WO2004017995A1 - 掻痒の予防および/または治療剤 - Google Patents
掻痒の予防および/または治療剤 Download PDFInfo
- Publication number
- WO2004017995A1 WO2004017995A1 PCT/IB2003/003475 IB0303475W WO2004017995A1 WO 2004017995 A1 WO2004017995 A1 WO 2004017995A1 IB 0303475 W IB0303475 W IB 0303475W WO 2004017995 A1 WO2004017995 A1 WO 2004017995A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- compound
- unsubstituted
- pruritus
- seq
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5082—Supracellular entities, e.g. tissue, organisms
- G01N33/5088—Supracellular entities, e.g. tissue, organisms of vertebrates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
Definitions
- the present invention relates to a prophylactic and / or therapeutic agent for pruritus comprising, as an active ingredient, a substance that suppresses the function related to GPR4 signal transduction.
- the present invention also relates to a nitrogen-containing tricyclic compound or a quaternary ammonium salt thereof, or a pharmaceutically acceptable salt thereof, which is useful as a prophylactic and / or therapeutic agent for pruritus.
- the present invention relates to a screening agent for the treatment of pruritus, which is based on a decrease in the number of bruising behaviors induced by sphingosylphosphorylcholine (SPC).
- SPC sphingosylphosphorylcholine
- GPCR4 a G protein-coupled receptor protein
- SPC sphingosylphosphorinolecoline
- LPC lysophosphatidinorecoline
- LPC another ligand of GPR4, accumulates in psoriatic lesions [Brit. Giannareo ob Pharmacolology (Br. J. Pharmacol.), 134, 398-402 (1995). Induction of edema, erythema and cell infiltration by intradermal administration to humans [Acta. Derm. Venereol., Vol. 80, pp. 242-246 (2000)] Power suggests involvement in dermatitis.
- 0GR—1 [Nichiya's Senole Biology (Nat. Cell Biol.), Volume 2, pp. 261–267 (2000)], G2A [Science ), 293, 702-705 (2001)]
- W002 / 24222 literally discloses the use of GPR4 antagonists for the treatment of atopic dermatitis.
- pruritus is caused by a wide variety of diseases, including various skin diseases (atopic dermatitis, eczema, dermatitis, prurigo, prurigo, xerosis, insect bite, Scabies, mycosis, skin pruritus, hypertrophic scar, psoriasis, bullous disease, drug eruption, etc., liver-biliary disease (primary biliary cirrhosis, cholestasis, cirrhosis, etc.), renal disease (long kidney) Insufficiency, etc.), endocrine 'metabolism disorders (diabetes, thyroid dysfunction, etc.), blood disorders
- skin diseases atopic dermatitis, eczema, dermatitis, prurigo, prurigo, xerosis, insect bite, Scabies, mycosis, skin pruritus, hypertrophic scar, psoriasis, bullous disease, drug eruption, etc.
- liver-biliary disease
- pruritus may be caused by pregnant women, drugs, etc. Pharmaceutical Journal), Vol. 37, No. 1 (issued on January 1, 2001), 73-77 and 79-82H) c
- a GPCR known as a constitutively active GPCR which, when overexpressed in cells, causes a signal to flow even in the absence of a ligand.
- the signal that flows in the absence of ligand is called constitutive activity.
- Constitutively active GPCRs include naturally occurring GPCRs and mutant GPCRs created by introducing mutations such as amino acid substitutions and deletions [Molecular Pharmacol. ), 57 volumes, 890 pages (2000), W098 / 46995].
- Antagonists that suppress the constitutive activity of GPCRs are called inversegonists.
- the object of the present invention is to provide a convenient object of the present invention.
- An object of the present invention is to provide an inhibitor of GPR4 signal transduction function containing a nitrogen-containing tricyclic compound or a quaternary ammonium salt thereof or a pharmaceutically acceptable salt thereof as an active ingredient. .
- Another object of the present invention is to provide a screening method for a therapeutic agent for pruritus, which is based on a decrease in the number of SPC-induced breaking actions.
- the present invention relates to the following (1) to (22).
- a prophylactic and / or therapeutic agent for pruritus comprising, as an active ingredient, a substance that suppresses a signal transmission function of a protein having the amino acid sequence of SEQ ID NO: 11.
- a preventive and / or therapeutic agent for pruritus comprising one of the above as an active ingredient.
- SEQ ID NOS: 11, 13 and 17 wherein one or more amino acids in the amino acid sequence according to any one of deletion, substitution or addition have an amino acid sequence, and An antibody that recognizes a protein having a function related to signal transmission of a protein having the amino acid sequence of SEQ ID NO: 11,
- R 1 is a substituted or unsubstituted heterocyclic group, -NR 5 R 6 (wherein R 5 and R 6 are the same or different and are hydrogen, substituted or unsubstituted lower alkyl, substituted Or unsubstituted cycloalkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aralkyl, or substituted or unsubstituted heterocyclic alkyl Or R 5 and R 6 together with an adjacent nitrogen atom form a substituted or unsubstituted heterocyclic group), -OR 7 (where R 7 is hydrogen, substituted or unsubstituted) Is unsubstituted lower alkyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstitute
- each R 5a and R 6a Ru 6 synonymous der - C0 2 R 7b (wherein, R 7b has the same meaning as the R 7), - N + R 5b R 6b R 8 (wherein, R 5b and R 6b have the same meanings as R 5 and R 6 , respectively, and R 8 represents lower alkyl, lower alkenyl, or aralkyl), formyl, carboxy, or cyano ,
- R 2 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or Represents an unsubstituted aralkyl, or a substituted or unsubstituted heterocyclic alkyl,
- n 0 or 1
- Z 1 and Z 2 are the same or different and are hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkynyl, substituted Or unsubstituted aryl, substituted or unsubstituted aralkyl, or substituted or unsubstituted heterocyclic alkyl, or Z 1 and Z 2 may be substituted together with two adjacent carbon atoms. Or a substituted or unsubstituted aromatic ring or a substituted or unsubstituted heterocyclic ring,
- Z 3 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted Represents a substituted heterocyclic group, substituted or unsubstituted aralkyl, or substituted or unsubstituted heterocyclic alkyl)] or a quaternary ammonium salt thereof, or a drug thereof.
- a physically acceptable salt is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted Represents a substituted heterocyclic
- the nitrogen-containing tricyclic compound or the quaternary ammonium salt thereof or the pharmacologically acceptable salt thereof As an active ingredient, the nitrogen-containing tricyclic compound or the quaternary ammonium salt thereof or the pharmacologically acceptable salt thereof according to any of the items (4) to (8).
- test compound a substance that reduces the number of ruptures induced by SPC
- the pruritus is pruritus associated with a skin disease, liver / biliary tract disease, renal disease, endocrine / metabolic disease, blood disease, malignant tumor, neurological disease, or a disease selected from AIDS (1) to (3).
- the prophylactic and / or therapeutic agent for pruritus according to any of (10) to (10).
- Pruritus is pruritus associated with skin disease, and the skin disease is atopic dermatitis, eczema, dermatitis, depraved measles, prurigo, xeroderma, insect bite, scabies, mycosis, skin pruritus
- a method for preventing and / or treating pruritus which comprises administering a therapeutically effective amount of a substance that suppresses a signal transmission function of a protein having the amino acid sequence of SEQ ID NO: 11.
- the present inventors have found a new finding that a substance that suppresses the signal transduction function of GPR4, which is a GPCR, is effective in preventing or treating pruritus, and completes the present invention. Reached.
- the present inventors have proposed a constitutionally active type
- GPR4 which is a GPCR. It has been found that substances that inhibit the activity are effective in preventing and / or treating pruritus.
- Substances that suppress the function of GPR4 related to signal transmission include substances that inhibit or suppress the expression of GPR4 itself, substances that inhibit the binding of ligand to GPR4, and signals generated by ligand binding to GPR4.
- FfiU inhibits transmission [eg, changes in intracellular cAMP concentration (increase or decrease), changes in intracellular Ca 2+ concentration (increase), mitogen-activated protein (MAP) kinase, etc.]
- Substances that suppress signal transduction caused by the constitutive activity of GPR4 for example, GPR4 inverse agonists and the like are included).
- the structure of the above substance is not particularly limited as long as it has these functions, and may have a known structure.
- GPR4 includes, for example, a protein having the amino acid sequence described in any one selected from SEQ ID NOS: 11, 13, and 17, or a protein selected from SEQ ID NOs: 11, 13, and 17 Signal transmission of a protein having an amino acid sequence in which at least one amino acid is deleted, substituted or added in the amino acid sequence according to any one of the above, and having the amino acid sequence described in SEQ ID NO: 11 Proteins having a function of
- SEQ ID NO: 11 having at least one amino acid deleted, substituted or added in the amino acid sequence described in any one of SEQ ID NOs: 1, 13 and 17, and having SEQ ID NO: 1
- a protein having a function related to signal transduction of a protein having the amino acid sequence described in 1 is described in [Molecular Cloning, A
- any of SEQ ID NOS: 11, 13, and 17 It can be obtained by introducing a site-specific mutation into MA encoding a protein having one of the amino acid sequences described above.
- the number of amino acids deleted, substituted or added is not particularly limited, but is preferably 1 to tens, preferably 1 to 20, more preferably 1 to 10, and more preferably:! ⁇ 5.
- the deletion, substitution or addition of one or more amino acid residues in the amino acid sequence according to any one of SEQ ID NOs: 11, 13 and 17 means that the amino acid sequence in the amino acid sequence At any one or more positions of the amino acid residue means that there is a deletion, substitution or addition of one or more amino acid residues, the deletion, substitution or addition may occur simultaneously,
- the amino acid residue to be substituted or added may be either natural or non-natural.
- Natural amino acid residues include L-alanine, L-asparagine, L-asparaginate, L-glutamine, L-glutamate, glycine, L-histidine, L-isoleucine, L- Leucine, L-lysine, L-arginine, L-methionine, L-phenylalanine, L-purine, L-serine, L-threonine, L-tryptophan, L-thymosine, L -Various and L-cysteine residues.
- Group A leucine, isoleucine, nonoleucine, / lin, nonolein / kurin, alanine, 2-aminoptanoic acid, methionine, 0-methylserine, tert-ptinoregulin, tert-ptinolelanine, Cyclohexanolane
- Group B aspartic acid, glutamic acid, isoaspartic acid, isoglutamic acid, 2-aminoaminoadipic acid, 2-aminosveric acid
- Group D lysine, arginine, orditin, 2,4-diaminobutanoic acid, 2,3-diaminopropionic acid
- Group E Proline, 3-hydroxyproline, 4-hydroxyproline
- Group F serine, threonine, homoserine
- Group G pheninoleanine, tyrosine
- a protein having an amino acid sequence in which one or more amino acid residues are deleted, substituted or added in the amino acid sequence according to any one of SEQ ID NOs: 11, 13, and 17, Having the amino acid sequence of SEQ ID NO: 11 In order to have a function related to signal transduction of a protein, the amino acid sequence thereof and the amino acid sequence of SEQ ID NO: 11 are at least 75% or more, usually at least 80%, preferably at least 90%, and more preferably Preferably have an identity of 95% or more.
- Examples of the substance that inhibits or suppresses the expression of GPR4 itself include, for example, 15 to 60 consecutive nucleotides selected from the nucleotide sequence of any one selected from SEQ ID NOS: 12, 14, and 18.
- An oligonucleotide having an complementary sequence of an oligonucleotide consisting of (hereinafter, also referred to as an antisense oligonucleotide); SEQ ID NO: 12, a base selected from any one selected from 14, 14 and 18
- Oligonucleotides that hybridize with the DNA having the sequence under stringent conditions and suppress the function related to signal transduction of the protein having the amino acid sequence of SEQ ID NO: 11, derivatives of these oligonucleotides hereinafter, referred to as Oligonucleotide derivatives
- the antisense 'oligonucleotide is an oligonucleotide consisting of a continuous 15 to 60 bases selected from the base sequence of any one selected from SEQ ID NOS: 12, 14, and 18.
- An antisense having a complementary sequence of nucleotides is not particularly limited as long as it is an oligonucleotide.
- Particularly preferred is an antisense 'oligonucleotide having the complementary sequence of the translation initiation region of the above-mentioned oligonucleotide.
- the antisense oligonucleotide is prepared by a conventional method based on the information on the nucleotide sequence of any one selected from SEQ ID NOs: 12, 14, and 18 or the nucleotide sequence of a fragment thereof. For example, it can be prepared by using a DNA synthesizer.
- Oligonucleotide derivatives include an oligonucleotide derivative in which the phosphoric acid ester bond in the oligonucleotide is converted into a phosphorothioate bond, and a phosphoric diester bond in the oligonucleotide.
- Oligonucleotide derivative converted to '- ⁇ 5' phosphoamidate bond Oligonucleotide derivative, oligonucleotide in which the bond between ribose and phosphoric acid diester in the oligonucleotide is converted to peptide nucleic acid bond
- Oligonucleotide derivative in which peracyl is substituted with C-5propenylperacyl and oligonucleotide derivative in which peracyl in oligonucleotide is substituted with C-5 thiazolylperacyl, in oligonucleotide
- Oligonucleotide derivative in which a cytosine is substituted with C-5 propynylcytosine Oligonucleotide derivative in which cytosine in oligonucleotide is substituted with phenoxazine-modified cytosine, origonucleic acid in which ribose in oligonucleotide is substituted with 2,
- DNA having the base sequence described in any one selected from SEQ ID NOs: 12, 14, and 18 and a nucleotide that hybridizes under stringent conditions are referred to as SEQ ID NOs: 12, 14, and Using a part or all of the DNA having the base sequence described in any one of 18 as a probe as a probe, the colony-hybridization method, the plaque hybridization method, the southern plot DNA obtained by using the hybridization method or the like.Specifically, 0.7 to 1.0 mol / 1 chloride is used by using a filter on which DNA derived from colonies or plaques is immobilized.
- a 0.1- to 2-fold concentration of SSC solution (the composition of a 1-fold concentration SSC solution is 150 mmolVl Sodium, 15 mmol / 1 citric acid Used consisting birds um), it can be mentioned DNA or the like can be identified by the child filter washed with 6 5 ° C conditions.
- Hybridization is based on Molecular 'Cloning 2nd Edition, Current' Protocols 'in' Molecular, No, Iology, DNA Cloning 1: Core Techniques, A Practical
- the nucleotide to be hybridized is, for example, 1% when calculated using the above BLAST, FASTA, etc.)
