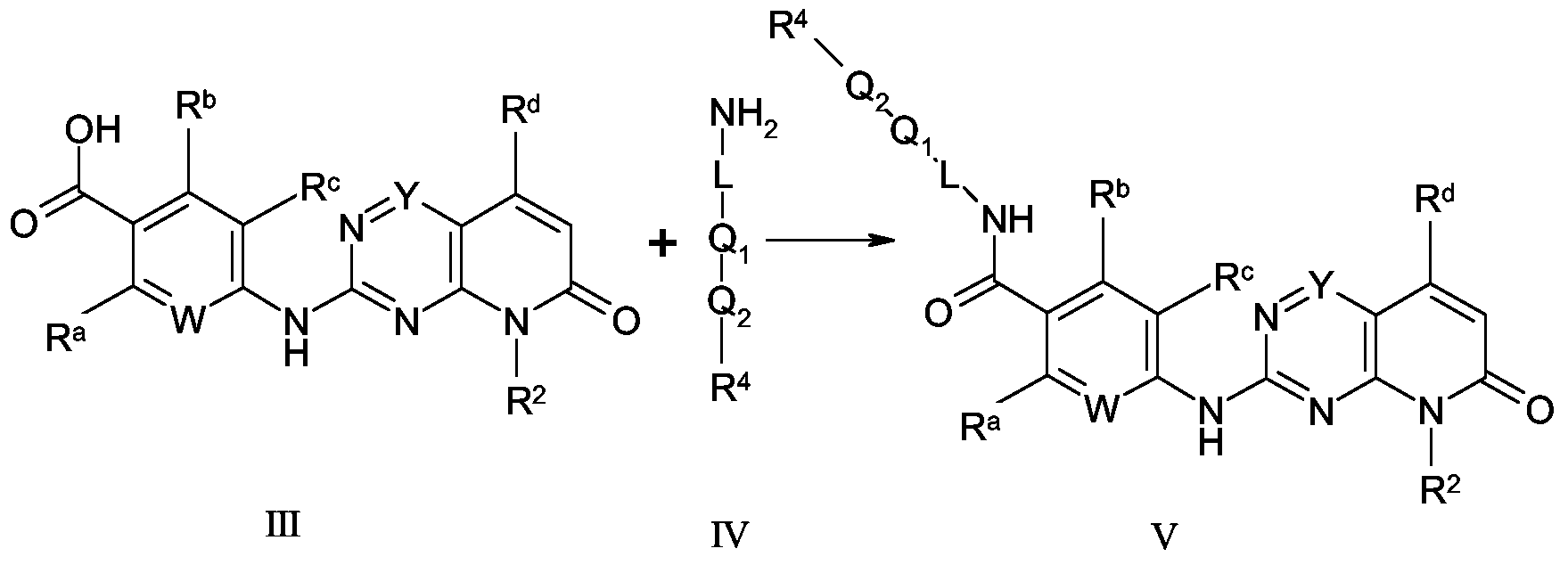
PTERIDINONE ALS PLK (POLO LIKE KINASE) INHIBITOREN
Die vorliegende Erfindung betrifft neue Pteridinone der allgemeinen Formel (1 )
(1) wobei die Reste L, Q1, Q2, X, Y, Ra, Rb, Rc, R1, R2, R3 und R4 die in den Ansprüchen und der Beschreibung genannten Bedeutungen haben, deren Isomere, Verfahren zur Herstellung dieser Pteridinone sowie deren Verwendung als Arzneimittel.
Hintergrund der Erfindung
Tumorzellen entziehen sich teilweise oder völlig der Regulation und Kontrolle durch den Organismus und zeichnen sich durch ein unkontrolliertes Wachstum aus. Dies beruht einerseits auf dem Verlust von Kontroll-Proteinen, wie z.B. Rb, pl6, p21 und p53 als auch auf der Aktivierung von so genannten Beschleunigern des Zellzykluses, den cyclin- abhängigen Kinasen (CDK' s).
Darüber hinaus wird auch für die Proteinkinase Aurora B eine essentielle Funktion beim Eintritt in die Mitose beschrieben. Aurora B phosphoryliert Histon H3 an SerlO und leitet damit die Chromosomenkondensation ein (Hsu et al. 2000, Cell 102:279-91). Ein spezifischer Zellzyklusarrest in der G2/M Phase kann aber auch z.B. durch Inhibition von spezifischen Phosphatasen wie z.B. Cdc25C (Russell andNurse 1986, Cell 45:145-53) ausgelöst werden. Hefen mit defektem Cdc25 Gen arretieren in der G2 Phase, während eine Überexpression von Cdc25 zu einem verfrühten Eintritt in die Mitosephase führt (Russell und Nurse 1987, Cell 49:559-67). Desweiteren kann ein Arrest in der G2/M Phase
auch durch Inhibition von bestimmten Motorproteinen, den so genannten Kinesinen wie z.B. Eg5 (Mayer et al. 1999, Science 286:971-4), oder durch Mikrotubuli stabilisierende oder destabilisierende Agentien (z.B. Colchicin, Taxol, Etoposid, Vinblastin, Vincristin) ausgelöst werden (Schiff und Horwitz 1980, Proc NatlAcad Sei U S A 77:1561-5).
Neben den Cyclin-abhängigen und den Aurora Kinasen spielen des weiteren die so genannten Polo-like Kinasen, eine kleine Familie von Serin/Threonin-Kinasen, eine wichtige Rolle bei der Regulation des eukaryontischen Zellzykluses. Bisher wurden die Polo-like Kinasen PLK-I, PLK-2, PLK-3 und PLK-4 in der Literatur beschrieben. Besonders für PLK-I wurde eine zentrale Rolle in der Regulation der Mitosephase gezeigt. PLK-I ist für die Reifung der Zentrosomen, für die Aktivierung der Phosphatase Cdc25C, sowie für die Aktivierung des Anaphase Promoting Complex verantwortlich (Glover et al. 1998, Genes Dev. 12:3777-87; Qian et al. 2001, MolBiol Cell. 12:1791-9). Die Injektion von PLK-I Antikörpern führt zu einem G2 Arrest in nicht transformierten Zellen, während Tumorzellen in der Mitosephase arretieren (Lane undNigg 1996, J Cell Biol. 135:1701- 13). Überexpression von PLK-I konnte für verschiedene Tumorarten, wie nicht kleinzelliges Lungenkarzinom, Plattenepithelkarzinom, Brust- und kolorektales Karzinom (Wolf et al. 1997, Oncogene 14 :543 -549; Knecht etal. 1999, Cancer Res. 59:2794 -2797; Wolf et al. 2000, Pathol. Res. Pract. 196:753 -759; Takahashi et al. 2003, Cancer Sei. 94: 148-52) gezeigt werden. Daher stellt diese Klasse von Proteinen ebenfalls einen interessanten Angriffspunkt zur therapeutischen Intervention proliferativer Krankheiten dar (Liu and Erikson 2003, Proc NatlAcad Sei USA 100:5789-5794).
Pteridinon-Derivate sind als Wirkstoffe mit antiproliferativer Wirkung aus dem Stand der Technik bekannt. WO 01/019825 und WO 03/020722 beschreiben die Verwendung von Pteridinonderivaten zur Behandlung von Tumorerkrankungen.
Die Resistenz vieler Tumorarten erfordert die Entwicklung neuer Arzneimittel zur Tumorbekämpfung. Daher ist es die Aufgabe der vorliegenden Erfindung neue Verbindungen mit antiproliferativer Wirkung bereitzustellen.
Detaillierte Beschreibung der Erfindung
Es wurde nun überraschenderweise gefunden, dass Verbindungen der allgemeinen Formel (I), worin die Reste L, Q1, Q2, X, Y, Z, Ra, Rb, Rc, R1, R2, R3 und R4 die nachstehend genannten Bedeutungen haben, als Inhibitoren spezifischer Zellzykluskinasen wirken. Somit können die erfindungsgemäßen Verbindungen beispielsweise zur Behandlung von Erkrankungen, die mit der Aktivität spezifischer Zellzykluskinasen in Zusammenhang stehen und durch exzessive oder anomale Zellproliferation charakterisiert sind, verwendet werden.
Die vorliegende Erfindung betrifft Verbindungen der allgemeinen Formel (1)
(1) worin
die gestrichelte Linie eine optionale Bindung darstellt, wobei
X N oder C-Re bedeutet, falls X und CR1 durch eine Doppelbindung verknüpft sind, oder
X -N-Rd bedeutet, falls X und CR1 durch eine Einfachbindung verknüpft sind,
Y N oder CH;
R1 ein Rest ausgewählt aus der Gruppe bestehend aus Wasserstoff, Halogen, =O,
-OR5, -C(=O)R6, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6 und Pseudohalogen,
oder ein gegebenenfalls einfach oder mehrfach substituierter Rest ausgewählt aus der Gruppe bestehend aus C1-6Alkyl, C2-6Alkenyl, C2-6Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(=O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -
NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen, oder sofern X und CR1 durch eine Einfachbindung verknüpft sind, auch die Gruppe O;
R2 Wasserstoff oder ein gegebenenfalls einfach oder mehrfach substituierter Rest ausgewählt aus der Gruppe bestehend aus C1-6Alkyl, C2-6Alkenyl, C2-6Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6,
-NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
R3 ein Rest ausgewählt aus der Gruppe bestehend aus Wasserstoff, Halogen, -OR5, -C(O)R5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5SO2R6, -N=CR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6 und Pseudohalogen, oder ein gegebenenfalls einfach oder mehrfach substituierter Rest ausgewählt aus der Gruppe bestehend aus C1-6Alkyl, C2-6Alkenyl, C2-6Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen:
L eine Bindung oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem C1-16-Alkyl, C2-16- Alkenyl und C2-16-Alkinyl, wobei der/die Substituent(en) gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(=O)R5, -C(O)OR5, -C(=O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6,
-NR5C(O)NR6R7, -NR5SO2R6, -N=CR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
Q1 und Q2 jeweils unabhängig voneinander eine Bindung oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem
C1-16-Alkyl, C2-16-Alkenyl, C2-16-Alkinyl, C3-10-Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituent(en) gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, R5, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -N=CR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6,
-NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
R4 Wasserstoff oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem C1-16Alkyl, C2-16Alkenyl, C2-16Alkinyl, C3- 10Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituent(en) gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus -NO2, R5, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
Ra, Rb, Rc, jeweils unabhängig voneinander ein Rest ausgewählt aus der Gruppe bestehend aus Wasserstoff, Halogen, -NO2, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -NOR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen; oder ein gegebenenfalls einfach oder mehrfach substituierter Rest ausgewählt aus der Gruppe bestehend aus C1-6Alkyl, C2-6 Alkenyl, C2-6Alkinyl ,
C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(=O)R5, -C(O)OR5, -C(=O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(=O)OR6, -NR5C(=O)NR6R7, -NR5SO2R6, -NOR5R6, -SSR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen und Pseudohalogen;
Rd Wasserstoff oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem C1-6Alkyl, C2-6Alkenyl, C2-6Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -N=CR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
Re ein Rest ausgewählt aus der Gruppe bestehend aus Wasserstoff, Halogen, Pseudohalogen oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem C1-6Alkyl, C2-6Alkenyl, C2-6Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die
Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NO2, -OR5, -C(O)R5, -C(O)OR5, -C(O)NR5R6, -NR5R6, -NR5C(O)R6, -NR5C(O)OR6, -NR5C(O)NR6R7, -NR5SO2R6, -N=CR5R6, -SR5, -SOR5, -SO2R5, -SO2NR5R6, -NR5SO2NR6R7, -OSO2NR5R6 und Pseudohalogen;
R5, R6 und R7 jeweils unabhängig voneinander Wasserstoff oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls einfach oder mehrfach substituiertem C1-5Alkyl, C2-5Alkenyl, C2-5Alkinyl, C3-10Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus C3-10Cycloalkyl, Aryl,
Heterocyclyl, Heteroaryl, Halogen, -NO2, -OR8, -C(=O)R8, -C(=O)OR8, -C(O)NR8R9, -NR8R9, -NR8C(=O)R9, -NR8C(=O)OR9, -NR8CC=O)NR9R1 °, -NR8C(=O)ONR9R10, -NR8SO2R9, -N=CR8R9, -SR8, -SOR8, -SO2R8, -SO2NR8R9, -NR8SO2NR9R10, -OSO2NR8R9 und Pseudohalogen;
R8, R9 und R10 jeweils unabhängig voneinander Wasserstoff oder ein Rest ausgewählt aus der Gruppe bestehend aus gegebenenfalls substituiertem C1-8Alkyl, C2-8Alkenyl, C2-8Alkinyl, C3-10Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituent(en) gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Halogen, -NH2, -OH und Pseudohalogen;
bedeuten, gegebenenfalls in Form ihrer Tautomeren, Racemate, Enantiomere, Diasteriomere und Gemische, sowie gegebenenfalls ihre pharmakologisch unbedenklichen Säureadditionssalze.
