WO1998012228A1 - Polymers containing polysaccharides such as alginates or modified alginates - Google Patents
Polymers containing polysaccharides such as alginates or modified alginates Download PDFInfo
- Publication number
- WO1998012228A1 WO1998012228A1 PCT/US1997/016890 US9716890W WO9812228A1 WO 1998012228 A1 WO1998012228 A1 WO 1998012228A1 US 9716890 W US9716890 W US 9716890W WO 9812228 A1 WO9812228 A1 WO 9812228A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alginate
- polymer
- modified
- cell
- chains
- Prior art date
Links
- 0 C*(C)c1ccncc1 Chemical compound C*(C)c1ccncc1 0.000 description 1
- FYNVARJNCVQAAI-UHFFFAOYSA-N OC(CC1)OC1=O Chemical compound OC(CC1)OC1=O FYNVARJNCVQAAI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G83/00—Macromolecular compounds not provided for in groups C08G2/00 - C08G81/00
- C08G83/002—Dendritic macromolecules
- C08G83/003—Dendrimers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/34—Muscles; Smooth muscle cells; Heart; Cardiac stem cells; Myoblasts; Myocytes; Cardiomyocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/39—Connective tissue peptides, e.g. collagen, elastin, laminin, fibronectin, vitronectin, cold insoluble globulin [CIG]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/14—Macromolecular materials
- A61L27/20—Polysaccharides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/36—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix
- A61L27/38—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix containing added animal cells
- A61L27/3839—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix containing added animal cells characterised by the site of application in the body
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/36—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix
- A61L27/38—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix containing added animal cells
- A61L27/3839—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix containing added animal cells characterised by the site of application in the body
- A61L27/3843—Connective tissue
- A61L27/3847—Bones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L27/54—Biologically active materials, e.g. therapeutic substances
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L27/58—Materials at least partially resorbable by the body
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H13/00—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids
- C07H13/02—Compounds containing saccharide radicals esterified by carbonic acid or derivatives thereof, or by organic acids, e.g. phosphonic acids by carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0084—Guluromannuronans, e.g. alginic acid, i.e. D-mannuronic acid and D-guluronic acid units linked with alternating alpha- and beta-1,4-glycosidic bonds; Derivatives thereof, e.g. alginates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L5/00—Compositions of polysaccharides or of their derivatives not provided for in groups C08L1/00 or C08L3/00
- C08L5/04—Alginic acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/20—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices containing or releasing organic materials
- A61L2300/23—Carbohydrates
- A61L2300/232—Monosaccharides, disaccharides, polysaccharides, lipopolysaccharides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/20—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices containing or releasing organic materials
- A61L2300/25—Peptides having up to 20 amino acids in a defined sequence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/20—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices containing or releasing organic materials
- A61L2300/252—Polypeptides, proteins, e.g. glycoproteins, lipoproteins, cytokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/20—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices containing or releasing organic materials
- A61L2300/252—Polypeptides, proteins, e.g. glycoproteins, lipoproteins, cytokines
- A61L2300/254—Enzymes, proenzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/412—Tissue-regenerating or healing or proliferative agents
- A61L2300/414—Growth factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/60—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a special physical form
- A61L2300/64—Animal cells
Definitions
- the invention relates to materials which contain polysaccharide chains, particularly alginate or modified alginate chains.
- the polysaccharide, particularly alginate or modified alginate, chains may be included as side chains or auxiliary chains from a backbone polymer chain, which may also be a polysaccharide. Further, the polysaccharide chains may be crosslinked between side chains, auxiliary chains and/or backbone chains.
- These materials are advantageously modified by covalent bonding thereto of a biologically active molecule for cell adhesion or other cellular interaction.
- the materials are particularly useful to provide polymeric matrices for many applications, such as in tissue engineering applications for bone or soft tissue replacement. For example, the loss of bony tissue is a central feature of many aspects of clinical dentistry (e.g.
- the materials described herein can be useful for repair or replenishment of lost bony tissue.
- the materials are also useful for drug delivery applications when the biologically active molecule is attached by a degradeable bond.
- Unmodified alginate a polysaccharide, has been previously utilized as a tissue engineering matrix in cell encapsulation and transplantation studies. It provides a useful matrix because cells can be immobilized within alginate with little cell trauma and alginate/cell mixtures can be transplanted in a minimally invasive manner.
- One aspect of this invention is to provide a matrix which combines specific cell adhesion ligands in the matrix such that high control over cell-matrix interactions, due to cell adhesion and matrix interactions, is attained.
- One embodiment of the invention is directed to polymers containing a polymer backbone to which is linked polysaccharide groups, particularly of alginates or modified alginates, which preferably are polymerized D-mannuronate and/or L- guluronate monomers.
- the polysaccharide, particularly alginate, groups are present as side chains on the polymer backbone which is intended to include side chains at the terminal end of the backbone, thus being a continuation of the main chain.
- the polymers provide synthetic modified polysaccharides and alginates exhibiting controllable properties depending upon the ultimate use thereof. Further, the invention is directed to processes for preparing such polymers and to the use of such polymers, for example, as cell transplantation matrices, preformed hydrogels for cell transplantation, non-degradable matrices for immunoisolated cell transplantation, vehicles for drug delivery, wound dressings and replacements for industrially applied alginates.
- Another embodiment of the invention is directed to polysaccharides, particularly alginates, which are modified by being crosslinked.
- the alginates may further be modified by covalent bonding thereto of a biologically active molecule for cell adhesion or other cellular interaction.
- Crosslinking of the alginate can particularly provide alginate materials with controlled mechanical properties and shape memory properties which greatly expand their range of use, for example, to tissue engineering applications where size and shape of the matrix is of importance.
- the modification of the crosslinked alginates with the biologically active molecules can provide a further three - dimensional environment which is particularly advantageous for cell adhesion, thus making such alginates further useful as cell transplantation matrices.
- modified alginates such as alginate backbone (i.e. unmodified alginate) or the above described side chain alginates or crosslinked alginates, modified by covalent bonding thereto of a biologically active molecule for cell adhesion or other cellular interaction, which is particularly advantageous for maintenance, viability and directed expression of desirable patterns of gene expression.
- the modified alginate polymers provide a three-dimensional environment which is particularly advantageous for cell adhesion.
- the invention is directed to processes for preparing such polymers and to the use of such polymers, for example, for forming gels or highly viscous liquids having cell adhesion properties particularly for cell transplantation matrices, such as injectable cell transplantation solutions and preformed hydrogels for cell transplantation.
- Organ or tissue failure remains a frequent, costly, and serious problem in health care despite advances in medical technology.
- Available treatments now include transplantation of organs from one individual to another, performing surgical reconstructing, use of mechanical devices (e.g. , kidney dialyzer) and drug therapy.
- mechanical devices e.g. , kidney dialyzer
- these treatments are not perfect solutions. Transplantation of organs is limited by the lack of organ donors, possible rejection and other complications.
- Mechanical devices cannot perform all functions of an organ, e.g., kidney dialysis can only help remove some metabolic wastes from the body.
- drug levels comparable to the control systems of the body is difficult to achieve. This is partially due to difficulties in controlling the drug level in vivo. Financially, the cost of surgical procedures is very high.
- tissue engineering is the application of the principles and methods of engineering and the life sciences toward the fundamental understanding of structure/function relationships in normal and pathological mammalian tissues and the development of biological substitutes to restore, maintain or improve function. It thus involves the development of methods to build biological substitutes as supplements or alternatives to whole organ or tissue transplantation .
- the use of living cells and/or extracellular matrix (ECM) components in the development of implantable parts or devices is an attractive approach to restore or to replace function.
- ECM extracellular matrix
- a small biopsy can be grown into a large tissue mass and, potentially, could be used to treat many patients.
- the increased tissue supply may reduce the cost of the therapy because early intervention is possible during the disease, and this may prevent the long-term hospital ⁇ zation which results as tissue failure progresses.
- the use of immunosuppression may also be avoided in some applications by using the patient's own cells.
- Alginate is a linear polysaccharide, isolated, for example, from brown sea algae, which forms a stable hydrogel in the presence of divalent cations (e.g. , Ba + + , Ca + + ) (Smidsrod et al (1990): Alginate as immobilization matrix for cells. TIBTECH, 8:71- 78.) Alginate is currently being used for the in vitro culture of some cells types, as an injectable cell delivery matrix, for immunoisolation based therapies, and as an enzyme immobilization substrate (Atala et al., 1993: Injectable alginate seeded with chondrocytes as a potential treatment for vesicoureteral reflux. J.
- Alginates occur naturally as copolymers of D-mannuronate (M) and L-guluronate (G) and have different monomer compositions when isolated from different natural sources.
- the block length of monomer units, overall composition and molecular weight of the alginate influence its properties.
- calcium alginates rich in G are stiff materials, (see Sutherland, IW (1991): Alginates. In Biomaterials. : Novel materials from biological sources.). It is theorized that gel formation is due primarily to the G- block, and that the M-block is essentially non-selective.
- the calcium ions would be selectively bound between sequences of polyguluronate residues and held between diaxially linked L-guluronate residues which are in the 'C 4 chair conformation.
- the calcium ions would thus be packed into the interstices between polyguluronate chains associated pairwise and this structure is named the "egg-box" sequence.
- the ability to form a junction zone depends on the length of the G-blocks in different alginates (Sutherland, supra.). Other advantages of alginates include their wide availability, low diffusional barrier for all nutrients and relative biocompatibility (Smidsrod et al., Trends in Biotech, 8:71-78, 1990).
- alginate hydrogels used with a cellular component are lack of inherent cell adhesion. Such is necessary for cell attachment and long term survival of most mammalian cell systems. While chrondrocytes and islets of Langerhans have been successfully transplanted using alginates, the absence of suitable cell adhesion by alginates practically limits their use to cartilage and islet cell applications. Most other cell types require attachment to an extracellular substrate to remain viable.
- Plasticity 5:245:255.
- a disadvantage of such systems is that conversion of the polymers from a liquid to a solid, gel or highly viscous system requires conditions which are detrimental to cell viability, e.g., use of organic solvents and/or elevated temperatures.
- alginate hydrogels used in biotechnology applications are stable, and this can present a limitation in the use of these materials (e.g., loss of calcium from gels leads to gel dissolution).
- alginate hydrogels have a limited range of physical properties due to the limited number of variables one can currently manipulate (i.e., alginate concentration, specific divalent cation used for gelling, and concentration of divalent cation). This limitation is especially evident when alginate is utilized as an injectable cell delivery vehicle in tissue engineering. It is not possible to obtain a pre-defined and desirable shape of the matrix following injection, and it is thus not possible to create a new tissue with a specific and desirable shape and size. This is especially important whenever the size and shape of the new tissue are critical to the function of the tissue, for example, in reconstruction of facial features such as nose or ears, or relining of joints.
- An object of the present invention was to design improved synthetic analogues of alginates, to provide a process for preparing such polymers and to provide compositions and methods utilizing such polymers, particularly in tissue engineering applications. It is further useful according to the invention to provide alginate- containing materials in which the gel stability is related to an additional variable besides cation binding from the divalent cations.
- the disadvantages of the previous systems can be avoided by providing an alginate which can be gelled or made highly viscous under mild conditions, i.e., in the presence of divalent metal cations such as Ca + + or Ba + + in aqueous systems, without requiring, for example, organic solvents and/or increased temperature.
- the invention provides polymers with side chains of polysaccharides in general which may not exhibit the gelling behavior of alginates, but which provide polysaccharides with controllable properties, such as degradation.
- These polymers may comprise a polymeric backbone section to which is covalently linked a polysaccharide side chain.
- Another embodiment provides a polymeric backbone section to which is bonded a side chain, preferably multiple side chains, of polymerized, optionally modified, D-mannuronate (M units) and/or L-guluronate (G units) monomers.
- M units D-mannuronate
- G units L-guluronate
- the modified alginates preferably maintain the mild gelling behavior of conventional alginates, but do not have the disadvantages discussed above.
- the linkage between the polymeric backbone section and the side chain(s) may be provided by difunctional or multifunctional linker compounds, by groups incorporated within the polymeric backbone section reactive with the polysaccharide units and/or by groups on the polysaccharide units or derivatives thereof reactive with groups on the polymeric backbone section.
- the polymers may advantageously further comprise biologically active molecules bonded thereto, particularly preferably bonded through the carboxylic acid groups on M and/or G units.
- the side chains are alginates, the biologically active molecules exhibit cell adhesion properties and the polymers are useful for cell transplantation.
- An advantageous aspect of these materials is the ability to provide a polymer analogous to alginates, but with high controllability of the properties, particularly when used for cell transplantation purposes.
- the chemical structures, functionality and sizes of the different parts of the polymer, i.e., the backbone, linker, side chain and, optionally, biologically active molecule(s) can be provided so as to control many properties of the polymer in physiological systems, such as, for example, degradeability, biocompatibility, organ or tissue specificity and affinity, cell adhesion, cell growth and cell differentiation, manner and rate of removal from the system, solubility and viscosity.
- polymeric backbone section there can be used any homo- or co-polymer which is compatible with the ultimate use and which has the appropriate functional groups such that it can be covalently linked directly or through a linker to the polysaccharide, particularly polymerized M and/or G units, or suitable modifications thereof.
- Any polymer meeting the above requirements is useful herein, and the selection of the specific polymer and acquisitions or preparation of such polymer would be conventionally practiced in the art. See The Biomedical Engineering Handbook, ed. Bronzino, Section 4, ed. Park.
- polymeric backbone section are, for example, poly(vinyl alcohols), poly(alkylene oxides) particularly poly(ethylene oxides), polypeptides, poly(amino acids), such as poly(lysine), poly(allylamines)(PAM), poly(acrylates), modified styrene polymers such as poly(4-aminomethylstyrene), polyesters, polyphosphazenes, pluronic polyols, polyoxamers, poly(uronic acids) and copolymers, including graft polymers thereof.
- the polymeric backbone section may be selected to have a wide range of molecular weights, generally from as low as 100 up to ten million.
- the occurrence and rate of degradeability of the polymer and the manner and rate of release from physiological systems of the polymer can be influenced.
- a high molecular weight non-degradable polymeric backbone section for instance having a molecular weight above about 100,000, will in general provide a more stable polymer which may be useful in, for example, non-degradable matrices for immunoisolated cell transplantation.
- a polymeric backbone section having a molecular weight of less than about 30,000 to 50,000 or one in which the backbone itself is degradable can be cleared through the kidneys and by other normal metabolic routes.
- Polymers with a degradable polymeric backbone section include those with a backbone having hydrolyzable groups therein, such as polymers containing ester groups in the backbone, for example, aliphatic polyesters of the poly( ⁇ -hydroxy acids) including poly(gIycolic acid) and poly(lactic acid).
- a degradable polymer for the backbone is a graft polymer of PEO (polyethylene oxide) and acetyl-aspartate shown by the following equation, wherein the first equation shows formation of the degradable polymer backbone, and the second schematic shows the attachment of side chains thereto:
- A DCC, HOBT, DMAP, DMF.
- B: X OBT or OSu, NMM, DMF.
- Copolymer content can be controlled by the block length of PEO and mixing in Ac-aspartate.
- the solubility, viscosity, biocompatibihty, etc., of the polymeric backbone section also is a consideration as to its effect on the desired properties of the final polymer product.
- the polymeric backbone section can be one which incorporates linkage sites for the polysaccharide side chains so that a separate linker group is not required.
- poly(amino acids) having free amino groups may be used for this purpose.
- linker group may be selected from any divalent moieties which are compatible with the ultimate use of the polymer and which provide for covalent bonding between the ' polymeric backbone section and the polysaccharide side chain(s).
- linker groups are conventionally known in the art for such purpose, and can be linked to the backbone in a conventional manner.