- a DNA having at least 75% or more homology with the DNA having the complementary sequence of the DNA having the base sequence of any one selected from 2, 14 and 18 is preferable, and more preferably 8 DNAs having a homology of 0% or more, more preferably DNAs having a homology of 95% or more.
- RNA and the like can be used, but DNA is preferably used.
- the antisense 'oligonucleotide or the antisense' oligo A nucleotide derivative or a nucleotide or a nucleotide derivative that hybridizes with a DNA having the nucleotide sequence of any one selected from SEQ ID NOs: 12, 14, and 18 under stringent conditions alone
- a vector for gene therapy such as a retrovirus vector, an adenovirus vector, or an adenovirus associated virus vector
- a drug formulated according to the usual method described below is used as a prophylactic and / or therapeutic agent for pruritus. Can also be used.
- a gene therapy vector When a gene therapy vector is used as the prophylactic and / or therapeutic agent, it can be produced by preparing the gene therapy vector and a base for use in the gene therapy agent [Nature Genet., 8, 42 (1994)].
- the above-mentioned base may be any base that is usually used for injections, for example, distilled water, sodium chloride or a salt solution such as a mixture of sodium chloride and an inorganic salt, or the like.
- examples thereof include sugar solutions such as mannitol, lactose, dextran, and pudose, amic acid solutions such as daricin and arginine, and mixed solutions of an organic acid solution or a salt solution and a dalcose solution.
- an osmotic pressure regulator, a ⁇ regulator, a vegetable oil such as sesame oil and soybean oil, or an auxiliary agent such as lecithin or a surfactant such as a nonionic surfactant is added to these bases.
- An injection may be prepared as a solution, suspension or dispersion. These injections can also be prepared as preparations for dissolution at the time of use by operations such as powdering and freeze-drying.
- the prophylactic and / or therapeutic agent can be used as it is in the case of a liquid, or in the case of a solid, immediately before administration, if necessary, dissolved in the above-mentioned sterilized base.
- Examples of the method of administration include a method of local administration so as to be absorbed at the treatment site of the patient.
- DNA can be transported to a target treatment site by a non-viral gene transfer method.
- Non-viral gene transfer methods include calcium phosphate coprecipitation [Virology, 52, 456-467 (1973); Science, 209, 1414-1422 (1980)], and microinjection [Proc. Natl. Acad. Sci. USA, 77, 5399-5403 (1980); Proc. Natl. Acad. Sci. USA, 77, 7380-7384 (1980); Cell, 27, 223-231 (1981); Nature, 294, 92-94 (1981)], liposome-mediated membrane fusion-mediated transfer [Proc. Natl. Acad. Sci. USA, 84, 7413-7417 (1987); Biochemistry, 28, 9508-9514 (1989); J. Biol. Chem., 264, 12126-12129 (1989); Hum.
- Examples of the substance that inhibits the binding of a ligand to GPR4 include an antibody that recognizes GPR4, a compound that has an antagonistic effect on GPR4, and the like.
- any antibody that recognizes GPR4 can be used, but an antibody that specifically recognizes GPR4 is preferable.
- the antibody may be a polyclonal antibody or a monoclonal antibody. Examples of such an antibody include a neutralizing antibody that recognizes GPR4.
- human chimeric antibodies, humanized antibodies and the like can also be used as the antibodies of the present invention.
- the above antibody can be prepared, for example, according to the following method.
- a polyclonal antibody can be prepared by administering a purified sample of GPR4 or a partial fragment thereof polypeptide or a peptide having an amino acid sequence of a part of GPR4 as an antigen to an animal.
- Animals to be administered include egrets, goats, rats, mice, hamsters and the like.
- the dose of the antigen is preferably 50 to 100 g per animal.
- a peptide When a peptide is used, it is desirable to use, as an antigen, a peptide obtained by covalently binding a peptide to a carrier protein such as keyhole limpet haemocyanin or bovine thyroglobulin.
- a peptide serving as an antigen can be synthesized by a peptide synthesizer.
- the administration of the antigen is performed 3 to 10 times every 1 to 2 weeks after the first administration.
- Plasma samples are collected from the fundus venous plexus 3 to 7 days after each administration, and the reaction of the serum with the antigen used for immunization is determined by enzyme immunoassay [enzyme immunoassay (ELISA): published by Medical Shoin ( 1976), AntiDodies-A Laboratory Manual, Cold spring harbor Laboratory (1988)].
- enzyme immunoassay enzyme immunoassay (ELISA): published by Medical Shoin ( 1976), AntiDodies-A Laboratory Manual, Cold spring harbor Laboratory (1988)].
- a polyclonal antibody can be obtained by obtaining serum from a non-human mammal whose serum has a sufficient antibody titer against the antigen used for immunization, and separating and purifying the serum. .
- Methods for separation and purification include centrifugation, salting out with 40-50% saturated ammonium sulfate, and caprinoleic acid precipitation [Antibodies, A Laboratory Manual, Cold Spring Harbor Laboratory, (1988)], or DEAE-Sepharose column, A method in which chromatography using an anion exchange power column, protein A or G-power column, gel filtration column, or the like is used alone or in combination.
- a rat whose serum shows a sufficient antibody titer is produced. Serve as a source of cells.
- the spleen is removed 3 to 7 days after the last administration of the antigenic substance to the rat showing the antibody titer.
- the spleen is shredded in a MEM medium (manufactured by Nissui Pharmaceutical), loosened with forceps, centrifuged at 1,200 rpm for 5 minutes, and the supernatant is discarded.
- MEM medium manufactured by Nissui Pharmaceutical
- the spleen cells in the obtained precipitate fraction are treated with Tris-ammonium chloride buffer (pH7.65) for 1 to 2 minutes to remove red blood cells, washed three times with MEM medium, and the resulting spleen cells are used as antibody-producing cells. Used as
- myeloma cells use cell lines obtained from mice or rats.
- 8-azaguanine-resistant mouse derived from BALB / c
- myeloma cell line P3-X63Ag8-U1 (hereinafter abbreviated as P3-U1) [Curr. Topics Microbiol. Immunol., 81, 1 (1978), Eur.J. Immunol., 6, 511 (1976)), SP2 / 0-Agl (SP-2) [Nature, 276, 269 (1978)], P3-X63-Ag8653 (653) [ J.
- a purified plate of GPR4 or its partial fragment polypeptide used as an antigen, or a peptide having a partial amino acid sequence of GPR4 is coated on an appropriate plate, and the hybridoma culture supernatant or The purified antibody obtained in (d) above is reacted as the first antibody, and the second antibody is an anti-rat or anti-mouse immunoglobulin antibody labeled with biotin, an enzyme, a chemiluminescent substance or a radioactive compound.
- a reaction according to the labeling substance is carried out, and those which specifically react with the polypeptide used as the antigen are selected as the hybridomas producing the monoclonal antibody used in the present invention.
- Cloying was repeated twice by the limiting dilution method using the hybridoma [the first time using HT medium (medium obtained by removing aminopterin from the HAT medium), and the second time using normal medium]. Those with a strong antibody titer are selected as the hybridoma strain producing the monoclonal antibody used in the present invention.
- mice or nude mice 8 to 10 weeks old which were treated with pristane [0.5 ml of 2,6,10,14-tetratramethinolepentadecane (Pristo) intraperitoneally and bred for 2 weeks]
- Intraperitoneally 5 to 20 ⁇ 10 6 hybridoma cells producing the monoclonal antibody used in the present invention obtained in (c) are injected intraperitoneally.
- Hybridomas develop ascites cancer between 10 and 21 S.
- the ascites is collected from the mouse with ascites tumor and centrifuged at 3,000 rpm for 5 minutes to remove solids.
- a monoclonal antibody can be purified and obtained in the same manner as the method used for the polyclonal antibody.
- the antibody subclass is determined using a mouse monoclonal antibody typing kit or a rat monoclonal antibody typing kit.
- Polypeptide amount is calculated Lowry method or from the absorbance at 2 8 0 nm.
- the above-mentioned preventive and / or therapeutic agent for pruritus containing an antibody recognizing GPR4 can be prepared as follows.
- compositions containing the antibody as an active ingredient can be administered alone.
- the active ingredient is usually mixed with one or more pharmacologically acceptable carriers, and the drug is produced by any method well known in the technical field of pharmaceuticals. It is desirable to provide it as a drug product.
- a sterile solution dissolved in water or an aqueous carrier such as an aqueous solution of salt, glycine, glucose, human albumin or the like is used.
- pharmacologically acceptable additives such as buffering agents and tonicity agents to bring the formulation solution closer to physiological conditions, such as sodium acetate, sodium chloride, sodium lactate, Potassium chloride, sodium citrate and the like can also be added.
- it can be stored after being freeze-dried and dissolved in an appropriate solvent before use.
- Dosage forms include tablets, injections and the like.
- Suitable formulations for oral administration include tablets and the like. Tablets include excipients such as lactose and mannitol; disintegrators such as starch; lubricants such as magnesium stearate; binders such as hydroxypropylcellulose; surfactants such as fatty acid esters; glycerin; It can be manufactured by using a plasticizer or the like as an additive.
- Formulations suitable for parenteral administration include injections and the like. For example, an injection is prepared using a carrier comprising a salt solution, a glucose solution or a mixture of both.
- the components exemplified as additives in oral preparations can be added.
- a substance that suppresses a function related to signal transduction caused by the constitutive activity of GPR4 can also be obtained by searching for a substance that can suppress signal transduction caused by the constitutive activity.
- Examples of the compound having an antagonistic action on GPR4 include a compound represented by the formula (I).
- the compound represented by the formula (I) is referred to as compound (I). The same applies to compounds of other formula numbers.
- each group of the compound (I) for example, a linear or branched alkyl having 1 to 10 carbon atoms may be used, and specifically, methinole, echinole, Propinole, isopropynole, petitinole, isoptinole, sec-butynole, tert-butylinole, pentinole, isopentinole, neopentyl, hexinole, heptyl, octyl, isoctyl, nonyl, decyl, etc.
- cycloalkyl examples include cycloalkyl having 3 to 8 carbon atoms, and specific examples thereof include cyclopropyl, cyclobutynole, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like. .
- Examples of the lower alkenyl include straight-chain, branched or cyclic alkenyl having 2 to 8 carbon atoms, and specific examples thereof include butyl, aryl, 1-propenyl, puttenolene, penteninole, Hexeninole, hepteninole, octeninole, cyclohexyl, 2,6-octactenyl, and the like.
- Examples of the lower alkynyl include straight-chain or branched alkynyl having 2 to 8 carbon atoms, and specific examples thereof include ethur, propynyl, butynyl, pentyl, hexynyl, heptyl, and octynyl. Is mentioned.
- Halogen represents each atom of fluorine, chlorine, bromine and iodine.
- Examples of the aryl and the aromatic ring formed by joining together two adjacent carbon atoms and removing one hydrogen atom include monocyclic groups having 6 to 14 carbon atoms, Examples thereof include bicyclic or tricyclic aryls, and specific examples include phenyl, naphthyl, indul, and anthral.
- alkylene portion of aralkyl and heterocyclic alkyl has the same meaning as the above-mentioned definition of lower alkyl (i) except that one hydrogen atom is removed.
- aryl moiety of aralkyl examples include, in addition to the groups defined in the above-mentioned aryl (vi), a bicyclic fused ring group in which the aryl is fused with a cycloalkyl, Specifically, there are indanil, 1,2,3,4-tetrahydrodronaphthyl, 6,7,8,9-tetrahydr-5H-benzocycloheptyl, and the like.
- Examples of the heterocyclic group and the heterocyclic group portion of the heterocyclic alkyl and the group obtained by removing one hydrogen atom from a heterocyclic ring formed by joining together two adjacent carbon atoms include nitrogen.
- Selected from atoms, oxygen atoms and sulfur atoms A 5- or 6-membered monocyclic heterocyclic group containing at least one atom, a bicyclic or tricyclic fused 3- to 8-membered ring composed of nitrogen, oxygen and sulfur atoms
- Examples include a condensed heterocyclic group containing at least one selected atom, such as pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, benzoimidazolyl, 2-oxobenzoimidazolyl, and benzotol.
- heterocyclic group formed together with the adjacent nitrogen atom for example, a 5- or 6-membered monocyclic heterocyclic group containing at least one nitrogen atom (the monocyclic heterocyclic group) Heterocyclic groups may contain other nitrogen, oxygen or sulfur atoms),
- a condensed heterocyclic group containing at least one nitrogen atom which is a bicyclic or tricyclic ring in which a 3- to 8-membered ring is condensed (the condensed heterocyclic group is other nitrogen atom, oxygen atom or sulfur which may contain an atom).
- pyridyl tetrahydropyridyl, indolinyl, isoindolinyl, pyrrolidinyl, thiazolidinyl, oxazolidinyl ⁇ , pyridino, Homopiperidino, Piperazinyl, Homopirazuranole, Monoreholino, Tiomonoreholino, ⁇ / Lehi Droazepi-Nore, Perhidroazosininole, Te Trahidroquinolyl, Tetrahidroisoquinolyl, Zodrazynol Indolyl, isoindolyl, prenyl, dihydroindolyl, pyrrolyl, dihydrinyl Lil, pyrazolyl, Application Benefits Azori Le, Te Torazoriru, Lee imidazolyl, etc. can be mentioned, et al. It is.
- Substituents in the substituted lower alkyl and the substituted lower alkyl are the same or different and include, for example, cycloalkyl, lower alkanoyl, lower alkoxy, aryloxy, substituted aryloxy having 1 to 3 substituents [the substituted aryloxy]
- substituents in the above are the same or different and include, for example, lower alkyl, lower alkoxy, lower alkoxycarbonyl, halogen, cyano, nitro, hydroxy, carboxy, amino, etc. having 1 to 3 substituents. I can do it.
- the lower alkyl has the same meaning as the lower alkyl (i)
- the halogen has the same meaning as the halogen (V)
- the lower alkyl part of the lower alkoxy and the lower alkoxycarbonyl has the same meaning as the lower alkyl (i).
- A) aralkyloxy, substituted aralkyloxy [Substituents in the substituted aralkyloxy are the same or different, for example, lower alkyl, lower alkoxy, lower alkoxycarbonyl, halogen, cyano, nitro having 1 to 3 substituents. , Hydroxy, carboxy, amino and the like.