Ein Aspekt der Erfindung sind Verbindungen der allgemeinen Formel (1), worin
Y CH; bedeutet.
Ein weiterer Aspekt der Erfindung sind Verbindungen der allgemeinen Formel (1), worin Rc ein Rest ausgewählt aus der Gruppe bestehend aus Wasserstoff, -F, -Cl, Methyl und
Ethyl bedeutet.
Ein anderer Aspekt der Erfindung sind Verbindungen der allgemeinen Formel (1), worin Ra und Rb jeweils unabhängig voneinander Wasserstoff oder Fluor; oder ein gegebenenfalls einfach oder mehrfach substituierter Rest ausgewählt aus der Gruppe bestehend aus C1-2Alkyl, C2Alkenyl, C2Alkinyl, C3-6Cycloalkyl, Aryl, Heterocyclyl und Heteroaryl, wobei der/die Substituenten gleich oder verschieden sein können und ausgewählt werden aus der Gruppe bestehend aus Wasserstoff,
Halogen, -NO2, -OR4, -C(=O)R4, -C(=O)OR4, -C(O)NR4R5, -NR4R5, -
NR4C(O)R5, -NR4C(=O)OR5, -NR4C(=O)NR5R6, -NR4SO2R5, -N=CR4R5, -SR4, - SOR5, -SO2R4, -SO2NR4R5, -NR4, -SO2NR4R5 , -OSO2NR4R5 und Pseudohalogen bedeuten.
Ein zusätzlicher Aspekt der Erfindung sind Verbindungen der allgemeinen Formel (1), worin Ra und Rb unabhängig voneinander Wasserstoff oder Fluor bedeuten und die übrigen Reste wie vorstehend erwähnt definiert sind.
Erfindungsgemäß sind auch Verbindungen der allgemeinen Formel (1) umfasst, worin R2 Isopropyl oder Cyclopentyl bedeutet und die übrigen Reste wie vorstehend erwähnt definiert sind.
Ein Aspekt der Erfindung ist die Verwendung von Verbindungen der allgemeinen Formel (1) als Arzneimittel.
Ein weiterer Aspekt der Erfindung ist die Verwendung von Verbindungen der allgemeinen Formel (1) als Arzneimittel mit antiproliferativer Wirkung.
Ein wesentlicher Aspekt der Erfindung ist die Verwendung von Verbindungen der allgemeinen Formel (1) zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen ausgewählt aus der Gruppe bestehend aus Krebs, bakteriellen und viralen Infektionen, Entzündungs- und Autoimmunerkrankungen, Chemotherapeutika induzierter Alopezie und Mukositis, kardiovaskulärer Erkrankungen, nephrologischen Erkrankungen, sowie chronisch und akut neurodegenerativen Erkrankungen.
Ein anderer Aspekt der Erfindung ist die Verwendung einer Verbindung der Formel (I) zur Herstellung eines Arzneimittels zur Inhibierung der Polo-like Kinasen.
Ein zusätzlicher Aspekt der Erfindung ist die Verwendung einer Verbindung der allgemeinen Formel (1) zur Herstellung eines Arzneimittels zur Inhibierung der Polo-like Kinase PLKl.
Ein Aspekt der Erfindung ist die Verwendung von Verbindungen der allgemeinen Formel (1) zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von auf Überexpression der Polo-like Kinasen, beruhenden Tumorerkrankungen.
Ein anderer Aspekt der Erfindung ist eine Methode zur Behandlung und/oder Prävention von Erkrankungen ausgewählt aus der Gruppe bestehend aus Krebs, bakteriellen und viralen Infektionen, Entzündungs- und Autoimmunerkrankungen, Chemotherapeutika induzierter Alopezie und Mukositis, kardiovaskulärer Erkrankungen, nephrologischen Erkrankungen, sowie chronisch und akut neurodegenerativen Erkrankungen, dadurch gekennzeichnet, dass man einem Patienten eine effektive Menge einer Verbindung der Formel (I) gemäß einem der Ansprüche 1 bis 6 verabreicht.
Ein zusätzlicher Aspekt der Erfindung sind pharmazeutische Zubereitungen, enthaltend als Wirkstoff eine oder mehrere Verbindungen der allgemeinen Formel (I) gemäß einem der Ansprüche 1 bis 6 gegebenenfalls in Kombination mit üblichen Hilfs- und/oder Trägerstoffen.
DEFINITIONEN
Wie hierin verwendet treffen folgenden Definitionen zu, falls nicht anders beschrieben.
Unter Alkyl-Substitutenten sind jeweils gesättigte, geradkettige oder verzweigte aliphatische Kohlenwasserstoffreste (Alkylrest) zu verstehen.
Die Alkenyl-Substituenten sind jeweils geradkettige oder verzweigte, ungesättigte Alkylreste, die mindestens eine Doppelbindung aufweisen.
Unter Alkinyl-Substituenten sind jeweils geradkettige oder verzweigte, ungesättigte Alkylreste, die mindestens eine Dreifachbindung aufweisen, zu verstehen.
Halogenalkyl bezieht sich auf Alkylreste, in denen ein oder mehrere Wasserstoffatome durch Halogenatome ersetzt sind. Halogenalkyl umfasst sowohl gesättigte Alkylreste als auch ungesättigte Alkenyl- und Alkinylreste, wie beispielsweise -CF3, -CHF2, -CH2F, -CF2CF35-CHFCF3, -CH2CF3, -CF2CH3, -CHFCH3, -CF2CF2CF3, -CF2CH2CH3, -CF=CF2, -CCl=CH2, -CBr=CH2, -CJ=CH2, -C≡C-CF3, -CHFCH2CH3 und -CHFCH2CF3.
Halogen bezieht sich auf Fluor-, Chlor-, Brom- und/oder Jodatome.
Unter Pseudohalogen sind folgende Reste zu verstehen: -OCN, -SCN, -CF3 und -CN.
Unter Cycloalkyl ist ein mono- oder bizyklischer Ring zu verstehen, wobei das Ringsystem ein gesättigter Ring aber auch ein ungesättigter, nichtaromatischer Ring sein kann, welcher gegebenenfalls auch Doppelbindungen enthalten kann, wie zum Beispiel Cyclopropyl, Cyclopropenyl, Cyclobutyl, Cyclobutenyl, Cyclopentyl, Cyclopentenyl, Cyclohexyl, Cyclohexenyl, Norbornyl, Norbornenyl, Spiro[5.5]undecan, Spiro[5.4]decan und Spiro[4.4]-nonan.
Aryl bezieht sich auf monozyklische oder bizyklische Ringe mit 6 - 12 Kohlenstoffatomen wie beispielsweise Phenyl und Naphthyl.
Unter Heteroaryl sind mono- oder bizyklische Ringe zu verstehen, welche anstelle eines oder mehrere Kohlenstoffatome ein oder mehrere, gleich oder verschiedene Heteroatome enthalten, wie z.B. Stickstoff-, Schwefel- oder Sauerstoffatome. Beispielsweise genannt seien Furyl, Thienyl, Pyrrolyl, Oxazolyl, Thiazolyl, Isoxazolyl, Isothiazolyl, Pyrazolyl, Imidazolyl, Triazolyl, Tetrazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidyl, Pyridazinyl, Pyrazinyl und Triazinyl. Beispiele für bizyklische Heteroarylreste sind Indolyl, Isoindolyl, Benzofuranyl, Benzothienyl, Benzoxazolyl, Benzothiazolyl,
Benzisoxazolyl, Benzisothiazolyl, Benzimidazolyl, Indazolyl, Isoquinolinyl, Quinolinyl, Quinoxalinyl, Cinnolinyl, Phthalazinyl, Quinazolinyl und Benzotriazinyl, Indolizinyl, Oxazolopyridinyl, Imidazopyridinyl, Naphthyridinyl, Indolinyl, Isochromanyl, Chromanyl, Tetrahydroisochinolinyl, Isoindolinyl, Isobenzotetrahydrofuranyl, Isobenzotetrahydrothienyl, Isobenzothienyl, Benzoxazolyl, Pyridopyridinyl,
Benzotetrahydrofuranyl, Benzotetrahydrothienyl, Purinyl, Benzodioxolyl, Triazinyl, Phenoxazinyl, Phenothiazinyl, Pteridinyl, Benzothiazolyl, Imidazopyridinyl, Imidazothiazolyl, Dihydrobenzisoxazinyl, Benzisoxazinyl, Benzoxazinyl, Dihydrobenzisothiazinyl, Benzopyranyl, Benzothiopyranyl, Coumarinyl, Isocoumarinyl, Chromonyl, Chromanonyl, Pyridinyl-N-oxid Tetrahydroquinolinyl, Dihydroquinolinyl, Dihydroquinolinonyl, Dihydroisoquinolinonyl, Dihydrocoumarinyl, Dihydroisocoumarinyl, Isoindolinonyl, Benzodioxanyl, Benzoxazolinonyl, Pyrrolyl-N- oxide, Pyrimidinyl-N-oxid, Pyridazinyl-N-oxid, Pyrazinyl-N-oxid, Quinolinyl-N-oxid, Indolyl-N-oxid, Indolinyl-N-oxid, Isoquinolyl-N-oxid, Quinazolinyl-N-oxid, Quinoxalinyl- N-oxid, Phthalazinyl-N-oxid, Imidazolyl-N-oxid, Isoxazolyl-N-oxid, Oxazolyl-N-oxid, Thiazolyl-N-oxid, Indolizinyl-N-oxid, Indazolyl-N-oxid, Benzothiazolyl-N-oxid, Benzimidazolyl-N-oxid, Pyrrolyl-N-oxid, Oxadiazolyl-N-oxid, Thiadiazolyl-N-oxid, Triazolyl-N-oxid, Tetrazolyl-N-oxid, Benzothiopyranyl-S-oxid und Benzothiopyranyl-^S- dioxid.