- chemistries useful for reacting with carboxylate groups are particularly useful in providing the linker or linkage site on the polymer; see Bronzino and Hermanson, cited below.
- the linker group may be selected to significantly affect the biodegradability of the polymer depending upon the extent of hydrolyzability of groups in the linker chain. Amino acid linkers and derivatives thereof are preferred due to the controllability of the degradation feature.
- amino acid linker groups such as glycine
- glycine will provide ester linkages which are readily hydrolyzable and, thus, facilitate degradation of the polymer in an aqueous environment
- amino alcohols provide an ether linkage which is significantly less degradable.
- Amino aldehydes are also useful linker groups.
- the substituent groups on the amino acids will also affect the rate of degradeability of the linkage.
- the linker group may also be varied in chain length depending upon the desired properties. Linkages providing, for example, from 1 to 10 atoms between the backbone and side chain, are preferred, although longer linkage chains are possible.
- the linker may be branched to provide multiple attachment sites for the side chains, for example, to provide a dendrite configuration such as shown in Example 5.
- the linker will be in the form of a residue of the linking compound without the group removed during bonding.
- the side chains are polysaccharides, preferably optionally modified alginate units, which enable the preparation of a gel or highly viscous liquid in the presence of a divalent metal, e.g., Ca + ⁇ or Ba + + .
- they are comprised of polymerized D-mannuronate (M) and/or L-guluronate (G) monomers, but, also encompass modified such monomers.
- the side chains are particularly preferably comprised of oligomeric blocks of M units, G units or M and G units.
- the molecular weight of each side chain or the number of units and length of such side chains is again a function of the desired ultimate properties of the polymer and selectability of this aspect is an advantageous feature of the invention.
- the molecular weight of the side chain may range from about 200 up to one million, and may contain, preferably 2 to 5,000 M and/or G units.
- higher molecular weight side chains e.g. above about 100,000, are generally useful when more stable polymers are desired and lower molecular weight side chains, e.g. , below about 30,000 to 50,000, are generally useful when biodegradable species capable of removal through the kidneys, or other normal functions, are desired.
- side chains In general, more side chains will result in a more rigid, compact polymer, and provide a more dense concentration of attached biologically active molecules, if present.
- the number of side chains is preferably from 1 to 100% of the reactive monomer units available on the backbone per polymer molecule. It is not necessary that every linker group or linkage site be provided with a side chain. For example, free linkers or linkage sites may be left to facilitate the solubility and/or compatibility of the polymer in its intended system. Additionally, free linkers or linkage sites may be provided to allow for the later addition of differently structured or proportioned alginate side chains or other side chains.
- the whole side chain or individual M and/or G units may be modified from the naturally occurring units.
- Naturally occurring M and G alginate units exhibit the same general chemical structure irrespective of their source, although, the distribution and proportions of M and G units will differ depending upon the source.
- Natural source alginates for example from seaweed or bacteria, can thus be selected to provide side chains with appropriate M and G units for the ultimate use of the polymer. Isolation of alginate chains from natural sources for use as the side chains herein can be conducted by conventional methods. See Biomaterials: Novel Materials from Biological Sources, ed. Byrum, Alginates chapter (ed. Sutherland), p. 309-331 (1991).
- synthetically prepared alginates having a selected M and G unit proportion and distribution prepared by synthetic routes analogous to those known in the art can be used as the side chains.
- natural or synthetic source alginates may be modified to provide M and G units with a modified structure as long as the polymers with modified side chains still provide a gel or highly viscous liquid by interaction of the alginate units with a divalent metal.
- the M and/or G units may be modified, for example, with polyalkylene oxide units of varied molecular weight such as shown for modification of polysaccharides in Spaltro (U.S. Pat. 5,490,978) with other alcohols such as glycols. Modification of the side chains with such groups generally will make the polymer more soluble, which generally will result in a less viscous gel. Such modifying groups can also enhance the stability of the polymer.
- Useful polysaccharides other than alginates include agarose and microbial polysaccharides such as those listed in Table 1 :
- N-neutral, A anionic and C-cationic
- the polymeric backbone section, linkages and side chains may be provided in a number of configurations which configuration will be a factor in the controllability of the polymer properties.
- the configuration of the polymeric backbone section, the number and location of linkage sites and the type and number of side chains will determine the configuration Examples of useful configurations are shown in Figure 1 although the invention is not limited to such configurations and further configurations using the three basic structural units can be provided according to the invention Especially preferred, however, are polymers having the branched configuration It is noted that the "side chains" of the linear polymers are on the terminal ends of the backbone, but are still considered side chains herein Further, the side chains may be present between sections of polymei backbone in an alternating block type configuration.
- One preferred embodiment is materials wherein the backbone itself is an alginate.
- the side chains may be polyguluronate derived from sodium alginate.
- a particular example involves cross-linking polyguluronate to itself, via a hydrolytically degradable bond, utilizing a bifunctional cross-linking molecule to form a cross-linked polymer.
- Dendritic polymers and comb polymers, as described below, can also be provided as such materials. These structures can provide a highly cross-linked polymer which would rapidly degrade to low molecular weight components and readily be cleared by the body. To achieve this goal, for example, polyaldehyde guluronate is reacted with hydrazine and sodium borohydride to afford polyhydrazino guluronate.
- the hydrazine groups on this alginate derived polymer are used to incorporate G-block chains via the their hemiacetal termini.
- This provides materials from naturally derived polysaccharides with hydrolyzable hydrazone linkages, hence, biocompatible and biodegradable. Hydrolysis of the hydrazone linkage in these materials will lead to short chain polysaccharides that can be excreted by the kidney. Further more, reduction of the hydrazone bond by borohydrides can form a chemically stable hydrazine bond that provide non-degradable materials.
- both biodegradable and non-degradable biomaterials can be derived from natural polysaccharides. Cells within the polymer are not damaged by the cross-linking reaction, indicating that these materials are useful for cell transplantation, for example.
- Dendrimers provide a particularly interesting backbone structure since they exhibit different properties from the corresponding linear polymers due to the difference in molecular shape and structures.
- Dendritic molecules can be provided as a backbone with handles to branch off a large number of functional groups in a compact region. Since polypeptides are biodegradable and their degradation products (i.e., the amino acids) are non-toxic, certain polypeptides (e.g. polylysines) can be used as dendritic handles.
- soluble polymer supports which combine the advantages of both solid phase and solution phase syntheses can be used to prepare the materials. The most typical soluble polymer supports utilized arc comprised of poly(ethylene oxide) (PEO).
- a further useful backbone structure is comb polymers which contain many side chains extending from a polymer backbone.
- Poly(vinyl alcohol) (PVA) provides a particularly useful backbone for comb polymers.
- the alcohol groups of the PVA can be esterified and subjected to the above-discussed carbodiimide linkage chemistry to provide the side chain linkages.
- the materials containing a polymer backbone may be prepared utilizing synthetic methods known in the art, some of which are discussed above, for example in the Biomedical Engineering Handbook, section 4,; see also Odian, Principles of Polymerization. Chapter 9, 2nd ed., (1970).
- polymeric backbone starting materials can be used which already contain suitable linkage sites, e.g. free amino groups such as certain poly(amino acids), or the polymers can be reacted with linker compounds to provide suitable linkage sites, particularly by the reaction of suitable sites on the polymeric backbone with amino acid derivatives, optionally with the amino groups being protected.
- some reactive sites on the backbone may be protected to prevent addition of the linker group if it is desired to keep such sites free or to subsequently provide such sites with different linker groups.
- This chemistry is conventional in the field of linker/polymer formation, especially involving ester, amide, ether and other covalent linkages; see, e.g., Bronzino and Hermanson, cited above.
- protective groups see, e.g., Vogel's Textbook of Practical Organic Chemistry, 5th ed. p. 550+ and 784+ .
- reaction with the side chain of M and/or G units is conducted, preferably through grafting by reductive amination of the reducing end of the side chain with the amino group of the linker, to produce the subject polymers.
- the side chains are provided as described above from natural sources or synthetically, and may have, optionally, the described modifications, they may be bonded as described above, or by other conventional methods.
- Another embodiment of synthetic analogues of alginate materials are those provided by covalent crosslinking of the alginate.
- This covalent crosslinking greatly expands the range of situations in which these materials are useful.
- One specific application of this modification is the development of matrices of the alginate with shape memory.
- the crosslinked alginate provides advantageous shape memory properties and compression resistance properties which make them particularly advantageous for use in forming cell transplantation matrices.
- Shape memory matrices are designed to "remember" their original dimensions and, following injection in the body in a compact form (e.g. , through a syringe) or other means of placement in the body or in other locations which they may find use, resume their original size and shape.
- the shape memory property of the alginate is provided by crosslinking thereof.
- Crosslinking can also improve the compression resistance and/or other mechanical properties of the alginate. Further, a crosslinked alginate can provide a degree of cell adhesion even without use of biologically active cell adhesion ligands. Gelling by divalent cations provides another means of increasing the viscosity and degree of structure of the alginate in addition to the crosslinking. Further, the crosslinked alginate may be covalently bonded to at least one cell adhesion ligand to provide for cell adhesion and maintenance of cell viability.
- alginate used for crosslinking are alginate chains which contain polymerized D-mannuronate (M) and/or L-guluronate (G) monomers, but the term "alginate” or "alginate chain” as used herein also is intended to encompass chains wherein such monomers are modified such as described below when they are compatible with the ultimate use and able to be crosslinked covalently.
- the alginate chain is particularly preferably comprised of oligomeric blocks of M units, G units, M and G units, or mixtures of such blocks.
- the general structure of an alginate linear copolymer of M and G units is demonstrated by the following general formula:
- the molecular weight of the alginate chain and, thus, the number of units and length of the chains may be selected dependent upon the desired properties of the polymer.
- the molecular weight of each chain may range from about 1,000 to one million, for example.
- Higher molecular weight chains, e.g. , above about 100,000, are generally useful when more stable alginate polymers are desired and lower molecular weight chains, e.g., below about 30,000 to 50,000, are generally useful when biodegradable species capable of removal through the kidneys or through other normal metabolic functions are desired.
- the distribution of M and G units also provides a controllability feature of the invention with a higher ratio of G units generally providing a stiffer alginate material which will hold its shape better.
- An alginate chain having a percentage of G units based on the total of M and G units of from 10 to 100% is particularly preferred. Increasing or decreasing the number of G units in the chain will also allow for increasing or decreasing, respectively, the rate of gelation of the alginate. Such may be of interest when the alginate is used in an injectable solution and the rate is controlled so that the solution will gel at the appropriate time after injection.
- the alginate chain or individual M and/or G units may also be modified from the naturally occurring units. Sources for the naturally occurring alginates and for modified alginates are described above in relation to the alginate side chains for the polymeric backbone embodiment described above.
- alginate starting material materials having a polymeric backbone to which is linked alginate side chains, as described above.
- the crosslinking may occur between side chains of the same backbone and/or between side chains of other backbones. It is also possible to have different types of alginate- containing materials with crosslinking provided between alginate sections or chains thereof. Mixtures of any of the above alginate starting materials may also be used.
- the crosslinking of the alginates is by action of a crosslinking agent to provide covalent bonding, through the crosslinking agent, from the carboxylic acid groups of the uronic acid of one alginate unit to the carboxylic acid group of the uronic acid of another alginate unit.
- a crosslinking agent to provide covalent bonding, through the crosslinking agent, from the carboxylic acid groups of the uronic acid of one alginate unit to the carboxylic acid group of the uronic acid of another alginate unit.
- Such crosslinking is preferably between alginate units from different alginate chains.
- crosslinking may also occur between alginate units of the same chain or, in the case where the alginates are side chains on a polymer backbone as described above, crosslinking may occur between different side chains on the same or differing polymer backbones.
- the crosslinking agent may be any suitable agent with at least two functional groups which are capable of covalently bonding to the carboxylic acid groups and/or alcohol groups of the alginate or modified groups therefrom.
- Crosslinking agents of higher functionality may also be used.
- polyamines such as bifunctional, trifunctional, star polymers or dendritic amines are useful and these can be made, for example, by conversion from corresponding polyols.
- Preferred crosslinking agents are those with at least two nitrogen-based functional groups such as, for example, diamine or dihydrazide compounds; non-limiting examples thereof being diamino alkanes, Jeffamine series compounds, adipic acid dihydrazide and putrescine.
- crosslinking agent is lysine, especially an ester thereof, particularly the methyl or ethyl ester.
- the crosslinking agent may also be selected to provide a more or less biodegradable or non-biodegradable bond such that the lifetime of the resulting crosslinked alginate material in its environment, e.g. in vivo, can be modified for the intended utility.
- ethylene glycol could be coupled with two N-(t-Boc)glycine using carbodiimide chemistry to yield 1 ,2-ethylene- (N,N'-di-t-Boc)giycine intermediate.
- This intermediate could be deprotected using trifluoroacetic acid in methylene chloride at various temperatures to yield 1,2-ethyleneglycoldigIycinate intermediate.
- This intermediate could be deprotected using trifluoroacetic acid in methylene chloride at various temperatures to yield 1,2-ethyleneglycoldiglycinate intermediate.
- other molecules with two terminal alcohol functional groups could be utilized.
- polyols including, e.g., (star shaped or dendritic) could be transformed into similar types of crosslinkers with biodegradable ester functional groups incorporated using parallel chemical pathways.
- the crosslinking is facilitated by an activator compound which reacts with the carboxylic acid group of the alginate unit to make it more reactive to the crosslinking agent.
- an activator compound which reacts with the carboxylic acid group of the alginate unit to make it more reactive to the crosslinking agent.
- Useful activators for making a carboxylic acid group more reactive to the crosslinking agent, particularly an amine functional group of the crosslinking agent, are known in the art.
- carbodiimides particularly water-soluble carbodiimides such as, for example, l-ethyl-3-(3-dimethylami.nopropyl)carbodiimide (EDC), l-cyclohexyI-3-(2- morpholinoethyl)carbodiimide (CMC), and CDI (carbodiimidazole).
- EDC l-ethyl-3-(3-dimethylami.nopropyl)carbodiimide
- CMC l-cyclohexyI-3-(2- morpholinoethyl)carbodiimide
- CDI carbodiimidazole
- a stabilizer for stabilizing the resulting activated group.
- useful stabilizers for the activator groups are known in the art.
- a useful stabilizer is 1 -hydroxybenzotriazole (HOBT) which stabilizes the activated group against hydrolysis.
- HOBT 1 -hydroxybenzotriazole
- Other useful stabilizers include N-hydroxysuccinimide and N- hydroxysulfylsuccinimide (sulfo-NHS).
- EDC activate carboxylic acid to yield O-acylisourea intermediate.
- This intermediate reacts with HOBT to form HOBT activated intermediate.
- Primary amino groups in lysine ethyl ester then couple the activated carboxyl groups of adjacent alginate molecules to form crosslinked alginates.
- the crosslinking can generally be conducted at room temperature and neutral pH conditions, however, the conditions may be varied to optimize the particular application and crosslinking chemistry utilized.
- pH of from 4.0 to 8.0 and temperatures from 0°C to room temperature (25 °C) are optimal and preferred.
- crosslinking chemistries can also be used. For example, using poly(ethylene glycol) (PEG) as a spacer in a crosslinking agent with an N-protected amino acid (see Example 12). Also, crosslinking of oxidized alginate can be conducted with adipic acid dihydrazide. The oxidation results in polyaldehyde alginates (limit oxidized alginates) for crosslinking (See Example 17). Additionally, crosslinking can be effected by light activation using photoreactive materials (See Example 26).