- the lower alkyl has the same meaning as the lower alkyl (i)
- the halogen has the same meaning as the halogen (V)
- the lower alkyl part of the lower alkoxy and the lower alkoxycarbol has the same meaning as the lower alkyl (i).
- substituents may be the same or different and include, for example, a hydroxy group having 1 to 3 substituents, halogen, etc.], a substituted lower alkanol, and the same or different substituents in the substituted lower alkanol.
- halogens having 1 to 3 substituents, etc. mono- or di-lower alkylamino, lower anorekyls Rehoniru, lower alk keno less Honoré alkylsulfonyl, lower Anorekokishi force Noreboniru ⁇ Mi Bruno, lower Arukanoiruami Roh, mono- or di-lower alkylamino Nokarubo sulfonyl, mono- or di-lower alkylamino Nokarubo - Ruokishi, heterocyclic group and the like.
- the lower alkyl moieties of ruthio, lower alkylsulfonyl, lower alkylsulfinyl, lower alkoxycarbonylamino and lower alkylamino have the above-mentioned aryl (vi), cycloalkyl (ii), halogen (V;), It has the same meaning as the heterocyclic group (ix) and the lower alkyl (i), and the alkylene portion of the aralkyloxy has the same meaning as the lower alkyl (i) obtained by removing one hydrogen atom.
- the lower alkyl moiety of the mono- or di-lower alkylamino, mono- or di-lower-alkylaminocarbonyl and mono- or di-lower-alkylaminocarbonyloxy is as defined above for the lower alkyl (i), respectively.
- the two lower alkyl moieties of the di-lower alkylaminocarbonyl group may be the same or different.
- (xii) a substituted aryl, a substituted aralkyl, a substituted cycloalkyl, a substituted lower alkenyl, a substituted lower alkynyl, a substituted heterocyclic group, a substituted heterocyclic alkyl, a substituted hetero atom formed together with an adjacent nitrogen atom
- the substituents in a ring group, a substituted aromatic ring each formed with two adjacent carbon atoms, and a substituted heterocyclic ring each formed with two adjacent carbon atoms are: In addition to the groups mentioned in the definition of the substituent (xi) in the above-mentioned substituted lower alkyl, lower alkyl, aryl, substituted aryl, aralkyl, substituted aralkyl, heterocyclic group, substituted heterocyclic group, heterocyclic group And alkyl, substituted heterocyclic alkyl and the like.
- the substituent in the substituted aryl and the substituted heterocyclic group formed together with the adjacent nitrogen atom is a lower alkyl [the lower alkyl is the same as the lower alkyl (i)] or Substituted lower alkyl [the lower alkyl has the same meaning as the above lower alkyl (i), and the substituents in the substituted lower alkyl may be the same or different and include, for example, halogen, hydroxy, Carboxy, lower alkoxycarbonyl and the like.
- the halogen is the same as the halogen (V)
- the lower alkyl part of the lower alkoxycarboyl is the same as the lower alkyl (i).
- the aryl portion of aralkyl is the above-mentioned lower alkyl (i), aryl (vi).
- the substituent in the substituted aryl, the substituted aralkyl, the substituted heterocyclic group, and the substituted heterocyclic alkyl may be the same or different, for example, a lower alkyl having 1 to 3 substituents [the lower alkyl is the lower alkyl. (The lower alkyl portion of the lower alkoxy is the same as the lower alkyl (i)), halogen [the halogen is the same as the halogen (V)], and the like. No.
- Examples of the quaternary ammonium salt of compound (I) include a lower alkyl halide (the lower alkyl and the halogen are the same as defined above) and an aralkyl halide ( Halogen and the aralkyl are as defined above, and quaternary ammonium salts to which hydroxy lower alkyl (the lower alkyl is as defined above) and the like are not particularly limited.
- a non-toxic, water-soluble salt is preferable.
- Acid salts such as organic acid salts, alkaline salts such as sodium salts and potassium salts, metallic salts such as metallic salts, magnesium salts, calcium salts and the like alkaline earth metal salts, aluminum salts, zinc salts, etc.
- Metal salts such as ammonium and tetramethylammonium, organic amine addition salts such as morpholine addition salt and piperidine addition salt, or glycine addition salt, phenylalanine addition salt and lysine And amino acid addition salts such as aspartic acid addition salt, aspartic acid addition salt and glutamic acid addition salt.
- R 2 , R 3 , R 4 , R 5 , R 6 , X and Y are each as defined above, represents lower alkyl, aryl or benzyl, and R 11 are the same or different.
- lower alkyl, cycloalkyl and halogen are as defined above, respectively.
- alkenyl portion of alkoxysulfonyloxy and alkylsulfonyloxy, and the aryl portion of aryloxysynolephonyloxy and arylsulfonyloxy have the same meanings as the lower alkyl and aryl, respectively.
- the heterocyclic group formed together with the adjacent nitrogen atom has the same meaning as described above.
- compound (Ilia) as a raw material and reacting it with 1 equivalent to a large excess of YH (wherein Y is as defined above), according to the method disclosed in JP-A-7-61983. As a result, compound (IV) can be obtained.
- the compound (Ilia) can be synthesized by the method disclosed in JP-A-7-61983 or a method analogous thereto.
- an acid such as hydrochloric acid, acetic acid, or trifluoroacetic acid into the reaction system as needed.
- the reaction is usually performed at a temperature between 0 ° C and the boiling point of the solvent used in the reaction, preferably at a temperature between room temperature and 80 ° C, and is completed in 5 minutes to 100 hours.
- the inert solvent for example, water, methanol, ethanol, acetic acid, trifluoroacetic acid, dichloroethane, cyclomouth honolem, tetrahydrofuran, dimethylinolecetamide, dimethinolehonolemamide, acetone, etc. are used alone. Alternatively, they can be used as a mixture, and preferably a mixed solvent of chloroform and acetic acid is used.
- Compound (Ic) can be produced from compound (Ib) by the following method. Manufacturing method 2
- the compound (I-c) can be obtained by reacting at a temperature between the boiling points of, preferably at room temperature for 1 to 48 hours.
- inert solvent examples include water, methanol, ethanol, benzene, tonolen, xylene, ethynole acetate, hexane, acetonitrinole, dichloromethane, dichloroethane, chlorophonolem, carbon tetrachloride, 4-Dioxane, tetrahydrofuran, dimethylacetamide, dimethylformamide, acetone, and the like can be used alone or in combination, and preferably ethyl acetate, dichloromethane, chloroform, and the like are used.
- Compound (I-b) can be produced from compound (I-c) by the method shown below.
- Compound (I-c) was used in an inert solvent in a reaction from 1 equivalent to a large excess of R 5 RH (wherein R 5 and R 6 are as defined above), usually from 1 to 10 ° C.
- the compound (Ib) can be obtained by reacting at a temperature between the boiling points of the solvents, preferably at a temperature between 20 ° C and 100 ° C for 1 to 100 hours.
- Inert solvents include, for example, water, methanol, ethanol, benzene, tonolen, xylene, ethynole acetate, hexane, acetonitrile, dichloromethane, dichloroethane, chloroform, carbon tetrachloride, 1, 4-Dioxane, tetrahydrofuran, dimethylacetamide, dimethylformamide, acetone, and the like are used alone or as a mixture, and preferably, chloroform, dimethylformamide and the like are used. Since the reaction usually proceeds well under basic conditions, it is desirable to add an appropriate base into the reaction system as needed.
- triethynoleamine, diisopropylethylamine, pyridine, N-methylmorpholine, potassium carbonate, sodium hydride, potassium hydride, calcium hydride, diisopropylethylamine, 1,8-Diazabicyclo [5.4.0] indene 7-ene or the like can be used, and among them, triaderamine is preferable.
- Compound (I-e) can be produced from compound (I-b) using compound (I-d) by the method shown below.
- R 14 and R 15 is the same or different and is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkynyl, substituted Or an unsubstituted aralkyl, or a substituted or unsubstituted heterocyclic alkyl, or a substituted or unsubstituted heterocyclic ring wherein R 14 and R 15 are taken together with the adjacent CH (CH 2 ) n N. May be formed.
- R 16 is hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted Represents arylalkyl, or substituted or unsubstituted aryl, and m represents an integer from 0 to 3.
- Lower alkyl, cycloalkyl, lower alkenyl, lower alkynyl, aralkyl, heterocyclic alkyl, and aryl are each as defined above, and their substituents are also defined as above.
- Examples of the substituted or unsubstituted heterocyclic ring formed by R 14 and R 15 together with the adjacent CH (CH 2 ) n N include tetrahydropyridine, pyrrolidine, pyridin, and homopyridin. Gin, piperazine, homopiperazine, monorephorin, thiomonoreolin, penolehydrazazepine, penolehydrozoazosin, tetrahydroquinoline, tetrahydroisoquinoline, and the like. It has the same meaning as the substituent of the heterocyclic group formed together with the nitrogen atom.
- Compound (Id) in an inert solvent is preferably 2-4 equivalents of lithium aluminum hydride, aluminum diisopropyl hydride, and preferably diisopropyl hydride.
- the compound (ie) can be obtained by treating for 10 minutes to 24 hours, preferably for 1 to 3 hours in the presence of a reducing agent such as aluminum salt.
- inert solvent for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, tonolen, xylene, tetrahydrofuran, getyl ether, etc. can be used alone or in combination.
- dichloromethane or toluene is used.
- Compound (Id) in an inert solvent usually at a temperature between 0 ° C and the boiling point of the solvent used in the reaction, preferably at a temperature between room temperature and 100 ° C, from 1 equivalent to a large excess of the appropriate salt Compound (If) can be obtained by treating for 1 to 48 hours, preferably 1 to 3 hours in the presence of a group.
- Suitable bases include, for example, sodium hydroxide, lithium hydroxide, calcium hydroxide, calcium carbonate, cesium carbonate, sodium methoxide, and the like, and preferred.
- the inert solvent include water, tetrahydrofuran, acetyl ether, methanol, ethanol, ethanol, ethanol, dichloromethane, dichloroethane, benzene, tonylene, xylene, and the like. They can be used alone or as a mixture, and preferably, tetrahydrofuran or methanol, or a mixed solvent thereof with water is used. From compound (I-c), compound (I-h) can be produced from compound (I-g) by the following method.
- alkyl in trialkylsilyltrialkyltin has the same meaning as the above lower alkyl.
- Examples of aluminum metal include sodium and potassium.
- Compound (I-g) is dissolved in an inert solvent at a temperature between 0 ° C and the boiling point of the solvent used in the reaction, preferably at a temperature between room temperature and 200 ° C, from 1 equivalent to a large excess, preferably 2 to 4 equivalents of TN 3 (where T is as defined above) and usually from catalyst to large excess to accelerate the reaction, preferably 0.5 to 2 equivalents of a suitable additive
- the compound (I-h) can be obtained by reacting for 1 to 200 hours, preferably 3 to 48 hours in the presence of.
- Suitable additives include, for example, silicon tetrachloride, lithium chloride, aluminum chloride, ammonium chloride, trialkyltin chloride, dialkyltin oxide, trialkylaluminum, triethylamine hydrochloride, Lietylamine.
- Examples include hydrobromide, sodium hydride, potassium tert-ptoxide, sodium hydroxide, zinc bromide, and the like, preferably ammonium chloride, dialkyltin oxide, and the like.
- inert solvent examples include water, acetonitrile, dimethylformamide, dimethylacetamide, N-methyl-2-pyrrolidone, dimethylsulfoxide, acetic acid, glacial acetic acid, tetrahydrofuran, benzene , Toluene, xylene and the like can be used alone or as a mixture, and dimethylformamide or toluene is preferably used.
- R 18 is a substituted or unsubstituted lower alkyl.
- Q represents an alkali metal or an alkaline earth metal
- p represents 1 if Q is an alkali metal
- p represents 2 if Q is an alkaline earth metal.
- the alkali metal is synonymous with the alkali metal, and examples of the alkaline earth metal include magnesium and calcium.
- inert solvent for example, dimethylacetamide, N-methyl-2-pyrrolidone, dimethylsulfoxide and the like can be used alone or in combination, and preferably dimethylsulfoxide is used.
- Compound (I-j) can be produced from compound (I-c) by the method shown below.
- Compound (I_c) is prepared in an inert solvent at a temperature between 0 ° C and the boiling point of the solvent used in the reaction, preferably at a temperature between 30 ° C and 80 ° C, from 1 equivalent to a large excess, preferably In the presence of 1 to 8 equivalents of R 7a SH (wherein R 7a is as defined above) and 1 to 3 equivalents, preferably 1 to 3 equivalents of a suitable base, for 1 to 100 hours, preferably 3 to Compound (Ij) can be obtained by reacting for ⁇ 72 hours.
- Suitable bases include, for example, triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine, potassium carbonate, sodium hydride, calcium hydride, calcium hydride, diisopropylethyl.
- Amine, 1,8-diazabicyclo [5.4.0] indene-7-ene and the like can be used, and among them, 1,8-diazabicyclo [5.4.0] indene-7-ene is preferable.
- inert solvent for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, toluene, xylene, ethyl acetate, dimethylinolacetamide, N-methyl-2-pyrrolidone, dimethyl sulfoxide, etc. are used alone. Or a mixture thereof, and preferably a form of black mouth is used.
- Compound (1-1) can be produced from compound (I-k) in compound (I-j) by the following method.
- Compound (1-1) can be produced by performing the same reaction as in step 6 of production method 5 using compound (I-k).
- Compound (Im) can be produced from compound (i-i) by the method shown below.
- R 2 , R 3 , R 4 , R 18 , n, X, and Y are each as defined above).
- Compound (I-in) can be produced by performing the same reaction as in Step 6 of Production Method 5 using compound (I-i).
- Compound (I-n) can be produced from compound (I-m) by the following method.
- R 7c represents a group obtained by removing hydrogen from the definition of R 7 above
- Suitable bases include, for example, lithium carbonate, sodium hydride, lithium hydride, calcium hydride, lower alkyl lithium, etc., and preferably sodium hydride, hydrogen hydride, etc. Is mentioned.
- the inert solvent include dichloromethane, chloroform, tetrachloride carbon, dichloroethane, benzene, toluene, xylene, ethyl acetate, dimethylinolenolemamide, dimethylacetoamide, and ⁇ -methinole-2.