Heterocyclyl bezieht sich auf 5 - 12 Kohlenstoffatome umfassende gesättigte oder ungesättigte, nicht aromatische mono-, bizyklische, überbrückte oder spirozyklische bizyklische Ringe, welche anstelle eines oder mehrere Kohlenstoffatome Heteroatome, wie Stickstoff, Sauerstoff oder Schwefel, tragen. Beispiele für solche Heterocyclylreste sind Tetrahydrofuranyl, Pyrrolidinyl, Pyrrolinyl, Imidazolidinyl, Imidazolinyl, Pyrazolidinyl, Pyrazolinyl, Piperidyl, Piperazinyl, Indolinyl, Isoindoliny, Morpholinyl, Thiomorpholinyl, Homomorpholinyl, Homopiperidyl, Homopiperazinyl, Thiomorpholinyl-S'-oxid, Thiomoφholinyl-5',1S'-dioxid, Tetrahydropyranyl, Piperidinyl, Tetrahydrothienyl, Homopiperidinyl, Homothiomorpholinyl-^S-dioxid, Oxazolidinonyl, Dihydropyrazolyl, Dihydropyrrolyl, Dihydropyrazinyl, Dihydropyridinyl, Dihydropyrimidinyl, Dihydrofuryl, Dihydropyranyl, Tetrahydrothienyl-S-oxid, Tetrahydrothienyl-^S-dioxid,
Homothiomorpholinyl-5-oxid, 2-Oxa-5-azabicyclo[2.2.1]heptan, 8-Oxa-3-aza- bicyclo[3.2.1 Joctan, 3,8-Diaza-bicyclo[3.2.1 Joctan, 2,5-Diaza-bicyclo[2.2.1 Jheptan, 3,8-Diaza-bicyclo[3.2.1]octan, 3,9-Diaza-bicyclo[4.2.1]nonan und 2,6-Diaza- bicyclo[3.2.2]nonan , 2,7-Diaza-spiro[3.5]nonan, 2,7-Diaza-spiro[4.4]nonan, 2,8-Diaza- spiro[4.5]decan und 3,9-Diaza-spiro[5.5]undecan.
Herstellung der erfindungsgemäßen Verbindungen:
Die Herstellung der erfindungsgemäßen Verbindungen kann nach den, im Folgenden beschriebenen Syntheseverfahren A bis C erfolgen, wobei die Substituenten der allgemeinen Formeln (I) bis (XVI) die zuvor genannten Bedeutungen haben. Diese Verfahren sind als Erläuterung der Erfindung zu verstehen, ohne selbige auf deren Gegenstand zu beschränken.
Analytik
Präperative Chromatographie:
Für die Mitteldruck Chromatographie (MPLC) wird Kieselgel der Firma Millipore (Bezeichnung: Granula Silica Si-60A 35-70μm) oder C-18 RP-Kieselgel der Firma
Macherey Nagel (Bezeichnung: Polygoprep 100-50 Cl 8) eingesetzt.
Für die präperative Hochdruck Chromatographie werden Säulen der Firma Waters
(Bezeichnung: XTerra Prep. MS C18, 5 μM, 30*100 mm oder Symmetrie C18, 5 μm,
19*100) verwendet.
Nuklear Magnet Resonanz (NMR) Spektroskopie:
Die Messung wird in deuteriertem Dimethylsulfoxid-d6 durchgeführt. Werden andere
Lösungsmittel verwendet sind diese explizit in den Beispielen oder in den Methoden vermerkt. Die Messwerte werden auf einer Delta-Scala in der Einheit ppm angegeben. Als Standard wird Tetramethylsilan verwendet. Die Messung erfolgt auf einem Avance 400
(400MHz-NMR-Spektrometer) von der Firma Bruker Biospin GmbH.
Massenspektroskopie / UV-Spektrometer:
Diese Daten werden mit Hilfe einer HPLC-MS Anlage (high Performance liquid chromatography mit Massendetektor) der Firma Agilent erzeugt.
Die Anlage ist so aufgebaut, dass anschließend an die Chromatographie (Säule: Zorbax SB-C8, 3,5 μm, 2, 1 *50, Fa. Agilent) ein Diodenarry-Detektor (Gl 315B von Fa. Agilent) und ein Massendetektor (1100 LS-MSD SL; G1946D; Fa. Agilent) in Reihe geschalten sind.
Die Anlage wird mit einem Fluß von 0,6 ml/min betrieben. Für einen Trennvorgang wird ein Gradient innerhalb von 3,5 min durchlaufen (Gradient Anfang: 95% Wasser und 5% Acetonitril; Gradient Ende: 5% Wasser und 95% Acetonitril; beiden Lösungsmitteln wird jeweils 0,1% Ameisensäure beigemischt).
Verfahren A
Stufe IA
Die Herstellung der Zwischenverbindung III erfolgt durch Substitution einer Abgangsgruppe LG, beispielsweise Halogen, SCN, Methoxy, Methansulfonyl, vorzugsweise Methansulfinyl oder Chlor, an einem heteroaromatischem System I durch ein Nukleophil II.
Schema IA
1 π m
Der Rest Rf entspricht entweder NH-L-Q1-Q2-R4 oder er repräsentiert Benzyloxy, Methoxy oder Hydroxy.
Es werden 1 Äquivalent der Verbindung I und 1 bis 2 Äquivalente der Verbindung II, in einem Lösungsmittel, beispielsweise 1,4-Dioxan, Tetrahydrofuran, N,N- Dimethylformamid oder JV, JV-Dimethylacetamid gerührt. Das Reaktionsgemisch wird 1 bis 5 Tage bei einer Temperatur von 15 - 25°C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand wird chromatographisch gereinigt.
Stufe 2A
Die Herstellung der Zwischenverbindung IV erfolgt durch Reduktion der Nitrogruppe an einem heteroaromatischem System III.
Schema 2A
Die Verbindung III wird in einem Lösungsmittel, beispielsweise Methanol, Ethanol, N,N- Dimethylformamid, Ethylacetat, Tetrahyrofuran oder Aceton gelöst. Man gibt einen Katalysator, beispielsweise Palladium auf Kohle, Palladiumhydroxid oder Raney-Nickel
zu. Diese Suspension wird in einen Autoklaven überfuhrt. Dieser wird mit einem Wasserstoffdruck von 2 bis 10 bar beaufschlagt. Man rührt 1 bis 5 Tage bei 20 bis 60°C. Anschließend wird der Katalysator abfiltriert und das Lösungsmittel im Vakuum entfernt. Alternativ kann man auch die obige Lösung mit Zinn(II)chlorid versetzen und 0,5-10 h bei 30 bis 100°C rühren. Nach wässriger Aufarbeitung wird die organische Phase im Vakuum eingeengt.
Stufe 3A
Die Herstellung der Zwischenverbindung VI erfolgt durch Kondensation eines Glyoxylat- Derivats V mit einer Verbindung IV. Schema 3A
Es werden 1 Äquivalent der Verbindung IV und 1 bis 2 Äquivalente der Verbindung V, in einem Lösungsmittel, beispielsweise 1,4-Dioxan, Tetrahydrofuran, JV,JV-Dimethylform- amid, Ethanol, Methanol oder JV, JV-Dimethylacetamid gerührt. Bei einer Temperatur von 15 bis 40°C werden 3 bis 7 Äquivalente einer Brönsted-Säure oder Lewis-Säure, beispielsweise Schwefelsäure, Essigsäure, Ameisensäure, Salzsäure, Aluminiumtrichlorid, Titantetrachlorid oder Ytterbium(III)triflathydrat zugegeben. Das Reaktionsgemisch wird 6 bis 36 h bei einer Temperatur von 50 bis 150°C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Stufe 4A
Verbindungen VI, deren Rest Rf Hydroxy darstellt, können direkt zur Herstellung der Endverbindungen VIII eingesetzt werden, wobei eine Verbindung VI mit einer Verbindung VII umgesetzt wird.
Verbindungen VI mit einem Rest Rf ungleich Hydroxy werden zuvor durch Hydrolyse oder ähnliche, dem Fachmann bekannte Verfahren in die Verbindungen überführt, bei denen der Rest Rf Hydroxy repräsentiert.
Schema 4A
VI vπ vm
Es werden 1 Äquivalent der Verbindung VI, 1 bis 1,5 Äquivalente der Verbindung VII und 1 bis 3 Äquivalente einer Base, beispielsweise Triethylamin oder Ethyldiisopropyl- amin in einem Lösungsmittel, beispielsweise 1,4-Dioxan, N,N-Dimethylformamid, N,N- Dimethylacetamid oder N-Methyl-2-pyrrolidinon gerührt. Bei einer Temperatur von 15 bis 25°C werden 1 bis 1,5 Äquivalente eines Kupplungsreagenzes, beispielsweise N,N- Dicyclohexylcarbodiimid, N,N-Diisopropylcarbodiimid, O-(Benzotriazol-l -y\)-N,N,N',N'- tetramethyluronium-tetrafluoroborat oder 1 -(3-N,N-Dimethylaminopropyl)-3- ethylcarbodiimid zugegeben. Das Reaktionsgemisch wird 4 bis 24 h bei einer Temperatur von 15 bis 25 °C gerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Verfahren B
Stufe IB
Die Herstellung der Zwischenverbindung III erfolgt durch Substitution einer Abgangsgruppe LG, beispielsweise Halogen, SCN, Methoxy, Methansulfonyl,
vorzugsweise Methansulfinyl oder Chlor, an einem heteroaromatischem System I durch ein Nukleophil II.
Schema IB
Es werden 1 Äquivalent der Verbindung I und 1 bis 1,5 Äquivalente der Verbindung II, in einem Lösungsmittel, beispielsweise 1 ,4-Dioxan, Tetrahydrofuran, Ethylacetat, N,N- Dimethylformamid, Acetonitril oder N,N-Dimethylacetamid gerührt.