- PEG poly(ethylene glycol)
- N-protected amino acid see Example 12
- crosslinking of oxidized alginate can be conducted with adipic acid dihydrazide. The oxidation results in polyaldehyde alginates (limit oxidized alginates) for crosslinking (See Example 17). Additionally, crosslinking can be effected by light activation using photoreactive materials (See Example 26).
- M cross-links
- M c may be modified by controlling the extent of cross-linking, or by varying the molecular weight of the cross- linking molecule (Simon et al. , Polymer 32:2577-2587, 1991 ). Both of these strategies may be utilized to alter the mechanical properties of the alginate gels. Covalent crosslinking has been achieved with several different approaches.
- cross-linking with lysine results in amide bond formation, which will provide stability and will degrade very slowly.
- PEG-crosslinkers contain an ester bond, which will be more labile to hydrolysis.
- cross-linking of oxidized alginate with adipic acid dihydrazide leads to a hydrazone bond.
- these materials may be both covalently and ionically (e.g. , calcium) cross-linked. This may prove advantageous in certain applications in which one desires a two-stage gelling.
- the polyaldehyde alginates described below will cross-link ionically very quickly (e.g. , minutes), while the covalent cross-linking reaction can be designed to occur very slowly (e.g. , hours).
- a surgeon could thus ionically cross-link these polymers to yield a solution which is amenable to injection via a syringe or endoscope, but is viscous enough (viscosity at this stage decreases with increasing extent of oxidation) so that it does not extravasate after being placed.
- the covalent cross-linking would subsequently harden the implanted material into a more rigid, non-flowable mass.
- Crosslinking of the alginate provides a more structured material, the extent of structuring being dependent, at least in part, on the extent of crosslinking.
- the extent of structuring of the alginate material will also depend, among other factors, upon the extent of gelling through action of the ionic bonding of the divalent metal cation, as discussed above, and upon the nature of the starting alginate material, which as discussed above may be varied, for example, to affect stiffness of the material.
- the crosslinked alginate material may run the spectrum through the following forms: a viscous liquid, a swellable gel, a non-swellable gel, a swollen polymer network or a solid matrix, for example.
- the extent of crosslinking is a function of the amount of crosslinking agent and crosslinking method used, i.e. , the molar percent of crosslinking agent per mole of crosslinkable alginate carboxylic acid groups.
- the alginate will be increased in viscosity as it is crosslinked.
- the extent of crosslinking will be dependent upon the ultimate use. For example, to provide gel materials which have super absorbency properties, it is useful to have a low crosslinking extent, for example, of about 1 to
- the extent of crosslinking is preferably from about 5% to 75%.
- the alginate is a viscous liquid when the crosslinking agent amount is about 25 mol% or less, a swellable gel when the amount is about 50% and a solid structure which maintains its size and shape when the amount is about 75% or higher.
- the crosslinking chemistry can be selected and optimized to control viscosity even at lower crosslinking extent.
- the crosslinking agent is used in a molar amount about equal (i.e., 100 mol%) to the number of crosslinkable alginate carboxylic acid (uronic acid) groups.
- the crosslinking can be conducted either before, after or simultaneously with the gelling by action of the divalent metal cations. It is preferred for certain applications that the crosslinking be conducted either before or simultaneously with the gelling by divalent cation so as to prevent problems with diffusion of the crosslinking agent to interior portions of the gelled material.
- This material may make an ideal two-stage gelling matrix.
- the extent of oxidation of the alginate in the first step of the synthesis controls the binding sites available for ionic gelling, and thus regulates the viscosity of calcium cross-linked gels.
- the covalent cross-linking reaction with adipic acid dihydrazide occurs over several hours, and thus can be used to harden the gel slowly.
- the ultimate mechanical properties of the matrix can be controlled by varying the extent of covalent crosslinking, and this will be a function of the adipic acid concentration. For example, a material largely insensitive to the time of ionic cross-linking time, and with a time frame for ionic cross-linking considerably shorter than that for covalent cross-linking can be designed.
- the crosslinked alginate is not gelled by action of divalent metal cations at all or is gelled by cations present in vivo only after the delivery of the crosslinked alginate into the biological system, e.g. , body.
- the extent of stiffness and matrix structure of the crosslinked alginate materials will be influenced both by the gelling by divalent cation and by the extent and nature of crosslinking.
- the ability to vary these and other factors provides great flexibility in designing a material which is particularly suited for its ultimate application.
- the matrix structure provided by the crosslinked alginates themselves can facilitate cell adhesion type properties, for example, due to trapping of cells in the matrix or action of a crosslinking agent, such as lysine.
- a crosslinking agent such as lysine.
- the crosslinked alginate as a matrix can be introduced for tissue engineering and the cell can migrate into the pores of the matrix in vivo. It is also advantageous, however, to provide the crosslinked alginates with biologically active molecules to facilitate cell adhesion or other biological interaction, as discussed below.
- the ligands may be added before, during or after crosslinking of the alginate and/or gelling by divalent cations.
- the polymers can be modified with biologically active molecules.
- Another aspect of the invention lies in modifying not only the above-discussed synthetic alginate analogues but also the base naturally occurring, modified or analogous alginate materials which are described herein. Even if the alginate or modified alginate material is not provided on a polymeric backbone and/or not crosslinked, the coupling of the alginate with certain biologically active molecules makes it very useful for tissue engineering and other applications.
- the polymeric backbone-containing and/or crosslinked alginates and the naturally occurring or modified base alginate materials can be modified with the cell adhesion active molecule(s), for example, by covalent bonding using amide chemistry between the amine groups of the biological molecules and a free carboxylic acid group of the uronic acid residues (of M and G units) of the alginate or other polysaccharide. If the material is crosslinked, bonded to a polymeric backbone and/or otherwise modified, free acid groups must remain to add cell adhesion groups. If the cell adhesion groups are added first, active groups for any subsequent crosslinking, polymer bonding or other modification must remain. Other chemistries can also be used to effect such bonding to the biologically active molecule.
- alginate or analogous materials can be modified to provide aldehyde groups thereon, which are reactive with the amino terminal of peptides to provide an imine bond which is reduced to a stable amine bond.
- aldehyde groups thereon, which are reactive with the amino terminal of peptides to provide an imine bond which is reduced to a stable amine bond.
- suitable cell adhesion molecules include known cell attachment peptides, proteoglycan attachment peptide sequences (see Table 2), biologically active proteoglycans (e.g. laminin and fibronectin) and other polysaccharides (e.g. , hyaluronic acid and chondroitin-6-sulfate).
- suitable biological molecules include peptide growth factors (such as EGF, VEGF, b-FGF, acidic FGF, platelet- derived growth factor, TGF or TGF- ⁇ ), and enzymes (Dominguez et al. , 1988: Carbodiimide coupling of ⁇ -galactosidase from Aspergillus oryzae to alginate. Enzyme
- the cell adhesion molecule bonded to the alginate chain are synthetic peptides containing the amino acid sequence arginine-glycine -aspartic acid (RGD) which is known as a cell attachment ligand and found in various natural extracellular matrix molecules. Further of interest is GREDVY (endothelial cell specific) peptide.
- RGD arginine-glycine -aspartic acid
- GREDVY endothelial cell specific peptide.
- the alginates with such a modification provide cell adhesion properties to the alginate analogue, natural alginate or modified alginate, particularly when used as a cell transplantation matrix, and sustains long-term survival of mammalian cell systems, as well as controlling cell growth and differentiation.
- Coupling of the cell adhesion molecules to the alginate can be conducted utilizing synthetic methods which are in general known to one of ordinary skill in the art.
- a particularly useful method is by formation of an amide bond between the carboxylic acid groups on the alginate chain and amine groups on the cell adhesion molecule.
- Other useful bonding chemistries include those discussed in Hermanson, Bioconjugate Techniques, p. 152-185 (1996), particularly by use of carbodiimide couplers, DCC and DIC (Woodward's Reagent K). Since many of the cell adhesion molecules are peptides, they contain a terminal amine group for such bonding.
- the amide bond formation is preferably catalyzed by l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), which is a water soluble enzyme commonly used in peptide synthesis.
- EDC l-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- EDC reacts with carboxylate moieties on the alginate backbone creating activated esters which are reactive towards amines.
- R-NH 2 represents any molecule with a free amine (i.e. lysine or any peptide sequence N-terminus).
- EDC may be used in conjunction with N- hydroxysuccinimide, N-hydroxysulfylsuccinimide or HOBT to facilitate amide bonding over competing reactions.
- the reaction conditions for this coupling chemistry can be optimized, for example, by variation of the reaction buffer, pH, EDC:uronic acid ratio, to achieve efficiencies of peptide incorporation between 65 and 75%, for example.
- the pH is about 6.5 to 7.5.
- the ionic concentration providing the buffer is preferably about 0.1 to 0.6 molar.
- the EDC:uronic acid groups molar ratio is preferably from 1 :50 to 20:50. When HOBT is used, the preferred molar ratio of EDC:HOBt:uronic acid is about 4:1 :4.
- the density of cell adhesion ligands, a critical regulator of cellular phenotype following adhesion to a biomaterial Massia and Hubbell, J. Cell Biol. 1 14:1089-1 100, 1991; Mooney et al., J Cell Phys. 151 :497-505, 1992; and Hansen et al., Mol. Biol. Cell 5:967-975, 1994
- the biological molecules useful for attachment to the above-described alginate materials are not, however, limited to those providing cell adhesion function.
- the polymer could be bound to a molecule with antiseptic function when used as a wound dressing, or which provides adhesion tissue specific gene expression, growth factors to enhance proliferation of cells in the environment or vascularation of the tissue or anti-inflammatory activity.
- the combination of the alginate and alginate analogue materials with cell adhesion ligands bonded thereto provides a unique three dimensional environment in which the cells interact through various forces for adhesion to the alginate which has many uses, particularly for tissue engineering applications.
- the cell adhesion ligands provide specific cell membrane receptor sites for the desired cells.
- the number, type and location of the cell adhesion ligands on the alginate or alginate analogue material will affect the cell adhesion and cell viability maintenance properties and such factors can be varied to suit the particular application.
- Such applications include tissue engineering methods applied to humans and animals.
- 10 12 to 10 "4 moles of adhesion molecules per milliliter of hydrated alginate are used; see Massia et al. , 3.
- cell adhesion ligands with differing cell adhesion ligands or other bioactive molecules may be utilized according to the invention.
- additional groups may be bonded at other sites on the alginate or to suitable sites on ligands already present on the alginate or alginate analogue material.
- the alginate having a polymeric backbone and/or being crosslinked or the natural or modified alginate or other polysaccharide, optionally with bioactive molecules, can create a synthetic extracellular environment for mammalian cells that is capable of performing the diverse functions of the natural extracellular matrix (ECM).
- ECM extracellular matrix
- the materials described herein will, thus, have application in the field of tissue engineering, biomaterials, and in the basic cell biological sciences for studying three dimensional cell interactions and tissue morphogenesis.
- the materials described herein are advantageous as a model system for creating a synthetic ECM capable of guiding cellular gene expression during in vitro or in vivo tissue formation.
- the natural ECM regulates cell growth and differentiation with features that allow the control of the mechanical and chemical environment around the cells (D.E. Ingber. Mechanochemical Switching between growth and Differentiation by Extracellular Matrix, in Principles of Tissue Engineering (Ed, Lanza, Langer and
- Unmodified alginate has been used as a cell immobilization material for many years due to the stable hydrogels formed with mild gelling conditions. However, the alginate acts only as a neutral agent suspending cells or cell aggregates in three dimensions.
- the alginate can be transformed into a dynamic, interactive matrix capable of guiding cellular gene expression in space and time.
- the ability to control the viscoela tic properties of the alginate is an integral aspect in guiding cellular gene expression (see M. Opas, Substratum Mechanics and Cell Differentiation. International Review of Cytology, Vol. 150, p. 119-137 (1994); and GF Oster, JD Murry, and AK Harris, Mechanical aspects of Mesenchymal
- Morphogenesis Journal of Embryology and Experimental Morphology, Vol. 78, p 83- 125 (1983)) and can be used in model in vitro cell culture systems and tissue engineering applications.
- Matrices play a central role in tissue engineering. Matrices are utilized to deliver cells to desired sites in the body, to define a potential space for the engineered tissue, and to guide the process of tissue development. Direct injection of cell suspension without matrices have been utilized in some cases, but it is difficult to control the placement of transplanted cells. In addition, the majority of mammalian cell types are anchorage dependent and will die if not provided with an adhesion substrate. Alginate materials in polymerized form and/or crosslinked and/or modified with bioactive molecules, as discussed above, can be advantageously used as matrices to achieve cell delivery with high loading and efficiency to specific sites.
- the first type of scaffolds, immunoprotective devices utilize a semipermeable membrane to limit communication between cells in the device and the host.
- the small pores in these devices e.g. , (d ⁇ 10 ⁇ m) allow low molecular weight proteins and molecules to be transported between the implant and the host tissue, but they prevent large proteins (e.g. , immunoglopulins) and host cells (e.g. , lymphocytes) of the immune system from entering the device and mediating rejection of the transplanted cells.
- large proteins e.g. , immunoglopulins
- host cells e.g. , lymphocytes
- open structures with large pore sized e.g. , (d > 10 ⁇ m) are typically utilized if the new tissue is expected to integrate with the host tissue.
- the morphology of the matrix can guide the structure of an engineered tissue, including the size, shape and vascularization of the tissue.
- the alginate, alginate analogue and modified alginate materials of the invention are useful for cell transplantation matrices. These materials can be used to provide such a matrix in any of several ways. For instance, when the matrix is desired to be a temporary matrix for replacement by natural tissue, the material can be designed for biodegradability and system release, for example, by providing hydrolyzable linkages, using relatively low molecular weight alginate chains, biodegradable crosslinking agents, biodegradeable polymer backbones and/or low molecular weight polymer backbone sections.
- non-hydrolyzable linkages alginate chains of higher molecular weight, non-degradable crosslinking agents and/or higher molecular weight polymer backbone sections can be used.
- the many ways in which the properties of the materials can be altered provides a high degree of controllability in providing materials which meet the requirements for the specific application.
- the matrices can be introduced to the body without cells, but cells will migrate into the matrix, in vivo, and regenerate therein.
- the alginate or analogue material can be provided in an injectable form, optionally bound to appropriate viable cells, after injection in which case endogenous divalent metal cation in the physiological system after injection causes gelation of the alginate portions of the material.
- divalent metal cations are added to the solution, for example as a calcium sulfate solution, just prior to injection.
- the material can be designed to control its rate of gelation to match the ultimate utility.
- injectable solutions can be utilized for delivery of cells to regenerate urologic tissues, for reconstructive surgery, skin replacement, other orthopedic applications or other tissue replacement or repair applications.
- the alginate-containing materials provide a highly structured, gelled or highly viscous matrix in which the cells are compatible and grow to achieve their intended function, such as tissue replacement, eventually replacing the matrix.
- the materials may act as analogs to natural glycosamine-glycans and proteoglycans of the extracellular matrix in the body.
- the materials can be used to implant a matrix which does not contain cells and subsequently the cells can be seeded into the matrix in vivo.
- the materials optionally may be provided, for example, as a gel, as a viscous solution, as a relatively rigid body, as preformed hydrogel, within a semi-permeable membrane, within microcapsules, etc. , and the polymer properties controlled as discussed above to facilitate such applications.
- the utility of the polymers for cell transplantation and tissue engineering is a significant advance in the art, particularly since it was previously considered not to be practical or possible to achieve such results with synthetic materials; see C. Ezzell, The Journal of NIH Research. July 1995, Vol. 7, p. 49-53.
- the materials are also advantageously useful as vehicles for drug delivery particularly for sustained release.
- the bioactive molecule i.e. , the drug
- it is useful that the bioactive molecule, i.e. , the drug be linked to the alginate polymer and/or analogue material by a degradeable bond chosen for controllable release in the system.
