WO2006078039A1 - L-amino acid producing microorganism and a method for producing l-amino acid - Google Patents
L-amino acid producing microorganism and a method for producing l-amino acid Download PDFInfo
- Publication number
- WO2006078039A1 WO2006078039A1 PCT/JP2006/301071 JP2006301071W WO2006078039A1 WO 2006078039 A1 WO2006078039 A1 WO 2006078039A1 JP 2006301071 W JP2006301071 W JP 2006301071W WO 2006078039 A1 WO2006078039 A1 WO 2006078039A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- strain
- amino acid
- bacterium
- type
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/24—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Enterobacteriaceae (F), e.g. Citrobacter, Serratia, Proteus, Providencia, Morganella, Yersinia
- C07K14/245—Escherichia (G)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/70—Vectors or expression systems specially adapted for E. coli
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
- C12P13/08—Lysine; Diaminopimelic acid; Threonine; Valine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
- C12P13/14—Glutamic acid; Glutamine
Definitions
- the present invention relates to the fermentation industry, and specifically to a method for producing an L-amino acid using a bacterium of the Enterobacteriaceae family, wherein the bacterium has been modified so as to not produce type I fimbrial adhesin protein.
- L-amino acids are employed as components of pharmaceuticals, animal feed additives, seasonings, and various other nutritive mixtures.
- L-amino acids are industrially produced by fermentation using L-amino-acid producing bacteria such as coryneform bacteria and bacteria of the genus Escherichia.
- L-amino-acid producing bacteria such as coryneform bacteria and bacteria of the genus Escherichia.
- artificially mutated strains of these bacteria that have been isolated from nature or transformants in which activity of an L-amino acid biosynthesis enzyme has been increased by genetic recombination have been employed as the L-amino acid producing bacteria.
- L-lysine In addition to methods of increasing the level of expression of enzymes which are L-amino acid biosynthesis pathways, another methods of increasing the ability to produce L-amino acids such as L-lysine have been developed. Such methods include improving energy efficiency by modifying the energy pathway (EP 1 170376 ), increasing the ability to produce nicotinamide adenine dinucleotide phosphoric acid by amplifying the nicotinamide nucleotide transhydrogenase gene (US 5,830,716), and inactivating genes related to flagella production (WO02/097089). Escherichia, Salmonella, and Bacillus bacteria have fimbriae.
- Fimbriae can be divided into types I to V which have no direct relation to sexual processes such as conjugation and gene transfer, and sexual fimbriae that are produced on the outer layers of donor bacteria and are essential to conjugate with recipient bacteria and to gene transfer (Shoji Mizushima, Kinichiro Miura, Bacterial Anatomy 129 (1979)).
- Type I fimbriae There are nine genes which contribute to the formation of type I fimbriae, and these genes form an operon.
- the fimH gene located downstream within the operon, encodes a type 1 fimbrial adhesin protein.
- Type I fimbrial adhesin protein does not contribute to the formation of fimbriae, but is known to specifically recognize the mannosylated proteins of the host.
- the capacity for bacterial aggregation can be enhanced by introducing an amino acid substitution into the gene which encodes the type I fimbrial adhesin protein (Mark A. Schenbri, Gunna Christiansen and Per Klemm: FimH-mediated autoaggregation of Escherichia coli: Molecular Microbiology (2001 ) 41 (6), 1419-1430).
- Objects of the present invention include enhancing the productivity of L-amino acid producing strains and providing a method for producing an L-amino acid.
- the above objects were achieved by finding that the bacterium which is modified so as to not produce type I fimbrial adhesion can improve L-amino acid productivity.
- Jt is a further object of the present invention to provide the bacterium as described above, wherein said bacterium has been modified so as to not produce type I fimbrial adhesin protein by inactivation of the gene encoding type I fimbrial adhesin protein on the chromosome.
- L-amino acid is selected from the group consisting of L-lysine, L-threonine, L-glutamic acid, and combinations thereof.
- It is a further object of the present invention to provide a method for producing an L-amino acid comprising cultivating the bacterium as described above in a medium, and collecting said L-amino acid from the medium.
- L-amino acid is selected from the group consisting of L-lysine, L-threonine, L-glutamic acid, and combinations thereof.
- Fig. 1 shows the structure of pM W l 18-attL-Tc-attR.
- the bacterium of the present invention is a bacterium having the ability to produce an L-amino acid and which belongs to the Enterobacteriaceae family.
- the bacterium of the present invention has been modified so as to not produce type I fimbrial adhesin protein.
- the ability to produce L-amino acids may include production of any single L-amino acid, or production of multiple L-amino acids.
- L-amino acids of the present invention include L-lysine, L-glutamic acid, L-threonine, L-valine, L-leucine, L-isoleucine, L-serine, L-aspartic acid, L-asparagine, L-glutamine, L-arginine, L-cysteine (cystine), L-methionine, L -phenylalanine, L-tryptophan, L-tyrosine, L-glycine, L-alanine, L-proline, L-ornithine, L-citrulline, and L-homoserine.
- L-lysine, L-threonine, and L-glutamic acid are particularly desirable.
- the phrase "ability to produce an L-amino acid” refers to the ability of a bacterium of the Enterobacteriaceae family to produce and cause accumulation of an L-amino acid in the bacterial cells or into a medium to a degree which permits its recovery from the bacterial cells or the medium when the bacterium is cultured in the medium.
- the bacterium which has this ability may originally have had the ability to produce an L-amino acid, or may be a bacterium, such as those set forth below, that has been modified by a mutation method or recombinant DNA technique to impart the ability to produce an L-amino acid.
- the bacterium of the present invention may also be one that has had the ability to produce an L-amino acid imparted to it by a modification which results in prevention of production of type I fimbrial adhesin production.
- the Enterobacteriaceae family includes bacteria belonging to the genus Escherichia, Enterobacter, Erwinia, Klebsiella, Pantoea, Photorhabdus, Povidencia, Salmonela, Serralia, Shigella, Morganella, and Yersinia, etc..
- NBCl National Center for Biotechnology Information
- the parent strain from the Enterobacteriaceae family employed for modification is desirably from the genus Escherichia, Enterobacter, or Pantoea.
- the parent strain from the genus Escherichia which is used to obtain the bacterium of the present invention is not specifically limited. Specifically, strains described by Neidhardt et al. may be employed (Neidhardt, F. C. et al., Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington D. C, 1029 table 1 ). Of these, one example is Escherichia coli. Specific examples of Escherichia coli are Escherichia coli W31 10 (ATCC 27325) and Escherichia coli MG 1655 (ATCC 47076), both of which are derived from the prototype wild-type strain K12.
- strains can be obtained from the American Type Culture Collection (P.O.Box 1549 Manassas, VA 20108 USA (703) 365-2700) using the assigned registration number (http://www.atcc.org (reference)). The registration number corresponding to each strain is listed at the American Type Culture Collection web site and catalog.
- Examples of strains from the genus Enterobacter are Enterobacter agglomerans and Enterobacter aerogenes.
- An example of a strain from the genus Pantoea is Pantoea ananatis.
- Enterobacter agglomerans has on occasion been reclassified as Panloeu agglomerans, Pantoea ananatis, or Pantoea stewartii. (lnt.J.Syst.Bacteriol.,43, 162-173)
- any bacterium classified in the Enterobacteriaceae family may be employed.
- Pantoea ananatis AJ 13355 (FERM BP-6614), AJ 13356 (FERM BP-6615), AJ 13601 (FERM BP-7207), or any derivative thereof may be employed to breed Pantoea ananatis by genetic engineering methods. When isolated, these strains were identified and deposited as Enterobacter agglomerans. As stated above, analysis by 16S rRNA base sequencing has caused them to be reclassified as Pantoea ananatis.
- an auxotrophic mutant, an analog-resistant strain, or a metabolic regulation mutant can be obtained, or a recombinant strain having enhanced expression of an L-amino acid biosynthetic enzyme can be created.
- methods that have been employed to breed coryneform bacteria and bacteria from the genus Escherichia can be used (See “Amino Acid Fermentation", the Japan Scientific Societies Press [Gakkai Shuppan Center], 1st Edition, published on May 30, 1986, pp.77-100).
- one or more properties such as an auxotrophic mutation, analog resistance, and a metabolic regulation mutation may be imparted.
- the activity of one or more L-amino acid biosynthetic enzymes may be enhanced.
- imparting properties such as an auxotrophic mutation, analog resistance, and a metabolic regulation mutation may be combined with the methods for enhancing an activity of one or more L-amino acid biosynthetic enzymes.
- An auxotrophic mutant strain, L-amino acid analog resistant strains, or metabolic regulation-mutated strain with the ability to produce an L-amino acid can be obtained by subjecting a parent strain or wild-type strain to a typical mutation treatment, such as exposure to X-rays or UV radiation, or treatment with a mutagenic agent such as N-methyI-N'-nitro-N-nitrosoguanidine(NTG), and selecting from among the obtained mutants those which exhibit an auxotrophic mutation, analog resistance, or a metabolic regulation-mutation and which also have the ability to produce an L-amino acid. Genetic recombination can be employed to enhance the L-amino acid biosynthetic enzyme activity of a bacterium which produces L-amino acids.
- L-lysine analog-resistant strains and metabolic regulation-mutated strains which are able to produce L-lysine include Escherichia coli strain AJ l 1442 (FERM BP- 1543, NRRL B-12185; see JP56-18596A and U.S. 4,346, 170) and Escherichia coli strain VL61 1 (JP 2000-189180A).
- Strain WC 196 (see WO96/ 17930) may also be employed as an Escherichia coli L-lysine producing strain.
- Strain WC 196 was originally bred by imparting resistance to S-(2-aminoethyl)cysteine (AEC) to strain W31 10, which is derived from Escherichia coli K-12.
- AEC S-(2-aminoethyl)cysteine
- the resulting strain was also designated as Escherichia coli strain AJ 13069 and deposited on December 6, 1994, accession number PERM P-14690, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1-1 -1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566). Then, the deposit was converted to an international deposit under the provisions of the Budapest Treaty on September 29, 1995, and received an accession number of FERM BP-5252.
- genes encoding proteins involved in L-lysine biosynthesis include, but are not limited to, genes encoding diaminopimelic acid pathway enzymes, such as the dihydrodipicolinate synthase gene (dapA), aspartokinase gene (lysC), dihydrodipicolinate reductase gene (dapB), diaminopimelate decarboxylase gene (lysA), diaminopimelate dehydrogenase gene (ddh) (WO96/40934), phosphenolpyruvate carboxylase gene (ppc) (JP60-87788), aspartate aminotransferase gene (aspC) (JP 6-102028), diaminopimelate epimerase gene (dapF) (JP2003- 135066), aspartate semialdehyde dehydrogena
- diaminopimelic acid pathway enzymes such as the dihydrodipicolinate synthase gene (dapA), asparto
- Lys biosynthetic enzymes In addition to methods of increasing the level of expression of Lys biosynthetic enzymes, methods of increasing the level of expression of the gene involved in energy efficiency (cyo) (EPI 170376 ), the nicotinamide nucleotide transhydrogenase gene (pntAB)(US 5,830,716), ybjE gene(WO2005/073390) have also been developed.
- the ability to produce L-lysine can be imparted by introducing a gene encoding a protein involved in L-lysine biosynthesis into a host as set forth below.
- a gene fragment encoding an L-lysine biosynthesis gene is ligated to a vector which is able to function in a host microorganism which has been employed to produce L-lysine.
- the chosen vector preferably is a multicopy-type vector, which is then used to transform the host. Since the number of copies of the genes encoding proteins involved in L-lysine biosynthesis increases in the transformed host cell, the expression level is increased and the activity of the enzymes is enhanced.
- genes encoding proteins involved in L-lysine biosynthesis are not specifically limited other than that they must be able to be expressed in the host microorganism. Examples include genes derived from Escherichia coli and coryneform bacteria. Since the genomic sequences of both Escherichia coli and Corynebacterium glutamicum are known, a primer can be synthesized based on the sequences of the genes, and PCR employing the chromosomal DNA of a microbe such as Escherichia coli K-12 can be employed to obtain the genes (Science 277 (533 I), 1453-1474 ( 1997) Proceedings of the 9th International Symposium on the Genetics of Industrial Microorganisms: 21 ).
- the plasmids which may be used for gene cloning may be capable of autonomous replication in the bacteria from the Enterobacteriaceae family; specific examples are pBR322, pTWV228 (Takara-Bio Co.), pMWl 19 (Nippon Gene Corp.), pUC19, pSTV29 (Takara-Bio Co.), and RSFlOlO (Gene vol. 75 (2), p. 271 -288, 1989). Phage DNA vectors may also be employed.
- the vector is digested with a restriction enzyme matched to the end of the DNA fragment containing the target gene.
- the ligation is usually conducted with a ligase such as T4 DNA ligase.
- the target genes may be introduced into a variety of separate vectors, or introduced into a single vector.
- the usual methods known to those skilled in the art can be employed to digest and ligate DNA, prepare chromosomal DNA, conduct PCR, prepare plasmid DNA, transform hosts, and determine oligonucleotides for use as primers. These methods are described in Sambrook, J., Fritsch, E.
- Enhancing expression of the genes encoding proteins involved in L-lysine biosynthesis can be achieved by introducing multiple copies of the target gene into the chromosomal DNA of the microorganism. Multiple copies of the target gene can be introduced into the chromosomal DNA of the microorganism by using a DNA in which multiple copies are present in chromosomal DNA as a target in homologous recombination. Such site-specific introduction of mutations based on genetic recombination using homologous recombination is already established. There is a method employing single strand DNA and a method employing a plasmid containing temperature-sensitive replication origin (U.S. Patent 6,303,383 or JP05-007491 A).
- Repetitive DNA and inverted repeats present on the ends of transposable elements can be employed as sequences in which multiple copies are preset in chromosomal DNA.
- multiple copies of the target gene can be introduced into a chromosome. With either method, as a result of increasing the number of copies of the target gene in the transformant, the enzymatic activity of L-lysine biosynthesis increases.
- L-lysine biosynthesis enzyme activity can be enhanced by replacing the expression regulatory sequence of the promoter of the target gene with a stronger one (JPO 1 -2 ] 5280A).
- a stronger one JPO 1 -2 ] 5280A.
- a paper by Goldstein Prokaryotic promoters in biotechnology. Biotechnol. Annu. Rev., 1995, 1 , 105-128 describes methods of evaluating the strength of promoters and gives examples of strong promoters.
- substitution of several nucleotides into the spacer between a ribosome binding site (RBS) and a start codon, particularly into the sequence immediately upstream from a start codon, is known to have a strong effect on mRNA translation efficiency.
- the expression adjustment regions of the promoters or the like of the target gene can be determined using promoter search vectors and gene analysis software such as GENETYX. Substitution of the expression regulating sequence can be conducted, for example, in the same manner as in the above-described gene substitution employing temperature-sensitive plasmids.
- the L-lysine producing bacterium of the present invention may have reduced or deficient activity of enzymes which catalyze reactions that branch off from the L-Iysine biosynthesis pathway and produce compounds other than L-lysine.
- enzymes which catalyze reactions that branch off from the L-Iysine biosynthesis pathway and produce compounds other than L-lysine.
- examples of such enzymes are homoserine dehydrogenase, lysine decarboxylase (cad A, l dcC), and malic enzyme.
- Strains in which the activity of these enzymes is reduced or eliminated are described in WO 95/23864, WO96/17930, and WO2005/010175.
- Examples of methods of reducing or eliminating the intracellular activity of an enzyme include mutating or deleting a gene encoding the enzyme in cells of a microorganism so that intracellular activity is reduced or eliminated as compared to a non-mutated strain. For example, this can be achieved by using recombination to inactivate the gene encoding the enzyme on the chromosome, or to modify an expression regulating sequence such as a promoter or the Shine-Dalgarno (SD) sequence. (WO95/34672,Biotechnol Prog 1999, 15,58-64)
- Enzymatic activity can also be decreased or eliminated by constructing a gene encoding a mutant enzyme which lacks a coding region, using homologus recombination to replace the normal gene on the chromosome with this gene, and introducing a transposon or IS factor into the gene.
- the following methods may be employed to introduce a mutation causing a decrease of, or eliminating, the above enzyme activity by gene recombination.
- a portion of the sequence of the targeted gene is modified, a mutant gene that does not produce a normally functioning enzyme is prepared, DNA containing this gene is used to transform a microorganism from the Enterobacteriaceae family, and the mutant gene is made to recombine with the gene on the chromosome, which results in replacing the target gene on the chromosome with the mutant gene.
- Such gene substitution using homologous recombination can be conducted by methods employing linear DNA, such as the method known as "Red-driven integration" (Proc. Natl. Acad. Sci. USA, 2000, vol. 97, No.
- the L-glutamic acid producing bacterium employed in the present invention can be a microorganism from the Enterobacteriaceae family that has been modified to enhance expression of a gene encoding an enzyme related to L-glutamic acid biosynthesis, for example.
- enzymes related to L-glutamic acid biosynthesis are glutamate dehydrogenase (also referred to as "GDH" hereinafter), glutamine synthetase, glutamate
- -I I - synthase isocitrate dehydrogenase, aconate hydratase, citrate synthase (also referred to as "CS" hereinafter), pyruvate carboxylase, pyruvate dehydrogenase, pyruvate kinase, phosphophenol pyruvate synthase, enolase, phosphoglyceromutase, phosphoglycerate kinase, glyceraldehyde-3-phosphate dehydrogenase, triosephosphate isomerase, fructose bisphosphate aldolase, phosphofructokinase, and glucose phosphate isomerase.
- these enzymes one or more from among CS, PEPC, and GDH are desirable, with all three being preferred.
- U.S. Patents 6,197,559 and 6,331 ,419 and European Patent 0999282 describe microbes from the Enterobacteriaceae family that have been modified by methods such as those set forth above to enhance expression of the citrate synthase gene, phosphenolpyruvate carboxylase gene, and/or glutamate dehydrogenase gene.
- a microorganism which has reduced or eliminated activity of enzymes which catalyze reactions that branch off from the L-glutamic acid biosynthesis pathway and produce compounds other than L-glutamic acid may also be employed as the microorganism from the Enterobacteriaceae family which has the ability to produce L-glutamic acid.
- Examples of enzymes which catalyze reactions that branch off from the L-glutamic biosynthesis pathway and produce compounds other than L-glutamic acid are: 2-oxoglutamate dehydrogenase, isocitrate lyase, phosphate acetyl transferase, acetate kinase, acetohydroxamate synthase, acetolactate synthase, formate acetyl transferase, lactate dehydrogenase, glutamate decarboxylase.
- 2-oxoglutamate dehydrogenase isocitrate lyase
- phosphate acetyl transferase acetate kinase
- acetohydroxamate synthase acetolactate synthase
- formate acetyl transferase lactate dehydrogenase
- glutamate decarboxylase the reduction or elimination of 2-oxoglutamate dehydrogenas
- U.S. Patents 6,197,559 and 6,331 ,419 describe methods of eliminating or reducing the 2-oxoglutamate dehydrogenase activity in a microrganism from the Enterobacteriaceae family.
- Specific examples of a bacterium from the Enterobacteriaceae family in which 2-oxoglutamate dehydrogenase activity has been eliminated or reduced are:
- Pantoea ananatis AJ 13355 (FERM BP-6614) Escherichia c ⁇ li AJ 12949 (FERM BP-4881 ).
- the preferred L-tryptophan producing microorganism employed in the present invention enhances the activity of one or more of the following proteins: anthranilate synthase, phosphoglycerate dehydrogenase, and tryptophan synthase.
- Anthranilate synthase and phosphoglycerate dehydrogenase are subject to feedback suppression by L-tryptophan and L-serine, respectively.
- their enzymatic activity can be enhanced by retaining a desensitized mutant enzyme.
- the anthranilate synthase gene (trpE) and/or the phosphoglycerate dehydrogenase gene (serA) can be mutated so as to not be subject to feedback inhibition, and the mutant gene obtained can be introduced into a microorganism of the Enterobacteriaceae family to obtain a microorganism retaining the desensitized enzyme.
- microbes are transformed strains obtained by introducing plasmid pGH5 having a mutant form of serA encoding desensitized phosphoglycerate dehydrogenase into Escherichia coli SV 164 which has retained a desensitized from of anthranilate synthase (WO94/08031).
- a microorganism into which a recombinant DNA containing a tryptophan operon has been introduced is a further example of a desirable L-tryptophan-producing bacteria.
- Escherichia coli into which a tryptophan operon containing a gene encoding desensitized anthranilate synthase has been introduced JP57-71397A and JP 62-244382A, and U.S. Patent 4,371 ,614).
- enhancement of the expression of the gene (trpBA) encoding tryptophan synthase also enhances or imparts L-tryptophan production capability.
- Tryptophan synthase contains ⁇ and ⁇ subunits encoded by trpA and trpB, respectively.
- L-tryptophan producing bacteria are Escherichia coli strain AGX17(pGX44) (NRRL B-12263), which requires L-phenylalanine and L-tyrosine, and strain AGX6(pGX50)aroP (NRRL B-12264), which retains a plasmid pGX50 containing a tryptophan operon (see U.S. Patent 4,371,614 for both of these).
- L-phenylalanine-producing strains derived from the K-12 strain include the AJ 12739 strain in which the tyrA gene and the tyrR gene are disrupted (tyrA::TnlO, tyrR) (VKPM B-8197), and a strain in which the yddG gene and the yedA gene, which are involved in phenylalanine excretion, are amplified (WO 03/044192).
- the strain which is modified to enhance genes which encode proteins involved in aromatic amino acids biosynthesis can be used as tryptophan and/or phenylalanine-producing strain, such genes include genes which encode proteins involved in a common pathway for aromatic acids, such as aroF, aroG, aroH, aroB, aroD, aroE, aroK, aroL, aroA, and aroC genes.
- a strain having 6-dimethylaminopurine resistance (JP 5-304969A) is desirable as the L-threonine producing bacterium employed in the present invention.
- Examples of recombinant bacteria of the genus Escherichia are a strain in which a mutation producing excessive L-threonine biosynthase has been introduced into the threonine biosynthesis gene and the gene has been amplified on a plasmid (JP l -29559AandJP5-227977A), a strain in which a threonine operon has been amplified with a plasmid (Japanese Patent Application Publication No.