Bei einer Temperatur von 15 bis 25°C werden 2 bis 2,5 Äquivalente einer Base, beispielsweise Kaliumcarbonat, Natriumcarbonat, Cäsiumcarbonat, N-Ethy\-N,N- diisopropylamin oder Triethylamin zugegeben. Das Reaktionsgemisch wird 12 bis 72 h bei einer Temperatur von 15 bis 25°C weitergerührt. Die unlöslichen Bestandteile werden abfiltriert und mit einem der oben genannten Lösungsmittel nachgewaschen. Anschließend wird das Lösungsmittel abdestilliert und der Rückstand wird chromatographisch gereinigt.
Stufe 2B Die Herstellung der Zwischenverbindung VI erfolgt durch Kondensation eines Oxalsäure¬ derivats mit einer Zwischenverbindung IV.
Schema 2B
IV V VI
Es werden 1 Äquivalent der Verbindung IV und 2 bis 2,5 Äquivalente einer Base, beispielsweise Kaliumcarbonat, Natriumcarbonat, Cäsiumcarbonat, N-Ethy\-N,N- diisopropylamin oder Triethylamin in einem Lösungsmittel, beispielsweise 1,4-Dioxan, Toluol, Tetrahydrofuran, Ethylacetat, N,N-Dimethylformamid, Acetonitril oder N,N- Dimethylacetamid gerührt. Bei einer Temperatur von -78 bis -30°C werden 1 bis 1,5 Äquivalente einer Verbindung V zugegeben. Das Reaktionsgemisch wird 3 bis 6 h bei einer Temperatur von -70 bis -30°C weitergerührt. Anschließend lässt man innerhalb von 5 bis 12 h auf 20°C aufwärmen und rührt danach für 6 bis 12 h bei 130°C. Nach Filtration der Reaktionslösung über Kieselgel wird das Lösungsmittel im Vakuum entfernt und der Rückstand in Wasser umkristallisiert.
Stufe 3B
Die Herstellung der Verbindung VIII erfolgt durch Substitution einer Abgangsgruppe LG, beispielsweise Halogen, SCN, Methoxy, Methansulfonyl, vorzugsweise Methansulfinyl oder Chlor an einem heteroaromatischem System VI durch ein Nukleophil VII.
Schema 3B
VI vπ viπ
Es werden 1 Äquivalent der Verbindung VI und 1 bis 3 Äquivalente der Verbindung VII in einem Lösungsmittel, beispielsweise 1,4-Dioxan, N,N-Dimethylformamid, N,N- Dimethylacetamid oder N-Methyl-2-pyrrolidinon gerührt. Bei einer Temperatur von 15 bis 40°C werden 1 bis 10 Äquivalente einer Mineralsäure, beispielsweise Schwefelsäure oder Salzsäure zugegeben. Das Reaktionsgemisch wird 12 bis 72 h bei einer Temperatur von 60 bis 120°C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Stufe 4B Verbindungen VIII deren Rest Rf Hydroxy darstellt können direkt zur Herstellung der
Endverbindungen X eingesetzt werden, wobei eine Verbindung VIII mit einer Verbindung IVX umgesetzt wird.
Verbindungen VIII mit einem Rest Rf ungleich Hydroxy werden zuvor durch Hydrolyse oder ähnliche, dem Fachmann bekannte Verfahren in die Verbindungen überführt, bei denen der Rest Rf Hydroxy repräsentiert.
Schema 4B
Es werden 1 Äquivalent der Verbindung VIII, 1 bis 1,5 Äquivalente der Verbindung IVX und 1 bis 3 Äquivalente einer Base, beispielsweise Triethylamin oder Ethyldiisopropyl- amin in einem Lösungsmittel, beispielsweise 1,4-Dioxan, N,N-Dimethylformamid, N,N- Dimethylacetamid oder N-Methyl-2-pyrrolidinon gerührt. Bei einer Temperatur von 15 bis 25°C werden 1 bis 1,5 Äquivalente eines Kupplungsreagenzes, beispielsweise N,N- Dicyclohexylcarbodiimid, N,N-Diisopropylcarbodiimid, O-(Benzotriazol-l -y\)-N,N,N',N'- tetramethyluronium-tetrafluoroborat oder 1 -(3-N,NDimethylaminopropyl)-3- ethylcarbodiimid zugegeben. Das Reaktionsgemisch wird für 4 bis 24 h bei einer Temperatur von 15 bis 25°C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Verfahren C
Stufe IC Die Verbindung I wird wie in WOOl 19825 beschrieben hergestellt.
Die Herstellung der Verbindung III erfolgt durch Substitution einer Abgangsgruppe LG, beispielsweise Halogen, SCN, Methoxy, Methansulfonyl, vorzugsweise Methansulfinyl oder Chlor, an einem heteroaromatischem System I durch ein Nukleophil II.
Schema IC
i π πi
Die Herstellung erfolgt analog zu WOOl 19825.
Stufe 2C
Verbindungen III deren Rest Rf Hydroxy darstellt können direkt zur Herstellung der Endverbindungen V eingesetzt werden, wobei eine Verbindung III mit einer Verbindung IV umgesetzt wird.
Verbindungen III mit einem Rest Rf ungleich Hydroxy werden zuvor durch Hydrolyse oder ähnlich dem Fachmann bekannte Verfahren in die Verbindungen überführt, bei denen der Rest Rf Hydroxy repräsentiert. Schema 2C
m IV V
Es werden 1 Äquivalent der Verbindung III, 1 bis 1,5 Äquivalente der Verbindung IV und 1 bis 3 Äquivalente einer Base, beispielsweise Triethylamin oder Ethyldiisopropylamin in einem Lösungsmittel, beispielsweise 1,4-Dioxan, N,N-Dimethylformamid, N,N- Dimethylacetamid oder N-Methyl-2-pyrrolidinon gerührt. Bei einer Temperatur von 15 bis 25°C werden 1 bis 1,5 Äquivalente eines Kupplungsreagenzes, beispielsweise N,N- Dicyclohexylcarbodiimid, N,N-Diisopropylcarbodiimid, O-(Benzotriazol-l -y\)-N,N,N',N'- tetramethyluronium-tetrafluoroborat oder 1 -(3-N,N-Dimethylaminopropyl)-3- ethylcarbodiimid zugegeben. Das Reaktionsgemisch wird 4 bis 24 h bei einer Temperatur von 15 bis 25 °C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Verfahren D
Stufe ID
Die Verbindung I wird, wie in WOO 170741 beschrieben hergestellt. Die Herstellung der Verbindung III erfolgt durch Substitution einer Abgangsgruppe LG, beispielsweise Halogen, SCN, Methoxy, Methansulfonyl, vorzugsweise Methansulfinyl oder Chlor, an einem heteroaromatischem System I durch ein Nukleophil II.
Schema ID
i π in
Die Herstellung erfolgt analog zu WOO 170741.
Stufe 2D
Verbindungen III deren Rest Rf Hydroxy darstellt können direkt zur Herstellung der
Endverbindungen V eingesetzt werden, wobei eine Verbindung III mit einer Verbindung
IV umgesetzt wird.
Verbindungen III mit einem Rest Rf ungleich Hydroxy werden zuvor durch Hydrolyse oder ähnlich dem Fachmann bekannte Verfahren in die Verbindungen überfuhrt, bei denen der Rest Rf Hydroxy repräsentiert.
Schema 2D
Es werden 1 Äquivalent der Verbindung III, 1 bis 1,5 Äquivalente der Verbindung IV und 1 bis 3 Äquivalente einer Base, beispielsweise Triethylamin oder Ethyldiisopropylamin in einem Lösungsmittel, beispielsweise 1,4-Dioxan, N,N-Dimethylformamid, N,N- Dimethylacetamid oder N-Methyl-2-pyrrolidinon gerührt. Bei einer Temperatur von 15 bis 25°C werden 1 bis 1,5 Äquivalente eines Kupplungsreagenzes, beispielsweise N,N- Dicyclohexylcarbodiimid, N,N-Diisopropylcarbodiimid, O-(Benzotriazol-l -y\)-N,N,N',N'- tetramethyluronium-tetrafluoroborat oder 1 -(3 -N,N-Dimethylaminopropyl)-3 - ethylcarbodiimid zugegeben. Das Reaktionsgemisch wird 4 bis 24 h bei einer Temperatur von 15 bis 25 °C weitergerührt. Danach wird das Lösungsmittel abdestilliert und der Rückstand chromatographisch gereinigt.
Methode 1
4-(8-Cyclopentyl-5-methyl-6,7-dioxo-5,6,7,8-tetrahvdro-pteridin-2-ylamino)-benzoesäure
a) 2-Chlor-N4-cyclopentyl-N5-methyl-pyrimidin-4,5-diamin
4,0 g (22,5 mmol) 2,4-Dichlor-5-methylamino-pyrimidin und 6,4 g (46,1 mmol)
Kaliumcarbonat werden in 100 ml Acetonitril vorgelegt. Anschließend werden 2,5 ml (24,7 mmol) Cyclopentylamin hinzu gegeben und der Ansatz 3 Tage bei 25°C gerührt. Die unlöslichen Bestandteile werden abfiltriert. Das Filtrat wird im Vakuum eingeengt und das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient Kieselgel und als Eluent wird ein Gemisch bestehend aus Cyclohexan und Essigester (2:1) verwendet. Ausbeute: 1 ,6 g (7, 1 mmol, 31 %) MS (ESI): 227 (M+H)+
b) 2-Chlor-8-cyclopentyl-5-methyl-5,8-diydropteridin-6,7-dion
2,4 g (10,6 mmol) 2-Chlor-N4-cyclopentyl-N5-methyl-pyrimidin-4,5-diamin in 80 ml Toluol und 0,08 ml N,N-Dimethylacetamid werden unter Argonatmosphäre und bei -65 °C mit 5,6 ml (32,3 mmol) N,N-Diisopropylethylamin und anschließend mit 1,5 ml (16,1 mmol) Methyloxalylchlorid versetzt. Es wird 3 h bei dieser Temperatur gerührt und danach lässt man innerhalb von 4 h auf 25°C aufwärmen. Anschließend wird 3 h refluxiert. Das Reaktionsgemisch wird über Kieselgel filtriert, mit Toluol gewaschen und anschließend im Vakuum eingeengt. Der Rückstand wird mit Wasser verrührt, abgesaugt, mit Wasser und anschließend mit Ether gewaschen und im Vakuum getrocknet.