- Other utilities of the materials which may or may not employ a bound biological molecule include, for example, wound dressings, wound healing matrix materials, matrices for in vitro cell culture studies, replacements for conventional industrial alginates used, for instance, as food thickening agents and as printing additives, for example to thicken inks, and similar uses wherein the above-described properties are desired.
- One particularly advantageous use of the crosslinked materials, not necessarily containing bioactive components, is as highly absorbent materials. Particularly, materials with a low extent of crosslinking, e.g. , about 1-20% crosslinking, have this utility. The absorbency property makes them useful, for example, in disposable diaper applications.
- the controllability of the properties of the synthetic polysaccharides according to the invention and the consistent reproduceability of such selected synthetic polysaccharides makes them particularly advantageous for many applications.
- a pentapeptide (GRGDY) was used as the model cell adhesion peptide.
- the N- terminal free amine provides a site for coupling to alginate, while the C-terminal tyrosine provides a site for iodination (forming 1 5 I-labeled peptide), which allows the coupling reaction to be quantitatively analyzed.
- the ratio of uronic acid: EDC: sulfo-NHS is constant, while only the peptide available for reaction is varied. This chemistry consistently gives 65 - 75 % coupling efficiency compared to available peptide as shown in Figure 2.
- the solution is allowed to react for 14 - 18 hours at which time hydroxyl amine is added to quench any unreacted activated sulfo-NHS- esters and reestablishing carboxylates.
- the solution is extensively dialyzed against water in 3500 MWCO dialysis tubing. Preliminary experiments utilizing 125 I-labeled GRGDY indicate ⁇ 0.5% of unreacted peptide remains after dialysis, suggesting a relatively pure alginate-peptide product.
- the dialyzed solution is sterile filtered, lyophilized and stored dry until use.
- a recently described technique (Klock et al. , Appl. Microbiol. Biotechnol. 40:638-643, 1994) can be used to detect any polyphenol contaminants in the alginate.
- alginate hydrogels will be done for two- dimensional cell culture experiments to save on reagents. This process is done under sterile conditions with all reactants in sterile filtered aqueous solutions. The reaction may be done in the above MES buffer or in diH 2 O with subsequent 10-fold loss of reaction efficiency. 125 I experiments show similar reaction efficiencies to the bulk modified alginate.
- Cross-linked alginate gels are cast between parallel glass plates with 2mm spacers and disks are punched out with circular punches. Ten to twelve disks are added to 50 ml centrifuge mbes with 40 mis reaction solution with the reactants at the same ratios as above. The disks are extensively washed in water, and then DMEM before being placed in 24-well plates for cell experiments.
- reaction conditions have been optimized by variation of the reaction buffer, pH, EDC.uronic acid ratio, and efficiencies of peptide incorporation between 65 and 75% are typically obtained.
- the density of cell adhesion ligands, a critical regulator of cellular phenotype following adhesion to a biomaterial can be readily varied over a 5-order of magnitude density range. Both surface coupling, as well as bulk coupling of alginate can be readily obtained with this approach.
- Oligo-guiuronate This scheme displays an example of the synthetic pathways that can be used for the synthesis of the graft copolymers. Oligo-guluronate was prepared by partial hydrolysis of alginate with 70% guluronate content. Hydrolysis occurred preferentially at the alternating region (M/G region), therefore, a mixmre of oligo-mannuronate and oligo-guluronate resulted. The latter was separated from the other oligomer by differential solubility at highly acidic conditions. See Penman A, Sanderson GR (1972): A method for the determination of uronic acid sequence in alginates.
- PVA poly (vinyl alcohol)
- the Boc protecting group was removed under acidic conditions and subsequent grafting of oligo-guluronate by reductive amination of the reducing end of carbohydrate with the amino group of glycine on PVA furnished the desired copolymers.
- Comb polymers PAM-Gul by Direct Linking of Backbone to Side Chains
- PEO Polyethylene glycol
- the dendritic polymers may form networks by coordination of calcium ions between side chains of two or more differing dendritic polymers.
- the linker group is hydrolyzable, and thus degradable.
- Dendritic polylysines as the polymeric backbone for incorporation of G-block alginate chains where prepared as shown in the following schematic.
- the amino groups of lysine was protected by Boc group while the carboxyl end was unprotected for peptide coupling which was achieved by DCC/HOBt chemistry.
- PEO (8000 Mw) was first coupled to hydroxysuccinamide ester of di-Boc protected lysine in dichloromethane.
- the ether-washed polymer was dissolved into 25% TFA in CH 2 C1 2 to remove Boc group. Precipitation into ether furnished the deprotected peptide ready for next cycle of coupling.
- Coupling of the corresponding free polyamines on the polymer support with excess DCC/HOBt -activated di-Boc-lysine furnished PEO-linked
- G-l , G-2 and G-3 dendrimers were established by TLC, elemental analysis and l3 C-NMR spectroscopy.
- PVA poly (vinyl alcohol)
- PVA belongs to a class of water-soluble polymers whose properties can be varied widely. PVA cannot be synthesized directly due to the instability of its monomer. Deacetylation of poly (vinyl acetate) through alcoholysis, hydrolysis or aminolysis leads to PVA's. The hydrophilicity and water solubility can be readily controlled by the extent of hydrolysis and molecular weight of the poly (vinyl acetate) ' used. PVA is not truly biodegradable, due to a lack of labile bonds, but PVA with a molecular weight ⁇ 20K can be cleared through the kidneys and has been used as drug delivery matrices and surgical prosthesis.
- DCC coupling method shown in the following schematic.
- the amino-protecting group (Boc) can be removed by utilization of trifluoroacetic acid in CH 2 C1 2 .
- the polymer is characterized by standard laboratory methods such as TLC, FT-IR, 'H-NMR and aqueous SEC.
- the amino functional ized PVA can be covalently coupled through reductive amination to oligoguluronate.
- Preparation 1 Five stock solutions of aqueous sodium alginates (2%) were prepared in Erlenmeyer flasks. Lysine ethyl ester was added to each solution to yield the following ratios: 0, 25, 50, 100, 150% lysine:uronic acid mole ratio. EDC and HOBT in twice the amount of moles of lysine each were added to each solution. The mixtures were thoroughly mixed and poured over petri dishes. Calcium sulfate powder was then added to gel the solutions for 24-48 hours. Circular discs were made from each batch, washed with distilled water, and lyophilized. The dimensions and weights of each disc was measured before and after lyophilization.
- Preparation 2 Several stock solutions of aqueous sodium alginates (2%) were prepared in Erlenmeyer flasks. The selected amount of lysine ethyl ester (0, 25, 50, 100, 150% lysine:uronic acid mole ratio) was added to each solution and stirred for 24 hours. Each solution was poured into a petri dish to form a layer 3 mm in diameter. Calcium sulfate powder was then added to the surface of the layers to induce gelling. After the gels hardened (24-48 hr.), circular disks were cut from all the gels. Each set of discs were transferred into a test tube.
- EDC and HOBT were then added (lysine:EDC:HOBT 1:2:2 mole ratio) to each tube.
- the disks were shaken for 24 hours and rinsed with distilled water. The dimensions and wet-weight of each disk were recorded. Each disc was then frozen, and lyophilized, and the dimensions and dry-weight of each disk were recorded afterwards.
- Alginate + lysine + EDC + HOBT maintained size and shape
- Cross-linked alginate gels (100% lysine content, EDC + HOBT to cross-link) were lyophilized and sterilized. Scanning electron microscopy examination of matrices revealed a highly porous material with large pores (Fig. 5). The matrices were placed in a suspension of smooth muscle cells in tissue culture medium (DMEM supplemented with 10% calf serum), and examined for cell infiltration. Observation of matrices with a microscope indicated that the cell suspension absorbed into the cross-linked alginate matrices, and this resulted in a distribution of cells throughout the matrices (not shown in figure).
- tissue culture medium DMEM supplemented with 10% calf serum
- the gels were cured for 3 hours between parallel plates about 2.5 mm apart. Disks were then punched out and added to either sodium citrate (to remove the calcium) or 0.1 M calcium chloride for 10 minutes prior to storage in distilled water. Gel stability with removal of calcium and mechanical testing wefe done with all conditions.
- Example 11 Alginate cross-linking with diamines is performed in a 0.1 M MES buffer of pH
- Poly (ethylene glycol) with molecular weights MW 200, 400, 600, and 1000 was obtained from Lancaster Synthesis Inc.
- PEG with MW 3400 and 1 ,3- Dicyclohexylcarbodiimide (DCC) were from Aldrich chemical company.
- the reaction solutions are cast between parallel glass plates for 12 - 16 hours and defined shapes may be cut from these hydrogel sheets. Defined shapes may also be formed by casting the reacting solution into a mold of the desired shape, and allowing the cross-linking reaction to occur.
- the resultant hydrogels are characterized for mechanical properties (elastic modulus, shear modulus, maximum true stress, maximum extension), and swelling properties.
- the methyl ester of lysine and modified amino terminated PEG of molecular weights 200, 400, 600, 1000 and 3400 was used to cross-link alginate in ratios of ammo arboxylic groups 3 - 50% .
- Example 13 Lysine cross-linked alginates.
- the alginate hydrogels were cross-linked with the methyl ester of lysine.
- the carbodiimide chemistry was optimized for maximum effective cross-linking at any given lysine content by varying pH, ionic strength of the reaction buffer, and the EDC :HOBt: uronic acid ratio.
- the mechanics of the hydrogel network can be controlled by varying the amount of lysine available for cross-linking ( Figure 6).
- the elastic modulus of the hydrogels increases with increasing lysine content up to 35% , but then decrease with additional lysine added.
- the mechanical properties including stiffness shown by the elastic modulus, as well as strength and elasticity, can be controlled by varying the amount of lysine available for reaction.
- the swelling capabilities of the lysine crosslinked hydrogels were determined by measuring the volume of water a known mass of crosslinked alginate absorbs.
- Swelling ability of lysine crosslinked alginate hydrogels decreases with increased crosslinking.
- Lightly cross-linked alginates absorb over 2000X their mass in water, suggesting they will be useful as superabsorbent materials (e.g., in disposable diapers).
- the more highly crosslinked gels absorbed approximately 70 times their weight in water, with intermediate crosslink densities ranging between these two extremes.
- the dry disks were then rewet with diH 2 O over 48 hours and weighed to determine if the lyophilized disks were able to absorb comparable amounts of water that they initially held.
- the lyophilized disks were able to absorb similar amounts of water that they initially held +/- about 10% .
- the alginate network phase separates to make a highly porous sponge.
- the hydrated network microstructure does not seem to reform immediately upon rehydration, as much of the water soaked up by the sponges may be removed by placing the sponge on a towel within the first few hours after rehydration. However, after 48 hours of soaking very little water may be removed from the from the structure when exposed to a drying towel suggesting the hydrated network structure returns.
- Lyophilized alginate matrices exhibit shape memory properties advantageous for certain applications.
- Lyophilized alginate matrices act as hydrophilic sponges when exposed to water, hydrating almost instantly.
- 50% lysine crosslinked sponges were compressed, rolled up into tight cylinders and delivered through 3mm ID silicone tubing to mimic endoscopic delivery on the laboratory bench.
- the rolled up matrices were pushed through the tubing with flexible teflon string (1.3 mm OD), and they returned to their initial shape and dimensions within seconds upon rehydration.
- the water absorption properties of the compressed disks are similar to noncompressed disks (in ⁇ & above swelling study) and absorb around 90% of the initial water content after 24 hours.
- Example 15 The molecular weight between cross-links in the alginate network was calculated directly from the shear modulus and swelling measurements (Peppas and Merrill, 3. Appl. Polym. Sci. 21: 1763-1770, 1977) to assess the density of functional cross-links (interchain). This number can then be compared to the total number of cross-links (intra- and interchain) determined chemically. Calculation of M ⁇ . was performed utilizing the relationship:
- G is the shear modulus
- R and T are the gas constant and temperature in Kelvin respectfully
- C r is the concentration of the polymer in the cross-linking solution
- M is the molecular weight between cross-links
- M COMP is the number average molecular weight of the native polymer.
- Q is the swelling ratio defined as
- v r being the volume fraction of the polymer in the unswollen cross-linked gel and v s is the volume fraction in the swollen gel.
- G is obtained from manipulation of the stress-strain data from compression testing by plotting stress versus -( ⁇ _-_l/ ⁇ 2 ) with
- a decrease in the modulus was found with further increase in the cross-linking molecule molecular weight.
- Varying the cross-link density (utilizing the PEG with 1000 molecular weight), again had a strong effect on the stiffness of the gels. Similar to the results witJi lysine, an increase in modulus was noted up to a certain cross-linking, but then decreased with additional cross-linking.
- Figure 9 showing elastic modulus vs cross-link density [%] utilizing the PEG of 1000 molecular weight as the cross-linking molecule. This decrease in modulus is again likely attributable to an increase in network defects at higher PEG diamine concentrations, including more dangling half- reacted PEGs which detract from the mechanics .
- Example 17 Cross-linked polyaldehyde alginates (PAA).
- PPA cross-linked polyaldehyde alginates
- An additional approach to covalently cross-link alginates is to oxidize alginate and cross-link it with a bifunctional cross-linker to form hydrogels.
- alginate, 1 was derivatized by sodium periodate oxidation, as shown below, at ambient temperatures to yield the limit-oxidized product, 2.
- the reaction was monitored by the appearance of the aldehydic symmetric vibrational band (carbonyl) via FTIR.
- Limit-oxidized alginate was then cross-linked via the aldehyde groups with adipic dihydrazide to form hydrogels, 3. This process was followed by the disappearance of the symmetric vibrational band at 1735 cm 1 .
- limit-oxidized alginates were cross-linked with adipic dihydrazide at various % w/w alginates.
- the compressive modulus of the resulting gels was measured and evaluated ( Figure 10).
- Gelling was set at 3% w/w alginates with a modulus of 200 kPa, and increased with the alginate percentage to reach 900 kPa at 10% w/w alginate content.
- This cross-linking procedure provides a wide range of control (700 kPa) over the mechanical strength of alginate-based biomaterials.
- the mechanical strength of cross-linked limit-oxidized alginates also depended on the concentration of the cross-linker as well as the calcium ion content in the final gel.
- the compressive modulus increased wim the concentration of adipic dihydrazide in the gel ( Figure 11). For example, at 25 mM adipic dihydrazide, the modulus was at 200 kPa and increased to 100 kPa at 150 mM. No difference was observed at higher concentration of the cross-linker. A significant increase of 250 kPa was also observed for the compressive modulus as the calcium ion concentration increased from 10 mM to 30 mM ( Figure 12).
- the polyguluronate sequence responsible for alginate gelation was isolated, derivatized and cross-linked to form hydrogels, analogous to the scheme shown in Example 17, but for an un-crosslinked material.
- sodium polyguluronate, 1 was isolated from alginates by acid hydrolysis following a modified procedure (Haug, A. ; Larsen, B.; Smidsrod, O. Acta Chem. Scand. 1966, 20, 183-190; and Haug, A.; Larsen, B.; Smidsrod, O. Acta Chem. Scand. 1967, 21, 691-704).
- the product was characterized by FTIR, H-NMR, and 13 C-NMR and correlated well with the reported characterizations (see also Penman, A.
- the compressive modulus then increased as the PAG content in the final gel increased to reach 880 kPa at 10 % w/w PAG. This was expected since the number of aldehydic functional groups increased with the PAG content in the gel. Hence, the efficiency of the cross-linker increases and results in a larger modulus. As a result, varying the % w/w PAG in the final gel, can provide a control over the elasticity as well as the strength of the corresponding gel. The mechanical strength of gels can be increased by increasing the degree of cross-linking. Hence, 6% w/w PAG was cross-linked at different concentrations of adipic dihydrazide and the compressive modulus was evaluated.