- a further example is Escherichia coli strain VKPM B-3996 (see U.S. Patent 5, 175,107). Strain VKPM B-3996 was deposited on November 19, 1987, with the Russian. National Collection of Industrial Microorganisms (VKPM), GNIl Genetika, and given deposit number VKPM B-3996. Strain VKPM B-3966 harbors a plasmid pVIC40 (WO90/04636), which is obtained by inserting a threonine biosynthesis gene (threonine operon: thrABC) into a broad host vector plasmid pAYC32 (see Chistorerdov, A. Y., Tsygankov, Y.D.
- pVlC40 feedback inhibition by L-threonine of the aspartokinase I homoserine dehydrogenase I encoded by thrA in the threonine operon is repressed.
- VKPM B-5318 strain (EP 0593792B) may also be used as an L-threonine-producing strain.
- VKPM B-5318 was deposited at the Russian National Collection of Industrial Microorganisms (VKPM), GNII Genetika(Dorozhny proezd. 1, Moscow 1 13545, Russian Federation), with a registration number of VKPM B-5318 on November 19, 1987.
- the VKPM B-5318 strain is autotroph to L-isoleucine,and the threonine operon encoding the threonine biosynthesis enzyme is located downstream Cl temparature-sensitive represser,PR-promoer and N end of Cro protein derived from ⁇ phage,moreover,the strain harbors plasmid DNA constructed so that the expression of threonine biosynthesis gene is regulated by psromoter and represser derived from ⁇ phage.
- the bacterium of the present invention is additionally modified to enhance expression of one or more of the following genes: the mutant thrA gene which codes for aspartokinase homoserine dehydrogenase I and is resistant to feedback inhibition by threonine; the thrB gene which codes for homoserine kinase; the thrC gene which codes for threonine synthase; the rhtA gene which codes for a putative transmembrane protein; the asd gene which codes for aspartate- ⁇ -semialdehyde dehydrogenase; and the aspC gene which codes for aspartate aminotransferase (aspartate transaminase);
- the mutant thrA gene which codes for aspartokinase homoserine dehydrogenase I and is resistant to feedback inhibition by threonine
- the thrB gene which codes for homoserine kinase
- the thrC gene which codes for
- the thrA gene which encodes aspartokinase homoserine dehydrogenase 1 of Escherichia coli has been elucidated (nucleotide positions 337 to 2799, GenBank accession NC_000913.2, gi: 49175990).
- the thrA gene is located between the thrL and thrB genes on the chromosome of E. coli K-12.
- the thrB gene which encodes homoserine kinase of Escherichia coli has been elucidated (nucleotide positions 2801 to 3733, GenBank accession NC_000913.2, gi: 49175990).
- the thrB gene is located between the thrA and thrC genes on the chromosome of E. coli K-12.
- the thrC gene which encodes threonine synthase of Escherichia coli has been elucidated (nucleotide positions 3734 to 5020, GenBank accession NC_OOO9I 3.2, gi: 49175990).
- the thrC gene is located between the thrB gene and the yaaX open reading frame on the chromosome of E. coli K-12. All three genes functions as a single threonine operon.
- the regulation sequence which encode attenuater and leader peptide present upstream threonine operon. To enhance the expression of threonine operon, a leader sequence and/or an attenuator is desirably removed from said operon.(WO2005/049808, WO2003/097839)
- a mutant thrA gene which codes for aspartokinase homoserine dehydrogenase I and is resistant to feedback inhibition by threonine, as well as, the thrB and thrC genes can be obtained as one operon from the well-known plasmid pVIC40 which is present in the threonine producing E. coli strain VKPM B-3996. Plasmid pVIC40 is described in detail in U.S. Patent No. 5,705,371.
- the rhtA gene exists at 18 min on the E. coli chromosome close to the glnHPQ operon, which encodes components of the glutamine transport system.
- the rhtA gene is identical to ORFl (ybi F gene, nucleotide positions 764 to 1651 , GenBank accession number AAA218541 , gi:440181) and located between the pexB and ompX genes.
- the unit expressing a protein encoded by the ORFl has been designated the rhtA gene (rht: resistance to homoserine and threonine).
- the asd gene of E. coli has already been elucidated (nucleotide positions 357251 1 to 3571408, GenBank accession NC_000913.1 , gi: 16131307), and can be obtained by PCR (polymerase chain reaction; refer to White, T.J. et al., Trends Genet., 5, 185 (1989)) utilizing primers prepared based on the nucleotide sequence of the gene.
- the asd genes of other microorganisms can be obtained in a similar manner.
- coli has already been elucidated (nucleotide positions 983742 to 984932, GenBank accession NC_000913.1 , gi: 16128895), and can be obtained by PCR.
- the aspC genes of other microorganisms can be obtained in a similar manner.
- L-histidine producing bacteria examples include strains of Escherichia coli FERM-P5038 and 5048, both of which incorporate a vector containing DNA encoding an L-histidine biosynthetic enzyme (JP56-005099A), a strain having the vector containing the amino acid export gene rht (European Patent Publication No. 1016710), and Escherichia coli strain 80, which is imparted with resistance to sulfaguanidine, D,L-l ,2,4-triazole-3-alanine, and streptomycin (VKPM B-7270 Russian Patent Publication No. 2,1 19,536).
- JP56-005099A a vector containing DNA encoding an L-histidine biosynthetic enzyme
- a strain having the vector containing the amino acid export gene rht European Patent Publication No. 1016710
- Escherichia coli strain 80 which is imparted with resistance to sulfaguanidine, D,
- a microorganism with enhanced expression of genes encoding enzymes of the L-histidine biosynthesis pathway may be employed as a microorganism which has the ability to produce L-histidine.
- enzymes involved in L-histidine biosynthesis are ATP phosphoribosyltransferase (hisG), phosphoribosyl AMP cyclohydrolase (hisl), phosphoribosyl ATP pyrophosphohydrolase (hisIE), phosphoribosylformimino-5-aminoimidazole carboxamide ribotide isomerase (hisA), amidotransferase (hisH), histidinolphosphate aminotransferase (hisC), histidinolphosphatase (hisB), and histidinoldehydrogenase (hisD).
- Desirable examples of the L-cysteine producing bacterium of the present invention are bacteria which have reduced cystathionine- ⁇ -lyase activity (JP2003-169668A) and Escherichia coli bacteria which retain serineacetyltransferase, the activity of which is decreased by feedback suppression by L-cysteine (JPl 1 -155571 A).
- L-proline producing bacterium of the present invention are Escherichia coli strain 702 (VKPMB-801 1 ), which is resistant to 3,4-dehydroxypropline and azatadine-2-carboxyIate, and strain 702ilvA (VKPMB-8012), which lacks the HvA of strain 702 (JP2002-300874A).
- Escherichia coli strains with resistance to ⁇ -methylmethionine, p-fluorophenylalanine, D-arginine, arginine hydroxamic acid, S-(2-aminoethyl)cysteine, ⁇ -methylserine, ⁇ -2-thienylalanine, or sulfaguanidine (JP56-106598A).
- Escherichia coli strain 237 US Patent Application No. 2003/058315
- which is an L-arginine producing bacterium harboring mutant N-acetylglutamate synthass is also a desirable L-arginine producing strain.
- Microorganisms with an enhanced level of expression of genes encoding enzymes related to L-arginine biosynthesis can also be employed as a microorganism having L-arginine production ability.
- enzymes related to L-arginine biosynthesis are one or more selected from among N-acetylglutamate synthase (argA), N-acetylglutamylphosphate reductase ⁇ argC), ornithine acetyltransferase (argj), N-acetylglutamate kinase (argB), acetylornithine transferase (argD), acetylomithine deacetylase (argE), ornithinecarbamoyltransferase (argF), argininosuccinate synthase (argG), argininosuccinate lyase (argff), and carbamoylphosphate synthase (carAB).
- argA N-acety
- L-leucine producing bacteria of the present invention include a bacterium of the genus Escherichia in which the branched chain amino acid transferase encoded by the HvE gene is inactivated and the activity of aromatic amino acid transaminase encoded by the tyrB gene is enhanced (JP2004-024259), Escherichia coli strain H-9068 (ATCC21530), Escherichia c ⁇ li strain H-9070 (FERM BP-4704), Escherichia coli strain 9072 (FERM BP-4706) which is resistant to 4-azoleucine or 5,5,5-trifluoroleucine (U.S. Patent No.
- the bacterium of the present invention may be improved by enhancing the expression of one or more genes involved in L-leucine biosynthesis.
- genes of the leuABCD operon which are preferably represented by a mutant leuA gene coding for isopropylmalate synthase freed from feedback inhibition by L-leucine (US Patent 6,403,342).
- the bacterium of the present invention may be improved by enhancing the expression of one or more genes coding for proteins which excrete L-amino acid from the bacterial cell. Examples of such genes include the b2682 and b2683 genes iygaZH genes) (EP 1239041 A2).
- L-isoleucine producing bacteria examples include Escherichia coli variants with resistance to 6-dimethylaminopurine (JP5-304969A), Escherichia coli mutants with resistance to L-isoleucine hydroxamate (JP5-130882A), Escherichia coli strains with resistance to thiaisoleucine (JP5-130882A), Escherichia coli mutants with resistance to DL-ethionine (JP5-130882A), and variants with resistance to arginine hydroxamate(JP5-130882A) with the ability to produce L-isoleucine.
- Examples of recombinant bacteria of the genus Escherichia are strains with genes encoding proteins involved in L-isoleucine biosynthesis, such as threonine deaminase or acetohydroxate synthase enhanced with plasmids (JP2-458A, 2-42988A, and 8-47397A).
- bacterium producing L-valine include, but are not limited to, strains which have been modified to overexpress the HvGMEDA operon (U.S. Patent No. 5,998,178). It is desirable to remove the region of the HvGMEDA operon which is required for attenuation so that expression of the operon is not attenuated by L-valine that is produced. Furthermore, the UvA gene in the operon is desirably disrupted so that threonine deaminase activity is decreased.
- parent strains for deriving L-valine-producing bacteria of the present invention include also include mutants having a mutation of amino-acyl t-RNA synthetase (U.S. Patent No. 5,658,766).
- E. coli VL 1970 which has a mutation in the ileS gene encoding isoleucine tRNA synthetase, can be used.
- E. coli VLl 970 has been deposited in the Russian National Collection of Industrial Microorganisms (VKPM) (Russia, 1 13545 Moscow, 1 Dorozhny Proezd.) on June 24, 1988 under accession number VKPM B-441 1.
- mutants requiring lipoic acid for growth and/or lacking H+-ATPase can also be used as parent strains (WO96/06926).
- the microorganism of the present invention can be obtained by modifying a microbe of the Enterobactehaceae family which has the ability to produce L-amino acid so that it cannot produce type I fimbrial adhesin protein, as set forth above.
- a microbe of the Enterobacteriaceae family of the present invention it makes no difference whether the ability to produce an L-amino acid is imparted first or a modification to prevent production of type 1 fimbrial adhesin protein is made first.
- a microbe from the Enterobacteriaceae family which has the ability to produce an L-amino acid may be modified so that it does not produce type I fimbrial adhesin protein, or a microbe from the Enterobacteriaceae family that no longer produces type I fimbrial adhesin protein may be imparted with the ability to produce an L-amino acid. It suffices for the microorganism of the present invention to be modified so that it does not normally produce type 1 fimbrial adhesin protein in the manner of wild-type strains or unmodified strains. However, it is desirable for the microbe of the present invention to have an ability to cause accumulation of an L-amino acid exceeding that of these strains.
- the "fimbriae” of the present invention are filamentous protrusions present on the outer cell membrane of microorganism from the Enterobacteriaceae family. Fimbriae can be divided into two types. One type is the fimbriae which has no direct relation to sexual processes such as conjugation and gene transfer, and the second type is sexual fimbriae that are produced on the outer layer of gene-donor bacteria and are essential to conjugation with gene-recipient bacteria.
- the "fimbriae” referred to in the present invention are the former, and are not directly related to sexual processes such as conjugation, gene transfer, or the like (Shoji Mizushima, Kinichiro Miura, Bacterial Anatomy 129 (1979)).
- type 1 fimbrial adhesion protein refers to a protein which belongs to a group of proteins which control the formation of fimbriae, and furthermore, the protein has a function blocking a coagulation of red corpuscles by D-mannose.
- type I fimbrial adhesion protein refers to when the quantity of type 1 fimbrial adhesin protein that is produced is lower than that in unmodified strains, and when the conformation of the protein is modified so that normal fimbriae cannot be produced by microbes from the Enterobacteriaceae family.
- wild-type strains such as Escherichia coli strain W31 10 (ATCC 27325), which is derived from the prototypical wild-type strain K 12, and Escherichia coli MG 1655 (ATCC 47076), are examples of control strains from the genus Escherichia.
- Confirmation of a decrease in the quantity of type 1 fimbrial adhesin protein which is produced, or the lack of production of type I fimbrial adhesin protein can be determined by the immunofluorescent antibody method, a decrease in the level of coagulation in the presence of D-mannose, or the lack of ability for coagulation (see the methods of Pallesen et al.: Microbiology 141 ; 2839-2848).
- the type I fimbrial adhesin protein (Seq. ID No. 2) from the genus Escherichia is encoded by the geney/wH(Seq. ID No. 1 ).
- Type I fimbrial adhesin proteins from other microbes of the Enterobacteriaceae family includey?wHhomologs; The homologs of ⁇ niH from Enterobacteriaceae can be obtained by searching for genes having high homology to the gene denoted by Seq. ID No. 1 with BLAST (http://blast.genome.jp).
- GenBank Accession number of the amino acid sequence of the type 1 fimbrial adhesin protein from Escherichia microbes and they?wH gene encoding this protein are given in Table I .
- Examples of the group of genes which encode proteins which control formation of fimbriae in the bacteria from genus Escherichia are fimB, fimE, fimA, fiml, fimC, fimD, fimF, fimG, and fim ⁇ .
- the amino acid sequences encoded by these genes, their gene sequences, and their GenBank Accession numbers are given in Table 1.
- the type I fimbrial adhesin protein is, from Escherichia coli, one which has the amino acid sequence of Seq. ID No. 2 in Table 1.
- the amino acid sequence of the type I fimbrial adhesion protein may include one or several amino acid substitutions, deletions, insertions, or additions .
- the term "one or several" means 1 to 20, desirably 1 to 10, and preferably 1 to 5.
- the above amino acid substitutions, deletions, insertions, or additions are conservative mutations that maintain the production and activity of type 1 fimbrial adhesion protein.
- conservative mutation means that when the substitution is an aromatic amino acid, substitution of Phe, Trp, and Tyr for each other; when the substitution is a hydrophobic amino acid, substitution of Leu, He, and VaI for each other; in the case of a polar amino acid, substitution of GIn and Asn for each other; in the case of a basic amino acid, substitution of Lys, Arg, and His for each other; in the case of an acidic amino acid, substitution of Asp and GIu for each other; and in the case of an amino acid having an hydroxyl group, substitution of Ser and Thr for each other.
- a representative conservative mutation is a conservative substitution.
- substitutions that are considered to be conservative substitutions are the substitution of Ala by Ser or Thr; substitution of Asp by Asn, GIu, or GIn; substitution of Cys by Ser or Ala; substitution of GIn by Asn, GIu, Lys, His, Asp, or Arg; substitution of GIu by Asn, GIn, Lys, or Asp; substitution of GIy by Pro; substitution of His By Asn, Lys, GIn, Arg, or Tyr; substitution of He by Leu, Met, VaI, or Phe; substitution of Leu by lie, Met, VaI, or Phe; substitution of Lys by Asn, GIu, GIn, His, or Arg; substitution of Met by He Leu, VaI, or Phe; substitution of Phe by Trp Tyr, Met, He or Leu; substitution of Ser by Thr or Ala; substitution of Thr by Ser or Ala; substitution of Trp by Phe or Tyr; substitution of Tyr by His Phe, or Trp; and substitution of VaI by Met, He,
- the gene encoding the type I fimbrial adhesin protein can be any DNA which hybridizes under stringent conditions with a probe prepared from the base sequence of SEQ. ID. No. l or a homologous gene of the SEQ ID No. l sequence.
- stringent conditions refers to conditions under which specific hybrids form and nonspecific hybrids do not form. By way of example, these are conditions under which highly homologous fragments of DNA, for example, DNA with a degree of homology of not less than 80%, preferably not less than 90%, and most preferably not less than 95% will hybridize.
- a further example is Southern blot hybridization washing conditions at a temperature and salt concentration corresponding to 6O 0 C, 1 x SSC, 0.1 percent SDS, preferably 0.1 x SSC, 0.1 percent SDS, and more preferably, 68 0 C, 0.1 x SSC, and 0.1 percent SDS, conducted one, two, or three times.
- the length of the probe can be suitably selected based on the hybridization conditions, but normally ranges from 100 bp to 1 Kbp.
- modified so as to not produce type I fimbrial adhesin protein means a modification that does not destroy the fimbriae, but causes type I fimbrial adhesin protein to not function normally.
- a modification may be the introduction of a mutation into the protein with a drug or the like, weakening adhesion, or the use of genetic engineering or the introduction of a mutation into the gene relating to the formation of type I fimbrial adhesin so as to reduce the level of production of type 1 fimbrial adhesin protein, or breed a bacterium that does not produce type 1 fimbrial adhesin protein.
- the transcription or translation of the gene encoding the type I fimbrial adhesin protein may be interfered with, resulting in either in the protein not being produced or being produced at a low level.
- a mutation may be introduced to the gene encoding the type I fimbrial adhesin protein on the chromosome and/or the region controlling expression of the gene, so that type 1 fimbrial adhesin proteins do not function properly.
- a mutation may be introduced into a gene encoding type 1 fimbrial adhesin protein as to reduce the level of expression of genes coding for type I fimbrial adhesin protein on the chromosome.
- modification so as to not produce type I fimbrial adhesin can be achieved by deleting the gene encoding type 1 fimbrial adhesin protein on the chromosome, or by modifying an expression regulating sequence such as a promoter or Shine-Dalgamo (SD) sequence.
- SD Shine-Dalgamo
- the/?/ «H gene shown in Table 1 is the chromosomal gene encoding type 1 fimbrial adhesin protein.
- the//wHgene can be deleted or a mutation can be introduced into the region encodingy ⁇ wH, as described above. This can also be achieved by introducing a mutation attenuating the expression of the/?/ «Hgene upstream from fimH; for example, a frame shift mutation or nonsense mutation can be introduced into one of the genes upstream from//mHthat are indicated in Table 1 , or an upstream gene can be partially deleted.
- a transposon or a gene imparting resistance to an antibiotic can be incorporated in the region encoded by fim ⁇ , or the mutation described in the Journal of Bacteriology, July, 2001 , 4099-4102 or the mutation described in Molecular Microbiology (2001) 41 (6), 1419-1430, can be incorporated.
- the present invention is not limited thereto.
- the target gene on the chromosome can be replaced with a mutated gene by introducing a mutation of the sequence of the target gene, preparing a mutant form of the gene that does not produce a properly functioning enzyme, transforming a microorganism from the Enterobacteriaceae family with DNA containing the gene, and introducing the mutation of the gene to recombine with the gene on the chromosome.
- a method employing linear DNA and a method employing a plasmid containing a temperature-sensitive replication origin U.S.
- Patent 6,303,383 or JP 05-007491 The introduction of a mutation at a specific site by gene substitution employing homologous recombination as set forth above can also be conducted using a plasmid which is not able to replicate on the chromosome.
- a gene encoding the type I fimbrial adhesin protein can be obtained by the following methods.
- Chromosomal DNA can be prepared from a microorganism belonging to the family Enterobacteriaceae, for example, by the method of Saito and Miura (see H. Saito and K. Miura, Biochem. Biophys. Acta, 72, 619 (1963); Book of Bioengineering Experiments, comp. by the Japan Bioengineering Society, p. 97-98, Baifukan, 1992).
- a gene encoding the type I fimbrial adhesin protein can be obtained by using a known database such as GenBank, preparing an oligonucleotide, and employing PCR. For example, it can be constructed by referring to the sequences of the genus Escherichia listed in Table 1.
- a gene encoding the type I fimbrial adhesin protein prepared as set forth above, or a portion of such a gene can be used for gene inactivation.
- the gene used for gene inactivation need only have a degree of homology adequate to occur homologous recombination with the gene encoding the typr* 1 fimbrial adhesin protein on the chromosomal DNA of a microbe of the Enterobacteriaceae family, such a homologous gene may also be employed.
- the term "degree of homology adequate to undergo homologous recombination” is desirably homology of 80% or more, preferably 90% or more, more preferably 95% or more, and particularly preferably, 97% or more.
- stringent conditions refers to conditions under which specific hybridization occurs but nonspecific hybridization does not.
- An example of such conditions is washing once, preferably two or three times, at a salt concentration corresponding to 6O 0 C, 1 x SSC, 0.1 percent SDS, preferably 0.1 x SSC and 0.1 percent SDS.
- the gene encoding type I fimbrial adhesin protein on the chromosome can be inactivated, for example, by deleting part of the sequence of the gene so it is not able to produce a properly functioning type 1 fimbrial adhesin protein, transforming a group of enterobacteria with DNA containing this gene, and causing the deficient gene to recombine with the gene on the chromosome.
- Such gene inactivation by gene substitution using homologous recombination is already established.
- There exist methods employing linear DNA such as the method developed by Datsenko and Wanner called "red-driven integration" (Proc. Natl. Acad. Sci. USA, 2000, vol. 97, No.
- a gene-disrupted strain can be constructed in one step by using a PCR product, which is obtained using synthetic oligonucleotides as primers which are designed to include part of a targeted gene at its 5' terminus, and part of an antibiotic resistance gene at its 3' terminus. Furthermore, the integrated antibiotic resistance gene can be removed by introducing attL and attR, which are attachment sites of lambda phage, and the PCR product, and combining the excision system derived from lambda phage with the red-driven integration method.
- a strain in which the targeted gene is disrupted and the antibiotic resistance gene is removed can be obtained by the following method.