Ausbeute: 2,33 g (8,3 mmol, 78%) MS (ESI): 303 (M+Na)+
c) 4-(8-Cyclopentyl-5-methyl-6,7-dioxo-5,6,7,8-tetrahydro-pteridin-2-ylamino)- benzoesäure
33 mg (0,12 mmol) 2-Chlor-8-cyclopentyl-5-methyl-5,8-diydropteridin-6,7-dion werden in 1 ml Dioxan gelöst und mit 21 mg 4-Aminobenzoesäure und 0,47 ml (0,94 mmol) 2 N Salzsäure versetzt. Der Ansatz wird 36 h refluxiert. Anschließend wird das Reaktionsgemisch mit Natriumhydrogencarbonat basisch gestellt und eingeengt. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient Kieselgel und als Eluent wird Ethanol verwendet. Ausbeute: 14 mg (0,04 mmol, 31%) MS (ESI): 404 (M+Na)+
Methode 2
4-(4-Amino-cvclohexyl)-morpholin
a) Dibenzyl-(4-morpholino-4-yl-cyclohexylVamin
3,9 g (30 mmol) 4-Dibenzylamino-cyclohexanon werden in 100 ml Dichlormethan gelöst und mit 3,9 g (45 mmol) Morpholin und 9,5 g (45 mmol) Natriumtriacetoxyborhydrid 12 h bei Raumtemperatur gerührt. Anschließend wird mit Wasser und Kaliumcarbonat versetzt, die organische Phase abgetrennt, getrocknet und im Vakuum das Lösungsmittel abgezogen. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient Kieselgel und als Eluent wird Essigester, dem 10% einer Mischung aus 90% Methanol und 10% gesättigter wässriger Ammoniak-Lösung zugefügt sind, verwendet. Die geeigneten Fraktionen werden gesammelt und im Vakuum eingeengt.
Ausbeute: 6,6 g (18 mmol, 60%) cis-Isomer 2 g (5,4 mmol, 18%) trans-Isomer
b) trans-4-Moφholino-4-yl-cyclohexylamin
7,2 g (16,4 mmol) trans-Dibenzyl-4-morpholino-cyclohexylamin werden in 100 ml
Methanol gelöst und an 1,4 g Palladium auf Kohle (10% Pd) bei 30 bis 50°C hydriert. Das
Lösungsmittel wird im Vakuum abgezogen und der Rückstand aus Ethanol und konz. HCl kristallisiert.
Ausbeute: 3,9 g (15,2 mmol, 93%)
Schmelzpunkt: 312 °C
Analog zu dieser Methode werden folgende Verbindungen hergestellt. Die dabei eingesetzten Amine sind kommerziell erhältlich oder ihre Herstellung ist literaturbekannt (JChem Soc 1948, 155, 157).
4-(8-Cvclopentyl-6-methyl-7-oxo-7,8-dihvdro-pteridin-2-ylamino)-3-methoxy- benzoesäure
a) 4-(4-Cvclopentylamino-5-nitro-pyrimidin-2-ylamino)-3 -methoxy- benzoesäurebenzylester
2,4 g (9,8 mmol) (2-Chloro-5-nitro-pyrimidin-4-yl)-cyclopentyl-amin und 4,36 g (14,8 mmol) 4-Amino-3-methoxybenzoesäurebenzylester werden in 12 ml Isopropanol suspendiert und 1 h unter Rückfluss erhitzt. Das Reaktionsgemisch wird auf 20°C abgekühlt und der entstehende Niederschlag abgesaugt. Dieses Rohprodukt wird über Säulenchromatographie gereinigt. Als Trägermaterial dient Kieselgel und als Eluent wird ein Gemisch bestehend aus Cyclohexan und Essigester (8:2) verwendet. Ausbeute: 2,4 g (5,1 mmol, 52%) MS (ESI): 464 (M+H)+
b) 4-(4-Cvclopentylamino-5-amino-pyrimidin-2-ylamino)-3-methoxy- benzoesäurebenzylester 2,4 g (5,1 mmol) 4-(4-Cyclopentylamino-5-nitro-pyrimidin-2-ylamino)-3-methoxy- benzoesäurebenzylester werden in 150 ml Ethylacetat auf 70°C erhitzt und mit 3,5 g (15,5 mmol) Zinn(II)chlorid Dihydrat versetzt. Es wird 30 min bei dieser Temperatur gerührt. Anschließend lässt man die Reaktionsmischung auf 20°C abkühlen und gibt 3 ml konzentrierten Ammoniak zu. Die unlöslichen Bestandteilen werden abfiltriert und die organische Phase abgetrennt. Die organische Phase wird getrocknet und im Vakuum eingeengt.
Ausbeute: 1 ,9 g (4,4 mmol, 86%) MS (ESI): 434 (M+H)+
c) 4-(8-Cyclopentyl-6-methyl-7-oxo-7,8-dihydro-pteridin-2-ylamino)-3-methoxy- benzoesäurebenzylester
300 mg (0,69 mmol) 4-(4-Cyclopentylamino-5-amino-pyrimidin-2-ylamino)-3-methoxy- benzoesäurebenzylester, 77 μl (0,69 mmol) 2-Oxo-propionsäureethylester und 71 mg (0,69 mmol) Ytterbiumtriflat Monohydrat werden in 6 ml Dioxan gelöst. Nach 6 h bei 120°C wird das Lösungsmittel unter Vakuum entfernt. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient Cl 8-RP-Kieselgel und es wird ein Gradient durchlaufen, der am Startpunkt aus 95% Wasser und 5% Acetonitril und am Endpunkt aus 2% Wasser und 98% Acetonitril besteht. Beiden Lösungsmitteln ist 0,1% Ameisensäure zugemischt. Ausbeute: 80 mg (0, 17 mmol, 24%) MS (ESI): 486 (M+H)+
d) 4-(8-Cyclopentyl-6-methyl-7-oxo-7, 8-dihvdro-pteridin-2-ylamino)-3 -methoxy- benzoesäure
80 mg (0,165 mmol) 4-(8-Cyclopentyl-6-methyl-7-oxo-7,8-dihydro-pteridin-2-ylamino)-3- methoxy-benzoesäurebenzylester werden in 440 μl 5 M dioxanischer Salzsäure und in
310 μl 6 M wässriger Salzsäure gelöst und 16 h unter Rückfluss erhitzt. Der entstehende
Niederschlag wird abfiltriert und getrocknet.
Ausbeute: 60 mg (0, 152 mmol, 92%)
MS (ESI): 396 (M+H)+
Methode 4
(S)-I -(Tetrahvdro-pwan-4-yl)-pyrrolidin-3 -ylamin
a) [(S)-I -(Tetrahydro-pyran-4-yl)-pyrrolidin-3 -vi] -carbaminsäure-fert-butylester
4 g (21,47 mmol) (S)-3-Boc-amino-pyrrolidin und 2,37g (23,62 mmol) Tetrahydro-pyran- 4-on werden in 50 ml Dichlormethan gelöst und mit 0,43 ml (7,51 mmol) Eisessig versetzt. Man lässt 1 h bei 25°C rühren und gibt anschließend 0,8 ml (13,96 mmol) Eisessig und portionsweise 9,1 g (42,94 mmol) Natriumtrisacetoxyborhydrid zu. Nach 2 h bei 25°C wird mit 50 ml gesättigter Natriumhydrogencarbonat-Lösung versetzt. Diese Mischung rührt man 4 h bei 25°C. Anschließend werden die Phasen getrennt. Die wässrige Phase wird nochmals mit 50 ml Dichlormethan extrahiert. Die vereinigten organischen Phasen werden getrocknet und das Lösungsmittel unter Vakuum entfernt. Ausbeute: 5,72 g (21 , 19 mmol, 98%)
b) (S)-I -(Tetrahvdro-pyran-4-yl)-pyrrolidin-3-ylamin
11,66 g (43,12 mmol) [(S)-l-(Tetrahydro-pyran-4-yl)-pyrrolidin-3-yl]-carbaminsäure-tert- butylester werden unter Wasserkühlung in 80 ml Trifluoressigsäure gelöst. Man last 1 h bei 25°C rühren und entfernt anschließend das Lösungsmittel unter Vakuum. Der Rückstand wird in einem Gemisch aus Ether und Isopropanol gelöst. Diese Lösung wird mit isopropanolischer Salzsäure versetzt. Dabei fällt ein Niederschlag aus. Dieser wird abgesaugt und getrocknet. Ausbeute: 9,47 g (38,97 mmol, 90%) MS (ESI): 171 (M+H)
+
Beispiel 1 4-(8-Cyclopentyl-5-methyl-6,7-dioxo-5,6,7,8-tetrahydro-pteridin-2-ylamino)-iV-(4- morpholin-4-yl-cvclohexyl)-benzamid
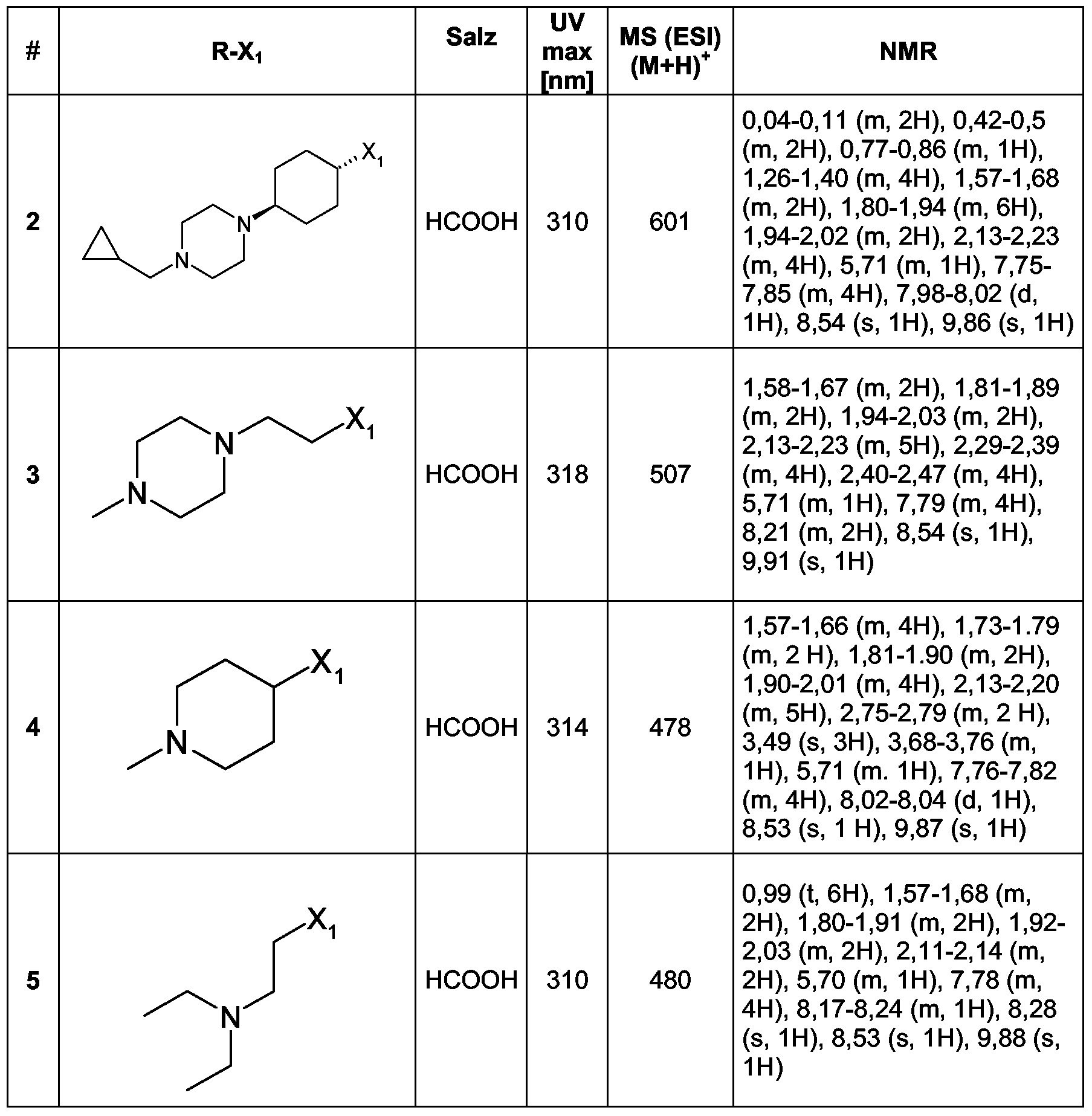
45 mg (0,119 mmol) 4-(8-Cyclopentyl-5-methyl-6,7-dioxo-5,6,7,8-tetrahydro-pteridin-2- ylamino)-benzoesäure (Methode 1), 166 μl (0,954 mmol) N-Ethyldiisopropylamin, 46 mg (0,143 mmol) O-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium-tetrafluorborat und 33 mg (0,179 mmol) trans-4-Morpholin-4-yl-cyclohexylamin (Methode 2) werden in 4 ml N,N-Dimethylformamid gelöst. Nach 15 h bei Raumtemperatur wird das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient C18-RP-Kieselgel und es wird ein Gradient durchlaufen der am Startpunkt aus 95% Wasser und 5% Acetonitril und am Endpunkt aus 2% Wasser und 98% Acetonitril besteht. Beiden Lösungsmitteln ist 0,1% Ameisensäure zugemischt. Die Verbindung wird als Formiat erhalten. Ausbeute: 34 mg (0,061 mmol; 51 %) UV max: 314 nm MS (ESI): 548 (M+H)
+ 1H-NMR: 1,23-1,41 (m, 4H), 1,58-1,68 (m, 2H), 1,80-1,93 (m, 6H), 1,94-2,03 (m, 2H), 2,13-2,23 (m, 2H), 5,71 (m, 1H), 7,74-7,82 (m, 4H), 7,97-8,01 (m, 1H), 8,18 (s, 1H), 8,54 (s, 1H), 9,86 (s, 1H)
Beispiele 2-10 Die folgenden Verbindungen sind über ein analoges Verfahren, wie in Beispiel 1 beschrieben, hergestellt. Das für die Darstellung des Amids eingesetzte Amin ist kommerziell erhältlich oder nach der in Methode 2 oder Methode 4 beschriebenen Vorschrift hergestellt.