- -Pyrrolidone, tetrahydrofuran, getinoleether and the like can be used alone or as a mixture, and chloroform is preferably used.
- Compound (I-ma) is prepared in an inert solvent at a temperature between 0 ° C and the boiling point of the solvent used in the reaction, preferably at a temperature between room temperature and 60 ° C, from 1 equivalent to a large excess, preferably
- the compound (I-o) can be obtained by treating in the presence of 3 to 6 equivalents of an appropriate oxidizing agent for 1 to 48 hours, preferably for 3 to 24 hours.
- Suitable oxidizing agents include, for example, manganese dioxide, chromic acid, pyridinum chromate, pyridinium chromate, potassium permanganate, sulfur trioxide, monopyridine, oxone And the like, and preferably manganese dioxide.
- Inert solvents include, for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, toluene, xylene, ethyl acetate, acetic acid, propionic acid, butyric acid, trifluoroacetic acid, water, Use pyrimidine, dimethinolehonoremamide, dimethylacetamide, N-methyl-2-pyrrolidone, 1,4-dioxane, tetrahydrofuran, getinoleatenole, etc. alone or in combination It is possible to use dimethylformamide, tetrahydrofuran, or the like.
- Compound (I-P) can be produced from compound (.) By the following method.
- Compound (1-0) was heated in an inert solvent at a temperature between 0 ° C and the boiling point of the solvent used in the reaction. , Preferably at a temperature between room temperature and 90 ° C, from 1 equivalent to a large excess, preferably 1 to 3 equivalents of hydroxylamin or its hydrochloride, sulfate, paratoluenesulfonate, etc. -Phenylcarbamylhydroxylamin or its hydrochloride, sulfate, paratoluenesulfonate, etc., or N-hydroxybenzamide, preferably hydroxylamine, for 1 to 48 hours, preferably 3 to 48 hours Compound -P) can be obtained by reacting for 24 hours. If necessary, 1 equivalent to a large excess, preferably 1 to 3 equivalents of a suitable dehydrating agent, or 1 equivalent to a large excess, preferably 2 to 6 equivalents of a suitable base, or microwaves Irradiation may be performed.
- Suitable dehydrating agents include, for example, acetic anhydride, phthalic anhydride, sodium hydrogensulfate, oxone, sodium formate, dialkyltin oxide, alumina, silica gel, sodium acetate, formamide , Pentoxide, iron (111) chloride, formic acid, acetic acid, propionic acid, oxychlorinated phosphorus, paratoluenesulfonic acid, etc., preferably acetic anhydride, phthalic anhydride, etc. .
- Suitable bases include, for example, triethylamine, pyridine, sodium hydride, hydrogen hydride, and the like, and preferably triethylamine or pyridine.
- Inert solvents include, for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, tonolene, xylene, nitrobenzene, acetonitrile, ethyl acetate, acetic acid, propionic acid, butyric acid, and triic acid.
- Compound (I-q) can be produced from compound (I-p) by the following method.
- Compound (I-q) can be produced by performing the same reaction as in step 7 of Production method 6 using compound (I- ⁇ ).
- Compound (I-r) can be produced from compound (I-c) by the following method.
- R 2 , RR 4 , R 5b , R 6b , R 8 , U, n, X and Y are as defined above, and Q a represents an alkali metal as defined above
- Inert solvents include, for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, tonolen, xylene, dimethinolehonolemamide, dimethylacetamide, N-methyl-2-pyrrolidone, 1,4-Dioxane, Te Trahi Drofuran or the like can be used alone or as a mixture, and dimethylformamide or the like is preferably used.
- Compound (Is) can be produced from compound (tri) by the following method.
- R 2 , RRT, n, X and Y are each as defined above).
- Compound (I-s) can be produced by performing the same reaction as in step 7 of Production method 6 using compound (I-r).
- Compound (I-t) can be produced from compound (tri) by the method shown below.
- Compound (I ⁇ ) can be produced by performing the same reaction as in Step 6 of Production method 5 using compound (Ir).
- Compound (Iu) can be produced from compound (Illb) by the following method.
- R 2, R 3, R 4, R 5, R 6, R 5b, R 6b, R 7c, R 8, R 9, R 10, R u, R 18, Q, p, U, X And Y are as defined above.
- Compound (V) can be produced by performing the same reaction as in step 8 of production method 7 using compound (Illb).
- Compound (VI) can be produced by performing the same reaction as in step 6 of production method 5 using compound (V).
- Compound (VII) can be produced by performing the same reaction as in step 13 of production method 12 using compound (VI). ⁇ Step 2 2>
- Compound (VII) in an inert solvent usually at a temperature between 0 ° C and 80 ° C, 2-4 equivalents of silver nitrate, silver oxide (1), silver oxide (11), chromic acid, chlorochromic acid Pyridinium, pyridinium dichromate, potassium permanganate, sodium periodate, sodium perchlorate, hydrogen peroxide, sodium chlorite
- the compound (VI II) is prepared by treating the mixture in the presence of an oxidizing agent such as, for example, silver nitrate or sodium perchlorate for 10 minutes to 24 hours, preferably for 1 to 4 hours. be able to. If necessary, add 0.1 to 4 equivalents of organic substances such as acetic acid or inorganic substances such as sulfuric acid, sodium dihydrogen phosphate, sulfamine phosphorus, and lutemium oxide as additives. You may.
- inert solvents examples include getyl ether, tetrahydrofuran, 1,4-dioxane, dimethylformamide, dimethylacetamide, dimethylsulfoxide, benzene, toluene / xylene, dichloromethane. And chlorophonolem, 1,2-dichloroethane, acetonitrile, ethyl acetate, methinole acetate, methylethyl ketone, hydrochloric acid, acetic acid, acetic anhydride, sulfuric acid, water, etc., and preferably acetonitrile, Water and the like can be mentioned, and these can be used alone or as a mixture.
- the compound (VIII) is reacted with 1 to 20 equivalents of a halogenating agent for 10 minutes to 24 hours in an inert solvent, usually at a temperature between 0 ° C and 80 ° C, preferably at room temperature, and then 1 equivalent Compound (IX) can be produced by reacting with a large excess of R ⁇ OH (wherein R 7 is as defined above).
- halogenating agent examples include thiol chloride, oxalyl chloride, and oxylin chloride, and preferably include thionyl chloride.
- examples of the inert solvent include dichloromethane, black form, tetrahydrofuran, dimethylformamide, dimethylacetamide, 1,4-dioxane, acetonitrile, benzene, toluene, and the like. Xylene and the like can be mentioned, and these can be used alone or as a mixture.
- the inert solvent preferably dimethlomethane is used.
- Compound (IX) is used to carry out the same reaction as in Step 2 of Production Method 1 Compound (X) can be produced.
- Compound (XI) can be produced by performing the same reaction as in step 3 of production method 2 using compound (X).
- Compound (I-u) can be produced by performing the same reaction as in step 1 of production method 1 using compound (XI).
- Compound (I-v) can be produced from compound (I-u) by the following method.
- Compound (I-v) can be produced by performing the same reaction as in Step 6 of Production Method 5 using compound (I-u).
- Compound (I-w) can be produced from compound (I-V) by the following method.
- the compound (IV) is reacted with 1 to 20 equivalents of a halogenating agent for 10 minutes to 24 hours in an inert solvent, usually at a temperature between 0 ° C and 80, preferably at room temperature, and then 1 equivalent
- the compound (Iw) can be produced by reacting with a large excess of R 5a R 6a NH (wherein R 5a and R 6a have the same meanings as described above). If necessary, one equivalent to a large excess of a suitable base may be added.
- Examples of the mouth-forming agent include thionyl chloride, oxalyl chloride, and oxylin chloride, and preferably thionyl chloride.
- Suitable bases include, for example, pyridine, triethylamine, diisopropylaminoethylamine, N-methylmorpholine and the like, preferably triethylamine.
- Examples of the inert solvent include dichloromethane, chloroform, tetrahydrofuran, dimethylformamide, dimethylacetamide, 1,4-dioxane, acetonitrile, benzene, toluene and xylene. These can be used alone or as a mixture. As the inert solvent, preferably methane is used.
- the compound (I- W ) In the production of the compound (I- W ), a technique commonly used in peptide chemistry can be used. That is, in an inert solvent of compound (Iv), 0.5 to 10 equivalents of an appropriate condensing agent and 1 to 10 equivalents of R 5a R 6a NH (wherein R 5a and K 6a are each as defined above)
- the compound (Iw) can be obtained usually by reacting at a temperature between 0 ° C and 50 ° C for 10 minutes to 70 hours.
- inert solvent examples include getyl ether, tetrahydrofuran, 1,4-dioxane, dimethylformamide, dimethylacetamide, dimethylsnoleoxide, benzene, tonolene, xylene, acetonitrinole, and acetic acid. Jet And tetrachlorofuran, dimethylformamide and the like, and preferably, tetrahydrofuran, dimethylformamide and the like.
- Suitable condensing agents include, for example, 1,3-dicyclohexylcarbodiimide, 1-ethyl-3- (3-dimethylaminopropyl) carposimid hydrochloride, 1-ethyl-3- (3-dimethylaminopropyl) Pill resin-bonded polystyrene resin (EDC resin).
- EDC resin Diethyl-3- (3-dimethylaminopropyl) Pill resin-bonded polystyrene resin
- N-hydroxysuccinic acid imide, 3,4-dihydroxy-13-hydroxy-4-oxo-1,2,3-benzotriazine, 1-hydroxybenzotriazole and the like are preferable. Additives such as 1-hydroxybenzotriazole can also be added.
- EDC resin can be produced by the method described in Tetrahedron Letters, Vol. 34, No. 48, p. 7685 (1993).
- Compound (I-y) can be produced from compound (I-X) in compound (I) by the following method.
- R 22 and R 23 are the same or different and are hydrogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted Represents a substituted cycloalkyl, a substituted or unsubstituted lower alkenyl, a substituted or unsubstituted lower alkynyl, a substituted or unsubstituted aralkyl, or a substituted or unsubstituted heterocyclic alkyl.
- lower alkyl, cycloalkyl, lower alkenyl, lower alkynyl, aralkyl, and heterocyclic alkyl have the same meanings as described above, and their substituents have the same meanings as described above.
- Step 2 9 A compound ( ⁇ - ⁇ ) in an inert solvent in an amount of 1 equivalent to a large excess, preferably ⁇ to ⁇ equivalent of 2 R 23 C0 (wherein K 22 and R 23 are as defined above), In the presence of 1 equivalent to a large excess, preferably 1 to 3 equivalents of a suitable reducing agent, usually at a temperature between -78 ° C and 100 ° C, preferably at a temperature between 0 ° C and 50.
- the compound (Iy) can be obtained by reacting for from minutes to 48 hours.
- Suitable reducing agents include, for example, sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride, and preferably sodium cyanoborohydride. Is mentioned.
- a catalyst amount to a solvent amount preferably 0.5 equivalent to a solvent amount, may be added to a suitable acid.
- suitable acids include, for example, formic acid, acetic acid, trifluoroacetic acid, propionic acid, hydrochloric acid and the like, and preferably acetic acid.
- Inert solvents include, for example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, tonolene, xylene, ethynoleate, 1,4-dioxane, dimethylformamide, dimethylacetamide, a
- Examples include, for example, cetonitrile, hexane, formic acid, acetic acid, trifluoroacetic acid, propionic acid, and hydrochloric acid, which can be used alone or in combination.
- tetrahydrofuran, acetic acid and the like are mentioned.
- the conversion of each functional group and the conversion of the functional group contained in the substituents in the compound (I) and the parent compound can be performed by a known method [for example, a comprehensive transgenic method].
- John's Daini / jR Comprehensive Organic l'rans format ions, sec edit edit on), R.C., Larock, John * Pilly Lee 'And' Sands' The method described in Incorporated (John Wiley & Sons Inc.) (1999)] can also be used.
- the compound (I) having a desired functional group at a desired position can be obtained by appropriately combining and carrying out the above methods.
- the isolation and purification of the intermediate product in the above-mentioned production method is carried out by appropriately combining the methods used in ordinary organic synthesis, for example, filtration, extraction, washing, drying, concentration, crystallization, seed chromatography, etc. Can be done. Further, purification can be performed by a purification method commonly used in a general parallel synthesis method, for example, a purification method using a scavenger resin or an ion exchange resin. Also smells intermediate Alternatively, it can be subjected to the next reaction without purification.
- the compounds (I) may exist as isomers such as positional isomers, geometric isomers, optical isomers or tautomers, and all possible isomers including these are included. Mixtures of the isomers in any ratio are encompassed by the present invention.
- the compound (I) may be purified as it is when a salt of compound (I) is obtained, and when compound (I) is obtained in a free form, compound (I) May be dissolved or suspended in an appropriate solvent, and an acid or a base may be added thereto for isolation and purification.
- the compound (I) or a pharmacologically acceptable salt thereof may be present in the form of an adduct with water or various solvents, and these adducts are also included in the present invention.
- the compound having antipruritic activity is
- Examples of mammals other than the above-mentioned humans include mice and the like, and the destructive behavior refers to the behavior in which the animal pushes its body with its hind limbs. More specific examples of the Starchy Jung method for the treatment of pruritus include, for example, the method described in Test Example 2 below.
- ⁇ _ is. CH 3 MS m / z 524 (M + H) +
- FIG. 1 shows the inhibitory effect of Compound 1 (oral administration) on SPC-induced pruritus.
- the symbols (# #, **) represent the following meanings, respectively.
- FIG. 2 shows the inhibitory effect of compound 3 (oral administration) on SPC-induced pruritus.
- the symbols (# #, **) represent the following meanings, respectively.
- Test example 1 GPR4 antagonism 10 5 cells per 1 ⁇ l of the GPR4 ats cells obtained in Reference Example 5 (the ats cells express GPR4 upon stimulation with 17
- the activity (antagonism) of the test compound was expressed by the inhibition rate calculated based on the counts (count per second) with and without addition of 17] 3-estradiol as shown in the following formula. IC 5.
- the value was calculated from the inhibition rate by the linear approximation analysis of the Logit-Log conversion method.
- A, B, and C represent the following meanings, respectively.
