシタビン及びプロカイン等のdnaメチルトランスフェラーゼ阻害剤との組合せ |
| JP2015071637A (ja) * | 2008-06-09 | 2015-04-16 | サイクラセル リミテッド | スパシタビン(cndac)と、デシタビン及びプロカイン等のdnaメチルトランスフェラーゼ阻害剤との組合せ |
| US9012458B2 (en) | 2010-06-25 | 2015-04-21 | Eisai R&D Management Co., Ltd. | Antitumor agent using compounds having kinase inhibitory effect in combination |
| US10226478B2 (en) | 2011-04-14 | 2019-03-12 | Cyclacel Limited | Dosage regimen for sapacitabine and decitabine in combination for treating acute myeloid leukemia |
| US8962650B2 (en) | 2011-04-18 | 2015-02-24 | Eisai R&D Management Co., Ltd. | Therapeutic agent for tumor |
| US9945862B2 (en) | 2011-06-03 | 2018-04-17 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| US11598776B2 (en) | 2011-06-03 | 2023-03-07 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| US9334239B2 (en) | 2012-12-21 | 2016-05-10 | Eisai R&D Management Co., Ltd. | Amorphous form of quinoline derivative, and method for producing same |
| US10517861B2 (en) | 2013-05-14 | 2019-12-31 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of endometrial cancer subjects to lenvatinib compounds |
| US10259791B2 (en) | 2014-08-28 | 2019-04-16 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10407393B2 (en) | 2014-08-28 | 2019-09-10 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10822307B2 (en) | 2014-08-28 | 2020-11-03 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11186547B2 (en) | 2014-08-28 | 2021-11-30 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1859797A1 (en) | 2007-11-28 |
| JPWO2006090931A1 (ja) | 2008-07-24 |
| US20090047365A1 (en) | 2009-02-19 |
| JP5106098B2 (ja) | 2012-12-26 |
| EP1859797A4 (en) | 2011-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006090931A1 (ja) | スルホンアミド化合物の抗癌剤との新規併用 | |
| ES2362898T3 (es) | Nuevo uso combinado de un compuesto de sulfonamina en el tratamiento de cáncer. | |
| TWI335919B (en) | Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors | |
| Sharma et al. | Prevention of MDR development in leukemia cells by micelle-forming polymeric surfactant | |
| CN111053768A (zh) | 用于治疗黑素瘤的药物组合 | |
| WO2006090928A1 (ja) | スルホンアミド化合物の血管新生阻害物質との新規併用 | |
| KR20210072043A (ko) | 소분자 mdm 2 단백질 분해제 | |
| EP2338486A1 (en) | 3-(indolyl)- or 3-(azaindolyl)-4-arylmaleimide derivatives for use in the treatment of colon and gastric adenocarcinoma | |
| JP7183371B2 (ja) | 抗腫瘍剤、抗腫瘍効果増強剤及び抗腫瘍用キット | |
| US20080286282A1 (en) | Novel Use of Sulfonamide Compound in Combination with Angiogenesis Inhibitor | |
| TW200911272A (en) | Potentiation of cancer chemotherapy | |
| WO2019043176A2 (en) | HISTONE-DEACETYLASE INHIBITOR IN COMBINATION WITH ANTIMETABOLITE AGENT FOR CANCER THERAPY | |
| CN109310650A (zh) | 用于减少过表达c-myc的癌症中的c-myc的化合物 | |
| KR20170046193A (ko) | 감소된 brca2 활성을 갖는 암 대상자의 치료를 위한 sns-595의 사용 방법 | |
| Zhang et al. | Discovery of YH677 as a cancer stemness inhibitor that suppresses triple-negative breast cancer growth and metastasis by regulating the TGFβ signaling pathway | |
| KR20110131190A (ko) | 3 고리성 피라졸로피리미딘 유도체 | |
| Warmann et al. | MDR1 modulators improve the chemotherapy response of human hepatoblastoma to doxorubicin in vitro | |
| JP2012509935A (ja) | L1camの活性または発現を抑制する物質および抗癌剤を含む抗癌用組成物 | |
| JP2010513287A (ja) | 癌治療のための組成物及び方法 | |
| EP4351564A1 (en) | Combination mcl-1 inhibitors with anti-cancer agents | |
| CN115996716A (zh) | Eif4a抑制剂组合 | |
| US11045449B2 (en) | 3-(5-fluoroindolyl)-4-arylmaleimide compounds and their use in tumor treatment | |
| AU2010336256B2 (en) | Antitumor Agent or Postoperative Adjuvant Chemotherapeutic Agent for Hepatocellular Carcinoma Treatment | |
| WO2021092893A1 (zh) | 二芳醚类化合物在制备抗肿瘤药物中的应用 | |
| Zetschok et al. | OXINDOLE SYNTHESIS VIA ORGANOCATALYTIC REACTIONS WITH THIOESTER ENOLATES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007504858 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11885129 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006715262 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006715262 Country of ref document: EP |