- a linear DNA cassette comprising an antibiotic resistance gene, attachment sites of lambda phage and a target gene is initially prepared. This is usually prepared by PCR using a suitably-prepared template.
- a template in which attL and attR (SEQ ID NO: 9 (GenBank accession No. M l 2458 and SEQ ID NO: 10 (GenBank accession No. M 12459)) are inserted at respective terminals of an antibiotic resistance gene is used as a template of the linear DNA cassette.
- the template may be plasmid for example pM W 1 18-attL-Tc-attR, pM W 1 18-attL Cm-attR, (Fig.l) , a gene inserted on a chromosome, or a synthetic oligonucleotide.
- the antibiotic resistance gene is preferably a chloramphenicol resistance gene, a streptomycin resistance gene, or an ampicillin resistance gene
- any antibiotic resistance gene can be used provided that the gene functions as an antibiotic resistance gene in Enterobacteriaceae family a and is different from a marker gene which may be contained in two helper plasmids as described below.
- the antibiotic resistance gene which is employed can be one whereby the expression is increased by replacing a promoter sequence and the like, or one in which a mutation is introduced in its structural gene sequence so that an enzyme activity is increased.
- the linear DNA cassette is prepared in the following order from the 5'terminus: (targeted gene 5' sequence)-(attL)-(antibiotic resistance gene)-(attR)-(targeted gene 3' sequence).
- the linear DNA cassette is integrated into the chromosome.
- pKD46 can be used as a helper plasmid for integrating the linear DNA cassette into chromosome.
- pKD46 include temperature-sensitive replication origin and ampicillin resistance gene, a 2,154 nt DNA fragment of lambda phage (GenBank/EMBL accession No. J02459, 31088-33241 ), which contains the genes (gamma, beta, and exo genes) encoding Red recombinase of the lambda Red homologous recombination system and which is under the control of the arabinose-inducible ParaB promoter.
- pKD46 can be introduced into a host by electroporation.
- the pKD46-amplified strain is cultured with arabinose.
- the linear DNA cassette is introduced at the logarithmic growth phase and incubated at a high temperature to obtain a gene-disrupted strain which is resistant to an antibiotic by the antibiotic resistance gene in the linear DNA cassette.
- the confirmation of the gene disruption can be made by PCR or measurement of the concentration of L-lysine or L-threonine produced by the strain.
- helper plasmid for excising the antibiotic resistance gene is then introduced.
- the helper plasmid harbors a gene encoding integrase (Int) (SEQ ID NO: 13, GenBank accession No. J02459. B [gi:215104]) and a gene encoding excisionase (Xis) (SEQ ID NO: 15, GenBank accession No. J02459 [gi:215104]) of lambda phage and shows temperature-sensitive replication.
- the antibiotic resistance gene between attL and attR is excised and as a result, a structure that contains only the attL or attR sequence remains on the chromosome. By incubating at a high temperature, the helper plasmid is lost. Thus, a strain in which the targeted gene is disrupted and the antibiotic gene is eliminated can be obtained.
- examples of methods of modification which preclude the production of type I fimbrial adhesin protein are subjecting a group of Enterobacteriaceae family to UV radiation or treating it with N-methyl-N'-nitro-N-nitrosoguanidine (NTG), nitrous acid, or some other mutagenic agent commonly employed in mutation treatments and selecting the strains that are unable to produce type I fimbrial adhesin protein.
- NTG N-methyl-N'-nitro-N-nitrosoguanidine
- nitrous acid or some other mutagenic agent commonly employed in mutation treatments and selecting the strains that are unable to produce type I fimbrial adhesin protein.
- the method for producing an L-amino acid of the present invention comprises culturing the microorganism of the present invention in a medium, causing the production and accumulation of L-amino acid in the culture or in the bacterial ceil, and recovering the L-amino acid from the culture or bacterial cell.
- the cultivation, collection, and purification of an L-amino acid from the medium and the like may be performed in a manner similar to conventional fermentation methods wherein an amino acid is produced using a bacterium.
- the medium employed can be one that is conventionally employed in the production of L-amino acids by fermentation.
- the usual medium contains a carbon source, nitrogen source, inorganic ions, and other organic components as needed.
- exemplary carbon sources include sugar such as glucose, sucrose, lactose, galactose, fructose, or starch hydrolysis product; alcohol such as glycerol or sorbitol; and organic acids such as fumaric acid, citric acid, or succinic acid.
- exemplary nitrogen sources include an inorganic ammonium salt such as ammonium sulfate, ammonium chloride, or ammonium phosphate; organic nitrogen such as a soybean hydrolysis product; ammonia gas; ammonia water; or the like.
- Other nutrients such as vitamin B 1 and L-homoserine or yeast extract can be employed in suitable quantities as a source of a trace organic nutrient.
- potassium phosphate magnesium sulfate, iron ions, manganese ions, and the like can be added. So long as the medium employed in the present invention contains a carbon source, nitrogen source, inorganic ions and, as needed, other trace organic components, it does not matter whether it is a natural or a synthetic medium.
- L-amino acid can be recovered from the fermentation solution by the usual ion-exchange resin method, precipitation method, and combinations of these with other known methods.
- the bacterial cell can be crushed by ultrasonic waves or the like, a supernatant can be centrifugally separated from the bacterial cell, and the supernatant can be subjected to ion-exchange resin to recover the L-amino acid.
- a liquid medium can be prepared under conditions which cause L-glutamic acid to precipitate, and the culture can be conducted while L-glutamic acid precipitates.
- An example of the conditions under which L-glutamic acid will precipitate is a pH of 5.0 to 4.0, preferably pH 4.5 to 4.0, more preferably pH 4.3 to 4.0, and particularly preferably, pH 4.0.
- the L-glutamic acid precipitating out into the culture solution can be collected by centrifugation, filtration, or the like. In that case, the L-glutamic acid dissolved in the medium can be crystallized and then separated.
- Example 1 Construction of a bacterium which is modified to not produce type I fimbrial adhesin protein.
- Lysine decarboxylase is encoded by the cadA gene (GenBank Accession No. NP_418555, Seq. ID No. 42) and the IdcC gene (GenBank Accession No. NP_414728, Seq. ID No. 44) (see International Application Publication WO96/17930).
- the parent strain employed here was strain WC 196.
- This strain was named Escherichia coli strain AJ 13069 and was deposited on December 6, 1994, as deposit number FERM P- 14690, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1 -1-1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566). This strain was converted on September 29, 1995, to an international deposit under the provisions of the Budapest Treaty and given deposit number FERM BP-5252.
- the genes cadA and IdcC encoding lysine decarboxylase were deleted by the method known as "Red-driven integration” developed by Datsenko and Wanner (Proc. Natl. Acad. Sci. USA, 2000, Vol. 97, No. 12, p 6640-6645) and by a ⁇ phage-derived excision system (J. Bacteriol. 2002 Sep; 184 (18): 5200-3. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex.
- pMWl 18-attL-Cm-attR was employed as PCR template.
- pMWl 18-attL-Cm-attR was a plasmid obtained by inserting the attL and attR genes - attachment sites of ⁇ phage - and the cat gene, a gene imparting antibiotic-resistance, into pMW l 18 (Takara-Bio Co.) in the order attL-cat-attR.
- the sequence of attL is shown by SEQ. ID No. 1 1 and the sequence of attR by SEQ. ID No.12.
- the amplified PCR product was purified on an agarose gel and introduced by electroporation into Escherichia coli strain WC 196, which contains plasmid pKD46 with a temperature-sensitive replication origin.
- Plasmid pKD46 (Proc. Natl. Acad. Sci. USA, 2000, Vol. 97, No. 12, p 6640-6645) includes a DNA fragment (GenBank/EMBL Accession No.
- Plasmid pKD46 was required to introduce the PCR product into the chromosome of strain WC 196.
- Competent cells for electroporation were prepared as follows.
- the Escherichia coli strain WC 196 that had been cultured overnight at 3O 0 C in LB medium containing 100 mg/L of ampicillin was diluted 100-fold with 5 mL of SOB medium (Molecular Cloning: Laboratory Manual, 2nd Ed., Sambrook J. et al., Cold Spring Harbor Laboratory Press (1989)) containing L-arabinose (1 mM) and ampicillin (20 mg/L).
- the diluted product was cultured until reaching an OD600 of about 0.6 at 3O 0 C, concentrated 100-fold, and readied for electroporation by washing three times in 10 percent glycerol.
- Electroporation was conducted using 70 ⁇ L of competent cells and about 100 ng ofthe PCR product. Following electroporation, 1 mL of SOC medium (Molecular Cloning: Laboratory Manual, 2nd Ed., Sambrook J. et al., Cold Spring Harbor Laboratory Press ( 1989)) was added and the cells were cultured for 2.5 hours at 37°C. They were then plate cultured on L-agar medium containing chloramphenicol (Cm, 25 mg/L) at 37°C and the Cm-resistant recombinants were selected. Next, to remove the pKD46 plasmid, cell were subcultured at 42°C on Cm-containing L-agar medium and the ampicillin resistance of the colonies obtained was tested.
- SOC medium Molecular Cloning: Laboratory Manual, 2nd Ed., Sambrook J. et al., Cold Spring Harbor Laboratory Press ( 1989)
- the ampicillin-sensitive strains without pKD46 were collected.
- the deletion of the cadA gene in mutants identified based on chloramphenicol resistance gene was confirmed by PCR.
- the cadA-deficient strain obtained was named strain WC 196 ⁇ cadA::att-cat.
- helper plasmid pMW-intxis-ts was employed to remove the att-cat genes that were inserted inside the cadA gene.
- pMW-intxis-ts is a plasmid which harbors a gene (SEQ. ID No. 13) encoding the integrase (Int) of the ⁇ phage and a gene (SEQ. ID No. 15) encoding the excisionase (Xis) of the ⁇ phage, and which has a temperature-sensative replication origin.
- pMW-intxis-ts recombination occurs due to the recognition of attL (SEQ. ID No. 1 1 ) and attR (SEQ. ID No. 12) on the chromosome, allowing gene excision between attL and attR, leaving behind only the attL or attR sequence on the chromosome.
- Competent cells of the strain WC I 96 ⁇ cadA::att-cat obtained as set forth above were prepared by ordinary methods, transformed with helper plasmid pMW-intxis-ts, and plate cultured on L-agar medium containing 50 mg/L of ampicillin at 3O 0 C. The ampicillin-resistant strains were selected.
- PCR template pMWl 18-attL-Cm-attR and helper plasmid pMW-intxis-ts were prepared as set forth below. (3-1) pMWl 18-attL-Cm-attR pMWl 18-attL-Cm-attR was constructed based on pMWl 18-attL-Tc-attR. The four following DNA fragments were spliced.
- BgI ll-EcoRI DNA fragment 120 bp (SEQ. ID No. 1 1) containing attL obtained by PCR amplification of a sequence corresponding to the chromosome of E. coli strain W3350 (ATCC 31278 containing ⁇ prophage) using primers in the form of oligonucleotides Pl and P2 (SEQ. JD NO. 17 and 18) (these primers contained additional BgI Il and EcoRI endonuclease recognition sites).
- Pstl-Hindlll DNA fragment (182 bp) (SEQ. ID No. 12) containing attR obtained by PCR amplification of a sequence corresponding to the chromosome of E. coli strain W3350 (containing ⁇ prophage) using primers in the form of oligonucleotides P3 and P4 (SEQ. ID Nos. 19 and 20) (these primers contained additional Pstl and HindIII endonuclease recognition sites).
- pMWl 18-ter_rrnB Bglll-HindlH large fragment (3,916 bp) of pMWl 18-ter_rrnB.
- pMWl 18-ter_rrn was a fragment obtained by ligation of the following three fragments: -A large fragment (2,359 bp) containing an Aatll-EcoRIpol fragment of pMWl 18 was obtained by cleaving pMWl 18 with EcoRI restriction endonuclease, processing with the Klenow fragment of DNA polymerase 1, and digesting with Aatll restriction endonuclease, -Aatll-Bgl ll small fragment (1 194 bp) of pUC19 containing the bla gene of ampicillin resistance (ApR), obtained by employing oligonucleotides P5 and P6 (SEQ. ID Nos. 21 and 22) as primers (these primers contained additional Aatll and BgI Il endonuclease recognition sites) in
- -Bg I II-Pstlpol small fragment (363 bp) of transcription terminator rrnB obtained by employing oligonucleotides P7 and P8 (SEQ. ID Nos. 23 and 24) as primers (these primers had additional BgI II and Pstl endonuclease identification sites) in PCR amplification of the region corresponding to the chromosome of E. coli strain MG 1655.
- pML-Tc-terJhrL Small EcoRI-Pstl fragment (1,388 bp) (SEQ. ID No. 29) of pML-Tc-terJhrL comprising a tetracycline resistance gene and transcription terminator ter_thrL.
- pML-Tc-ter-thrL was obtained as follows.
- pML-MCS MoI Biol (Mosk). 2005 Sep-Oct;39(5):823-3 I ) was digested with Xbal and BamHI restriction endonucleases and the large fragment (3342 bp) was spliced to an Xbal-BamHI fragment (68 bp) containing terminator ter thrL.
- the region of the Xbal-BamHI fragment corresponding to the chromosome of E. coli strain MG1655 was amplified by PCR using oligonucleotides P9 and PlO (SEQ. ID Nos. 25 and 26) as primers (these primers contained additional Xbal and BamHI endonuclease recognition sites).
- the ligated reaction product was named plasmid pML-ter thrL.
- pML-ter_thrL was cleaved with Kpnl and Xbal restriction endonucleases, treated with the Klenow fragment of DNA polymerase I, and then ligated to the small EcoRI-Van911 fragment (1 ,317 bp) of pBR322 containing a tetracycline resistance gene (pBR322 was treated with the Klenow fragment of DNA polymerase I using EcoRJ and Van91 I restriction endonucleases).
- the product of the ligation reaction was named plasmid pML-Tc-ter_thrL.
- pMW I 18-attL-Tc-attR was obtained as set forth above.
- pMWI 18-attL-Cm-attR was constructed by ligation the large BamHI-Xbal fragment (4,413 bp) of pM Wl 18-attL-Tc-attR, promoter PA2 (the initial promoter of T7 phage), the cat gene of chloramphenicol resistance (CmR), and BgI Il-Xbal artificial DNA fragment ( 1 , 162 bp) comprising the transcription terminator terjhrL and attR.
- the artificial DNA fragment (SEQ. ID No. 30) was obtained as follows.
- the pML-MSC (MoI Biol (Mosk). 2005 Sep-Oct;39(5):823-31 ; Biotechnologiya (Russian) No. 5: 3-20.) was cleaved with Kpnl and Xbal restriction endonucleases and ligated to the small Kpnl-Xbal fragment (120 bp) containing promoter PA2 (the initial promoter of T7 phage).
- the Kpnl-Xbal fragment was obtained by PCR amplification of the region corresponding to T7 phage DNA using oligonucleotides Pl 1 and Pl 2 (SEQ. ID Nos. 27 and 28) as primers (these primers had additional Kpnl and Xbal endonuclease recognition sites).
- the product of the splicing reaction was named plasmid pML-PA2-MCS.
- the Xbal site was removed from pML-PA2-MCS.
- the product obtained was called plasmid pML-PA2-MCS(XbaI-).
- the small Bgl ll-Hindlll fragment (928 bp) of pML-PA2-MCS(Xbal-) containing promoter PA2 (the initial promoter of T7 phage) and the cat gene imparting resistance to chloramphenicol (CmR) was ligated to the small Hindlll-Hindlll fragment (234 bp) of pMWl 18-attL-Tc-attR containing transcription terminator ter thrL and attR.
- the targeted artificial DNA fragment (1,156 bp) was obtained by PCR amplification of the splicing reaction mixture with primers in the form of oligonucleotides P9 and P4 (SEQ. ID Nos. 25 and 20) (these primers contained additional Hindlll and Xbal endonuclease recognition sites).
- the first fragment was comprised of nt 37168 to 38046 (SEQ. ID No. 39), containing CI repressor, promoters Prm and Pr, and the leader sequence of the cro gene. This fragment was obtained by amplification using oligonucleotides Pl ' and P2' (SEQ. ID NO. 31 and 32) as primers.
- the second fragment was comprised of nt 27801 to 29100 (SEQ. ID NO. 40), containing the xis-int genes of ⁇ phage. This fragment was obtained by amplification using oligonucleotides P3' and P4' (SEQ. ID NO. 33 and 34) as primers. All the primers include suitable endonuclease recognition sites.
- the PCR amplification fragment containing cl repressor that was obtained was cleaved with CIaI restriction endonuclease, treated with the Klenow fragment of DNA polymerase I, and digested with EcoRI restriction endonuclease.
- the second PCR amplified fragment was digested with EcoRl and Pstl restriction endonucleases.
- plasmid pMWPIaclacl-ts was digested with BgI Il endonuclease, treated with the Klenow fragment of DNA polymerase I, and cleaved with Pstl restriction endonuclease.
- the vector fragment of pMWPladlacl-ts was eluted from agarose gel and ligated to the cut PCR amplified fragment.
- Plasmid pMWPIaclacl-ts was derived from pMWPIaclacl, which comprised the following components: 1) a Bgl ll-Hindlll artificial DNA fragment comprising Pl acUV5 promoter and the lacl gene controlled by the RBS of bacteriophage T7 gene 10; 2) an Aatll-Bgl ll fragment containing an ampicillin resistance (ApR) gene, obtained by PCR amplification of the region corresponding to plasmid pUC19 using oligonucleotides P5' and P6' (SEQ. ID Nos.
- ApR ampicillin resistance
- primers (these primers contained additional AatII and BgI lI endonuclease recognition sites); 3) an Aatll-Bgl II fragment containing the Aatll-Pvul fragment of recombinant plasmid pMWl 18-ter_rrnB.
- Plasmid pMWl 18-ter_rrnB was constructed in the following manner.
- the region corresponding to the chromosome of E. coli strain MG1655 was amplified by PCR using oligonucleotides P7' and P8' (SEQ. ID Nos. 37 and 38) containing suitable endonuclease recognition sites as primers, yielding a Pstl-Hindlll fragment containing terminator ter rrnB.
- P7' and P8' SEQ. ID Nos. 37 and 38
- The/?mHgene was deleted from the WC196 ⁇ cadA ⁇ ldcC strain by the procedure of (1 ) above; the primers of SEQ. ID Nos. 7 and 8 were employed to delete the fimH gene.
- This obtained strain did not produce type 1 t ⁇ mbrial adhesion protein, and was designated WC196 ⁇ cadA ⁇ IdcC ⁇ fimH::Cm.
- Strains WC196 ⁇ cadA ⁇ IdcC and WC196 ⁇ cadA ⁇ IdcC ⁇ fimH::Cm were transformed by the usual methods with plasmid Lys production-plasmid pCABD2 earring dapA, dapB, and lysC genes (WOO 1/53459), obtaining strains
- These strains were cultured at 37 0 C in L medium containing 20 mg/L of streptomycin to a final OD600 of about 0.6.
- a 40 percent glycerol solution was added in a quantity equal to that of the culture solution, the mixture was stirred, and suitable amouts were poured out and stored at -80 0 C. This was referred to as glycerol stock.
- the glycerol stocks of these strains were melted, 100 ⁇ L amounts were uniformly plated onto L plates containing 20 mg/L of streptomycin, and the strain were cultured for 24 hours at 37°C. About 1/8 of the bacterial cells obtained on each plate was inoculated onto a 20 mL fermentation culture containing 20 mg/L of streptomycin in a 500 mL Sakaguchi flask and cultured for 48 hours at 37°C in a reciprocating shaking incubator. After culturing, the amount of L-lysine that had accumulated in the medium was measured by a ordinary method (Sakura Seiki, Biotech Analyzer AS210). The composition of the fermentation medium is shown below (unit: g/L).
- the medium was adjusted to pH 7.0 with KOH and sterilized for lO min at 1 15°C in an autoclave (the glucose and MgSO4-7H2O were separately sterilized).
- a 30 g/L quantity of CaCO3 (that had been dry sterilized for 2 hours at 180 0 C) was added.
- 20 mg/L amounts of streptomycin was added as antibiotic.
- Culturing was conducted for 48 hours under conditions of a temperature of 37 0 C with stirring at 1 15 rpm.
- B-5318 was employed as the L-threonine-producing parent strain which does not produce type I fimbrial adhesin protein .
- Strain B-5318 was deposited on November 19, 1987, under registration number VKPM B-5318 with the Russian National Collection of Industrial Microorganisms (VKPM), GNII Genetika.
- the strain which does not produce type I fimbrial adhesin was constructed from the L-threonine producing bacterium by the same method as in Example 1 by employing synthetic oligonucleotides SEQ. ID Nos. 7 and 8. Specifically, the following fimH deficient strain was constructed.
- B-5318 ⁇ fimH-cat was derived from strain B-5318 by method ( I ) in Example 1.
- Strain B-5318 and strain B-5318 ⁇ fimH-cat were cultured for 24 hours at 37 0 C on L agar medium containing 20 mg/L of streptomycin sulfate and L agar medium containing 20 mg/L of streptomycin sulfate and 25 mg/L of chloramphenicol, respectively.
- One-fifth of the bacterial cell was collected from each of the plates and inoculated onto 50 mL of LB liquid medium containing the above-stated antibiotics and the bacteria were precultured for 3.5 hours at a temperature of 39 0 C at 144 rpm.
- strain B5318 20 mg/L of streptomycin sulfate salt was added, and for strain B-53 l 8 ⁇ fimH-cat, 20 mg/L of streptomycin sulfate salt and 25 mg/L of chloramphenicol were added.
- Escherichia coli strain AJ 12949 was employed as the L-glutamic acid-producing parent strain which does not produce type 1 fimbrial adhesin protein .
- Strain AJ 12949 was deposited on December 28, 1993, as depositary number FERM P- 14039, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1-1-1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566), and converted on November 1 1 , 1994, to an international deposit under the provisions of the the Budapest Treaty, and given a deposit number FERM BP-4881.