4-(8-Cvclopentyl-5-methyl-6,7-dioxo-5,6,7,8-tetrahvdro-pteridin-2-ylamino)-3-methoxy- JV-(I -methyl-piperidin-4- vP-benzamid
30 mg (0,11 mmol) 2-Chlor-8-cyclopentyl-5-methyl-5,8-dihydropteridin-6,7-dion (Methode 1) werden in 0.3 ml Isoamylalkohol suspendiert und mit 28 mg (0.11 mmol) 4- Amino-3-methoxy-7V-(l-methyl-piperidin-4-yl)-benzoesäureamid (J Pharm Sei. 1989, 78(10):829-32) und 25 mg (0.15 mmol) p-Toluolsulfonsäure für 30 min bei 140°C erhitzt. Das Reaktionsgemisch wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient C18-RP-Kieselgel und es wird ein Gradient durchlaufen der am Startpunkt aus 95% Wasser und 5% Acetonitril und am Endpunkt aus 2% Wasser und 98% Acetonitril besteht. Beiden Lösungsmitteln ist 0,1% Ameisensäure zugemischt. Die Verbindung wird als Formiat erhalten.
Ausbeute: 15 mg (0,030 mmol; 28 %)
UV max: 322 nm
MS (ESI): 508 (M+H)+
1H-NMR: 1,52-1,69 (m, 4H), 1,73-1,98 (m, 6 H), 2,00-2,26 (m, 7H), 2,79-2,88 (m, 2H), 3,48 (s, 3H), 3,71-3,83 (m, 1H), 3,94 (s, 3H), 5,65 (m, 1H), 7,49-7,54
(m, 2H), 8,08-8,14 (m, 1H), 8,17-8,22 (m, 2H), 8,25 (s, 1H), 8,51 (s, 1H)
Beispiele 12-13
Die folgenden Verbindungen sind über ein analoges Verfahren, wie in Beispiel 11 beschrieben, hergestellt.
Das für die Darstellung der Verbindungen eingesetzte Anilin wird nach literaturbekannten Verfahren hergestellt (J Pharm Sei. 1989, 78(10):829-32; BioorgMed Chem Lett. 2003 13(3):369-373 oder J Med Chem. 1990, 33(ll):3072-78).
4-(8-Cvclopentyl-6-methyl-7-oxo-7,8-dihydro-pteridin-2-ylamino)-3-methoxy-iV-(l- methyl-piperidin-4-ylVbenzamid
70 mg (0,177 mmol) von 4-(8-Cyclopentyl-6-methyl-7-oxo-7,8-dihydro-pteridin-2- ylamino)-3-methoxy-benzoesäure (Methode 3), 1,3 ml (10 mmol) Triethylamin, 74 mg (0,230 mmol) O-(Benzotriazol-l-yl)-N,N,N',N'-tetramethyluronium-tetrafluorborat und 20 mg (0,177 mmol) 1-Methyl-piperidin werden in 3 ml Dichlormethan gelöst. Nach 5 h bei 25°C wird das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient C18-RP-Kieselgel und es wird ein Gradient durchlaufen der am Startpunkt aus 95% Wasser und 5% Acetonitril und am Endpunkt aus 2% Wasser und 98% Acetonitril besteht. Beiden Lösungsmitteln ist 0,1% Ameisensäure zugemischt. Die Verbindung wird als Formiat erhalten. Ausbeute: 12 mg ( mmol; 14 %) UV max: 363 nm MS (ESI): 492 (M+H)+ 1H-NMR: 1,53-1,68 (m, 4H), 1,74-1,91 (m, 6H), 1,96-2,04 (m, 2H), 2,14-2,25 (m,
5H), 2,36 (s, 3H), 2,78-2,85 (m, 2H), 3,91 (s, 3H), 5,66 (m, 1H), 7,51-7,56 (m, 2H), 8,04 (d, 1H), 8,17 (d, 1H), 8,26 (s, 1H), 8,76 (s, 1H), 8,77 (s, 1H)
Beispiel 15
Die folgende Verbindung wird analog zu Beispiel 14 hergestellt.
Das für die Darstellung des Amids eingesetzte Amin ist kommerziell erhältlich oder ist nach der in Methode 2 beschriebenen Vorschrift hergestellt.
Beispiel 16
4-(8-Cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-dlpyrimidin-2-ylamino)-iV- propyl-benzamid
27 mg (0,088 mmol) 8-Cyclopentyl-2-methansulfonyl-5-methyl-8H-pyrido[2,3- d]pyrimidin-7-on wird in 0,5 ml 2-Butanol gelöst und mit 23 mg (0,1 mmol) A- Aminobenzoesäure-N-propylamid Ηydrochlorid versetzt.
Nach 15 h bei 100°C wird das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird durch Säulenchromatographie gereinigt. Als Trägermaterial dient C18-RP-Kieselgel und es wird ein Gradient durchlaufen der am Startpunkt aus 95% Wasser und 5% Acetonitril und am Endpunkt aus 5% Wasser und 95% Acetonitril besteht. Beiden Lösungsmitteln ist 0,1%
Ameisensäure zugemischt.
Ausbeute: 11 mg (mmol; 31%)
UV max: 350 nm
MS (ESI): 406 (M+H)+
1H-NMR: 0,83-0,94 (m, 3H), 1,47-1,69 (m, 4H), 1,73-1,86 (m, 2H), 1,89-2,03 (m,
2H), 2,19-2,32 (m, 2H), 2,39 (s, 3H), 3,16-3,26 (m, 2H), 5,81-5,93 (m, 1H), 6,21-6,26 (m, 1H), 7,77-7,87 (m, 4H), 8,26-8,35 (m, 1H), 8,87 (s, 1H), 10,18 (s, 1H)
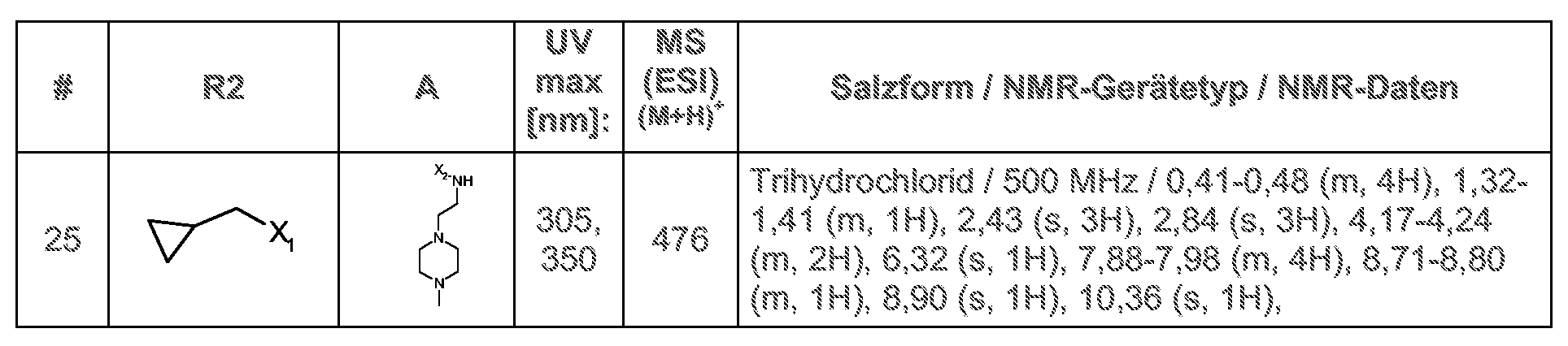
Beispiele 17-25 Die folgenden Verbindungen sind über ein analoges Verfahren, wie in Beispiel 14 beschrieben, hergestellt. Die Herstellung des Benzoesäure-Derivats ist in Methode 5 beschrieben. Das für die Darstellung des Amids eingesetzte Amin ist kommerziel erhältlich .
Beispiele26-40 Die folgenden Verbindungen sind über ein analoges Verfahren, wie in Beispiel 14 beschrieben, hergestellt. Die Herstellung des Benzoesäure-Derivats ist in Methode 5 beschrieben. Das für die Darstellung des Amids eingesetzte Amin ist kommerziel erhältlich oder in Methode 6 beschrieben.