- cross-linked polyaldehyde guluronates can also be controlled by the amount of calcium ions present in the these materials.
- PAG 6% w/w
- adipic dihydrazide 150 mM
- the compressive modulus of the resulting gels increased with increasing calcium concentrations to an optimal value of 600 kPa at 40 mM calcium chloride (See Figure 15), wherein me open block, ⁇ , is sodium chloride and the closed block, ⁇ , is calcium chloride). Above this concentration, there was no statistical differences in the compressive modulus.
- the modulus was 600 kPa at 10 % w/w PAG compared to 880 kPa for the original gel condition. This was expected since it is well known that the reactivity of hydrazide groups with aldehydes is optimal at lower pHs. Under acidic conditions, aldehydes are protonated and, hence, are more susceptible to nucleophilic attack by the hydrazide groups. At neutral to basic conditions however, slower kinetics are in effect and a longer time interval is required for the completion of the reaction. This results in a lower degree of cross-linking which directly causes a decrease in the compressive modulus.
- the degree of cross-linking in these materials can also be controlled by varying the degree of oxidation of the polyguluronate chains (see Painter, T.; Larsen, B. Carbohyd. Res. 1969, 10, 186-187; Painter, T,; Larsen, B. Acta Chem. Scand. 1970, 24, 813-833; and, Ishak, M. F.; Painter, T. Acta Chem. Scand. 1971, 25, 3875-3877). This provided another control over the number of aldehydic units on the polyguluronate strand that are available for cross-linking.
- polyguluronate was oxidized using various amounts of sodium periodate and cross-linked at 10% w/w PAG with adipic dihydrazide at the optimal concentration of 150 mM in 24 well plates. All materials gelled, starting at 20 % theoretical oxidation with a compressive modulus of 500 kPa (See Fig.17). The modulus increased with the percentage of oxidation of polyguluronate to reach a maximum value of 1000 kPa at 100% theoretical oxidation.
- VEGF vascular endothelial growth factor
- Polyaldehyde guluronate was also cross-linked with adipic dihydrazide in the absence of calcium chloride at 10% w/w PAG. A slower release of 125 I-VEGF from these materials was observed in both cases, with and with out heparin. In the presence of heparin, VEGF was initially released at a rate of 4%/day for 5 days, followed by a slower release at a rate of 0.2%/day for 14 days (See Fig. 19), wherein the open block, ⁇ , is with heparin and the closed block, ⁇ , is without heparin).
- VEGF was released at a rate of 6% /day for five days, followed by 0.8 %/day for the next 14 days.
- a cell adhesion ligand, GRGDY was coupled to sodium poly(guluronate) and PAG with the same type of EDC chemistry utilized for coupling to alginate.
- trace 125 I-GRGDY was mixed with the adhesion peptide and the mixture was dialyzed against double distilled water. The dialyzate was counted in a liquid scintillation counter to determine the amount of radioactive material present.
- EDC electroactive material
- Different chemistry can also be used to couple this ligand to PAG.
- This approach utilizes the reductive amination coupling of the amine functional groups on the terminus of the peptide and the aldehydic group abundant in the PAG material. Essentially, the amine reacted with the aldehyde to form a labile imine bond which was reduced using sodium cyanoborohydride (NaCNBH 3 ) to form a stable amine linkage. The degree of incorporation of the peptide into the PAG material was 65 % . The imine bond between the amino terminal of the peptide and the aldehyde group of PAG formed with this reaction is likely also forming when the EDC chemistry is utilized to couple me peptide.
- This new coupling chemistry can also be used to chemically bind other peptides and proteins (e.g. , growth factors) or drugs to PAG and limit-oxidized alginates.
- other peptides and proteins e.g. , growth factors
- drugs e.g., drugs to PAG and limit-oxidized alginates.
- this reaction results in the formation of a labile bond that will degrade and release the bound molecule.
- This will allow drugs to be released slowly from the matrices, and this release will be chemically controlled, diffusion controlled or controlled by both processes.
- This bond can be easily reduced using sodium cyanoborohydride to yield a very stable bond if one wishes the bound molecule to remain bound (e.g., cell adhesion ligand). Any growth factor having pendant amino groups can be coupled with this reaction.
- Pharmaceutical drugs that could potentially be used will have amino groups available from the imine bond formation according to the scheme below for example.
- growth hormones and drugs could be modified to incorporate free hydrazines, hydrazides, or semicarbazides groups that can form hydrazone or semicarbazone linkages respectively. This will allow for a different release of the drug or hormone and consequently provide another level of control over the rate of release.
- PEG hydrazide cross-linkers PAG can be crosslinked with adipic acid dihydrazide.
- Dihydrazide cross-linkers can be synthesized with various lengths starting with a polyethylene glycol core.
- Poly(ethylene glycol), PEG, with molecular weights of 200, 400, 1000, and 3400 can be reacted with succinic anhydride in the presence of N,N-dimethylamino pyridine to form poly(ethylene glycol disuccinate) anhydride
- These dihydrazides have a non-degradable core with ether linkages.
- controlling the molecular weights of the starting PEG polymers will yield PEG dihydrazides with various chain lengths.
- These dihydrazides can be used to cross-link PAG and form materials with only one degradable linkage which is the hydrazone bond. This bond can be further stabilized by borohydride reduction to yield non-degradable materials.
- PAG polymers can be crosslinked with dihydrazides to form non-degradable materials, materials with hydrazone degradable bonds, or materials with hydrazone and ester bonds both of which are degradable. This approach will provide materials with various rates of degradation.
- by selecting the appropriate length of PEG polymers used the mechanical properties of the resulting cross-linked biomaterial can be controlled.
- a photopolymerizable polyguluronate can be synthesized from hydrazido acrylate monomers coupled to G-block polyguluronate via the aldehydic terminus. These materials can then be injected into the desired site and polymerized photochemical ly to form hydrogels. Hydrazido acrylate can be synthesized starting with acryloyl chloride and t-butylcarbazate to form the protected hydrazido acrylate.
- Non-degradable Biodegradable Deprotection using trfiluoro acetic acid, TFA will afford the desired monomer.
- This methodology provides a means to deliver ' G-block into the desired site and polymerize it afterwards via photoinduced free-radical polymerization.
- Hydrazides react with aldehydes as in the case of PAG.
- the hydrazides react with the hemiacetal terminal of G-block to form an acrylic hydrazone terminal on the G-block chain. Hydrazido aery lates were chosen because of the ease of incorporating these functional groups in G-blocks.
- These monomers can be copolymerized with acrylic acid, acrylamide, MMA, HEMA, HPMA, allyl amine, dimethylallyl amine, or other monomers with similar functionality.
- the degree of G-block incorporation can be controlled by varying the percentages of co-monomer used.
- the pyrrolidine unit formed after the polymerization restricts the mobility of the polymer due to its cyclic structure and renders the backbone more rigid.
- the stiffness of the backbone is then controlled by the percentage of diallyldimethyl ammonium chloride units incorporated.
- Another approach for the synthesis of these polymers is to prepolymerize hydrazido acrylates with diallyl dimethyl ammonium chloride and allyldimethyl ammonium chloride monomers.
- a common approach to control the mechanical strength of poly acrylates and derivatives is by cross-linking these polymers with acrylate-based cross-linkers (Naghash et al. , Polymer, 1187-1196, 1997; Dietz and Peppas, Polymer, 3767- 3781, 1997).
- Ethyleneglycol dimethacrylate, hexaethyleneglycol dimethacrylate, and other bifunctional and multifunctional cross-linkers can crosslink G-block- hydrazido acrylate monomers.
- Dendritic polymers can be provided by coupling of G-block to PEG-lysine dendritic polymers . It is well known that the hemiacetal terminus of monosaccharides and polysaccharides is in equilibrium with the open aldehyde form.
- reaction of amines with aldehydes is also in an equilibrium.
- the imine bond formed between the amine and the aldehyde is reduced which shifts the equilibrium to form more aldehydes and drive the reaction to completion.
- This process is sluggish, however, and a faster and more efficient method is needed to couple G-block to different polymers.
- hydrazide functional groups can be used to achieve this coupling. Hydrazines, hydrazides, and semicarbazides react with aldehydes to form imine-like bonds. This process does not depend on the presence of borohydrides.
- Hydrazide moieties are more nucleophilic than amines and can attack electrophilic centers like carbonyl groups much faster.
- sodium cyanoborohydride could eventually be used later to induce stability on the hydrazone or semicarbazone linkage.
- Dendrimers with reactive functional groups on the terminals for G-block coupling can be synthesized using a similar method to the one used for lysine dendrimers and the end groups modified to semicarbazide terminals.
- Comb polymers Monosaccharides and oligosaccharides have been successfully incorporated into the backbone of polyacrylamides (Callstrom and Bednarski, RS
- Low molecular weight poly(vinyl alcohol), PVA, canl be modified by reaction with succinic anhydride in the presence of N,N-dimethyl aminopyridine, DMAP, to afford poly(vinylsuccinate) intermediate.
- the ester bond thus formed between PVA and the succinate group is a biodegradable bond susceptible to enzymatic cleavages in biological systems.
- Hydrazine can then be coupled to this intermediate via DCC coupling to form poly(vinylhydrazidosuccinate). Therefore, the degree of hydrazide incorporation can be controlled in the final product by controlling the number of succinic anhydride molecules used in the previous reaction. The control over the degree of hydrazide incorporation is crucial since this will dictate the degree of G-block incorporation in the next step.
- This linkage can be further stabilized by reduction with sodium cyanoborohydride.
- the degree of G-block incorporation in the final material will exhibit a direct relationship with the gelling properties as well as the strength of the hydrogel formed.
- the reactive hydrazido groups provide a means to attach G-block to the polymer backbone via the hemiacetal terminus.
- the amide bond formed between the poly (ally lamine) and the succinate group is a non-degradable bond.
- Polyaldehyde guluronate, PAG was allowed to react with hydrazine and sodium borohydride to afford the polyhydrazino guluronate derivative.
- the hydrazine groups on this alginate derived polymer are used to incorporate G-block chains via their hemiacetal termini.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Dermatology (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Transplantation (AREA)
- Zoology (AREA)
- Polymers & Plastics (AREA)
- Cell Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Vascular Medicine (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Botany (AREA)
- Developmental Biology & Embryology (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Virology (AREA)
- Genetics & Genomics (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Materials Engineering (AREA)
- Cardiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Materials For Medical Uses (AREA)
- Medicinal Preparation (AREA)
Abstract
Description



















































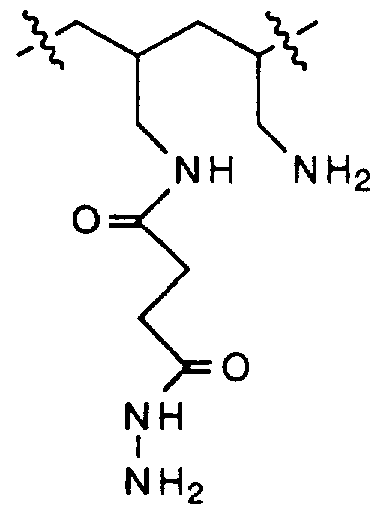



Claims
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69739085T DE69739085D1 (en) | 1996-09-19 | 1997-09-19 | POLYMERS CONTAIN POLYSACCHARIDES SUCH AS ALGINATES OR MODIFIED ALGINATES |
| AU44930/97A AU733716B2 (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginates or modified alginates |
| CA002266581A CA2266581C (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginates or modified alginates |
| JP51497398A JP4335316B2 (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginate or modified alginate |
| EP97943460A EP0927196B1 (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginates or modified alginates |
| US09/147,900 US6642363B1 (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginates or modified alginates |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2646796P | 1996-09-19 | 1996-09-19 | |
| US2636296P | 1996-09-19 | 1996-09-19 | |
| US60/026,362 | 1996-09-19 | ||
| US60/026,467 | 1996-09-19 | ||
| US4156597P | 1997-03-21 | 1997-03-21 | |
| US60/041,565 | 1997-03-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998012228A1 true WO1998012228A1 (en) | 1998-03-26 |
Family
ID=27362748
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/016890 WO1998012228A1 (en) | 1996-09-19 | 1997-09-19 | Polymers containing polysaccharides such as alginates or modified alginates |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US6642363B1 (en) |
| EP (1) | EP0927196B1 (en) |
| JP (2) | JP4335316B2 (en) |
| AT (1) | ATE413415T1 (en) |
| AU (1) | AU733716B2 (en) |
| CA (1) | CA2266581C (en) |
| DE (1) | DE69739085D1 (en) |
| ES (1) | ES2317657T3 (en) |
| WO (1) | WO1998012228A1 (en) |
Cited By (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999052560A1 (en) * | 1998-04-13 | 1999-10-21 | Massachusetts Institute Of Technology | Comb copolymers for regulating cell-surface interactions |
| WO1999058656A2 (en) * | 1998-05-13 | 1999-11-18 | The Regents Of The University Of Michigan | Sustained dna delivery from structural matrices |
| WO2000021572A2 (en) * | 1998-10-09 | 2000-04-20 | The University Of Michigan | Hydrogels and water soluble polymeric carriers for drug delivery |
| WO2000049135A2 (en) * | 1999-01-21 | 2000-08-24 | Advanced Medical Solutions Limited | Substrate for cell growth |
| EP1035136A1 (en) * | 1999-02-26 | 2000-09-13 | Seiko Epson Corporation | Process for the manufacture of polyguluronic acids |
| WO2001013967A1 (en) * | 1999-08-20 | 2001-03-01 | Andre Beaulieu | Solid wound healing formulations containing fibronectin |
| WO2001066164A1 (en) * | 1999-04-22 | 2001-09-13 | Eidgenossisch Technische Hochschule Zurich | Modified protein matrices |
| JP2002529549A (en) * | 1998-11-11 | 2002-09-10 | アクイジティオ ソチエタ ペル アツィオニ | Crosslinking method of carboxylated polysaccharide |