- mice After subcutaneously administering SPC (50 ⁇ g / site) to the back of a ddY male mouse (3 to 4 weeks old) (SPC administration group), the mouse was transferred to an ataryl observation cage (7.5 X 7.5 X). 15 cm), and the behavior of the mouse was photographed using a video camera for 60 minutes in an unmanned environment. The video was regenerated, and the number of hind limb breaking actions (scratching behav ior) was visually counted.
- the negative control group saline-administered group received saline (0.1 tnL / site) instead of SPC.
- each of Compound 1 and Compound 3 300 mg / kg was suspended in an ice solution of 0.5 weight / volume (w / v)% methylcellulose (MC), and the SPC was administered for 1 hour. Oral administration before.
- a 0.5 w / v% MC aqueous solution (10 mL / kg) was orally administered 1 hour before administration of the SPC and the physiological saline, respectively.
- the test was performed with 10 animals per group.
- the inhibition rate of the SPC-induced breaking action by the compound was determined by the following formula.
- a and B represent the following meanings, respectively.
- ddY mice (4 weeks old) Compound 48/80 (10 ⁇ g / site) was subcutaneously administered to the back (Compound 48 / ⁇ 0 administration group), and the mice were then placed in an Acryl observation cage (7.5 x 7.5 x 15). cm), and the behavior of the mouse was photographed using a video camera for 60 minutes in an unmanned state. The video was played and the number of scratching behaviors by the hind limbs was visually counted. In the negative control group (saline administration group), saline (0.1 mL / site) was administered instead of Compound 48/80.
- Compound 1 and Compound 3 were suspended in a 0.5 w / vttC aqueous solution, respectively, and orally administered before Compound 48/80 administration 1 B Terama.
- a 0.5 w / v% MC aqueous solution was orally administered 1 hour before the administration of Compound 48/80 and the physiological saline solution, respectively.
- the test was performed with 10 animals in each group.
- the inhibition rate of compound 48 / 80-induced rupture behavior by the compound was determined by the following formula.
- the number of rupture behaviors in the group treated with Compound 48/80 was 35, which was higher than the number of rupture behaviors in the negative control group (7).
- the number of rupture behaviors of the compound 1 administration group was 16 times, and the number of rupture behaviors of the compound 48/80 administration group was reduced by 54% in the compound 1 administration group.
- the number of rupture behaviors in the group treated with Compound 48/80 was 54, which was higher than that in the negative control group (13).
- the number of rupture behaviors of the compound 3 administration group was 26 times, and the number of rupture behaviors of the compound 48/80 administration group was suppressed by 52% in the compound 3 administration group.
- Test Example 4 Effect on pruritus in hapten repeated application chronic dermatitis model
- the hapten repeated application chronic dermatitis model was prepared by the method of Kitagaki et al., “Journal of Investigative Dermatology,” Vol. 105, pp. 749—755 (1995 Year) ”with some modifications.
- oxazolone manufactured by Sigma-Aldrich
- acetone manufactured by Kanto Chemical Co., Ltd.
- An oxazolone-acetone solution (antigen solution) was used.
- 10 L of the antigen solution was applied to the shaved rostral back of a BALB / c male mouse ( 6 weeks old) to sensitize the mouse, and after 7 days (day 0), 10 / XL antigen solution was applied to the same site. was repeatedly applied for 2 or 3 days until day 16 to create a chronic dermatitis model.
- Compound 1 was suspended in a 0.5 w / v% MC aqueous solution at a concentration of 10 and 30 mg / mL, respectively, and orally administered at 10 mL / kg one hour before application of the antigen solution on day 16.
- a control group was also prepared in which only a 0.5 w / v% MC aqueous solution was orally administered.
- mice on Day 16 An analysis of the pruritic response of mice on Day 16 was carried out using a method described in Kuraishi et al.'S method, "Ichiguchi Bian. Sheena nanole * off 'Fugyo'-Makonichi (European Journal of Pharmacology), Vol. Modified.
- mice For adapting the mouse to a new environment, and allowed to stand for one hour in a cage for acrylic observation (7 .5 x 8 X 15 cm ). Immediately after the antigen solution was applied to the rostral back (Dayl6), it was returned to the cage, and the behavior was photographed in an unmanned environment using an 8 mm video camera recorder. Video playback was used to observe the rupture behavior for 1 hour after application of the antigen solution. The number of rupture behaviors of the hind limb to and around the application site was counted. The mice exhibited several very fast continuous crushing actions per second, and this series of actions was regarded as one crushing action.
- the number of rupture behaviors for the compound 1 administration group was 138 ⁇ 33 (mean soil standard error) at 100 mg / kg and 2 ⁇ 1 at 300 mg / kg, and the number of rupture behaviors for the control group (264 ⁇ 29) (**: P ⁇ 0.01, ***: P ⁇ 0.001, Dunnett test).
- Test Example 5 Analysis of GPR4 mRNA expression in mouse skin and dorsal root ganglion (DRG)
- GPR4 mRNA in mouse skin and dorsal root ganglion was performed by reverse transcriptase polymerase chain reaction (RT-PCR) using the fj method. The following genetic experiments were performed using molecular cloning (Molecular Cloning). ).
- Primers corresponding to the nucleotide sequence were set and synthesized (Invito Jidone).
- cDNA was prepared from SUPERSCRIPT Preampli cation System (Invitrogen) according to the manual.
- Re trasncriptase was used as a negative control to confirm the contamination of the genome.
- GPR4 Since GPR4 is expressed in the skin, substances that suppress the function related to signal transduction of a protein having the amino acid sequence of SEQ ID NO: 11 include keratotic skin disease, psoriasis group (psoriasis vulgaris, pustular psoriasis) , Psoriatic erythroderma, psoriatic arthritis), ichthyosis group (ichthyosis vulgaris, vesicular congenital ichthyosis erythroderma, non-vesicular congenital ichthyosis erythroderma), hoof keratosis Pityriasis piliforma / erythematous keratosis, Darie's disease, palmar pustulosis, oral leukoplakia, oral papilloma, oral lichen planus, amyloid lichen, pemphigus group, pemphigus It is suggested that it is useful as a therapeutic agent for
- GPR4 is expressed in DRG
- a substance that suppresses the signal transduction function of the protein having the amino acid sequence of SEQ ID NO: 11 may be used postoperatively or It is suggested that it is also useful as a therapeutic agent for cancer pain, headache, toothache, menstrual pain, ear pain, sore throat, trauma pain, symptomatic neuralgia and the like.
- the medicament according to the present invention comprises a nitrogen-containing tricyclic compound represented by the formula (I) or a quaternary ammonium salt thereof or a pharmacologically acceptable salt thereof, and a hydrate and a solvate thereof. It is characterized by containing a substance selected from the group as an active ingredient.
- the above-mentioned substance as an active ingredient may be administered as it is, but generally, it contains the above-mentioned substance as an active ingredient and one or more pharmaceutical additives. It is desirable to administer it in the form of a pharmaceutical composition.
- Such a pharmaceutical composition can be produced according to a method known or commonly used in the field of pharmacology itself.
- the medicament according to the present invention in the form of a pharmaceutical composition may contain one or more active ingredients of another medicament.
- the medicament of the present invention is applicable to mammals including human.
- the administration route of the medicament of the present invention is not particularly limited, and the most effective administration route for treatment and / or prevention can be appropriately selected from either oral administration or parenteral administration such as intravenous administration. it can.
- Examples of formulations suitable for oral administration include, for example, tablets and the like, and examples of formulations suitable for parenteral administration include, for example, injections.
- excipients such as lactose and mannite
- disintegrants such as starch
- lubricants such as magnesium stearate
- binders such as hydroxypropyl cellulose
- fatty acid esters A surfactant such as glycerin can be used.
- preparations suitable for parenteral administration can be prepared preferably using an aqueous medium isotonic with human blood.
- injection The agent can be prepared as a solution, suspension, or dispersion using an aqueous medium selected from a salt solution, a pudose solution, a mixture of salt water and a glucose solution, and the like, with appropriate auxiliaries according to a conventional method. it can.
- parenteral preparations for example, one or more selected from diluents, flavors, preservatives, excipients, disintegrants, lubricants, binders, surfactants, plasticizers, etc. Can also be used.
- the dose and frequency of administration of the medicament of the present invention are not particularly limited, and the kind, administration route, treatment and Z or prevention purpose of the active ingredient, age and weight of the patient, nature and severity of symptoms It can be appropriately selected according to various conditions such as. For example, it is preferable to administer 0.1 to 100 mgZ kg per adult daily in 3 to 4 divided doses. However, these dosages and the number of administrations vary depending on the various conditions described above.
- JP-A-7 61983 2 _ (2 Echiru 5, 7-dimethyl-3 H- I Mi Dazo [4, 5-b] pyridinium Jin one 3- Irumechiru) one 10, 1 1-dihydrazide mud over 5H —Dibenzo
- [b, f] azepine (30.0 g, 78.4 mmol) was dissolved in a mixed solvent of chloroform (300 mL) and acetic acid (300 mL), and 1-methylbiperazine (23.6 g, 236 mmol) and formaldehyde (37% aqueous solution, 7.64 g, 94.1 mmol) were added, heated to 60 ° C and stirred for 18 hours. After confirming the progress of the reaction by thin-layer chromatography, a saturated aqueous sodium hydrogen carbonate solution was added under ice cooling, and the mixture was extracted with ethyl acetate.
- Example 2 Compound 2 ⁇ 2- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-13-ylmethyl) -18- (1,2,5,6-tetra) Synthesis of hydropyridin-1- (1-methyl) -1-10,11-dihydro-5H-dibenzo [b, f] ylzepin ⁇
- Example 3 Compound 3 ⁇ 2- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] Synthesis of pyridin-3-ylmethyl) 8_ (pyrrolidine-1-ylmethyl) —10,11-dihydro-5H-dibenzo [b, f] azepine ⁇
- [b, f] Azepin (19 mg, 0.050 mmol) was dissolved in a mixed solvent of a click Rorohonoremu (0.30 mL) and ft acid (0.30 mL), corresponding R 6 NH click throat Holm solution (lO mol / L , 0.15 tiiL) and formaldehyde (37% aqueous solution, 0.005 mL) were added. Heated to C and stirred for 20 hours. After confirming the progress of the reaction by thin-layer chromatography, the solvent was distilled off, and the residue was dissolved in lip-mouth form and washed twice with water.
- Example 6 Compound 1 3 ⁇ 8- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-1-3-ylmethyl) -1 10,11 dihydro-1 Synthesis of 5H-dibenzo [b, f] azepine-2-ylmethyl] -1-methylpidinidine Dissolve Compound 3 (11.4 g, 24.5 mmol) obtained in Example 3 in dichloromethane (200 mL) Then, methyl iodide (1.98 mL, 31.8 mmol) was added, and the mixture was stirred at room temperature for 10 hours.
- Example 9 Compound 16 6 ⁇ 1— [8- ( 2 Echiru 5, 7 - dimethyl 3 H- Imidazo [4, 5-b] pyridinium Jin one 3- Irumechiru) one 10, 11- dihydric chondroitinase 5H- dibenzo [b, f] Azepin one 2- Inoremechinore] Piperi Jin Of 4-Ethyl olevonic acid ester ⁇
- Example 10 Compound 17 7 2- (N- [8- (2-ethyl-5,7-dimethinoley 3H-imidazo [4,5-b ] Pyridin-3- (inomethyl) -1 10,11-dihydro 51-1-dibenzo [b, f] azepine-12-inolemethinole] -1-N-methylamino) ethanol Synthesis of aluminum hydride Lithium (15.7 mg, 0.38 mmol) was suspended in tetrahydrofuran (0.3 mL) and dissolved in tetrahydrofuran (0.9 mL) with stirring under ice-cooling.
- Done Objects 1 5 (126 mg, 0.253 mmol) and the mixture was stirred for 1.5 hours at room temperature. After confirming the progress of the reaction by thin-layer chromatography, water (0.016 mL), a 2 mol / L aqueous sodium hydroxide solution (0.016 mL), and water (0.048 mL) were sequentially added dropwise with stirring. The precipitate was filtered, and the residue obtained by concentrating the filtrate was purified by NH-silica gel chromatography (elution solvent: ethyl acetate) to obtain Compound 17 (47.6 mg, 0.101 mmol, yield 40%). Got.
- Example 11 1 Compound 18 ⁇ 1 1- [8- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridine-13-ylmethyl) -1 10,11-Dihydrochloride 1-5H-Dibenzo Synthesis of [b, f] azepine-12-ylmethyl] pyridine-14-yl ⁇ methanol> Compound 16 was used in place of compound 15 in the same manner as in Example 10, yield 50%. Thus, compound 18 was obtained.
- Example 13 Compound 20 0 ⁇ 1— [8— (2-ethyl-5,7-dimethyl-3H—imidazo [4,5-b] pyridine ” One 3-irmethyl) one 10, 11-dihydro_5H-dibenzo
- Example 14 Compound 21 (N- [8- (2-ethyl-5,7-dimethyl-3H-imidazo) [4,5-b] pyridin-3-ylmethyl) -1,10-dihydro-5H-dibenzo [b, f] azepine-12-ylmethyl] -1-N-methylaminoacetonitrile> Synthesis of
- Example 15 Compound 22 (N — [8— (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1,10-dihydro-5H-dibenzo Synthesis of [b, f] azepine-12-ylmethyl] -1-N— [2- (pyrrolidine-1-yl) ethyl] amine / 2 fumarate ⁇
- reaction solution was cooled to room temperature, sodium triacetoxyborohydride (464 mg, 2.19 mmol) was added, and the mixture was stirred at room temperature for 12 hours.
- Ethyl acetate and a 1 mol / L aqueous sodium hydroxide solution were added to the reaction solution, and the organic layer was dried over anhydrous magnesium sulfate.
- Example 16 Compound 2 3 ⁇ N— [8— (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-1-ylmethyl) 10,11 dihydro 5H-dibenzo [b, f] azepine Synthesis of 2-ylmethyl] —N— (2-methoxethyl) amine monofumarate
- Example 17 Compound 24 4 2— ⁇ [8- (2-ethyl-5,7— Dimethyl-3H-imidazo [4,5-b] pyridin-3-inolemethyl) -1 10,11 dihydro 5H-dibenzo [b, f] azepine-1-2-ylmethyl: ⁇ aminoethanol 0.5 fumaric acid Salt> 2 using 2-ethanol Amin instead of (pyro lysine one 1 one I le) Echiruami down, in the same manner as in Step 2 of Example 1 5, in 39% yield 2- ⁇ [8 - (2 —Ethyl-5,7_Dimethyl-3H—Imidazo [4,5-b] pyridin-13-ylmethyl) -1 10,11 Dihydro-1-5H—Dibenzo [b, f] azepine-12-ylmethyl] a Mino ethanol was obtained.