Biologische Eigenschaften
Wie durch DNA-Färbung mit darauf folgender FACS Analyse gezeigt werden konnte, ist die, durch die erfindungsgemäßen Verbindungen bewirkte, Proliferationsinhibition vor allem durch einen Arrest der Zellen in der G2/M Phase des Zellzyklus vermittelt. Die Zellen arretieren abhängig von dem verwendeten Zelltyp für eine bestimmte Zeitspanne in dieser Zellzyklus Phase, bevor der programmierte Zelltod eingeleitet wird. Ein Arrest in der G2/M Phase des Zellzyklus kann z.B. durch die Inhibition spezifischer
Zellzykluskinasen ausgelöst werden. Auf Grund ihrer biologischen Eigenschaften eignen sich die erfindungsgemäßen Verbindungen der allgemeinen Formel I, deren Isomere und deren physiologisch verträgliche Salze zur Behandlung von Erkrankungen, die durch exzessive oder anomale Zellproliferation charakterisiert sind.
Zu solchen Erkrankungen gehören beispielsweise: Virale Infektionen (z.B. HIV und Kaposi Sarkoma); Entzündung und Autoimmun-Erkrankungen (z.B. Colitis, Arthritis, Alzheimer Erkrankung, Glomerulonephritis und Wund-Heilung); bakterielle, fungale und/oder parasitäre Infektionen; Leukämien, Lymphome und solide Tumore; Haut- Erkrankungen (z.B. Psoriasis); Knochen-Erkrankungen; kardiovaskuläre Erkrankungen
(z.B. Restenose und Hypertrophie). Ferner sind sie nützlich als Schutz von proliferierenden Zellen (z.B. Haar-, Intestinal-, Blut- und Progenitor-Zellen) gegen DNA-Schädigung durch Strahlung, UV-Behandlung und/oder zytostatischer Behandlung (Davis et al., 2001). Die neuen Verbindungen können zur Prävention, Kurz- oder Langzeitbehandlung der vorstehend genannten Erkrankungen, auch in Kombination mit anderen Wirkstoffen, die für dieselben Indikationen Verwendung finden, z.B. Zytostatika, Steroide oder Antikörper, verwendet werden.
Beispiel PLK-I Kinascassay
Rekombinantes humanes und an seinem N-terminalen Ende mit GST verbundenes PLKl Enzym wird aus Bakulovirus infizierten Insektenzellen (Sf21) isoliert. Die Reinigung erfolgt durch Affinitätschromatographie an Glutathion Sepharose Säulen.
4x107 Sf21 Zellen (Spodoptera frugiperda) in 200 ml Sf-900 II Serum freien
Insektenzellmedium (Life Technologies) werden in eine Spinnerflasche ausgesät. Nach 72 Stunden Inkubation bei 27°C und 70 rpm werden IxIO8 Sf21 Zellen in insgesamt 180 ml Medium in eine neue Spinnerflasche ausgesät. Nach weiteren 24 Stunden werden 20 ml rekombinanter Baculovirus Stammsuspension zugesetzt und die Zellen 72 Stunden bei 27°C bei 70 rpm kultiviert. 3 Stunden vor dem Ernten wird Okadainsäure zugesetzt
(Calbiochem, Endkonzentration 0,1 μM) und die Suspension weiter inkubiert. Die Zellzahl wird bestimmt, die Zellen abzentrifugiert (5 Minuten, 4°C, 800 rpm) und Ix mit PBS (8 g NaCM. 0,2 g KCl/l, 1,44 g Na2HPO4/l, 0,24 g KH2PO4/1) gewaschen. Nach nochmaligem Abzentrifugieren wird das Pellet in flüssigem Stickstoff Schock gefroren. Danach wird das Pellet rasch aufgetaut und in eiskaltem Lysispuffer (50 mM HEPES pH 7,5, 10 mM
MgCl2, 1 mM DTT, 5 μg/ml Leupeptin, 5 μg/ml Aprotinin, 100 μM NaF, 100 μM PMSF, 10 mM ß-Glycerolphosphat, 0.1 mM Na3VO4, 30 mM 4-Nitrophenylphosphate) zu IxIO8 Zellen/ 17,5 ml resuspendiert. Die Zellen werden 30 Minuten auf Eis lysiert. Nach dem Entfernen der Zelltrümmer durch Zentrifugation (4000 rpm, 5 Minuten) wird der klare Überstand mit Glutathion Sepharosebeads versetzt (1 ml resuspendierte und gewaschene Beads für 50 ml Überstand) und 30 Minuten bei 4°C auf einem Rotationsbrett inkubiert.
Danach werden die Beads mit Lysispuffer gewaschen und das rekombinante Protein mit 1 ml Elutionspuffer/ ml resuspendierte Beads (Elutionspuffer: 100 mM Tris/HCl pH=8,0, 120 mM NaCl, 20 mM reduziertes Glutathion (Sigma G-4251), 10 mM MgCl2, 1 mM DTT) von den Beads eluiert. Die Proteinkonzentration wird mittels Bradford Assay bestimmt.
Assay
In einem Napf einer 96-Loch Rundbodenplatte (Fa. Greiner bio-one, PS-Microtiterplatte Nr.650101 ) werden folgende Komponenten zusammengefügt:
- 10 μl zu testende Verbindung in variabler Konzentration (z.B. beginnend bei 300 μM, und Verdünnung in 1 :3) in 6% DMSO, 0,5 mg/ml Casein (Sigma C-5890), 60 mM ß-Glycerophosphat, 25 mM MOPS ρH=7,0, 5 mM EGTA, 15 mM MgCl2, 1 mM DTT
- 20 μl Substratlösung (25 mM MOPS ρH=7,0, 15 mM MgCl2, 1 mM DTT, 2,5 mM EGTA, 30 mM ß-Glycerophosphat, 0,25 mg/ml Casein)
- 20 μl Enzymverdünnung (1:100 Verdünnung des Enzymstocks in 25 mM MOPS pH=7,0, 15 mM MgCl2, 1 mM DTT)
-10 μl ATP Lösung (45 μM ATP mit 1,1 IxIO6 Bq/ml gamma-P33-ATP).
Durch Zusatz der ATP Lösung wird die Reaktion gestartet und 45 Minuten bei 30 °C unter leichtem Schütteln (650 rpm auf IKA Schüttler MTS2) durchgeführt. Die Reaktion wird durch Zusatz von 125 μl eiskalter 5%iger TCA pro Napf gestoppt und mindestens 30 Minuten auf Eis inkubiert. Das Präziptitat wird durch Ernten auf Filterplatten (96-well- Microtiter-Filterplatte: UniFilter-96, GF/B; Fa. Packard; Nr.6005177) übertragen, dann viermal mit l%iger TCA gewaschen und bei 60°C getrocknet. Nach Zugabe von 35μl Szintillationslösung (Ready-Safe; Beckmann) pro Napf wird die Platte mit Sealing-tape zugeklebt und die präzipitierte Menge P33 mit dem Wallac Betacounter gemessen. Die Messdaten werden mit der Standard Graphpad Software (Levenburg-Marquard Algorhythmus) ausgewertet.
Die Wirkung der erfindungsgemäßen Verbindungen wird im Cytotoxizitätstest an kultivierten humanen Tumorzellen und/oder in einer FACS-Analyse, beispielsweise an
HeLa S3 -Zellen, bestimmt. Die Verbindungen zeigen in beiden Testmethoden eine gute bis sehr gute Wirksamkeit, d.h. beispielsweise einen EC50-Wert im HeLa S3- Cytotoxizitätstest kleiner 5 μmol/L, in der Regel kleiner 1 μmol/L.
Messung der Cytotoxizität an kultivierten humanen Tumorzellen
Zur Messung der Cytotoxizität an kultivierten humanen Tumorzellen werden
Zellen der zervikalen Carcinoma Tumorzell-Linie HeLa S3 (erhalten von American Type Culture Collection (ATCC)) in Ham's F12 Medium (Life Technologies) und 10% fötalem Rinderserum (Life Technologies) kultiviert und in der log- Wachstumsphase geerntet. Anschließend werden die HeLa S3 Zellen in 96-well Platten (Costar) mit einer Dichte von 1000 Zellen pro well eingebracht und über Nacht in einem Inkubator (bei 37°C und 5 % CO2) inkubiert, wobei auf jeder Platte 6 wells nur mit Medium gefüllt werden (3 wells zur Mediumkontrolle, 3 wells zur Inkubation mit reduzierten AlamarBlue Reagenz). Die Wirksubstanzen werden in verschiedenen Konzentrationen (gelöst in DMSO; DMSO- Endkonzentration: 0.1%) zu den Zellen zugegeben (jeweils als Dreifachbestimmung). Nach 72 Stunden Inkubation werden zu jedem well 20 μl AlamarBlue Reagenz (AccuMed International) zugesetzt, und die Zellen für weitere 5-7 Stunden inkubiert. Zur Kontrolle wird zu 3 wells je 20 μl reduziertes AlamarBlue Reagenz gegeben (AlamarBlue Reagenz, das für 30 min autoklaviert wird). Nach Inkubation wird der Farbumsatz des AlamarBlue Reagenz in den einzelnen wells in einem Perkin Eimer Fluoreszenzspektrophotometer bestimmt (Exitation 530 nm, Emission 590 nm, Slits 15, Integrate time 0.1). Die Menge an umgesetzten AlamarBlue Reagenz repräsentiert die metabolische Aktivität der Zellen. Die relative Zellaktivität wird in Prozent der Kontrolle (HeLa S3 Zellen ohne Inhibitor) berechnet und die Wirkstoffkonzentration, die die Zellaktivität zu 50% hemmt (IC50) abgeleitet. Die Werte werden hierbei aus dem Mittelwert von drei Einzelbestimmungen - unter Korrektur des Leerwertes (Mediumkontrolle)- berechnet.
FACS- Analyse
Propidium Iodid (PI) bindet stöchiometrisch an doppelsträngige DNA, und ist damit geeignet den Anteil an Zellen in der Gl, S, und G2/M Phase des Zellzykluses auf der Basis des zellulären DNA Gehaltes zu bestimmen. Zellen in der GO und Gl Phase haben einen diploiden DNA Gehalt (2N), während Zellen in der G2 oder Mitose einen 4N DNA Gehalt haben.