| JP2003502389A (en) * | 1999-06-18 | 2003-01-21 | オークエスト,インコーポレイテッド | Hyaluronic acid-sulfated polysaccharide conjugate for injection |
| US6748954B2 (en) * | 2000-10-27 | 2004-06-15 | The Regents Of The University Of Michigan | Drug release from polymer matrices through mechanical stimulation |
| US6991652B2 (en) | 2000-06-13 | 2006-01-31 | Burg Karen J L | Tissue engineering composite |
| US7008476B2 (en) * | 2003-06-11 | 2006-03-07 | Az Electronic Materials Usa Corp. | Modified alginic acid of alginic acid derivatives and thermosetting anti-reflective compositions thereof |
| WO2007011743A2 (en) | 2005-07-14 | 2007-01-25 | Lipothera, Inc. | Sustained release enhanced lipolytic formulation for regional adipose tissue treatment |
| US7241730B2 (en) | 1998-08-27 | 2007-07-10 | Universitat Zurich | Enzyme-mediated modification of fibrin for tissue engineering: fibrin formulations with peptides |
| WO2008006658A1 (en) * | 2006-07-14 | 2008-01-17 | Fmc Biopolymer As | Hydrogels containing low molecular weight alginates and biostructures made therefrom |
| WO2008048770A1 (en) | 2006-10-17 | 2008-04-24 | Lipothera, Inc. | Methods, compositions, and formulations for the treatment of thyroid eye disease |
| WO2008098109A2 (en) * | 2007-02-08 | 2008-08-14 | Northwestern University | Hydrogel compositions |
| WO2009069131A1 (en) * | 2007-11-27 | 2009-06-04 | Hadasit Medical Research Services & Development Ltd. | Alginate biomaterials for the treatment of hepatic disorders |
| WO2011073229A1 (en) * | 2009-12-17 | 2011-06-23 | Glaxo Group Limited | Compositions comprising alginates with high guluronic acid/mannuronic acid ratio for use in the treatment of dentine hypersensitivity |
| WO2011070529A3 (en) * | 2009-12-10 | 2011-11-10 | Universidade Do Minho | Dextrin hydrogel for biomedical applications |
| US8133881B2 (en) | 2003-01-13 | 2012-03-13 | Shire Llc | Carbohydrate conjugates to prevent abuse of controlled substances |
| US20120064588A1 (en) * | 2009-05-22 | 2012-03-15 | Hiroyuki Nagaoka | Protein complex having activity catalyzing asymmetric oxidation reaction and process for producing the same |
| WO2012106658A1 (en) | 2011-02-03 | 2012-08-09 | Northeastern University | Methods and compositions for highly specific capture and release of biological materials |
| WO2012149358A1 (en) * | 2011-04-28 | 2012-11-01 | President And Fellows Of Harvard College | Injectable preformed macroscopic 3-dimensional scaffolds for minimally invasive administration |
| US8728456B2 (en) | 2009-07-31 | 2014-05-20 | President And Fellows Of Harvard College | Programming of cells for tolerogenic therapies |
| EP2796101A1 (en) | 2013-04-23 | 2014-10-29 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a cross-linked gel for encapsulating kidney stones and/or kidney stone fragments |
| EP2796100A1 (en) | 2013-04-23 | 2014-10-29 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gelling system for the removal of kidney stone fragments |
| KR101465994B1 (en) * | 2005-05-27 | 2014-11-27 | 로이어 바이오메디칼 인코포레이티드 | Bioresorbable polymer matrices and methods of making and using the same |
| US8932583B2 (en) | 2005-12-13 | 2015-01-13 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US8993538B2 (en) | 2003-05-05 | 2015-03-31 | Ben Gurion University Of The Negev Research And Development Authority | Injectable cross-linked polymeric preparations and uses thereof |
| US9012399B2 (en) | 2008-05-30 | 2015-04-21 | President And Fellows Of Harvard College | Controlled release of growth factors and signaling molecules for promoting angiogenesis |
| DE202013012275U1 (en) | 2013-04-23 | 2015-12-18 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a cross-linked gel for enclosing kidney stones and / or kidney stone fragments |
| DE202013012287U1 (en) | 2013-04-23 | 2016-01-18 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gel-forming system for removing kidney stone fragments |
| US9297005B2 (en) | 2009-04-13 | 2016-03-29 | President And Fellows Of Harvard College | Harnessing cell dynamics to engineer materials |
| US9351945B1 (en) | 2015-02-27 | 2016-05-31 | John Daniel Dobak, III | Reduction of adipose tissue |
| US9370558B2 (en) | 2008-02-13 | 2016-06-21 | President And Fellows Of Harvard College | Controlled delivery of TLR agonists in structural polymeric devices |
| US9452132B2 (en) | 2009-05-27 | 2016-09-27 | Neothetics, Inc. | Methods for administration and formulations for the treatment of regional adipose tissue |
| US9486512B2 (en) | 2011-06-03 | 2016-11-08 | President And Fellows Of Harvard College | In situ antigen-generating cancer vaccine |
| US9597531B2 (en) | 2010-11-24 | 2017-03-21 | Neothetics, Inc. | Selective, lipophilic, and long-acting beta agonist monotherapeutic formulations and methods for the cosmetic treatment of adiposity and contour bulging |
| US9603894B2 (en) | 2010-11-08 | 2017-03-28 | President And Fellows Of Harvard College | Materials presenting notch signaling molecules to control cell behavior |
| US9610328B2 (en) | 2010-03-05 | 2017-04-04 | President And Fellows Of Harvard College | Enhancement of skeletal muscle stem cell engraftment by dual delivery of VEGF and IGF-1 |
| US9675561B2 (en) | 2011-04-28 | 2017-06-13 | President And Fellows Of Harvard College | Injectable cryogel vaccine devices and methods of use thereof |
| US9687455B2 (en) | 2014-08-14 | 2017-06-27 | John Daniel Dobak | Sodium tetradecyl sulfate formulations for treatment of adipose tissue |
| US9693954B2 (en) | 2010-06-25 | 2017-07-04 | President And Fellows Of Harvard College | Co-delivery of stimulatory and inhibitory factors to create temporally stable and spatially restricted zones |
| EP3101064A4 (en) * | 2014-01-31 | 2017-09-13 | Seikagaku Corporation | Diamine crosslinking agent, acidic polysaccharide crosslinked body, and medical material |
| US9770535B2 (en) | 2007-06-21 | 2017-09-26 | President And Fellows Of Harvard College | Scaffolds for cell collection or elimination |
| US9821045B2 (en) | 2008-02-13 | 2017-11-21 | President And Fellows Of Harvard College | Controlled delivery of TLR3 agonists in structural polymeric devices |
| EP3290047A1 (en) | 2013-04-05 | 2018-03-07 | Claudia Zylberberg | Matrix metalloproteinases and uses thereof |
| US9937249B2 (en) | 2012-04-16 | 2018-04-10 | President And Fellows Of Harvard College | Mesoporous silica compositions for modulating immune responses |
| US9993497B2 (en) | 2012-12-30 | 2018-06-12 | Hadasit Medical Research Services And Development Ltd. | Use of alginate compositions in preventing or reducing liver damage caused by a hepatotoxic agent |
| US10022486B2 (en) | 2011-06-24 | 2018-07-17 | Gearbox, Llc | Device, system, and method including micro-patterned cell treatment array |
| CN110201234A (en) * | 2019-06-26 | 2019-09-06 | 山东百多安医疗器械有限公司 | A kind of used in tissue engineering prostatic cell micro-capsule and preparation method thereof |
| CN110401637A (en) * | 2019-06-28 | 2019-11-01 | 中南民族大学 | Trust method based on name in a kind of name data network |
| US10647959B2 (en) | 2011-04-27 | 2020-05-12 | President And Fellows Of Harvard College | Cell-friendly inverse opal hydrogels for cell encapsulation, drug and protein delivery, and functional nanoparticle encapsulation |
| US10682400B2 (en) | 2014-04-30 | 2020-06-16 | President And Fellows Of Harvard College | Combination vaccine devices and methods of killing cancer cells |
| US10739338B2 (en) | 2014-03-24 | 2020-08-11 | Qt Holdings Corp | Shaped articles including hydrogels and methods of manufacture and use thereof |
| WO2021158105A1 (en) | 2020-02-03 | 2021-08-12 | Mosa Meat B.V. | Hydrogels for cultured meat production |
| US11150242B2 (en) | 2015-04-10 | 2021-10-19 | President And Fellows Of Harvard College | Immune cell trapping devices and methods for making and using the same |
| CN113571628A (en) * | 2020-04-29 | 2021-10-29 | 香港大学 | Stretchable ionic hydrogels with high thermal potential for low grade heat collection |
| US11202759B2 (en) | 2010-10-06 | 2021-12-21 | President And Fellows Of Harvard College | Injectable, pore-forming hydrogels for materials-based cell therapies |
| US20210393850A1 (en) * | 2018-11-07 | 2021-12-23 | Universiteit Maastricht | Bioinks |
| WO2022047354A1 (en) | 2020-08-31 | 2022-03-03 | The Board Of Trustees Of The University Of Illinois | Alginate-coated mesenchymal stromal and progenitor cells and methods for using the same |
| US11555177B2 (en) | 2016-07-13 | 2023-01-17 | President And Fellows Of Harvard College | Antigen-presenting cell-mimetic scaffolds and methods for making and using the same |
| US11752238B2 (en) | 2016-02-06 | 2023-09-12 | President And Fellows Of Harvard College | Recapitulating the hematopoietic niche to reconstitute immunity |
| US11786457B2 (en) | 2015-01-30 | 2023-10-17 | President And Fellows Of Harvard College | Peritumoral and intratumoral materials for cancer therapy |
| US12029829B2 (en) | 2016-03-22 | 2024-07-09 | President And Fellows Of Harvard College | Biocompatible adhesives and methods of use thereof |
| US12097306B2 (en) | 2016-12-13 | 2024-09-24 | Takeda Pharmaceutical Company Limited | Conformal coating of biological surfaces |
Families Citing this family (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7427602B1 (en) * | 1998-05-13 | 2008-09-23 | The Regents Of The University Of Michigan | Sustained DNA delivery from structural matrices |
| JP2003527822A (en) | 1998-08-27 | 2003-09-24 | マサチューセッツ インスティテュート オブ テクノロジー | Rationally designed heparinases from heparinases I and II |
| WO2000065521A2 (en) | 1999-04-23 | 2000-11-02 | Massachusetts Institute Of Technology | System and method for polymer notation |
| ES2230318T3 (en) * | 2000-05-31 | 2005-05-01 | Mnemoscience Gmbh | THERMOPLASTIC MATERIALS WITH MEMORY FORM AND POLYMER NETWORKS FOR FABRIC ENGINEERING. |
| WO2005046447A2 (en) * | 2003-11-10 | 2005-05-26 | Symphony Medical, Inc. | Method to control ventricular rate in atrial fibrillation patients |
| EP1552818A1 (en) * | 2004-01-07 | 2005-07-13 | Shin-Etsu Chemical Co., Ltd. | Release sustaining composition and sustained release preparation |
| US20050220882A1 (en) * | 2004-03-04 | 2005-10-06 | Wilson Pritchard | Materials for medical implants and occlusive devices |
| DE102004019241A1 (en) * | 2004-04-16 | 2005-11-03 | Cellmed Ag | Injectable cross-linked and uncrosslinked alginates and their use in medicine and aesthetic surgery |
| WO2006031388A2 (en) * | 2004-08-20 | 2006-03-23 | Hyperbranch Medical Technology, Inc. | Dentritic polymers, crosslinked gels, and their uses in orthopedic applications |
| CA2583373C (en) * | 2004-10-12 | 2013-09-03 | Fmc Biopolymer As | Self-gelling alginate systems and uses thereof |
| DE102004055729A1 (en) * | 2004-11-18 | 2006-05-24 | Cellmed Ag | Production of two or more layered microcapsules |
| US9427496B2 (en) | 2005-02-18 | 2016-08-30 | Drexel University | Method for creating an internal transport system within tissue scaffolds using computer-aided tissue engineering |
| EP2377884A1 (en) | 2005-05-10 | 2011-10-19 | Neoloch Aps | Neuritogenic peptides |
| US20070020755A1 (en) * | 2005-07-07 | 2007-01-25 | Teresa Woodruff | Stage specific follicle maturation systems |
| KR100718329B1 (en) * | 2005-09-08 | 2007-05-14 | 광주과학기술원 | Polysaccharide-functionalized nanoparticle, drug delivery system for controlled release, and method of preparing the same |
| US20070248675A1 (en) * | 2005-09-08 | 2007-10-25 | Gwangju Institute Of Science And Technology | Composite comprising polysaccharide-functionalized nanoparticle and hydrogel matrix, a drug delivery system and a bone defect replacement matrix for sustained release comprising the same, and the preparation method thereof |
| ATE499111T1 (en) * | 2005-12-16 | 2011-03-15 | Oxthera Inc | COMPOSITIONS AND METHODS FOR OXALATE REDUCTION |
| AU2007224137A1 (en) * | 2006-03-01 | 2007-09-13 | Fmc Biopolymer As | Biodegradable foam |
| WO2007146319A2 (en) * | 2006-06-13 | 2007-12-21 | Symphony Medical, Inc. | Methods and apparatus for using polymer-based beads and hydrogels for cardiac applications |
| EP2029727B1 (en) * | 2006-06-16 | 2012-04-11 | FMC Biopolymer AS | Alginate coated, collagen matrix cellular device, preparative methods, and uses thereof. |
| EP2503332A3 (en) * | 2006-08-09 | 2013-11-13 | Sumitomo Bakelite Company, Ltd. | Sugar chain-capturing substance and use thereof |
| WO2008098220A1 (en) * | 2007-02-08 | 2008-08-14 | University Of Washington | Biocompatile amino acid anhydride polymers |
| US20090010983A1 (en) * | 2007-06-13 | 2009-01-08 | Fmc Corporation | Alginate Coated, Polysaccharide Gel-Containing Foam Composite, Preparative Methods, and Uses Thereof |
| EP2167144A4 (en) | 2007-06-13 | 2012-11-21 | Fmc Corp | Biopolymer based implantable degradable devices |
| KR20100044173A (en) * | 2007-06-13 | 2010-04-29 | 에프엠씨 코포레이션 | Peptide linked cell matrix materials for stem cells and methods of using the same |
| ES2322637B1 (en) * | 2007-06-26 | 2010-03-05 | Universidad Del Pais Vasco | ALGINATE MICROPARTICLES MODIFIED WITH RGD AS A DRUG RELEASE SYSTEM. |
| CN103463638A (en) * | 2007-08-28 | 2013-12-25 | Fmc有限公司 | Delayed self-gelling alginate systems and uses thereof |
| EP2210917A1 (en) * | 2007-11-01 | 2010-07-28 | Osaka City University | -1,3-glucan-derived polyaldehyde/polyamine hydrogel |
| EP2082755A1 (en) * | 2008-01-16 | 2009-07-29 | CellMed AG | Monolithic alginate implants networked in situ |
| US8668863B2 (en) | 2008-02-26 | 2014-03-11 | Board Of Regents, The University Of Texas System | Dendritic macroporous hydrogels prepared by crystal templating |
| CA2722425A1 (en) * | 2008-04-28 | 2009-11-05 | Surmodics, Inc. | Poly-.alpha.(1-4)glucopyranose-based matrices with hydrazide crosslinking |
| US8252317B2 (en) * | 2009-02-26 | 2012-08-28 | General Electric Company | Metal alginate hydrogel and articles thereof |
| CA2780294C (en) | 2009-11-09 | 2018-01-16 | Spotlight Technology Partners Llc | Polysaccharide based hydrogels |
| CA2780274C (en) | 2009-11-09 | 2018-06-26 | Spotlight Technology Partners Llc | Fragmented hydrogels |
| US8197849B2 (en) * | 2010-02-12 | 2012-06-12 | National Health Research Institutes | Cross-linked oxidated hyaluronic acid for use as a vitreous substitute |
| TWI398275B (en) * | 2010-07-12 | 2013-06-11 | Agricultural Res Inst | Skin wound dressing and manufacturing method thereof |
| US20120064097A1 (en) * | 2010-07-20 | 2012-03-15 | Washburn Newell R | Enhanced Binding of Pro-Inflammatory Cytokines by Polysaccharide-Antibody Conjugates |
| WO2012048283A1 (en) | 2010-10-08 | 2012-04-12 | Board Of Regents, The University Of Texas System | One-step processing of hydrogels for mechanically robust and chemically desired features |