- Strain AJ12949 ⁇ fimH-cat was derived from strain AJ 12949 (BP-4881 ). Strain AJ 12949 and strain AJ 12949 ⁇ fimH-cat were cultured for 24 hours at 37 0 C in L agar medium containing 50 mg/L of ampicillin and L agar solution containing 50 mg/L of ampicillin and 25 mg/L of chloramphenicol, respectively. One-eighth of the bacterial cells was collected from each plate and inoculated onto 2O mL of fermentation medium containing 50 mg/L of ampicillin in a 500 mL Sakaguchi flask and cultured for 24 hours at 37 0 C in a reciprocating shaking incubator. Following culturing, the amount of glutamic acid accumulated in the medium was measured by a known method (Sakura Seiki, Biodex Analyzer AS210).
- composition of the fermentation medium is given below (unit: g/L)
- the medium was adjusted to pH 7.0 with KOH and sterilized for 10 min at I l 5 0 C in an autoclave (the glucose and MgSO4'7H2O were separately sterilized).
- a 50 g/L quantity of CaCO3 (that had been dry sterilized for 2 hours at 18O 0 C) was added.
- a 50 mg/L quantity of ampicillin was added as an antibiotic for strain AJ12949, and a 50 mg/L quantity of ampicillin and a 50 mg/L quantity of chloramphenicol were added for AJ12949 ⁇ f ⁇ mH-cat.
- Pantoea ananatis strain AJ 13601 can also be employed as the L-glutamic acid-producing parent strain which does not produce type I fimbrial adhesin protein.
- Pantoea ananatis strain AJ 13601 was deposited on August 18, 1999, as depositary number FERM P-17516, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology, Ministry of Economy, Trade and Industry (1 -1 -3 Higashi, Tsukuba, lbaraki Prefecture, Postal Code 305-8566). This strain was converted on July 6, 2000, to a deposit under the provisions of the Budapest Treaty under deposit number FERM BP-7207.
- a strain that does not produce type I fimbrial adhesin can be constructed from the L-glutamic acid-producing microbe by the same method as in Example 1 using synthetic nucleotides having SEQ. ID Nos. 7 and 8.
- a mutant strain can also be constructed by introducing a temperature-sensitive plasmid having a deleted gene encoding type 1 fimbrial adhesin protein.
- the strain not producing type I fimbrial adhesin protein can be cultured in an L-glutamic acid production medium in a reciprocating shaking incubator. Following culturing, the amount of L-glutamic acid which had accumulated in the medium is measured by a ordinary method to confirm an increase in accumulated L-glutamic acid. This method permits the obtaining of a strain that does not produce type I fimbrial adhesin protein and has an enhanced ability to produce L-glutamic acid.
Abstract
An L-amino acid producing bacterium of the Enterobacteriaceae family is described, wherein the bacterium has been modified so as to not produce type I fimbrial adhesin protein is cultured in a medium to produce and excrete said L-amino acid in the medium, and collecting said L-amino acid from the medium.
Description
DESCRIPTION
L-AMlNO ACID PRODUCING MICROORGANISM AND A METHOD FOR PRODUCING L-AMINO ACID
Technical Field
The present invention relates to the fermentation industry, and specifically to a method for producing an L-amino acid using a bacterium of the Enterobacteriaceae family, wherein the bacterium has been modified so as to not produce type I fimbrial adhesin protein. L-amino acids are employed as components of pharmaceuticals, animal feed additives, seasonings, and various other nutritive mixtures.
Background Art
L-amino acids are industrially produced by fermentation using L-amino-acid producing bacteria such as coryneform bacteria and bacteria of the genus Escherichia. To enhance productivity, artificially mutated strains of these bacteria that have been isolated from nature or transformants in which activity of an L-amino acid biosynthesis enzyme has been increased by genetic recombination have been employed as the L-amino acid producing bacteria. (US 5,661 ,012 US 6,040,160, US 5,827,698;US 5,932,453, and WO01/53459)
In addition to methods of increasing the level of expression of enzymes which are L-amino acid biosynthesis pathways, another methods of increasing the ability to produce L-amino acids such as L-lysine have been developed. Such methods include improving energy efficiency by modifying the energy pathway (EP 1 170376 ), increasing the ability to produce nicotinamide adenine dinucleotide phosphoric acid by amplifying the nicotinamide nucleotide transhydrogenase gene (US 5,830,716), and inactivating genes related to flagella production (WO02/097089).
Escherichia, Salmonella, and Bacillus bacteria have fimbriae. Fimbriae can be divided into types I to V which have no direct relation to sexual processes such as conjugation and gene transfer, and sexual fimbriae that are produced on the outer layers of donor bacteria and are essential to conjugate with recipient bacteria and to gene transfer (Shoji Mizushima, Kinichiro Miura, Bacterial Anatomy 129 (1979)).
There are nine genes which contribute to the formation of type I fimbriae, and these genes form an operon. The fimH gene, located downstream within the operon, encodes a type 1 fimbrial adhesin protein. Type I fimbrial adhesin protein does not contribute to the formation of fimbriae, but is known to specifically recognize the mannosylated proteins of the host. Furthermore, it is known that the capacity for bacterial aggregation can be enhanced by introducing an amino acid substitution into the gene which encodes the type I fimbrial adhesin protein (Mark A. Schenbri, Gunna Christiansen and Per Klemm: FimH-mediated autoaggregation of Escherichia coli: Molecular Microbiology (2001 ) 41 (6), 1419-1430).
However, there is no information between the effect of introducing a mutation which causes reduction in the adhesion activity of type I fimbrial adhesin protein and L-amino acid production.
SUMMARY OF THE INVENTION
Objects of the present invention include enhancing the productivity of L-amino acid producing strains and providing a method for producing an L-amino acid. The above objects were achieved by finding that the bacterium which is modified so as to not produce type I fimbrial adhesion can improve L-amino acid productivity.
It is an object of the present invention to provide an L-amino acid producing bacterium of the Enterobacteriaceae family, wherein the bacterium has been modified so as to not produce type I fimbrial adhesin protein.
It is a further object of the present invention to provide the bacterium as described above, wherein said bacterium has been modified so as to not produce type I
fimbrial adhesin protein by introducing a mutation into the gene encoding type I fimbrial adhesin protein on the chromosome and/or into the region regulating expression thereof.
It is a further object of the present invention to provide the bacterium as described above, wherein said bacterium has been modified so as to not produce type I fimbrial adhesin protein by attenuation of the gene encoding type 1 fimbrial adhesin protein.
Jt is a further object of the present invention to provide the bacterium as described above, wherein said bacterium has been modified so as to not produce type I fimbrial adhesin protein by inactivation of the gene encoding type I fimbrial adhesin protein on the chromosome.
It is a further object of the present invention to provide the bacterium as described above, wherein the gene encoding type I fimbrial adhesin protein is fimH.
It is a further object of the present invention to provide the bacterium as described above, wherein said gene encoding type I fimbrial adhesin protein is selected from the group consisting of: (a) a DNA comprising the nucleotide sequence ofSEQ JD NO:1 , and (b) a DNA that is able to hybridize with the complementary strand of the nucleotide sequence of SEQ ID NO: I , or a probe prepared from said nucleotide sequence under stringent conditions, and wherein the DNA encodes the type I fimbrial adhesin protein.
It is a further object of the present invention to provide the bacterium as described above, wherein said bacterium of the Enterobacteriaceae family is selected from the group consisting of Escherichia, Pantoea, and Enterobacter.
It is a further object of the present invention to provide the bacterium as described above, wherein said L-amino acid is selected from the group consisting of L-lysine, L-threonine, L-glutamic acid, and combinations thereof.
It is a further object of the present invention to provide a method for producing an L-amino acid comprising cultivating the bacterium as described above in a medium, and collecting said L-amino acid from the medium.
It is a further object of the present invention to provide the method as described
above, wherein said L-amino acid is selected from the group consisting of L-lysine, L-threonine, L-glutamic acid, and combinations thereof.
BRIEF DESCRIPTION OF THE DRAWINGS Fig. 1 shows the structure of pM W l 18-attL-Tc-attR. Fig. 2 shows the structure of pMWl 18-attL-Cm-attR Fig. 3 shows the structure of pMW-intxis-ts.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS <1> The bacterium of the present invention
The bacterium of the present invention is a bacterium having the ability to produce an L-amino acid and which belongs to the Enterobacteriaceae family. The bacterium of the present invention has been modified so as to not produce type I fimbrial adhesin protein. The ability to produce L-amino acids may include production of any single L-amino acid, or production of multiple L-amino acids. Examples of produced L-amino acids of the present invention include L-lysine, L-glutamic acid, L-threonine, L-valine, L-leucine, L-isoleucine, L-serine, L-aspartic acid, L-asparagine, L-glutamine, L-arginine, L-cysteine (cystine), L-methionine, L -phenylalanine, L-tryptophan, L-tyrosine, L-glycine, L-alanine, L-proline, L-ornithine, L-citrulline, and L-homoserine. L-lysine, L-threonine, and L-glutamic acid are particularly desirable.
The phrase "ability to produce an L-amino acid" refers to the ability of a bacterium of the Enterobacteriaceae family to produce and cause accumulation of an L-amino acid in the bacterial cells or into a medium to a degree which permits its recovery from the bacterial cells or the medium when the bacterium is cultured in the medium. The bacterium which has this ability may originally have had the ability to produce an L-amino acid, or may be a bacterium, such as those set forth below, that has been modified by a mutation method or recombinant DNA technique to impart the ability to produce an L-amino acid. The bacterium of the present invention may also be one that has had the ability to produce an L-amino acid imparted to it by a modification which results in
prevention of production of type I fimbrial adhesin production.
The Enterobacteriaceae family includes bacteria belonging to the genus Escherichia, Enterobacter, Erwinia, Klebsiella, Pantoea, Photorhabdus, Povidencia, Salmonela, Serralia, Shigella, Morganella, and Yersinia, etc.. Specifically, bacteria of the Enterobacteriaceae family which have been classified in the database of the National Center for Biotechnology Information (NCBl) can be employed (http://www.ncbi. n Im. nih.gov/htbin-post/Taxonomy/wgetorg?mode=Tree&id= 1236&lvl=3&keep= l &srchmode=l&unlock). Of these, the parent strain from the Enterobacteriaceae family employed for modification is desirably from the genus Escherichia, Enterobacter, or Pantoea.
The parent strain from the genus Escherichia which is used to obtain the bacterium of the present invention is not specifically limited. Specifically, strains described by Neidhardt et al. may be employed (Neidhardt, F. C. et al., Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington D. C, 1029 table 1 ). Of these, one example is Escherichia coli. Specific examples of Escherichia coli are Escherichia coli W31 10 (ATCC 27325) and Escherichia coli MG 1655 (ATCC 47076), both of which are derived from the prototype wild-type strain K12.
These strains can be obtained from the American Type Culture Collection (P.O.Box 1549 Manassas, VA 20108 USA (703) 365-2700) using the assigned registration number (http://www.atcc.org (reference)). The registration number corresponding to each strain is listed at the American Type Culture Collection web site and catalog. Examples of strains from the genus Enterobacter are Enterobacter agglomerans and Enterobacter aerogenes. An example of a strain from the genus Pantoea is Pantoea ananatis. In recent years, based on 16S rRNA base sequencing analysis, Enterobacter agglomerans has on occasion been reclassified as Panloeu agglomerans, Pantoea ananatis, or Pantoea stewartii. (lnt.J.Syst.Bacteriol.,43, 162-173) For the present invention, any bacterium classified in the Enterobacteriaceae family, whether under genus Enterobacter or Pantoea, may be employed. Strains Pantoea ananatis AJ 13355 (FERM BP-6614), AJ 13356 (FERM BP-6615), AJ 13601 (FERM BP-7207), or any derivative thereof may be
employed to breed Pantoea ananatis by genetic engineering methods. When isolated, these strains were identified and deposited as Enterobacter agglomerans. As stated above, analysis by 16S rRNA base sequencing has caused them to be reclassified as Pantoea ananatis.
Methods of imparting the ability to produce an L-amino acid to a bacterium of the Enterobacteriaceae family will be described below.
To impart the ability to produce an L-amino acid, an auxotrophic mutant, an analog-resistant strain, or a metabolic regulation mutant can be obtained, or a recombinant strain having enhanced expression of an L-amino acid biosynthetic enzyme can be created. Conventionally, methods that have been employed to breed coryneform bacteria and bacteria from the genus Escherichia can be used (See "Amino Acid Fermentation", the Japan Scientific Societies Press [Gakkai Shuppan Center], 1st Edition, published on May 30, 1986, pp.77-100). Here, in the breeding of L-amino acid producing bacteria, one or more properties such as an auxotrophic mutation, analog resistance, and a metabolic regulation mutation may be imparted. When a recombinant strain is created, the activity of one or more L-amino acid biosynthetic enzymes may be enhanced. Furthermore, imparting properties such as an auxotrophic mutation, analog resistance, and a metabolic regulation mutation may be combined with the methods for enhancing an activity of one or more L-amino acid biosynthetic enzymes.
An auxotrophic mutant strain, L-amino acid analog resistant strains, or metabolic regulation-mutated strain with the ability to produce an L-amino acid can be obtained by subjecting a parent strain or wild-type strain to a typical mutation treatment, such as exposure to X-rays or UV radiation, or treatment with a mutagenic agent such as N-methyI-N'-nitro-N-nitrosoguanidine(NTG), and selecting from among the obtained mutants those which exhibit an auxotrophic mutation, analog resistance, or a metabolic regulation-mutation and which also have the ability to produce an L-amino acid. Genetic recombination can be employed to enhance the L-amino acid biosynthetic enzyme activity of a bacterium which produces L-amino acids.
Specific examples of L-lysine analog-resistant strains and metabolic
regulation-mutated strains which are able to produce L-lysine include Escherichia coli strain AJ l 1442 (FERM BP- 1543, NRRL B-12185; see JP56-18596A and U.S. 4,346, 170) and Escherichia coli strain VL61 1 (JP 2000-189180A). Strain WC 196 (see WO96/ 17930) may also be employed as an Escherichia coli L-lysine producing strain. Strain WC 196 was originally bred by imparting resistance to S-(2-aminoethyl)cysteine (AEC) to strain W31 10, which is derived from Escherichia coli K-12. The resulting strain was also designated as Escherichia coli strain AJ 13069 and deposited on December 6, 1994, accession number PERM P-14690, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1-1 -1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566). Then, the deposit was converted to an international deposit under the provisions of the Budapest Treaty on September 29, 1995, and received an accession number of FERM BP-5252.
Furthermore, enzymatic activity relating to L-lysine biosynthesis can be enhanced to construct a bacterium which produces L-lysine. Examples of genes encoding proteins involved in L-lysine biosynthesis include, but are not limited to, genes encoding diaminopimelic acid pathway enzymes, such as the dihydrodipicolinate synthase gene (dapA), aspartokinase gene (lysC), dihydrodipicolinate reductase gene (dapB), diaminopimelate decarboxylase gene (lysA), diaminopimelate dehydrogenase gene (ddh) (WO96/40934), phosphenolpyruvate carboxylase gene (ppc) (JP60-87788), aspartate aminotransferase gene (aspC) (JP 6-102028), diaminopimelate epimerase gene (dapF) (JP2003- 135066), aspartate semialdehyde dehydrogenase gene (asd) (WO00/61723), and genes encoding aminoadipic acid pathway enzymes, such as the homoaconitate hydratase gene (JP2000-157276).
In addition to methods of increasing the level of expression of Lys biosynthetic enzymes, methods of increasing the level of expression of the gene involved in energy efficiency (cyo) (EPI 170376 ), the nicotinamide nucleotide transhydrogenase gene (pntAB)(US 5,830,716), ybjE gene(WO2005/073390) have also been developed.
For example, the ability to produce L-lysine can be imparted by introducing a gene encoding a protein involved in L-lysine biosynthesis into a host as set forth below.
That is, a gene fragment encoding an L-lysine biosynthesis gene is ligated to a vector which is able to function in a host microorganism which has been employed to produce L-lysine. The chosen vector preferably is a multicopy-type vector, which is then used to transform the host. Since the number of copies of the genes encoding proteins involved in L-lysine biosynthesis increases in the transformed host cell, the expression level is increased and the activity of the enzymes is enhanced.
The genes encoding proteins involved in L-lysine biosynthesis are not specifically limited other than that they must be able to be expressed in the host microorganism. Examples include genes derived from Escherichia coli and coryneform bacteria. Since the genomic sequences of both Escherichia coli and Corynebacterium glutamicum are known, a primer can be synthesized based on the sequences of the genes, and PCR employing the chromosomal DNA of a microbe such as Escherichia coli K-12 can be employed to obtain the genes (Science 277 (533 I), 1453-1474 ( 1997) Proceedings of the 9th International Symposium on the Genetics of Industrial Microorganisms: 21 ). The plasmids which may be used for gene cloning may be capable of autonomous replication in the bacteria from the Enterobacteriaceae family; specific examples are pBR322, pTWV228 (Takara-Bio Co.), pMWl 19 (Nippon Gene Corp.), pUC19, pSTV29 (Takara-Bio Co.), and RSFlOlO (Gene vol. 75 (2), p. 271 -288, 1989). Phage DNA vectors may also be employed.
To ligate the target gene to the above-described vector to prepare recombinant DNA, the vector is digested with a restriction enzyme matched to the end of the DNA fragment containing the target gene. The ligation is usually conducted with a ligase such as T4 DNA ligase. The target genes may be introduced into a variety of separate vectors, or introduced into a single vector. The usual methods known to those skilled in the art can be employed to digest and ligate DNA, prepare chromosomal DNA, conduct PCR, prepare plasmid DNA, transform hosts, and determine oligonucleotides for use as primers. These methods are described in Sambrook, J., Fritsch, E. F., and Maniatis, T., "Molecular
Cloning A Laboratory Manual, Second Edition", Cold Spring Harbor Laboratory Press, (1989). Any method which achieves adequate transformation efficiency can be used to introduce recombinant DNA that has been prepared as set forth above into the microbe. An example is the electroporation method (Canadian Journal of Microbiology, 43. 197 (1997)).
Enhancing expression of the genes encoding proteins involved in L-lysine biosynthesis can be achieved by introducing multiple copies of the target gene into the chromosomal DNA of the microorganism. Multiple copies of the target gene can be introduced into the chromosomal DNA of the microorganism by using a DNA in which multiple copies are present in chromosomal DNA as a target in homologous recombination. Such site-specific introduction of mutations based on genetic recombination using homologous recombination is already established. There is a method employing single strand DNA and a method employing a plasmid containing temperature-sensitive replication origin (U.S. Patent 6,303,383 or JP05-007491 A). Repetitive DNA and inverted repeats present on the ends of transposable elements can be employed as sequences in which multiple copies are preset in chromosomal DNA. Alternatively, as disclosed in EP0332488, multiple copies of the target gene can be introduced into a chromosome. With either method, as a result of increasing the number of copies of the target gene in the transformant, the enzymatic activity of L-lysine biosynthesis increases.
In addition to the above-described genetic amplification, L-lysine biosynthesis enzyme activity can be enhanced by replacing the expression regulatory sequence of the promoter of the target gene with a stronger one (JPO 1 -2 ] 5280A). For example, 1 ac promoter, trp promoter, trc promoter, tac promoter, lambda phage PR promoter, lambda phage PL promoter, tet promoter are all known as strong promoters. Substitution with these promoters enhances the enzymatic activity by increasing expression of the target gene. A paper by Goldstein (Prokaryotic promoters in biotechnology. Biotechnol. Annu. Rev., 1995, 1 , 105-128) describes methods of evaluating the strength of promoters and gives examples of strong promoters.
This can also be achieved by modifying factors relating to regulation of
expression of the target gene, such as operators and repressors (Hamilton et al,; J Bacteriol. l 989 Sep; 171(9): 4617-22). As is disclosed in WO00/18935, it is possible to introduce a substitution of several bases into the promoter region of a target gene to increase its strength.
Furthermore, the substitution of several nucleotides into the spacer between a ribosome binding site (RBS) and a start codon, particularly into the sequence immediately upstream from a start codon, is known to have a strong effect on mRNA translation efficiency. The expression adjustment regions of the promoters or the like of the target gene can be determined using promoter search vectors and gene analysis software such as GENETYX. Substitution of the expression regulating sequence can be conducted, for example, in the same manner as in the above-described gene substitution employing temperature-sensitive plasmids.
Furthermore, the L-lysine producing bacterium of the present invention may have reduced or deficient activity of enzymes which catalyze reactions that branch off from the L-Iysine biosynthesis pathway and produce compounds other than L-lysine. Examples of such enzymes are homoserine dehydrogenase, lysine decarboxylase (cad A, l dcC), and malic enzyme. Strains in which the activity of these enzymes is reduced or eliminated are described in WO 95/23864, WO96/17930, and WO2005/010175.
Examples of methods of reducing or eliminating the intracellular activity of an enzyme include mutating or deleting a gene encoding the enzyme in cells of a microorganism so that intracellular activity is reduced or eliminated as compared to a non-mutated strain. For example, this can be achieved by using recombination to inactivate the gene encoding the enzyme on the chromosome, or to modify an expression regulating sequence such as a promoter or the Shine-Dalgarno (SD) sequence. (WO95/34672,Biotechnol Prog 1999, 15,58-64)
This can also be achieved by introducing an amino acid substitution (missense mutation) into the region of the gene encoding the enzyme on the chromosome, introducing a stop codon (nonsense mutation), introducing or deleting one or two bases to create a frame shift mutation, or partially deleting a portion or a region of the gene, or the
entire gene (Journal of Biological Chemistry 272: 861 1 -8617 ( 1997), Journal of Antimicrobial Chemotherapy 200 46, 793-79. J. biological Chemistry vol 272 NO.13 pp861 1-8617). Enzymatic activity can also be decreased or eliminated by constructing a gene encoding a mutant enzyme which lacks a coding region, using homologus recombination to replace the normal gene on the chromosome with this gene, and introducing a transposon or IS factor into the gene.