Für eine PI-Färbung werden beispielsweise IxIO6 HeLa S3 Zellen auf eine 75 cm2 Zellkulturflasche ausgesät, nach 24 h wird entweder 0.1 % DMSO als Kontrolle zugesetzt, bzw. die Substanz in verschiedenen Konzentrationen (in 0.1% DMSO). Die Zellen werden für 24 h mit der Substanz bzw. mit DMSO inkubiert, bevor die Zellen 2 x mit PBS gewaschen und dann mit Trypsin /EDTA abgelöst werden. Die Zellen werden zentrifugiert (1000 Upm, 5 min, 4°C), und das Zellpellet 2 x mit PBS gewaschen, bevor die Zellen in 0.1 ml PBS resuspendiert werden. Anschließend werden die Zellen für 16 Stunden bei 4°C oder alternativ für 2 Stunden bei -20°C mit 80% Ethanol fixiert. Die fixierten Zellen werden zentrifugiert (1000 Upm, 5min, 4°C), mit PBS gewaschen und anschließend nochmals zentrifugiert. Das Zellpellet wird in 2 ml 0.25% Triton X-100 in PBS resuspendiert, und 5 min auf Eis inkubiert, bevor 5 ml PBS zugeben werden und erneut zentrifugiert wird. Das Zellpellet wird in 350 μl PI Färbelösung (0.1 mg/ml RNase A (Sigma, No. R-4875), 10 μg/ml Prodrom Iodid (Sigma, No. P-4864) in 1 x PBS) resuspendiert. Die Zellen werden für 20 min im Dunkeln mit dem Färbepuffer inkubiert, bevor sie in Probenmessgefäße für das FACS Scan überführt werden. Die DNA Messung erfolgte in einem Becton Dickinson FACS Analyzer, mit einem Argonlaser (500 mW, Emission 488 nm), und dem DNA Cell Quest Programm (BD). Die logarithmische PI Fluoreszenz wird mit einem band-pass Filter (BP 585/42) bestimmt. Die Quantifizierung der Zellpopulationen in den einzelnen Zellzyklusphasen erfolgte mit dem ModFit LT Programm von Becton Dickinson.
Entsprechend werden die erfindungsgemäßen Verbindungen auch auf weiteren Tumorzellen getestet. Beispielsweise sind diese Verbindungen auf Karzinomen verschiedenster Gewebe (z. Bsp. Brust (MCF7); Colon (HCTl 16), Kopf-Hals (FaDu),
Leber (HepG2), Lunge (NCI-H460), Magen (NCI-N87), Pankreas (BxPC-3), Prostata (DU145)), Sarkome (z. Bsp. SK-UT-IB, Saos-2), Leukämien und Lymphome (z. Bsp. HL- 60, THP-I, Raji, Jurkat, GRANTA-519) und anderen Tumoren (z. Bsp. Melanome (BRO), Gliome (U- 87MG)) aktiv und könnten in solchen Indikationen eingesetzt werden. Dies belegt die breite Anwendbarkeit der erfindungsgemäßen Verbindungen zur Behandlung verschiedenster Tumortypen.
Die Verbindungen der allgemeinen Formel (I) können allein oder in Kombination mit anderen erfindungsgemäßen Wirkstoffen, gegebenenfalls auch in Kombination mit weiteren pharmakologisch aktiven Wirkstoffen, zur Anwendung gelangen.
Geeignete Anwendungsformen sind beispielsweise Tabletten, Kapseln, Zäpfchen, Lösungen, - insbesondere Lösungen zur Injektion (s.c, i.V., i.m.) und Infusion - Säfte, Emulsionen oder dispersible Pulver. Hierbei soll der Anteil der pharmazeutisch wirksamen Verbindung(en) jeweils im Bereich von 0,1 - 90 Gew.-%, bevorzugt 0,5 - 50 Gew.-% der Gesamtzusammensetzung liegen, d.h. in Mengen die ausreichend sind, um den unten angegebenen Dosierungsbereich zu erreichen. Die genannten Dosen können, falls erforderlich, mehrmals täglich gegeben werden.
Entsprechende Tabletten können beispielsweise durch Mischen des oder der Wirkstoffe mit bekannten Hilfsstoffen, beispielsweise inerten Verdünnungsmitteln, wie Calciumcarbonat, Calciumphosphat oder Milchzucker, Sprengmitteln, wie Maisstärke oder Alginsäure, Bindemitteln, wie Stärke oder Gelatine, Schmiermitteln, wie Magnesiumstearat oder Talk, und/oder Mitteln zur Erzielung des Depoteffektes, wie Carboxymethylcellulose, Celluloseacetatphthalat, oder Polyvinylacetat erhalten werden. Die Tabletten können auch aus mehreren Schichten bestehen.
Entsprechend können Dragees durch Überziehen von analog den Tabletten hergestellten Kernen mit üblicherweise in Drageeüberzügen verwendeten Mitteln, beispielsweise Kollidon oder Schellack, Gummi arabicum, Talk, Titandioxid oder Zucker, hergestellt werden. Zur Erzielung eines Depoteffektes oder zur Vermeidung von Inkompatibilitäten kann der Kern auch aus mehreren Schichten bestehen. Desgleichen kann auch die
Drageehülle zur Erzielung eines Depoteffektes aus mehreren Schichten bestehen, wobei die oben bei den Tabletten erwähnten Hilfsstoffe verwendet werden können.
Säfte der erfindungsgemäßen Wirkstoffe beziehungsweise Wirkstoffkombinationen können zusätzlich noch ein Süßungsmittel, wie Saccharin, Cyclamat, Glycerin oder Zucker sowie ein geschmacksverbesserndes Mittel, z.B. Aromastoffe, wie Vanillin oder Orangenextrakt, enthalten. Sie können außerdem Suspendierhilfsstoffe oder Dickungsmittel, wie Natriumcarboxymethylcellulose, Netzmittel, beispielsweise Kondensationsprodukte von Fettalkoholen mit Ethylenoxid, oder Schutzstoffe, wie p-Hydroxybenzoate, enthalten.
Injektions- und Infusionslösungen werden in üblicher Weise, z.B. unter Zusatz von Isotonantien, Konservierungsmitteln, wie p-Hydroxybenzoate, oder Stabilisatoren, wie Alkalisalzen der Ethylendiamintetraessigsäure, gegebenenfalls unter Verwendung von Emulgiermitteln und /oder Dispergiermitteln, wobei beispielsweise bei der Verwendung von Wasser als Verdünnungsmittel gegebenenfalls organische Lösemittel als Lösevermittler bzw. Hilfslösemittel eingesetzt werden können, hergestellt und in Injektionsflaschen oder Ampullen oder Infusionsflaschen abgefüllt.
Die eine oder mehrere Wirkstoffe beziehungsweise Wirkstoffkombinationen enthaltenden Kapseln können beispielsweise hergestellt werden indem man die Wirkstoffe mit inerten Trägern, wie Milchzucker oder Sorbit, mischt und in Gelatinekapseln einkapselt. Geeignete Zäpfchen lassen sich beispielsweise durch Vermischen mit dafür vorgesehenen Trägermitteln, wie Neutralfetten oder Polyäthylenglykol beziehungsweise dessen Derivaten, herstellen.
Als Hilfsstoffe seien beispielsweise Wasser, pharmazeutisch unbedenkliche organische Lösemittel, wie Paraffine (z.B. Erdölfraktionen), Öle pflanzlichen Ursprungs (z.B. Erdnuss- oder Sesamöl), mono- oder polyfunktionelle Alkohole (z.B. Ethanol oder Glycerin), Trägerstoffe wie z.B. natürliche Gesteinsmehle (z.B. Kaoline, Tonerden,
Talkum, Kreide) synthetische Gesteinsmehle (z.B. hochdisperse Kieselsäure und Silikate),
Zucker (z.B. Rohr-, Milch- und Traubenzucker) Emulgiermittel (z.B. Lignin, Sulfitablaugen, Methylcellulose, Stärke und Polyvinylpyrrolidon) und Gleitmittel (z.B. Magnesiumstearat, Talkum, Stearinsäure und Natriumlaurylsulfat) erwähnt.
Die Applikation erfolgt in üblicher Weise, vorzugsweise oral oder transdermal, insbesondere bevorzugt oral. Im Falle der oralen Anwendung können die Tabletten selbstverständlich außer den genannten Trägerstoffen auch Zusätze, wie z.B. Natriumeitrat, Calciumcarbonat und Dikalziumphosphat zusammen mit verschiedenen Zuschlagstoffen, wie Stärke, vorzugsweise Kartoffelstärke, Gelatine und dergleichen enthalten. Weiterhin können Gleitmittel, wie Magnesiumstearat, Natriumlaurylsulfat und Talkum zum Tablettieren mitverwendet werden. Im Falle wässriger Suspensionen können die Wirkstoffe außer den oben genannten Hilfsstoffen mit verschiedenen Geschmacksaufbesserern oder Farbstoffen versetzt werden.
Für den Fall der parenteralen Anwendung können Lösungen der Wirkstoffe unter Verwendung geeigneter flüssiger Trägermaterialien eingesetzt werden. Die Dosierung für die intravenöse Anwendung liegt bei 1 - 1000 mg pro Stunde, vorzugsweise zwischen 5 - 500 mg pro Stunde.
Trotzdem kann es gegebenenfalls notwendig sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit vom Körpergewicht bzw. der Art des Applikationsweges, vom individuellen Verhalten gegenüber dem Medikament, der Art von dessen Formulierung und dem Zeitpunkt bzw. Intervall, zu welchen die Verabreichung erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Formulierungsbeispiele illustrieren die vorliegende Erfindung ohne sie jedoch in ihrem Umfang zu beschränken:
Pharmazeutische Formulierungsbeispiele
A) Tabletten pro Tablette
Wirkstoff 100 mg
Milchzucker 140 mg
Maisstärke 240 mg
Polyvinylpyrrolidon 15 mg
Magnesiumstearat 5 mg
500 mg
Der fein gemahlene Wirkstoff, Milchzucker und ein Teil der Maisstärke werden miteinander vermischt. Die Mischung wird gesiebt, danach mit einer Lösung von Polyvinylpyrrolidon in Wasser befeuchtet, geknetet, feucht granuliert und getrocknet. Das Granulat, der Rest der Maisstärke und das Magnesiumstearat werden gesiebt und miteinander vermischt. Das Gemisch wird zu Tabletten geeigneter Form und Größe verpresst.
B) Tabletten pro Tablette
Wirkstoff 80 mg
Milchzucker 55 mg
Maisstärke 190 mg
Mikrokristalline Cellulose 35 mg
Polyvinylpyrrolidon 15 mg
Natrium-carboxymethylstärke 23 mg
Magnesiumstearat 2 mg
400 mg
Der fein gemahlene Wirkstoff, ein Teil der Maisstärke, Milchzucker, mikrokristalline Cellulose und Polyvinylpyrrolidon werden miteinander vermischt, die Mischung gesiebt und mit dem Rest der Maisstärke und Wasser zu einem Granulat verarbeitet, welches getrocknet und gesiebt wird. Dazu gibt man die Natriumcarboxymethylstärke und das Magnesiumstearat, vermischt und verpresst das Gemisch zu Tabletten geeigneter Größe.
C) Ampullenlösung
Wirkstoff 50 mg
Natriumchlorid 50 mg
Aqua pro inj. 5 ml
Der Wirkstoff wird bei Eigen-pH oder gegebenenfalls bei pH 5,5 - 6,5 in Wasser gelöst und mit Natriumchlorid als Isotonans versetzt. Die erhaltene Lösung wird pyrogenfrei filtriert und das Filtrat unter aseptischen Bedingungen in Ampullen abgefüllt, die anschließend sterilisiert und zugeschmolzen werden. Die Ampullen enthalten 5 mg, 25 mg und 50 mg Wirkstoff.