- the compound 24 was obtained as a fumarate in the same manner as in Example 1.
- Example 19 Compound 26 ⁇ N- [8- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1 10,11-dihydro) Synthesis of 5H-dibenzo [b, f] azepine-2-inolemethyl] —N-methyl-1-N_ (2H-tetorazol-5-ylmethyl) amine ⁇
- the compound 13 (667 mg, 1.10 mmol) obtained in Example 6 was dissolved in chloroform (11 mL), and the N-methyl-N— (2-trityl-2H— Tetrazol-5-inolemethinole) amine (390 mg, 1.10 mmol) and triethylamine (0.31 mL, 2.3 mmol) were added, and the mixture was stirred at 60 ° C overnight.
- the reaction solution was cooled to room temperature, saturated aqueous sodium bicarbonate was added, and the mixture was extracted three times with chloroform. The organic layers were combined, washed with brine, dried over anhydrous magnesium sulfate, and concentrated.
- Example 20 Compound 2 7 ⁇ 2- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1-8- [4- (2H-tetrazol-5-yl) -piperidin-1 Synthesis of 1-ylmethyl] -1 10,11-dihydro_5H-dibenzo [b, f] T-zepin ⁇
- Yo-Ii-Dani (10,11-dihidro 5H-dibenzo [b, f] azepine-12-ylmethyl) -11-methylbiperidinidine (0.015 g, 0.050 mmol) was dissolved in dimethylformamide (0.50 mL).
- a solution of the corresponding YH (wherein Y is as defined above) in chloroform (1.0 mmol / L, 0.060 mL) and lithium hydroxide monohydrate (0.070 g) were added, and the mixture was added at room temperature for 20 minutes. Stirred for hours.
- Example 22 Compound 1 2 5 ⁇ [8- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyri) Synthesis of 5- (methyl-3-methyl) -1,5-methyl-10,11-dihydro-5H-dibenzo [b, f] azepine-12-yl] acetic acid ⁇
- Example 23 Compound 9 2 ⁇ acetic acid [8- (2-ethyl-5,7-dimethyl) — Synthesis of 3H-imidazo [4,5-b] pyridin-3- (inolemethyl) -1,10,11-dihydro 5H-dibenzo [b, f] azepine-12-ylmethyl] ester ⁇
- Example 24 Compound 93 3 [[8— (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1-10,11 dihidro 5H-dibenzo [b, f] azepine-1-2-inole Methanol synthesis
- Example 2 3 compound obtained in 9 2 (2.79 g, 6.14 mmol ) was suspended in Tetorahi mud furan (6 1 mL), diisocyanato Li Umume Tokishido / methanol solution (28%, 6.2 mL, 31 mmol) and In addition, the mixture was stirred at room temperature for 3.5 hours. After confirming the progress of the reaction by thin-layer chromatography, water was added to the reaction solution, followed by stirring at room temperature for 0.5 hour. The precipitated crystals were collected by filtration, dried under reduced pressure, suspended in ethanol, stirred for 1 hour under reflux with heating, and further stirred for 1 hour at room temperature. The precipitated crystals were collected by filtration and dried under reduced pressure to obtain Compound 93 (2.04 g, 4.95 mmol, yield 81%).
- Example 26 Compound 95 5 ⁇ 2-aryloxymethyl-8- (2-ethyl-1, 7-dimethinolee 3H Synthesis of —imidazo [4,5-b] pyridin-3- (inolemethyl) -1-10,11 dihidro 5H-dibenzo [b, f] azepine ⁇
- Example 27 Compound 96 ⁇ 2- (2-Ethyl_5,7-dimethyl-3H-imidazo [4,5-b] pyridine Synthesis of 1-3-ylmethyl) -1-8- (2-methoxyethoxymethyl) 10,11-dihydro-5H-dibenzo [b, f] azepine ⁇
- Example 30 Compound 9 9 ⁇ 2-benzyloxymethyl-8- (2-ethyl-5,7-dimethyl-1H-imi) Synthesis of Dazo [4,5-b] pyridin-3-ylmethyl) -1 10,11 dihydro 5H-dibenzo [b, f] azepine ⁇ Compound 99 was obtained in a yield of 78% in the same manner as in Example 25 except that benzyl alcohol was used instead of methanol.
- Example 31 1 Compound 100 ⁇ 2- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-inolemethinole) -18- (2-phenyl) Synthesis of 1,11-dihydro-5H-dibenzo [b, f] azepine ⁇
- Compound 100 was obtained in a yield of 38% in the same manner as in Example 25 except that 2-phenylethanol was used instead of methanol.
- Example 32 Compound 101 ⁇ 2— (2-ethyl-5,7-dimethyl-3H-imidazo [4, Synthesis of 5-b] pyridin-3-ylmethyl) -8- (pyridin-2-ylmethoxymethyl) -10,11-dihydro-15H-dibenzo [b, f] azepine ⁇
- Compound 101 was obtained in a yield of 65% in the same manner as in Example 25 except that pyridin-2-ylmethanol was used instead of methanol.
- Example 15 8- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridine_3-inoremethinole) -1,10-dihydro-1 obtained in Step 1 of 5 5H dibenzo [b, f] azepine-12-carboaldehyde (650 mg, 1.58 mmol) was suspended in acetonitrile (16 mL), and hydroxylamine hydrochloride (153 mg, 2.38 mmol) , Triethylamine (0.331 mL, 2.38 mmol) and phthalic anhydride (328 mg,
- Example 3 7 Compound 10 6 ⁇ 2- (2-Ethyl_5,7_dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1-8- (2H-tetrazol-5-lemethyl) -1 10,11-Dihidro 511 -Dibenzo [b, f] azepine ⁇ Synthesis
- Example 36 Using compound 105 obtained in Example 36, compound 106 was obtained in a yield of 76% in the same manner as in the latter stage of Example 20.
- Example 38 Compound 107 ⁇ [8- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-13-lemethyl) -1 10,11-dihydro 5H —Synthesis of dibenzo [b, f] azepine-1-yl] acetic acid ⁇
- Example 39 Compound 108 ⁇ [8- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl) -1 10,11-dihydro-1 Synthesis of 5H-dibenzo [b, f] azepine-2-ylmethylsulfanyl] acetic acid methyl ester ⁇ Compound 13 (1.04 g, 1.71 mmol) obtained in Example 6 was added to a closed mouth form (17 mL).
- acetic acid (10,11-dihydro_5H_dibenzo [b, f] azepine-12-inolemethinole) obtained in step 1 was suspended in methanol (110 mL).
- a sodium methoxide / methanol solution (38%, 1.14 mL, 5.36 mmol) was added, and the mixture was stirred at room temperature for 1 hour.
- the reaction solution was concentrated, and a saturated saline solution and chloroform were added to the residue, and the mixture was extracted three times with chloroform.
- the organic layers were combined, dried over anhydrous magnesium sulfate, and concentrated.
- 1_ (8-ethoxycarbonyl 10,11 dihydro-5H
- Example 42 Compound 11 1 ⁇ 8- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridine-13-ylmethyl) -1 10,11-dihydro-5H-dibenzo [b, f] azepine-2 Synthesis of rubonic acid
- Example 43 Compound 111 2 [[8- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyri) Of 1,3-dihydro-5H-dibenzo [b, f] azepine-12-yl] (4-methylpiperazine-1-inole) methanone ⁇
- Example 4 Compound 1 1 3 ⁇ [8— (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-3-3-inoremethino) 1-10,11-dihydro-1 5H-dibenzo [b, f] azepine Synthesis of 2-yl] (pyrrolidine-1-yl) methanone
- Example 4 5 Compound 1 1 4 ⁇ [8— (2-ethyl-5 , 7-Dimethyl-3H-imidazo [4,5-b] pyridin-3-ylmethyl-1-10,11 dihydro-5H-dibenzo [b, f] azepine-1-2-yl] (4-hydroxypiper Synthesis of dino) methanone
- Example 46 Compound 1 15 ⁇ 8- (2-ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin_3-ylmethyl) -1 10,11 dihydro — Synthesis of 5H—dibenzo [b, f] azepine-1—2-carboxylic acid (2-hydroxyxethyl) amide
- Example 47 Compound 1 16 ⁇ 8- (2-Ethyl-5,7-dimethyl-3H-imidazo) [4,5-b] Pyridin-3-ylmethyl) -1,10,11 dihydro-5H-dibenzo [b, f] azepine-1-2-carburonic acid [2- (pyrrolidine-1-1-) Le) ethyl] amide ⁇
- Example 48 Compound 1 17 ⁇ [8— (2-ethyl-5,7-dimethyl-3H-imidazo [4 , 5-b] Pyridin-13-ylmethyl-1-10,11-dihydro 5H-dibenzo [b, f] azepine-12-yl] (morpholino) methanone ⁇
- Example 49 Compound 1 18 ⁇ 8— (2-ethyl-5,7-dimethyl-3H-imidazo [ Synthesis of 4,5-b] pyridin-3-ylmethyl) -10,11 dihydro-5H-dibenzo [b, f] azepine-1-bis bis (2-hydroxyxetyl) amide
- Example 52 Compound 1 2 1 ⁇ 1-1- [8- (2-ethyl-5,7-dimethyl-31-tomidazo [4,5-b] pyridine) Synthesis of 13-ylmethyl) -5-methyl-10,11 dihydro 5H dibenzo [b, f] azepine-12-ylmethyl] piperidine-14
- Example 53 Compound 122 2 ⁇ 2- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b ] Pyridin-3- (inolemethyl) -1-5-methyl-8_ [4- (2H-tetrazo-l-5-yl) piperidinomethinole]-10,11-dihidro 5H-dibenzo [b, f] azepine ⁇ 1 Synthesis of hydrochloride ⁇
- Example 54 Compound 1 2 3 ⁇ 1-one [8- (2-ethyl-5,7- Dimethyl-3H-midazo [4,5-b] pyridin-3-inolemethyl) _5-Methinolay 10, 11-Dihidro Synthesis of 5H-dibenzo [b, f] azepine-12-ylmethyl] piperidine-41-ylmethano
- Example 5 1 obtained in 2 steps 1 [8- (2-Echiru -5, 7-dimethyl ⁇ 3 H - Lee Midazo [4, 5-b] pyridinium Jin one 3- Irumechiru) Single 5- methyl- 10,11-dihydroquinone 511-dibenzo [b, f] azepine-12-inolemethyl] pi-zin-vinyl-4-ethyl ether ester (0.61 g, 1.08 mmol) was added to dichloromethane (10 mL), cooled to -78 ° C and stirred.
- Example 55 Compound 1 2 4 [2- (2-Ethyl-5,7-dimethyl-3H-imidazo [4,5-b] pyridin-1-3-ylmethyl) _5 Synthesis of —Methyl-8— (211-Tetrazol-5-yl) -1 10,11—Dihydro_5H—Dibenzo [b, f] azepine]
- 2-tritinole 2H-tetrazole-5-inoremetanol nore (2.00 g, 5.84 mmol)
- N-methinole 2_nitrobenzenesnolephone amide (1.64 g, 7.59 mmol)
- triphenylinolephosphine (1.53 g, 5.84 mmol) was dissolved in a mixed solvent of tetrahydrofuran (30 mL) and toluene (20 mL), and a solution of acetyldiazolate in getyltoluene (40%, 2.65 mL, 5.84 mmol) was added. And stirred overnight.
- pSV2bsr (manufactured by Kaken Pharmaceutical Co., Ltd.) was digested with PvuII and EcoRI, and treated with Klenow to obtain a 2.6 kb PvuII (blunt-ended) 1 ⁇ RI (blunt-ended) fragment.
- Gal4-ER chimeric gene [Cell, vol. 54, p. 199 (1988), Procedurals 'ob-the-national' academy 'ob' science 'op-the-united-de-step' ER a AF2 in pM (distributed by Prof. Shigeaki Kato of the University of Tokyo) containing 'America (Proc. Natl. Acad. Sci. USA), Vol. 90, p. 1657 (1993)] After digestion with [Klenow], ill (blunt end) (blunt end) fragments were obtained.
- Plasmid pGERbsrR2 was constructed by combining the S (blunt end) -EcoRI (blunt end) fragment from pSV2bsr and the ⁇ 11 (blunt end) -Ndel (blunt end) fragment from ERctAF2 in pM described above. did. pGERbsrR2 can express a chimeric protein (Gal4-ER) of the DNA binding domain of transcription factor Gal4p derived from yeast (Saccharomyces cerevisiae) and the ligand binding domain of estrogen receptor.
- Gal4-ER chimeric protein
- pcDNA3 (Invitrogen) was digested with Xhol and treated with Klenow to obtain an Xhol (flat end) fragment. By binding the fragments, pcDNA3 from which the first cleavage site was eliminated was constructed. The pcDNA3 from which the ⁇ I cleavage site had been eliminated was digested with, and treated with Klenow to obtain a (blunt-ended) fragment. By ligating the fragments, pcDNA3 from which and the cleavage site were eliminated was constructed. The plasmid was cut with & gill, and treated with Klenow to obtain a 3 ⁇ 4rill (blunt end) fragment.
- pAMoERC3Sc Japanese Unexamined Patent Publication No. 05-336963 was digested with Xhol and Nsil, and treated with Klenow to obtain a 2.2 kb 1 (blunt end) (blunt end) fragment containing the oriP sequence.
- Plasmid pcDNA3— or iP is obtained by joining the Bglll (blunt end) fragment derived from pcDNA3 and the 1 (blunt end) _ (flat end) fragment derived from pAMoERC3Sc in which the above cleavage site and Emil cleavage site have been eliminated. was created.
- pcDNA3-oriP was digested with ⁇ l ⁇ I and Hindlll to obtain an Xhol-Hindlll fragment.
- pSE01uc2 W098 / 14474 was digested with Xhol and Ncol, it was treated with Klenow to obtain a 1 (blunt-ended) -Ncol (blunt-ended) fragment containing the ympicillin resistance gene.
- the plasmid pASdl-lucl was constructed by ligating the fragments. After cutting pASdl-lucl with ⁇ 1 and Hindlll, a 0.1 kb Xhol-Hindlll fragment was obtained.
- Xhol-Hindlll fragment derived from pcDNA3-oriP and the Xhol-Hindlll fragment derived from pASdl-lucl were ligated to construct plasmid pcDNA3-oriP-Sdl.
- pcDNA3-oriP-Sdl was digested with Xhol and Kpnl to obtain an Xhol-Kpnl fragment.
- Four types of DNAs having the nucleotide sequences represented by SEQ ID NOs: 1, 2, 3, and 4 were synthesized using a DNA synthesizer. The synthetic DNAs are mixed and annealed to form a double strand with a poly A added signal.
- Each of the synthesized primates was phosphorylated using T4 polynucleotide kinase, and then mixed and annealed to obtain double-stranded DNA.
- Plasmid pcDNA3-oriP-Sdl-pA was constructed by binding the double-stranded DNA to an Xhol-Kpnl fragment derived from pcDNA3_oriP_Sdl.
- pcDNA3-oriP-Sdl-pA was digested with ⁇ I and treated with Klenow to obtain an Xhol (blunt-ended) fragment.
- pFR_luc manufactured by Stratagene
- Hindlll and BamHI it was treated with Klenow to obtain a 0.14 kb iidlll (blunt-end) fragment (blunt-end) fragment.
- the Xhol (blunt end) fragment derived from pcDNA3-oriP-Sdl-pA and the iUjidlll-giHI fragment derived from pFR-luc were ligated to prepare a plasmid pAGalSdl.
- pAGalSdl contains a promoter having a sequence obtained by repeating Gal4p response element (UASG) five times.
- USG Gal4p response element
- P SE01uc2 (W098 / 14474) is digested with Hindlll and Sacl and treated with Klenow to obtain a 1.7 kb iiiLdlll (smooth powder) containing the firefly 'no luciferase gene. End) One ⁇ £ l (blunt end) fragment was obtained.
- Plasmid pAGalSdl-luc was constructed by ligating the Hindlll (blunt end) -I ⁇ (blunt end) fragment from pSE0luc2 and the ⁇ RI (blunt end) fragment from pAGalSdl.
- the expression plasmid pAGal9-luc having oriP of the Epstein 'Bir' virus was digested with Hindlll and Sacl to obtain a 6.9 kb Hindlll-Sacl fragment containing oriP.
- pAMo-d a JP 2001-211885) was digested with Hindlll and Sacl, were obtained Hindlll- Sacl fragment containing the tetracycline re down resistance gene (Tc R).
- pAMo-nd Japanese Patent Application Laid-Open No. 2001-211885 was cut with Hindlll and Sacl to obtain a iiidlll- ⁇ I fragment containing a tetracycline resistance gene.
- Gal-ER chimeric transcription factor expression plasmid pGERbsrR2 is reduced to 1 / zg // i
- TE buffer 10 mmol / L Tris-HCl (pH 8.0), 1 mmol / L ethylenediamine tetraacetic acid
- Namalwa KJM-1 cells are serum-free adapted B cell lines that express the EBNA-1 gene.
- the transformant cells the RPMI1640'ITPSG medium [RPMI1640 medium 8 ml (manufactured by Nissui Ltd. Kusurisha), 1/40 volume of 7. 5% NaHC0 3, 3% 200mmol / L L- glutamine solution (I Nbi DOO 0.5% penicillin / streptomycin solution (Invitrogen ⁇ -genene earth, 5,000 units / ml penicin, 5,000 ⁇ g / ml streptmycin), 10 mmol / L N-2-hydroxyxyl piperazine-N'_2-ethanesulfonic acid
- plasticidin S (Blasticidin S) (KK-400 : manufactured by Kaken Pharmaceutical Co., Ltd.) to 2.0 / ig / ml and dispense into a 96-well plate (500-2000 cells / well).
- P GERbsrR2 acquired many stable transformants ⁇ integrated into a chromosome DNA (single clones).
- Each transformant was subcultured on RPMI1640.ITPSG medium containing 2.0 g / ml of plasticidin S.
- pAGalSdl-luc was introduced by an electroporation method and cultured for 2 days. After culturing, 17 ⁇ -estradiol ( ⁇ 8875: manufactured by Sigma) (final concentration: lOnmol / L) was added, and after further culturing for 24 hours, firefly luciferase activity was measured.
- pACREpluc a reporter plasmid capable of expressing the firefly luciferase gene under the control of the cAMP response element (CRE), was constructed by the following method.
- pACREpluc carries the hygromycin resistance gene oriP of Epstein-Per.
- pAMo (Jananore 'ob' biologica cane. Chemistry (J. Biol. Chem.), 268 vol., 22782 M (1993), ⁇ IJ name pAMoPRC3Sc (JP-A-5-336963)] was partially digested with Clal A DNA fragment that had been cleaved off was obtained. The DNA fragment was partially digested with Mlul to obtain a 9.5 kb Clal-Mlul fragment. pAGE248 [Journal of Noviological Chemistry (J. Biol. Chem.), vol. 269, p.
- Plasmid pAGal9h was constructed by ligating the Sail-Hindlll fragment derived from pAGal9-luc and the Xhol_Hindlll fragment derived from pAMoh. After pBluescriptll KS + (Toyobo) is cut with Sail and Xhol, it is dephosphorylated using phosphatase (Alkaline Phosphatase E. coli C75, Takara Shuzo), and the Sall-Xhol fragment containing the ampicillin resistance gene is removed. Obtained. A double-stranded DNA containing two CRE sequences was prepared by annealing synthetic oligonucleotides having the nucleotide sequences represented by SEQ ID NOS: 5 and 6.
- pBS-CREI is a plasmid in which the double-stranded DNA is integrated in a direction in which a Sail cleavage site and an I cleavage site are regenerated, and each has one of the above-mentioned cleavage sites.
- pBS-CREI was cut with Seal and Sail to obtain a Seal-Sail fragment containing ColEl ori.
- the Xhol fragment and the Scal-Sall fragment derived from pBS_CREI were ligated to construct pBS-CREII containing four CRE sequences.
- Seal-Xhol fragment was obtained.
- pBS-CREII was cut with Seal and Sail to obtain a Scal-Sall fragment containing ColEl ori.
- the pBS-CREII-derived Seat Xhol fragment and Seal-Sail fragment were ligated to construct pBS-CREIV containing eight CRE sequences. Cut pBS-CREIV with Seal and Xhol and contain ori of phage f1
- Scal-Xhol fragment was obtained.
- pBS-CREIV was cut with Seal and Sail to obtain a Scal-Sall fragment containing ColEl ori.
- a pBS-CREIV-derived Seat Xhol fragment and a Scal-Sall fragment were ligated to construct pBS-CREVIII containing 16 CRE sequences.
- pBS-CREVIII was digested with Xhol, treated with Klenow, and digested with Hindlll to obtain an ndlll-1 (blunt end) fragment containing 16 CREs.
- pAGalSdl was cut with Mlul and Hindlll to obtain a 1. kb Mlul-Hindlll fragment.
- pAGal9h was digested with ⁇ 1, treated with Klenow, and further digested with ⁇ _ ⁇ to obtain a 1 (blunt end) — iiiJ fragment.
- a Hindlll-Xhol (blunt end) fragment derived from pBS-CREVIII, a MluI_HindIII fragment derived from pAGalSdl, and an Xbal (blunt end) -Mlul fragment derived from pAGal9h were ligated to construct a plasmid pACREh.
- Hindlll (blunt end) -Xhol fragment containing CRE and ⁇ dlll (blunt end) 1 ⁇ I fragment containing firefly luciferase gene were obtained. Acquired each.
- Hindlll (blunt end) -Xhol fragments derived from pACREluc a plasmid pACRElucH in which the Hindlll site upstream of the CRE sequence in pACREluc had disappeared was constructed.
- pG-Enhancer vector (Promega) was digested with Hindlll and Hpal to obtain a Hindlll-Hpal fragment containing the luc + gene (modified firefly luciferase gene).
- pACRElucH was digested with Notl, Klenow-treated, and further digested with ndlll to obtain a Hindlll-Notl (blunt end) fragment containing CRE.
- a plasmid pACREpluc was constructed by ligating a Hindlll-Hpal fragment derived from the GL3-Enhancer vector and a Hindlll-Notl (blunt end) fragment derived from pACRElucH.
- Reference Example 4 Construction of GPR4-induced expression plasmid
- Single-stranded cDNA was synthesized by First-Strand Synthesis System for RT-PCR (manufactured by Gipcone Earth). Using 5 ⁇ l of a solution obtained by diluting the single-stranded cDNA 250-fold with water as type ⁇ , using synthetic DNA having the sequence shown in SEQ ID NOs: 7 and 8 as a primer specific to the GPR4 gene, PCR was performed. GPR4 cDNA was obtained.
- the sequence of the GPR4 gene-specific primer can be obtained from the sequence information of the GPR4 gene (GenBank accession number:
- PCR is a thermal cycler
- the amplified GPR4 cDNA fragment was cut with Hindi II and ⁇ I, which cut the sequence designed on the primer.
- the fragment containing GPR4 cDNA was recovered by agarose gel electrophoresis.
- GPR4-induced expression plasmid pAGal9_GPR4 (2 ⁇ g) and reporter plasmid pACREpluc (2 ⁇ g) were co-transfected into KJMGER8 of 6 ⁇ 10 6 cells by the above electroporation method.
- the transformant strain were suspended in RPMI1640 ⁇ ITPSG culture locations 8 ml, in C0 2 incubator primary, cultured for 24 hours at 37 ° C. After cultivation, add Plasticidin S (2.0 ⁇ g / ml), hygromycin B (300 ⁇ g / ml) and dieneticin (500 g / ml), and further culture for 14 days to obtain a stable transformant (GPR4 assay). Cells).
- the transformant was subcultured in RPMI 1640 • ITPSG medium containing plasticidin S (2.0 ⁇ g / ml), hygromycin ⁇ (300 ⁇ g / ml) and dieneticin (500 ⁇ g / ml).
- control plasmid pAGal9-nd (2 ⁇ g) and reporter plasmid pACREpluc (2 ⁇ g) were co-transfected into KJMGER8 to obtain a stable transformant (referred to as control cell).
- Reference Example 6 Cloning of DNA encoding human GPR4 homolog from mouse
- polypeptide having the amino acid sequence represented by SEQ ID NO: 13 was a mouse human GPR4 homolog (mouse GPR4).
- the DNA encoding mouse GPR4 can be designed and synthesized based on the nucleotide sequence represented by SEQ ID NO: 14, using a mouse cDNA library that can be prepared for sale or prepared by a known method. Nucleotides can be obtained by PCR using primer sets. Reference Example 7: Cloning of DNA encoding human GPR4 homolog from rat
- RNA prepared from rat lung-derived mRNA was used as a type II, and the components described below were used.
- DNTP dATP, dGTP, dCTP, dTTP
- Taq Gold PerkinElmer 2.5 units per unit so that the concentration is 200 umol / L IX Taq Gold (Mg plus) Buffer (PerkinElmer)
- a reaction solution containing 40 / iL was prepared, and PCR was performed under the following conditions.
- Plasmid pT7RG was obtained by a conventional method from a transformant obtained by transforming Escherichia coli JM109 strain using the obtained recombinant plasmid DNA. Plasmid P T7RG result of determining the total base sequence of the PT7RG contained the cDNA of about 1. ikb having the nucleotide sequence represented by SEQ ID NO: 18.
- SEQ ID NO: 17 shows the amino acid sequence of the polypeptide encoded by the DNA consisting of the nucleotide sequence represented by SEQ ID NO: 18.
- the ⁇ A amino acid sequences were compared analysis program [GENETYX WIN ver. 5 ⁇ 0 ( soft Tuea Ltd.)] human with, and the Amino acid sequence of mouse GPR4, respectively is 93.0%, 99.2% Was found.
- a tablet having the following composition is prepared by a conventional method.
- Formulation Compound 1 20 mg
- a prophylactic and / or therapeutic agent for pruritus a nitrogen-containing cyclic compound having a prophylactic and / or Z or therapeutic effect on pruritus, comprising as an active ingredient a substance that suppresses the function related to GPR4 signal transduction.
- the quaternary ammonium salt or a pharmacologically acceptable salt thereof, a nitrogen-containing tricyclic compound having an inhibitory effect on the function of GPR4 related to signal transmission, or the quaternary ammonium salt or a quaternary ammonium salt or a salt thereof A prophylactic and / or therapeutic agent for pruritus containing a pharmacologically acceptable salt, a nitrogen-containing tricyclic compound or a quaternary ammonium salt thereof, or a pharmacologically acceptable salt thereof as an active ingredient.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Cell Biology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Tropical Medicine & Parasitology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pathology (AREA)
- Microbiology (AREA)
- Toxicology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description































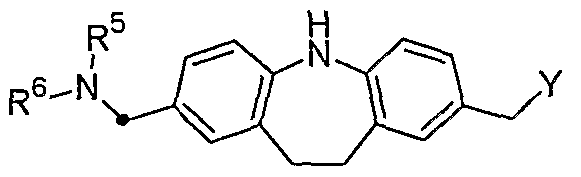
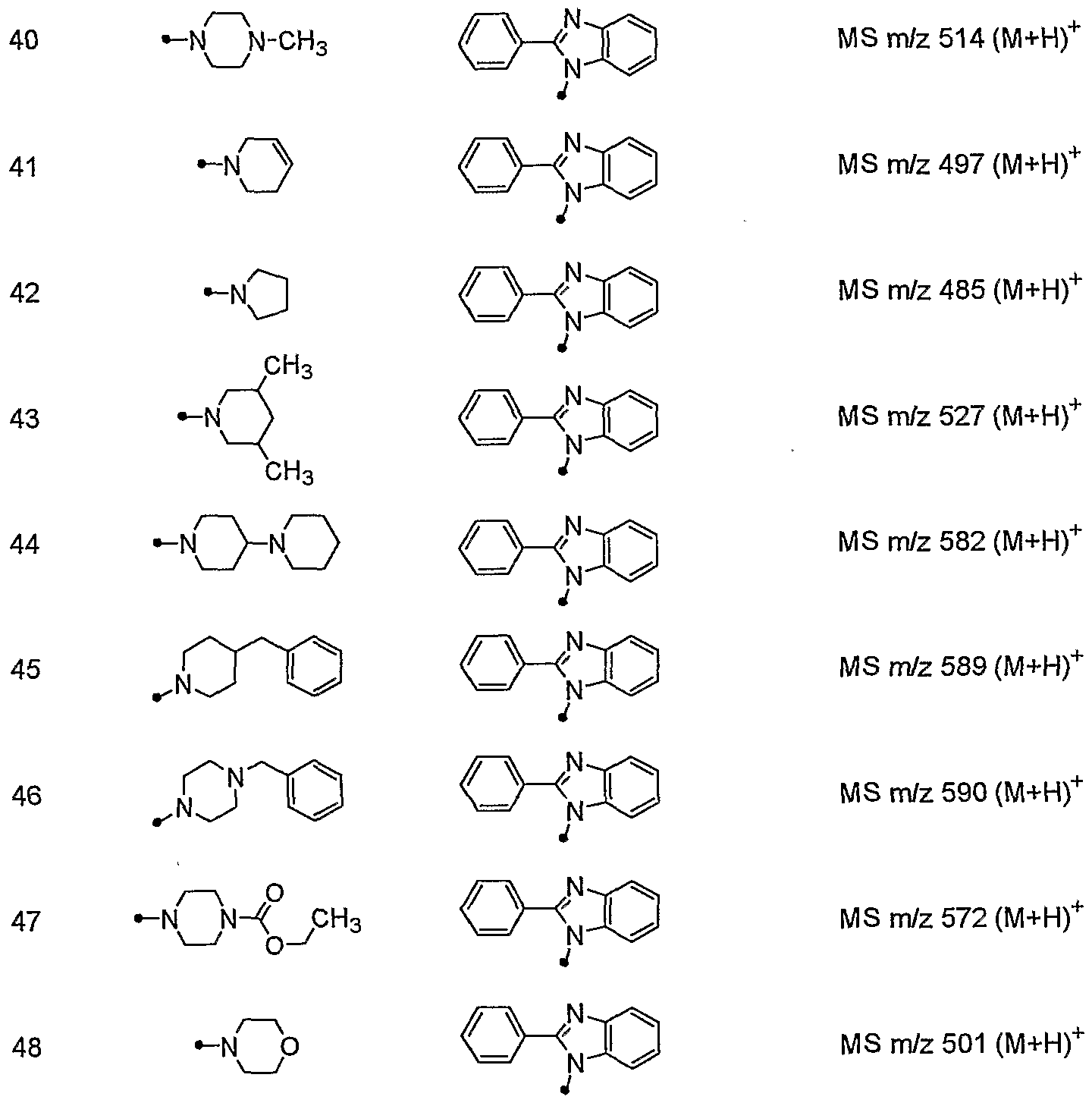
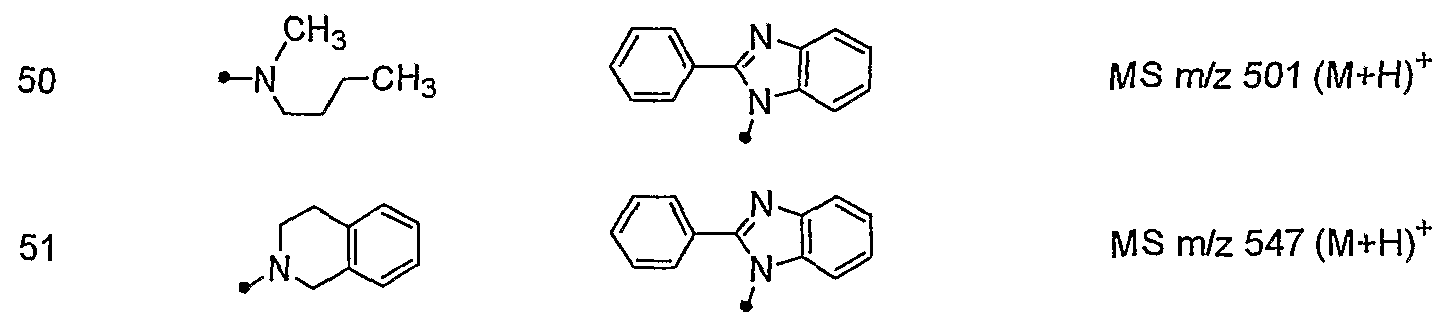























Claims

Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004530443A JPWO2004017995A1 (ja) | 2002-08-22 | 2003-08-22 | 掻痒の予防および/または治療剤 |
| CA002496464A CA2496464A1 (en) | 2002-08-22 | 2003-08-22 | Agent for prevention and/or treatment of itching |
| AU2003262230A AU2003262230A1 (en) | 2002-08-22 | 2003-08-22 | Preventive and/or therapeutic drugs for itch |
| EP03792564A EP1547616A1 (en) | 2002-08-22 | 2003-08-22 | Preventive and/or therapeutic drugs for itch |
| US10/525,324 US20060252679A1 (en) | 2002-08-22 | 2003-08-22 | Preventive and/or trerapeutic drgs for itch |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002-241522 | 2002-08-22 | ||
| JP2002241522 | 2002-08-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004017995A1 true WO2004017995A1 (ja) | 2004-03-04 |
Family
ID=31943995
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2003/003475 WO2004017995A1 (ja) | 2002-08-22 | 2003-08-22 | 掻痒の予防および/または治療剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20060252679A1 (ja) |
| EP (1) | EP1547616A1 (ja) |
| JP (1) | JPWO2004017995A1 (ja) |
| AU (1) | AU2003262230A1 (ja) |
| CA (1) | CA2496464A1 (ja) |
| WO (1) | WO2004017995A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004093912A1 (ja) * | 2003-04-23 | 2004-11-04 | Kyowa Hakko Kogyo Co. Ltd. | 好中球性炎症疾患の予防および/または治療剤 |
| WO2005082904A1 (ja) * | 2004-02-26 | 2005-09-09 | Kyowa Hakko Kogyo Co., Ltd. | 好中球性炎症疾患の予防及び/または治療剤 |
| US7250417B2 (en) | 2003-07-02 | 2007-07-31 | Sugen Inc. | Arylmethyl triazolo- and imidazopyrazines as c-Met inhibitors |
| WO2008096829A1 (ja) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | 3環系化合物 |
| KR101210008B1 (ko) | 2004-11-05 | 2012-12-07 | (주)아모레퍼시픽 | 스핑고실포스포릴콜린 또는 그 유도체의 용도 |
| US8486980B2 (en) | 2008-08-06 | 2013-07-16 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compound |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2552804T3 (es) * | 2005-05-04 | 2015-12-02 | Evotec Ag | Compuestos heterocíclicos condensados, y sus composiciones y usos |
| WO2007023880A1 (ja) * | 2005-08-24 | 2007-03-01 | Kyowa Hakko Kogyo Co., Ltd. | ケモカイン産生阻害剤 |
| JP2010513246A (ja) * | 2006-12-15 | 2010-04-30 | ノバルティス アーゲー | Gpr4の阻害 |
| US8207139B2 (en) * | 2009-09-15 | 2012-06-26 | Li Yang | Function of GPR4 in vascular inflammatory response to acidosis and related methods |
| US8748435B2 (en) | 2011-04-01 | 2014-06-10 | Novartis Ag | Pyrazolo pyrimidine derivatives |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0325755A1 (en) * | 1987-12-14 | 1989-08-02 | Kyowa Hakko Kogyo Co., Ltd. | Tricyclic compounds |
| EP0549352A2 (en) * | 1991-12-27 | 1993-06-30 | Kyowa Hakko Kogyo Co., Ltd. | Tricyclic compounds as angiotensin II antagonists |
| JPH0940662A (ja) * | 1995-05-24 | 1997-02-10 | Kyowa Hakko Kogyo Co Ltd | 三環式化合物 |
| WO2002024222A2 (en) * | 2000-09-20 | 2002-03-28 | The Cleveland Clinic Foundation | Ligands for g protein coupled receptors and methods of using them |
| WO2002061087A2 (en) * | 2000-12-19 | 2002-08-08 | Lifespan Biosciences, Inc. | Antigenic peptides, such as for g protein-coupled receptors (gpcrs), antibodies thereto, and systems for identifying such antigenic peptides |
-
2003
- 2003-08-22 JP JP2004530443A patent/JPWO2004017995A1/ja not_active Abandoned
- 2003-08-22 WO PCT/IB2003/003475 patent/WO2004017995A1/ja not_active Application Discontinuation
- 2003-08-22 AU AU2003262230A patent/AU2003262230A1/en not_active Abandoned
- 2003-08-22 CA CA002496464A patent/CA2496464A1/en not_active Abandoned
- 2003-08-22 US US10/525,324 patent/US20060252679A1/en not_active Abandoned
- 2003-08-22 EP EP03792564A patent/EP1547616A1/en not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0325755A1 (en) * | 1987-12-14 | 1989-08-02 | Kyowa Hakko Kogyo Co., Ltd. | Tricyclic compounds |
| EP0549352A2 (en) * | 1991-12-27 | 1993-06-30 | Kyowa Hakko Kogyo Co., Ltd. | Tricyclic compounds as angiotensin II antagonists |
| JPH0940662A (ja) * | 1995-05-24 | 1997-02-10 | Kyowa Hakko Kogyo Co Ltd | 三環式化合物 |
| WO2002024222A2 (en) * | 2000-09-20 | 2002-03-28 | The Cleveland Clinic Foundation | Ligands for g protein coupled receptors and methods of using them |
| WO2002061087A2 (en) * | 2000-12-19 | 2002-08-08 | Lifespan Biosciences, Inc. | Antigenic peptides, such as for g protein-coupled receptors (gpcrs), antibodies thereto, and systems for identifying such antigenic peptides |
Non-Patent Citations (3)
| Title |
|---|
| "The Merck Manual of Diagnosis and Therapy Seventeenth Edition", 1999, MERCK RESEARCH LABORATORIES, article BEERS, M.H.: "Section 10 Dermatologic Disorders, Dermatitis", pages: 786 - 793, XP002974909 * |
| HEIBER,M ET AL.: "ISOLATION OF THREE NOVEL HUMAN ENCODING G PROTEIN-COUPLED RECEPTORS.", DNA CELL BIOL., vol. 14, no. 1, 1995, pages 25 - 35, XP002066093 * |
| MAHADEVANM M.S. ET AL: "ISOLATION OF A NOVEL G PROTEIN-COUPLED RECEPTOR (GPR4) LOCALIZED TO CHROMOSOME 19Q13.3.", GENOMICS, vol. 30, 1995, pages 84 - 88, XP002920408 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004093912A1 (ja) * | 2003-04-23 | 2004-11-04 | Kyowa Hakko Kogyo Co. Ltd. | 好中球性炎症疾患の予防および/または治療剤 |
| US7250417B2 (en) | 2003-07-02 | 2007-07-31 | Sugen Inc. | Arylmethyl triazolo- and imidazopyrazines as c-Met inhibitors |
| WO2005082904A1 (ja) * | 2004-02-26 | 2005-09-09 | Kyowa Hakko Kogyo Co., Ltd. | 好中球性炎症疾患の予防及び/または治療剤 |
| EP1724271A1 (en) * | 2004-02-26 | 2006-11-22 | Kyowa Hakko Kogyo Co., Ltd. | Preventive and/or therapeutic agent for neutrophilic inflammation disease |
| EP1724271A4 (en) * | 2004-02-26 | 2013-01-23 | Kyowa Hakko Kirin Co Ltd | PREVENTIVE AND / OR THERAPEUTIC AGENT AGAINST NEUTROPHIC INFLAMMATORY DISEASE |
| KR101210008B1 (ko) | 2004-11-05 | 2012-12-07 | (주)아모레퍼시픽 | 스핑고실포스포릴콜린 또는 그 유도체의 용도 |
| WO2008096829A1 (ja) | 2007-02-07 | 2008-08-14 | Kyowa Hakko Kirin Co., Ltd. | 3環系化合物 |
| US8242151B2 (en) | 2007-02-07 | 2012-08-14 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compounds |
| US8486980B2 (en) | 2008-08-06 | 2013-07-16 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compound |
| US9475805B2 (en) | 2008-08-06 | 2016-10-25 | Kyowa Hakko Kirin Co., Ltd. | Tricyclic compound |
Also Published As
| Publication number | Publication date |
|---|---|
| US20060252679A1 (en) | 2006-11-09 |
| CA2496464A1 (en) | 2004-03-04 |
| AU2003262230A1 (en) | 2004-03-11 |
| JPWO2004017995A1 (ja) | 2005-12-08 |
| EP1547616A1 (en) | 2005-06-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11912718B2 (en) | Solid forms of a thienopyrimidinedione ACC inhibitor and methods for production thereof | |
| TWI330180B (en) | Triazole derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 | |
| CN110511219B (zh) | 苯基取代的二氢萘啶类化合物及其用途 | |
| TW201425300A (zh) | 用以合成甲狀腺激素類似物及其多形體之方法 | |
| RU2765718C1 (ru) | Замещенное производное дигидропирролопиразола | |
| EA024967B1 (ru) | Полициклический lpaантагонист и его применение | |
| JPH10510281A (ja) | イミダゾール誘導体 | |
| WO2004093912A1 (ja) | 好中球性炎症疾患の予防および/または治療剤 | |
| CN112707854B (zh) | 吡咯酰胺类化合物及其用途 | |
| JPWO2004054617A1 (ja) | 中枢疾患の予防および/または治療剤 | |
| WO2004017995A1 (ja) | 掻痒の予防および/または治療剤 | |
| WO2004017994A1 (ja) | 喘息の予防および/または治療剤 | |
| US20220185815A1 (en) | Pyrrolopyrazole derivative | |
| JP2018523698A (ja) | (4s)−および(4r)−4−(4−シアノ−2−メトキシフェニル)−5−エトキシ−2,8−ジメチル−1,4−ジヒドロ−1,6−ナフチリジン−3−カルボキサミドの代謝産物の調製方法およびその使用 | |
| JP2007063261A (ja) | X線照射による肺障害の予防及び/または治療剤 | |
| BR112017026994B1 (pt) | Composto, e, composição farmacêutica. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004530443 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2496464 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006252679 Country of ref document: US Ref document number: 10525324 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003792564 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003792564 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10525324 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003792564 Country of ref document: EP |