For example, the following methods may be employed to introduce a mutation causing a decrease of, or eliminating, the above enzyme activity by gene recombination. A portion of the sequence of the targeted gene is modified, a mutant gene that does not produce a normally functioning enzyme is prepared, DNA containing this gene is used to transform a microorganism from the Enterobacteriaceae family, and the mutant gene is made to recombine with the gene on the chromosome, which results in replacing the target gene on the chromosome with the mutant gene. Such gene substitution using homologous recombination can be conducted by methods employing linear DNA, such as the method known as "Red-driven integration" (Proc. Natl. Acad. Sci. USA, 2000, vol. 97, No. 12, p 6640-6645), and by methods employing a plasmid containing a temperature-sensitive replication (Proc. Natl. Acad. Sci. USA, 2000, vol. 97, No. 12, p 6640-6645, U.S. Patent 6,303,383 or JP05-007491 A). Furthermore, the incorporation of a site-specific mutation by gene substitution using homologous recombination such as set forth above can also be conducted with a plasmid lacking the ability to replicate in the host.
The above-described methods of enhancing and decreasing the activity of enzymes relating to L-lysine biosynthesis can also be applied to the breeding of bacteria producing other L-amino acids. Methods of breeding bacteria producing other L-amino acids will be described below.
The L-glutamic acid producing bacterium employed in the present invention can be a microorganism from the Enterobacteriaceae family that has been modified to enhance expression of a gene encoding an enzyme related to L-glutamic acid biosynthesis, for example. Examples of enzymes related to L-glutamic acid biosynthesis are glutamate dehydrogenase (also referred to as "GDH" hereinafter), glutamine synthetase, glutamate
-I I -
synthase, isocitrate dehydrogenase, aconate hydratase, citrate synthase (also referred to as "CS" hereinafter), pyruvate carboxylase, pyruvate dehydrogenase, pyruvate kinase, phosphophenol pyruvate synthase, enolase, phosphoglyceromutase, phosphoglycerate kinase, glyceraldehyde-3-phosphate dehydrogenase, triosephosphate isomerase, fructose bisphosphate aldolase, phosphofructokinase, and glucose phosphate isomerase. Among these enzymes, one or more from among CS, PEPC, and GDH are desirable, with all three being preferred.
For example, U.S. Patents 6,197,559 and 6,331 ,419 and European Patent 0999282 describe microbes from the Enterobacteriaceae family that have been modified by methods such as those set forth above to enhance expression of the citrate synthase gene, phosphenolpyruvate carboxylase gene, and/or glutamate dehydrogenase gene. A microorganism which has reduced or eliminated activity of enzymes which catalyze reactions that branch off from the L-glutamic acid biosynthesis pathway and produce compounds other than L-glutamic acid may also be employed as the microorganism from the Enterobacteriaceae family which has the ability to produce L-glutamic acid. Examples of enzymes which catalyze reactions that branch off from the L-glutamic biosynthesis pathway and produce compounds other than L-glutamic acid are: 2-oxoglutamate dehydrogenase, isocitrate lyase, phosphate acetyl transferase, acetate kinase, acetohydroxamate synthase, acetolactate synthase, formate acetyl transferase, lactate dehydrogenase, glutamate decarboxylase. Among these, the reduction or elimination of 2-oxoglutamate dehydrogenase activity is desirable.
U.S. Patents 6,197,559 and 6,331 ,419 describe methods of eliminating or reducing the 2-oxoglutamate dehydrogenase activity in a microrganism from the Enterobacteriaceae family. Specific examples of a bacterium from the Enterobacteriaceae family in which 2-oxoglutamate dehydrogenase activity has been eliminated or reduced are:
Pantoea ananatis AJ 13601 (FERM BP-7207)
Klebsiella planticola A J 13410 (FERM BP-6617)
Pantoea ananatis AJ 13355 (FERM BP-6614)
Escherichia cυli AJ 12949 (FERM BP-4881 ).
Strain AJ 12949 was deposited on December 28, 1993, as depository number FERM P-14039, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1 -1 -1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566), and converted to a international deposit under the provisions of the Budapest Treaty on November 1 1 , 1994, and given accession number FERM BP-4881.
Strain AJl 3601 was deposited at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology, Ministry of International Trade and Industry on August 18, 1999 and given an accession number of FERM P- 17516. The deposition was converted to an international deposition under the provisions of Budapest Treaty on July 6, 2000 and given an accession number of FERM BP-7207.
The preferred L-tryptophan producing microorganism employed in the present invention enhances the activity of one or more of the following proteins: anthranilate synthase, phosphoglycerate dehydrogenase, and tryptophan synthase. Anthranilate synthase and phosphoglycerate dehydrogenase are subject to feedback suppression by L-tryptophan and L-serine, respectively. Thus, their enzymatic activity can be enhanced by retaining a desensitized mutant enzyme. Specifically, for example, the anthranilate synthase gene (trpE) and/or the phosphoglycerate dehydrogenase gene (serA) can be mutated so as to not be subject to feedback inhibition, and the mutant gene obtained can be introduced into a microorganism of the Enterobacteriaceae family to obtain a microorganism retaining the desensitized enzyme. Specific examples of such microbes are transformed strains obtained by introducing plasmid pGH5 having a mutant form of serA encoding desensitized phosphoglycerate dehydrogenase into Escherichia coli SV 164 which has retained a desensitized from of anthranilate synthase (WO94/08031).
A microorganism into which a recombinant DNA containing a tryptophan operon has been introduced is a further example of a desirable L-tryptophan-producing
bacteria. One specific example is Escherichia coli into which a tryptophan operon containing a gene encoding desensitized anthranilate synthase has been introduced (JP57-71397A and JP 62-244382A, and U.S. Patent 4,371 ,614). Among the tryptophan operons, enhancement of the expression of the gene (trpBA) encoding tryptophan synthase also enhances or imparts L-tryptophan production capability. Tryptophan synthase contains α and β subunits encoded by trpA and trpB, respectively.
Further examples of L-tryptophan producing bacteria are Escherichia coli strain AGX17(pGX44) (NRRL B-12263), which requires L-phenylalanine and L-tyrosine, and strain AGX6(pGX50)aroP (NRRL B-12264), which retains a plasmid pGX50 containing a tryptophan operon (see U.S. Patent 4,371,614 for both of these).
Examples of L-phenylalanine-producing strains derived from the K-12 strain include the AJ 12739 strain in which the tyrA gene and the tyrR gene are disrupted (tyrA::TnlO, tyrR) (VKPM B-8197), and a strain in which the yddG gene and the yedA gene, which are involved in phenylalanine excretion, are amplified (WO 03/044192).
The strain which is modified to enhance genes which encode proteins involved in aromatic amino acids biosynthesis can be used as tryptophan and/or phenylalanine-producing strain, such genes include genes which encode proteins involved in a common pathway for aromatic acids, such as aroF, aroG, aroH, aroB, aroD, aroE, aroK, aroL, aroA, and aroC genes.
A strain having 6-dimethylaminopurine resistance (JP 5-304969A) is desirable as the L-threonine producing bacterium employed in the present invention. Examples of recombinant bacteria of the genus Escherichia are a strain in which a mutation producing excessive L-threonine biosynthase has been introduced into the threonine biosynthesis gene and the gene has been amplified on a plasmid (JP l -29559AandJP5-227977A), a strain in which a threonine operon has been amplified with a plasmid (Japanese Patent Application Publication No. Hei 2-109985), and a strain in which a gene encoding pyruvate carboxylase and a gene encoding nicotinamide nucleotide transhydrogenase have been amplified (JP2002-51787A).
A further example is Escherichia coli strain VKPM B-3996 (see U.S. Patent
5, 175,107). Strain VKPM B-3996 was deposited on November 19, 1987, with the Russian. National Collection of Industrial Microorganisms (VKPM), GNIl Genetika, and given deposit number VKPM B-3996. Strain VKPM B-3966 harbors a plasmid pVIC40 (WO90/04636), which is obtained by inserting a threonine biosynthesis gene (threonine operon: thrABC) into a broad host vector plasmid pAYC32 (see Chistorerdov, A. Y., Tsygankov, Y.D. Plasmid, 1986, 16, 161-167) having a streptomycin-resistance marker. In pVlC40, feedback inhibition by L-threonine of the aspartokinase I homoserine dehydrogenase I encoded by thrA in the threonine operon is repressed.
The Escherichia coli VKPM B-5318 strain (EP 0593792B) may also be used as an L-threonine-producing strain. VKPM B-5318 was deposited at the Russian National Collection of Industrial Microorganisms (VKPM), GNII Genetika(Dorozhny proezd. 1, Moscow 1 13545, Russian Federation), with a registration number of VKPM B-5318 on November 19, 1987. The VKPM B-5318 strain is autotroph to L-isoleucine,and the threonine operon encoding the threonine biosynthesis enzyme is located downstream Cl temparature-sensitive represser,PR-promoer and N end of Cro protein derived from λphage,moreover,the strain harbors plasmid DNA constructed so that the expression of threonine biosynthesis gene is regulated by psromoter and represser derived from λphage.
Preferably, the bacterium of the present invention is additionally modified to enhance expression of one or more of the following genes: the mutant thrA gene which codes for aspartokinase homoserine dehydrogenase I and is resistant to feedback inhibition by threonine; the thrB gene which codes for homoserine kinase; the thrC gene which codes for threonine synthase; the rhtA gene which codes for a putative transmembrane protein; the asd gene which codes for aspartate-β-semialdehyde dehydrogenase; and the aspC gene which codes for aspartate aminotransferase (aspartate transaminase);
The thrA gene which encodes aspartokinase homoserine dehydrogenase 1 of Escherichia coli has been elucidated (nucleotide positions 337 to 2799, GenBank accession NC_000913.2, gi: 49175990). The thrA gene is located between the thrL and thrB genes on
the chromosome of E. coli K-12. The thrB gene which encodes homoserine kinase of Escherichia coli has been elucidated (nucleotide positions 2801 to 3733, GenBank accession NC_000913.2, gi: 49175990). The thrB gene is located between the thrA and thrC genes on the chromosome of E. coli K-12. The thrC gene which encodes threonine synthase of Escherichia coli has been elucidated (nucleotide positions 3734 to 5020, GenBank accession NC_OOO9I 3.2, gi: 49175990). The thrC gene is located between the thrB gene and the yaaX open reading frame on the chromosome of E. coli K-12. All three genes functions as a single threonine operon. The regulation sequence which encode attenuater and leader peptide present upstream threonine operon. To enhance the expression of threonine operon, a leader sequence and/or an attenuator is desirably removed from said operon.(WO2005/049808, WO2003/097839)
A mutant thrA gene which codes for aspartokinase homoserine dehydrogenase I and is resistant to feedback inhibition by threonine, as well as, the thrB and thrC genes can be obtained as one operon from the well-known plasmid pVIC40 which is present in the threonine producing E. coli strain VKPM B-3996. Plasmid pVIC40 is described in detail in U.S. Patent No. 5,705,371.
The rhtA gene exists at 18 min on the E. coli chromosome close to the glnHPQ operon, which encodes components of the glutamine transport system. The rhtA gene is identical to ORFl (ybi F gene, nucleotide positions 764 to 1651 , GenBank accession number AAA218541 , gi:440181) and located between the pexB and ompX genes. The unit expressing a protein encoded by the ORFl has been designated the rhtA gene (rht: resistance to homoserine and threonine). Also, it was revealed that the rh(A23 mutation is an A-for-G substitution at position -1 with respect to the ATG start codon (ABSTRACTS of the 17th International Congress of Biochemistry and Molecular Biology in conjugation with Annual Meeting of the American Society for Biochemistry and Molecular Biology, San Francisco, California August 24-29, 1997, abstract No. 457, EP 1013765 A).
The asd gene of E. coli has already been elucidated (nucleotide positions 357251 1 to 3571408, GenBank accession NC_000913.1 , gi: 16131307), and can be obtained by PCR (polymerase chain reaction; refer to White, T.J. et al., Trends Genet., 5,
185 (1989)) utilizing primers prepared based on the nucleotide sequence of the gene. The asd genes of other microorganisms can be obtained in a similar manner. Also, the aspC gene of E. coli has already been elucidated (nucleotide positions 983742 to 984932, GenBank accession NC_000913.1 , gi: 16128895), and can be obtained by PCR. The aspC genes of other microorganisms can be obtained in a similar manner.
Examples of L-histidine producing bacteria that are desirable for use in the present invention are strains of Escherichia coli FERM-P5038 and 5048, both of which incorporate a vector containing DNA encoding an L-histidine biosynthetic enzyme (JP56-005099A), a strain having the vector containing the amino acid export gene rht (European Patent Publication No. 1016710), and Escherichia coli strain 80, which is imparted with resistance to sulfaguanidine, D,L-l ,2,4-triazole-3-alanine, and streptomycin (VKPM B-7270 Russian Patent Publication No. 2,1 19,536).
A microorganism with enhanced expression of genes encoding enzymes of the L-histidine biosynthesis pathway may be employed as a microorganism which has the ability to produce L-histidine. Examples of enzymes involved in L-histidine biosynthesis are ATP phosphoribosyltransferase (hisG), phosphoribosyl AMP cyclohydrolase (hisl), phosphoribosyl ATP pyrophosphohydrolase (hisIE), phosphoribosylformimino-5-aminoimidazole carboxamide ribotide isomerase (hisA), amidotransferase (hisH), histidinolphosphate aminotransferase (hisC), histidinolphosphatase (hisB), and histidinoldehydrogenase (hisD).
Desirable examples of the L-cysteine producing bacterium of the present invention are bacteria which have reduced cystathionine-β-lyase activity (JP2003-169668A) and Escherichia coli bacteria which retain serineacetyltransferase, the activity of which is decreased by feedback suppression by L-cysteine (JPl 1 -155571 A). Desirable examples of L-proline producing bacterium of the present invention are Escherichia coli strain 702 (VKPMB-801 1 ), which is resistant to 3,4-dehydroxypropline and azatadine-2-carboxyIate, and strain 702ilvA (VKPMB-8012), which lacks the HvA of strain 702 (JP2002-300874A).
Examples of a bacterium which produces L-arginine are Escherichia coli strains
with resistance to α-methylmethionine, p-fluorophenylalanine, D-arginine, arginine hydroxamic acid, S-(2-aminoethyl)cysteine, α-methylserine, β-2-thienylalanine, or sulfaguanidine (JP56-106598A). Escherichia coli strain 237 (US Patent Application No. 2003/058315), which is an L-arginine producing bacterium harboring mutant N-acetylglutamate synthass, is also a desirable L-arginine producing strain. This strain was deposited on April 10, 2000, with the Russian National Collection of Industrial Microorganisms (VKPM), GNII Genetika under number VKPM B-7925 and was converted on May 18, 2001 to an International Deposit under the Budapest Treaty. A derivative of strain 237 that produces L-arginine and has enhanced acetic acid utilizing capability, Escherichia coli strain 382 (EPl 170358A) can also be employed. Escherichia coli strain 382 was deposited on April 10, 2000, with the Russian National Collection of Industrial Microorganisms (VKPM) as deposit number VKPM B-7926.
Microorganisms with an enhanced level of expression of genes encoding enzymes related to L-arginine biosynthesis can also be employed as a microorganism having L-arginine production ability. Examples of enzymes related to L-arginine biosynthesis are one or more selected from among N-acetylglutamate synthase (argA), N-acetylglutamylphosphate reductase {argC), ornithine acetyltransferase (argj), N-acetylglutamate kinase (argB), acetylornithine transferase (argD), acetylomithine deacetylase (argE), ornithinecarbamoyltransferase (argF), argininosuccinate synthase (argG), argininosuccinate lyase (argff), and carbamoylphosphate synthase (carAB).
L-leucine producing bacteria of the present invention include a bacterium of the genus Escherichia in which the branched chain amino acid transferase encoded by the HvE gene is inactivated and the activity of aromatic amino acid transaminase encoded by the tyrB gene is enhanced (JP2004-024259), Escherichia coli strain H-9068 (ATCC21530), Escherichia cυli strain H-9070 (FERM BP-4704), Escherichia coli strain 9072 (FERM BP-4706) which is resistant to 4-azoleucine or 5,5,5-trifluoroleucine (U.S. Patent No. 5,744,331 ) strains of Escherichia coli in which feedback inhibition of isopropylmalate synthase by L-leucine has been repressed (European Patent No. 1067191 ), and Escherichia coli strain AJ l 1478 which is resistant to β-2-thienylalanine and β-hydroxyleucine (U.S.
Patent 5,763,231).
The bacterium of the present invention may be improved by enhancing the expression of one or more genes involved in L-leucine biosynthesis. Examples include genes of the leuABCD operon, which are preferably represented by a mutant leuA gene coding for isopropylmalate synthase freed from feedback inhibition by L-leucine (US Patent 6,403,342). In addition, the bacterium of the present invention may be improved by enhancing the expression of one or more genes coding for proteins which excrete L-amino acid from the bacterial cell. Examples of such genes include the b2682 and b2683 genes iygaZH genes) (EP 1239041 A2).
Examples of L-isoleucine producing bacteria are Escherichia coli variants with resistance to 6-dimethylaminopurine (JP5-304969A), Escherichia coli mutants with resistance to L-isoleucine hydroxamate (JP5-130882A), Escherichia coli strains with resistance to thiaisoleucine (JP5-130882A), Escherichia coli mutants with resistance to DL-ethionine (JP5-130882A), and variants with resistance to arginine hydroxamate(JP5-130882A) with the ability to produce L-isoleucine. Examples of recombinant bacteria of the genus Escherichia are strains with genes encoding proteins involved in L-isoleucine biosynthesis, such as threonine deaminase or acetohydroxate synthase enhanced with plasmids (JP2-458A, 2-42988A, and 8-47397A).
An example of a bacterium producing L-valine include, but are not limited to, strains which have been modified to overexpress the HvGMEDA operon (U.S. Patent No. 5,998,178). It is desirable to remove the region of the HvGMEDA operon which is required for attenuation so that expression of the operon is not attenuated by L-valine that is produced. Furthermore, the UvA gene in the operon is desirably disrupted so that threonine deaminase activity is decreased.
Examples of parent strains for deriving L-valine-producing bacteria of the present invention include also include mutants having a mutation of amino-acyl t-RNA synthetase (U.S. Patent No. 5,658,766). For example, E. coli VL 1970, which has a mutation in the ileS gene encoding isoleucine tRNA synthetase, can be used. E. coli VLl 970 has been deposited in the Russian National Collection of Industrial
Microorganisms (VKPM) (Russia, 1 13545 Moscow, 1 Dorozhny Proezd.) on June 24, 1988 under accession number VKPM B-441 1.
Furthermore, mutants requiring lipoic acid for growth and/or lacking H+-ATPase can also be used as parent strains (WO96/06926).
The microorganism of the present invention can be obtained by modifying a microbe of the Enterobactehaceae family which has the ability to produce L-amino acid so that it cannot produce type I fimbrial adhesin protein, as set forth above. When breeding a microbe of the Enterobacteriaceae family of the present invention, it makes no difference whether the ability to produce an L-amino acid is imparted first or a modification to prevent production of type 1 fimbrial adhesin protein is made first. Furthermore, a microbe from the Enterobacteriaceae family which has the ability to produce an L-amino acid may be modified so that it does not produce type I fimbrial adhesin protein, or a microbe from the Enterobacteriaceae family that no longer produces type I fimbrial adhesin protein may be imparted with the ability to produce an L-amino acid. It suffices for the microorganism of the present invention to be modified so that it does not normally produce type 1 fimbrial adhesin protein in the manner of wild-type strains or unmodified strains. However, it is desirable for the microbe of the present invention to have an ability to cause accumulation of an L-amino acid exceeding that of these strains.
The "fimbriae" of the present invention are filamentous protrusions present on the outer cell membrane of microorganism from the Enterobacteriaceae family. Fimbriae can be divided into two types. One type is the fimbriae which has no direct relation to sexual processes such as conjugation and gene transfer, and the second type is sexual fimbriae that are produced on the outer layer of gene-donor bacteria and are essential to conjugation with gene-recipient bacteria. The "fimbriae" referred to in the present invention are the former, and are not directly related to sexual processes such as conjugation, gene transfer, or the like (Shoji Mizushima, Kinichiro Miura, Bacterial Anatomy 129 (1979)).
In the present invention, the phrase "type 1 fimbrial adhesion protein" refers to a protein which belongs to a group of proteins which control the formation of fimbriae, and
furthermore, the protein has a function blocking a coagulation of red corpuscles by D-mannose.
The phrase "modified so as not to produce type I fimbrial adhesion protein" refers to when the quantity of type 1 fimbrial adhesin protein that is produced is lower than that in unmodified strains, and when the conformation of the protein is modified so that normal fimbriae cannot be produced by microbes from the Enterobacteriaceae family. For example, wild-type strains such as Escherichia coli strain W31 10 (ATCC 27325), which is derived from the prototypical wild-type strain K 12, and Escherichia coli MG 1655 (ATCC 47076), are examples of control strains from the genus Escherichia. Confirmation of a decrease in the quantity of type 1 fimbrial adhesin protein which is produced, or the lack of production of type I fimbrial adhesin protein can be determined by the immunofluorescent antibody method, a decrease in the level of coagulation in the presence of D-mannose, or the lack of ability for coagulation (see the methods of Pallesen et al.: Microbiology 141 ; 2839-2848).
The type I fimbrial adhesin protein (Seq. ID No. 2) from the genus Escherichia is encoded by the geney/wH(Seq. ID No. 1 ). Type I fimbrial adhesin proteins from other microbes of the Enterobacteriaceae family includey?wHhomologs; The homologs of βniH from Enterobacteriaceae can be obtained by searching for genes having high homology to the gene denoted by Seq. ID No. 1 with BLAST (http://blast.genome.jp).
The GenBank Accession number of the amino acid sequence of the type 1 fimbrial adhesin protein from Escherichia microbes and they?wH gene encoding this protein are given in Table I . Examples of the group of genes which encode proteins which control formation of fimbriae in the bacteria from genus Escherichia are fimB, fimE, fimA, fiml, fimC, fimD, fimF, fimG, and fimΗ. The amino acid sequences encoded by these genes, their gene sequences, and their GenBank Accession numbers are given in Table 1.
Table

Since there may be some differences in DNA sequences between the genera or strains of the Enterobacteήaceae family, the gene encoding the type I fimbrial adhesin protein can be any DNA which hybridizes under stringent conditions with a probe prepared from the base sequence of SEQ. ID. No. l or a homologous gene of the SEQ ID No. l sequence. The term "stringent conditions" refers to conditions under which specific
hybrids form and nonspecific hybrids do not form. By way of example, these are conditions under which highly homologous fragments of DNA, for example, DNA with a degree of homology of not less than 80%, preferably not less than 90%, and most preferably not less than 95% will hybridize. A further example is Southern blot hybridization washing conditions at a temperature and salt concentration corresponding to 6O0C, 1 x SSC, 0.1 percent SDS, preferably 0.1 x SSC, 0.1 percent SDS, and more preferably, 680C, 0.1 x SSC, and 0.1 percent SDS, conducted one, two, or three times. The length of the probe can be suitably selected based on the hybridization conditions, but normally ranges from 100 bp to 1 Kbp.
The phrase "modified so as to not produce type I fimbrial adhesin protein" means a modification that does not destroy the fimbriae, but causes type I fimbrial adhesin protein to not function normally. Such a modification may be the introduction of a mutation into the protein with a drug or the like, weakening adhesion, or the use of genetic engineering or the introduction of a mutation into the gene relating to the formation of type I fimbrial adhesin so as to reduce the level of production of type 1 fimbrial adhesin protein, or breed a bacterium that does not produce type 1 fimbrial adhesin protein. For example, in microorganisms of the Enterobacteήaceae family that do not produce type I fimbrial adhesin protein, the transcription or translation of the gene encoding the type I fimbrial adhesin protein may be interfered with, resulting in either in the protein not being produced or being produced at a low level. A mutation may be introduced to the gene encoding the type I fimbrial adhesin protein on the chromosome and/or the region controlling expression of the gene, so that type 1 fimbrial adhesin proteins do not function properly. Furthermore, a mutation may be introduced into a gene encoding type 1 fimbrial adhesin protein as to reduce the level of expression of genes coding for type I fimbrial adhesin protein on the chromosome.
Specifically, modification so as to not produce type I fimbrial adhesin can be achieved by deleting the gene encoding type 1 fimbrial adhesin protein on the chromosome, or by modifying an expression regulating sequence such as a promoter or Shine-Dalgamo (SD) sequence. This can also be achieved by introducing an amino acid substitution
(missense mutation) into the coding region of the type I fimbrial adhesin gene on the chromosome, introducing a stop codon (nonsense mutation), adding or deleting one or two bases to create a frame shift mutation, or partially deleting a portion, a region, or the entire gene (Journal of Biological Chemistry 272: 861 1 -8617 ( 1997), Proceedings of the National Academy of Sciences, USA 95, 551 1-5515 (1998), Journal of Biological Chemistry 266, 20833-20839 (1991)).
Furthermore, they?mH gene, located downstream within the operon, modification so as to not produce type I fimbrial adhesin can also be achieved by introducing the mutation into the gene in table 1 located upstream fnnH gene.
In Escherichia coli, it is possible to identify the/?/«H gene shown in Table 1 as the chromosomal gene encoding type 1 fimbrial adhesin protein. To prevent the production of type I fimbrial adhesin protein, the//wHgene can be deleted or a mutation can be introduced into the region encodingyϊwH, as described above. This can also be achieved by introducing a mutation attenuating the expression of the/?/«Hgene upstream from fimH; for example, a frame shift mutation or nonsense mutation can be introduced into one of the genes upstream from//mHthat are indicated in Table 1 , or an upstream gene can be partially deleted. More particularly, as is described further below in the embodiments, a transposon or a gene imparting resistance to an antibiotic can be incorporated in the region encoded by fimΗ, or the mutation described in the Journal of Bacteriology, July, 2001 , 4099-4102 or the mutation described in Molecular Microbiology (2001) 41 (6), 1419-1430, can be incorporated. However, the present invention is not limited thereto.
For example, the following methods can be employed to introduce the above-described mutations by genetic recombination. The target gene on the chromosome can be replaced with a mutated gene by introducing a mutation of the sequence of the target gene, preparing a mutant form of the gene that does not produce a properly functioning enzyme, transforming a microorganism from the Enterobacteriaceae family with DNA containing the gene, and introducing the mutation of the gene to recombine with the gene on the chromosome. Such site-specific incorporation of mutations by gene substitution employing homologous recombination is already established. There exists a method
employing linear DNA and a method employing a plasmid containing a temperature-sensitive replication origin (U.S. Patent 6,303,383 or JP 05-007491 ). The introduction of a mutation at a specific site by gene substitution employing homologous recombination as set forth above can also be conducted using a plasmid which is not able to replicate on the chromosome.
Specifically, a gene encoding the type I fimbrial adhesin protein can be obtained by the following methods. Chromosomal DNA can be prepared from a microorganism belonging to the family Enterobacteriaceae, for example, by the method of Saito and Miura (see H. Saito and K. Miura, Biochem. Biophys. Acta, 72, 619 (1963); Book of Bioengineering Experiments, comp. by the Japan Bioengineering Society, p. 97-98, Baifukan, 1992). A gene encoding the type I fimbrial adhesin protein can be obtained by using a known database such as GenBank, preparing an oligonucleotide, and employing PCR. For example, it can be constructed by referring to the sequences of the genus Escherichia listed in Table 1.
A gene encoding the type I fimbrial adhesin protein prepared as set forth above, or a portion of such a gene, can be used for gene inactivation. However, since the gene used for gene inactivation need only have a degree of homology adequate to occur homologous recombination with the gene encoding the typr* 1 fimbrial adhesin protein on the chromosomal DNA of a microbe of the Enterobacteriaceae family, such a homologous gene may also be employed. Here, the term "degree of homology adequate to undergo homologous recombination" is desirably homology of 80% or more, preferably 90% or more, more preferably 95% or more, and particularly preferably, 97% or more. If the gene is a DNA that will hybridize with the above gene under stringent conditions, homologous recombination will occur. The term "stringent conditions" refers to conditions under which specific hybridization occurs but nonspecific hybridization does not. An example of such conditions is washing once, preferably two or three times, at a salt concentration corresponding to 6O0C, 1 x SSC, 0.1 percent SDS, preferably 0.1 x SSC and 0.1 percent SDS.
The gene encoding type I fimbrial adhesin protein on the chromosome can be
inactivated, for example, by deleting part of the sequence of the gene so it is not able to produce a properly functioning type 1 fimbrial adhesin protein, transforming a group of enterobacteria with DNA containing this gene, and causing the deficient gene to recombine with the gene on the chromosome. Such gene inactivation by gene substitution using homologous recombination is already established. There exist methods employing linear DNA such as the method developed by Datsenko and Wanner called "red-driven integration" (Proc. Natl. Acad. Sci. USA, 2000, vol. 97, No. 12, p 6640-6645), and methods employing plasmids containing temperature-sensitive replication (U.S. Patent 6,303,383 or Japanese Patent Application Publication No. Hei 05-007491 ). Such gene destruction by gene substitution employing homologous recombination as set forth above can also be conducted using a plasmid which is not able to replicate in a host. Examples of deficient genes which have been modified so to not be able to produce type I fimbrial adhesin protein are genes from which all or a part of the region of Seq. ID No. 1 has been deleted, genes incorporating missense mutations, genes into which transposons or marker genes have been inserted, genes incorporating nonsense mutations, and genes incoφorating frame shift mutations.
In addition, a method based on a combination of the method called "red-driven integration" and an excision system derived from lambdn phage (J. Bacteriol. 2002 Sep; 184(18): 5200-3) Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. Cho EH, Gumport RI, Gardner JF.) can be used as the method for disrupting a gene on a chromosome (WO2005/010175).
According to the red-driven integration method, a gene-disrupted strain can be constructed in one step by using a PCR product, which is obtained using synthetic oligonucleotides as primers which are designed to include part of a targeted gene at its 5' terminus, and part of an antibiotic resistance gene at its 3' terminus. Furthermore, the integrated antibiotic resistance gene can be removed by introducing attL and attR, which are attachment sites of lambda phage, and the PCR product, and combining the excision system derived from lambda phage with the red-driven integration method.
Specifically, a strain in which the targeted gene is disrupted and the antibiotic
resistance gene is removed can be obtained by the following method. A linear DNA cassette comprising an antibiotic resistance gene, attachment sites of lambda phage and a target gene is initially prepared. This is usually prepared by PCR using a suitably-prepared template.
A template in which attL and attR (SEQ ID NO: 9 (GenBank accession No. M l 2458 and SEQ ID NO: 10 (GenBank accession No. M 12459)) are inserted at respective terminals of an antibiotic resistance gene is used as a template of the linear DNA cassette. The template may be plasmid for example pM W 1 18-attL-Tc-attR, pM W 1 18-attL Cm-attR, (Fig.l) , a gene inserted on a chromosome, or a synthetic oligonucleotide.
While the antibiotic resistance gene is preferably a chloramphenicol resistance gene, a streptomycin resistance gene, or an ampicillin resistance gene, any antibiotic resistance gene can be used provided that the gene functions as an antibiotic resistance gene in Enterobacteriaceae family a and is different from a marker gene which may be contained in two helper plasmids as described below. To easily confirm the acquisition of the antibiotic resistance, the antibiotic resistance gene which is employed can be one whereby the expression is increased by replacing a promoter sequence and the like, or one in which a mutation is introduced in its structural gene sequence so that an enzyme activity is increased. The linear DNA cassette is prepared in the following order from the 5'terminus: (targeted gene 5' sequence)-(attL)-(antibiotic resistance gene)-(attR)-(targeted gene 3' sequence).
The linear DNA cassette is integrated into the chromosome. As a helper plasmid for integrating the linear DNA cassette into chromosome, pKD46 can be used (Proc. Natl. Acad. Sci. USA, 2000, 97, 6640-6645). pKD46 include temperature-sensitive replication origin and ampicillin resistance gene, a 2,154 nt DNA fragment of lambda phage (GenBank/EMBL accession No. J02459, 31088-33241 ), which contains the genes (gamma, beta, and exo genes) encoding Red recombinase of the lambda Red homologous recombination system and which is under the control of the arabinose-inducible ParaB promoter. pKD46 can be introduced into a host by electroporation. The pKD46-amplified
strain is cultured with arabinose. The linear DNA cassette is introduced at the logarithmic growth phase and incubated at a high temperature to obtain a gene-disrupted strain which is resistant to an antibiotic by the antibiotic resistance gene in the linear DNA cassette. The confirmation of the gene disruption can be made by PCR or measurement of the concentration of L-lysine or L-threonine produced by the strain.
A helper plasmid for excising the antibiotic resistance gene is then introduced. The helper plasmid harbors a gene encoding integrase (Int) (SEQ ID NO: 13, GenBank accession No. J02459. B [gi:215104]) and a gene encoding excisionase (Xis) (SEQ ID NO: 15, GenBank accession No. J02459 [gi:215104]) of lambda phage and shows temperature-sensitive replication. By introduction of the helper plasmid, recombination occurs due to recognition of attL (SEQ ID NO: 1 1) and attR (SEQ ID NO: 12) on the chromosome. The antibiotic resistance gene between attL and attR is excised and as a result, a structure that contains only the attL or attR sequence remains on the chromosome. By incubating at a high temperature, the helper plasmid is lost. Thus, a strain in which the targeted gene is disrupted and the antibiotic gene is eliminated can be obtained.
In addition to the above-described gene manipulation, examples of methods of modification which preclude the production of type I fimbrial adhesin protein are subjecting a group of Enterobacteriaceae family to UV radiation or treating it with N-methyl-N'-nitro-N-nitrosoguanidine (NTG), nitrous acid, or some other mutagenic agent commonly employed in mutation treatments and selecting the strains that are unable to produce type I fimbrial adhesin protein. <2> Method for Producing L-amino acids
The method for producing an L-amino acid of the present invention comprises culturing the microorganism of the present invention in a medium, causing the production and accumulation of L-amino acid in the culture or in the bacterial ceil, and recovering the L-amino acid from the culture or bacterial cell. In the present invention, the cultivation, collection, and purification of an L-amino acid from the medium and the like may be performed in a manner similar to conventional fermentation methods wherein an amino acid is produced using a bacterium.
The medium employed can be one that is conventionally employed in the production of L-amino acids by fermentation. The usual medium contains a carbon source, nitrogen source, inorganic ions, and other organic components as needed. Here, exemplary carbon sources include sugar such as glucose, sucrose, lactose, galactose, fructose, or starch hydrolysis product; alcohol such as glycerol or sorbitol; and organic acids such as fumaric acid, citric acid, or succinic acid. Exemplary nitrogen sources include an inorganic ammonium salt such as ammonium sulfate, ammonium chloride, or ammonium phosphate; organic nitrogen such as a soybean hydrolysis product; ammonia gas; ammonia water; or the like. Other nutrients such as vitamin B 1 and L-homoserine or yeast extract can be employed in suitable quantities as a source of a trace organic nutrient. In addition, as needed, small amounts of potassium phosphate, magnesium sulfate, iron ions, manganese ions, and the like can be added. So long as the medium employed in the present invention contains a carbon source, nitrogen source, inorganic ions and, as needed, other trace organic components, it does not matter whether it is a natural or a synthetic medium.
Culturing can be conducted for 1 to 7 days under aerobic conditions at a temperature of 24 to 370C and at a pH of 5 to 9. Inorganic and organic acidic and alkaline substances as well as ammonia gas can be used to adjust the pH. L-amino acid can be recovered from the fermentation solution by the usual ion-exchange resin method, precipitation method, and combinations of these with other known methods. When L-amino acid accumulates within the bacterial cell, the bacterial cell can be crushed by ultrasonic waves or the like, a supernatant can be centrifugally separated from the bacterial cell, and the supernatant can be subjected to ion-exchange resin to recover the L-amino acid.
A liquid medium can be prepared under conditions which cause L-glutamic acid to precipitate, and the culture can be conducted while L-glutamic acid precipitates. An example of the conditions under which L-glutamic acid will precipitate is a pH of 5.0 to 4.0, preferably pH 4.5 to 4.0, more preferably pH 4.3 to 4.0, and particularly preferably, pH 4.0. When culturing is conducted under conditions causing L-glutamic acid to precipitate, the L-glutamic acid precipitating out into the culture solution can be collected by
centrifugation, filtration, or the like. In that case, the L-glutamic acid dissolved in the medium can be crystallized and then separated.
The present invention is further described below with reference to the following non-limiting examples.
Examples
Example 1 : Construction of a bacterium which is modified to not produce type I fimbrial adhesin protein.
<l-]> Construction of a strain in which the genes cadA and IdcC encoding lysine decarboxylase are destroyed
First, a strain that does not produce lysine decarboxylase was constructed. Lysine decarboxylase is encoded by the cadA gene (GenBank Accession No. NP_418555, Seq. ID No. 42) and the IdcC gene (GenBank Accession No. NP_414728, Seq. ID No. 44) (see International Application Publication WO96/17930). The parent strain employed here was strain WC 196. This strain was named Escherichia coli strain AJ 13069 and was deposited on December 6, 1994, as deposit number FERM P- 14690, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1 -1-1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566). This strain was converted on September 29, 1995, to an international deposit under the provisions of the Budapest Treaty and given deposit number FERM BP-5252.
The genes cadA and IdcC encoding lysine decarboxylase were deleted by the method known as "Red-driven integration" developed by Datsenko and Wanner (Proc. Natl. Acad. Sci. USA, 2000, Vol. 97, No. 12, p 6640-6645) and by a λ phage-derived excision system (J. Bacteriol. 2002 Sep; 184 (18): 5200-3. Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. Cho EH, Gumport RI, Gardner JF.).( WO2005/010175) Based on the "Red-driven integration" method, a synthetic oligonucleotide designed with a part of the target gene on its 5' terminal end and
a part of a antibiotic resistance gene on its 3' terminal end was employed as a primer to obtain a PCR product, which was then employed to construct a strain having an inactivated gene in a single step. By further combining this with an excision system derived from λ phage, it was possible to remove from the gene-inactivated strain the integrated antibiotic resistance gene. (1) Disruptionof the cadA gene
A plasm id pMWl 18-attL-Cm-attR was employed as PCR template. pMWl 18-attL-Cm-attR was a plasmid obtained by inserting the attL and attR genes - attachment sites of λ phage - and the cat gene, a gene imparting antibiotic-resistance, into pMW l 18 (Takara-Bio Co.) in the order attL-cat-attR. The sequence of attL is shown by SEQ. ID No. 1 1 and the sequence of attR by SEQ. ID No.12.
A synthetic oligonucleotide with Seq. ID Nos. 3 and 4 on its 5' terminal, corresponding to a part of the cadA gene, and sequences corresponding to the both ends of attL and attR on its 3'terminal, was employed as a primer to conduct PCR. The amplified PCR product was purified on an agarose gel and introduced by electroporation into Escherichia coli strain WC 196, which contains plasmid pKD46 with a temperature-sensitive replication origin. Plasmid pKD46 (Proc. Natl. Acad. Sci. USA, 2000, Vol. 97, No. 12, p 6640-6645) includes a DNA fragment (GenBank/EMBL Accession No. J02459, Nos. 31088 to 33241) having a total of 2,154 bases of λ-phage containing genes (γ, β, and exo genes) encoding the Red recombinase of the λ Red homologous recombination system controlled by an arabinose-derived ParaB promoter. Plasmid pKD46 was required to introduce the PCR product into the chromosome of strain WC 196.
Competent cells for electroporation were prepared as follows. The Escherichia coli strain WC 196 that had been cultured overnight at 3O0C in LB medium containing 100 mg/L of ampicillin was diluted 100-fold with 5 mL of SOB medium (Molecular Cloning: Laboratory Manual, 2nd Ed., Sambrook J. et al., Cold Spring Harbor Laboratory Press (1989)) containing L-arabinose (1 mM) and ampicillin (20 mg/L). The diluted product was cultured until reaching an OD600 of about 0.6 at 3O0C, concentrated 100-fold, and readied
for electroporation by washing three times in 10 percent glycerol. Electroporation was conducted using 70 μL of competent cells and about 100 ng ofthe PCR product. Following electroporation, 1 mL of SOC medium (Molecular Cloning: Laboratory Manual, 2nd Ed., Sambrook J. et al., Cold Spring Harbor Laboratory Press ( 1989)) was added and the cells were cultured for 2.5 hours at 37°C. They were then plate cultured on L-agar medium containing chloramphenicol (Cm, 25 mg/L) at 37°C and the Cm-resistant recombinants were selected. Next, to remove the pKD46 plasmid, cell were subcultured at 42°C on Cm-containing L-agar medium and the ampicillin resistance of the colonies obtained was tested. The ampicillin-sensitive strains without pKD46 were collected. The deletion of the cadA gene in mutants identified based on chloramphenicol resistance gene was confirmed by PCR. The cadA-deficient strain obtained was named strain WC 196ΔcadA::att-cat.
The above-described helper plasmid pMW-intxis-ts was employed to remove the att-cat genes that were inserted inside the cadA gene. pMW-intxis-ts is a plasmid which harbors a gene (SEQ. ID No. 13) encoding the integrase (Int) of the λ phage and a gene (SEQ. ID No. 15) encoding the excisionase (Xis) of the λ phage, and which has a temperature-sensative replication origin. By introducing pMW-intxis-ts, recombination occurs due to the recognition of attL (SEQ. ID No. 1 1 ) and attR (SEQ. ID No. 12) on the chromosome, allowing gene excision between attL and attR, leaving behind only the attL or attR sequence on the chromosome.
Competent cells of the strain WC I 96ΔcadA::att-cat obtained as set forth above were prepared by ordinary methods, transformed with helper plasmid pMW-intxis-ts, and plate cultured on L-agar medium containing 50 mg/L of ampicillin at 3O0C. The ampicillin-resistant strains were selected.
Next, to remove the pMW-intxis-ts plasmid, cells were subcultured at 420C on L agar medium. The ampicillin resistance and chloramphenicol resistance of the colonies obtained were tested and a strain sensitive to chloramphenicol and ampicillin, which was a cadA-deleted strain which had removed the att-cat and pM W-intxis-ts, was collected. This strain was named WC 196ΔcadA.
(2) Strain WC196ΔcadA and deletion of the ldcC gene
Deletion of the ldcC gene in strain WC196ΔcadA was conducted using primers with SEQ. ID NO. 5 and 6 to destroy the ldcC by the above-described method. This resulted in a strain in which cadA and ldcC had been deleted, and was designated WC196ΔcadAΔIdcC. (3) Preparation of PCR template and helper plasmid
PCR template pMWl 18-attL-Cm-attR and helper plasmid pMW-intxis-ts were prepared as set forth below. (3-1) pMWl 18-attL-Cm-attR pMWl 18-attL-Cm-attR was constructed based on pMWl 18-attL-Tc-attR. The four following DNA fragments were spliced.
1) BgI ll-EcoRI DNA fragment (120 bp) (SEQ. ID No. 1 1) containing attL obtained by PCR amplification of a sequence corresponding to the chromosome of E. coli strain W3350 (ATCC 31278 containing λ prophage) using primers in the form of oligonucleotides Pl and P2 (SEQ. JD NO. 17 and 18) (these primers contained additional BgI Il and EcoRI endonuclease recognition sites).
2) Pstl-Hindlll DNA fragment (182 bp) (SEQ. ID No. 12) containing attR obtained by PCR amplification of a sequence corresponding to the chromosome of E. coli strain W3350 (containing λ prophage) using primers in the form of oligonucleotides P3 and P4 (SEQ. ID Nos. 19 and 20) (these primers contained additional Pstl and HindIII endonuclease recognition sites).