| US9095558B2 (en) | 2010-10-08 | 2015-08-04 | Board Of Regents, The University Of Texas System | Anti-adhesive barrier membrane using alginate and hyaluronic acid for biomedical applications |
| WO2013025763A2 (en) * | 2011-08-15 | 2013-02-21 | President And Fellows Of Harvard College | Tissue engineering using injectable, oxidized alginate hydrogels |
| WO2013106852A1 (en) | 2012-01-13 | 2013-07-18 | President And Fellows Of Harvard College | Controlled delivery of tlr agonists in structural polymeric devices |
| US9480747B2 (en) | 2012-01-24 | 2016-11-01 | Bvw Holding Ag | Class of anti-adhesion hydrogels with healing aspects |
| US11565027B2 (en) | 2012-12-11 | 2023-01-31 | Board Of Regents, The University Of Texas System | Hydrogel membrane for adhesion prevention |
| WO2015076843A1 (en) * | 2013-11-25 | 2015-05-28 | Halliburton Energy Services, Inc. | Viscosity enhancer |
| JP6787789B2 (en) | 2014-04-04 | 2020-11-18 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Refillable drug delivery device and how to use it |
| US11065362B2 (en) | 2014-06-12 | 2021-07-20 | President And Fellows Of Harvard College | Viscoelastic hydrogels with fast stress relaxation |
| US10376592B2 (en) | 2015-06-29 | 2019-08-13 | University of Pittsburgh—of the Commonwealth System of Higher Education | Stapled acid-sensitive endosome disrupting alginates |
| US9790467B2 (en) | 2015-09-22 | 2017-10-17 | Qt Holdings Corp | Methods and compositions for activation or expansion of T lymphocytes |
| JP7025021B2 (en) | 2015-10-26 | 2022-02-24 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Reduced and oxidized polysaccharides and methods of their use |
| WO2017165389A2 (en) * | 2016-03-24 | 2017-09-28 | Millennium Pharmaceuticals, Inc. | Alginate hydrogel compositions |
| US10668190B2 (en) | 2016-05-03 | 2020-06-02 | Bvw Holding Ag | Multiphase gel |
| US11591564B2 (en) * | 2016-12-16 | 2023-02-28 | University of Pittsburgh—of the Commonwealth System of Higher Education | Peptide conjugated hydrogel substrate for the maintenance and expansion of human pluripotent stem cells |
| US11634512B2 (en) | 2017-01-27 | 2023-04-25 | Cornell University | Zwitterionically modified polymers and hydrogels |
| WO2018165327A1 (en) | 2017-03-08 | 2018-09-13 | Alafair Biosciences, Inc. | Hydrogel medium for the storage and preservation of tissue |
| KR102123508B1 (en) * | 2018-10-01 | 2020-06-16 | 한국원자력연구원 | The method for manufacturing of electroconductive micro-needle patch and electroconductive micro-needle patch manufactured thereby |
| WO2020123470A1 (en) * | 2018-12-10 | 2020-06-18 | The Regents Of The University Of California | Salivary gland regeneration |
| DE102020116108A1 (en) | 2020-06-18 | 2021-12-23 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung eingetragener Verein | Process for the production of carrier particles for the cultivation of biological cells, carrier particles and their application |
| US20230181644A1 (en) | 2021-10-28 | 2023-06-15 | Lyell Immunopharma, Inc. | Methods of generating cells |
| WO2024077174A1 (en) | 2022-10-05 | 2024-04-11 | Lyell Immunopharma, Inc. | Methods for culturing nr4a-deficient cells |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5110605A (en) * | 1990-08-21 | 1992-05-05 | Oramed, Inc. | Calcium polycarbophil-alginate controlled release composition and method |
| US5429821A (en) * | 1992-05-29 | 1995-07-04 | The Regents Of The University Of California | Non-fibrogenic high mannuronate alginate coated transplants, processes for their manufacture, and methods for their use |
| US5622718A (en) * | 1992-09-25 | 1997-04-22 | Keele University | Alginate-bioactive agent conjugates |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1550819A (en) * | 1975-06-04 | 1979-08-22 | Nat Res Dev | Polymeric support for biogically active materials |
| DE3370026D1 (en) | 1983-06-15 | 1987-04-09 | Gist Brocades Nv | Biologically active conjugates and their preparation and use |
| DE3886051T2 (en) * | 1987-09-28 | 1994-03-24 | Jolla Cancer Res Found | SELECTION OF CELLS WITH IMPROVED CELL ADHESION PROPERTIES. |
| IT1219587B (en) * | 1988-05-13 | 1990-05-18 | Fidia Farmaceutici | SELF-CROSS-LINKED CARBOXYLY POLYSACCHARIDES |
| NO171069C (en) | 1990-05-29 | 1993-01-20 | Protan Biopolymer As | COVALENT CIRCUIT, STRONGLY SWELLING ALKALIMETAL AND AMMONIUM ALGINATE GELS, AND PROCEDURES FOR PREPARING THEREOF |
| US5227298A (en) | 1990-08-17 | 1993-07-13 | The Trustees Of Columbia University In The City Of New York | Method for microencapuslation of cells or tissue |
| US5605938A (en) * | 1991-05-31 | 1997-02-25 | Gliatech, Inc. | Methods and compositions for inhibition of cell invasion and fibrosis using dextran sulfate |
| JPH0581A (en) * | 1991-06-21 | 1993-01-08 | Koken Co Ltd | Carrier for cell culture and culture |
| JPH07503943A (en) * | 1991-10-29 | 1995-04-27 | クローバー コンソリデイテッド,リミテッド | Cross-linked polysaccharides, polycations and lipids useful for encapsulation and drug release |
| US6197346B1 (en) * | 1992-04-24 | 2001-03-06 | Brown Universtiy Research Foundation | Bioadhesive microspheres and their use as drug delivery and imaging systems |
| ATE162397T1 (en) | 1993-02-12 | 1998-02-15 | Mayo Foundation | COMPOSITION OF MICROPARTICLES IN COMPRESSED PHASE AND METHOD |
| IT1268955B1 (en) * | 1994-03-11 | 1997-03-18 | Fidia Advanced Biopolymers Srl | ACTIVE ESTERS OF CARBOXYL POLYSACCHARIDES |
| JP3107726B2 (en) * | 1994-05-13 | 2000-11-13 | 株式会社クラレ | Water-swellable polymer gel |
| DE69530553T2 (en) | 1994-05-13 | 2004-03-25 | KURARAY CO., LTD, Kurashiki | MEDICAL POLYMER GEL |
| US5863551A (en) * | 1996-10-16 | 1999-01-26 | Organogel Canada Ltee | Implantable polymer hydrogel for therapeutic uses |
-
1997
- 1997-09-19 US US09/147,900 patent/US6642363B1/en not_active Expired - Lifetime
- 1997-09-19 CA CA002266581A patent/CA2266581C/en not_active Expired - Fee Related
- 1997-09-19 AU AU44930/97A patent/AU733716B2/en not_active Ceased
- 1997-09-19 ES ES97943460T patent/ES2317657T3/en not_active Expired - Lifetime
- 1997-09-19 AT AT97943460T patent/ATE413415T1/en not_active IP Right Cessation
- 1997-09-19 EP EP97943460A patent/EP0927196B1/en not_active Expired - Lifetime
- 1997-09-19 DE DE69739085T patent/DE69739085D1/en not_active Expired - Lifetime
- 1997-09-19 WO PCT/US1997/016890 patent/WO1998012228A1/en active Application Filing
- 1997-09-19 JP JP51497398A patent/JP4335316B2/en not_active Expired - Fee Related
-
2009
- 2009-03-11 JP JP2009057966A patent/JP5111416B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5110605A (en) * | 1990-08-21 | 1992-05-05 | Oramed, Inc. | Calcium polycarbophil-alginate controlled release composition and method |
| US5429821A (en) * | 1992-05-29 | 1995-07-04 | The Regents Of The University Of California | Non-fibrogenic high mannuronate alginate coated transplants, processes for their manufacture, and methods for their use |
| US5622718A (en) * | 1992-09-25 | 1997-04-22 | Keele University | Alginate-bioactive agent conjugates |
Cited By (128)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999052560A1 (en) * | 1998-04-13 | 1999-10-21 | Massachusetts Institute Of Technology | Comb copolymers for regulating cell-surface interactions |
| US6150459A (en) * | 1998-04-13 | 2000-11-21 | Massachusetts Institute Of Technology | Comb polymers for regulating cell surface interactions |
| US6399700B2 (en) | 1998-04-13 | 2002-06-04 | Massachusetts Institute Of Technology | Comb copolymers for regulating cell-surface interactions |
| US6207749B1 (en) | 1998-04-13 | 2001-03-27 | Massachusetts Institute Of Technology | Comb copolymers for regulating cell-surface interactions |
| WO1999058656A2 (en) * | 1998-05-13 | 1999-11-18 | The Regents Of The University Of Michigan | Sustained dna delivery from structural matrices |
| WO1999058656A3 (en) * | 1998-05-13 | 2000-01-06 | Univ Michigan | Sustained dna delivery from structural matrices |
| US7241730B2 (en) | 1998-08-27 | 2007-07-10 | Universitat Zurich | Enzyme-mediated modification of fibrin for tissue engineering: fibrin formulations with peptides |
| WO2000021572A2 (en) * | 1998-10-09 | 2000-04-20 | The University Of Michigan | Hydrogels and water soluble polymeric carriers for drug delivery |
| EP1374908A3 (en) * | 1998-10-09 | 2004-03-24 | The Regents Of The University Of Michigan | Polymer-drug conjugates comprising hydrazide linkers |
| EP1374908A2 (en) * | 1998-10-09 | 2004-01-02 | The Regents Of The University Of Michigan | Polymer-drug conjugates comprising hydrazide linkers |
| WO2000021572A3 (en) * | 1998-10-09 | 2000-11-30 | Univ Michigan | Hydrogels and water soluble polymeric carriers for drug delivery |
| US7186413B2 (en) | 1998-10-09 | 2007-03-06 | The Regents Of The University Of Michigan | Hydrogels and water soluble polymeric carriers for drug delivery |
| JP2002529549A (en) * | 1998-11-11 | 2002-09-10 | アクイジティオ ソチエタ ペル アツィオニ | Crosslinking method of carboxylated polysaccharide |
| JP2002536974A (en) * | 1999-01-21 | 2002-11-05 | アドバンスト・メディカル・ソリューションズ・リミテッド | Cell proliferation |
| WO2000049135A3 (en) * | 1999-01-21 | 2000-12-14 | Adv Med Solutions Ltd | Substrate for cell growth |
| WO2000049135A2 (en) * | 1999-01-21 | 2000-08-24 | Advanced Medical Solutions Limited | Substrate for cell growth |
| US6586589B1 (en) | 1999-02-26 | 2003-07-01 | Seiko Epson Corporation | Process for treating alginic acid to produce polyguluronic acids or concentrated solutions for use in producing polyguluronic acids |
| EP1035136A1 (en) * | 1999-02-26 | 2000-09-13 | Seiko Epson Corporation | Process for the manufacture of polyguluronic acids |
| WO2001066164A1 (en) * | 1999-04-22 | 2001-09-13 | Eidgenossisch Technische Hochschule Zurich | Modified protein matrices |
| JP2003502389A (en) * | 1999-06-18 | 2003-01-21 | オークエスト,インコーポレイテッド | Hyaluronic acid-sulfated polysaccharide conjugate for injection |
| WO2001013967A1 (en) * | 1999-08-20 | 2001-03-01 | Andre Beaulieu | Solid wound healing formulations containing fibronectin |
| GB2368523B (en) * | 1999-08-20 | 2004-05-12 | Andre Beaulieu | Solid wound healing formulations containing fibronectin |
| GB2368523A (en) * | 1999-08-20 | 2002-05-08 | Andre Beaulieu | Solid wound healing formulations containing fibronectin |
| US6991652B2 (en) | 2000-06-13 | 2006-01-31 | Burg Karen J L | Tissue engineering composite |
| US6748954B2 (en) * | 2000-10-27 | 2004-06-15 | The Regents Of The University Of Michigan | Drug release from polymer matrices through mechanical stimulation |
| US8133881B2 (en) | 2003-01-13 | 2012-03-13 | Shire Llc | Carbohydrate conjugates to prevent abuse of controlled substances |
| US8993538B2 (en) | 2003-05-05 | 2015-03-31 | Ben Gurion University Of The Negev Research And Development Authority | Injectable cross-linked polymeric preparations and uses thereof |
| US9006213B2 (en) | 2003-05-05 | 2015-04-14 | Ben Gurion University Of The Negev Research And Development Authority | Injectable cross-linked polymeric preparations and uses thereof |
| US7008476B2 (en) * | 2003-06-11 | 2006-03-07 | Az Electronic Materials Usa Corp. | Modified alginic acid of alginic acid derivatives and thermosetting anti-reflective compositions thereof |
| KR101465994B1 (en) * | 2005-05-27 | 2014-11-27 | 로이어 바이오메디칼 인코포레이티드 | Bioresorbable polymer matrices and methods of making and using the same |
| KR101536897B1 (en) * | 2005-05-27 | 2015-07-15 | 로이어 바이오메디칼 인코포레이티드 | Bioresorbable polymer matrices and methods of making and using the same |
| WO2007011743A2 (en) | 2005-07-14 | 2007-01-25 | Lipothera, Inc. | Sustained release enhanced lipolytic formulation for regional adipose tissue treatment |
| US9452147B2 (en) | 2005-07-14 | 2016-09-27 | Neothetics, Inc. | Lipolytic methods |
| US9707192B2 (en) | 2005-07-14 | 2017-07-18 | Neothetics, Inc. | Lipolytic methods |
| EP2397034A1 (en) | 2005-07-14 | 2011-12-21 | Lithera, Inc. | Sustained Release Enhanced Lipolytic Formulation for Regional Adipose Tissue Treatment |
| US9370498B2 (en) | 2005-07-14 | 2016-06-21 | Neothetics, Inc. | Methods of using lipolytic formulations for regional adipose tissue treatment |
| US11096997B2 (en) | 2005-12-13 | 2021-08-24 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US10149897B2 (en) | 2005-12-13 | 2018-12-11 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US9446107B2 (en) | 2005-12-13 | 2016-09-20 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US9132210B2 (en) | 2005-12-13 | 2015-09-15 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US8932583B2 (en) | 2005-12-13 | 2015-01-13 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| US10137184B2 (en) | 2005-12-13 | 2018-11-27 | President And Fellows Of Harvard College | Scaffolds for cell transplantation |
| WO2008006658A1 (en) * | 2006-07-14 | 2008-01-17 | Fmc Biopolymer As | Hydrogels containing low molecular weight alginates and biostructures made therefrom |
| WO2008048770A1 (en) | 2006-10-17 | 2008-04-24 | Lipothera, Inc. | Methods, compositions, and formulations for the treatment of thyroid eye disease |
| WO2008098109A3 (en) * | 2007-02-08 | 2008-10-30 | Univ Northwestern | Hydrogel compositions |
| WO2008098109A2 (en) * | 2007-02-08 | 2008-08-14 | Northwestern University | Hydrogel compositions |
| US10695468B2 (en) | 2007-06-21 | 2020-06-30 | President And Fellows Of Harvard College | Scaffolds for cell collection or elimination |
| US9770535B2 (en) | 2007-06-21 | 2017-09-26 | President And Fellows Of Harvard College | Scaffolds for cell collection or elimination |
| US9387222B2 (en) | 2007-11-27 | 2016-07-12 | Hadasit Medical Research Services And Development Ltd. | Alginate biomaterials for the treatment of hepatic disorders |
| WO2009069131A1 (en) * | 2007-11-27 | 2009-06-04 | Hadasit Medical Research Services & Development Ltd. | Alginate biomaterials for the treatment of hepatic disorders |
| US10328098B2 (en) | 2007-11-27 | 2019-06-25 | Hadasit Medical Research Services And Development Ltd. | Alginate biomaterials for the treatment of hepatic disorders |
| US10328133B2 (en) | 2008-02-13 | 2019-06-25 | President And Fellows Of Harvard College | Continuous cell programming devices |
| US10568949B2 (en) | 2008-02-13 | 2020-02-25 | President And Fellows Of Harvard College | Method of eliciting an anti-tumor immune response with controlled delivery of TLR agonists in porous polymerlc devices |
| US9821045B2 (en) | 2008-02-13 | 2017-11-21 | President And Fellows Of Harvard College | Controlled delivery of TLR3 agonists in structural polymeric devices |
| US9370558B2 (en) | 2008-02-13 | 2016-06-21 | President And Fellows Of Harvard College | Controlled delivery of TLR agonists in structural polymeric devices |
| US10258677B2 (en) | 2008-02-13 | 2019-04-16 | President And Fellows Of Harvard College | Continuous cell programming devices |
| US9539309B2 (en) | 2008-05-30 | 2017-01-10 | President And Fellows Of Harvard College | Controlled release of growth factors and signaling molecules for promoting angiogenesis |