3) Bglll-HindlH large fragment (3,916 bp) of pMWl 18-ter_rrnB. pMWl 18-ter_rrn was a fragment obtained by ligation of the following three fragments: -A large fragment (2,359 bp) containing an Aatll-EcoRIpol fragment of pMWl 18 was obtained by cleaving pMWl 18 with EcoRI restriction endonuclease, processing with the Klenow fragment of DNA polymerase 1, and digesting with Aatll restriction endonuclease, -Aatll-Bgl ll small fragment (1 194 bp) of pUC19 containing the bla gene of ampicillin resistance (ApR), obtained by employing oligonucleotides P5 and P6 (SEQ. ID Nos. 21 and 22) as primers (these primers contained additional Aatll and BgI Il endonuclease
recognition sites) in PCR amplification of the sequence corresponding to the pUC 19 plasmid,
-Bg I II-Pstlpol small fragment (363 bp) of transcription terminator rrnB obtained by employing oligonucleotides P7 and P8 (SEQ. ID Nos. 23 and 24) as primers (these primers had additional BgI II and Pstl endonuclease identification sites) in PCR amplification of the region corresponding to the chromosome of E. coli strain MG 1655.
4) Small EcoRI-Pstl fragment (1,388 bp) (SEQ. ID No. 29) of pML-Tc-terJhrL comprising a tetracycline resistance gene and transcription terminator ter_thrL. pML-Tc-ter-thrL was obtained as follows. pML-MCS (MoI Biol (Mosk). 2005 Sep-Oct;39(5):823-3 I ) was digested with Xbal and BamHI restriction endonucleases and the large fragment (3342 bp) was spliced to an Xbal-BamHI fragment (68 bp) containing terminator ter thrL. The region of the Xbal-BamHI fragment corresponding to the chromosome of E. coli strain MG1655 was amplified by PCR using oligonucleotides P9 and PlO (SEQ. ID Nos. 25 and 26) as primers (these primers contained additional Xbal and BamHI endonuclease recognition sites). The ligated reaction product was named plasmid pML-ter thrL. pML-ter_thrL was cleaved with Kpnl and Xbal restriction endonucleases, treated with the Klenow fragment of DNA polymerase I, and then ligated to the small EcoRI-Van911 fragment (1 ,317 bp) of pBR322 containing a tetracycline resistance gene (pBR322 was treated with the Klenow fragment of DNA polymerase I using EcoRJ and Van91 I restriction endonucleases). The product of the ligation reaction was named plasmid pML-Tc-ter_thrL. pMW I 18-attL-Tc-attR was obtained as set forth above. pMWI 18-attL-Cm-attR was constructed by ligation the large BamHI-Xbal fragment (4,413 bp) of pM Wl 18-attL-Tc-attR, promoter PA2 (the initial promoter of T7 phage), the cat gene of chloramphenicol resistance (CmR), and BgI Il-Xbal artificial DNA fragment ( 1 , 162 bp) comprising the transcription terminator terjhrL and attR. The artificial DNA fragment (SEQ. ID No. 30) was obtained as follows.
The pML-MSC (MoI Biol (Mosk). 2005 Sep-Oct;39(5):823-31 ;
Biotechnologiya (Russian) No. 5: 3-20.)) was cleaved with Kpnl and Xbal restriction endonucleases and ligated to the small Kpnl-Xbal fragment (120 bp) containing promoter PA2 (the initial promoter of T7 phage). The Kpnl-Xbal fragment was obtained by PCR amplification of the region corresponding to T7 phage DNA using oligonucleotides Pl 1 and Pl 2 (SEQ. ID Nos. 27 and 28) as primers (these primers had additional Kpnl and Xbal endonuclease recognition sites). The product of the splicing reaction was named plasmid pML-PA2-MCS.
The Xbal site was removed from pML-PA2-MCS. The product obtained was called plasmid pML-PA2-MCS(XbaI-).
The small Bgl ll-Hindlll fragment (928 bp) of pML-PA2-MCS(Xbal-) containing promoter PA2 (the initial promoter of T7 phage) and the cat gene imparting resistance to chloramphenicol (CmR) was ligated to the small Hindlll-Hindlll fragment (234 bp) of pMWl 18-attL-Tc-attR containing transcription terminator ter thrL and attR. The targeted artificial DNA fragment (1,156 bp) was obtained by PCR amplification of the splicing reaction mixture with primers in the form of oligonucleotides P9 and P4 (SEQ. ID Nos. 25 and 20) (these primers contained additional Hindlll and Xbal endonuclease recognition sites). (3-2) pMW-intxis-ts
Initially, two DNA fragments were amplified using λ phage DNA (Fermentas) as template. The first fragment was comprised of nt 37168 to 38046 (SEQ. ID No. 39), containing CI repressor, promoters Prm and Pr, and the leader sequence of the cro gene. This fragment was obtained by amplification using oligonucleotides Pl ' and P2' (SEQ. ID NO. 31 and 32) as primers. The second fragment was comprised of nt 27801 to 29100 (SEQ. ID NO. 40), containing the xis-int genes of λ phage. This fragment was obtained by amplification using oligonucleotides P3' and P4' (SEQ. ID NO. 33 and 34) as primers. All the primers include suitable endonuclease recognition sites.
The PCR amplification fragment containing cl repressor that was obtained was cleaved with CIaI restriction endonuclease, treated with the Klenow fragment of DNA polymerase I, and digested with EcoRI restriction endonuclease. The second PCR
amplified fragment was digested with EcoRl and Pstl restriction endonucleases.
Additionally, plasmid pMWPIaclacl-ts was digested with BgI Il endonuclease, treated with the Klenow fragment of DNA polymerase I, and cleaved with Pstl restriction endonuclease. The vector fragment of pMWPladlacl-ts was eluted from agarose gel and ligated to the cut PCR amplified fragment. (Biotechnologiya (Russian) No. 5: 3-20.)
Plasmid pMWPIaclacl-ts was derived from pMWPIaclacl, which comprised the following components: 1) a Bgl ll-Hindlll artificial DNA fragment comprising Pl acUV5 promoter and the lacl gene controlled by the RBS of bacteriophage T7 gene 10; 2) an Aatll-Bgl ll fragment containing an ampicillin resistance (ApR) gene, obtained by PCR amplification of the region corresponding to plasmid pUC19 using oligonucleotides P5' and P6' (SEQ. ID Nos. 35 and 36) as primers (these primers contained additional AatII and BgI lI endonuclease recognition sites); 3) an Aatll-Bgl II fragment containing the Aatll-Pvul fragment of recombinant plasmid pMWl 18-ter_rrnB.
Plasmid pMWl 18-ter_rrnB was constructed in the following manner. The region corresponding to the chromosome of E. coli strain MG1655 was amplified by PCR using oligonucleotides P7' and P8' (SEQ. ID Nos. 37 and 38) containing suitable endonuclease recognition sites as primers, yielding a Pstl-Hindlll fragment containing terminator ter rrnB. Before ligation, the pMW 1 18 and the ter rrnB fragment (complement ; SEQ. ID No. 41) were restricted with Pvul and Pstl, respectively, treated with the Klenow fragment of DNA polymerase to blunt the ends, and digested with Aatll or Hindlll endonuclease. To construct a pMWPIaclacl-ts mutant, the Aatll-EcoRV fragment of the plasmid pMWPIaclacl was replaced with the Aatll-EcoRV fragment of plasmid pMAN997 containing the par, ori, and repAts genes of the pSClOI replicon. (Applied and Environmental Microbiology, June 2005, p. 3228-32) <l -2> Construction of a strain which does not produce type I fimbrial adhesin protein (strain deleting the fimH gene: WCI 96ΔcadAΔIdcCΔfimH strain) from WCl96ΔcadAΔIdcC
The/?mHgene was deleted from the WC196ΔcadAΔldcC strain by the procedure of (1 ) above; the primers of SEQ. ID Nos. 7 and 8 were employed to delete the
fimH gene. This obtained strain did not produce type 1 tϊmbrial adhesion protein, and was designated WC196ΔcadAΔIdcCΔfimH::Cm.
Strains WC196ΔcadAΔIdcC and WC196ΔcadAΔIdcCΔfimH::Cm were transformed by the usual methods with plasmid Lys production-plasmid pCABD2 earring dapA, dapB, and lysC genes (WOO 1/53459), obtaining strains
WC 196ΔcadAΔldcC/pCABD2 and WC196ΔcadAΔIdcCΔfιmH::Cm/pCABD2. These strains were cultured at 370C in L medium containing 20 mg/L of streptomycin to a final OD600 of about 0.6. A 40 percent glycerol solution was added in a quantity equal to that of the culture solution, the mixture was stirred, and suitable amouts were poured out and stored at -800C. This was referred to as glycerol stock.
Example 2
Evaluation of the L-lysine production of a strain which does not produce type 1 fimbrial adhesin protein
The glycerol stocks of these strains were melted, 100 μL amounts were uniformly plated onto L plates containing 20 mg/L of streptomycin, and the strain were cultured for 24 hours at 37°C. About 1/8 of the bacterial cells obtained on each plate was inoculated onto a 20 mL fermentation culture containing 20 mg/L of streptomycin in a 500 mL Sakaguchi flask and cultured for 48 hours at 37°C in a reciprocating shaking incubator. After culturing, the amount of L-lysine that had accumulated in the medium was measured by a ordinary method (Sakura Seiki, Biotech Analyzer AS210). The composition of the fermentation medium is shown below (unit: g/L).
Glucose 40
(NH4)2SO4 24
K2HPO4 1.0
MgSO4-7H2O 1.0
FeSO4-7H2O 0.01
MnSO4-5H2O 0.01
Yeast extract 2.0
Up to 1 L
The medium was adjusted to pH 7.0 with KOH and sterilized for lO min at 1 15°C in an autoclave (the glucose and MgSO4-7H2O were separately sterilized). A 30 g/L quantity of CaCO3 (that had been dry sterilized for 2 hours at 1800C) was added. 20 mg/L amounts of streptomycin was added as antibiotic. Culturing was conducted for 48 hours under conditions of a temperature of 370C with stirring at 1 15 rpm.
The results are given in Table 2 (OD is the absorbance at 600 nm by the bacterial mass diluted 26-fold, Lys (g/L) is the amount of L-lysine accumulated in the flask, and the yield (%) is the L-lysine yield from sugar). As will be shown in Table 2, strain WC196ΔcadAΔldcCΔfιmH::Cm/pCABD2 accumulated more L-lysine than strain WC 196ΔcadAΔldcC::Cm/pCABD2, from which the fimH gene had not been deleted. Table 2

Example 3
Evaluation of the L-threonine production of a strain which does not produce type I fimbrial adhesin protein
B-5318 was employed as the L-threonine-producing parent strain which does not produce type I fimbrial adhesin protein . Strain B-5318 was deposited on November 19, 1987, under registration number VKPM B-5318 with the Russian National Collection of Industrial Microorganisms (VKPM), GNII Genetika. The strain which does not produce type I fimbrial adhesin was constructed from the L-threonine producing bacterium by the
same method as in Example 1 by employing synthetic oligonucleotides SEQ. ID Nos. 7 and 8. Specifically, the following fimH deficient strain was constructed.
B-5318ΔfimH-cat was derived from strain B-5318 by method ( I ) in Example 1. Strain B-5318 and strain B-5318ΔfimH-cat were cultured for 24 hours at 370C on L agar medium containing 20 mg/L of streptomycin sulfate and L agar medium containing 20 mg/L of streptomycin sulfate and 25 mg/L of chloramphenicol, respectively. One-fifth of the bacterial cell was collected from each of the plates and inoculated onto 50 mL of LB liquid medium containing the above-stated antibiotics and the bacteria were precultured for 3.5 hours at a temperature of 390C at 144 rpm.
Following preculturing, a quantity of preculture solution amounting to 10 percent of the volume of the main culture medium was inoculated into a one-liter jar fermenter containing 300 mL of main culture medium and the main culture was cultivated at 400C at pH 7.0. The composition of the main culture medium is shown below. Composition of Main Culture Medium Glucose l OO g/L
Yeast extract 1.8 g/L
FeSO4-7H2O 18 mg/L
MnSO4-4H2O 18 mg/L
KH2PO4 1 g/L ,
MgSO4-7H2O 0.36 g/L
(NH4)2SO4 4.5 g/L
NaCI 0.6 g/L
For strain B5318, 20 mg/L of streptomycin sulfate salt was added, and for strain B-53 l 8ΔfimH-cat, 20 mg/L of streptomycin sulfate salt and 25 mg/L of chloramphenicol were added.
Ammonia gas was added to adjust the culture to pH 7.0. When the glucose was exhausted, culturing was stopped and liquid chromatography was employed to measure the quantity of L-threonine. The results are shown in Table 3. OD is the absorbance at 600 nm by the bacterial mass diluted 101 -fold, Thr (g/L) is the amount of L-threonine which had
accumulated in the flask, and the yield (%) is the L-threonine yield from sugar.
Use of the B-5318ΔfimH-cat strain, which lacked the fimH gene, enhanced the yield of
L-threonine compared with the B-5318 strain, which served as a control.
Table 3
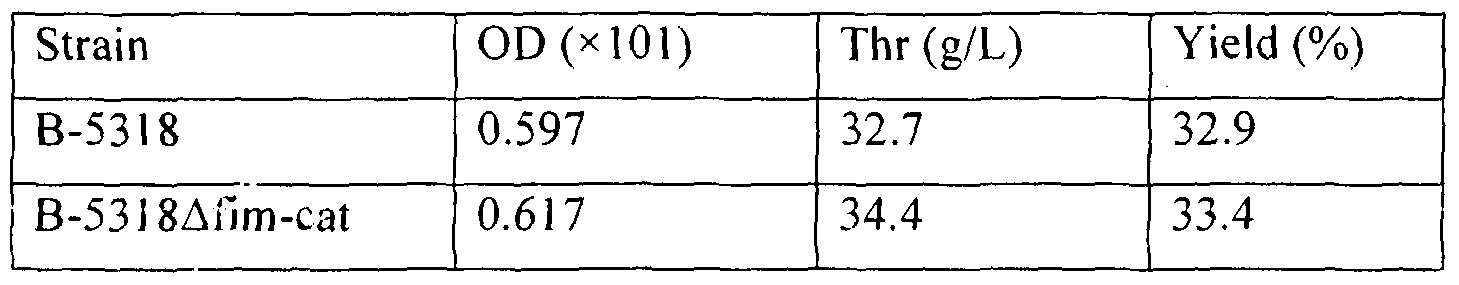
Example 4
Evaluation of the L-glutamic acid production of a strain which does not produce type I fimbrial adhesin protein
<4-l> Evaluation of the L-glutamic acid production of a Escherichia col\ strain which does not produce type 1 fimbrial adhesin protein
Escherichia coli strain AJ 12949 was employed as the L-glutamic acid-producing parent strain which does not produce type 1 fimbrial adhesin protein . Strain AJ 12949 was deposited on December 28, 1993, as depositary number FERM P- 14039, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology (currently the International Patent Organism Depositary, National Institute of Industrial Science and Technology, an Independent Administrative Institution, Central 6, 1-1-1 Higashi, Tsukuba, Ibaraki, Japan Postal Code 305-8566), and converted on November 1 1 , 1994, to an international deposit under the provisions of the the Budapest Treaty, and given a deposit number FERM BP-4881.
Culturing was conducted in the following specific manner. Strain AJ12949ΔfimH-cat was derived from strain AJ 12949 (BP-4881 ). Strain AJ 12949 and strain AJ 12949ΔfimH-cat were cultured for 24 hours at 370C in L agar medium containing 50 mg/L of ampicillin and L agar solution containing 50 mg/L of ampicillin and 25 mg/L of chloramphenicol, respectively. One-eighth of the bacterial cells was collected from each
plate and inoculated onto 2O mL of fermentation medium containing 50 mg/L of ampicillin in a 500 mL Sakaguchi flask and cultured for 24 hours at 370C in a reciprocating shaking incubator. Following culturing, the amount of glutamic acid accumulated in the medium was measured by a known method (Sakura Seiki, Biodex Analyzer AS210).
The composition of the fermentation medium is given below (unit: g/L)
Glucose 40
Yeast extract 2
FeSO4-7H2O 0.01
MnSO4-4H2O 0.01
KH2PO4 1
MgSO4-7H2O 1
(NH4)2SO4 20
Thiamine hydrochloride 0.01
The medium was adjusted to pH 7.0 with KOH and sterilized for 10 min at I l 50C in an autoclave (the glucose and MgSO4'7H2O were separately sterilized). A 50 g/L quantity of CaCO3 (that had been dry sterilized for 2 hours at 18O0C) was added. A 50 mg/L quantity of ampicillin was added as an antibiotic for strain AJ12949, and a 50 mg/L quantity of ampicillin and a 50 mg/L quantity of chloramphenicol were added for AJ12949ΔfϊmH-cat.
The results are given in Table 4 (OD is the absorbance at 600 nm by the bacterial mass diluted 26-fold, GIu (g/L) is the amount of L-glutamine accumulated in the flask, and the yield (%) is the L-glutamic acid yield from sugar). Strain AJ 12949ΔfimH-cat, the strain that did not produce type I fimbrial adhesin protein, accumulated more L-glutamic acid than strain AJ 12949, from whichy/wHwas not deleted.
Table 4

<4-2> Evaluation of the L-glutamic acid production of a Pantoea ananatis strain which does not produce type 1 fimbrial adhesin protein
Pantoea ananatis strain AJ 13601 can also be employed as the L-glutamic acid-producing parent strain which does not produce type I fimbrial adhesin protein. Pantoea ananatis strain AJ 13601 was deposited on August 18, 1999, as depositary number FERM P-17516, at the National Institute of Bioscience and Human-Technology, Agency of Industrial Science and Technology, Ministry of Economy, Trade and Industry (1 -1 -3 Higashi, Tsukuba, lbaraki Prefecture, Postal Code 305-8566). This strain was converted on July 6, 2000, to a deposit under the provisions of the Budapest Treaty under deposit number FERM BP-7207. A strain that does not produce type I fimbrial adhesin can be constructed from the L-glutamic acid-producing microbe by the same method as in Example 1 using synthetic nucleotides having SEQ. ID Nos. 7 and 8. A mutant strain can also be constructed by introducing a temperature-sensitive plasmid having a deleted gene encoding type 1 fimbrial adhesin protein.
The strain not producing type I fimbrial adhesin protein can be cultured in an L-glutamic acid production medium in a reciprocating shaking incubator. Following culturing, the amount of L-glutamic acid which had accumulated in the medium is measured by a ordinary method to confirm an increase in accumulated L-glutamic acid. This method permits the obtaining of a strain that does not produce type I fimbrial adhesin protein and has an enhanced ability to produce L-glutamic acid.
Claims
1 . An L-amino acid producing bacterium of the Enterobacteriaceae family, wherein the bacterium has been modified so as to not produce type I fimbrial adhesin protein.
2. The bacterium according to claim 1 , wherein said bacterium has been modified so as to not produce type 1 fimbrial adhesin protein by introducing a mutation into the gene encoding type I fimbrial adhesin protein on the chromosome and/or into the region regulating expression thereof.
3. The bacterium according to claim 1 , wherein said bacterium has been modified so as to not produce type I fimbrin adhesin protein by attenuation of the gene encoding type I fimbrial adhesin protein.
4. The bacterium according to claim 1 , wherein said bacterium has been modified so as to not produce type 1 fimbrial adhesion protein by inactivation of the gene encoding type 1 fimbrial adhesin protein on the chromosome.
5. The bacterium according to claim 1 , wherein the gene encoding type 1 fimbrial adhesin protein 
6. The bacterium according to claim 1 , wherein said gene encoding type 1 fimbrial adhesin protein is selected from the group consisting of:
(a) a DNA comprising the nucleotide sequence of SEQ ID NO: 1 , and (b) a DNA that is able to hybridize with the complementary strand of the nucleotide of SEQ ID NO: 1 , or a probe prepared from said nucleotide sequence under stringent conditions, and wherein the DNA encodes the type 1 fimbrial adhesin protein.
7. The bacterium according to claim 1 wherein said bacterium of the Enterobacteriaceae family is selected from the group consisting of Escherichia, Pantoea, and Enterobacter.
8. The bacterium according to claim I , wherein said L-amino acid is selected from the group consisting of L-lysine, L-threonine, L-glutamic acid, and combinations thereof.
9. A method for producing an L-amino acid, comprising -cultivating the bacterium according to any of claim 1 to 8 in a medium, and -collecting said L-amino acid from the medium.
10. The method according to claim 10, wherein said L-amino acid is selected from the group consisting of L-lysine. L-threonine, L-glutamic acid, and combinations thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06701432A EP1838726A1 (en) | 2005-01-18 | 2006-01-17 | L-amino acid producing microorganism and a method for producing l-amino acid |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005009826 | 2005-01-18 | ||
| JP2005-009826 | 2005-01-18 | ||
| JP2005120222 | 2005-04-18 | ||
| JP2005-120222 | 2005-04-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006078039A1 true WO2006078039A1 (en) | 2006-07-27 |
Family
ID=36570316
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/301071 WO2006078039A1 (en) | 2005-01-18 | 2006-01-17 | L-amino acid producing microorganism and a method for producing l-amino acid |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US7547531B2 (en) |
| EP (1) | EP1838726A1 (en) |
| WO (1) | WO2006078039A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007100009A1 (en) | 2006-03-03 | 2007-09-07 | Ajinomoto Co., Inc. | Method for production of l-amino acid |
| WO2007125954A1 (en) | 2006-04-28 | 2007-11-08 | Ajinomoto Co., Inc. | Microorganism capable of producing l-amino acid, and process for production of l-amino acid |
| WO2009011354A1 (en) * | 2007-07-19 | 2009-01-22 | Ajinomoto Co., Inc. | Method of producing l-amino acid |
| WO2009028294A1 (en) * | 2007-08-27 | 2009-03-05 | Ajinomoto Co., Inc. | Mutant lipase and use thereof |
| WO2009031565A1 (en) | 2007-09-04 | 2009-03-12 | Ajinomoto Co., Inc. | Amino acid-producing microorganism and method of producing amino acid |
| WO2009088049A1 (en) | 2008-01-10 | 2009-07-16 | Ajinomoto Co., Inc. | Method for production of desired substance by fermentation process |
| WO2009093703A1 (en) | 2008-01-23 | 2009-07-30 | Ajinomoto Co., Inc. | Method of producing l-amino acid |
| WO2009107631A1 (en) | 2008-02-25 | 2009-09-03 | 味の素株式会社 | Process for production of 5'-guanylic acid |
| WO2010027045A1 (en) | 2008-09-08 | 2010-03-11 | 味の素株式会社 | Microorganism capable of producing l-amino acid, and method for producing l-amino acid |
| JP2010098998A (en) * | 2008-10-23 | 2010-05-06 | Toray Ind Inc | Method for producing 1,5-pentane diamine |
| WO2010061890A1 (en) | 2008-11-27 | 2010-06-03 | 味の素株式会社 | Process for producing l-amino acid |
| EP2202299A1 (en) | 2008-12-22 | 2010-06-30 | Ajinomoto Co., Inc. | A method for producing L-lysine |
| WO2010074209A1 (en) * | 2008-12-26 | 2010-07-01 | 味の素株式会社 | Mutant lipase and use thereof |
| WO2011013707A1 (en) | 2009-07-29 | 2011-02-03 | 味の素株式会社 | Method for producing l-amino acid |
| WO2011024555A1 (en) | 2009-08-28 | 2011-03-03 | 味の素株式会社 | Process for production of l-amino acid |
| EP2295546A2 (en) | 2009-08-10 | 2011-03-16 | Ajinomoto Co., Inc. | Method for producing 5'-guanylic acid |
| WO2011055710A1 (en) | 2009-11-06 | 2011-05-12 | 味の素株式会社 | Method for producing l-amino acid |
| EP2559754A2 (en) | 2011-08-18 | 2013-02-20 | Ajinomoto Co., Inc. | Method for producing an L-amino acid using a bacterium of the family enterobacteriaceae having enhanced expression of the flagella formation and motility cascade genes |
| WO2013051685A1 (en) | 2011-10-07 | 2013-04-11 | 味の素株式会社 | Mutant γ-glutamyltransferase, and method for producing γ-glutamylvalylglycine or salt thereof |
| WO2014025023A1 (en) | 2012-08-10 | 2014-02-13 | 味の素株式会社 | METHOD FOR PRODUCING γ-GLUTAMYL-VALYL-GLYCINE CRYSTAL |
| EP2796560A1 (en) | 2013-04-23 | 2014-10-29 | Ajinomoto Co., Inc. | A method for producing an L-amino acid using a bacterium of the family Enterobacteriaceae having attenuated expression of the yjjK gene |
| WO2015030019A1 (en) | 2013-08-30 | 2015-03-05 | Ajinomoto Co.,Inc. | A METHOD FOR PRODUCING AN L-AMINO ACID USING A BACTERIUM OF THE FAMILY ENTEROBACTERIACEAE HAVING ATTENUATED EXPRESSION OF THE znuACB GENE CLUSTER |
| WO2015041265A1 (en) | 2013-09-17 | 2015-03-26 | 味の素株式会社 | Method for producing l-amino acid from seaweed-derived biomass |
| WO2015050276A1 (en) | 2013-10-02 | 2015-04-09 | Ajinomoto Co.,Inc. | A method for producing an l-amino acid using a bacterium of the family enterobacteriaceae having attenuated expression of a phosphate transporter-encoding gene |
| WO2015050234A1 (en) | 2013-10-02 | 2015-04-09 | 味の素株式会社 | Ammonia control apparatus and ammonia control method |
| WO2015060314A1 (en) | 2013-10-21 | 2015-04-30 | 味の素株式会社 | Method for producing l-amino acid |
| WO2015122544A1 (en) | 2014-02-14 | 2015-08-20 | Ajinomoto Co.,Inc. | A METHOD FOR PRODUCING AN L-AMINO ACID USING A BACTERIUM OF THE FAMILY ENTEROBACTERIACEAE HAVING OVEREXPRESSED THE yajL GENE |
| EP3098319A1 (en) | 2015-05-28 | 2016-11-30 | Ajinomoto Co., Inc. | A method for producing an l-amino acid using a bacterium of the family enterobacteriaceae having an attenuated expression of a gsha gene |
| WO2017146195A1 (en) | 2016-02-25 | 2017-08-31 | Ajinomoto Co., Inc. | A method for producing l-amino acids using a bacterium of the family enterobacteriaceae overexpressing a gene encoding an iron exporter |
| WO2018030507A1 (en) * | 2016-08-10 | 2018-02-15 | 味の素株式会社 | Production method for l-amino acid |
| CN108531439A (en) * | 2018-04-16 | 2018-09-14 | 南京工业大学 | A kind of Recombinant organism and its construction method and application |
| CN108795916A (en) * | 2018-07-16 | 2018-11-13 | 南京工业大学 | A kind of lysine decarboxylase mutant, its encoding gene and its expression and application |
| WO2020138178A1 (en) | 2018-12-27 | 2020-07-02 | Ajinomoto Co., Inc. | Method for producing basic l-amino acids or salts thereof by fermentation of an enterobacteriaceae bacterium |
| WO2020171227A1 (en) | 2019-02-22 | 2020-08-27 | Ajinomoto Co., Inc. | METHOD FOR PRODUCING L-AMINO ACIDS USING A BACTERIUM BELONGING TO THE FAMILY Enterobacteriaceae HAVING OVEREXPRESSED ydiJ GENE |
| WO2020204179A1 (en) | 2019-04-05 | 2020-10-08 | Ajinomoto Co., Inc. | Method of producing l-amino acids |
| WO2021060438A1 (en) | 2019-09-25 | 2021-04-01 | Ajinomoto Co., Inc. | Method for producing l-amino acids by bacterial fermentation |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4427878B2 (en) | 1999-08-20 | 2010-03-10 | 味の素株式会社 | Method for producing L-glutamic acid by fermentation method with precipitation |
| JP4599726B2 (en) | 2001-02-20 | 2010-12-15 | 味の素株式会社 | Method for producing L-glutamic acid |
| JP4894134B2 (en) * | 2003-07-29 | 2012-03-14 | 味の素株式会社 | Methods for determining metabolic fluxes that affect substance production |
| CN101243177B (en) * | 2004-01-30 | 2012-12-05 | 味之素株式会社 | L-amino acid-producing microorganism and method for producing L-amino acid |
| US7344874B2 (en) * | 2004-03-04 | 2008-03-18 | Ajinomoto Co., Inc. | L-glutamic acid-producing microorganism and a method for producing L-glutamic acid |
| US7482140B2 (en) * | 2004-06-15 | 2009-01-27 | Ajinomoto Co., Inc. | L-tyrosine-producing bacterium and a method for producing L-tyrosine |
| US7205132B2 (en) * | 2004-09-10 | 2007-04-17 | Ajinomoto Co., Inc. | L-glutamic acid-producing microorganism and a method for producing L-glutamic acid |
| US7915018B2 (en) | 2004-10-22 | 2011-03-29 | Ajinomoto Co., Inc. | Method for producing L-amino acids using bacteria of the Enterobacteriaceae family |
| US7794989B2 (en) * | 2004-12-28 | 2010-09-14 | Ajinomoto Co., Inc. | L-glutamic acid-producing microorganism and a method for producing L-glutamic acid |
| JP2008283863A (en) | 2005-08-26 | 2008-11-27 | Ajinomoto Co Inc | L-amino acid-producing bacterium and method for producing l-amino acid |
| WO2007037460A1 (en) | 2005-09-27 | 2007-04-05 | Ajinomoto Co., Inc. | An l-amino acid-producing bacterium and a method for producing l-amino acids |
| EP1929028A1 (en) | 2005-09-27 | 2008-06-11 | Ajinomoto Co., Inc. | An l-amino acid-producing bacterium and a method for producing l-amino acids |
| WO2007119574A2 (en) | 2006-03-23 | 2007-10-25 | Ajinomoto Co., Inc. | A method for producing an l-amino acid using bacterium of the enterobacteriaceae family with attenuated expression of a gene coding for small rna |
| JP2009060791A (en) | 2006-03-30 | 2009-03-26 | Ajinomoto Co Inc | L-amino acid-producing bacterium and method for producing l-amino acid |
| JP2010110217A (en) * | 2007-02-22 | 2010-05-20 | Ajinomoto Co Inc | L-amino acid-producing microorganism and method for producing l-amino acid |
| JP2010226956A (en) * | 2007-07-23 | 2010-10-14 | Ajinomoto Co Inc | Method for producing l-lysine |
| JP2013074795A (en) | 2010-02-08 | 2013-04-25 | Ajinomoto Co Inc | MUTANT rpsA GENE AND METHOD FOR PRODUCING L-AMINO ACID |
| JP2014036576A (en) | 2010-12-10 | 2014-02-27 | Ajinomoto Co Inc | Method for producing l-amino acids |
| TWI748408B (en) * | 2020-04-14 | 2021-12-01 | 中國石油化學工業開發股份有限公司 | Recombinant microorganism and method for producing 1,5-diaminopentane |
| CN111850010B (en) * | 2020-06-08 | 2021-04-09 | 黑龙江伊品生物科技有限公司 | dapB gene modified recombinant strain and construction method and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0796912A1 (en) * | 1994-12-09 | 1997-09-24 | Ajinomoto Co., Inc. | Novel lysine decarboxylase gene and process for producing l-lysine |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6720139B1 (en) * | 1999-01-27 | 2004-04-13 | Elitra Pharmaceuticals, Inc. | Genes identified as required for proliferation in Escherichia coli |
| ATE417094T1 (en) | 1999-12-24 | 2008-12-15 | Ajinomoto Kk | METHOD FOR PRODUCING L-AMINO ACID |
| JP4380029B2 (en) | 2000-07-05 | 2009-12-09 | 味の素株式会社 | Manufacturing method of substances using microorganisms |
| CA2414460A1 (en) * | 2000-07-07 | 2002-01-17 | Medimmune, Inc. | Fimh adhesin proteins and methods of use |
| JP2002330763A (en) | 2001-05-02 | 2002-11-19 | Ajinomoto Co Inc | Method for producing objective substance by fermentation |
| US7335496B2 (en) | 2003-06-05 | 2008-02-26 | Ajinomoto Co., Inc. | Method for producing target substance |
| CN101243177B (en) | 2004-01-30 | 2012-12-05 | 味之素株式会社 | L-amino acid-producing microorganism and method for producing L-amino acid |
| US7344874B2 (en) | 2004-03-04 | 2008-03-18 | Ajinomoto Co., Inc. | L-glutamic acid-producing microorganism and a method for producing L-glutamic acid |
| US7300776B2 (en) | 2004-04-26 | 2007-11-27 | Ajinomoto Co., Inc. | L-amino acid-producing bacterium and a method for producing L-amino acid |
| US7482140B2 (en) | 2004-06-15 | 2009-01-27 | Ajinomoto Co., Inc. | L-tyrosine-producing bacterium and a method for producing L-tyrosine |
-
2006
- 2006-01-17 EP EP06701432A patent/EP1838726A1/en not_active Withdrawn
- 2006-01-17 WO PCT/JP2006/301071 patent/WO2006078039A1/en active Application Filing
- 2006-01-17 US US11/275,562 patent/US7547531B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0796912A1 (en) * | 1994-12-09 | 1997-09-24 | Ajinomoto Co., Inc. | Novel lysine decarboxylase gene and process for producing l-lysine |
Non-Patent Citations (1)
| Title |
|---|
| ORNDORFF P E ET AL: "Immunoglobulin-mediated agglutination of and biofilm formation by Escherichia coli K-12 require the type 1 pilus fiber", INFECTION AND IMMUNITY, vol. 72, no. 4, April 2004 (2004-04-01), pages 1929 - 1938, XP002384921, ISSN: 0019-9567 * |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007100009A1 (en) | 2006-03-03 | 2007-09-07 | Ajinomoto Co., Inc. | Method for production of l-amino acid |
| WO2007125954A1 (en) | 2006-04-28 | 2007-11-08 | Ajinomoto Co., Inc. | Microorganism capable of producing l-amino acid, and process for production of l-amino acid |
| WO2009011354A1 (en) * | 2007-07-19 | 2009-01-22 | Ajinomoto Co., Inc. | Method of producing l-amino acid |
| WO2009028294A1 (en) * | 2007-08-27 | 2009-03-05 | Ajinomoto Co., Inc. | Mutant lipase and use thereof |
| WO2009031565A1 (en) | 2007-09-04 | 2009-03-12 | Ajinomoto Co., Inc. | Amino acid-producing microorganism and method of producing amino acid |
| EP2749652A2 (en) | 2008-01-10 | 2014-07-02 | Ajinomoto Co., Inc. | A method for producing a target substance by fermentation |
| WO2009088049A1 (en) | 2008-01-10 | 2009-07-16 | Ajinomoto Co., Inc. | Method for production of desired substance by fermentation process |
| WO2009093703A1 (en) | 2008-01-23 | 2009-07-30 | Ajinomoto Co., Inc. | Method of producing l-amino acid |
| WO2009107631A1 (en) | 2008-02-25 | 2009-09-03 | 味の素株式会社 | Process for production of 5'-guanylic acid |
| WO2010027045A1 (en) | 2008-09-08 | 2010-03-11 | 味の素株式会社 | Microorganism capable of producing l-amino acid, and method for producing l-amino acid |
| JP2010098998A (en) * | 2008-10-23 | 2010-05-06 | Toray Ind Inc | Method for producing 1,5-pentane diamine |
| WO2010061890A1 (en) | 2008-11-27 | 2010-06-03 | 味の素株式会社 | Process for producing l-amino acid |
| EP2202299A1 (en) | 2008-12-22 | 2010-06-30 | Ajinomoto Co., Inc. | A method for producing L-lysine |
| WO2010074209A1 (en) * | 2008-12-26 | 2010-07-01 | 味の素株式会社 | Mutant lipase and use thereof |
| JP2012044869A (en) * | 2008-12-26 | 2012-03-08 | Ajinomoto Co Inc | Mutant lipase and application thereof |
| WO2011013707A1 (en) | 2009-07-29 | 2011-02-03 | 味の素株式会社 | Method for producing l-amino acid |
| EP2295546A2 (en) | 2009-08-10 | 2011-03-16 | Ajinomoto Co., Inc. | Method for producing 5'-guanylic acid |
| WO2011024555A1 (en) | 2009-08-28 | 2011-03-03 | 味の素株式会社 | Process for production of l-amino acid |
| WO2011055710A1 (en) | 2009-11-06 | 2011-05-12 | 味の素株式会社 | Method for producing l-amino acid |
| EP2559754A2 (en) | 2011-08-18 | 2013-02-20 | Ajinomoto Co., Inc. | Method for producing an L-amino acid using a bacterium of the family enterobacteriaceae having enhanced expression of the flagella formation and motility cascade genes |
| WO2013024904A1 (en) | 2011-08-18 | 2013-02-21 | Ajinomoto Co.,Inc. | A method for producing an l-amino acid using a bacterium of the family enterobacteriaceae having enhanced expression of the flagella formation and motility cascade genes |
| EP2559754A3 (en) * | 2011-08-18 | 2013-03-20 | Ajinomoto Co., Inc. | Method for producing an L-amino acid using a bacterium of the family enterobacteriaceae having enhanced expression of the flagella formation and motility cascade genes |
| US9284584B2 (en) | 2011-08-18 | 2016-03-15 | Ajinomoto Co., Inc. | Method for producing an L-amino acid using a bacterium of the family Enterobacteriaceae having enhanced expression of the flagella formation and motility cascade genes |
| WO2013051685A1 (en) | 2011-10-07 | 2013-04-11 | 味の素株式会社 | Mutant γ-glutamyltransferase, and method for producing γ-glutamylvalylglycine or salt thereof |
| WO2014025023A1 (en) | 2012-08-10 | 2014-02-13 | 味の素株式会社 | METHOD FOR PRODUCING γ-GLUTAMYL-VALYL-GLYCINE CRYSTAL |
| EP2796560A1 (en) | 2013-04-23 | 2014-10-29 | Ajinomoto Co., Inc. | A method for producing an L-amino acid using a bacterium of the family Enterobacteriaceae having attenuated expression of the yjjK gene |
| WO2015030019A1 (en) | 2013-08-30 | 2015-03-05 | Ajinomoto Co.,Inc. | A METHOD FOR PRODUCING AN L-AMINO ACID USING A BACTERIUM OF THE FAMILY ENTEROBACTERIACEAE HAVING ATTENUATED EXPRESSION OF THE znuACB GENE CLUSTER |
| WO2015041265A1 (en) | 2013-09-17 | 2015-03-26 | 味の素株式会社 | Method for producing l-amino acid from seaweed-derived biomass |
| WO2015050276A1 (en) | 2013-10-02 | 2015-04-09 | Ajinomoto Co.,Inc. | A method for producing an l-amino acid using a bacterium of the family enterobacteriaceae having attenuated expression of a phosphate transporter-encoding gene |
| WO2015050234A1 (en) | 2013-10-02 | 2015-04-09 | 味の素株式会社 | Ammonia control apparatus and ammonia control method |
| WO2015060314A1 (en) | 2013-10-21 | 2015-04-30 | 味の素株式会社 | Method for producing l-amino acid |
| WO2015122544A1 (en) | 2014-02-14 | 2015-08-20 | Ajinomoto Co.,Inc. | A METHOD FOR PRODUCING AN L-AMINO ACID USING A BACTERIUM OF THE FAMILY ENTEROBACTERIACEAE HAVING OVEREXPRESSED THE yajL GENE |
| EP3098319A1 (en) | 2015-05-28 | 2016-11-30 | Ajinomoto Co., Inc. | A method for producing an l-amino acid using a bacterium of the family enterobacteriaceae having an attenuated expression of a gsha gene |
| WO2017146195A1 (en) | 2016-02-25 | 2017-08-31 | Ajinomoto Co., Inc. | A method for producing l-amino acids using a bacterium of the family enterobacteriaceae overexpressing a gene encoding an iron exporter |
| WO2018030507A1 (en) * | 2016-08-10 | 2018-02-15 | 味の素株式会社 | Production method for l-amino acid |
| US11198894B2 (en) | 2016-08-10 | 2021-12-14 | Ajinomoto Co., Inc. | Method of producing an l-amino acid involving a carotenoid biosynthesis enzyme |
| CN108531439A (en) * | 2018-04-16 | 2018-09-14 | 南京工业大学 | A kind of Recombinant organism and its construction method and application |
| CN108531439B (en) * | 2018-04-16 | 2020-04-07 | 南京工业大学 | Escherichia coli genetic engineering bacterium and construction method and application thereof |
| CN108795916A (en) * | 2018-07-16 | 2018-11-13 | 南京工业大学 | A kind of lysine decarboxylase mutant, its encoding gene and its expression and application |
| CN108795916B (en) * | 2018-07-16 | 2020-02-21 | 南京工业大学 | Lysine decarboxylase mutant, coding gene thereof, expression and application thereof |
| WO2020138178A1 (en) | 2018-12-27 | 2020-07-02 | Ajinomoto Co., Inc. | Method for producing basic l-amino acids or salts thereof by fermentation of an enterobacteriaceae bacterium |
| WO2020171227A1 (en) | 2019-02-22 | 2020-08-27 | Ajinomoto Co., Inc. | METHOD FOR PRODUCING L-AMINO ACIDS USING A BACTERIUM BELONGING TO THE FAMILY Enterobacteriaceae HAVING OVEREXPRESSED ydiJ GENE |
| WO2020204179A1 (en) | 2019-04-05 | 2020-10-08 | Ajinomoto Co., Inc. | Method of producing l-amino acids |
| WO2021060438A1 (en) | 2019-09-25 | 2021-04-01 | Ajinomoto Co., Inc. | Method for producing l-amino acids by bacterial fermentation |
Also Published As
| Publication number | Publication date |
|---|---|
| US20060160191A1 (en) | 2006-07-20 |
| US7547531B2 (en) | 2009-06-16 |
| EP1838726A1 (en) | 2007-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7547531B2 (en) | L-amino acid producing microorganism which has been modified to inactive the fimH gene, and a method for producing I-amino acid | |
| US9644009B2 (en) | L-amino acid-producing bacterium and a method for producing an L-amino acid | |
| JP5598329B2 (en) | Microorganism producing L-amino acid and method for producing L-amino acid | |
| US7919284B2 (en) | L-amino acid producing microorganism and a method for producing an L-amino acid | |
| CN101405398B (en) | Method for producing L-amino acid | |
| US8354255B2 (en) | L-amino acid-producing bacterium and a method for producing L-amino acids | |
| WO2008075483A1 (en) | Process for production of l-amino acid | |
| EP1928899B1 (en) | An l-amino acid-producing bacterium and a method for producing an l-amino acid | |
| EP1963486A1 (en) | L-amino acid producing bacterium and method of producing l-amino acid | |
| EP1979486A1 (en) | L-amino acid producing bacterium and method of producing l-amino acid | |
| US7871801B2 (en) | L-amino acid-producing bacterium and a method for producing L-amino acids | |
| US10787691B2 (en) | Method for producing L-amino acid | |
| CN103732736B (en) | The enterobacteriaceae lactobacteriaceae of the expression of flagellum formation and the mobility cascade gene with enhancing is used to produce the amino acid whose method of L- | |
| WO2021060438A1 (en) | Method for producing l-amino acids by bacterial fermentation | |
| CN101273138A (en) | An L-amino acid-producing bacterium and a method for producing L-amino acids | |
| WO2020171227A1 (en) | METHOD FOR PRODUCING L-AMINO ACIDS USING A BACTERIUM BELONGING TO THE FAMILY Enterobacteriaceae HAVING OVEREXPRESSED ydiJ GENE | |
| JP2007117076A (en) | L-amino acid producing bacteria and method for producing l-amino acid | |
| JP2007117077A (en) | L-amino acid-producing bacteria and method for producing the l-amino acid | |
| CN101273139A (en) | An L-amino acid-producing bacterium and a method for producing L-amino acids | |
| WO2008044714A1 (en) | Process for the preparation of l-threonine employing a bacterium of the enterobacteriaceae family with enhanced mdte and mdtf expression | |
| JP2009240161A (en) | Method for producing l-amino acid | |
| US20210317487A1 (en) | Method for Producing Basic L-Amino Acids or Salts Thereof By Fermentation of an Enterobacteriaceae Bacterium | |
| MX2007008641A (en) | A method for producing an l-amino acid using a bacterium of the enterobacteriaceae family having a pathway of glycogen biosynthesis disrupted. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006701432 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006701432 Country of ref document: EP |