| US9012399B2 (en) | 2008-05-30 | 2015-04-21 | President And Fellows Of Harvard College | Controlled release of growth factors and signaling molecules for promoting angiogenesis |
| US9297005B2 (en) | 2009-04-13 | 2016-03-29 | President And Fellows Of Harvard College | Harnessing cell dynamics to engineer materials |
| US9631205B2 (en) | 2009-05-22 | 2017-04-25 | Sanyo Foods Co., Ltd. | Protein complex having activity catalyzing asymmetric oxidation reaction and process for producing the same |
| US9556457B2 (en) | 2009-05-22 | 2017-01-31 | Sanyo Foods Co., Ltd. | Protein complex having activity catalyzing asymmetric oxidation reaction and process for producing the same |
| US8852904B2 (en) * | 2009-05-22 | 2014-10-07 | Sanyo Foods Co., Ltd. | Protein complex having activity catalyzing asymmetric oxidation reaction and process for producing the same |
| US20120064588A1 (en) * | 2009-05-22 | 2012-03-15 | Hiroyuki Nagaoka | Protein complex having activity catalyzing asymmetric oxidation reaction and process for producing the same |
| US9452132B2 (en) | 2009-05-27 | 2016-09-27 | Neothetics, Inc. | Methods for administration and formulations for the treatment of regional adipose tissue |
| US8728456B2 (en) | 2009-07-31 | 2014-05-20 | President And Fellows Of Harvard College | Programming of cells for tolerogenic therapies |
| US9381235B2 (en) | 2009-07-31 | 2016-07-05 | President And Fellows Of Harvard College | Programming of cells for tolerogenic therapies |
| US10080789B2 (en) | 2009-07-31 | 2018-09-25 | President And Fellows Of Harvard College | Programming of cells for tolerogenic therapies |
| WO2011070529A3 (en) * | 2009-12-10 | 2011-11-10 | Universidade Do Minho | Dextrin hydrogel for biomedical applications |
| US9205103B2 (en) | 2009-12-10 | 2015-12-08 | Universidade Do Minho | Dextrin hydrogel for biomedical applications |
| US9084903B2 (en) | 2009-12-17 | 2015-07-21 | Glaxo Group Limited | Compositions comprising alginates with high guluronic acid/mannuronic acid ratio for use in the treatment of dentine hypersensitivity |
| WO2011073229A1 (en) * | 2009-12-17 | 2011-06-23 | Glaxo Group Limited | Compositions comprising alginates with high guluronic acid/mannuronic acid ratio for use in the treatment of dentine hypersensitivity |
| US9610328B2 (en) | 2010-03-05 | 2017-04-04 | President And Fellows Of Harvard College | Enhancement of skeletal muscle stem cell engraftment by dual delivery of VEGF and IGF-1 |
| US9693954B2 (en) | 2010-06-25 | 2017-07-04 | President And Fellows Of Harvard College | Co-delivery of stimulatory and inhibitory factors to create temporally stable and spatially restricted zones |
| US11202759B2 (en) | 2010-10-06 | 2021-12-21 | President And Fellows Of Harvard College | Injectable, pore-forming hydrogels for materials-based cell therapies |
| US9603894B2 (en) | 2010-11-08 | 2017-03-28 | President And Fellows Of Harvard College | Materials presenting notch signaling molecules to control cell behavior |
| US9597531B2 (en) | 2010-11-24 | 2017-03-21 | Neothetics, Inc. | Selective, lipophilic, and long-acting beta agonist monotherapeutic formulations and methods for the cosmetic treatment of adiposity and contour bulging |
| KR20140016896A (en) * | 2011-02-03 | 2014-02-10 | 노스이스턴 유니버시티 | Methods and compositions for highly specific capture and release of biological materials |
| KR101922741B1 (en) | 2011-02-03 | 2019-02-20 | 노스이스턴 유니버시티 | Methods and compositions for highly specific capture and release of biological materials |
| US9927334B2 (en) | 2011-02-03 | 2018-03-27 | Northeastern University | Methods, compositions and devices employing alginic acid hydrogels for highly specific capture and release of biological materials |
| WO2012106658A1 (en) | 2011-02-03 | 2012-08-09 | Northeastern University | Methods and compositions for highly specific capture and release of biological materials |
| EP2670856A1 (en) * | 2011-02-03 | 2013-12-11 | Northeastern University | Methods and compositions for highly specific capture and release of biological materials |
| EP2670856A4 (en) * | 2011-02-03 | 2014-12-03 | Univ Northeastern | Methods and compositions for highly specific capture and release of biological materials |
| US10647959B2 (en) | 2011-04-27 | 2020-05-12 | President And Fellows Of Harvard College | Cell-friendly inverse opal hydrogels for cell encapsulation, drug and protein delivery, and functional nanoparticle encapsulation |
| US9675561B2 (en) | 2011-04-28 | 2017-06-13 | President And Fellows Of Harvard College | Injectable cryogel vaccine devices and methods of use thereof |
| US10045947B2 (en) | 2011-04-28 | 2018-08-14 | President And Fellows Of Harvard College | Injectable preformed macroscopic 3-dimensional scaffolds for minimally invasive administration |
| WO2012149358A1 (en) * | 2011-04-28 | 2012-11-01 | President And Fellows Of Harvard College | Injectable preformed macroscopic 3-dimensional scaffolds for minimally invasive administration |
| AU2019201669B2 (en) * | 2011-04-28 | 2021-04-01 | President And Fellows Of Harvard College | Injectable preformed macroscopic 3-dimensional scaffolds for minimally invasive administration |
| AU2017204078B2 (en) * | 2011-04-28 | 2019-04-04 | President And Fellows Of Harvard College | Injectable preformed macroscopic 3-dimensional scaffolds for minimally invasive administration |
| US9486512B2 (en) | 2011-06-03 | 2016-11-08 | President And Fellows Of Harvard College | In situ antigen-generating cancer vaccine |
| US10406216B2 (en) | 2011-06-03 | 2019-09-10 | President And Fellows Of Harvard College | In situ antigen-generating cancer vaccine |
| US10610635B2 (en) | 2011-06-24 | 2020-04-07 | Gearbox Llc | Device, system, and method including micro-patterned cell treatment array |
| US10022486B2 (en) | 2011-06-24 | 2018-07-17 | Gearbox, Llc | Device, system, and method including micro-patterned cell treatment array |
| US9937249B2 (en) | 2012-04-16 | 2018-04-10 | President And Fellows Of Harvard College | Mesoporous silica compositions for modulating immune responses |
| US11278604B2 (en) | 2012-04-16 | 2022-03-22 | President And Fellows Of Harvard College | Mesoporous silica compositions comprising inflammatory cytokines comprising inflammatory cytokines for modulating immune responses |
| US9993497B2 (en) | 2012-12-30 | 2018-06-12 | Hadasit Medical Research Services And Development Ltd. | Use of alginate compositions in preventing or reducing liver damage caused by a hepatotoxic agent |
| US10500226B2 (en) | 2012-12-30 | 2019-12-10 | Hadasit Medical Research Services And Development Ltd. | Alginate compositions and uses thereof |
| EP3290047A1 (en) | 2013-04-05 | 2018-03-07 | Claudia Zylberberg | Matrix metalloproteinases and uses thereof |
| DE202013012287U1 (en) | 2013-04-23 | 2016-01-18 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gel-forming system for removing kidney stone fragments |
| EP2796100A1 (en) | 2013-04-23 | 2014-10-29 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gelling system for the removal of kidney stone fragments |
| DE202013012275U1 (en) | 2013-04-23 | 2015-12-18 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a cross-linked gel for enclosing kidney stones and / or kidney stone fragments |
| EP2796101A1 (en) | 2013-04-23 | 2014-10-29 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a cross-linked gel for encapsulating kidney stones and/or kidney stone fragments |
| WO2014173467A1 (en) | 2013-04-23 | 2014-10-30 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gel-forming system for removing urinary calculi and fragments thereof |
| WO2014173468A1 (en) | 2013-04-23 | 2014-10-30 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a crosslinked gel for surrounding urinary calculi and/or fragments thereof |
| US9925311B2 (en) | 2013-04-23 | 2018-03-27 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Kit for producing a crosslinked gel for surrounding urinary calculi and/or fragments thereof |
| US10232079B2 (en) | 2013-04-23 | 2019-03-19 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Gel-forming system for removing urinary calculi and fragments thereof |
| EP3101064A4 (en) * | 2014-01-31 | 2017-09-13 | Seikagaku Corporation | Diamine crosslinking agent, acidic polysaccharide crosslinked body, and medical material |
| US10919840B2 (en) | 2014-01-31 | 2021-02-16 | Seikagaku Corporation | Diamine crosslinking agents, crosslinked acidic polysaccharides and medical materials |
| US10294195B2 (en) | 2014-01-31 | 2019-05-21 | Seikagaku Corporation | Diamine crosslinking agents, crosslinked acidic polysaccharides and medical materials |
| US10739338B2 (en) | 2014-03-24 | 2020-08-11 | Qt Holdings Corp | Shaped articles including hydrogels and methods of manufacture and use thereof |
| US10682400B2 (en) | 2014-04-30 | 2020-06-16 | President And Fellows Of Harvard College | Combination vaccine devices and methods of killing cancer cells |
| US11998593B2 (en) | 2014-04-30 | 2024-06-04 | President And Fellows Of Harvard College | Combination vaccine devices and methods of killing cancer cells |
| US9687455B2 (en) | 2014-08-14 | 2017-06-27 | John Daniel Dobak | Sodium tetradecyl sulfate formulations for treatment of adipose tissue |
| US11786457B2 (en) | 2015-01-30 | 2023-10-17 | President And Fellows Of Harvard College | Peritumoral and intratumoral materials for cancer therapy |
| US9351945B1 (en) | 2015-02-27 | 2016-05-31 | John Daniel Dobak, III | Reduction of adipose tissue |
| US9844520B2 (en) | 2015-02-27 | 2017-12-19 | John Daniel Dobak, III | Reduction of adipose tissue |
| US11065210B2 (en) | 2015-02-27 | 2021-07-20 | 10Xbio, Llc | Reduction of adipose tissue |
| US10485767B2 (en) | 2015-02-27 | 2019-11-26 | John Daniel Dobak, III | Reduction of adipose tissue |
| US11150242B2 (en) | 2015-04-10 | 2021-10-19 | President And Fellows Of Harvard College | Immune cell trapping devices and methods for making and using the same |
| US11752238B2 (en) | 2016-02-06 | 2023-09-12 | President And Fellows Of Harvard College | Recapitulating the hematopoietic niche to reconstitute immunity |
| US12029829B2 (en) | 2016-03-22 | 2024-07-09 | President And Fellows Of Harvard College | Biocompatible adhesives and methods of use thereof |
| US11555177B2 (en) | 2016-07-13 | 2023-01-17 | President And Fellows Of Harvard College | Antigen-presenting cell-mimetic scaffolds and methods for making and using the same |
| US12097306B2 (en) | 2016-12-13 | 2024-09-24 | Takeda Pharmaceutical Company Limited | Conformal coating of biological surfaces |
| US20210393850A1 (en) * | 2018-11-07 | 2021-12-23 | Universiteit Maastricht | Bioinks |
| CN110201234A (en) * | 2019-06-26 | 2019-09-06 | 山东百多安医疗器械有限公司 | A kind of used in tissue engineering prostatic cell micro-capsule and preparation method thereof |
| CN110401637A (en) * | 2019-06-28 | 2019-11-01 | 中南民族大学 | Trust method based on name in a kind of name data network |
| WO2021158105A1 (en) | 2020-02-03 | 2021-08-12 | Mosa Meat B.V. | Hydrogels for cultured meat production |
| CN113571628A (en) * | 2020-04-29 | 2021-10-29 | 香港大学 | Stretchable ionic hydrogels with high thermal potential for low grade heat collection |
| WO2022047354A1 (en) | 2020-08-31 | 2022-03-03 | The Board Of Trustees Of The University Of Illinois | Alginate-coated mesenchymal stromal and progenitor cells and methods for using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2266581C (en) | 2007-03-13 |
| ES2317657T3 (en) | 2009-04-16 |
| JP2001524136A (en) | 2001-11-27 |
| JP4335316B2 (en) | 2009-09-30 |
| EP0927196B1 (en) | 2008-11-05 |
| DE69739085D1 (en) | 2008-12-18 |
| JP2009120855A (en) | 2009-06-04 |
| AU733716B2 (en) | 2001-05-24 |
| JP5111416B2 (en) | 2013-01-09 |
| US6642363B1 (en) | 2003-11-04 |
| AU4493097A (en) | 1998-04-14 |
| EP0927196A4 (en) | 2000-12-13 |
| CA2266581A1 (en) | 1998-03-26 |
| ATE413415T1 (en) | 2008-11-15 |
| EP0927196A1 (en) | 1999-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6642363B1 (en) | Polymers containing polysaccharides such as alginates or modified alginates | |
| Pandit et al. | Periodate oxidized hyaluronic acid-based hydrogel scaffolds for tissue engineering applications | |
| Deng et al. | Alginate modification via click chemistry for biomedical applications | |
| Varghese et al. | Hydrogels for musculoskeletal tissue engineering | |
| Baroli | Hydrogels for tissue engineering and delivery of tissue-inducing substances | |
| JP4667486B2 (en) | Water-soluble elastin crosslinking agent | |
| US6165489A (en) | Crosslinked collagen compositions for in situ administration | |
| Laurencin et al. | Biologically active chitosan systems for tissue engineering and regenerative medicine | |
| US5324775A (en) | Biologically inert, biocompatible-polymer conjugates | |
| KR100674177B1 (en) | Cross-linked hyaluronic acids and medical uses thereof | |
| EP2150282B1 (en) | Compositions and methods for scaffold formation | |
| AU2005257078B2 (en) | Hydrogels of hyaluronic acid and alpha, beta-polyaspartylhydrazide and their biomedical and pharmaceutical uses | |
| CN107708675A (en) | The composition and kit of pseudoplastic behavior microgel matrix | |
| JPH08502082A (en) | Biocompatible polymer conjugate | |
| KR100737954B1 (en) | Injectable hydrogels based on hyaluonic acid for tissue regeneration | |
| EP1806367A2 (en) | Polymers containing polysaccharides such as alginates or modified alginates | |
| AU2005291306B2 (en) | Covalent grafting of hydrophobic substances on collagen | |
| Kim et al. | Three-dimensional porous collagen/chitosan complex sponge for tissue engineering | |
| Kesharwani et al. | Tissue regeneration properties of hydrogels derived from biological macromolecules: A review | |
| Jin | In-situ forming biomimetic hydrogels for tissue regeneration | |
| Sarkar et al. | Modified alginates in drug delivery | |
| Metters et al. | Biodegradable hydrogels: tailoring properties and function through chemistry and structure | |
| WO2005116084A1 (en) | Process | |
| AU6185801A (en) | Polymers containing polysaccharides such as alginates or modified alginates | |
| AU6185901A (en) | Polymers containing polysaccharides such as alginates or modified alginates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US US US UZ VN YU ZW AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2266581 Country of ref document: CA Ref country code: JP Ref document number: 1998 514973 Kind code of ref document: A Format of ref document f/p: F Ref country code: CA Ref document number: 2266581 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1997943460 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09147900 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997943460 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |